Embed Size (px)
Citation preview

E L S E V I E R Biochimica et Biophysica Acta 1253 (1995) 103-111
BR Biochi~ic~a et Biophysica ~.ta
Specific dinucleoside polyphosphate cleaving enzymes from chromaffin cells: a fluorimetric study
Antonio Ramos, Pedro Rotllfin *
Departamento de Bioqulmica y Biologla Molecular, Universidad de La Laguna, 38206 La Laguna, Tenerife, Canary Islands, Spain
Received 13 April 1995; revised 12 July 1995; accepted 13 July 1995
Abstract
This article presents a fluorimetric study of the main properties of the enzymes dinucleoside tetraphosphate (asymmetrical) hydrolase or dinucleoside tetraphosphatase (Ap4Aase, EC 3.6.1.17) and dinucleoside triphosphate hydrolase or dinucleoside triphosphatase (Ap3Aase, EC 3.6.1.29), botla present in adrenal medulla cytosolic extracts. Diethenoadenosine polyphosphates, E-(Ap,A), are used as artificial fluorogenic substrates. Ap4Aase exhibits a molecular mass around 20 kDa and neutral optimum pH (7.0-7.5). It requires Mg 2+ and preferentially hydrolyzes substrates with four phosphate groups. K m for E-(Ap4A) is 1.3 ~,M and K i for AP4A and GpaG are 1 and 0.2 /xM respectively. K m for Ap4A determined by HPLC is 1.6 /xM. E-(ApsA) and e-(Ap6A) are hydrolyzed at reduced rates. This enzyme is inhibited by Zn 2+, F - and very strongly by Ap4 and E-Ap4. Ca 2+ cannot replace Mg 2÷, but behaves as inhibitor in its presence. The substrate analogs dinucleoside triphosphates Ap3A, Gp3G, mTGp3G and mVGp3 A and the periodate-oxidized nucleotides o-(ApnA), oE-(Ap4A), o-Ap4 and oe-Ap4 behave as inhibitors. Ap3Aase exhibits a molecular mass around 30 kDa and neutral optimum pH (7.0-7.5). It requires Mg 2+ or Ca 2÷, but retains a low measurable activity around 10% in the absence of these divalent cations. It only hydrolyzes substrates with three phosphate groups. K m for e-(AP3A) is I 1 /zM and K~ for Ap3A and GP3G are 20 and 22 IzM, respectively. K m for Ap3A determined by HPLC is 16 /zM. m7GP3G and mTGp3A are also good substrates for triphosphatase.
Keywords: Dinucleoside polyphosphate; Diadenosine polyphosphate; Diethenoadenosine polyphosphate; Fluorogenic substrate; Dinucleoside tetraphos- phate (asymmetrical) hydrolase; Dinucleoside triphosphate hydrolase; (Chromaffin cell)
1. Introduct ion
Dinucleoside polyphosphates, Np,N', constitute a fam- ily of nucleotides containing two nucleoside moieties (N,N') joined in 5'-5' linkage by a polyphosphate chain
(Pn) containing n >_ 2 members. It has been reported that some compounds of this family, e.g., AP4A and Ap5A are powerful inhibitors of adenylate kinase [1] and adenosine kinase [2], while Gp4G behaves as a potent GMP reductase activator [3]. Some Np,N' have been found in both pro-
Abbreviations: Ap,A, Adenc,sine (5') polyphospho (5') adenosine or diadenosine polyphosphates; Ap2 A, adenosine (5') diphospho (5') adenosine or diadenosine pyrophosphate; AP3A, adenosine (5') triphospho (5') adenosine or diadenosine triphosphate; ApaA, adenosine (5') tetraphospho (5') adenosine or diadenosine tetraphosphate; ApsA, adenosine (5') pentaphospho (5') adenosine or diadenosine pentaphosphate; Ap6A, adenosine (5') hexaphospho (5') adenosine or diadenosine hexaphosphate; Ap3Aase, dinucleoside triphosphate hydrolase (EC 3.6.1.29); ApaAase, dinucleoside tetraphosphate hydrolase (EC 3.6.1.17); Ap4 and Aps, adenosine 5'-tetraphosphate and pentaphosphate respectively; GP2G, guanosine (5') diphospho (5') guanosine or diguanosine pyrophosphate; Gp3G, guanosine (5') triphospho (5') guanosine or diguanosine triphosphate; Gp4G, guanosine (5') tetraphospho (5') guanosine or diguanosine tetraphosphate; Gp.,G, guanosine (5') pentaphospho (5') guanosine or diguanosine pentaphosphate; Np, N', dinucleoside polyphosphate; mTGp3G, 7-methylguanosine (5') triphospho (5') guanosine; mTGp3A, 7-methylguanosine (5') triphospho (5') adenosine; e stands for 'etheno' (30). E-Ado, E-AMP, e-ADP, E-ATP and e-Ap4 and e-Ap5 refer to the l,N6-etheno derivatives of adenosine, AMP, ADP, ATP, Ap4 and Aps, respectively; e-(Apn A), l.N6-ethenoadenosine (5') polyphospho (5') 1,N6-ethenoadenosine or diethenoadenosine polyphosphates; e-(Ap~ A), e-(Ap3 A), e-(Ap4 A), e-(ApsA) and e-(Ap6A) refer to the corresponding derivatives of Ap, A (n = 2-6) containing both Ado moieties substituted by E-Ado; o-(Ap~A) and oe-(Ap,A), periodate oxidized Ap~A and e-(Apn), respectively; o-Ap4 and oe-Ap4 , periodate oxidized Ap4 and e-mp4, respectively ; RP-HPLC, reverse-phase high-performance liquid chromatography.
* Corresponding author. Fax: + 34 22 630095. I Preliminary results of this work were presented as abstracts at the IV Portuguese-Spanish Biochemistry Congress, Portugal 1991 and at the 7th
International Symposium on Chromaffin Cell Biology and Pharmacology, Canada, 1993.
0167-4838/95/$09.50 © 1995 Elsevier Science B.V. All rights reserved SSD1 0167-4838(95)00154--9

104 A. Ramos, P. Rotll(m / Biochimica et Biophysica Acta 1253 (1995) 103-111
and eukaryotic cells but their precise biological roles are still poorly understood; current knowledge on these com- pounds has been recently updated [4].
Gp4G, Gp3G, Gp3A and Gp2G were the first com- pounds of this family discovered in living materials during the 1960's in the encysted embryos of the crustacean Artemia and related organisms, [5]. Ap4 A, the most repre- sentative member of the diadenosine polyphosphate fam- ily, was found as a product of the 'in vitro' action of some aminoacyl-tRNA synthetases [6] but its presence has been established in many cells [7]. Depending on the biological system investigated, Ap4A has been proposed as playing roles in the initiation of DNA replication, in DNA repair, and in cell responses to environmental stresses [8,9].
