Embed Size (px)
Citation preview

POLYMER LETTERS VOL. 8, PP. 573-584(1970)
THE CATIONIC POLYMERIZATION OF “END0”-DICYCLOPEN- TADIENE, 1,2-DMYDRO-“ENDO”-DICYCLOPENTADIENE AND 9.1 0-DIHYDRO-“ENDO”-DICYCLOPENTADIENE
Synopsis
The polymerizations of “endo”dicyc1opentadiene (I), 1,2-dihydro-“endo”- dicyclopentadiene (11) and 9,1O-dihydro-“endo”-dicyclopentadiene (111), in- itiated by cationic systems, have been investigated under different conditions. Linear polymers with% in the range 1300-4500 have been obtained with (I) and (11): poly-(I) shows less than one double bond for structural unit. In the case of (111) only oligomers were observed. Copolymerization runs show a reac- tivity of (111) lower than (11). Spectroscopic and chemical analyses of the polymers obtained indicate that besides the norbornene double bond, also the cyclopentenic unsaturation, present in (I) and (111), reacts.
Some evidences for a hydride shift in cyclopentene ring, after the attack of the initiator, were collected. In the case of poly-(I) the main enchainment re- sulted from the norbornene double bond opening, but about 30% of nortricyclene repeating unit was also observed.
INTRODUCTION
Dicyclopentadiene has been widely used in the random terpolymerization with ethylene and propylene induced by’ziegler-Natta catalysts (1). In these polymerizations the diolefine reacts through the norbornene double bond open- ing, whereas the cyclopentene double bond prevailingly does not react (2).
Studies on the homopolymerization of dicyclopentadiene are not reported in the literature. Only in a paper on the ring-opening polymerization of cyclic olefms, it has been reported that “endo”-dicyclopentadiene (I) gives gelled polymers with mixed Al and W halides (3). We have observed that strong cationic initiators can polymerize (I) to soluble products having l o w m . The study of the cationic polymerization of (I) shows a particular interest be- cause in the same molecule there are two types of unsaturations, the norbornenic and the cyclopentenic double bond which can undergo cationic attack.
Monomers with these unsaturations have been subjected to cationic polymer- ization and the structure of the resulting polymers was elucidated (4). The complexity of the structure in the polymers obtained from (I) has induced us to
573 el970 John Wiley 81 Sons, Inc.

POLYMER LETTERS 574
investigate the 1,2-dihydro-“endo”-dicyclopentadiene (11) and the 9,lO-dihydro “endo”-dicyclopentadiene (111) homopolymerization and their copolymerization under the saye conditions.
Experimental
Preparation of Reagents
“Endo”-dicyclopentadiene (I) (Fluka A G) was purified by crystallization and distillation under vacuum with Podbelniak Hypercal column: VPC purity was 99.5%, the main impurity being (11) = 0.4%.
1,2-dihydro-“endo”-dicyclopentadiene (11) was prepared as previously de- scribed (5). The product was distilled twice under vacuum and after in a Nester-Faust spinning band column: VPC purity was 98%- the main impurity being (I) = 1.5%. Furthermore, traces of 1,2-dihydro-“exo”-dicyclopentadiene were present.
9,1O-dihydro-“endo”dicyclopentadiene (111) was obtained according to ref. (6). After distillation under vacuum the product was 99.5% (WC) pure.
CH2C12 was purified and dried according to ref. (7). n-Heptane and nitrobenzene were washed in usual manner and distilled
Et2A1C1, EtA1C12, A1C13, TiC14, SnCl4, BF3.0Et2 (pure grade reagents) under nitrogen: the former over LiA1H4 and the latter over BaO.
were distilled or sublimated under vacuum before use. (CH3)3CC 1 (Fluka AG purissimum), washed with NaHC03 solution, was dried over MgS04 and fractionally distilled. The purity was checked by VPC and UV analyses. BF3 (Fluka AG) was used as a CH2C12 solution stored at -78OC: the concentra- tion was obtained by a titration method (8).
Monomers, catalysts and solvents were handled under nitrogen atmosphere.
