Embed Size (px)
Citation preview

DOI: 10.1542/pir.32-4-1352011;32;135Pediatrics in Review
Deborah M. ConsoliniThrombocytopenia in Infants and Children
http://pedsinreview.aappublications.org/content/32/4/135located on the World Wide Web at:
The online version of this article, along with updated information and services, is
Pediatrics. All rights reserved. Print ISSN: 0191-9601. Boulevard, Elk Grove Village, Illinois, 60007. Copyright © 2011 by the American Academy of published, and trademarked by the American Academy of Pediatrics, 141 Northwest Pointpublication, it has been published continuously since 1979. Pediatrics in Review is owned, Pediatrics in Review is the official journal of the American Academy of Pediatrics. A monthly
at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

Thrombocytopenia in Infants and ChildrenDeborah M. Consolini,
MD*
Author Disclosure
Dr Consolini has
disclosed no financial
relationships relevant
to this article. This
commentary does
contain a discussion
of an unapproved/
investigative use of a
commercial
product/device.
Objectives After completing this article, readers should be able to:
1. Explain the relationship between platelet count and bleeding risk.2. State the two basic underlying pathologic mechanisms that may lead to clinically
significant thrombocytopenia.3. Describe the typical presentation and natural history of immune (idiopathic) thrombo-
cytopenic purpura (ITP) in children.4. List the features of the complete blood count and peripheral blood smear that suggest
a serious disorder associated with decreased platelet production.5. Discuss the treatment modalities that have been proven to be effective in raising the
platelet count to a safe level in children who have ITP and are experiencing significantbleeding manifestations.
IntroductionPlatelets are essential for maintaining the integrity of the vascular endothelium andcontrolling hemorrhage from small-vessel injury through the formation of platelet plugs(primary hemostasis). More extensive injury and involvement of larger blood vesselsrequires, in addition to platelets, the participation of the coagulation system to provide afirm, stable, fibrin clot (secondary hemostasis). Thrombocytopenia, defined as a plateletcount of less than 150�103/�L (150�109/L), is the most common cause of defectiveprimary hemostasis that can lead to significant bleeding in children.
Thrombocytopenia should be suspected when a child presents with a history of easybruising or bleeding, particularly mucosal or cutaneous bleeding. However, the mostcommon office presentation of a patient who has isolated thrombocytopenia is the
unexpected discovery of a low platelet count when a com-plete blood count (CBC) is obtained for unrelated reasons.
Thrombocytopenia can be caused by one of two mecha-nisms: decreased production of platelets or increased de-struction or removal of platelets from the circulation. Man-agement of thrombocytopenia should be guided by anunderstanding of its cause and clinical course. The principalmanagement goal in all patients who have thrombocytopeniais to maintain a safe platelet count to prevent significantbleeding. What constitutes a safe platelet count in a specificpatient varies, depending on the cause of the thrombocyto-penia and consideration of all other aspects of hemostasis, aswell as the patient’s expected level of activity.
Platelet Count and Bleeding RiskPlatelets are nonnucleated, cellular fragments produced bythe megakaryocytes of the bone marrow. As the megakaryo-cyte matures, the cytoplasm fragments, and large numbers ofplatelets are released into the circulation. Once released, thelife span of platelets is about 7 to 10 days, after which they areremoved from the circulation by cells of the monocyte-
*Assistant Professor of Pediatrics, Thomas Jefferson University, Philadelphia, PA; Diagnostic Referral Division, A.I. duPontHospital for Children, Wilmington, DE.
Abbreviations
CBC: complete blood countDIC: disseminated intravascular coagulationHIV: human immunodeficiency virusHUS: hemolytic-uremic syndromeICH: intracranial hemorrhageIg: immunoglobulinIGIV: immune globulin intravenousITP: immune thrombocytopenic purpuraKMS: Kasabach-Merritt syndromeMPV: mean platelet volumeNAIT: neonatal alloimmune thrombocytopeniaNEC: necrotizing enterocolitisPBS: peripheral blood smearSLE: systemic lupus erythematosusTTP: thrombotic thrombocytopenic purpuraWAS: Wiscott-Aldrich syndrome
Article hematology
Pediatrics in Review Vol.32 No.4 April 2011 135 at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from
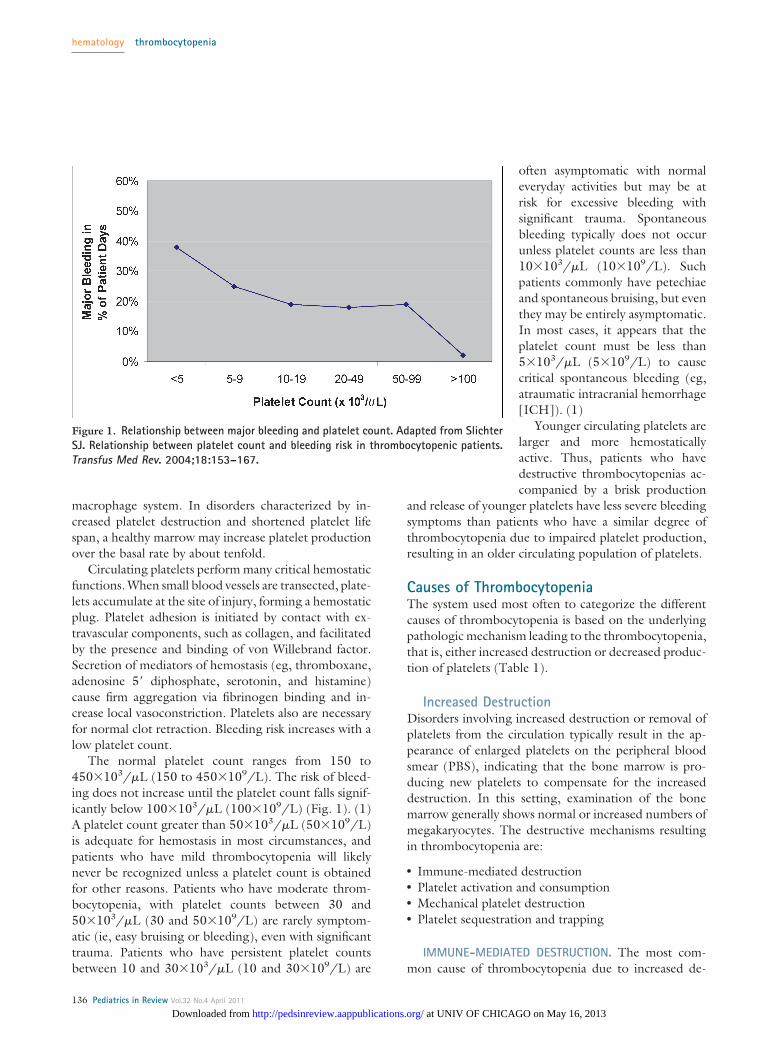
macrophage system. In disorders characterized by in-creased platelet destruction and shortened platelet lifespan, a healthy marrow may increase platelet productionover the basal rate by about tenfold.
Circulating platelets perform many critical hemostaticfunctions. When small blood vessels are transected, plate-lets accumulate at the site of injury, forming a hemostaticplug. Platelet adhesion is initiated by contact with ex-travascular components, such as collagen, and facilitatedby the presence and binding of von Willebrand factor.Secretion of mediators of hemostasis (eg, thromboxane,adenosine 5� diphosphate, serotonin, and histamine)cause firm aggregation via fibrinogen binding and in-crease local vasoconstriction. Platelets also are necessaryfor normal clot retraction. Bleeding risk increases with alow platelet count.
The normal platelet count ranges from 150 to450�103/�L (150 to 450�109/L). The risk of bleed-ing does not increase until the platelet count falls signif-icantly below 100�103/�L (100�109/L) (Fig. 1). (1)A platelet count greater than 50�103/�L (50�109/L)is adequate for hemostasis in most circumstances, andpatients who have mild thrombocytopenia will likelynever be recognized unless a platelet count is obtainedfor other reasons. Patients who have moderate throm-bocytopenia, with platelet counts between 30 and50�103/�L (30 and 50�109/L) are rarely symptom-atic (ie, easy bruising or bleeding), even with significanttrauma. Patients who have persistent platelet countsbetween 10 and 30�103/�L (10 and 30�109/L) are
often asymptomatic with normaleveryday activities but may be atrisk for excessive bleeding withsignificant trauma. Spontaneousbleeding typically does not occurunless platelet counts are less than10�103/�L (10�109/L). Suchpatients commonly have petechiaeand spontaneous bruising, but eventhey may be entirely asymptomatic.In most cases, it appears that theplatelet count must be less than5�103/�L (5�109/L) to causecritical spontaneous bleeding (eg,atraumatic intracranial hemorrhage[ICH]). (1)
Younger circulating platelets arelarger and more hemostaticallyactive. Thus, patients who havedestructive thrombocytopenias ac-companied by a brisk production
and release of younger platelets have less severe bleedingsymptoms than patients who have a similar degree ofthrombocytopenia due to impaired platelet production,resulting in an older circulating population of platelets.
Causes of ThrombocytopeniaThe system used most often to categorize the differentcauses of thrombocytopenia is based on the underlyingpathologic mechanism leading to the thrombocytopenia,that is, either increased destruction or decreased produc-tion of platelets (Table 1).
Increased DestructionDisorders involving increased destruction or removal ofplatelets from the circulation typically result in the ap-pearance of enlarged platelets on the peripheral bloodsmear (PBS), indicating that the bone marrow is pro-ducing new platelets to compensate for the increaseddestruction. In this setting, examination of the bonemarrow generally shows normal or increased numbers ofmegakaryocytes. The destructive mechanisms resultingin thrombocytopenia are:
● Immune-mediated destruction● Platelet activation and consumption● Mechanical platelet destruction● Platelet sequestration and trapping
IMMUNE-MEDIATED DESTRUCTION. The most com-mon cause of thrombocytopenia due to increased de-
Figure 1. Relationship between major bleeding and platelet count. Adapted from SlichterSJ. Relationship between platelet count and bleeding risk in thrombocytopenic patients.Transfus Med Rev. 2004;18:153–167.
hematology thrombocytopenia
136 Pediatrics in Review Vol.32 No.4 April 2011
at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

