Embed Size (px)
DESCRIPTION
a
Citation preview

of March 3, 2012This information is current as
http://www.jimmunol.org/content/184/7/3718doi:10.4049/jimmunol.0903613February 2010;
2010;184;3718-3724; Prepublished online 24J Immunol Salameh and Xiaojing MaXiaoyan Kang, Ha-Jeong Kim, Michelle Ramirez, Sarah 1 in Macrophages Engulfing Apoptotic CellsTranscription via Poly(ADP-Ribose) PolymerasePolymorphism Mediates Allele-Specific
1082 A > G−The Septic Shock-Associated IL-10
References
http://www.jimmunol.org/content/184/7/3718.full.html#related-urlsArticle cited in:
http://www.jimmunol.org/content/184/7/3718.full.html#ref-list-1, 17 of which can be accessed free at:cites 55 articlesThis article
Subscriptions http://www.jimmunol.org/subscriptions
is online atThe Journal of ImmunologyInformation about subscribing to
Permissions http://www.aai.org/ji/copyright.html
Submit copyright permission requests at
Email Alerts http://www.jimmunol.org/etoc/subscriptions.shtml/
Receive free email-alerts when new articles cite this article. Sign up at
Print ISSN: 0022-1767 Online ISSN: 1550-6606.Immunologists, Inc. All rights reserved.
by The American Association ofCopyright ©2010 9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
on March 3, 2012
ww
w.jim
munol.org
Dow
nloaded from

The Journal of Immunology
The Septic Shock-Associated IL-10 21082 A > GPolymorphism Mediates Allele-Specific Transcription viaPoly(ADP-Ribose) Polymerase 1 in Macrophages EngulfingApoptotic Cells
Xiaoyan Kang,* Ha-Jeong Kim,* Michelle Ramirez,† Sarah Salameh,‡ and
Xiaojing Ma*,x
The biallelic IL-10 single nucleotide polymorphism at 21082 of the promoter region linked to individual variation in cytokine
inducibility has been strongly implicated in several pathological conditions including the development of, and outcomes in, septic
shock during pneumococcal infection, acute respiratory distress syndrome, and cardiac dysfunction. However, the molecular basis
of the single nucleotide polymorphism-mediated variable IL-10 production levels has not been explored. In this study, we report
that the21082G > A alleles in the promoter region of the human IL-10 gene physically interact with a nuclear protein in an allele-
specific manner that results in different levels of IL-10 transcription. This protein has been identified as poly(ADP-ribose) poly-
merase 1 (PARP-1). We show that PARP-1 acts as a transcription repressor, and its DNA-binding activity is strongly regulated in
macrophages that engulf apoptotic cells but not stimulated with LPS. These findings unveil a novel role of PARP-1 in the
regulation of IL-10 production in an allele-dependent way, which determines individual susceptibility to sepsis-induced inflam-
matory pathology and the immunological sequelae in a physiological process in which clearance of infection-induced apoptotic
cells by professional phagocytes triggers the cytokine synthesis. The Journal of Immunology, 2010, 184: 3718–3724.
Interleukin-10 is an important anti-inflammatory cytokine thatmodulates proinflammatory cytokines, such as TNF-a, as wellas synthesis of NO, apoptosis of inflammatory cells, and sup-
pression of macrophage activation (1). IL-10 attenuates the proin-flammatory response in bacterial sepsis and reduces others (2).However, excess of IL-10 induces immunosuppression in sepsis (2)and increases mortality by impairing bacterial clearance in pneu-mococcal pneumonia (3). In humans, elevated circulating IL-10 hasbeen associated with septic shock (4), severity of injury (5–10) andmortality (11, 12). In acute respiratory distress syndrome (ARDS),the studies have been mixed. Lower levels of IL-10 were found inpatients withARDS comparedwith critically ill non-ARDS patients(13). High plasma IL-10 but low bronchoalveolar lavage concen-tration of IL-10 correlated with increased mortality in ARDS (14,15). Susceptibility for invasive pneumococcal disease has also been
associated with the mannose-binding lectin gene, but no geneticlinkage has been found for sepsis severity (16).The elimination of apoptotic cells and cell bodies by phagocytes
represents an evolutionarily conserved means to prevent exposure ofsurrounding tissue to potentially cytotoxic, immunogenic, or infl-ammatory cellular content (17, 18). Resolution of inflammation de-pends not only on the removal of apoptotic cells but also on activesuppression of inflammatory mediator production. Aberrations in ei-ther mechanism are associated with chronic inflammatory conditionsand autoimmune disorders (15, 19, 20). Uptake of apoptotic cells byphagocytes is thought to suppress autoimmune responses through therelease of anti-inflammatory cytokines IL-10, TGF-b, platelet acti-vating factor, and PGE2 and inhibition of proinflammatory cytokinesTNF-a, GM-CSF, IL-12, the b form of pro-IL-1, and IL-18 (21–23).A systemic infectious insult is often associated with subsequent
hyporesponsiveness to endotoxin and an increased risk of late noso-comialinfectioninsomepatients.Forexample,immediatelyfollowingcardiac surgery, many patients become relatively refractory to LPSstimulation.Onestudyfoundthat in thesepatients, stimulatedcytokineproduction in whole blood was lowest in cases with the highestpostoperative plasma IL-10 levels. Those patients in whom thewholeblood response to endotoxin was maintained over the first 48 h weremore likely to have an uncomplicated short stay (24).It has been reported that 50–75% of the variation in IL-10 pro-
duction is genetically controlled (25, 26). Single nucleotide poly-morphisms (SNPs) have been associated with different cytokineproduction (27). There are three major SNPs in the human IL-10promoter region: 21082G . A, 2819C . T, and 2592C . A, re-spectively. Individuals homozygous for theG allele (21082GG) havehigher circulating IL-10, higher expression of IL-10 mRNA, andgreater production of IL-10 after in vitro stimulation (27–29). It wasshown that theAallele of the21082 polymorphism is associatedwithlower IL-10 production and sepsis susceptibility in patients, whereas
*Department of Microbiology and Immunology, Weill Cornell Medical College,†Department of Pediatrics, Critical Care Medicine, Weill Cornell Medical Collegeand New York Presbyterian Hospital, and xProgram in Immunology and MicrobialPathogenesis, Weill Graduate School of Medical Sciences, Cornell University, NewYork, NY 10065; and ‡Weill Cornell Medical College in Qatar, Doha, Qatar
Received for publication November 12, 2009. Accepted for publication January 20,2010.
This work was supported byNational Institutes of Health Grant R01 AI045899 (to X.M).
Address correspondence and reprint requests to Dr. Xiaojing Ma, Weill MedicalCollege of Cornell University, 1300 York Avenue, New York, NY 10065. E-mailaddress: [email protected]
Abbreviations used in this paper: 3-AB, 3-aminobenzamide; AC, apoptotic cell;ARDS, acute respiratory distress syndrome; C, control IgG; ChIP, chromatin immu-noprecipitation; CLP, cecal ligation and puncture; CV, control vector; FP, free probe;Mf/AC, ratio of macrophage versus AC; NU-1025, 8-hydroxy-2-methylquinazolin-4-[3H]one; PARP-1, poly(ADP-ribose) polymerase 1; SNP, single nucleotide poly-morphism; WT, wild-type.
