Embed Size (px)
Citation preview

& Rearrangement Reactions
Direct Observation of Carbamoylnitrenes
Hongmin Li,[a] Huabin Wan,[a] Zhuang Wu,[a] Dingqing Li,[a] Didier B¦gu¦,[b] Curt Wentrup,*[c]
and Xiaoqing Zeng*[a]
Abstract: As the prototype Curtius rearrangement reaction,carbamoyl azide decomposes into aminoisocyanate and mo-
lecular nitrogen. However, the key intermediate carbamoyl-nitrene was previously undetected, even though the decom-
position of carbamoyl azides has been studied frequentlysince its discovery in the 1890s. Upon ArF laser (l= 193 nm)
photolysis, the stepwise decomposition of the two simplest
carbamoyl azides H2NC(O)N3 and Me2NC(O)N3, isolated in
solid noble gas matrices, occurs with the formation of thecorresponding carbamoylnitrenes H2NC(O)N and Me2NC(O)N.
Both triplet species are characterized for the first time bycombining matrix-isolation IR spectroscopy and quantum-
chemical calculations. Subsequent visible-light irradiationscause efficient rearrangement of these nitrenes into the re-spective aminoisocyanates.
Introduction
As one of the most well-known reactions in chemistry, the Cur-
tius rearrangement describes the decomposition of acyl azides(i.e. , RC(O)N3) into isocyanates (i.e. , RNCO) with the loss of N2
on either heating or irradiation.[1] Due to the close similarity inthe molecular structures of the azides, similar Curtius-type rear-rangements have been suggested for phosphoryl azide (i.e. ,
R2P(O)N3),[2] sulfuryl azide (i.e. , RS(O)2N3),[3] and sulfinyl azide(i.e. , RS(O)N3),[4] all of which can, in principle, extrude N2 and
furnish the imines RP(O)NR, O2SNR, and OSNR, respectively.Due to the broad applications of these a-oxo-azides and thepotential utility of the highly reactive a-oxo-nitrenes as photo-labeling intermediates, the underlying mechanisms of the rear-
rangement including the structures and reactivities of the ni-trenes have been the focus of numerous experimental andtheoretical studies.[5] Two distinct pathways, namely a concert-ed and a stepwise pathway, have been suggested. Generally,the in situ chemical trapping reaction in solution and the
direct detection of the corresponding a-oxo-nitrenes (i.e. ,
RC(O)N, R2P(O)N, RS(O)2N, or RS(O)N) by using various spectro-
scopic methods, such as time-resolved and matrix-isolationspectroscopy methods,[2–4] have provided the most convincing
evidence for the occurrence of the stepwise decompositionpathways of the respective azides.
Carbamoyl azides are useful reagents in synthetic chemis-try.[6] The study of the decomposition of carbamoyl azidesdates back to work in the 1890s by Curtius and Burkhardt,[7]
who found that phenylcarbamoyl azide decomposes into N2
and a complex product mixture on heating. Further studies on
the decomposition of arylcarbamoyl azides confirm the forma-tion of both isocyanates and the formal nitrene-trapping prod-ucts in solution.[8] To directly detect the key intermediates,matrix-isolation studies on the photolysis of the two simplest
carbamoyl azides, H2NC(O)N3[9, 10] and Me2NC(O)N3,[11, 12] have
been performed. The photolysis of matrix-isolated H2NC(O)N3
by xenon-lamp UV irradiation through Corning filters (UV trans-mitting, visible l= 440–660 nm cut-out filters) or by usinga low-pressure mercury lamp (l= 254 and 185 nm) resulted in
the formation of H2NNCO, H2NN, CO, and N2, whereas the pho-tolysis of matrix-isolated Me2NC(O)N3 with a low-pressure mer-
cury lamp (l= 254 nm filter) or a xenon lamp (l= 280 nmfilter) led to Me2NNCO, Me2NN, Me¢Me, HNCO, CO, and N2.[12]
The failure to capture the carbamoylnitrenes at cryogenic tem-
peratures suggests that the decomposition of both carbamoylazides occurs in a concerted path without nitrene intermedi-
ates. This is similar to the case of the parent acyl azideHC(O)N3, where no nitrene but only HNCO was found ina solid argon matrix after photolysis with an ArF laser (l=
193 nm).[13] Indeed, quantum-chemical calculations on the po-tential energy surface (PES) of the decomposition of HC(O)N3
support the preference of the concerted decomposition path-way, with the transition state of the stepwise decomposition
to HC(O)N being higher in energy than that of the concertedroute.[14]
[a] H. Li, H. Wan, Z. Wu, D. Li, Prof. Dr. X. ZengThe Key Lab of Health Chemistry andMolecular Diagnosis of Suzhou, College of ChemistryChemical Engineering and Materials ScienceSoochow University, Suzhou 215123 (P.R. China)E-mail : [email protected]
[b] Prof. Dr. D. B¦gu¦Institut des Sciences Analytiques et dePhysico-Chimie pour l’Environnement et les Mat¦riaux (IPREM)Universit¦ de Pau et des Pays de l’Adour, 64000 Pau (France)
[c] Prof. Dr. C. WentrupSchool of Chemistry and Molecular BiosciencesThe University of Queensland, 4072, Brisbane, Queensland (Australia)E-mail : [email protected]
Supporting information for this article is available on the WWW underhttp ://dx.doi.org/10.1002/chem.201600824.
