Embed Size (px)
Citation preview

CHILDHOOD TUMOURS
Dr Bello JM

Introduction
• Both benign and malignant tumours occur in childhood.
• Benign tumours are more common than malignant tumours but they are generally of little immediate consequence
• Childhood cancer comprise 2% of all malignant tumours but they are the leading cause of death in this age group

Introduction • Cancers of childhood differ from those in adults in
the following respects:• 1) Sites. Childhood Cancers more commonly
pertain to haematopoietic system, neural tissue and soft tissues – Cancer in adults are most commonly of epithelial origin
eg the lung, breast, prostate, colon and skin.• 2) Genetic basis. Many of paediatric cancers have
underlying genetic abnormalities.• 3) Regression. Foetal and neonatal malignancies
have a tendency to regress spontaneously or to mature.

• 4) Histologic features. They are usually have a primitive or embryonal appearance rather than pleomorphic-anaplastic histologic appearance.
• 5. Management. Many of paediatric malignant tumours are curable by chemotherapy and/or radiotherapy but may develop second malignancy.

BURKITT LYMPHOMA

BURKITT LYMPHOMA
• Burkitt lymphoma (BL) is one of the most rapidly growing malignancies known.
• It was first described in African children, predominantly presenting as jaw tumour
• It comprises about 30% of childhood NHLs

Epidemiology
• Three distinct variants of Burkitt lymphoma are recognised:
–endemic,– sporadic and –immunodeficiency-associated.
• In equatorial Africa , endemic BL is the most common childhood malignancy.
• Its peak incidence is in 4- to 7-year-olds, and it commonly involves the jaw, other facial bones and the abdominal viscera.

• The sporadic variant occurs worldwide and mainly affects children and young adults.
• Unlike endemic BL, sporadic BL often presents as an abdominal mass involving the ileocecum.
• Immunodefficiency associated BL mainly occurs in HIV-infected people and may be the initial manifestation of AIDS.
• Like most other B-cell lymphomas, BL afflicts males more than females.

Molecular pathogenesis• All cases are associated with translocations that
upregulate expression of c-MYC oncogene on chromosome 8, – either by placing it close to IgH i.e {t(8;14)} or – IgL i.e. [t(2;8 for κ) or t(8;22 for λ)].
• In these cases, expression of MYC gene driven by the Ig heavy-chain promoter leads to uncontrolled tumor cell growth

• EBV is present in virtually all cases of endemic BL but is responsible for less than 30% of sporadic and immunodeiciency-related cases.
• Many patients experience prodromal polyclonal B-cell activation caused by bacterial, viral or parasitic infections (e.g., malaria).

CLINICAL FEATURES AND PATHOLOGY
• BL typically produces extranodal tumors rather than lymphadenopathy.
• All variants of this lymphoma have a high risk for CNS involvement.
• The classic presentation for endemic BL is a destructive tumor in the jaws or other facial bones.
• Patients with sporadic BL typically present with abdominal masses.
• All types may involve ovaries, kidneys and breast.


Morphology
• BL cells are medium sized and lack significant cytologic atypia.
• Tissue sections reveal abundant mitotic figures, relecting the extremely high proliferative rate in this tumor.
• Macrophages ingesting the cellular debris of apoptotic tumor cells are scattered throughout the tumor, imparting a “starry sky” microscopic appearance to the tumor

Starry sky appearance in BL

• Because of its high proliferative rate, BL responds to intensive chemotherapy.
• Thus, up to 90% of people with early stage disease and 60%–80% of those with high-stage disease may be cured.
• Children and young adults with BL tend to fare better than adults.
• Tumor lysis syndrome can occur when therapy begins, as a result of rapid tumor cell death.
• This is a potentially lethal complication of treatment

Wilm’s tumor
Nephroblastoma

• This is an embryonic tumour derived from primitive renal epithelial and mesenchymal components.
• It is the most common abdominal malignant tumour of young children

• The peak incidence is between 2 and 5 years of age, and 95% of tumors occur before the age of 10 years.
• Approximately 5% to 10% of Wilms tumors involve both kidneys, either – simultaneously (synchronous) or – one after the other (metachronous).
• Bilateral Wilms tumors have a median age of onset approximately 10 months earlier than tumors restricted to one kidney– These patients are presumed to harbor a germline
mutation in one of the Wilms tumor–predisposing genes.

• The risk of Wilms tumor is increased in association with at least four recognizable groups of congenital malformations associated with distinct chromosomal loci.
• 1) WAGR syndrome,– characterized by Wilms tumor, aniridia, genital anomalies,
and mental retardation. – Their lifetime risk is approximately 33%. – Individuals with WAGR syndrome carry constitutional
(germline) deletions of 11p13.– In this location are WT1 and PAX6 genes • Patients with deletions restricted to PAX6, with normal WT1
function, develop sporadic aniridia, but they are not at increased risk for Wilms tumors.

The presence of germline WT1 deletions
in WAGR syndrome represents the “first hit”;
the development of Wilms tumor in these patients frequently correlates with the occurrence of a
nonsense or frameshift mutation in the second WT1 allele (“second hit”).

2) Denys-Drash syndrome, • characterized by GONADAL DYSGENESIS (male
pseudohermaphroditism) and EARLY-ONSET NEPHROPATHY leading to renal failure.
• Associated with ~90% risk for Wilms tumor• As in patients with WAGR, these patients also
demonstrate germline abnormalities in WT1– dominant-negative missense mutation in the gene

II. Denys-Drash syndrome
• In addition to Wilms tumors, these individuals are also at increased risk for developing germ cell tumors called
GONADOBLASTOMAS, almost certainly a consequence of disruption in normal gonadal development.