Copyright� 2010 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/10/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0903613
on March 3, 2012
ww
w.jim
munol.org
Dow
nloaded from

the G allele is associated with increased mortality in severe sepsis(30). Another study demonstrated that the IL-10 intermediate/highproducer genotype (21082 G-allele carrier) was associated witha lower risk of death among patients with acute renal failure whorequire dialysis (31). It was reported that individuals with geneticpredisposition for increased IL-10 inducibility, as determined by theIL-10 21082 polymorphism, have a higher risk of severe pneumo-coccal infection leading to septic shock (32). A nested case-controlepidemiology study in patients with ARDS and controls who wereadmitted to an intensive care unit with sepsis, trauma, aspiration,or massive transfusions revealed that the high IL-10–producing21082GG genotype is associated with variable odds for ARDS de-velopment depending on age, being associated with lower mortality,and organ failure (33). It was reported that in patients with acutepancreatitis, those that developed further into septic shock showeda significantly higher prevalence of the 21082G allele than thosewithout shock (34).ThepreponderanceofclinicaldataontheassociationoftheIL-102
1082 SNP with bacterial septic shock and the sequelae is contrastedwith an almost total lack of understanding of the molecular basis ofthe SNP-associated variability in IL-10 levels in different in-dividuals. We undertook the current study to address a key question:how does the 21082 SNP affect IL-10 gene expression at the mo-lecular level inmacrophages responding tomicrobial challenges andto apoptotic cells?
Materials and MethodsCells and reagents
Mousemonocytic cell lineRAW264.7 andhumanMonoMac-6 cell linewereobtained from American Type Culture Collection (Manassas, VA). Humanmonocytes were derived from blood buffy coats purchased commerciallyfrom the NewYork Blood Center (NewYork, NY). Cells were maintained inRPMI 1640 with 10% FBS, 100 U/ml penicillin, 100 mg/ml streptomycin,and 200 mM L-glutamine. Abs against poly(ADP-ribose) polymerase 1(PARP-1) were purchased from Santa Cruz Biotechnologies (Santa Cruz,CA). LPSwas fromSigma-Aldrich (St. Louis,MO; catalog numberL-3129).3-Aminobenzamide (3-AB), 8-hydroxy-2-methylquinazolin-4-[3H]one(NU-1025), and fisetin were from Sigma-Aldrich. These compounds weredissolved inDMSOat 50mM, and aliquotswere stored in the dark at220˚C.
Reporter plasmids
ThehumanIL-10promoter-luciferaseconstruct(pIL-10[21105/+36]/pGL2basicvector) was cloned by PCR amplification of human genomic DNA from humanmonocytes. PCR product was cut using BamHI plus EcoRVand then ligated tothe pGL2-basic vector (cut by SmaI plus BaglII). Initially, SNPs obtained were21082 A, 2819 T, and 2592 T. To obtain the SNPs of interest, site-directedmutagenesis utilizing the Quikchange XL Site-Directed Mutagenesis Kit (Stra-tagene, La Jolla, CA) was performed. First, we mutated the2592 T to an A andhad21082 A,2819 T, and2592 A. Also, we made21082 G/2819 T/2592 Tfrom the initial construct (ATT).Subsequently,wemade21082G/2819C/2592Tand21082G/2819C/2592C.Finally,wemade21082A/2819C/2592Cbychanging the21082 G back to A.
Transient transfection and measurement of luciferase activity
Transfection of RAW264.7 cells with plasmids containing full-length hIL-10promoter was performed using electroporation followed by luciferase assay.Cells were collected, washed once with RPMI 1640 medium, and resus-pended in the same medium at a concentration of 103 106 cells/condition. Atotal of 700 btl cell suspension and 300 Ixg DNAwere placed in a 0.45-cmelectroporation cuvette (Gene Pulser 165-2088; Bio-Rad, Hercules, CA), andelectroporation was carried out at 975 mFD and 300 V (0.4 capacitance).Transfected cells were collected and resuspended to 53 106/ml in RPMI 1640plus 10% FBS, and chloroquine was added to a final concentration of 10 mM.Cells were placed inwells (2ml/well) of a 24-well plate and incubated for 16 hat 37˚C in a 5% CO2 atmosphere. Next, cells were treated with 1.2% DMSOfor ∼7 h; appropriate stimuli were added for 24 h. Poststimulation, cellswere harvested and lysed with 13 lysis buffer and vigorous shaking. Lysateswere used for the luciferase assay. All statistical analyses were performedwith two-tailed Student t test. Data were considered significant if p, 0.05.
Induction of apoptosis
Jurkat T cells were the source of apoptotic cells. Staurosporine (0.5 mg/ml)(Cayman Chemical, Ann Arbor, MI) was added at (0.5 ng/ml) to T cellsresuspended at 4 3 106 cells/ml with complete RPMI 1640 (Life Technol-ogies/Invitrogen, Carlsbad, CA). Postincubation for 6 h at 37˚C in thepresence of 5% CO2, the cells were harvested and washed three times withincomplete RPMI 1640. At this time, ∼65% of the population was AnnexinV-positive (early apoptotic) and propidium iodide-negative as determined byFACS staining. Cell viability by trypan blue staining was .90%.
Nuclear extract preparation and EMSA
Nuclear extracts and EMSAwere performed as described by Schreiber et al(11). The probe sequences were as follows: TTCTTTGGGAG/AGGGGA-AGTA (the SNP is in boldface).
DNA pulldown assay and PAGE analysis
Complementary biotinylated oligonucleotides encompassing the 21082A/G-binding site, TTCTTTGGGAG/AGGGGAAGTA (the critical SNPA or G is inboldface), were synthesized and annealed to form dsDNA. Biotinylated dsDNA(2mg)was conjugated to 100ml streptavidin-boundmagnetic beads (Dynabeads,M280; Dynal, Invitrogen) in binding/washing buffer (10 mM Tris–HCl [pH 8],1mMEDTA,and0.1MNaCl) for 30min at room temperature.ConjugatedDNAwas collectedwith amagnetic particle concentrator.DNA-conjugated beadswerethenblockedwith 0.5%BSA inTGEDNbuffer (120mMTris–HCl [pH8], 1mMEDTA, 0.1MNaCl, 1 mMDTT, 0.1%Triton X-100, and 10% glycerol) at roomtemperature for 1 h. Beadswerewashed once in TGEDNbuffer and resuspendedin 50 ml TGEDN. Ten-microliter beads conjugated to 2 mg DNAwere equili-brated with TGEDN buffer and incubated with 500 mg RAW264.7 cell nuclearextracts and 20 mg herring sperm DNA (Sigma-Aldrich) at 41˚C for 2 h. Beadswere washed in TGEDN buffer, and boundmaterials were eluted in 20 ml of thesame buffer supplemented with 0.5% SDS and 1 M NaCl. Eluted proteins wereseparated by 10% or 12% SDS-polyacrylamide gel. The gel was visualized byCoomassie staining.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assaywas performed by following theprotocol in the ChIPAssay kit (Upstate Biotechnology, Lake Placid, NY). Thepresenceof a selectedDNAsequenceof the human IL-10 genewas assessedbyPCR.The primers usedwere: sense,21217/21198, 59-CAACTGGCTCCCC-TTACCTT-39; and antisense, 2998/2979, 59-ACCTCCTATCCAGCCTC-CAT-39, yielding a 239-bp product. As a negative control, a separate region ofthe human IL-10 promoter was also included in the ChIP experiment. It islocated between23158 and22947 upstream of the21217/2979 region. Thepair of PCR primers used in this control were: sense, 59-AGTGAGAAGG-CAGGCACCTA-39; and antisense, 59-ATCCCCCACTGGAAAAATTC-39,yielding a 212-bp product. The PCR cycles were as follows: 94˚C for 4 min,1 cycle; 94˚C for 30 s, 54˚C for 30 s, and 72˚C for 30 s, 32 cycles; and 72˚C for7 min, 1 cycle.
