Embed Size (px)
Citation preview

REVIEWPreclinical and Clinical Development ofCyclin-Dependent Kinase Modulators
Adrian M. Senderowicz, Edward A. Sausville
In the last decade, the discovery and cloning of the cyclin-dependent kinases (cdks), key regulators of cell cycle pro-gression, have led to the identification of novel modulators ofcdk activity. Initial experimental results demonstrated thatthese cdk modulators are able to block cell cycle progression,induce apoptotic cell death, promote differentiation, inhibitangiogenesis, and modulate transcription. Alteration of cdkactivity may occur indirectly by affecting upstream path-ways that regulate cdk activity or directly by targeting thecdk holoenzyme. Two direct cdk modulators, flavopiridoland UCN-01, are showing promising results in early clinicaltrials, in which the drugs reach plasma concentrations thatcan alter cdk activity in vitro. Although modulation of cdkactivity is a well-grounded concept and new cdk modulatorsare being assessed for clinical testing, important scientificquestions remain to be addressed. These questions includewhether one or more cdks should be inhibited, how cdkinhibitors should be combined with other chemotherapyagents, and which cdk substrates should be used to assess thebiologic effects of these drugs in patients. Thus, modulationof cdk activity is an attractive target for cancer chemo-therapy, and several agents that modulate cdk activity are inor are approaching entry into clinical trials. [J Natl CancerInst 2000;92:376–87]
INTRODUCTION TO CELL CYCLE REGULATION
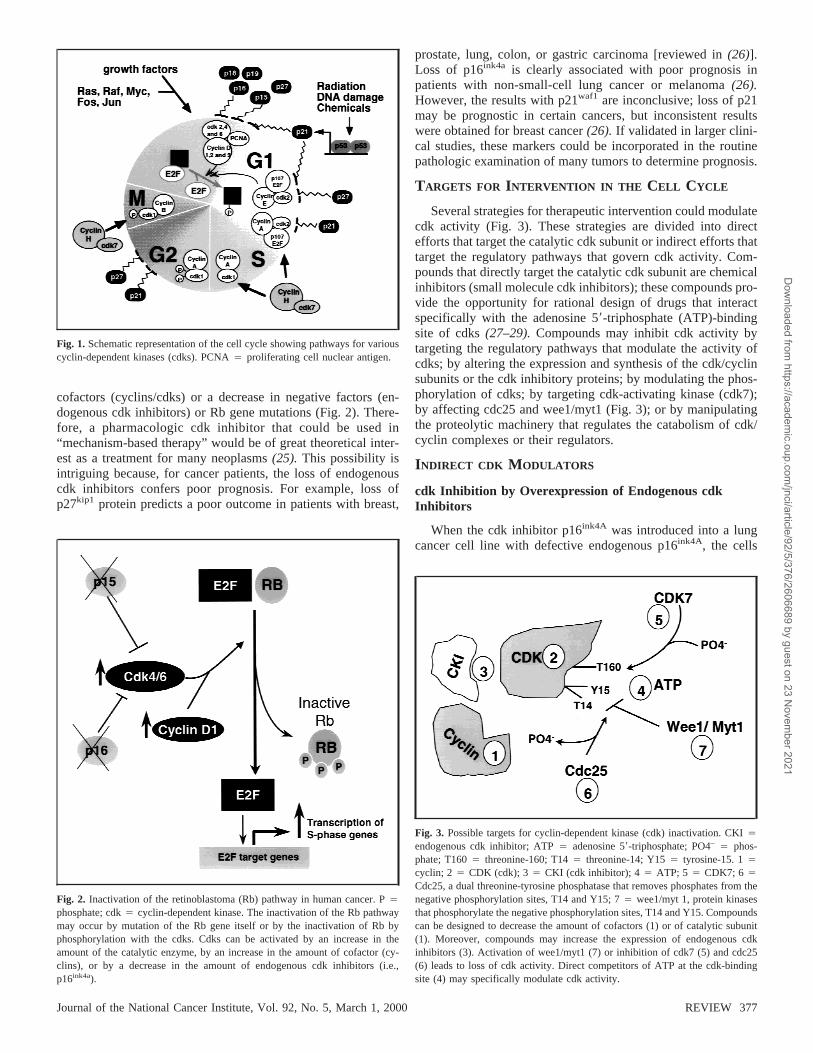
After activation of several mitogenic signaling cascades, cellstraverse the cell cycle in several tightly controlled phases (Fig.1). G1 phase separates M and S phases. In this period, cellscommit to enter the cell cycle and prepare to duplicate theirDNA (1). After G1 phase, cells enter S phase, the period of DNAsynthesis (genome duplication). After S phase, cells enter G2
phase, the period in which cells can repair errors that mightoccur during DNA duplication and thus prevent passing theseerrors to daughter cells. During G2 phase, cells prepare to enterM phase, the period in which chromatids and then daughter cellsseparate. After M phase, cells can enter G1 phase again or enterG0 phase, a replicatively quiescent phase. In G0 phase, the cellsusually have a diploid amount of DNA, which represents thedifferentiated functioning cell not committed to the cell cycle.
The progression of cells from G1 to S phase is accompaniedby the phosphorylation of the retinoblastoma gene product (Rbprotein), a tumor suppressor gene active in the control of G1
phase(2,3). Phosphorylation of Rb protein by serine/threoninekinases known as cyclin-dependent kinases (cdks) inactivatesRb (4). The cdks, key regulators of the cell cycle, consist ofcatalytic subunits that form complexes with proteins known ascyclins. There are at least nine cdks (cdk1–cdk9)(4–7).The cdksthat are clearly involved in cell cycle control are cdk1 through
cdk7. Although structurally related to cdk1 through cdk7, cdk8and cdk9 are important transcriptional regulators(5,6).There areat least 15 cyclins (cyclin A through cyclin T)(8–10).Cyclinexpression varies during the cell cycle, and indeed the periodicexpression of different cyclins defines the start of each phase ofthe cell cycle and also marks the transitions between the variousphases. Cyclins and their cognate cdk catalytic subunits nonco-valently form 1 : 1 complexes to produce the cdk holoenzyme.The holoenzyme is activated by the phosphorylation of specificresidues in the cdk catalytic subunit. This phosphorylation canbe catalyzed by cdk7/cyclin H, which is also known as thecdk-activating kinase(11,12).
Specific cdks operate in distinct phases of the cell cycle.Complexed with their respective D-type cyclin partners, cdk4and cdk6 are responsible for the cell’s progression through G1
phase (Fig. 1). A complex of cdk2 and cyclin E is responsible forthe cell’s progression from G1 phase to S phase. A complex ofcdk2 and cyclin A is required for the cell’s progression throughS phase, and a complex of cdk1 (also known as cdc2) and cyclinB is required for mitosis(1). These complexes are in turn regu-lated by a stoichiometric combination with small inhibitory pro-teins called endogenous cdk inhibitors. The INK4 (inhibitor ofcdk4) family includes p16ink4a, p15ink4b, p18ink4c, and p19ink4d,and its members specifically inhibit cyclin D-associated kinases.Members of the kinase inhibitor protein family p21waf1, p27Kip1,and p57kip2 bind and inhibit the activity of complexes of cyclinE and cdk2 and complexes of cyclin A and cdk2(13–15).Al-though members of the kinase inhibitor protein family wereinitially thought to exclusively regulate G1 and S phases, severalreports(16–18)demonstrated that these proteins can also regu-late the G2/M-phase transition.
DNA synthesis (S phase) begins with the cdk4- and/or cdk6-mediated phosphorylation of Rb protein (which is complexedwith the transcriptional factor E2F). Phosphorylated Rb is re-leased from its complex with E2F. The released E2F then pro-motes the transcription of numerous genes required for the cellto progress through S phase, including thymidylate synthase anddihydrofolate reductase, among others(2,19,20).Additional in-formation about cell cycle regulation can be found in severalreviews(21–24).
The vast majority of human cancers have abnormalities insome component of the Rb pathway (Fig. 2) because of hyper-activation of cdks resulting from the overexpression of positive
Affiliation of authors:DTP Clinical Trials Unit, Developmental TherapeuticsProgram, Division of Cancer Treatment and Diagnosis, National Cancer Insti-tute, Bethesda, MD.
Correspondence to:Adrian M. Senderowicz, M.D., National Institutes ofHealth, Bldg. 10, Rm. 6N113, Bethesda, MD 20892 (e-mail: [email protected]).
See“Notes” following “References.”
376 REVIEW Journal of the National Cancer Institute, Vol. 92, No. 5, March 1, 2000
Dow
nloaded from https://academ
ic.oup.com/jnci/article/92/5/376/2606689 by guest on 23 N
ovember 2021
cofactors (cyclins/cdks) or a decrease in negative factors (en-dogenous cdk inhibitors) or Rb gene mutations (Fig. 2). There-fore, a pharmacologic cdk inhibitor that could be used in“mechanism-based therapy” would be of great theoretical inter-est as a treatment for many neoplasms(25). This possibility isintriguing because, for cancer patients, the loss of endogenouscdk inhibitors confers poor prognosis. For example, loss ofp27kip1 protein predicts a poor outcome in patients with breast,
prostate, lung, colon, or gastric carcinoma [reviewed in(26)].Loss of p16ink4a is clearly associated with poor prognosis inpatients with non-small-cell lung cancer or melanoma(26).However, the results with p21waf1 are inconclusive; loss of p21may be prognostic in certain cancers, but inconsistent resultswere obtained for breast cancer(26). If validated in larger clini-cal studies, these markers could be incorporated in the routinepathologic examination of many tumors to determine prognosis.
TARGETS FOR INTERVENTION IN THE CELL CYCLE
Several strategies for therapeutic intervention could modulatecdk activity (Fig. 3). These strategies are divided into directefforts that target the catalytic cdk subunit or indirect efforts thattarget the regulatory pathways that govern cdk activity. Com-pounds that directly target the catalytic cdk subunit are chemicalinhibitors (small molecule cdk inhibitors); these compounds pro-vide the opportunity for rational design of drugs that interactspecifically with the adenosine 58-triphosphate (ATP)-bindingsite of cdks(27–29).Compounds may inhibit cdk activity bytargeting the regulatory pathways that modulate the activity ofcdks; by altering the expression and synthesis of the cdk/cyclinsubunits or the cdk inhibitory proteins; by modulating the phos-phorylation of cdks; by targeting cdk-activating kinase (cdk7);by affecting cdc25 and wee1/myt1 (Fig. 3); or by manipulatingthe proteolytic machinery that regulates the catabolism of cdk/cyclin complexes or their regulators.
INDIRECT CDK MODULATORS
cdk Inhibition by Overexpression of Endogenous cdkInhibitors
When the cdk inhibitor p16ink4A was introduced into a lungcancer cell line with defective endogenous p16ink4A, the cells
Fig. 1. Schematic representation of the cell cycle showing pathways for variouscyclin-dependent kinases (cdks). PCNA4 proliferating cell nuclear antigen.