The implication of Ap4A and Ap3A in the platelet function arose from the discovery of these dinucleotides as dense body components co-stored together with ATP, ADP and serotonin in human platelets [10,11] and from their effects on platelet aggregation [12,13]. The presence of ApsA and Ap6A in platelets and their implication in the control of blood pressure has been reported very recently [14]. The finding of Ap4A, ApsA and Ap6A in the chro- maffin granules [15,16] and of Ap4A and ApsA in synap- tic vesicles from rat brain and Torpedo electric organ [17,18] has raised the question of the involvement of diadenosine polyphosphates in neurosecretory functions as neurotransmitters or extracellular neural modulators. In this way, the exocytotic release of ApnA by chromaffin cells [19], their effects on catecholamine secretion [20] as well as the existence of Ap4A binding sites on these cells have been reported [21].
In mammals, intracellular enzymes which specifically hydrolyze Ap4A and Ap3A at the phosphate chain, as well as serum enzymes cleaving Ap4 A have been characterized [22,23]. Attention has been also directed towards ectoen- zymes able to hydrolytically degrade AP3A and Ap4A in endothelial [24,25] and chromaffin cells [26]. All these enzymes must be relevant in regulating intra- and extracel- lular levels of Ap, A and hence in the control of the physiological functions of these dinucleotides.
We have recently shown [27] that the fluorogenic derivative of Ap4A di(1,N6-ethenoadenosine) tetraphos- phate, E-(Ap4A), is hydrolyzed by chromaffin cell cytoso- lic extracts and that F - ion, currently believed to be a strong specific inhibitor of asymmetrical diadenosine te- traphosphate hydrolases [28], inhibits cytosolic activity present in chromaffin cells [29]. In this paper we report a characterization of the specific enzymatic systems present in chromaffin cells, dinucleoside tetraphosphate (asymmet- rical) hydrolase and dinucleoside triphosphate hydrolase, both involved in intracellular hydrolysis of the putative neurotransmitters diadenosine polyphosphates, making use of the corresponding fluorogenic derivatives di( l ,N 6- ethenoadenosine) polypbosphates, ¢-(Ap, A), as useful substrate analogs.
2. Materials and methods
2.1. Materials
Adenosine and guanosine derived nucleotides including periodate oxidized ApnA were from Sigma (St. Louis, MO, USA). E-Ado, E-ATP, e-ADP, E-AMP were also from Sigma. mVGP3 G, mVGp3 A, alkaline phosphatase and Crotalus durissus phosphodiesterase were from Boehringer (Mannheim, Germany). Molecular weight markers for gel filtration (size exclusion) chromatography: Blue dextran, bovine serum albumin, carbonic anhydrase and cy- tochrome c were from Sigma. Sephacryl S-200 HR and prepacked Cibacron blue affinity columns were from Phar- macia (Uppsala, Sweden). 2-Chloroacetaldehyde 45% aqueous solution was from Merck (Darmstadt, Germany). All other products were reagent grade.
2.2. Preparation and characterization o f e-(Ap, A) com- pounds
At the initial stages of this work, e-(Ap,A) (n = 2-6) were prepared according to the method recently published for E-(Ap4A) [27]; essentially this involved the treatment of precursors ApnA with 2-chloroacetaldehyde [30] and chomatographic purification of the e-(Ap~A) formed from the reaction mixture. Actually, we have slightly modified the procedure as follows: once the reaction was terminated, the reaction mixture was fortified with 1 M potassium phosphate buffer and methanol (or acetonitrile) up to the same concentration of these components in the mobile phase used in the preparative RP-HPLC. Then, 0.5-1.0 ml samples of the resulting mixture were applied in the preparative column and peaks corresponding to the e- (ApnA) compounds of interest collected. The HPLC col- lected fractions were additionally purified by ion-exchange chromatography to eliminate phosphate and organic sol- vents and minor potential impurities in a DEAE-Sephacel column (20 x 200 mm) equilibrated in 0.1 M ammonium bicarbonate and eluted by a linear gradient 0.1-0.7 M of the same salt; elution was monitored at 280 nm with a UV-1 Pharmacia detector and the collected fractions were lyophilized and stored at -70°C until use.
The confirmation of purity and structure of e-(Ap, A) compounds and the characterization of spectral properties were done as described for e-(Ap4 A) by Rotll~n et al. [27]. A Shimadzu UV 160A spectrophotometer (Tokyo, Japan) and a Hitachi F-2000 spectrofluorimeter (Tokyo) were used for spectral characterizations. All ~-(Ap, A) com- pounds showed typical UV absorption spectra with max- ima at 265 and 274 nm and shoulders at 260 and 310 nm. The molar absorption coefficients in 50 mM Tris-HCl (pH 8.0), 4 mM MgC12 for e-(ApnA), n = 2-6, were respec- tively as follows: 9.4, 9.5, 9.7, 10.0 and 9.8. 10 3 M -1
cm -l at 265 nm and 8.7, 8.9, 9.1, 9.4, and 9.4- 10 3 M -1

A. Ramos, P. Rotll6n / Biochimica et Biophysica Acta 1253 (1995) 103-111 105
cm- l at 275 nm. These compounds showed typical fluo- rescence spectra with excitation maxima at 280 and 307 nm and one emission maximum at 410 nm. The fluores- cence emission is strikingly less than that of the isolated e-Ado 5'-phosphate moietie, s and it increases the longer the polyphosphate chain is. Enzymatic hydrolysis of the polyphosphate chain induces substantial emission fluores- cence increases of 16, 9, 7, 6 and 5-fold, respectively, for E-(Ap~A), n = 2-6, in 50 mM Tris-HCl (pH 8.0), 4 mM MgCI 2 .
E-Ap4 was obtained by 2-chloroacetaldehyde treatment of Ap4 according to above mentioned procedures. E-Aps was obtained from ~-(Ap6 A) through controlled hydrolysis by Crotalus durissus phosphodiesterase and RP-HPLC separation of the released s-Aps.
2.3. Preparation o f oxidized nucleotides
The 2',3'-dialdehyde derivatives of e-(Ap3A), e- (Ap4A), E-(ApsA, Ap4 and e-Ap4 were obtained essen- tially as described for the preparation of periodate oxidized ATP [31]. Briefly, neutral aqueous solutions (0.5 ml) of each nucleotide (0.2 mM) were incubated with NalO 4 at final concentrations of 0.4 mM (for the mononucleotides) or 0.8 mM (for the dinucleotides) and incubated at 4°C in the dark for 60 min. Glycerol to consume excess periodate was then added and the incubation maintained for 30 min. RP-HPLC of the reaction mixture demonstrated a total conversion of the nucleotides into their corresponding oxidized derivatives. These derivatives eluted as peaks of considerably reduced retention relative to their respective parent compounds. Both oE-(Ap3A) and oe-(Ap4A) were substrates of C. durissus phosphodiesterase, hydrolysis also producing a marked increase in fluorescence emission.
