Embed Size (px)
Citation preview

Movement Disorders and Extrapyramidal System
Sibel Ertan, MDDept. of Neurology

Definition Neurologic syndromes in which there is either an excess of movement, or a paucity of voluntary and automatic movements unrelated to weakness or
spasticity.

Most movement disorders are associated with pathologic alterations in the
basal ganglia or their connections.

But disorders of the• Cerebellum or its pathways• Cerebral cortex• Thalamus• Brain stem• Spinal cord• Peripheral nerves may also cause several movementdisorders.
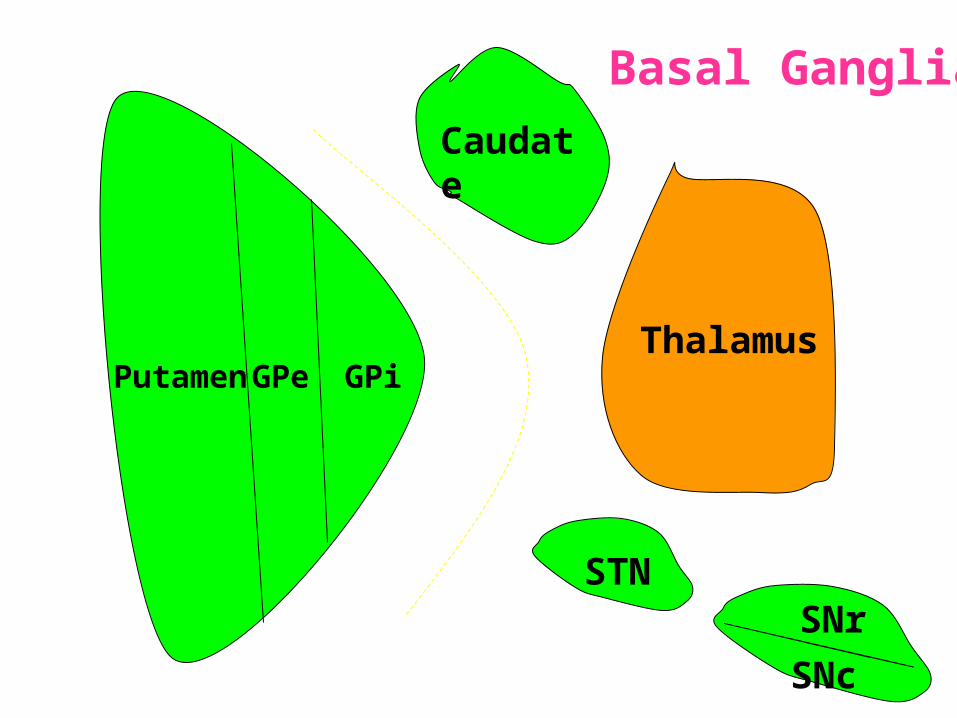
PutamenThalamus
STNSNr
GPiGPe
Caudate
SNc
Basal Ganglia
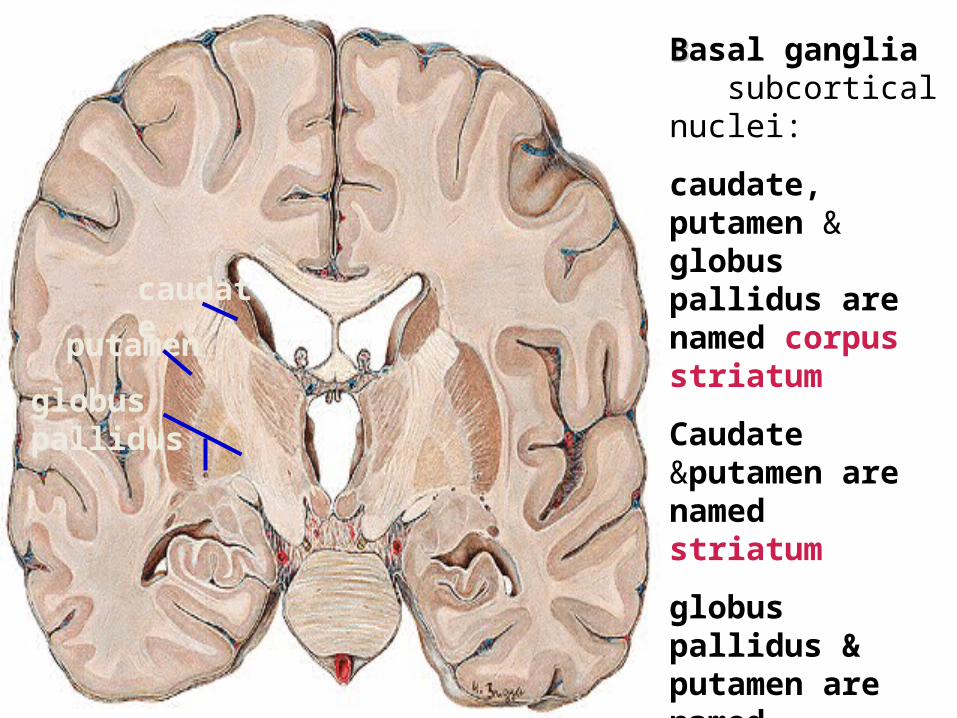
BBasal ganglia subcortical nuclei:
caudate, putamen & globus pallidus are named corpus striatum
Caudate &putamen are named striatum
globus pallidus & putamen are named lentiform nuclei
caudate putame
nglobus pallidus

Definition
The term extrapyramidal system, coined by British neurologist Kinnier Wilson, refers to the basal ganglia and an array of brain stem nuclei (red nucleus, reticular formation etc.)
to which they are connected.