ResultsDifferential transcriptional activities of21082A and21082G–IL-10 promoter haplotypes
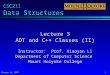
To understand the regulation of the IL-10 gene transcription viathe 21082A/G SNP in macrophages, we used a well-establishedtransient transfection system in the murine macrophage-like cell lineRAW264.7 and a human IL-10 promoter-luciferase reporter constructcontaining the region between21105 and +30 upstream of the IL-10transcription initiation site. Two versions of this construct were en-gineered to reflect the21082 SNPof interest:21082A and21082G,respectively. These constructs were completely identical otherwise.Following transient transfection of the two reporter constructs,RAW264.7 cells were stimulated with apoptotic cells in differentamounts or with LPS; luciferase activity was measured afterward. Asshown in Fig. 1, the21082A and21082G haplotype IL-10 promoterconstructs exhibited different responses to apoptotic cells (ACs) andto LPS in that ∼61–65%more activities were seen with the21082Gpromoter over those of the21082A promoter in response to ACs (at1:1 ratio of phagocytes to ACs) and to LPS, respectively. As theamount ofACs increased to 1:2and1:3 ratios, the responsesof the twopromoters became more equal. It is noteworthy that the basal
The Journal of Immunology 3719
on March 3, 2012
ww
w.jim
munol.org
Dow
nloaded from
transcriptional activities of the two haplotype promoters also differedby∼63%, suggesting that the inherent differenceof the twopromotersis intrinsic independently of stimulation, and cellular stimulationmerely amplifies the inherent differences.
Differential DNA binding to 21082 SNP by a novel NF
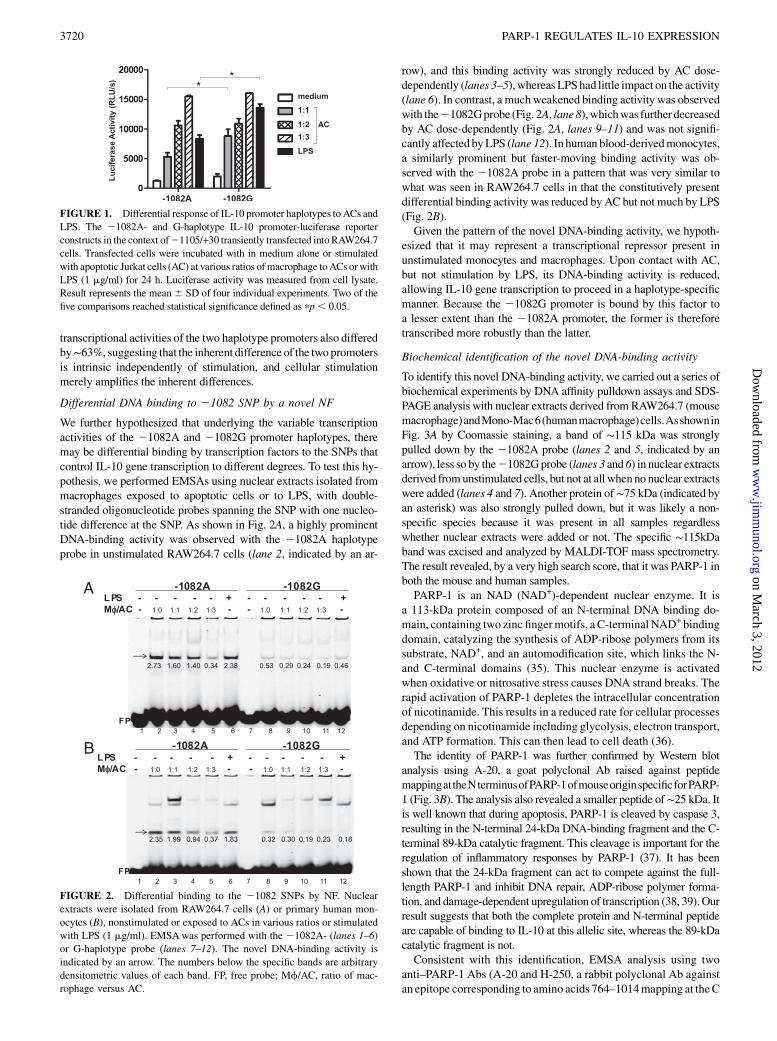
We further hypothesized that underlying the variable transcriptionactivities of the 21082A and 21082G promoter haplotypes, theremay be differential binding by transcription factors to the SNPs thatcontrol IL-10 gene transcription to different degrees. To test this hy-pothesis, we performed EMSAs using nuclear extracts isolated frommacrophages exposed to apoptotic cells or to LPS, with double-stranded oligonucleotide probes spanning the SNP with one nucleo-tide difference at the SNP. As shown in Fig. 2A, a highly prominentDNA-binding activity was observed with the 21082A haplotypeprobe in unstimulated RAW264.7 cells (lane 2, indicated by an ar-
row), and this binding activity was strongly reduced by AC dose-dependently (lanes 3–5),whereasLPShad little impact on the activity(lane 6). In contrast, a muchweakened binding activity was observedwith the21082Gprobe (Fig. 2A, lane8),whichwas further decreasedby AC dose-dependently (Fig. 2A, lanes 9–11) and was not signifi-cantly affected byLPS (lane 12). In human blood-derivedmonocytes,a similarly prominent but faster-moving binding activity was ob-served with the 21082A probe in a pattern that was very similar towhat was seen in RAW264.7 cells in that the constitutively presentdifferential binding activity was reduced byAC but not much by LPS(Fig. 2B).Given the pattern of the novel DNA-binding activity, we hypoth-
esized that it may represent a transcriptional repressor present inunstimulated monocytes and macrophages. Upon contact with AC,but not stimulation by LPS, its DNA-binding activity is reduced,allowing IL-10 gene transcription to proceed in a haplotype-specificmanner. Because the 21082G promoter is bound by this factor toa lesser extent than the 21082A promoter, the former is thereforetranscribed more robustly than the latter.
