Embed Size (px)
Citation preview

Splice Site Mutation in the Human Protein C Gene Associated With Venous Thrombosis: Demonstration of Exon Skipping by Ectopic Transcript Analysis
By Bent Lind, Wouter W. van Solinge, Marianne Schwartz, and Sixtus Thorsen
Heterozygosity for a G + C mutation converting the highly conserved Gln'" (CAG) to His (CAC) was identified at the last nucleotide of exon 7 of the protein C gene in two family members with deep vein thrombosis. As the nucleotide is a part of the 5' splice site of intron G, it was examined how the mutation affected splicingof protein C pre-mRNA. Rele- vant protein C cDNA fragments were amplified with poly- merase chain reaction after reverse transcription of ec- topic mRNA from peripheral blood lymphocytes. Southern blot analysis and nucleotide sequencing of these frag- ments showed a fragment (A) corresponding to correctly spliced mRNA originating from the normal allele and a frag-
HE VITAMIN K-dependent plasma glycoprotein pro- T tein C is the precursor of the anticoagulant serine pro- teinase-activated protein C.'" It is synthesized by hepato- cytes. The cDNA4 and the encoding protein C have been cloned and completely sequenced. The 1 1.2-kb-long gene is located on chromosome no. 2 and made up of 9 exons and 8 introns. Mature protein C (amino acid residues I through 419; molecular weight [M,], ~ 5 6 , 0 0 0 ) is orga- nized in three structural domain^.',^ From the NH,-ter- minus they are the Gla-domain, the epidermal growth fac- tor domain, and the serine proteinase domain. Thrombin complexed with the endothelial cell membrane protein, thrombomodulin, activates protein C by cleaving the Arg'69-Le~'70 peptide bond in the NH,-terminal part of the serine proteinase region.',337 Activated protein C exerts its anticoagulant function by proteolytic inactivation of the blood coagulation cofactors Va and VIIIa in the presence of protein S.
Protein C deficiency is an autosomally inherited dis- Homozygotes or compound heterozygotes for pro-
tein C deficiency that have undetectable levels of protein C activity in plasma suffer massive thrombotic complications during the neonatal period. Heterozygotes may develop ve- nous thromboembolic disease, usually starting after the age of 15 years (prevalence about 1 in 15,000 individuals). De- fects in the protein C gene result in a decreased plasma concentration of normal protein C (type 1 defect, most fre- quent) or in the secretion of an abnormal dysfunctional molecule (type 2 defect). A diversity of mutations responsi- ble for hereditary protein C deficiency have been identified. Most of these are missense or nonsense mutations in the protein coding region of the protein C gene."
Precursor mRNA splicing is the process by which introns are removed and exons joined to form mature mRNA.".l2 Splicing occurs within the spliceosome, a complex assembly of small nuclear (sn)RNAs and associated proteins. Con- served sequences at the exon-intron and intron-exon junc- tions denoted the 5' and 3' splice site consensus sequences, respectively, are essential for efficient and accurate splicing. Quite a number of splice site mutations have been held re- sponsible for genetic diseases.' ' , I 3 These lesions abolish or reduce formation of correctly spliced mRNA or lead to pro- duction of mRNA with skipped exons or other abnormal transcripts.
ment (B) corresponding to a truncated mRNA lacking exon 7, originating from the mutant allele. A third fragment (C) lacking exons 7 and 8 was identified in both affected and unaffected family members, as well as in normal controls. Analysis of human liver protein C mRNA indicated that the ectopic lymphocyte mRNA was qualitatively representa- tive for the tissue-specific mRNA. In conclusion, evidence is provided showing that the mutation abolishes formation of correctly spliced mRNA. This agrees with the observa- tion that the mutation results in a type 1 protein C defi- ciency. 0 1993 by The American Society of Hematology.
The present work describes a missense mutation of the protein C gene associated with deep venous thrombosis (DVT). It affects the 5' splice site of intron G at position - 1. The influence of the mutation on precursor mRNA splicing was studied using polymerase chain reaction (PCR)-ampli- fied cDNA obtained by reverse transcription of protein C mRNA isolated from peripheral lymphocytes. Lympho- cytes produce trace levels of ectopic mRNA for a number of tissue-specific proteins.I4l6 These transcripts have been proven to represent a useful material in investigating the qualitative consequences of gene defect at the mRNA level. l6
MATERIALS AND METHODS
Clinical material. Subjects under study were the proband, her parents, and her only sibling, a sister. Complete anamnesis was taken by one of the investigators (B.L.). Thrombotic events were corroborated by hospital records when possible. The presence of risk factors associated with the thromboembolic events were noted. The study was in accordance with the Helsinki Declaration of 1975 as revised in 1983 and approved by the Regional Ethical Commit- tee.
Blood samples. Blood was obtained between 8 AM and 10 AM from healthy individuals and from patients at least 3 months after they had suffered their last thrombotic attack. Blood for determina- tion of functional plasminogen activator inhibitor- 1 (PAI- 1) was collected in 0. I vol platelet-stabilizing anticoagulant (0.1 1 mol/L citric acid, 15 mmol/L theophylline, 3.7 mmol/L adenosine, 0.198 mmol/L dipyridamole, pH 5.0). Blood for other hemostatic parame-
From the Section for Hemostasis and Thrombosis, Department of Clinical Biochemistry, and the Section of Clinical Genetics, Depart- ment of Pediatrics, Rigshospitalet, Copenhagen, Denmark.
Submitted April 5, 1993; accepted June 22, 1993. Supported by grantsfrom the Clinical Research Foundation, Uni-
versity of Copenhagen, and the Novo Foundation. Address reprint requests to Bent Lind, MD, Section for Hemosta-
sis and Thrombosis, Department of Clinical Biochemistry KB 3-01- 1. Rigshospitalet, Blegdamsvej 9, DK-2100 Copenhagen, Denmark.
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. section I734 solely to indicate this fact. 0 I993 by The American Society of Hematology. 0006-4971/93/8208-0017$3.00/0
Blood, Vol82, No 8 (October 15), 1993: pp 2423-2432 2423
For personal use only.on October 3, 2017. by guest www.bloodjournal.orgFrom

2424 LlND ET AL
ters was collected in 0.1 vol 105.4 mmol/L Na,-citrate. Plasma was separated within 1 hour by centrifugation at 4°C and 3,500gfor 20 minutes (PAI-1) or 10 minutes. It was stored at -80°C until ana- lyzed. Blood for isolation of DNA and RNA was collected in tubes containing K,-EDTA at a final concentration of 3.9 mmol/L and stored at -20°C until use in the case of DNA. Blood for isolation of RNA was processed immediately. Lymphocytes were used as a source of RNA. These cells were isolated from blood using Lym- phoprep (NycoMed AS, Oslo, Norway), according to the manufac- turer's instructions.
This was obtained from a recipient of a liver trans- plant. The specimens were excised after removal of the liver and immediately put on dry ice. The macroscopic appearance of the specimens were normal. The liver transplantation was performed due to Budd-Chian syndrome associated with polycythemia vera.
Reagents for immunoassays. Buffers used were coating buffer (2.46 mmol/L NaH,P04, 7.47 mmol/L Na,HP04, 145 mmol/L NaCl, pH 7.2), wash buffer (2.46 mmol/L NaH,PO,, 7.47 mmol/L Na,HPO,, 500 mmol/L NaCl, 0.1% [vol/vol] Tween-20, pH 7.2), and assay buffer (wash buffer supplemented with 1 mmol/L Na,- EDTA and 2.5 g/L bovine albumin [Sigma, St Louis, MO; cata- logue no. A 45031). Antibodies used were monoclonal mouse IgG (F47A2) and biotinylated monoclonal IgG against human protein C (F25A2) (Novo Nordisk, Bagsvaerd, Denmark). Rabbit lgs against human protein C (A 370) and human protein S (A 384), horseradish peroxidase-conjugated rabbit Igs against human pro- tein S (P 419), and horseradish peroxidase-conjugated swine lgs against rabbit Igs (P 2 17) were from DAKO (Glostrup, Denmark).