struction of platelets in infants and children is animmune-mediated process. Autoantibodies, drug-dependent antibodies, or alloantibodies may mediateplatelet destruction through interaction with plateletmembrane antigens, leading to increased platelet clear-ance from the circulation.
Primary ITP is an acquired immune-mediated disor-der characterized by isolated thrombocytopenia in theabsence of any obvious initiating or underlying cause.(2)(3) Formerly, the abbreviation ITP stood for idio-pathic thrombocytopenic purpura. The new terminologyreflects the current understanding of the immune-
mediated nature of the disease and the absence or mini-mal signs of bleeding in most cases. The platelet countnow used to define cases of ITP is less than 100�103/�L(100�109/L). (2) The term secondary ITP refers toimmune-mediated thrombocytopenias that are due to anunderlying disease or to drug exposure. (2)(3) The dis-tinction between primary and secondary ITP is clinicallyrelevant both in regard to prognosis and treatment.
ITP is the most common immune-mediated throm-bocytopenia in children, with an annual incidence ofsymptomatic cases estimated to be between 3 and 8 casesper 100,000 children. Pediatric patients who developITP usually present between the ages of 2 and 10 years,with a peak incidence at 2 to 5 years. There does notappear to be a significant sex bias in childhood ITP.(4)(5)
The typical case of symptomatic childhood ITP ischaracterized by the sudden appearance of bruising ormucocutaneous bleeding in an otherwise healthy child,often after a preceding viral illness. An increased risk ofITP is also associated with measles, mumps, rubella im-munization, which accounts for perhaps 50% of all ITPcases during the second year after birth. This form of ITPtends to be transient and rarely is the bleeding severe.
The history should reveal no systemic symptoms suchas fever, weight loss, or bone pain. Other than muco-cutaneous bleeding, patients should appear well. Nolymphadenopathy or hepatosplenomegaly should bepresent. If one or more of these findings are present,another diagnosis should be strongly considered. Other-wise, the diagnosis of ITP can be made based on twocriteria: 1) isolated thrombocytopenia with otherwisenormal blood counts and PBS and 2) no clinically appar-ent associated conditions that may cause thrombocyto-penia.
The severity of bleeding symptoms in childhood ITPis proportionate to the degree of thrombocytopenia.Serious bleeding requiring transfusion is uncommon.The presenting platelet count is usually less than20�103/�L (20�109/L). This value probably is due topatients who have higher platelet counts, which are muchless likely to lead to bleeding, never coming to medicalattention. Children who have ITP and platelet countsgreater than 30�103/�L (30�109/L) usually have fewor no symptoms and require no treatment other thanrestriction of activity and avoidance of medications thathave antiplatelet or anticoagulant activity. For thosewhose platelet counts are below 30�103/�L (30�109/L), treatment recommendations are based on the pres-ence and severity of associated bleeding symptoms or therisk thereof. (4)(6)
Table 1. Causes ofThrombocytopeniaIncreased Platelet Destruction
● Immune-mediated–Immune thrombocytopenic purpura–Neonatal alloimmune thrombocytopenia–Neonatal autoimmune thrombocytopenia–Autoimmune diseases–Drug-induced
● Platelet activation/consumption–Disseminated intravascular coagulation–Hemolytic-uremic syndrome–Thrombotic thrombocytopenic purpura–Kasabach-Merritt syndrome–Necrotizing enterocolitis–Thrombosis
● Mechanical platelet destruction● Platelet sequestration
–Chronic liver disease–Type 2B and platelet-type von Willebrand disease–Malaria
Decreased Platelet Production
● Infection● Cyanotic congenital heart disease● Bone marrow failure or infiltrate
–Acute lymphoblastic leukemia and othermalignancies
–Acquired aplastic anemia–Fanconi pancytopenia
● Nutritional deficiencies● Genetically impaired thrombopoiesis
–Thrombocytopenia with absent radii syndrome–Congenital amegakaryocytic thrombocytopenia–Wiskott-Aldrich syndrome–X-linked thrombocytopenia with thalassemia–Giant platelet disorders–Bernard-Soulier syndrome–May-Hegglin/Fechtner/Epstein and Sebastiansyndromes
hematology thrombocytopenia
Pediatrics in Review Vol.32 No.4 April 2011 137 at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

ITP is now classified by duration into newly diag-nosed, persistent (3- to 12-month duration), andchronic (�12-month duration). (2)(3) Whereas ITP inadults typically has an insidious onset and follows achronic course, ITP in children is usually short-lived,with about two thirds of patients making full and sus-tained recoveries within 6 months of presentation, withor without treatment. (2)(3) Children who have persis-tent or chronic ITP or who manifest any atypical featuresshould be referred to or discussed with a hematologistexperienced in the assessment and treatment of childrenwho have ITP.
Neonatal alloimmune thrombocytopenia (NAIT) isan uncommon syndrome whose estimated incidence inthe general population is 1 in 1,000 to 5,000 births. Thecondition is manifested by an isolated, transient, butpotentially severe thrombocytopenia in the neonate dueto platelet destruction by maternal alloantibodies. NAIToccurs when fetal platelets contain an antigen inheritedfrom the father that the mother lacks. Fetal platelets thatcross the placenta into the maternal circulation triggerthe production of maternal immunoglobulin (Ig) Gantiplatelet antibodies against the foreign antigen. Theseantibodies recross the placenta into the fetal circulationand destroy the body’s platelets, resulting in fetal andneonatal thrombocytopenia (analogous to hemolytic dis-ease of the newborn). In contrast to Rh sensitization,NAIT often develops in the first pregnancy of an at-riskcouple. The most serious complication of NAIT is ICH,which occurs in approximately 10% to 20% of affectednewborns, with up to 50% of these events occurring inutero. (7) The mother of a newborn who has NAIT isasymptomatic, although she may have a history of previ-ously affected pregnancies. Affected newborns typicallypresent with signs consistent with severe thrombocyto-penia, including petechiae, bruising, and bleeding, butare otherwise healthy. Platelet counts are often less than10�103/�L (10�109/L). The platelet count typicallyfalls in the first few days after birth, but then rises over thenext 1 to 4 weeks as the alloantibody concentrationdeclines. In families who have an affected infant, the rateof recurrence is as high as 75% to 90%, and thrombocy-topenia in the second affected child is always as or moresevere than in the first. (7)
Neonatal autoimmune thrombocytopenia also canoccur. This disorder is mediated by maternal antibodiesthat react with both maternal and infant platelets. Thispathologic mechanism occurs in maternal autoimmunedisorders, including ITP and systemic lupus erythemato-sus (SLE). The diagnosis usually is apparent from themother’s medical history and maternal thrombocytope-
nia. Mothers of infants who manifest unexplained neo-natal thrombocytopenia should be investigated for thepresence of an autoimmune disorder because neonatalthrombocytopenia can sometimes be the presenting signof maternal disease. Clinical signs are consistent withisolated thrombocytopenia, as in NAIT.
The risk of severe thrombocytopenia and ICH isgreater in alloimmune than in autoimmune neonatalthrombocytopenia. Ninety percent of infants born tomothers who have ITP have safe (�50�103/�L[50�109/L]) or normal platelet counts. (8) The risk forsevere thrombocytopenia in the infant generally corre-lates with the severity of ITP in the mother. Neonatalthrombocytopenia may be predicted if the mother hashad a splenectomy for treatment of chronic refractoryITP, the mother’s platelet count has been less that50�103/�L (50�109/L) at some time during the preg-nancy, or an older sibling has had neonatal thrombocy-topenia. (8) Platelet counts of infants born to motherswho have ITP often decrease sharply during the firstseveral days after birth. The nadir typically occurs at 2 to5 days of age, and infants should be monitored closelyduring this time. (8)
Autoimmune diseases such as SLE can present inchildhood with isolated immune-mediated thrombocy-topenia, and the true diagnosis may not be apparent for aprolonged period of time. Autoimmune diseases occurmore commonly in older children and have an insidiousonset of symptoms and persistent thrombocytopeniabeyond 6 months from the time of presentation. In-frequently, other autoimmune-mediated cytopeniaspresent coincidentally with thrombocytopenia. In partic-ular, Evans syndrome is characterized by a Coombs(direct antiglobulin test)-positive hemolytic anemia inassociation with immune-mediated thrombocytopenia.Antibody-mediated thrombocytopenia also occurs withantiphospholipid antibody syndrome and autoimmunelymphoproliferative syndrome.
Drug-induced thrombocytopenia is a rare cause ofthrombocytopenia in children. Medications begunwithin the past month are more likely to be the cause ofthe thrombocytopenia than medications that have beentaken for longer periods of time. Drug-induced throm-bocytopenia is typically caused by drug-dependent anti-bodies formed against a new antigen on the plateletsurface that is created by drug binding to a plateletsurface protein. Heparin-induced thrombocytopenia,which can be associated with severe thrombosis, is due tothe formation of antibodies against the heparin-plateletfactor 4 complex. The platelet count in heparin-inducedthrombocytopenia is usually only moderately decreased.
hematology thrombocytopenia
138 Pediatrics in Review Vol.32 No.4 April 2011
at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

