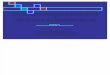
Esclerosis lateral amiotrófica i maneig multidisciplinar
M.A. Rubio
Mayo 2017
Servicio de Neurología.
Hospital del Mar. Parc de Salut Mar

0
20
40
60
80
100
120
140
160
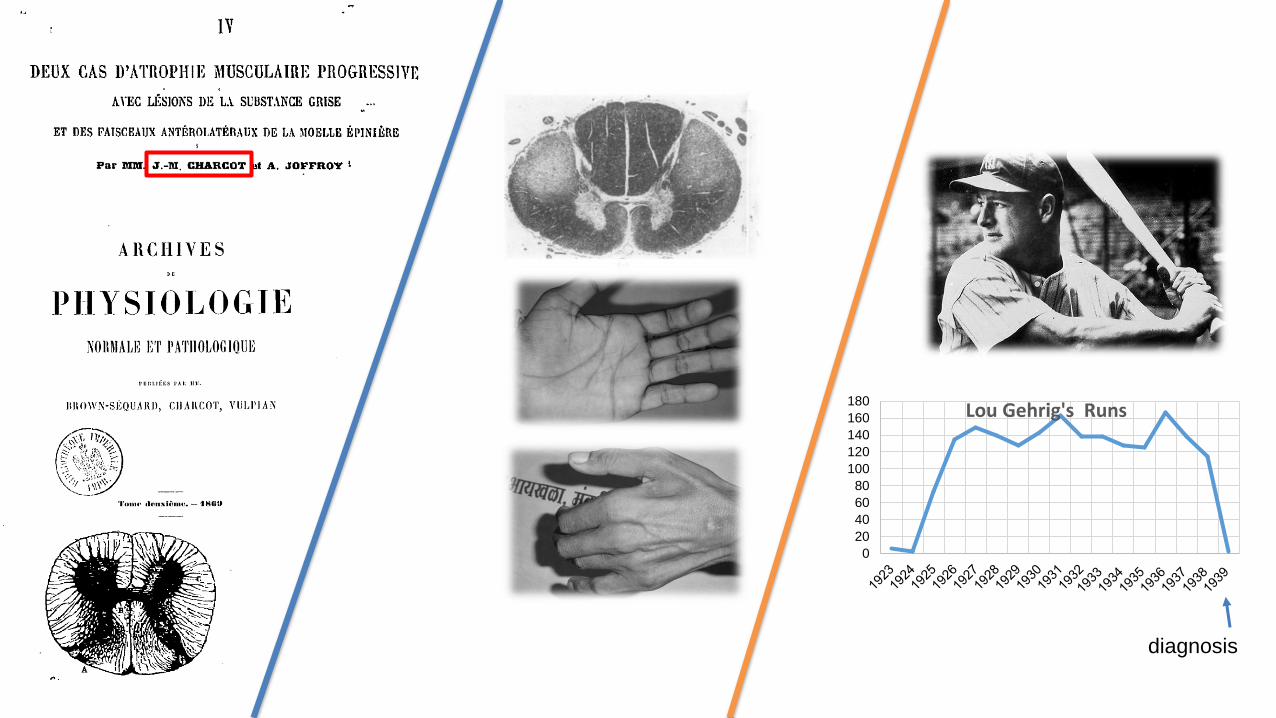
180Lou Gehrig's Runs
diagnosis
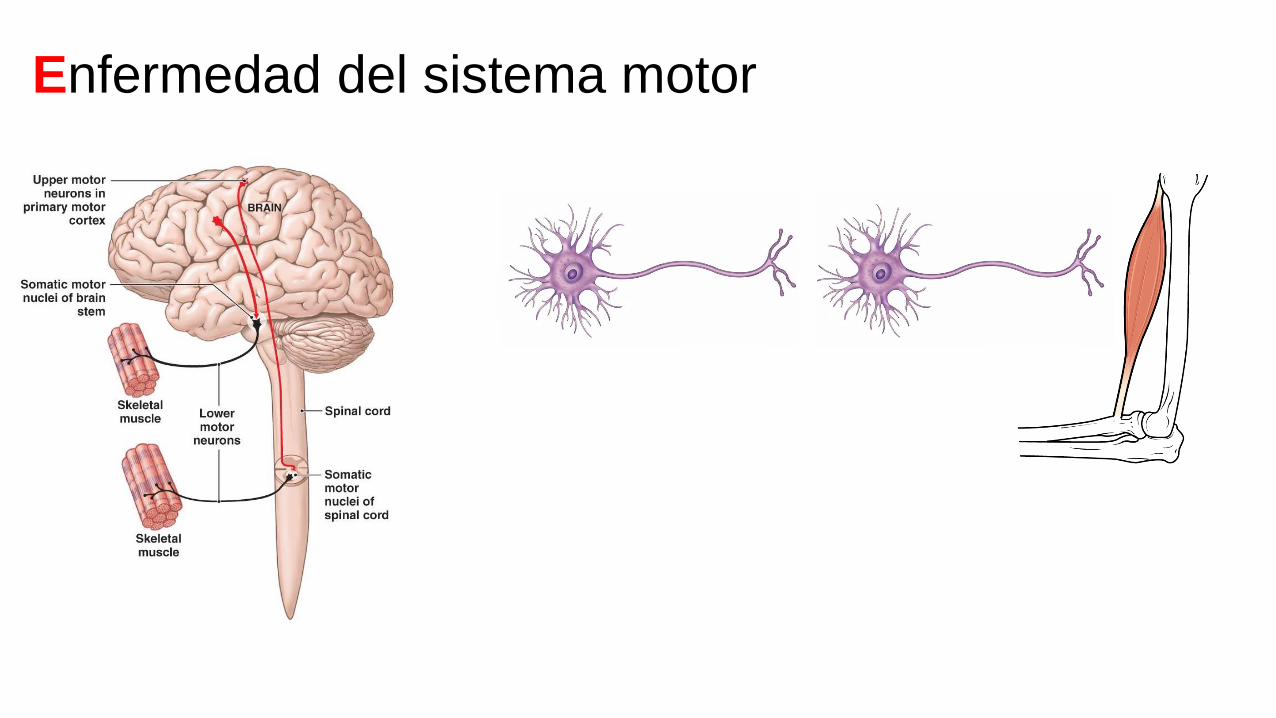
Enfermedad del sistema motor
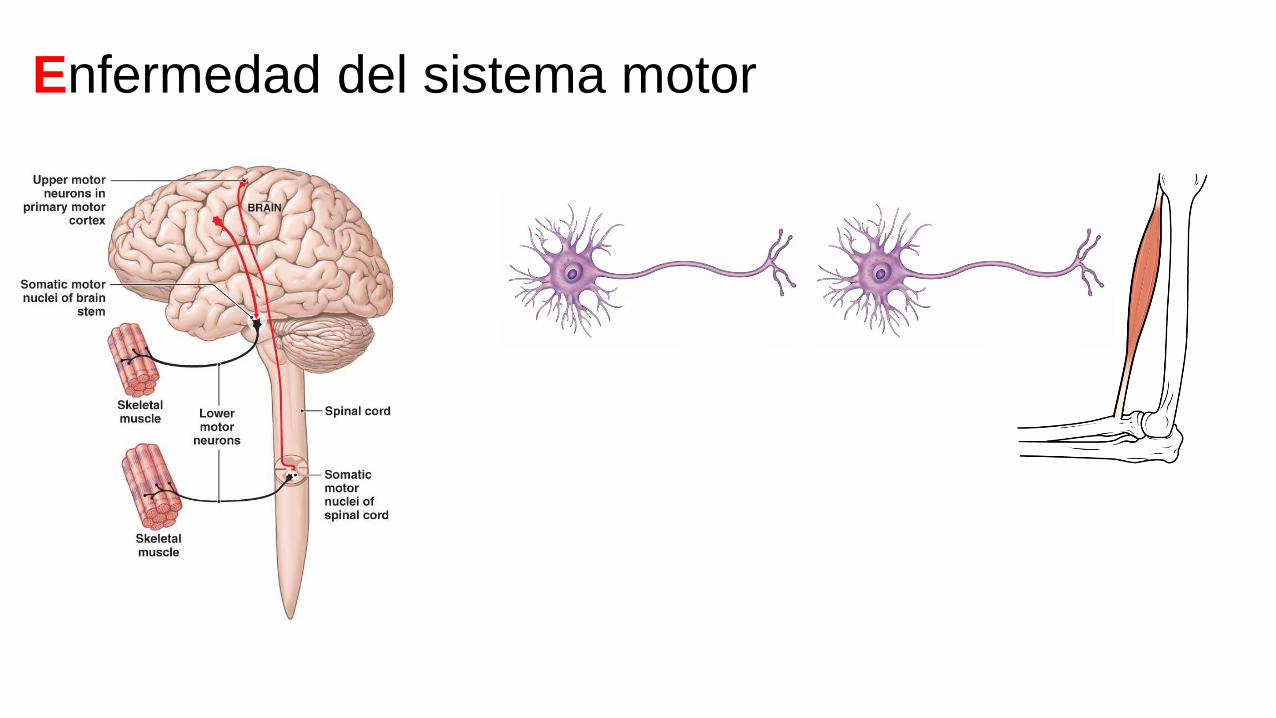