2.4. HPLC instrumentation and analysis conditions
The chromatographic system used has previously been detailed [29]. For analytical purposes involving e-deriva- tives a tzBondapak C-18 column (4 X 300 mm, 10 /zm particle) (Waters) eluted with 0.1 M KH2P04 (pH 6.0), 10% methanol or 4% acetonitrile and /zBondapak phenyl column (4 X 300 mm, 10 /zm particle) (Waters) eluted with 0.1 M KH2PO 4 (pH 6.0), 5% methanol or 2% acetonitrile were used at flow rates of t.5 ml /min. Prepar- ative chromatography of ,.~-(ApnA) compounds was per- formed on a tzBondapak C-18 column (8 X 300 mm, 20 /zm particle) (Waters) eluted with 0.1 M KH2PO 4 (pH 6.0), 10% methanol or 4% acetonitrile at 4.5-5.0 ml/min. Details on the preparation of mobile phases, standards and samples as well as on the elution profiles for the separation of ApnA and e-(ApnA) compounds from their enzymatic degradation products have been published [29,32].
Chromatographic assays, to determine K m and Vrnax of dinucleoside tetraphosphatase on Ap4A and of dinucleo- side triphosphatase on Ap3A were performed as follows:
samples from each partially purified enzyme were appro- priately diluted and incubated in Eppendorf tubes at 37°C in a standard medium composed of Tris-HCl 50 mM (pH 7.5), 4 mM MgCI 2 and bovine serum albumin 0.5 mg/ml . Substrate concentrations ranged from 0.2 to 10 ~M ApaA and from 2 to 40/~M Ap3A according to the enzyme to be assayed. Final volumes of reaction mixtures were 200 /zl. 25 /xl aliquots were taken off from the reaction mixture every 2 min and immediately transferred to ice-cold tubes containing 25 ILl of mobile phase fortified with 200 /xM zinc acetate (a common inhibitor of both hydrolases) to stop the enzymatic reaction. Two 20 /xl aliquots of this final mixture were then injected into the chromatograph and initial reaction rates of Ap4A and Ap3A hydrolysis were calculated from peak areas corresponding to the nucleotide product AMP. A Nova-Pak C-18 column (4 × 150 mm, 4 /zm particle) (Waters) eluted with 0.1 M KH2PO 4 (pH 6.0), 2% acetonitrile at a flow rate of 1.0 ml /min was used for these assays.
2.5. Partial purification o f cytosolic hydrolases
All purification steps were carried out at 0-5°C. Fresh adrenochromaffin tissue (35 g) carefully dissected from bovine adrenal glands was homogenized in 70 ml of 10 mM Tris-HCl (pH 7.5), 0.15 M KCI in a Potter homoge- nizer and the obtained homogenate centrifuged at 100000 X g for 60 min. To the cytosolic extract so obtained, saturated ammonium sulfate was added up to 50% satura- tion, the mixture was stirred for 30 min and then cen- trifuged at 10000 X g for 20 min to collect the super- natant. Solid ammonium sulfate up to 90% saturation was added to the supernatant, stirring was maintained for 30 min and the precipitated protein collected after centrifuga- tion at 10000 x g for 20 min. The collected protein was redissolved in homogenization buffer up to a volume of 10 ml, and applied onto a Sephacryl $200 HR column (26 x 900 mm) equilibrated with the homogenization buffer and eluted at 3.0 ml/min.
Fractions containing hydrolytic activity on E-(Ap3A) and E-(Ap4A) were combined and applied to a 1 ml prepacked Cibacron blue affinity chromatography column equilibrated in 10 mM Tris-HC! (pH 7.5), 0.5 mM EDTA. The enzyme solution was recirculated through the column at 0.5 ml /min for 20 h; after which both hydrolytic activities were undetectable in the recirculating solution. The column was washed with 5 ml of equilibrating buffer and elution of e-(Ap4A) hydrolase was initiated by addi- tion of 80 /xM Ap4, MgCI 2 to the elution buffer [33]; E-(Ap3A) hydrolase was then eluted by a 0.2-2 M KC! gradient in the elution buffer. Activity corresponding to the 20 kDa enzyme practically disappeared after the affinity chromatography step. However, activity corresponding to the 30 kDa enzyme was recovered as a fraction with undetectable protein but enzyme activity was unstable.
Consequently, the active fractions obtained after the gel

106 A. Ramos, P. Rotlldn / Biochimica et Biophysica Acta 1253 (1995) 103-111
filtration step were used for the enzyme characterization studies. Specific enzymatic activities in the 100000 × g supernatants were 0.4 and 3.0 m U / m g protein for the triphosphatase and tetraphosphatase, respectively. In the reconstituted active fractions after the gel filtration step specific activities for the triphosphatase and tetraphos- phatase reached values of 8.3 and 135 m U / m g protein, respectively.
2.6. Fluorimetric enzyme assays and calculation of kinetic parameters
Enzyme assays were performed in a standard medium composed of Tris-HCl 50 mM (pH 7.5), 4 mM MgCI 2, bovine serum albumin 0.5 mg/ml , appropriate enzyme amount and saturating substrate concentrations according to the enzyme to be assayed. It was verified that enzyme activity was a linear function of protein amount and of incubation time. When the effects of substrate concentra- tion, pH, ions or inhibitors on enzyme activity were stud- ied the standard reaction mixture was accordingly modi- fied. Reaction progress was followed by means of the increase of fluorescence emission at 410 nm using excita- tion at 305 nm and bandpass widths of 10 or 20 nm. A Hitachi F-2000 spectrofluorimeter was used throughout.
Continuous fluorimetric assays and calculations of ki- netic parameters using the integrated form of the Michaelis-Menten equation were essentially as described by Wierzchowski et al. [34] except that the reaction mix- ture was fortified with 5 units of alkaline phosphatase to avoid enzyme inhibition by accumulation of the released enzyme products E-Ado 5'-phosphates. Omission of alka- line phosphatase resulted in anomalous non-linear plots of the integrated Michaelis-Menten equation. Increase of flu- orescence emission is only associated with the initial hy- drolytic e-(ApnA) cleavage producing e-Ado 5'-phos- phates, not with the subsequent hydrolysis of these moi- eties down to e-Ado.
Enzymes were assayed in a final cuvette volume of 1 ml under continuous stirring at 37°C. Because undesirable photodecomposition of E-derivatives was noted when con- tinuously exposed to the excitation beam (leading to non- fluorescent degradation products as demonstrable by RP- HPLC), the fluorescence measurements were usually taken every 50 or 100 s allowing the reaction to proceed in the dark between consecutive measurements with the aid of the spectrofluorimeter shutter.
Fluorescence intensity measurements were converted into substrate concentrations according to the formula:
[ s ] , = [ S ] o - - lf-lo
where [S] o and [S], are the initial and time t substrate concentrations respectively and I o, I, and If are the initial, time t and final fluorescence once all substrate was con-
sumed. If was taken after addition of 1 /xl containing 1 mU of C. durissus phosphodiesterase to assure a complete degradation of the substrate and an accurate measure of If. To calculate K m and Vma x data for substrate concentration and time were analyzed and plotted according to the integrated form of the Michaelis-Menten equation:
[S]o 1 Vm~ x 1 ( [ S ] o - [S],) - - - - - ( l l )
In [S], t K m K m t
Inhibition constants for ApnA, alternative substrates which competitively inhibit hydrolysis of E-(ApnA) were determined under conditions of first order kinetics, e.g. at initial substrate concentrations < 0.2 K m. Substrate con- centration and reaction time data were analyzed and plot- ted according to the equation:
In[S], = ln[S]o - kt
derived from [S], = [Sloe kt, the typical form of a first order equation where k = Vm ax /K m and (Vmax//KmX1 +
[ I ] /K i) in the absence and presence of inhibitor, respec- tively.
