
7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 1/14
Zaman Muscle Pathology
Review the normal structure and function of the myocyte and neuromuscular junction.
Motor units
o Motor neuron, peripheral axon, distal neuromuscular junction and skeletal muscle.
o 100-200 myofibers innervated by the same motor neuron and all in the unit are of the same type
(type I or II). Therefore, histochemical type is determined by the nerve supply.
Myosite = Muscle cells. Composed of myofibrils.
o Myofibrils are in turn, made up of sarcomeres
o Dystrophin
Central member of the dystrophin glycoprotein complex – connects the actin cytoskeleton
with the muscle fiber membrane and its external environment.
Describe the procedure for muscle biopsy
Must have a rational Ddx – Ddx is important to determine procedure and studies to be done. Also time,
which muscle, number of specimens and handling.
2 types of specimens
o Fresh – Histo studies in all patients and immunofluorescence in selected patients.
o Fixed – In formalin – routine microscopy and EM – reserved for special situations. Sample is removedusing Rayport clamp i.e
Used to evaluate patients with neuromuscular disease and occasionally to assess pts with myopathy. It is
also involved in the diagnosis of neuropathic disease, mainly in the distinction of an atypical neurogenic
disorder from a primary myopathic one .
Studies in a frozen sample include, in addition to H&E

7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 2/14
o NADH, Myofiber stains (ATPase, type I and II), mitochondrial stain (Gomori), PAS (glycogen/vessels),
Fat stain (Sudan black, oil red, O lipids, metabolic, mitochondria and myofibers – fresh specimens
used).
Paraffin sections are usually stained with H&E – longitudinal and cross section.
o Useful for evaluating inflammation, myopathies and vasculitis – i.e – processes with non-uniform
distribution.
o Stains Organism stains
Elastic stains (vasculitis)
Immunohistochemistry – inflammatory cells
FISH – Viruses
Congo red or thioflavin S – Amyloid (MC)
Indications for Biopsy ***
o Myopathies – Including inflammatory (tx can alter histopathology findings, so is best before tx if risk is
high enough to justify it. Repeat biopsy is indicated to evaluate pt with known inflammatorymyopathy who’s still weak after steroid therapy.
o Other disorders with therapeutic options less definitive than those for inflammatory myopathies. i.e
palliative tx, clinical trials, genetic counseling, prognosis.
o One common indication is to distinguish between myopathy and neuropathy . Classic presentations
are distinct but in practice, their H&P and lab findings often overlap. They can also coexist, making
dx more difficult.
Not indicated in
o Dystrophinopathies – Genetic testing is more commonly used.
o Myotonic dystrophy – genetic testing.
o Periodic paralyses – genetic
o Endocrine myopathies (i.e cortisol deficiency – can dx with electrodiagnostic work up vs biopsy).
Other diagnostic means
o Labs
CK – Useful but not definitive in differentiation neuropathy vs myopathy. >1000 = muscle
disease, 200-600 = either but less likely in myopathy patients.
Serum aldolase – More helpful in dx myopathy. Longer half life in serum vs CK therefore more
dx when CK is normal.o Electrodiagnosis
Helpful to determine if disease is neuropathic, myopathic or mixed.
Changes in N conduction and muscle AP – Neurogenic.
Shows different findings in neurogenic vs myopathic disorders.
Avoid in muscle that will undergo biopsy – it damages muscle and will affect biopsy
interpretation for 1-2 months. If pt has suspected myopathy, needle EMG should be done
one only 1 side.

7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 3/14
7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 4/14
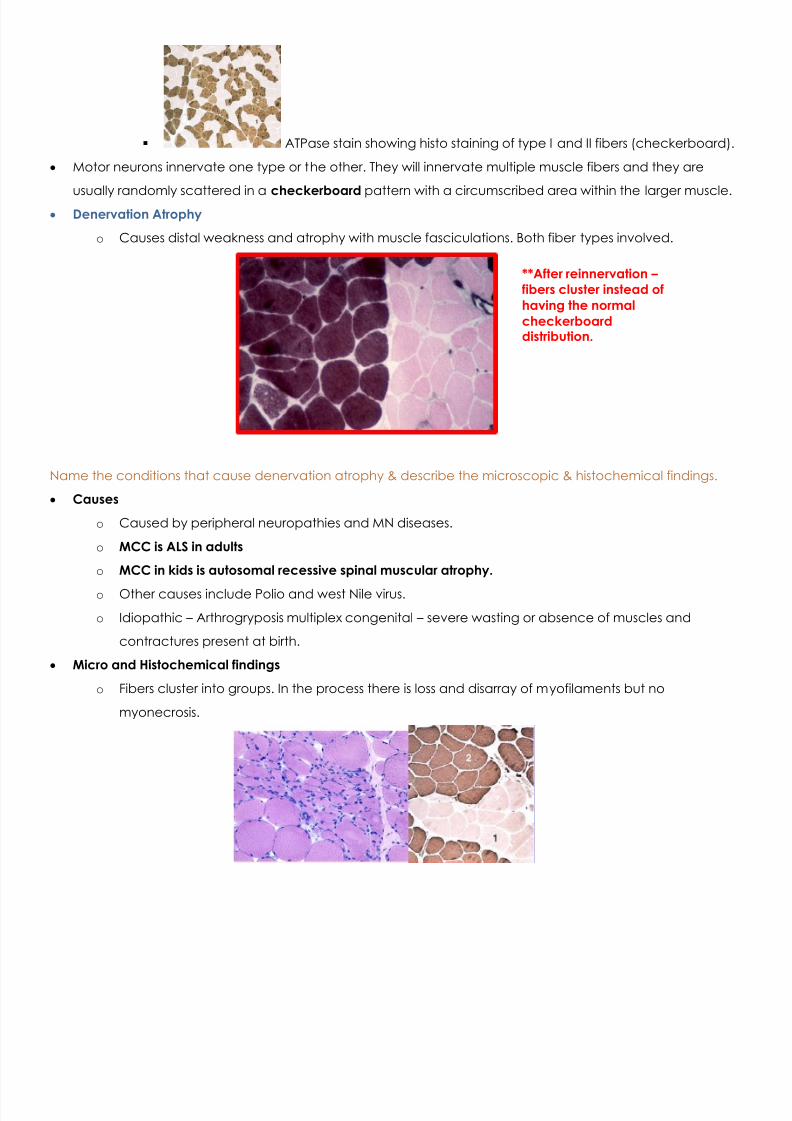
ATPase stain showing histo staining of type I and II fibers (checkerboard).
