Embed Size (px)
Citation preview

The Effect of Histamine and Cyclic Adenosine Monophosphate on Myosin LightChain Phosphorylation in Human Umbilical Vein Endothelial CellsAlan B. Moy, Sandra S. Shasby, Brooke D. Scott, and D. Michael ShasbyDepartment of Medicine, University of Iowa College of Medicine, and Veterans Administration Hospital, Iowa City, Iowa 52242
Abstract
Histamine causes adjacent endothelial cells to retract fromeach another. We examined phosphorylation of the 20-kDmyosin light chain (MLC20) in human umbilical vein endothe-lial cells (HUVECs) exposed to histamine to determine if wecould find evidence to support the hypothesis that retraction ofthese cells in response to histamine represents an actomyosin-initiated contraction of the endothelial cytoskeleton. Wefoundthat MLC20 in HUVECswas constitutively phosphorylatedwith - 0.2 mol phosphate/mol MLC20. Histamine increasedMLC20 phosphorylation by 0.18±0.05 mol phosphate/molMLC20. This peak increase in phosphorylation occurred 30 safter initiating histamine exposure, persisted through 90 s, andreturned to control levels by 5 min. Agents that increase HU-VECcAMPprevent cell retraction in response to histamine.An increase in HUVECcAMPdecreased MLC20phosphoryla-tion by 0.18±0.02 mol phosphate/mol MLC20and preventedthe increase in MLC20phosphorylation after exposure to hista-mine. Tryptic peptide maps of phosphorylated myosin lightchain indicated that myosin light chain kinase phosphorylatedMLC20 in HUVECsunder basal, cAMP-, and histamine-sti-mulated conditions. Phosphoaminoacid analysis of the mono-phosphorylated peptide indicated that, in contrast to smoothmuscle cells, ser " and thr 8 monophosphorylation occurs inHUVECs. On the basis of our results, modulation of myosinlight chain kinase activity may be an important regulatory stepin the control of endothelial barrier function. (J. Clin. Invest.1993. 92:1198-1206.) Key words: myosin * phosphorylation.endothelial * histamine * cAMP
Introduction
Edema caused by molecules such as histamine is associatedwith the separation of adjacent endothelial cells from one an-other ( 1-8). Although there are multiple reports documentingthis phenomenon, the mechanism of cell separation remainsincompletely understood.
Agents that disrupt actin filaments or that chelate calciumand breakdown calcium-dependent cell-cell and cell-substrateadhesion cause edema and separation of adjacent endothelialcells (6, 7, 9). These data demonstrate that separation may
Address correspondence and reprint requests to Dr. Michael S. Shasby,Department of Internal Medicine, University of Iowa College of Medi-cine, Iowa City, IA 52242.
Received for publication 12 October 1992 and in revised form 2April 1993.
result from loss of tethering between cells and between cells andsubstrate. They also suggest that constitutive centripetal intra-cellular cytoskeletal tension opposes these normal tetheringforces, and unopposed expression of a constitutive tension mayaccount for cell retraction when tethering forces are released.
Others have suggested that the separation of adjacent endo-thelial cells from each other involves initiation of an activecontraction mediated by actin and myosin (10, 11). Supportfor this hypothesis comes from two observations. First, Wysol-merski and Lagunoff( 12) found that retraction of endothelialcells exposed to histamine was prevented if the cells were de-pleted of ATP before exposure to histamine. Second, 100 gMcalcium, ATP, myosin light chain kinase (MLCK), I and cal-modulin caused phosphorylation of the 20-kD myosin lightchain (MLC20) and contraction of the cytoskeleton of endothe-lial cells after their cell membranes were removed with deter-gents(l0, 11, 13).
Endothelial cell retraction in response to histamine is welldocumented (4). Whenhistamine binds to an HI receptor onan endothelial cell it initiates a signal transduction cascade thatresults in an increase in cell calcium and diacylglycerol (DAG)(2). The increase in cell calcium and DAGcould stimulate anacute increase in MLC20phosphorylation by MLCK(calciumdependent) or by protein kinase C (PKC) (calcium and DAGdependent). The acute increase in light chain phosphorylationcould then increase actomyosin contraction and centripetaltension ( 14-16).
In the experiments described in this manuscript we investi-gated the effects of histamine on phosphorylation of MLC20inhuman umbilical vein endothelial cells (HUVECs). Wewereinterested to determine if MLC20was constitutively phosphor-ylated, if histamine acutely increased MLC20phosphorylationin HUVECs, and, if so, how much phosphorylation of MLC20increased. Because MLC20 phosphorylation is mediated byboth MLCKand PKCin other nonmuscle cells, we also con-structed tryptic peptide maps of the phosphorylated MLC20todirectly determine which kinase was responsible for MLC20phosphorylation in HUVECs, constitutively and after stimula-tion with histamine ( 15). Wepreviously found that increasingcell cAMPprevented HUVECsfrom retracting in response tohistamine, but the increase in cAMPdid not prevent the in-crease in cell calcium (2). In other cells, one of the effects ofincreased cell cAMP is to inhibit MLC20 phosphorylation byMLCK( 17 ). Hence, we were also interested in determining ifincreased cell cAMPprevents increased MLC20phosphoryla-tion in histamine-stimulated HUVECs. If it does, it would, inpart, explain how cAMPprevents the response of HUVECstohistamine.
1. Abbreviations used in this paper: DAG, diacylglycerol; HUVEC,human umbilical vein endothelial cells; MLC20, 20-kD myosin lightchain kinase; MLCK, myosin light chain kinase; PKC, protein kinase C.
1198 A. B. Moy, S. S. Shasby, B. D. Scott, and D. M. Shasby
J. Clin. Invest.© The American Society for Clinical Investigation, Inc.0021-9738/93/09/1 198/09 $2.00Volume 92, September 1993, 1198-1206

Methods
Materials. Tissue culture supplies were obtained from the CancerCenter, University of Iowa. Fetal bovine serum was obtained fromHyclone Laboratories, Inc. (Logan, UT). Polyclonal rabbit IgG anti-myosin antibody against human platelet whole myosin was obtainedfrom Biomedical Technologies, Inc. (Stoughton, MA). Protein A,Staphylococcus aureus cell suspension was obtained from CalbiochemCorp. (San Diego, CA). Rat brain PKCwas obtained from Calbio-chem Corp. Histamine, theophylline, 8-bromo cAMP, DL-histidine,DL-glutamic acid, phosphoserine, and phosphothreonine were ob-tained from Sigma Chemical Co. (St. Louis, MO). L-l-p-tosylamino-2-phenylethyl chloromethyl ketone (TPCK) trypsin was from Worth-ington Biochemical Corp. (Freehold, NJ). [32P]Orthophosphate andy-[32PIATP were obtained from NewEngland Nuclear(Boston, MA).[35S]Methionine was obtained from ICN Radiochemicals (Irvine,CA). Transwells were purchased through Costar Corp. (Cambridge,MA). Smooth muscle MLCKand turkey gizzard MLCwere gifts fromDr. James Sellers, National Heart, Lung, and Blood Institute (Be-thesda, MD). All other chemicals were reagent grade.
Cell culturing. Cultured HUVECswere prepared by collagenasetreatment of freshly obtained umbilical veins as described (2). Forexperiments designed to measure the amount of phosphorylatedMLC20 and for some of the experiments designated for peptide map-ping, harvested cells were plated on 25-mm diameter polycarbonatefilters (0.8-Mm pore size) precoated with 30 Mg/ml of fibronectin as
described (2). Alternatively, harvested cells were plated on 60-mmdiameter tissue culture plates (Costar Corp.) precoated with 1%gelatinfor some of the peptide mapping experiments. There was no differencein the peptide maps of cells grown on micropore filters when comparedwith cells grown on tissue culture plastic. However, basal phosphoryla-tion was higher ( - 0.2 mol phosphate/mol light chain) in cells grownon tissue culture plastic than in cells grown on micropore filters (n > 25for each). All cells were cultured in medium 199 and supplementedwith 20% fetal bovine serum, basal medium Eagle vitamins and aminoacids, glucose (5 mM), glutamine (2 mM), penicillin (100 Mu/ml), andstreptomycin (100 Mg/ml). All studies were performed on primarycultures that were 2 d postconfluent at the time of study. Cultures were
identified as endothelial cells by their characteristic uniform morphol-ogy, uptake of acetylated LDL ( - 99%of cells), and by indirect immu-nofluorescent staining for factor VIII (- 97% of cells). Because thecells are not visible on the polycarbonate filters, for each experiment, a
control well was plated and examined for morphology to be certain thatthe morphology was consistent with that of cells identified as endothe-lial cells by these staining techniques.
