Embed Size (px)
Citation preview

Steroid-resistant AsthmaCellular Mechanisms Contributing to Inadequate Response to Glucocorticoid Therapy
Ellen R. Sher,t Donald Y. M. Leung,*§ Wendy Surs,* Jeffrey C. Kam, *George Zieg, * Alan K. Kamada, * and Stanley J. Szefler* hIIDivisions of4Allergy-Immunology and Clinical Pharmacology, Departments of *Pediatrics and tMedicine, National Jewish Center forImmunology and RespiratoryfMedicine, Denver, Colorado 80206; and Departments of§Pediatrics and I"Pharmacol(gy, UniversitY ofColorado Health Sciences Center, Denver, Colorado 80262
Abstract
The current study examined whether alterations in glucocorti-coid receptor (GR) binding contribute to poor response to glu-cocorticoid therapy in asthma. 29 asthma patients with forcedexpiratory volume in 1 s (FEV 1) < 70% predicted were stud-ied. Patients were classified as steroid sensitive (SS) if theirmorning FEVI increased > 30% after a I-wk course of oralprednisone 20 mg twice daily and steroid resistant (SR) if theyfailed to increase > 15%. PBMCobtained from these twogroups, 17 SR and 12 SS, as well as 12 normal controls wereanalyzed.
SRpatients had two distinguishable GRbinding abnormali-ties: 15 of the 17 SR patients demonstrated a significantly re-duced GRbinding affinity, as compared with SS patients (P= 0.0001 ) and normal controls (P = 0.0001). This defect waslocalized to T cells and reverted to normal after 48 h in culturemedia. However, incubation with a combination of IL-2 andIL-4 sustained this abnormality. The other two SRpatients hadan abnormally low GRnumber with normal binding affinitythat was not limited to T cells. Furthermore, GRnumber failedto normalize after incubation in media alone or IL-2 and IL4.Therefore, SRasthma may be due to more than one abnormal-ity, the majority related to a reversible cytokine-induced reduc-tion in GRbinding affinity and the second related to an irrevers-ible reduction in GRnumber. These findings may have impor-tant implications for the design of alternative treatmentapproaches for recalcitrant asthma. (J. Clin. Invest. 1994.93:33-39.) Key words: steroid-resistant asthma * glucocorti-coid receptors - glucocorticoids * cytokines * immune activation
Introduction
Recent studies have demonstrated the importance of airwayinflammation and immune activation in the pathogenesis ofasthma ( 1, 2). Glucocorticoids are the most potent antiinflam-matory therapy commonly used in this disease (3, 4). Certainasthma patients, however, fail to respond to combined sys-temic and inhaled glucocorticoid treatment despite very highdoses over extended treatment periods (5, 6). Many of these
Address correspondence to Donald Y. M. Leung, M.D., Ph.D., Na-tional Jewish Center for Immunology and Respiratory Medicine, 1400Jackson Street, Room K926, Denver, CO80206.
Received/for publication IO May, 1993 and in revised form 13 Au-gust 1993.
patients continue treatment with glucocorticoids despite theonset of serious adverse effects and poor clinical response.These patients require alternative approaches to treatment. It istherefore important to understand the mechanisms underlyingthis apparent steroid resistance.
Recent studies indicate that steroid-resistant (SR)' asthmais associated with a failure of glucocorticoids to inhibit their invitro T cell proliferation and cytokine secretion (7, 8). Moreimportantly, T cells from peripheral blood of SR asthmatics,but not steroid-sensitive (SS) asthmatics, are persistently acti-vated despite continued treatment with aggressive steroid ther-apy (9). Glucocorticoids bind to a specific intracellular recep-tor to inhibit activation of T cells by various stimuli ( 10, 11 ). Itis therefore possible that the poor glucocorticoid response inSR asthma is due to an alteration in glucocorticoid receptor(GR) number or binding affinity. In this study, we examinedGRbinding parameters in PBMCfrom SRasthmatics to deter-mine whether abnormalities in GRbinding may contribute totheir inadequate response to glucocorticoid therapy.
Methods
Patient selection. Patients with a diagnosis of asthma, based on Ameri-can Thoracic Society criteria ( 12), were selected for evaluation. Theywere included if they had a morning prebronchodilator forced expira-tory volume in I s (FEV,) < 70% of predicted values and a . 15%increase in FEV, after two inhalations of albuterol (90 ug per actua-tion). Patients were excluded if they had evidence for other types oflung disease, pregnancy, suspected noncompliance with medical care,or concurrent therapy with medications that alter glucocorticoid metab-olism, such as anticonvulsants or erythromycin. A complete set of pul-monary function tests with lung volume, methacholine bronchial chal-lenge, and diffusion capacity/total lung capacity ratio were obtained ifthe diagnosis of asthma required confirmation. Informed consent, ap-proved by the National Jewish Institutional Review Board, was ob-tained from all patients before their entry into this study.
Patients were classified as SS or SR based on their prebronchodila-tor morning FEV, and their response to a course of oral prednisone.Asthmatic patients were defined as SR if they failed to improve theirmorning prebronchodilator FEV, by 2 15% after a l-wk course ofprednisone at a minimum oral dose of 40 mg/d ( 13). Patients wereclassified as SS if they had an increase in baseline FEV, of 30% orgreater. All SR asthma patients had glucocorticoid pharmacokineticstudies to exclude those patients with an abnormality in prednisoneabsorption or metabolism ( 14).
Cell isolation. Peripheral blood was collected in heparinized sy-ringes and PBMCwere isolated by gradient centrifugation (Ficoll-Pa-que®; Pharmacia LKB Biotechnology, Inc., Piscataway, NJ). Furtherisolation for the T cell component was performed by sheep red cell (E)
1. Abbreviations used in this paper: AP- 1, activation protein- 1; FEV,,forced expiratory volume in I s; GR, glucocorticoid receptor; GRE,glucocorticoid-responsive elements; SR. steroid resistant; SS, steroidsensitive.