• Despite the importance of WT1 in NEPHROGENESIS and its unequivocal role as a TUMOR SUPPRESSOR GENE, only about 10% of patients with sporadic (nonsyndromic) Wilms tumors demonstrate WT1 mutations, suggesting that the majority of these tumors arise from MUTATIONS IN OTHER GENES.

3) Beckwith-Wiedemann syndrome
characterized by 1. ENLARGEMENT OF BODY ORGANS (organomegaly),2. MACROGLOSSIA, 3. HEMIHYPERTROPHY, 4. OMPHALOCELE, and 5. ABNORMAL LARGE CELLS IN THE ADRENAL CORTEX
(adrenal cytomegaly). • The chromosomal region implicated in BWS has been
localized to band 11p15.5 (“WT2”), distal to the WT1 locus.

• In addition to Wilms tumors, patients with BWS are also at increased risk for developing
HEPATOBLASTOMA, • PANCREATOBLASTOMA, • ADRENOCORTICAL TUMORS, and
RHABDOMYOSARCOMAS.

Nephrogenic Rests
• Nephrogenic rests are putative precursor lesions of Wilms tumors and are seen in the renal parenchyma adjacent to approximately
25% to 40% of unilateral tumors;
this frequency rises to nearly 100% in cases of bilateral Wilms tumors.

Morphology
• Grossly, Wilms tumor tends to present as a large, solitary, well-circumscribed mass, although 10% are either bilateral or multicentric at the time of diagnosis.
• On cut section, The tumor is soft, homogeneous, and tan to gray with occasional foci of hemorrhage, cyst formation, and necrosis.

• Microscopically, Wilms tumors attempt to recapitulate different stages of nephrogenesis.
• It is characterised by 3 cell types:– BLASTEMAL,– STROMAL, and– EPITHELIAL.

• Sheets of small blue cells with few distinctive features characterize the BLASTEMAL component.
• EPITHELIAL DIFFERENTIATION is usually in the form of abortive tubules or glomeruli.
• STROMAL CELLS are usually fibrocytic or myxoid in nature, although skeletal muscle differentiation is not uncommon.

• Rarely, other heterologous elements are identified, including
• Squamous or mucinous epithelium, • Smooth muscle, • Adipose tissue, • Cartilage, • Osteoid and• Neurogenic tissue
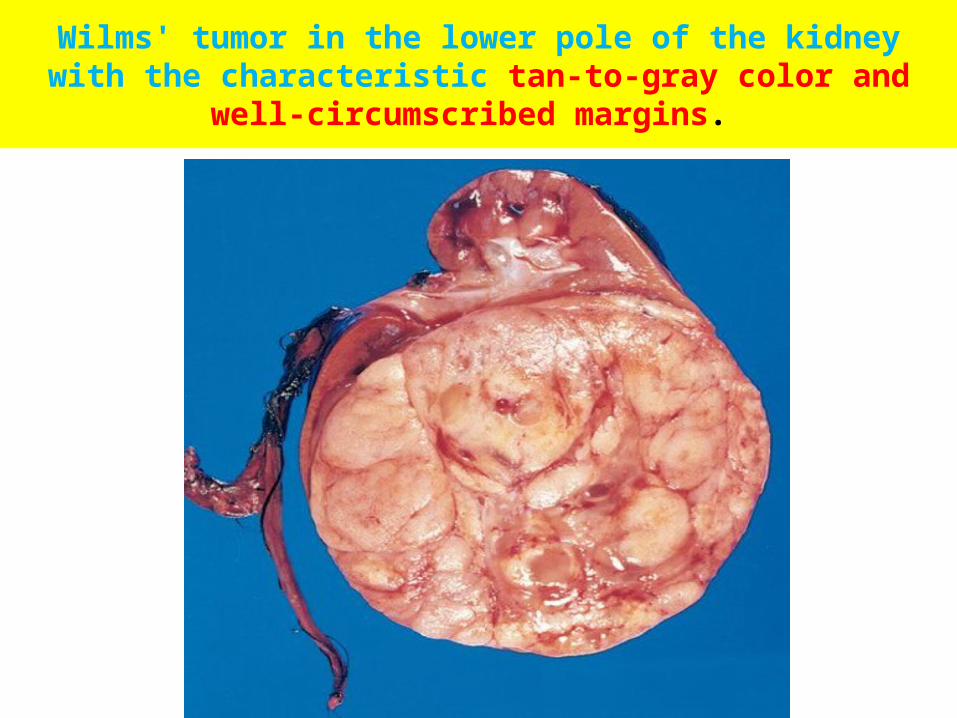
Wilms' tumor in the lower pole of the kidney with the characteristic tan-to-gray color and well-circumscribed margins.


Clinical Features.
• Abdominal mass• Hematuria, • Pain in the abdomen, • Intestinal obstruction, • Hypertension• In a considerable number of these patients,
pulmonary metastases are present at the time of primary diagnosis.

Prognosis• Cure Rate 85 %• Anaplastic histology remains a critical
determinant of adverse prognosis.• Even anaplasia restricted to the kidney (i.e.,
without extra-renal spread) confers an increased risk of recurrence and death.

• Molecular parameters that correlate with adverse prognosis include
• loss of genetic material on chromosomes 11q and 16q, and
• gain of chromosome 1q in the tumor cells.

• Along with the increased survival of individuals with Wilms tumor have come reports of an increased relative risk of developing second primary tumors, including
• Bone and soft-tissue SARCOMAS,• Leukemia and Lymphomas, and
• Breast cancers.


• No racial or gender predisposition.• Bilateral in 25 to 35% cases.• Age of diagnosis is 18 months.• Unilateral cases around 24 months & bilateral
before 12 months.





