Enfermedad del sistema motor
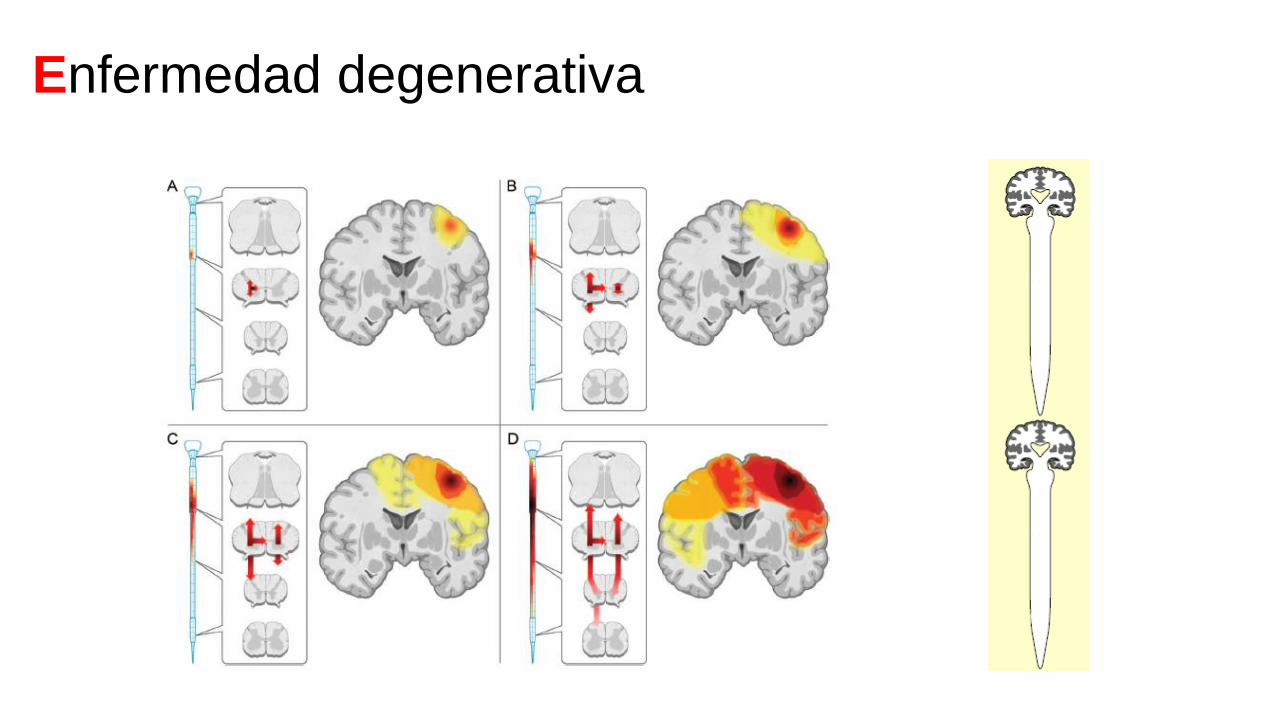
Enfermedad degenerativa
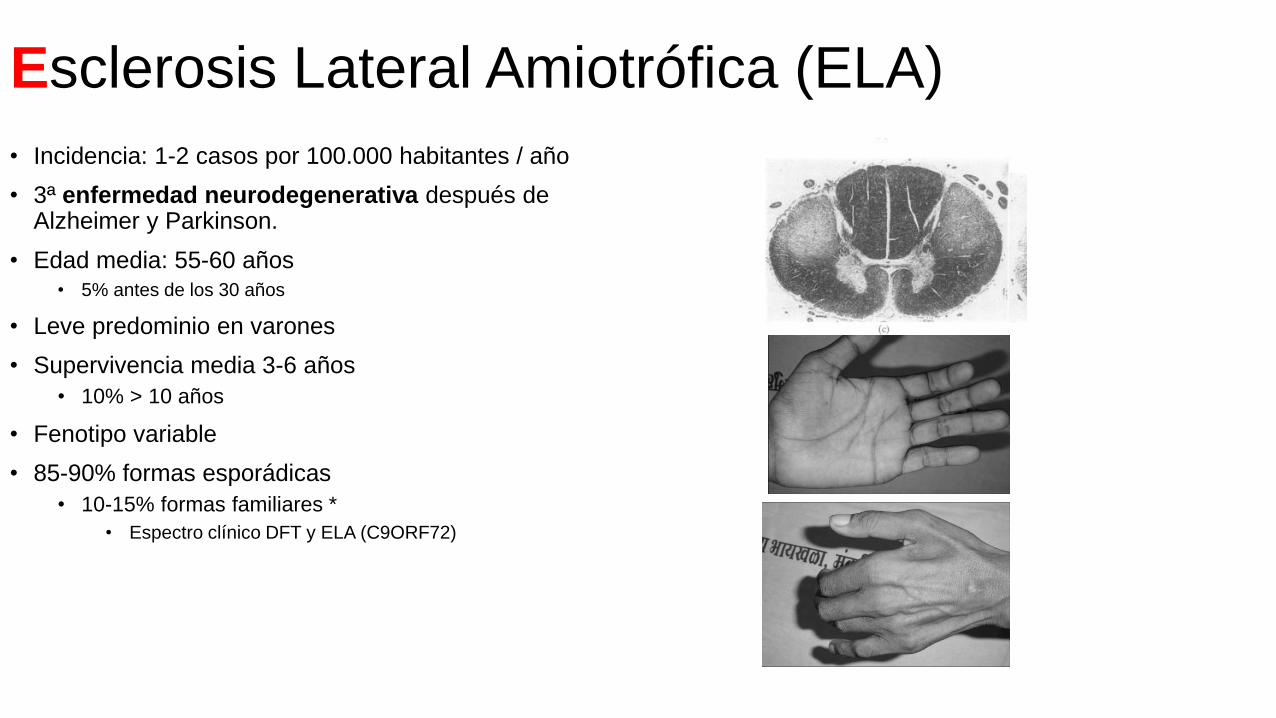
Esclerosis Lateral Amiotrófica (ELA)
• Incidencia: 1-2 casos por 100.000 habitantes / año
• 3ª enfermedad neurodegenerativa después de Alzheimer y Parkinson.
• Edad media: 55-60 años• 5% antes de los 30 años
• Leve predominio en varones