Fig. 2. Inactivation of the retinoblastoma (Rb) pathway in human cancer. P4
phosphate; cdk4 cyclin-dependent kinase. The inactivation of the Rb pathwaymay occur by mutation of the Rb gene itself or by the inactivation of Rb byphosphorylation with the cdks. Cdks can be activated by an increase in theamount of the catalytic enzyme, by an increase in the amount of cofactor (cy-clins), or by a decrease in the amount of endogenous cdk inhibitors (i.e.,p16ink4a).
Fig. 3. Possible targets for cyclin-dependent kinase (cdk) inactivation. CKI4
endogenous cdk inhibitor; ATP4 adenosine 58-triphosphate; PO4– 4 phos-phate; T1604 threonine-160; T144 threonine-14; Y154 tyrosine-15. 14
cyclin; 2 4 CDK (cdk); 34 CKI (cdk inhibitor); 44 ATP; 5 4 CDK7; 6 4
Cdc25, a dual threonine-tyrosine phosphatase that removes phosphates from thenegative phosphorylation sites, T14 and Y15; 74 wee1/myt 1, protein kinasesthat phosphorylate the negative phosphorylation sites, T14 and Y15. Compoundscan be designed to decrease the amount of cofactors (1) or of catalytic subunit(1). Moreover, compounds may increase the expression of endogenous cdkinhibitors (3). Activation of wee1/myt1 (7) or inhibition of cdk7 (5) and cdc25(6) leads to loss of cdk activity. Direct competitors of ATP at the cdk-bindingsite (4) may specifically modulate cdk activity.
Journal of the National Cancer Institute, Vol. 92, No. 5, March 1, 2000 REVIEW 377
Dow
nloaded from https://academ
ic.oup.com/jnci/article/92/5/376/2606689 by guest on 23 N
ovember 2021

were arrested in G1 phase(30). This effect occurs only in cellswith functional Rb(31). When both p53 and p16ink4a were in-troduced into cells, apoptotic cell death followed(32). Whenp21waf1 or p27kip1 were introduced,in vitro andin vivoantitumoreffects and G1/S arrest were observed(33,34).When p27 wasintroduced in several preclinical tumor models, apoptotic celldeath was induced, but the relationship between the putative cdkinhibition of p27kip1 and its induction of apoptotic cell death isunclear(35,36).
Several small molecule chemical inhibitors of cdks appear tomodulate the expression of cdk inhibitors. For example, lovas-tatin blocks cells in G1 phase by induction of p21waf1and p27kip1,which leads to the loss of cdk activity(37,38). Rapamycinblocks lymphocytes in G1 phase by preventing the interleukin2-stimulated degradation of p27kip1 (39). However, it is unclearwhether the cell cycle inhibitory effects of these small moleculechemical inhibitors can be solely explained by the induction ofp27kip1.
cdk Inhibition by Peptidomimetic-Based Approaches
Another strategy to block cell cycle progression by loss ofcdk activity is to use small peptides that mimic the effects ofendogenous cdk inhibitors. Several carriers have been tested thatintroduce peptides into cells, including a 16-residue segmentderived from theDrosophila antenappedia protein. When this16-residue transmembrane carrier was linked to the third ankyrinrepeat of the p16ink4A protein and inserted into cells, Rb-dependent G1 arrest and cell senescence were observed(40).Hybrids containing the antenappedia peptide and differentp21waf1 peptides were constructed. In a breast-derived cell line,the chimera containing the carboxyl-terminal peptide of p21,amino acids 141–160, had a higher specificity for cdk4/cyclin Dthan for cdk2/cyclin E and arrested the cells in G1 phase(41). Incontrast,in vitro the chimera containing amino-terminal peptidesof p21, amino acids 17–33 and 63–77, inhibited both cdk1 andcdk2, and cells transduced with this chimera were arrested in allphases of the cell cycle(42).
Another approach to inhibiting cdk activity is to developpeptides that bind to cdks and inhibit cdk kinase activity(43,44).Colas et al.(43) demonstrated that a 20-amino acid peptide,identified by use of a combinatorial library, specifically bindscdk2 and inhibits its activity at low nanomolar concentrationsinvitro. This peptide could act by blocking the interaction of thecatalytic subunit with substrates or cyclin cofactors(43).Chen etal. (44) have shown that 8-amino acid peptides derived from theputative cyclin-cdk2-binding region of p21waf1 (PVKRRLFG)and E2F1 (PVKRRLDL) linked to N-terminal residues derivedfrom human immunodeficiency virus Tat protein or antennape-dia protein can block cells in S phase. This effect was associatedwith a loss of cdk2 activity. Although all of the cells tested withthese chimeras showed clear evidence of G1/S-phase arrest, im-mortalized/transformed cells were more prone to apoptotic celldeath.
cdk Inhibition by Depletion in cdk/Cyclin Subunits
Antisense technology has been used to deplete messengerRNAs (mRNAs) for cdk and/or cyclins(45–47).When cyclinD1 was depleted from tumor cell lines, a substantial antiprolif-erative effect was observed that was synergistic with differentstandard chemotherapeutic agents(45).
Several compounds can inhibit tumor progression by the
modulation of cdk/cyclin subunits. In breast carcinoma celllines, antiestrogens, such as tamoxifen, inhibit the expression ofcyclin D and other cell cycle-related proteins and inhibit cdkactivity (48). In breast carcinoma cell lines, retinoids, such asall-trans-retinoic acid and 9-cis-retinoic acid, inhibit the expres-sion of multiple cell cycle regulators, including cyclin D1, cyclinD3, cdk2, and cdk4(49). In some model systems, rapamycin, aninhibitor of FKBP (FK-506-binding protein)/mTOR (mamma-lian target of rapamycin), was also associated with a decline incyclin D1 protein(39). Although treatment of cells with any ofthese compounds may lead to the decline of cyclin proteins andthe perturbation of other cell cycle-related proteins, it is unclearhow these compounds act. Perhaps the changes result from adirect interaction between the drug and the pathways that regu-late the production of cyclin/cdk or result from the G1-phasearrest and/or Rb dephosphorylation, which are observed withthese compounds.
cdk Inhibition by Modulation of Proteasomal Machinery
Sequential turnover of certain cell cycle regulators, includingcyclins and p27kip1, is mediated by the 20S proteasome, whichpromotes proteolytic degradation through the ubiquitin/proteasome pathway. Increased turnover of cyclins with the as-sociated loss of cdk activity may lead to cell cycle arrest with orwithout apoptotic cell death. Inhibiting 20S proteasome-mediated degradation could lead to accumulation of cdk inhibi-tors, such as p27ink4a, and to cell cycle arrest with or withoutapoptotic cell death(50). An important unresolved issue is thenet effect and/or specificity of modulating proteasomal path-ways. Nonspecific proteasome modulation could alter many sig-naling pathways (by the accumulation of proteins that activate orinhibit cdks) and thus could have a final effect on cells that isdifficult to predict.
cdk Activation by Modulation of UpstreamPhosphatases/Kinases
The abrogation (overriding) of intact cell cycle checkpointsby upstream phosphatases and/or kinases could induce the “in-appropriate acceleration” of certain phases of the cell cycle. Forexample, when the G2-phase checkpoint is activated by geno-toxic stress (i.e.,g-irradiation), the G2 period is extended toallow DNA repair. In the presence of agents that abrogate (over-ride) this checkpoint, premature mitosis occurs, resulting inapoptotic cell death(51). Thus, abolishing the G2 checkpointmight sensitize cells to agents that would normally cause cells topause or arrest in G2 phase(51,52).
DIRECT CDK MODULATORS (SMALL MOLECULAR
CDK INHIBITORS )
Chemical (small molecular) cdk inhibitors can be subdividedinto the following eight families (Fig. 4): 1) purine derivatives(isopentenyladenine, 6-dimethylaminopurine, olomoucine, ros-covitine, CVT-313, and purvalanol and its derivatives), 2) bu-tyrolactone I, 3) flavopiridols (flavopiridol and deschloroflavo-piridol), 4) staurosporines (staurosporine and UCN-01), 5)toyocamycin, 6) 9-hydroxyellipticine, 7) polysulfates (suramin),and 8) paullones. Not all small molecular cdk inhibitors arespecific for cdks. In fact, staurosporine, UCN-01, suramin, 6-di-methylaminopurine, and isopentenyladenine are relatively non-specific protein kinase inhibitors. In contrast, flavopiridol, bu-
378 REVIEW Journal of the National Cancer Institute, Vol. 92, No. 5, March 1, 2000
Dow
nloaded from https://academ
ic.oup.com/jnci/article/92/5/376/2606689 by guest on 23 N
ovember 2021

tyrolactone I, olomoucine, roscovitine, CVT-313, paullones, andpurvalanol derivatives are clearly more selective for cdks. Bu-tyrolactone I, olomoucine, roscovitine, CVT-313, purvalanol,and paullone derivatives are relatively selective for cdk1 andcdk2 but are relatively inactive for cdk4 and cdk6. Flavopiridolcan inhibit all cdks tested(53–55)
Olomoucine, Roscovitine, and Other Purine Derivatives
The first cdk inhibitor discovered was dimethylaminopurine(56).This compound was initially shown to inhibit mitosis in seaurchin embryos without inhibiting protein synthesis. Later, di-methylaminopurine was shown to inhibit cdk1 activity (IC50
[concentration that inhibits activity by 50%]4 120mM) but tobe relatively nonspecific(27). Isopentenyladenine, a derivativeof dimethylaminopurine, was somewhat more potent and selec-tive for the cdks (IC50 4 55 mM) (57). Other active purinederivatives have been identified in screening campaigns formore specific and potent cdk inhibitors. Olomoucine potentlyinhibited cdk1 and cdk2 activities (IC50 4 7 mM) (27,57).Ros-covitine, a derivative of olomoucine, is a more potent cdk in-hibitor (IC50 values for cdk1/cdk24 0.7 mM) (27).
The crystal structures of cdk2 complexed with isopentenyl-adenine, olomoucine, or roscovitine showed that all three inhibi-tors bind to the ATP site(29,58). CVT-313, another purineanalogue, was identified by use of a combinatorial library strat-egy and the crystal structure of cdk2. Similar to previous ana-logues, CVT-313 was specific for cdk1 and cdk2 with IC50
values of 4.2 and 1.5mM, respectively(59).A combinatorial approach was then used to modify the purine
scaffold of 2-fluoro-6-chloropurine, and several compounds thatpotently and specifically inhibited cdc2 and cdk2 were identi-fied. Four novel compounds (purvalanol-A, purvalanol-B, com-pound 52, and compound 52E) were characterized through abattery ofin vitro kinases experiments(60).The crystal structureof purvalanol-B complexed with cdk2 showed that purvalanol-Bbound to the ATP-binding site resembles the binding of olo-
moucine to cdk2. The more membrane-permeable purvalanol-Awas tested on the National Cancer Institute’s (NCI’s) 60-cell-line anticancer drug screen panel. The average IC50 value ofpurvalanol-A was 2mM, demonstrating that it was a more activeantiproliferative agent than purvalanol-B(60).Cell cycle studiesof purvalanol-A on human fibroblasts showed that it arrestedcells in G1/S phase and G2/M phase, compatible with the puta-tive inhibitory properties in cdk1 and cdk2, respectively(60).