Although this condition is more commonly seen inadults, heparin-induced thrombocytopenia has beendescribed in children. Other drugs commonly used inpediatrics that can cause thrombocytopenia include car-bamazepine, phenytoin, valproic acid, trimethoprim/sulfamethoxazole, and vancomycin. Support for the di-agnosis of drug-induced thrombocytopenia is providedby resolution of the thrombocytopenia within approxi-mately 1 week of withdrawal of the offending drug.
PLATELET ACTIVATION AND CONSUMPTION. In pa-tients who experience disseminated intravascular co-agulation (DIC) and the microangiopathic disordershemolytic-uremic syndrome (HUS) and thromboticthrombocytopenic purpura (TTP), thrombocytopeniaoccurs because of systemic platelet activation, aggrega-tion, and consumption (Table 2). More localized plateletactivationandconsumptioncontributestothethrombocy-topenia seen in Kasabach-Merritt syndrome (KMS), ne-crotizing enterocolitis (NEC), and thrombosis in infantsand neonates. In infants who have KMS, thrombocyto-penia results from shortened platelet survival caused bysequestration of platelets and coagulation activa-tion in large vascular malformations of the trunk, extrem-ities, or abdominal viscera. Cutaneous vascular lesionsare noted at birth in approximately 50% of patients.Detection of visceral lesions requires imaging studies. Allpatients have severe thrombocytopenia, hypofibrinogen-emia, elevated fibrin degradation products, and fragmen-tation of red blood cells on PBS.
NEC is a syndrome of gastrointestinal necrosis thatoccurs in 2% to 10% of infants whose birthweights areless than 1,500 g. Thrombocytopenia is a frequent find-ing and can result in significant bleeding. In the earlystages of NEC, declining platelet counts correlate withthe presence of necrotic bowel and worsening disease.The primary mechanism of thrombocytopenia appearsto be platelet destruction, although the destruction isnot caused by laboratory-detectable DIC in most cases.Thrombosis in infants and neonates is often accompa-nied by thrombocytopenia. A thromboembolic disordershould be considered if the thrombocytopenia cannot beexplained by other conditions.
MECHANICAL PLATELET DESTRUCTION. The use of ex-tracorporeal therapies, such as extracorporeal membraneoxygenation, cardiopulmonary bypass, hemodialysis, andapheresis, is associated with mechanical destruction ofplatelets, which may result in thrombocytopenia. Ex-change transfusion also may reduce platelet number byloss in the exchange effluent. Severe ongoing hemor-
rhage requiring rapid and repeated red blood cell trans-fusions may lead to thrombocytopenia due to a “washout” phenomenon.
PLATELET SEQUESTRATION AND TRAPPING. Aboutone third of the platelet mass is normally sequestered inthe spleen at any given time. A greater proportion ofplatelets are sequestered in patients who experience hy-persplenism, reducing the number of circulating plateletsand leading to thrombocytopenia. The survival of plate-lets in persons who have hypersplenism is normal or nearnormal. It is the pooling and unavailability of platelets“trapped” in the spleen that is the problem. Leukopeniaor anemia also may accompany a low platelet countcaused by hypersplenism. Conditions in this categoryinclude:
● Chronic liver disease with portal hypertension andcongestive splenomegaly. Occasionally, isolated throm-bocytopenia may be the initial manifestation of thistype of chronic liver disease. The platelet count istypically in the range of 50 to 100�103/�L (50 to100�103/L) and usually does not represent a clinicallyimportant problem.
● Type 2B and platelet-type von Willebrand disease.Thrombocytopenia in this disorder is caused by in-creased removal of platelets from the circulation. In-creased binding between larger von Willebrand factormultimers and platelets leads to the formation of smallplatelet aggregates that are cleared from the circula-tion, resulting in a lower platelet count.
● Malaria with hypersplenism. This diagnosis should beconsidered in any child who has fever, splenomegaly,thrombocytopenia, and a history of recent travel to anendemic area.
Decreased ProductionImpaired platelet production may be due to loss of bonemarrow space from infiltration, suppression or failure ofcellular elements, or a defect in megakaryocyte develop-ment and differentiation. In this setting, examination ofthe bone marrow generally shows decreases in the num-ber of megakaryocytes. Causes of marrow dysfunctioninclude:
● Infection● Cyanotic heart disease● Bone marrow failure or infiltration● Nutritional deficiencies● Genetic defects
hematology thrombocytopenia
Pediatrics in Review Vol.32 No.4 April 2011 139 at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

Tab
le2.
Diso
rder
sof
Syst
emic
Plat
elet
Acti
vati
onan
dCo
nsum
ptio
nPa
thop
hysio
logy
Clin
ical
Feat
ures
Diag
nosis
Trea
tmen
tPr
ogno
sis
Diss
emin
ated
intr
avas
cula
rco
agul
atio
n(D
IC)
Path
olog
icac
tivat
ion
ofco
agul
atio
nm
echa
nism
s,re
sulti
ngin
exte
nsiv
em
icro
vasc
ular
thro
mbo
sisle
adin
gto
ische
mia
and
end-
orga
nda
mag
e.
Cons
umpt
ion
ofpl
atel
ets
and
coag
ulat
ion
fact
ors
and
activ
atio
nof
fibrin
olys
isre
sult
inhe
mor
rhag
e.
Com
plic
atio
nof
unde
rlyin
gill
ness
essu
chas
seps
is,as
phyx
ia,m
econ
ium
aspi
ratio
n,se
vere
resp
irato
rydi
stre
sssy
ndro
me,
exte
nsiv
etr
aum
a.
Sym
ptom
sin
clud
ebl
eedi
ng,
hypo
tens
ion,
incr
ease
dva
scul
arpe
rmea
bilit
y,an
dsh
ock.
yTh
rom
bocy
tope
nia
yFr
agm
ente
dRB
Csy
Prol
onge
dPT
,aPT
Ty
Decr
ease
dpl
asm
afib
rinog
eny
Incr
ease
dFD
Ps
Only
effe
ctiv
eth
erap
yis
reve
rsal
ofun
derly
ing
caus
e.
Tem
poriz
ing
mea
sure
sin
clud
e:•
Plat
elet
tran
sfus
ions
for
blee
ding
,sev
ere
thro
mbo
cyto
peni
a•
FFP
tore
plen
ishco
agul
atio
nan
dan
tithr
ombo
ticfa
ctor
s
Varie
s,de
pend
ing
onth
eun
derly
ing
diso
rder
and
the
exte
ntof
the
intr
avas
cula
rth
rom
bosis
,but
rega
rdle
ssof
caus
eis
ofte
ngr
im,w
ith10
%to
50%
mor
talit
y.
Hem
olyt
ic-u
rem
icsy
ndro
me
(HUS
)M
ost
case
spr
eced
edby
episo
deof
bloo
dydi
arrh
eadu
eto
vero
toxi
n-pr
oduc
ing
Esch
eric
hia
coli
O157
H7.
Vero
toxi
nen
ters
bloo
dstr
eam
,at
tach
esto
glom
erul
aren
doth
eliu
m,a
ndin
activ
ates
the
met
allo
prot
ease
ADAM
TS13
,re
spon
sible
for
clea
ving
very
larg
em
ultim
ers
ofvW
F.Un
clea
ved
vWF
initi
ates
plat
elet
aggr
egat
ion
and
activ
atio
n,re
sulti
ngin
netw
ork
ofm
icro
thro
mbi
inki
dney
sth
atle
adto
frag
men
tatio
nof
RBCs
,co
nsum
ptio
nof
plat
elet
s,an
dtis
sue
ische
mia
.
Less
com
mon
form
with
out
diar
rhea
poss
ibly
due
tofa
ctor
Hde
ficie
ncy
with
unco
ntro
lled
com
plem
ent
activ
atio
nan
dth
rom
bosis
afte
rm
inor
endo
thel
iali
njur
y.
Seen
pred
omin
antly
inch
ildre
n.Us
ually
prec
eded
byep
isode
ofbl
oody
diar
rhea
.
Onse
tof
illne
ssch
arac
teriz
edby
pallo
ran
dol
igur
iaw
ithth
rom
bocy
tope
nia
and
labo
rato
ryev
iden
ceof
mic
roan
giop
athi
che
mol
ytic
anem
iaan
dAR
F.
yTh
rom
bocy
tope
nia
yFr
agm
ente
dRB
Csy
Incr
ease
dBU
N,c
reat
inin
ey
Nor
mal
PT,a
PTT
yN
orm
alpl
asm
afib
rinog
eny
Nor
mal
FDPs
Supp
ortiv
eca
re.
Dial
ysis,
asne
eded
,unt
ilev
iden
ceof
reco
verin
gre
nal
func
tion.
Antib
iotic
trea
tmen
tno
tre
com
men
ded
beca
use
itm
ayst
imul
ate
furt
her
vero
toxi
npr
oduc
tion.
Plat
elet
tran
sfus
ions
may
wor
sen
outc
ome.
Ifdi
agno
stic
unce
rtai
nty
betw
een
HUS
and
TTP
beca
use
ofne
urol
ogic
orot
her
nonr
enal
invo
lvem
ent,
plas
ma
exch
ange
reco
mm
ende
d.
Mor
talit
yra
teis
5%to
10%
.Mos
tsu
rviv
ors
reco
ver
with
out
maj
orco
nseq
uenc
es.S
mal
lpr
opor
tion
ofpa
tient
sde
velo
psch
roni
cre
nal
insu
ffici
ency
.
Thro
mbo
ticth
rom
bocy
tope
nic
purp
ura
(TTP
)M
ost
com
mon
lyas
soci
ated
with
inhi
bitio
nof
ADAM
TS13
byau
toan
tibod
ies.
Asin
HUS,
with
out
prop
ercl
eava
geof
vWF,
spon
tane
ous
coag
ulat
ion
occu
rs.
InTT
P,a
netw
ork
ofm
icro
thro
mbi
inm
ultip
leor
gan
syst
ems
resu
ltsin
clin
ical
and
labo
rato
ryfin
ding
s.Ra
rer
form
calle
dUp
shaw
-Sch
ulm
ansy
ndro
me
isa
gene
tical
lyin
herit
eddy
sfun
ctio
nof
ADAM
TS13
.
•Th
rom
bocy
tope
nia
•M
icro
angi
opat
hic
hem
olyt
ican
emia
•AR
F•
Neu
rolo
gic
sym
ptom
s•
Feve
r
ARF
and
neur
olog
icsy
mpt
oms
may
not
bepr
esen
tin
itial
ly.F
ever
unco
mm
on.O
nly
thro
mbo
cyto
peni
aan
dm
icro
angi
opat
hic
hem
olyt
ican
emia
with
out
othe
rap
pare
ntca
use
requ
ired
tosu
spec
tdi
agno
sis.
yTh
rom
bocy
tope
nia
yFr
agm
ente
dRB
Csy
Elev
ated
LDH
yIn
crea
sed
BUN
,cre
atin
ine
yN
orm
alPT
,aPT
Ty
Nor
mal
plas
ma
fibrin
ogen
yN
orm
alFD
Psy
Decr
ease
d(<
5%of
norm
al)
ADAM
TS13
activ
ityy
Dete
ctab
lein
hibi
tors
ofAD
AMTS
13
Daily
plas
ma
exch
ange
usin
gFF
Pre
duce
san
ti-AD
AMTS
13an
tibod
ies
and
repl
enish
esco
ncen
trat
ions
ofth
een
zym
e.M
aybe
need
edfo
r1
to8
wee
ksbe
fore
labo
rato
ryva
lues
norm
alize
.
Upsh
aw-S
chul
man
synd
rom
epa
tient
sre
ceiv
eFF
Pev
ery
2to
3w
eeks
tom
aint
ain
adeq
uate
conc
entr
atio
nsof
func
tioni
ngen
zym
e.
Mor
talit
yra
te�
95%
for
untr
eate
dca
ses
but
10%
to20
%fo
rpa
tient
str
eate
dea
rlyw
ithpl
asm
aex
chan
ge.
Appr
oxim
atel
y30
%of
patie
nts
expe
rienc
ea
rela
pse
with
in10
year
sof
the
first
atta
ck.
aPT
T�
activ
ated
part
ialt
hrom
bopl
astin
time,
AR
F�ac
ute
rena
lfai
lure
,BU
N�
bloo
dur
eani
trog
en,F
DP�
fibri
nde
grad
atio
npr
oduc
t,FF
P�fr
esh
froz
enpl
asm
a,L
DH
�la
ctat
ede
hydr
ogen
ase,
PT�
prot
hrom
bin
time,
RB
C�
red
bloo
dce
ll,vW
F�vo
nW
illeb
rand
fact
or
hematology thrombocytopenia
140 Pediatrics in Review Vol.32 No.4 April 2011
at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