Striatum (caudate+putamen) is the principle receptive structure of the basal
ganglia.
Globus pallidus is the principle output structure of the basal ganglia.
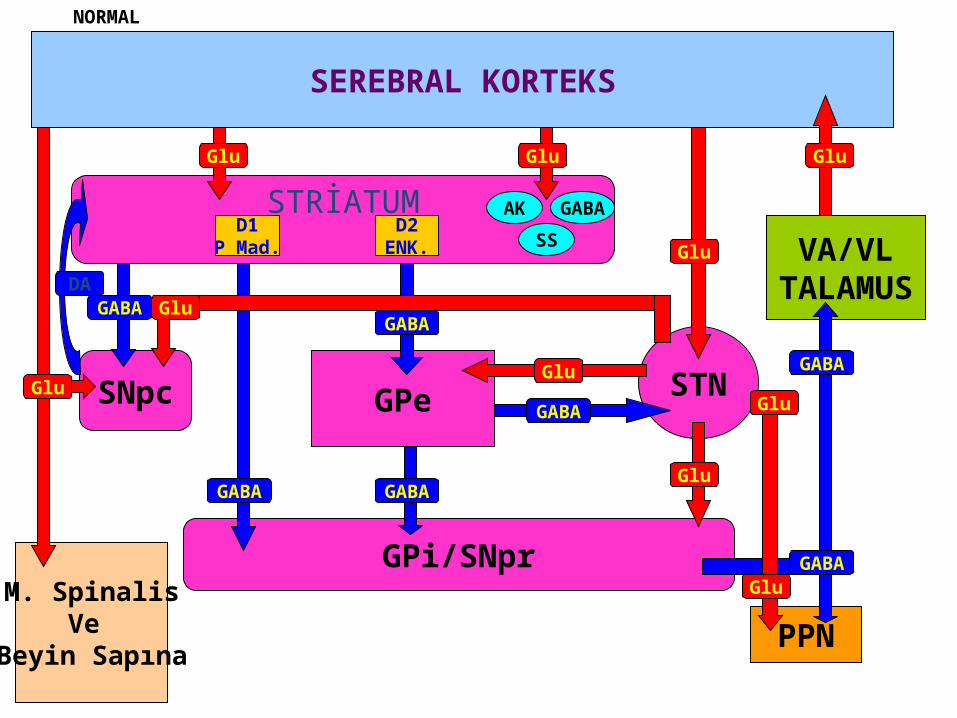
STRİATUM
SEREBRAL KORTEKS
Glu
D1P Mad.
D2ENK.
AK
SS
GABA
Glu
GPi/SNpr
GABA
SNpc
M. SpinalisVe
Beyin Sapına
GABA
Glu
DA
GPe
GABA
GABA
STNGABA
Glu
PPN
VA/VLTALAMUS
Glu
Glu
GABA
Glu
GABA
Glu
Glu
Glu
NORMAL
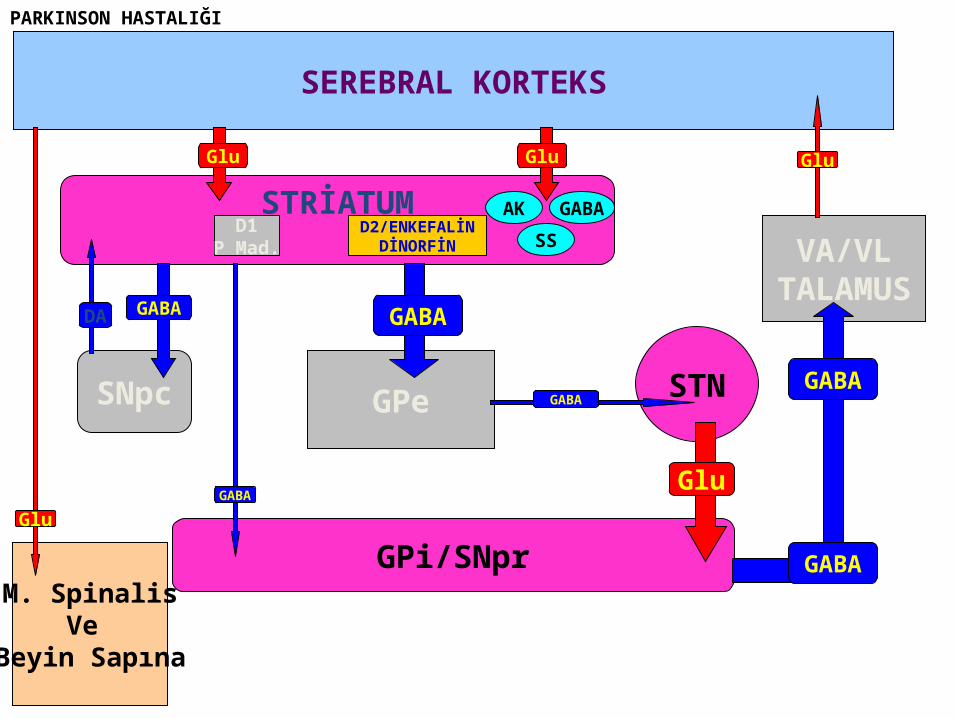
STRİATUM
SEREBRAL KORTEKS
Glu
D1P Mad.
D2/ENKEFALİNDİNORFİN
AK
SS
GABA
Glu
GPi/SNpr
GABA
SNpc
M. SpinalisVe
Beyin Sapına
GABA
Glu
GPe STNGABA
VA/VLTALAMUS
Glu
GABA
Glu
GABA
GABADA
PARKINSON HASTALIĞI
STRİATUM
SEREBRAL KORTEKS
Glu
D1P Mad.
D2ENK.
AK
SS
GABA
Glu
GPi/SNpr
GABA
SNpc
M. SpinalisVe
Beyin Sapına
GABA
Glu
DA
GPe
GABA
STNGABA
VA/VLTALAMUS
GABA
Glu
Glu
GABA
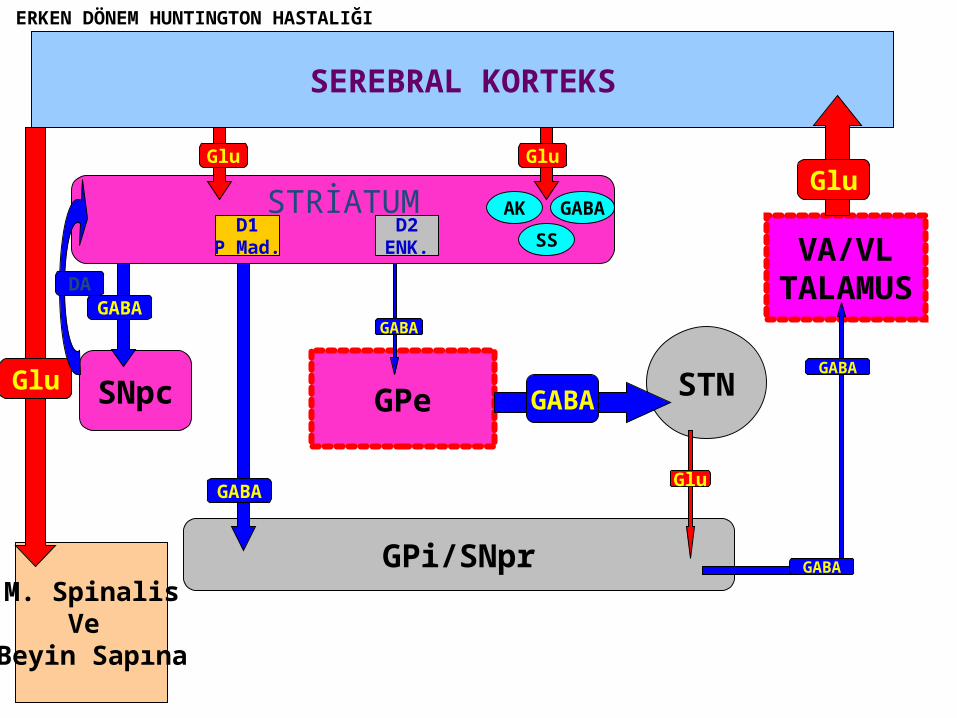
ERKEN DÖNEM HUNTINGTON HASTALIĞI

• Diseases of the basal ganglia are associated with abnormal involuntary movements that typically occur at rest and disappear in sleep.
• They are generally divided into two categories: Hyperkinetic and hypokinetic
• The hyperkinetic variety is seen in such disorders as chorea, athetosis, ballism, dystonia, tremor, and tics.
• The hypokinetic variety is seen largely in Parkinson’s disease and Parkinson plus syndromes
• Following anatomic loci for pathology are agreed on:- Substantia nigra in Parkinson’s disease- Caudate nucleus in chorea- Subthalamic nucleus in ballism- Caudate or lentiform nucleus (especially putamen) in dystonia

Hypokinetic Disorders

• ParkinsonismSix cardinal features:
1. Tremor at rest2. Rigidity3. Bradykinesia-hypokinesia4. Flexed posture
5. Loss of postural reflexes6. Freezing phenomenon

• Tremor, rigidity, and flexed posture are referred positive phenomena.
• Bradykinesia, loss of postural reflexes, and freezing are negative phenomena.

Rest Tremor• 4-5 Hz• Present in the extremities, almost always distally• Classic “pill-rolling” tremor involves the thumb
and the forefinger• Rest tremor disappears with action but
reemerges as the limb maintain a posture.• Rest tremor is also common in the lips, chin, and
tongue• Rest tremor of the hands increases with walking• Stress worsens the tremor