Polymerization Procedure
Polymerization runs were carried out in three-necked flasks, equipped with mechanical stirrer, flamed under dry nitrogen and placed in a thermostatic bath. The reagents were added by means of glass syringes in the order: solvent, mono- mer, and catalyst. The catalyst was generally preformed at room temperature during five minutes and slowly added to the cooled monomer solution. The reaction was stopped with a few drops of methanol and the polymer recovered by pouring the reaction mixture in an excess of methanol. The polymer was purified by dissolving it in CHC13; then it was filtered and dried under vacuum. Oligomers (oily-waxy products) were looked for by distilling under vacuum the

POLYMER LETTERS 575
polymerization solution after removing the solid polymer. The same procedure was adopted in the copolymerization experiments: the
conversion was tested also by W C quantitative analyses of the unreacted mon- omers (column EAS 5%, 5 mt, T = 1 10°C, carrier H2).
Analyses
The IR spectra were measured with a Perkin-Elmer 125 mod. spectrophoto- meter.
The NMR spectra (referred to TMs) were recorded on c s 2 or C6D6 SOlUtiOnS at room temperature on a Varian A-60 spectrometer.
The vapour phase chromatograph-mass spectrometer (WC-MS) LKB-9000 was used for the M W evaluation.
The MW of the polymers were determined by vapour pressure osmometry (Mechrolab mod. 302) on benzene solution at 37OC. Intrinsic viscosities were obtained from measurements in toluene at 3OoC with Ubbelhode viscometer.
The polymer chlorine content was determined by a colorimetric method (9). The determination of unsaturation content of poly-(I) was carried out ac-
cording to ref.(lO). The method was tested on monomer (111) solutions: the deviation was - 3%. Indication of side reactions, taking place on polymer solu- tion (1 l), was obtained in the case of poly - (11) because iodine consumption corresponding to a 10% of unsaturation was observed.
+
All X-ray diagrams were obtained with Ni-filtered CuKa radiation.
Results
Polymerization of (I)
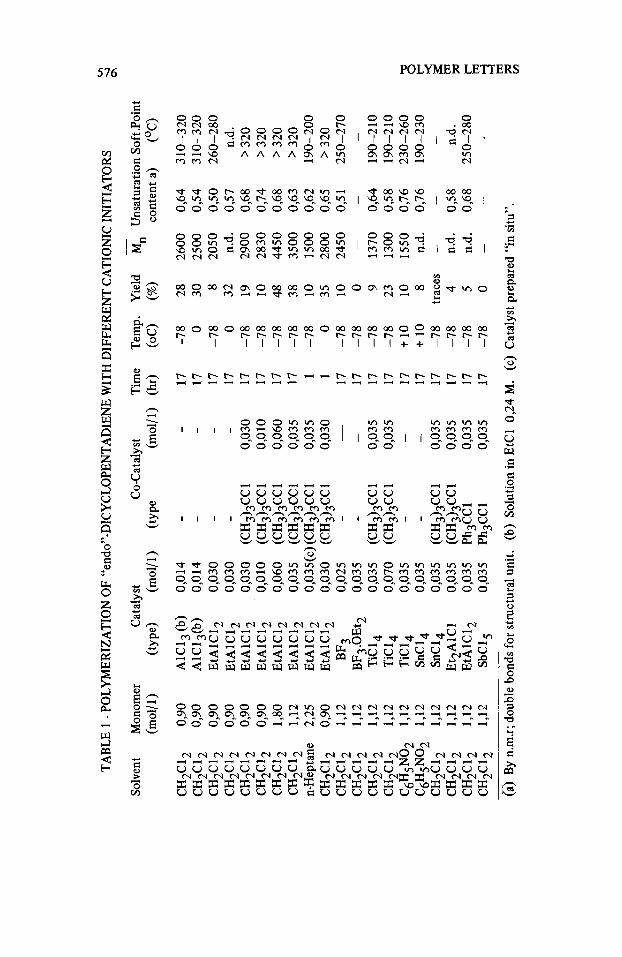
Table 1 collects the results of the cationic polymerization of (I) in different conditions. Only the strongest Lewis acids are able to give solid polymers with good yields. The polymer is in form of a white powder, amorphous at X-ray examination, completely soluble in CS2, aromatic and halogenated solvents, less soluble or insoluble in ketons and paraffinic solvents.
No crosslinked polymers have been detecfed whereas oligomers, soluble in methanol, were present only in traces ( < 2% on the monomer).
system about one C1 atom is present for one macromolecule, while for the Et- A1C12-(CH3)3CC1 system one C1 atom is present for 6-10 macromolecules, depending on the experimental conditions.
Intrinsic viscosity determinations on polymer samples obtained in different conditions give indication of low m.
Evaluation of M n “via” osmometry gives values ofmn ranging between 1300 and 4500. By plotting log [q] against log M n (Fig. 1) a linear relationship from
Softening temperatures range between 180 and >32OoC depending o n E n .
