Embed Size (px)
Citation preview

The Effects of Helicobacter pylori Infection on Dendritic Cells
by
David Rizzuti
A thesis submitted in conformity with the requirements for the degree of Master of Science
Department of Physiology University of Toronto
© Copyright by David Rizzuti (2012)

ii
The Effects of Helicobacter pylori Infection on Dendritic Cells
David Rizzuti
Master of Science
Department of Physiology
University of Toronto
2012
Abstract
Helicobacter pylori infects the human gastric mucosa and induces a chronic gastritis that does
not eliminate the pathogen. H. pylori infection is the primary risk factor for gastric cancer
development. The host immune system is crucial for both bacterial elimination and tumor
surveillance. The interaction between H. pylori and dendritic cells (DC), key orchestrators of the
host immune response, was examined.
H. pylori activated the Signal Transducer and Activator of Transcription 3 (STAT3) pathway,
assayed using immunoblotting, in bone marrow-derived DCs. This blunted DC maturation,
determined by CD86 and MHC II expression, in an autocrine/paracrine IL-10-dependent
pathway. H. pylori infection stimulated the production of cytokines including IL-10, IL-12p40,
and TNF-α as determined using ELISA and multiplex analysis. DC co-culture with T cells did
not drive regulatory T cell development. These results demonstrate that H. pylori blunts DC
maturation, which may play an important role in promoting bacterial persistence and disease.

iii
Acknowledgments
There are many people that I would like to acknowledge for making this thesis possible.
My supervisor, Dr. Nicola Jones, thank you for your incredible mentorship and support. I could
not have asked for anything more from a supervisor; thank you for believing in me and giving
me the motivation to succeed when I needed it. Thank you for helping me grow, not just as a
scientist, but as a person. You were committed to helping me realize my potential and your
concern for my well-being is what made you truly stand out.
My supervisory committee members, Dr. Dana Philpott and Dr. Anthony Gramolini, thank you
for your feedback and guidance. You provided me with the ideal combination of support and
motivation to succeed in this project.
To my lab members, thank you for making this research such an enjoyable experience. Esther,
thank you for your support and advice, and teaching me the lab skills and techniques I needed to
succeed. Dana, thank you for being there with invaluable advice whenever I needed it.
Christiane, the morning and afternoon coffee breaks were essential and incredible; thank you for
helping me stay motivated. Mika, Ted, Dave, Emma, and Heidi, thanks for being awesome
members of the lab and making it an enjoyable environment. Ilya, thanks for being a great friend
and for all your advice and support in everything, from the experiments to the applications.
Ramzi, thanks for teaching me everything I know about flow cytometry, and for the priceless
advice. You were an excellent model for teaching and I follow your example when showing
others new protocols. Kelsey, thanks for keeping the ice cold. Everyone else in the SickKids Cell
Biology department, thanks for making this a great time.
My family, especially my parents Donna and Luciano, and brother Daniel, thank you for
supporting me throughout this endeavor and helping me through the tough times. To all my
friends, especially Vince, Dustin, Theo, Looch, and Champ, you guys were always there for me.
Last but most importantly, I want to thank the Lord for watching over me through every step of
this journey. Nothing would be possible without Him.

iv
Table of Contents
Acknowledgments .......................................................................................................................... iii
Table of Contents ........................................................................................................................... iv
List of Abbreviations ..................................................................................................................... vi
List of Figures ................................................................................................................................. x
Chapter 1 Introduction .................................................................................................................... 1
1.1 Helicobacter pylori ............................................................................................................. 1
1.1.1 Gastric Mucosal Infection and Colonization .......................................................... 1
1.1.2 Disease Pathogenesis .............................................................................................. 2
1.1.3 Virulence Factors .................................................................................................... 5
1.2 The Immune Response ...................................................................................................... 10
1.2.1 The Dendritic Cell ................................................................................................. 11
1.2.2 T Cell Phenotypes ................................................................................................. 14
1.2.3 Dendritic Cell Responses to H. pylori .................................................................. 17
1.2.4 H. pylori-altered DCs: potential role in disease .................................................... 22
1.2.5 Signal Transducer and Activator of Transcription 3 (STAT3) ............................. 23
1.3 Summary ........................................................................................................................... 26
Chapter 2 Materials and Methods ................................................................................................. 28
2.1 Bone Marrow-Derived Dendritic Cells ............................................................................. 28
2.2 Treatment/Infection ........................................................................................................... 28
2.3 Western Blotting ............................................................................................................... 29
2.4 Densitometric analysis ...................................................................................................... 30
2.5 Flow Cytometry ................................................................................................................ 30
2.6 ELISA Cytokine Analysis ................................................................................................. 30
2.7 Multiplex Bead-based Luminex Assay ............................................................................. 30

v
2.8 OT-II Transgenic Mouse CD4+ Splenocytes ................................................................... 31
2.9 STAT3 KO Mice ............................................................................................................... 31
2.10 DC:T Cell Coincubation .................................................................................................. 31
2.11 Statistical Analysis ........................................................................................................... 32
Chapter 3 Results .......................................................................................................................... 33
3.1 H. pylori infection induces STAT3 phosphorylation in dendritic cells ............................ 33
3.2 H. pylori upregulates expression of DC maturation markers ........................................... 35
3.3 Activated dendritic cells secrete a specific cytokine profile in response to H. pylori
infection ............................................................................................................................ 38
3.4 H. pylori induces more pronounced maturation in STAT3 KO dendritic cells ................ 41
3.5 IL-10 secretion by dendritic cells in response to H. pylori infection activates STAT3
to blunt maturation ............................................................................................................ 44
3.6 H. pylori-infected dendritic cells do not drive regulatory T cell development ................. 47
Chapter 4 Discussion .................................................................................................................... 50
Chapter 5 Future Directions .......................................................................................................... 57
References ..................................................................................................................................... 59

vi
List of Abbreviations
A. lwoffi: Acinetobacter lwoffi
APC: antigen-presenting cells
APCa: allophcocyanin
BMDC: bone marrow-derived dendritic cell
BSA: bovine serum albumin
cag PAI: cag pathogenicity island
CagA: cytotoxin-associated gene A
CD: cluster of differentiation
DAMP: danger-associated molecular pattern
DC: dendritic cell
DNA: deoxyribonucleic acid
E. coli: Escherichia coli
ELISA: enzyme-linked immunosorbent assay
EPIYA: glutamate-proline-isoleucine-tyrosine-alanine
ERK 1: extracellular signal-regulated kinase 1
ERK 2: extracellular signal-regulated kinase 2
FB: flow cytometry buffer
FITC: flourescein isothiocyanate
GEEC: GPI-AP-enriched early endosomal compartments

vii
GERD: gastroesophageal reflux disease
GI: gastrointestinal
GM-CSF: granulocyte macrophage colony-stimulating factor
GPI-AP: glycosylphosphatidylinositol-anchored proteins
H. pylori: Helicobacter pylori
IBD: inflammatory bowel disease
IFN: interferon
IL: interleukin
JAK: Janus kinase
JAM-A: junctional adhesion molecule
KO: knockout
LFA-1: lymphocyte function-associated antigen
LPS: lipopolysaccharide
MALT: mucosal-associated lymphoid tissue
MHC: major histocompatibility complex
MOI: multiplicity of infection
NLR: Nod-like receptor
OVA: ovalbumin peptide
PAMP: pathogen-associated molecular pattern
PBS: phosphate-buffered saline

viii
P-CagA: phosphorylated CagA
PD: programmed death
PE: phycoerythrin
PFA: paraformaldehyde
PRR: pattern recognition receptor
PSTAT3: phosphorylated STAT3
RIPA: radioimmunoprecipitation assay
RPMI: Roswell Park Memorial Institute
SDS: sodium dodecyl sulfate
SH2: Src-Homology 2
STAT3: signal transducer and activator of transcription 3
T4SS: type IV secretion system
TAA: tumor-associated antigen
TBST: tris-buffered saline supplemented with tween
Tc: cytotoxic T cell
TCR: T cell receptor
TGF: transforming growth factor
Th: helper T cell
Th1: type 1 helper T cell
Th17: Th17 helper T cell

ix
Th2: type 2 helper T cell
TLR: Toll-like receptor
TNF: tumor necrosis factor
Treg: regulatory T cell
VacA: vacuolating cytotoxin A
VEGF: vascular endothelial growth factor
WT: wildtype
ZO-1: zonula occludens 1

x
List of Figures
Figure 1.1-1: Gastric cancer model as proposed by Correa. ........................................................... 4
Figure 1.1-2: CagA signaling pathways. ......................................................................................... 6
Figure 1.2-1: The immunological synapse .................................................................................... 13
Figure 1.2-2: T cell differentiation ................................................................................................ 15
Figure 1.2-3: Potential dendritic cell - H. pylori interactions ....................................................... 18
Figure 1.2-4: STAT3 signaling pathway. ..................................................................................... 25
Figure 3.1-1: H. pylori activates the STAT3 pathway in dendritic cells ...................................... 34
Figure 3.2-1: H. pylori induces expression of dendritic cell maturation markers ........................ 36
Figure 3.3-1: Dendritic cells secrete a specific cytokine profile upon infection with H. pylori ... 40
Figure 3.4-1: STAT3 KO DCs express enhanced levels of maturation markers in response to H.
pylori infection .............................................................................................................................. 43
Figure 3.5-1: α-IL-10 antibody inhibits H. pylori-induced STAT3 phosphorylation in BMDCs to
promote enhanced expression of maturation markers .................................................................. 46
Figure 3.6-1: H. pylori infection does not induce dendritic cells to produce CD4+ FoxP3
+ T cells
....................................................................................................................................................... 49
Figure 4-1: Potential model of dendritic cell responses to H. pylori ............................................ 54

1
Chapter 1 Introduction
1.1 Helicobacter pylori
1.1.1 Gastric Mucosal Infection and Colonization
Helicobacter pylori (H. pylori) is a microaerophilic, spiral-shaped, Gram-negative bacterium that
selectively infects the gastric mucosa of almost half of the human population (Cover and Blaser,
2009; Kusters et al., 2006). Infection usually occurs early in childhood and colonization
generally persists for the lifetime of the host. Infected individuals can develop several disease
phenotypes, including simple gastritis, peptic ulcers, and gastric cancer (Wroblewski et al.,
2010). Indeed, H. pylori was recognized as a Type I carcinogen in 1994 and infection with this
pathogen remains the primary risk factor for gastric cancer development (Amieva and El-Omar,
2008; Kusters et al., 2006; Peek and Crabtree, 2006; Uemura et al., 2001).
H. pylori possess numerous adaptations that facilitate successful colonization of the human
gastric epithelium. The generation of cytosolic and cell-surface urease permits transient survival
in the highly acidic gastric lumen (Amieva and El-Omar, 2008). The urease enzyme functions to
convert urea into NH3 and CO2, which buffer the acidic environment of the gastric lumen.
Several polar flagella and a spiral shape facilitate mobility of the bacteria in the highly motile
gastric environment, which is constantly churning and secreting mucus. This mobility, combined
with a chemotaxis receptor (TlpB) that detects extracellular pH, allows H. pylori to orient and
remain close to the epithelial layer where the pH is almost neutral (Amieva and El-Omar, 2008;
Croxen et al., 2006). Several specialized adhesion molecules allow the bacteria to adhere to the
epithelium and further facilitate colonization and persistence. The outer-membrane protein BabA
binds to Lewis B blood group antigens which are expressed on gastric epithelial cells, while the
adhesin SabA binds to sialyl-Lewis-X, a glycoprotein expressed on the epithelial cell surface
(Amieva and El-Omar, 2008; Linden et al., 2002; Montecucco and Rappuoli, 2001). H. pylori
uses these diverse factors in concert to orient, localize, and adhere to the gastric epithelial
surface, which facilitates the translocation of bacterial proteins into epithelial cells and grants the

2
bacteria access to cellular nutrients that are necessary for survival. This creates a remarkable
niche that facilitates bacterial survival and contributes to life-long infection and persistence.
1.1.2 Disease Pathogenesis
Helicobacter pylori infection is associated with three main disease phenotypes. All infected
individuals will develop gastritis (a chronic mild inflammation in the gastric mucosa); 80-90% of
these individuals will remain asymptomatic and will not develop any serious gastrointestinal (GI)
complications as a result of this H. pylori infection. This phenotype has therefore been termed
the “simple gastritis phenotype” and is found in the overwhelming majority of infected
individuals (Amieva and El-Omar, 2008). Approximately ten percent of individuals infected with
H. pylori develop a “duodenal ulcer phenotype” characterized by an antral-predominant gastritis
accompanied by high levels of acid secretion (Cover and Blaser, 2009). Patients with this
phenotype, which is particularly prevalent in Western societies, are predisposed to the
development of duodenal ulcers (Kusters et al., 2006).
Approximately 1-2% of individuals infected with H. pylori will develop cancer, including gastric
adenocarcinoma and mucosal-associated lymphoid tissues (MALT) lymphomas (Wroblewski et
al., 2010). These are associated with the third disease phenotype, known as the “gastric cancer
phenotype”, which is characterized by decreased levels of acid production and a corpus-
predominant gastritis (El-Omar et al., 1997). The H. pylori-mediated chronic gastritis is thought
to be the primary stage in the transformation from a normal gastric mucosa through a series of
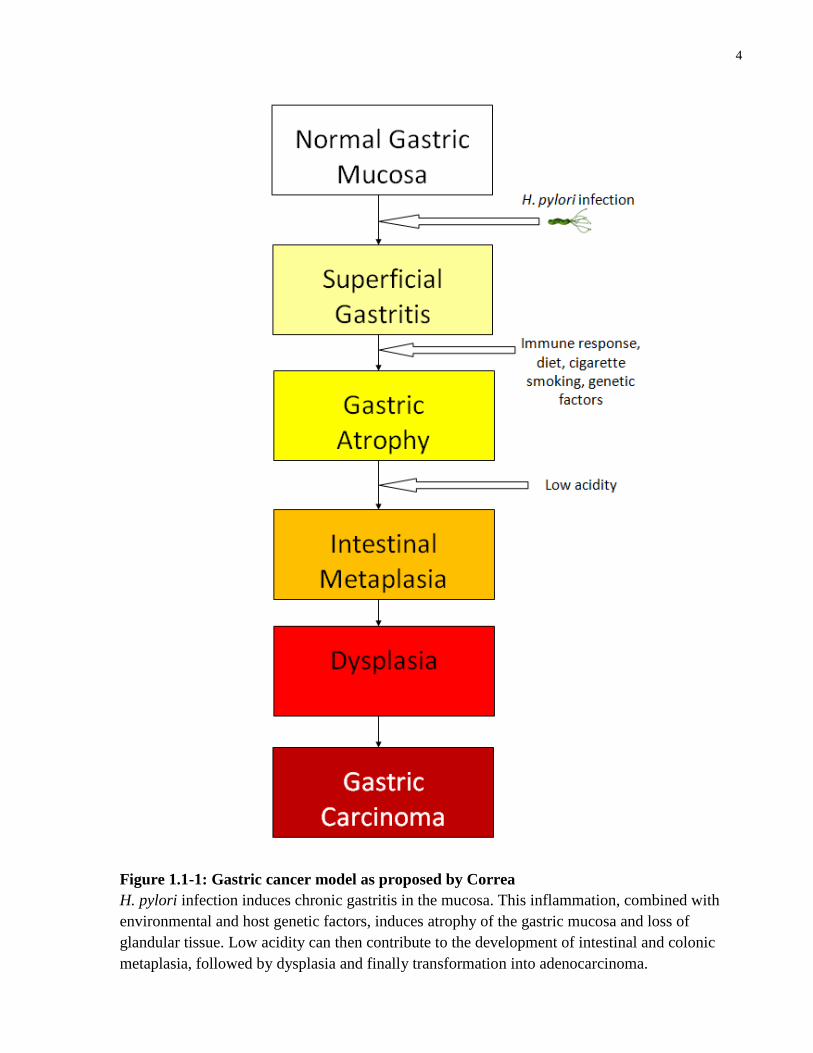
definite histological and functional stages which culminates in gastric adenocarcinoma, as
proposed by Correa (Correa, 1992, 2004) (Figure 1.1-2). Briefly, the transformation begins with
chronic gastritis that proceeds into atrophic gastritis and inflammatory destruction of the mucosa.
Several factors, including H. pylori infection, inflammatory responses, dietary and lifestyle
habits, and genetic susceptibility, are thought to contribute to this progression. Intestinal
metaplasia followed by dysplasia and finally transformation to adenocarcinoma are the later
stages of this transformation (Correa and Houghton, 2007). However the exact mechanisms of H.
pylori-mediated carcinogenesis have yet to be fully elucidated, and bacterial, host, and
environmental factors appear to play complex and often synergistic roles in mediating the cancer
risk associated with H. pylori infection (Amieva and El-Omar, 2008). The goal of this study is
not to directly examine the carcinogenic potential of H. pylori, rather to examine the interaction

3
between H. pylori and the host immune response. Subversion or modulation of the host immune
response may play an important role in bacterial persistence and simultaneously contribute to
disease pathogenesis by inhibiting immune responses to developing cancerous cells.
4
Figure 1.1-1: Gastric cancer model as proposed by Correa
H. pylori infection induces chronic gastritis in the mucosa. This inflammation, combined with
environmental and host genetic factors, induces atrophy of the gastric mucosa and loss of
glandular tissue. Low acidity can then contribute to the development of intestinal and colonic
metaplasia, followed by dysplasia and finally transformation into adenocarcinoma.

5
1.1.3 Virulence Factors
Pathogenesis of H. pylori strains vary considerably largely due to variations in two major
bacterial factors: the cytotoxin-associated gene A (CagA) toxin, an injected protein that is
encoded by the cag pathogenicity island (cag PAI), and the vacuolating-cytotoxin A (VacA),
which is toxin is secreted by the bacteria and induces the formation of large vacuoles in cultured
epithelial cells (Akopyants et al., 1998; Terebiznik et al., 2006). The molecular and cellular
mechanisms of action of these two major H. pylori virulence factors have garnered much
attention and are described below.
1.1.3.1 cag PAI and CagA
The cag PAI is a 40-kb deoxyribonucleic acid (DNA) sequence in the H. pylori genome that
contains approximately 27 to 31 genes, the majority of which (at least 18) code for a bacterial
type IV secretion system (T4SS). This T4SS acts as a “molecular syringe” to transfer the
bacterial effector protein CagA into the host cell cytoplasm (Akopyants et al., 1998; Odenbreit et
al., 2000). CagA is a 120-140kDa protein, encoded by the cagA gene; CagA+ strains of H. pylori
confer a much higher risk for the development of gastric cancer to their hosts (Parsonnet et al.,
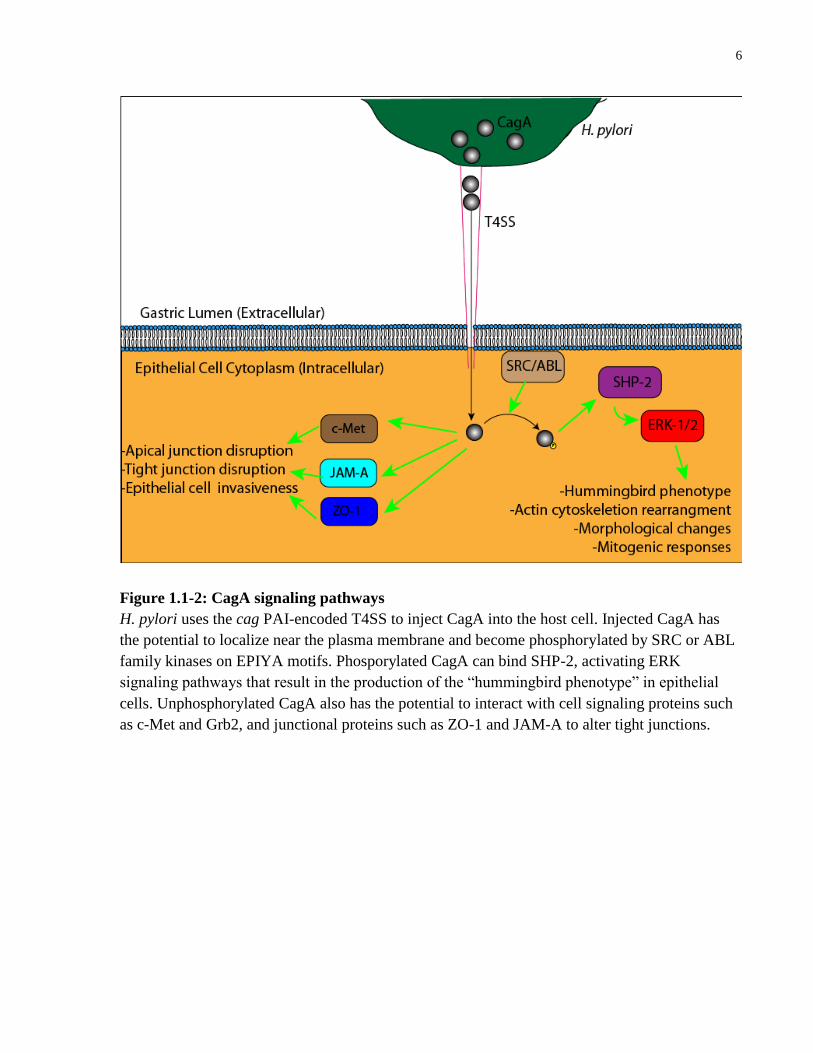
1997; Robinson et al., 2007). Once CagA is injected into the host cell, it localizes near the
plasma membrane, and becomes phosphorylated via host c-Src and Abl family kinases (Amieva
and El-Omar, 2008; Selbach et al., 2002; Tammer et al., 2007) (Figure 1.1-3). The
phosphorylation occurs on the tyrosine residue of glutamate-proline-isoleucine-tyrosine-alanine
(EPIYA) motifs, which are located near the C-terminus of the CagA protein (Naito et al., 2006).
Upon phosphorylation of the EPIYA motifs, the CagA protein will interact with multiple
intracellular effector molecules. Phosphorylated CagA (P-CagA) interacts with the tyrosine
phosphatase SHP-2, inducing activation of the extracellular signal-regulated kinases 1 and 2
(ERK 1/2), which culminates in morphological changes such as cell migration, elongation, and
scattering, collectively known as the “hummingbird phenotype” (Moese et al., 2004; Robinson et
al., 2007; Yamazaki et al., 2003).
6
Figure 1.1-2: CagA signaling pathways
H. pylori uses the cag PAI-encoded T4SS to inject CagA into the host cell. Injected CagA has
the potential to localize near the plasma membrane and become phosphorylated by SRC or ABL
family kinases on EPIYA motifs. Phosporylated CagA can bind SHP-2, activating ERK
signaling pathways that result in the production of the “hummingbird phenotype” in epithelial
cells. Unphosphorylated CagA also has the potential to interact with cell signaling proteins such
as c-Met and Grb2, and junctional proteins such as ZO-1 and JAM-A to alter tight junctions.