Also, assays were performed in smaller volumes (50- 200 /zl) in Eppendorf tubes in continuous and discontinu- ous mode. In the former, fluorescence readings were taken every 5-15 min after diluting 10 /zl aliquots of the reaction mixture in the fluorimeter cuvette with 1 ml of phosphate buffer 20 mM (pH 7.5); in the discontinuous mode, protein and incubation time were fixed for a maxi- mal substrate consumption of 10% in 5-15 min and the fluorescence readings taken at fixed times as before. Under these conditions a considerable saving of substrate and enzyme preparation was achieved.
In all cases, one unit of e-(Apn A)ase activity is defined as the amount of enzyme hydrolyzing 1 /zmol of e-(ApnA) at 37°C.
2.7. Protein determination
Protein concentrations were determined by the colori- metric method of Bradford [35].
3. Results
3.1. Identification of e-(ApnA) cleaving activities: sub- strate and reaction products, molecular mass and sensitiv- ity to inhibitors
We have previously shown [29] that cytosolic extracts from chromaffin cells contain at least one fluoride sensi- tive hydrolytic activity able to asymmetrically cleave e- (ApnA), however activity present in cytosolic extracts could not be totally inhibited by fluoride even at a concen- tration as high as 10 mM. This suggests the presence of other hydrolytic enzymes.
Gel filtration chromatography of cytosolic extracts from
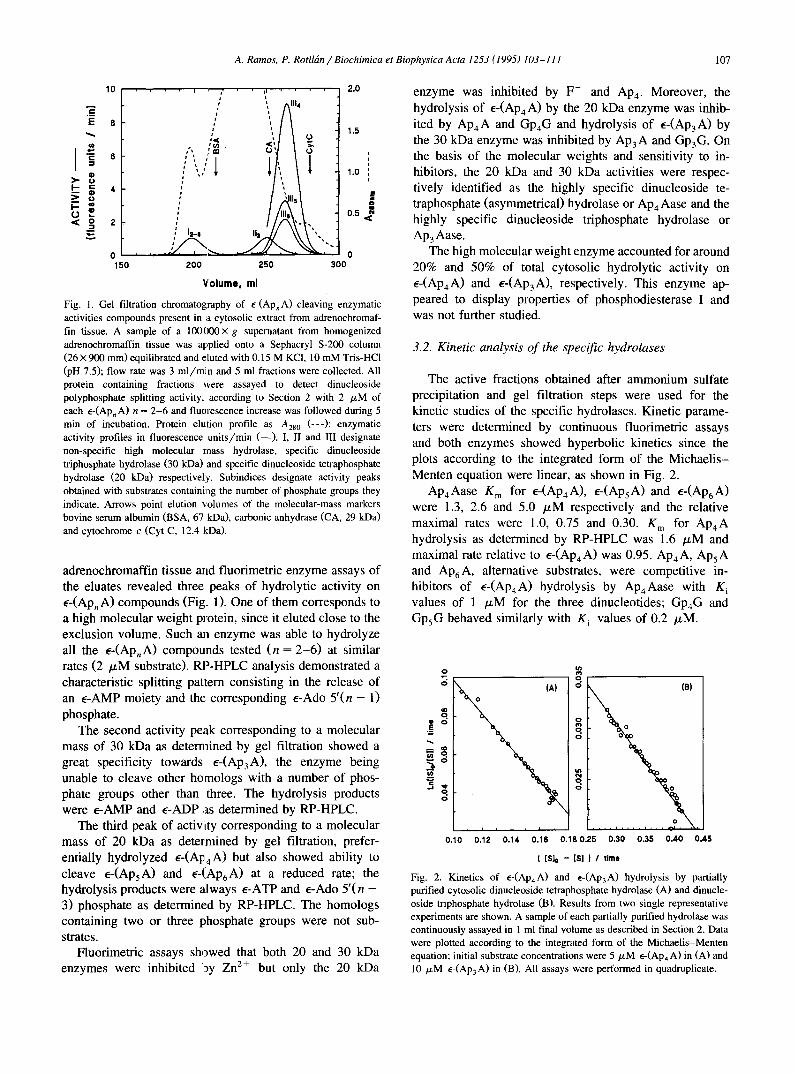
A. Ramos, P. Rotlldm / Biochimica et Biophysica A cta 1253 (1995) 103-111 107
10 . . . . , , ; , , ,, . . . . 2,0
E e / " 1.5
° ' [ ~- ~ ',,' 1.0
___g 4 ___o i I- o ." 0.5,~
-- 12-e ~
1 5 0 2 0 0 2 5 0 3 0 0
V o l u m e , ml
Fig. I. Gel filtration chromatography of e-(Ap~A) cleaving enzymatic activities compounds present in a cytosolic extract from adrenochromaf- fin tissue. A sample of a 100000× g supernatant from homogenized adrenochromaffin tissue was applied onto a Sephacryl S-200 column (26×900 mm) equilibrated and eluted with 0.15 M KCI, 10 mM Tris-HC1 (pH 7.5); flow rate was 3 ml/min and 5 ml fractions were collected. All protein containing fractions were assayed to detect dinucleoside polyphosphate splitting activity, according to Section 2 with 2 /~M of each e-(Ap,A) n = 2-6 and fluorescence increase was followed during 5 min of incubation. Protein elution profile as A28 o (---); enzymatic activity profiles in fluorescence units/min ( - - ) . I, II and III designate non-specific high molecular mass hydrolase, specific dinucleoside triphosphate hydrolase (30 kDa) and specific dinucleoside tetraphosphate hydrolase (20 kDa) respectively. Subindices designate activity peaks obtained with substrates containing the number of phosphate groups they indicate. Arrows point elution volumes of the molecular-mass markers bovine serum albumin (BSA, 67 kDa), carbonic anhydrase (CA, 29 kDa) and cytochrome c (Cyt C, 12.4 t'dDa).
adrenochromaffin tissue and fluorimetric enzyme assays of the eluates revealed three peaks of hydrolytic activity on e-(Ap, A) compounds (Fig. 1). One of them corresponds to a high molecular weight protein, since it eluted close to the exclusion volume. Such ml enzyme was able to hydrolyze all the e-(ApnA) compounds tested (n = 2-6) at similar rates (2 /~M substrate). RP-HPLC analysis demonstrated a characteristic splitting pattern consisting in the release of an E-AMP moiety and the corresponding E-Ado 5'(n - 1) phosphate.