The chlorine content of poly(1) has shown that with the TiC14-(CH3)3CC1
TABL
E 1
- PO
LYM
ERIZ
ATI
ON
OF “endo”-DICYCLOPENTADIENE WIT
H D
IFFE
REN
T CA
TIO
NIC
INIT
IATO
RS
-
Solv
ent
Mon
omer
C
atal
yst
Co-
Cat
alys
t Ti
me
Tem
p.
Yie
ld
Mn
Uns
atur
atio
n Sof
t.Poi
nt
(mol
l 1)
(type
) (m
ow)
(type
(m
ol/l)
(h
r)
(OC)
(%
I co
nten
t a)
(OC
)
CH2C
12
CH2C
12
CH2C
12
CH2C
12
CH2C
12
CH2C
12
CH2C
12
CH2C
1 2
CH2C
12
CH
2Cl2
CH
2C 12
CH
2C 1 2
C
H2C
l2
‘gH
gN02
‘g
HgN
02
CH2C
12
CH2C
12
CH2C
12
CH2C
12
n-H
epta
ne
0,90
0,
90
0,90
0,
90
0,90
0,
90
1,80
1,
12
2,25
0,
90
1,12
1,
12
1,12
1,
12
1,12
1,
12
1,12
1,
12
1,12
1,
12
AlC
13 (b
) A1
C 1 3
(b)
EtA
l C12
Et
AlC
12
EtA
lC12
Et
AlC
12
EtA
lClZ
Et
AlC
12
EtA
lC 12
Et
AlC
12
BF3
.OEt
2 BF
3
Tic
1 T
ic 1
Tic
1 Sn
C 1
SnC
14
Et2A
lCl
EtA
lC 12
Sb
C15
0,01
4 -
0,O 1
4 -
0,03
0 -
0,03
0 -
0,03
0 (C
H&
CCl
0,01
0 (C
H3)
3CC1
0,
060
(CH
3)3C
C1
0,03
5 (C
H3)
3CC1
0,
035(
~) (CH
3)3C
C1
0,03
0 (C
H3)
3CC1
0,
025
- 0,
035
- 0,
035
(CH
&CC
l 0,
070
(CH
&CC
l 0,
035
- 0,
035
- 0,
035
(CH
&CC
l 0,
035
(CH
3)3C
C1
0,03
5 Ph
3CC
l 0,
035
Ph3C
C1
- 0,
030
0,01
0 0,
060
0,03
5 0,
035
0,03
0 -
-
0,03
5 0,
035
-
0,03
5 0,
035
0,03
5 0,
035
17
-78
28
17
0 30
17
-7
8 8
17
0 32
17
-7
8 19
17
-7
8 10
17
-7
8 48
17
-7
8 38
1
-78
10
1 0
35
17
-78
10
17
-78
0 17
-7
8 9
17
-78
23
17
+10
10
17
+10
8
17
-78
trace
s 17
-7
8 4
17
-78
5 17
-7
8 0
2600
25
00
2050
n.
d.
2900
28
30
4450
35
00
1500
28
00
2450
1370
13
00
1550
n.
d.
n.d.
n.
d.
-
-
0,64
0,
54
0,50
03
7 0,
68
0,74
0,
68
0,63
0,
62
0,65
0,
5 1
0,64
0,
58
0,76
0,
76
-
0,58
0,
68
310-
320
3 10-
320
260-
280
n.d.
>
320
> 32
0 >
320
> 32
0
> 32
0 25
0-27
0
190-
200
190-
210
190-
210
230-
260
2 19
0-23
0 -
M
56
n.d.
