Embed Size (px)
Citation preview

| 944 | haematologica | 2009; 94(7)
Original Article
Funding: this research wasfunded by Fondazione Cittàdella Speranza and by MIUR(Ministero Istruzione Universitàe Ricerca).
Acknowledgments: the authorswould like to thank S. Disaròand Dr. C. Frasson (Dept. ofPediatrics, University of Padova,Italy) for the flow cytometryanalysis; and Dr. Leonard M.Neckers (Urologic OncologyBranch, NCI, Bethesda, MD,USA) for critical review ofthe manuscript.
Manuscript received onDecember 19, 2008. Revisedversion arrived on February 12,2009. Manuscript accepted onMarch 2, 2009.
Correspondence: Paolo Bonvini, Ph.D.,Clinica di OncoematologiaPediatrica, AziendaOspedaliera-Università diPadova, via Giustiniani 3,35128 Padova, Italy.E-mail: [email protected]
The online version of this articlecontains a supplementaryappendix.
BackgroundThe loss of cell cycle regulation due to abnormal function of cyclin-dependent kinases(cdk) occurs in tumors and leads to genetic instability of chemotherapy-resistant cells. Inthis study, we investigated the effect of the cdk inhibitor flavopiridol in anaplastic largecell lymphomas, in which unrestrained proliferation depends on NPM-ALK tyrosinekinase activity.
Design and MethodsEffects of flavopiridol were examined in ALK-positive and -negative anaplastic large celllymphoma cells by means of immunoblotting and immunofluorescence analyses to assesscdk expression and activity, quantitative real time reverse transcriptase polymerase chainreaction to measure drug-induced changes in transcription, and FACS analyses to monitorchanges in proliferation and survival.
ResultsTreatment with flavopiridol resulted in growth inhibition of anaplastic large cell lym-phoma cells, along with accumulation of subG1 cells and disappearance of S phase with-out cell cycle arrest. Consistent with flavopiridol activity, phosphorylation at cdk2, cdk4,cdk9 sites on RB and RNA polymerase II was inhibited. This correlated with induction ofcell death through rapid mitochondrial damage, inhibition of DNA synthesis, and down-regulation of anti-apoptotic proteins and transcripts. Notably, flavopiridol was less activein ALK-positive cells, as apoptosis was observed at higher concentrations and later timepoints, and resistance to treatment was observed in cells maintaining NPM-ALK signaling.NPM-ALK inhibition affected proliferation but not survival of anaplastic large cell lym-phoma cells, whereas it resulted in a dramatic increase in apoptosis when combined withflavopiridol.
ConclusionsThis work provides the first demonstration that targeting cdk is effective against anaplas-tic large cell lymphoma cells, and proves the critical role of NPM-ALK in the regulation ofresponsiveness of tumor cells with cdk dysregulation.
Key words: anaplastic large cell lymphoma, NPM-ALK, cell cycle.
Citation: Bonvini P, Zorzi E, Mussolin L, Monaco G, Pigazzi M, Basso G, and Rosolen A. Theeffect of the cyclin-dependent kinase inhibitor flavopiridol on anaplastic large cell lymphoma cellsand relationship with NPM-ALK kinase expression and activity. Haematologica 2009;94:944-955.doi:10.3324/haematol.2008.004861
©2009 Ferrata Storti Foundation. This is an open-access paper.
The effect of the cyclin-dependent kinase inhibitor flavopiridol on anaplastic large cell lymphoma cells and relationship with NPM-ALK kinase expression and activityPaolo Bonvini, Elisa Zorzi, Lara Mussolin, Giovanni Monaco, Martina Pigazzi, Giuseppe Basso, and Angelo Rosolen
Clinica di Oncoematologia Pediatrica, Azienda Ospedaliera-Università di Padova, Italy
ABSTRACT

Introduction
Cyclin-dependent kinases (cdk) are a family ofserine/threonine kinases that, in complex with cyclins,regulate cell cycle progression. Unlike cyclins, whichfluctuate to ensure proper timing of complex formation,cdk remain stable during the cell cycle but their activa-tion is transient due to rapid downregulation of cyclins orchanges in their phosphorylation state.1 Four of the cdkso far identified are active during the cell cycle (cdk1,cdk2, cdk4 and cdk6), one acts as an activating kinase forthe others (cdk7), and three are involved in transcription-al regulation (cdk7, cdk8 and cdk9). In association withcyclins D, E and A, cdk4/6 and cdk2 govern G1- and S-phase progression respectively, through sequential phos-phorylation of retinoblastoma tumor suppressor protein(RB) and E2F1 transcription factor,2 whereas the cyclinB1-cdk1 complex promotes G2-phase traversal and exitfrom mitosis.3 In complex with cyclins H and T, cdk7 andcdk9, instead, act as direct regulators of the transcription-al machinery, promoting initiation and elongation of nas-cent transcripts through phosphorylation of the RNApolymerase II enzyme.4 Inhibition of cdk7/9 primarilyaffects general transcription and cell survival, whereasinhibition of cdk1, 2 and 4/6 accounts mostly for growtharrest or delay in phase progression.5 Deregulated cdkactivity is a hallmark of human cancer, and a variety ofgenetic and epigenetic events, such as overexpression ofcyclins, diminished levels of cdk inhibitors or gain-of-function mutations in cdk, have been described to causeoveractivity of these enzymes and provide a selectivegrowth advantage in tumor cells.6-8 This renders cdk suit-able targets for anti-cancer therapy and has promptedgreat interest in the development of specific inhibitors.9,10
Flavopiridol is a semisynthetic ATP-competitiveinhibitor of cdk,11,12 capable of inducing either cell cyclearrest or apoptosis, through inhibition of cdk 1, 2 and4/6, or cdk7 and 9, but without a direct effect on proteinstability.13,14 The primary response of tumor cells toflavopiridol is cytostatic growth arrest with delayedcytotoxicity, but cell cycle arrest and apoptosis mayoccur concomitantly, and cell death can be the preferen-tial response. However, due to effects on multiple cellu-lar targets and actions beyond cdk inhibition, flavopiri-dol has antitumor activity on a variety of cancer cells,but lacks an univocal mechanism of action that explainsselective cell killing. Here, we studied the effect offlavopiridol and examined its potential mechanism ofaction on proliferation and survival of anaplastic largecell lymphomas (ALCL), a subset of T-cell lymphomascharacterized by chromosomal translocations involvingthe ALK gene, which gives rise to the fusion oncopro-tein NPM-ALK, characterized by constitutive activetyrosine kinase activity.15,16 NPM-ALK signals through amultitude of downstream survival pathways(JAK/STAT, PI3K/AKT, RAS/ERK and JNK) and isresponsible for the enhanced transcription and expres-sion of several anti-apoptotic molecules, cell-cycle regu-lators, ribosomal proteins and transcription factors, aswell as for the inactivation of their correspondinginhibitors (RB, p21WAF, p27Kip).17 However, little is known
about the effects of simultaneous interruption of sur-vival signaling and cell cycle regulatory pathways on thebehavior of ALCL cells, and cdk inhibitors have notbeen studied in ALCL nor have they been shown tomodulate NPM-ALK signaling. We, therefore, studiedthe effects of flavopiridol on these aspects.
Design and Methods
Cell cultureHuman ALK-positive ALCL cell lines Karpas299,
SUDHL1, and ALK-negative FE-PD cells were main-tained in RPMI 1640 medium containing 15% heat-inactivated fetal calf serum (FCS), 2 mmol/L glutamine,100 U/mL penicillin, and 100 µg/mL streptomycinunder standard tissue-culture conditions.
Reagents and antibodiesThe cdk inhibitor flavopiridol (NSC 649890) was
obtained from the Developmental TherapeuticsProgram (National Cancer Institute, NIH, Bethesda,MD, USA), dissolved in dimethylsulfoxide (DMSO) andstored at -80°C until use. WHI-154 was purchased fromCalbiochem (Calbiochem, USA). Antibodies were pur-chased from Cell Signaling (PARP; E2F1; RB; cyclin B1;cdk2; cdk4: cdk7, cdk9; STAT3Y705; Akt and AktS472; JNKand JNKT183/Y185; ERK1/2 and ERK1/2T202/Y204; p38α andp38αT180/Y182; NPM-ALKY664) (Cell Signaling Technology,Inc., USA); SIGMA (γ-tubulin; RBS780; RBS612; RBT821)(SIGMA-Aldrich Co., USA); Calbiochem (cyclin E)(Oncogene Research Products, USA); Santa Cruz (Mcl-1; Bax [N20]; cytochrome-c [7H8.2C12]; cyclin D3; RNAPol II; STAT3) (Santa Cruz Biotechnology, Inc., USA);BD Transduction laboratories (p21WAF and p27KIP) (BDBiosciences Pharmingen, USA); Upstate (Bax [6A7]; Bak;cyclin A) (Upstate Biotechnology, NY, USA); Alexis (cas-pase-3) (Axxora Life Science, USA); Covance (RNA PolIISer2 [H5]) (Covance, CA, USA). Caspase inhibitor z-vad-fmk was purchased from Biomol (Biomol InternationalLP, USA). PMSF was purchased from SIGMA (SIGMA-Aldrich Co., USA), whereas leupeptin and aprotininprotease inhibitors were obtained from CAPPEL (ICNBiomedicals Inc., USA). DAPI nucleic acid stain, fluo-rophore-conjugated goat anti-rabbit Alexa488 and goatanti-mouse Alexa546 antibodies were bought fromMolecular Probes (Molecular Probes Inc., USA).Horseradish peroxidase-conjugated sheep anti-mouseand donkey anti-rabbit antibodies were purchased fromGE Healthcare (GE Healthcare Bio-Sciences AB,Uppsala, Sweden), as were protein A-sepharose beadsand protein G-sepharose Fast-FlowTM beads. The BCAprotein assay was from PIERCE (Pierce Chemical Co.,USA) while western blot chemiluminescence reagentswere purchased from Chemicon (ChemiconInternational, Inc., USA). Nitrocellulose and PVDFmembranes were from Schleicher & Schuell. All of theother chemicals used were purchased from SIGMA.