Coating of microtiter plates for assays of protein C and protein S. Coating of wells in 96-well microtiter plates (NUNC, Roskilde, Denmark) with antibodies in coating buffer was performed as previ- ously described17 using 100 pL of 10 mg/L monoclonal mouse IgG against human protein C or 100 pL of rabbit Ig against human protein S diluted 1:300.
Assay of protein C. The functional and antigen concentrations of protein C in plasma were analyzed by Staclot protein C and Asserachrom protein C, respectively (both Diagnostica Stago, As- nPres, France). The antigen concentration was also analyzed by an enzyme-linked immunosorbent assay (ELISA) using two different monoclonal antibodies (MoAbs). In brief, the assay was performed at room temperature as follows. Fifty microliters of plasma diluted 200-fold in assay buffer and 50 pL of 2.6 mg/L biotinylated mono- clonal protein C antibody in the same buffer were added to the antibody-coated wells. After 45 minutes of incubation, the wells were washed four times in wash buffer. This was followed by the addition of 100 pL of 1 mg/L horseradish peroxidase avidin D (Vec- tor Laboratories, Burlingame, CA) in wash buffer to each well. After 30 minutes of incubation, the wells were washed again as described above. The final steps for measuring peroxidase activity were the same as described for the assay of urokinase-type plasmin- ogen activator-PAL 1 (u-PA-PAL 1) complex." The assay was Cali- brated against protein C control plasma (Diagnostica Stago; cata- logue no. 0408). The specificity and accuracy of the assay were tested in the following ways. Analysis of plasma immunodepleted of protein C (Diagnostica Stago; catalogue no. 040 1) gave rise to an absorbance value that did not differ from that of the assay buffer. The recovery (mean t SEM [range]) of purified protein C (kindly supplied by Dr Bjorn Dahlback, Malmo, Sweden) was 82% f 5% (55% to 1 12%) when added to 15 plasma samples at a concentration of 5 mg/L. Finally, analyses of 6 plasma samples serially diluted from 1 : I 0 0 to 1 : 1,600 gave concentration estimates that were inde- pendent of the degree of dilution. The intra-assay and inter-assay variations were 3.0% and lo%, respectively, when estimated at nor- mal levels of protein C.
Liver tissue.
Assay ofprotein S. The plasma concentration of total and non- protein-bound protein S were analyzed by ELISA at room tempera- ture. In brief, total protein S was analyzed by the addition of 100-pL plasma samples diluted 800-fold in assay buffer (supplemented with 30 g/L polyethylene glycol [PEG]-6000) to the antibody- coated wells. After 15 minutes of incubation, the wells were washed four times in wash buffer. Then, 100 pL of peroxidase-conjugated polyclonal anti-protein S diluted 5,000-fold in assay buffer was added to each well. After 40 minutes of incubation, the wells were washed and peroxidase activity was measured as described for the assay of protein C. In the assay of nonprotein-bound protein S, the complex between protein S and C4b-binding protein was precipi- tated by PEG-6000, as described by Malm et a1." The supernatant was diluted 500-fold and analyzed as described for total protein S. Assays of both total and nonprotein-bound protein S were cali- brated against total protein S in protein S control plasma (Diagnos- tics Stago; catalogue no. 0808). The specificity and accuracy were tested in the following ways. Analysis of plasma immunodepleted of protein S (Diagnostica Stago; catalogue no. 0448) gave rise to an absorbance value that did not differ from that of the assay buffer. The recovery (mean & SEM [range]) of purified protein S (kindly supplied by Dr Bjorn Dahlback) was 117% + 4% (84% to 132%) when added to 10 plasma samples at a concentration of 30 mg/L. Finally, analyses of 7 plasma samples serially diluted 1:400 to 1 :6,400 gave concentration estimates that were independent of the degree of dilution. The intra-assay and inter-assay variations were 2.7% and 3.8% for total protein Sand 2.8% and 13% for nonprotein- bound protein S, respectively, when estimated at normal levels of protein S.
Other biochemical assays. The plasma concentrations of an- tithrombin 111 and plasminogen were analyzed by Coatest an- tithrombin and Coatest plasminogen (Chromogenix AB, Miilndal, Sweden), respectively, using the ACL 300 Coagulation System (In- strumentation Laboratory, Milan, Italy). PAI- 1 was analyzed as pre- viously d e ~ r i b e d . ' ~ The following clotting assays were performed by using the ACL 300 or ACL 300 Research Coagulation System. Activated partial thromboplastin time (APTT) and coagulation fac- tors I1 + VI1 + X (functional) were analyzed by Automated APTT (Organon Teknika, Durham, NC) and PT-SSI (NycoMed AS), re- spectively. Fibrinogen was analyzed by IL Test PT-Fibrinogen (In- strumentation Laboratory) using citrated plasma for calibration. The concentration of fibrinogen in the calibrator was determined spectrophotometrically at 280 nm, as previously reported.'' The thrombin time was determined by using 5.2 X lo3 NIH units/L bovine thrombin (Leo Pharmaceuticals, Ballerup, Denmark) in Im- idazole buffer (32 mmol/L Imidazole, I8 mmol/L Imidazole-HC1, 132 mmol/L NaCI, 1 g/L bovine albumin, 0.01% [vol/vol] Tween- 80, pH 7.4). The program TT (extended acquisition time) on the ACL 300 or ACL 300 Research Coagulation System was selected.
Agarosegel electrophoresis and Western blotting. Electrophore- sis in 1 % (wt/vol) agarose gel was performed as described by Johans- sonzo using 75 mmol/L barbiturate buffer, pH 8.6, containing 1.9 mmol/L CaCl,. The application volume was 10 pL Na,-citrate sta- bilized plasma. Western blotting was performed at room tempera- ture as follows. After electrophoresis, proteins in the gel were trans- ferred to a nitrocellulose membrane (BioRad, Richmond, CA; catalogue no. 162-0 1 13) as described by Brosstad et a1.*' Remaining sites were blocked by incubation of the nitrocellulose membrane for 1 hour in a solution of nonfat dry milk powder (30 g/L) in distilled water. This was followed by washing four times, each time for 10 minutes in Tris buffer (20 mmol/L Tris, 500 mmol/L NaCI, 0.05% [vol/vol] Tween-20, adjusted with HCI to pH 7.5). The blocked and washed nitrocellulose membrane was then incubated at least 16 hours with rabbit Ig against protein C diluted 2,000-fold in Tris
For personal use only.on October 3, 2017. by guest www.bloodjournal.orgFrom

SPLICE SITE MUTATION IN HUMAN PROTEIN C GENE 2425
buffer. The washing procedure was repeated and the membrane was subsequently incubated for 2 hours with peroxidase-conjugated swine Igs against rabbit Igs diluted 1,000-fold in Tris buffer. After washing five times for 10 minutes with Tris buffer and an additional wash for 10 minutes with 50 mmol/L Na-acetate, 26 mmol/L ace- tate acid, pH 5.0, protein C was visualized by incubation of the nitrocellulose membrane for I O minutes in 250 mg/L 3,3’-diamino- benzidine-tetrahydrocloride (DAB-fizzing-tablets; Kem-En-Tec A/ S, Copenhagen, Denmark), 0.03% (vol/vol) H202 in distilled water. All incubations above were performed under continuous shaking.