• Supervivencia media 3-6 años
• 10% > 10 años
• Fenotipo variable
• 85-90% formas esporádicas
• 10-15% formas familiares *
• Espectro clínico DFT y ELA (C9ORF72)
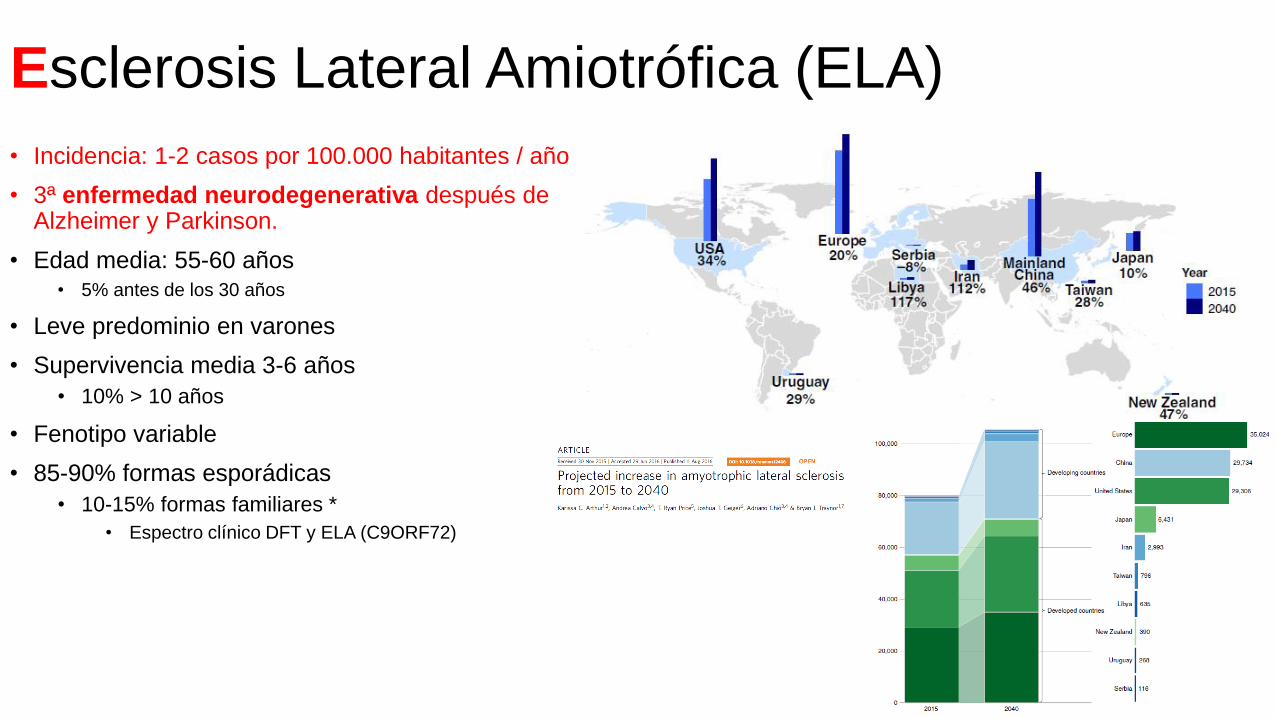
Esclerosis Lateral Amiotrófica (ELA)
• Incidencia: 1-2 casos por 100.000 habitantes / año
• 3ª enfermedad neurodegenerativa después de Alzheimer y Parkinson.