Cell viability assay ALCL cell viability was assessed by MTT. Briefly,
0.1×106/mL cells were seeded in 96-well plates. The
The effect of flavopiridol on ALCL
haematologica | 2009; 94(7) | 945 |

P. Bonvini et al.
| 946 | haematologica | 2009; 94(7)
cells were grown in the presence or absence of the drugat 37°C for up to 72 h and reduction of the MTT salt wasmeasured every 24 h at 540 nM. MTT salt (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide;SIGMA Co., USA) was added for 4 h. Values representthe mean (±SD) of triplicate cultures of three independ-ent experiments. The median-effect dose (IC50) was cal-culated using CalcuSyn software (Calcusyn, Biosoft,MO, USA) and applying Chou’s median-effect equation.
Cell lysis, immunoblotting and immunoprecipitationALCL cells were treated with flavopiridol or left
untreated as indicated. The cells were then washedtwice in ice-cold 1x phosphate-buffered saline (PBS) andlysed by addition of TritonX-100 sample buffer (10 mMTris-HCl [pH 7.5]; 130 mM NaCl; 1% TritonX-100; 5mM EDTA; 1 mg/mL BSA; 20 mM sodium phosphate[pH 7.5]; 10 mM sodium pyrophosphate [pH 7.0]; 25mM glycerophosphate; 1 mM sodium orthovanadate; 10mM sodium molybdate; 1 mM PMSF; 20 µg/mL leu-peptin; 20 µg/mL aprotinin). The lysates were clarifiedby high-speed centrifugation, and 30 µg of lysate werefractionated by sodium dodecylsulfate polyacrylamidegel electrophoresis (SDS-PAGE) and transferred to nitro-cellulose. To assess processing and cleavage of caspase 3flavopiridol-treated and untreated cells were lysed with200 µL of urea-buffer [62.5 mM Tris-HCl, (pH 6.8); 6 Murea; 10% glycerol; 2% SDS; 5% β-mercaptoethanol].Samples were prepared for western blotting as describedabove, and normalized for the expression of γ-tubulin.Proteins were visualized by chemiluminescence using acommercial kit (Chemicon). Immunoprecipitation wasperformed as described previously. Briefly, the cells werelysed as above, and 0.5 mg of protein lysates were pre-cipitated, overnight at 4°C, with 1 µg specific primaryantibodies. The immunocomplexes were adsorbed onto30 µL protein G sepharose beads, incubated at 4°C for120 min and then resuspended in sample buffer beforefractionation by SDS gel electrophoresis. Western blot-ting was performed as above.
Assessment of apoptosisAfter flavopiridol treatment and induction of apopto-
sis, 0.5×106 cells were harvested and washed with tem-perate PBS. The cells were resuspended in 1 mL of 1Xannexin-binding buffer (10 mM HEPES, pH 7.4; 140 mMNaOH; 2.5 mM CaCl2), stained with 5 µL annexin-V-flu-orescein isothiocyanate (FITC) and 5 µl of 5 µg/mL pro-pidium iodide (PI), and then incubated for 15 min atroom temperature in the dark (Immunostep Research,Spain). The apoptotic cells were determined using aBeckam Coulter FC500 flow cytometer. Both earlyapoptotic (annexin-V-positive, PI-negative) and late(annexin-V-positive and PI-positive) apoptotic cells wereincluded in cell death determinations.
BrdU analysisFlavopiridol-treated and untreated cells (1×106) were
pulse-labeled with 10 µM BrdU for 45 min before collec-tion (FITC BrdU Flow Kit, BD Pharmingen). Briefly, afterBrdU labeling the cells were washed in 1X DPBS, fixedin 4% paraformaldehyde and permeabilized with
saponin detergent for 30 min on ice, according to theinstructions provided with the kit. The cells were thentreated with DNase for 1 h at 37°C, to expose incorpo-rated BrdU, washed with 1X DPBS and stained withFITC-conjugated anti-BrdU antibody (1/50), for 20 minat room temperature. At the end, total DNA was stainedwith 7-amino-actinomycin D (7-AAD) fluorescent dyefor 15 min at room temperature in the dark, and then,once resuspended in staining buffer, analyzed by two-color flow cytometry.
Real-time polymerase chain reactionALCL cells were untreated or treated with 200 nM
flavopiridol for the indicated time periods. After treat-ment, the cells were lysed, and total RNA was isolatedusing Trizol (Invitrogen, Carlshad, CA, USA) followingthe manufacturer’s instructions. Contaminating DNAwas removed with a DNA-freeTM Kit (AMBION Inc.,USA) according to the manufacturer’s instructions. Onemicrogram of RNA was reverse transcribed usingSuperScript II reverse transcriptase (Invitrogen) and ran-dom hexamers. The following primers and probesequences were designed using Primer Express version2.0 (Applied Biosystems) for TaqMan- based quantita-tive real-time polymerase chain reaction (RQ-PCR)experiments: human Mcl-1, forward 5’-TAAGGA-CAAAACGGGACTGG-3’, reverse 5’-ACATTCCT-GATGCCACCTTCTAG-3’,Taqman probe 5’-FAM-CTGGGATGGGTTTGTGGAGTTCTTCCA-TAMRA-3’. The thermal cycler conditions were 2 min at 50°C foruracil N-glycosylase treatment and 10 min at 95°C forinactivation of uracil N-glycosylase and activation ofAmpliTaq Gold Polymerase, followed by 50 cycles of 15seconds at 95°C and 1 min at 60°C. All reactions wereperformed on the ABI Prism 7000 Sequence DetectionSystem (Applied Biosystems, CA, USA). To compareRQ-PCR assays from different runs, the threshold wasset at 0.1. To identify the most appropriate endogenouscontrol gene for the quantification of Mcl-1 expressionin ALCL cell lines, we conducted a preliminary expres-sion analysis of 11 house-keeping genes in these celllines, by RQ-PCR using 5’ nuclease technology, andHuman TaqMan® pre-developed assay reagent endoge-nous controls (ABI, Foster City, CA, USA). 18S was thebest candidate control gene because its expressionshowed the lowest variability across the test samples.Each sample was tested in triplicate, and Mcl1 mRNAlevels were normalized to that of 18s rRNA.
Subcellular fractionationLysates were obtained by resuspending ALCL cells in
digitonin-lysis buffer [250 mM sucrose; 20 mM Hepes,(pH 7.4); 5 mM MgCl2; 10 mM KCl; 1 mM EDTA; 0.05%digitonin; 1 mM EGTA; 1 mM PMSF, 20 µg/mL apro-tinin, and 20 µg/mL leupeptin]. After a 20-minute incu-bation on ice, the cells were centrifuged for 10 min at13,000 g, and the supernatant (mitochondria-free,cytosolic fraction) was recovered and frozen at -20°C forsubsequent use. Enriched mitochondrial pellets werewashed in ice-cold 1xPBS and then lysed in CHAPSO-buffer (5 mM MgCl2; 137 mM KCl; 1 mM EDTA; 1 mMEGTA; 1% CHAPSO [3-([3-cholamidopropyl]dimethy-

lammonio)-2-hydroxy-1-propanesulfonate]; 1.4 µMpepstatin; 1 mM PMSF, 20 µg/mL aprotinin, and 20µg/mL leupeptin). Samples were then clarified as aboveand supernatants isolated. Proteins from both fractionswere then resolved by 12-15% SDS-PAGE and westernblotted as described previously.
Cell cycle analysis and cell sortingCell cycle analysis was performed on ALCL cells treat-
ed with 200 nM Flavopiridol or left untreated (DMSO).The cells were washed in ice-cold 1x PBS, fixed in cold70% ethanol, pelleted and resuspended in staining buffer(3.8 mM sodium citrate, 0.5 mg/mL RNase, 0.01 mg/mLPI) and incubated on ice, according to the manufacturer’sinstructions (Coulter DNA PrepTM Reagents kit; BeckamCoulter Inc., USA). Samples were analyzed on aBeckham Coulter FC500 flow cytometer. DNA his-tograms were analyzed using MultiCycle® for Windows(Phoenix Flow Systems, USA). To purify viable cells after24h-treatment with flavopiridol, Karpas299 were ana-lyzed in a FACSVantage fluorescence-activated cell sorter(Becton Dickinson), and forward (FSCA-A, Y axes) versusside scatter (SSC-A, X axes) dot plot analysis performed.As shown in Figure 5B, 30×103 events were analyzedafter flavopiridol treatment, and the viable cells weresorted out from the apoptotic cell population or the cel-lular debris. The cells were then stained with PI for DNAcontent analysis or processed for immunofluorescence asdescribed previously. Cell cycle analysis and immunoflu-orescence studies were done in parallel in intact cellssorted from untreated controls (DMSO).
Fluorescence microscopyALCL cells were treated with 200 nM flavopiridol or
left untreated (DMSO). The cells were then spottedonto 12-well multitest slides (ICN Biomedicals, Inc.,USA), fixed in 3.7% formaldehyde, permeabilized in0.2% TritonX-100 and blocked in 100 mM glycin, fol-lowed by incubation in 10% FCS-PBS. Permeabilizedcells were incubated for 1 h at 37°C with the specificprimary antibodies indicated and, after washing in PBS,incubated with fluorophore-conjugated secondary anti-bodies Alexa488 or Alexa546, at a 1:1000 dilution of 2mg/mL stock. Cells were then washed in PBS, mountedonto slides with 1:1 PBS/glycerol, with the addition ofDAPI nucleic acid stain (1:1000). The cells wereobserved at 63x/0.75 NA in a Leica DMBL microscope.Images were acquired with a Leica DC 300F digitalcamera and prepared for reproduction with LeicaIM1000 software (Leica Mycrosystem Ltd., Germany).