Purification of human platelet myosin MLC20. Human plateletMLC20 was purified from whole platelet myosin using the proceduredescribed by Daniel and Adelstein ( 18). Preparations were used if con-
taminating kinases were not present. Whole-platelet myosin was puri-fied according to Sellers et al. (19). Whole-platelet myosin was not a
suitable substrate for generating in vitro peptide mapstandards becauseit contained contaminating endogenous kinases.
Isotopic labeling and stimulation. Cells designated for quantitatingMLC20phosphorylation were grown on micropore filters and were la-beled with 1.5 ml [35S]methionine (555 MiCi/ml) in M199 with 10%fetal bovine serum for 48 h at 37°C and 5% CO2. Labeling in < 10%fetal bovine serum increased basal phosphorylation of MLC20.
Cells designated for peptide mapping were grown on either micro-pore filters or tissue culture plates. Confluent cells were washed threetimes with a phosphate-free buffer with the following composition(mM): 119 NaCl, 5 KC1, 5.6 glucose, 0.4 MgCl2, 1 CaCl2, 25 Pipes(pH7.2), and were then labeled with 3 ml of 300-400 MCi/ml [32p]-orthophosphate for 2 h at 37°C without CO2.
Labeled cells were exposed to either buffer, histamine (I01- mol/liter), or a mixture of agents used to increase cell cAMP(8-bromo-cAMP, l0-4 mol/liter, forskolin, 2 X Il0- mol/liter, and theophylline,l0-3 mol/liter) with or without histamine. There was no difference in
MLC20phosphorylation between cells exposed to the mixture of agents
to increase cAMPversus 8-bromo-cAMP alone, and these groups werecombined for analysis.
Reactions were terminated by snap freezing on a dry ice methanolbath and the cells were lysed by thawing in 1 ml of a buffer containing1% NP-40, 100 mMsodium pyrophosphate, 250 mMNaCl, 50 mMNaF, 5 mMEGTA, 0.1 mMPMSF, 10 gg/ml leupeptin, 15 mMfl-mercaptoethanol, and 20 mMTris-HCI (pH 7.9). The lysate was sedi-ment at l00,000 g for 5 min at 4VC, and the supernatant was incubatedwith 20 Ml of rabbit anti-human platelet myosin antibody (2 mg/ml)at 4VC for 1 h. 50 Ml of a prewashed S. aureus cell suspension (Pansor-bin) was then added to the suspension, which was incubated for anadditional 30 min at 4VC (20). The mixture was centrifuged (100,000g for 1 min) and the pellet was washed with 0.5 ml of the lysing bufferand recentrifuged. The pellet was then washed with 0.5 ml of a 50:50mixture of lysing buffer and PBS. The pellet was resuspended in 200 Mlof SDSsample buffer for SDS-PAGEor 35 Ml of urea-lysing buffer fortwo-dimensional electrophoresis.
Isoelectric focusing. The unphosphorylated and phosphorylatedMLC20 isoforms were separated by two-dimensional electrophoresis asdescribed by Ludowyke et al. ( 15) but with the following modifica-tions. The sample was suspended in 35 Ml of a buffer containing 9.5 Murea (ultrapure urea; Boehringer Mannheim Corp., Indianapolis, IN),NP-40, 0.04% pharmalyte (pH 4.5-5.4), 0.04% pharmalyte (pH4-6.5) and 0.1 MDTT. Tube gels were prepared using 1.5 mmi.d. x180-mm glass tubes. For 10 tubes, 2.78 g of ultra pure urea was mixedwith 1.5 ml of H20, 0.83 ml of 30%:0.8% acrylamide/bisacrylamide,0.28 ml of pharmalyte (pH 4-6.5), 0.28 ml of pharmalyte (pH 4.5-5.4), and 0.28 ml of CHAPSbuffer (100 mgCHAPS, 0.3 ml H20, and0.03 ml NP-40). Gels were drawn to a height of 150 mm. Gels werepolymerized with 5 ul of TEMEDand 10 Ml ammonium persulfate.Isoelectric focusing was performed using a cell (model 175; Bio-RadLaboratories (Richmond, CA).
The anolyte contained 0.01 Mglutamic acid and the catholyte con-tained 0.01 Mhistidine. Gels were prefocused at 25°C at 200 V for 2 hfollowed by 500 V for 5 h. 30-,Ml samples were loaded onto the gel,covered with 10 Ml of an overlaying buffer, which contained 8 Murea,2%pharmalyte (pH 3-10), and 0.5% pharmalyte (pH 4.5-5.4). Catho-lyte buffer was layered over the overlaying buffer. The samples werethen electrophoresed at 2,500 V for an additional 32,000 V h.
Second Dimension SDS-PAGE. Tube gels were extracted andrinsed with transfer solution (3% SDS, 0.07 MTris-Base (pH 6.7), and0.03 mg/ml bromphenol blue), layered onto a polyacrylamide/bisac-rylamide slab gel ( 1 5%:0.4%), and sealed to the slab gel with hot agar(2.3% SDS, 0.062 MTris-Base (pH 6.8), 0.5% agarose, and 5% 3-mer-captoethanol). The gels were run at 4°C for 18 to 20 h at 200 V. Gelswere treated with En3hance (Dupont Pharmaceuticals Inc., Wilming-ton, DE) to accelerate autoradiograph development. Autoradiographswere developed using Kodak X-OMATARfilm. The relative isoformdistribution was quantitated using an Image Quant laser densitometer(Molecular Dynamics, Sunnyvale, CA).
Two-dimensional peptide mapping of myosin tryptic peptides. Im-munoprecipitate from 32P-labeled HUVECswas dissolved in SDSsam-ple buffer as above and separated by an 8-15% polyacrylamide gradientgel, and electrophoresed at 200 V for 4 to 5 h at 21 'C. The 32P-labeledMLC20 was cut from the wet gel into small pieces and washed threetimes for 30 min with 40% methanol, 10% acetic acid, and 50% water,and then an additional three times with 10% methanol for the sametime period. The washed gel slices were subsequently dried under nitro-gen and then digested with 1 ml of 60 Mg/ml of TPCK-trypsin in 50mMammonium bicarbonate at 37°C for 4 h. The digest was collectedand stored at 4°C. The remaining gel was further digested with 1 ml of30 Mg/ ml trypsin for an additional 20 h. Digests were pooled and lyoph-ilized. Peptides were resuspended in 1 ml of water and lyophilized.
Digest was resuspended with 30 Ml of electrophoresis buffer (aceticacid/formic acid/water, 9:3:88), spotted on a plastic 20 x 20 X 0.20mmsilica P 60 TLC plate (EM Science, Gibbstown, NJ). Peptideswere electrophoresed toward the cathode at 900 V at 4VC for 45 minusing the technique described by Gracy (21). Peptide migration was
Histamine and cAMPAlter Myosin Phosphorylation in Endothelial Cells 1199

monitored by the movement of acid fuschin dye toward the anode.Plates were then dried and subjected to ascending chromatographyusing 1-butanol/acetic acid/pyridine/water (49:8:38:30). Plates werethen dried, wrapped in cellophane, and autoradiographs were devel-oped using Kodak X-Omat AR film.
Phosphoamino acid analysis. Peptides that were generated fromtwo-dimensional TLC maps were scraped from the TLC plate andeluted with 50 mMNH4HCO3. The peptide was then lyophilized,washed with water, and lyophilized again to remove the ammoniumbicarbonate. Peptides were subjected to acid hydrolysis in 6 N HCOat120'C for 3 h and then relyophilized. Dry residue was washed withwater and relyophilized. Labeled phosphoamino acids were resus-pended in electrophoresis buffer (acetic acid/formic acid/water,9:3:88), which contained 2 ,g/,gl of phosphothreonine and phospho-serine, and were then spotted onto a TLC plate. Amino acids weresubjected to electrophoresis at 700 V for 2 h and 50 min at 4VC. La-beled phosphoamino acids were detected by autoradiography and iden-tified by comparing the corresponding migration of the phosphoaminoacid standards, which were stained with ninhydrin.
Measurement of intracellular cAMP. HUVECsplated on polycar-bonated filters were exposed to control media (M199 without serum),histamine ( I0-' M), or cAMPmixture (forskolin [2 X I0-5 MJ andtheophylline [ 10 -3 M]). After the indicated time intervals, the mediumwas aspirated and the cells were extracted twice with 1 ml of ethanol.The extracts were combined and centrifuged at 3,000 g for 15 min at4°C. The supernates were dried under nitrogen and reconstituted in thesample buffer provided (cAMP 3H Assay System; Amersham Corp.,Arlington Heights, IL). The rest of the analysis system was conductedas described in the directions for the assay system.