25
0-28
0 -
(a)
By n
.m.r;
dou
ble b
onds
for s
truct
ural
uni
t. (b
) So
lutio
n in
EtC
l 0,
24 M
. (c
) C
atal
yst p
repa
red
“in
situ
”
POLYMER LETTERS 577
3.1 3.2 3.3 3.4 3.5 3.6 3.7 log M"
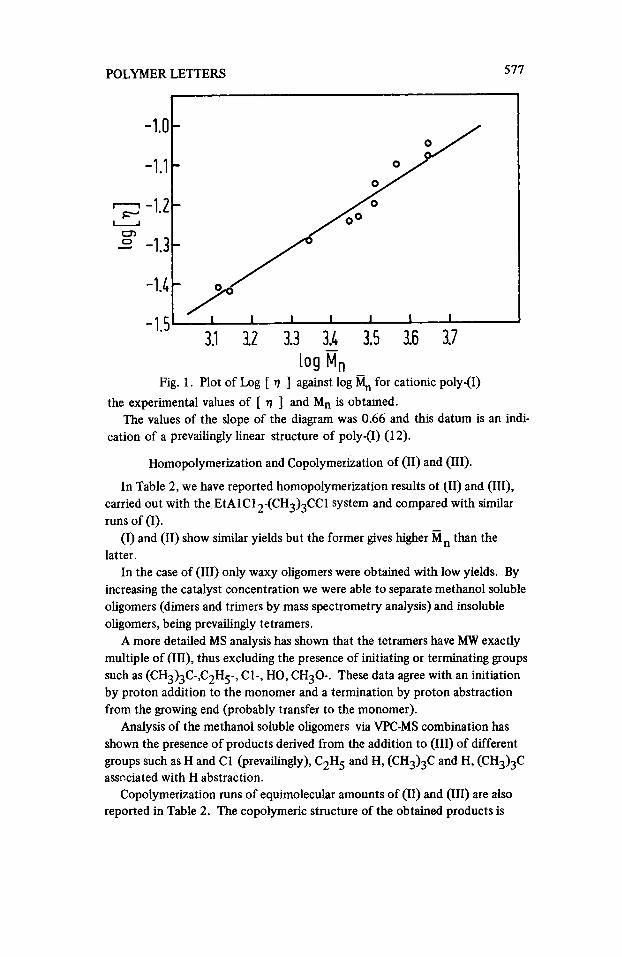
Fig. 1. Plot of Log [ rl 3 against log % for cationic poly-(I) the experimental values of [ q ] and Mn is obtained.
cation of a prevailingly linear structure of poly-(I) (1 2). The values of the slope of the diagram was 0.66 and this datum is an indi-
Homopolymerization and Copolymerization of (11) and (111).
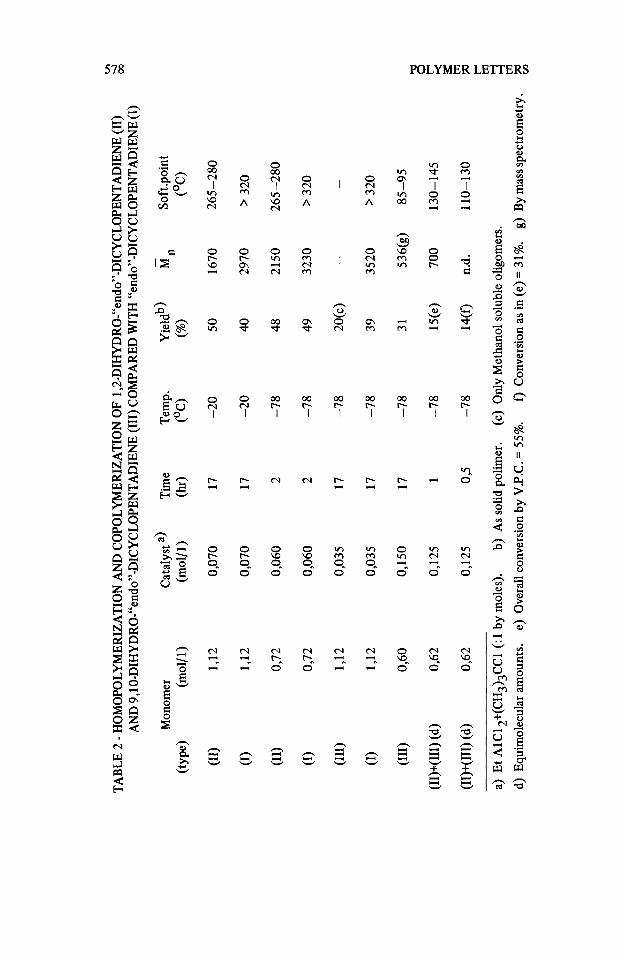
In Table 2, we have reported homopolymerization results of (11) and (111), carried out with the EtAlC12-(CH3)3CC1 system and compared with similar runs of (I).
(I) and (11) show similar yields but the former gives higher 8, than the latter.
In the case of (111) only waxy oligomers were obtained with low yields. By increasing the catalyst concentration we were able to separate methanol soluble oligomers (dimers and trimers by mass spectrometry analysis) and insoluble oligomers, being prevailingly tetramers.