INFECTION. Thrombocytopenia due to infections notassociated with evidence of DIC is usually caused bybone marrow suppression. In some cases, increased de-struction due to an infection-induced immune-mediatedprocess or splenomegaly and reticuloendothelial hyper-activity may compound the problem of bone marrowsuppression. The most common infectious agents asso-ciated with thrombocytopenia due to bone marrow sup-pression are Epstein-Barr virus, cytomegalovirus, parvo-virus, varicella virus, and rickettsiae. In most cases, thethrombocytopenia is transient, with recovery within aperiod of weeks. Thrombocytopenia is a common find-ing in patients who are infected with the human immu-nodeficiency virus (HIV), in whom both platelet destruc-tion and impaired production appear to play roles indecreasing the platelet count.
CYANOTIC HEART DISEASE. Cyanotic congenital heartdisease is associated with thrombocytopenia. The cause isunclear, but the mechanism appears to involve decreasedproduction of megakaryoctes.
BONE MARROW FAILURE OR INFILTRATION. Throm-bocytopenia associated with anemia and leukopenia (ie,pancytopenia) suggests general bone marrow dysfunc-tion or infiltration. Serious disorders such as leukemia orother malignancy, hemophagocytic lymphohistiocytosis,acquired aplastic anemia, myelodysplasia, and inheritedbone marrow failure syndromes such as Fanconi pancy-topenia syndrome and dyskeratosis congenita can presentwith pancytopenia. General bone marrow dysfunctioncan also be caused by exposure to chemotherapeuticagents or radiation.
Acute lymphoblastic leukemia is the most commonchildhood leukemia. Affected children usually have otherclinical and laboratory findings besides thrombocytope-nia. These manifestations include systemic symptomssuch as fever, bone pain, and weight loss as well ashepatosplenomegaly, lymphadenopathy, leukocytosis,and anemia.
Acquired aplastic anemia is a rare disorder caused byprofound, almost complete bone marrow failure. Specificsymptoms associated with acquired aplastic anemia varybut can include fever, fatigue, dizziness, weakness, head-aches, and episodes of excessive bleeding. Pancytopeniais often seen on presentation. Based on the response ofapproximately 50% of patients to immunosuppressivemedications, including antithymocyte globulin, cyclo-sporine, high-dose corticosteroids, and cyclophosph-amide, most cases are now believed to be due to an
immune-mediated destruction of hematopoietic stemcells.
Fanconi pancytopenia syndrome is a rare autosomalrecessive disorder. The mean age at diagnosis of thepancytopenia is approximately 6 to 9 years, but thecondition may be recognized earlier by characteristiccongenital malformations that are present in 60% to 70%of affected patients. The most common malformationsare hypopigmented macules, cafe-au-lait macules, abnor-malities of the thumbs, microcephaly, and urogenitalabnormalities. Short stature of prenatal onset may also beseen.
NUTRITIONAL DEFICIENCIES. Folate, vitamin B12, andiron deficiencies have been associated with thrombocy-topenia. Folate and vitamin B12 deficiencies impair bonemarrow production, usually resulting in pancytopenia.Iron deficiency, which can cause either thrombocytosisor thrombocytopenia, appears to impair a late stage ofthrombopoiesis. Thrombocytosis and iron deficiencymay be a response to gastrointestinal blood loss.
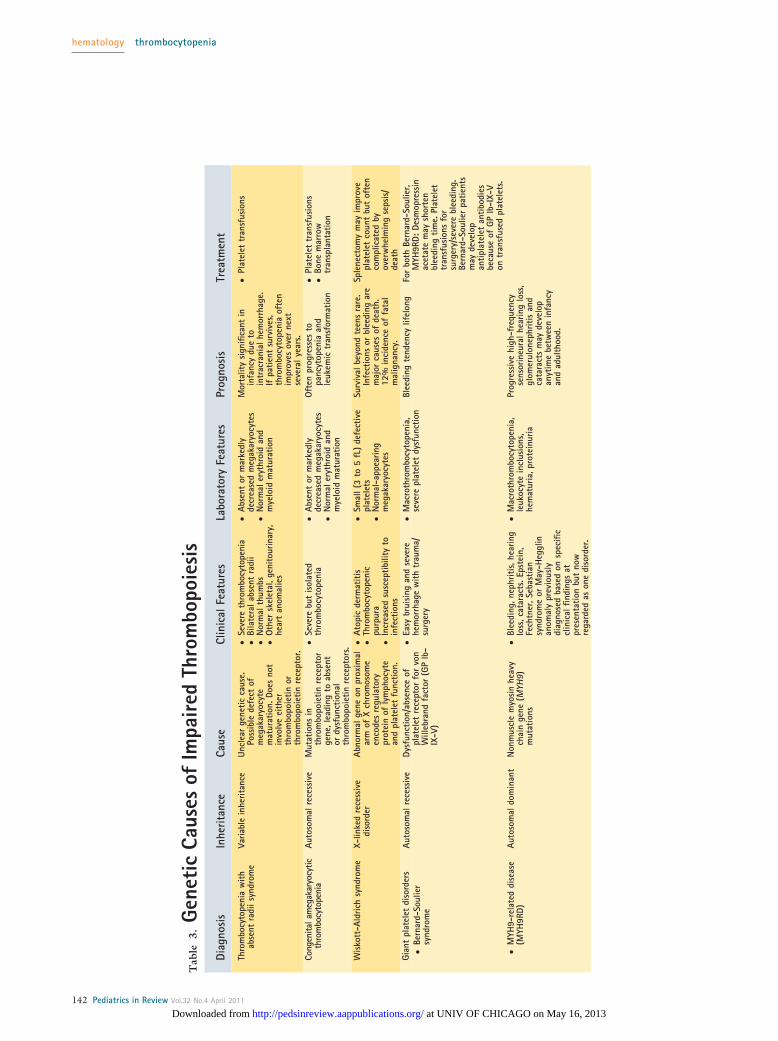
GENETIC CAUSES OF IMPAIRED THROMBOPOIESIS. Alarge number of rare inherited diseases present withreduced platelet count, and many involve impaired plate-let function as well. These conditions arise from geneticdefects of the megakaryocyte lineage that result in im-paired thrombopoiesis. The consideration of congenitalthrombocytopenia should be greater in patients whohave a prolonged history of asymptomatic abnormalplatelet counts or a family history of thrombocytopenia.Some patients born with congenital thrombocytopeniaare followed for many years with the presumptive diag-nosis of ITP until another family member is discoveredto have a low platelet count. Table 3 outlines the geneticcauses of impaired thrombopoiesis.
Clinical ManifestationsChildren who have thrombocytopenia may be asymp-tomatic or symptomatic. In asymptomatic patients,thrombocytopenia is often detected unexpectedly on aCBC obtained for another clinical issue. Symptomaticpatients generally present with mucosal or cutaneousbleeding.
Mucosal bleeding usually manifests as epistaxis, gin-gival bleeding or extensive oral mucous membranebleeding (“wet purpura”), hematuria, or in postpubertalfemales, excessive menstrual bleeding. The presence of“wet purpura” is widely perceived as a risk factor forpotentially life-threatening hemorrhage.
Cutaneous bleeding usually appears as petechiae or
hematology thrombocytopenia
Pediatrics in Review Vol.32 No.4 April 2011 141 at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from
Tab
le3.
Gene
tic
Caus
esof
Impa
ired
Thro
mbo
poie
sis
Diag
nosi
sIn
herit
ance
Caus
eCl
inic
alFe
atur
esLa
bora
tory
Feat
ures
Prog
nosi
sTr
eatm
ent
Thro
mbo
cyto
peni
aw
ithab
sent
radi
isyn
drom
eVa
riabl
ein
herit
ance
Uncl
ear
gene
ticca
use.
Poss
ible
defe
ctof
meg
akar
yocy
tem
atur
atio
n.Do
esno
tin
volv
eei
ther
thro
mbo
poie
tinor
thro
mbo
poie
tinre
cept
or.
●Se
vere
thro
mbo
cyto
peni
a●
Bila
tera
labs
ent
radi
i●
Nor
mal
thum
bs●
Oth
ersk
elet
al,g
enito
urin
ary,
hear
tan
omal
ies
●Ab
sent
orm
arke
dly
decr
ease
dm
egak
aryo
cyte
s●
Nor
mal
eryt
hroi
dan
dm
yelo
idm
atur
atio
n
Mor
talit
ysi
gnifi
cant
inin
fanc
ydu
eto
intr
acra
nial
hem
orrh
age.
Ifpa
tient
surv
ives
,th
rom
bocy
tope
nia
ofte
nim
prov
esov
erne
xtse
vera
lyea
rs.
●Pl
atel
ettr
ansf
usio
ns
Cong
enita
lam
egak
aryo
cytic
thro
mbo
cyto
peni
aAu
toso
mal
rece
ssiv
eM
utat
ions
inth
rom
bopo
ietin
rece
ptor
gene
,lea
ding
toab
sent
ordy
sfun
ctio
nal
thro
mbo
poie
tinre
cept
ors.
●Se
vere
but
isol
ated
thro
mbo
cyto
peni
a●
Abse
ntor
mar
kedl
yde
crea
sed
meg
akar
yocy
tes
●N
orm
aler
ythr
oid
and
mye
loid
mat
urat
ion
Oft
enpr
ogre
sses
topa
ncyt
open
iaan
dle
ukem
ictr
ansf
orm
atio
n
●Pl
atel
ettr
ansf
usio
ns●
Bone
mar
row
tran
spla
ntat
ion
Wis
kott
-Ald
rich
synd
rom
eX-
linke
dre
cess
ive
diso
rder
Abno
rmal
gene
onpr
oxim
alar
mof
Xch
rom
osom
een
code
sre
gula
tory
prot
ein
ofly
mph
ocyt
ean
dpl
atel
etfu
nctio
n.
●At
opic
derm
atiti
s●
Thro
mbo
cyto
peni
cpu
rpur
a●
Incr
ease
dsu
scep
tibili
tyto
infe
ctio
ns
●Sm
all(
3to
5fL
)de
fect
ive
plat
elet
s●
Nor
mal
-app
earin
gm
egak
aryo
cyte
s
Surv
ival
beyo
ndte
ens
rare
.In
fect
ions
orbl
eedi
ngar
em
ajor
caus
esof
deat
h.12
%in
cide
nce
offa
tal
mal
igna
ncy.
Sple
nect
omy
may
impr
ove
plat
elet
coun
tbu
tof
ten
com
plic
ated
byov
erw
helm
ing
seps
is/
deat
hGi
ant
plat
elet
diso
rder
s●
Bern
ard-
Soul
ier
synd
rom
e
Auto
som
alre
cess
ive
Dysf
unct
ion/
abse
nce
ofpl
atel
etre
cept
orfo
rvo
nW
illeb
rand
fact
or(G
PIb
-IX
-V)
●Ea
sybr
uisi
ngan
dse
vere
hem
orrh
age
with
trau
ma/
surg
ery
●M
acro
thro
mbo
cyto
peni
a,se
vere
plat
elet
dysf
unct
ion
Blee
ding
tend
ency
lifel
ong
For
both
Bern
ard-
Soul
ier,
MYH
9RD:
Desm
opre
ssin
acet
ate
may
shor
ten
blee
ding
time.
Plat
elet
tran
sfus
ions
for
surg
ery/
seve
rebl
eedi
ng.
Bern
ard-
Soul
ier
patie
nts
may
deve
lop
antip
late
let
antib
odie
sbe
caus
eof
GPIb
-IX-
Von
tran
sfus
edpl
atel
ets.
●M
YH9-
rela
ted
dise
ase
(MYH
9RD)
Auto
som
aldo
min
ant
Non
mus
cle
myo
sin
heav
ych
ain
gene
(MYH
9)m
utat
ions
●Bl
eedi
ng,n
ephr
itis,
hear
ing
loss
,cat
arac
ts.E
pste
in,
Fech
tner
,Seb
astia
nsy
ndro
me
orM
ay-H
eggl
inan
omal
ypr
evio
usly
diag
nose
dba
sed
onsp
ecifi
ccl
inic
alfin
ding
sat
pres
enta
tion
but
now
rega
rded
ason
edi
sord
er.
●M
acro
thro
mbo
cyto
peni
a,le
ukoc
yte
incl
usio
ns,
hem
atur
ia,p
rote
inur
ia
Prog
ress
ive
high
-fre
quen
cyse
nsor
ineu
ralh
earin
glo
ss,
glom
erul
onep
hriti
san
dca
tara
cts
may
deve
lop
anyt
ime
betw
een
infa
ncy
and
adul
thoo
d.
hematology thrombocytopenia
142 Pediatrics in Review Vol.32 No.4 April 2011
at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