In vitro phosphorylation assays. Standards for tryptic peptide mapsof MLC20were generated in vitro from turkey gizzard MLC20or plate-let MLC20 phosphorylated with either smooth muscle MLCKor ratbrain PKC. Phosphorylation of MLCby MLCKwas carried out in avolume of 200 1d using 20 mMTris-HCl (pH 7.6), 5 mMMgCI2, 200MMCaCI2, I mM[,y-32P]ATP (400-1,000 cpm/pmol), 2 MMcalmo-dulin, 7 Mg/ml smooth muscle MLCK, and 3.25 mg/ml turkey gizzardsmooth muscle MLC20or 50 ug/ml human platelet MLC20. The reac-tion was initiated with ATP at 25°C for 10 min and terminated byprecipitating the assay mixture with 40 ul 100% TCA. Samples weremicrocentrifuged, and the precipitate was washed three times with I mlof 10% TCA. The pellet was resuspended with 200 Ml of SDS-samplebuffer and 0. 1%bromphenol blue and made alkaline with 1 NNaOH.
Phosphorylation of MLC20by PKCwas carried out in a final vol-umeof 400 Ml using 20 mMTris-HCl (pH 7.6), 5 mMMgCl2, 200 AMCaC12, 1 mM[,y-32P]ATP (400-1,000 cpm/pmol), 50 Mg/ml phos-phatidylserine, 0.8 Mg/ml 1,3 diolein, and 2.7 Mg/ml PKCat 30°C for60 min. Phospholipids were stored in chloroform and evaporatedunder a constant stream of nitrogen. Phospholipids were then resus-pended in Tris buffer and sonicated at 4°C for 5 min using a sonicator(model 200; Branson Ultrasonics Corp., Danbury, CT). The reactionwas initiated with the labeled ATP.
Calculating the stoichiometry ofMLC20phosphorylation. Quantita-tive phosphorylation of MLC20was determined using laser densitome-try of autoradiographs made from the two-dimensional gels of 35S-la-
35S-Iabelled
w
CLch0(0)
IEF
beled MLC20immunoprecipitated from the cells. There were two phos-phorylated isoforms of MLC20 (designated A and B) and eachphosphorylated isoform could exist as an unphosphorylated (A or B),monophosphorylated (A' or B'), or diphosphorylated (A" or B") iso-form. The volume (V) of each phosphorylation state of each isoformwas integrated on the densitometer. The volume of each phosphoryla-tion state for each of the isoforms was then expressed as a fraction (fxn)of the total radioactivity for that isoform, for example, fraction A= V(A)/V(A) + V(A') + V(A"). Fractions for B isoforms were calcu-lated similarly. Phosphorylation stoichiometry was then calculated bythe following formula: mol phosphate/mol isoform A = Fxn A' + 2X Fxn A" and mol phosphate/mol isoform B = Fxn B' + 2 X Fxn B".
Statistical analysis. Data are reported as means±standard errors(SE). Comparisons between two groups were made using the Student'st test. Comparisons between more than two groups were made usinganalysis of variance and individual groups comparisons were done us-ing a Tukey honest significance difference test for post hoc compari-sons of means. Differences were considered significant at the P c 0.05level.
Results
Identification of MLCisoforms in HUVECsMLCisoforms from HUVECswere separated by two-dimen-sional gel electrophoresis. Fig. 1 A is an autoradiograph of atwo-dimensional gel of immunoprecipitated MLCfrom un-stimulated HUVECslabeled with [35S]methionine. Isoformswere observed at three molecular masses, 20, 19.7, and 16 kD.The most basic of the 20- and 19.7-kD isoforms were unphos-phorylated as they were not detectable in 32P-labeled cells (Fig.1 B). Similarly, the 16-kD isoforms were unphosphorylated asthey were also not observed in 32P-labeled cells (Fig. 1 B).Kawamoto et al. ( 14) also found 20- and 19.7-kD phosphory-lated isoforms and a 1 6-kD isoform in platelets. Unlike humanplatelet and smooth muscle MLC, the unphosphorylated Aand B isoforms of HUVECsare focused adjacent to each other( 14, 22). Also, the 16-kD isoform existed at only one isoelec-tric point in human platelets whereas in HUVECsthere are two1 6-kD isoforms with distinct isoelectric points (Fig. 1 A).
The 20- and the 19.7-kD isoforms each focused at threedistinct isoelectric points, corresponding to the unphosphory-lated, monophosphorylated, and diphosphorylated states ofeach isoform. The unphosphorylated isoforms were focusedtoward the basic end where isoform B had a higher molecularmass and more acidic isoelectric point than isoform A. Thecorresponding mono- and diphosphorylated isoforms of A andB were focused toward the acidic end and were arbitrarily desig-nated A', B' and A", B", respectively (Fig. 1 A). Weconfirmedthe identity of the unphosphorylated isoforms in 35S-labeledcells by depleting cells of ATPusing antimycin Aand deoxyglu-cose (23). ATPdepletion generated two isoforms, which corre-
Figure 1. (A) Autoradiograph ofP-labelled immunoprecipitated MLCisoforms
from 35-labeled HUVECs. The iso-forms were separated by two-di-
B' B mensional gel electrophoresis. Iso-forms existed in unphosphorylated
4 $. r(A and B), monophosphorylatedA'mXA' (A' and B'), and diphosphorylated
a - IS# d4 (A" and B") states with distinct iso-electric points. (B) Autoradiograph
44
-Soof immunoprecipitated MLCiso-forms from _P-labeled HUVECs.
IEF - See text for explanation.
1200 A. B. Moy, S. S. Shasby, B. D. Scott, and D. M. Shasby
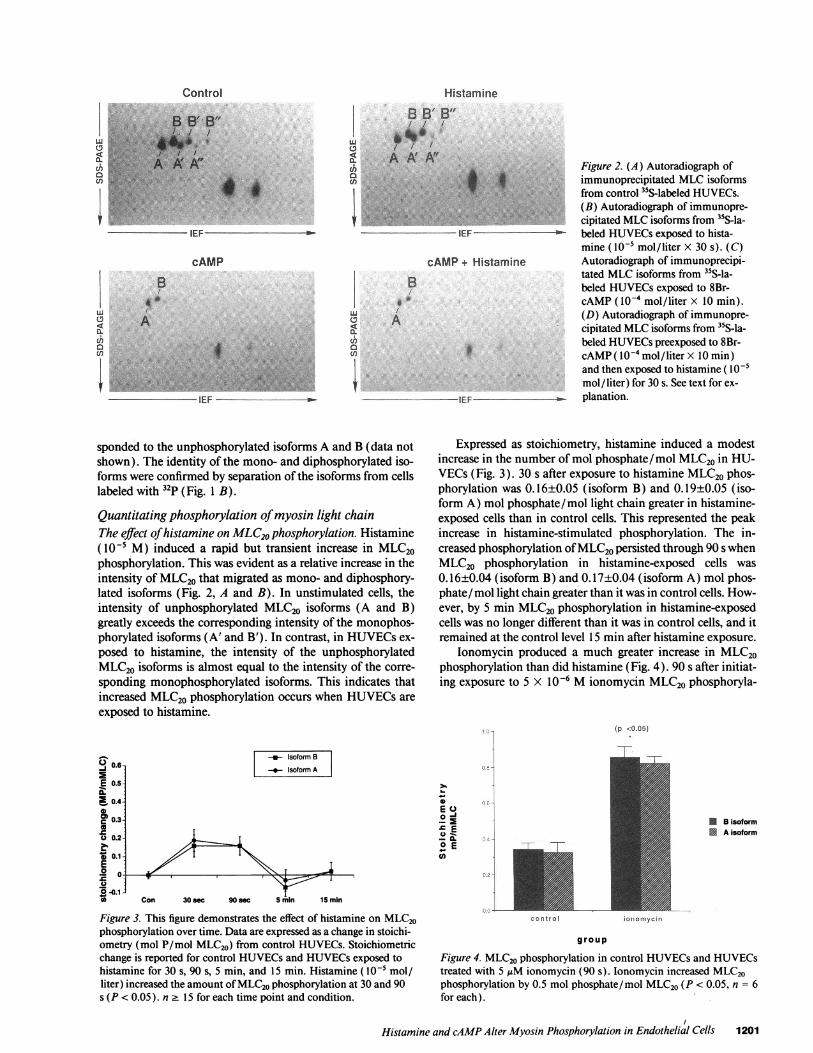
Histamine
B B' B"
LJ / / /
g AA'A"C')en
uc.CCaCCl
IEF
cAMP+ Histamine
B
A
IEF- -
Figure 2. (A) Autoradiograph ofimmunoprecipitated MLCisoformsfrom control 35S-labeled HUVECs.(B) Autoradiograph of immunopre-cipitated MLCisoforms from "S-la-beled HUVECsexposed to hista-mine ( 10-5 mol/liter X 30 s). (C)Autoradiograph of immunoprecipi-tated MLCisoforms from "S-la-beled HUVECsexposed to 8Br-cAMP( I0-4 mol/liter x 10 min).(D) Autoradiograph of immunopre-cipitated MLCisoforms from 3"S-la-beled HUVECspreexposed to 8Br-cAMP( I0 - mol/liter X 10 min)and then exposed to histamine ( IO05mol/liter) for 30 s. See text for ex-planation.
sponded to the unphosphorylated isoforms A and B (data notshown). The identity of the mono- and diphosphorylated iso-forms were confirmed by separation of the isoforms from cellslabeled with 32p (Fig. 1 B).