7
CagA has also been shown to exert phosphorylation-independent effects, where the N-terminus
targets the protein to epithelial cell junctions (Bagnoli et al., 2005). Unphosphorylated CagA
forms induce profound effects on the apical junctional complex, interacting with multiple
cellular proteins, including c-Met, E-cadherin, and Grb2. This results in mitogenic and
proinflammatory responses, ablation of cell polarity, disruption of adherens junctions, and
activation of matrix metalloproteases, which induces epithelial cell invasiveness (Amieva et al.,
2003; Oliveira et al., 2006; Wroblewski et al., 2010). Unphosporylated CagA can also alter the
epithelial cell tight junction, interacting with the junctional adhesion molecule A (JAM-A) and
zonula occludens 1 (ZO-1) to alter the development of tight junctions. In addition, our lab has
shown that CagA activates the signal transducer and activator of transcription 3 in a
phosphorylation independent manner (Bronte-Tinkew et al., 2009). Collectively, these studies
show that phosphorylation of CagA is not a requirement for induction of epithelial cell changes.
H. pylori CagA+ strains are associated with higher incidences of gastric cancer (Blaser et al.,
1995; Wu et al., 2003), peptic ulcers (Nomura et al., 2002), metaplasia, and inflammation
(Amieva and El-Omar, 2008; Brandt et al., 2005; Wirth et al., 1998) than CagA- strains. As
described above, CagA has the ability to induce profound morphological changes and increase
cell proliferation (Peek et al., 1997), and the potential to interfere with numerous cell functions
and pathways. The specific molecular mode of action of CagA in the colonization of the human
stomach, however, remains to be determined. Studies have found phosphorylated CagA in
gastric biopsies from infected individuals, indicating that it is indeed injected into the cell and
phosphorylated (Yamazaki et al., 2003). It is not currently known which pathways described
above are actually activated in human H. pylori infection; indeed, although CagA can induce
phosphorylation-independent effects in cultured epithelial cells, the presence of
unphosphorylated CagA has not yet been demonstrated within host cells. Additionally, the host
cells and proteins that are the actual targets of CagA insertion have not been delineated, and the
advantages gained by the bacteria from CagA insertion are ambiguous. The mechanisms by
which CagA induces carcinogenesis have not been fully elucidated; while animal models and
cell culture experiments have uncovered potential oncogenic molecular pathways and
epidemiological studies have linked CagA unequivocally with increased cancer risk, many
questions remain unanswered. Studies often produce conflicting data, owing to different
experimental techniques, strains, and models. While transgenic mice expressing CagA develop

8
carcinomas (Ohnishi et al., 2008), this occurred without an inflammatory reaction; in human H.
pylori infection as well as Mongolian gerbil models (Shibata et al., 2006), CagA is associated
with increased inflammation, and this effect is associated with the development of cancer.
Further confounding all of these issues is the fact that the vast majority of patients infected with
CagA+ strains of H. pylori remain cancer-free and asymptomatic. Clearly much remains to be
discovered about CagA, its targets and mechanisms of action, and the factors that contribute to
CagA-associated disease progression in H. pylori -infected patients.
1.1.3.2 VacA
The second major H. pylori virulence factor is the vacuolating cytotoxin A (VacA) – an 88-kDa
secreted toxin that was initially found in vitro to induce the production of large vacuoles in
epithelial cells (Leunk et al., 1988). The presence of VacA has also been associated with an
increased risk for H. pylori-induced carcinogenesis (Robinson et al., 2007) VacA is encoded by
the vacA gene, and genetic diversity in this gene among strains results in variations in phenotype,
cancer risk, and toxic effects (Amieva and El-Omar, 2008; de Sablet et al., 2011).
The secreted VacA protein can be disassociated into p33 N-terminal and p55 C-terminal subunits
upon exposure to either basic or acidic conditions (Cover and Blanke, 2005), but assembly into a
large (6- or 7-subunit) oligomeric complex is necessary for the formation of an anion-selective
membrane channel which is crucial for the biological activity of VacA (Dhar et al., 2003). The
VacA membrane channel increases transcellular permeability and releases anions, urea,
bicarbonate, and other nutrients from the cell cytosol to support bacterial growth (Montecucco
and de Bernard, 2003). The complex is endocytosed into GPI-AP-enriched early endosomal
compartments (GEECs). Here the anion channel conducts chloride ions into the endosome
(Gauthier et al., 2007) resulting in osmotic swelling of the compartment, inducing the large
characteristic vacuoles.
In addition to the induction of vacuoles, VacA can induce multiple effects on different host cell
types. The p33 subunit targets the cellular mitochondria, forms pores in the inner membrane and
changes the inner membrane potential, and induces apoptosis (programmed cell death) (Cover
and Blanke, 2005; Cover et al., 2003; Galmiche et al., 2000). VacA induces the release of
cytochrome c from the mitochondria, resulting in the activation of caspase 3, culminating in the
induction of apoptosis (Galmiche et al., 2000; Willhite and Blanke, 2004; Yamasaki et al., 2006).

9
A novel mechanism of VacA-induced apoptosis has been demonstrated, whereby the toxin
fragments the mitochondrial network via recruitment of dynamin-related protein 1 (Jain et al.,
2011). This activates the Bcl-2-associated X protein, resulting in apoptosis. VacA has also been
recently shown to induce autophagy in gastric epithelial cells (Terebiznik et al., 2009).
Autophagy is an important cellular mechanism that plays a role in the maintenance of cellular
homeostasis as well as innate immunity (Raju and Jones, 2010).
VacA has the potential to modulate the host immune system by interacting with several types of
immune cells, including CD4+ T cells. VacA can inhibit T cell proliferation, activation, and
antigen presentation, thus suppressing T-cell-mediated immunity (Basso et al., 2008;
Boncristiano et al., 2003; Gebert et al., 2003). A recent study implicated the protein kinase C
family in uptake of VacA into T cells (Sewald et al., 2011). VacA has also been shown to bind
CD18 (the β2 integrin subunit) on T cells and cause cellular vacuolization using the lymphocyte
function-associated antigen (LFA-1) (Sewald et al., 2008). VacA also affects immune antigen-
presenting cells (APCs), inhibiting Ii-dependent antigen presentation (Boncristiano et al., 2003;
Molinari et al., 1998). Furthermore it has been shown to inhibit B cell activation and
proliferation, and induce B cell apoptosis (Singh et al., 2006; Torres et al., 2007). VacA can also
exert proinflammatory effects, inducing release of TNF-α and IL-6 release from mast cells
(Cover and Blanke, 2005). Clearly VacA is a multifunctional toxin that can exert profound
effects on the host immune response. VacA induces multiple responses from diverse host cell
types in vitro; although the precise actions of VacA in vivo remain to be elucidated.
1.1.3.3 Lipopolysaccharide (LPS)
Lipopolysaccharide (LPS) is a structural component of the cell wall found in Gram-negative
bacteria such as H. pylori and E. coli that typically induces a strong inflammatory response. LPS
is recognized by the innate immune receptor TLR4 to induce production of proinflammatory
cytokines (Lu et al., 2008). However H. pylori LPS does not induce inflammatory responses in
epithelial cells, even at high concentrations, and may not be recognized by host TLR4 (Backhed
et al., 2003). Furthermore H. pylori LPS has been shown to possess 103 times less endotoxin
activity as compared to LPS from other Gram-negative bacteria such as E. coli (Cunningham et
al., 1996; Perez-Perez et al., 1995). It has also been shown to bind trefoil factor 1 in the gastric
mucosa to potentially promote colonization (Reeves et al., 2008). Therefore the low

10
immunogenicity of H. pylori LPS represents a potential mechanism by which the pathogen is
able to evade the host immune response.
1.2 The Immune Response
Chronic gastritis is a hallmark of H. pylori infection. Despite the presence of this low-level
inflammatory response, the infection persists for the lifetime of the host, which suggests that H.
pylori is able to evade or subvert the host immune response. The causative role that H. pylori
infection plays in the development of gastric cancer further emphasizes the bacteria’s potential
immune-moderating effects, as a lack of tumor immunosurveillance during infection may be
involved(Strioga et al., 2012). Therefore, the interaction between the bacterium and the host
immune response is an area of intense research interest.
The host immune response is composed of several components, including the innate immune
response and the adaptive immune response. The innate response is a rapid, generalized response
to distinct molecular patterns that are conserved among microorganisms (Murphy et al., 2012).
These patterns are known as Pathogen-Associated Molecular Patterns (PAMPs), and innate
immunity uses Pattern Recognition Receptors (PRRs) to recognize these PAMPs (Murphy et al.,
2012). These PRRs include the Toll-like Receptor (TLR) family, a group of receptors that
recognize components such as bacterial lipopolysaccharide or unmethylated DNA in
extracellular and endosomal compartments (Rad et al., 2009). Nod-like receptors (NLRs) are a
more recently-discovered type of PRR that recognize PAMPs in intracellular compartments such
as the cytosol; these include Nod1 and Nod2 which recognize a component of the bacterial cell
wall, peptidoglycan (Fritz et al., 2007; Girardin et al., 2003). These PAMPs are often highly
conserved among microorganisms and are not found in the normal host; therefore these PRRs
provide the host immune system with a quick and generalized way of discriminating “self” vs.
“non-self” (Murphy et al., 2012).
The adaptive immune response is a precise and targeted response to specific antigens. This
adaptive response includes the recruitment, activation, and proliferation of different classes of
immune cells, and thus is more delayed than the innate immune response (Murphy et al., 2012).
Briefly, antigen-presenting cells (APCs) utilize innate PRRs and other receptors to detect foreign
bodies, invaders, and Danger-Associated Molecular Patterns (DAMPs) such as cytokines and
peptides released from apoptotic, necrotic, or tumor cells (Murphy et al., 2012). These APCs,

11
which include immune cells such as macrophages and dendritic cells (DCs), are the sentinels of
the immune system; once they detect something awry in the extracellular environment they
activate the suitable effector cells of the immune system (including T cells and B cells) to deal
with it appropriately (Murphy et al., 2012). Dendritic cells are particularly specialized APCs;
they express the broadest repertoire of PRRs and are capable of inducing powerful effector
immune responses to antigens as well as immunoregulatory and tolerant responses to self-
peptides and commensal organisms (Rad et al., 2009). Thus DCs are key moderators of the
adaptive immune response and represent an ideal target for H. pylori’s immune-modulating
effects. This thesis is focused on the interaction between H. pylori and this decisive immune cell.
1.2.1 The Dendritic Cell
As dendritic cells are the most potent APCs in the host immune system, their primary role is to
initiate adaptive immune responses by detecting, processing, and presenting antigens to T cells in
order to activate these effector cells appropriately (Lenahan and Avigan, 2006; Murphy et al.,
2012). DCs express high levels of the type I transmembrane protein CD11c, making it a suitable
marker for DC detection. Immature dendritic cells are found throughout the body at the sites of
antigen capture such as the gastric lamina propria. DCs use PRRs and processes such as
phagocytosis, macropinocytosis, and membrane ruffling to constantly sample the extracellular
environment, vigilantly searching for antigenic material (Murphy et al., 2012). Upon encounter
with an antigen, DCs internalize the antigen and undergo a maturation process to present
peptides of the antigen to T cells. The DCs enlarge, grow characteristic dendrites, and migrate to
draining lymph nodes, where the population of naïve T cells awaits their antigen presentation.
Within the DC, antigens are internalized into endosomes that fuse with lysosomes to degrade the
contents into antigenic peptides. The peptides are loaded onto Major Histocompatibility
Complex (MHC) molecules and the MHC-peptide complex is transported to the cell surface. The
MHC class I complex can then synapse with a T Cell Receptor (TCR) combined with a CD8
molecule on the surface of a CD8+ Cytotoxic T cell (Tc), while the MHC class II complex
synapses with a TCR combined with a CD4 molecule on the surface of a CD4+ Helper T cell
(Th) (Murphy et al., 2012). This MHC II-TCR interaction is the first signal needed for the DC to
activate the naïve Th cell, and the TCR must recognize the specific antigen loaded onto the MHC
II complex. The second signal is accomplished when the DC costimulatory complex of CD80/86

12
and CD40 contacts the CD28 and CD40L receptors on the T cell. Together these interactions
form the immunological synapse (Figure 1.2-1). In order to achieve these appropriate
interactions, part of the DC maturation process involves the upregulation of CD40, CD80, and
CD86 expression on the cell surface. Thus immature DCs express low levels of cell surface
MHC II and CD40/80/86, and increase expression of these molecules upon maturation. When the
DC presents the MHC II-antigen complex as well as the costimulatory complex to the CD4+ T
cell with the appropriate TCR, it activates the T cell and induces its proliferation, producing an
army of effector cells that will specifically target the antigen that was presented by the DC
(Murphy et al., 2012).
In addition to their role in activating immune responses to pathogenic microorganisms, DCs also
play a very important role in tumor surveillance (Steinman and Banchereau, 2007). DCs
recognize tumor associated antigens and mature in the tumor microenvironment (Lin et al.,
2010). DCs have the ability to induce immune responses towards these tumor associated
antigens, activating T cells to target tumor cells (Lin et al., 2010). Therefore dendritic cells play a
key role in immune surveillance, as they alert the immune system to danger signals from foreign
invaders as well as native cells that have been transformed into malignant problems. DCs
activate appropriate immune responses to deal with the threat accordingly.
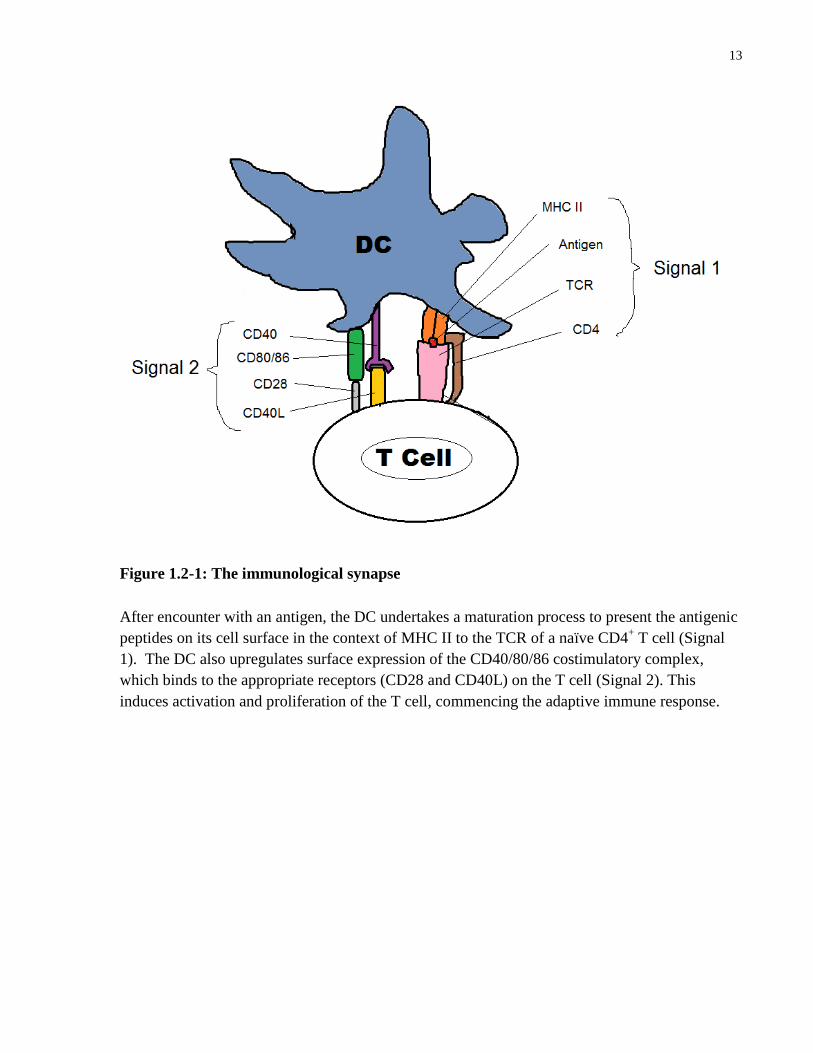
13
Figure 1.2-1: The immunological synapse
After encounter with an antigen, the DC undertakes a maturation process to present the antigenic
peptides on its cell surface in the context of MHC II to the TCR of a naïve CD4+ T cell (Signal
1). The DC also upregulates surface expression of the CD40/80/86 costimulatory complex,
which binds to the appropriate receptors (CD28 and CD40L) on the T cell (Signal 2). This
induces activation and proliferation of the T cell, commencing the adaptive immune response.

14
1.2.2 T Cell Phenotypes
DCs are not only important activators of the adaptive immune response; they also moderate
immune responses by dictating the phenotype of T cell that develops upon activation. As
described above, DCs can present antigen in the context of the MHC class I molecule to CD8+
cytotoxic T cells, priming them to proliferate and recognize virally infected cells (Murphy et al.,
2012). DCs also present antigen in the context of MHC class II molecules to naïve CD4+ helper
T cells, and it is through these CD4+ T cells that the DC’s true immunomodulating potential is
revealed. Different types of cytokines are released by DCs depending upon the type and context
of the antigen that the DC encounters. This cytokine secretion is the third signal that DCs
provide to the T cell to stimulate proliferation, differentiation, and initiation of adaptive T cell
responses. These cytokines will drive the development of different CD4+ T cell phenotypes, each
of which produces a different adaptive immune response (Figure 1.2-2).
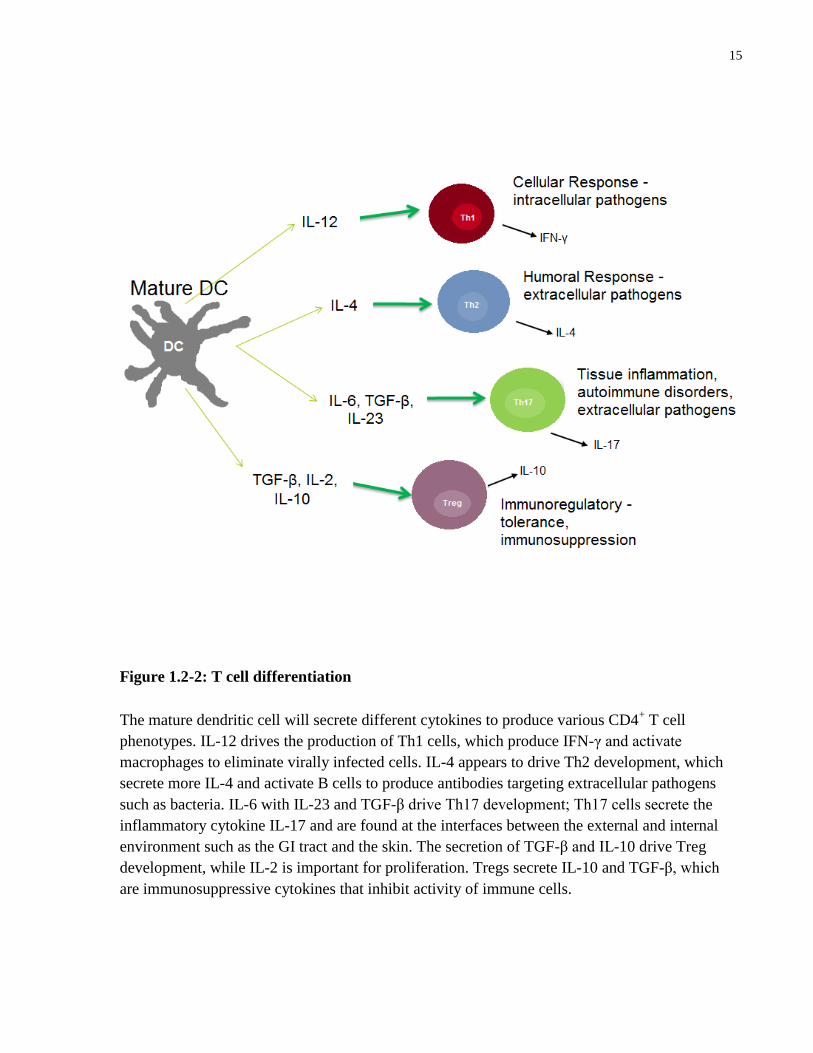
15
Figure 1.2-2: T cell differentiation
The mature dendritic cell will secrete different cytokines to produce various CD4+ T cell
phenotypes. IL-12 drives the production of Th1 cells, which produce IFN-γ and activate
macrophages to eliminate virally infected cells. IL-4 appears to drive Th2 development, which
secrete more IL-4 and activate B cells to produce antibodies targeting extracellular pathogens
such as bacteria. IL-6 with IL-23 and TGF-β drive Th17 development; Th17 cells secrete the
inflammatory cytokine IL-17 and are found at the interfaces between the external and internal
environment such as the GI tract and the skin. The secretion of TGF-β and IL-10 drive Treg
development, while IL-2 is important for proliferation. Tregs secrete IL-10 and TGF-β, which
are immunosuppressive cytokines that inhibit activity of immune cells.