Chem. Eur. J. 2016, 22, 7856 – 7862 Ó 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim7856
Full PaperDOI: 10.1002/chem.201600824

Recently, we have shown that reactive a-oxo-nitrenes can begenerated efficiently through the irradiation of a-oxo-azides
with an ArF laser in solid noble gas matrices.[15] For instance,the ArF laser photolysis of the matrix-isolated acyl azide
FC(O)N3 enabled the direct observation of the carbonyl nitreneFC(O)N,[15a] which was not observed in a previous study of thephotolysis in solid matrices by using a mercury lamp (l>200 nm).[16] The use of the short-pulse ArF laser has the distinctadvantage of reducing secondary irradiation of the photolabile
a-oxonitrenes by the unwanted UV or visible light from themercury or xenon lamps.
Herein, we revisit the photolysis of H2NC(O)N3 andMe2NC(O)N3 in solid, cryogenic matrices. The formation of the
two simplest carbamoyl nitrenes, that is, H2NC(O)N andMe2NC(O)N, in the triplet ground states has been firmly estab-
lished by combining matrix-isolation IR spectroscopy and
quantum-chemical calculations.
Results and Discussion
Prior to describing the matrix-isolation experiments, theground-state potential energy surfaces for the decomposition
of H2NC(O)N3 (1 a) and Me2NC(O)N3 (1 b) are computationallyexplored with the B3LYP/6-311 + + G(3df,3pd) and CBS-QB3
methods. Both methods consistently predict that the energiesof the transition state (TS2) for the stepwise decomposition of
singlet H2NC(O)N3 (1 a) through the nitrene intermediate
H2NC(O)N (2 a) are almost identical with those for the concert-ed decomposition (TS4) (Scheme 1 and Figure 1).
For Me2NC(O)N3 (1 b) the TS2 is higher in energy than TS4,by 2.7 kcal mol¢1 at the CBS-QB3 level. Such small energy dif-ferences imply that the Curtius rearrangement of the two
azides to the aminoisocyanates 3 may occur through bothpathways; in other words, the stepwise route through the elu-
sive carbamoyl nitrene intermediates 2 should be at leasta competing process. It should be noted that there are moder-
ate activation barriers of about 15 kcal mol¢1 for the intramo-
lecular rearrangements of the two singlet nitrenes to the re-spective isocyanates through the transition states TS3
(Figure 1).It is noteworthy that, although acylnitrenes usually have
closed-shell singlet ground states with partial oxazirine struc-tures,[5] the carbamoylnitrenes studied here have triplet (T, 3A’’)
ground states at all the computational levels examined (i.e. ,CBS-QB3, B3LYP, CASSCF(8,6), and CASPT2). For H2NC(O)N the
closed-shell singlet (CSS, 1A) state lies 4–11 kcal mol¢1 higherthan the triplet state depending on the method. The open-
shell singlet (OSS, 1A’’) state is predicted to lie 28 kcal mol¢1
higher at both the CASSCF(8,6)/6-311 + + G** and CASPT2/6-311 + G**//CASSCF(8,6)/6-311 + + G** levels (Figure 2) but only
approximately 1 kcal mol¢1 higher at the CBS-QBS and B3LYPlevels. For Me2NC(O)N the energy gap between the CSS and T
states was estimated to be 8 kcal mol¢1 with the CASPT2method, and the OSS state is about 27 kcal mol¢1 higher in
energy. Because single-determinant DFT methods are unrelia-
ble for open-shell systems, the CASSCF/CASPT2 structures andenergies are more realistic. It may then be surmised that any
initially generated singlet carbamoylnitrene (either at the CSSor OSS state) may either relax to the lower-energy triplet
ground state through intersystem crossing or rearrange intothe corresponding isocyanate.
Like other acylnitrenes, the CSS state has partial oxazirine
structure, with an OCN angle of 95.08 and an N-O distance of1.87 æ in the CSS state H2NC(O)N, whereas, in the triplet state
H2NC(O)N the OCN angle is 119.58 and the N-O distance is2.26 æ. For Me2NC(O)N these two states exhibit similar structur-al differences. The triplet and the open-shell singlet stateshave normal nitrene structures (Figure 2). The question of the
nature of the CSS states—nitrene or oxazirine—will be dis-cussed further below in conjunction with the infrared spectraldata.
The photolysis experiments were performed by irradiatingthe matrix-isolated R2NC(O)N3 (R = H, CH3) in solid N2, Ar, and
Ne at 2.7 K with an ArF laser. For H2NC(O)N3, an optimal laserenergy of 5 MJ was used for 5 min, after which nearly half of
the azide had vanished. The IR difference spectrum, obtained
by subtracting the spectra recorded after and before the pho-tolysis, is presented as the lower trace A), in Figure 3. By refer-
ring to the previously reported IR spectrum of H2NNCO in solidAr,[10] its production after the laser photolysis can be ascer-
tained by the characteristic IR bands at n= 2263.7 and2215.7 cm¢1 (denoted with “c” in Figure 3 A) for the antisym-
Scheme 1. Decomposition of the investigated azides through nitrene inter-mediates.
Figure 1. Calculated free energy profile (in [kcal mol¢1]) for the decomposi-tion of R2NC(O)N3 [R = H (1 a), R = CH3 (1 b)] at the CBS-QB3 level.