Mechanisms of Steroid-resistant Asthma 33
J. Clin. Invest.© The American Society for Clinical Investigation, Inc.0021-9738/94/01/0033/07 $2.00Volume 93, January 1994, 33-39

rosetting (Colorado Serum Co., Denver, CO), and the (E-) non-T cellpopulation was purified further by lysis with anti-CD3 (Ortho Diag-nostic Systems, Inc., Raritan, NJ) and rabbit complement (GibcoLabs., Grand Island, NY). This procedure yielded an E (+) fraction> 97% T cell purity and < 1% B cells. The E (-) fraction contained< 5%T cells.
All blood samples were collected between 7 and 8 a.m., before medi-cations and at least 24 h after any oral glucocorticoid therapy. A totalblood sample of 80 ml was required for a standard binding assay and400 ml for assay that required isolation of purified T cells.
Glucocorticoid receptor binding analysis. [3H ] dexamethasone(Amersham Corp., Arlington Heights, IL) radioligand binding assayand Scatchard analysis, based on the method of Crabtree et al. ( 15),were used to measure nuclear and cytosolic GRbinding parameters inPBMCfrom normal donors and asthma patients. Cells (2 x 106) wereincubated in RPMI 1640 (Gibco Labs.) at 37°C for 1 h with 10 differ-ent concentrations of [3H]dexamethasone in duplicate ranging from0.8 to 400 nM in the presence and absence of 1,000-fold excess ofunlabeled dexamethasone. Measurements of glucocorticoid bound tonuclear receptors were obtained by hypotonic lysis of one set of PBMCwith 1.5 mMMgCl2 at 3YC for 30 min followed by centrifugation at12,000 g for 4 min. The supernatant was removed to isolate the nuclearfraction for radioligand binding analysis. Cytosolic receptors were ob-tained after hypotonic lysis of the other set of PBMCwith 1.5 mMMgCl2 containing dextran-coated charcoal at 3°C for 30 min. The cellswere then centrifuged at 12,000 g for 4 min and 100 ,ul of supernatantwas removed for cytosolic receptor binding parameters. Analyzing thetwo fractions, we assumed that GRbinding is saturable while nonspe-cific binding is nonsaturable. For measurement of nonspecific binding,a single measurement with a solution of 20 nM [3H ] dexamethasoneand 2 uM of unlabeled dexamethasone (Sigma Chemical Co., St.Louis, MO) was used.
All values obtained for both cytoplasmic and nuclear-bound gluco-corticoid were corrected for nonsaturable binding for each respectiveconcentration. Saturation binding analysis was performed assuming alinear binding plot of the bound divided by the free [ 3H ]-dexamethasone concentration versus the amount bound and extrapo-lating to the amount bound at an infinite free hormone concentration.A least-squares linear regression fit was used to define the binding pa-rameters, specifically, receptor sites per cell and binding affinity.
Reversibility and cytokine incubation protocols. PBMCfrom nor-mal donors and SRasthma patients were isolated and resuspended at aconcentration of I x 106 cells/ml in RPMI 1640 medium (GibcoLabs.) containing 10% heat-inactivated fetal calf serum (HycloneLabs., Logan, UT). Cells were incubated in the absence and presenceof IL-2 (50 U/ml; Cetus Corp., Emeryville, CA) and/or IL-4 (50 U/ml; gift of Dr. Paul Trotta, Schering-Plough Research Institute, Bloom-field, NJ) for 48 h at 37°C in 5% CO2. PBMCwere then analyzed forGRbinding parameters.
Results
Patient characteristics. Patient characteristics are summarizedin Table I. SRand SS asthmatics were similar for all parameterswith two exceptions. First, although the SR asthmatics had ahigher baseline FEV1 (P = 0.025) before the prednisone courseas compared with the SS asthma patients, their FEVy aftertreatment with prednisone was significantly lower than SS asth-matics (P = 0.0001 ). Second, 12 of the 17 SRasthma patientsreceived maintenance oral prednisone (mean daily dose = 24mg) at the time of their GRassay as compared with none of theSS asthma patients. Of note, the majority of SRpatients (type ISR asthma, see below) developed cushingoid features duringprednisone therapy. In contrast, one of the patients subse-quently labeled type II SR asthma (see below) maintained anormal plasma cortisol concentration (12 ,gg/dl) and did not
Table I. Patient Characteristics
SS Asthma SR AsthmaParameter Patients Patients
Number of subjects 12 17Age (yr)* 24 24Sex (mr/f) 8/4 12/5FEVI before BD (percent predicted)* 47 58FEV, after BD (percent predicted)* 69 74FEV, after steroid burst 82 57Duration of asthma (yr)* 16 19Inhaled steroids (yes/no) 6/6 14/3Systemic steroids (yes/no) 0/12 12/5Systemic steroid doset N/A 24Atopy (positive/negative) 11/1 16/1
* Mean value of patients before burst.Mean daily prednisone dose in milligrams. BD, bronchodilator dose.
develop cushingoid features despite continuous treatment withoral prednisone (20 mg daily). The other type II SR asthmapatient did not receive maintenance prednisone therapy be-cause of poor clinical response.
PBMCglucocorticoid receptor binding parameters in ste-roid-resistant asthma patients. Representative Scatchard plotsare presented in Fig. 1 from PBMC[3H Idexamethasone radio-ligand binding data from a normal donor, a patient with SRasthma, and a patient with SS asthma. PBMCGRbindingparameters were examined in three study groups: 17 SRasthma patients, 12 SS asthma patients, and 12 normal non-asthmatic controls. Each study group showed distinctive bind-ing patterns for their respective nuclear GR. Nonspecific [3H I-dexamethasone binding was low in all three study groups(2.6±0.5 vs 5.2±1.6 vs 3.5±1.0, respectively, for normals, SSasthmatics, and SR asthmatics) and no significant difference(P = 0.2, ANOVA) was observed in nonspecific bindingamong the three groups.
0.08
0.06
(V
Wo
co
0.04
0.02
0.002500
[3H] Dexamethasone bound (cpm)Figure 1. Scatchard analysis of [3H ] dexamethasone radioligandbinding data of PBMCfrom a normal donor, an SS asthmatic, and atype I SRasthmatic. Glucocorticoid receptor number is determinedby the intercept of the x axis and is normalized for the number of cellsand the dissociation constant is determined by the reciprocal of thelinear slope.