Rest tremor

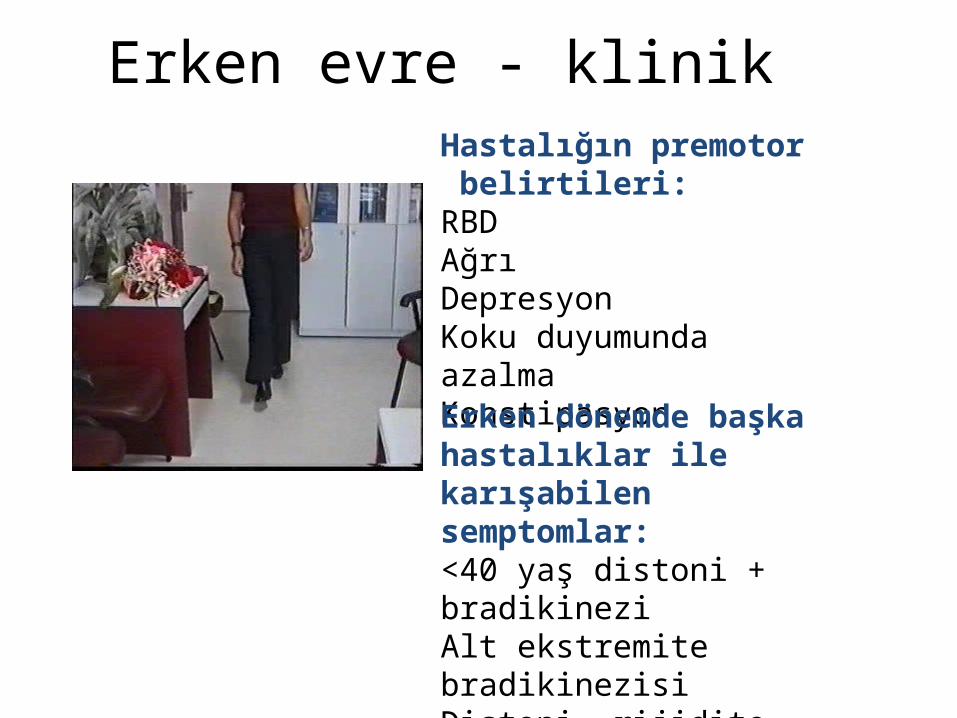
Erken evre - klinikHastalığın premotor belirtileri:RBDAğrıDepresyonKoku duyumunda azalmaKonstipasyonErken dönemde başka hastalıklar ile karışabilen semptomlar:<40 yaş distoni + bradikineziAlt ekstremite bradikinezisiDistoni, rijidite, ağrı

Rigidity
• Increased resistance (muscle tone) to passive movement elicited when the examiner moves the patient’s limbs, neck or trunk
• Equal in all directions• The underlying tremor may cause
“cogwheeling”

Flexed posture
• Commonly begins in the arms and spreads to involve the entire body
• Striatal hand• Striatal toe• Lateral tilting of the trunk is common

Posture

Bradykinesia
• Slowness of movement, difficulty in initiating a movement, and loss of automatic movement
• Hypokinesia is the reduction in amplitude of movement

Bradykinesia

Loss of postural reflexes
• Pulltest is positive• With progress of the disease frequent fallings• The patient collapses into the chair on
attempting to sit down (sitting en bloc)

Freezing
• Inability to perform active movements (motor block)
• Often involves the legs when walking but can also involve eyelid opening,speaking and writing.

Freezing

Freezing

The many causes of parkinsonism are
divided into four categories:
1.Idiopathic (%77.7)
2. Parkinson-plus syndromes (%12.2)
3. Symptomatic (secondary) (%8.2)
4. Heredodegenerative diseases (%0.6)

1.Idiopathic (%77.7)- %10 genetic- %90 unknown
2. Parkinson-plus syndromes (%12.2)- Multisiystem atrophy- Alzheimer’s disease- Lewy body dementia- Progressive supranuclear palsy- Lytigo-bodig (ALS-dementia-Parkinsonism
3. Symptomatic (secondary) (%8.2)- Toxic- Metabolic- Drug induced- Lesions- Infections
4. Heredodegenerative diseases (%0.6)

The core biochemical pathology in parkinsonism is decreased dopaminergic neurotransmission in the basal ganglia.
• Degeneration of the nigrostriatal dopamine system• Degeneration of the striatum with loss of dopamine
receptors• Drug induced parkinsonism as the result of blockade of
dopamine receptors.
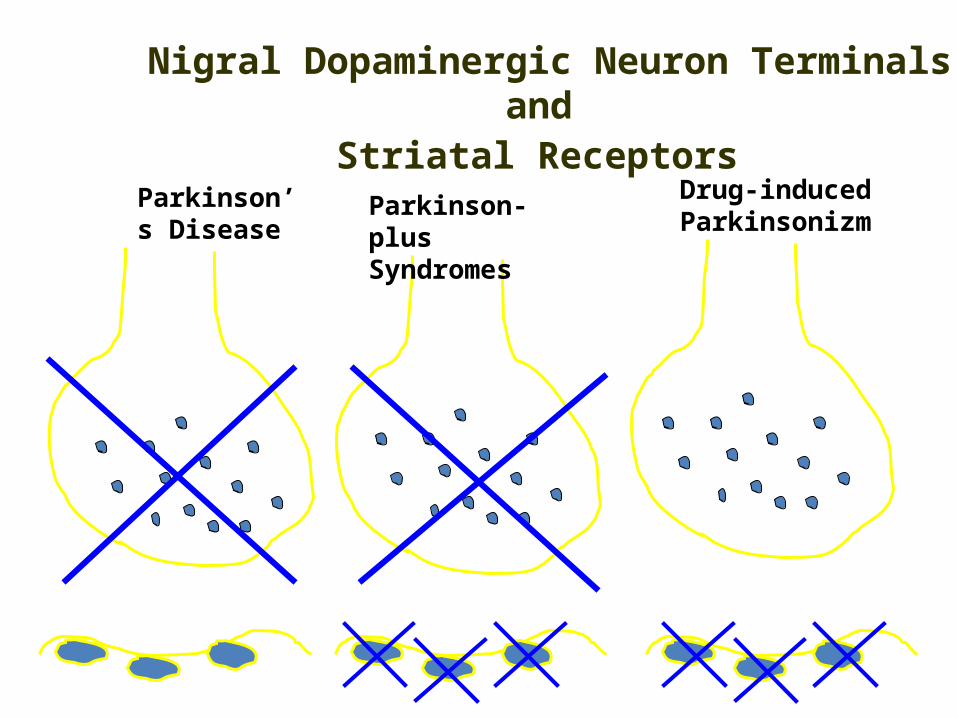
Parkinson’s Disease
Parkinson-plus Syndromes
Drug-induced Parkinsonizm
Nigral Dopaminergic Neuron Terminals and
Striatal Receptors

Parkinson’s disease – HistoryParkinson’s disease – History
1817: James Parkinson
“Shaking Palsy”
1960: Dopaminergic neuronal
loss in substantia nigra
1960: Levodopa
1982: MPTP (ABD, California)
(toxic substance in
synthetic heroin)
After 1982: Experimental
models
2005: Etiopathogenesis ?

Parkinson’s disease (primary parkinsonism)Parkinson’s disease (primary parkinsonism)
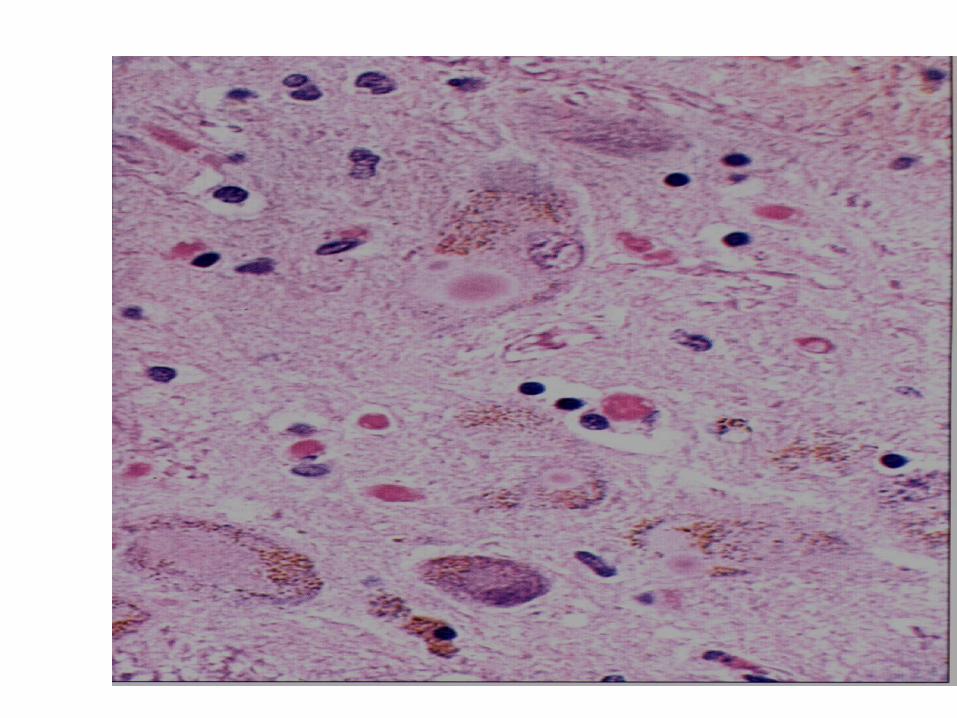
• Degeneration of the neuromelanin-containing neurons in the brain stem, especially in substantia nigra pars compacta and in the locus ceruleus.
• Many of the surviving neurons contain eosinophilic cytoplasmic inclusions known as Lewy bodies.
• By the time symptoms appear, the substantia nigra already has lost about 60% of dopaminergic neurons and the dopamine content in the striatum is about 80% less than normal.