Mitochondrial membrane permeabilization assayTo measure changes in mitochondrial transmembrane
potential (∆Ψm), ALCL cells were treated with 200 nMflavopiridol, in the presence or in the absence of 30 µMz-vad-fmk caspase inhibitor. The cells (1×106) were thenharvested, washed and incubated for 20 min at roomtemperature with 40 nM 3,3-dihexyloxacarbocyanine(DiOC6 Molecular Probes) and analyzed by flowcytometry, with excitation and emission settings of 488nm and 525 nm, respectively. The percentage of cellsexhibiting low fluorescence, reflecting loss of inner
mitochondrial membrane potential, was determined bycomparison with untreated controls.
Results
Flavopiridol inhibits growth and induces mitochondrial apoptosis in anaplastic large cell lymphoma cells in vitro
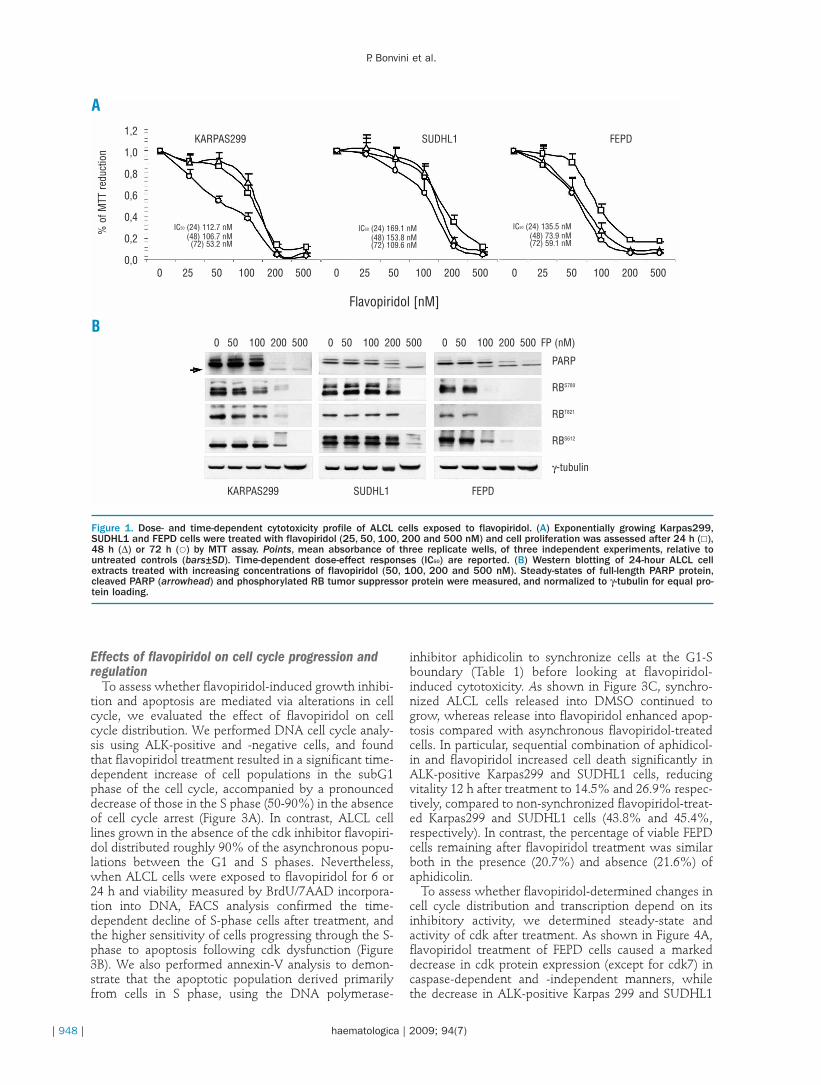
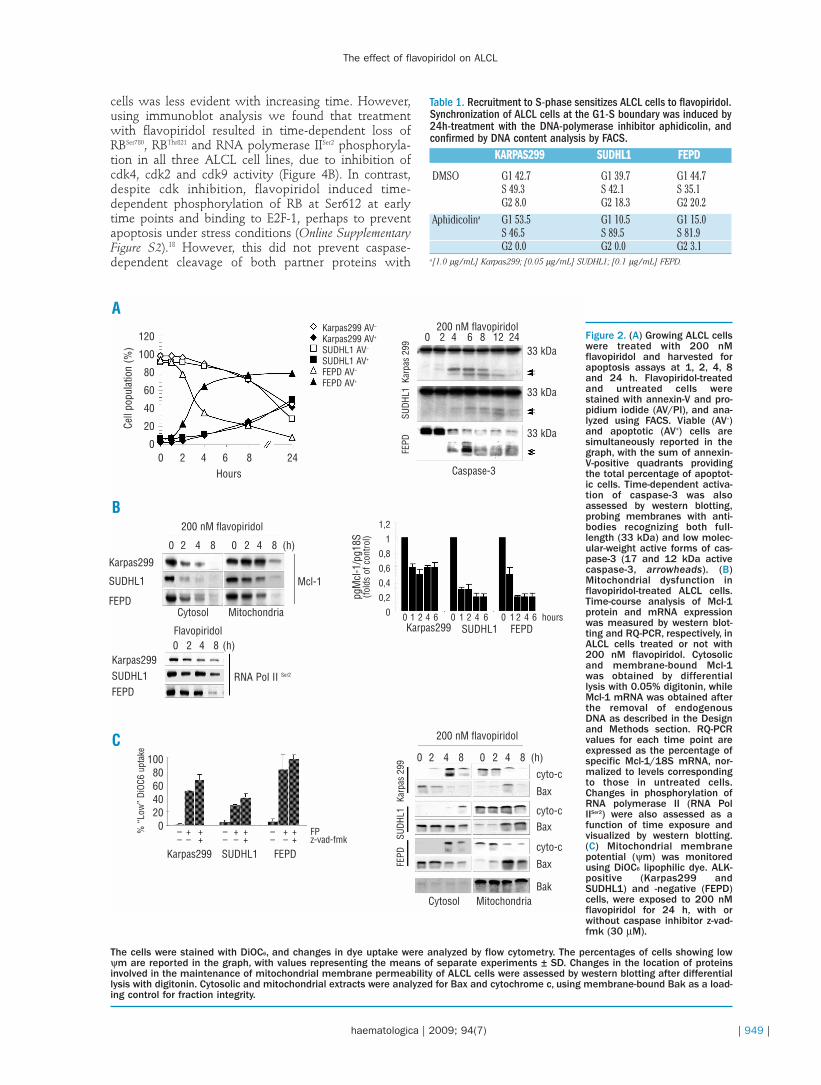
We first investigated the dose- and time-dependenteffect of treatment with flavopiridol (25-500 nM) on thegrowth of human ALK-positive (Karpas299, SUDHL1)and -negative (FEPD) ALCL cells by MTT assay, assess-ing viability every 24 h for 3 days. Treatment withflavopiridol decreased viability in a dose- and time-dependent manner (112.7nM≤IC50≤169.1 nM at 24 h;73.9 nM≤IC50≤153.8 nM at 48 h; 53.2 nM≤IC50≤109.6nM at 72 h), with maximal inhibition at 24 h with dosesof 200 nM or higher, and more pronounced effects atlater time points (Figure 1A). Dose- and time-dependentdifferences among ALK-positive and -negative cells ingrowth and survival were observed at lower concentra-tions of flavopiridol, with ALK-negative FEPD cellsbeing the most sensitive among all, as confirmed byPARP cleavage analysis, a hallmark of apoptosis, andretinoblastoma (RB) state of phosphorylation, a well-known cdk downstream target, 24 h after treatment(Figure 1B). Based on these data, we selected 200 nMflavopiridol as an effective cytotoxic concentration forour study, assessing drug response as a function of timeof exposure. Annexin-V staining was performed todetermine apoptotic cells following drug treatment, andwe found that apoptosis increased rapidly in flavopiri-dol-treated cells, ranging between 20% and 60% after 8h (up to 50-90% at 24 h), with cell-type specific differ-ences in timings of caspase-3 activation and activity asshown by immunoblot analysis (Figure 2A).
Modulation of caspase activation in flavopiridol-treat-ed cells is determined by release of cytochrome c intothe cytosol, which results from dysfunction of mito-chondrial homeostasis via loss of membrane permeabil-ity after downregulation of Bcl-2 family proteins. Mcl-1plays a significant role in maintaining mitochondrialouter membrane permeability; we, therefore, studiedthe effect of treatment with flavopiridol on constitutiveMcl-1 expression and transcription in ALCL cells. Theimmunoblot analysis revealed a significant, time-dependent decrease in Mcl-1 expression in all three celllines following treatment with flavopiridol; thisdecrease strictly correlated with Mcl-1 transcriptionalinhibition following cdk9-dependent RNA polymeraseII dephosphorylation (Figure 2B). Under these condi-tions the number of cells exhibiting low uptake oflipophilic dye DiOC6 increased markedly after flavopiri-dol treatment, being up to 80% in ALK-negative FEPDcells, and up to 30-50% in ALK-positive Karpas 299 andSUDHL1 cells (Figure 2C, graph). This was observedboth in the presence and absence of z-vad-fmk caspaseinhibitor, and correlated with accumulation of Bax andcytochrome c into mitochondria and the cytosol,respectively, in cells undergoing apoptosis (Figure 2Cand Online Supplementary Figure S1).