A more detailed MS analysis has shown that the tetramers have MW exactly multiple of (III), thus excluding the presence of initiating or terminating groups such as (CH3)3C-,C2H5-, C1-, HO, CH3O-. These data agree with an initiation by proton addition to the monomer and a termination by proton abstraction from the growing end (probably transfer to the monomer).
Analysis of the methanol soluble oligomers via VPC-MS combination has shown the presence of products derived from the addition to (111) of different groups such as H and C1 (prevailingly), C2H5 and H, (CH3)3C and H, (CH&C associated with H abstraction.
Copolymerization runs of equimolecular amounts of (11) and (111) are also reported in Table 2. The copolymeric structure of the obtained products is
VI 4
00
TABL
E 2
- HO
MO
POLY
MER
IZA
TIO
N A
ND
CO
POLY
MER
IZA
TIO
N O
F 1,2-DIHYDRO-“endo”-DICYCLOPENTADIENE
(11)
AN
D 9,10-DIHYDRO-“endo”-DICYCLOPENTADIENE
(111)
COM
PARE
D W
ITH
“endo”-DICYCLOPENTADIENE (I
)
Cat
alys
t a)
(mol
/ 1)
0,07
0
0,07
0
0,06
0
0,06
0
0,03
5
0,03
5
0,15
0
0,12
5
0,12
5
Tim
e (W
17
17 2 2 17
17
17 1 0 95
Tem
p.
(OC
)
-20
-20
-78
-78
-78
-78
-78
-78
-78
-
Mn
1670
2970
2150
3230
-
3520
53
w
700
n.d.
Soft
.poi
nt
(OC)
265-
280
> 32
0
265-
280
> 32
0 -
> 32
0
85-9
5
130-
145
110-
130
a)
Et A
1C12
t(CH
3)3C
C1
(:1 b
y m
oles
). d)
Equ
imol
ecul
ar am
ount
s. b)
As s
olid
pol
imer
. (c
) O
nly
Met
hano
l sol
uble
olig
omer
s. f)
Con
vers
ion
as in
(e) =
3 1%
. g)
By
mas
s spe
ctro
met
ry.
m
e) O
vera
ll co
nver
sion
by V
.P.C
. = 5
5%.

POLYMER LETTERS 579
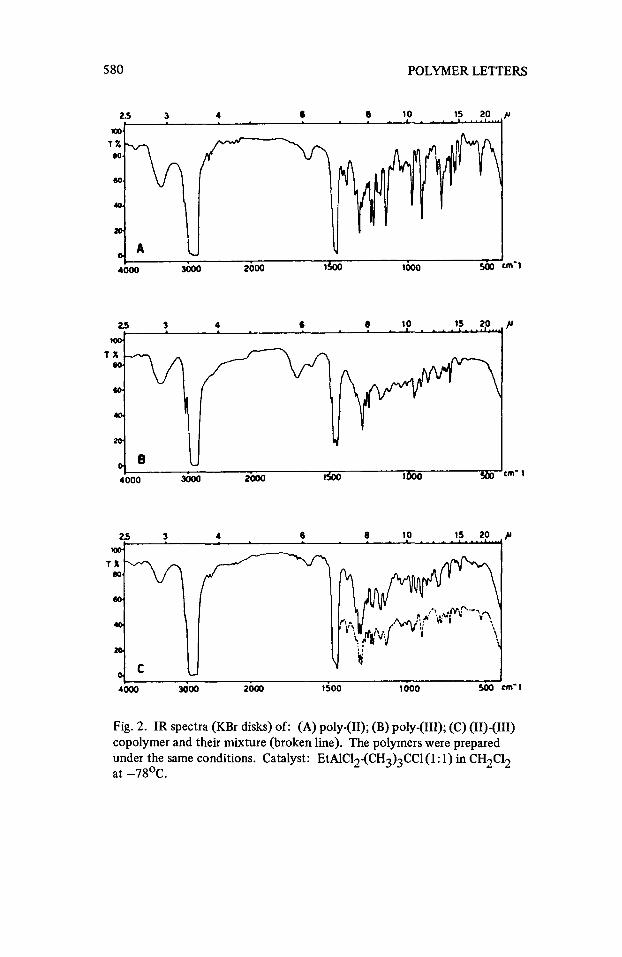
supported by comparison of the softening points (Table 2) of poly-(11), poly- (111), their mixtures and the expected copolymer. The X-ray diffraction inten- sity diagram for (11)-(111) copolymers shows a maximum corresponding to an angle different from that of pure poly-(11) and poly-(111) (13). Furthermore, the IR spectra of Figure 2-C show a coalescence of bands in the ranges 780-950 and 1220-1270 cm-’ in agreement with the previous conclusion.