Motor neurons innervate one type or the other. They will innervate multiple muscle fibers and they are
usually randomly scattered in a checkerboard pattern with a circumscribed area within the larger muscle. Denervation Atrophy
o Causes distal weakness and atrophy with muscle fasciculations. Both fiber types involved.
Name the conditions that cause denervation atrophy & describe the microscopic & histochemical findings.
Causes
o Caused by peripheral neuropathies and MN diseases.
o MCC is ALS in adults
o MCC in kids is autosomal recessive spinal muscular atrophy.
o Other causes include Polio and west Nile virus.
o Idiopathic – Arthrogryposis multiplex congenital – severe wasting or absence of muscles and
contractures present at birth.
Micro and Histochemical findings
o Fibers cluster into groups. In the process there is loss and disarray of myofilaments but no
myonecrosis.
**After reinnervation – fibers cluster instead ofhaving the normalcheckerboarddistribution.
7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 5/14
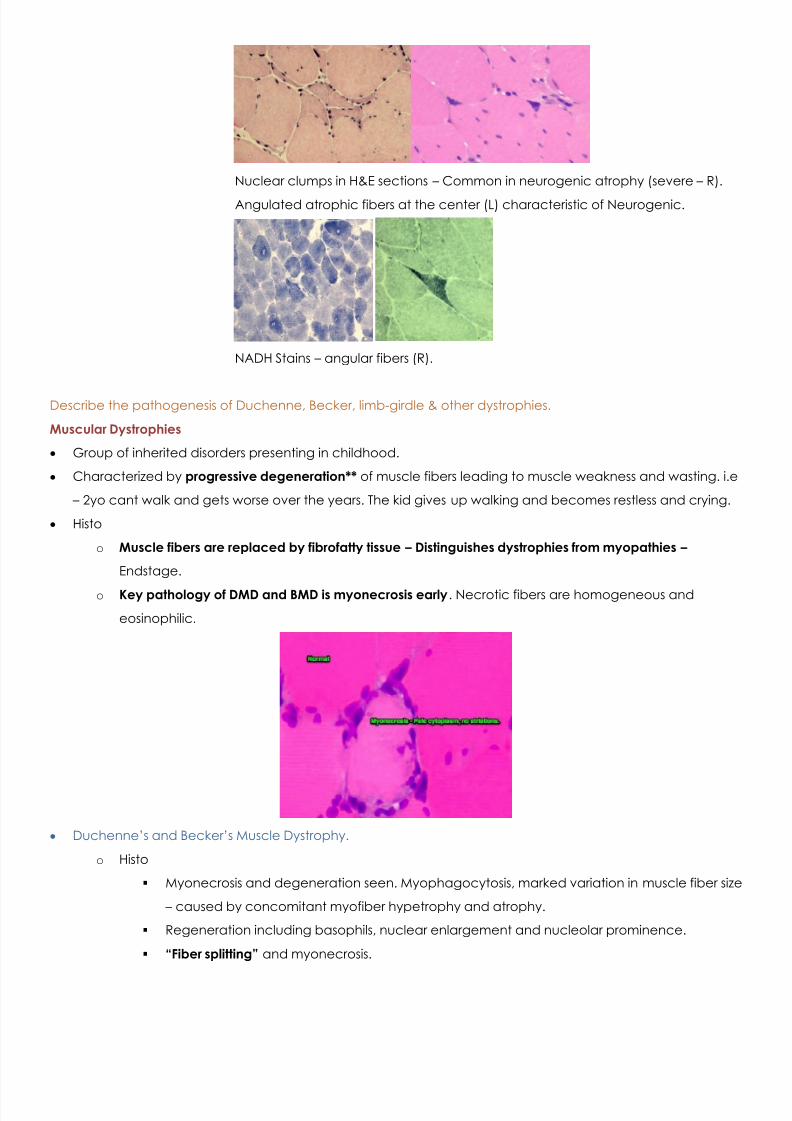
Nuclear clumps in H&E sections – Common in neurogenic atrophy (severe – R).
Angulated atrophic fibers at the center (L) characteristic of Neurogenic.
NADH Stains – angular fibers (R).
Describe the pathogenesis of Duchenne, Becker, limb-girdle & other dystrophies.
Muscular Dystrophies
Group of inherited disorders presenting in childhood.
Characterized by progressive degeneration** of muscle fibers leading to muscle weakness and wasting. i.e
– 2yo cant walk and gets worse over the years. The kid gives up walking and becomes restless and crying.
Histo
o Muscle fibers are replaced by fibrofatty tissue – Distinguishes dystrophies from myopathies –
Endstage.
o Key pathology of DMD and BMD is myonecrosis early . Necrotic fibers are homogeneous and
eosinophilic.