The effect of flavopiridol on ALCL
haematologica | 2009; 94(7) | 947 |
P. Bonvini et al.
| 948 | haematologica | 2009; 94(7)
Effects of flavopiridol on cell cycle progression andregulation
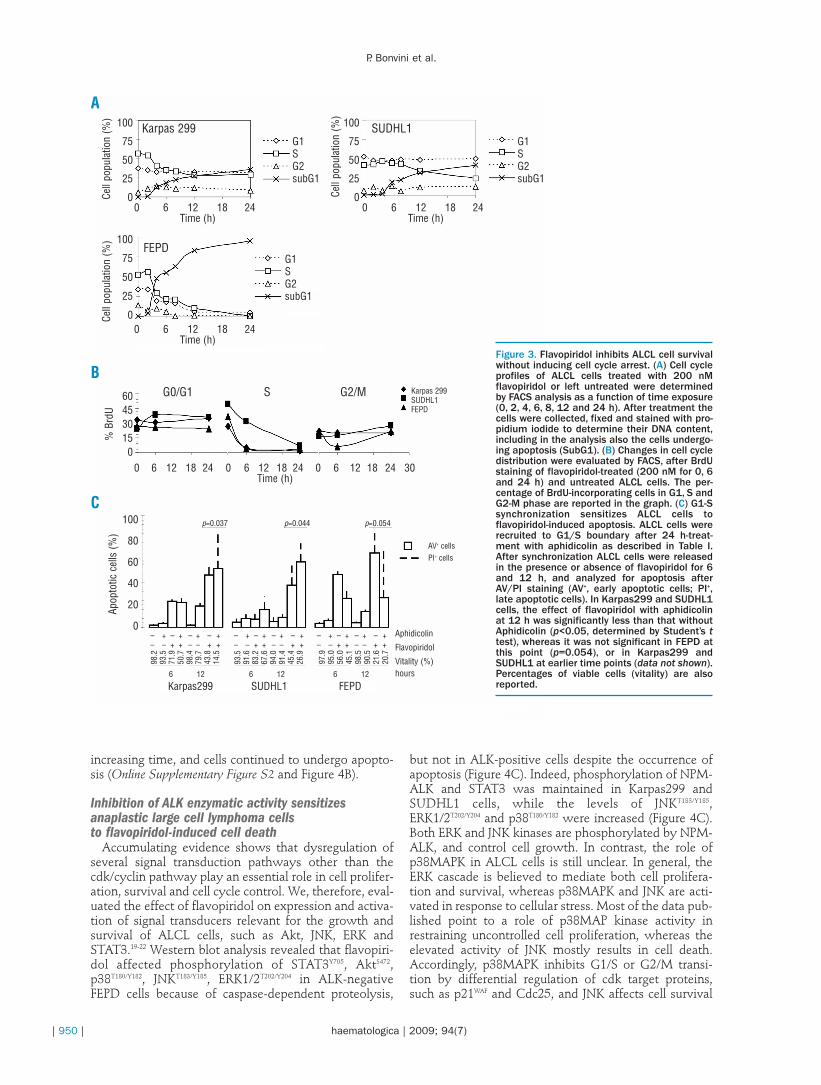
To assess whether flavopiridol-induced growth inhibi-tion and apoptosis are mediated via alterations in cellcycle, we evaluated the effect of flavopiridol on cellcycle distribution. We performed DNA cell cycle analy-sis using ALK-positive and -negative cells, and foundthat flavopiridol treatment resulted in a significant time-dependent increase of cell populations in the subG1phase of the cell cycle, accompanied by a pronounceddecrease of those in the S phase (50-90%) in the absenceof cell cycle arrest (Figure 3A). In contrast, ALCL celllines grown in the absence of the cdk inhibitor flavopiri-dol distributed roughly 90% of the asynchronous popu-lations between the G1 and S phases. Nevertheless,when ALCL cells were exposed to flavopiridol for 6 or24 h and viability measured by BrdU/7AAD incorpora-tion into DNA, FACS analysis confirmed the time-dependent decline of S-phase cells after treatment, andthe higher sensitivity of cells progressing through the S-phase to apoptosis following cdk dysfunction (Figure3B). We also performed annexin-V analysis to demon-strate that the apoptotic population derived primarilyfrom cells in S phase, using the DNA polymerase-
inhibitor aphidicolin to synchronize cells at the G1-Sboundary (Table 1) before looking at flavopiridol-induced cytotoxicity. As shown in Figure 3C, synchro-nized ALCL cells released into DMSO continued togrow, whereas release into flavopiridol enhanced apop-tosis compared with asynchronous flavopiridol-treatedcells. In particular, sequential combination of aphidicol-in and flavopiridol increased cell death significantly inALK-positive Karpas299 and SUDHL1 cells, reducingvitality 12 h after treatment to 14.5% and 26.9% respec-tively, compared to non-synchronized flavopiridol-treat-ed Karpas299 and SUDHL1 cells (43.8% and 45.4%,respectively). In contrast, the percentage of viable FEPDcells remaining after flavopiridol treatment was similarboth in the presence (20.7%) and absence (21.6%) ofaphidicolin.
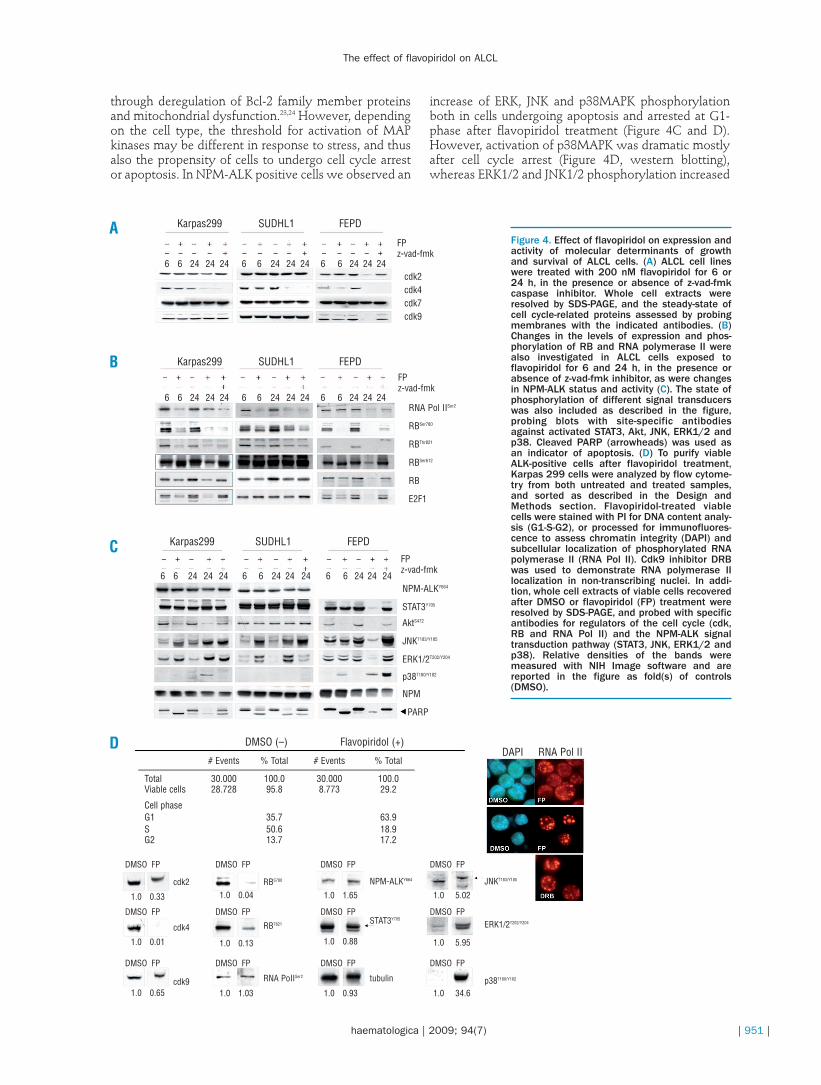
To assess whether flavopiridol-determined changes incell cycle distribution and transcription depend on itsinhibitory activity, we determined steady-state andactivity of cdk after treatment. As shown in Figure 4A,flavopiridol treatment of FEPD cells caused a markeddecrease in cdk protein expression (except for cdk7) incaspase-dependent and -independent manners, whilethe decrease in ALK-positive Karpas 299 and SUDHL1
Figure 1. Dose- and time-dependent cytotoxicity profile of ALCL cells exposed to flavopiridol. (A) Exponentially growing Karpas299,SUDHL1 and FEPD cells were treated with flavopiridol (25, 50, 100, 200 and 500 nM) and cell proliferation was assessed after 24 h (£),48 h (∆) or 72 h (�) by MTT assay. Points, mean absorbance of three replicate wells, of three independent experiments, relative tountreated controls (bars±SD). Time-dependent dose-effect responses (IC50) are reported. (B) Western blotting of 24-hour ALCL cellextracts treated with increasing concentrations of flavopiridol (50, 100, 200 and 500 nM). Steady-states of full-length PARP protein,cleaved PARP (arrowhead) and phosphorylated RB tumor suppressor protein were measured, and normalized to γ-tubulin for equal pro-tein loading.
A
B
FEPDSUDHL1KARPAS299
IC50 (24) 112.7 nM(48) 106.7 nM(72) 53.2 nM
IC50 (24) 169.1 nM(48) 153.8 nM(72) 109.6 nM
IC50 (24) 135.5 nM(48) 73.9 nM(72) 59.1 nM
% o
f MTT
redu
ctio
n
FEPDSUDHL1KARPAS299
0 50 100 200 500 0 50 100 200 500 0 50 100 200 500 FP (nM)
PARP
RBS780
RBT821
RBS612
γ-tubulin
0 25 50 100 200 500
1,2
1,0
0,8
0,6
0,4
0,2
0,00 25 50 100 200 500 0 25 50 100 200 500
Flavopiridol [nM]
cells was less evident with increasing time. However,using immunoblot analysis we found that treatmentwith flavopiridol resulted in time-dependent loss ofRBSer780, RBThr821 and RNA polymerase IISer2 phosphoryla-tion in all three ALCL cell lines, due to inhibition ofcdk4, cdk2 and cdk9 activity (Figure 4B). In contrast,despite cdk inhibition, flavopiridol induced time-dependent phosphorylation of RB at Ser612 at earlytime points and binding to E2F-1, perhaps to preventapoptosis under stress conditions (Online SupplementaryFigure S2).18 However, this did not prevent caspase-dependent cleavage of both partner proteins with
The effect of flavopiridol on ALCL
haematologica | 2009; 94(7) | 949 |
Table 1. Recruitment to S-phase sensitizes ALCL cells to flavopiridol.Synchronization of ALCL cells at the G1-S boundary was induced by24h-treatment with the DNA-polymerase inhibitor aphidicolin, andconfirmed by DNA content analysis by FACS.