The second activity peak corresponding to a molecular mass of 30 kDa as determined by gel filtration showed a great specificity towards e-(Ap3A), the enzyme being unable to cleave other homologs with a number of phos- phate groups other than three. The hydrolysis products were e-AMP and e-ADP as determined by RP-HPLC.
The third peak of activity corresponding to a molecular mass of 20 kDa as dete[mined by gel filtration, prefer- entially hydrolyzed e-(Al:,4 A) but also showed ability to cleave e-(ApsA) and e-(Ap6A) at a reduced rate; the hydrolysis products were ~Jways e-ATP and E-Ado 5'(n - 3) phosphate as determined by RP-HPLC. The homologs containing two or three phosphate groups were not sub- strates.
Fluorimetric assays showed that both 20 and 30 kDa enzymes were inhibited 19y Zn 2+ but only the 20 kDa
enzyme was inhibited by F- and AP4. Moreover, the hydrolysis of E-(ApaA) by the 20 kDa enzyme was inhib- ited by Ap4A and GP4G and hydrolysis of e-(mp3A) by the 30 kDa enzyme was inhibited by Ap3A and Gp3G. On the basis of the molecular weights and sensitivity to in- hibitors, the 20 kDa and 30 kDa activities were respec- tively identified as the highly specific dinucleoside te- traphosphate (asymmetrical) hydrolase or mp4 Aase and the highly specific dinucleoside triphosphate hydrolase or Ap3Aase.
The high molecular weight enzyme accounted for around 20% and 50% of total cytosolic hydrolytic activity on e-(ApaA) and e-(Ap3A), respectively. This enzyme ap- peared to display properties of phosphodiesterase I and was not further studied.
3.2. Kinetic analysis o f the specific hydrolases
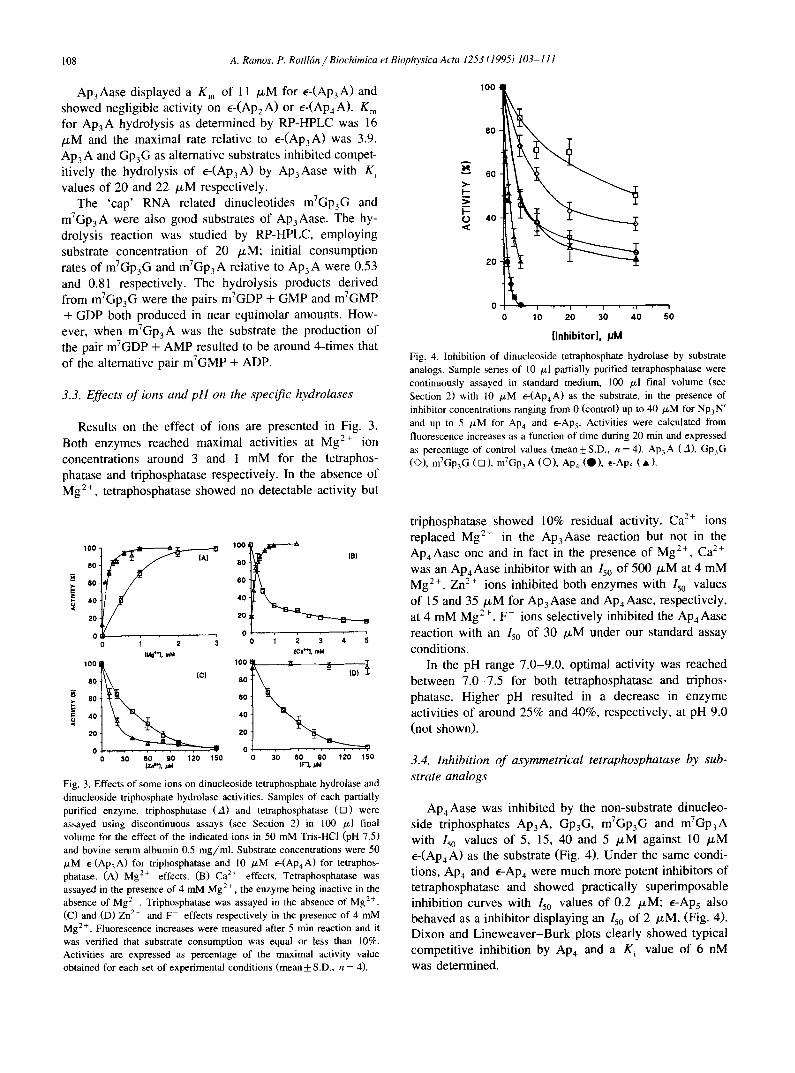
The active fractions obtained after ammonium sulfate precipitation and gel filtration steps were used for the kinetic studies of the specific hydrolases. Kinetic parame- ters were determined by continuous fluorimetric assays and both enzymes showed hyperbolic kinetics since the plots according to the integrated form of the Michaelis- Menten equation were linear, as shown in Fig. 2.
Ap4Aase K m for e-(Ap4A), e-(ApsA) and e-(Ap6A) were 1.3, 2.6 and 5.0 /xM respectively and the relative maximal rates were 1.0, 0.75 and 0.30. K m for ApaA hydrolysis as determined by RP-HPLC was 1.6 /zM and maximal rate relative to e-(Ap4A) was 0.95. Ap4A, ApsA and AP6A, alternative substrates, were competitive in- hibitors of e-(Ap4A) hydrolysis by ApaAase with K i values of 1 ~M for the three dinucleotides; Gp4G and GpsG behaved similarly with K i values of 0.2 /xM.
i i I I
O
d (B)
0.10 0 .12 0 .14 0 .16 0 .15 0 .25 0 .30 0 .35 0 . 4 0 0 .45
( [S]o - [S] ) / t ime
Fig. 2. Kinetics of e-(Ap4A) and e-(Ap3A) hydrolysis by partially purified cytosolic dinucleoside tetraphosphate hydrolase (A) and dinucle- oside triphosphate hydrolase (B). Results from two single representative experiments are shown. A sample of each partially purified hydrolase was continuously assayed in 1 ml final volume as described in Section 2. Data were plotted according to the integrated form of the Michaelis-Menten equation; initial substrate concentrations were 5 tzM e-(Apa A) in (A) and 10 ~M e-(Ap3A) in (B). All assays were performed in quadruplicate.
108 A. Ramos, P. Rotlldn / Biochimica et Biophysica Acta 1253 (1995) 103-111
Ap3Aase displayed a K m of 11 /zM for E-(Ap3A) and showed negligible activity on e-(Ap2A) or e-(Ap4A). K m for AP3A hydrolysis as determined by RP-HPLC was 16 /xM and the maximal rate relative to E-(Ap3A) was 3.9. Ap3 A and Gp3G as alternative substrates inhibited compet- itively the hydrolysis of e-(Ap3A) by Ap3Aase with K i values of 20 and 22 /zM respectively.