superficial ecchymoses. Patients who have thrombocyto-penia may also have persistent, profuse bleeding fromsuperficial cuts. Petechiae, the pinhead-sized, red, flat,discrete lesions caused by extravasation of red cells fromskin capillaries and often occurring in crops in dependentareas, are highly characteristic of decreased platelet num-ber or function. Petechiae are nontender and do notblanch under pressure. They are asymptomatic and notpalpable and should be distinguished from small telangi-ectasias and vasculitic (palpable) purpura. Purpura de-scribes purplish discolorations of the skin due to thepresence of confluent petechiae. Ecchymoses are non-tender areas of bleeding into the skin that are typicallysmall, multiple, and superficial and can develop withoutnoticeable trauma. Ecchymoses often have a variety ofcolors due to the presence of extravasated blood (red orpurple) and the ongoing breakdown of heme pigment inthe extravasated blood by skin macrophages (green, yel-low, or brown).
This pattern of bleeding differs from that of patientswho have disorders of coagulation factors, such as hemo-philia. Patients who have thrombocytopenia tend to haveless deep bleeding into muscles or joints, more bleedingafter minor cuts, less delayed bleeding, and less postsur-gical bleeding. In addition, patients who have coagula-tion factor disorders tend not to have petechiae. Al-though rare, bleeding into the central nervous system isthe most common cause of death due to thrombocyto-penia. When such bleeding occurs, it is often preceded bya history of head trauma.
EvaluationA thorough history and physical examination and judi-cious use of laboratory testing can lead to the appropriatediagnosis in most patients (Fig. 2). Patients should bequestioned about past and current bleeding symptoms,including bruising with little or no trauma, nosebleeds,blood in the urine or stool, gum bleeding, bleeding withsurgical or dental procedures, or excessive menstrualbleeding. Duration and onset of the bleeding symptomsmay help determine whether the cause is acquired orcongenital. If thrombocytopenia is due to an acquiredcause, the onset of symptoms may be linked to a specifictrigger (eg, infection). Congenital thrombocytopeniashould be considered in patients who have a prolongedhistory of asymptomatic abnormal platelet counts or afamily history of thrombocytopenia.
In children who have suspected or known thrombo-cytopenia, the skin, gingivae, and oral cavity should beexamined carefully for evidence of bleeding. In hospital-ized patients, careful examination for bleeding should be
performed at the site of indwelling catheters, drains, andincisions or areas of previous trauma. Table 4 lists the“red flags” in the history and physical examination ofchildren who have thrombocytopenia that should lead toconsideration of diagnoses other than ITP.
Laboratory EvaluationThe laboratory evaluation of thrombocytopenia beginswith a CBC and evaluation of the PBS. Although a dyingskill for most general practitioners, the ability to assessthe PBS accurately is invaluable in the evaluation ofchildren who have thrombocytopenia or other hemato-logic abnormalities. Consultation with a hematopatholo-gist or experienced laboratory technologist may be use-ful.
The CBC should be examined closely for the plateletcount and mean platelet volume (MPV) as well as forevidence of any other cytopenias (anemia or leukopenia).A platelet count that does not make sense clinicallyshould be confirmed before undertaking extensive eval-uation to be sure that thrombocytopenia exists and thefinding is not due to artifact or laboratory error. Spuriousthrombocytopenia can be caused by improper collectionor inadequate anticoagulation of the blood sample, re-sulting in platelet clumps that are counted as leukocytesby automated cell counters. Once thrombocytopeniahas been confirmed, an MPV that is significantly higherthan normal suggests one of the macrothrombocyto-penia syndromes. A mildly elevated MPV is consistentwith a destructive cause. A low MPV is typically seen inpatients who have Wiscott-Aldrich syndrome (WAS)gene mutations.
The PBS should be examined to estimate the plateletnumber (1 platelet/high power field�platelet count of�10�103/�L [10�109/L]), determine the plateletmorphology and the presence or absence of plateletclumping, and assess whether there are associated whiteand red blood cell changes. Large platelets suggest eitheran ongoing platelet destructive process leading to theproduction of younger and larger platelets or the pres-ence of a congenital macrothrombocytopenia syndrome.Small platelets in the appropriate clinical setting suggestWAS. The presence of schistocytes suggests a micro-angiopathic process such as DIC, HUS, or TTP. Sphero-cytes suggest autoimmune hemolytic anemia coupledwith immune-mediated thrombocytopenia (Evans syn-drome).
Other tests may be useful in determining the cause ofthe thrombocytopenia but are generally performed basedon suggestive findings from the initial history and phys-ical examination and laboratory testing. A positive direct
hematology thrombocytopenia
Pediatrics in Review Vol.32 No.4 April 2011 143 at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

Coombs test suggests an autoimmune process in a pa-tient who has evidence of hemolysis as well as spherocyteson the PBS. For patients who have persistant or chronicITP, antinuclear antibody, serum immunoglobulins(IgG, IgA, IgM), and antiphospholipid antibodiesshould be considered. Fibrin degradation products andfibrinogen measurements are useful to diagnose intravas-cular coagulation.
If the PBS results are consistent with a microangio-pathic process, additional tests should be considered,including serum lactate dehydrogenase and creatinine toassess for HUS or TTP. HIV testing should be consid-ered because thrombocytopenia may be the initial diseasemanifestation in as many as 10% of patients who haveHIV infection. If clinical suspicion or local prevalence ishigh, tests to identify hepatitis C viral infection or Heli-cobacter pylori infection should also be considered.
Screening tests for inherited disorders associated withthrombocytopenia should be considered in patients whoexperience chronic thrombocytopenia, especially in thepresence of short stature or other congenital anomalies.
Platelet antibodies can be detected by a variety ofassays. Although many of these assays have high sensitiv-ity, they lack specificity and are not indicated or per-formed routinely to confirm the diagnosis of acute ITP inchildren.
A bone marrow examination is not necessary in mostcases of isolated unexplained thrombocytopenia in chil-dren. In general, a bone marrow examination is indicatedwhen clinical signs or symptoms suggest either marrowinfiltration or failure. These findings include pancytope-nia; the presence of blasts on the PBS; and the presenceof systemic symptoms such as fever, fatigue, weight loss,or bone pain. A bone marrow examination also is indi-
Figure 2. Diagnostic algorithm for thrombocytopenia. CAMT�congenital amegakaryocytic thrombocytopenia, DIC�disseminatedintravascular coagulation, HUS�hemolytic-uremic syndrome, TAR�thrombocytopenia with absent radii syndrome, TTP�thromboticthrombocytopenic purpura, WAS�Wiscott-Aldrich syndrome.
hematology thrombocytopenia
144 Pediatrics in Review Vol.32 No.4 April 2011
at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