KARPAS299 SUDHL1 FEPD
DMSO G1 42.7 G1 39.7 G1 44.7S 49.3 S 42.1 S 35.1G2 8.0 G2 18.3 G2 20.2
Aphidicolina G1 53.5 G1 10.5 G1 15.0S 46.5 S 89.5 S 81.9G2 0.0 G2 0.0 G2 3.1
a[1.0 µg/mL] Karpas299; [0.05 µg/mL] SUDHL1; [0.1 µg/mL] FEPD.
Figure 2. (A) Growing ALCL cellswere treated with 200 nMflavopiridol and harvested forapoptosis assays at 1, 2, 4, 8and 24 h. Flavopiridol-treatedand untreated cells werestained with annexin-V and pro-pidium iodide (AV/PI), and ana-lyzed using FACS. Viable (AV–)and apoptotic (AV+) cells aresimultaneously reported in thegraph, with the sum of annexin-V-positive quadrants providingthe total percentage of apoptot-ic cells. Time-dependent activa-tion of caspase-3 was alsoassessed by western blotting,probing membranes with anti-bodies recognizing both full-length (33 kDa) and low molec-ular-weight active forms of cas-pase-3 (17 and 12 kDa activecaspase-3, arrowheads). (B)Mitochondrial dysfunction inflavopiridol-treated ALCL cells.Time-course analysis of Mcl-1protein and mRNA expressionwas measured by western blot-ting and RQ-PCR, respectively, inALCL cells treated or not with200 nM flavopiridol. Cytosolicand membrane-bound Mcl-1was obtained by differentiallysis with 0.05% digitonin, whileMcl-1 mRNA was obtained afterthe removal of endogenousDNA as described in the Designand Methods section. RQ-PCRvalues for each time point areexpressed as the percentage ofspecific Mcl-1/18S mRNA, nor-malized to levels correspondingto those in untreated cells.Changes in phosphorylation ofRNA polymerase II (RNA PolIISer2) were also assessed as afunction of time exposure andvisualized by western blotting.(C) Mitochondrial membranepotential (ψm) was monitoredusing DiOC6 lipophilic dye. ALK-positive (Karpas299 andSUDHL1) and -negative (FEPD)cells, were exposed to 200 nMflavopiridol for 24 h, with orwithout caspase inhibitor z-vad-fmk (30 µM).
The cells were stained with DiOC6, and changes in dye uptake were analyzed by flow cytometry. The percentages of cells showing lowψm are reported in the graph, with values representing the means of separate experiments ± SD. Changes in the location of proteinsinvolved in the maintenance of mitochondrial membrane permeability of ALCL cells were assessed by western blotting after differentiallysis with digitonin. Cytosolic and mitochondrial extracts were analyzed for Bax and cytochrome c, using membrane-bound Bak as a load-ing control for fraction integrity.
120
100
80
60
40
20
0
100806040200
1,2
1
0,8
0,6
0,4
0,2
0
0 2 4 6 8 24Hours
200 nM flavopiridol
200 nM flavopiridol
Cytosol Mitochondria
Karpas299 AV–
Karpas299 AV+
SUDHL1 AV–
SUDHL1 AV+
FEPD AV–
FEPD AV+
Flavopiridol
0 2 4 8 0 2 4 8 (h)
0 2 4 8 0 2 4 8 (h)
0 1 2 4 6 0 1 2 4 6 0 1 2 4 6 hours
200 nM flavopiridol0 2 4 6 8 12 24
Caspase-3
33 kDa
33 kDa
cyto-c
cyto-c
cyto-c
Bak
Bax
Bax
Bax
33 kDa
FEPD
SUDH
L1Ka
rpas
299
FEPD
SUDH
L1Ka
rpas
299
FEPDSUDHL1Karpas299
0 2 4 8 (h)
Mcl-1
RNA Pol II Ser2
Cytosol
Karpas299
SUDHL1
FEPD
Karpas299SUDHL1FEPD
Karpas299 SUDHL1 FEPD
Mitochondria
Cell
popu
latio
n (%
)%
“Lo
w”
DiOC
6 up
take
– + + – + + – + + FP– – + – – + – – + z-vad-fmk
pgM
cl-1
/pg1
8S(fo
lds
of c
ontro
l)
A
B
C
P. Bonvini et al.
| 950 | haematologica | 2009; 94(7)
increasing time, and cells continued to undergo apopto-sis (Online Supplementary Figure S2 and Figure 4B).
Inhibition of ALK enzymatic activity sensitizesanaplastic large cell lymphoma cells to flavopiridol-induced cell death
Accumulating evidence shows that dysregulation ofseveral signal transduction pathways other than thecdk/cyclin pathway play an essential role in cell prolifer-ation, survival and cell cycle control. We, therefore, eval-uated the effect of flavopiridol on expression and activa-tion of signal transducers relevant for the growth andsurvival of ALCL cells, such as Akt, JNK, ERK andSTAT3.19-22 Western blot analysis revealed that flavopiri-dol affected phosphorylation of STAT3Y705, AktS472,p38T180/Y182, JNKT183/Y185, ERK1/2T202/Y204 in ALK-negativeFEPD cells because of caspase-dependent proteolysis,
but not in ALK-positive cells despite the occurrence ofapoptosis (Figure 4C). Indeed, phosphorylation of NPM-ALK and STAT3 was maintained in Karpas299 andSUDHL1 cells, while the levels of JNKT183/Y185,ERK1/2T202/Y204 and p38T180/Y182 were increased (Figure 4C).Both ERK and JNK kinases are phosphorylated by NPM-ALK, and control cell growth. In contrast, the role ofp38MAPK in ALCL cells is still unclear. In general, theERK cascade is believed to mediate both cell prolifera-tion and survival, whereas p38MAPK and JNK are acti-vated in response to cellular stress. Most of the data pub-lished point to a role of p38MAP kinase activity inrestraining uncontrolled cell proliferation, whereas theelevated activity of JNK mostly results in cell death.Accordingly, p38MAPK inhibits G1/S or G2/M transi-tion by differential regulation of cdk target proteins,such as p21WAF and Cdc25, and JNK affects cell survival
Figure 3. Flavopiridol inhibits ALCL cell survivalwithout inducing cell cycle arrest. (A) Cell cycleprofiles of ALCL cells treated with 200 nMflavopiridol or left untreated were determinedby FACS analysis as a function of time exposure(0, 2, 4, 6, 8, 12 and 24 h). After treatment thecells were collected, fixed and stained with pro-pidium iodide to determine their DNA content,including in the analysis also the cells undergo-ing apoptosis (SubG1). (B) Changes in cell cycledistribution were evaluated by FACS, after BrdUstaining of flavopiridol-treated (200 nM for 0, 6and 24 h) and untreated ALCL cells. The per-centage of BrdU-incorporating cells in G1, S andG2-M phase are reported in the graph. (C) G1-Ssynchronization sensitizes ALCL cells toflavopiridol-induced apoptosis. ALCL cells wererecruited to G1/S boundary after 24 h-treat-ment with aphidicolin as described in Table I.After synchronization ALCL cells were releasedin the presence or absence of flavopiridol for 6and 12 h, and analyzed for apoptosis afterAV/PI staining (AV+, early apoptotic cells; PI+,late apoptotic cells). In Karpas299 and SUDHL1cells, the effect of flavopiridol with aphidicolinat 12 h was significantly less than that withoutAphidicolin (p<0.05, determined by Student’s ttest), whereas it was not significant in FEPD atthis point (p=0.054), or in Karpas299 andSUDHL1 at earlier time points (data not shown).Percentages of viable cells (vitality) are alsoreported.
0 6 12 18 24Time (h)
0 6 12 18 24Time (h)
G1SG2subG1
Cell
popu
latio
n (%
)
Cell
popu
latio
n (%
)Ce
ll po
pula
tion
(%)
% B
rdU
G1SG2subG1
100
75
50
25
0
100
75
50
25
0
100
75
50
25
0
604530150
100
80
60
40
20
0
G1SG2subG1
G0/G1 S G2/M
SUDHL1Karpas 299
FEPD
0 6 12 18 24Time (h)
0 6 12 18 24 0 6 12 18 24 0 6 12 18 24 30Time (h)
Karpas299 SUDHL1 FEPD
Apop
totic
cel
ls (%
)
Karpas 299SUDHL1FEPD
AV+ cellsPI+ cells
Aphidicolin
Flavopiridol
Vitality (%)hours
p=0.037
– + – + – + – +– – + + – – + +
– + – + – + – +– – + + – – + +
6 12 6 12 6 12
– + – + – + – +– – + + – – + +
98.2
93.5
71.9
50.7
98.4
79.7
43.8
14.5
93.5
91.6
83.2
67.6
94.0
91.4
45.4
26.9
97.9
95.0
56.0
45.1
98.5
90.5
21.6
20.7
p=0.044 p=0.054
A
B
C
through deregulation of Bcl-2 family member proteinsand mitochondrial dysfunction.23,24 However, dependingon the cell type, the threshold for activation of MAPkinases may be different in response to stress, and thusalso the propensity of cells to undergo cell cycle arrestor apoptosis. In NPM-ALK positive cells we observed an
increase of ERK, JNK and p38MAPK phosphorylationboth in cells undergoing apoptosis and arrested at G1-phase after flavopiridol treatment (Figure 4C and D).However, activation of p38MAPK was dramatic mostlyafter cell cycle arrest (Figure 4D, western blotting),whereas ERK1/2 and JNK1/2 phosphorylation increased
The effect of flavopiridol on ALCL
haematologica | 2009; 94(7) | 951 |
Figure 4. Effect of flavopiridol on expression andactivity of molecular determinants of growthand survival of ALCL cells. (A) ALCL cell lineswere treated with 200 nM flavopiridol for 6 or24 h, in the presence or absence of z-vad-fmkcaspase inhibitor. Whole cell extracts wereresolved by SDS-PAGE, and the steady-state ofcell cycle-related proteins assessed by probingmembranes with the indicated antibodies. (B)Changes in the levels of expression and phos-phorylation of RB and RNA polymerase II werealso investigated in ALCL cells exposed toflavopiridol for 6 and 24 h, in the presence orabsence of z-vad-fmk inhibitor, as were changesin NPM-ALK status and activity (C). The state ofphosphorylation of different signal transducerswas also included as described in the figure,probing blots with site-specific antibodiesagainst activated STAT3, Akt, JNK, ERK1/2 andp38. Cleaved PARP (arrowheads) was used asan indicator of apoptosis. (D) To purify viableALK-positive cells after flavopiridol treatment,Karpas 299 cells were analyzed by flow cytome-try from both untreated and treated samples,and sorted as described in the Design andMethods section. Flavopiridol-treated viablecells were stained with PI for DNA content analy-sis (G1-S-G2), or processed for immunofluores-cence to assess chromatin integrity (DAPI) andsubcellular localization of phosphorylated RNApolymerase II (RNA Pol II). Cdk9 inhibitor DRBwas used to demonstrate RNA polymerase IIlocalization in non-transcribing nuclei. In addi-tion, whole cell extracts of viable cells recoveredafter DMSO or flavopiridol (FP) treatment wereresolved by SDS-PAGE, and probed with specificantibodies for regulators of the cell cycle (cdk,RB and RNA Pol II) and the NPM-ALK signaltransduction pathway (STAT3, JNK, ERK1/2 andp38). Relative densities of the bands weremeasured with NIH Image software and arereported in the figure as fold(s) of controls(DMSO).