• Edad media: 55-60 años• 5% antes de los 30 años
• Leve predominio en varones
• Supervivencia media 3-6 años
• 10% > 10 años
• Fenotipo variable
• 85-90% formas esporádicas
• 10-15% formas familiares *
• Espectro clínico DFT y ELA (C9ORF72)
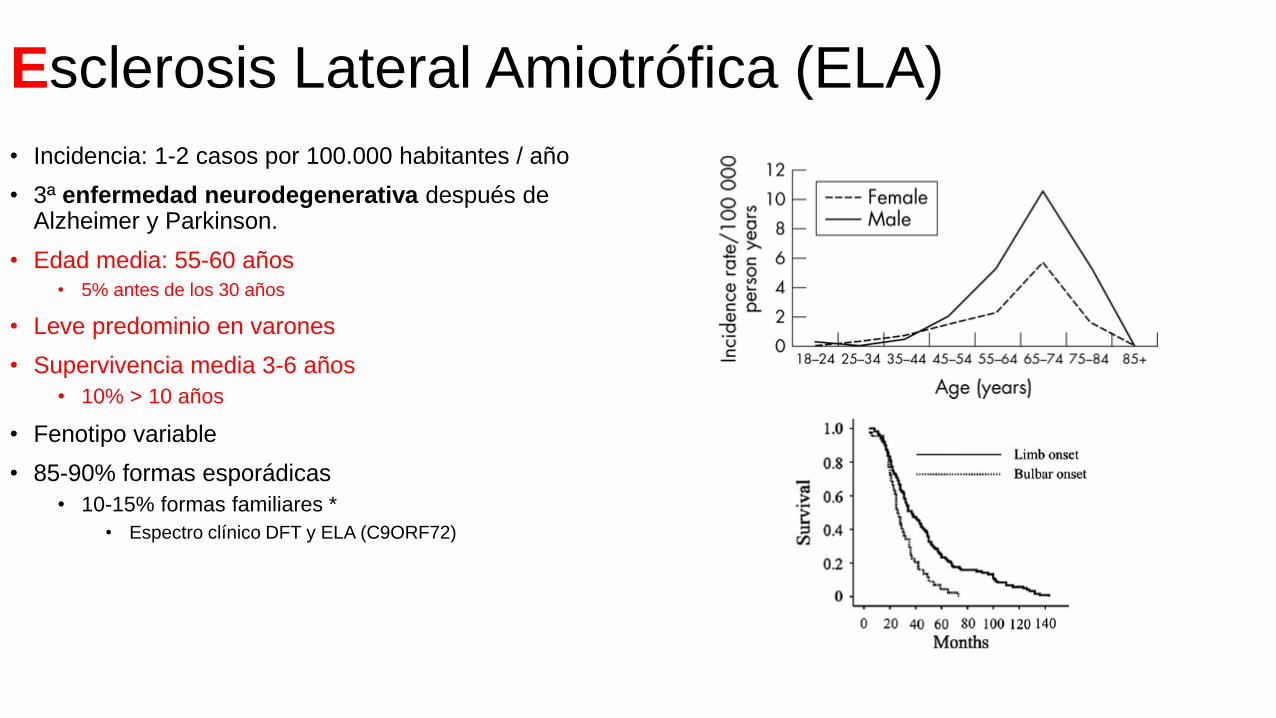
Esclerosis Lateral Amiotrófica (ELA)
• Incidencia: 1-2 casos por 100.000 habitantes / año
• 3ª enfermedad neurodegenerativa después de Alzheimer y Parkinson.