Biochemical identification of the novel DNA-binding activity
To identify this novel DNA-binding activity, we carried out a series ofbiochemical experiments by DNA affinity pulldown assays and SDS-PAGE analysis with nuclear extracts derived from RAW264.7 (mousemacrophage)andMono-Mac6(humanmacrophage)cells.AsshowninFig. 3A by Coomassie staining, a band of ∼115 kDa was stronglypulled down by the 21082A probe (lanes 2 and 5, indicated by anarrow), less so by the21082Gprobe (lanes 3 and 6) in nuclear extractsderived fromunstimulated cells, but not at all whenno nuclear extractswere added (lanes 4 and 7). Another protein of∼75 kDa (indicated byan asterisk) was also strongly pulled down, but it was likely a non-specific species because it was present in all samples regardlesswhether nuclear extracts were added or not. The specific ∼115kDaband was excised and analyzed by MALDI-TOF mass spectrometry.The result revealed, by a very high search score, that it was PARP-1 inboth the mouse and human samples.PARP-1 is an NAD (NAD+)-dependent nuclear enzyme. It is
a 113-kDa protein composed of an N-terminal DNA binding do-main, containing two zinc fingermotifs, a C-terminal NAD+ bindingdomain, catalyzing the synthesis of ADP-ribose polymers from itssubstrate, NAD+, and an automodification site, which links the N-and C-terminal domains (35). This nuclear enzyme is activatedwhen oxidative or nitrosative stress causes DNA strand breaks. Therapid activation of PARP-1 depletes the intracellular concentrationof nicotinamide. This results in a reduced rate for cellular processesdepending on nicotinamide including glycolysis, electron transport,and ATP formation. This can then lead to cell death (36).The identity of PARP-1 was further confirmed by Western blot
analysis using A-20, a goat polyclonal Ab raised against peptidemappingattheNterminusofPARP-1ofmouseoriginspecificforPARP-1 (Fig. 3B). The analysis also revealed a smaller peptide of∼25 kDa. Itis well known that during apoptosis, PARP-1 is cleaved by caspase 3,resulting in the N-terminal 24-kDa DNA-binding fragment and the C-terminal 89-kDa catalytic fragment. This cleavage is important for theregulation of inflammatory responses by PARP-1 (37). It has beenshown that the 24-kDa fragment can act to compete against the full-length PARP-1 and inhibit DNA repair, ADP-ribose polymer forma-tion, and damage-dependent upregulation of transcription (38, 39). Ourresult suggests that both the complete protein and N-terminal peptideare capable of binding to IL-10 at this allelic site, whereas the 89-kDacatalytic fragment is not.Consistent with this identification, EMSA analysis using two
anti–PARP-1 Abs (A-20 and H-250, a rabbit polyclonal Ab againstan epitope corresponding to amino acids 764–1014mapping at theC
FIGURE 1. Differential response of IL-10 promoter haplotypes to ACs and
LPS. The 21082A- and G-haplotype IL-10 promoter-luciferase reporter
constructs in the context of21105/+30 transiently transfected intoRAW264.7
cells. Transfected cells were incubated with in medium alone or stimulated
with apoptotic Jurkat cells (AC) at various ratios ofmacrophage toACs orwith
LPS (1 mg/ml) for 24 h. Luciferase activity was measured from cell lysate.
Result represents the mean6 SD of four individual experiments. Two of the
five comparisons reached statistical significance defined as pp, 0.05.
FIGURE 2. Differential binding to the 21082 SNPs by NF. Nuclear
extracts were isolated from RAW264.7 cells (A) or primary human mon-
ocytes (B), nonstimulated or exposed to ACs in various ratios or stimulated
with LPS (1 mg/ml). EMSAwas performed with the 21082A- (lanes 1–6)
or G-haplotype probe (lanes 7–12). The novel DNA-binding activity is
indicated by an arrow. The numbers below the specific bands are arbitrary
densitometric values of each band. FP, free probe; Mf/AC, ratio of mac-
rophage versus AC.
3720 PARP-1 REGULATES IL-10 EXPRESSION
on March 3, 2012
ww
w.jim
munol.org
Dow
nloaded from
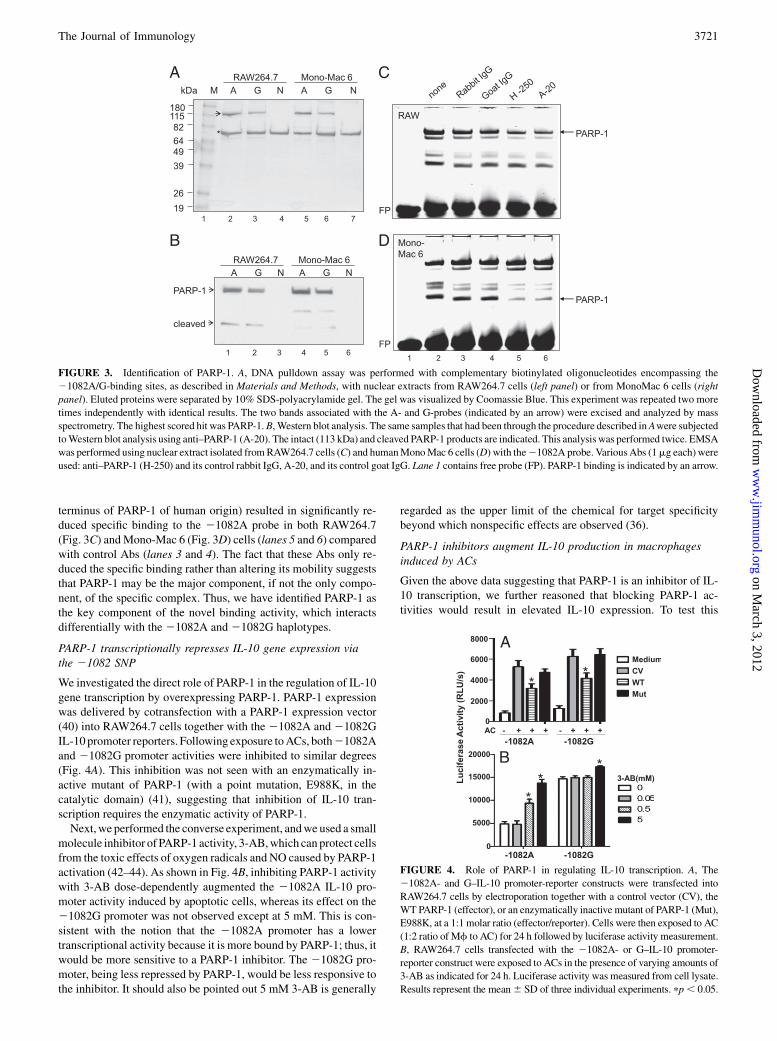
terminus of PARP-1 of human origin) resulted in significantly re-duced specific binding to the 21082A probe in both RAW264.7(Fig. 3C) andMono-Mac 6 (Fig. 3D) cells (lanes 5 and 6) comparedwith control Abs (lanes 3 and 4). The fact that these Abs only re-duced the specific binding rather than altering its mobility suggeststhat PARP-1 may be the major component, if not the only compo-nent, of the specific complex. Thus, we have identified PARP-1 asthe key component of the novel binding activity, which interactsdifferentially with the 21082A and21082G haplotypes.
PARP-1 transcriptionally represses IL-10 gene expression viathe 21082 SNP
We investigated the direct role of PARP-1 in the regulation of IL-10gene transcription by overexpressing PARP-1. PARP-1 expressionwas delivered by cotransfection with a PARP-1 expression vector(40) into RAW264.7 cells together with the 21082A and 21082GIL-10 promoter reporters. Following exposure toACs, both21082Aand 21082G promoter activities were inhibited to similar degrees(Fig. 4A). This inhibition was not seen with an enzymatically in-active mutant of PARP-1 (with a point mutation, E988K, in thecatalytic domain) (41), suggesting that inhibition of IL-10 tran-scription requires the enzymatic activity of PARP-1.Next,we performed the converse experiment, andwe used a small
molecule inhibitor of PARP-1 activity, 3-AB,which can protect cellsfrom the toxic effects of oxygen radicals and NO caused by PARP-1activation (42–44). As shown in Fig. 4B, inhibiting PARP-1 activitywith 3-AB dose-dependently augmented the 21082A IL-10 pro-moter activity induced by apoptotic cells, whereas its effect on the21082G promoter was not observed except at 5 mM. This is con-sistent with the notion that the 21082A promoter has a lowertranscriptional activity because it is more bound by PARP-1; thus, itwould be more sensitive to a PARP-1 inhibitor. The 21082G pro-moter, being less repressed by PARP-1, would be less responsive tothe inhibitor. It should also be pointed out 5 mM 3-AB is generally
regarded as the upper limit of the chemical for target specificitybeyond which nonspecific effects are observed (36).