A
B
C
D
Karpas299 SUDHL1 FEPD
Karpas299 SUDHL1 FEPD
Karpas299 SUDHL1 FEPD
DMSO (–) Flavopiridol (+)
# Events % Total # Events % Total
Total 30.000 100.0 30.000 100.0Viable cells 28.728 95.8 8.773 29.2
Cell phaseG1 35.7 63.9S 50.6 18.9G2 13.7 17.2
DMSO FP
1.0 0.33
cdk2 RBS780
RBT821
RNA PoIISer2 tubulin
STAT3Y705
NPM-ALKY664 JNKT183/Y185
ERK1/2T202/Y204
p38T180/Y182
cdk4
cdk9
1.0 0.04
1.0 0.13
1.0 1.03 1.0 0.93
1.0 0.88
1.0 1.65 1.0 5.02
1.0 5.95
1.0 34.6
1.0 0.01
1.0 0.65
DMSO FP
DMSO FP
DMSO FP
DMSO FP
DMSO FP
DMSO FP
DMSO FP
DMSO FP
DMSO FP
DMSO FP
DMSO FP
DAPI RNA Pol II
6 6 24 24 24 6 6 24 24 24 6 6 24 24 24
FPz-vad-fmk
FPz-vad-fmk
FPz-vad-fmk
cdk2cdk4cdk7cdk9
RNA Pol IISer2
RBSer780
RBThr821
RBSer612
RB
E2F1
NPM-ALKY664
STAT3Y705
AktS472
JNKT183/Y185
ERK1/2T202/Y204
p38T180/Y182
NPM
PARP
6 6 24 24 24 6 6 24 24 24 6 6 24 24 24
6 6 24 24 24 6 6 24 24 24 6 6 24 24 24

P. Bonvini et al.
| 952 | haematologica | 2009; 94(7)
mostly in apoptotic cells (Figure 4C). Besides, downreg-ulation of cdk activity in G1-arrested cells was stillobserved, as was the accumulation of inactive RNApolymerase II in dot-like structures typical of transcrip-tionally inhibited cells,25 suggesting that flavopiridoluptake and inhibitory activity were unaffected (Figure4D, western blotting and immunofluorescence).Whether the phosphorylation of MAP kinases isinvolved in the regulation of flavopiridol-induced apop-tosis, or simply a secondary response to deregulation ofsignaling, is not known. However, when used in combi-nation with flavopiridol, p38 MAPK inhibitor,SB202190, but not JNK and ERK inhibitors SP600125and PD98059, caused a robust increase in annexin V
staining, suggesting a possible negative effect of phos-phorylated p38 MAPK on flavopiridol-mediated apopto-sis (unpublished data).
To assess whether flavopiridol-induced apoptosis wasNPM-ALK dependent, we next examined the role ofNPM-ALK on drug response, measuring apoptosis in thepresence or absence of the ALK small-molecule inhibitorWHI-154.26,27 As shown in Figure 5A, expression ofNPM-ALKY664 in Karpas299 cells was inhibited by WHI-154 in a dose-dependent manner, under conditions thatreduced stability of the total protein without cytotoxiceffects (see below). Inhibition of key signaling proteinswas concentration-dependent and showed a strong cor-relation with NPM-ALK inactivation. As expected, phos-
Figure 5. WHI-154 inhibitor-mediated suppression of NPM-ALK tyrosine kinase activity. (A)To evaluate expression of phos-pho-NPM-ALK (NPM-ALKY664), aswell as changes in the levels ofexpression and degree of phos-phorylation of NPM-ALK-relatedor -unrelated downstream signal-ing proteins, ALK-positive and -negative, Karpas299 and FEPDcells respectively, were treatedwith 5, 10, 20 and 50 µM WHI-154 for 24 h, or left untreated.Total cell lysates were preparedas described in the Design andMethods section, and resolved bySDS-PAGE. Blots were probed forthe proteins indicated, and rela-tive densities of the bands weremeasured with NIH Image soft-ware and normalized to untreat-ed controls. (B) Combined treat-ment with flavopiridol and WHI-154 markedly enhances thedownregulation of proteinsinvolved in growth and survival ofALCL cells. Karpas299 cells wereexposed to 200 nM flavopiri-dol±20 µM WHI-154 for 24 h.Thirty mg of proteins of wholecell lysates from treated anduntreated samples were resolvedby SDS-PAGE and analyzed bywestern blotting for the proteinsindicated in the figure. Blotswere stripped and probed for γ-tubulin to ensure equivalentloading and transfer. Karpas299cells, untreated or treated withflavopiridol or WHI-154 were alsostained with PI for DNA contentanalysis of diploid and sub-diploid cells. (C) Striking potenti-ation of apoptosis in ALCL cellsco-exposed to flavopiridol andWHI-154. Karpas299 and FEPDcells were treated with WHI-154for 12 h, prior to addition offlavopiridol (200 nM) for the indi-cated time periods (6 and 12 h).After treatment the cells werestained with annexin-V and pro-pidium iodide for apoptosisanalysis by FACS. Percentages ofearly apoptotic (AV+), late apop-totic (PI+) and viable cells (vitali-ty) are reported.
C
B
A NPM-ALKY664
RB
RBT821
CDK2Fold
cha
nge
CDK9
CDK4
RBS612
RBS780
STAT3Y7052,01,00,0
2,01,00,0
2,01,00,0
2,01,00,0
2,01,00,0
2,01,00,0
2,01,00,0
2,01,00,0
2,01,00,0
2,01,00,0
CDK7
Karpas299
0 5 10 20 50 0 5 10 20 50 µM
FEPD Karpas299
Karpas299
0 5 10 20 50
WHI-154FP
[µM] WHI-154[200 mM] FP
FEPD6 h 12h
Karpas2996 h 12h
Vitality (%)
WHI-154FP
cdk2cdk4cdk7cdk9
RBS780
RBS612
RB
NPM-ALKNPM-ALKY664
STAT3Y705
JNKT183/Y185
ERK1/2T202/Y204
PARPγ-tubulin
DNA ContentDNA Content
4.9% WHI-P1540.0%
30.8%
720600480360240120
0
720600480360240120
0
Cell
num
ber
Cell
num
ber
Apop
totic
cel
ls (%
)
720600480360240120
0
Cell
num
ber
0 32 64 96 128 160 192 224 256
DNA Content0 32 64 96 128 160 192 224 256
0 32 64 96 128 160 192 224 256
DMSO
G1 43.8%S 44.6%G2 11.5%
FP
G1 37.9%S 20.0%G2 10.0%
AV+ cellsPI+ cells
10 20 10 20 10 20 10 20 10 20 10 20 10 20 10 20
100
80
60
40
20
0
G1 69.6%S 11.8%G2 13.7%
0 5 10 20 50 µM
FEPD
97.3
96.0
95.1
89.2
53.5
47.7
97.6
96.0
95.5
56.5
28.3
30.5
96.9
94.5
93.5
54.2
48.5
54.1
96.5
95.3
94.6
25.9
15.3
24.8

phorylation of STAT3 was strongly impaired, but so toowas the expression of cdk2, cdk4 and cdk-phosphory-lated RB, whereas cdk7 and cdk9 levels were mostlyunaffected (Figure 5A). In contrast, changes of proteinexpression in ALK-negative FEPD cells treated withWHI-154 were not significant. At concentrations forwhich complete inhibition of NPM-ALK autophospho-rylation was observed, WHI-154 caused G1 cell cyclearrest in the absence of apoptosis in ALK-positiveKarpas299 cells, whereas perturbations of the cell cycleby flavopiridol reflected the induction of apoptosis pre-viously demonstrated (Figure 5B). The advantage of acombination of these drugs was, therefore, investigated,and compared to the ability of single agents to perturbthe expression of proteins critical for signaling(STAT3Y705, JNKT183/Y185, ERK1/2T202/Y204), growth (cyclin Aand B1, cdk2 and 4, p27Kip, RBS780 and RBS612) and survival(PARP). As shown by immunoblot analysis, upon co-administration of WHI-154 with flavopiridol, all regula-tors of proliferation and survival were totally depletedfrom ALCL cells, including active NPM-ALKY664, activat-ed STAT3Y705, JNKT183/Y185 and ERK1/2T202/Y204, and apoptosiswas strongly induced as shown by PARP cleavageanalysis (Figure 5B). When cells were stained withannexin-V and examined by FACS, apoptosis wasenhanced in WHI-154/flavopiridol-treated Karpas299cells, but not in ALK-negative FEPD cells (Figure 5C).When used alone, WHI-154 did not cause apoptosis ineither ALK-positive or -negative ALCL cells. WhenWHI-154 was added to flavopiridol, however, flavopiri-dol-dependent apoptosis increased in Karpas299 cellsfrom 11% to ~50% after 6 h, and from 44% to ~70%12 h post-treatment, indicating the role of NPM-ALK inmodulating tumor cell responsiveness to cdk inhibition.Conversely, the combined treatment had minimaleffects in FEPD cells, and apoptosis was mainly due toflavopiridol (~45% and ~75%, at 6 and 12h in flavopiri-dol-treated cells; ~48% and ~80% in flavopiridol/WHI-154-treated cells).