Quantitating phosphorylation of myosin light chainThe effect of histamine on MLC20phosphorylation. Histamine(10-5 M) induced a rapid but transient increase in MLC20phosphorylation. This was evident as a relative increase in theintensity of MLC20 that migrated as mono- and diphosphory-lated isoforms (Fig. 2, A and B). In unstimulated cells, theintensity of unphosphorylated MLC20 isoforms (A and B)greatly exceeds the corresponding intensity of the monophos-phorylated isoforms (A' and B'). In contrast, in HUVECsex-
posed to histamine, the intensity of the unphosphorylatedMLC20 isoforms is almost equal to the intensity of the corre-
sponding monophosphorylated isoforms. This indicates thatincreased MLC20 phosphorylation occurs when HUVECsare
exposed to histamine.
_ Isoform B
Isoform A
0.5
2 0.4
C 0.3
0.2
O 0.1
00-
Con 30 sec 90 sec 5 min 15 min
Figure 3. This figure demonstrates the effect of histamine on MLC20phosphorylation over time. Data are expressed as a change in stoichi-ometry (mol P/mol MLC20) from control HUVECs. Stoichiometricchange is reported for control HUVECsand HUVECsexposed tohistamine for 30 s, 90 s, 5 min, and 15 min. Histamine (l0-' mol/liter) increased the amount of MLC20phosphorylation at 30 and 90s (P < 0.05). n 2 15 for each time point and condition.
Expressed as stoichiometry, histamine induced a modestincrease in the number of mol phosphate/mol MLC20 in HU-VECs (Fig. 3). 30 s after exposure to histamine MLC20phos-phorylation was 0.16±0.05 (isoform B) and 0.19±0.05 (iso-form A) mol phosphate/mol light chain greater in histamine-exposed cells than in control cells. This represented the peakincrease in histamine-stimulated phosphorylation. The in-creased phosphorylation of MLC20persisted through 90 s whenMLC20 phosphorylation in histamine-exposed cells was
0.16±0.04 (isoform B) and 0.17±0.04 (isoform A) mol phos-phate/mol light chain greater than it was in control cells. How-ever, by 5 min MLC20phosphorylation in histamine-exposedcells was no longer different than it was in control cells, and itremained at the control level 15 min after histamine exposure.
lonomycin produced a much greater increase in MLC20phosphorylation than did histamine (Fig. 4). 90 s after initiat-ing exposure to 5 X 10-6 Mionomycin MLC20phosphoryla-
- (p <0.05)
S.
E Q
° E
o 4
I
c o n t r o
B isoformA isoform
ionomycin
group
Figure 4. MLC20phosphorylation in control HUVECsand HUVECstreated with 5 1IM ionomycin (90 s). Ionomycin increased MLC20phosphorylation by 0.5 mol phosphate/mol MLC20 (P < 0.05, n = 6for each).
Histamine and cAMPAlter Myosin Phosphorylation in Endothelial Cells 1201
Control
.. :-
"T'14.";..V..,
w ..hire
OA%CLCha
if-'t
4_
cAMP
B
A
IEF
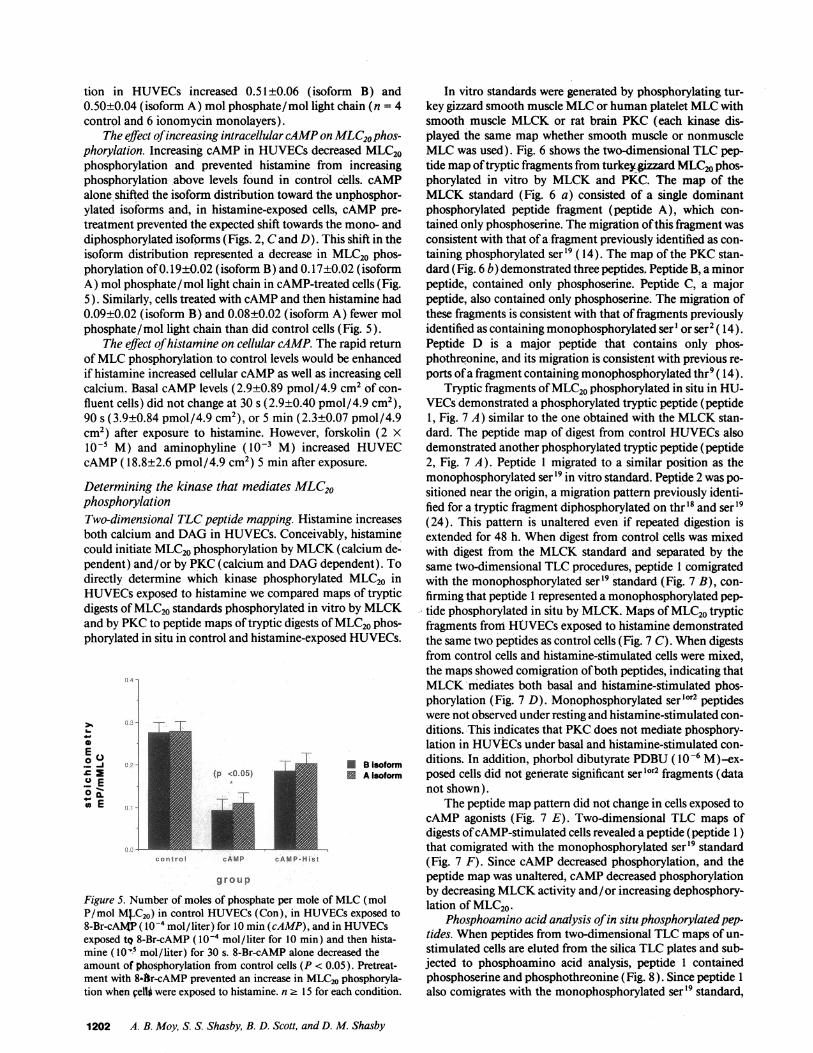
tion in HUVECs increased 0.51±0.06 (isoform B) and0.50±0.04 (isoform A) mol phosphate/mol light chain (n = 4control and 6 ionomycin monolayers).
The effect of increasing intracellular cAMPon MLC20phos-phorylation. Increasing cAMP in HUVECsdecreased MLC20phosphorylation and prevented histamine from increasingphosphorylation above levels found in control cells. cAMPalone shifted the isoform distribution toward the unphosphor-ylated isoforms and, in histamine-exposed cells, cAMP pre-treatment prevented the expected shift towards the mono- anddiphosphorylated isoforms (Figs. 2, Cand D). This shift in theisoform distribution represented a decrease in MLC20 phos-phorylation of 0.19±0.02 (isoform B) and 0.17±0.02 (isoformA) mol phosphate/mol light chain in cAMP-treated cells (Fig.5). Similarly, cells treated with cAMPand then histamine had0.09±0.02 (isoform B) and 0.08±0.02 (isoform A) fewer molphosphate/mol light chain than did control cells (Fig. 5).
The effect of histamine on cellular cAMP. The rapid returnof MLCphosphorylation to control levels would be enhancedif histamine increased cellular cAMPas well as increasing cellcalcium. Basal cAMPlevels (2.9±0.89 pmol/4.9 cm2 of con-fluent cells) did not change at 30 s (2.9±0.40 pmol/4.9 cm2),90 s (3.9±0.84 pmol/4.9 cm2), or 5 min (2.3±0.07 pmol/4.9cm2) after exposure to histamine. However, forskolin (2 Xl0-5 M) and aminophyline (l0-' M) increased HUVECcAMP(18.8±2.6 pmol/4.9 cm2) 5 min after exposure.