The 'cap' RNA related dinucleotides mTGp3 G and m7Gp3 A were also good substrates of AP3Aase. The hy- drolysis reaction was studied by RP-HPLC, employing substrate concentration of 20 /xM; initial consumption rates of mTGp3 G and mTGp3A relative to Ap3A were 0.53 and 0.81 respectively. The hydrolysis products derived from m7Gp3G were the pairs mTGDP + GMP and mTGMP + GDP both produced in near equimolar amounts. How- ever, when mTGP3 A was the substrate the production of the pair m7GDP + AMP resulted to be around 4-times that of the alternative pair m7GMP + ADP.
3.3. Effects of ions and pH on the specific hydrolases
Results on the effect of ions are presented in Fig. 3. Both enzymes reached maximal activities at Mg 2+ ion concentrations around 3 and 1 mM for the tetraphos- phatase and triphosphatase respectively. In the absence of Mg 2+, tetraphosphatase showed no detectable activity but
100
80
2 0 J.
ok 0
w 60
I--
m
I-- 0 40 ,,<
i i i , i i
10 20 30 40 50
[Inhibitor]. gM
Fig. 4. Inhibition of dinucleoside tetraphosphate hydrolase by substrate analogs. Sample series of 10 /xl partially purified tetraphosphatase were continuously assayed in standard medium, 100 /xl final volume (see Section 2) with 10 /xM e-(Ap4A) as the substrate, in the presence of inhibitor concentrations ranging from 0 (control) up to 40 p,M for Np3N' and up to 5 /zM for Ap4 and E-Aps. Activities were calculated from fluorescence increases as a function of time during 20 min and expressed as percentage of control values (mean+S.D., n = 4). Ap3A (A), Gp3G (O), mTGP3 G ( n ) , mTGp3 A (O), Ap4 (O), e-Ap 5 ( * ) .
,oo, .o ,.,
40 40
20 20
o 0 0 1 2 3 0 1 2 3 4 5
100 I
80 _=
60
i,o 2 0
[Mg'~l, mM
0 . . , - . . . . • • , • • ,
30 60 90 120 150
[CI'~, mM
4 O
2
0 30 60 90 120 150 I N ,
Fig. 3. Effects of some ions on dinucleoside tetraphosphate hydrolase and dinucleoside triphosphate hydrolase activities. Samples of each partially purified enzyme, triphosphatase (A) and tetraphosphatase (rn) were assayed using discontinuous assays (see Section 2) in 100 /xl final volume for the effect of the indicated ions in 50 mM Tris-HCl (pH 7.5) and bovine serum albumin 0.5 mg /ml . Substrate concentrations were 50 /xM E-(Ap3A) for triphosphatase and 10 /xM e-(ApaA) for tetraphos- phatase. (A) Mg 2+ effects. (B) Ca 2÷ effects. Tetraphosphatase was assayed in the presence of 4 mM Mg 2÷, the enzyme being inactive in the absence of Mg 2+. Triphosphatase was assayed in the absence of Mg 2÷. (C) and (D) Zn 2÷ and F - effects respectively in the presence of 4 mM Mg 2+. Fluorescence increases were measured after 5 min reaction and it was verified that substrate consumption was equal or less than 10%. Activities are expressed as percentage of the maximal activity value obtained for each set of experimental conditions (mean _+ S.D., n = 4).
triphosphatase showed 10% residual activity. C a 2+ ions replaced Mg 2+ in the Ap3Aase reaction but not in the Ap4Aase one and in fact in the presence of Mg 2+, Ca 2+ was an A p 4 Aase inhibitor with an 150 of 500/xM at 4 mM Mg 2+. Zn 2+ ions inhibited both enzymes with 150 values of 15 and 35/xM for AP3Aase and AP4Aase, respectively, at 4 mM Mg 2+. F- ions selectively inhibited the Ap4Aase reaction with an 150 of 30 /zM under our standard assay conditions.
In the pH range 7.0-9.0, optimal activity was reached between 7.0-7.5 for both tetraphosphatase and triphos- phatase. Higher pH resulted in a decrease in enzyme activities of around 25% and 40%, respectively, at pH 9.0 (not shown).
3.4. Inhibition of asymmetrical tetraphosphatase by sub- strate analogs
Ap4Aase was inhibited by the non-substrate dinucleo- side triphosphates Ap3A, Gp3G, mVGp3 G and m7Gp3 A with Iso values of 5, 15, 40 and 5 /xM against 10 ~M e-(Ap4 A) as the substrate (Fig. 4). Under the same condi- tions, Ap4 and e-AP4 were much more potent inhibitors of tetraphosphatase and showed practically superimposable inhibition curves with 150 values of 0.2 /xM; E-Ap5 also behaved as a inhibitor displaying an 15o of 2 /zM, (Fig. 4). Dixon and Lineweaver-Burk plots clearly showed typical competitive inhibition by Ap4 and a K i value of 6 nM was determined.

A. Ramos, P. Rotll6n / Biochimica et Biophysica Acta 1253 (1995) 103-111 109
3.5. Effects o f periodate oxidized nucleotides on the spe- cific hydrolases
Incubation of 20 /~IV[ oe- (Ap3A) with triphosphatase did not result in a noticeable fluorescence increase and hydrolysis of 20 /~M e--(Ap3A) by the enzyme was not significantly affected by the presence of 20 /zM oe- (Ap3A). Similarly, incubation of 10 /xM oe- (Ap4A) with tetraphosphatase resulted in a negligible fluorescence in- crease; however, the oxidized dinucleotide (10 /xM) markedly inhibited the splitting of 10 /zM e-(ApaA) by the hydrolase (Fig. 5A). Both oxidized substrates, oE- (Ap4A) and o-(Ap4A), behaved as effective inhibitors of Ap4Aase displaying 150 'values around 2 /~M with 10 /zM e-(Ap4A) as the substrate. Similarly, both o-Ape and
o e-Ap4 inhibited the hydrolase with 150 values around 2 /~M, inhibition being completely reversed by alkaline phosphatase (not shown).
Inhibition of Ap4Aase samples incubated in the pres- ence of o-Ap4 (10 /zM at 37°C for 30 min) and then treated by sodium borohydride (1 mM at 37°C for 30 min) was again completely reversed by alkaline phosphatase, Fig. 5B. This indicated that o-Ape was unable to bind covalently to the enzyme binding site and failed to produce irreversible inactivation. Also, o - (ApaA) and o~-(ApaA) apparently failed to produce irreversible Ap4 Aase inactiva- tion, since there were no differences in activity between samples incubated with o-(Ap4A) or o e-(Ap4A) whether followed by sodium borohydride treatment or not.