• Edad media: 55-60 años• 5% antes de los 30 años
• Leve predominio en varones
• Supervivencia media 3-6 años
• 10% > 10 años
• Fenotipo variable
• 85-90% formas esporádicas
• 10-15% formas familiares *
• Espectro clínico DFT y ELA (C9ORF72)
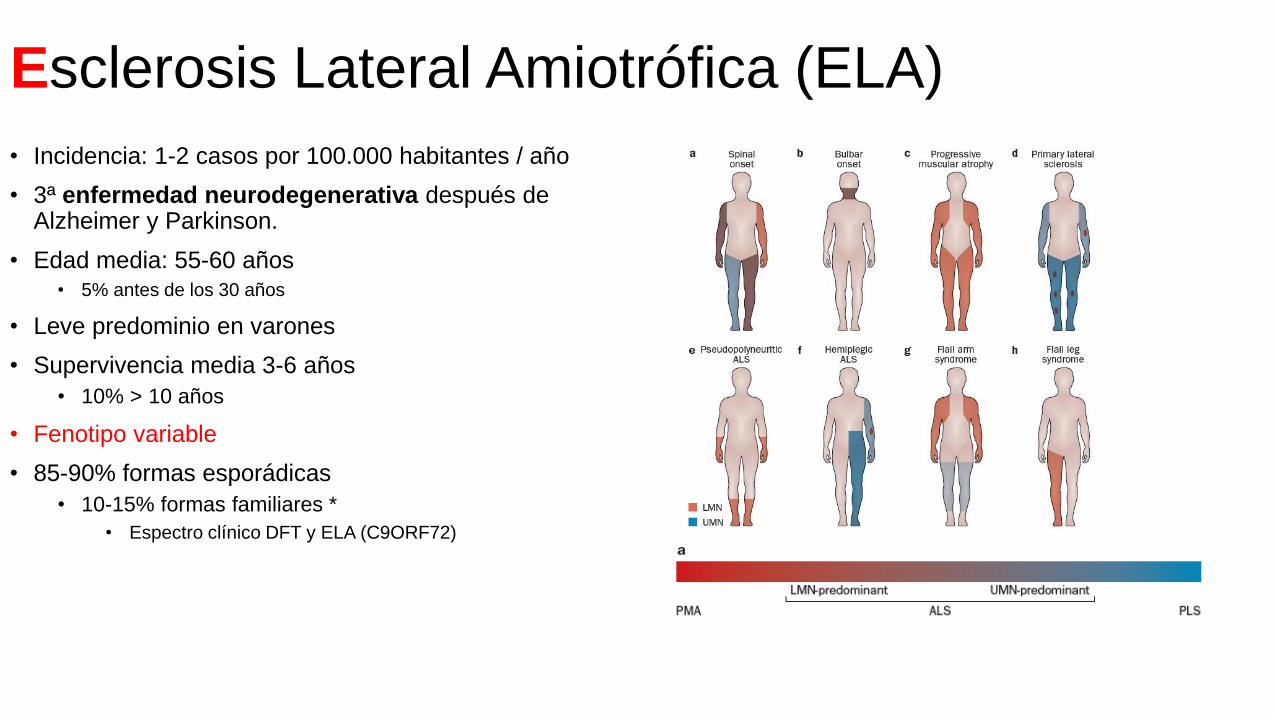
Esclerosis Lateral Amiotrófica (ELA)
• Incidencia: 1-2 casos por 100.000 habitantes / año
• 3ª enfermedad neurodegenerativa después de Alzheimer y Parkinson.
• Edad media: 55-60 años• 5% antes de los 30 años
• Leve predominio en varones
• Supervivencia media 3-6 años
• 10% > 10 años
• Fenotipo variable
• 85-90% formas esporádicas
• 10-15% formas familiares *
• Espectro clínico DFT y ELA (C9ORF72)
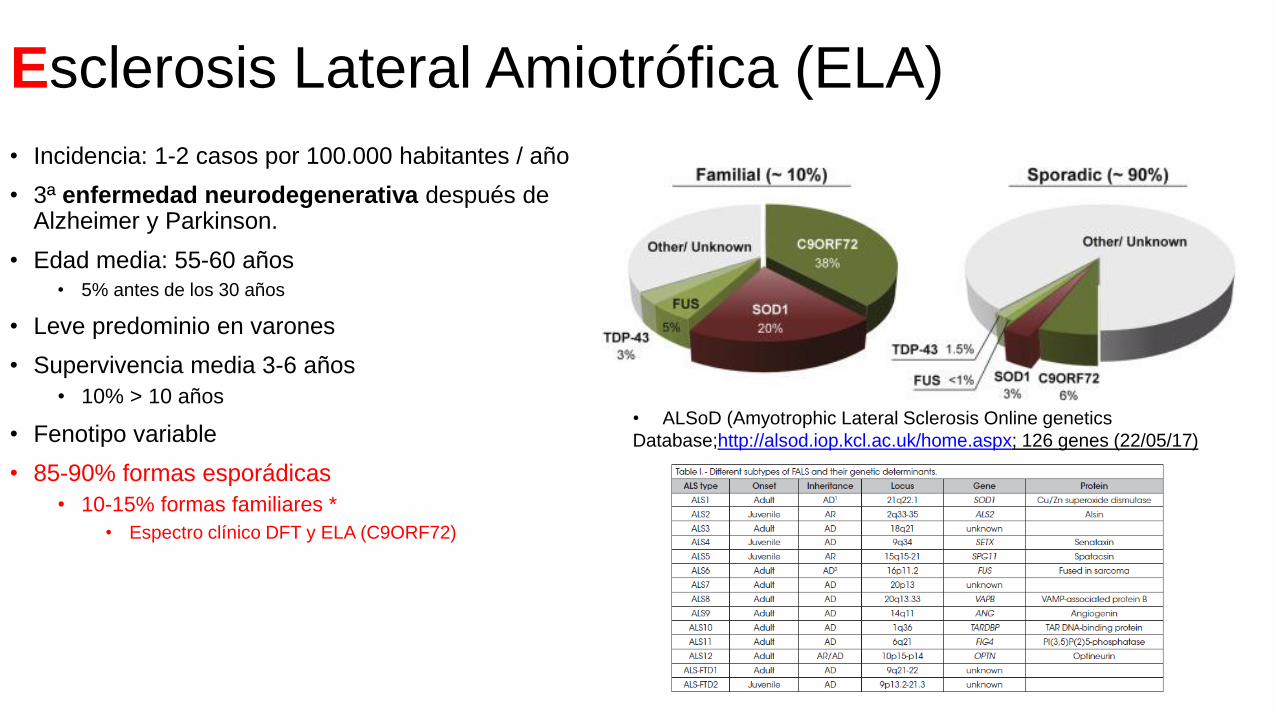
Esclerosis Lateral Amiotrófica (ELA)
• Incidencia: 1-2 casos por 100.000 habitantes / año
• 3ª enfermedad neurodegenerativa después de Alzheimer y Parkinson.