Parkinson’s disease (primary parkinsonism)Parkinson’s disease (primary parkinsonism)
• PD makes up approximately 80% of cases of parkinsonism.
• Mean age at onset in both sexes is 55 years
(range:20-80).• Over 60 yrs of age risk of Parkinson’s disease is %1• Male/female = 3/2.• Prevalence 160/100.000 and incidence 20/100.000/yr.• The cause of PD is unknown.

Parkinson’s DiseaseParkinson’s Disease Unilateral onset Frequent initial sympton: unilateral
upper ektremity tremor Bilateral involvement All over the course of the disease
asymmetric involvement

Clinical Staging of PH According Clinical Staging of PH According to Hoehn-Yahr Scaleto Hoehn-Yahr Scale
0- Asymptomatic1- Unilateral involvement2- Bilateral involvement3- Involvement of postural reflexes, imbalance and falls Mild-moderate morbidity4- Needs continious support5- Bedridden

Parkinson’s Disease - SymptomatologyParkinson’s Disease - Symptomatology Tremor: Static postural, 3-7 Hz, (hand, arm, foot, leg, tongue, chin, lip) Bradykinesia - Hypomimia - Micrography Rijidity Disarthria (hipophoni, palilali) Gait disorder (short steps) Motor blocks (freezing) Loss of associated movements of the arms Flexed posture

Disphagia Loss of postural reflexes, falls Pain, sensory complaints Autonomic findings: Constipation, postural
hypotension, sweeting, urinary incontinans, empotans
Depression: ~% 50 of cases Dementia: Mild, % 20-40 of cases
Parkinson’s Disease - SymptomatologyParkinson’s Disease - Symptomatology

Diagnosis of PDDiagnosis of PD
Based on clinical findings and signs
No radiologic “marker” No laboratory “marker”

Parkinson’s disease (primary parkinsonism)Parkinson’s disease (primary parkinsonism)TreatmentTreatment
• Treatment is aimed at controlling symptoms because no drug or surgicl approach unequivocally prevents progression of PD.
• Treatment is lifelong.• Treatment includes pharmacotherapy, physiotherapy
and surgery.

Motor semptomların semptomatik tedavisi• Dopaminerjik ajanlar
– Levodopa• Levodopa + karbidopa• Levodopa + benserazid• COMT inhibitörleri* (entakapon,
tolkapon) – Dopamin agonistleri
• Non-ergo† – Pramipeksol– Ropinirol– Rotigotin– Piribedil– Apomorfin
• Ergot deriveleri– Bromokriptin– Pergolid– Kabergolin– Dihidroergokriptin– Lisurid
– Selektif MAO-B‡ inhibitörleri• Selejilin• Rasajilin
• Non-dopaminerjik ajanlar– Antikolinerjik ajanlar:
• Triheksifenidil• Benztropin
– NMDA§ antagonistleri• Amantadin
* Katekol-O- metil transferaz inhibitörleri ; her zaman L-dopa ile birlikte kullanılırlar
† Apomorfinin subkutan enjeksiyon formları var ve L-dopa ile ilişkili motor fluktuasyonlarda kullanılır
‡ Monoamin oksidaz tip B§ N-metil-D-aspartat
Schapira AHV, Olanow CW. In: Principles of Treatment in Parkinson’s Disease; 2005.
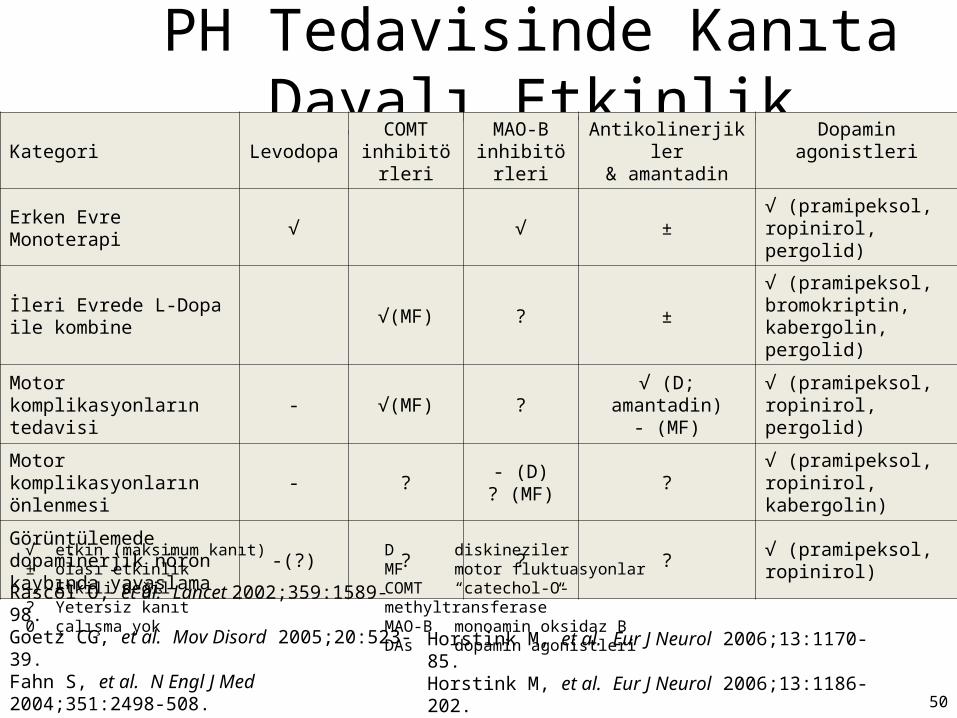
PH Tedavisinde Kanıta Dayalı EtkinlikKategori Levodopa
COMT inhibitörleri
MAO-B inhibitörleri
Antikolinerjikler& amantadin
Dopamin agonistleri
Erken Evre Monoterapi √ √ ±√ (pramipeksol, ropinirol, pergolid)
İleri Evrede L-Dopa ile kombine
√(MF) ? ±√ (pramipeksol, bromokriptin, kabergolin, pergolid)
Motor komplikasyonların tedavisi
- √(MF) ?√ (D; amantadin)
- (MF)√ (pramipeksol, ropinirol, pergolid)
Motor komplikasyonların önlenmesi
- ?- (D)
? (MF)?
√ (pramipeksol, ropinirol, kabergolin)
Görüntülemede dopaminerjik nöron kaybında yavaşlama
-(?) ? ? ?√ (pramipeksol, ropinirol)
√ etkin (maksimum kanıt)± olası etkinlik- Etkili değil? Yetersiz kanıt0 çalışma yok
D diskinezilerMF motor fluktuasyonlarCOMT “catechol-O-methyltransferase”MAO-B monoamin oksidaz BDAs dopamin agonistleri
Rascol O, et al. Lancet 2002;359:1589-98.Goetz CG, et al. Mov Disord 2005;20:523-39.Fahn S, et al. N Engl J Med 2004;351:2498-508.
Horstink M, et al. Eur J Neurol 2006;13:1170-85.Horstink M, et al. Eur J Neurol 2006;13:1186-202. 50
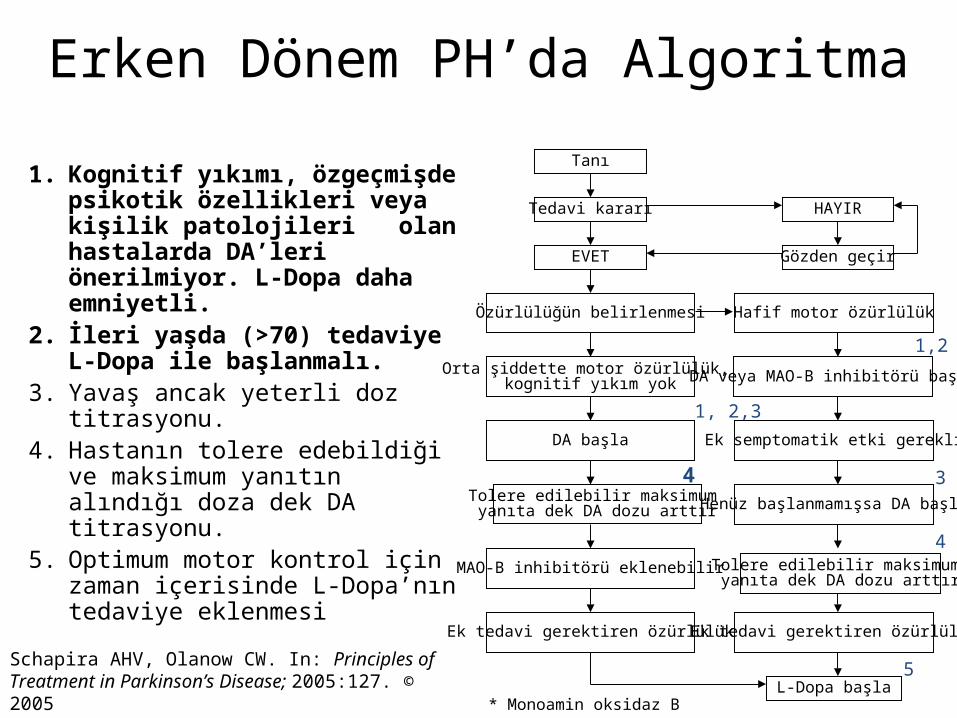
Erken Dönem PH’da Algoritma
1. Kognitif yıkımı, özgeçmişde psikotik özellikleri veya kişilik patolojileri olan hastalarda DA’leri önerilmiyor. L-Dopa daha emniyetli.
2. İleri yaşda (>70) tedaviye L-Dopa ile başlanmalı.
3. Yavaş ancak yeterli doz titrasyonu.4. Hastanın tolere edebildiği ve maksimum
yanıtın alındığı doza dek DA titrasyonu.5. Optimum motor kontrol için zaman
içerisinde L-Dopa’nın tedaviye eklenmesi
Schapira AHV, Olanow CW. In: Principles of Treatment in Parkinson’s Disease; 2005:127. © 2005
Tanı
Tedavi kararı
EVET
Özürlülüğün belirlenmesi
Orta şiddette motor özürlülük, kognitif yıkım yok
DA başla
Tolere edilebilir maksimum yanıta dek DA dozu arttır
MAO-B inhibitörü eklenebilir
Ek tedavi gerektiren özürlülük
HAYIR
Gözden geçir
Hafif motor özürlülük
DA veya MAO-B inhibitörü başla
Ek semptomatik etki gerekli
Henüz başlanmamışsa DA başla
Ek tedavi gerektiren özürlülük
L-Dopa başla
1, 2,3
1,2
3
4
5
* Monoamin oksidaz B
Tolere edilebilir maksimum yanıta dek DA dozu arttır
4