34 Sher, Leung, Surs, Kam, Zieg, Kamada, and Szefler
so8
* No steroidso Inhaled steroids alone* Oral + Inhaled steroidsO Oral steroids alone
0
o1 Is0 i00 9 0f
I"__ ......... .. .. ...''.2 .... .1 '.
T~~Oa_ .. _...
NL SS SR.Type I SR.Type 11(n=12) In=12) (n=15) (n=2I
15000 9
'10000
-
Esfln2
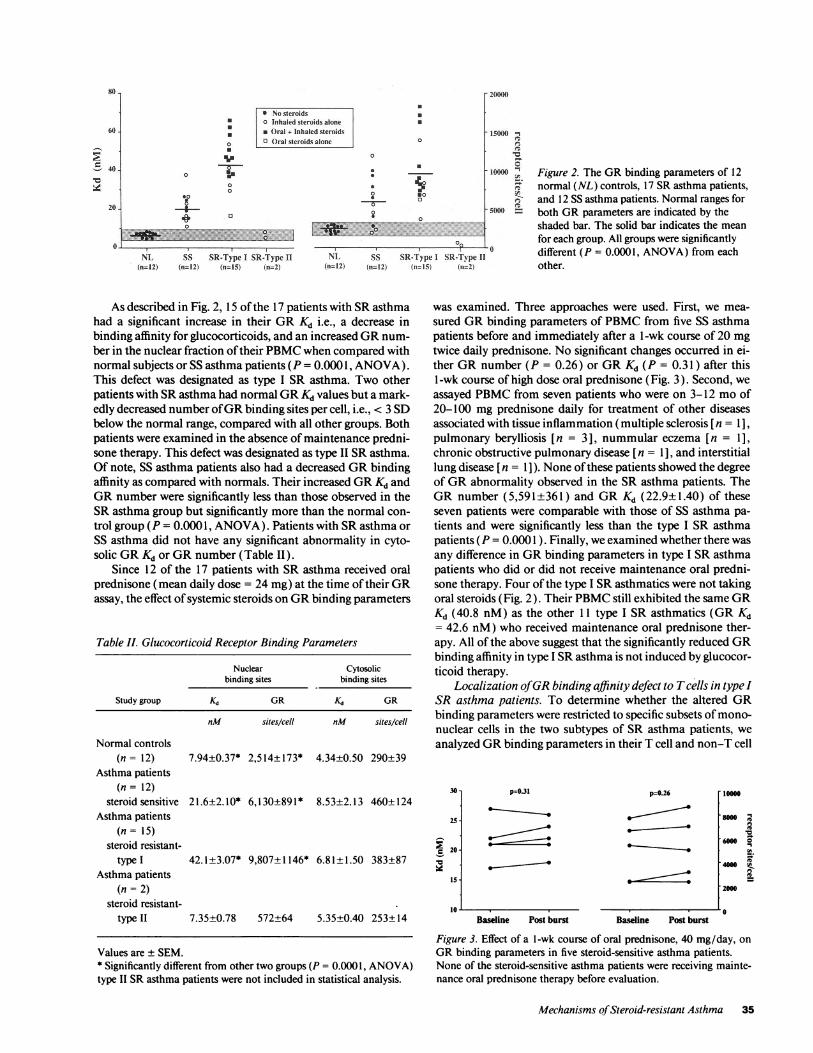
Figure 2. The GRbinding parameters of 12normal (NL) controls, 17 SRasthma patients,and 12 SS asthma patients. Normal ranges forboth GRparameters are indicated by theshaded bar. The solid bar indicates the meanfor each group. All groups were significantlydifferent (P = 0.0001, ANOVA) from eachother.
As described in Fig. 2, 15 of the 17 patients with SRasthmahad a significant increase in their GRKd i.e., a decrease inbinding affinity for glucocorticoids, and an increased GRnum-ber in the nuclear fraction of their PBMCwhen compared withnormal subjects or SS asthma patients (P = 0.0001, ANOVA).This defect was designated as type I SR asthma. Two otherpatients with SRasthma had normal GRKd values but a mark-edly decreased number of GRbinding sites per cell, i.e., < 3 SDbelow the normal range, compared with all other groups. Bothpatients were examined in the absence of maintenance predni-sone therapy. This defect was designated as type II SRasthma.Of note, SS asthma patients also had a decreased GRbindingaffinity as compared with normals. Their increased GRKd andGRnumber were significantly less than those observed in theSRasthma group but significantly more than the normal con-trol group (P = 0.0001, ANOVA). Patients with SRasthma orSS asthma did not have any significant abnormality in cyto-solic GRKd or GRnumber (Table II).
Since 12 of the 17 patients with SR asthma received oralprednisone (mean daily dose = 24 mg) at the time of their GRassay, the effect of systemic steroids on GRbinding parameters
Table II. Glucocorticoid Receptor Binding Parameters
Nuclear Cytosolicbinding sites binding sites
Study group Kd GR XS GR
nM sites/cell nM sites/cell
Normal controls(n = 12) 7.94±0.37* 2,514±173* 4.34±0.50 290±39
Asthma patients(n= 12)
steroid sensitive 21.6±2.10* 6,130±891* 8.53±2.13 460±124Asthma patients
(n= 15)steroid resistant-
type I 42.1±3.07* 9,807+1146* 6.81±1.50 383±87Asthma patients
(n = 2)steroid resistant-
type II 7.35±0.78 572±64 5.35±0.40 253±14
Values are ± SEM.* Significantly different from other two groups (P = 0.0001, ANOVA)type II SRasthma patients were not included in statistical analysis.
was examined. Three approaches were used. First, we mea-
sured GRbinding parameters of PBMCfrom five SS asthmapatients before and immediately after a -wk course of 20 mg
twice daily prednisone. No significant changes occurred in ei-ther GRnumber (P = 0.26) or GRKd (P = 0.31 ) after this1 -wk course of high dose oral prednisone (Fig. 3). Second, we
assayed PBMCfrom seven patients who were on 3-12 mo of20-100 mg prednisone daily for treatment of other diseasesassociated with tissue inflammation (multiple sclerosis [ n = 1],pulmonary berylliosis [n = 3], nummular eczema [n = 1],chronic obstructive pulmonary disease [ n = 1], and interstitiallung disease [ n = 1 ]). None of these patients showed the degreeof GRabnormality observed in the SR asthma patients. TheGRnumber (5,591±361) and GRKd (22.9±1.40) of theseseven patients were comparable with those of SS asthma pa-tients and were significantly less than the type I SR asthmapatients (P = 0.000 1 ). Finally, we examined whether there was
any difference in GRbinding parameters in type I SRasthmapatients who did or did not receive maintenance oral predni-sone therapy. Four of the type I SRasthmatics were not takingoral steroids (Fig. 2). Their PBMCstill exhibited the same GRKd (40.8 nM) as the other 11 type I SR asthmatics (GR Kd= 42.6 nM) who received maintenance oral prednisone ther-apy. All of the above suggest that the significantly reduced GRbinding affinity in type I SRasthma is not induced by glucocor-ticoid therapy.