Duchenne’s and Becker’s Muscle Dystrophy.
o Histo
Myonecrosis and degeneration seen. Myophagocytosis, marked variation in muscle fiber size
– caused by concomitant myofiber hypetrophy and atrophy.
Regeneration including basophils, nuclear enlargement and nucleolar prominence.
“Fiber splitting” and myonecrosis.
7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 6/14
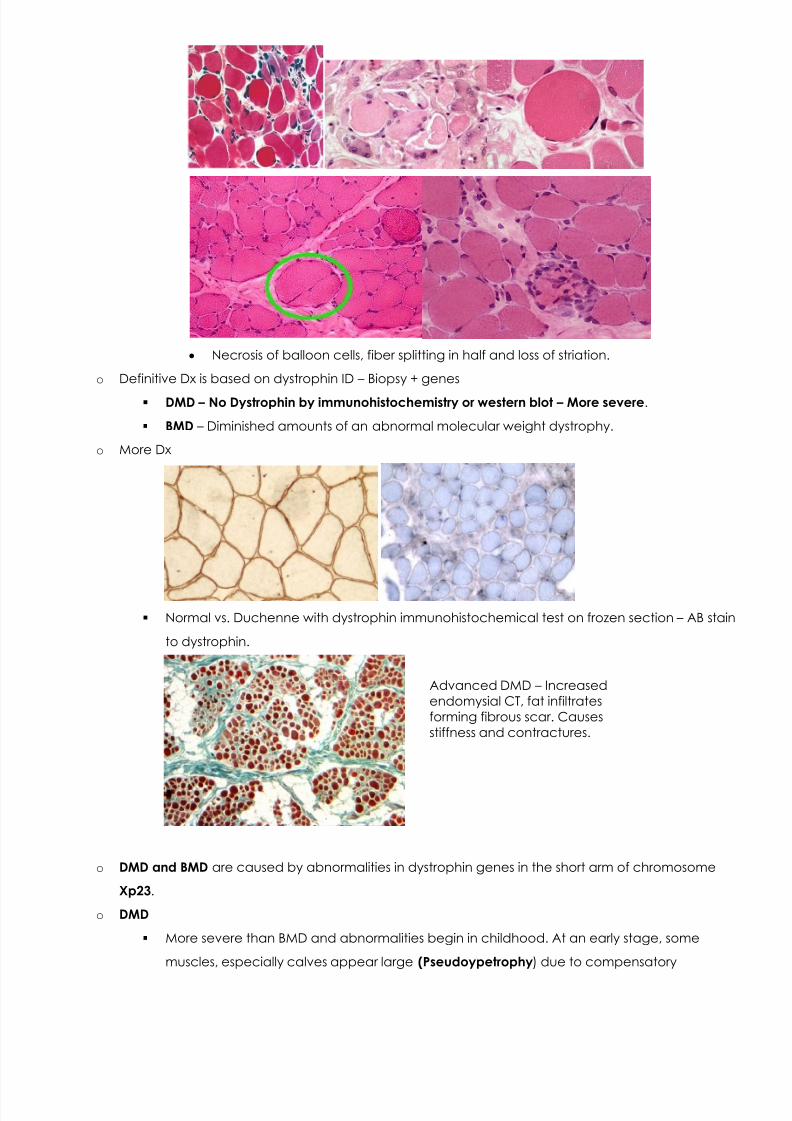
Necrosis of balloon cells, fiber splitting in half and loss of striation.
o Definitive Dx is based on dystrophin ID – Biopsy + genes
DMD – No Dystrophin by immunohistochemistry or western blot – More severe .
BMD – Diminished amounts of an abnormal molecular weight dystrophy.
o More Dx
Normal vs. Duchenne with dystrophin immunohistochemical test on frozen section – AB stain
to dystrophin.
o DMD and BMD are caused by abnormalities in dystrophin genes in the short arm of chromosome
Xp23 .
o DMD
More severe than BMD and abnormalities begin in childhood. At an early stage, some
muscles, especially calves appear large (Pseudoypetrophy ) due to compensatory
Advanced DMD – Increasedendomysial CT, fat infiltratesforming fibrous scar. Causesstiffness and contractures.

7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 7/14
hypetrophy of normal myofibers and increased fat (myonecrosis and regeneration. More
muscle is lost as disease progresses.
Patients are in wheel chairs by age 12 and death occurs at end of 2 nd decade – respiratory
insufficiency and other complications.
o BMD
Symptoms begin later in life and the disease is more protracted. Some patients have a nearly
normal lifespan. Mutations can cause dilated cardiomyopathy and can be fatal. 20-40% of females with
dystrophin mutations have mild muscle disease or dilated L ventricle.
Limb Girdle Dystrophy
o MC in the proximal musculature of trunk and limbs (like X-linked dystrophies).
o 6 dominant and 10 recessive subtypes (90% LGMDs).
o Less frequent vs. dystrinopathies and are milder. Begin in adolescence or adulthood and have a
slower progression.o A subset known as Severe Childhood Autosomal Recessive Muscular Dystrophy (SCARMD) is almost
as severe as Duchenne’s.
Discuss the genetics and key pathological changes of myotonic dystrophy.
o Genetics
Autosomal Dominant – Transmitted from the mother.
2 genetic forms
DM1 – Distal weakness. CTG trinucleotide expansion of the DMPK (dystrophin myotonica
protein kinase) on chromosome I9q13. Lots of repeats, the more repeats the earlier the onset
of sxs. Repeats increased with generations.