Determining the kinase that mediates MLC20phosphorylationTwo-dimensional TLCpeptide mapping. Histamine increasesboth calcium and DAGin HUVECs. Conceivably, histaminecould initiate MLC20phosphorylation by MLCK(calcium de-pendent) and/or by PKC(calcium and DAGdependent). Todirectly determine which kinase phosphorylated MLC20 inHUVECsexposed to histamine we compared maps of trypticdigests of MLC20standards phosphorylated in vitro by MLCKand by PKCto peptide maps of tryptic digests of MLC20phos-phorylated in situ in control and histamine-exposed HUVECs.
U.
E
o a,2n E*0
.w E
T(p <0.05)
77~
control cAMP cAMP-Hist
g r o u p
Figure 5. Number of moles of phosphate per mole of MLC(molP/mol M.C20) in control HUVECs(Con), in HUVECsexposed to8-Br-cAMP ( l0-4 mol/liter) for 10 min (cAMP), and in HUVECsexposed tQ 8-Br-cAMP ( IO-4 mol/ liter for 10 min) and then hista-mine ( I0 -' mol /liter) for 30 s. 8-Br-cAMP alone decreased theamount. of phosphorylation from control cells (P < 0.05). Pretreat-ment with 8-.r-cAMP prevented an increase in MLC20phosphoryla-tion when Delli were exposed to histamine. n 2 15 for each condition.
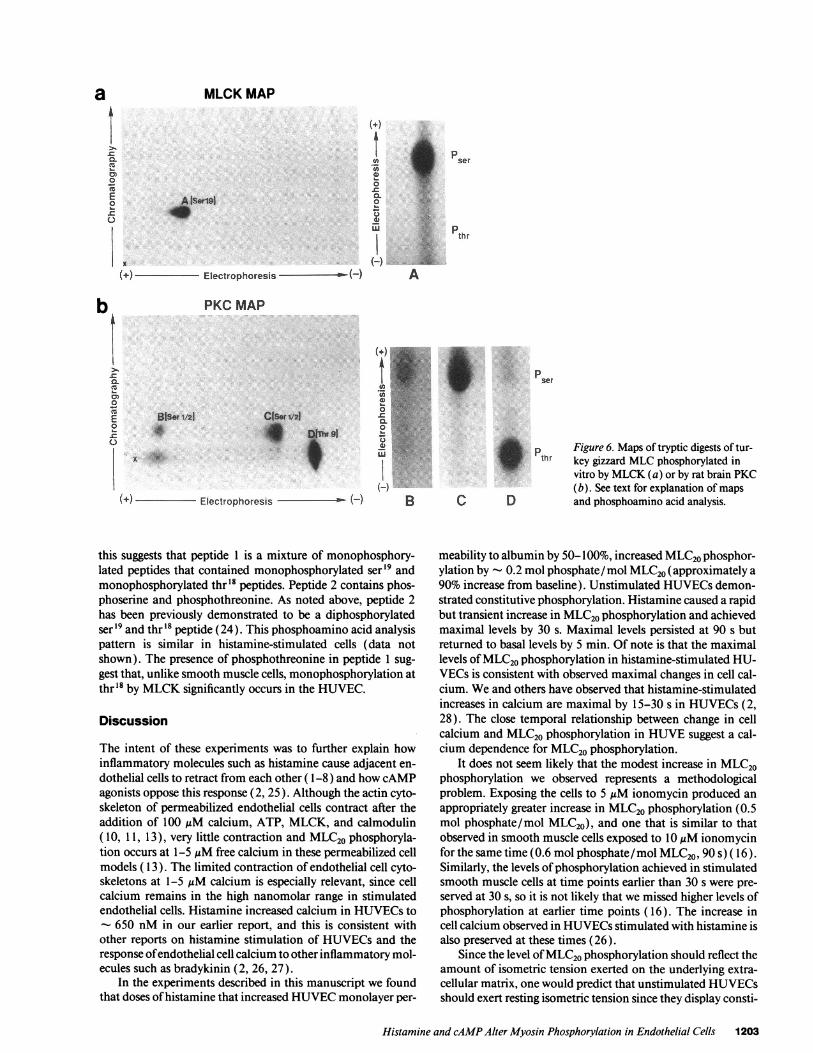
In vitro standards were generated by phosphorylating tur-key gizzard smooth muscle MLCor human platelet MLCwithsmooth muscle MLCKor rat brain PKC (each kinase dis-played the same map whether smooth muscle or nonmuscleMLCwas used). Fig. 6 shows the two-dimensional TLC pep-tide mapof tryptic fragments from turkeygizzard MLC20phos-phorylated in vitro by MLCKand PKC. The map of theMLCKstandard (Fig. 6 a) consisted of a single dominantphosphorylated peptide fragment (peptide A), which con-tained only phosphoserine. The migration of this fragment wasconsistent with that of a fragment previously identified as con-taining phosphorylated ser'9 (14). The map of the PKCstan-dard (Fig. 6 b) demonstrated three peptides. Peptide B, a minorpeptide, contained only phosphoserine. Peptide C, a majorpeptide, also contained only phosphoserine. The migration ofthese fragments is consistent with that of fragments previouslyidentified as containing monophosphorylated ser' or ser2 ( 14).Peptide D is a major peptide that contains only phos-phothreonine, and its migration is consistent with previous re-ports of a fragment containing monophosphorylated thr9 ( 14).
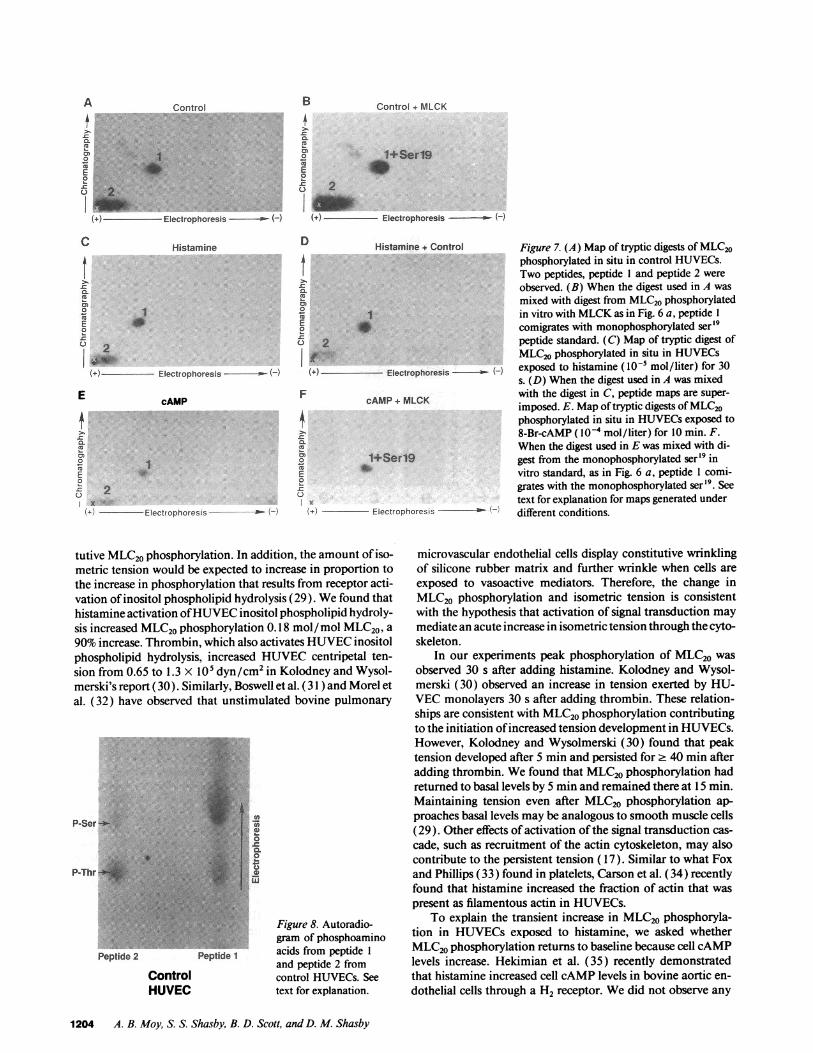
Tryptic fragments of MLC20phosphorylated in situ in HU-VECsdemonstrated a phosphorylated tryptic peptide (peptide1, Fig. 7 A) similar to the one obtained with the MLCKstan-dard. The peptide map of digest from control HUVECsalsodemonstrated another phosphorylated tryptic peptide (peptide2, Fig. 7 A). Peptide 1 migrated to a similar position as themonophosphorylated ser '9 in vitro standard. Peptide 2 was po-sitioned near the origin, a migration pattern previously identi-fied for a tryptic fragment diphosphorylated on thr'8 and ser'9(24). This pattern is unaltered even if repeated digestion isextended for 48 h. When digest from control cells was mixedwith digest from the MLCKstandard and separated by thesame two-dimensional TLC procedures, peptide 1 comigratedwith the monophosphorylated ser'9 standard (Fig. 7 B), con-firming that peptide 1 represented a monophosphorylated pep-tide phosphorylated in situ by MLCK. Maps of MLC20trypticfragments from HUVECsexposed to histamine demonstratedthe same two peptides as control cells (Fig. 7 C). Whendigestsfrom control cells and histamine-stimulated cells were mixed,the maps showed comigration of both peptides, indicating thatMLCKmediates both basal and histamine-stimulated phos-phorylation (Fig. 7 D). Monophosphorylated senor2 peptideswere not observed under resting and histamine-stimulated con-ditions. This indicates that PKCdoes not mediate phosphory-lation in HUVECsunder basal and histamine-stimulated con-ditions. In addition, phorbol dibutyrate PDBU(_10-6M)ex-posed cells did not generate significant sernr2 fragments (datanot shown).