Chem. Eur. J. 2016, 22, 7856 – 7862 www.chemeurj.org Ó 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim7857
Full Paper

metric NCO stretching vibration. Traces of CO (n= 2144.6 cm¢1
in Ne matrix) were also formed, whereas the IR bands of the
remaining fragment H2NN[9] could not be clearly identified dueto overlap with the strong IR bands of the azide precursor (de-
noted with “a” in Figure 3 A).Importantly, another new species exhibiting a distinguishable
IR band at n= 1644.4 cm¢1 was also formed, with a positionquite close to the absorptions of water molecules (n= 1630.9,1614.4, and 1606.3 cm¢1). However, this band was not ob-
served in the previous photolysis experiments of H2NC(O)N3
with the xenon or low-pressure mercury lamps.[9, 10] Its frequen-cy is very close to that of the predicted strongest IR band forthe nitrene intermediate H2NC(O)N in its triplet ground state
(Table 1). Considering an intense visible absorption transitionat l= 490 nm (oscillator strength f = 0.01, Table S1 in the Sup-
porting Information) for the triplet nitrene, predicted with the
time-dependent (TD)-B3LYP/6-311 + + G(3df,3pd) method, thematrix containing the laser photolysis products was subjected
to blue light irradiation (l= (440�200) nm, 20 W). The result-ing IR difference spectrum is shown in Figure 3 B, which nicely
agrees with the calculated IR spectrum of H2NC(O)N in the trip-let state (Figure 3 C). Upon visible-light irradiation, the species
“b” (Figure 2 A) associated with the band at n= 1644.4 cm¢1
and other weaker ones at n= 3566.8, 3447.4, 1587.8, and1319.0 cm¢1 disappeared completely. As a result, the IR bands
of H2NC(O)N3 (a) and H2NNCO (c) appeared, which stronglysupports the assignment of the new species as the carbamoyl-
nitrene H2NC(O)N (2 a). The reformation of azide during thevisible-light irradiation of the corresponding triplet nitrene in
the presence of N2 in the same matrix cages has been ob-
served frequently.[15a, 18]
According to the calculations, the strongest IR band ob-
served at n= 1644.4 cm¢1 is due mainly to the CO stretchingvibration of H2NC(O)N in the triplet state, that is, compound
2 a (T). The two weaker bands at n= 3566.8 and 3447.4 cm¢1
are attributed to the antisymmetric and symmetric NH2 stretch-
es. They are slightly redshifted compared to those of the azide
precursor at n= 3587.7 and 3469.5 cm¢1, respectively. The HNHand CNH bending modes occur at n= 1584.5 and 1331.6 cm¢1,
respectively. The C¢Nnitrene stretch predicted near n= 900 cm¢1
was not observed due to its very low IR intensity (calculated tobe 0.5 km mol¢1). As for the higher-energy closed-shell singletH2NC(O)N, the predicted IR frequencies for the CO (n=
1361 cm¢1) and C¢Nnitrene (n= 1795 cm¢1) stretching vibrationsdeviate substantially from the observations (Table 1). For theOSS state of compound 2 a a low frequency of n= 1534 cm¢1
is predicted, which makes it unlikely that this state is being ob-served. In order to verify the validity of the DFT calculations of
the IR spectra, they were repeated at the CASSCF(8,6) level(harmonic frequencies only, Table 1, second column, and Ta-
bles S3 and S4 in the Supporting Information). The calculated
B3LYP and CASSCF wavenumbers are in broad agreement, buta scaling factor of approximately 0.9 is required for the har-
monic CASSCF calculations, whereas no scaling factor isneeded for the anharmonic vibrations at the DFT level.
The production of H2NC(O)N upon the ArF laser photolysisof H2NC(O)N3 followed by rearrangement into H2NNCO under
Figure 2. Calculated molecular structures (bond lengths in [æ] and angles in[8] in italics) and relative energies (in parentheses, in [kcal mol¢1]) ofR2NC(O)N [R = H (2 a), R = CH3 (2 b)] at the CASSCF(8,6)/6-311 + + G** levelfor the triplet (T), closed-shell singlet (CSS), and open-shell singlet (OSS)states. The relative energies calculated at the CASPT2/6-311 + G**//CASSCF(8,6)/6-311 + + G** level are given in bold (in parentheses, in [kcalmol¢1]).
Figure 3. A) Ne-matrix mid-IR difference spectrum (n= 3650–1100 cm¢1,transmittance T) showing the decomposition of H2NC(O)N3 (a) under ArFlaser irradiation (l= 193 nm, 5 mJ, 3 Hz, 5 min). B) Ne-matrix mid-IR differ-ence spectrum showing the conversion from H2NC(O)N (b) to H2NNCO (c)and H2NC(O)N3 (a) under subsequent irradiation with blue light(l= (440�20) nm, 20 W, 2 min), the intensities of the bands have been mul-tiplied by fifty times for clarity. C) Calculated IR spectrum of H2NC(O)N (b) atthe B3LYP/6-311 + + G(3df,3pd) level with harmonic frequencies scaled[17] bya factor of 0.9679. The IR bands marked by asterisks correspond to H2O mol-ecules due to changes of matrix sites upon photolysis.