16
DCs can induce different inflammatory immune responses, which are dependent on the type of
cytokines secreted, to deal with pathogenic microorganisms and antigens, or they can induce
tolerogenic responses to prevent immune reactions to commensal bacteria and self-peptides. The
release of the cytokine interleukin-12 (IL-12) from the DC with appropriate antigen and
costimulatory presentation will drive the development of type 1 helper T cells (Th1) and the
cellular immune response (Murphy et al., 2012). Th1 cells secrete the cytokine interferon-gamma
(IFN-γ) and activate macrophages and cytotoxic T cells to kill virally infected cells (Murphy et
al., 2012). The cellular immune response is adept at dealing with intracellular pathogens such as
viruses. The cytokine IL-4 is crucial for the development of the Th2 helper T cell phenotype,
which drives the humoral immune response. Th2 cells activate B cells to secrete antibodies;
these antibodies bind to extracellular pathogens such as bacteria and activate the complement
system to kill the invading microorganism (Murphy et al., 2012).
The release of the cytokines IL-6, IL-23, and transforming growth factor beta (TGF-β) from the
DC stimulates the transcription factor RORγt in the naïve T cells and drives the Th17 helper T
cell phenotype (Dong, 2008; Manel et al., 2008). Th17 cells are proinflammatory and are found
at the interfaces between the external and internal environment, such as the lining of the GI tract
and the skin. They secrete the inflammatory cytokine IL-17, and recruit inflammatory immune
cells such as neutrophils to deal with extracellular pathogens such as bacteria and fungi (Ivanov
et al., 2006). Excessive activity of Th17 cells appears to play a significant role in the
pathogenesis of multiple inflammatory and immune-mediated diseases, including inflammatory
bowel disease (IBD), rheumatoid arthritis, and asthma (Tesmer et al., 2008).
Additionally DCs can drive development of an inducible regulatory T cell (Treg) phenotype to
prevent immune reactivity to auto-antigens and commensal bacteria. Treg development is
regulated by the forkhead transcription factor FoxP3. Tregs express FoxP3 in their nucleus and
CD25 on the cell surface (Chen et al., 2003). The exact cytokine profile driving Treg
development has not been fully elucidated, but the release of TGF-β as well as IL-10 from the
DC appears to be required (Chaudhry et al., 2011; Fu et al., 2004; Lan et al., 2006); while IL-2
(Malek, 2003) and more recently IL-18 (Oertli et al., 2012) have been implicated in Treg
proliferation and skewing. Tregs are immunoregulatory T cells that inhibit the proliferation,
activation, and activity of other immune cells including APCs and effector T cells, effectively
suppressing the immune response by contact-dependent and contact-independent mechanisms

17
(Curotto de Lafaille and Lafaille, 2009). These inducible Tregs produce immunosuppressive
cytokines such as TGF-β and IL-10 to inhibit immune cells (Corthay, 2009; O'Garra et al., 2004).
IL-10 particularly has potent immunoregulatory effects, blocking the maturation of DCs and
inhibiting the antigen-presenting functions of other APCs (Corinti et al., 2001; Enk et al., 1993).
Thus inducible Tregs can effectively suppress the immune response by inhibiting other immune
cells. Tregs play important roles in preventing autoimmune disease and shutting down the
immune response after the offending organism has been successfully eliminated (Mills and
McGuirk, 2004).
This ability to drive the differentiation of multiple distinct T cell phenotypes, each of which
produces a vastly different immune response, makes dendritic cells key orchestrators of the
immune system. DCs have the ability to profoundly modulate the immune response, initiating
both powerful effector responses towards threats (such as pathogens and tumor cells) and
tolerant regulatory responses towards self-peptides and commensal organisms.
1.2.3 Dendritic Cell Responses to H. pylori
H. pylori has the ability to evade or subvert the host immune response, inducing a chronic
gastritis that is insufficient to eliminate the pathogen and potentially inhibiting immune tumor
surveillance to play a causative role in the development of gastric cancer. As dendritic cells are
key modulators of the immune response, they are ideal targets for H. pylori’s immune-
manipulating effects. Dendritic cells are present in the gastric mucosa of H. pylori-infected
individuals, residing in the lamina propria just beneath the epithelial cell layer (Kao et al., 2006).
DCs extend long dendritic projects through epithelial-apical junctions in order to directly take up
bacteria (Necchi et al., 2009) (Figure 1.2-3). Furthermore H. pylori can be found in the lamina
propria as well as inside vacuoles in gastric epithelial cells (Dubois and Boren, 2007; Necchi et
al., 2007; Terebiznik et al., 2006) thus allowing exposure to DCs. Several studies have begun to
assess the effects of H. pylori on dendritic cell maturation, activation, and induction of T cell
responses. However these limited studies to date have produced conflicting results.
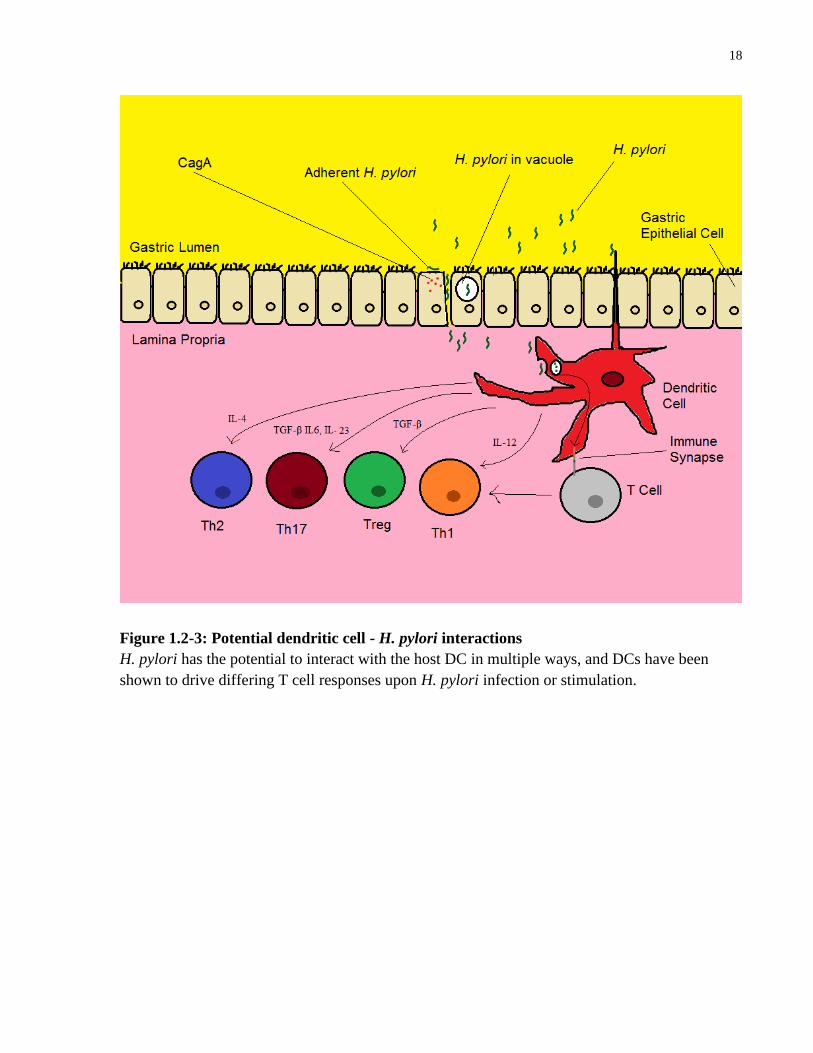
18
Figure 1.2-3: Potential dendritic cell - H. pylori interactions
H. pylori has the potential to interact with the host DC in multiple ways, and DCs have been
shown to drive differing T cell responses upon H. pylori infection or stimulation.

19
A study by Rad et al. found that H. pylori lysates induced maturation of murine bone marrow-
derived dendritic cells (BMDCs) as determined by MHC class II, CD80, and CD86 upregulation
(Rad et al., 2007). The BMDCs secreted IL-12 and IL-10 in response to the H. pylori lysates. A
later study by Rad et al. also implicated specific PRRs, including TLR2, TLR4, and TLR9, in the
DC recognition of H. pylori (Rad et al., 2009).
Kranzer et al. found that H. pylori induced maturation of human DCs as measured by
upregulation of CD80, CD83, CD86, and MHC II surface expression (Kranzer et al., 2004). H.
pylori also induced production of IL-6, IL-8, IL-10, and IL-12 in the DCs (Kranzer et al., 2004).
Importantly, in this study these effects were similar to the effects of stimulating the DCs with
lipopolysaccharide (LPS) from Escherichia coli (E. coli) bacteria, a potent activator of DC
maturation (Kranzer et al., 2004). The H. pylori-stimulated DCs produced less IL-12 than the
LPS-stimulated DCs, although the functional effect on T cell phenotype was not examined. In
further studies, this group showed that the virulence factors cag PAI and VacA were not required
for H. pylori-induced DC maturation, activation, and DC-mediated T cell activation (Kranzer et
al., 2005).
Guiney et al. utilized DCs derived from human peripheral blood monocytes and found that H.
pylori induced secretion of IL-12 with no production of IL-10 (Guiney et al., 2003). The authors
suggest that this would polarize a Th1 phenotype, although no functional assays were conducted.
Interestingly, in this study the H. pylori-stimulated DCs produced less IL-12 than DCs stimulated
with another bacteria, Salmonella enterica (Guiney et al., 2003).
A comprehensive study by Bimczok et al. found increased numbers of DCs in H. pylori-infected
patients as compared to uninfected controls (Bimczok et al., 2010). The DCs from H. pylori-
infected patients also had increased surface expression of the MHC II component HLA-DR, as
well as the maturation markers CD83 and CD86 (Bimczok et al., 2010). This study also
examined the effect of H. pylori infection on gastric DCs isolated from uninfected control
subjects, as well as peripheral monocyte-derived DCs. In this study H. pylori induced maturation
of the gastric DCs and the monocyte-derived DCs, as assessed by upregulation of CD80, CD83,
CD86, and CD11c cell surface expression (Bimczok et al., 2010). Interestingly, H. pylori
induced the gastric DCs to secrete IL-6, IL-8, and IL-10, but did not induce secretion of IL-12
(Bimczok et al., 2010). Despite the lack of secretion of this important Th1-stimulating cytokine,

20
the H. pylori-pulsed gastric DCs induced T cells to secrete IFN-γ, which is consistent with Th1
responses. Therefore the authors concluded that H. pylori activates human gastric DCs to induce
a predominant Th1 response (Bimczok et al., 2010).
Another study utilizing human monocyte-derived dendritic cells found that H. pylori upregulated
expression of the maturation markers CD40, CD80, CD86, and the MHC II in DCs (Chang et al.,
2012). This study demonstrated that H. pylori induced secretion of IL-10 and TNF-α from these
DCs, and specific PRRs such as TLR2 and TLR4 were implicated in this secretion (Chang et al.,
2012). Interestingly, the authors found that monocyte-derived DCs that were produced from
gastric cancer patients did not secrete IL-10 in response to H. pylori stimulation, and speculated
that this defective IL-10 response contributes to the gastric cancer seen in H. pylori patients
(Chang et al., 2012).
Kao et al. found that H. pylori induced secretion of IL-12 and IL-10 from murine BMDCs (Kao
et al., 2006). However they also demonstrated much less secretion of IL-12 and IL-10 in the H.
pylori-infected BMDCs compared to BMDCs stimulated with another gastritis-causing pathogen,
Acinetobacter lwoffi (A. lwoffi) (Kao et al., 2006). The authors found H. pylori-stimulated
BMDCs induced the release of IFN-γ and TNF-α from T cells, but to a lesser extent than A.
lwoffi-stimulated BMDCs. Furthermore they found that H. pylori could effectively inhibit the
release of IL-12 but not IL-10 from the BMDCs stimulated with A. lwoffi (Kao et al., 2006).
These results led the authors to suggest that H. pylori can have inhibitory effects on the dendritic
cell, causing defects in the host immune response (Kao et al., 2006). A more recent study by the
same group found that H. pylori induced BMDCs to polarize a Treg polarized response,
maintaining high levels of Tregs and decreasing Th17 development in a TGF-β and IL-10
dependent manner (Kao et al., 2010). In this study the authors suggest that this DC-induced Treg
polarization and Th17 suppression facilitates H. pylori persistence and immune escape (Kao et
al., 2010). In contrast, a study by Khamri et al. found that H. pylori stimulated human monocyte-
derived DCs to secrete IL-23 and IFN-γ, which induced the production of IL-17-secreting Th17
cells (Khamri et al., 2010). The authors found that the cag PAI and an intact T4SS were required
for this DC-mediated IL-17 production (Khamri et al., 2010).
Finally, a recent study showed that H. pylori induces the development of tolerogenic DCs which
stimulate naïve T cells to express Foxp3; these DCs were capable of preventing airway

21
hyperresponsiveness in a murine experimental model of asthma (Oertli et al., 2012). In this study
H. pylori did not induce maturation of BMDCs as assessed by CD40, CD80, and CD86 cell
surface expression but induced secretion of IL-10 from these immature DCs. Indeed the authors
showed that H. pylori inhibited LPS-induced maturation of DCs in a contact dependent manner.
H. pylori also inhibited LPS-induced secretion of IL-6 and IL-12 (Oertli et al., 2012). The
authors demonstrated that H. pylori induces the BMDCs to drive Foxp3+ Treg development and
that the release of TGF-β and IL-18 from the DC was necessary to drive this Treg conversion
(Oertli et al., 2012). Supporting these results are studies that show H. pylori-specific Tregs can
suppress memory T-cell responses to H. pylori in infected individuals (Lundgren et al., 2003).
This stimulation of immunosuppressive Tregs by H. pylori also decreases gastric tumor
immunosurveillance and immune response to tumor antigens (Groux, 2003). Tregs have been
shown to contribute to myeloma-related immune dysfunction, and the accumulation of Tregs in
various human carcinomas is associated with a poor prognosis (Braga et al., 2012; Ladoire et al.,
2011). Furthermore the balance between immunosuppressive Tregs and highly inflammatory
Th17 cells is perturbed in many human cancers, indicating that these classes play a key role in
the potential development or suppression of cancer (Braga et al., 2012).
Thus the studies to date assessing H. pylori and dendritic cells contain some conflicting results.
Methodological differences can explain some of these paradoxical results. Different strains of H.
pylori are used in these studies; these strains often differ in terms of pathogenicity owing to
differences in expression of virulence factors such as VacA, CagA, and the cag PAI itself.
Furthermore the “dendritic cells” used in the studies vary quite widely, as some studies use
murine bone marrow-derived dendritic cells, others use human peripheral monocyte-derived
DCs, which still others take pre-differentiated DCs from the gastric mucosa. Among the cultured
DCs, even slight differences in culture conditions can affect the phenotype of the DC before it
has even been exposed to the H. pylori. Finally, the use of different controls, and the comparison
of H. pylori’s effects to those of different bacteria can explain differences in the presentation of
some results. However despite these concessions, it is clear that the effects of H. pylori infection
on DCs remain unclear, and the mechanisms by which the bacteria evades or moderates the host
immune response are still being elucidated.

22
1.2.4 H. pylori-altered DCs: potential role in disease
Additional evidence for the ability of H. pylori to affect the host immune system can be found in
recent epidemiological studies which link H. pylori infection with protection against the
development of several autoimmune and immune-mediated diseases. As H. pylori has infected
and coevolved with human hosts for at least 50,000 years; the presence of this bacterium has
likely influenced the evolution and development of modern human physiology and immunology
(Atherton and Blaser, 2009). Yet H. pylori colonization is presently detected in fewer than 6% of
children in developed countries; this eradication of an immunological influence has been
epidemiologically correlated with an increase in the occurrence of several disease pathologies. H.
pylori eradication has been associated with an increase in gastroesophageal reflux disease
(GERD), Barrett’s oesophagus, and the development of esophageal adenocarcinoma (Atherton
and Blaser, 2009). Additionally H. pylori infection has been negatively associated with
esophageal eosinophilia, an allergic inflammatory condition (Dellon et al., 2011).
An inverse association between H. pylori colonization and asthma has also been described (Chen
and Blaser, 2007). Recently, a study attempting to elucidate the mechanisms behind this
correlation used mouse models of allergen-induced asthma (Arnold et al., 2011a). In this study,
mice were infected with H. pylori as newborns or as adults; neonatal mice infected with H. pylori
displayed a reduction in multiple asthma indicators. Significantly, this protection was associated
with both impaired maturation of DCs that infiltrated the lungs and accumulation of Tregs. This
protection was abrogated by antibiotic elimination of H. pylori and abolished by systemic Treg
depletion. Furthermore, adoptive transfer of purified Tregs was sufficient to confer protection to
uninfected recipient mice (Arnold et al., 2011a). More recently the same group found that H.
pylori causes DCs to drive Treg differentiation and induce immune tolerance (Oertli et al., 2012).
Interestingly the authors showed that H. pylori exposure was able to prevent DCs from inducing
asthma in a DC-mediated experimental model of asthma (Oertli et al., 2012). Furthermore Tregs
from H. pylori-infected mice were able to prevent asthma in this model.
H. pylori infection has also been found to be inversely correlated with the incidence of
Inflammatory Bowel Disease (IBD) (Luther et al., 2010). A recent study demonstrated that H.
pylori DNA had an increase in immunoregulatory DNA sequences and that incubation of DCs
with this DNA reduced proinflammatory cytokine expression as compared to E. coli DNA

23
(Luther et al., 2011). Furthermore, the H. pylori DNA was able to suppress the inflammatory
response caused by the E. coli DNA. In addition, administration of H. pylori DNA reduced
dextran sodium sulphate-induced colitis in a murine model while E. coli DNA did not. This
presents an intriguing mechanism for the negative correlation between H. pylori infection and
IBD, and demonstrates a new pathway by which H. pylori DNA can alter host immunity.
Collectively these studies further demonstrate that H. pylori can modulate the host immune
system. In some cases this negatively impacts the host, allowing for persistent infection and
decreasing immune tumor surveillance to cause cancer. However it also appears to play a
beneficial role in the prevention of other immune-mediated disorders and pathologies.
Importantly DCs may play a potential role in mediating these effects.
1.2.5 Signal Transducer and Activator of Transcription 3 (STAT3)
The mechanisms by which H. pylori evades and modulates the host immune response remain
unclear. One potential mechanism by which H. pylori may modulate the host immune response is
through activation of the Signal Transducer and Activator of Transcription 3 (STAT3) pathway.
STAT3 is a member of the STAT family, signal transducer proteins that are activated by the
tyrosine phosphorylation cascade in response to ligand binding. STAT3 is a cytoplasmic protein
that acts as a signal messenger and transcription factor to induce cellular responses to cytokines
and growth factors. Importantly hyperactivation of STAT3 is found in many human cancers and
is correlated with both tumor progression and poor prognosis (Lin et al., 2010). Recent work
form our laboratory indicates that during H. pylori infection in vivo, STAT3 is activated in
gastric epithelial cells in a CagA-dependent manner. (Bronte-Tinkew et al., 2009). STAT3
activation was also found in a subset of infiltrating immune cells (Bronte-Tinkew et al., 2009).
Therefore H. pylori possesses the ability to manipulate and activate this crucial signaling cascade
whose abberrant activation is found in many human cancers.
1.2.5.1 STAT3 Signaling
STAT3 is activated via a tyrosine phosphorylation cascade in response to exctracellular ligand
binding. Ligands that activate the STAT3 pathway include cytokines such as IL-10 and IL-6 and
growth factors such as Vascular Endothelial Growth Factor (VEGF) (Lin et al., 2010). The
ligand (e.g. IL-6) binds to its receptor (IL-6R) on the cell surface (Figure 1.2-4). The IL-6R then

24
dimerizes with a gp130 subunit, resulting in the phosphorylation and activation of associated
Janus Kinase 2 (JAK2) enzymes (Kim et al., 2007). JAK2 is a tyrosine kinase which
phosphorylates tyrosine residues on the cytoplasmic tail of the gp130 subunit (Greenlund et al.,
1995). The phosphorylated tyrosines recruit cytoplasmic STAT3 monomers to the gp130
subunit. The STAT3 monomers are subsequently phosphorylated on their Y705 residues, and
dimerize through Src-Homology 2 domain (SH2) phosphotyrosine interactions. The
phosphorylated STAT3 dimer (PSTAT3) is then translocated into the nucleus via nuclear
localization signals (Reich and Liu, 2006). Inside the nucleus DNA binding domains permit
binding to promoter regions of target genes which are involved in cell maturation, growth,
proliferation, angiogenesis and immune function (Kim et al., 2007; Kisseleva et al., 2002; Levy
and Darnell, 2002). Importantly, STAT3 is implicated in tumorigenesis and cancer progression.
Abberant, constitutive STAT3 activation has been found in a majority of both hematopoeitic and
epithelial cancers as well as tumour cells (Ernst and Putoczki, 2011; Wang et al., 2004).
Constitutive STAT3 activation in diverse human cancers enhances tumor proliferation,
angiogenesis, metastasis, and survival, and inhibits tumor apoptosis as well as antitumor
immunity (Wang et al., 2004; Yu et al., 2009).
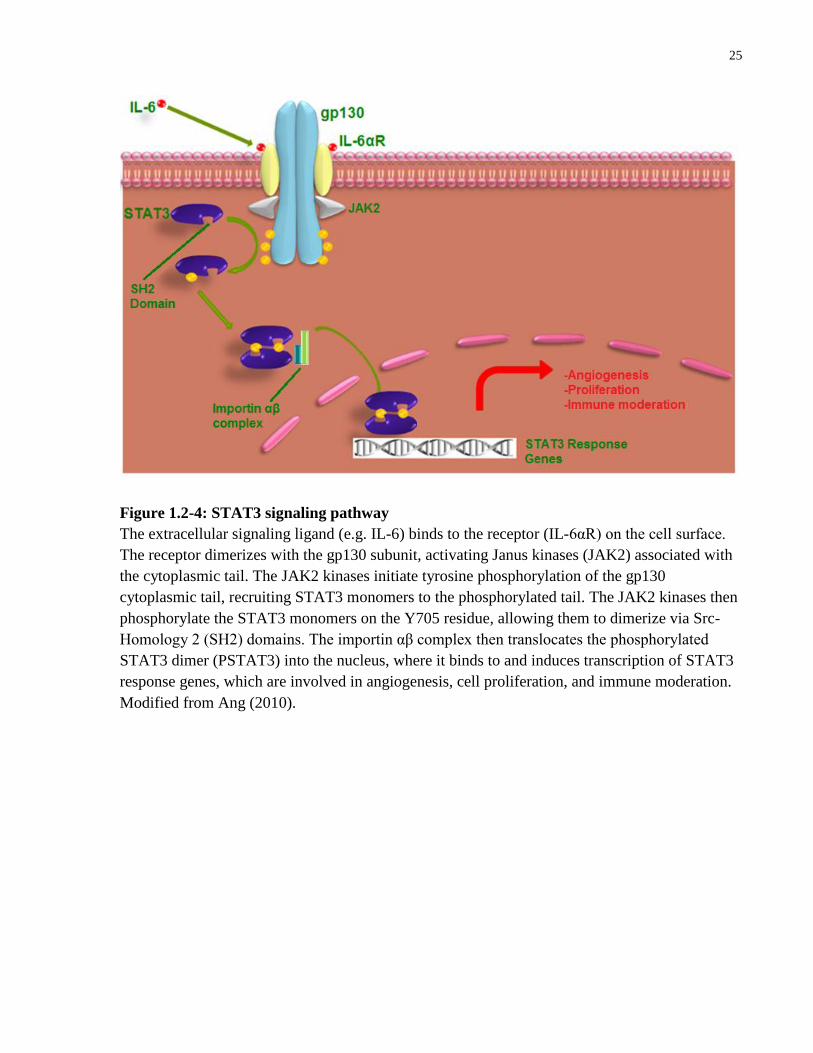
25
Figure 1.2-4: STAT3 signaling pathway
The extracellular signaling ligand (e.g. IL-6) binds to the receptor (IL-6αR) on the cell surface.
The receptor dimerizes with the gp130 subunit, activating Janus kinases (JAK2) associated with
the cytoplasmic tail. The JAK2 kinases initiate tyrosine phosphorylation of the gp130
cytoplasmic tail, recruiting STAT3 monomers to the phosphorylated tail. The JAK2 kinases then
phosphorylate the STAT3 monomers on the Y705 residue, allowing them to dimerize via Src-
Homology 2 (SH2) domains. The importin αβ complex then translocates the phosphorylated
STAT3 dimer (PSTAT3) into the nucleus, where it binds to and induces transcription of STAT3
response genes, which are involved in angiogenesis, cell proliferation, and immune moderation.
Modified from Ang (2010).