The peptide mappattern did not change in cells exposed tocAMP agonists (Fig. 7 E). Two-dimensional TLC maps ofdigests of cAMP-stimulated cells revealed a peptide (peptide 1)that comigrated with the monophosphorylated ser'9 standard(Fig. 7 F). Since cAMPdecreased phosphorylation, and thepeptide map was unaltered, cAMPdecreased phosphorylationby decreasing MLCKactivity and/or increasing dephosphory-lation of MLC20.
Phosphoamino acid analysis of in situ phosphorylated pep-tides. Whenpeptides from two-dimensional TLC maps of un-stimulated cells are eluted from the silica TLC plates and sub-jected to phosphoamino acid analysis, peptide 1 containedphosphoserine and phosphothreonine (Fig. 8). Since peptide 1also comigrates with the monophosphorylated ser'9 standard,
1202 A. B. Moy, S. S. Shasby, B. D. Scott, and D. M. Shasby
MLCKMAP
L
5 AIser191
4,
(+) Electrophoresis -' .-(-
PKCMAP
PserL
F)
BiSer1/21
x
CIse1J21
w 9~~~~1 S(+) Electrophoresis -(-)
this suggests that peptide 1 is a mixture of monophosphory-lated peptides that contained monophosphorylated ser'9 andmonophosphorylated thr'8 peptides. Peptide 2 contains phos-phoserine and phosphothreonine. As noted above, peptide 2has been previously demonstrated to be a diphosphorylatedser19 and thr'8 peptide (24). This phosphoamino acid analysispattern is similar in histamine-stimulated cells (data notshown). The presence of phosphothreonine in peptide 1 sug-gest that, unlike smooth muscle cells, monophosphorylation atthr'8 by MLCKsignificantly occurs in the HUVEC.
Discussion
The intent of these experiments was to further explain howinflammatory molecules such as histamine cause adjacent en-dothelial cells to retract from each other ( 1-8) and how cAMPagonists oppose this response (2, 25). Although the actin cyto-skeleton of permeabilized endothelial cells contract after theaddition of 100 ,M calcium, ATP, MLCK, and calmodulin( 10, 11, 13), very little contraction and MLC20phosphoryla-tion occurs at 1-5 ,uM free calcium in these permeabilized cellmodels ( 13 ). The limited contraction of endothelial cell cyto-skeletons at 1-5 1AM calcium is especially relevant, since cellcalcium remains in the high nanomolar range in stimulatedendothelial cells. Histamine increased calcium in HUVECsto
- 650 nM in our earlier report, and this is consistent withother reports on histamine stimulation of HUVECsand theresponse of endothelial cell calcium to other inflammatory mol-ecules such as bradykinin (2, 26, 27).
In the experiments described in this manuscript we foundthat doses of histamine that increased HUVECmonolayer per-
l § \ Ail_;
+s.'' ^.:.'.,_ :y':::, _F
._ :., mr _@ a:ijX ,3;'
..,-.s::'.E s$:.
Q 1. o_ ,.,.'.'Q - E
.S ,.*: ix
...(-)
B C
pthr
D
Figure 6. Maps of tryptic digests of tur-key gizzard MLCphosphorylated invitro by MLCK(a) or by rat brain PKC(b). See text for explanation of mapsand phosphoamino acid analysis.
meability to albumin by 50-100%, increased MLC20phosphor-ylation by - 0.2 mol phosphate/ mol MLC20(approximately a90%increase from baseline). Unstimulated HUVECsdemon-strated constitutive phosphorylation. Histamine caused a rapidbut transient increase in MLC20phosphorylation and achievedmaximal levels by 30 s. Maximal levels persisted at 90 s butreturned to basal levels by 5 min. Of note is that the maximallevels of MLC20phosphorylation in histamine-stimulated HU-VECs is consistent with observed maximal changes in cell cal-cium. Weand others have observed that histamine-stimulatedincreases in calcium are maximal by 15-30 s in HUVECs(2,28). The close temporal relationship between change in cellcalcium and MLC20phosphorylation in HUVEsuggest a cal-cium dependence for MLC20phosphorylation.
It does not seem likely that the modest increase in MLC20phosphorylation we observed represents a methodologicalproblem. Exposing the cells to 5 ,M ionomycin produced anappropriately greater increase in MLC20phosphorylation (0.5mol phosphate/mol MLC20), and one that is similar to thatobserved in smooth muscle cells exposed to 10 ,M ionomycinfor the same time (0.6 mol phosphate/mol MLC20, 90 s) ( 16).Similarly, the levels of phosphorylation achieved in stimulatedsmooth muscle cells at time points earlier than 30 s were pre-served at 30 s, so it is not likely that we missed higher levels ofphosphorylation at earlier time points ( 16). The increase incell calcium observed in HUVECsstimulated with histamine isalso preserved at these times (26).
Since the level of MLC20phosphorylation should reflect theamount of isometric tension exerted on the underlying extra-cellular matrix, one would predict that unstimulated HUVECsshould exert resting isometric tension since they display consti-
Histamine and cAMPAlter Myosin Phosphorylation in Endothelial Cells 1203
a
.19Cb
CCC
I
iC.
b
pser
pthr
(A)
.;
:IAT
A Control B Control + MLCK
Cu
o 1 0 1+Serl9E WE0
.)0 2--top- (- ) --duo(+) Electrophoresis - w H- W()Electrophoresis ()
C Histamine
1E
soCC
M;_.
i__0 2
(4.) Electrophoresis a-
E cAMP
0.rCL
En2° I0
20
x1+i Electrophoresis -- (-
D
.00.iu
ECL0
2
F
0..C02
I f. ..
.c
Cu
E0
.0z-
Histamine + Control
I
Electrophoresis - - ()
cAMP+ MLCK
1+Serl90
E(+) Electrophoresis e ll-
Figure 7. (A) Map of tryptic digests of MLC20phosphorylated in situ in control HUVECs.Two peptides, peptide 1 and peptide 2 wereobserved. (B) When the digest used in A wasmixed with digest from MLC20phosphorylatedin vitro with MLCKas in Fig. 6 a, peptide 1comigrates with monophosphorylated ser'9peptide standard. (C) Map of tryptic digest ofMLC20phosphorylated in situ in HUVECsexposed to histamine ( I0 mol/liter) for 30s. (D) Whenthe digest used in A was mixedwith the digest in C, peptide maps are super-imposed. E. Mapof tryptic digests of MLC20phosphorylated in situ in HUVECsexposed to8-Br-cAMP ( l0-4 mol/liter) for 10 min. F.When the digest used in E was mixed with di-gest from the monophosphorylated ser'9 invitro standard, as in Fig. 6 a, peptide 1 comi-grates with the monophosphorylated ser 9. Seetext for explanation for maps generated underdifferent conditions.
tutive MLC20phosphorylation. In addition, the amount of iso-metric tension would be expected to increase in proportion tothe increase in phosphorylation that results from receptor acti-vation of inositol phospholipid hydrolysis (29). Wefound thathistamine activation of HUVECinositol phospholipid hydroly-sis increased MLC20phosphorylation 0.18 mol/mol MLC20, a90% increase. Thrombin, which also activates HUVECinositolphospholipid hydrolysis, increased HUVECcentripetal ten-sion from 0.65 to 1.3 x 105 dyn/cm2 in Kolodney and Wysol-merski's report (30). Similarly, Boswell et al. (3 1 ) and Morel etal. (32) have observed that unstimulated bovine pulmonary
P-Ser-.-
P-Thr it'
Peptide 2 Peptide 1
ControlHUVEC
Figure 8. Autoradio-gram of phosphoaminoacids from peptide 1and peptide 2 fromcontrol HUVECs. Seetext for explanation.
microvascular endothelial cells display constitutive wrinklingof silicone rubber matrix and further wrinkle when cells areexposed to vasoactive mediators. Therefore, the change inMLC20 phosphorylation and isometric tension is consistentwith the hypothesis that activation of signal transduction maymediate an acute increase in isometric tension through the cyto-skeleton.