Chem. Eur. J. 2016, 22, 7856 – 7862 www.chemeurj.org Ó 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim7858
Full Paper

subsequent blue-light irradiation described for Ne matrices
above was also observed in solid N2 and Ar matrices (Figur-
es S1 and S2 in the Supporting Information).The successful generation of H2NC(O)N prompted us to also
revisit the photolysis of Me2NC(O)N3 (1 b),[10–12] even thoughthe PES analysis (Figure 1) suggests that the concerted decom-
position pathway may be slightly more favorable. The IR differ-ence spectrum obtained after the irradiation of Me2NC(O)N3 (d)
in a solid nitrogen matrix is shown in Figure 4 A. The Curtius
rearrangement product Me2NNCO (3 b)[10, 12] (peak “f”) can beclearly identified by its characteristic IR band at n=
2225.8 cm¢1. Importantly, a much weaker band at n=
1618.4 cm¢1 is also observed, and its intensity decreases on
prolonged laser irradiation. The position of this new band is
quite close to that of triplet state H2NC(O)N (2 a) at n=
1643.7 cm¢1 in a solid N2 matrix, which means that triplet state
Me2NC(O)N (2 b) is the likely carrier of this new band.The TD-B3LYP/6-311 + + G(3df,3pd) calculations indicate that
triplet state Me2NC(O)N should be sensitive to visible light witha predicted intense transition at l= 627 nm (f = 0.0208,
Table S1 in the Supporting Information). Thus, the matrix con-
taining the laser photolysis products was irradiated withyellow light (l= (570�20) nm, 20 W). As expected, the band
at n= 1618.4 cm¢1 and several other weaker ones (labeled “e”in Figure 4 B) vanished completely. Similarly to the situation de-
scribed for the parent molecule 1 a, both the azide precursor1 b and the Curtius-rearrangement product, that is, the aminoi-socyanate 3 b, were formed. Therefore, these new bands are
reasonably assigned to the missing nitrene intermediateMe2NC(O)N (2 b) in its triplet ground state. This is further sup-ported by the good agreement with the predicted IR spectrum(Figure 4 C), which permits the assignments of the characteris-
tic CO and C¢Nnitrene stretching vibrations at n= 1618.4 and994.4 cm¢1, respectively. Unexpectedly, another two less in-
tense but distinct bands at n= 1605.7 and 1584.0 cm¢1 were
also found to be associated with the nitrene spectrum; theymay arise from combination of the fundamentals and gain in-
tensity from the strong band at n= 1618.4 cm¢1 through Fermicoupling. The absence of new IR bands in the range n= 1800–
1700 cm¢1 rules out the presence of the closed-shell singletstate compound 2 b, for which a strong band above n=
1700 cm¢1 is predicted (Tables 2 and S4 in the Supporting In-
formation). For reasons of symmetry, it is not possible to calcu-late the open-shell singlet state compound 2 b at the DFT
level, but the CASSCF(8,6) calculation indicates that the stron-gest vibration would appear at significantly lower wavenum-
ber, that is, n= 1565 cm¢1 (Tables 2 and S4 in the SupportingInformation).
Table 1. Calculated and experimentally observed IR wavenumbers in [cm¢1] and intensities in [km mol¢1] for H2NC(O)N (2 a).
Calculated[a] Observed[b] Assignments[c]
open-shell singlet (OSS) closed-shell singlet (CSS) triplet (T) N2 matrix Ar matrix Ne matrix
3522 (51) 3507 (52) 3569 (76) 3527 (51) 3543.4 (w) 3548.1 (m) 3566.8 (w) n1, nasymNH2
3410 (55) 3409 (45) 3456 (77) 3418 (49) 3425.1 (w) 3430.1 (m) 3447.4 (w) n2, nsymNH2
1596 (226) 1612 (196) 1795 (321) 1618 (261) 1643.7 (s) 1638.0 (s) 1644.4 (s) n3, nCN++nCO1534 (18) 1556 (42) 1564 (44) 1549 (8) 1584.5 (vw) 1587.8 (vw) n4, dNH2
1289 (78) 1218 (104) 1361 (40) 1295 (74) 1331.6 (m) 1320.0 (m) 1319.0 (m) n5, dHNC++nNCN1041 (12) 1082 (36) 1014 (25) 1048 (11) n6, dHNC901 (1) 853 (1) 980 (6) 883 (<1) n7, n(C¢N)688 (27) 634 (172) 611 (21) 677 (37) 683.4 (w) 674.4 (vw) n8, 1OCN2
518 (15) 534 (124) 462 (29) 520 (14) n9, wNH2++nCN471 (1) 471 (48) 426 (12) 456 (<1) n10, sOC(NH2)403 (11) 453 (13) 420 (79) 404 (10) n11, s
204 (35) 272 (29) 239 (130) 211 (66) n12, 1NH2
[a] Normal font: calculated anharmonic, unscaled IR frequencies and intensities (in parentheses) at the B3LYP/6-311 + + G(3df,3pd) level of theory. Secondcolumn in italics : for the OSS harmonic wavenumbers at the CASSCF(8,6)/6-311 + + G** level scaled by a factor of 0.9. [b] Observed band positions of themost intense matrix sites in different matrices at 2.7 K. The relative band intensities are given in parentheses: s = strong, m = medium, w = weak, vw = veryweak. [c] Tentative assignments based on calculated vibrational displacement vectors for the triplet state. See text, Figure 6 and Figure S6 in the Support-ing Information. For the CASSCF-calculated frequencies of all species, see Table S3 in the Supporting Information.
Figure 4. A) N2-matrix IR difference spectrum (n = 2400–650 cm¢1, transmit-tance T) showing the decomposition of Me2NC(O)N3 (d) under ArF laser irra-diation (l= 193 nm, 3 mJ, 3 Hz, 7 min). B) N2-matrix IR difference spectrumshowing the conversion from Me2NC(O)N (e) to Me2NNCO (f) andMe2NC(O)N3 (d) under the subsequent irradiation with yellow light(l= (570�20) nm, 20 W, 2 min), the intensities of the bands have been mul-tiplied by fifteen times for clarity. C) Calculated IR spectrum of Me2NC(O)N(e) at the B3LYP/6-311 + + G(3df,3pd) level with harmonic frequencies scaledby a factor of 0.9679.