Paullones
With the use of the antiproliferativein vitro profile of flavo-piridol in NCI’s anticancer drug screen panel and the computa-tional algorithm COMPARE, several members of the paullonefamily were identified(61). Kenpaullone (NSC 664704) po-tently inhibited cdk1/cyclin B (IC50 4 0.4 mM), cdk2/cyclin A(IC50 4 0.68mM), cdk2/cyclin E (IC50 4 7.5 mM), and cdk5/p35 (IC50 4 0.85mM) but had much lower activity toward otherkinases(28). Kenpaullone competitively inhibits the binding ofATP, with an apparentKi (i.e., inhibitory constant) for cdk1/cyclin B of about 2.5mM. Molecular modeling studies demon-strated that kenpaullone may bind to the ATP-binding site withresidue contacts similar to other cdk2 inhibitors(28). Cell cycleeffects of kenpaullone were characterized with the MCF10Abreast epithelial cell line. Cells were synchronized in G0/G1
phase by serum starvation and then stimulated to re-enter the cellcycle in the presence of vehicle or kenpaullone at its approxi-mate IC50 concentration (30mM). Twenty hours later, vehicle-treated cells entered S phase. However, cells exposed to ken-paullone were arrested at the G1/S boundary. A similar effectwas obtained with another paullone analogue, 10-bromo-paullone (NSC 672234)(28).
CONSEQUENCES OF CDK INHIBITION
Cell Cycle Arrest
Initial cell cycle studies by van den Heuvel and Harlow(62)demonstrated that ectopic expression of cdk1-dominant negative
Fig. 4. Chemical structures ofsmall molecular cdk inhibitors.R 4 residual group.
Journal of the National Cancer Institute, Vol. 92, No. 5, March 1, 2000 REVIEW 379
Dow
nloaded from https://academ
ic.oup.com/jnci/article/92/5/376/2606689 by guest on 23 N
ovember 2021

alleles was able to block U2OS osteosarcoma cell lines at theG2/M-phase boundary. In contrast, expression of dominant nega-tive alleles of cdk2 or cdk3 blocked cells in S phase(62). Thus,roles for each cdk in human cell cycle began to be assigned.
Ectopic expression of endogenous cdk inhibitors (such asp16, p21, or p27) or peptidomimetics derived from p21, p16, orE2F1, as described above in detail, demonstrates the feasibilityof using this method to arrest cells in the cell cycle.
As described above, several small molecular cdk inhibitors,including roscovitine, olomoucine, purvalanol, and flavopiridol,arrest cells at either the G1/S- or the G2/M-phase boundaries(53,59,60,63,64).It is unclear why these agents arrest some cellsin G1/S phase and other cells in G2/M phase or both.
Apoptotic Cell Death
The antiproliferative effects of olomoucine, flavopiridol, androscovitine were accompanied by the induction of apoptotic celldeath in certain cell types(64–66).In one study(66), the abilityof flavopiridol and olomoucine to induce apoptotic cell deathvaried, depending on the growth status of the cells. That is,flavopiridol or olomoucine protected postmitotic nondividingPC12 neuronal cells from apoptotic cell death after the with-drawal of nerve growth factor. However, flavopiridol did notprotect cycling PC12 cells from apoptotic cell death after thewithdrawal of nerve growth factor(66). Similarly, cdk4 andcdk6 proteins from dominant negative alleles, but not cdk2 orcdk3 proteins from dominant negative alleles, protected neuronsfrom apoptotic cell death after the withdrawal of nerve growthfactor (67). Thus, susceptibility of PC12 cells to flavopiridol-and olomucine-induced apoptotic cell death may vary, depend-ing on the growth state of the cells.
HeLa cervical carcinoma cells treated with staurosporine andtumor necrosis factor-a were protected from apoptotic cell deathby cdc2, cdk2, or cdk3 encoded by dominant negative alleles.However, only cdk2 encoded by a dominant negative allele pro-tected cells from apoptotic cell death induced by ectopic expres-sion of topoisomerase-IIa (68).Finally, certain apoptotic stimuliinduce the caspase-mediated cleavage in endogenous cdk inhibi-tors (p21/p27) or cdk-inhibitory proteins (wee1 and cdc27) lead-ing to activation of cdks(69–71).Thus, cell cycle arrest and/orapoptosis induced by the inhibition of cdks depends on severalfactors, including the mechanism of inhibition, the type of cells,and the proliferation status of the cells.
Differentiation
During differentiation, cells exit the cell cycle and lose cdk2activity. Lee et al.(72) tested whether the chemical cdk2 inhibi-tors flavopiridol and roscovitine could induce a differentiatedphenotype by exposing NCI-H358 lung carcinoma cells to acdk2 antisense construct, flavopiridol, or roscovitine. They ob-served that all three cdk2 inhibitors could induce mucinous dif-ferentiation with the loss of cdk2 activity.
When U937 myelomonocytic leukemia cells were treatedwith aminopurvalanol, the cells acquired a phenotype character-istic of differentiated macrophages. Moreover, aminopurvalanol,a potent inhibitor of cdk1 and cdk2, appeared to arrest cells atthe G2/M boundary and then to induce apoptotic cell death(73).Other investigators(74)observed a similar phenomenon; ectopicexpression of p21waf1 or p27kip1 resulted in a “differentiatedphenotype” with cells arrested in G1 or G2 phases and a 4Namount of DNA.
Transcriptional Effects
To compare the effects of several chemical cdk inhibitors onthe expression of complementary DNA in yeast cells, Gray et al.(60) incubatedSaccharomyces cerevisiaewith compound 52and flavopiridol (each at 25mM ) for 2 hours and measuredmRNA by oligonucleotide array methods. Two percent to 3% ofthe 6200 yeast genes examined showed a greater than twofoldchange in transcript levels in the presence of these agents. More-over, almost 50% of affected transcripts were affected by bothcompound 52 and flavopiridol. These genes fell into distinctgroups, including genes that regulate progression of cell cycle,genes that regulate phosphate and cellular energy metabolism,and genes that regulate guanosine 58-triphosphate (GTP)- orATP-binding proteins. However, more than 40% of the changesin mRNA were not concordant between flavopiridol and com-pound 52. These discrepancies might be explained 1) by thebroad cdk-inhibitory activity of flavopiridol compared with theselective cdk2/cdk1-inhibitory activities of compound 52, 2) bythe different intracellular concentrations achieved by these in-hibitors, 3) by the distinctive molecular structures of these in-hibitors, or 4) from their putative effects on other cellular targets.
PRECLINICAL PHARMACOLOGY OF FLAVOPIRIDOL
Of the cdk inhibitors, flavopiridol has advanced the farthesttoward clinical applications.
Mechanism of Action
Flavopiridol, also known as L86-8275 or HMR 1275, is asemisynthetic flavonoid derived from rohitukine, an alkaloidisolated from a plant indigenous to India (Fig. 4). Table 1 con-tains a summary of the most important preclinicalin vitro effectsof flavopiridol. Initially, flavopiridol displayed modest activityin vitro as an inhibitor of tyrosine kinase of the epidermal growthfactor receptor and an inhibitor of protein kinase A (IC50 4 21and 122mM, respectively)(75). However, when flavopiridolwas tested in the NCI’s 60-cell-line anticancer drug screenpanel, its IC50 was 66 nM. This concentration is about 1000times lower than the concentration required to inhibit proteinkinase A and the tyrosine kinase of the epidermal growth factorreceptor(75). The antiproliferative effect was not associatedwith the presence of the epidermal growth factor receptor(75,76).Flavopiridol was shown to arrest cells in G1 phase or atthe G2/M boundary, raising the possibility that flavopiridol may
Table 1.Pharmacologic effects of flavopiridol*
Effect IC50, nM Reference Nos.
Growth inhibition, NCI DTP screen 66 (75,76)cdk inhibition 40–200 (53,54,77,78)Apoptosis 100–1000 (65,66,84–89)Cell cycle arrest 100–300 (53,67,87,89)Cyclin D1 depletion 100–300 (83)Differentiation 100–300 (72)VEGF depletion 50–100 (93)Sensitization to standard chemotherapies 100–300 (88,94,95)Epidermal growth factor receptor tyrosine
kinase inhibition21 000 (75)
Protein kinase A inhibition 122 000 (75)
*IC50 4 concentration that inhibits growth or activity by 50%; NCI4
National Cancer Institute; DTP4 Developmental Therapeutics Program; VEGF4 vascular endothelial growth factor.
380 REVIEW Journal of the National Cancer Institute, Vol. 92, No. 5, March 1, 2000
Dow
nloaded from https://academ
ic.oup.com/jnci/article/92/5/376/2606689 by guest on 23 N
ovember 2021

inhibit cdk2 and cdk1(76). Studies using purified cdks showedthat flavopiridol inhibits the activities of cdk1, cdk2, and cdk4;this inhibition is competitively blocked by ATP, with aKi of 41nM (53,54,76–78).The crystal structure of the complex of des-chloroflavopiridol and cdk2 showed that flavopiridol binds tothe ATP-binding pocket, with the benzopyran occupying thesame region as the purine ring of ATP(79). This observationwas concordant with our earlier biochemical studies with flavo-piridol (54). Flavopiridol inhibits all cdks thus far examined(IC50 4 approximately 100 nM), but it inhibits cdk7 (cdk-activating kinase) less potently (IC50 4 approximately 300 nM)(53,54,77).
In addition to directly inhibiting cdks, flavopiridol causes adecrease in the level of cyclin D1, an oncogene that is overex-pressed in many human neoplasias. Neoplasms that overexpresscyclin D1 have a poor prognosis(80–82).When MCF-7 humanbreast carcinoma cells were incubated with flavopiridol, levelsof cyclin D1 protein decreased within 3 hours(83). This effectwas followed by a decline in the levels of cyclin D3 with noalteration in the levels of cyclin D2 and cyclin E, the remainingG1 cyclins, and then 2 hours later, loss of cdk4 activity occurred.Thus, depletion of cyclin D1 appears to lead to the loss of cdk4activity (83). The depletion of cyclin D1 is caused by depletionof cyclin D1 mRNA, not by shortening the half-life of the pro-tein. Depletion of cyclin D1 mRNA was associated with a spe-cific decline in cyclin D1 promoter, measured by a luciferasereporter assay(83). The transcriptional repression of cyclin D1observed after treatment with flavopiridol is consistent with theeffects of flavopiridol on yeast cells (see above) and underscoresthe conserved effect of flavopiridol on eukaryotic cyclin tran-scription (60). In summary, flavopiridol can induce cell cyclearrest by at least three mechanisms: 1) by direct inhibition of cdkactivities by binding to the ATP-binding site; 2) by prevention ofthe phosphorylation of cdks at threonine-160/161 by inhibitionof cdk7/cyclin H (77,78); and 3) for G1-phase arrest, by a de-crease in the amount of cyclin D1, an important cofactor forcdk4 and cdk6 activation.