• Edad media: 55-60 años• 5% antes de los 30 años
• Leve predominio en varones
• Supervivencia media 3-6 años
• 10% > 10 años
• Fenotipo variable
• 85-90% formas esporádicas
• 10-15% formas familiares *
• Espectro clínico DFT y ELA (C9ORF72)
• ALSoD (Amyotrophic Lateral Sclerosis Online genetics
Database;http://alsod.iop.kcl.ac.uk/home.aspx; 126 genes (22/05/17)
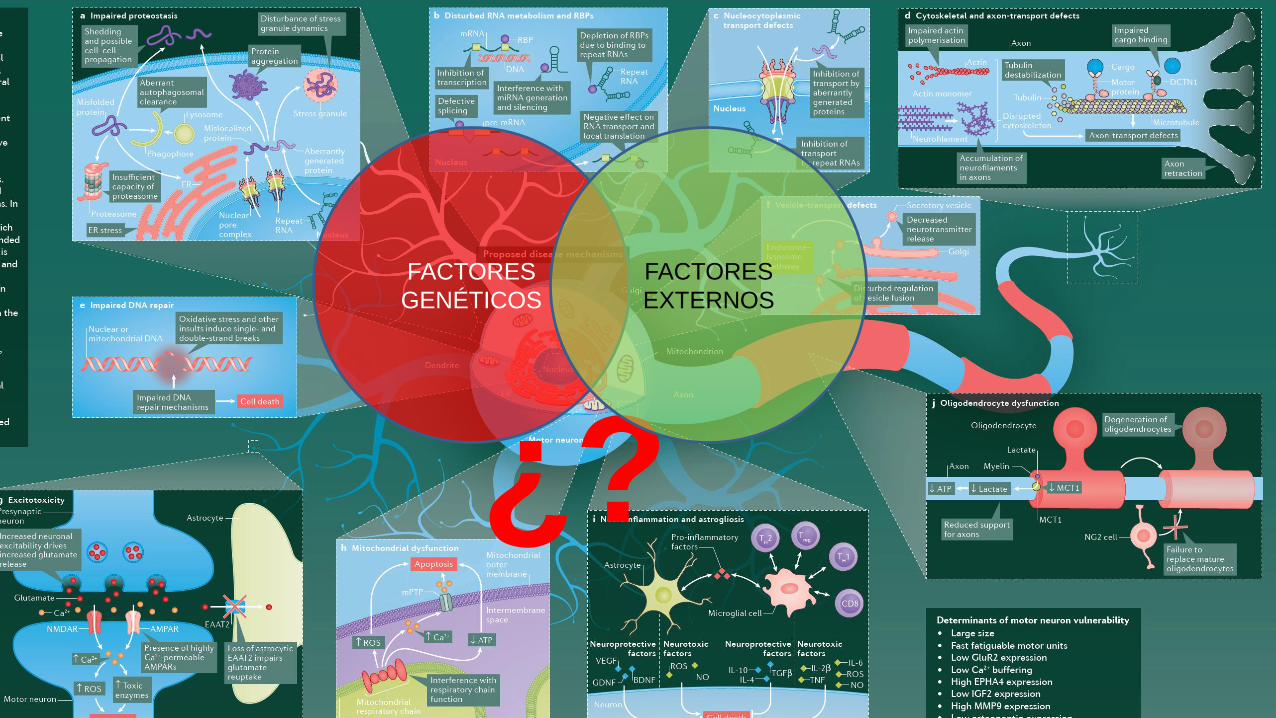
¿?
FACTORES
GENÉTICOS
FACTORES
EXTERNOS
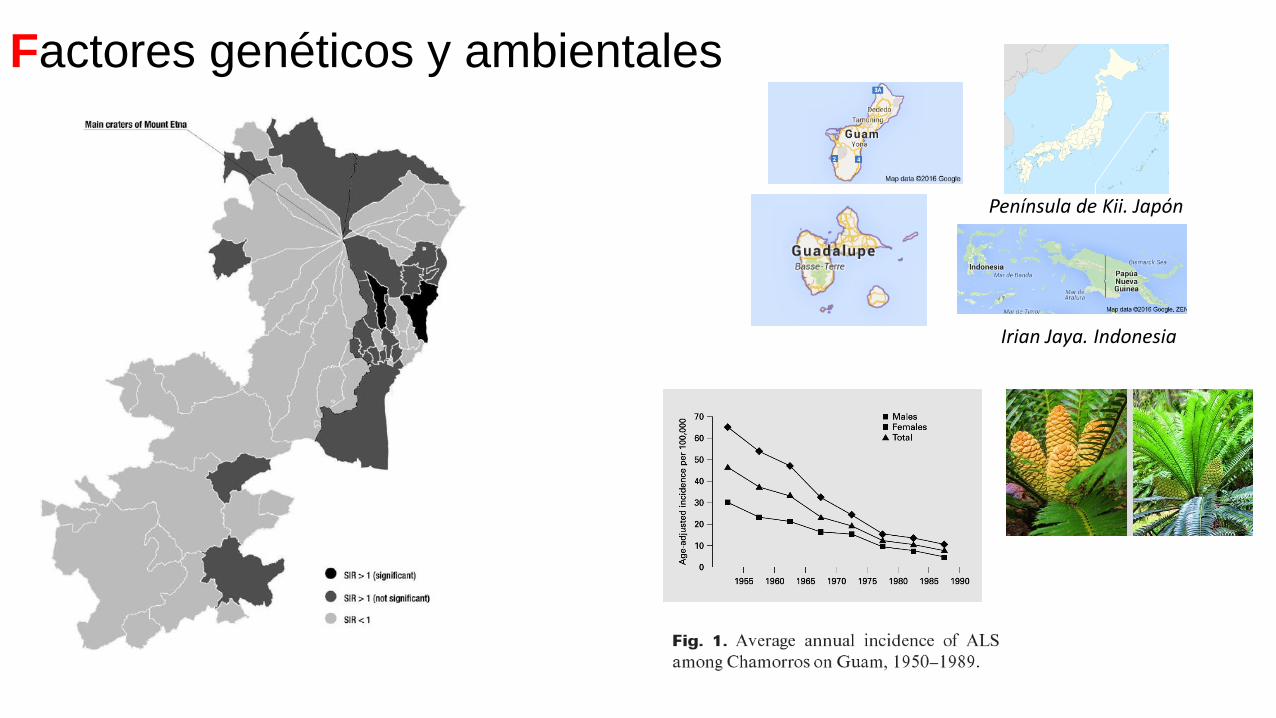
Factores genéticos y ambientales
Península de Kii. Japón
Irian Jaya. Indonesia

DEBILIDADATROFIA
FASCICULACIONES
PÉRDIDA DE DESTREZA
Signos y síntomas
PROPAGACIÓN DE LOS SÍNTOMAS
Extremidades
• Distal > Proximal
• Extremidades superiores
• Extremidades inferiores
• Musculatura cervical
• Disartria Anartria; incapacidad para la comunicación
verbal.