cated for unexplained, chronic, stable thrombocytope-nia, even when the presumed diagnosis is ITP, or if thesubsequent clinical course is inconsistent with the naturalhistory of ITP because of the development of new clinicalsigns or symptoms or laboratory abnormalities.
TreatmentManagement of thrombocytopenia in an individual pa-tient should be guided by an understanding of its causeand predicted clinical course. Correction of the causemay not be possible (eg, congenital thrombocytopenias)or may not be necessary (eg, mild-to-moderate ITP).The principal management goal in all patients who havethrombocytopenia is to maintain a safe platelet count toprevent significant bleeding, not to achieve a normalplatelet count. What constitutes a safe platelet count in aparticular patient varies, depending on the cause of thethrombocytopenia and consideration of all other aspectsof hemostasis. For patients who have significant bleedingsymptoms, treatment is essential. Asymptomatic or min-imally symptomatic thrombocytopenia may be treated ifthe thrombocytopenia is severe or the perceived risk ofbleeding is great.
Activity RestrictionsWhen moderate-to-severe thrombocytopenia is recog-nized, implementing reasonable precautions to minimizebleeding risk is recommended. These steps includetrauma precautions (eg, avoidance of contact sports anduse of a helmet while cycling) and avoidance of medica-tions that have antiplatelet or anticoagulant activity (in-cluding aspirin-containing preparations, ibuprofen, andother nonsteroidal anti-inflammatory drugs).
Invasive ProceduresA platelet count greater than 50�103/�L (50�109/L)provides safety for most invasive procedures. If the risksof potentially serious bleeding are believed to be severe,a platelet count of greater than 100�103/�L (100�109/L) is often required by surgeons or anesthesiolo-gists. For common minor procedures, such as toothextractions, a platelet count of 30 to 50�103/�L(50�109/L) often is sufficient. (1) For patients whohave lower platelet counts, some measure to increase theplatelet count immediately before the procedure may berequired. A short course of corticosteroids (prednisone2 mg/kg per day for 1 week) or a single dose of immuneglobulin intravenous (IGIV) (1 g/kg) is often sufficientto increase the platelet count acutely for proceduralhemostasis. Platelet transfusions can be used in urgentsituations. Although platelet survival in the circulation of
patients who have destructive thrombocytopenias maynot be normal, platelet transfusion nearly always providesprompt, satisfactory hemostasis, even if only for a shortduration.
Emergency Management of Critical BleedingPatients who have severe thrombocytopenia and criticalbleeding require immediate transfusion of platelets re-gardless of the cause of the thrombocytopenia. ICH isthe most serious consequence of severe thrombocyto-penia. Early diagnostic imaging should be consideredfor patients who have severe thrombocytopenia andneurologic signs or symptoms to identify ICH. Forpatients who have unstable or progressive ICH, emer-gency craniotomy may be necessary. For patients whohave ITP with life-threatening bleeding, in addition toplatelets, adjunctive treatment with high doses of corti-costeroid (intravenous methylprednisolone 30 mg/kg
Table 4. Red Flags Suggesting aDiagnosis Other Than ImmuneThrombocytopenic PurpuraHistory
● Fever● Bone pain● Weight loss● Fatigue● Recent history of infections or vaccinations● Past medical history of diseases associated with
thrombocytopenia (eg, autoimmune disorders,cirrhosis)
● Dietary history suggestive of iron, vitamin B12, orfolate deficiency
● Exposure to medications known to be associatedwith thrombocytopenia
● Travel history to an endemic area for malaria
Physical Examination
● Lymphadenopathy● Splenomegaly● Joint swelling● Short stature● Limb defects, including radial agenesis and thumb
abnormalities● Cataracts● Sensorineural hearing loss● Oral leukoplakia● Dystrophic nails● Eczema in male patient● Frequent infections● Superficial hemangiomas
hematology thrombocytopenia
Pediatrics in Review Vol.32 No.4 April 2011 145 at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

Tab
le5.
Init
ialT
reat
men
tfo
rN
ewly
Diag
nose
dIm
mun
eTh
rom
bocy
tope
nic
Purp
ura
(ITP)
Wit
hSi
gnifi
cant
Blee
ding
orRi
skof
Blee
ding
Trea
tmen
tM
echa
nism
ofAc
tion
Resp
onse
Rate
Toxi
citie
sCo
mm
ents
Cort
icos
tero
ids
Dosi
ngva
ries
from
pred
niso
ne2
mg/
kgpe
rda
yor
ally
for
2to
4w
eeks
tohi
gh-d
ose
met
hylp
redn
isol
one
30m
g/kg
per
day
(max
imum
,1g)
intr
aven
ousl
yfo
r3
to7
days
●Re
duce
san
tibod
ypr
oduc
tion
●Re
duce
sre
ticul
oend
othe
lial
syst
emph
agoc
ytos
isof
antib
ody-
coat
edpl
atel
ets
●Im
prov
esva
scul
arin
tegr
ity●
Impr
oves
plat
elet
prod
uctio
n
Upto
75%
achi
eve
plat
elet
resp
onse
,dep
endi
ngon
dose
;res
pons
eto
ther
apy
usua
llyw
ithin
2to
7da
ys
●Be
havi
oral
chan
ges
●Sl
eep
dist
urba
nce
●In
crea
sed
appe
tite
●W
eigh
tga
in●
Gast
ritis
/gas
troi
ntes
tinal
hem
orrh
age
●Im
mun
osup
pres
sion
●Po
orlin
ear
grow
th
●N
ocu
rativ
ebe
nefit
know
n●
Oft
en,a
drop
inpl
atel
etco
unt
afte
rst
eroi
dsdi
scon
tinue
d●
Repe
ated
cour
ses
may
bene
cess
ary
ifsi
gnifi
cant
blee
ding
sym
ptom
spe
rsis
tor
recu
rIm
mun
eGl
obul
inIn
trav
enou
s(IG
IV)
Sing
ledo
se1
g/kg
Unkn
own
and
likel
ym
ultif
acto
rial.
Theo
ries
incl
ude:
●Re
ticul
oend
othe
lials
yste
mbl
ocka
de/in
hibi
tion
●Au
toan
tibod
yne
utra
lizat
ion
byan
ti-id
ioty
pean
tibod
ies
●In
crea
sed
auto
antib
ody
clea
ranc
edu
eto
com
petit
ive
inhi
bitio
nof
imm
unog
lobu
linFc
rece
ptor
>80
%ac
hiev
epl
atel
etre
spon
se;r
espo
nse
toth
erap
yus
ually
with
in24
hour
s
●N
ause
a/vo
miti
ng●
Seve
rehe
adac
he●
Feve
r●
Chill
s●
Mor
epr
onou
nced
inol
der
patie
nts
●N
ocu
rativ
ebe
nefit
know
n●
30%
have
sign
ifica
ntdr
opin
plat
elet
coun
tin
2to
6w
eeks
●IG
IVap
pear
sto
impr
ove
plat
elet
coun
tsm
ore
relia
bly
than
cort
icos
tero
ids
orno
trea
tmen
t.So
me
expe
rts
belie
veth
atin
patie
nts
who
fail
tosh
owan
ypl
atel
etris
ew
ithIG
IV,t
hedi
agno
sis
ofIT
Psh
ould
bere
cons
ider
edan
dbo
nem
arro
was
sess
men
tst
rong
lyco
nsid
ered
Anti-
Rho(
D)im
mun
egl
obul
inSi
ngle
dose
50to
75�
g/kg
Spec
ific
red
bloo
dce
llan
tibod
ies
coat
red
bloo
dce
lls,w
hich
are
then
take
nup
byth
ere
ticul
oend
othe
lial
syst
emin
plac
eof
antib
ody-
coat
edpl
atel
ets
50%
to77
%ac
hiev
epl
atel
etre
spon
se,d
epen
ding
ondo
se;u
pto
50%
resp
ond
with
in24
hour
s
●H
emol
ysis
(can
bese
vere
)●
Diss
emin
ated
intr
avas
cula
rco
agul
atio
n●
Rena
lfai
lure
(rar
e)●
Hea
dach
e,fe
ver,
chill
sle
ssco
mm
onth
anw
ithIG
IV
●W
ithhi
gher
dose
s,is
com
para
ble
toIG
IV●
Onl
yef
fect
ive
inch
ildre
nw
hoar
eRh
o(D)
-pos
itive
.Sho
uld
only
begi
ven
ifhe
mog
lobi
n>
10g/
dL(1
00g/
L)an
dCo
ombs
-neg
ativ
e.Tr
eate
dpa
tient
sm
ust
bew
atch
edfo
rpr
esen
cean
dse
quel
aeof
sign
ifica
nthe
mol
ysis
.IGI
Vis
gene
rally
pref
erre
d.
hematology thrombocytopenia
146 Pediatrics in Review Vol.32 No.4 April 2011
at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from
Tab
le6.
Trea
tmen
tO
ptio
nsfo
rCh
ildre
nW
hoH
ave
Refr
acto
ryIm
mun
eTh
rom
bocy
tope
nic
Purp
ura
Wit
hSi
gnifi
cant
Blee
ding
Sym
ptom
sTr
eatm
ent
Mec
hani
smof
Actio
nRe
spon
seRa
teTo
xici
ties
Com
men
ts
Sple
nect
omy
Redu
ces
dest
ruct
ion
ofbo
thau
tolo
gous
and
tran
sfus
edpl
atel
ets
Usua
llyra
ises
the
plat
elet
coun
tw
ithin
hour
s;60
%to
70%
achi
eve
ape
rman
ent
rem
issi
on
●In
trao
pera
tive
blee
ding
●Li
fe-t
hrea
teni
ngin
fect
ions
Even
thos
eno
tde
mon
stra
ting
rise
inpl
atel
etco
unt
may
bem
ore
resp
onsi
veto
prev
ious
lyfa
iled
med
ical
ther
apie
s.H
igh-
dose
met
hylp
redn
isol
one
30m
g/kg
per
day
(max
imum
,1g)
for
3da
ysfo
llow
edby
20m
g/kg
per
day
for
4da
ys
●Re
duce
san
tibod
ypr
oduc
tion
●Re
duce
sre
ticul
oend
othe
lial
syst
emph
agoc
ytos
isof
antib
ody-
coat
edpl
atel
ets
●Im
prov
esva
scul
arin
tegr
ity●
Impr
oves
plat
elet
prod
uctio
n
>60
%to
100%
achi
eve
plat
elet
resp
onse
;re
spon
seto
ther
apy
usua
llyw
ithin
2to
7da
ys
●Be
havi
oral
chan
ges
●In
crea
sed
appe
tite
●Ga
strit
is/g
astr
oint
estin
alhe
mor
rhag
e●
Imm
unos
uppr
essi
on●
Poor
linea
rgr
owth
●De
crea
sed
bone
min
eral
izat
ion
Ifre
peat
edco
urse
sne
cess
ary,
alte
rnat
ive
ther
apie
ssh
ould
beco
nsid
ered
.
Sing
leor
com
bina
tion
regi
men
s:cy
clos
porin
A,az
athi
oprin
e,vi
ncris
tine,
cycl
opho
spha
mid
e,da
nazo
l�
pred
niso
ne,i
mm
une
glob
ulin
intr
aven
ous
(IGIV
)
Imm
unos
uppr
essi
onvi
acy
toto
xic
effe
cts
�70
%ac
hiev
epl
atel
etre
spon
se;r
espo
nse
toth
erap
yva
ries
but
gene
rally
days
tom
onth
s
Usua
ladv
erse
effe
cts
ofin
divi
dual
agen
tsEx
perie
nce
inch
ildre
nlim
ited.
Ritu
xim
ab37
5m
g/m
2pe
rdo
sein
trav
enou
sly
wee
kly
for
4w
eeks
Not
com
plet
ely
defin
ed.
Poss
ibly
,sou
rce
ofpa
thog
enic
antib
odie
s(B
lym
phoc
ytes
)re
mov
edby
sele
ctiv
ede
stru
ctio
nof
CD20
-pos
itive
cells
,re
sulti
ngin
decr
ease
dan
tibod
ypr
oduc
tion.
Atle
ast
30%
com
plet
ere
spon
sera
tela
stin
gan
aver
age
of13
mon
ths;
resp
onse
isus
ually
prom
pt(1
to2
wee
ks)
but
may
take
upto
8w
eeks
.Del
aym
aybe
resu
ltof
time
need
edto
clea
rci
rcul
atin
gpa
thog
enic
antib
odie
s.
Usua
llyw
ellt
oler
ated
with
man
agea
ble
infu
sion
-re
late
dad
vers
eef
fect
s.H
ypog
amm
aglo
bulin
emia
isin
freq
uent
.IGI
V40
0m
g/kg
ever
y3
to4
wee
ksm
aybe
adm
inis
tere
dto
mai
ntai
nno
rmal
conc
entr
atio
ns.
Prog
ress
ive
mul
tifoc
alle
ukoe
ncep
halo
path
yha
sbe
enre
port
ed,b
utca
usal
rela
tions
hip
not
confi
rmed
.
Ifth
rom
bocy
tope
nia
recu
rs,
seco
ndco
urse
appe
ars
tobe
safe
and
effe
ctiv
e.
Thro
mbo
poie
tinre
cept
orag
onis
tsRo
mip
lost
im1
to10
�g/
kgsu
bcut
aneo
usly
wee
kly
Eltr
ombo
pag
25to
75m
gor
ally
daily
Rom
iplo
stim
,asu
bcut
aneo
usth
rom
bopo
iesi
s-st
imul
atin
gFc
pept
ide
fusi
onpr
otei
n,an
del
trom
bopa
g,an
oral
lyac
tive,
nonp
eptid
eth
rom
bopo
ietin
rece
ptor
agon
ist,
act
byst
imul
atin
gpl
atel
etpr
oduc
tion.
Ashi
ghas
80%
inad
ult
patie
nts.
Stud
ies
inch
ildre
nex
trem
ely
limite
d.Us
ecu
rren
tlyre
stric
ted
tore
frac
tory
patie
nts
who
have
sign
ifica
ntbl
eedi
ngsy
mpt
oms.
●Th
rom
bosi
s●
Incr
ease
dco
ncen
trat
ions
ofbo
nem
arro
wre
ticul
in
No
effe
cton
unde
rlyin
gpa
thol
ogic
mec
hani
sm,a
ndre
boun
dth
rom
bocy
tope
nia
whe
ndr
ugs
are
stop
ped
isco
mm
on.U
sed
toke
eppl
atel
ets
insa
fera
nge
(50
to20
0�10
3/�
L[5
0to
200�
109/L
]).V
ery
expe
nsiv
e,an
dlo
ng-t
erm
expe
rienc
elim
ited.
hematology thrombocytopenia
Pediatrics in Review Vol.32 No.4 April 2011 147 at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