In our experiments peak phosphorylation of MLC20 wasobserved 30 s after adding histamine. Kolodney and Wysol-merski (30) observed an increase in tension exerted by HU-VECmonolayers 30 s after adding thrombin. These relation-ships are consistent with MLC20phosphorylation contributingto the initiation of increased tension development in HUVECs.However, Kolodney and Wysolmerski (30) found that peaktension developed after 5 min and persisted for 2 40 min afteradding thrombin. Wefound that MLC20phosphorylation hadreturned to basal levels by 5 min and remained there at 15 min.Maintaining tension even after MLC20 phosphorylation ap-proaches basal levels may be analogous to smooth muscle cells(29). Other effects of activation of the signal transduction cas-cade, such as recruitment of the actin cytoskeleton, may alsocontribute to the persistent tension ( 17 ). Similar to what Foxand Phillips (33) found in platelets, Carson et al. (34) recentlyfound that histamine increased the fraction of actin that waspresent as filamentous actin in HUVECs.
To explain the transient increase in MLC20 phosphoryla-tion in HUVECsexposed to histamine, we asked whetherMLC20phosphorylation returns to baseline because cell cAMPlevels increase. Hekimian et al. (35) recently demonstratedthat histamine increased cell cAMPlevels in bovine aortic en-dothelial cells through a H2 receptor. Wedid not observe any
1204 A. B. Moy, S. S. Shasby, B. D. Scott, and D. M. Shasby

significant increase in cell cAMP from baseline in HUVECsexposed to histamine.
As noted above, histamine increases both calcium and dia-cylglycerol in HUVECs. Hence, histamine could activate cal-cium-calmodulin-dependent kinases (MLCK) or DAG-de-pendent kinases (PKC). Since PKC-mediated MLCphosphor-ylation has been associated with platelet and basophilactivation, it was of interest to us to determine which kinasewas responsible for phosphorylation of MLC20 in HUVECs,both at rest and after stimulation with histamine ( 14, 15). Wefound that both basal and histamine-stimulated MLC20phos-phorylation in HUVECswere mediated by MLCK, and wefound no evidence for MLCphosphorylation by PKCin HU-VECs, even when the cells were stimulated with PDBU.
Wepreviously found that increasing HUVECcAMPde-creased basal permeability and prevented histamine from in-creasing the permeability of HUVECmonolayers but did notprevent the increase in cell calcium (2). In the current experi-ments we found that increasing HUVECcAMPmarkedly re-duced phosphorylation of MLC20 and prevented an increasewith histamine. Hence, the effect of cAMPon histamine stimu-lation of MLC20 phosphorylation correlates with our earlierreport on in vitro permeability (2). We also observed thatMLCKmediates MLC20phosphorylation under cAMP-stimu-lated conditions. Our data suggest that A kinase modulatesMLC20phosphorylation indirectly by either decreasing the ba-sal activity of MLCKor enhancing phosphatase activity ( 17,36). Conti and Adelstein (37) has shown that A kinase canphosphorylate and decrease MLCKactivity. PhosphorylatedMLCKbinds poorly to the calcium-calmodulin complex, andcontraction is prevented even when signal transduction is acti-vated. On the basis of our results, it is conceivable that modula-tion of MLCKactivity may be an important regulatory step inthe control of endothelial barrier function.
In reports from others and ourselves, cAMP reduces thebasal permeability of monolayers of endothelial cells (2, 25,38). Whether a decrease in the level of MLC20phosphorylationenhances endothelial barrier function is unclear. Using siliconrubber matrix, other investigators have shown that increasingcell cAMP induces matrix relaxation (31, 32). If centripetalisometric tension opposes tethering forces that link cells andsubstrate together, then, perhaps, a decrease in MLC20 phos-phorylation could decrease opposing tension on tetheringforces which, in turn, could enhance adhesive forces betweenadjacent cells and cells to substrate. This could enhance barrierfunction. However, other effects of A kinase activation inde-pendent of MLCphosphorylation could also contribute to theenhanced barrier function, and it is not yet certain if cAMPprevents the response to histamine only by reducing MLC20phosphorylation or whether there are also other effects ofcAMPthat may prevent the increase in permeability (2).
Earlier reports of MLCK-mediated phosphorylation ofMLC20had identified ser 9 as the preferred site of MLCKphos-phorylation both in situ and in vitro in smooth muscle (24, 39,40). The thr'8 site has been shown to be diphosphorylated onlyunder extreme in vitro conditions (39). However, phosphoa-mino acid analysis of the monophosphorylated peptide fromHUVECs demonstrated phosphothreonine, suggesting thatthr'8 may be an acceptable monophosphorylation site in HU-VECs. It is unclear what effect monophosphorylation at thr'8would have on the actin-stimulated myosin ATPase activitycompared with monophosphorylation at ser'9. Diphosphory-
lation at thr'8 has been shown to augment and to have no effecton actomyosin contraction (24, 40-42). Protein sequencing ofthe threonine phosphorylated fragment will be necessary toconfirm the identity of the monophosphorylated thr 8 peptide.
The isoelectric focusing patterns of the MLC20 isoformsfrom HUVECswere similar to, but not the same as, those re-ported in human platelets and smooth muscle ( 14, 22). HU-VECs demonstrated two 1 6-kD isoforms whereas in plateletsthere was only one 16-kD isoform. Also the isoform pattern ofthe 20-kD isoforms is unique. Other work has suggested thatMLCsof nonmuscle cells may be encoded by different genesthan those in smooth muscle cells (43). Our own observationswould suggest that there are subtle differences among the iso-forms in smooth muscle, platelets, and endothelial cells, andthese differences could contribute to differences in functionalresponse.
In summary, doses of histamine that increase the permeabil-ity of monolayers of HUVECscaused a modest increase inMLC20phosphorylation in HUVECs, and the phosphorylationwas mediated by MLCK. The amount of histamine-stimulatedphosphorylation was consistent with the increase in HUVECcalcium that occurs with histamine stimulation, and furtherincreases in cell calcium with ionomycin caused an appropri-ately greater increase in MLC20phosphorylation. Increases incell cAMPreduced basal phosphorylation of MLC20and pre-vented the histamine-stimulated increase in MLC20phosphor-ylation. Although these data are consistent with the hypothesisthat MLC20phosphorylation contributes to retraction of HU-VECs stimulated with histamine, it does not rule out othermechanisms that may regulate cell retraction during inflamma-tion independent of actomyosin contraction.
Acknowledgments
Dr. Moy is a recipient of a Training Fellowship Grant from the Ameri-can Heart Association-Iowa Affiliate and the National Heart, Lung,and Blood Institute. This work was completed during Dr. D. M.Shasby's tenure as a Clinical Investigator and Dr. Scott's tenure as aResearch Associate of the Veterans Administration. The work was alsosupported by National Institutes of Health grant HL-33540 and Ameri-can Lung Research grant 35131.
References
1. Albertine, K. H., J. Weiner-Kronish, K. Koike, and N. C. Staub. 1984.Quantification of damage to lung microvessels in anesthetized sheep. J. Appl.Physiol. 57:1360-1368.
2. Carson, M., S. Shasby, and D. M. Shasby. 1989. Histamine and inositolphosphate accumulation in endothelium: cAMPand G-protein. Am. J. Physiol.257:L259-L264.
3. Laposata, M., D. Dovnarsky, and H. Shin. 1983. Thrombin-induced gapformation in confluent endothelial cell monolayers in vitro. Blood. 62:549-556.
4. Majno, G., and G. Palade. 1961. Studies on inflammation. 1. Effect ofhistamine and serotonin on vascular permeability: an electron microscopic study.J. Biophys. Biochem. Cytol. 11:571-605.
5. Shasby, D. M., S. Lind, S. Shasby, J. Goldsmith, and G. Hunninghake.1985. Reversible oxidant-induced increases in albumin transfer across culturedendothelium: alterations in cell shape and calcium homeostasis. Blood. 65:605-614.