Parkinson’s disease (primary parkinsonism)Parkinson’s disease (primary parkinsonism)TreatmentTreatment
Therapeutic choices for Parkinson’s disease
Medications
Dopamine precursor: levodopa (LD)±carbidopa or benserazide
Dopamine agonists: bromocriptine, pergolide, pramipexole,
ropinirole, apomorphine, cabergoline, pribedil.
Catecholamine-O-methyl transferase inhibitors:tolcapone and
entacapone.
Dopamine releaser, NMDA receptor antagonist: Amantadine.
Monoamine oxidase type B inhibitor: selegiline. rasagiline
Anticholinergics:trihexyphenidyl, benztropine, biperidene...
Antihistaminics:diphenhydramine, orphenadrine, phenindamine

Parkinson’s disease (primary parkinsonism)Parkinson’s disease (primary parkinsonism)TreatmentTreatment
Therapeutic choices for Parkinson’s disease
Surgery
Ablative surgery:Thalamotomy, pallidotomy.
Restorative surgery:Embryonic dopaminergic tissue
transplantation
Deep brain stimulation:Thalamic stimulation, pallidal
stimulation, subthalamic stimulation, PPN

Parkinson’s disease (primary parkinsonism)Parkinson’s disease (primary parkinsonism)TreatmentTreatment
• LD is the most effective drug, BUT 75% of patients have serious complications after 5 years of LD therapy.
• Younger patients, in particular, are more likely to show response fluctuations.
• DOPA-SPARING STRATEGY:Other antiparkinsonian drugs should be used first to delay the introduction of LD.
• Selegiline delays the need for LD therapy by an average of 9 months.

Adverse events and side Adverse events and side effects of L-Dopaeffects of L-Dopa
Noisea, vomiting Postural hypotension Psychosis: Hallucinations
delusions

Late Complications of L-DopaLate Complications of L-Dopa
Motor Complications1. “wearing-off” End of dose
phenomenon (predictable)
2. “On-off” fluctuations (unpredictable)
3. Dyskinesias:
- Chorea (“on” period)
- Dystonia (“on” or “of” period)