26
1.2.5.2 STAT3 Activation in Dendritic Cells
STAT3 activation in dendritic cells is a potential mechanism by which the DC response is
disrupted by H. pylori, leading to immune disruption and potentially carcinogenesis. Current
evidence suggests that DC maturation is inhibited by STAT3 activation, while inhibition of
STAT3 in dendritic cells promotes DC maturation (Lin et al., 2010; Melillo et al., 2010). STAT3
activation is often associated with tolerogenic functions (Barton, 2006; Cheng et al., 2003;
Nefedova et al., 2005). Indeed many tumors secrete high levels of the growth factor VEGF; this
VEGF activates STAT3 in DCs, preventing their maturation and thus decreasing the host
immune tumor surveillance (Lin et al., 2010). Iwata-Kajihara et al. demonstrated that STAT3-
depleted DCs were more resistant to cancer-cell derived inhibitory factors (Iwata-Kajihara et al.,
2011). These STAT3-deficient BMDCs were more potent activators of T cells and had a high
capacity to induce Th1 responses (Iwata-Kajihara et al., 2011). STAT3 signaling is also
necessary for Th17 development (Harris et al., 2007). This highly inflammatory STAT3-
dependent Th17 response has been shown to be pro-carcinogenic (Wang et al., 2009). Therefore
it appears STAT3 activation in dendritic cells supports a pro-tumorigenic environment.
Furthermore STAT3 activation in DCs prevents their maturation and activation, which decreases
immune surveillance and represents a potential mechanism by which the host immune response
can be manipulated.
1.3 Summary
H. pylori is a gastric pathogen that has the ability to alter the host immune response. Infection
with H. pylori persists for the lifetime of the host despite the induction of chronic gastritis, a low-
level inflammatory immune response. The bacterium is a type I carcinogen and plays a causative
role in gastric cancer, potentially by inhibiting host tumor immunosurveillance. Epidemiologic
evidence indicates that H. pylori infection is negatively correlated with several autoimmune and
immune-mediated diseases, supporting a role for the bacteria in altering immune responses.
Dendritic cells are ideal targets for the pathogen’s immune-altering efforts as they play a key role
in modulating the host’s immune response. DCs can drive powerful effector responses to
pathogenic organisms and necrotic/tumor cells as well as tolerant regulatory responses to
commensal organsisms and self-antigens. Therefore the hypothesis that H. pylori may
manipulate this key immune orchestrator in order to evade the immune response and persist in

27
the host is both intriguing and plausible. Mechanistically the activation of STAT3 in dendritic
cells has been shown to prevent DC maturation and inhibit DC-driven effector T cell responses
(Iwata-Kajihara et al., 2011; Melillo et al., 2010). STAT3 activation may decrease tumor
immunosurveillance by DCs to contribute to cancer development (Lin et al., 2010).
H. pylori activates STAT3 in both epithelial cells and infiltrating immune cells (Bronte-Tinkew
et al., 2009). Thus, H. pylori could activate STAT3 in DCs to modify DC maturation/activation
and thereby alter immune responses to promote persistence and inhibit DC tumor
immunosurveillance.
Hypothesis
H. pylori activates signal transducer and activator of transcription 3 (STAT3) in dendritic cells to
promote chronic infection and disease.
Specific Aims
This hypothesis will be addressed using several specific aims.
Aim 1: Determine whether H. pylori activates STAT3 in bone marrow derived dendritic cells
and characterize the phenotype of DCs.
Aim 2: Determine the effect of STAT3 activation on DCs during H. pylori infection.
Aim 3: Determine the mechanism of STAT3 activation in DCs during H. pylori infection.
Aim 4: Determine how H. pylori infected dendritic cells modulate T cell development.

28
Chapter 2 Materials and Methods
2
2.1 Bone Marrow-Derived Dendritic Cells
Bone Marrow-Derived Dendritic Cells (BMDCs) were obtained and cultured from C57BL6
wildtype (WT) mice, 6-8 weeks old. BMDCs were also obtained from STAT3 CD11c-Cre
knockout (KO) mice or littermate controls (Melillo et al., 2010). Bone marrow was extracted
from the femur and tibia and seeded at 2 x 105cells/ml with DC media (Lutz and Rossner, 2007).
DC media contains: Roswell Park Memorial Institute (RPMI)-1640 media, 10% fetal bovine
serum, 1% L-glutamine, 1% penicillin/streptomycin, 1X sodium pyruvate, 1X non-essential
amino acids, and 0.1% 2-mercaptoethanol. Recombinant murine granulocyte macrophage
colony-stimulating factor (GM-CSF) was added at a concentration of 40ng/ml, to stimulate
dendritic cell growth (Lutz and Rossner, 2007). DCs were seeded on bacteriological petri dishes
with 10ml DC media supplemented with GM-CSF for 3 days at 37°C in 5% CO2. On day 3, an
additional 10ml of DC media supplemented with GM-CSF was added to each plate. On day 6
and day 8 10ml of the solution from each plate was removed, centrifuged to pellet floating cells,
and resuspended with 10ml of fresh DC media supplemented with GM-CSF per plate. On day
10, the floating, non-adherent cell population (immature DCs) was harvested, counted, and
plated in 6-well tissue culture plates in antibiotic-free DC media to be used for
treatment/infection.
2.2 Treatment/Infection
H. pylori strain 7.13 was grown on Columbia blood agar plates (supplemented with 5% sheep
blood) under microaerophilic conditions (5% O2, 10% CO2, 85% N2) for 72h (Zheng and Jones,
2003). Bacteria were then collected using sterile inoculation loops and transferred into a bacterial
flask containing Brucella broth supplemented with 10% fetal bovine serum for 24 hours. Bacteria
were then harvested by centrifugation, resuspended in antibiotic-free DC media, and added to
immature BMDCs at a multiplicity of infection (MOI) of 10:1. Treated cells were incubated at
37°C for 20 hours. The optimal infection time was previously determined using a series of time-
course experiments (Ang, 2010). A set of untreated DCs served as control cells. A set of DCs

29
was treated with IL-6 [100ng/ml] to serve as a positive control for STAT3 activation, and a
negative control for DC maturation (Niemand et al., 2003). The optimal dosage of IL-6 was
determined by a dose-response experiment performed previously (Ang, 2010). An additional set
of DCs was treated with E. coli lipopolysaccharide (LPS; Sigma Aldrich) [1ng/ml] to serve as a
positive control for DC maturation (Foti et al., 1999). Treatment of DCs with a neutralizing rat-
anti-mouse IL-10 antibody (eBioscience, San Diego, CA, USA) [0.5µg/ml or 1µg/ml] was
conducted to examine the effects of IL-10 on the DC maturation and STAT3 activation. Rat-anti-
IgG antibody (BioLegend, San Diego, CA, USA) [1µg/ml] served as a control antibody to ensure
the specificity of the IL-10 antibody-mediated effects. After 20 hours of incubation the cells
were harvested for analysis. Western blotting was used to examine STAT3 phosphorylation and
activation. Flow cytometry using αCD11c, αCD86, and αMHC class II fluorophores was used to
examine the maturation status of the dendritic cells. ELISA assays and multiplex bead assays
will be used to examine the cytokine release profile of the dendritic cells.
2.3 Western Blotting
After 20h of treatment/infection, cells were harvested by vigorous pipetting and washed with
phosphate-buffered saline (PBS; Sigma Aldrich). Cells were lysed using 100µl of a
radioimmunoprecipitation assay (RIPA) buffer cocktail supplemented with protease inhibitors on
ice. Lysates were centrifuged, supernatants collected and stored at -20°C (Bronte-Tinkew et al.,
2009). Lysates (supplemented with 10% laemelli buffer) were loaded onto 12% SDS-
polyacrylamide gels and run at 110V at room temperature for 1.5h. Separated proteins were
transferred onto nitrocellulose membrane (BioTrance, NT; Pall Corp., All Arbor, MI, USA) at
30V for 8h at 4°C (Bronte-Tinkew et al., 2009). Membranes were then blocked with 5% skim
milk made using Tris-buffered saline supplemented with tween (TBST) for 30min, followed by
overnight incubation at 4°C using rabbit-anti-PSTAT3 antibody (1:200; Cell Signalling MA,
USA) and mouse-anti-actin antibody (1:10,000) in TBST milk. Blots were then incubated using
corresponding horseradish peroxidase-conjugated secondary antibodies (1:2,000 and 1:10,000
for the PSTAT3 and actin primary antibodies, respectively) for 1h at room temperature (Bronte-
Tinkew et al., 2009). Bands were then visualized by chemiluminescence using Kodak Biomax
MR film or FluorChem E Imager (Proteinsimple, Santa Clara, CA, USA).

30
2.4 Densitometric analysis
Densitometry was performed utilizing the FlourChem FCII software. Densities of PSTAT3 and
actin bands were measured for each treatment and expressed as a ratio of PSTAT3/actin, and
then normalized to the control treatment.
2.5 Flow Cytometry
After 20h of treatment/infection, BMDCs were harvested by vigorous pipetting and centrifuged
at 350g for 10min. Supernatants were collected and stored at -20°C to examine cytokine release
profile. Cells were rinsed using flow cytometry buffer (FB) (PBS supplemented with 1% bovine
serum albumin (BSA)), and then incubated with anti-CD16/32 (FC receptor block; eBioscience,
San Diego CA USA) [25µg/µl] for 15min on ice, to block non-specific antibody binding (Oliver
et al., 1999). DCs were then incubated with flourescein isothiocyanate (FITC)-labelled αCD11c,
phycoerythrin (PE)-labelled αCD86, and allophcocyanin (APCa)-labelled αMHC class II
antibodies (all from eBioscience) for 30min. DCs were then washed twice with FB before
fixation using 1x paraformadehyde (PFA, made with PBS) for 20min. Cells were subsequently
washed with FB and analyzed using a LSR-II flow cytometer (Becton Dickson, San Jose, CA,
USA). Flow cytometry data was then analyzed utilizing FlowJo software (Tree Star Inc,
Ashland, OR, USA).
2.6 ELISA Cytokine Analysis
BMDCs were infected for 20h as described above. Supernatants were collected and stored at -
80°C. Supernatants were assayed for IL-6, IL-10, IL-12p40 and TNF-α using
specific ELISAs as per the manufacturer’s instructions (R&D systems, MN, USA).
2.7 Multiplex Bead-based Luminex Assay
Supernatants from BMDCs were assayed for IL-2, IL-4, IL-5, IL-10, IL-12, IL-17, and IFN-γ
using the Th1/Th2 Cytokine Mouse 6-Plex Panel kit plus the IL-17 Mouse Singleplex Bead
Luminex Assay Kit as per the manufacturer’s instructions (Invitrogen).

31
2.8 OT-II Transgenic Mouse CD4+ Splenocytes
OT-II transgenic mice were kindly donated by the laboratory of Dr. Tania Watts or purchased
from Jackson Laboratory. These transgenic mice express the mouse alpha-chain and beta-chain T
cell receptor that pairs with the CD4 coreceptor and is specific for chicken ovalbumin 323-339 in
the context of the MHC class II molecule (Barnden et al., 1998). Consequently all CD4+
splenocytes (i.e. T cells) harvested from these animals will be activated when presented with the
ovalbumin peptide by APCs (such as DCs) with appropriate co-receptors and cytokine
stimulation (Barnden et al., 1998). This circumvents the need for antigen specificity when
working with a model of T cell activation by APCs.
2.9 STAT3 KO Mice
STAT3 KO mice were kindly donated by the laboratory of Dr. Tak Mak or Dr. Christian
Schindler. These DC-specific STAT3 KO mice were generated using a Cre-flox system. Briefly,
Stat3flox/flox
mice, in a 129/C57BL/6J mixed background were crossed seven generations onto
C57BL/6J background, and then with CD11c-BAC-Cre transgenic mice (Melillo et al., 2010).
STAT3 expression was examined via immunoblotting to confirm knockout.
2.10 DC:T Cell Coincubation
Sets of DCs were loaded with 10ng/ml or 100ng/ml ovalbumin peptide 323-339 (OVA) with or
without H. pylori for 20h. DCs were then counted and incubated with splenocytes from OT-II
transgenic mice (at a ratio of 1:2) which were harvested and magnetically sorted for the CD25-
CD4+ population according to the manufacturer’s instructions (Miltenyi Biotec, Bergisch
Gladbach, Germany), in DC media with or without recombinant murine IL-2 [10ng/ml] and
recombinant human TGF-β1 [10ng/ml] (Oertli et al., 2012). The coculture was incubated for 72h
at 37°C in 5% CO2. Cells were then harvested, supernatants collected to examine cytokine
release using ELISA-based assays, and flow cytometry performed to examine T cell phenotype
using αCD4 and αFoxP3 fluorophores (Oertli et al., 2012).

32
2.11 Statistical Analysis
All experiments were performed at least 3 times, and statistical significance (P values) was
calculated using a one-way ANOVA to compare the means ± SE for independent treatment
groups. A P value of less than 0.05 was deemed statistically significant.

33
Chapter 3 Results
3
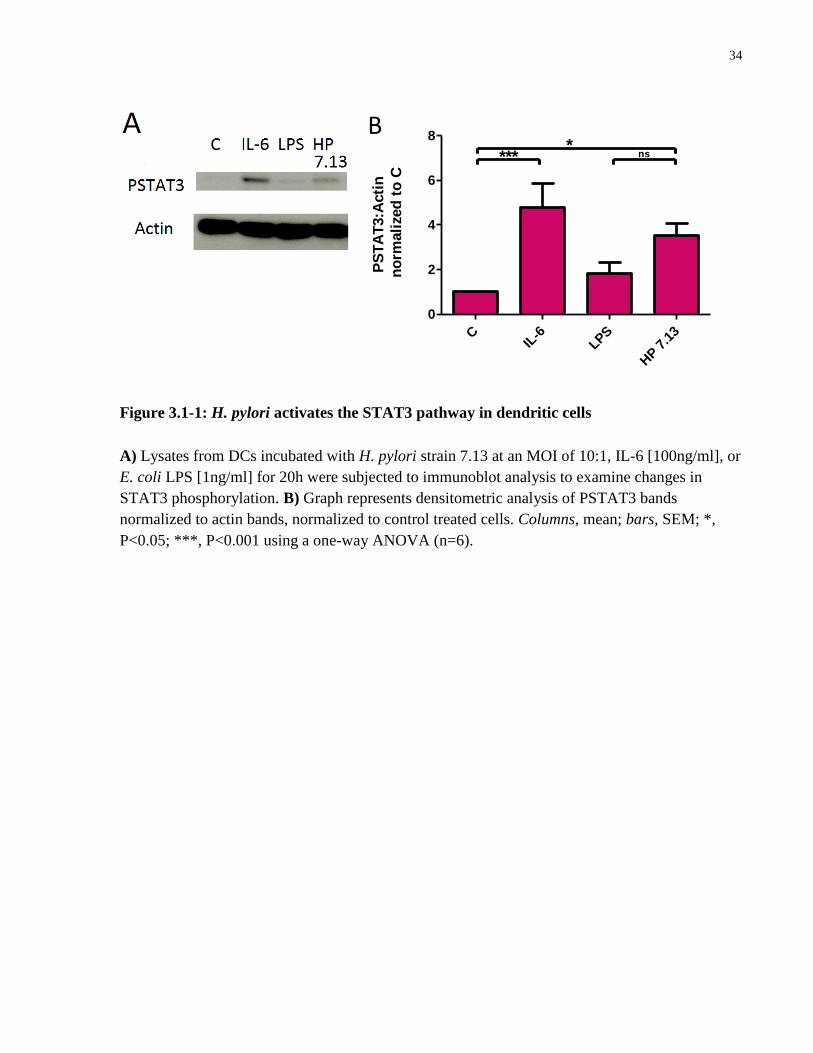
3.1 H. pylori infection induces STAT3 phosphorylation in dendritic cells
To determine whether H. pylori induces activation of the STAT3 pathway in DCs, expression of
phosphorylated STAT3 (PSTAT3) was measured in dendritic cell lysates using western blotting.
Cultured BMDCs were incubated with H. pylori strain 7.13 at an MOI of 10:1, treated with IL-6
(a known activator of STAT3), or treated with E. coli LPS (a known inducer of DC maturation)
for 20h. Immunoblot analysis of these cell lysates revealed phosphorylation of STAT3 in DCs
treated with H. pylori and IL-6 compared to uninfected control and LPS-treated cells (Figure
3.1-1A, B).
34
CIL
-6LPS
HP 7
.13
0
2
4
6
8
****
B
PS
TA
T3:A
cti
n
no
rmali
zed
to
C
ns
Figure 3.1-1: H. pylori activates the STAT3 pathway in dendritic cells
A) Lysates from DCs incubated with H. pylori strain 7.13 at an MOI of 10:1, IL-6 [100ng/ml], or
E. coli LPS [1ng/ml] for 20h were subjected to immunoblot analysis to examine changes in
STAT3 phosphorylation. B) Graph represents densitometric analysis of PSTAT3 bands
normalized to actin bands, normalized to control treated cells. Columns, mean; bars, SEM; *,
P<0.05; ***, P<0.001 using a one-way ANOVA (n=6).