Another important action of flavopiridol is the selective in-duction of apoptotic cell death. Hematopoietic cell lines areoften quite sensitive to flavopiridol-induced apoptotic cell death(65,84–86),but the mechanism(s) by which flavopiridol inducesapoptosis have not yet been elucidated. Flavopiridol does notmodulate topoisomerase I/II activity(65). In certain hematopoi-etic cell lines, neither BCL-2/BAX nor p53 appeared to be af-fected(65,85),whereas, in other systems, BCL-2 may be inhib-ited (86). It is unclear whether the putative flavopiridol-inducedinhibition of cdk activity is required for induction of apoptosis.
Cell cycle arrest, but again with clear evidence of apoptoticcell death, was observed with a panel of head and neck squa-mous cell carcinoma cell lines, including a cell line (HN30) thatis refractory to several DNA-damaging agents, such asg-irra-diation and bleomycin(87). The apoptotic effect was indepen-dent of p53 status and was associated with the depletion ofcyclin D1 (87). These findings have been corroborated in otherpreclinical models(88–90).Efforts to understand flavopiridol-induced apoptosis are under way.
To determine whether flavopiridol has antiangiogenic prop-erties, Brusselbach et al.(91) incubated human umbilical veinendothelial cells (HUVECs) with flavopiridol and observedapoptotic cell death even in cells that were not cycling. In an-other report, Kerr et al.(92) tested flavopiridol in anin vivo
angiogenesis model and found that flavopiridol decreased bloodvessel formation in the mouse Matrigel model of angiogenesis.Melillo et al. (93) demonstrated that, at low nanomolar concen-trations, flavopiridol blunted the induction of vascular endothe-lial growth factor (VEGF) by hypoxia in human monocytes. Thiseffect was caused by a decreased stability of VEGF mRNA,which paralleled the decline in VEGF protein. Thus, the antitu-mor activity of flavopiridol may be supplemented by antiangio-genic effects. Whether the various antiangiogenic actions of fla-vopiridol result from its interaction with a cdk target or othertargets requires further study.
To test for synergistic effects with other compounds, cyto-toxic assays of flavopiridol in combination with standard che-motherapeutic agents were performed(94,95). Synergistic ef-fects in A549 lung carcinoma cells were demonstrated whentreatment with flavopiridol followed treatment with paclitaxel,cytarabine, topotecan, doxorubicin, or etoposide. In contrast, asynergistic effect was observed with 5-fluorouracil only whencells were treated with flavopiridol for 24 hours before additionof 5-fluorouracil. Synergistic effects with cisplatin were notschedule dependent(95). However, Chien et al.(88) failed todemonstrate a synergistic effect between flavopiridol and cis-platin and/org-irradiation in bladder carcinoma models.
Several questions about the antiproliferative activity of fla-vopiridol remain unanswered. Why does treatment with flavo-piridol cause some cells to arrest at the G1/S-phase boundary andother cells to arrest at the G2/M-phase boundary? What role doesthe depletion of D-type cyclins play in flavopiridol-induced G1/Sarrest? What is the relationship between cdk inhibition andapoptotic cell death, and what are the targets for flavopiridol-induced apoptotic cell death?
Antitumor Effect in Preclinical Models
Experiments using colorectal (Colo205) and prostate (LnCap/DU-145) carcinoma xenograft models in which flavopiridol wasadministered frequently over a protracted period demonstratedthat flavopiridol is cytostatic(75). This demonstration led toclinical trials in humans of flavopiridol administered as a 72-hour continuous infusion every 2 weeks(96) (see below).
Subsequent studies in some models of human leukemia/lymphoma xenografts demonstrated that flavopiridol adminis-tered intravenously as a bolus rendered animals tumor free,whereas flavopiridol administered as an infusion only delayedtumor growth(84). In head and neck (HN-12) xenografts, whenflavopiridol was administered as an intraperitoneal bolus daily at5 mg/kg for 5 days, a substantial growth delay was observed(87). Again, apoptotic cell death and cyclin D1 depletion wereobserved in tissues from xenografts treated with flavopiridol atpeak plasma concentrations of 5–8mM (84). Based on theseresults, the feasibility of a phase I trial with the administration offlavopiridol as a 1-hour infusion is currently being explored atthe NCI (see below).
Preclinical Pharmacokinetics and Toxicology
Murine plasma concentration–time profiles for flavopiridolexhibited biexponential behavior with meana andb half-livesof 16.4 and 201.0 minutes, respectively. The mean total-bodyplasma clearance was 22.6 mL/minute per kg, and the mean oralbioavailability after bolus intragavage was approximately 20%.Pharmacokinetic studies in dogs(75) had very similar results.
The metabolism of flavopiridol was investigated in isolated
Journal of the National Cancer Institute, Vol. 92, No. 5, March 1, 2000 REVIEW 381
Dow
nloaded from https://academ
ic.oup.com/jnci/article/92/5/376/2606689 by guest on 23 N
ovember 2021

liver perfusion models. Flavopiridol was glucuronidated in theliver, and then this flavopiridol metabolite excreted in the biliarytract. This property underlies flavopiridol’s propensity to un-dergo enterohepatic circulation(97,98). Preclinical pharmaco-logic and toxicologic evaluations have identified dose-limitingtoxic effects as reversible hematopoietic and gastrointestinal ef-fects.
HUMAN CLINICAL TRIALS OF FLAVOPIRIDOL
Two clinical trials of flavopiridol given as a 72-hour continu-ous infusion every 2 weeks have been completed(96,99).In theNCI phase I trial of infusional flavopiridol, 76 patients weretreated. Dose-limiting toxicity was secretory diarrhea with amaximal tolerated dose of 50 mg/m2 per day for 3 days.
In the presence of antidiarrheal prophylaxis (a combination ofcholestyramine and loperamide), patients tolerated higher doses,defining a second maximal tolerated dose, 78 mg/m2 per day for3 days. The dose-limiting toxicity observed at the higher doselevel was reversible hypotension and a substantial proinflamma-tory syndrome (fever, fatigue, local tumor pain, and modulationof acute-phase reactants)(96).
Tumors in one patient with non-Hodgkin’s lymphoma, onepatient with colon cancer, and one patient with kidney cancerdecreased in size (minor responses4 shrinkage of <50%) formore than 6 months. Moreover, one patient with refractory renalcancer achieved a partial response (shrinkage of >50% ofmasses)(96).Of 14 patients who received flavopiridol for morethan 6 months, five patients received flavopiridol for more than1 year and one patient received flavopiridol for more than 2years (96). This potential “disease stabilization,” which mayhave been noted in this trial, is consistent with preclinical mod-els, where tumor stasis is observed. Appropriate measurementsof cytostatic effects are necessary to confirm that cdk inhibitionmight be related to this clinical outcome. Plasma concentrationsof 300–500 nM flavopiridol, which inhibit cdk activityin vitro,were safely achieved during our trial(96).
In a complementary phase I trial also exploring the use of a72-hour continuous infusion of flavopiridol every 2 weeks,Thomas et al.(99) found that the dose-limiting toxicity is diar-rhea, corroborating the experience of the NCI. Moreover, plasmaconcentrations of 300–500 nM flavopiridol were also observed.It is interesting that there was one patient in this trial withrefractory gastric cancer that had metastasized to the liver whowas initially treated surgically and subsequently failed to re-spond to one treatment regimen of 5-fluorouracil. When treatedwith flavopiridol, this patient achieved a sustained complete re-sponse without any evidence of disease for more than 2 yearsafter treatment was completed.
In September 1998, we began the first phase I trial of a daily1-hour infusion of flavopiridol for 5 consecutive days every 3weeks. This dose schedule was based on our antitumor resultsobserved in leukemia/lymphoma and head and neck xenograftstreated with flavopiridol (see above). At this time, 27 patientshave been treated in this phase I trial. The recommended phaseII dose is 37.5 mg/m2 per day for 5 consecutive days. Dose-limiting toxic effects observed at 52.5 mg/m2 per day are nausea/vomiting, neutropenia, fatigue, and diarrhea. Other (non-dose-limiting) side effects are “local tumor pain” and anorexia(Senderowicz AM: unpublished results). To reach higher flavo-piridol concentrations, the protocol was amended to administerflavopiridol for 3 days only. Higher peak plasma flavopiridol
concentrations (approximately 4mM) may be obtained with thisschedule (Senderowicz AM: unpublished results).
A phase I trial testing the combination of paclitaxel and fla-vopiridol demonstrated good tolerability with a dose-limitingpulmonary toxicity(100).
Phase II trials of flavopiridol given as a 72-hour continuousinfusion to patients with chronic lymphocytic leukemia, non-small-cell lung cancer, non-Hodgkin’s lymphoma, or colon,prostate, gastric, head and neck, or kidney cancer, etc., and phaseI trials of flavopiridol administered on novel schedules and incombination with standard chemotherapeutic agents are beingexplored(101–104).
Several important clinical questions remain to be answered inthese trials. Is flavopiridol an “effective” anticancer agent?Which is the best schedule for flavopiridol monotherapy? Whatis the best method to combine flavopiridol and other agents?Which is the most reliable pharmacodynamic parameter to fol-low in patients? How should “stable disease” be defined in phaseII trials?
PRECLINICAL PHARMACOLOGY OF UCN-01
Staurosporine is a nonspecific protein kinase inhibitor thatarrests cell cycle progression in transformed and nontransformedcells at 1–100 nM (105). At similar concentrations, staurospo-rine inhibits many protein and tyrosine kinases(105). Severalanalogues of staurosporine have been evaluated to identify com-pounds with greater specificity for protein kinases.
Mechanism of Action
One staurosporine analogue, UCN-01 (7-hydroxystaurospo-rine; Fig. 4), has potent activity against several protein kinase Cisoenzymes, particularly the Ca2+-dependent protein kinase Cwith an IC50 of about 30 nM. UCN-01 has lower potency againstthe novel Ca2+-independent protein kinases C (IC50 4 approxi-mately 500 nM) and no effect against the atypical protein kinasesC (106–108),similar to the activity of staurosporine. In additionto its effects on protein kinase C, UCN-01 has antiproliferativeactivity in several human tumor cell lines(109–113).In contrast,another highly selective potent protein kinase C inhibitor, GF109203X, has a modest antiproliferative activity, despite a simi-lar capacity to inhibit protein kinase Cin vitro (110).Thus, theseresults suggest that the antiproliferative activity of UCN-01 isprobably not explained solely by inhibition of protein kinase C.UCN-01 moderately inhibited the activity of immunoprecipi-tated cdk1 (cdc2) and cdk2 (IC50 4 300–600 nM). However,when intact cells were exposed to UCN-01, “inappropriate ac-tivation” of the same kinases occurred(110).