Isolation of genomic DNA. Genomic DNA was isolated from peripheral blood cells as except that 1 vol of whole blood was diluted with 3.5 vol cold buffer and that the sodium dodecyl sulfate (SDS)-disrupted nuclear pellet was incubated for I6 to 18 hours at 4°C with 40 pg/mL proteinase K (Boehringer Mann- heim, Mannheim, Germany). Deproteinizing, precipitation, and quality control of DNA was performed as described by Miller et al.23
Southern blot analysis of genomic DNA. Ten micrograms of genomic DNA from the proband and the same amount from a nonrelated normal control were digested with Msp I or Taq I (both from Promega, Madison, WI) according to the manufacturer’s in- structions. The digests were electrophoresed as described24 using a 0.7% (wt/vol) HSB agarose gel (Litex A/S, Vallensbzk, Denmark) and an electrophoresis buffer containing 90 mmol/L Tris-borate, 2.5 mmol/L Na2-EDTA, pH 8.3. Transfer of DNA to a Hybond-N nylon membrane (Amersham International, Amersham, UK), hy- bridization with protein C cDNA4 labeled with [(u-~*P]~CTP (Amersham International) by nick translation, and subsequent washing were performed as described.24
PCR ofgenomic DNA. All nine exons, splicejunctions, and the putative promotor region in the protein C gene were amplified by PCR using the DNA Thermal Cycler from Perkin-Elmer Cetus (Norwalk, CT). Primers (Table 1) were derived from the genomic sequence’ and were purchased from the Technical University of Denmark (Lyngby, Denmark). PCR amplification of exons 1,2,3, 7,8, and 9 was performed in 100 pL volumes containing 10 mmol/ L Tris-HC1, pH 8.3, 50 mmol/L KCl, 1.5 mmol/L MgCI,, 0.01% (wt/vol) gelatine, 0.2 mmol/L dNTP (dATP, dCTP, dGTP, dTTP Boehringer Mannheim), I pmol/L of each primer, 0.5 to 1.5 pg genomic DNA, and 2.5 U Taq DNA polymerase (Stratagene, La Jolla, CA). The samples were overlaid with 100 pL mineral oil and subjected to denaturation (94°C for 2 minutes) followed by 30 cy- cles of denaturation (96°C for I minute), annealing (exon 1,67”C; exons 2, 3, and 9, 65°C; exons 7 and 8, 63°C; all for I minute), extension (72°C for 1 minute) and subsequently followed by an elongation period (72°C for 5 minutes). PCR amplification of exons 4, 5, and 6 was performed in 100 pL volumes containing 67 mmol/L Tris-HC1, pH 8.8,6.7 mmol/L MgCl,, 10 mmol/L 2-mer- captoethanol, 6.7 pmol/L Na,-EDTA, 16.6 mmol/L (NH4)2S04, 0.1 vol glycerol, 0.25 mmol/L dNTP (dATP, dCTP, dGTP, and dTTP; Boehringer Mannheim), 0.6 pmol/L ofeach primer, and 0.5 to 1.5 pg genomic DNA. The samples were overlaid with 100 pL mineral oil and denatured at 94°C for 8 minutes. After the addition of 2.5 U AmpliTaq DNA polymerase (Perkin-Elmer Cetus), the samples were subjected to 30 cycles of denaturation (94°C for 1 minute), annealing (exons 4 and 5 , 68°C; exon 6, 57°C; all for 1 minute), and extension (72°C for 2 minutes). Controls without in- put DNA were routinely included to exclude amplification of con- taminating DNA. Five microliters of each of the reaction products were electrophoresed in 3% (wt/vol) NuSieve GTG agarose gels (FMC Bioproducts, Rockland, ME) using 40 mmol/L Tris-acetate, 1 mmol/L Na2-EDTA, pH 8.1, as electrophoresis buffer and marker V (Boehringer Mannheim) as size marker.24 DNA was stained by ethidium bromide as described.24
Table 1. Oligonucleotide Primers for PCR Amplification and Nucleotide Sequencing
Exon Name Nucleotide No.‘ Sequence ( 5 - 31
Exon 1 P1,l tt - 1647 to - 1628 CTTGGTAGGCAGAGGTGGGC P2,lt
Exon2 P1.2t P2,2t*
Exon3 P1,3t+ P2,3t
Exon P1.45tt 4 and 5 P1’,45t
P2.45t
P2.6tS
P2,7t*
P2,8t*
P2,9t* Ps65 Pa9+§ Ps6i911 Pa89 Pa9i*§ Pagsllll Pagull
Exon6 P1.6t
Exon7 P1,7t
Exon8 P1.8t
Exon 9 P1.9t
-1 163 to -1 183
198 t o 178 1156 to 1176 163710 1617 2921 to 2940 2937 to 2957 3289 to 3270 3160to3179 3555 to 3536 60 1 4 to 6034 6349 to 6029 7046 to 7067 7384 to 7363 8320 to 8339 9072 to 9052 3386 to 3405 8886 to 8867 3414 to 3433 7256 to 7237 8678 to 8659 8434 to 841 5 8487 to 8468
-105 to -85 CAAGTGGGGTGGGGAGAATGG GCCCTTTCATTCCGCTTCCAC CATTCCCATCTGCCCCTCACC GGGAGGAAAAGAGCAGAGGAC CATCCTAATCGCTCCACTCAG ACACCGGCTGCAGGAGCCTG CCTGACGCTGCCCGCTCTCTC TGCTGGTGCCGCGCCCCCAA ATCGGCAGCTTCAGCTGCGA CTCCCTAGAAACCCTCCTGA GAACCCTGCACTGTGGCAAAG TCTGCACCCTGAGCATAGCTG GGAACCCAGGAAAGTGCATATG CTTCTGTGGAGCTCAAGAAGCC CTCAGGAAAGTGCCACTGGG GATGGAAGGACAGAACAGCAG CTGGCGGCGCTGTAGCTGTG CGTAGTTGTGAAGGAGCCCA TACAAGCTGGGGGACGACCT CAAGCCTGACAAGGAGCTTC GATGAAGTTGAGGACGAAGG ATGTCCAGGTCCAGCTCCCA TGTCATTGTCGGTGGTGCTC
* Nucleotide numbers as in Foster et aL5 t Primers for amplification of genomic DNA. * Primers for sequencing of amplified genomic DNA. 9 Primers for amplification of cDNA. I1 Primers for sequencing of amplified cDNA. 7 Oligonucleotide used as exon 9-specific probe.
Restriction enzyme site analysis of exon 7. Five microliters of PCR-amplified exon 7 was digested with Mae I1 (Boehringer Mann- heim) according to the manufacturer’s instructions and analyzed by electrophoresis in a 3% (wt/vol) NuSieve GTG agarose gel as de- scribed for PCR-amplified genomic DNA.
Isolation of total RNA from lymphocytes and liver tissue. Total cellular RNA was isolated according to Chirgwin et al,” with minor modifications. Briefly, the cells were homogenized in 5 mol/L guanidinium thiocyanate, and total cellular RNA was pelleted through a cushion of 5.7 mol/L CsCl by ultracentrifugation. The RNA pellet was resuspended in 20 mmol/L Tris-HC1, 1 mmol/L Na,-EDTA, 0.2% SDS containing 200 pg/mL autodigested protein- ase K (Boehringer Mannheim) and incubated for I hour at 37°C. After phenol/chloroform extraction, the RNA was ethanol-precipi- tated and redissolved in waterF4
Reverse transcription of total RNA and PCR amplification ofpro- rein C cDNA segments. This was performed by using a GeneAmp RNA PCR kit (Perkin-Elmer Cetus) according to the instructions of the manufacturer. Briefly, I pg of total RNA was reverse tran- scribed using random hexamers as primers. Then, 50 pmol of each of the oligonucleotide primers (Table 1) per 100 pL reaction vol- ume was added. Subsequently, the samples were subjected to dena- turation (95OC for 5 minutes) and 30 cycles of denaturation (94°C for 1 minute), annealing (57°C for 1 minute), and extension (72°C for 1 minute). This was followed by an elongation period (72°C for 7 minutes). Controls without input RNA as well as controls in which the reverse transcription step was omitted were routinely included to exclude amplification of contaminating DNA.