DM2 – Proximal weakness. Caused by a CCTG expansion of ZNF9 (Zinc finger protein 9) gene
on 3q21. Protein disorder.
Weakness, stiffness – more pronounced in facial and distal muscles .
Atrophy and weakness of facial muscles, ptosis and frontal baldness. Characteristic facial
appearance. Myotonia (spasms) occur spontaneously or elicited by mild stimulation. Pt wont let go
of your hand. May have cataracts, arrhythmias, testicular atrophy and diabetes. Progressive
weakness.o Pathological changes
Biopsy shows atrophy of type I fibers , profusion of central nuclei (normally they are under
sarcolemma) and ring fibers . None of the changes are diagnostic alone but combination strongly
suggests myotonic dystrophy.
7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 8/14
Describe 3 congenital myopathies & discuss the differential diagnosis of neonatal hypotonia.
o Most frequent these dystrophies are of Laminin a2 . Other groups are caused by mutations of enzymes that
glycosylate alpha dystroglycan.
o Normal vs. congenital dystrophy with low laminin.
o Many CMD patients have neuronal migration defects because of dystroglycan. Main defects are visual
abnormalities.
o CM are primary muscle disorders. Most are mutations of contractile and structural proteins in the sarcoplasm
and inclusions can be seen . Many myopathies and inclusion types. MC – nemaline, centronuclear, central
core and myofibrillar.
Nemaline (Rod shape with a-actinin – component of Z bands), centronuclear (Small myofibers with
central nuclei and central areas without contractile filaments).
o Kids look funny and have severe hypotonia. CM are rare.
o DDx of neonatal hypotonia includes
Perinatal asphyxia
Metabolic Disorders
Congenital CNS abnormalities
Spinal muscular atrophy (Werdnig-Hoffman disease) .
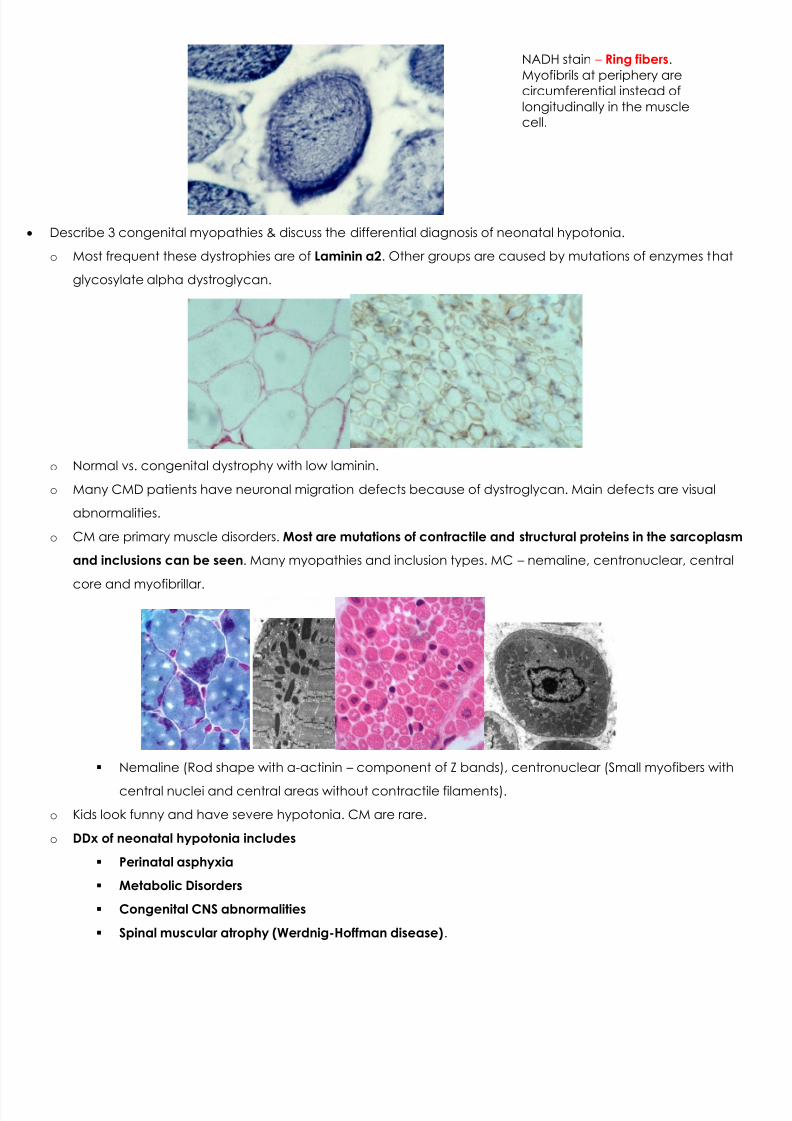
NADH stain – Ring fibers .Myofibrils at periphery arecircumferential instead oflongitudinally in the musclecell.