Experimental evidence suggests that DNA damage leads tocell cycle arrest to allow DNA repair. In cells where the G1-phase checkpoint is not active because of p53 inactivation, ir-radiated cells accumulate in G2 phase because the G2 checkpointis mediated by the inactivation of cyclin B/cdc2 by wee1 kinase(Fig. 5, A). In contrast, UCN-01 (Fig. 5, B) induces the activa-tion of cdc2/cyclin B and thus promotes cells to enter earlymitosis with the onset of apoptotic cell death. These effectscould be partially explained by the inactivation of wee1, thekinase that negatively regulates the G2/M-phase transition oractivation of cdc25 phosphatase(114).Thus, although UCN-01at high concentrations can directly inhibit cdksin vitro, UCN-01can modulate cellular upstream regulators at much lower con-centration, leading to inappropriate cdc2 activation by acting on
382 REVIEW Journal of the National Cancer Institute, Vol. 92, No. 5, March 1, 2000
Dow
nloaded from https://academ
ic.oup.com/jnci/article/92/5/376/2606689 by guest on 23 N
ovember 2021

targets that remain to be defined. DNA-damaging agents notonly can activate the G2-phase checkpoint but also can activatethe S-phase checkpoint(115).Studies from other groups suggestthat UCN-01 not only is able to abrogate the G2 checkpointinduced by DNA-damaging agents but also, in some circum-stances, is able to abrogate the DNA damage-induced S-phasecheckpoint(115).
Another pharmacologic feature of UCN-01 is the increasedcytotoxicity in cells containing mutated p53 genes(51). In CA-46 Burkitt’s lymphoma and HT-29 colon carcinoma cell linescarrying mutated p53 genes, cytotoxicity results when these cellsare exposed to UCN-01. Compared with the isogenic wild-typeMCF-7 cell line, the MCF-7 cell line with no endogenous p53because of the ectopic expression of E6, a human papillomavirustype-16 protein, showed enhanced cytotoxicity when treatedwith a DNA-damaging agent, such as cisplatin, and UCN-01.The synergistic effect of UCN-01 is enhanced with many che-motherapeutic agents, including mitomycin C, 5-fluorouracil,carmustine, and camptothecin, among others(116–122).There-fore, it is possible that combining UCN-01 with these and otheragents could improve its therapeutic index.
Another interesting aspect of UCN-01 is its ability to arrestcells in G1 phase of the cell cycle(109,112,113,123–125).Hu-man epidermoid carcinoma A431 cells contain mutated p53.When incubated with UCN-01, these cells were arrested in G1
phase, Rb was hypophosphorylated, and p21waf1 and p27kip1
accumulated(113). However, in another report(125), the anti-proliferative effect of UCN-01 was not dependent on the func-tional status of Rb. Thus, the G1 arrest observed with UCN-01 isapparently independent of the status of p53 and Rb. Furtherstudies on the putative target(s) for UCN-01 in the G1-phasearrest of cells are under way.
Courage et al.(126) found that UCN-01-resistant A549 lungcarcinoma cells were sensitive to two other protein kinase Cinhibitors, CGP 41251 and Ro 31-8220, and were marginallyresistant (about twofold) to etoposide. These UCN-01-resistantcells had lost several protein kinase C isoenzymes, protein ki-nase Ca, «, and u. However, the levels of these isoenzymesreturned to baseline when these resistant cells were cultured in
UCN-01-free medium. Thus, as described above, it is unlikelythat the protein kinase C family of signaling proteins is the onlytarget for UCN-01 cytotoxicity(110,126).
Although many important questions have been answered,several questions remain. What is the real target for UCN-01 inG1/S phase-arrested cells? Does protein kinase C play any rolein UCN-01-induced cell cycle arrest and/or apoptotic cell death?What is the real target for the G2 checkpoint abrogation?
Spectrum of In Vivo Antitumor Activity
UCN-01 administered by an intravenous or intraperitonealroute displayed antitumor activity in xenograft model systemswith breast carcinoma (MCF-7), renal carcinoma (A498), andleukemia (MOLT-4 and HL-60) cell lines (Senderowicz AM:unpublished results). The antitumor effect was greater whenUCN-01 was given over a longer period. This requirement for alonger period of treatment was also observed inin vitro models,with highest antitumor activity observed when UCN-01 waspresent for 72 hours(109).Thus, a clinical trial using a 72-hourcontinuous infusion every 2 weeks was conducted (describedbelow).
Preclinical Pharmacokinetics and Toxicology
Pharmacokinetics and toxicologic studies using severalschedules were done in rats and dogs. When beagle dogs weregiven a 72-hour continuous infusion of UCN-01, local (site ofinjection) and gastrointestinal toxic effects were dose-limitingand a steady-state plasma concentration of 330 nM UCN-01 wasachieved. Pharmacokinetic parameters were a volume of distri-bution (6.09 L/kg), a total clearance of 0.6 L/kg per hour with ab half-life of about 12 hours(127).
PHASE I CLINICAL TRIALS OF UCN-01
We recently completed the first phase I trial of UCN-01 inhumans(128). Clinical features of UCN-01 observed includedthe unexpectedly long half-life (approximately 30 days). Thishalf-life was 100 times longer than the half-life observed inpreclinical models, which was probably caused by the avid bind-
Fig. 5. Abrogation (overriding) of the G2-phase checkpoint byUCN-01. Cells with wild-type p53 arrest in G1 phase.A) Afterexposure to genotoxic stress (i.e.,g-irradiation), tumor cell lineswith mutated p53 arrest in G2 phase of the cell cycle by inhi-bition of cyclin B/cdc2.B) After exposure to genotoxic stress(i.e.,g-irradiation), tumor cell lines with mutated p53 incubatedwith UCN-01 do not arrest in G2 phase and progress to M phaseas a result of inappropriate activation of cyclin B/cdc2, leadingto premature mitosis and apoptosis.
Journal of the National Cancer Institute, Vol. 92, No. 5, March 1, 2000 REVIEW 383
Dow
nloaded from https://academ
ic.oup.com/jnci/article/92/5/376/2606689 by guest on 23 N
ovember 2021

ing of UCN-01 toa1-acid glycoprotein(129,130).Other clinicalfeatures were the relative lack of myelotoxicity or gastrointes-tinal toxicity (prominent side effects observed in animal mod-els), despite very high plasma concentrations achieved (35–50mM) (128–131).Dose-limiting toxic effects were nausea/vomiting (amenable to standard antiemetic treatments), symp-tomatic hyperglycemia associated with an insulin-resistancestate, and pulmonary toxicity characterized by substantial hyp-oxemia without obvious radiologic changes. The recommendedphase II dose of UCN-01 given on a 72-hour continuous infusionschedule was 42.5 mg/m2 per day(131). One patient with re-fractory metastatic melanoma developed a partial response thatlasted 8 months. Tumors in a few patients with leiomyosarcoma,non-Hodgkin’s lymphoma, or lung cancer were stabilized (ù6months) (131). The concentration of “free” UCN-01 was as-sessed in saliva. At the recommended phase II dose, concentra-tions of free UCN-01 that may cause G2 checkpoint abrogationcan be achieved. Table 2 compares important clinical and phar-macologic features of the cdk modulators flavopiridol and UCN-01. Future trials are being explored in which UCN-01 would begiven by infusion for shorter periods(132) and/or in combina-tion with DNA-damaging agents.
SUMMARY
With knowledge of the role of cdks in cell cycle regulationand the discovery that approximately 90% of all neoplasias areassociated with “cdk hyperactivation” leading to the inactivationof the Rb pathway(2), novel cdk inhibitors are being developed.The first two modulators of cdk function tested in clinical trials,flavopiridol and UCN-01, have been observed to reach plasmaconcentrations that can modulate cdk-related functions. Futureclinical trials should determine what is the best schedule foradministering chemical cdk inhibitors, should determine what isthe best combination of chemical cdk inhibitors and standardchemotherapeutic agents, and should demonstrate cdk modula-tion in tumor samples from patients treated with cdk inhibitors.
REFERENCES
(1) Sherr CJ. Cancer cell cycles. Science 1996;274:1672–7.(2) Hatakeyama M, Weinberg RA. The role of RB in cell cycle control. Prog
Cell Cycle Res 1995;1:9–19.(3) Morgan DO, Fisher RP, Espinoza FH, Farrell A, Nourse J, Chamberlin H,
et al. Control of eukaryotic cell cycle progression by phosphorylation ofcyclin-dependent kinases. Cancer J Sci Am 1998;4 Suppl 1:S77–83.
(4) Morgan DO. Cyclin-dependent kinases: engines, clocks, and micropro-cessors. Annu Rev Cell Dev Biol 1997;13:261–91.
(5) Rickert P, Seghezzi W, Shanahan F, Cho H, Lees E. Cyclin C/CDK8 is anovel CTD kinase associated with RNA polymerase II. Oncogene 1996;12:2631–40.
(6) Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates itshigh-affinity, loop-specific binding to TAR RNA. Cell 1998;92:451–62.
(7) Grana X, De Luca A, Sang N, Fu Y, Claudio PP, Rosenblatt J, et al.PITALRE, a nuclear CDC2-related protein kinase that phosphorylates theretinoblastoma proteinin vitro. Proc Natl Acad Sci U S A 1994;91:3834–8.
(8) MacLachlan TK, Sang N, Giordano A. Cyclins, cyclin-dependent kinasesand cdk inhibitors: implications in cell cycle control and cancer. Crit RevEukaryot Gene Expr 1995;5:127–56.
(9) Edwards MC, Wong C, Elledge SJ. Human cyclin K, a novel RNA poly-merase II-associated cyclin possessing both carboxy-terminal domain ki-nase and Cdk-activating kinase activity. Mol Cell Biol 1998;18:4291–300.
(10) Peng J, Zhu Y, Milton JT, Price DH. Identification of multiple cyclinsubunits of human P-TEFb. Genes Dev 1998;12:755–62.
(11) Kaldis P, Russo AA, Chou HS, Pavletich NP, Solomon MJ. Human andyeast Cdk-activating kinases (CAKs) display distinct substrate specifici-ties. Mol Biol Cell 1998;9:2545–60.
(12) Tassan JP, Schultz SJ, Bartek J, Nigg EA. Cell cycle analysis of theactivity, subcellular localization, and subunit composition of human CAK(CDK-activating kinase). J Cell Biol 1994;127:467–78.