Southern blot analysis of PCR-ampliJied protein C cDNA seg- ments preparedfrom RNA. Twenty microliters of PCR-amplified cDNA segments were separated in a 3% (wt/vol) NuSieve 3: 1 aga- rose gel (FMC BioProducts) using 40 mmol/L Tris-acetate, pH 8.1, I mmol/L Na,-EDTA as electrophoresis buffer.24 Southern blot analysis was performed as described for genomic DNA, using the
For personal use only.on October 3, 2017. by guest www.bloodjournal.orgFrom

2426 LlND ET AL
exon 9-specific oligonucleotide, Pa9s (Table I) , as a probe. The probe was 5’-end-labeled using T4 polynucleotide kinase (Pharma- cia LKB Biotechnology, Uppsala, Sweden) and [y-”P]ATP (Amer- sham International). The sizes of the cDNA fragments on the film were estimated by interpolation on a plot of the log,,, (number of basepairs) of specific markers versus their migration distance. The size marker was prepared by Nco I and Msp I restriction enzyme cleavage of PCR-amplified exon 9 from genomic DNA from a nor- mal subject. Partial digestion with Nco I (Boehringer Mannheim) according to the instruction of the manufacturer results in an un- cleaved fragment (753 bp) and two additional fragments (605 bp and 484 bp, respectively). Complete digestion with Msp I (Boeh- ringer Mannheim) results in one fragment (231 bp).
Second-run PCR ofprotein C cDNA segments. To sequence the PCR-amplified cDNA segments derived from lymphocyte or hepa- tocyte RNA, 2 pL of cDNA PCR product was amplified as de- scribed for PCR-amplified genomic DNA using nested primers (Ta- ble l).
Purification and nucleotide sequencing of PCR-amplified protein C DNA segments. Amplified genomic DNA segments or ampli- fied cDNA segments derived from lymphocyte or hepatocyte RNA were electrophoresed as described for analysis of PCR-amplified genomic DNA products and isolated from the gel using Prep-A- Gene DNA purification kit (BioRad). Single-stranded DNA for nu- cleotide sequencing was prepared by asymmetric PCR using the same reaction conditions as described for PCR of genomic DNA except that in each reaction one of the two primers was in excess (primer concentrations 0.5 pmol/L and 0.0 1 pmol/L, respectively); the template DNA used was 50 ng ofisolated double-stranded DNA and the concentration of each dNTP was decreased to 0.05 mmol/ L. To remove excess of dNTP and to concentrate the single- stranded DNA in the asymmetric PCR product it was filtered through Ultrafree MC 30.000 NMWL filter units (Millipore, Bed- ford, MA). Nucleotide sequencing was performed using Sequenase kit Version 2.0 (US Biochemicals, Cleveland, OH) and relevant oligonucleotides (Table 1).
Numbering system for amino acids and nucleotides. The amino acids and the nucleotides were numbered according to Foster et al.’ Exons and introns were assigned the numbers 1 through 9 and the letters A through H, respectively, according to Plutzky et aL6
RESULTS
Clinical and biochemical characterization. The pro- band is a female born in 197 I . At 17 years of age she had a spontaneous attack of phlebography-verified DVT affecting the veins in the proximal half of the left thigh. The father, born in 1940, has suffered several spontaneous attacks of phlebography-verified DVT in the lower extremities and one attack of scintigraphy-verified pulmonary embolism since the age of 33. On the left side, the deep veins on crus were affected; on the right side, the deep veins on crus and thigh were affected, as well as the superficial veins on crus. Neither the proband nor the father has any concurrent dis- ease. The mother and the sister of the proband have no history of thromboembolism or any other disease.
In agreement with the clinical data, the plasma concen- tration of protein C antigen was markedly reduced in both the proband and her father when protein C was analyzed by ELISA using two different MoAbs (Table 2). The same re- sult was obtained when the plasma of the father was ana- lyzed by ELISA using polyclonal antibodies (Asserachrom) (data not shown). The functional concentration of protein
C was analyzed in the father. It was reduced to the same extent as the antigen concentration (Table 2), indicating that only normal protein C is synthesized and secreted, but at a reduced rate (type 1 deficiency). This is consistent with the finding that protein C in the plasma of the father mi- grated as a homogeneous band with the same mobility as protein C from normal subjects when analyzed by agarose gel electrophoresis followed by Western blotting (not shown). The antigen and functional concentrations of pro- tein C were within normal range in both the mother and the sister of the proband (Table 2).
The plasmas of the proband and her affected father were analyzed for hemostatic parameters other than protein C that may predict an increased risk of thromboembolism. These parameters were the anticoagulant proteins an- tithrombin I11 and protein S (total and nonprotein-bound), fibrinogen, thrombin time, plasminogen, PAI- I , and APTT. These parameters were all within normal range.
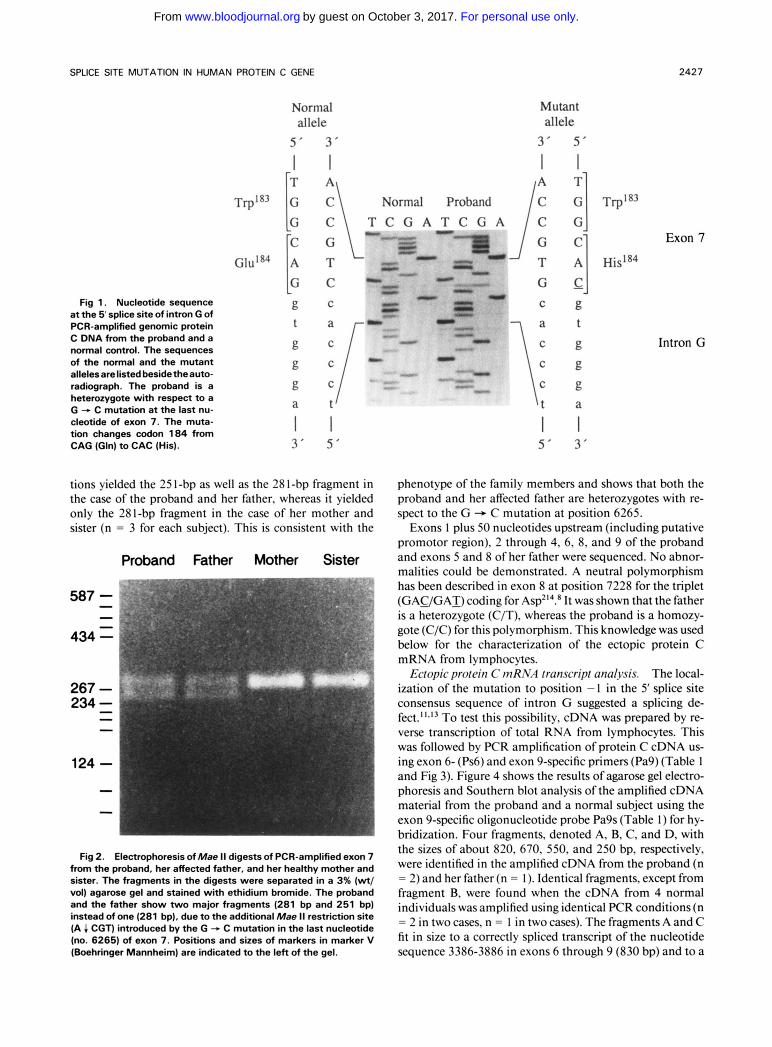
Gene analysis. Southern blot analysis of Msp I - or Tag I-digested genomic DNA probed with protein C cDNA gave the same results with material from the proband and nor- mal subjects (data not shown), indicating that no gross ab- normalities were present in the protein C gene of the pro- band. Sequence analysis of exon 7 plus the adjacent splice junctions from the protein C gene of the proband showed a G + C mutation in one of the alleles in the last nucleotide (no. 6265) of exon 7 (Fig 1). The mutation converts the normal Gln’84codon (CAG) to His (CAC) in the NH,-termi- nal part of the serine proteinase region.