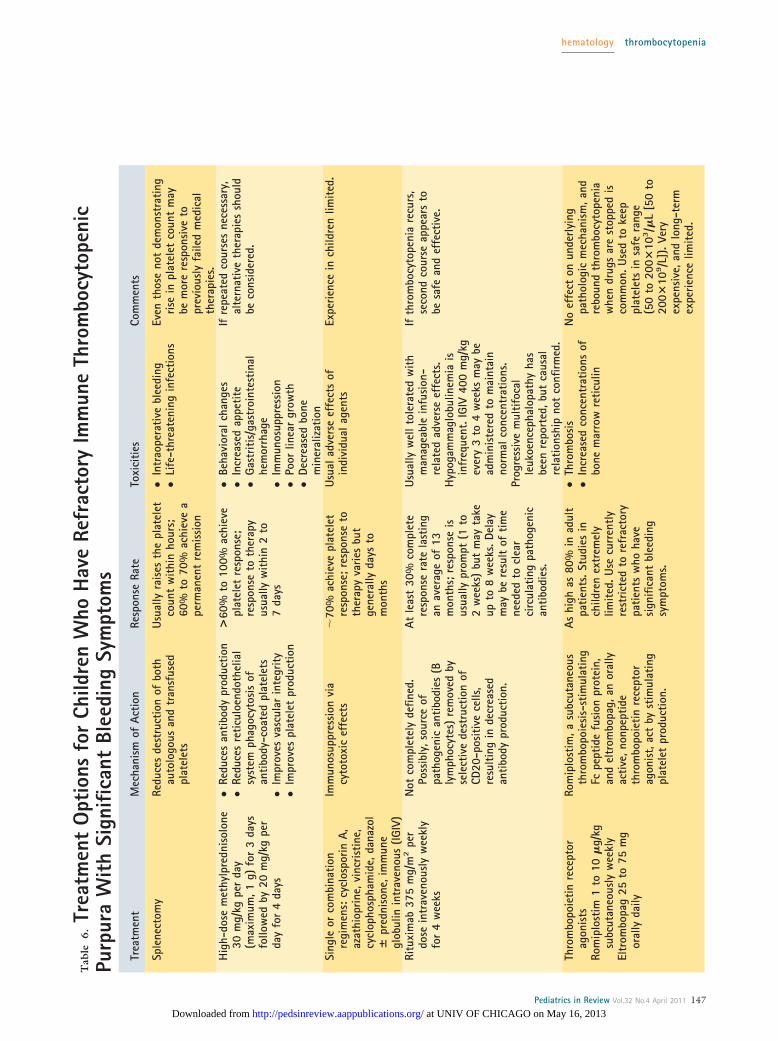
up to 1 g/day for 3 days) and a single dose of IGIV(1 g/kg) is also appropriate. Emergency splenectomymay be considered in cases of refractory ITP accompa-nied by life-threatening hemorrhage.
Thrombocytopenia Associated With OtherCytopenias
Patients who have pancytopenia with systemic symptomsor significant findings on examination should be evalu-ated carefully in a timely manner because they are atincreased risk for a serious disorder that may requireurgent intervention. Consultation with a pediatric hema-tologist should be strongly considered. Treatment of theidentified underlying primary disorder guides subse-quent management. For such patients, maintenance of asafe platelet count may be only a small part of the overalltreatment plan.
Isolated ThrombocytopeniaThe most likely diagnosis in an otherwise healthy childwho has isolated thrombocytopenia is ITP. Most patientsdo not have serious bleeding (including those whoseplatelet counts are �10�103/�L [10�109/L]). ICH isextremely rare, with an incidence of 0.1% to 0.5%. Al-though treatments for childhood ITP may reduce theseverity and duration of the initial thrombocytopenicepisode and presumably the risk of bleeding, they do notappear to affect the eventual recovery rate. Up to twothirds of children who have ITP recover within 6 monthsof presentation with or without treatment. (3)(5)
Most experts agree that pharmacologic interventionis not generally needed for children who have mild-to-moderate thrombocytopenia (platelet counts �30�103/�L] 30�109/L]) because they are unlikely to haveserious bleeding. Exceptions to this policy include chil-dren who have concomitant or preexisting conditionsthat increase their risk for severe bleeding and childrenundergoing procedures likely to include blood loss. (6)
For patients whose platelet counts are less than30�103/�L (30�109/L), treatment recommendationsare based on the presence and severity of associatedbleeding or the risk thereof. Although there is no definedmeans to predict which children who have ITP will sufferfrom an ICH, retinal hemorrhages and extensive muco-sal bleeding or “wet purpura” have been reported toprecede and possibly predict spontaneous ICH. (4)Thus, any individual who has ITP and actual or obviouspotential for significant bleeding requires immediatetreatment, regardless of the platelet count.
When therapy is indicated, the primary treatmentoptions for the newly diagnosed patient are corticoste-
roids, IGIV, and anti-Rho(D) immune globulin (Table5). Several studies have shown that the duration ofsymptomatic thrombocytopenia is shortened by any ofthese three interventions compared with no treatment.All ITP therapies are temporizing interventions intendedfor rapid reversal of a real or perceived risk for significanthemorrhage. They do not need to be continued untilnormal platelet counts are reached. Therapy is targetedto increase the platelet count above a threshold (usually�20�103/�L [20�109/L]) that stops bleeding oreliminates the risk of serious bleeding. (6) Platelet trans-fusions are indicated in patients who have ITP only inthe setting of life-threatening bleeding, such as ICH.Because of accelerated platelet destruction in ITP, plate-let transfusions result in a relatively limited rise in theplatelet count of very short duration (measured in hoursor even minutes) that may be adequate for the immediatehemostasis required in the setting of ICH but otherwiseis of no benefit.
Almost all children who develop ITP are treated in theambulatory setting. Patients who require pharmacologicintervention with IGIV or high-dose intravenous corti-costeroids are usually hospitalized for an average of 1 to3 days. Platelet counts are monitored once or twiceweekly, depending on the clinical situation and severityof the thrombocytopenia. When recovery of plateletcounts is detected, the interval between platelet countassessment may be lengthened. Monitoring should con-tinue until the platelet count has returned to normal andis stable.
Approximately 20% to 30% of children who presentwith ITP eventually develop chronic ITP, defined aspersistent thrombocytopenia beyond 12 months fromthe time of presentation. Patients who have chronic ITPare usually clinically indistinguishable from those whohave acute ITP at presentation. Children younger than10 years of age are more likely to have remissions thanolder patients. Children whose bleeding manifestationslast more than 14 days are substantially more likely todevelop chronic ITP.
All children who have persistent (3 to 12 months) orchronic (�12 months) ITP should have their cases re-viewed and managed by a pediatric hematologist. Indi-viduals who have chronic ITP should undergo evaluationthat includes bone marrow examination to exclude othercauses of thrombocytopenia. In chronic ITP, plateletcounts tend to range between 20 and 75�103/�L(20 and 75�109/L); consequently, many patients re-quire no or only intermittent treatment for episodes ofsignificant bleeding or increased risk of bleeding.
A small percentage of pediatric patients who have ITP
hematology thrombocytopenia
148 Pediatrics in Review Vol.32 No.4 April 2011
at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