By determining the relative concentration of the monomers (WC) at the beginning and at the end of the copolymerization reaction, it can be calculated that about 1,7 molecules of (11) have reacted for one molecule of (111). This agrees with the higher reactivity of the norbornene than the cyclopentene double bond of (I) towards electrophilic agents (14).
Spectroscopic Characterization
IR spectra of (I), (11), (111) homopolymers and (11)-(111) copolymers, pre- pared under the same conditions, are reported in Figure 2. The corresponding NMR’spectra are in Figure 2. The corresponding NMR spectra are in Figure 3.
No unsaturations are evident in the spectra of poly-(11) whereas the band at 1308 cm-’, typical of [2.2.1] -bicycloheptane structure, is present (4a). These data, together with the general characteristics of the IR spectra of the polymer, which are similar to the polynorbornene(4) and to the poly-5-methyl- “endo”-norbornene-2 ones(l5), suggest a polymerization through norbornene double bond opening. The methanol insoluble fraction of poly-(111) shows practically the absence of unsaturations and the presence of a [2.2.1] -bicycle- heptane structure (band at 1292 ciii’). A low content of probably terminal double bonds (3040 cm-l) is also present: this datum agrees with the mass spectrometry results which suggest a termination reaction through proton abstraction. Two diffuse signals are present in the NMR spectra centered at 7.84 and 8.58~, the former assigned to - H protons and the latter to -CH- protons(l6).The ratio of their intensities is 0,5 whereas the calculated one, for the structure resulting from the cyclopentene double bond opening, is 0,75. This datum seems to indicate a main polymerization mechanism ;‘via” hydride shift as represented by the structures (A) or (B) for which the -C;H/-CHT ratio is 0,4.
c 2
In the carbonium ion (A) a partial lack of planarity can be observed which could b’e balanced by a distortion of the poly-cyclic system. Both the struc- tures (A) and (B) show high steric hindrance in the enchantment of the repeat units of (111) as previously discussed for the cyclopentene and norbornene
5 80 POLYMER LETTERS
mJ T X a0
60
2.5 3 4 6 0 10
i'=9 A I
1- 1000 a cm'l
P
I
4000 3000 2000 1500 1000
Fig. 2. IR spectra (KBr disks) of (A) poly(I1); (B) poly-(III); (C) (II)-(III) copolymer and their mixture (broken line). The polymers were prepared under the same conditions. Catalyst: EtAlC12-(CH3)3CCl(l: 1) in CH2C12 at -78OC.
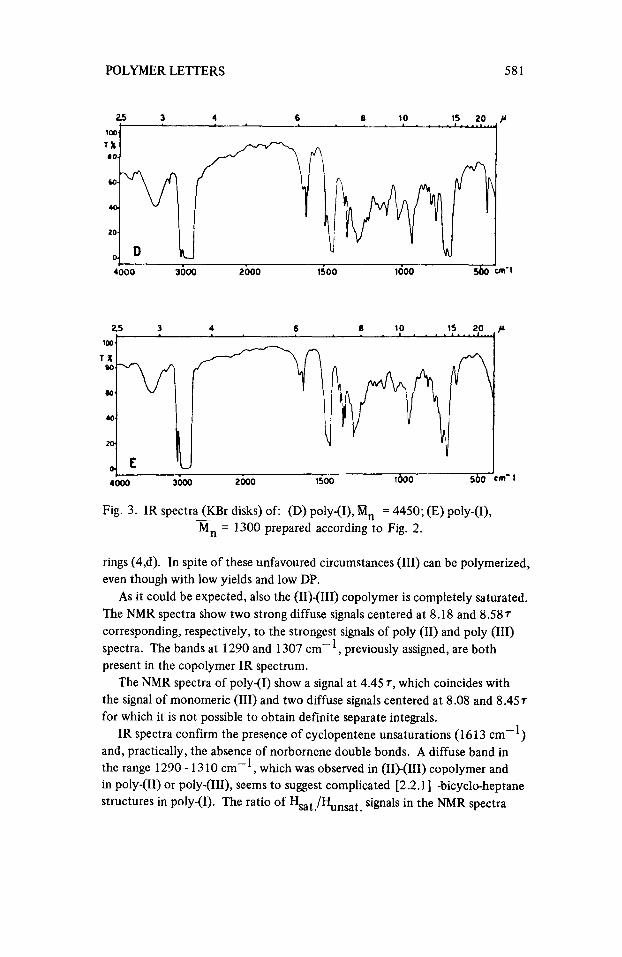
POLYMER LETTERS 58 1
3 4 6 e 10 15 20 P
25 3 4 6 e 10
loo.