35
3.2 H. pylori upregulates expression of DC maturation markers
The effect of H. pylori infection on dendritic cell maturation was next assessed. BMDCs were
untreated (C) or treated with IL-6 [100ng/ml], E. coli LPS [1ng/ml], or Helicobacter pylori strain
7.13 at a MOI of 10:1 and incubated for 20h. Flow cytometry was performed using a BD LSR-II
flow cytometer to quantify and analyze the expression of the DC maturation markers CD86 and
MHC class II. Expression of the DC-specific marker CD11c was used to select a pure dendritic
cell population (Figure 3.2-1A). H. pylori infection increased the proportion of MHC IIhi
CD86hi
BMDCs to a similar extent as treatment with E. coli LPS, whereas treatment with the STAT3
activator IL-6 did not increase the expression of these maturation markers compared to untreated
(control) cells (Figure 3.2-1B, C).
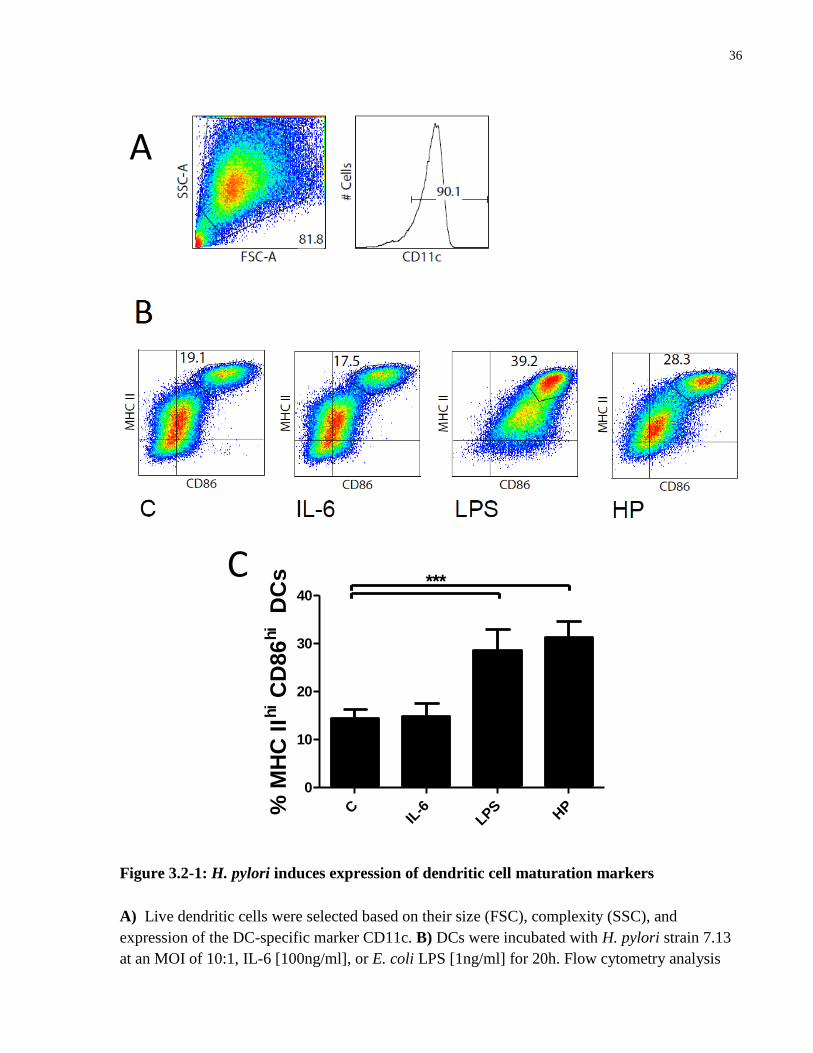
36
A
CIL
-6LPS
HP
0
10
20
30
40***
% M
HC
IIh
i CD
86
hi
DC
s
Figure 3.2-1: H. pylori induces expression of dendritic cell maturation markers
A) Live dendritic cells were selected based on their size (FSC), complexity (SSC), and
expression of the DC-specific marker CD11c. B) DCs were incubated with H. pylori strain 7.13
at an MOI of 10:1, IL-6 [100ng/ml], or E. coli LPS [1ng/ml] for 20h. Flow cytometry analysis
C

37
was used to quantify the population expressing high levels of the maturation markers CD86 and
MHC class II. Cells in the gated population in the upper right quadrant of each graph represent
the DCs with high expression of both markers (i.e. mature DCs). C) Quantification of gated
CD86hi
MHC IIhi
DCs. Columns, mean; bars, SEM; ***, P<0.001 using a one-way ANOVA
(n=6).

38
3.3 Activated dendritic cells secrete a specific cytokine profile in response to H. pylori infection
As the cytokines released from the DC dictate the CD4+ helper T cell phenotype that develops,
the cytokine release profile from H. pylori-infected DCs was examined. The culture supernatants
from control, H. pylori, LPS, and IL-6 treated BMDCs were subjected to Luminex multiplex
assays which profiled levels of the following cytokines: IL-2, IL-4, IL-5, IL-10, IL-12, IL-17,
and IFN-γ. Treatment with H. pylori increased the levels of IL-10 and IL-12 in the BMDC
culture supernatants (Figure 3.3-1A). H. pylori infection also increased the ratio of IL-10:IL-12
released by the BMDC. In contrast treatment with proinflammatory LPS increased the IL12:IL-
10 ratio (Figure 3.3-1B). These results were confirmed with ELISA-based assays which revealed
significant increases in IL-10 and TNF-α release from BMDCs in response to H. pylori infection,
as well as non-significant IL-12p40 increases (Figure 3-3C).
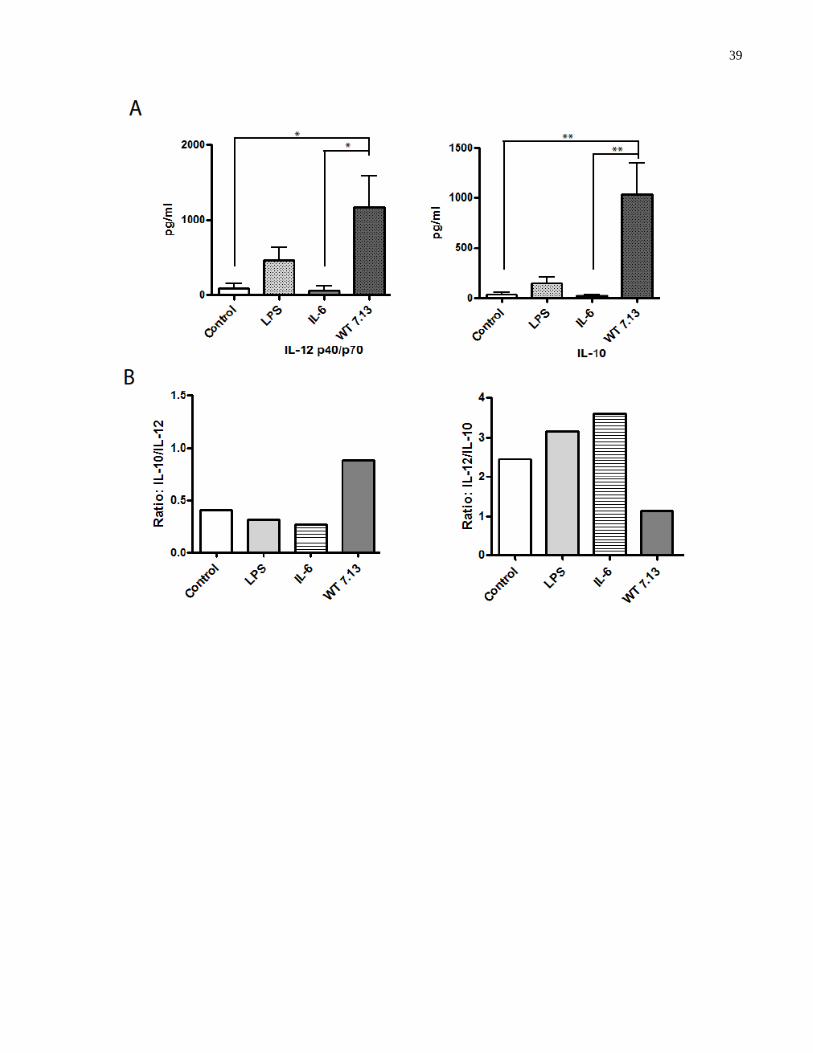
39
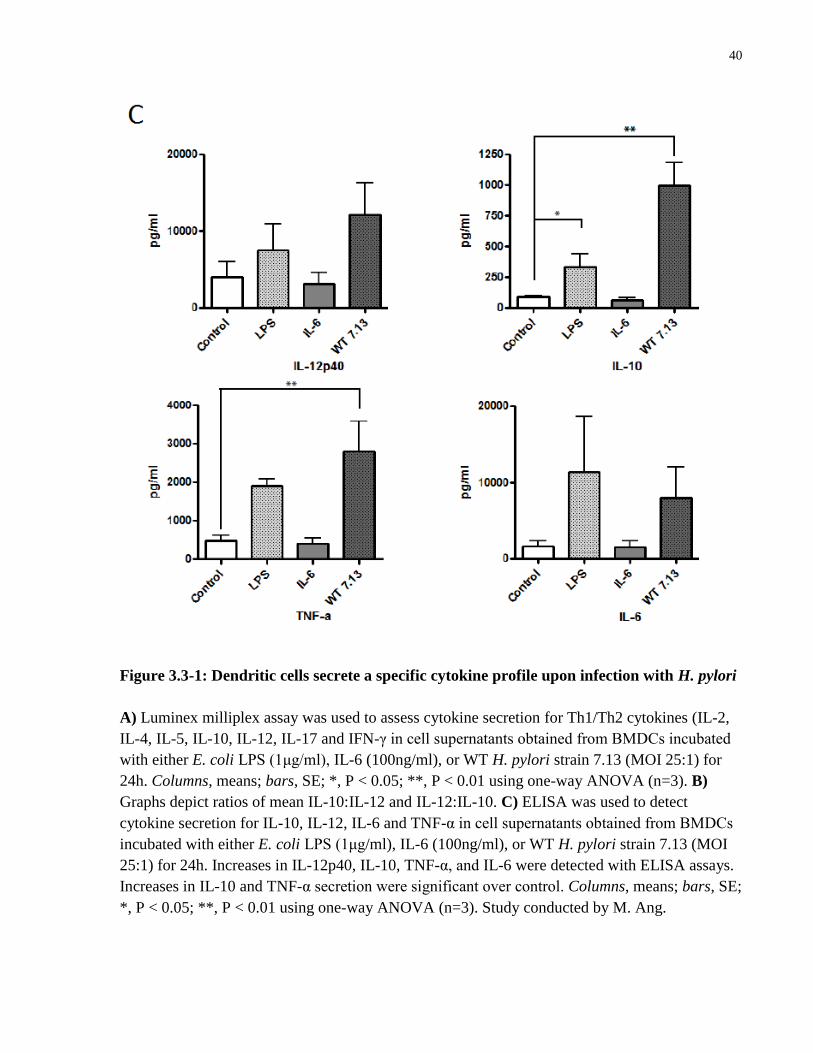
40
Figure 3.3-1: Dendritic cells secrete a specific cytokine profile upon infection with H. pylori
A) Luminex milliplex assay was used to assess cytokine secretion for Th1/Th2 cytokines (IL-2,
IL-4, IL-5, IL-10, IL-12, IL-17 and IFN-γ in cell supernatants obtained from BMDCs incubated
with either E. coli LPS (1μg/ml), IL-6 (100ng/ml), or WT H. pylori strain 7.13 (MOI 25:1) for
24h. Columns, means; bars, SE; *, P < 0.05; **, P < 0.01 using one-way ANOVA (n=3). B)
Graphs depict ratios of mean IL-10:IL-12 and IL-12:IL-10. C) ELISA was used to detect
cytokine secretion for IL-10, IL-12, IL-6 and TNF-α in cell supernatants obtained from BMDCs
incubated with either E. coli LPS (1μg/ml), IL-6 (100ng/ml), or WT H. pylori strain 7.13 (MOI
25:1) for 24h. Increases in IL-12p40, IL-10, TNF-α, and IL-6 were detected with ELISA assays.
Increases in IL-10 and TNF-α secretion were significant over control. Columns, means; bars, SE;
*, P < 0.05; **, P < 0.01 using one-way ANOVA (n=3). Study conducted by M. Ang.

41
3.4 H. pylori induces more pronounced maturation in STAT3 KO dendritic cells
I next elucidated the effect of H. pylori-induced STAT3 activation in dendritic cells by utilizing
BMDCs derived from dendritic cell-specific STAT3 KO mice (kindly provided by the laboratory
of Dr. Tak Mak). A CD11c-Cre STAT3-flox system was used to generate Cre+ flox
+ BMDCs
that lack STAT3 expression. Cre- sex-matched littermates provided control STAT3
+ BMDCs.
Cre+ (STAT3 KO) and Cre
- (WT) BMDCs were treated with IL-6 [100ng/ml], E. coli LPS
[1ng/ml], no treatment (C), or Helicobacter pylori strains 7.13 and 60190 at a MOI of 10:1 and
incubated for 20h. Sets of STAT3 KO and WT DCs were then lysed and immunoblotting
performed to determine STAT3 expression and activation. STAT3 KO (Cre+) DCs lacked
STAT3 expression (Figure 3.4-1A). In parallel, DCs from each condition were stained with
αCD11c, αCD86, and αMHC class II fluorophores and flow cytometry analysis was performed
to quantify the expression. H. pylori infection induced significantly more maturation as assessed
by expression of maturation markers in the STAT3 KO dendritic cells as compared to the WT
DCs (Figure 3.4-1B, C).
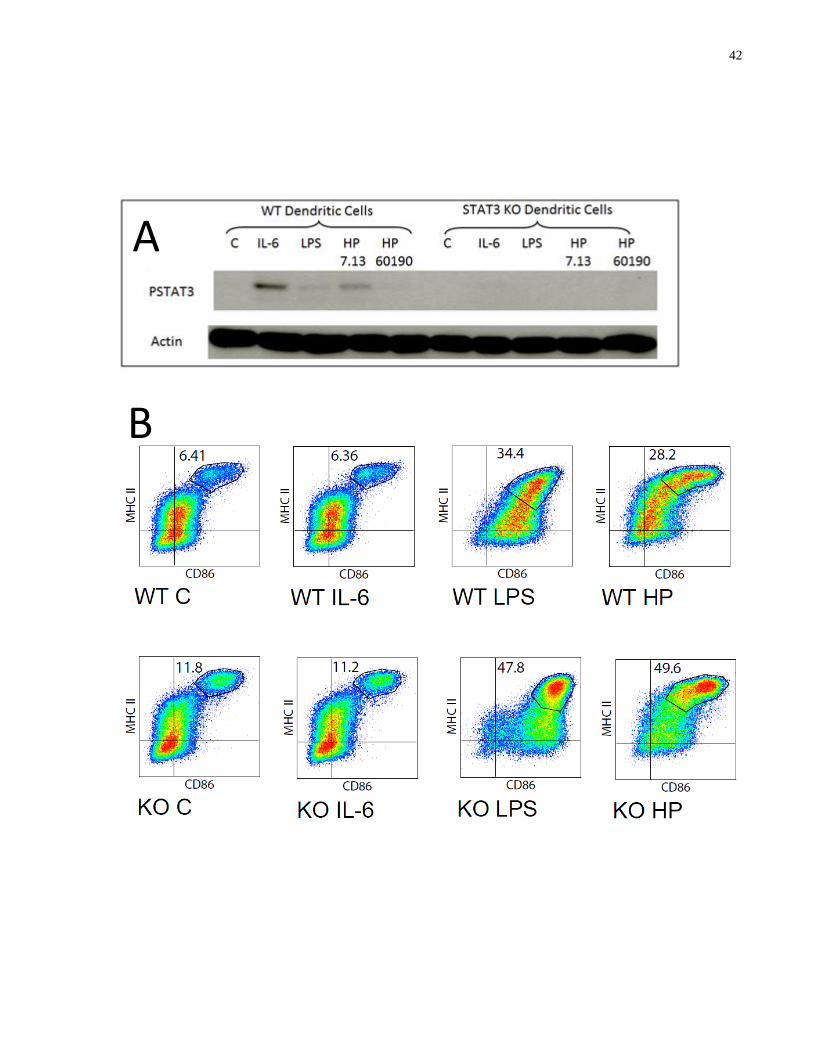
42
B
A
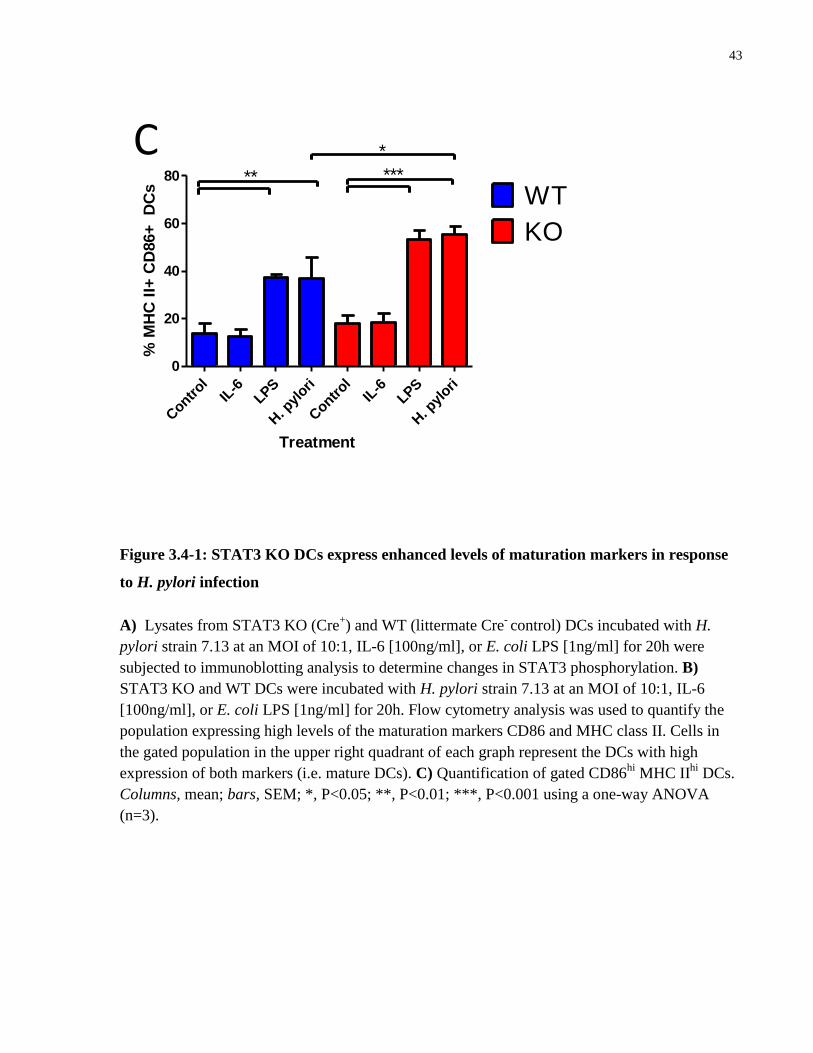
43
Contr
olIL
-6LPS
H. p
ylori
Contr
olIL
-6LPS
H. p
ylori
0
20
40
60
80 ** ***
WT
KO
*C
Treatment
% M
HC
II+
CD
86+
D
Cs
Figure 3.4-1: STAT3 KO DCs express enhanced levels of maturation markers in response
to H. pylori infection
A) Lysates from STAT3 KO (Cre+) and WT (littermate Cre
- control) DCs incubated with H.
pylori strain 7.13 at an MOI of 10:1, IL-6 [100ng/ml], or E. coli LPS [1ng/ml] for 20h were
subjected to immunoblotting analysis to determine changes in STAT3 phosphorylation. B)
STAT3 KO and WT DCs were incubated with H. pylori strain 7.13 at an MOI of 10:1, IL-6
[100ng/ml], or E. coli LPS [1ng/ml] for 20h. Flow cytometry analysis was used to quantify the
population expressing high levels of the maturation markers CD86 and MHC class II. Cells in
the gated population in the upper right quadrant of each graph represent the DCs with high
expression of both markers (i.e. mature DCs). C) Quantification of gated CD86hi
MHC IIhi
DCs.
Columns, mean; bars, SEM; *, P<0.05; **, P<0.01; ***, P<0.001 using a one-way ANOVA
(n=3).

44
3.5 IL-10 secretion by dendritic cells in response to H. pylori infection activates STAT3 to blunt maturation
To determine whether the IL-10 released by the dendritic cells in response to H. pylori infection
was responsible for STAT3 phosphorylation, BMDCs were incubated with a neutralizing α-IL-
10 antibody [0.5 and 1µg/ml], in the presence or absence of H. pylori to examine the effects of
the IL-10 released by the dendritic cells. An isotype control IgG antibody was used to ensure the
specificity of the IL-10 antibody. BMDCs were lysed and immunoblotting performed to examine
phosphorylation of STAT3. Treatment with the α-IL-10 antibody inhibited the STAT3
phosphorylation induced by H. pylori in the BMDCs (Figure 3.5-1A, B). Importantly, α-IL-10
treatment also enhanced expression of the maturation markers MHC class II and CD86 in
BMDCs in response to H. pylori infection as compared to BMDCs treated with H. pylori alone
(Figure3-5C). Treatment with the isotype control IgG antibody affected neither the STAT3
phosphorylation induced in the BMDCs by H. pylori (Figure 3.5-1A, B) nor the maturation of
the BMDCs in response to the H. pylori (Figure 3.5-1C), demonstrating that this was an IL-10-
specific effect.
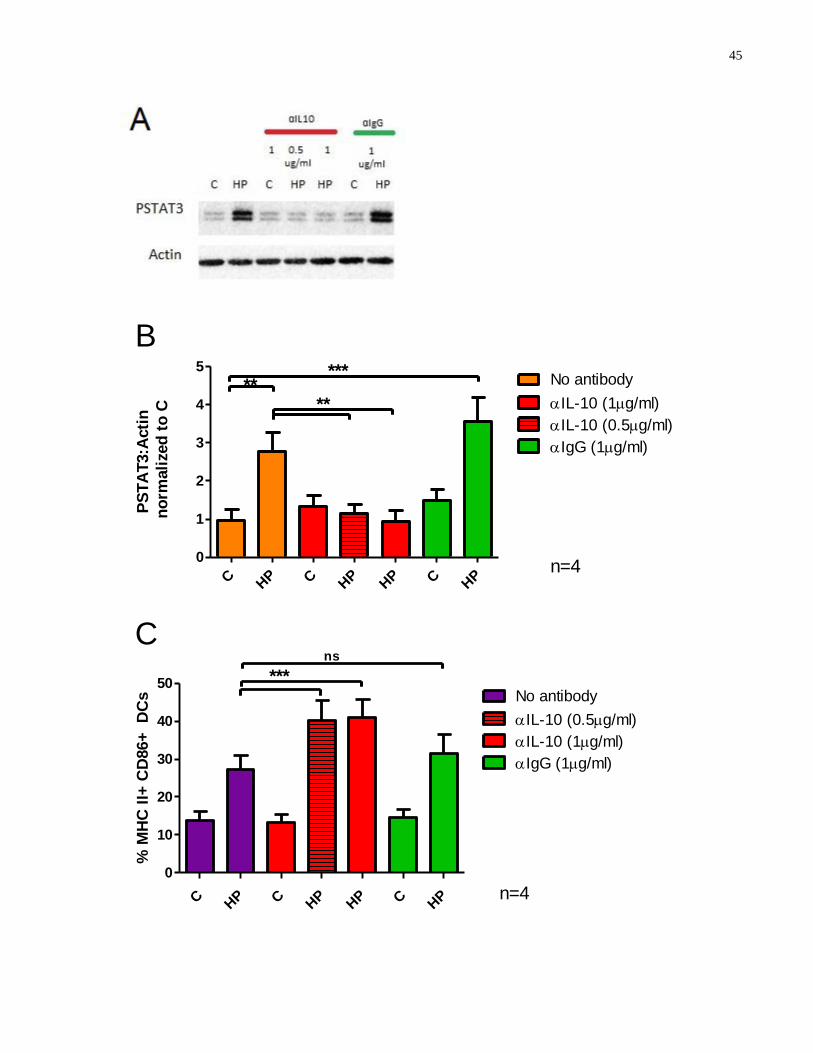
45
C HP C H
PHP C H
P
0
1
2
3
4
5
n=4
B
****
***No antibody
IL-10 (1g/ml)
IL-10 (0.5g/ml)
IgG (1g/ml)
PS
TA
T3:A
cti
n
no
rmali
zed
to
C
C HP C H
PHP C H
P
0
10
20
30
40
50 ***
ns
n=4
IL-10 (1g/ml)
IL-10 (0.5g/ml)
No antibody
IgG (1g/ml)
C
% M
HC
II+
CD
86+
D
Cs

46
Figure 3.5-1: α-IL-10 antibody inhibits H. pylori-induced STAT3 phosphorylation in
BMDCs to promote enhanced expression of maturation markers
A) Lysates from BMDCs treated with functional grade α-IL-10 antibody [0.5 or 1µg/ml] or
isotype control IgG antibody [1µg/ml], in the presence or absence of H. pylori, were subjected to
immunoblotting analysis to determine STAT3 phosphorylation levels. B) Densitometric analysis
was used to quantify the changes in STAT3 phosphorylation levels. Columns, mean; bars, SEM;
**, P<0.01; ***, P<0.001 using a one-way ANOVA (n=4). C) BMDCs were treated with
functional grade α-IL-10 antibody [0.5 or 1µg/ml] or isotype control IgG antibody [1µg/ml], in
the presence or absence of H. pylori at an MOI of 10:1 for 20h. Flow cytometry analysis was
used to quantify the population expressing high levels of the maturation markers CD86 and
MHC class II. Graph represents quantification of the gated CD86hi
MHC IIhi
DCs (i.e. mature
DCs). Columns, mean; bars, SEM; ***, P<0.001 using a one-way ANOVA (n=4).