7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 9/14
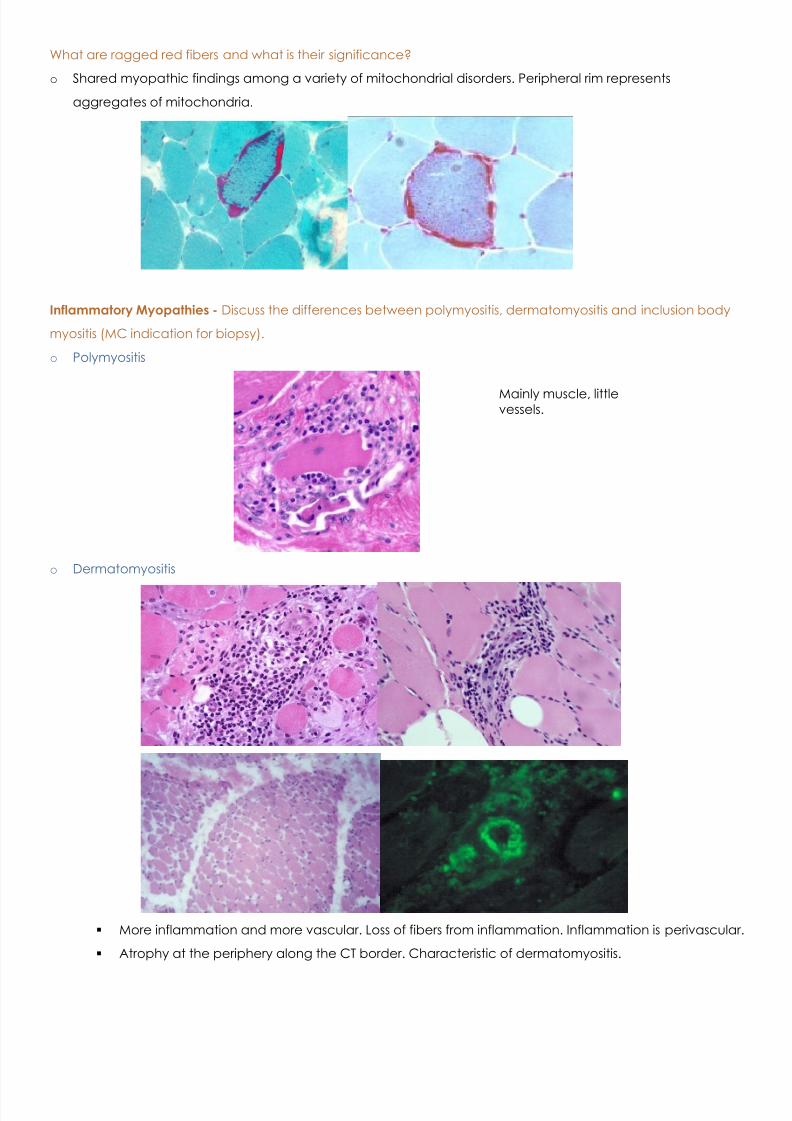
What are ragged red fibers and what is their significance?
o Shared myopathic findings among a variety of mitochondrial disorders. Peripheral rim represents
aggregates of mitochondria.
Inflammatory Myopathies - Discuss the differences between polymyositis, dermatomyositis and inclusion body
myositis (MC indication for biopsy).
o Polymyositis
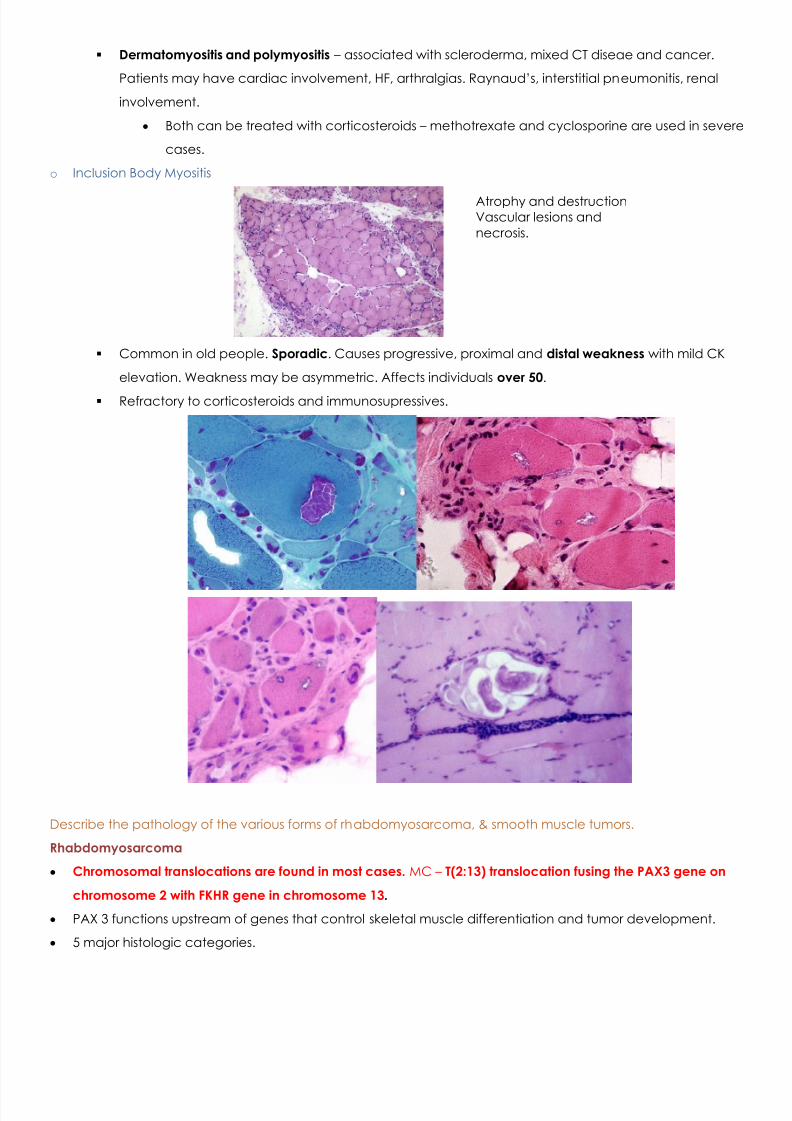
o Dermatomyositis
More inflammation and more vascular. Loss of fibers from inflammation. Inflammation is perivascular.