Exon 7 plus adjacent splice junctions (nucleotide no. 6014-6349, 336 bp) from normal subjects contain a single Mae I1 restriction site (A 4 CGT, nucleotides no. 6294- 6297) in intron G, the cleavage of which gives rise to one major 281-bp fragment. The G + C mutation at position 6265 creates an additional Mae I1 restriction site (nucleo- tides no. 6264-6267) at the boundary between exon 7 and intron G, the cleavage of which gives rise to one major 25 1- bp fragment. Figure 2 shows that Mae I1 restriction site analysis of PCR-amplified exon 7 plus adjacent splicejunc-
Table 2. Clinical and Biochemical Parameters
Plasma Concentration (arbitrarv units/LJ
Age at First Protein C Protein C Year of Thrombotic Antigen Functional
Subject BiKh Event (0.63-1.46) (0.70-1.30)
- Proband (9) 197 1 17 0.43 Father 1940 33 0.43 0.48 Mother 1944 Nil 1.01 1.14 Sister 1967 Nil 1.11 1.16
Normal ranges are shown in parentheses (95% reference interval). Protein C antigen was analyzed by ELISA using two different MoAbs for catching and detecting protein C, respectively. Protein C functional was analyzed by Staclot protein C (Diagnostica Stago). Samples from the proband and her father were collected at a stage in which they were without medication with vitamin K antagonist. The concentration of co- agulation factor II + VI1 + X functional was within the normal range in all samples
For personal use only.on October 3, 2017. by guest www.bloodjournal.orgFrom
SPLICE SITE MUTATION IN HUMAN PROTEIN C GENE
Trp183
G1uIg4
Fig 1. Nucleotide sequence at the 5 splice site of intron G of PCR-amplified genomic protein C DNA from the proband and a normal control. The sequences of the normal and the mutant allelesarelisted beside theauto- radiograph. The proband is a heterozygote with respect to a G - C mutation at the last nu- cleotide of exon 7. The muta- tion changes codon 184 from CAG (Gln) to CAC (His).
Normal allele
3' 3'
I 1
12 Normal Proband T C G A T C G A C
T L- C ' C
2428 LlND ET AL
Normal protein C cDNA
e : Ps6 Pa9 : 4 F w n t A (830 bp) - Y
cDNA lacking exon 7 (patient specific)
1 6
cDNA lacking exon 7 and 8 Pa9u 1
: Ps61’
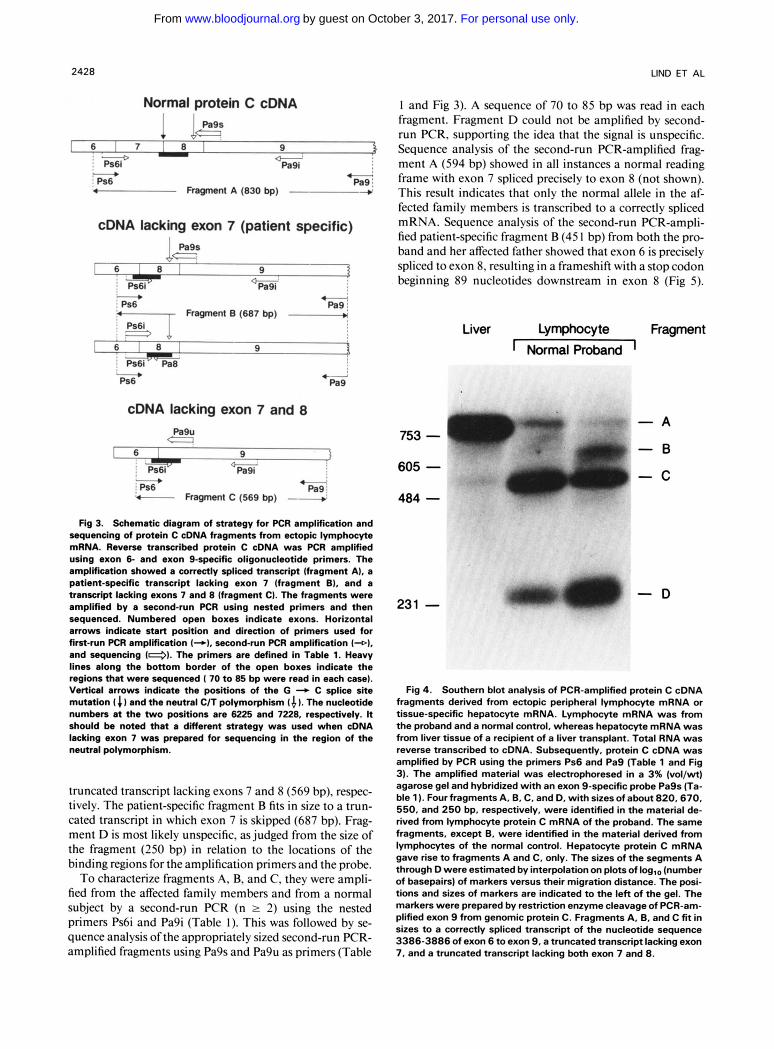
Fig 3. Schematic diagram of strategy for PCR amplification and sequendng of protein C cDNA fragments from ectopic lymphocyte mRNA. Reverse transcribed protein C cDNA was PCR amplified using exon 6- and exon 9-specific oligonucleotide primers. The amplification showed a correctly spliced transcript (fragment A), a patient-specific transcript lacking exon 7 (fragment E), and a transcript lacking exons 7 and 8 (fragment C). The fragments were amplified by a second-run PCR using nested primers and then sequenced. Numbered open boxes indicate exons. Horizontal arrows indicate start position and direction of primers used for first-run PCR amplification (+I, second-run PCR amplification ( 4 1 , and sequencing (4). The primers are defined in Table 1. Heavy lines along the bottom border of the open boxes indicate the regions that were sequenced ( 70 to 85 bp were read in each case). Vertical arrows indicate the positions of the G + C splice site mutation I C I and the neutral C/T polymorphism (4 I. The nucleotide numbers at the two positions are 6225 and 7228, respectively. It should be noted that a different strategy was used when cDNA lacking exon 7 was prepared for sequencing in the region of the neutral polymorphism.
truncated transcript lacking exons 7 and 8 (569 bp), respec- tively. The patient-specific fragment B fits in size to a trun- cated transcript in which exon 7 is skipped (687 bp). Frag- ment D is most likely unspecific, as judged from the size of the fragment (250 bp) in relation to the locations of the binding regions for the amplification primers and the probe.
To characterize fragments A, B, and C, they were ampli- fied from the affected family members and from a normal subject by a second-run PCR (n 2 2) using the nested primers Ps6i and Pa9i (Table I). This was followed by se- quence analysis of the appropriately sized second-run PCR- amplified fragments using Pa9s and Pa9u as primers (Table
I and Fig 3). A sequence of 70 to 85 bp was read in each fragment. Fragment D could not be amplified by second- run PCR, supporting the idea that the signal is unspecific. Sequence analysis of the second-run PCR-amplified frag- ment A (594 bp) showed in all instances a normal reading frame with exon 7 spliced precisely to exon 8 (not shown). This result indicates that only the normal allele in the af- fected family members is transcribed to a correctly spliced mRNA. Sequence analysis of the second-run PCR-ampli- fied patient-specific fragment B (45 I bp) from both the pro- band and her affected father showed that exon 6 is precisely spliced to exon 8, resulting in a frameshift with a stop codon beginning 89 nucleotides downstream in exon 8 (Fig 5).