• Disfagia (tanto para sólidos como líquidos)
• Pérdida de peso
• Malnutrición
• Necesidad de nutrición mediante gastrostomía o sonda nasogástrica.
• Riesgo de aspiraciones
• Exceso de salivación
• ‘Síndrome pseudobulbar’, con labilidad emocional y bostezos
frecuentes.
Región Bulbar
• Disnea, primero a grandes esfuerzos, y posteriormente a esfuerzos más
moderados y leves.
• Raramente es la manifestación inicial (5%).
• A menudo no expresan disnea a esfuerzos, y cuando lo hacen ya existe
una marcada debilidad de la musculatura respiratoria.
• Importancia en la evaluación de la función respiratoria incluso en
pacientes aparentemente asintomáticos.
• Signos y síntomas que pueden indicar la presencia de hipoventilación:
nicturia, cefalea matutina, ortopnea, hipersomnia diurna o cambios de
humor e irritabilidad.
Musculatura respiratoria

• Síntomas sensitivos subjetivos hasta en un 50% de pacientes; 10% tendrán signos objetivos de afectación sensitiva.
• Afectación cognitiva, llegando hasta un 50% según algunos estudios; apatía, desinhibición, comportamiento esterotipados…
• En los últimos años hay una creciente evidencia que relaciona la ELA con la demencia fronto-temporal. Mutaciones en gen responsable también de demencia fronto-temporal y ELA asociada a demencia fronto-temporal.
• Problemas cognitivos habitualmente preceden a los motores.
Síntomas no motores
Diagnóstico
• No existe un marcador biológico que permita el diagnóstico.
• Criterios Diagnósticos
• Presencia de determinadas características clínicas
• Exclusión de otras enfermedades• Problemas medulares o radiculares
• Problemas sistema nervioso central (tumores)
• Otras enfermedades: Neuropatías inmunomediadas, miastenia gravis, miopatías

Diagnóstico
Criterios diagnósticos se van revisando para detectar cuanto antes y al
mayor número de pacientes criterios: El Escorial (1994), posteriormente se
revisaron (2000), y en 2008 (consenso de Awaji-shima).
Existen diferentes “grados de evidencia diagnóstica”:
ELA ‘posible’ Corresponde a las formas parciales. Puede mantenerse en el
tiempo como una forma parcial o evolucionar con el tiempo a una ELA
‘completa’
ELA ‘probable’ No ELA ‘completa’….pero casi. Con el tiempo evolucionará a
una ELA ‘completa’
ELA ‘definida’ ELA ‘completa’

•Curable ≠ Tratable
• Complicaciones respiratorias
• Problemas nutricionales
• Broncoaspiración
• Aparatos ortopédicos
• Sialorrea
• Depresión, labilidad emocional
• Dolor
• Estreñimiento
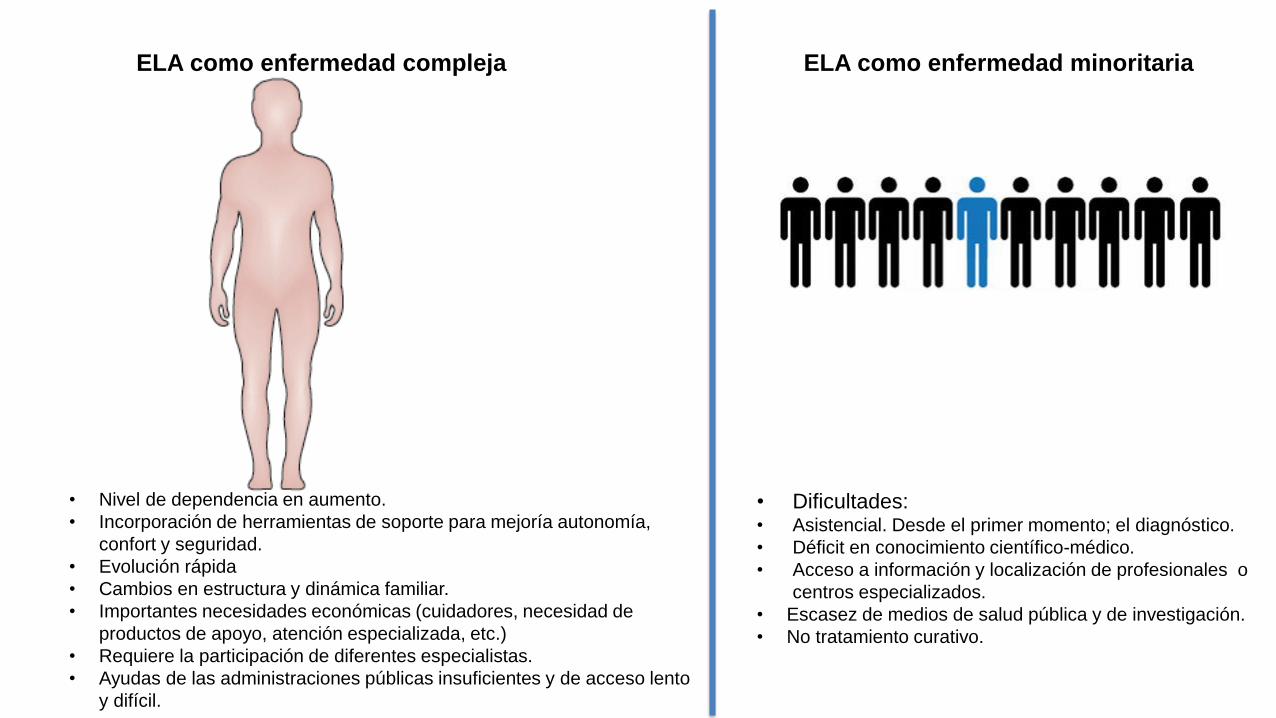
ELA como enfermedad compleja ELA como enfermedad minoritaria
• Dificultades:• Asistencial. Desde el primer momento; el diagnóstico.