and significant hemorrhagic symptoms demonstrate re-sistance to both corticosteroids and IGIV. Managementof such so-called refractory ITP is difficult (Table 6).
Pulse intermittent high-dose corticosteroids and sple-nectomy have been the mainstays of treatment in thesesituations, although not without significant risks. Newertreatment modalities that may be useful include ritux-imab, a chimeric anti-CD20 monoclonal antibody, andthe thrombopoietin receptor agonists romiplostim andeltrombopag, although studies of these agents in chil-dren are limited (9)(10)(11) and not approved by theUnited States Food and Drug Administration for chil-dren younger than 18 years. The recent use of thrombo-poietin receptor agonists reflects a new paradigm in thetreatment of ITP, with the focus not on reducing plateletconsumption through immune modulation or suppres-sion but on increasing platelet production.
References1. Slichter SJ. Relationship between platelet count and bleedingrisk in thrombocytopenic patients. Transfus Med Rev. 2004;18:153–1672. Rodeghiero F, Stasi R, Gemsheimer T, et al. Standardization ofterminology, definitions and outcome criteria in immune thrombo-cytopenic purpura of adults and children: report from an interna-tional working group. Blood. 2009;113:2386–23933. Provan D, Stasi R, Newland A, et al. International consensusreport on the investigation and management of primary immunethrombocytopenia. Blood. 2010;115:168–1864. Medeiros D, Buchanan GR. Current controversies in the man-agement of idiopathic thrombocytopenic purpura during child-hood. Pediatr Clin North Am. 1996;43:757–7725. Kuhne T, Buchanan GR, Zimmerman S, et al. A prospectivecomparative study of 2540 infants and children with newly diag-nosed idiopathic thrombocytopenic purpura (ITP) from the In-tercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605–6086. George JN, Woolf SH, Raskob GE, et al. Idiopathic thrombo-cytopenic purpura: a practice guideline developed by explicitmethods for the American Society of Hematology. Blood. 1996;88:3–407. Bussel JB, Zabusky MR, Berkowitz RL, et al. Fetal alloimmunethrombocytopenia. N Engl J Med. 1997;337:22–268. Payne SD, Resnik R, Moore TR, et al. Maternal characteristicsand risk of severe neonatal thrombocytopenia and intracranial hem-orrhage in pregnancies complicated by autoimmune thrombocyto-penia. Am J Obstet Gynecol. 1997;177:149–1559. Wang J, Wiley JM, Luddy R, et al. Chronic immune thrombo-cytopenic purpura in children: assessment of rituximab treatment.J Pediatr. 2005;146:217–22110. Bennett CM, Rogers ZR, Kinnamon DD, et al. Prospectivephase I/II study of rituximab in childhood and adolescentchronic immune thrombocytopenic purpura. Blood. 2006;107:2639–264211. Bussel JB, Kuter DJ, Pullarkat V, et al. Safety and efficacy oflong-term treatment with romiplostim in thrombocytopenic pa-tients with chronic ITP. Blood. 2009;113:2161–2171
Summary• Thrombocytopenia should be suspected in any child
presenting with a history of easy bruising orbleeding or petechiae, but it also may present as anincidental finding in an asymptomatic individual.
• Thrombocytopenia may be caused by either increaseddestruction or removal of platelets from thecirculation or decreased production of platelets.
• Destructive mechanisms resulting inthrombocytopenia include immune-mediateddestruction, platelet activation and consumption,mechanical platelet destruction, and plateletsequestration or trapping.
• Impaired platelet production may be due to bonemarrow infiltration, suppression, or failure or defectsin megakaryocyte development and differentiation.
• A thorough history and physical examination andjudicious use of laboratory testing can lead to theappropriate diagnosis in most patients who havethrombocytopenia.
• Childhood ITP generally presents with the suddenappearance of bruising, bleeding, or petechiae in anotherwise healthy child.
• The diagnosis of ITP can be made using two criteria:1) isolated thrombocytopenia with otherwise normalblood counts and peripheral blood smear and 2) noclinically apparent associated conditions that maycause thrombocytopenia.
• Further evaluation, including bone marrowassessment, should be considered in patients whohave atypical clinical or laboratory features atpresentation; thrombocytopenia lasting more than 6months; or a subsequent clinical course that isinconsistent with the natural history of ITP,including failure to respond to usually effectivetherapies.
• Management of thrombocytopenia should be guidedby an understanding of its cause and clinical course,with the principal goal in all patients being tomaintain a safe platelet count to prevent significantbleeding.
• For childhood ITP, pharmacologic intervention,including corticosteroids, IGIV, and anti-Rho(D)immune globulin, has been shown to raise theplatelet count more quickly than no therapy and isrecommended for children who have or at risk forsevere or life-threatening bleeding, based on strongevidence.
• ITP in children usually is short-lived, with at leasttwo thirds of patients making a full and sustainedrecovery within 6 months of presentation, with orwithout treatment.
hematology thrombocytopenia
Pediatrics in Review Vol.32 No.4 April 2011 149 at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

PIR QuizQuiz also available online at http://pedsinreview.aappublications.org.
1. A 4-year-old boy is brought to the office with a 3-week history of bruising. He has had no othercomplaints. He has mild bruising but no petechiae and no mucosal bleeding. His physical examinationfindings are otherwise normal. Laboratory results include a white blood cell count of 8.4�103/�L(8.4�109/L), hemoglobin of 13.4 g/dL (134 g/L), and platelet count of 31�103/�L (31�109/L). The mostappropriate management is:
A. High-dose intravenous corticosteroids.B. Intravenous anti-D.C. Intravenous gamma globulin.D. Observation.E. Oral corticosteroids.
2. A 4-year-old girl presents with a 5-week history of bruising but is otherwise well. On physical examination,the only abnormal finding is increased bruising and scattered petechiae. Her platelet count is 29�103/�L(29�109/L), hemoglobin is 9.5 g/dL (95 g/L), and white blood cell count is 2.1�103/�L (2.1�109/L). Themost appropriate next step is to:
A. Administer antibiotics.B. Observe the child.C. Obtain antiplatelet antibodies.D. Obtain Ebstein-Barr virus titers.E. Perform a bone marrow aspiration.
3. An 18-month-old girl has a blood count performed at a health supervision visit. The child is well and herphysical examination findings are normal. The laboratory calls because the platelet count is 1�103/�L(1�109/L). The hemoglobin is 13.5 g/dL (135 g/L), white blood cell count is 7.6�103/�L (7.6�109/L), andabsolute neutrophil count is 3.9x103/�L (3.9�109/L). The most appropriate next step is to:
A. Measure immunoglobulins, antinuclear antibody, and antiphospholipid antibodies.B. Measure prothrombin and partial thromboplastin times.C. Perform a bone marrow aspiration.D. Refer to a pediatric hematologist.E. Repeat the platelet count.
4. A 6-year-old boy who has known chronic immune thrombocytopenic purpura is involved in a motor vehicleaccident and arrives in the emergency department unresponsive. Emergency computed tomography scan ofthe head reveals a large subdural hematoma. The child’s blood type is A-negative. The platelet count is1�103/�L (1�109/L). You administer a platelet transfusion and high doses of intravenousmethylprednisolone and begin intravenous gamma globulin. During the emergency craniotomy, it is difficultto control the bleeding. The most appropriate next therapy is:
A. Anti-D immune globulin.B. Cyclophosphamide.C. Emergency splenectomy.D. Plasmapheresis.E. Rituximab (anti-CD 20 monoclonal antibody).
hematology thrombocytopenia
150 Pediatrics in Review Vol.32 No.4 April 2011
at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

CorrectionsThe caption for Figure 2 in the article entitled “Focus on Diagnosis: Urine Electrolytes” inthe February issue of the journal (Pediatr Rev. 2011;32:65–68) is incorrect. The correctcaption should read, “A graphic illustration of a positive urine anion gap, with the numberof unmeasured anions exceeding the number of unmeasured cations. When this situationoccurs in the context of metabolic acidosis, it is consistent with renal tubular acidosis,indicating an impaired ability to excrete protons in the urine as ammonium.” We regret theerror.
The caption for Figure 1 in the article entitled “Sacral Dimples” in the March issue ofthe journal (Pediatr Rev. 2011;32:109–114) is incorrect. The correct caption should read,“Solitary dimple whose location is greater than 2.5 cm above the anus indicated the needfor further evaluation. . . .” We regret the error.
5. A 7-year-old girl presents with a 3-day history of bruising and an episode of epistaxis lasting 30 minutes.On physical examination, the only abnormalities are scleral icterus, widespread bruising, and cutaneous aswell as mucosal petechiae. Laboratory results include a platelet count of 3�103/�L (3�109/L), hemoglobinof 7.8 g/dL (78 g/L), white blood cell count of 12.9�103/�L (12.9�109/L), absolute neutrophil count of8.8�103/�L (8.8�109/L), and mean corpuscular volume of 86 fL. Urinalysis is negative for red blood cells.The most appropriate next study is:
A. Antiplatelet antibodies.B. Bone marrow aspirate.C. Direct antiglobulin (Coombs) test.D. Flow cytometry on peripheral blood.E. Serum blood urea nitrogen and creatinine assessment.
hematology thrombocytopenia
Pediatrics in Review Vol.32 No.4 April 2011 151 at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from

DOI: 10.1542/pir.32-4-1352011;32;135Pediatrics in Review
Deborah M. ConsoliniThrombocytopenia in Infants and Children
ServicesUpdated Information &
http://pedsinreview.aappublications.org/content/32/4/135including high resolution figures, can be found at:
References
http://pedsinreview.aappublications.org/content/32/4/135#BIBLat: This article cites 11 articles, 5 of which you can access for free
Subspecialty Collections
vascular_-_other_multisystem_disorders_subhttp://pedsinreview.aappublications.org/cgi/collection/collagen_Collagen Vascular & Other Multisystem Disordersogy:musculoskeletal_disorders_subhttp://pedsinreview.aappublications.org/cgi/collection/rheumatolRheumatology/Musculoskeletal Disordersof_blood_neoplasmshttp://pedsinreview.aappublications.org/cgi/collection/disorders_Disorders of Blood/Neoplasmsorders_subhttp://pedsinreview.aappublications.org/cgi/collection/blood_disBlood Disordersy:oncology_subhttp://pedsinreview.aappublications.org/cgi/collection/hematologHematology/Oncologyvascular_other_multisystem_disordershttp://pedsinreview.aappublications.org/cgi/collection/collagen_Collagen Vascular and Other Multisystem Disordersfollowing collection(s): This article, along with others on similar topics, appears in the
Permissions & Licensing
/site/misc/Permissions.xhtmltables) or in its entirety can be found online at: Information about reproducing this article in parts (figures,
Reprints/site/misc/reprints.xhtmlInformation about ordering reprints can be found online:
at UNIV OF CHICAGO on May 16, 2013http://pedsinreview.aappublications.org/Downloaded from





