47
3.6 H. pylori-infected dendritic cells do not drive regulatory T cell development
The functional effect of H. pylori infection on dendritic cell activation and phenotype was next
examined. STAT3 KO and WT BMDCs were incubated with H. pylori for 20h prior to 6h
treatment with penicillin-streptomycin to kill any remaining extracellular bacteria. These
BMDCs were then loaded with differing concentrations of the ovalbumin peptide (OVA;
residues 323-339) for 6h. BMDCs were then co-incubated with naïve CD4+ CD25
- splenocytes
from OT-II transgenic mice at a ratio of 1:2. Recombinant murine IL-2 [10ng/ml] (R&D
Systems, Minneapolis, MN, USA) and recombinant human TGF-β1 [10ng/ml] (PeproTech,
Rocky Hill, NJ, USA) were added to drive regulatory T cell development (Kryczek et al., 2007;
Malek, 2003). Cells were cultured for 72h before they were harvested, supernatants collected,
and cells were stained with αCD4 and αFoxP3 fluorophores to examine regulatory T cell (Treg)
development. Loading BMDCs with a low dose of the ovalbumin antigen (10ng/ml) combined
with addition of the recombinant cytokines IL-2 and TGF-β1 was sufficient to drive CD4+
FoxP3+ Treg development (Figure 3.6-1A). Furthermore STAT3 KO BMDCs induced
development of lower levels of CD4+ FoxP3
+ Tregs (Figure 3.6-1B). However both STAT3 KO
and WT BMDCs treated with H. pylori did not induce production of CD4+ FoxP3
+ Tregs
(Figure 3.6-1C, D).
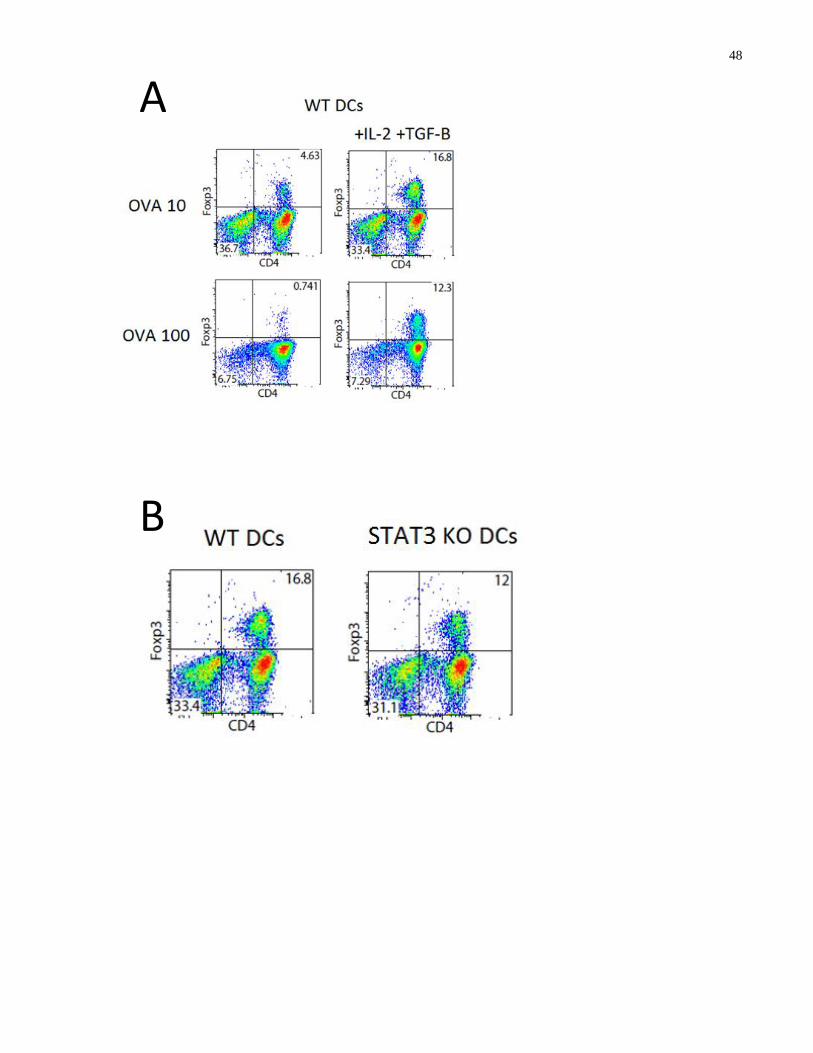
48
A
B
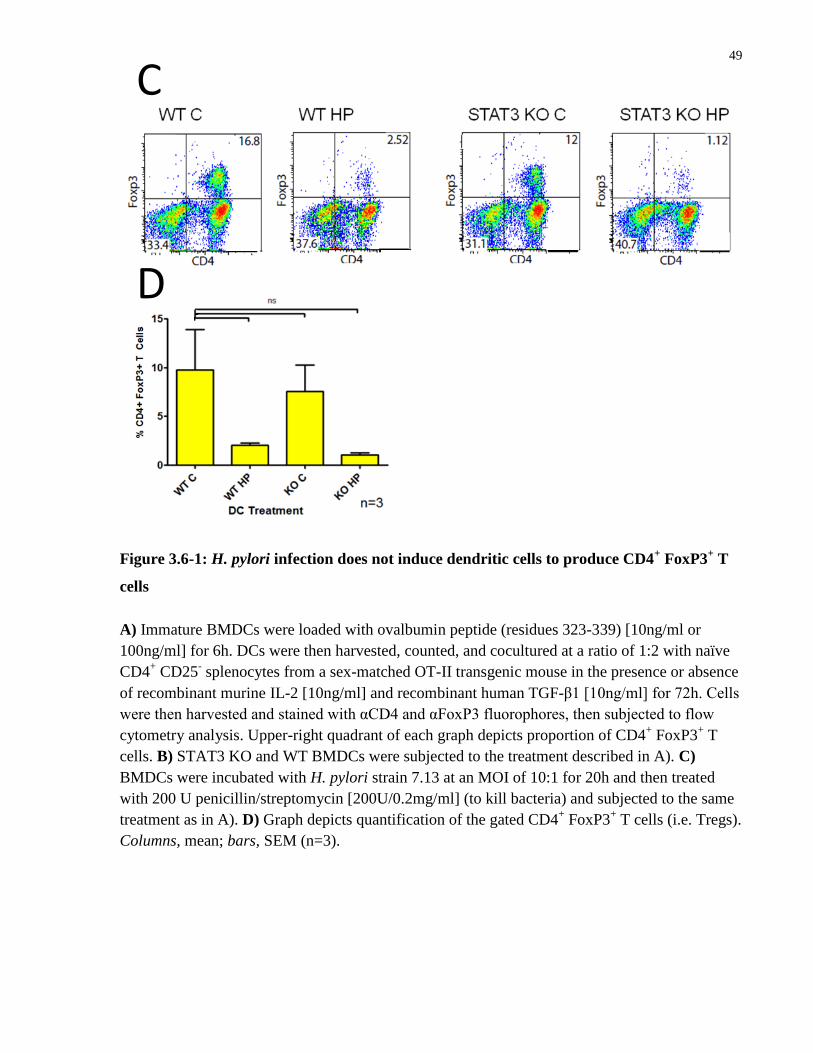
49
Figure 3.6-1: H. pylori infection does not induce dendritic cells to produce CD4+ FoxP3
+ T
cells
A) Immature BMDCs were loaded with ovalbumin peptide (residues 323-339) [10ng/ml or
100ng/ml] for 6h. DCs were then harvested, counted, and cocultured at a ratio of 1:2 with naïve
CD4+ CD25
- splenocytes from a sex-matched OT-II transgenic mouse in the presence or absence
of recombinant murine IL-2 [10ng/ml] and recombinant human TGF-β1 [10ng/ml] for 72h. Cells
were then harvested and stained with αCD4 and αFoxP3 fluorophores, then subjected to flow
cytometry analysis. Upper-right quadrant of each graph depicts proportion of CD4+ FoxP3
+ T
cells. B) STAT3 KO and WT BMDCs were subjected to the treatment described in A). C)
BMDCs were incubated with H. pylori strain 7.13 at an MOI of 10:1 for 20h and then treated
with 200 U penicillin/streptomycin [200U/0.2mg/ml] (to kill bacteria) and subjected to the same
treatment as in A). D) Graph depicts quantification of the gated CD4+ FoxP3
+ T cells (i.e. Tregs).
Columns, mean; bars, SEM (n=3).
C
D

50
Chapter 4 Discussion
4
The ability to alter and evade the host immune response is crucial for the survival and persistence
of H. pylori in the host gastric mucosa. As dendritic cells are key orchestrators of the host
immune response, they are ideal targets for the pathogen’s immune-manipulating efforts. The
interaction between H. pylori and dendritic cells is beginning to be elucidated, but the
mechanisms by which H. pylori affects DC function are still being determined.
This study demonstrated for the first time that H. pylori activated the STAT3 pathway in
dendritic cells. This STAT3 activation serves to blunt DC maturation, as H. pylori infection
induced more maturation (as assessed by expression of the maturation markers MHC II and
CD86) in STAT3 KO DCs as compared to control DCs. This is consistent with current literature
indicating that STAT3 is a negative regulator of DC maturation and function (Iwata-Kajihara et
al., 2011; Lin et al., 2010; Melillo et al., 2010). The H. pylori-mediated STAT3 activation in
dendritic cells represents a potential mechanism by which H. pylori manipulates the immune
system to persist and promote carcinogenesis. STAT3 activation in DCs is often associated with
tolerogenic functions (Barton, 2006; Cheng et al., 2003; Nefedova et al., 2005); therefore this H.
pylori-induced STAT3 activation could potentially induce tolerant DCs that suppress immune
responses to the bacteria instead of inducing effector immune responses. This represents a novel
potential mechanism by which the bacterium evades the host immune response to persist in the
gastric mucosa and may also help to explain the causative role of H. pylori in gastric cancer
development. Current evidence indicates that STAT3 activation in DCs contributes to tumor
progression by preventing DC maturation and thus decreasing the host tumor
immunosurveillance (Lin et al., 2010). Indeed it was demonstrated that STAT3-depleted DCs
were more resistant to cancer-cell derived inhibitory factors (Iwata-Kajihara et al., 2011).
Therefore the activation of STAT3 in DCs by H. pylori may potentially inhibit the DC responses
to tumors that develop in the gastric mucosa while simultaneously inhibiting the DC responses to
the bacterium.

51
In this study IL-6 was used as a control to drive STAT3 activation in the dendritic cells, and
while H. pylori induced STAT3 activation, it was not as pronounced at the IL-6-mediated
STAT3 activation (Figure 3.1-1). As IL-6 is a ligand that binds to its receptor to induce STAT3
activation, a strong induction of STAT3 phosphorylation upon treatment with this pure ligand is
expected. However live H. pylori has numerous virulence factors, secreted proteins, and
adhesion molecules, and interacts with the DC in multiple ways. Therefore it is not surprising
that the potency of the DC STAT3 activation by this live organism is slightly lower than that of
the pure ligand IL-6.
In response to H. pylori infection DC maturation is blunted and the dendritic cells secrete a
specific cytokine profile, including IL-10, the IL-12p40 subunit, TNF-α, and an increased
IL10:IL-12p40 ratio. IL-10 is a multifunctional cytokine that acts as an autocrine and paracrine
inhibitor of DC maturation and function (Corinti et al., 2001). IL-10 also activates the STAT3
pathway (Lin et al., 2010). Therefore an IL-10-neutralizing antibody was used to determine if the
H. pylori-mediated IL-10 secretion was acting as an autocrine/paracrine activator of STAT3.
Treatment with the neutralizing antibody completed abrogated the H. pylori-mediated STAT3
activation in the DCs. Furthermore anti-IL-10 treatment resulted in increased maturation of DCs
in response to H. pylori infection, indicating that this IL-10 autocrine/paracrine activation of
STAT3 is responsible for the blunted DC maturation.
IL-10 has been shown to possess immunoregulatory potential and induce the differentiation of
FoxP3+ Tregs (Lan et al., 2006). Furthermore a recent study demonstrated that H. pylori-infected
murine BMDCs drove the development of FoxP3+ Tregs that had potent immunoregulatory
capacities (Oertli et al., 2012). Therefore we co-incubated our H. pylori-infected dendritic cells
with naïve CD4+ T cells and examined the ensuing FoxP3 expression in an effort to replicate
their results. However, we found that H. pylori-infected DCs did not drive FoxP3 expression in T
cells. Methodological differences can potentially explain the differing results, as the BMDCs
were grown using slightly differing protocols. Furthermore Oertli et al. utilized the pre-mouse
clinical isolate strain PMSS1, while our study used the 7.13 mouse-adapted strain. Importantly
the 7.13 strain has been shown to be highly carcinogenic and injects significant amounts of the
CagA toxin into cells (Franco et al., 2008). Interestingly, recent evidence indicates that tolerant
responses (mediated by Tregs) rather than immunity (mediated by effector T cells) are protective
against H. pylori-induced gastric cancer precursor lesions (Arnold et al., 2011b). Therefore it is

52
entirely plausible that a carcinogenic strain of H. pylori (7.13) would not drive Treg development
while a non-carcinogenic strain will induce tolerant Treg responses.
Indeed the full complement of cytokines released by DCs in response to H. pylori infection likely
play an important role in pathogenesis. TNF-α is a pleiotropic cytokine which, while primarily
known to be proinflammatory, has also been shown to play immunoregulatory roles. In fact
dendritic-cell derived TNF-α has been shown to be necessary for the development of IL-10-
producing CD4+ T cells (Hirata et al., 2010). Furthermore TNF-α was found to upregulate
Programmed Death-1 (PD-1) levels on monocytes to cause IL-10 production, which effectively
inhibited T cell expansion and function (Said et al., 2010). These studies demonstrate that TNF-α
has clear immunoregulatory potential, therefore it is plausible that H. pylori-induced DC
secretion of TNF-α may act in an immunoregulatory manner, complementing the IL-10 secretion
and STAT3 activation to dampen the immune response and promote persistence.
The IL-12 cytokine is composed of a p40 (40 kDa) and a p35 (35kDa) subunit; the biologically
active form consists of two covalently linked chains of p40 and p35 and is known as IL-12p70
(Oppmann et al., 2000). The assays used in this study detected the secretion of the IL-12p40
subunit from the DC in response to H. pylori infection. Importantly the p40 subunit is shared by
another member of the IL-12 cytokine family: IL-23. IL-23 is composed of the p40 subunit and
another 19 kDa subunit, p19 (Oppmann et al., 2000), and can mediate pro-carcinogenic immune
responses (Kortylewski et al., 2009). IL-23 induces the development of Th17 cells (Iwakura and
Ishigame, 2006). Interestingly Th17 cells are proinflammatory and STAT3 signaling has been
shown to play a role in Th17 development (Harris et al., 2007). The highly inflammatory
STAT3-dependent Th17 response has been shown to be pro-carcinogenic (Wang et al., 2009).
Therefore the potential implications of H. pylori-mediated DC secretion of IL-23 for disease
pathogenesis are intriguing and require further study.
Taken together, these results present a potential model of H. pylori persistence and pathogenesis.
H. pylori induce the secretion of IL-10 from the dendritic cell, which acts in an autocrine and
paracrine fashion to activate STAT3 in DCs, blunting maturation. This inhibits the development
of proper effector T cell and other immune responses, facilitating bacterial persistence. However
the STAT3 activation in the DCs may also decreases DC tumor immunosurveillance. Therefore
the manipulation of the DC by H. pylori is a double-edged sword: it induces the secretion of

53
proinflammatory mediators which drive carcinogenesis, and also inhibits DC tumor
immunosurveillance to prevent recognition and DC-mediated immune elimination of the
developing tumor cells.
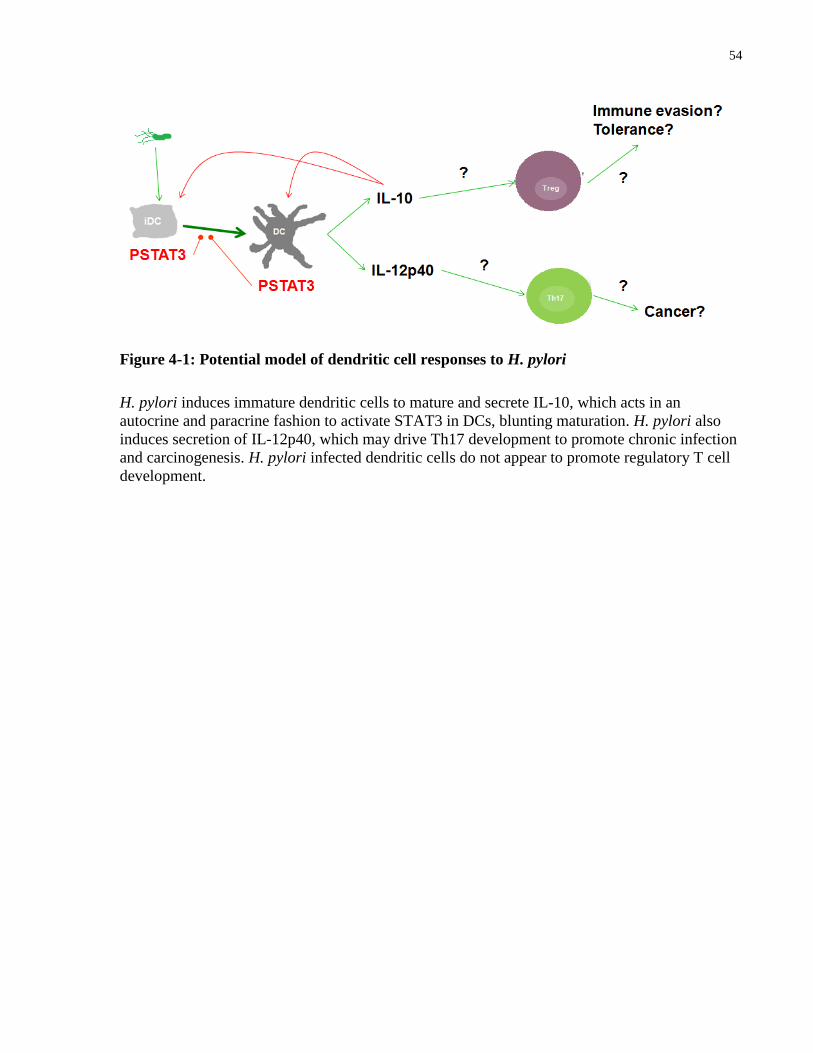
54
Figure 4-1: Potential model of dendritic cell responses to H. pylori
H. pylori induces immature dendritic cells to mature and secrete IL-10, which acts in an
autocrine and paracrine fashion to activate STAT3 in DCs, blunting maturation. H. pylori also
induces secretion of IL-12p40, which may drive Th17 development to promote chronic infection
and carcinogenesis. H. pylori infected dendritic cells do not appear to promote regulatory T cell
development.