PARP-1 inhibitors augment IL-10 production in macrophagesinduced by ACs
Given the above data suggesting that PARP-1 is an inhibitor of IL-10 transcription, we further reasoned that blocking PARP-1 ac-tivities would result in elevated IL-10 expression. To test this
FIGURE 3. Identification of PARP-1. A, DNA pulldown assay was performed with complementary biotinylated oligonucleotides encompassing the
21082A/G-binding sites, as described in Materials and Methods, with nuclear extracts from RAW264.7 cells (left panel) or from MonoMac 6 cells (right
panel). Eluted proteins were separated by 10% SDS-polyacrylamide gel. The gel was visualized by Coomassie Blue. This experiment was repeated two more
times independently with identical results. The two bands associated with the A- and G-probes (indicated by an arrow) were excised and analyzed by mass
spectrometry. The highest scored hit was PARP-1.B, Western blot analysis. The same samples that had been through the procedure described inAwere subjected
toWestern blot analysis using anti–PARP-1 (A-20). The intact (113 kDa) and cleaved PARP-1 products are indicated. This analysis was performed twice. EMSA
was performed using nuclear extract isolated fromRAW264.7 cells (C) and humanMonoMac 6 cells (D) with the21082A probe. VariousAbs (1mg each) were
used: anti–PARP-1 (H-250) and its control rabbit IgG, A-20, and its control goat IgG. Lane 1 contains free probe (FP). PARP-1 binding is indicated by an arrow.
FIGURE 4. Role of PARP-1 in regulating IL-10 transcription. A, The
21082A- and G–IL-10 promoter-reporter constructs were transfected into
RAW264.7 cells by electroporation together with a control vector (CV), the
WT PARP-1 (effector), or an enzymatically inactive mutant of PARP-1 (Mut),
E988K, at a 1:1 molar ratio (effector/reporter). Cells were then exposed to AC
(1:2 ratio of Mf to AC) for 24 h followed by luciferase activity measurement.
B, RAW264.7 cells transfected with the 21082A- or G–IL-10 promoter-
reporter construct were exposed to ACs in the presence of varying amounts of
3-AB as indicated for 24 h. Luciferase activity was measured from cell lysate.
Results represent the mean6 SD of three individual experiments. pp, 0.05.
The Journal of Immunology 3721
on March 3, 2012
ww
w.jim
munol.org
Dow
nloaded from
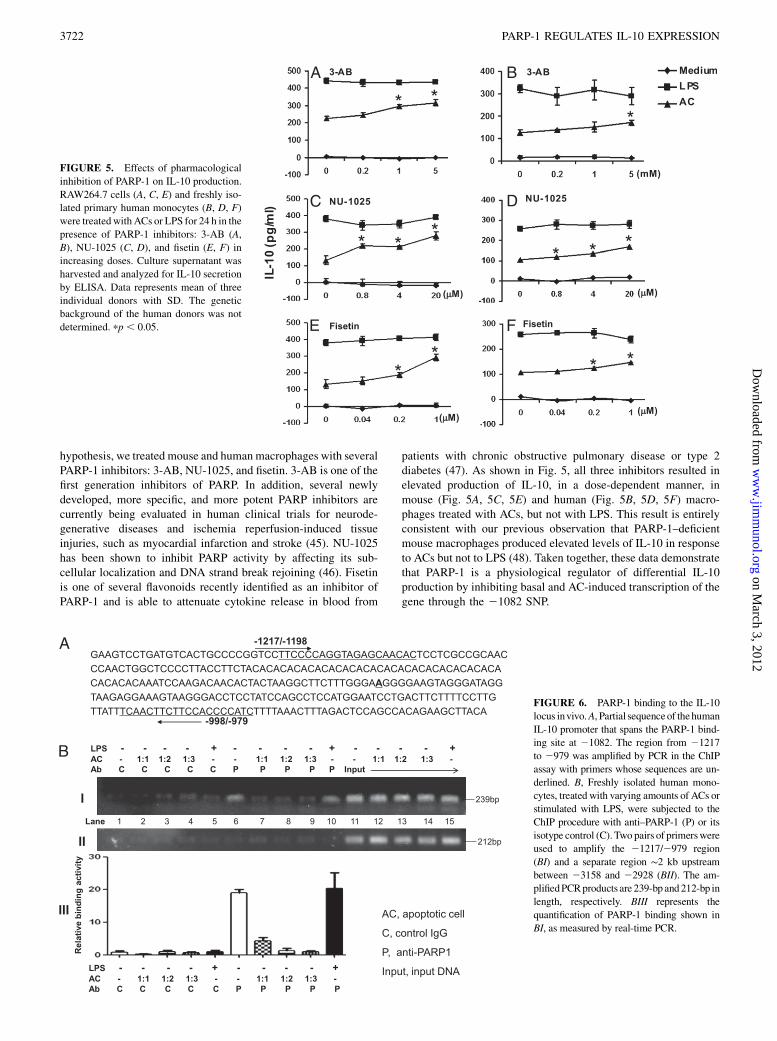
hypothesis, we treated mouse and humanmacrophages with severalPARP-1 inhibitors: 3-AB, NU-1025, and fisetin. 3-AB is one of thefirst generation inhibitors of PARP. In addition, several newlydeveloped, more specific, and more potent PARP inhibitors arecurrently being evaluated in human clinical trials for neurode-generative diseases and ischemia reperfusion-induced tissueinjuries, such as myocardial infarction and stroke (45). NU-1025has been shown to inhibit PARP activity by affecting its sub-cellular localization and DNA strand break rejoining (46). Fisetinis one of several flavonoids recently identified as an inhibitor ofPARP-1 and is able to attenuate cytokine release in blood from
patients with chronic obstructive pulmonary disease or type 2diabetes (47). As shown in Fig. 5, all three inhibitors resulted inelevated production of IL-10, in a dose-dependent manner, inmouse (Fig. 5A, 5C, 5E) and human (Fig. 5B, 5D, 5F) macro-phages treated with ACs, but not with LPS. This result is entirelyconsistent with our previous observation that PARP-1–deficientmouse macrophages produced elevated levels of IL-10 in responseto ACs but not to LPS (48). Taken together, these data demonstratethat PARP-1 is a physiological regulator of differential IL-10production by inhibiting basal and AC-induced transcription of thegene through the 21082 SNP.
FIGURE 5. Effects of pharmacological
inhibition of PARP-1 on IL-10 production.
RAW264.7 cells (A, C, E) and freshly iso-
lated primary human monocytes (B, D, F)
were treatedwithACs or LPS for 24 h in the
presence of PARP-1 inhibitors: 3-AB (A,
B), NU-1025 (C, D), and fisetin (E, F) in
increasing doses. Culture supernatant was
harvested and analyzed for IL-10 secretion
by ELISA. Data represents mean of three
individual donors with SD. The genetic
background of the human donors was not
determined. pp , 0.05.
FIGURE 6. PARP-1 binding to the IL-10
locus invivo.A, Partial sequenceof thehuman
IL-10 promoter that spans the PARP-1 bind-
ing site at 21082. The region from 21217
to 2979 was amplified by PCR in the ChIP
assay with primers whose sequences are un-
derlined. B, Freshly isolated human mono-
cytes, treated with varying amounts of ACs or
stimulated with LPS, were subjected to the
ChIP procedure with anti–PARP-1 (P) or its
isotype control (C). Two pairs of primerswere
used to amplify the 21217/2979 region
(BI) and a separate region ∼2 kb upstream
between 23158 and 22928 (BII). The am-
plifiedPCRproducts are239-bp and212-bp in
length, respectively. BIII represents the
quantification of PARP-1 binding shown in
BI, as measured by real-time PCR.