(13) Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators ofG1-phase progression. Genes Dev 1999;13:1501–12.
(14) LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, ChouHS, et al. New functional activities for the p21 family of CDK inhibitors.Genes Dev 1997;11:847–62.
(15) Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, et al.The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators ofcyclin D-dependent kinases in murine fibroblasts. EMBO J 1999;18:1571–83.
(16) Bates S, Ryan KM, Phillips AC, Vousden KH. Cell cycle arrest and DNAendoreduplication following p21Waf1/Cip1 expression. Oncogene 1998;17:1691–703.
(17) Dulic V, Stein GH, Far DF, Reed SI. Nuclear accumulation of p21Cip1 atthe onset of mitosis: a role at the G2/M-phase transition. Mol Cell Biol1998;18:546–57.
(18) Niculescu AB 3rd, Chen X, Smeets M, Hengst L, Prives C, Reed SI.Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycletransitions: pRb is a critical determinant in blocking DNA replication andin preventing endoreduplication [published erratum appears in Mol CellBiol 1998;18:1763]. Mol Cell Biol 1998;18:629–43.
(19) Zhang HS, Postigo AA, Dean DC. Active transcriptional repression by theRb-E2F complex mediates G1 arrest triggered by p16INK4a, TGFbeta,and contact inhibition. Cell 1999;97:53–61.
Table 2.Phase I trials with cdk modulators
Flavopiridol (96) UCN-01 (131)
Schedule 72-h continuous infusion every 2 wk 72-h continuous infusion every 4 wk (cycle 1) followedby 36-h continuous infusion every 4 wk (cycles 2 orhigher)
Dose-limiting toxicity (maximaltolerated dose)
Diarrhea (50 mg/m2 per day for 3 days)Hypotension (78 mg/m2 per day for 3 days)
Nausea/vomiting, hyperglycemia, and hypoxemia (42.5mg/m2 per day for 3 days)
Other toxic effects Anorexia, proinflammatory syndrome Headache, myalgias
Suggestion of activity Non-Hodgkin’s lymphoma and renal, colon,gastric, or prostate cancer
Melanoma, non-Hodgkin’s lymphoma, or leiomyosarcoma
Median plasma concentration atmaximal-tolerated dose
271 nM (50 mg/m2 per day for 3 days)344 nM (78 mg/m2 per day for 3 days)
Total 4 36.4 mM (42.5 mg/m2 per day for 3 days)Free* 4 111 nM (42.5 mg/m2 per day for 3 days)
Plasma half-life, h 11.6 588
*Free 4 concentration of UCN-01 in saliva.
384 REVIEW Journal of the National Cancer Institute, Vol. 92, No. 5, March 1, 2000
Dow
nloaded from https://academ
ic.oup.com/jnci/article/92/5/376/2606689 by guest on 23 N
ovember 2021

(20) Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev1998;12:2245–62.
(21) DelSal G, Loda M, Pagano M. Cell cycle and cancer: critical events at theG1 restriction point. Crit Rev Oncog 1996;7:127–42.
(22) Grana X, Reddy EP. Cell cycle control in mammalian cells: role of cyc-lins, cyclin dependent kinases (CDKs), growth suppressor genes and cy-clin-dependent kinase inhibitors (CKIs). Oncogene 1995;11:211–9.
(23) Pardee AB. Multiple molecular levels of cell cycle regulation. J CellBiochem 1994;54:375–8.
(24) Pines J. Cyclins and cyclin-dependent kinases: theme and variations. AdvCancer Res 1995;66:181–212.
(25) Sausville EA, Zaharevitz D, Gussio R, Meijer L, Louarn-Leost M, KunickC, et al. Cyclin-dependent kinases: initial approaches to exploit a noveltherapeutic target. Pharmacol Ther 1999;82:285–92.
(26) Tsihlias J, Kapusta L, Slingerland J. The prognostic significance of alteredcyclin-dependent kinase inhibitors in human cancer. Annu Rev Med 1999;50:401–23.
(27) Meijer L, Kim SH. Chemical inhibitors of cyclin-dependent kinases.Methods Enzymol 1997;283:113–28.
(28) Zaharevitz DW, Gussio R, Leost M, Senderowicz AM, Lahusen T, Ku-nick C, et al. Discovery and initial characterization of the paullones, anovel class of small-molecule inhibitors of cyclin-dependent kinases. Can-cer Res 1999;59:2566–9.
(29) De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M, Kim SH.Inhibition of cyclin-dependent kinases by purine analogues: crystal struc-ture of human cdk2 complexed with roscovitine. Eur J Biochem 1997;243:518–26.
(30) Jin X, Nguyen D, Zhang WW, Kyritsis AP, Roth JA. Cell cycle arrest andinhibition of tumor cell proliferation by the p16INK4 gene mediated by anadenovirus vector. Cancer Res 1995;55:3250–3.
(31) Chintala SK, Fueyo J, Gomez-Manzano C, Venkaiah B, Bjerkvig R, YungWK, et al. Adenovirus-mediated p16/CDKN2 gene transfer suppressesglioma invasionin vitro. Oncogene 1997;15:2049–57.
(32) Sandig V, Brand K, Herwig S, Lukas J, Bartek J, Strauss M. Adenovirallytransferred p16INK4/CDKN2 and p53 genes cooperate to induce apop-totic tumor cell death. Nat Med 1997;3:313–9.
(33) Craig C, Wersto R, Kim M, Ohri E, Li Z, Katayose D, et al. A recom-binant adenovirus expressing p27Kip1 induces cell cycle arrest and loss ofcyclin-Cdk activity in human breast cancer cells. Oncogene 1997;14:2283–9.
(34) Eastham JA, Hall SJ, Sehgal I, Wang J, Timme TL, Yang G, et al.In vivogene therapy with p53 or p21 adenovirus for prostate cancer. Cancer Res1995;55:5151–5.
(35) Katayose Y, Kim M, Rakkar AN, Li Z, Cowan KH, Seth P. Promotingapoptosis: a novel activity associated with the cyclin-dependent kinaseinhibitor p27. Cancer Res 1997;57:5441–5.
(36) Wang X, Gorospe M, Huang Y, Holbrook NJ. p27Kip1 overexpres-sion causes apoptotic death of mammalian cells. Oncogene 1997;15:2991–7.
(37) Lee SJ, Ha MJ, Lee J, Nguyen P, Choi YH, Pirnia F, et al. Inhibition ofthe 3-hydroxy-3-methylglutaryl-coenzyme A reductase pathway inducesp53-independent transcriptional regulation of p21(WAF1/CIP1) in humanprostate carcinoma cells. J Biol Chem 1998;273:10618–23.
(38) Gray-Bablin J, Rao S, Keyomarsi K. Lovastatin induction of cyclin-dependent kinase inhibitors in human breast cells occurs in a cell cycle-independent fashion. Cancer Res 1997;57:604–9.
(39) Hashemolhosseini S, Nagamine Y, Morley SJ, Desrivieres S, Mercep L,Ferrari S. Rapamycin inhibition of the G1 to S transition is mediated byeffects on cyclin D1 mRNA and protein stability. J Biol Chem 1998;273:14424–9.
(40) Fahraeus R, Paramio JM, Ball KL, Lain S, Lane DP. Inhibition of pRbphosphorylation and cell-cycle progression by a 20-residue peptide de-rived from p16CDKN2/INK4A. Curr Biol 1996;6:84–91.
(41) Ball KL, Lain S, Fahraeus R, Smythe C, Lane DP. Cell-cycle arrest andinhibition of Cdk4 activity by small peptides based on the carboxy-terminal domain of p21WAF1. Curr Biol 1997;7:71–80.
(42) Bonfanti M, Taverna S, Salmona M, D’Incalci M, Broggini M. p21WAF1-derived peptides linked to an internalization peptide inhibit human cancer cellgrowth. Cancer Res 1997;57:1442–6.
(43) Colas P, Cohen B, Jessen T, Grishina I, McCoy J, Brent R. Genetic
selection of peptide aptamers that recognize and inhibit cyclin-dependentkinase 2. Nature 1996;380:548–50.
(44) Chen YN, Sharma SK, Ramsey TM, Jiang L, Martin MS, Baker K, et al.Selective killing of transformed cells by cyclin/cyclin-dependent kinase 2antagonists. Proc Natl Acad Sci U S A 1999;96:4325–9.
(45) Kornmann M, Arber N, Korc M. Inhibition of basal and mitogen-stimulated pancreatic cancer cell growth by cyclin D1 antisense is asso-ciated with loss of tumorigenicity and potentiation of cytotoxicity to cis-platinum. J Clin Invest 1998;101:344–52.
(46) Wang MB, Billings KR, Venkatesan N, Hall FL, Srivatsan ES. Inhibitionof cell proliferation in head and neck squamous cell carcinoma cell lineswith antisense cyclin D1. Otolaryngol Head Neck Surg 1998;119:593–9.
(47) Driscoll B, Wu L, Buckley S, Hall FL, Anderson KD, Warburton D.Cyclin D1 antisense RNA destabilizes pRb and retards lung cancer cellgrowth. Am J Physiol 1997;273:L941–9.
(48) Watts CK, Sweeney KJ, Warlters A, Musgrove EA, Sutherland RL. An-tiestrogen regulation of cell cycle progression and cyclin D1 gene expres-sion in MCF-7 human breast cancer cells. Breast Cancer Res Treat 1994;31:95–105.
(49) Zhou Q, Stetler-Stevenson M, Steeg PS. Inhibition of cyclin D expressionin human breast carcinoma cells by retinoidsin vitro. Oncogene 1997;15:107–15.
(50) Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, LazarusDD, et al. Proteasome inhibitors: a novel class of potent and effectiveantitumor agents. Cancer Res 1999;59:2615–22.
(51) Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, O’Connor P.UCN-01: a potent abrogator of G2 checkpoint function in cancer cellswith disrupted p53. J Natl Cancer Inst 1996;88:956–65.
(52) Roberge M, Berlinck RG, Xu L, Anderson HJ, Lim LY, Curman D, et al.High-throughput assay for G2 checkpoint inhibitors and identification ofthe structurally novel compound isogranulatimide. Cancer Res 1998;58:5701–6.
(53) Carlson BA, Dubay MM, Sausville EA, Brizuela L, Worland PJ. Flavo-piridol induces G1 arrest with inhibition of cyclin-dependent kinase(CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res 1996;56:2973–8.
(54) Losiewicz MD, Carlson BA, Kaur G, Sausville EA, Worland PJ. Potentinhibition of CDC2 kinase activity by the flavonoid L86-8275. BiochemBiophys Res Commun 1994;201:589–95.
(55) Singh SS, Sausville EA, Senderowicz AM. Cyclin D1 and Cdk6 are thetargets for flavopiridol-mediated G1 block in MCF10A breast epithelialcell line [abstract]. Proc Am Assoc Cancer Res 1999;40:28.