Chem. Eur. J. 2016, 22, 7856 – 7862 www.chemeurj.org Ó 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim7859
Full Paper
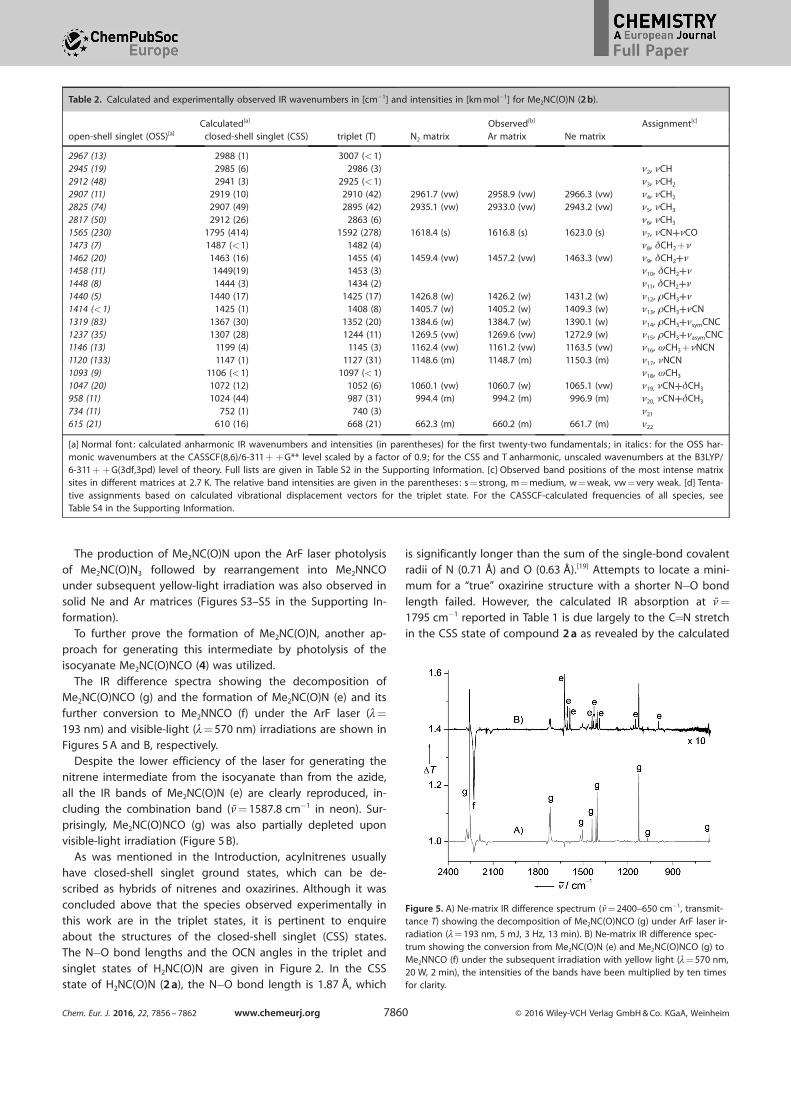
The production of Me2NC(O)N upon the ArF laser photolysis
of Me2NC(O)N3 followed by rearrangement into Me2NNCOunder subsequent yellow-light irradiation was also observed in
solid Ne and Ar matrices (Figures S3–S5 in the Supporting In-formation).
To further prove the formation of Me2NC(O)N, another ap-proach for generating this intermediate by photolysis of theisocyanate Me2NC(O)NCO (4) was utilized.
The IR difference spectra showing the decomposition ofMe2NC(O)NCO (g) and the formation of Me2NC(O)N (e) and its
further conversion to Me2NNCO (f) under the ArF laser (l=
193 nm) and visible-light (l= 570 nm) irradiations are shown in
Figures 5 A and B, respectively.Despite the lower efficiency of the laser for generating the
nitrene intermediate from the isocyanate than from the azide,all the IR bands of Me2NC(O)N (e) are clearly reproduced, in-cluding the combination band (n= 1587.8 cm¢1 in neon). Sur-
prisingly, Me2NC(O)NCO (g) was also partially depleted uponvisible-light irradiation (Figure 5 B).
As was mentioned in the Introduction, acylnitrenes usuallyhave closed-shell singlet ground states, which can be de-
scribed as hybrids of nitrenes and oxazirines. Although it was
concluded above that the species observed experimentally inthis work are in the triplet states, it is pertinent to enquire
about the structures of the closed-shell singlet (CSS) states.The N¢O bond lengths and the OCN angles in the triplet and
singlet states of H2NC(O)N are given in Figure 2. In the CSSstate of H2NC(O)N (2 a), the N¢O bond length is 1.87 æ, which
is significantly longer than the sum of the single-bond covalent
radii of N (0.71 æ) and O (0.63 æ).[19] Attempts to locate a mini-mum for a “true” oxazirine structure with a shorter N¢O bond
length failed. However, the calculated IR absorption at n=
1795 cm¢1 reported in Table 1 is due largely to the C=N stretch
in the CSS state of compound 2 a as revealed by the calculated
Table 2. Calculated and experimentally observed IR wavenumbers in [cm¢1] and intensities in [km mol¢1] for Me2NC(O)N (2 b).