• Déficit en conocimiento científico-médico.
• Acceso a información y localización de profesionales o
centros especializados.
• Escasez de medios de salud pública y de investigación.
• No tratamiento curativo.
• Nivel de dependencia en aumento.
• Incorporación de herramientas de soporte para mejoría autonomía,
confort y seguridad.
• Evolución rápida
• Cambios en estructura y dinámica familiar.
• Importantes necesidades económicas (cuidadores, necesidad de
productos de apoyo, atención especializada, etc.)
• Requiere la participación de diferentes especialistas.
• Ayudas de las administraciones públicas insuficientes y de acceso lento
y difícil.

• Evidencia acumulada de que la atención multidisciplinar aumenta la supervivencia
(Irlanda, Holanda, Italia, EEUU).
• Causas aumento de supervivencia no del todo conocidas: acceso a más ayudas,
tiempos para procedimientos más cortos, acuden pacientes más jóvenes, menos
hospitalizaciones.
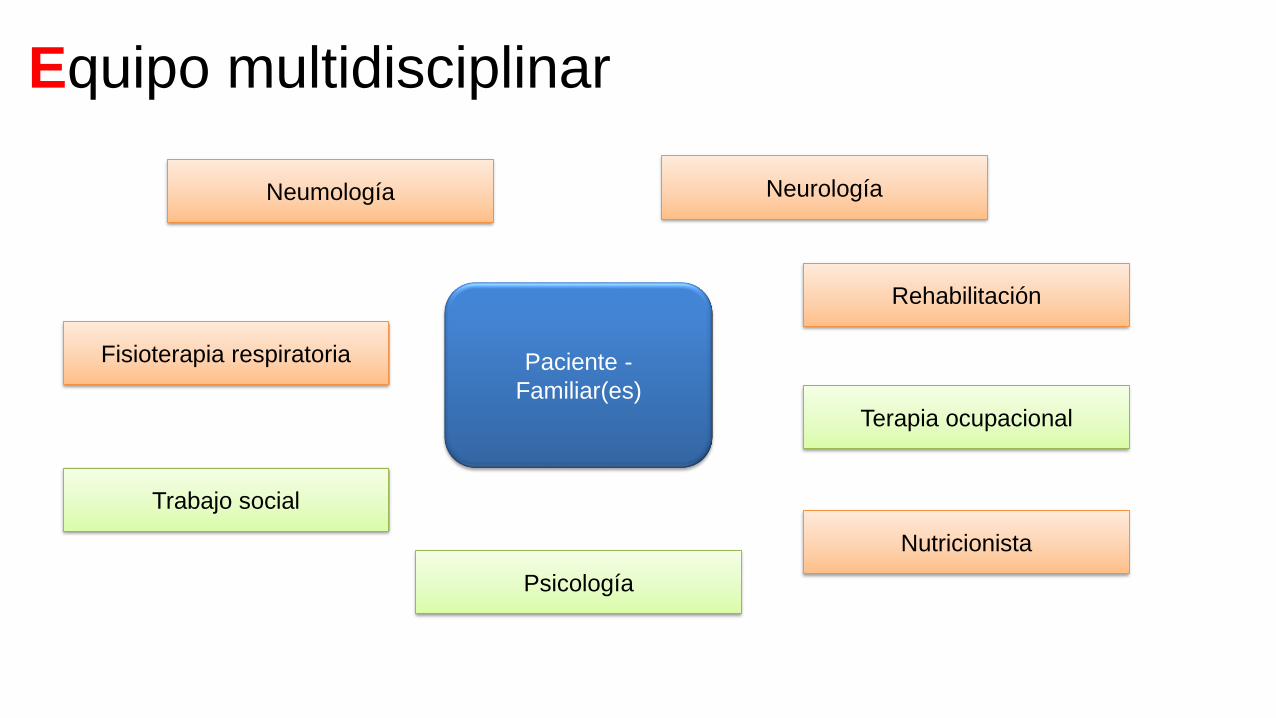
Equipo multidisciplinar
Paciente -
Familiar(es)
Neurología
Rehabilitación
Terapia ocupacional
Nutricionista
Psicología
Trabajo social
Fisioterapia respiratoria
Neumología

Recommended