Localization of GRbinding affinity defect to Tcells in type ISR asthma patients. To determine whether the altered GRbinding parameters were restricted to specific subsets of mono-
nuclear cells in the two subtypes of SR asthma patients, we
analyzed GRbinding parameters in their T cell and non-T cell
30
25
c 20
is-
p=0.31
Baseline Post burst
p=0.26 10000
8000
fo
26000 0
E92000
Baseline Post burst
Figure 3. Effect of a l-wk course of oral prednisone, 40 mg/day, on
GRbinding parameters in five steroid-sensitive asthma patients.None of the steroid-sensitive asthma patients were receiving mainte-nance oral prednisone therapy before evaluation.
Mechanisms of Steroid-resistant Asthma 35
60 -
40
20.
000
.0
0
4-
NL SS SR-Type I SR-Type 11(n=12) (n=12) (n= 15) (n=2)
0
.illqft a..00
20000
%ue =
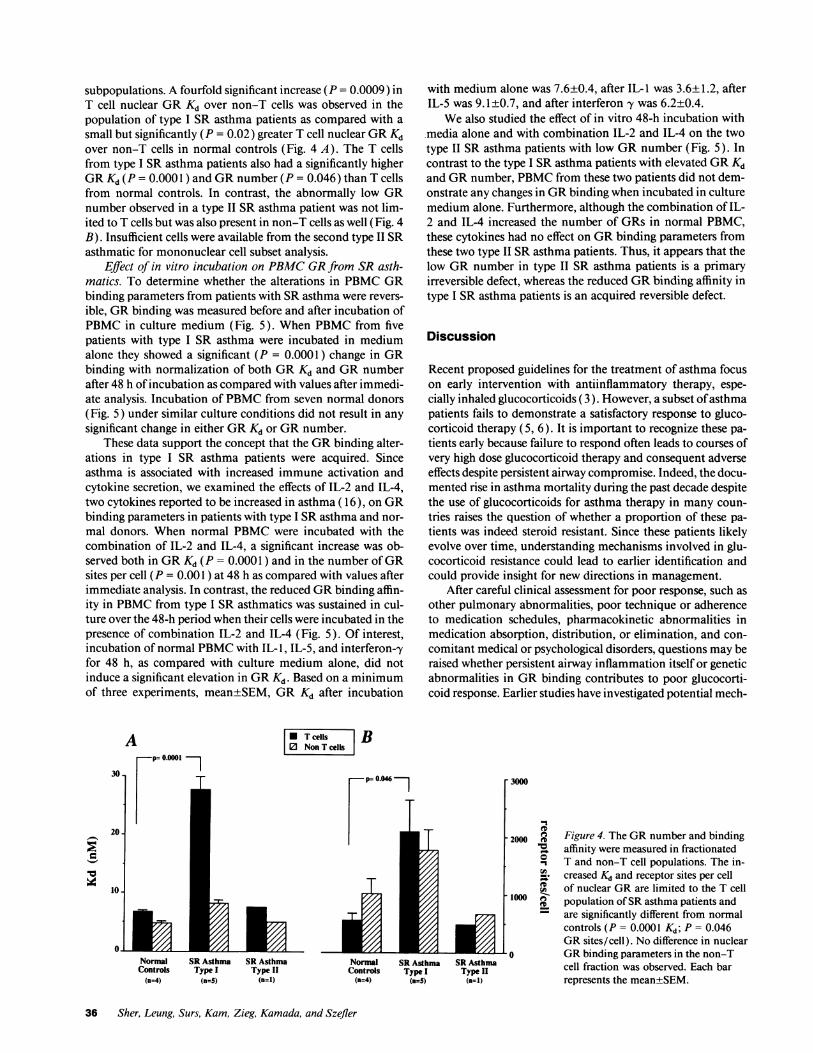
subpopulations. A fourfold significant increase (P = 0.0009) inT cell nuclear GRKd over non-T cells was observed in thepopulation of type I SR asthma patients as compared with asmall but significantly (P = 0.02) greater T cell nuclear GRKdover non-T cells in normal controls (Fig. 4 A). The T cellsfrom type I SR asthma patients also had a significantly higherGRKd (P = 0.0001 ) and GRnumber (P = 0.046) than T cellsfrom normal controls. In contrast, the abnormally low GRnumber observed in a type II SR asthma patient was not lim-ited to T cells but was also present in non-T cells as well (Fig. 4B). Insufficient cells were available from the second type II SRasthmatic for mononuclear cell subset analysis.