Calculated[a] Observed[b] Assignment[c]
open-shell singlet (OSS)[a] closed-shell singlet (CSS) triplet (T) N2 matrix Ar matrix Ne matrix
2967 (13) 2988 (1) 3007 (<1)2945 (19) 2985 (6) 2986 (3) n2, nCH2912 (48) 2941 (3) 2925 (<1) n3, nCH2
2907 (11) 2919 (10) 2910 (42) 2961.7 (vw) 2958.9 (vw) 2966.3 (vw) n4, nCH2
2825 (74) 2907 (49) 2895 (42) 2935.1 (vw) 2933.0 (vw) 2943.2 (vw) n5, nCH3
2817 (50) 2912 (26) 2863 (6) n6, nCH3
1565 (230) 1795 (414) 1592 (278) 1618.4 (s) 1616.8 (s) 1623.0 (s) n7, nCN++nCO1473 (7) 1487 (<1) 1482 (4) n8, dCH2 +n
1462 (20) 1463 (16) 1455 (4) 1459.4 (vw) 1457.2 (vw) 1463.3 (vw) n9, dCH2++n
1458 (11) 1449(19) 1453 (3) n10, dCH2++n
1448 (8) 1444 (3) 1434 (2) n11, dCH2++n
1440 (5) 1440 (17) 1425 (17) 1426.8 (w) 1426.2 (w) 1431.2 (w) n12, 1CH3++n
1414 (<1) 1425 (1) 1408 (8) 1405.7 (w) 1405.2 (w) 1409.3 (w) n13, 1CH3++nCN1319 (83) 1367 (30) 1352 (20) 1384.6 (w) 1384.7 (w) 1390.1 (w) n14, 1CH3++nsymCNC1237 (35) 1307 (28) 1244 (11) 1269.5 (vw) 1269.6 (vw) 1272.9 (w) n15, 1CH3++nasymCNC1146 (13) 1199 (4) 1145 (3) 1162.4 (vw) 1161.2 (vw) 1163.5 (vw) n16, wCH3 + nNCN1120 (133) 1147 (1) 1127 (31) 1148.6 (m) 1148.7 (m) 1150.3 (m) n17, nNCN1093 (9) 1106 (<1) 1097 (<1) n18, wCH3
1047 (20) 1072 (12) 1052 (6) 1060.1 (vw) 1060.7 (w) 1065.1 (vw) n19, nCN++dCH3
958 (11) 1024 (44) 987 (31) 994.4 (m) 994.2 (m) 996.9 (m) n20, nCN++dCH3
734 (11) 752 (1) 740 (3) n21
615 (21) 610 (16) 668 (21) 662.3 (m) 660.2 (m) 661.7 (m) n22
[a] Normal font: calculated anharmonic IR wavenumbers and intensities (in parentheses) for the first twenty-two fundamentals; in italics : for the OSS har-monic wavenumbers at the CASSCF(8,6)/6-311+ + G** level scaled by a factor of 0.9; for the CSS and T anharmonic, unscaled wavenumbers at the B3LYP/6-311 + + G(3df,3pd) level of theory. Full lists are given in Table S2 in the Supporting Information. [c] Observed band positions of the most intense matrixsites in different matrices at 2.7 K. The relative band intensities are given in the parentheses: s = strong, m = medium, w = weak, vw = very weak. [d] Tenta-tive assignments based on calculated vibrational displacement vectors for the triplet state. For the CASSCF-calculated frequencies of all species, seeTable S4 in the Supporting Information.
Figure 5. A) Ne-matrix IR difference spectrum (n= 2400–650 cm¢1, transmit-tance T) showing the decomposition of Me2NC(O)NCO (g) under ArF laser ir-radiation (l= 193 nm, 5 mJ, 3 Hz, 13 min). B) Ne-matrix IR difference spec-trum showing the conversion from Me2NC(O)N (e) and Me2NC(O)NCO (g) toMe2NNCO (f) under the subsequent irradiation with yellow light (l= 570 nm,20 W, 2 min), the intensities of the bands have been multiplied by ten timesfor clarity.
Chem. Eur. J. 2016, 22, 7856 – 7862 www.chemeurj.org Ó 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim7860
Full Paper

vibrational displacement vectors shown in Figure 6 and by theanalysis of the vibrational wavefunctions reported in Tables 1
and 2. The C¢O stretch occurs at n= 1361 cm¢1 and is stronglymixed with a deformation mode. These data indicate a high
degree of C=N double-bond character and C¢O single-bond
character as expected for an oxazirine. A comparison of the vi-brational displacement vectors in compounds 2 a (T) and 2 a(CSS) is shown in Figure S6 in the Supporting Information.
In the open-shell singlet and the triplet states there are no
such C=N double-bond stretches but only a weak C¢N singlebond stretch n7 at approximately n= 900 cm¢1 (Table 1 and
Figure S6 in the Supporting Information).
Conclusion
The photolysis of the carbamoyl azides H2NC(O)N3 andMe2NC(O)N3 has been revisited in solid N2, Ar, and Ne matrices
by using a l= 193 nm ArF excimer laser. The key nitrene inter-mediates H2NC(O)N and Me2NC(O)N in their triplet groundstates both of which were missing in the earlier matrix-isola-tion studies, have been generated and characterized bymatrix-isolation IR spectroscopy and quantum-chemical calcu-
lations. The direct observation of the carbamoylnitrenes duringthe photolysis of the respective azide precursors, followed byefficient rearrangement into the corresponding aminoisocya-nates, for the first time clearly supports a stepwise photochem-ical decomposition pathway, as proposed initially by Curtiusand Burkhardt in the 1890s.
Experimental Section
Caution! Covalent azides are potentially hazardous and explosive.Although we have not experienced any incident during this work,safety precautions (face shields, leather gloves, and protectiveleather clothing) are recommended for handling the azides.
Sample preparation : The carbamoyl azides H2NC(O)N3[20] and
Me2NC(O)N3[21] were prepared according to published protocols.