753 - 605 - 484 -
231 -
Liver Lymphocyte Fragment 1 Normal Proband I
- A
- 8
- c
- D
Fig 4. Southem blot analysis of PCR-amplified protein C cDNA fragments derived from ectopic peripheral lymphocyte mRNA or tissue-specific hepatocyte mRNA. Lymphocyte mRNA was from the proband and a normal control, whereas hepatocyte mRNA was from liver tissue of a recipient of a liver transplant. Total RNA was reverse transcribed to cDNA. Subsequently, protein C cDNA was amplified by PCR using the primers Ps6 and Pa9 (Table 1 and Fig 3). The amplified material was electrophoresed in a 3% (vol/wt) agarose gel and hybridized with an exon 9-specific probe Pa9s (Ta- ble l) . Four fragments A, B, C, and D, with sizes of about 820,670, 550, and 250 bp, respectively, were identified in the material de- rived from lymphocyte protein C mRNA of the proband. The same fragments, except B, were identified in the material derived from lymphocytes of the normal control. Hepatocyte protein C mRNA gave rise to fragments A and C, only. The sizes of the segments A through D were estimated by interpolation on plots of log,, (number of basepairs) of markers versus their migration distance. The posi- tions and sizes of markers are indicated to the left of the gel. The markers were prepared by restriction enzyme cleavage of PCR-am- plified exon 9 from genomic protein C. Fragments A, B, and C fit in sizes to a correctly spliced transcript of the nucleotide sequence 3386-3886 of exon 6 to exon 9, a truncated transcript lacking exon 7, and a truncated transcript lacking both exon 7 and 8.
For personal use only.on October 3, 2017. by guest www.bloodjournal.orgFrom

SPLICE SITE MUTATION IN HUMAN PROTEIN C GENE 2429
Sequence analysis of the second-run PCR-amplified frag- ment C (333 bp) from the two affected family members as well as from the normal subject showed that exon 6 is pre- cisely spliced to exon 9 without any shift in the reading frame.
Fragment B is specific for the proband and her father, suggesting that it originates from the mutant allele. To con- firm the allelic origin of fragment B (lacking exon 7), it was assumed that the father is a heterozygote (T/C), whereas the proband is a homozygote (C/C) with respect to a neutral polymorphism at position 7228 in exon 8. Assuming a link- age between the mutation in exon 7 and the neutral poly- morphism in exon 8, it was deduced that position 7228 in exon 8 on the mutant allele from the father would be a C. Fragment B from the father was amplified by a second-run PCR (n = 3) using the nested primers Ps6i and Pa8 (Table I and Fig 3). Sequence analysis of this fragment ( I58 bp) us- ing Ps6i as a primer confirmed that position 7228 in exon 8 of the father is a C and not a T (Fig 3). This finding indicates that the patient-specific truncated fragment B originates from the mutant allele.
Comparison oreDNA t ranscripts ofprof f i n C mRNA.fiom lj~mphocytes and liver tissue. To investigate whether the ectopic protein C mRNA from lymphocytes was representa- tive for the tissue-specific mRNA from hepatocytes, protein C cDNA was generated from RNA isolated from liver tissue removed from a recipient of a liver transplant. The same procedure was followed as described for preparation of pro- tein C PCR products from lymphocyte RNA. Agarose gel electrophoresis and Southern blot analysis of the amplified material showed the presence of two fragments of the same sizes (about 820 and 550, respectively) as fragments A and C derived from ectopic protein C mRNA (Fig 4). The two
Fig 5. Nucleotide sequence at the exon 6-exon 8 boundary of the second-run PCR-amplified cDNA fragment B. The fragment was derived from ec- topic lymphocyte protein C mRNA of the affected father (for strategy see Fig 3, upper diagram of cDNA lacking exon 7). Exon 6 is precisely joined to exon 8, which results in a frameshift with a stop codon 89 nucleotides downstream in exon 8.
fragments were amplified by second-run PCR and se- quenced (n = 2). Identical results were obtained asdescribed for the ectopic transcripts, ie, the large fragment represented the correctly spliced transcript and the small fragment repre- sented the transcript, in which exon 6 was precisely spliced to exon 9 (not shown). It should be noted that the correctly spliced transcript was dominant in the tissue-specific-de- rived material, whereas the transcript lacking exons 7 and 8 was dominant in the ectopic-derived material.
DISCUSSION
This study describes a missense mutation in the protein C gene associated with a type 1 protein C deficiency and DVT. The affected family members are heterozygotes and the in- heritance is compatible with an autosomal dominant trait. The mutation (G + C) is located in the last nucleotide (no. 6265) of exon 7 and results in a change of the codon for Gln'84 (CAG) to His (CAC) in the NH,-terminal part of the serine proteinase region of protein C.
The mutation is most likely the cause of the protein C deficiency associated with a thrombotic tendency. Firstly, Southern blot analysis ofgenomic DNA and sequence analy- sis of all exons plus adjacent splice junctions and putative promotor region of the protein C gene did not show other abnormalities in the proband or the father. Secondly, the mutation (G6265 + C) is localized to a nucleotide sequence (CCC TGG CAG6265 in human protein C) in human and animal serine proteinases that codes for the highly con- served amino acid sequence P r ~ ' ~ ~ - T r p ' ~ ~ - G l n ' ~ ~ (residues are numbered as in human protein C). These serine protein- ases include, apart from human protein C,5 bovine protein C:6 human factor Vll,27 human and bovine factor IX,28*29 human and bovine factor X.'.31 human and bovine
3'
I G Proband /
T c G A / i C C
I 5 '
5'
I C
A Exon 6
"I T Exon 8
------- G ~ 1 ~ 1 3 7
G ~1~~~~
I "I T
3'
For personal use only.on October 3, 2017. by guest www.bloodjournal.orgFrom

2430
human factor XI,” human plasma pre- kallikrein,” human tissue-type plasminogen activator,36 human pla~minogen,~’ human complement component Clr,” and human and canine pre~hymotrypsinogen.’~.~ Thirdly, in a recently published database of mutations in protein C deficiency, the same mutation is listed as a homo- zygote case with plasma protein C activity less than 1%. Fourthly, the mutation is localized to position - 1 of the 5’ splice site consensus sequence of intron G.
The localization of the mutation (G + C) to position - I in the 5’ splice site of the intron G suggests defective pre- mRNA ~plicing.”.’~ The frequency of occurrence of G at this position in 5’ splice sites of vertebrate genes is 77%, whereas the frequency of the mutated C is 870.” To delin- eate how the 5’ splice site mutation affected gene expression, amplified segments of cDNA transcripts of ectopic protein C mRNA from lymphocytes were analyzed. The results showed that correctly spliced protein C mRNA could be demonstrated for the normal allele, but not for the mutant allele. In addition, transcript analysis showed that the mu- tant allele specifically gave rise to a truncated mRNA lack- ing exon 7 (Figs 3 and 6). Collectively, the results of analysis ofectopic mRNA from lymphocytes indicate that the muta- tion abolishes or at least drastically reduces the formation of correctly spliced mRNA.
An essential reaction during splicing is the base pairing of the 5’ splice site consensus sequence (5’-CAGgtaagt-3‘) with the complementary sequence on U I snRNA in the spliceo- some.”*’* A consensus value reflecting the specificity ofthis base pairing reaction can be calculated according to Shapiro and Senapathy.4’ The mutation reduces the consensus value for the 5’ splice site of intron G from 80.3 to 66.8 (values for the 5’splice sites of normal protein Care in the range of 74.8 to 9 1 . 1 ). The 5‘ splice site consensus value for intron F is 91.1. The respective consensus values for the mutated 5’ splice site and the 5’ splice site of intron Fare in good agree- ment with the experimental findings that the 5’ splice site of intron F is used in favor of the mutated 5’ splice site of intron G, resulting in the skipping of exon 7.