? 4000 3000 2000 1500 tboo
Fig. 3. IR spectra (KBr disks) of: (D) poly-(I), 8, = 4450; (E) poly-(I), - Mn = 1300 prepared according to Fig. 2.
rings (4,d). In spite of these unfavoured circumstances (111) can be polymerized, even though with low yields and low DP.
As it could be expected, also the (11)-(111) copolymer is completely saturated. The NMR spectra show two strong diffuse signals centered at 8.18 and 8.587 corresponding, respectively, to the strongest signals of poly (11) and poly (111) spectra. The bands at 1290 and 1307 cm-', previously assigned, are both present in the copolymer IR spectrum.
The NMR spectra of poly-(I) show a signal at 4.45 7, which coincides with the signal of monomeric (111) and two diffuse signals centered at 8.08 and 8.457 for which it is not possible to obtain definite separate integrals.
IR spectra confirm the presence of cyclopentene unsaturations (1613 cm-l) and, practically, the absence of norbornene double bonds. A diffuse band in the range 1290- 13 10 cm-', which was observed in (11)-(111) copolymer and in poly-(11) or poly-(HI), seems to suggest complicated [2.2.1] -bicycle-heptane structures in poly-(I). The ratio of Hsat./Ylnsat signals in the NMR spectra
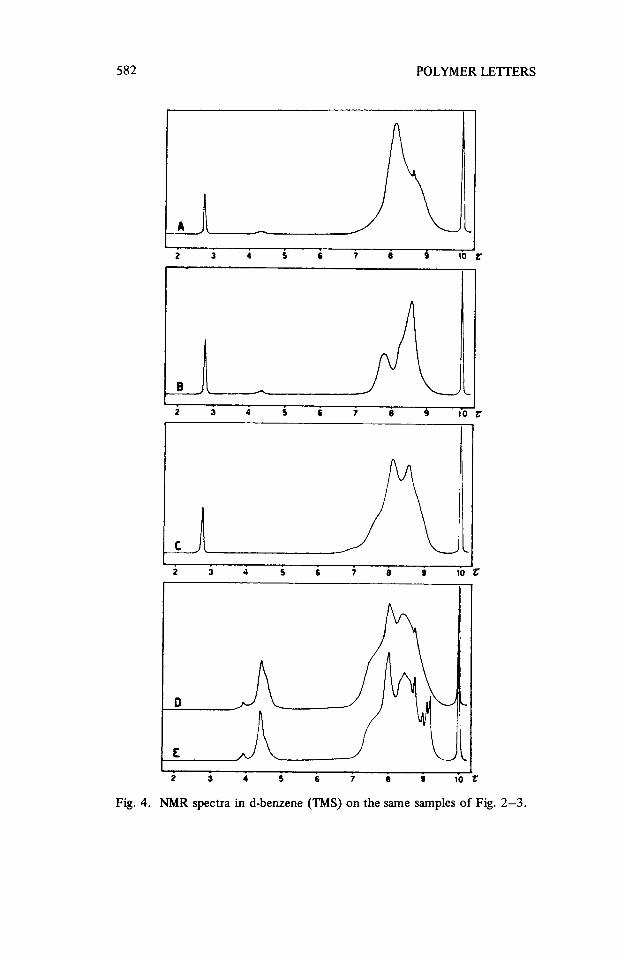
582
B / \
L
A
POLYMER LETTERS
I
L
2 3 4 5 6 7 8 9 10 c
I
2 3 4 5 6 7 8 9 10 t
Fig. 4. NMR spectra in d-benzene (TMS) on the same samples of Fig. 2-3.

POLYMER LETTERS 583
of poly-(I) is generally between 8 and 10. This ratio should be 5 for a poly-(I) derived from a simple norbornene double bond opening. This datum agrees with chemical determinations of unsaturations which are about 70% of the theoretical value.