55
While this study provides a reductionist examination of the interaction between H. pylori and the
dendritic cell, it is important to recognize the limitations of an in vitro model of host-pathogen
interaction. In the infected host the bacterium does not encounter DCs in isolation; rather it
interacts with a plethora of host cells including the gastric epithelial cell and likely other immune
cells such as macrophages, neutrophils, and natural killer cells (Alvarez-Arellano et al., 2007;
Jones et al., 1999; Lewis et al., 2011; Yun et al., 2005). These interactions will likely influence
the DC and immune response to the bacteria. Indeed preliminary data from our laboratory
suggests that exosomes (intercellular communicative vesicles) secreted by H. pylori-infected
epithelial cells can influence DC maturation (data not shown). Therefore while the in vitro
interaction between H. pylori and cultured murine dendritic cells is an appropriate method for
studying this pathogen, it is important to remember that these interactions take place within the
context of the full host-pathogen relationship. It has been shown that gastric stromal factors can
condition the DC response to H. pylori, suppressing H. pylori-stimulated DC activation
(Bimczok et al., 2011). This demonstrates that while in vitro assays are useful for examining the
interaction between pathogens and isolated cell types, the true in vivo host-pathogen interaction
is likely conditioned by numerous factors and may significantly differ from the in vitro results in
the laboratory. Therefore this STAT3 activation in DCs may be validated in human H. pylori
infection. Gastric tissue sections from H. pylori-infected patients can be assayed for the presence
of STAT3+ CD11c
+ dendritic cells and compared to tissue sections from uninfected control
subjects. This would validate these in vitro findings in human H. pylori infection and support the
hypothesis that STAT3 activation in DCs is a mechanism by which this pathogen manipulates
the host immune response.
While the generation of murine bone marrow-derived dendritic cells provides a good tool to
study the interaction between H. pylori and immature DCs, there are other methods for
generating cultured dendritic cells. One alternative DC culture method involves the
differentiation of harvested peripheral blood monocytes into DCs (Guiney et al., 2003). The use
of DCs derived from human peripheral blood monocytes would allow the validation of H. pylori-
induced STAT3 activation in human DCs. Additionally gastric dendritic cells can be harvested
from the gastric lamina propria of subjects and infected with H. pylori (Bimczok et al., 2010).
This would allow for the study of host-generated DCs, which may be more pertinent to human

56
infection than DCs cultured from precursors in a Petri dish. However despite these limitations
bone marrow-derived dendritic cells remain an inexpensive, non-invasive, valuable tool to study
the interaction between H. pylori and DCs. Other models can be used to validate findings and
explore their relevance in infection.
There are various strains of H. pylori that differ on many levels, such as pathogenicity and
presence of virulence factors. Some strains express a functional cag PAI and inject the CagA
toxin, others express the cag PAI and construct a functional T4SS but do not produce CagA,
while still others do not express the cag PAI at all. Strains also differ in expression of VacA and
the outer membrane protein OipA. Strains that express the intact T4SS and inject the CagA toxin
are more carcinogenic than strains that do not express these factors, while the presence of VacA
and OipA is also associated with pathological outcomes (Franco et al., 2008). This study used the
laboratory mouse-adapted H. pylori strain 7.13, which injects large amounts of CagA into host
cells (Franco et al., 2008). The necessity of the virulence factors for STAT3 activation in the
DCs is not examined in this study, nor is the relevance for other H. pylori strains. Despite these
limitations, this study describes a novel interaction H. pylori and the dendritic cell, an important
moderator of the host immune response.
In conclusion, these results demonstrated that H. pylori infection induces the activation of the
STAT3 signaling pathway in dendritic cells. Importantly IL-10 secreted by the DC acted as an
autocrine/paracrine activator of STAT3 to blunt DC maturation. Infected DCs also secreted a
specific cytokine profile in response to H. pylori infection, which does not appear to drive Treg
development. Overall this study describes a novel mechanism by which H. pylori alters the host
immune response to facilitate persistence, a mechanism that may also contribute to the
carcinogenic potential of this pathogen.

57
Chapter 5 Future Directions
The results of this study provide some exciting insights into the interaction between H. pylori
and dendritic cells. They also provide multiple future directions to further examine the effect of
H. pylori infection on the host immune response. The cytokines released from the STAT3 KO
BMDCs infected with H. pylori still need to be fully elucidated and should provide clues as to
the specific effect that the STAT3 activation has on the DC cytokine release profile. Particularly
it will help to elucidate whether STAT3 activation induces a change in the ratio of the pro-
versus anti-inflammatory cytokines that are released. Furthermore the presence of other
cytokines such as IL-18 and TGF-β in the H. pylori-treated DC supernatants was not examined
and could be explored in the future. This is pertinent since the Oertli paper found IL-18 to be
critical for the induction of Foxp3+ Treg development by H. pylori-treated DCs (Oertli et al.,
2012). Similarly TGF-β is a critical cytokine in the development of Tregs as well as Th17 cells,
so the presence of this cytokine should be examined to shed light on the T cell responses being
driven by the H. pylori-infected DCs. The cytokines released from the DC:T cell cocultures will
also help elucidate the phenotype of T cell which is being produced by the dendritic cell in
response to H. pylori infection.
The mechanisms by which H. pylori induces IL-10 secretion that activates STAT3 will be
interesting to elucidate. Preliminary data indicates that STAT3 activation in the dendritic cells
occurs independently of the presence of the CagA toxin (data not shown). Furthermore a heat-
stable molecule appears to play a role (data not shown). Previous studies have implicated TLR2,
TLR4, and TLR9 in recognition of H. pylori by DCs, which lead to secretion of proinflammatory
cytokines (Rad et al., 2009). Therefore the use of genetically engineered KO mice that lack
expression of these innate immune PRRs will help elucidate the role that they play in the IL-10
secretion and STAT3 activation shown by DCs in response to H. pylori infection. Interestingly
the intracellular PRR Nod1 has been shown to recognize H. pylori peptidoglycan that is injected
into the host cell by the cag PAI-encoded T4SS (Grubman et al., 2010; Viala et al., 2004).
Furthermore Nod1 has been shown to work synergistically with TLR2 and TLR4 to induce the
secretion of IL-10 from DCs (Fritz et al., 2007). Therefore it is possible that Nod1 signaling in

58
response to H. pylori plays a role in the dendritic cell secretion of IL-10, which leads to the
autocrine/paracrine activation of STAT3. The use of Nod1-specific KO mice will help to
elucidate the role of this H. pylori-sensing innate immune receptor.
Furthermore correlation of STAT3 activation in DCs with human disease progression will
strongly support the role of this pathway in H. pylori pathogenesis. To this end we have access to
paraffin-embedded gastric tissues sections from H. pylori patients at the different precancerous
stages, including gastritis, atrophy, metaplasia, dysplasia, and adenocarcinoma, as well as control
human tissue sections. These sections will be subjected to immunohistochemical and
immunofluorescent staining to examine the expression of phosphorylated (activated) STAT3 in
CD11c+ dendritic cells. If increases in the number of CD11c
+ PSTAT3
+ dendritic cells are seen
in the progression from gastritis through atrophy, metaplasia, dysplasia, and carcinoma, it would
strongly support the hypothesis that STAT3 activation in the DC plays an important role in H.
pylori carcinogenesis.
Overall this study has demonstrated a novel mechanism by which H. pylori manipulates the
dendritic cell, a key orchestrator of the immune response, to promote immune evasion and
bacterial persistence. These future studies will further elucidate the mechanisms by which H.
pylori induces the STAT3 activation in the DC, and help to delineate the role of this activation in
the pathogenesis of the bacteria.

59
References
Akopyants, N.S., Clifton, S.W., Kersulyte, D., Crabtree, J.E., Youree, B.E., Reece, C.A.,
Bukanov, N.O., Drazek, E.S., Roe, B.A., and Berg, D.E. (1998). Analyses of the cag
pathogenicity island of Helicobacter pylori. Mol Microbiol 28, 37-53.
Alvarez-Arellano, L., Camorlinga-Ponce, M., Maldonado-Bernal, C., and Torres, J. (2007).
Activation of human neutrophils with Helicobacter pylori and the role of Toll-like receptors 2
and 4 in the response. FEMS Immunol Med Microbiol 51, 473-479.
Amieva, M.R., and El-Omar, E.M. (2008). Host-bacterial interactions in Helicobacter pylori
infection. Gastroenterology 134, 306-323.
Amieva, M.R., Vogelmann, R., Covacci, A., Tompkins, L.S., Nelson, W.J., and Falkow, S.
(2003). Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA.
Science 300, 1430-1434.
Ang, M. (2010). 'The effect of Helicobacter pylori on innate immunity', MSc thesis, University
of Toronto.
Arnold, I.C., Dehzad, N., Reuter, S., Martin, H., Becher, B., Taube, C., and Muller, A. (2011a).
Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of
regulatory T cells. J Clin Invest 121, 3088-3093.
Arnold, I.C., Lee, J.Y., Amieva, M.R., Roers, A., Flavell, R.A., Sparwasser, T., and Muller, A.
(2011b). Tolerance rather than immunity protects from Helicobacter pylori-induced gastric
preneoplasia. Gastroenterology 140, 199-209.
Atherton, J.C., and Blaser, M.J. (2009). Coadaptation of Helicobacter pylori and humans: ancient
history, modern implications. J Clin Invest 119, 2475-2487.
Backhed, F., Rokbi, B., Torstensson, E., Zhao, Y., Nilsson, C., Seguin, D., Normark, S., Buchan,
A.M., and Richter-Dahlfors, A. (2003). Gastric mucosal recognition of Helicobacter pylori is
independent of Toll-like receptor 4. J Infect Dis 187, 829-836.
Bagnoli, F., Buti, L., Tompkins, L., Covacci, A., and Amieva, M.R. (2005). Helicobacter pylori
CagA induces a transition from polarized to invasive phenotypes in MDCK cells. Proc Natl Acad
Sci U S A 102, 16339-16344.
Barnden, M.J., Allison, J., Heath, W.R., and Carbone, F.R. (1998). Defective TCR expression in
transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of
heterologous regulatory elements. Immunol Cell Biol 76, 34-40.
Barton, B.E. (2006). STAT3: a potential therapeutic target in dendritic cells for the induction of
transplant tolerance. Expert Opin Ther Targets 10, 459-470.

60
Basso, D., Zambon, C.F., Letley, D.P., Stranges, A., Marchet, A., Rhead, J.L., Schiavon, S.,
Guariso, G., Ceroti, M., Nitti, D., et al. (2008). Clinical relevance of Helicobacter pylori cagA
and vacA gene polymorphisms. Gastroenterology 135, 91-99.
Bimczok, D., Clements, R.H., Waites, K.B., Novak, L., Eckhoff, D.E., Mannon, P.J., Smith,
P.D., and Smythies, L.E. (2010). Human primary gastric dendritic cells induce a Th1 response to
H. pylori. Mucosal Immunol 3, 260-269.
Bimczok, D., Grams, J.M., Stahl, R.D., Waites, K.B., Smythies, L.E., and Smith, P.D. (2011).
Stromal Regulation of Human Gastric Dendritic Cells Restricts the Th1 Response to
Helicobacter pylori. Gastroenterology 141, 929-938.
Blaser, M.J., Perez-Perez, G.I., Kleanthous, H., Cover, T.L., Peek, R.M., Chyou, P.H.,
Stemmermann, G.N., and Nomura, A. (1995). Infection with Helicobacter pylori strains
possessing cagA is associated with an increased risk of developing adenocarcinoma of the
stomach. Cancer Res 55, 2111-2115.
Boncristiano, M., Paccani, S.R., Barone, S., Ulivieri, C., Patrussi, L., Ilver, D., Amedei, A.,
D'Elios, M.M., Telford, J.L., and Baldari, C.T. (2003). The Helicobacter pylori vacuolating toxin
inhibits T cell activation by two independent mechanisms. J Exp Med 198, 1887-1897.
Braga, W.M., Atanackovic, D., and Colleoni, G.W. (2012). The Role of Regulatory T Cells and
TH17 Cells in Multiple Myeloma. Clin Dev Immunol 2012, 293479.
Brandt, S., Kwok, T., Hartig, R., Konig, W., and Backert, S. (2005). NF-kappaB activation and
potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl
Acad Sci U S A 102, 9300-9305.
Bronte-Tinkew, D.M., Terebiznik, M., Franco, A., Ang, M., Ahn, D., Mimuro, H., Sasakawa, C.,
Ropeleski, M.J., Peek, R.M., Jr., and Jones, N.L. (2009). Helicobacter pylori cytotoxin-
associated gene A activates the signal transducer and activator of transcription 3 pathway in vitro
and in vivo. Cancer Res 69, 632-639.
Chang, L.L., Wang, S.W., Wu, I.C., Yu, F.J., Su, Y.C., Chen, Y.P., Wu, D.C., Kuo, C.H., and
Hung, C.H. (2012). Impaired dendritic cell maturation and IL-10 production following H. pylori
stimulation in gastric cancer patients. Appl Microbiol Biotechnol 96, 211-220.
Chaudhry, A., Samstein, R.M., Treuting, P., Liang, Y., Pils, M.C., Heinrich, J.M., Jack, R.S.,
Wunderlich, F.T., Bruning, J.C., Muller, W., et al. (2011). Interleukin-10 signaling in regulatory
T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34, 566-578.
Chen, W., Jin, W., Hardegen, N., Lei, K.J., Li, L., Marinos, N., McGrady, G., and Wahl, S.M.
(2003). Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells
by TGF-beta induction of transcription factor Foxp3. J Exp Med 198, 1875-1886.
Chen, Y., and Blaser, M.J. (2007). Inverse associations of Helicobacter pylori with asthma and
allergy. Arch Intern Med 167, 821-827.

61
Cheng, F., Wang, H.W., Cuenca, A., Huang, M., Ghansah, T., Brayer, J., Kerr, W.G., Takeda,
K., Akira, S., Schoenberger, S.P., et al. (2003). A critical role for Stat3 signaling in immune
tolerance. Immunity 19, 425-436.
Corinti, S., Albanesi, C., la Sala, A., Pastore, S., and Girolomoni, G. (2001). Regulatory activity
of autocrine IL-10 on dendritic cell functions. J Immunol 166, 4312-4318.
Correa, P. (1992). Human gastric carcinogenesis: a multistep and multifactorial process--First
American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res
52, 6735-6740.
Correa, P. (2004). The biological model of gastric carcinogenesis. IARC Sci Publ, 301-310.
Correa, P., and Houghton, J. (2007). Carcinogenesis of Helicobacter pylori. Gastroenterology
133, 659-672.
Corthay, A. (2009). How do regulatory T cells work? Scand J Immunol 70, 326-336.
Cover, T.L., and Blanke, S.R. (2005). Helicobacter pylori VacA, a paradigm for toxin
multifunctionality. Nat Rev Microbiol 3, 320-332.
Cover, T.L., and Blaser, M.J. (2009). Helicobacter pylori in health and disease. Gastroenterology
136, 1863-1873.
Cover, T.L., Krishna, U.S., Israel, D.A., and Peek, R.M., Jr. (2003). Induction of gastric
epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res 63, 951-957.
Croxen, M.A., Sisson, G., Melano, R., and Hoffman, P.S. (2006). The Helicobacter pylori
chemotaxis receptor TlpB (HP0103) is required for pH taxis and for colonization of the gastric
mucosa. J Bacteriol 188, 2656-2665.
Cunningham, M.D., Seachord, C., Ratcliffe, K., Bainbridge, B., Aruffo, A., and Darveau, R.P.
(1996). Helicobacter pylori and Porphyromonas gingivalis lipopolysaccharides are poorly
transferred to recombinant soluble CD14. Infect Immun 64, 3601-3608.
Curotto de Lafaille, M.A., and Lafaille, J.J. (2009). Natural and adaptive foxp3+ regulatory T
cells: more of the same or a division of labor? Immunity 30, 626-635.
de Sablet, T., Piazuelo, M.B., Shaffer, C.L., Schneider, B.G., Asim, M., Chaturvedi, R., Bravo,
L.E., Sicinschi, L.A., Delgado, A.G., Mera, R.M., et al. (2011). Phylogeographic origin of
Helicobacter pylori is a determinant of gastric cancer risk. Gut 60, 1189-1195.
Dellon, E.S., Peery, A.F., Shaheen, N.J., Morgan, D.R., Hurrell, J.M., Lash, R.H., and Genta,
R.M. (2011). Inverse Association of Esophageal Eosinophilia With Helicobacter pylori Based on
Analysis of a US Pathology Database. Gastroenterology.
Dhar, S.K., Soni, R.K., Das, B.K., and Mukhopadhyay, G. (2003). Molecular mechanism of
action of major Helicobacter pylori virulence factors. Mol Cell Biochem 253, 207-215.

62
Dong, C. (2008). TH17 cells in development: an updated view of their molecular identity and
genetic programming. Nat Rev Immunol 8, 337-348.
Dubois, A., and Boren, T. (2007). Helicobacter pylori is invasive and it may be a facultative
intracellular organism. Cell Microbiol 9, 1108-1116.
El-Omar, E.M., Oien, K., El-Nujumi, A., Gillen, D., Wirz, A., Dahill, S., Williams, C., Ardill,
J.E., and McColl, K.E. (1997). Helicobacter pylori infection and chronic gastric acid
hyposecretion. Gastroenterology 113, 15-24.
Enk, A.H., Angeloni, V.L., Udey, M.C., and Katz, S.I. (1993). Inhibition of Langerhans cell
antigen-presenting function by IL-10. A role for IL-10 in induction of tolerance. J Immunol 151,
2390-2398.
Ernst, M., and Putoczki, T.L. (2011). Stat3 - Linking inflammation to (gastrointestinal)
tumourigenesis. Clin Exp Pharmacol Physiol.
Foti, M., Granucci, F., Aggujaro, D., Liboi, E., Luini, W., Minardi, S., Mantovani, A., Sozzani,
S., and Ricciardi-Castagnoli, P. (1999). Upon dendritic cell (DC) activation chemokines and
chemokine receptor expression are rapidly regulated for recruitment and maintenance of DC at
the inflammatory site. Int Immunol 11, 979-986.
Franco, A.T., Johnston, E., Krishna, U., Yamaoka, Y., Israel, D.A., Nagy, T.A., Wroblewski,
L.E., Piazuelo, M.B., Correa, P., and Peek, R.M., Jr. (2008). Regulation of gastric carcinogenesis
by Helicobacter pylori virulence factors. Cancer Res 68, 379-387.
Fritz, J.H., Le Bourhis, L., Sellge, G., Magalhaes, J.G., Fsihi, H., Kufer, T.A., Collins, C., Viala,
J., Ferrero, R.L., Girardin, S.E., et al. (2007). Nod1-mediated innate immune recognition of
peptidoglycan contributes to the onset of adaptive immunity. Immunity 26, 445-459.
Fu, S., Zhang, N., Yopp, A.C., Chen, D., Mao, M., Zhang, H., Ding, Y., and Bromberg, J.S.
(2004). TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25 - precursors. Am J
Transplant 4, 1614-1627.
Galmiche, A., Rassow, J., Doye, A., Cagnol, S., Chambard, J.C., Contamin, S., de Thillot, V.,
Just, I., Ricci, V., Solcia, E., et al. (2000). The N-terminal 34 kDa fragment of Helicobacter
pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J
19, 6361-6370.
Gauthier, N.C., Monzo, P., Gonzalez, T., Doye, A., Oldani, A., Gounon, P., Ricci, V., Cormont,
M., and Boquet, P. (2007). Early endosomes associated with dynamic F-actin structures are
required for late trafficking of H. pylori VacA toxin. J Cell Biol 177, 343-354.
Gebert, B., Fischer, W., Weiss, E., Hoffmann, R., and Haas, R. (2003). Helicobacter pylori
vacuolating cytotoxin inhibits T lymphocyte activation. Science 301, 1099-1102.
Girardin, S.E., Boneca, I.G., Carneiro, L.A., Antignac, A., Jehanno, M., Viala, J., Tedin, K.,
Taha, M.K., Labigne, A., Zahringer, U., et al. (2003). Nod1 detects a unique muropeptide from
gram-negative bacterial peptidoglycan. Science 300, 1584-1587.

63
Greenlund, A.C., Morales, M.O., Viviano, B.L., Yan, H., Krolewski, J., and Schreiber, R.D.
(1995). Stat recruitment by tyrosine-phosphorylated cytokine receptors: an ordered reversible
affinity-driven process. Immunity 2, 677-687.
Groux, H. (2003). Type 1 T-regulatory cells: their role in the control of immune responses.
Transplantation 75, 8S-12S.
Grubman, A., Kaparakis, M., Viala, J., Allison, C., Badea, L., Karrar, A., Boneca, I.G., Le
Bourhis, L., Reeve, S., Smith, I.A., et al. (2010). The innate immune molecule, NOD1, regulates
direct killing of Helicobacter pylori by antimicrobial peptides. Cell Microbiol 12, 626-639.
Guiney, D.G., Hasegawa, P., and Cole, S.P. (2003). Helicobacter pylori preferentially induces
interleukin 12 (IL-12) rather than IL-6 or IL-10 in human dendritic cells. Infect Immun 71, 4163-
4166.
Harris, T.J., Grosso, J.F., Yen, H.R., Xin, H., Kortylewski, M., Albesiano, E., Hipkiss, E.L.,
Getnet, D., Goldberg, M.V., Maris, C.H., et al. (2007). Cutting edge: An in vivo requirement for
STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol 179,
4313-4317.
Hirata, N., Yanagawa, Y., Satoh, M., Ogura, H., Ebihara, T., Noguchi, M., Matsumoto, M.,
Togashi, H., Seya, T., Onoe, K., et al. (2010). Dendritic cell-derived TNF-alpha is responsible
for development of IL-10-producing CD4+ T cells. Cell Immunol 261, 37-41.
Ivanov, II, McKenzie, B.S., Zhou, L., Tadokoro, C.E., Lepelley, A., Lafaille, J.J., Cua, D.J., and
Littman, D.R. (2006). The orphan nuclear receptor RORgammat directs the differentiation
program of proinflammatory IL-17+ T helper cells. Cell 126, 1121-1133.
Iwakura, Y., and Ishigame, H. (2006). The IL-23/IL-17 axis in inflammation. J Clin Invest 116,
1218-1222.
Iwata-Kajihara, T., Sumimoto, H., Kawamura, N., Ueda, R., Takahashi, T., Mizuguchi, H.,
Miyagishi, M., Takeda, K., and Kawakami, Y. (2011). Enhanced cancer immunotherapy using
STAT3-depleted dendritic cells with high Th1-inducing ability and resistance to cancer cell-
derived inhibitory factors. J Immunol 187, 27-36.
Jain, P., Luo, Z.Q., and Blanke, S.R. (2011). Helicobacter pylori vacuolating cytotoxin A (VacA)
engages the mitochondrial fission machinery to induce host cell death. Proc Natl Acad Sci U S A
108, 16032-16037.
Jones, N.L., Day, A.S., Jennings, H.A., and Sherman, P.M. (1999). Helicobacter pylori induces
gastric epithelial cell apoptosis in association with increased Fas receptor expression. Infect
Immun 67, 4237-4242.
Kao, J.Y., Rathinavelu, S., Eaton, K.A., Bai, L., Zavros, Y., Takami, M., Pierzchala, A., and
Merchant, J.L. (2006). Helicobacter pylori-secreted factors inhibit dendritic cell IL-12 secretion:
a mechanism of ineffective host defense. Am J Physiol Gastrointest Liver Physiol 291, G73-81.