(56) Meijer L, Pondaven P. Cyclic activation of histone H1 kinase during seaurchin egg mitotic divisions. Exp Cell Res 1988;174:116–29.
(57) Rialet V, Meijer L. A new screening test for antimitotic compounds usingthe universal M phase-specific protein kinase, p34cdc2/cyclin Bcdc13,affinity-immobilized on p13suc1-coated microtitration plates. AnticancerRes 1991;11:1581–90.
(58) Schulze-Gahmen U, Brandsen J, Jones HD, Morgan DO, Meijer L, VeselyJ, et al. Multiple modes of ligand recognition: crystal structures of cyclin-dependent protein kinase 2 in complex with ATP and two inhibitors,olomoucine and isopentenyladenine. Proteins 1995;22:378–91.
(59) Brooks EE, Gray NS, Joly A, Kerwar SS, Lum R, Mackman RL, et al.CVT-313, a specific and potent inhibitor of CDK2 that prevents neointi-mal proliferation. J Biol Chem 1997;272:29207–11.
(60) Gray NS, Wodicka L, Thunnissen AM, Norman TC, Kwon S, EspinozaFH, et al. Exploiting chemical libraries, structure, and genomics in thesearch for kinase inhibitors. Science 1998;281:533–8.
(61) Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, RubinsteinL, et al. Display and analysis of patterns of differential activity ofdrugs against human tumor cell lines: development of mean graph andCOMPARE algorithm. J Natl Cancer Inst 1989;81:1088–92.
(62) van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinasesin cell cycle control. Science 1993;262:2050–4.
(63) Buquet-Fagot C, Lallemand F, Montagne MN, Mester J. Effects of olo-mucine, a selective inhibitor of cyclin-dependent kinases, on cell cycleprogression in human cancer cell lines. Anticancer Drugs 1997;8:623–31.
(64) Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, et al.
Journal of the National Cancer Institute, Vol. 92, No. 5, March 1, 2000 REVIEW 385
Dow
nloaded from https://academ
ic.oup.com/jnci/article/92/5/376/2606689 by guest on 23 N
ovember 2021

Biochemical and cellular effects of roscovitine, a potent and selectiveinhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur JBiochem 1997;243:527–36.
(65) Parker BW, Kaur G, Nieves-Neira W, Taimi M, Kolhagen G, Shimizu T,et al. Early induction of apoptosis in hematopoietic cell lines after expo-sure to flavopiridol. Blood 1998;91:458–65.
(66) Park DS, Farinelli SE, Greene LA. Inhibitors of cyclin-dependent kinasespromote survival of post-mitotic neuronally differentiated PC12 cells andsympathetic neurons. J Biol Chem 1996;271:8161–9.
(67) Park DS, Morris EJ, Greene LA, Geller HM. G1/S cell cycle blockers andinhibitors of cyclin-dependent kinases suppress camptothecin-inducedneuronal apoptosis. J Neurosci 1997;17:1256–70.
(68) Meikrantz W, Schlegel R. Suppression of apoptosis by dominant negativemutants of cyclin-dependent protein kinases. J Biol Chem 1996;271:10205–9.
(69) Gervais JL, Seth P, Zhang H. Cleavage of CDK inhibitor p21(Cip1/Waf1)by caspases is an early event during DNA damage-induced apoptosis. JBiol Chem 1998;273:19207–12.
(70) Levkau B, Koyama H, Raines EW, Clurman BE, Herren B, Orth K, et al.Cleavage of p21Cip1/Waf1 and p27Kip1 mediates apoptosis in endothe-lial cells through activation of Cdk2: role of a caspase cascade. Mol Cell1998;1:553–63.
(71) Zhou BB, Li H, Yuan J, Kirschner MW. Caspase-dependent activation ofcyclin-dependent kinases during Fas-induced apoptosis in Jurkat cells.Proc Natl Acad Sci U S A 1998;95:6785–90.
(72) Lee HR, Chang TH, Tebalt MJ 3rd, Senderowicz AM, Szabo E. Inductionof differentiation accompanies inhibition of cdk2 in a non-small cell lungcancer cell line. Int J Oncol 1999;15:161–6.
(73) Rosania GR, Merlie J Jr, Gray N, Chang YT, Schultz PG, Heald R. Acyclin-dependent kinase inhibitor inducing cancer cell differentiation: bio-chemical identification using Xenopus egg extracts. Proc Natl Acad SciU S A 1999;96:4797–802.
(74) Liu M, Subramanyam YV, Baskaran N. Preparation and analysis ofcDNA from a small number of hematopoietic cells. Methods Enzymol1999;303:45–55.
(75) Sedlacek HH, Czech J, Naik R, Kaur G, Worland P, Losiewicz M, et al.Flavopiridol (L86-8275, NSC-649890), a new kinase inhibitor for tumortherapy. Int J Oncol 1996;9:1143–68.
(76) Kaur G, Stetler-Stevenson M, Sebers S, Worland P, Sedlacek H, Myers C,et al. Growth inhibition with reversible cell cycle arrest of carcinoma cellsby flavone L86–8275. J Natl Cancer Inst 1992;84:1736–40.
(77) Carlson B, Pearlstein R, Naik R, Sedlacek H, Sausville E, Worland P.Inhibition of CDK2, CDK4 and CDK7 by flavopiridol and structuralanalogs [abstract]. Proc Am Assoc Cancer Res 1996;37:424.
(78) Worland PJ, Kaur G, Stetler-Stevenson M, Sebers S, Sartor O, SausvilleEA. Alteration of the phosphorylation state of p34cdc2 kinase by theflavone L86–8275 in breast carcinoma cells. Correlation with decreasedH1 kinase activity. Biochem Pharmacol 1993;46:1831–40.
(79) De Azevedo WF Jr, Mueller-Dieckmann HJ, Schulze-Gahmen U, Wor-land PJ, Sausville E, Kim SH. Structural basis for specificity and potencyof a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc NatlAcad Sci U S A 1996;93:2735–40.
(80) Michalides R, van Veelen N, Hart A, Loftus B, Wientjens E, Balm A.Overexpression of cyclin D1 correlates with recurrence in a group offorty-seven operable squamous cell carcinomas of the head and neck.Cancer Res 1995;55:975–8.
(81) Gansauge S, Gansauge F, Ramadani M, Stobbe H, Rau B, Harada N, et al.Overexpression of cyclin D1 in human pancreatic carcinoma is associatedwith poor prognosis. Cancer Res 1997;57:1634–7.
(82) Fredersdorf S, Burns J, Milne AM, Packham G, Fallis L, Gillett CE, et al.High level expression of p27(kip1) and cyclin D1 in some human breastcancer cells: inverse correlation between the expression of p27(kip1) anddegree of malignancy in human breast and colorectal cancers. Proc NatlAcad Sci U S A 1997;94:6380–5.
(83) Carlson B, Lahusen T, Singh S, Loaiza-Perez A, Worland PJ, Pestell R,et al. Downregulation of cyclin D1 by transcriptional repression in MCF-7human breast carcinoma cells induced by flavopiridol. Cancer Res 1999;59:4634–41.
(84) Arguello F, Alexander M, Sterry JA, Tudor G, Smith EM, Kalavar NT, etal. Flavopiridol induces apoptosis of normal lymphoid cells, causes im-
munosuppression, and has potent antitumor activityin vivoagainst humanleukemia and lymphoma xenografts. Blood 1998;91:2482–90.
(85) Byrd JC, Shinn C, Waselenko JK, Fuchs EJ, Lehman TA, Nguyen PL, etal. Flavopiridol induces apoptosis in chronic lymphocytic leukemia cellsvia activation of caspase-3 without evidence of bcl-2 modulation or de-pendence on functional p53. Blood 1998;92:3804–16.
(86) Konig A, Schwartz GK, Mohammad RM, Al-Katib A, Gabrilove JL. Thenovel cyclin-dependent kinase inhibitor flavopiridol downregulates Bcl-2and induces growth arrest and apoptosis in chronic B-cell leukemia lines.Blood 1997;90:4307–12.
(87) Patel V, Senderowicz AM, Pinto D Jr, Igishi T, Raffeld M, Quintanilla-Martinez L, et al. Flavopiridol, a novel cyclin-dependent kinase inhibitor,suppresses the growth of head and neck squamous cell carcinomas byinducing apoptosis. J Clin Invest 1998;102:1674–81.
(88) Chien M, Astumian M, Liebowitz D, Rinker-Schaeffer C, Stadler W.Invitro evaluation of flavopiridol, a novel cell cycle inhibitor, in bladdercancer. Cancer Chemother Pharmacol 1999;44:81–7.
(89) Schrump DS, Matthews W, Chen GA, Mixon A, Altorki NK. Flavopiridolmediates cell cycle arrest and apoptosis in esophageal cancer cells. ClinCancer Res 1998;4:2885–90.
(90) Bible KC, Kaufmann SH. Flavopiridol: a cytotoxic flavone that inducescell death in noncycling A549 human lung carcinoma cells. Cancer Res1996;56:4856–61.
(91) Brusselbach S, Nettelbeck DM, Sedlacek HH, Muller R. Cell cycle-independent induction of apoptosis by the anti-tumor drug flavopiridol inendothelial cells. Int J Cancer 1998;77:146–52.
(92) Kerr JS, Wexler RS, Mousa SA, Robinson CS, Wexler EJ, Mohamed S,et al. Novel small molecule alpha v integrin antagonists: comparativeanti-cancer efficacy with known angiogenesis inhibitors. Anticancer Res1999;19:959–68.
(93) Melillo G, Sausville EA, Cloud K, Lahusen T, Varresio L, SenderowiczA. Flavopiridol, a protein kinase inhibitor, down-regulates hypoxic induc-tion of vascular endothelial growth factor expression in human mono-cytes. Cancer Res 1999;59:5433–7.
(94) Schwartz G, Farsi K, Maslak P, Kelsen D, Spriggs D. Potentiation ofapoptosis by flavopiridol in mitomycin-C-treated gastric and breast cancercells. Clin Cancer Res 1997;3:1467–72.
(95) Bible KC, Kaufmann SH. Cytotoxic synergy between flavopiridol (NSC649890, L86-8275) and various antineoplastic agents: the importance ofsequence of administration. Cancer Res 1997;57:3375–80.
(96) Senderowicz AM, Headlee D, Stinson SF, Lush RM, Kalil N, Villalba L,et al. Phase I trial of continuous infusion flavopiridol, a novel cyclin-dependent kinase inhibitor, in patients with refractory neoplasms. J ClinOncol 1998;16:2986–99.
(97) Jager W, Zembsch B, Wolschann P, Pittenauer E, Senderowicz AM,Sausville E, et al. Metabolism of the anticancer drug flavopiridol, a newinhibitor of cyclin dependent kinases, in rat liver. Life Sci 1998;62:1861–73.