Atrophy at the periphery along the CT border. Characteristic of dermatomyositis.
Mainly muscle, littlevessels.
7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 10/14
Dermatomyositis and polymyositis – associated with scleroderma, mixed CT diseae and cancer.
Patients may have cardiac involvement, HF, arthralgias. Raynaud’s, interstitial pn eumonitis, renal
involvement.
Both can be treated with corticosteroids – methotrexate and cyclosporine are used in severe
cases.
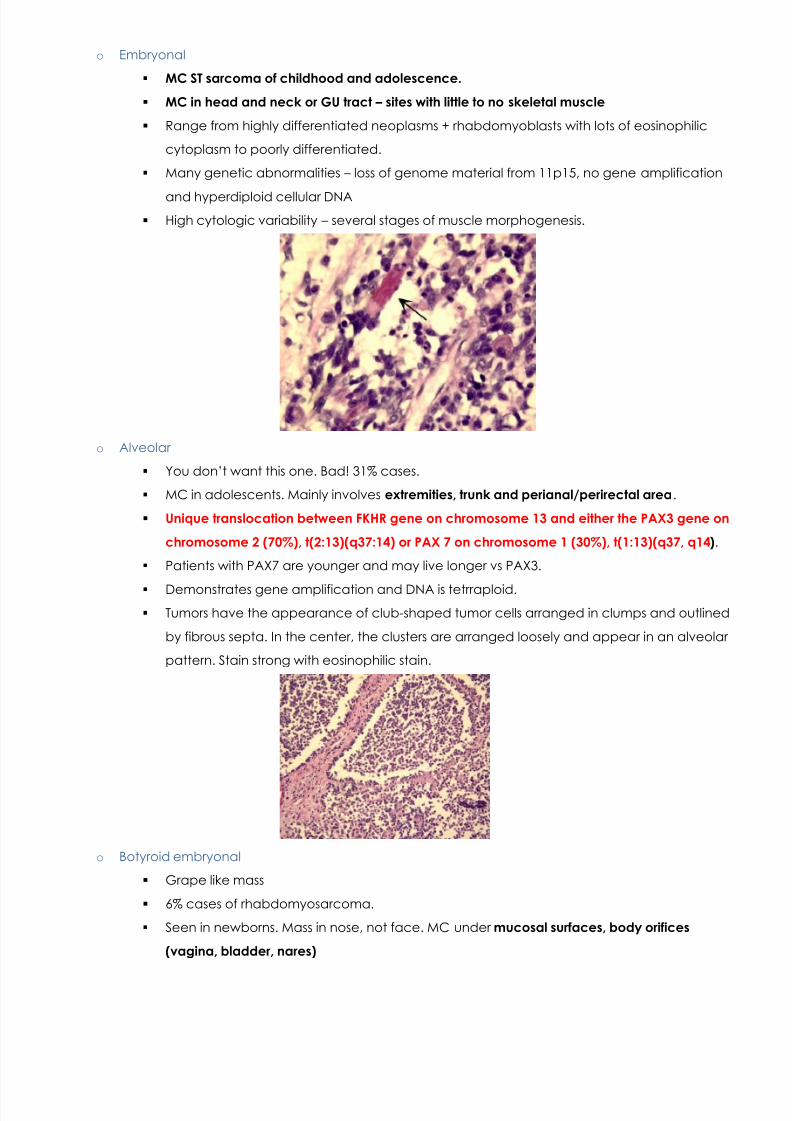
o Inclusion Body Myositis
Common in old people. Sporadic . Causes progressive, proximal and distal weakness with mild CK
elevation. Weakness may be asymmetric. Affects individuals over 50 .
Refractory to corticosteroids and immunosupressives.
Describe the pathology of the various forms of rhabdomyosarcoma, & smooth muscle tumors.
Rhabdomyosarcoma
Chromosomal translocations are found in most cases. MC – T(2:13) translocation fusing the PAX3 gene on
chromosome 2 with FKHR gene in chromosome 13 .
PAX 3 functions upstream of genes that control skeletal muscle differentiation and tumor development.
5 major histologic categories.
Atrophy and destructionVascular lesions andnecrosis.
7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 11/14
o Embryonal
MC ST sarcoma of childhood and adolescence.
MC in head and neck or GU tract – sites with little to no skeletal muscle
Range from highly differentiated neoplasms + rhabdomyoblasts with lots of eosinophilic
cytoplasm to poorly differentiated.
Many genetic abnormalities – loss of genome material from 11p15, no gene amplification
and hyperdiploid cellular DNA High cytologic variability – several stages of muscle morphogenesis.
o Alveolar
You don’t want this one. Bad! 31% cases.
MC in adolescents. Mainly involves extremities, trunk and perianal/perirectal area .
Unique translocation between FKHR gene on chromosome 13 and either the PAX3 gene on
chromosome 2 (70%), t(2:13)(q37:14) or PAX 7 on chromosome 1 (30%), t(1:13)(q37, q14 ).
Patients with PAX7 are younger and may live longer vs PAX3.
Demonstrates gene amplification and DNA is tetrraploid. Tumors have the appearance of club-shaped tumor cells arranged in clumps and outlined
by fibrous septa. In the center, the clusters are arranged loosely and appear in an alveolar
pattern. Stain strong with eosinophilic stain.