3722 PARP-1 REGULATES IL-10 EXPRESSION
on March 3, 2012
ww
w.jim
munol.org
Dow
nloaded from

Endogenous PARP-1 binding to the IL-10 locus
To demonstrate if PARP-1 could bind the Il10 gene in vivo weperformed ChIP assays in human PBMC-derived monocytes. Fol-lowing the immunoprecipitation step, specific primers were used toamplify a small region (from21217 to2979) covering the21082site as outlined in Fig. 6A. As shown in Fig. 6B, in samples treatedwith the control IgG (C), no binding activity was detected (Fig. 6BI,lanes 1–5), whereas in anti–PARP-1–treated samples (P), a specificand constitutive binding activity was observed (lane 6), which wasreduced in cells exposed to ACs (lanes 7–9). LPS treatment did notalter the constitutive binding activity (lane 10), and an unrelatedregion ∼2 kb upstream of the21082 site (49) showed little bindingunder the same experimental conditions (Fig. 6BII). The quantifi-cation of the relative binding activities measured by real-time PCRis shown in Fig. 6BIII. The data show that PARP-1 indeed bindsselectively and specifically to IL-10 invivo around the21082 site ina constitutive manner in monocytes and that exposure to ACs, butnot to LPS, reduces this binding.
DiscussionThis study has identified PARP-1 as a critical NF that determines dif-ferential IL-10 gene transcription in a21082G. A allele-dependentmanner in monocytes/macrophages that encounter/ingest ACs. Spe-cifically, PARP-1 appears to act as a transcriptional repressor witha direct, preferential binding for theA-allele than theG-allele, resultingin lesser expression of the former. This mechanism occurs both con-stitutively and in macrophages that engulf ACs, but not in response toLPS. In other words, individuals carrying the A- or G-allele of21082,respectively, have intrinsicallydifferent abilities toproduce IL-10 in thesteady state and in response toACs induced either intrinsically, such asin normal cellular turnover, or extrinsically, such as during an infection.In severe sepsis andhemorrhage,PARP-1activationhasemergedas
one of the central mechanisms of systemic inflammation, endothelialdysfunction, peripheral vascular failure, and reduction of cardiaccontractility. For example, there is evidence of significant PARP ac-tivation in the hearts of septic patients with impaired cardiac functionand that PARP activation may be partly responsible for the cardiacdepression seen in humans with severe sepsis (50). In a murine cecalligation and puncture (CLP) model of septic shock, treatment withtempol, a low-molecular-weight membrane-permeable radical scav-enger, caused a marked reduction in PARP activity in the lung andkidney glomeruli, accompanied by improved mesenteric arterialblood flow (51). PARP-deficient mice subjected to CLP had signifi-cantly lower plasma levels of TNF-a and IL-6, and they exhibiteda reduced degree of organ inflammation, indicated by decreasedmyeloperoxidase activity in the gut and lung. These effects were as-sociated with a significant improvement in the survival of CLP inPARP-deficient mice (52). Pseudomonas aeruginosa is commonlyassociated with nosocomial pneumonia. Ileal mucosal injury may beinducedby severe lung infection. It has been shown in a ratmodel ofP.aeruginosa-induced septic shock that pharmacological inhibition ofPARP-1 can reducegut inflammation and limit bacterial translocation(53). These studies demonstrate that innovative therapeutic strategiesbased on the pharmacological inhibition of PARP-1 catalytic activitymight provide benefits by preventing tissue injury, organ dysfunction,and lethality associated with these conditions.It iswellestablishedthatduringsepticshock,peroxynitrite-mediated
DNA strand-breaks activate PARP-1, resulting in cellular energeticsuppression and cell dysfunction. Our study, however, uncovers anadditional, novel, and immunologic mechanism (i.e., PARP-1 activa-tion leads to suppression of IL-10 transcription and production inageneticallydifferentialmanner).Thisfindingexplainstheindividuallyvariable susceptibility to sepsis associated with the IL-10 alleles in
a physiological process where infection-induced apoptosis occurs.Studies in recent years have suggested that dysregulated apoptoticimmune cell death may play a role in contributing to the immunedysfunction and multiple organ failure observed during sepsis (54).Lymphocytes are particularly prone to dysregulated apoptotic celldeath. Loss of lymphocytes is detrimental to the survival of septicanimals, as documented by the observation that RAG-deficient micearemarkedlymore susceptible to lethal effects of polymicrobial septicchallenge than their wild-type (WT) controls (54). Lymphocyte apo-ptosis occurs following the onset of experimental sepsis in the thymus,spleen, and GALTs. One of the hypotheses arising from these ob-servations is that the overt apoptotic loss of lymphocytes in the septicindividual reduces the number of functional immune cells available toneutralize the lethal effects of septic challenge. This notion is stronglysupported by the observation that if lymphocyte apoptosis is blockedvia the restricted overexpression of Bcl-2, the mortality in the CLPmodel of sepsis in mice is largely abrogated (54). Alternatively, orconcurrently, excessive ACs during an infection might induce largeamounts of immunosuppressive cytokines, such as IL-10 and TGF-b,that incapacitate the immune system in its response to the presence ofthe infectious pathogen, leading to immunological paralysis. The cy-tokine response in this process, especially the IL-10 response inphagocytes, is genetically determined to a large extent via the in-teraction with PARP-1, as we have shown in this article, resulting inindividual variability and susceptibility. It is worth pointing out that inthis study, we find overexpression ofWTPARP-1, but not the PARP-1mutant lacking catalytic activity, partially inhibits IL-10 transcription(Fig. 4A). We do not expect PARP-1 overexpression to inhibit IL-10transcription completely because ACs stimulate IL-10 transcriptionvia Pbx-1b binding to a separate site on the IL-10 promoter, as weshowed previously (55). PARP-1 does not act on Pbx-1b, blocking itstranscriptional activity, and PARP-1 merely fine-tunes the level of IL-10 transcription according to the21082 allelic status.In summary, our study has uncovered a potentially novel path-
way that involves apoptosis of lymphocytes during sepsis, theclearance of these apoptotic bodies, and the production of IL-10 ina 21082 A . G allele-specific manner via PARP-1, a critical in-flammatory molecule in sepsis and other pathologies. The studyprovides a mechanistic basis for the individual variability in IL-10production and in differential susceptibility to sepsis. It has strongimplications in the development of therapeutic targeting strategiesin sepsis that are sensitive to each patient’s genetic underpinning.
DisclosuresThe authors have no financial conflicts of interest.
References1. Oberholzer, A., C. Oberholzer, and L. L. Moldawer. 2002. Interleukin-10: a com-
plex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit. Care Med. 30 (1 Suppl): S58–S63.
2. Steinhauser, M. L., C. M. Hogaboam, S. L. Kunkel, N. W. Lukacs, R. M. Strieter,and T. J. Standiford. 1999. IL-10 is a major mediator of sepsis-induced im-pairment in lung antibacterial host defense. J. Immunol. 162: 392–399.
3. van der Poll, T., A. Marchant, C. V. Keogh, M. Goldman, and S. F. Lowry. 1996.Interleukin-10 impairs host defense in murine pneumococcal pneumonia.J. Infect. Dis. 174: 994–1000.
4. Marchant, A., J. Deviere, B. Byl, D. De Groote, J. L. Vincent, and M. Goldman.1994. Interleukin-10 production during septicaemia. Lancet 343: 707–708.