Discussion
Cyclin-dependent kinases are attractive targets fordrug development since their activity, required for thecorrect timing and ordering of the cell cycle, is fre-quently deregulated in cancer.28 Numerous small mole-cule inhibitors of cdk have been identified and proveneffective in treating tumors, based on the increasedsensitivity of the transformed cells to changes in thelevels and activity of cdk.29,30 However the conse-quences of cdk inactivation are complex and can resultin disparate outcomes depending on the tumor typeand the genetic context that drives their expression.Besides, cdk inhibition per se is not sufficient for selec-tive tumor cell killing, and relies on changes in theactivity of signal transduction pathways critical forsurvival, or in the balance of pro- and anti-apoptoticsignals.31 Competitive inhibitors of both cell-cycle andtranscriptional cdk exert a more potent antitumoractivity in different preclinical models than com-pounds with selective cdk inhibitory activity, due per-
haps to the functional compensation between cdkfamily members.32-35 Tumor cells engineered to silencecdk2 do not necessarily arrest or die, as they may com-pensate for lower cdk2 activity with cdk1 or cdk4/6,while cell-cycle arrest caused by combined depletionof cdk1 and cdk2 likely results in cell death, occurringeven more rapidly if other cdk are concomitantlyimpaired.36-39 As ALCL cells proliferate rapidly and cellcycle deregulation has recently been shown to con-tribute to lymphomagenesis, we investigated whethertargeting the cyclin/cdk axis could inhibit tumorgrowth, and assessed this hypothesis using the multi-ple cdk inhibitor flavopiridol, alone or combined withNPM-ALK kinase inhibitors.
ALCL is a highly aggressive subtype of non-Hodgkin’s lymphoma characterized by expression andconstitutive activity of the NPM-ALK tyrosine kinase.NPM-ALK controls functional activation of bothupstream regulators (RAS, ERK1/2, JNK and AKT) anddownstream effector proteins (Myc, c-Jun, Fos, andSTAT3/5) of ALCL signaling, and has direct or indirectactivity on growth, survival, migration and cell shap-ing.17 Consistently, a differential expression and activa-tion of various regulatory proteins, including D-typecyclins, cyclins A and E, as well as cdk inhibitors p21WAF,p27Kip and RB has been observed in NPM-ALK-express-ing cells, as a result of enhanced AP-1, STAT3 or MYCtranscriptional activity by JNK and ERK kinases, orreduced FOXO3A activity by AKT.27,40-42 We provedhere that cdk inactivation leads to mitochondrial dam-age, caspase activation and apoptosis in ALCL cells, intime- and dose-dependent manners, through inhibitionof DNA synthesis and RNA transcription, selectivekilling of S-phase cells, and the engagement of differentgrowth-inhibitory pathways. Our study revealed thatALCL cells do not display cell cycle arrest in the pres-ence of flavopiridol, but undergo preferential death ofthe S-phase population at drug concentrations that cor-relate with cdk inhibition. They stall in G1 or G2-Mphase when apoptosis is prevented by z-vad-fmk cas-pase inhibitor, whereas they die rapidly when allowedto enter S phase after recruitment to G1/S boundary.Due to flavopiridol inhibitory activity, RB tumor sup-pressor protein was dephosphorylated at cdk sitesSer780 and Thr821, and associated with E2F1 earlyafter drug addition, before caspase-dependent cleavage.Cdk inhibition also reduced phosphorylation and tran-scriptional activity of RNA polymerase II, causing aconcomitant decrease in protein expression, likely dueto degradation of the enzyme.
The transcripts most sensitive to reduced RNA poly-merase II phosphorylation are those with short half-lives, including transcripts encoding anti-apoptotic pro-teins. Depletion of the corresponding proteins inresponse to the inhibition of transcriptional cdks (i.e.cdk9) may induce cell death and, in some instances,may sensitize cells to other apoptotic stimuli. In thiscontext, flavopiridol caused strong inhibition of Mcl-1transcription and expression in ALCL cells, whichresulted in the collapse of mitochondrial membranepotential. When Mcl-1 expression was abolished, pro-apoptotic Bax protein accumulated at mitochondria,
The effect of flavopiridol on ALCL
haematologica | 2009; 94(7) | 953 |

P. Bonvini et al.
| 954 | haematologica | 2009; 94(7)
and release of cytochrome c into the cytoplasm correlat-ed with caspase-3 activation.
Known advantages of cdk inhibition are unrestrainedE2F-1 activity during S phase which leads to aberrantexpression of pro-apoptotic genes and predisposes cellsto death, as well as reduced regulatory phosphorylationof RNA polymerase II which affects short half-life tran-scripts of rapidly turned-over anti-apoptotic proteins.43-
45 Nonetheless, problems of this approach can be factorsthat favor cell cycle arrest over apoptosis, such as func-tional RB, EGFR and AKT, which impede E2F1-mediat-ed apoptosis, or JNK and ERK1/2 kinases, which affecttranscription and expression of anti-apoptotic proteins.We, therefore, looked at additional events contributingto flavopiridol antitumor activity, assessing the steady-state of primary downstream mediators of NPM-ALKtransforming activity in ALCL, whose inhibitionimproves flavopiridol cytotoxicity as recentlyshown.46,47 We found that ALCL cells treated withflavopiridol alone did not display a significant decline inNPM-ALK protein expression and activity, and exhibit-ed a pronounced activation of ERK1/2 and JNK kinases,not observed in ALK-negative ALCL cells. Consistently,differences in the extent and time of flavopiridol-induced apoptosis were observed between the ALK-negative and -positive cell lines, with the latter alsoshowing viable cells after treatment. These cells, whenisolated, were found to maintain NPM-ALK status andactivity despite the downregulation of cdk activity,which ruled out any lack of flavopiridol activity becauseof differential drug saturation or efflux, though suggest-ing a context-dependent model able to modulate drugeffectiveness and sensitivity in ALK-positive cells.48
The possibility that the interruption of NPM-ALK sig-naling could increase the response to flavopiridol wasinvestigated by targeting NPM-ALK, as shown with
other oncogenic kinases.30,49,50 Our study demonstratedthat the cytoreductive activity of the NPM-ALK small-molecule inhibitor WHI-154 resulted in downregulationof cell cycle-related proteins, including cdk, as well as innear-complete depletion of NPM-ALK-activated down-stream signal proteins. This led to cell cycle arrest in theabsence of apoptosis when WHI-154 was used as a sin-gle agent, whereas it caused an increase in cell deathwhen administered with the cdk-inhibitor flavopiridol.The onset of apoptosis was extremely fast and robustwith the combination of those two drugs, perhaps dueto enhanced efficacy on the molecular determinantscontrolling proliferation and survival of ALCL cells.Pharmacological interruption of NPM-ALK signalingdramatically lowered the ALCL threshold for caspaseactivation by flavopiridol, proving the critical role of theoncogenic kinase in preventing drug-induced apoptosis.These cells became particularly vulnerable when cellcycle and survival signal events were simultaneouslydisrupted, strengthening the hypothesis that targetingcyclin/cdk signaling is an effective anti-tumor approachagainst ALCL, and supporting the evidence that, incombination with NPM-ALK inhibition, this strategy iseven more promising in ALK-positive malignancies.
Authorship and Discloures
PB: performed and designed the research, analyzedthe data and wrote the paper. EZ, GM and MP: per-formed the research. LM: performed the research andcontributed to analysis of the data. GB: co-ordinated thefluorocytometric analysis and provided critical evalua-tion of results. AR: designed and co-ordinated theresearch, analyzed the data and wrote the paper.
The authors reported no potential conflicts of interest.
References
1. Vermeulen K, Van Bockstaele DR,Berneman ZN. The cell cycle: areview of regulation, deregulationand therapeutic targets in cancer.Cell Prolif 2003;36:131-49.
2. Novak B, Tyson JJ, Gyorffy B,Csikasz-Nagy A. Irreversible cell-cycle transitions are due to systems-level feedback. Nat Cell Biol 2007;9:724-8.
3. Massague J. G1 cell-cycle controland cancer. Nature 2004;432:298-306.
4. Oelgeschlager T. Regulation of RNApolymerase II activity by CTDphosphorylation and cell cycle con-trol. J Cell Physiol 2002;190:160-9.
5. Shapiro GI. Cyclin-dependentkinase pathways as targets for can-cer treatment. J Clin Oncol 2006;24:1770-83.
6. Berthet C, Kaldis P. Cell-specificresponses to loss of cyclin-depend-ent kinases. Oncogene 2007;26:4469-77.
7. Besson A, Dowdy SF, Roberts JM.
CDK inhibitors: cell cycle regulatorsand beyond. Dev Cell 2008;14:159-69.
8. Swanton C. Cell-cycle targeted ther-apies. Lancet Oncol 2004;5:27-36.
9. Malumbres M, Pevarello P, BarbacidM, Bischoff JR. CDK inhibitors incancer therapy: what is next?Trends Pharmacol Sci 2008;29:16-21.
10. Senderowicz AM. Small-moleculecyclin-dependent kinase modula-tors. Oncogene 2003;22:6609-20.
11. Sedlacek HH. Mechanisms of actionof flavopiridol. Crit Rev OncolHematol 2001;38:139-70.
12. Shapiro GI. Preclinical and clinicaldevelopment of the cyclin-depend-ent kinase inhibitor flavopiridol.Clin Cancer Res 2004;10:4270s-5s.
13. Grant S, Dent P. Gene profiling andthe cyclin-dependent kinaseinhibitor flavopiridol: what’s in aname? Mol Cancer Ther 2004;3:873-5.
14. Chen R, Keating MJ, Gandhi V,Plunkett W. Transcription inhibitionby flavopiridol: mechanism ofchronic lymphocytic leukemia cell
death. Blood 2005;106:2513-9.15. Bischof D, Pulford K, Mason DY,
Morris SW. Role of the nucleophos-min (NPM) portion of the non-Hodgkin’s lymphoma-associatedNPM-anaplastic lymphoma kinasefusion protein in oncogenesis. MolCell Biol 1997;17:2312-25.