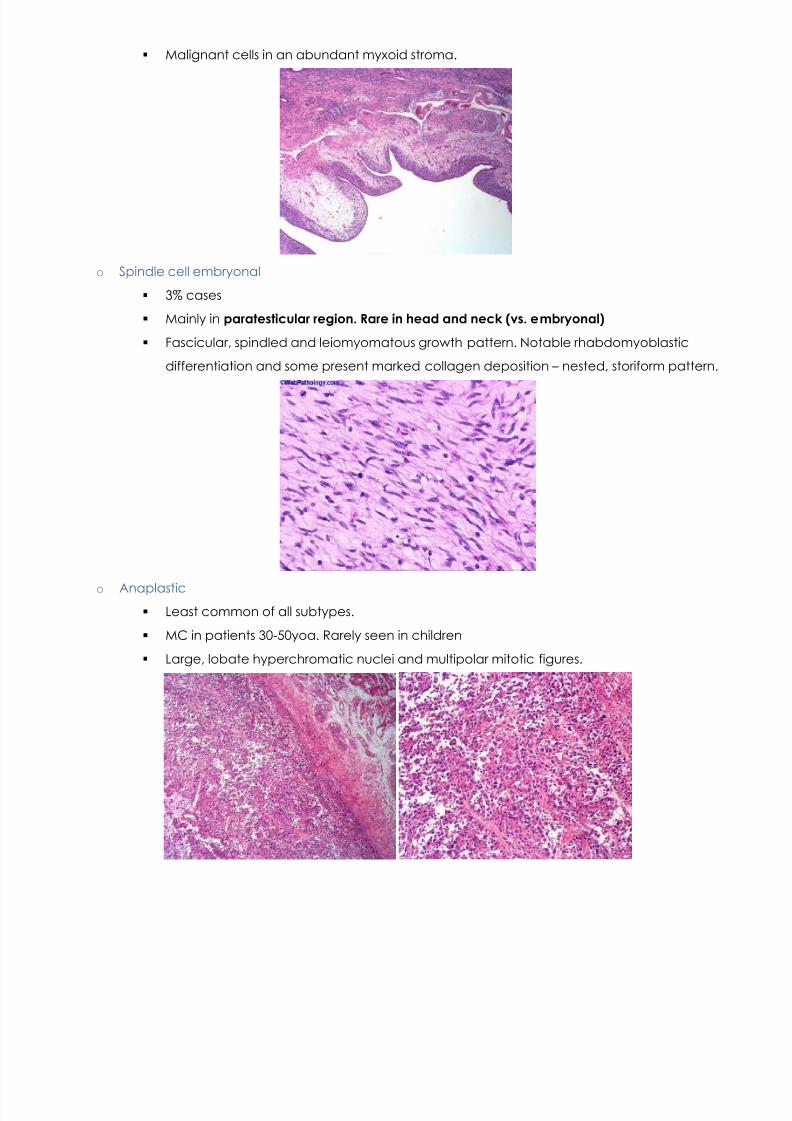
o Botyroid embryonal
Grape like mass
6% cases of rhabdomyosarcoma.
Seen in newborns. Mass in nose, not face. MC under mucosal surfaces, body orifices
(vagina, bladder, nares)
7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 12/14
Malignant cells in an abundant myxoid stroma.
o Spindle cell embryonal
3% cases
Mainly in paratesticular region. Rare in head and neck (vs. embryonal)
Fascicular, spindled and leiomyomatous growth pattern. Notable rhabdomyoblastic
differentiation and some present marked collagen deposition – nested, storiform pattern.
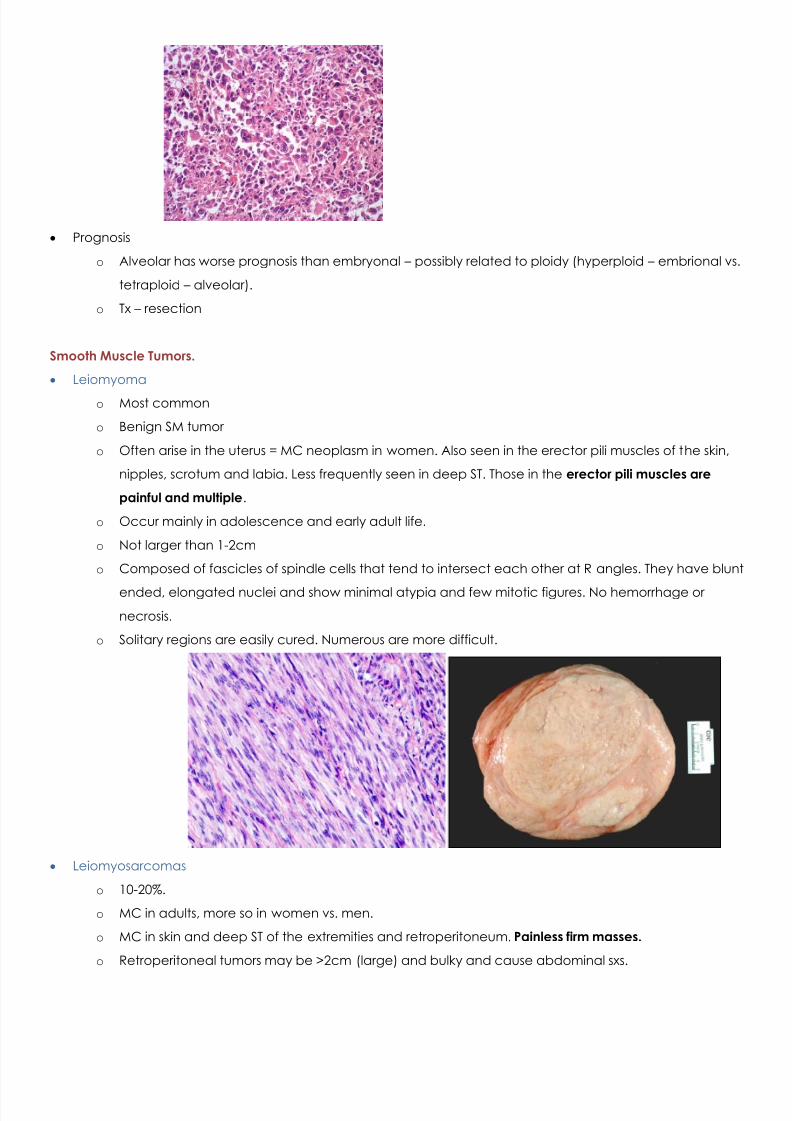
o Anaplastic Least common of all subtypes.