6. Shasby, D. M., S. Shasby, J. Sullivan, and M. Peach. 1982. Role of endothe-lial cell cytoskeleton in control of endothelial permeability. Circ. Res. 51:657-661.
7. Shasby, D. M., and S. Shasby. 1986. Effects of calcium on transendothelialalbumin transfer and electrical resistance. J. Appl. Physiol. 60:71-79.
8. Wysolmerski, R., and D. Lagunoff. 1985. The effect of ethchlorvynol oncultured endothelial cells-a model for the study of the mechanism of increasedvascular permeability. Am. J. Pathol. 11 9:505-512.
9. Nicolaysen, G. 1971. Intravascular concentrations of calcium and magne-
Histamine and cAMPAlter Myosin Phosphorylation in Endothelial Cells 1205

sium ions and edema-formation in isolated lungs. Acta Physiol. Scand. 81:325-339.
10. Schnittler, H., A. Wilke, T. Gress, N. Suttorp, and D. Drenckhahn. 1990.Role of actin and myosin in the control of paracellular permeability in pig, rat andhuman vascular endothelium. J. Physiol. 431:379-401.
11. Wysolmerski, R., and D. Lagunoff. 1990. Involvement of myosin lightchain kinase in endothelial cell retraction. Proc. Natl. Acad. Sci. USA. 87:16-20.
12. Wysolmerski, R., and D. Lagunoff. 1988. Inhibition of endothelial cellretraction by ATPdepletion. Am. J. Pathol. 132:28-37.
13. Wysolmerski, R., and D. Lagunoff. 1991. Regulation of permeabilizedendothelial cell retraction by myosin phosphorylation. Am. J. Physiol. 261:C32-C40.
14. Kawamoto, S., A. Bengur, J. Sellers, and R. Adelstein. 1989. In situ phos-phorylation of human platelet myosin heavy and light chains by protein kinase C.J. Biol. Chem. 264:2258-2265.
15. Ludowyke, R., I. Peleg, M. Beaven, and R. Adelstein. 1989. Antigen-in-duced secretion of histamine and the phosphorylation of myosin by protein ki-nase C in rat basophilic leukemic cells. J. Biol. Chem. 264:12492-12501.
16. Taylor, D., and J. Stull. 1988. Calcium dependence of myosin light chainphosphorylation in smooth muscle cells. J. Biol. Chem. 263:14456-14462.
17. Lamb, N., A. Fernandez, M. Conti, R. Adelstein, D. Glass, W. Welch, andJ. Feramisco. 1988. Regulation of actin microfilament integrity in living non-muscle cells by cAMP-dependent protein kinase and the myosin light chain ki-nase. J. Cell Biol. 106:1955-1971.
18. Daniel, J., and R. Adelstein. 1976. Isolation and properties of plateletmyosin light chain kinase. Biochemistry 15:2370-2377.
19. Sellers, J., M. Soboeiro, K. Fraust, A. Bengur, and E. Harvey. 1988. Prepa-ration and characterization of heavy meromyosin and subfragment 1 from verte-brate cytoplasmic myosin. Biochemistry. 27:6977-6982.
20. Isaacs, W., and A. Fulton. 1987. Cotranslational assembly of myosinheavy chain in developing cultured skeletal muscle. Proc. Natl. Acad. Sci. USA.84:6174-6178.
21. Gracy, R. 1977. Two dimensional thin layer methods. Methods Enzymol.47:195-204.
22. Singer, H. 1990. Protein kinase Cactivation and myosin light chain phos-phorylation in 32P-labeled arterial smooth muscle. Am. J. Physiol. 259:C63 1-C639.
23. Wilson, J., M. Winter, and D. Shasby. 1990. Oxidants, ATP depletion,and endothelial permeability to macromolecules. Blood. 76:2578-2582.
24. Haeberle, J., T. Sutton, and B. Trockman. 1988. Phosphorylation of twosites on smooth muscle myosin: effects on contraction of glycerinated vascularsmooth muscle. J. Biol. Chem. 263:4424-4429.
25. Stelzner, T., J. Weil, and R. O'Brien. 1989. Role of cyclic adenosinemonophosphate in the induction of endothelial barrier properties. J. Cell. Phys-iol. 139:157-166.
26. Jacob, R., J. Meritt, T. Hallam, and T. Rink. 1988. Repetitive spikes incytoplasmic calcium evoked by histamine in human endothelial cells. Nature(Lond.). 335:40-45.
27. Schilling, W. 1989. Effect of membrane potential on cytosolic calcium ofbovine aortic endothelial cells. Am. J. Physiol. 257:H778-H784.
28. Rotrosen, D., and J. Gallin. 1986. Histamine type I receptor occupancyincreases endothelial cytosolic calcium, reduces F-actin, and promotes albumindiffusion across cultured endothelial monolayers. J. Cell. Biol. 103:2379-2387.
29. Hai, C., and R. Murphy. 1988. Sr2" activates cross-bridge phosphoryla-tion and latch state in smooth muscle. Am. J. Physiol. 255:C401-C407.
30. Kolodney, M., and R. Wysolmerski. 1992. Isometric contraction by fibro-blasts and endothelial cells in tissue culture: a quantitative study. J. Cell Biol.117:73-82.
31. Boswell, C., G. Majno, I. Joris, and K. Ostrom. 1992. Acute endothelialcell contraction in vitro: a comparision with vascular smooth muscle cells andfibroblasts. Microvasc. Res. 43:178-191.
32. Morel, N., A. Dodge, W. Patton, I. Herman, H. Hechtman, and D.Shepro. 1989. Pulmonary microvascular endothelial cell contractility on siliconerubber substrate. J. Cell. Physiol. 141:653-659.
33. Fox, J., and D. Phillips. 1982. Role of phosphorylation in mediating theassociation of myosin with the cytoskeletal structures of human platelets. J. Biol.Chem. 257:4120-4126.
34. Carson, M., S. Shasby, S. Lind, and D. M. Shasby. 1992. Histamine,actin-gelsolin binding and polyphosphoinositides in human umbilical vein endo-thelial cells. Am. J. Physiol. (Lung Cell Mol. Physiol. 7) 263:L664-L669.
35. Hekimian, G., S. Cote, J. V. Sande, and J. M. Boeynaems. 1992. H2receptor-mediated responses of aortic endothelial cells to histamine. Am. J. Phys-iol. 262:H220-H224.
36. DeLanerolle, P., M. Nishikawa, D. Yost, and R. Adelstein. 1984. In-creased phosphorylation of myosin light chain kinase after an increase in cyclicAMPin intact smooth muscle. Science (Wash. DC). 223:1415-1417.
37. Conti, M., and R. Adelstein. 1980. Phosphorylation by cyclic adenosine3':5'-monophosphate dependent protein kinase regulates myosin light chain ki-nase. Fed. Proc. 39:1569-1573.
38. Langeler, E., and V. VanHinsbergh. 1991. Norepinephrine and iloprostimprove barrier function of human endothelial cell monolayers: role of cAMP.Am. J. Physiol. 260:C1052-C1059.
39. Ikebe, M., and D. Hartshorne. 1985. Phosphorylation of smooth musclemyosin at two distinct sites by myosin light chain kinase. J. Biol. Chem.260:10027-10031.
40. Umemoto, S., A. Bengur, and J. Sellers. 1989. Effect of multiple phos-phorylation of smooth muscle and cytoplasmic myosin in an in vitro motilityassay. J. Biol. Chem. 264:1431-1436.
41. Ikebe, M. 1989. Phosphorylation of a second site for myosin light chainkinase on platelet myosin. Biochemistry 28:8750-8755.
42. Itoh, K., T. Hara, and N. Shibata. 1992. Diphosphorylation of plateletmyosin by myosin light chain kinase. Biochim. Biophys. Acta. 1133:286-292.
43. Kumar, C., S. Mohan, P. Zavodny, S. Narula, and P. Leibowitz. 1989.Characterization and differential expression of human vascular smooth musclemyosin light chain 2 isoform in nonmuscle cells. Biochemistry. 28:4027-4035.
1206 A. B. Moy, S. S. Shasby, B. D. Scott, and D. M. Shasby






![KB Id - UNT Digital Library/67531/metadc332161/... · 1-[bis(hydroxymethyl)amino]-3-tris(hydroxymethyl)propane adenosine 3',5'-monophosphate adenosine 31,5'-monophosphate dependent](https://img.pdfslide.us/doc/110x75/60bf6195247f5a484a422257/kb-id-unt-digital-library-67531metadc332161-1-bishydroxymethylamino-3-trishydroxymethylpropane.jpg)