600 -
SO0 -
g 400 - i; 0 ¢¢ • 300 - 0 g h .
o 2(1o - i i
loo o
(A) 150 -I (B)
'oot i 0 10 20 30
0 10 20 30 ;s A 6'° ;s ;0 Time, rain Time, rain
Fig. 5. Effects of periodate-oxidized nucleotides on dinucleoside te- traphosphate hydrolase. Results of single representative experiments are shown. Experiments were performed in triplicate. (A) Two 10/zl samples of partially purified enzyme were diluted in standard assay medium: the first (control, O) was fortified with 10 /xM e-(Ap4A) and the second (0 ) with 10 /~M oe-(Ap4A). Final reaction mixtures were 100 /zl and were incubated at 37°C; reaction progress was followed by reading fluorescence from 10 /~1 aliquots taken every 15 min as described in Section 2. Immediately after the fourth aliquot was taken up, oe-(Ap4A) was added to the control tube and e-(Ap4A) to the second, additions being made in a negligible volttme to give 10 /xM final concentration in the remaining reaction mixtures. Fluorescence readings were again con- tinued at 15 min intervals. A negligible enzyme activity is noted on oe-(Ap4A) but it markedly reduced ~-(Ap4A) hydrolysis; a similar effect was obtained when oe-Ap4 was used instead of o¢-(ApaA). (B) Three 10 ~1 aliquots of partially purified enzyme were treated as follows. One of them (control, ©) was assayed as the control in A with 10/xM e-(AP4A) as the substrate. Fluorescence readings were taken every 10 min. The two remaining 10 /xl enzyme aliquats were appropriately diluted with stan- dard assay medium and mixed with Ap4 (A) or o-Ap4 ( • ) up to 12 /xM, and incubated at 37°C for 30 rain. Then sodium borohydride (2.5 /zl, 1 mM) was added, incubation being continued for 30 min. Afterwards, alkaline phosphatase was added (1 /zl, 20 units) and incubation continued for another 10 min. At this sta=,e both incubation mixtures were 90 /xl. After alkaline phosphatase treatment, I0 /xl of e-(Ap4A) were added to both tubes to give 10 /xM substrate in a 100 p,1 reaction mixture, as in the control assays. Fluorescence readings were taken after substrate addition every 10 min. Graphs present fluorescence increases with respect to 0 reaction time. It is noted that after incubation with phosphatase, reaction rate in the sample treated with o-Ap4 +borohydride does not differ from that treated with Ap4 +borohydride nor these from the control.
4 . D i s c u s s i o n
It follows from the present work that adrenomedullary chromaffin cells contain enzymatic systems able to hydro- lyze A p , A intracellularly, molecules for which roles as putative neurotransmitters have recently been proposed
[36]. In 1985 Wierzchowski et al. [34] proved the usefulness
of the fluorogenic derivative of Ap2 A, e-(Ap2 A), to assay a potato nucleotide pyrophosphatase and snake venom phosphodiesterase I. They suggested the use of the corre- sponding fluorogenic analogs of Ap3A and mP4A for the study of specific dinucleoside polyphosphate hydrolases. The prediction was essentially correct and the results reported here show that in addition to the chromaffin cell A p , A hydrolyzing ectoenzyme [37], specific cytosolic en- zymes able to hydrolyze A p , A may also be effectively studied through a fluorimetric approach, based on the use of e - (Ap ,A) compounds as very suitable artificial fluoro-
genic substrates. The enzymatic activities displaying high specificity to-
wards E-(Ap4A) and ~-(Ap3A) were identified as the dinucleoside tetraphosphate (asymmetrical) hydrolase and dinucleoside triphosphate hydrolase, respectively.
For tetraphosphatase, a ratio of Vmax/Km for e-(AP4 A) to Vmax/g m for Ap4A equal to 1.3 indicates that e-(AP4A) is an excellent substrate for the enzyme. Adrenomedullary Ap4Aase presents many similarities to the enzyme from human blood cells and other mammalian tissues [22,33] considering K m values, Mg 2÷ dependence, molecular mass and inhibition by Zn 2+, F - and Ap4. Interestingly, human placental enzyme exhibits a higher K m and is not inhibited by Zn 2+ [38]; such observation opens the possi- bility of existence of tissue-specific Ap4Aases, however, studies directed towards testing this possibil i ty remain to be performed. Other tetraphosphatases like that present in lupin [39] and Anemia [40] are not inhibited by Zn 2+ while S. pombe enzyme is not inhibited by fluoride [41].
Adrenomedullary tetraphosphatase was inhibited by NpaN' and the most powerful inhibitors were mTGP3 A and

110 A. Ramos, P. Rotllrn / Biochimica et Biophysica Acta 1253 (1995) 103-111
Ap3m with K i values around 0.5 /xM, slightly lower than K m values for e - (ApaA) and mp4m. Lazewska et al. [38] have reported similar findings concerning the effects of Np3N' on the human placental enzyme.
For the triphosphatase the ratio of Vmax/K m for e- (Ap3A) to Vmax/K m for A P 3 A was 0.37, indicating a
slight preference of the enzyme for Ap3A rather than for e-(Ap3A). General properties of adrenomedullary Ap3 Aase found in this work (Kin, molecular mass, Mg 2÷ depen- dence and inhibition by Zn 2+) indicate many similarities with the enzyme from varied eukaryotic organisms. Indeed the molecular mass of 30 kDa is close to the Ap3Aases from rat organs, lupin or even E. Coli [42,22] but clearly far from the enzyme found in Anemia, 115 kDa [43].
Both m7GP3 G and mTGp3 A were also good substrates for Ap3Aase and were hydrolyzed at rates of around 50 and 80% respectively when compared to AP3A hydrolysis. In the former substrate, the presence of the methyl group on a guanine ring does not appreciably modify the prefer- ence of the enzyme to cleave the substrate since the two possible product pairs m7GMP + GDP and m7GDP + GMP are released in approximately equimolar amounts. In the case of mTGp3 A, however, there is a favored cleavage of the substrate, releasing preferentially the pair m7GDP + AMP rather than the m7GMP + ADP pair. These results suggest that the chromaffin triphosphatase binds and pref- erentially cleaves the adenylate moiety of the substrate more than the guanylate moiety; in fact rat or Artemia triphosphatases exhibit higher relative velocities on Ap3A than on GP3G, [22,43].
The oxidized nucleotides oe-(Ap4A), o-(AP4A), o-AP4 and o e-Ap4 markedly inhibited the hydrolysis of e-(Ap4 A); however, these inhibitors were apparently unable to bind covalently to the binding site even after prolonged treat- ment of the inhibited enzyme with sodium borohydride. Such observation very probably indicates the lack of lysine able to react with the aldehyde groups generated in perio- date-oxidized nucleotides, thus forming a Schiff base, re- ducible by sodium borohydride at the adrenomedullary mp4 Aase substrate binding site.
It is worth noting that periodate oxidation of the te- traphosphatase substrate produces a effective inhibitor while periodate oxidation of the triphosphatase substrate produces only a non-substrate derivative. This suggests that intact ribose rings appear not to be essential for binding of substrate to the enzyme and that ribose hydrox- yls may play a catalytic role in the case of tetraphos- phatase. However, for triphosphatase, intact substrate ri- bose rings appear to be essential for binding to the en- zyme, since oxidized substrate does not behave as a sub- strate or an inhibitor.