PD- Motor Complication

L-Dopa induced motor complications “on” period
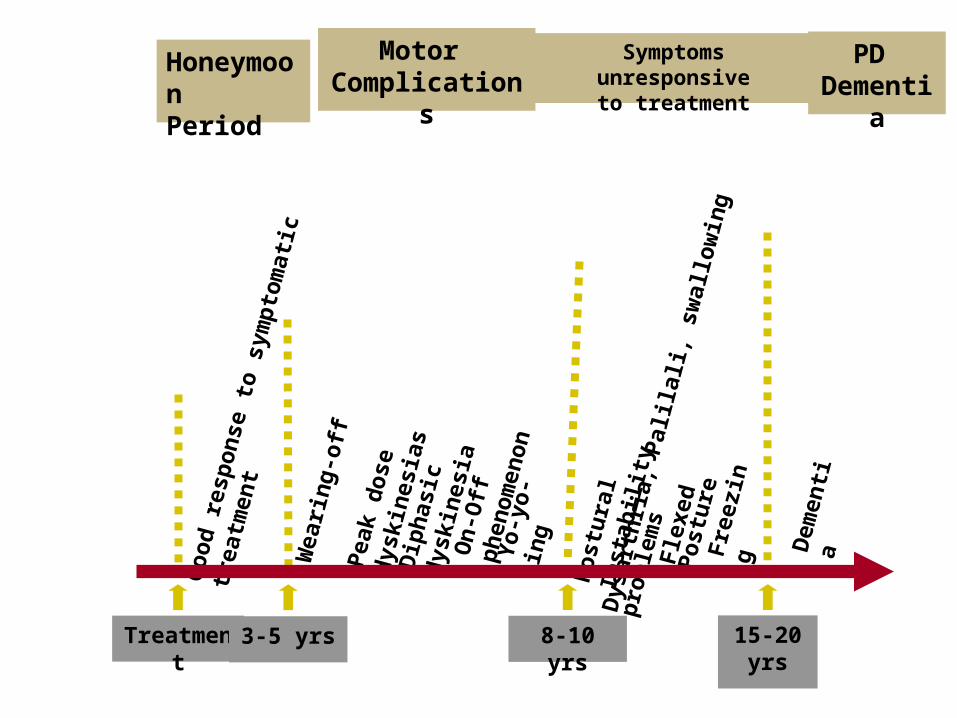
HoneymoonPeriod
Motor Complication
s
Treatment
Symptoms unresponsiveto treatment
PD Dementi
a
3-5 yrs
8-10 yrs
15-20 yrs
Wea
rin
g-
off
P
eak
do
se
dys
kin
esia
sD
iph
asic
d
yski
nes
iaO
n-O
ff
ph
eno
men
on
Yo
-yo
-in
gGo
od
res
po
nse
to
sym
pto
mat
ic
trea
tmen
t
Po
stu
ral
Inst
abili
tyD
ysar
thri
a, P
alila
li, s
wal
low
ing
pro
ble
ms
Fle
xed
P
ost
ure
Fre
ezin
g Dem
enti
a
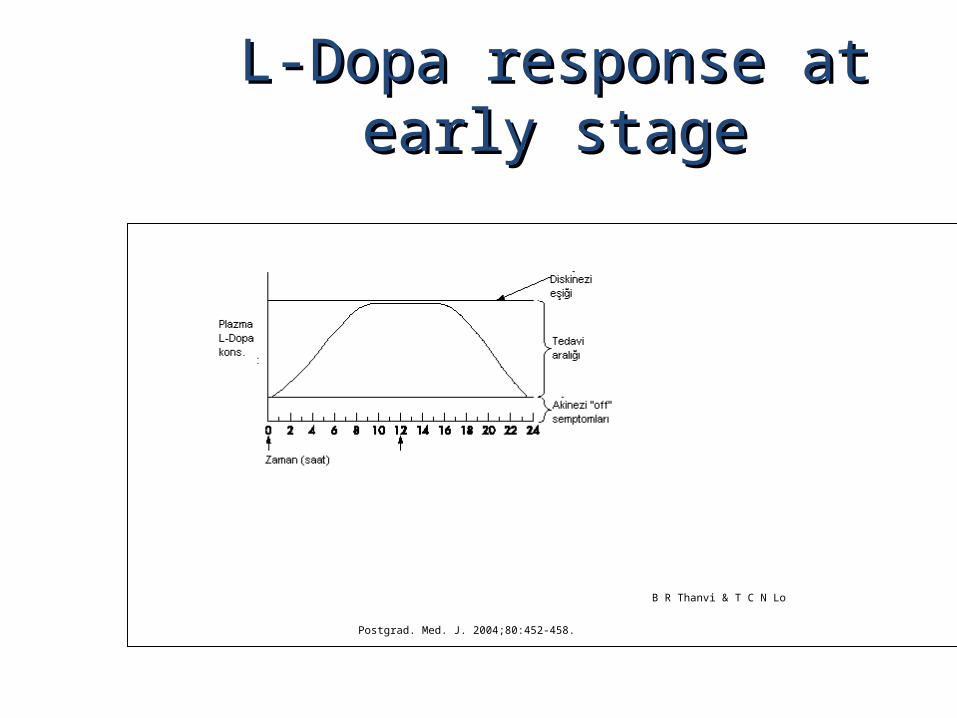
L-Dopa response at early stageL-Dopa response at early stage
B R Thanvi & T C N Lo
Postgrad. Med. J. 2004;80:452-458.
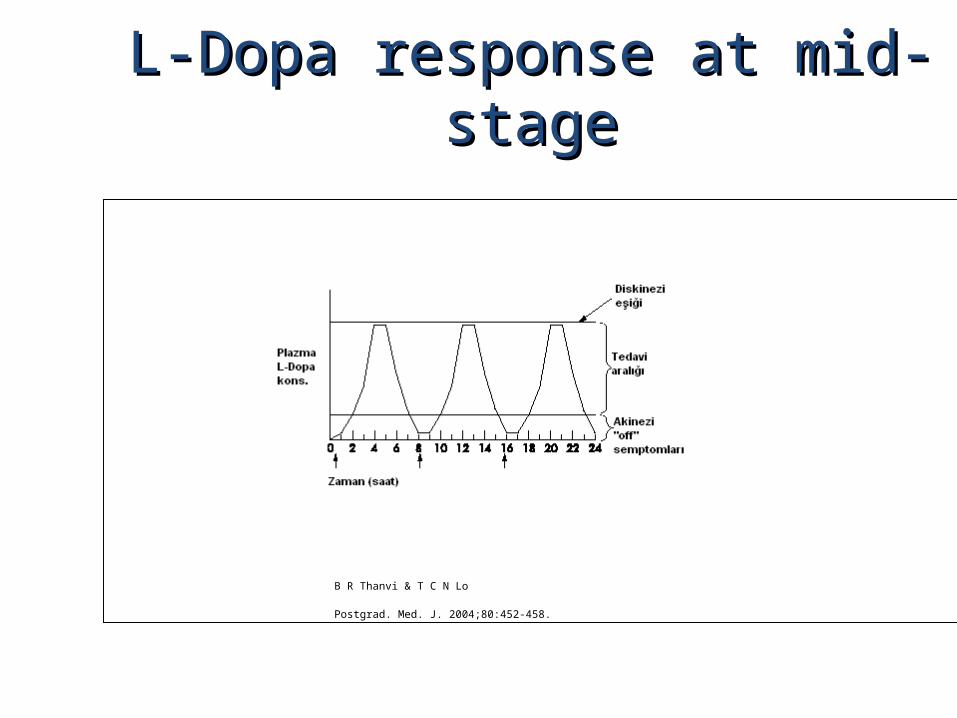
L-Dopa response at mid-stageL-Dopa response at mid-stage
B R Thanvi & T C N Lo
Postgrad. Med. J. 2004;80:452-458.
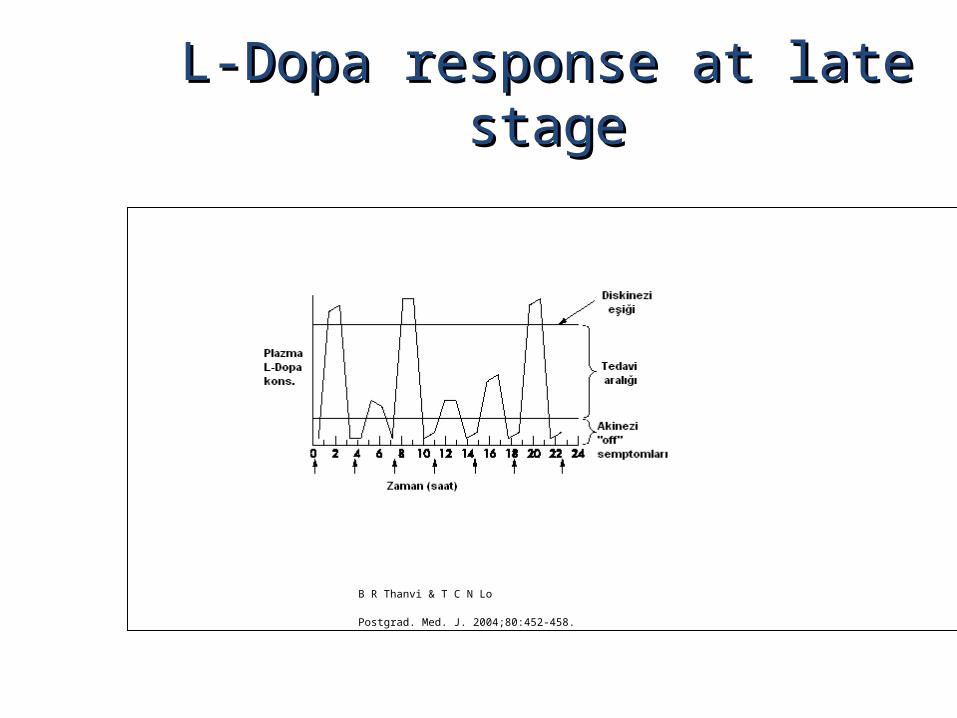
L-Dopa response at late stageL-Dopa response at late stage
B R Thanvi & T C N Lo
Postgrad. Med. J. 2004;80:452-458.