The purity of the compounds was checked by gas-phase IR spec-troscopy. The carbamoyl isocyanate Me2NC(O)NCO was prepared
according to reference [22] with modification. Briefly, it was pre-pared by the slow addition of 1,1-dimethylurea to a solution ofoxalyl chloride in CH2Cl2. The mixture was stirred for 5 min in anice–water bath. The solvent and byproducts (mainly HCl) werepumped off at room temperature for 30 min. Then, the residuewas warmed to 60 8C and the volatile product Me2NC(O)NCO wascollected in a cold trap (¢45 8C) as colorless liquid. The purity ofMe2NC(O)NCO was checked by IR spectroscopy.
Matrix isolation and photolysis : Matrix IR spectra were recordedon a FTIR spectrometer (Bruker 70 V) in a reflectance mode byusing a transfer optic. A KBr beam splitter and MCT detector wereused in the mid-IR region (n= 4000–650 cm¢1). For each spectrum,200 scans at a resolution of 0.5 cm¢1 were co-added.
The gaseous sample was mixed by passing a flow of matrix gas(Ne, Ar, N2) through a U-trap (ca. ¢50 8C) containing approximately30 mg of the azide. Then the mixture (azide/matrix gas �1:1000estimated) was passed through an aluminum oxide furnace (o.d.2.0 mm, i.d. 1.0 mm), which can be heated over a length of approx-imately 25 mm by a tantalum wire (o.d. 0.4 mm, resistance 0.4 W),deposited (2 mmol h¢1) at a high vacuum onto the gold-plated Cublock matrix support (2.7 K) in a high vacuum (�10¢6 Pa). Photoly-sis was performed by using an ArF excimer laser (Gamlaser EX5/250, 193 nm) and a high-power flashlight (Boyu T648, 20 W) withadjustable sources of blue, white, yellow, and violet light.
Quantum-chemical calculations : Structures and IR frequencies ofstationary points were calculated by using the DFT B3LYPmethod[23] with the 6-311 + + G(3df, 3pd) basis set. Accurate rela-tive energies of the species were further calculated by using thecomplete basis set (CBS-QB3).[24] Local minima were confirmed byvibrational frequency analysis, and transition states were furtherconfirmed by intrinsic reaction coordinate (IRC) calculations.[25] Allthe calculations were performed by using the Gaussian 09 softwarepackage.[26] The CASSCF (N = 8, M = 6)/6-311 + + G** calculationswere also performed with the Gaussian package (N= number ofelectrons; M = number of the orbitals in the active space). TheCASPT2 calculations were performed based on the CASSCF (8,6)/6-311 + + G** optimized structures with the MOLPRO program.[27]
Acknowledgements
This work was supported by the National Natural ScienceFoundation of China (21372173 and 21422304), the Beijing Na-
tional Laboratory for Molecular Sciences (20140128), the Priori-ty Academic Program Development of Jiangsu Higher Educa-
tion Institutions (PAPD), the University of Queensland, and theM¦socentre de Calcul Intensif Aquitain.
Keywords: azides · Curtius rearrangement · IR spectroscopy ·matrix isolation · nitrenes
[1] T. Curtius, Ber. Dtsch. Chem. Ges. 1890, 23, 3023 – 3033.[2] a) S. Vyas, S. Muthukrishnan, J. Kubicki, R. D. McCulla, G. Burdzinski, M.
Sliwa, M. S. Platz, C. M. Hadad, J. Am. Chem. Soc. 2010, 132, 16796 –16804; b) R. D. McCulla, G. A. Gohar, C. M. Hadad, M. S. Platz, J. Org.Chem. 2007, 72, 9426 – 9438.
[3] a) J. Kubicki, H. L. Luk, Y. Zhang, S. Vyas, H. L. Peng, C. M. Hadad, M. S.Platz, J. Am. Chem. Soc. 2012, 134, 7036 – 7044; b) X. Q. Zeng, H. Beck-ers, H. Willner, J. Am. Chem. Soc. 2013, 135, 2096 – 2099; c) D. S. Breslow,E. I. Edwards, E. C. Linsay, H. Omura, J. Am. Chem. Soc. 1976, 98, 4268 –4275; d) W. Lwowski, E. Scheiffele, J. Am. Chem. Soc. 1965, 87, 4359 –4365.
Figure 6. Calculated vibrational modes of the CSS state nitrene 2 a at theB3LYP/6-311 + + G(3df,3pd) level (see Table 1 and Figure S6 in the Support-ing Information).
Chem. Eur. J. 2016, 22, 7856 – 7862 www.chemeurj.org Ó 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim7861
Full Paper

[4] a) T. J. Maricich, V. L. Hoffman, J. Am. Chem. Soc. 1974, 96, 7770 – 7781;b) Z. Wu, D. Q. Li, H. M. Li, B. F. Zhu, H. L. Sun, J. S. Francisco, X. Q.Zeng, Angew. Chem. Int. Ed. 2016, 55, 1507 – 1510; Angew. Chem. 2016,128, 1529 – 1532; c) T. J. Maricich, C. N. Angeletakis, J. Org. Chem. 1984,49, 1931 – 1934.
[5] a) Nitrenes and Nitrenium Ions (Wiley Series of Reactive Intermediates inChemistry and Biology), Vol. 6 (Eds. : D. E. Falvey, A. D. Gudmundsdottir),Wiley, New York, 2013 ; b) N. P. Gritsan, M. S. Platz, W. T. Borden, TheStudy of Nitrenes by Theoretical Methods in Theoretical Methods in Photo-chemistry (Ed. : A. Kutateladze), Taylor and Francis, Boca Raton, 2005 ;c) Reactive Intermediate Chemistry (Eds. : R. A. Moss, M. S. Platz, M. Jo-nes, Jr.), Wiley-Interscience, New York, 2004.