5. Neidhardt, R., M. Keel, U. Steckholzer, A. Safret, U. Ungethuem, O. Trentz, and W.Ertel. 1997. Relationship of interleukin-10 plasma levels to severity of injury andclinical outcome in injured patients. J. Trauma 42: 863–870; discussion 870–871.
6. Igonin, A. A., V. W. Armstrong, M. Shipkova, N. B. Lazareva, V. G. Kukes, andM. Oellerich. 2004. Circulating cytokines as markers of systemic inflammatoryresponse in severe community-acquired pneumonia. Clin. Biochem. 37: 204–209.
7. Rodrıguez-Gaspar, M., F. Santolaria, A. Jarque-Lopez, E. Gonzalez-Reimers,A. Milena, M. J. de la Vega, E. Rodrıguez-Rodrıguez, and J. L. Gomez-Sirvent.2001. Prognostic value of cytokines in SIRS general medical patients. Cytokine15: 232–236.
The Journal of Immunology 3723
on March 3, 2012
ww
w.jim
munol.org
Dow
nloaded from

8. Friedman, G., S. Jankowski, A. Marchant, M. Goldman, R. J. Kahn, and J. L. Vincent.1997. Blood interleukin 10 levels parallel the severity of septic shock. J. Crit. Care 12:183–187.
9. Gomez-Jimenez, J., M. C. Martın, R. Sauri, R. M. Segura, F. Esteban, J. C. Ruiz,X. Nuvials, J. L. Boveda, R. Peracaula, and A. Salgado. 1995. Interleukin-10 andthe monocyte/macrophage-induced inflammatory response in septic shock. J.Infect. Dis. 171: 472–475.
10. Derkx, B., A. Marchant, M. Goldman, R. Bijlmer, and S. van Deventer. 1995.High levels of interleukin-10 during the initial phase of fulminant meningococcalseptic shock. J. Infect. Dis. 171: 229–232.
11. Monneret, G., M. E. Finck, F. Venet, A. L. Debard, J. Bohe, J. Bienvenu, andA. Lepape. 2004. The anti-inflammatory response dominates after septic shock:association of low monocyte HLA-DR expression and high interleukin-10concentration. Immunol. Lett. 95: 193–198.
12. Simmons, E. M., J. Himmelfarb, M. T. Sezer, G. M. Chertow, R. L. Mehta,E. P. Paganini, S. Soroko, S. Freedman, K. Becker, D. Spratt, et al; PICARDStudy Group. 2004. Plasma cytokine levels predict mortality in patients withacute renal failure. Kidney Int. 65: 1357–1365.
13. Armstrong, L., and A. B. Millar. 1997. Relative production of tumour necrosisfactor alpha and interleukin 10 in adult respiratory distress syndrome. Thorax 52:442–446.
14. Parsons, P. E., M. Moss, J. L. Vannice, E. E. Moore, F. A. Moore, and J. E. Repine.1997. Circulating IL-1ra and IL-10 levels are increased but do not predict thedevelopment of acute respiratory distress syndrome in at-risk patients. Am. J. Re-spir. Crit. Care Med. 155: 1469–1473.
15. Donnelly, S. C., R. M. Strieter, P. T. Reid, S. L. Kunkel, M. D. Burdick, I. Armstrong,A. Mackenzie, and C. Haslett. 1996. The association between mortality rates anddecreased concentrations of interleukin-10 and interleukin-1 receptor antagonist inthe lung fluids of patients with the adult respiratory distress syndrome. Ann. Intern.Med. 125: 191–196.
16. Roy, S., K. Knox, S. Segal, D. Griffiths, C. E. Moore, K. I. Welsh, A. Smarason,N. P. Day, W. L. McPheat, D. W. Crook, and A. V. Hill; Oxford PneumoccocalSurveillance Group. 2002. MBL genotype and risk of invasive pneumococcaldisease: a case-control study. Lancet 359: 1569–1573.
17. Savill, J., and V. Fadok. 2000. Corpse clearance defines the meaning of celldeath. Nature 407: 784–788.
18. Albert, M. L., S. F. Pearce, L. M. Francisco, B. Sauter, P. Roy, R. L. Silverstein,and N. Bhardwaj. 1998. Immature dendritic cells phagocytose apoptotic cells viaalphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes.J. Exp. Med. 188: 1359–1368.
19. Grigg, J. M., J. S. Savill, C. Sarraf, C. Haslett, and M. Silverman. 1991. Neutrophilapoptosis and clearance from neonatal lungs. Lancet 338: 720–722.
20. Cox, G., J. Crossley, and Z. Xing. 1995. Macrophage engulfment of apoptoticneutrophils contributes to the resolution of acute pulmonary inflammationin vivo. Am. J. Respir. Cell Mol. Biol. 12: 232–237.
21. Fadok, V. A., D. L. Bratton, A. Konowal, P.W. Freed, J. Y.Westcott, and P.M. Henson.1998. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatorycytokine production through autocrine/paracrine mechanisms involving TGF-beta,PGE2, and PAF. J. Clin. Invest. 101: 890–898.
22. Voll, R. E., E. A. Roth, I. Girkontaite, H. Fehr, M. Herrmann, H. M. Lorenz, andJ. R. Kalden. 1997. Histone-specific Th0 and Th1 clones derived from systemiclupus erythematosus patients induce double-stranded DNA antibody production.Arthritis Rheum. 40: 2162–2171.
23. Xu, W., A. Roos, N. Schlagwein, A. M. Woltman, M. R. Daha, and C. van Kooten.2006. IL-10-producing macrophages preferentially clear early apoptotic cells.Blood 107: 4930–4937.
24. Allen, M. L., J. A. Hoschtitzky, M. J. Peters, M. Elliott, A. Goldman, I. James,and N. J. Klein. 2006. Interleukin-10 and its role in clinical immunoparalysisfollowing pediatric cardiac surgery. Crit. Care Med. 34: 2658–2665.
25. Westendorp, R. G., J. A. Langermans, T. W. Huizinga, A. H. Elouali, C. L. Verweij,D. I. Boomsma, J. P. Vandenbroucke, and J. P. Vandenbrouke. 1997. Genetic in-fluence on cytokine production and fatal meningococcal disease. Lancet 349: 170–173.
26. Reuss, E., R. Fimmers, A. Kruger, C. Becker, C. Rittner, and T. Hohler. 2002.Differential regulation of interleukin-10 production by genetic and environ-mental factors—a twin study. Genes Immun. 3: 407–413.
27. Turner, D. M., D. M. Williams, D. Sankaran, M. Lazarus, P. J. Sinnott, andI. V. Hutchinson. 1997. An investigation of polymorphism in the interleukin-10gene promoter. Eur. J. Immunogenet. 24: 1–8.
28. Suarez, A., P. Castro, R. Alonso, L. Mozo, and C. Gutierrez. 2003. Interindividualvariations in constitutive interleukin-10 messenger RNA and protein levels andtheir association with genetic polymorphisms. Transplantation 75: 711–717.
29. Galley, H. F., P. R. Lowe, R. L. Carmichael, and N. R. Webster. 2003. Genotypeand interleukin-10 responses after cardiopulmonary bypass. Br. J. Anaesth. 91:424–426.
30. Stanilova, S. A., L. D. Miteva, Z. T. Karakolev, and C. S. Stefanov. 2006. In-terleukin-10-1082 promoter polymorphism in association with cytokine pro-duction and sepsis susceptibility. Intensive Care Med. 32: 260–266.
31. Jaber, B. L., M. Rao, D. Guo, V. S. Balakrishnan, M. C. Perianayagam,R. B. Freeman, and B. J. Pereira. 2004. Cytokine gene promoter polymorphismsand mortality in acute renal failure. Cytokine 25: 212–219.