The absence of correctly spliced mRNA from the mutant allele is compatible with the observations that the genetic defect reduces the antigen and functional concentrations of protein C to the same level (type 1 deficiency) and that no abnormal protein C can be demonstrated by agarose gel electrophoresis and Western blotting. Skipping of exon 7 in the patient-specific truncated mRNA results in precise splicing of exon 6 to exon 8 and in a frameshift with a stop codon (TGA) beginning 89 nucleotides downstream in exon 8. Translation of this transcript would result in a trun- cated protein that lacks most ofthe serine proteinase region. Skipping of exons 7 and 8 in the truncated mRNA obtained from both patients and normal subjects results in precise in-frame splicing of exon 6 to exon 9. Translation of this transcript would result in a truncated protein lacking 87 internal amino acid residues, including three cysteine resi- dues involved in the formation of two disulfide bridges. If the above-mentioned two transcribed protein C variants are formed, they are most likely retained and proteolytically degraded in the endoplasmic reticulum, as reported for
3’
I Father
T C G A
LlND ET AL
~ 1 ~ 2 1 5
Asp214
5 ’ Fig 6. Nucleotide sequence in the region of a neutral polymor-
phism ( C m at position no. 7228 in exon 8 of the protein C gene. Sequence analysis was performed on the second-run PCR-ampli- fied patient-specific truncated fragment B (lacking skipped exon 7 ) from the affected father. The fragment was derived from ectopic protein C mRNA from lymphocytes (for strategy see Fig 3. lower diagram of cDNA lacking exon 7) . The father is a heterozygote with respect to the neutral polymorphism. The truncated transcript (fragment B) showed a C and not a Ta t the position of the polymor- phism. As the C is present in the mutant allele, the result indicates that the truncated transcript originates from this allele.
other structural abnormal protein^!^.^^ However, it can not be excluded entirely that the truncated protein C variants are secreted but have escaped immunologic detection.
Lymphocytes were used as a source of mRNA to study pre-mRNA splicing due to the lack of access to specimens of liver tissue (the site of protein C synthesis) from the affected family members. Analysis of segments of amplified cDNA transcripts of protein C mRNA from liver tissue (removed from the recipient of a liver transplant) showed the presence of the same protein C mRNA transcripts as in normal lym- phocytes, namely the correctly spliced transcript and a mis- spliced variant lacking exons 7 and 8 (Fig 3). The patient- specific mRNA lacking exon 7 could not be demonstrated in the liver tissue. These results are in agreement with those of Chelly et all6 indicating that ectopic protein C mRNA transcripts can be used to study the qualitative conse- quences of gene defects. The correctly spliced mRNA tran- script was dominant compared with the variant lacking
For personal use only.on October 3, 2017. by guest www.bloodjournal.orgFrom

SPLICE SITE MUTATION IN HUMAN PROTEIN C GENE 243 1
exons 7 and 8 in material derived from liver tissue. The inverse was the case in material derived from normal lym- phocytes (Fig 4). This demonstrates that it is not possible to qualitatively extrapolate the results obtained with lympho- cytes to those obtained with liver tissue. We cannot deter- mine from our results whether the relative amounts of the amplified cDNA fragments reflect the original amounts of the normal and truncated protein C mRNA transcripts in the studied tissues or they reflect different efficiency in the reverse transcription reaction and PCR amplifi~ation!~,~~
Two other G + C mutations at position -1 within a 5’ splice site have been characterized. These are in the human j3-globin gene causing j3’ thala~semia~~ and in the XP group A complementing gene causing group A xerodermia pig- mentosum!’ In vitro studies of the first mutation showed drastic inhibition of the normal splice site and activation of several cryptic splice sites. With respect to the second muta- tion, no normally spliced mRNA from the mutant allele was detectable, but cryptic splice sites were activated. None of the two reported G + C mutations were associated with skipping of an upstream exon, as in the case of this study. Skipping of the upstream exon has been described in G + A and G + T mutations at position - 1 of the 5‘ splice site of ( I ) the j3-hexominidase A gene causing Tay-Sachs ( 2 ) a collagen gene causing Ehlers-Danlos syndrome type VII,49350 (3) the antithrombin I11 gene causing recurrent ve- nous thrombosis,” and (4) purine nucleoside phosphory- lase defi~iency.~~
In summary, a G + C mutation at position -1 of the 5‘ site consensus sequence of intron G in the protein C gene is described. The results indicate that the mutation abolishes or drastically reduces correct splicing of mRNA and leads to formation of a misspliced mRNA lacking exon 7. The gene defect can explain the phenotype of the proband and her affected father, namely a reduced plasma concentration of normal protein C (type 1 defect) associated with DVT.
ACKNOWLEDGMENT
We express our gratitude to Donald Foster for providing us with the protein C cDNA and to Berit Madsen and Kirsten Culmsee for their excellent technical assistance.
REFERENCES 1. Esmon CT: The roles of protein C and thrombomodulin in
the regulation of blood coagulation. J Biol Chem 264:4743, 1989 2. Kiesel W: Human plasma protein C. Isolation, characteriza-
tion, and mechanism of activation by a-thrombin. J Clin Invest 64:761, 1979
3. Stenflo J: The biochemistry of protein C, in Bertina RM (ed): Protein C and Related Proteins. Biochemical and Clinical Aspects. Edinburgh, UK, Churchill Livingstone, 1988, p 2 I
4. Foster D, Davie EW Characterization of a cDNA coding for human protein C. Proc Natl Acad Sci USA 81:4766, 1984
5. Foster DC, Yoshitake S, Davie EW: The nucleotide sequence of the gene for human protein C. Proc Natl Acad Sci USA 82:4673, 1985
6. Plutzky J, Hoskins JA, Long GL, Crabtree G R Evolution and organization of the human protein C gene. Proc Natl Acad Sci USA 83546, 1986
7. Preissner KT: Biological relevance of the protein C system
and laboratory diagnosis of protein C and protein S deficiencies. Clin Sci 78:351, 1990
8. Reitsma PH, Poort SR, Allaart CF, Bri&t E, Bertina RM: The spectrum of genetic defects in a panel of 40 Dutch families with symptomatic protein C deficiency type I: Heterogeneity and founder effects. Blood 78390, 1991
9. Allaart CF, Poort SR, Rosendaal FR, Reitsma PH, Bertina RM, Briet E: Increased risk of venous thrombosis in carriers of hereditary protein C deficiency defect. Lancet 34 1: 134, 1993
10. Reitsma PH, Poort SR, Gandrille S, Long GL, Sala N, Cooper D N Protein C deficiency: A database of mutations. Thromb Haemost 69:77, 1993
11. Padgett RA, Grabowski PJ, Konarska MM, Seiler S, Sharp PA: Splicing of messenger RNA precursors. Annu Rev Biochem 55:1119, 1986
12. Green MR: Biochemical mechanisms of constitutive and reg- ulated pre-mRNA splicing. Annu Rev Cell Biol 7559, 1991
13. Krawczak M, Reiss J, Cooper DN: The mutational spectrum of single base-pair substitutions in mRNA splice junctions of hu- man genes: Causes and consequences. Hum Genet 90:41, 1992
14. Chelly J, Concordet JP, Kaplan JC, Kahn A: Illegitimate transcription: Transcription of any gene in any cell type. Proc Natl Acad Sci USA 86:2617, 1989
15. Sarkar G, Sommer SS: Access to a messenger RNA sequence or its protein product is not limited by tissue or species specificity. Science 244:331, 1989