(98) Lush R, Stinson S, Senderowicz A, Hill K, Feuer J, Headlee D, et al.Flavopiridol pharmacokinetics suggest entoerohepatic circulation. ClinPharmacol Ther 1997;61:145.
(99) Thomas J, Cleary J, Tutsch K, Arzoomanian R, Alberti D, Simon K, et al.Phase I clinical and pharmacokinetic trial of flavopiridol [abstract]. ProcAm Assoc Cancer Res 1997;38:222.
(100) Schwartz G, Kaubisch A, Saltz L, Ilson D, O’Reilly E, Barazzuol J, et al.Phase I trial of sequential paclitaxel and the cyclin-dependent kinaseinhibitor flavopiridol [abstract]. Proc ASCO 1999;18:160a.
(101) Wright J, Blatner GL, Cheson BD. Clinical trials referral resource. Clini-cal trials of flavopiridol. Oncology (Huntingt) 1998;12:1018, 23–4.
(102) Werner J, Kelsen D, Karpeh M, Inzeo D, Barazzuol J, Sugarman A, et al.The cyclin-dependent kinase inhibitor flavopiridol is an active and unex-pectedly toxic agent in advanced gastric cancer [abstract]. Proc ASCO1998;17:234a.
(103) Shapiro G, Patterson A, Lynch C, Lucca J, Anderson I, Boral A, et al. Aphase II trial of flavopiridol in patients with stage IV non-small cell lungcancer [abstract]. Proc ASCO 1999;18:522a.
(104) Bennett S, Mani S, O’Reilly S, Wright J, Schilsky R, Vokes E, et al. PhaseII trial of flavopiridol in metastatic colorectal cancer: preliminary results[abstract]. Proc ASCO 1999;18:277a.
(105) Tamaoki T. Use and specificity of staurosporine, UCN-01, and calphostinC as protein kinase inhibitors. Methods Enzymol 1991;201:340–7.
386 REVIEW Journal of the National Cancer Institute, Vol. 92, No. 5, March 1, 2000
Dow
nloaded from https://academ
ic.oup.com/jnci/article/92/5/376/2606689 by guest on 23 N
ovember 2021

(106) Takahashi I, Kobayashi E, Asano K, Yoshida M, Nakano H. UCN-01, aselective inhibitor of protein kinase C fromStreptomyces.J Antibiot (To-kyo) 1987;40:1782–4.
(107) Takahashi I, Saitoh Y, Yoshida M, Sano H, Nakano H, Morimoto M, etal. UCN-01 and UCN-02, new selective inhibitors of protein kinase C. II.Purification, physico-chemical properties, structural determination andbiological activities. J Antibiot (Tokyo) 1989;42:571–6.
(108) Seynaeve CM, Kazanietz MG, Blumberg PM, Sausville EA, Worland PJ.Differential inhibition of protein kinase C isozymes by UCN-01, a stau-rosporine analogue. Mol Pharmacol 1994;45:1207–14.
(109) Seynaeve CM, Stetler-Stevenson M, Sebers S, Kaur G, Sausville EA,Worland PJ. Cell cycle arrest and growth inhibition by the protein kinaseantagonist UCN-01 in human breast carcinoma cells. Cancer Res 1993;53:2081–6.
(110) Wang Q, Worland PJ, Clark JL, Carlson BA, Sausville EA. Apoptosis in7-hydroxystaurosporine-treated T lymphoblasts correlates with activationof cyclin-dependent kinases 1 and 2. Cell Growth Differ 1995;6:927–36.
(111) Akinaga S, Gomi K, Morimoto M, Tamaoki T, Okabe M. Antitumoractivity of UCN-01, a selective inhibitor of protein kinase C, in murineand human tumor models. Cancer Res 1991;51:4888–92.
(112) Akinaga S, Nomura K, Gomi K, Okabe M. Effect of UCN-01, a selectiveinhibitor of protein kinase C, on the cell-cycle distribution of humanepidermoid carcinoma, A431 cells. Cancer Chemother Pharmacol 1994;33:273–80.
(113) Akiyama T, Yoshida T, Tsujita T, Shimizu M, Mizukami T, Okabe M, etal. G1 phase accumulation induced by UCN-01 is associated with dephos-phorylation of Rb and CDK2 proteins as well as induction of CDK in-hibitor p21/Cip1/WAF1/Sdi1 in p53-mutated human epidermoid carci-noma A431 cells. Cancer Res 1997;57:1495–501.
(114) Yu L, Orlandi L, Wang P, Orr M, Senderowicz AM, Sausville EA, et al.UCN-01 abrogates G2 arrest through a cdc2-dependent pathway that isassociated with inactivation of the Wee1Hu kinase and activation of theCdc25C phosphatase. J Biol Chem 1998;273:33455–64.
(115) Bunch RT, Eastman A. 7-Hydroxystaurosporine (UCN-01) causes redis-tribution of proliferating cell nuclear antigen and abrogates cisplatin-induced S-phase arrest in Chinese hamster ovary cells. Cell Growth Differ1997;8:779–88.
(116) Akinaga S, Nomura K, Gomi K, Okabe M. Enhancement of antitumoractivity of mitomycin C in vitro and in vivo by UCN-01, a selectiveinhibitor of protein kinase C. Cancer Chemother Pharmacol 1993;32:183–9.
(117) Bunch RT, Eastman A. Enhancement of cisplatin-induced cytotoxicity by7-hydroxystaurosporine (UCN-01), a new G2-checkpoint inhibitor. ClinCancer Res 1996;2:791–7.
(118) Hsueh CT, Kelsen D, Schwartz GK. UCN-01 suppresses thymidylatesynthase gene expression and enhances 5-fluorouracil-induced apoptosisin a sequence-dependent manner. Clin Cancer Res 1998;4:2201–6.
(119) Husain A, Yan XJ, Rosales N, Aghajanian C, Schwartz GK, Spriggs DR.UCN-01 in ovary cancer cells: effective as a single agent and in combi-nation with cis-diamminedichloroplatinum(II)independent of p53 status.Clin Cancer Res 1997;3:2089–97.
(120) Pollack IF, Kawecki S, Lazo JS. Blocking of glioma proliferationin vitroand in vivo and potentiating the effects of BCNU and cisplatin: UCN-01,a selective protein kinase C inhibitor. J Neurosurg 1996;84:1024–32.
(121) Shao RG, Cao CX, Shimizu T, O’Connor PM, Kohn KW, Pommier Y.Abrogation of an S-phase checkpoint and potentiation of camptothecincytotoxicity by 7-hydroxystaurosporine (UCN-01) in human cancer celllines, possibly influenced by p53 function. Cancer Res 1997;57:4029–35.
(122) Tsuchida E, Urano M. The effect of UCN-01 (7-hydroxystaurosporine), apotent inhibitor of protein kinase C, on fractionated radiotherapy or dailychemotherapy of a murine fibrosarcoma. Int J Radiat Oncol Biol Phys1997;39:1153–61.
(123) Akiyama T, Shimizu M, Okabe M, Tamaoki T, Akinaga S. Differentialeffects of UCN-01, staurosporine and CGP 41 251 on cell cycle progres-sion and CDC2/cyclin B1 regulation in A431 cells synchronized at Mphase by nocodazole. Anticancer Drugs 1999;10:67–78.
(124) Kawakami K, Futami H, Takahara J, Yamaguchi K. UCN-01, 7-hydroxyl-staurosporine, inhibits kinase activity of cyclin-dependent kinases andreduces the phosphorylation of the retinoblastoma susceptibility geneproduct in A549 human lung cancer cell line. Biochem Biophys ResCommun 1996;219:778–83.
(125) Shimizu E, Zhao MR, Nakanishi H, Yamamoto A, Yoshida S, Takada M,et al. Differing effects of staurosporine and UCN-01 on RB protein phos-phorylation and expression of lung cancer cell lines. Oncology 1996;53:494–504.
(126) Courage C, Bradder SM, Jones T, Schultze-Mosgau MH, Gescher A.Characterisation of novel human lung carcinoma cell lines selected forresistance to anti-neoplastic analogues of staurosporine. Int J Cancer1997;73:763–8.
(127) Kurata N, Kuwabara T, Tanii H, Fuse E, Akiyama T, Akinaga S, et al.Pharmacokinetics and pharmacodynamics of a novel protein kinase in-hibitors, UCN-01. Cancer Chemother Pharmacol 1999;44:12–8.
(128) Senderowicz AM, Headlee D, Lush R, Bauer K, Figg W, Murgo A, S, etal. Phase I trial of infusional UCN-01, a novel protein kinase inhibitor, inpatients with refractory neoplasms [abstract]. In: 10th National CancerInstitute–European Organization for Research on Treatment of CancerSymposium Proceedings. Dordrecht (The Netherlands): Kluwer Aca-demic Publishers; 1998. p. 111.
(129) Sausville EA, Lush RD, Headlee D, Smith AC, Figg WD, Arbuck SG, etal. Clinical pharmacology of UCN-01: initial observations and compari-son to preclinical models. Cancer Chemother Pharmacol 1998;42 Sup-pl:S54–9.
(130) Fuse E, Tanii H, Kurata N, Kobayashi H, Shimada Y, Tamura T, et al.Unpredicted clinical pharmacology of UCN-01 caused by specific bindingto human alpha1-acid glycoprotein. Cancer Res 1998;58:3248–53.
(131) Senderowicz AM, Headlee D, Lush R, Bauer K, Figg W, Murgo AS, et al.Phase I trial of infusional UCN-01, a novel protein kinase inhibitor, inpatients with refractory neoplasms [abstract]. Proc ASCO 1999;18:159a.
(132) Tamura T, Sasaki Y, Minami H, Fujii H, Ito K, Igarashi T, et al. Phase Istudy of UCN-01 by 3-hour infusion [abstract]. Proc ASCO 1999;18:159a.
NOTES
Editor’s note:E. A. Sausville is a participant in the National Cancer Institute’sCooperative Research and Development Agreement with Hoechst MarianRousell that manufactures flavopiridol.
We are indebted to colleagues from the Laboratory of Biological Chemistryand Developmental Therapeutics Program Clinical Trials Unit, National CancerInstitute, for all the encouragement and heavy work provided for these projects.We also want to acknowledge the National Cancer Institute and the NationalInstitutes of Health staff for their contributions in these clinical trials. Finally, wewant to acknowledge the patients and their relatives for their willingness toparticipate in our clinical trials and contribute to the advancement of medicine.
Manuscript received July 15, 1999; revised December 7, 1999; accepted De-cember 15, 1999.
Journal of the National Cancer Institute, Vol. 92, No. 5, March 1, 2000 REVIEW 387
Dow
nloaded from https://academ
ic.oup.com/jnci/article/92/5/376/2606689 by guest on 23 N
ovember 2021





