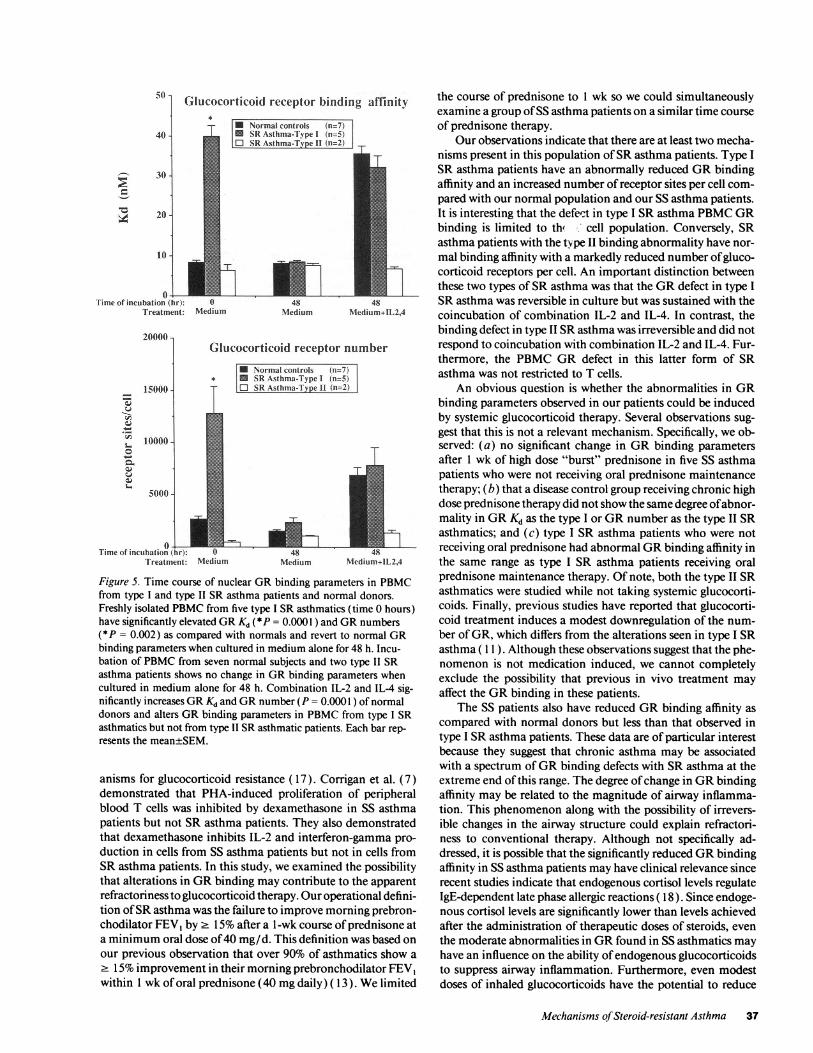
Effect of in vitro incubation on PBMCGRfrom SR asth-matics. To determine whether the alterations in PBMCGRbinding parameters from patients with SRasthma were revers-ible, GRbinding was measured before and after incubation ofPBMCin culture medium (Fig. 5). When PBMCfrom fivepatients with type I SR asthma were incubated in mediumalone they showed a significant (P = 0.0001) change in GRbinding with normalization of both GRKd and GRnumberafter 48 h of incubation as compared with values after immedi-ate analysis. Incubation of PBMCfrom seven normal donors(Fig. 5) under similar culture conditions did not result in anysignificant change in either GRKd or GRnumber.
These data support the concept that the GRbinding alter-ations in type I SR asthma patients were acquired. Sinceasthma is associated with increased immune activation andcytokine secretion, we examined the effects of IL-2 and IL-4,two cytokines reported to be increased in asthma ( 16), on GRbinding parameters in patients with type I SRasthma and nor-mal donors. When normal PBMCwere incubated with thecombination of IL-2 and IL-4, a significant increase was ob-served both in GRKd (P = 0.0001 ) and in the number of GRsites per cell (P = 0.001 ) at 48 h as compared with values afterimmediate analysis. In contrast, the reduced GRbinding affin-ity in PBMCfrom type I SR asthmatics was sustained in cul-ture over the 48-h period when their cells were incubated in thepresence of combination IL-2 and IL-4 (Fig. 5). Of interest,incubation of normal PBMCwith IL- 1, IL-5, and interferon-yfor 48 h, as compared with culture medium alone, did notinduce a significant elevation in GRKd. Based on a minimumof three experiments, mean±SEM, GRKd after incubation
A 0 Tcells B10 NonTcells|
with medium alone was 7.6±0.4, after IL-I was 3.6±1.2, afterIL-5 was 9.1±0.7, and after interferon y was 6.2±0.4.
Wealso studied the effect of in vitro 48-h incubation withmedia alone and with combination IL-2 and IL-4 on the twotype II SR asthma patients with low GRnumber (Fig. 5). Incontrast to the type I SRasthma patients with elevated GRKdand GRnumber, PBMCfrom these two patients did not dem-onstrate any changes in GRbinding when incubated in culturemedium alone. Furthermore, although the combination of IL-2 and IL-4 increased the number of GRs in normal PBMC,these cytokines had no effect on GRbinding parameters fromthese two type II SRasthma patients. Thus, it appears that thelow GRnumber in type II SR asthma patients is a primaryirreversible defect, whereas the reduced GRbinding affinity intype I SR asthma patients is an acquired reversible defect.
Discussion
Recent proposed guidelines for the treatment of asthma focuson early intervention with antiinflammatory therapy, espe-cially inhaled glucocorticoids (3). However, a subset of asthmapatients fails to demonstrate a satisfactory response to gluco-corticoid therapy (5, 6). It is important to recognize these pa-tients early because failure to respond often leads to courses ofvery high dose glucocorticoid therapy and consequent adverseeffects despite persistent airway compromise. Indeed, the docu-mented rise in asthma mortality during the past decade despitethe use of glucocorticoids for asthma therapy in many coun-tries raises the question of whether a proportion of these pa-tients was indeed steroid resistant. Since these patients likelyevolve over time, understanding mechanisms involved in glu-cocorticoid resistance could lead to earlier identification andcould provide insight for new directions in management.
After careful clinical assessment for poor response, such asother pulmonary abnormalities, poor technique or adherenceto medication schedules, pharmacokinetic abnormalities inmedication absorption, distribution, or elimination, and con-comitant medical or psychological disorders, questions may beraised whether persistent airway inflammation itself or geneticabnormalities in GRbinding contributes to poor glucocorti-coid response. Earlier studies have investigated potential mech-
r 3000
2000 eb
0
U)_.
'1000 m
Normal SRAsthma SR Asthma Normal SRAsthma SRAsthmaControls Type I Type II Controls Type I Type II
(n=4) (n=S) (n=1) (n=4) (,=S) (n=1)
Figure 4. The GRnumber and bindingaffinity were measured in fractionatedT and non-T cell populations. The in-creased Kd and receptor sites per cellof nuclear GRare limited to the T cellpopulation of SRasthma patients andare significantly different from normalcontrols (P = 0.0001 Kd; P = 0.046GRsites/cell). No difference in nuclearGRbinding parameters in the non-Tcell fraction was observed. Each barrepresents the mean±SEM.