16. Morris SW, Kirstein MN, ValentineMB, Dittmer KG, Shapiro DN,Saltman DL, Look AT. Fusion of akinase gene, ALK, to a nucleolar pro-tein gene, NPM, in non-Hodgkin’slymphoma. Science 1994;263:1281-4.
17. Chiarle R, Voena C, Ambrogio C,Piva R, Inghirami G. The anaplasticlymphoma kinase in the pathogene-sis of cancer. Nat Rev Cancer 2008;8:11-23.
18. Inoue Y, Kitagawa M, Taya Y.Phosphorylation of pRB at Ser612by Chk1/2 leads to a complexbetween pRB and E2F-1 after DNAdamage. Embo J 2007;26:2083-93.
19. Dai Y, Rahmani M, Grant S.Proteasome inhibitors potentiateleukemic cell apoptosis induced bythe cyclin-dependent kinase

inhibitor flavopiridol through aSAPK/JNK- and NF-κB-dependentprocess. Oncogene 2003;22:7108-22.
20. Gomez LA, de Las Pozas A, Perez-Stable C. Sequential combination offlavopiridol and docetaxel reducesthe levels of X-linked inhibitor ofapoptosis and AKT proteins andstimulates apoptosis in humanLNCaP prostate cancer cells. MolCancer Ther 2006;5:1216-26.
21. Kim DM, Koo SY, Jeon K, Kim MH,Lee J, Hong CY, Jeong S. Rapidinduction of apoptosis by combina-tion of flavopiridol and tumornecrosis factor (TNF)-alpha or TNF-related apoptosis-inducing ligand inhuman cancer cell lines. Cancer Res2003;63:621-6.
22. Takada Y, Sethi G, Sung B, AggarwalBB. Flavopiridol suppresses tumornecrosis factor-induced activation ofactivator protein-1, c-Jun N-terminalkinase, p38 mitogen-activated pro-tein kinase (MAPK), p44/p42MAPK, and Akt, inhibits expressionof antiapoptotic gene products, andenhances apoptosis throughcytochrome c release and caspaseactivation in human myeloid cells.Mol Pharmacol 2008;73:1549-57.
23. Kim BS, Yoon KH, Oh HM, Choi EY,Kim SW, Han WC, et al. Involve-ment of p38 MAP kinase during ironchelator-mediated apoptotic celldeath. Cell Immunol 2002; 220:96-106.
24. Shukla S, Gupta S. Apigenin-induced cell cycle arrest is mediatedby modulation of MAPK, PI3K-Akt,and loss of cyclin D1 associatedretinoblastoma dephosphorylationin human prostate cancer cells. CellCycle 2007;6:1102-14.
25. Bregman DB, Du L, van der Zee S,Warren SL. Transcription-dependentredistribution of the large subunit ofRNA polymerase II to discretenuclear domains. J Cell Biol 1995;129:287-98.
26. Sudbeck EA, Liu XP, Narla RK,Mahajan S, Ghosh S, Mao C, et al.Structure-based design of specificinhibitors of Janus kinase 3 as apop-tosis-inducing antileukemic agents.Clin Cancer Res 1999;5:1569-82.
27. Marzec M, Kasprzycka M, PtasznikA, Wlodarski P, Zhang Q, Odum N,et al. Inhibition of ALK enzymaticactivity in T-cell lymphoma cellsinduces apoptosis and suppressesproliferation and STAT3 phosphory-lation independently of Jak3. LabInvest 2005;85:1544-54.
28. Malumbres M, Barbacid M. Cellcycle kinases in cancer. Curr OpinGenet Dev 2007;17:60-5.
29. Hahntow IN, Schneller F, OelsnerM, Weick K, Ringshausen I, Fend F,et al. Cyclin-dependent kinase
inhibitor roscovitine induces apop-tosis in chronic lymphocyticleukemia cells. Leukemia 2004;18:747-55.
30. Wang L, Wang J, Blaser BW,Duchemin AM, Kusewitt DF, Liu T,et al. Pharmacologic inhibition ofCDK4/6: mechanistic evidence forselective activity or acquired resist-ance in acute myeloid leukemia.Blood 2007;110:2075-83.
31. Collins I, Garrett MD. Targeting thecell division cycle in cancer: CDKand cell cycle checkpoint kinaseinhibitors. Curr Opin Pharmacol2005;5:366-73.
32. Gao N, Kramer L, Rahmani M, DentP, Grant S. The three-substitutedindolinone cyclin-dependent kinase2 inhibitor 3-[1-(3H-imidazol-4-yl)-meth-(Z)-ylidene]-5-methoxy-1,3-dihydro-indol-2-one (SU9516) killshuman leukemia cells via down-reg-ulation of Mcl-1 through a transcrip-tional mechanism. Mol Pharmacol2006;70:645-55.
33. Jackman KM, Frye CB, Hunger SP.Flavopiridol displays preclinicalactivity in acute lymphoblasticleukemia. Pediatr Blood Cancer2008;50:772-8.
34. Smith ME, Cimica V, Chinni S,Challagulla K, Mani S, Kalpana GV.Rhabdoid tumor growth is inhibit-ed by flavopiridol. Clin Cancer Res2008; 14:523-32.
35. Zhang C, Lundgren K, Yan Z,Arango ME, Price S, Huber A, et al.Pharmacologic properties of AG-012986, a pan-cyclin-dependentkinase inhibitor with antitumor effi-cacy. Mol Cancer Ther 2008;7:818-28.
36. Martín A, Odajima J, Hunt SL,Dubus P, Ortega S, Malumbres M,Barbacid M. Cdk2 is dispensable forcell cycle inhibition and tumor sup-pression mediated by p27(Kip1) andp21(Cip1). Cancer Cell 2005;7:591-8.
37. Berthet C, Kaldis P. Cdk2 and Cdk4cooperatively control the expressionof Cdc2. Cell Div 2006;1:10.
38. Cai D, Latham VM Jr, Zhang X,Shapiro GI. Combined depletion ofcell cycle and transcriptional cyclin-dependent kinase activities inducesapoptosis in cancer cells. Cancer Res2006;66:9270-80.
39. Malumbres M, Sotillo R, SantamaríaD, Galán J, Cerezo A, Ortega S, et al.Mammalian cells cycle without theD-type cyclin-dependent kinasesCdk4 and Cdk6. Cell 2004;118:493-504.
40. Marzec M, Kasprzycka M, Liu X,Raghunath PN, Wlodarski P, WasikMA. Oncogenic tyrosine kinaseNPM/ALK induces activation of theMEK/ERK signaling pathway inde-pendently of c-Raf. Oncogene 2007;
26:813-21.41. Staber PB, Vesely P, Haq N, Ott RG,
Funato K, Bambach I, et al. Theoncoprotein NPM-ALK of anaplasticlarge-cell lymphoma induces JUNBtranscription via ERK1/2 and JunBtranslation via mTOR signaling.Blood 2007;110:3374-83.
42. Vega F, Medeiros LJ, Leventaki V,Atwell C, Cho-Vega JH, Tian L, et al.Activation of mammalian target ofrapamycin signaling pathway con-tributes to tumor cell survival inanaplastic lymphoma kinase-posi-tive anaplastic large cell lymphoma.Cancer Res 2006;66:6589-97.
43. Jiang J, Matranga CB, Cai D, LathamVM Jr, Zhang X, Lowell AM, et al.Flavopiridol-induced apoptosis dur-ing S phase requires E2F-1 and inhi-bition of cyclin A-dependent kinaseactivity. Cancer Res 2003;63:7410-22.
44. Litz J, Carlson P, Warshamana-Greene GS, Grant S, Krystal GW.Flavopiridol potently induces smallcell lung cancer apoptosis during Sphase in a manner that involvesearly mitochondrial dysfunction.Clin Cancer Res 2003;9:4586-94.
45. Matranga CB, Shapiro GI. Selectivesensitization of transformed cells toflavopiridol-induced apoptosis fol-lowing recruitment to S-phase.Cancer Res 2002;62:1707-17.
46. Lee YK, Isham CR, Kaufman SH,Bible KC. Flavopiridol disruptsSTAT3/DNA interactions, attenu-ates STAT3-directed transcription,and combines with the Jak kinaseinhibitor AG490 to achieve cytotox-ic synergy. Mol Cancer Ther 2006;5:138-48.
47. Yu C, Rahmani M, Dai Y, Conrad D,Krystal G, Dent P, Grant S. Thelethal effects of pharmacologicalcyclin-dependent kinase inhibitorsin human leukemia cells proceedthrough a phosphatidylinositol 3-kinase/Akt-dependent process.Cancer Res 2003;63:1822-33.
48. Chau BN, Wang JY. Coordinatedregulation of life and death by RB.Nat Rev Cancer 2003;3:130-8.
49. Phillip CJ, Stellrecht CM, Nimma-napalli R, Gandhi V. Targeting METtranscription as a therapeutic strate-gy in multiple myeloma. CancerChemother Pharmacol 2009;63:587-97.
50. Sambol EB, Ambrosini G, Geha RC,Kennealey PT, Decarolis P,O’Connor R, et al. Flavopiridol tar-gets c-KIT transcription and inducesapoptosis in gastrointestinal stromaltumor cells. Cancer Res 2006;66:5858-66.
The effect of flavopiridol on ALCL
haematologica | 2009; 94(7) | 955 |













![Elevated Cyclins and Cyclin-dependent Kinase Activity in ...[CANCER RESEARCH 58, 2042-2049, May I, 1998] Elevated Cyclins and Cyclin-dependent Kinase Activity in the Rhabdomyosarcoma](https://img.pdfslide.us/doc/110x75/5e4e63ca3358114ff2317f00/elevated-cyclins-and-cyclin-dependent-kinase-activity-in-cancer-research-58.jpg)