32. Schaaf, B. M., F. Boehmke, H. Esnaashari, U. Seitzer, H. Kothe, M. Maass, P. Zabel,and K. Dalhoff. 2003. Pneumococcal septic shock is associated with the interleukin-10-1082 gene promoter polymorphism. Am. J. Respir. Crit. Care Med. 168: 476–480.
33. Gong, M. N., B. T. Thompson, P. L. Williams, W. Zhou, M. Z. Wang, L. Pothier,and D. C. Christiani. 2006. Interleukin-10 polymorphism in position -1082 andacute respiratory distress syndrome. Eur. Respir. J. 27: 674–681.
34. Zhang, D. L., H. M. Zheng, B. J. Yu, Z. W. Jiang, and J. S. Li. 2005. Associationof polymorphisms of IL and CD14 genes with acute severe pancreatitis andseptic shock. World J. Gastroenterol. 11: 4409–4413.
35. Oliver, F. J., J. Menissier-de Murcia, C. Nacci, P. Decker, R. Andriantsitohaina,S. Muller, G. de la Rubia, J. C. Stoclet, and G. de Murcia. 1999. Resistance toendotoxic shock as a consequence of defective NF-kappaB activation in poly(ADP-ribose) polymerase-1 deficient mice. EMBO J. 18: 4446–4454.
36. Szabo, C., and V. L. Dawson. 1998. Role of poly(ADP-ribose) synthetase ininflammation and ischaemia-reperfusion. Trends Pharmacol. Sci. 19: 287–298.
37. Petrilli, V., Z. Herceg, P. O. Hassa, N. S. Patel, R. Di Paola, U. Cortes, L. Dugo,H. M. Filipe, C. Thiemermann, M. O. Hottiger, et al. 2004. Noncleavable poly(ADP-ribose) polymerase-1 regulates the inflammation response in mice. J. Clin.Invest. 114: 1072–1081.
38. Yung, T. M., and M. S. Satoh. 2001. Functional competition between poly(ADP-ribose) polymerase and its 24-kDa apoptotic fragment in DNA repair and tran-scription. J. Biol. Chem. 276: 11279–11286.
39. D’Amours, D., F. R. Sallmann, V. M. Dixit, and G. G. Poirier. 2001. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic pro-teases: implications for apoptosis. J. Cell Sci. 114: 3771–3778.
40. Hassa, P. O., M. Covic, S. Hasan, R. Imhof, and M. O. Hottiger. 2001. Theenzymatic and DNA binding activity of PARP-1 are not required for NF-kappa Bcoactivator function. J. Biol. Chem. 276: 45588–45597.
41. Rolli, V., M. O’Farrell, J. Menissier-de Murcia, and G. de Murcia. 1997. Randommutagenesis of the poly(ADP-ribose) polymerase catalytic domain revealsamino acids involved in polymer branching. Biochemistry 36: 12147–12154.
42. Heller, B., Z. Q. Wang, E. F. Wagner, J. Radons, A. Burkle, K. Fehsel, V. Burkart,and H. Kolb. 1995. Inactivation of the poly(ADP-ribose) polymerase gene affectsoxygen radical and nitric oxide toxicity in islet cells. J. Biol. Chem. 270: 11176–11180.
43. Nosseri, C., S. Coppola, and L. Ghibelli. 1994. Possible involvement of poly(ADP-ribosyl) polymerase in triggering stress-induced apoptosis. Exp. Cell Res.212: 367–373.
44. Virag, L., P. Bai, I. Bak, P. Pacher, J. G. Mabley, L. Liaudet, E. Bakondi, P. Gergely,M. Kollai, and C. Szabo. 2004. Effects of poly(ADP-ribose) polymerase inhibitionon inflammatory cell migration in a murine model of asthma. Med. Sci. Monit. 10:BR77–BR83.
45. Jagtap, P., and C. Szabo. 2005. Poly(ADP-ribose) polymerase and the therapeuticeffects of its inhibitors. Nat. Rev. Drug Discov. 4: 421–440.
46. Ryabokon, N. I., A. Cieslar-Pobuda, and J. Rzeszowska-Wolny. 2009. Inhibitionof poly(ADP-ribose) polymerase activity affects its subcellular localization andDNA strand break rejoining. Acta Biochim. Pol. 56: 243–248.
47. Weseler, A. R., L. Geraets, H. J. Moonen, R. J. Manders, L. J. van Loon,H. J. Pennings, E. F. Wouters, A. Bast, and G. J. Hageman. 2009. Poly (ADP-ribose) polymerase-1-inhibiting flavonoids attenuate cytokine release in bloodfrom male patients with chronic obstructive pulmonary disease or type 2 di-abetes. J. Nutr. 139: 952–957.
48. Chung, E. Y., J. Liu, Y. Zhang, and X. Ma. 2007. Differential expression inlupus-associated IL-10 promoter single-nucleotide polymorphisms is mediatedby poly(ADP-ribose) polymerase-1. Genes Immun 8: 577–589.
49. Cao, S., J. Liu, L. Song, and X. Ma. 2005. The protooncogene c-Maf is an es-sential transcription factor for IL-10 gene expression in macrophages. J. Im-munol. 174: 3484–3492.
50. Soriano, F. G., A. C. Nogueira, E. G. Caldini, M. H. Lins, A. C. Teixeira, S. B. Cappi,P. A. Lotufo, M. M. Bernik, Z. Zsengeller, M. Chen, and C. Szabo. 2006. Potentialrole of poly(adenosine 59-diphosphate-ribose) polymerase activation in the patho-genesis of myocardial contractile dysfunction associated with human septic shock.Crit. Care Med. 34: 1073–1079.
51. Yuksel, B. C., S. E. Serdar, A. Tuncel, N. Uzum, O. Ataoglu, A. Atan, S. Hengirmen,A. B. Iskit, and M. O. Guc. 2009. Effect of tempol, a membrane-permeable radicalscavenger, on mesenteric blood flow and organ injury in a murine cecal ligation andpuncture model of septic shock. Eur. Surg. Res. 43: 219–227.
52. Soriano, F. G., L. Liaudet, E. Szabo, L. Virag, J. G. Mabley, P. Pacher, andC. Szabo. 2002. Resistance to acute septic peritonitis in poly(ADP-ribose)polymerase-1-deficient mice. Shock 17: 286–292.
53. Lobo, S.M., S. R. Orrico,M.M. Queiroz, G. S. Cunrath, G. S. Chibeni, L.M. Contrin,P. M. Cury, E. A. Burdmann, A. M. de Oliveira Machado, P. Togni, et al. 2005.Pneumonia-induced sepsis and gut injury: effects of a poly-(ADP-ribose) polymeraseinhibitor. J. Surg. Res. 129: 292–297.
54. Hotchkiss, R. S., K. W. Tinsley, and I. E. Karl. 2003. Role of apoptotic cell deathin sepsis. Scand. J. Infect. Dis. 35: 585–592.
55. Chung, E. Y., J. Liu, Y. Homma, Y. Zhang, A. Brendolan, M. Saggese, J. Han,R. Silverstein, L. Selleri, and X. Ma. 2007. Interleukin-10 expression in mac-rophages during phagocytosis of apoptotic cells is mediated by homeodomainproteins Pbx1 and Prep-1. Immunity 27: 952–964.
3724 PARP-1 REGULATES IL-10 EXPRESSION
on March 3, 2012
ww
w.jim
munol.org
Dow
nloaded from