[6] a) P. Feng, X. Sun, Y. Su, X. Li, L. H. Zhang, X. Shi, N. Jiao, Org. Lett. 2014,16, 3388 – 3391; b) J. Paz, C. P¦rez-Balado, B. Iglesias, L. MuÇoz, J. Org.Chem. 2010, 75, 8039 – 8047; c) E. Garc�a-Egido, M. Fern�ndez-Su�rez, L.MuÇoz, J. Org. Chem. 2008, 73, 2909 – 2911; d) X. Q. Li, W. K. Wang, C.Zhang, Adv. Synth. Catal. 2009, 351, 2342 – 2350.
[7] T. Curtius, A. Burkhardt, J. Prakt. Chem. 1898, 58, 205 – 233.[8] a) E. Lieber, R. L. Minnis, Jr. , C. N. R. Rao, Chem. Rev. 1965, 65, 377 – 384;
b) V. J. Bauer, W. J. Fanshawe, S. R. Safir, J. Org. Chem. 1966, 31, 3440 –3441; c) W. Lwowski, S. Kanemasa, R. A. Murray, V. T. Ramakrishnan, T. K.Thiruvengadam, K. Yoshida, A. Subbaraj, J. Org. Chem. 1986, 51, 1719 –1723; d) M. Kurz, W. Reichen, Tetrahedron Lett. 1978, 19, 1433 – 1436.
[9] A. P. Sylwester, P. B. Dervan, J. Am. Chem. Soc. 1984, 106, 4648 – 4650.[10] J. H. Teles, G. Maier, Chem. Ber. 1989, 122, 745 – 748.[11] W. Lwowski, R. A. Mauriac, M. Thompson, J. Org. Chem. 1975, 40, 2608 –
2612.[12] T. Pasinszki, M. Krebsz, G. Tarczay, C. Wentrup, J. Org. Chem. 2013, 78,
11985 – 11991.[13] X. Q. Zeng, E. Bernhardt, H. Beckers, K. Banert, M. Hagedorn, H. Liu,
Angew. Chem. Int. Ed. 2013, 52, 3503 – 3506; Angew. Chem. 2013, 125,3587 – 3591.
[14] K. Banert, C. Berndt, M. Hagedorn, H. Liu, T. Anacker, J. Friedrich, G.Rauhut, Angew. Chem. Int. Ed. 2012, 51, 4718 – 4721; Angew. Chem.2012, 124, 4796 – 4800.
[15] For examples, see: a) X. Q. Zeng, H. Beckers, H. Willner, D. Grote, W.Sander, Chem. Eur. J. 2011, 17, 3977 – 3984; b) X. Q. Zeng, H. Beckers, H.Willner, P. Neuhaus, D. Grote, W. Sander, Chem. Eur. J. 2009, 15, 13466 –13473; c) X. Q. Zeng, H. Beckers, H. Willner, P. Neuhaus, D. Grote, W.Sander, J. Phys. Chem. A 2015, 119, 2281 – 2288.
[16] J. Jacobs, B. Jìlicher, G. Schatte, H. Willner, H. G. Mack, Chem. Ber. 1993,126, 2167 – 2176.
[17] M. P. Andersson, P. Uvdal, J. Phys. Chem. A 2005, 109, 2937 – 2941.
[18] H. F. Bettinger, H. Bornemann, J. Am. Chem. Soc. 2006, 128, 11128 –11134.
[19] P. Pyykkç, M. Atsumi, Chem. Eur. J. 2009, 15, 12 770 – 12 779.[20] J. Thiele, O. Stange, Liebigs Ann. Chem. 1894, 283, 1 – 46.[21] E. A. Allan, R. F. Hobson, L. W. Reeves, K. N. Shaw, J. Am. Chem. Soc.
1972, 94, 6604 – 6611.[22] V. P. Kozyukov, V. F. Mironov, Zhurnal Obshchei Khimii. 1983, 53, 1434 –
1435.[23] A. D. Becke, J. Chem. Phys. 1993, 98, 5648 – 5652.[24] J. A. Montgomery Jr. , M. J. Frisch, J. W. Ochterski, G. A. Petersson, J.
Chem. Phys. 2000, 112, 6532 – 6542.[25] a) K. Fukui, Acc. Chem. Res. 1981, 14, 363 – 368; b) H. P. Hratchian, H. B.
Schlegel, J. Chem. Theory Comput. 2005, 1, 61 – 69.[26] Gaussian 09, Revision A.2, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E.
Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Men-nucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian,A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara,K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O.Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr. , J. E. Peralta, F. Ogliaro,M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Ko-bayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyen-gar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B.Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann,O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin,K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg,S. Dapprich, A. D. Daniels, ©. Farkas, J. B. Foresman, J. V. Ortiz, J. Cio-slowski, D. J. Fox, Gaussian, Inc. , Wallingford CT, 2009.
[27] a) H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schìtz, WIREsComput. Mol. Sci. 2012, 2, 242 – 253; b) MOLPRO, version 2012.1, a pack-age of ab initio programs, H.-J. Werner, P. J. Knowles, G. Knizia, F. R.Manby, M. Schìtz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G.Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A.Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C.Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Kçppl, Y. Liu,A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura,A. Nicklass, D. P. O’Neill, P. Palmieri, D. Peng, K. Pflìger, R. Pitzer, M.Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M.Wang, 2012, see: http://www.molpro.net.
Received: March 7, 2016
Published online on April 23, 2016
Chem. Eur. J. 2016, 22, 7856 – 7862 www.chemeurj.org Ó 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim7862
Full Paper





