Clinical Features ExcludingIdiopathic PD
••Stepwise progression Stepwise progression of of parkinsonian parkinsonian symptoms and historysymptoms and historyof of recurrent strokerecurrent stroke
••History History of of recurrent head traumarecurrent head trauma••History History of of encephalitisencephalitis••Oculogyric crisisOculogyric crisis••Neuroleptic useNeuroleptic use••Sustained remissionSustained remission••Unilateral involvement after Unilateral involvement after 3 3 years years of of symptomsymptom--onset onset
••Supranuclear gaze palsySupranuclear gaze palsy

Clinical Features ExcludingIdiopathicPD
••CerebellarCerebellarfindingsfindings••Early Early severe severe autonomic symptomsautonomic symptoms••Early Early severe severe dementiadementia••BabinskiBabinskisignsign••Communicating hydrocephalus and Communicating hydrocephalus and basal ganglionic lesionsbasal ganglionic lesions
••NegativeNegativeresponseresponsetotoLL--DopaDopa••MPTP MPTP intoxicationintoxication

MMuultisltisyystem Atrostem Atrophyphy Degenerative, sDegenerative, sporadiporadic,c, rapid progressiverapid progressive Falls at early stages, severe dysarthriaFalls at early stages, severe dysarthria Significant symmetrySignificant symmetry Mild or absent tremorMild or absent tremor
Age at onset of the disease Age at onset of the disease 30 years 30 years
CombinationCombination ParkinsonizmParkinsonizm: : L-Dopa response is poor/absentL-Dopa response is poor/absent CCerebellarerebellar symptoms symptoms PyPyramidal ramidal symptomssymptoms Autonomic dysfunctionAutonomic dysfunction

MSA - P
MSA-C

MSA
•MRI (sensitivity%88-%60, specifisity%93-%100)
–Putaminalhypointensity
–In the outer border of putamenhyperintensity
–Atrophy of the cerebellum, middle cerebellar peduncles, midbrain
–Increased signal intensity in pons(hot crossbun)
Orta hat “raphe” ve transverspontinliflerde sinyal artışı, tegmentum, piramidal traktusve superiorserebellarpedinkületkilenmiyor
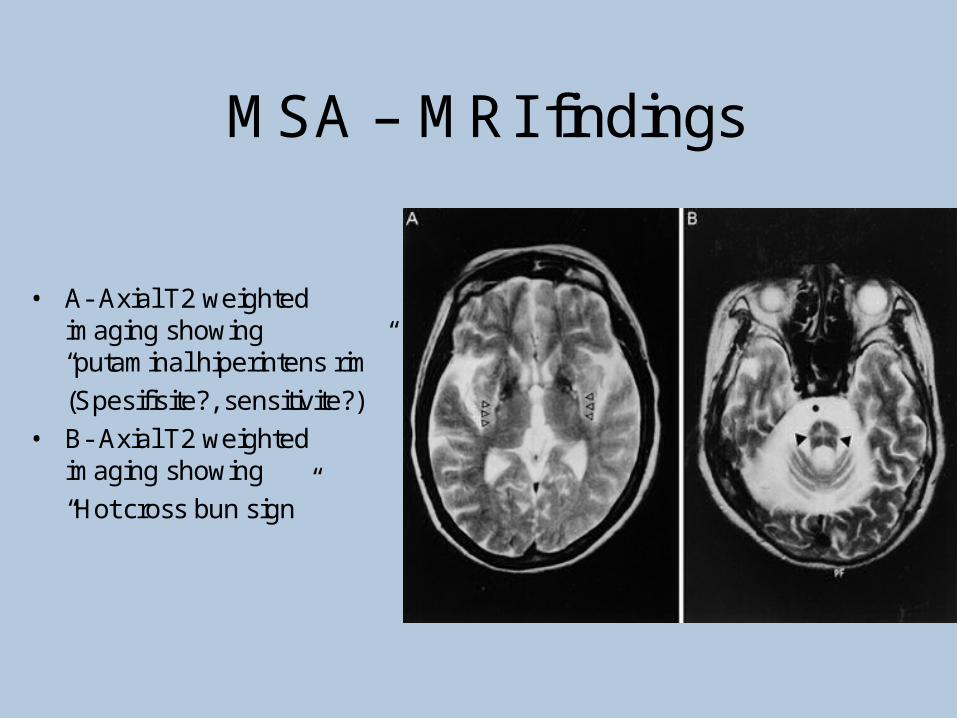
MSA – MRI findings
• A- Axial T2 weightedimaging showing“putaminal hiperintens rim”
(Spesifisite?, sensitivite?)
• B- Axial T2 weightedimaging showing
“Hot cross bun sign”
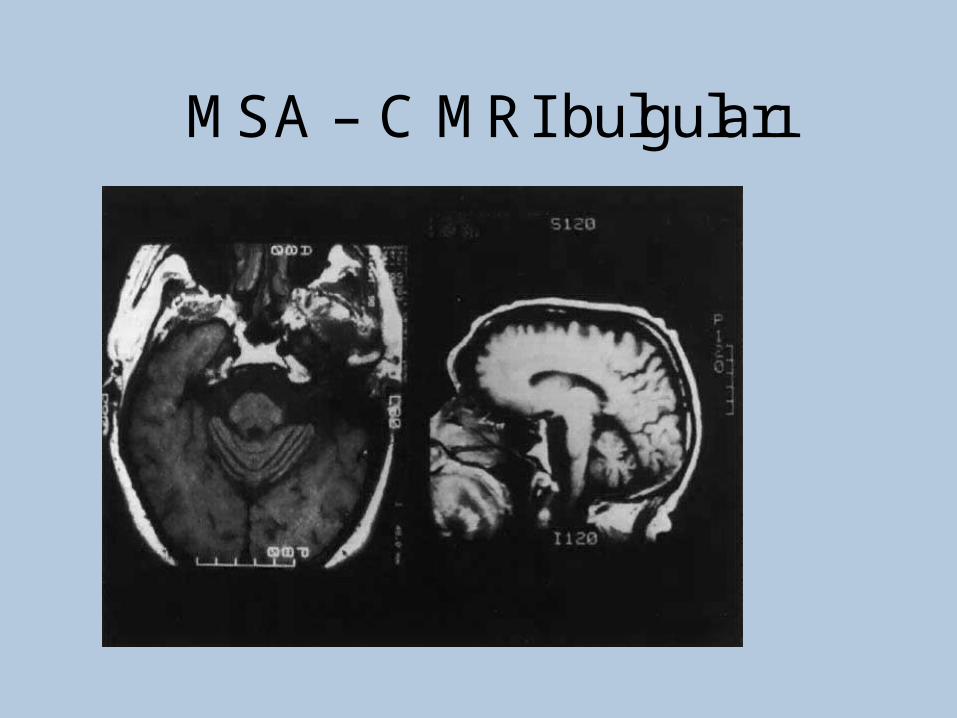
MSA – C MRI bulguları

Progressive supranuclear palsy (PSP)

PSP
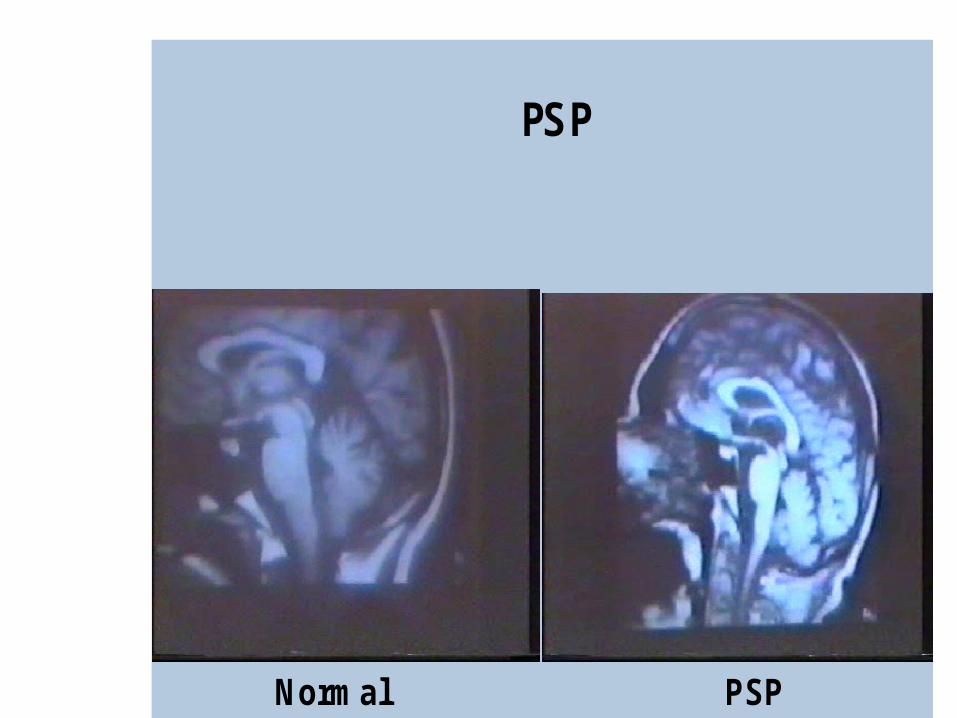
PSP
Normal PSP

Secondary Parkinsonism - CO Intoxication

Lower body parkinsonism

Hyperkinetic Disorders

– Tremor
* Involuntary oscillations of a body part produced by alternating or synchronous contractions of reciprocally innervated muscles.
* Physiological tremor These tremors are very small amplitude and are
demonstrable only by means of accelerometer. Enhanced physiological tremor: medical conditions, drugs, anxiety, fear…

* Essential tremor ET Typically a postural tremor (4-12 Hz) but may be
accentuated by goal-directed movements. The site of involvement in most cases is the hands and it is frequently asymmetric initially.
* Parkinsonian tremor Tremor at rest, at a frequency of 4-5 Hz, is the
most characteristic and the most prominent type of tremor in PD, but postural and kinetic tremor are also frequently seen. Onset of the tremor is usually in one of the hands; rarely, it may begin in the legs.
* Intention tremor Rhythmic involuntary oscillations that undergo
exacerbation as the hand or foot approaches the target of a voluntary movement. It indicates involvement of the cerebellum or its connections.