64
Kao, J.Y., Zhang, M., Miller, M.J., Mills, J.C., Wang, B., Liu, M., Eaton, K.A., Zou, W., Berndt,
B.E., Cole, T.S., et al. (2010). Helicobacter pylori immune escape is mediated by dendritic cell-
induced Treg skewing and Th17 suppression in mice. Gastroenterology 138, 1046-1054.
Khamri, W., Walker, M.M., Clark, P., Atherton, J.C., Thursz, M.R., Bamford, K.B., Lechler,
R.I., and Lombardi, G. (2010). Helicobacter pylori stimulates dendritic cells to induce
interleukin-17 expression from CD4+ T lymphocytes. Infect Immun 78, 845-853.
Kim, D.J., Chan, K.S., Sano, S., and Digiovanni, J. (2007). Signal transducer and activator of
transcription 3 (Stat3) in epithelial carcinogenesis. Mol Carcinog 46, 725-731.
Kisseleva, T., Bhattacharya, S., Braunstein, J., and Schindler, C.W. (2002). Signaling through
the JAK/STAT pathway, recent advances and future challenges. Gene 285, 1-24.
Kortylewski, M., Xin, H., Kujawski, M., Lee, H., Liu, Y., Harris, T., Drake, C., Pardoll, D., and
Yu, H. (2009). Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor
microenvironment. Cancer Cell 15, 114-123.
Kranzer, K., Eckhardt, A., Aigner, M., Knoll, G., Deml, L., Speth, C., Lehn, N., Rehli, M., and
Schneider-Brachert, W. (2004). Induction of maturation and cytokine release of human dendritic
cells by Helicobacter pylori. Infect Immun 72, 4416-4423.
Kranzer, K., Sollner, L., Aigner, M., Lehn, N., Deml, L., Rehli, M., and Schneider-Brachert, W.
(2005). Impact of Helicobacter pylori virulence factors and compounds on activation and
maturation of human dendritic cells. Infect Immun 73, 4180-4189.
Kryczek, I., Wei, S., Zou, L., Altuwaijri, S., Szeliga, W., Kolls, J., Chang, A., and Zou, W.
(2007). Cutting edge: Th17 and regulatory T cell dynamics and the regulation by IL-2 in the
tumor microenvironment. J Immunol 178, 6730-6733.
Kusters, J.G., van Vliet, A.H., and Kuipers, E.J. (2006). Pathogenesis of Helicobacter pylori
infection. Clin Microbiol Rev 19, 449-490.
Ladoire, S., Martin, F., and Ghiringhelli, F. (2011). Prognostic role of FOXP3+ regulatory T
cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol
Immunother 60, 909-918.
Lan, Y.Y., Wang, Z., Raimondi, G., Wu, W., Colvin, B.L., de Creus, A., and Thomson, A.W.
(2006). "Alternatively activated" dendritic cells preferentially secrete IL-10, expand
Foxp3+CD4+ T cells, and induce long-term organ allograft survival in combination with
CTLA4-Ig. J Immunol 177, 5868-5877.
Lenahan, C., and Avigan, D. (2006). Dendritic cell defects in patients with cancer: mechanisms
and significance. Breast Cancer Res 8, 101.
Leunk, R.D., Johnson, P.T., David, B.C., Kraft, W.G., and Morgan, D.R. (1988). Cytotoxic
activity in broth-culture filtrates of Campylobacter pylori. J Med Microbiol 26, 93-99.

65
Levy, D.E., and Darnell, J.E., Jr. (2002). Stats: transcriptional control and biological impact. Nat
Rev Mol Cell Biol 3, 651-662.
Lewis, N.D., Asim, M., Barry, D.P., de Sablet, T., Singh, K., Piazuelo, M.B., Gobert, A.P.,
Chaturvedi, R., and Wilson, K.T. (2011). Immune evasion by Helicobacter pylori is mediated by
induction of macrophage arginase II. J Immunol 186, 3632-3641.
Lin, A., Schildknecht, A., Nguyen, L.T., and Ohashi, P.S. (2010). Dendritic cells integrate
signals from the tumor microenvironment to modulate immunity and tumor growth. Immunol
Lett 127, 77-84.
Linden, S., Nordman, H., Hedenbro, J., Hurtig, M., Boren, T., and Carlstedt, I. (2002). Strain-
and blood group-dependent binding of Helicobacter pylori to human gastric MUC5AC
glycoforms. Gastroenterology 123, 1923-1930.
Lu, Y.C., Yeh, W.C., and Ohashi, P.S. (2008). LPS/TLR4 signal transduction pathway. Cytokine
42, 145-151.
Lundgren, A., Suri-Payer, E., Enarsson, K., Svennerholm, A.M., and Lundin, B.S. (2003).
Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell
responses to H. pylori in infected individuals. Infect Immun 71, 1755-1762.
Luther, J., Dave, M., Higgins, P.D., and Kao, J.Y. (2010). Association between Helicobacter
pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the
literature. Inflamm Bowel Dis 16, 1077-1084.
Luther, J., Owyang, S.Y., Takeuchi, T., Cole, T.S., Zhang, M., Liu, M., Erb-Downward, J.,
Rubenstein, J.H., Chen, C.C., Pierzchala, A.V., et al. (2011). Helicobacter pylori DNA decreases
pro-inflammatory cytokine production by dendritic cells and attenuates dextran sodium sulphate-
induced colitis. Gut 60, 1479-1486.
Lutz, M.B., and Rossner, S. (2007). Factors influencing the generation of murine dendritic cells
from bone marrow: the special role of fetal calf serum. Immunobiology 212, 855-862.
Malek, T.R. (2003). The main function of IL-2 is to promote the development of T regulatory
cells. J Leukoc Biol 74, 961-965.
Manel, N., Unutmaz, D., and Littman, D.R. (2008). The differentiation of human T(H)-17 cells
requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat
Immunol 9, 641-649.
Melillo, J.A., Song, L., Bhagat, G., Blazquez, A.B., Plumlee, C.R., Lee, C., Berin, C., Reizis, B.,
and Schindler, C. (2010). Dendritic cell (DC)-specific targeting reveals Stat3 as a negative
regulator of DC function. J Immunol 184, 2638-2645.
Mills, K.H., and McGuirk, P. (2004). Antigen-specific regulatory T cells--their induction and
role in infection. Semin Immunol 16, 107-117.

66
Moese, S., Selbach, M., Kwok, T., Brinkmann, V., Konig, W., Meyer, T.F., and Backert, S.
(2004). Helicobacter pylori induces AGS cell motility and elongation via independent signaling
pathways. Infect Immun 72, 3646-3649.
Molinari, M., Salio, M., Galli, C., Norais, N., Rappuoli, R., Lanzavecchia, A., and Montecucco,
C. (1998). Selective inhibition of Ii-dependent antigen presentation by Helicobacter pylori toxin
VacA. J Exp Med 187, 135-140.
Montecucco, C., and de Bernard, M. (2003). Molecular and cellular mechanisms of action of the
vacuolating cytotoxin (VacA) and neutrophil-activating protein (HP-NAP) virulence factors of
Helicobacter pylori. Microbes Infect 5, 715-721.
Montecucco, C., and Rappuoli, R. (2001). Living dangerously: how Helicobacter pylori survives
in the human stomach. Nat Rev Mol Cell Biol 2, 457-466.
Murphy, K., Travers, P., Walport, M., and Janeway, C. (2012). Janeway's immunobiology (New
York, Garland Science).
Naito, M., Yamazaki, T., Tsutsumi, R., Higashi, H., Onoe, K., Yamazaki, S., Azuma, T., and
Hatakeyama, M. (2006). Influence of EPIYA-repeat polymorphism on the phosphorylation-
dependent biological activity of Helicobacter pylori CagA. Gastroenterology 130, 1181-1190.
Necchi, V., Candusso, M.E., Tava, F., Luinetti, O., Ventura, U., Fiocca, R., Ricci, V., and Solcia,
E. (2007). Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic
lesions, and cancer by Helicobacter pylori. Gastroenterology 132, 1009-1023.
Necchi, V., Manca, R., Ricci, V., and Solcia, E. (2009). Evidence for transepithelial dendritic
cells in human H. pylori active gastritis. Helicobacter 14, 208-222.
Nefedova, Y., Cheng, P., Gilkes, D., Blaskovich, M., Beg, A.A., Sebti, S.M., and Gabrilovich,
D.I. (2005). Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J Immunol 175,
4338-4346.
Niemand, C., Nimmesgern, A., Haan, S., Fischer, P., Schaper, F., Rossaint, R., Heinrich, P.C.,
and Muller-Newen, G. (2003). Activation of STAT3 by IL-6 and IL-10 in primary human
macrophages is differentially modulated by suppressor of cytokine signaling 3. J Immunol 170,
3263-3272.
Nomura, A.M., Perez-Perez, G.I., Lee, J., Stemmermann, G., and Blaser, M.J. (2002). Relation
between Helicobacter pylori cagA status and risk of peptic ulcer disease. Am J Epidemiol 155,
1054-1059.
O'Garra, A., Vieira, P.L., Vieira, P., and Goldfeld, A.E. (2004). IL-10-producing and naturally
occurring CD4+ Tregs: limiting collateral damage. J Clin Invest 114, 1372-1378.
Odenbreit, S., Puls, J., Sedlmaier, B., Gerland, E., Fischer, W., and Haas, R. (2000).
Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion.
Science 287, 1497-1500.

67
Oertli, M., Sundquist, M., Hitzler, I., Engler, D.B., Arnold, I.C., Reuter, S., Maxeiner, J.,
Hansson, M., Taube, C., Quiding-Jarbrink, M., et al. (2012). DC-derived IL-18 drives Treg
differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J
Clin Invest.
Ohnishi, N., Yuasa, H., Tanaka, S., Sawa, H., Miura, M., Matsui, A., Higashi, H., Musashi, M.,
Iwabuchi, K., Suzuki, M., et al. (2008). Transgenic expression of Helicobacter pylori CagA
induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci U S A 105,
1003-1008.
Oliveira, M.J., Costa, A.C., Costa, A.M., Henriques, L., Suriano, G., Atherton, J.C., Machado,
J.C., Carneiro, F., Seruca, R., Mareel, M., et al. (2006). Helicobacter pylori induces gastric
epithelial cell invasion in a c-Met and type IV secretion system-dependent manner. J Biol Chem
281, 34888-34896.
Oliver, A.M., Grimaldi, J.C., Howard, M.C., and Kearney, J.F. (1999). Independently ligating
CD38 and Fc gammaRIIB relays a dominant negative signal to B cells. Hybridoma 18, 113-119.
Oppmann, B., Lesley, R., Blom, B., Timans, J.C., Xu, Y., Hunte, B., Vega, F., Yu, N., Wang, J.,
Singh, K., et al. (2000). Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with
biological activities similar as well as distinct from IL-12. Immunity 13, 715-725.
Parsonnet, J., Friedman, G.D., Orentreich, N., and Vogelman, H. (1997). Risk for gastric cancer
in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 40, 297-301.
Peek, R.M., Jr., and Crabtree, J.E. (2006). Helicobacter infection and gastric neoplasia. J Pathol
208, 233-248.
Peek, R.M., Jr., Moss, S.F., Tham, K.T., Perez-Perez, G.I., Wang, S., Miller, G.G., Atherton,
J.C., Holt, P.R., and Blaser, M.J. (1997). Helicobacter pylori cagA+ strains and dissociation of
gastric epithelial cell proliferation from apoptosis. J Natl Cancer Inst 89, 863-868.
Perez-Perez, G.I., Shepherd, V.L., Morrow, J.D., and Blaser, M.J. (1995). Activation of human
THP-1 cells and rat bone marrow-derived macrophages by Helicobacter pylori
lipopolysaccharide. Infect Immun 63, 1183-1187.
Rad, R., Ballhorn, W., Voland, P., Eisenacher, K., Mages, J., Rad, L., Ferstl, R., Lang, R.,
Wagner, H., Schmid, R.M., et al. (2009). Extracellular and intracellular pattern recognition
receptors cooperate in the recognition of Helicobacter pylori. Gastroenterology 136, 2247-2257.
Rad, R., Brenner, L., Krug, A., Voland, P., Mages, J., Lang, R., Schwendy, S., Reindl, W.,
Dossumbekova, A., Ballhorn, W., et al. (2007). Toll-like receptor-dependent activation of
antigen-presenting cells affects adaptive immunity to Helicobacter pylori. Gastroenterology 133,
150-163 e153.
Raju, D., and Jones, N.L. (2010). Methods to monitor autophagy in H. pylori vacuolating
cytotoxin A (VacA)-treated cells. Autophagy 6, 138-143.

68
Reeves, E.P., Ali, T., Leonard, P., Hearty, S., O'Kennedy, R., May, F.E., Westley, B.R.,
Josenhans, C., Rust, M., Suerbaum, S., et al. (2008). Helicobacter pylori lipopolysaccharide
interacts with TFF1 in a pH-dependent manner. Gastroenterology 135, 2043-2054, 2054 e2041-
2042.
Reich, N.C., and Liu, L. (2006). Tracking STAT nuclear traffic. Nat Rev Immunol 6, 602-612.
Robinson, K., Argent, R.H., and Atherton, J.C. (2007). The inflammatory and immune response
to Helicobacter pylori infection. Best Pract Res Clin Gastroenterol 21, 237-259.
Said, E.A., Dupuy, F.P., Trautmann, L., Zhang, Y., Shi, Y., El-Far, M., Hill, B.J., Noto, A.,
Ancuta, P., Peretz, Y., et al. (2010). Programmed death-1-induced interleukin-10 production by
monocytes impairs CD4+ T cell activation during HIV infection. Nat Med 16, 452-459.
Selbach, M., Moese, S., Hauck, C.R., Meyer, T.F., and Backert, S. (2002). Src is the kinase of
the Helicobacter pylori CagA protein in vitro and in vivo. J Biol Chem 277, 6775-6778.
Sewald, X., Gebert-Vogl, B., Prassl, S., Barwig, I., Weiss, E., Fabbri, M., Osicka, R.,
Schiemann, M., Busch, D.H., Semmrich, M., et al. (2008). Integrin subunit CD18 Is the T-
lymphocyte receptor for the Helicobacter pylori vacuolating cytotoxin. Cell Host Microbe 3, 20-
29.
Sewald, X., Jimenez-Soto, L., and Haas, R. (2011). PKC-dependent endocytosis of the
Helicobacter pylori vacuolating cytotoxin in primary T lymphocytes. Cell Microbiol 13, 482-
496.
Shibata, W., Hirata, Y., Maeda, S., Ogura, K., Ohmae, T., Yanai, A., Mitsuno, Y., Yamaji, Y.,
Okamoto, M., Yoshida, H., et al. (2006). CagA protein secreted by the intact type IV secretion
system leads to gastric epithelial inflammation in the Mongolian gerbil model. J Pathol 210, 306-
314.
Singh, M., Prasad, K.N., Saxena, A., and Yachha, S.K. (2006). Helicobacter pylori induces
apoptosis of T- and B-cell lines and translocates mitochondrial apoptosis-inducing factor to
nucleus. Curr Microbiol 52, 254-260.
Steinman, R.M., and Banchereau, J. (2007). Taking dendritic cells into medicine. Nature 449,
419-426.
Strioga, M., Schijns, V., Powell, D.J., Jr., Pasukoniene, V., Dobrovolskiene, N., and Michalek, J.
(2012). Dendritic cells and their role in tumor immunosurveillance. Innate Immun.
Tammer, I., Brandt, S., Hartig, R., Konig, W., and Backert, S. (2007). Activation of Abl by
Helicobacter pylori: a novel kinase for CagA and crucial mediator of host cell scattering.
Gastroenterology 132, 1309-1319.
Terebiznik, M.R., Raju, D., Vazquez, C.L., Torbricki, K., Kulkarni, R., Blanke, S.R., Yoshimori,
T., Colombo, M.I., and Jones, N.L. (2009). Effect of Helicobacter pylori's vacuolating cytotoxin
on the autophagy pathway in gastric epithelial cells. Autophagy 5, 370-379.

69
Terebiznik, M.R., Vazquez, C.L., Torbicki, K., Banks, D., Wang, T., Hong, W., Blanke, S.R.,
Colombo, M.I., and Jones, N.L. (2006). Helicobacter pylori VacA toxin promotes bacterial
intracellular survival in gastric epithelial cells. Infect Immun 74, 6599-6614.
Tesmer, L.A., Lundy, S.K., Sarkar, S., and Fox, D.A. (2008). Th17 cells in human disease.
Immunol Rev 223, 87-113.
Torres, V.J., VanCompernolle, S.E., Sundrud, M.S., Unutmaz, D., and Cover, T.L. (2007).
Helicobacter pylori vacuolating cytotoxin inhibits activation-induced proliferation of human T
and B lymphocyte subsets. J Immunol 179, 5433-5440.
Uemura, N., Okamoto, S., Yamamoto, S., Matsumura, N., Yamaguchi, S., Yamakido, M.,
Taniyama, K., Sasaki, N., and Schlemper, R.J. (2001). Helicobacter pylori infection and the
development of gastric cancer. N Engl J Med 345, 784-789.
Viala, J., Chaput, C., Boneca, I.G., Cardona, A., Girardin, S.E., Moran, A.P., Athman, R.,
Memet, S., Huerre, M.R., Coyle, A.J., et al. (2004). Nod1 responds to peptidoglycan delivered
by the Helicobacter pylori cag pathogenicity island. Nat Immunol 5, 1166-1174.
Wang, L., Yi, T., Kortylewski, M., Pardoll, D.M., Zeng, D., and Yu, H. (2009). IL-17 can
promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med 206, 1457-1464.
Wang, T., Niu, G., Kortylewski, M., Burdelya, L., Shain, K., Zhang, S., Bhattacharya, R.,
Gabrilovich, D., Heller, R., Coppola, D., et al. (2004). Regulation of the innate and adaptive
immune responses by Stat-3 signaling in tumor cells. Nat Med 10, 48-54.
Willhite, D.C., and Blanke, S.R. (2004). Helicobacter pylori vacuolating cytotoxin enters cells,
localizes to the mitochondria, and induces mitochondrial membrane permeability changes
correlated to toxin channel activity. Cell Microbiol 6, 143-154.
Wirth, H.P., Beins, M.H., Yang, M., Tham, K.T., and Blaser, M.J. (1998). Experimental
infection of Mongolian gerbils with wild-type and mutant Helicobacter pylori strains. Infect
Immun 66, 4856-4866.
Wroblewski, L.E., Peek, R.M., Jr., and Wilson, K.T. (2010). Helicobacter pylori and gastric
cancer: factors that modulate disease risk. Clin Microbiol Rev 23, 713-739.
Wu, A.H., Crabtree, J.E., Bernstein, L., Hawtin, P., Cockburn, M., Tseng, C.C., and Forman, D.
(2003). Role of Helicobacter pylori CagA+ strains and risk of adenocarcinoma of the stomach
and esophagus. Int J Cancer 103, 815-821.
Yamasaki, E., Wada, A., Kumatori, A., Nakagawa, I., Funao, J., Nakayama, M., Hisatsune, J.,
Kimura, M., Moss, J., and Hirayama, T. (2006). Helicobacter pylori vacuolating cytotoxin
induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and
cell death, independent of vacuolation. J Biol Chem 281, 11250-11259.
Yamazaki, S., Yamakawa, A., Ito, Y., Ohtani, M., Higashi, H., Hatakeyama, M., and Azuma, T.
(2003). The CagA protein of Helicobacter pylori is translocated into epithelial cells and binds to
SHP-2 in human gastric mucosa. J Infect Dis 187, 334-337.

70
Yu, H., Pardoll, D., and Jove, R. (2009). STATs in cancer inflammation and immunity: a leading
role for STAT3. Nat Rev Cancer 9, 798-809.
Yun, C.H., Lundgren, A., Azem, J., Sjoling, A., Holmgren, J., Svennerholm, A.M., and Lundin,
B.S. (2005). Natural killer cells and Helicobacter pylori infection: bacterial antigens and
interleukin-12 act synergistically to induce gamma interferon production. Infect Immun 73,
1482-1490.
Zheng, P.Y., and Jones, N.L. (2003). Helicobacter pylori strains expressing the vacuolating
cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining TACO
(coronin 1) protein. Cell Microbiol 5, 25-40.





