36 Sher, Leung, Surs, Kam, Zieg, Kamada, and Szefler
o - (
40 -
I--, 30-
10 20-
l0 -
03Time of incubation (hr):
Treatment:
20000 1
w
- ~
l
1-
c)
15000 -
10000 -
5000 -
Glucocorticoid receptor binding affinity
* Normal controls (n=7)* SRAsthma-Ti pe I (n=5)E SR Asthma-Tspe It (n=2) T
Mediumei d 8
Medium M1edium+1L2,4
Glucocorticoid receptor number
t* Normal controls (n=7)
SSR Asthma-Type I (n=5O SR Asthma-k v pe II n=2)
T
Time of incubation (fhr): 0Treatment: Medium Medium Niedium+11.2,4
Figure 5. Time course of nuclear GRbinding parameters in PBMCfrom type I and type II SR asthma patients and normal donors.Freshly isolated PBMCfrom five type I SRasthmatics (time 0 hours)have significantly elevated GRKd (*P = 0.0001 ) and GRnumbers(*P = 0.002) as compared with normals and revert to normal GRbinding parameters when cultured in medium alone for 48 h. Incu-bation of PBMCfrom seven normal subjects and two type II SRasthma patients shows no change in GRbinding parameters whencultured in medium alone for 48 h. Combination IL-2 and IL-4 sig-nificantly increases GRKd and GRnumber (P = 0.0001 ) of normaldonors and alters GRbinding parameters in PBMCfrom type I SRasthmatics but not from type II SRasthmatic patients. Each bar rep-resents the mean±SEM.
anisms for glucocorticoid resistance ( 17). Corrigan et al. (7)demonstrated that PHA-induced proliferation of peripheralblood T cells was inhibited by dexamethasone in SS asthmapatients but not SR asthma patients. They also demonstratedthat dexamethasone inhibits IL-2 and interferon-gamma pro-duction in cells from SS asthma patients but not in cells fromSRasthma patients. In this study, we examined the possibilitythat alterations in GRbinding may contribute to the apparentrefractoriness to glucocorticoid therapy. Our operational defini-tion of SRasthma was the failure to improve morning prebron-chodilator FEVy by 2 15% after a 1-wk course of prednisone ata minimum oral dose of 40 mg/d. This definition was based onour previous observation that over 90% of asthmatics show a2 15% improvement in their morning prebronchodilator FEV,within 1 wk of oral prednisone (40 mgdaily) ( 13 ). Welimited
the course of prednisone to 1 wk so we could simultaneouslyexamine a group of SS asthma patients on a similar time courseof prednisone therapy.
Our observations indicate that there are at least two mecha-nisms present in this population of SRasthma patients. Type ISR asthma patients have an abnormally reduced GRbindingaffinity and an increased number of receptor sites per cell com-pared with our normal population and our SS asthma patients.It is interesting that the defect in type I SR asthma PBMCGRbinding is limited to the cell population. Conversely, SRasthma patients with the type II binding abnormality have nor-mal binding affinity with a markedly reduced number of gluco-corticoid receptors per cell. An important distinction betweenthese two types of SR asthma was that the GRdefect in type ISR asthma was reversible in culture but was sustained with thecoincubation of combination IL-2 and IL-4. In contrast, thebinding defect in type II SRasthma was irreversible and did notrespond to coincubation with combination IL-2 and IL-4. Fur-thermore, the PBMCGR defect in this latter form of SRasthma was not restricted to T cells.
An obvious question is whether the abnormalities in GRbinding parameters observed in our patients could be inducedby systemic glucocorticoid therapy. Several observations sug-gest that this is not a relevant mechanism. Specifically, we ob-served: (a) no significant change in GRbinding parametersafter 1 wk of high dose "burst" prednisone in five SS asthmapatients who were not receiving oral prednisone maintenancetherapy; (b) that a disease control group receiving chronic highdose prednisone therapy did not show the same degree ofabnor-mality in GRKd as the type I or GRnumber as the type II SRasthmatics; and (c) type I SR asthma patients who were notreceiving oral prednisone had abnormal GRbinding affinity inthe same range as type I SR asthma patients receiving oralprednisone maintenance therapy. Of note, both the type II SRasthmatics were studied while not taking systemic glucocorti-coids. Finally, previous studies have reported that glucocorti-coid treatment induces a modest downregulation of the num-ber of GR, which differs from the alterations seen in type I SRasthma ( 11 ). Although these observations suggest that the phe-nomenon is not medication induced, we cannot completelyexclude the possibility that previous in vivo treatment mayaffect the GRbinding in these patients.
The SS patients also have reduced GRbinding affinity ascompared with normal donors but less than that observed intype I SRasthma patients. These data are of particular interestbecause they suggest that chronic asthma may be associatedwith a spectrum of GRbinding defects with SRasthma at theextreme end of this range. The degree of change in GRbindingaffinity may be related to the magnitude of airway inflamma-tion. This phenomenon along with the possibility of irrevers-ible changes in the airway structure could explain refractori-ness to conventional therapy. Although not specifically ad-dressed, it is possible that the significantly reduced GRbindingaffinity in SS asthma patients may have clinical relevance sincerecent studies indicate that endogenous cortisol levels regulateIgE-dependent late phase allergic reactions ( 18). Since endoge-nous cortisol levels are significantly lower than levels achievedafter the administration of therapeutic doses of steroids, eventhe moderate abnormalities in GRfound in SS asthmatics mayhave an influence on the ability of endogenous glucocorticoidsto suppress airway inflammation. Furthermore, even modestdoses of inhaled glucocorticoids have the potential to reduce
Mechanisms of Steroid-resistant Asthma 37

nocturnal plasma cortisol concentrations, further compromis-ing the availability of endogenous cortisol during the criticalnighttime period when pulmonary function is lowest in pa-tients with severe asthma ( 19, 20). Consequently, inhaled glu-cocorticoids may provide symptomatic relief while incom-pletely resolving the inflammatory process. Thus, an under-standing of the mechanisms which underlie SR asthma mayhave important implications for controlling inflammation inmilder forms of asthma.