CONCLUSIONS
The aforereferred results can be interpreted, in analogy with (11)-(111) copoly- merization results, by the assumption that bothnorbornene and, in part, CyClO- pentene double bonds of (I) are reactive with cationic systems. When cyclopen- tene double bond is attacked, an hydride shift from tertiary C atom in position 5 could take place and the resulting carbonium ion could react with norbornene double bond. The result would be a nortricyclene structure according to:
This hypothesis could be supported by IR bands at 789 and 739 (weak) cm-’ and by a multiplet in the NMR spectra of poly-(I) at 9.05 7 . This signal is evident only in the lower MW polymers. Both the bands have been attributed to mono-substituted nortricyclene structures (17) or simple nortricyclene rings
Owing to the reactivity of both double bonds of (I), branched polymers could be obtained. Nevertheless the high value of the exponent a in the Mark- Houwink relationship is in agreement with a prevailingly linear structure of
(16).
POlY-(I)*
Note added in proof
When this work was completed, a paper [ T. Corner, R.G. Foster and P. Hepworth, Polymer, 10, (S), 393 (1969)l appeared on the structure of polymers derived from “endo’TA) and “exo” dicyclopentadiene (B). In the case of poly-(A), obtained with BF3.0Et2 at room temperature, an “exo” structure of cyclopentene rings was observed. This conclusion could be drawn also for the poly-(A) prepared under our conditions as shown by spectroscopic analyses (Fig. 2-3). In the same paper a less than one unsaturation (0,8) for structural unit was found, in agreement with our results.

584
References
POLYMER LETTERS
(1) a. E. K. Cladding, B.S. Fisher, J.W. Collette, Ind. Eng. Chem., Prod. Res. Develop. - 1, 65 (1962).
b. R.E. Cunningham, J. Polymer Sci. A-3, - 3157 (1965). (2) a. G.Sartori, A.Valvassori, S.Faina, Atti Accad. Naz. Lincei, 35 - (8),
565 (1963); Rubber Chem. Technol. 38, 620, (1965).
J. - 1, 121 (1965). b. W. Cooper, D.E. Eaves, M.ETTunnicliffe, G. Vaughan, Europ. Polym.
(3) P.R. Marshall, B.J. Ridgewell, Europ. Polym. J. 5, 29 (1969). (4) a. G. Sartori, F. Ciampelli, N. Cameli, Chem.Inc(Milan)45, 1478 (1963).
b. J.P. Kennedy, H.S. Makowski, J. Macromol. Sci. (Chez) A-1, - (3),
c. J.Boor, E.A. Youngman, M. Dimbat, Makromol. Chem. 90, - 26 (1966). d. J.P. Kennedy, M.S. Makowski, J . Polymer Sci. C 2 2 4 7 (1968).
(5) S.J. Cristol, W.K. Seifert, S.B. Soloway, J.Am. Chem. SOC. 82, - 2351
(6) S.Cesca, M.L. Santostasi, W.Marconi, M.Greco, Ann. Chim. (Rome) 55, -
(7) M.S.C. Flett,P.H.Plesch, J.Chem. SOC. 1952, 3355. (8) C.F. Swinehart, A.R. Bumblis and H.F. G k , Anal. Chem. - 19,28 (1947). (9) J.G. Bergmann, JSanik, Anal. Chem. 29,241 (1957).
345 (1967).
(1960).
682 (1 965).
(1 0) S.C. Gallo, H.K. Wiese, J.F. Nelson, Ind-Eng . Chem. 40, 1277 (1 948). (1 1) T.S. Lee, I.M. Kolthoff, M.A. Mairs, J.Polymer Sci. 3 x 6 (1948). (12) B.H. Zimm, R.W. Kilb, J. Polymer Sci. 37, 19 (1959). (1 3) G.Crespi, A. Valvassori, G. Sartori “Cop~ymerization” ed. G.E. Ham,
(14) H.E. Bruson, T.W. Riener, J.Am. Chem. SOC. 67, 723 (1945); 68, - 8
(15) A>riola, S. Cesca, unpublished results. (16) R.H. Wiley, W.H. Rivera, T.H. Crawford,M.F. Bray, J.Polymer Sci.
(17) H.Hart, R.A. Martin, J. Org. Chem. 24, 1267 (1959).
Interscience, New York, 1964, p. 23 1.
(1946); 70, 2809 (1948).
61, - S 38 (1962).
SNAM PROGETTI S.p.A. Laboratori Studi Ricerche San Donato Milanese, 20097, Milan, Italy
Received May 15, 1970
Sebastiano Cesca Aldo Priola Giuliano Santi





