MC in patients 30-50yoa. Rarely seen in children
Large, lobate hyperchromatic nuclei and multipolar mitotic figures.
7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 13/14
Prognosis
o Alveolar has worse prognosis than embryonal – possibly related to ploidy (hyperploid – embrional vs.
tetraploid – alveolar).
o Tx – resection
Smooth Muscle Tumors.
Leiomyoma
o Most common
o Benign SM tumor
o Often arise in the uterus = MC neoplasm in women. Also seen in the erector pili muscles of the skin,
nipples, scrotum and labia. Less frequently seen in deep ST. Those in the erector pili muscles are
painful and multiple .
o Occur mainly in adolescence and early adult life.
o Not larger than 1-2cm
o Composed of fascicles of spindle cells that tend to intersect each other at R angles. They have blunt
ended, elongated nuclei and show minimal atypia and few mitotic figures. No hemorrhage or
necrosis.
o Solitary regions are easily cured. Numerous are more difficult.
Leiomyosarcomas
o 10-20%.
o MC in adults, more so in women vs. men.
o MC in skin and deep ST of the extremities and retroperitoneum. Painless firm masses.
o Retroperitoneal tumors may be >2cm (large) and bulky and cause abdominal sxs.
7/31/2019 Zaman Muscle Pathology
http://slidepdf.com/reader/full/zaman-muscle-pathology 14/14
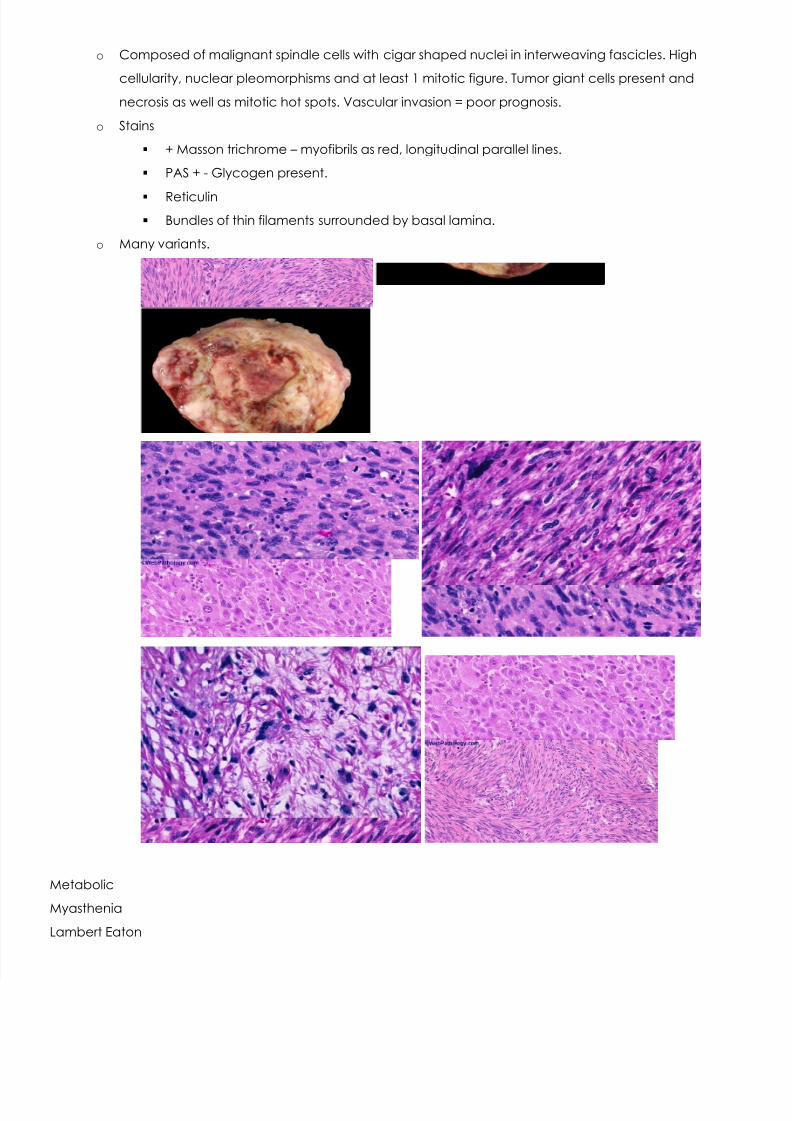
o Composed of malignant spindle cells with cigar shaped nuclei in interweaving fascicles. High
cellularity, nuclear pleomorphisms and at least 1 mitotic figure. Tumor giant cells present and
necrosis as well as mitotic hot spots. Vascular invasion = poor prognosis.
o Stains
+ Masson trichrome – myofibrils as red, longitudinal parallel lines.
PAS + - Glycogen present.
Reticulin Bundles of thin filaments surrounded by basal lamina.
o Many variants.
Metabolic
Myasthenia
Lambert Eaton
Recommended

