Since there is only one GRgene, our observations suggestthat type I SRasthma, which accounts for the large majority ofpatients with SR asthma, is acquired and restricted to T cells,whereas type II SR asthma is a form of primary cortisol resis-tance and is not limited to T cells. Wedemonstrated that thereduced GRbinding affinity can be induced in normal PBMCwith cytokines, i.e., combination IL-2 and IL-4. This raises theintriguing hypothesis that SRasthma may be the end result ofpoorly controlled asthma and ongoing immune activation. Ithas been previously demonstrated that PHA-induced T cellproliferation and cytokine production by PBMCfrom patientswith SR asthma are poorly inhibited by the addition of dexa-methasone or methylprednisolone in vitro (8, 9). Recently,however, we reported on several SR patients whose asthmacame under control with the combination of troleandomycinand methylprednisolone therapy, resulting in normalization ofT cell sensitivity in vitro to the inhibitory effects of methylpred-nisolone on T lymphocyte proliferation (8). Furthermore, cy-closporin A, a drug whose major action is inhibition of T cellproliferation and cytokine secretion, was reported to improvethe clinical symptoms of several patients with SRasthma (9).These observations support the hypothesis that ongoingasthma inflammation and cytokine secretion may contributeto the acquired GRdefect found in type I SR asthma.
The actual mechanisms by which cytokines or immune ac-tivation might induce a decrease in GRbinding affinity areunknown. One possible insight may relate to the marked differ-ence between the nuclear and cytosolic GRbinding parametersof PBMCfrom SRasthma or normal PBMCtreated with com-bination IL-2 and IL-4. In this regard, it is well known that theGRchanges its structure and/or conformation when translo-cated between the nucleus and cytosol.
At a cellular level, glucocorticoids exert their biological ef-fects by freely penetrating the plasma membrane and bindingto a specific intracellular receptor, i.e., the GR( 11). The unli-ganded receptor is thought to be a heteromer composed of asingle steroid- and DNA-binding subunit and two 90-kD heat-shock proteins. The binding of glucocorticoid to its receptorresults in the dissociation of the 90-kD heat-shock protein sub-units and exposure of the DNA-binding site on the receptor.This activated GRcomplex then translocates into the nucleusand regulates transcription by binding to specific DNA se-quences called glucocorticoid-responsive elements (GRE).Many of the glucocorticoid-inducible genes which have beenidentified are characterized by a cluster of multiple GREsup-stream of their promoter and enhancer regions. The inductionor repression of GRtarget genes ultimately results in the al-tered expression of glucocorticoid-regulated proteins (21).This latter action appears to be mediated via interaction of themodulatory domain of the GRwith transcriptional factors,such as activation protein- 1 (AP- 1). Overexpression of AP- 1interferes with the function of the modulatory domain of theGR (22). Since T cells from SR asthmatics are chronically
activated and cytokines can induce elevated AP- 1 levels (23),the latter observation may provide a plausible explanation forthe nuclear localization of the GRdefect in SRasthma.
Primary cortisol resistance is a rare, but well described syn-drome reported in humans and nonhuman primates (24, 25).The clinical syndrome is usually familial and characterized byelevated total plasma cortisol concentrations and the absenceof signs and symptoms of Cushing's syndrome. The mecha-nisms for end organ glucocorticoid resistance in the variousreported kindreds are heterogenous and have been demon-strated to be due to reduced GRnumber, decreased bindingaffinity for glucocorticoid, or poor DNAbinding of the GRtoGRE(26). Of note, our two type II SRasthma patients had lowGRnumber that was not restricted to their T cells. Clinically,one of these patients presented many features consistent withprimary cortisol resistance including the abilities to maintainnormal plasma cortisol concentrations and to remain free ofglucocorticoid adverse effects despite daily prednisone therapyin doses exceeding 20 mg/day.
In summary, we found that patients with SRasthma havealterations in PBMCGRnumber or binding affinity. With theincreasing use of systemic steroids to treat airway inflamma-tion in asthma, it is likely that more of these patients will beidentified. While these patients may respond to extremely highdose glucocorticoid therapy, recent studies have identified sev-eral promising treatment regimens as alternatives to systemicglucocorticoid therapy. These include cyclosporin, troleando-mycin, methotrexate, gold, and intravenous gammaglobulintherapy (27). Characteristic of these treatments is the variableresponse observed among individual patients. Our observa-tions suggest that the variability in molecular mechanisms con-tributing to glucocorticoid resistance may influence responseto therapeutic intervention. Continued research is needed todefine the mechanisms of action for ongoing immune activa-tion and the correlation to response to individual treatmentstrategies. An understanding of the mechanisms by which glu-cocorticoids fail to resolve inflammation in asthma may pro-vide important insight into the pathogenesis of asthma, espe-cially as related to progressive deterioration, and may result inthe rational design of innovative treatment approaches.
Acknowledaments
The authors thank Drs. Allen Adinoff, Lee Newman, and TalmadgeKing for referring patients to our study. We also thank MaureenPlourd-Sandoval for assistance in preparing this manuscript.
This work was supported in part by U. S. Public Health Servicegrants HL-36577 and AR-41256.
References
1. Wilson, J. W., R. Djukanovic, P. H. Howarth, and S. T. Holgate. 1992.Lymphocyte activation in bronchoalveolar lavage and peripheral blood in atopicasthma. Am. Rev. Respir. Dis. 145:958-960.
2. Walker, C., E. Bode, L. Boer, T. T. Hansel, K. Blaser, and J. Virchow, Jr.1992. Allergic and nonallergic asthmatics have distinct patterns of T-cell activa-tion and cytokine production in peripheral blood and bronchoalveolar lavage.Am. Rev. Respir. Dis. 146:109-115.
3. International Consensus Report on Diagnosis and Management ofAsthma. U. S. Department of Health and HumanServices, Public Health Service,National Institutes of Health, Publication No. 92-3091, June 1992.
4. Szefler, S. J. 1991. Glucocorticoid therapy for asthma: clinical pharmacol-ogy. J. Allergy Clin. Immunol. 88:147-165.
5. Schwartz, H. J., F. C. Lowell, and J. C. Melby. 1968. Steroid resistance inbronchial asthma. Ann. Intern. Med. 69(3):493-499.