16. Chelly J, Gilgenkrantz H, Hugnot JP, Hamard G, Lambert M, Rtcan D, Akli S, Cometto M, Kahn A, Kaplan J C Illegitimate transcription. Application to the analysis of truncated transcripts of the dystrophin gene in nonmuscle cultured cells from Duchenne and Becker patients. J Clin Invest 88:1161, 1991
17. Philips M, Juul A-G, Selmer J, Lind B, Thorsen S: A specific immunologic assay for functional plasminogen activator inhibitor I in plasma-Standardized measurements of the inhibitor and re- lated parameters in patients with venous thromboembolic disease. Thromb Haemost 68:486, 1992
18. Malm J, Laurel1 M, Dahlback B: Changes in the plasma lev- els of vitamin K-dependent proteins C and S and of C4b-binding protein during pregnancy and oral contraception. Br J Haematol 68:437, 1988
19. Thorsen S, Miillertz S, Suenson E, Kok P: Sequence of for- mation of molecular forms of plasminogen and plasmin-inhibitor complexes in plasma activated by urokinase or tissue-type plasmin- ogen activator. Biochem J 223: 179, I984
20. Johansson BG: Agarose gel electrophoresis. Scand J Clin Lab Invest 29:7, 1972 (suppl 124)
2 I . Brosstad F, Kjenniksen I, Renning B, Stormorken H: Visual- ization of von Willebrand factor multimers by enzyme-conjugated secondary antibodies. Thromb Haemost 55:276, 1986
22. Kunkel LM, Smith KD, Boyer SH, Borgaonkar DS, Wachtel SS, Miller OJ, Breg WR, Jones HW, Rary J, Rary JM: Analysis of human Y-chromosome-specific reiterated DNA in chromosome variants. Proc Natl Acad Sci USA 74:1245, 1977
23. Miller SA, Dykes DD, Polesky H F A simple salting out pro- cedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215, 1988
24. Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory, 1989
25. Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ: Iso- lation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 185294, 1979
26. Long GL, Belegaje RM, MacGillivray RTA: Cloning and sequencing of liver cDNA coding for bovine protein C. Proc Natl Acad Sci USA 815653, 1984
For personal use only.on October 3, 2017. by guest www.bloodjournal.orgFrom

2432 LlND ET AL
27. OHara PJ, Grant FJ, Haldeman BA, Gray CL, Insley MY, Hagen FS, Murray MJ: Nucleotide sequence of the gene coding for human factor VII, a vitamin K-dependent protein participating in blood.coagulation. Proc Natl Acad Sci USA 84:5 158, 1987
28. Yoshitake S, Schach BG, Foster DC, Davie EW, Kurachi K Nucleotide sequence of the gene for human factor IX (antihemo- philic factor B). Biochemistry 24:3736, 1985
29. Katayama K, Ericsson LH, Enfield DL, Walsh KA, Neurath H, Davie EW, Titani K Comparison of amino acid sequence of bovine coagulation factor IX (Christmas factor) with that of other vitamin K-dependent plasma proteins. Proc Natl Acad Sci USA 76:4990, 1979
30. Leytus SP, Foster DC, Kurachi K, Davie EW: Gene for hu- man factor X: A blood coagulation factor whose gene organization is essentially identical with that of factor IX and protein C. Bio- chemistry 25:5098, 1986
3 1. Fung MR, Campbell RM, MacGillivray RTA: Blood coagu- lation factor X mRNA encodes a single polypeptide chain contain- ing a prepro leader sequence. Nucleic Acids Res 12448 1, 1984
32. Degen SJF, Davie EW: Nucleotide sequence of the gene for human prothrombin. Biochemistry 26:6 165, 1987
33. MacGillivray RTA, Davie EW: Characterization of bovine prothrombin mRNA and its translation product. Biochemistry 23:1626, 1984
34. Asakai R, Davie EW, Chung DW: Organization of the gene for human factor XI. Biochemistry 26:7221, 1987
35. Chung DW, Fujikawa K, McMullen BA, Davie EW: Human plasma prekallikrein, a zymogen to a serine protease that contains four tandem repeats. Biochemistry 25:2410, 1986
36. Degen SJF, Rajput B, Reich E: The human tissue plasmino- gen activator gene. J Biol Chem 261:6972, 1986
37. Petersen TE, Martzen MR, Ichinose A, Davie E W Charac- terization of the gene for human plasminogen, a key proenzyme in the fibrinolytic system. J Biol Chem 265:6104, 1990
38. Journet A, Tosi M: Cloning and sequencing of full-length cDNA encoding the precursor of human complement component Clr. Biochem J 240:783, 1986
39. Tomita N, Izumoto Y, Horii A, Doi S, Yokouchi H, Ogawa M, Mori T, Matsubara K Molecular cloning and nucleotide se- quence of human pancreatic prechymotrypsinogen cDNA. Bio- chem Biophys Res Commun 158569, 1989
40. Pinsky SD, LaForge KS, Luc V, Scheele G: Identification of cDNA clones encoding secretory isoenzyme forms: Sequence deter- mination of canine pancreatic prechymotrypsinogen 2 mRNA. Proc Natl Acad Sci USA 80:7486, 1983
41. Shapiro MB, Senapathy P: RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implica- tions in gene expression. Nucleic Acids Res 15:7155, 1987
42. Klausner RD, Sitia R: Protein degradation in the endoplas- mic reticulum. Cell 62:6 1 I , 1990
43. Lodish H F Transport of secretory and membrane glycopro- teins from the rough endoplasmic reticulum to the Golgi. J Biol Chem 263:2107, 1988
44. Siebert PD, Larrick JW: Competitive PCR. Nature 359557, 1992
45. Diviacco S, Norio P, Zentilin L, Menzo S, Clementi M, Bia- monti G, Riva s, Falaschi A, Giacca M: A novel procedure for quantitative polymerase chain reaction by coamplification of com- petitive templates. Gene I22:3 13, 1992
46. Vidaud M, Gattoni R, Stevenin J, Vidaud D, Amselem S, Chibani J, Rosa J, Goossens M: A 5’ splice-region G C mutation in exon 1 of the human @-globin gene inhibits pre-mRNA splicing: A mechanism for @+-thalassemia. Proc Natl Acad Sci USA 86:1041, 1989
47. Satokata I, Tanaka K, Yuba S , Okada Y: Identification of splicing mutations of the last nucleotides of exons, a nonsense mu- tation, and a missense mutation of the XPAC gene as causes of group A xeroderma pigmentosum. Mutat Res 273:203, 1992
48. Akli S, Chelly J, Mezard C, Candy S , Kahn A, Poenaru L: A “G’ to “A” mutation at position - I of a 5’ splice site in a late infantile form of Tay-Sachs disease. J Biol Chem 265:7324, 1990
49. Weil D, DAlessio M, Ramirez F, de Wet W, Cole WG, Chan D, Bateman J F A base substitution in the exon of a collagen gene causes alternative splicing and generates a structurally abnormal polypeptide in a patient with Ehlers-Danlos syndrome type VII. EMBO J 8:1705, 1989
50. DAlessio M, Ramirez F, Blumberg BD, Wirtz MK, Rao VH, Godfrey MD, Hollister D W Characterization of a COLl AI splicing defect in a case of Ehlers-Danlos syndrome type VII: Fur- ther evidence of molecular homogeneity. Am J Hum Genet 49:400, 1991
5 I . Berg L-P, Grundy CB, Thomas F, Millar DS, Green PJ, Slomski R, Reiss J, Kakkar VV, Cooper DN: De novo splice site mutation in the antithrombin III (AT3) gene causing recurrent ve- nous thrombosis: Demonstration of exon skipping by ectopic tran- script analysis. Genomics 13:1359, 1992
52. Andrews LG, Markert M L Exon skipping in purine nucleo- side phosphorylase mRNA processing leading to severe immunode- ficiency. J Biol Chem 267:7834, 1992
For personal use only.on October 3, 2017. by guest www.bloodjournal.orgFrom

1993 82: 2423-2432
B Lind, WW van Solinge, M Schwartz and S Thorsen transcript analysisvenous thrombosis: demonstration of exon skipping by ectopic Splice site mutation in the human protein C gene associated with
http://www.bloodjournal.org/content/82/8/2423.full.htmlUpdated information and services can be found at:
Articles on similar topics can be found in the following Blood collections
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American
For personal use only.on October 3, 2017. by guest www.bloodjournal.orgFrom





