Essential tremor

Goal directed tremor

– Chorea (“dance”)
Characterized by sudden, frequent involuntary, arrhythmic, purposeless, and quick jerks of the trunk, extremities, and head associated with facial grimaces. They are usually distal and of low amplitude. Causes of chorea are hereditary, autoimmune, vascular, metabolic, toxic, inflammatory or drug induced.
– Athetosis (“without position”)
Slow, writhing, continuous, wormlike movements of the distal parts of the extremities, chiefly the fingers, which show bizarre posturing.

Generalized chorea

Huntington disease
• Progressive hereditary disorder that usually appears during adult life• Characterized by movement disorder (usually chorea), dementia, personality
disorder• Caudate nucleus and putamen are severly involved • GABAergic efferents projecting to lateral globus pallidus are lost chorea• Later striatal efferents to medial pallidum are lost parkinsonism, dystonia• Prevalence 4-8/100.000• 4p16.3, CAG-repeat disorder (N:11-34) (HD:37-86)• Huntingtin protein• Trinucleotide repeat is unstable in gametes• More juvenil cases of HD when an individual inherits the gene from father• Onset 35-40 yr (5-70)• Movement disorder, personality disorder, mental deterioration (course over a
period of 15 yr)

– Ballism (“jump or throw”)Sudden, quick, continuous, unusually violent, and
flinging in nature.Usually confined to the contralateral vascular lesion
in the subthalamic nucleus.
– Dystonia (“bad tone”)Twisting, slow, contorting, involuntary movement,
that is somewhat sustained and often repetitive. Dystonia can involve any part of the body. Dystonia is classified as (1) focal, (2) segmental (cranial
muscles (Meige syndrome), cranio-cervical dystonia, (3) multifocal (2 or more noncontiguous parts), (4) hemidystonia, (5) generalized (segmental crural + an other part of the body)

Classification of Torsion Dystonia• By age of onset
– Childhood onset, 0-12 yr– Adolescent onset, 13-20 yr– Adult onset, >20 yr
• By distribution– Focal– Segmental– Generalized– Hemidystonia
• By etiology– Primary (familial, sporadic)– Dystonia-plus – Secondary dystonia– Heredodegenerative diseases

Primary Dystonias• Pure dystonia except tremor• Familial and nonfamilial sporadic types• Most primary dystonias are sporadic, adult onset focal or segmental
dystonias• DYT 1 (Oppenheim dystonia)
– Onset 12.5±8.2– In %95 of patients symptoms begin in an arm or leg and the disorder spreads
to the neck or larynx– AD, 9q34.1, Torsin A protein– Penetrance rate 30%, 40%

Dystonic storm


Lingual dystonia

Generalized dystonia, chorea, spasmodic dysphonia (adductor)

Dystonic tremor


Generalized dystonia, myoclonus

Wilson disease Wilson disease (Hepatolenticular degeneration)(Hepatolenticular degeneration)
• Autosomal recessive disorder with the gene being located on the long arm of chromosome 13.
• The gene encodes a copper transporting P-type ATPase that is expressed in liver and kidney
• Two fundamental defects:
1.reduced biliary transport of copper,
2.impaired formation of plasma ceruloplasmin• Free Cu in serum is increased• Overflow of copper from the liver produces accumulation
in other organs, mainly in brain, kidney, and cornea .

Wilson disease Wilson disease (Hepatolenticular degeneration)(Hepatolenticular degeneration)
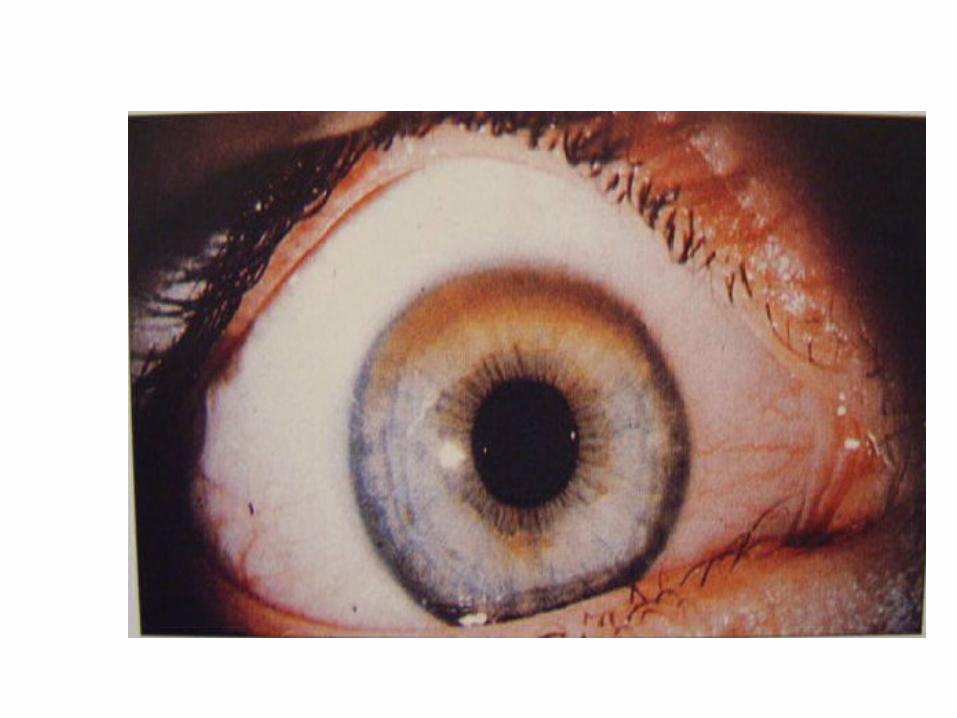
• In cornea, copper is deposited close to the endothelial surface of the Descement membrane (Kayser-Fleischer ring; most important diagnostic feature)
• Symptoms begin between the ages of 11 and 25 years• Wilson disease is a disorder of motor function; there are
no sensory symptoms and reflex alterations.• Symptoms of basal ganglia damage usually predominate
but cerebellar symptoms may occasionally be in the foreground.
• Tremors and rigidity are the most common early signs.• Seizures can occur at any stage of the disease.

Wilson disease Wilson disease (Hepatolenticular degeneration)(Hepatolenticular degeneration)
TreatmentTreatment• Initial phase of the treatment (toxic copper levels are
brought under control)
Penicillamine
Ammonium tetrathiomolybdate
Triethylene tetramine dihydrohloride (trientine)• Maintenance therapy
Zinc acetate
Trientine + Zinc acetate


![\SETOVI\ERTAN-BEGA.SET\SOUND file:///C|/SETOVI/ERTAN-BEGA-REPORT/ERTAN-BEGA.SET_PCG_REPORT.html[12/28/2015 11:31:03 PM] KORG PA Manager - SOUND Report ERTAN-BEGA.SET User 1 Soun](https://img.pdfslide.us/doc/110x75/5afcd8547f8b9a864d8cb40f/setoviertan-begasetsound-filecsetoviertan-bega-reportertan-begasetpcgreporthtml12282015.jpg)



