38 Sher, Leung, Surs, Kam, Zieg, Kamada, and Szefler

6. Carmichael, J., I. C. Paterson, P. Diaz, G. K. Crompton, A. B. Kay, andI. W. B. Grant. 1981. Corticosteroid resistance in chronic asthma. Br. Med. J.282:1419-1422.
7. Corrigan, C. J., P. H. Brown, N. C. Barnes, S. J. Szefler, J.-J. Tsai, A. J.Frew, and A. B. Kay. 1991. Glucocorticoid resistance in chronic asthma: gluco-corticoid pharmacokinetics, glucocorticoid receptor characteristics and inhibi-tion of peripheral blood T-cell proliferation by glucocorticoid in vitro. Am. Rev.Respir. Dis. 144:1016-1025.
8. Alvarez, J., W. Surs, D. Y. M. Leung, D. Ikle, E. W. Gelfand, and S. J.Szefler. 1992. Steroid resistant asthma: immunologic and pharmacologic fea-tures. J. Allergy Clin. Immunol. 89:714-72 1.
9. Corrigan, C. J., P. H. Brown, N. C. Barnes, J.-J. Tsai, A. J. Frew, and A. B.Kay. 1991. Glucocorticoid resistance in chronic asthma: peripheral blood T lym-phocyte activation and comparison of the T lymphocyte inhibitory effects ofglucocorticoids and cyclosporin A. Am. Rev. Respir. Dis. 144:1026-1032.
10. Gustafsson, J.-A., J. Carlstedt-Duke, L. Poellinger, S. Okret, A.-C. Wik-strom, M. Bronnegird, M. Gillner, Y. Dong, K. Fuxe, A. Cintra, et al. 1987.Biochemistry, molecular biology, and physiology of the glucocorticoid receptor.Endocr. Rev. 8:185-234.
1 1. Munck, A., D. B. Mendel, L. I. Smith, and E. Orti. 1990. Glucocorticoidreceptors and actions. Am. Rev. Respir. Dis. 141:S2-SIO.
12. American Thoracic Society. 1987. Standards for the diagnosis and care ofpatients with chronic obstructive pulmonary disease (COPD) and asthma. Am.Rev. Respir. Dis. 136:225-244.
13. Kamada, A. K., D. Y. M. Leung, M. C. Gleason, M. R. Hill, and S. J.Szefler. 1992. High-dose systemic glucocorticoid therapy in the treatment ofasthma: a case of resistance and patterns of response. J. Allergy Clin. Immunol.90:685-687.
14. Hill, M. R., S. J. Szefler, B. D. Ball, M. Bartoszek, and M. Brenner. 1990.Monitoring glucocorticoid therapy: a pharmacokinetic approach. Clin. Pharma-col. & Ther. 48:390-398.
15. Crabtree, G. R., K. A. Smith, and A. Munck. 1981. Glucocorticoid recep-tors. In Methods of Hematology. Vol. 2. The Leukemic Cell. D. Catovsky, I.Chanarin, E. Beutler, E. B. Brown, and A. Jacobs, editors. Churchill Livingstone,Inc., NewYork. 252-269.
16. Robinson, D. S., Q. Hamid, S. Ying, A. Tsicopoulos, J. Barkans, A. M.
Bentley, C. Corrigan, S. R. Durham, and A. B. Kay. 1992. Predominant TH2-likebronchoalveolar T-lymphocyte population in atopic asthma. N. Engl. J. Med.326:298-304.
17. Kamada, A. K., D. Y. M. Leung, and S. J. Szefler. 1992. Steroid resistancein asthma: our current understanding. Ped. Pulmonol. 14:180-186.
18. Herrscher, R. F., C. Kasper, and T. J. Sullivan. 1992. Endogenous cortisolregulates immunoglobulin E-dependent late phase reactions. J. C/in. Invest.90:596-603.
19. Law, C. M., J. W. Honour, J. L. Marchant, M. A. Preece, and J. 0.Warner. 1986. Nocturnal adrenal suppression in asthmatic children taking in-haled beclomethasone dipropionate. Lancet. i:942-944.
20. Tabachnik, E., and Z. Zadid. 1991. Diurnal cortisol secretion duringtherapy with inhaled beclomethasone dipropionate in children with asthma. J.Pediatr. 118:294-297.
21. Diamond, M. I., J. N. Miner, S. K. Yoshinaga, and K. R. Yamamoto.1990. Transcription factor interactions: selectors of positive or negative regula-tion from a single DNAelement. Science (Wash. DC) 249:1266-1272.
22. Yang-Yen, H.-F., J.-C. Chambard, Y.-L. Sun, T. Smeal, T. J. Schmidt, J.Drouin, and M. Karin. 1990. Transcriptional interference between c-Jun and theglucocorticoid receptor: mutual inhibition of DNAbinding due to direct protein-protein interaction. Cell. 62:1205-1215.
23. Jain, J., P. G. McCaffrey, V. E. Valge-Archer, and A. Rao. 1992. Nuclearfactor of activated T cells contains Fos and Jun. Nature (Lond.) 356:801-804.
24. Brandon, D. D., A. J. Markwick, G. P. Chrousos, and D. L. Loriaux. 1989.Glucocorticoid resistance in humans and nonhuman primates. Cancer Res.49:2203-2213.
25. Chrousos, G. P., A. Vingerhoeds, D. Brandon, C. Eil, M. Pugeat, M.DeVroede, D. L. Loriaux, and M. B. Lipsett. 1982. Primary cortisol resistance inman. A glucocorticoid receptor-mediated disease. J. Clin. Invest. 69:1261-1269.
26. Hurley, D. M., D. Accili, C. A. Stratakis, M. Karl, N. Vamvakopoulos, E.Rorer, K. Constantine, S. I. Taylor, and G. P. Chrousos. 1991. Point mutationcausing a single amino acid substitution in the hormone binding domain of theglucocorticoid receptor in familial glucocorticoid resistance. J. Clin. Invest.87:680-686.
27. Szefler, S. J. 1992. Anti-inflammatory therapy in allergic disorders. Med.C/in. North Am. 76:953-975.
Mechanisms of Steroid-resistant Asthma 39





