Although at present considerable progress related to the molecular properties of these highly specific hydrolases has been achieved [22,44] our knowledge of the physio- logical significance of these enzymes remains still at a speculative stage. Some proposals suggest roles related to
the regulation of nuclear division for the tetraphosphatase [45]; however, the presence of NpnN' and related highly specific catabolic enzymes in non-replicating systems like platelets or neural cells suggest a wider spectrum of func- tions for these enzymes.
NpnN' are produced in the vast majority of organisms in a rather unspecific way as 'secondary' products of enzymes whose catalytic cycle involves a nucleotidyl-en- zyme intermediate, usually a adenylate-enzyme intermedi- ate [46]. Only the brine shrimp Artemia sp. and other related Branchiopoda contain specific enzymes for Gp,G biosynthesis [47,48]. In sharp contrast to the lack of spe- cific enzymes for NpnN' biosynthesis, there is a widely distributed catabolic family of highly specific hydrolases acting on Np,N'.
Taking account that Ap4A and ApsA are extremely potent inhibitors of adenosine kinase [2] and adenylate kinase [1], these dinucleotides could be considered toxic metabolites able to block adenosine recycling into ATP, if accumulated in the cytosol. A way to avoid such a block- ade and trapping of adenosine and energy-rich phosphate bonds could be the existence of high affinity and speci- ficity cleaving enzymes such as Ap4 Aase.
A similar proposal may be formulated for the triphos- phatase: an effective removal of NP3N' (Ap3A, Gp3G, and likely 'cap' related structures derived from RNA catabolism) may be important to prevent not only the accumulation of these dinucleotides but also any inhibition that NP3N' could exert on asymmetrical tetraphosphatase (38 and this work), thus producing a blockade in adenosine recycling through tetraphosphatase inhibition. In short, it is suggested that specific dinucleoside polyphosphate hydro- lases may, in addition to other functions, play a role as intracellular 'cleaning' enzymes thus preventing accumula- tion of potentially toxic metabolites produced rather un- specifically but able to interfere with ATP synthesis.
Acknowledgements
This work was supported by grant 91/108 from the Gobierno Autrnomo de Canarias. Thanks are also given for a grant from the Rectorado de la Universidad de La Laguna.
References
[1] Lienhard, G.E. and Secemski, I.I. (1973) J. Biol. Chem. 248, 1121-1123.
[2] Rotll~n, P. and Miras-Portugal M.T. (1985) Eur. J. Biochem. 151, 365-371.
[3] Renart, M.F., Renart, J., Sillero, M.A.G. and Sillero, A. (1976) Biochemistry 15, 4962-4966.
[4] McLennan, A.G. (Ed.) (1992) Ap4A and other dinucleoside polyphosphates, CRC Press, Boca Rat6n, FL.
[5] Warner, A.H. (1992), in Ap4A and Other Dinucleoside Polyphos-

A. Ramos, P. Rotll6n / Biochimica et Biophysica Acta 1253 (1995) 103-111 111
phates (McLennan, A.G., ed.), pp. 275-303, CRC Press, Boca Rat6n, FL.
[6] Zamenick, P.C., Stephenson, M.L., Janeway, C.M. and Randerath, K. (1966) Biochem. Biophys. Res. Commun. 24, 98-103.
[7] Garrison, P.N. and Barnes, L.D. (1992) in Ap4A and Other Dinucle- oside Polyphosphates (McLennan, A.G., ed.), pp. 29-61, CRC Press, Boca Rat6n, FL.
[8] Kitzler, J.W., Farr, S.B. and Ames, B.N. (1992) in ApnA and Other Dinucleoside Polyphosphales (McLennan, A.G., ed.), pp. 135-149, CRC Press, Boca Rat6n, FL.
[9] Remy, P. (1992) in ApaA and Other Dinucleoside Polyphosphates (McLennan, A.G., ed.), pp. 151-204, CRC Press, Boca Rat6n, FL.
[10] Flodgaard, H. and Klenow H. (1982) Biochem. J. 208, 737-742. [11] Liithje, J. and Ogilvie, A. (1983) Biochem. Biophys. Res. Commun.
115, 253-262. [12] Liithje, J. and Ogilvie, A. (1984) Biochem. Biophys. Res. Commun.
118, 704-707. [13] Louie, S., Kim, B.K. and Zamenick, P. (1988) Thromb. Res. 49,
557-566. [14] Schliiter, H., Offers, E., Briiggemann, G., Giet, M., Tepel, M.,
Nordhoff, E., Karas, M., Spieker, C., Witzel, H. and Zidek W. (1994) Nature 367, 186-188.
[15] Rodrlguez, A., Torres, M., Delicado, E.G. and Miras-Portugal M.T. (1988) J. Neurochem. 51, 11696-1703.
[16] Pintor, J., Rotll~n, P., Tor~'es, M. and Miras-Portugal (1992) Anal. Biochem. 200, 296-300.
[17] Pintor, J. Diaz-Rey, M.A., Torres, M. and Miras-Portugal M.T. (1992) Neurosci. Lett. 136, 141-144.
[18] Pintor, J., Kowalewski, H.J., Torres, M., Miras-Portugal, M.T. and Zimmermann H. (1992) Neurosci. Res. Commun. 10, 9-14.
[19] Pintor, J., Torres, M. and lvliras-Portugal M.T. (1991) Life Sci. 48, 2317-2324.
[20] Castro, E., Torres, M., Miras-Portugal, M.T. and Gonz~ilez M.P. (1990) Br. J. Pharmacol. 100, 360-364.
[21] Pintor, J., Torres, M., Castro, E. and Miras-Portugal, M.T. (1991) Br. J. Pharmacol. 103, 1989-1984.
[22] Guranowski, A. and Sillero, A. (1992) in Ap4 A and Other Dinucleo- side Polyphosphates (McLennan, A.G., ed.), pp. 81-133, CRC Press, Boca Rat6n, FL.
[23] Ogilvie, A., (1992) in ApnA and Other Dinucleoside Polyphosphates (McLennan, A.G., ed.), pp 229-275, CRC Press, Boca Rat6n, FL.
[24] Goldman, S.J., Gordon, E.L. and Slakey, L.L. (1986) Circ. Res. 59, 362-366.
[25] Ogilvie, A., Liithje, J., Pohl, U. and Busse, R. (1989) Biochem J. 259, 97-103.
[26] Rodrlguez-Pascual, F., Torres, M., Rotll~.n, P. and Miras-Portugal M.T. (1992) Arch. Biochem. Biophys. 297, 176-183.
[27] Rotllfin, P., Ramos, A., Pintor, J. and Miras-Portugal, M.T. (1991) FEBS Lett. 280, 371-374.
[28] Guranowski, A. (1990) FEBS Lett. 262, 205-208. [29] Ramos, A., Guerra, M. and Rotll~in, P. (1991) Chromatographia 32,
350-356. [30] Tolman, G.L., Barrio, J.R. and Leonard, N.J. (1974) Biochemistry
13, 4869-4878. [31] Easterbrook-Smith, S.B., Wallace, J.C. and Keech, D.B. (1976)
Eur. J. Biochem. 62, 125-130. [32] Rotllfin, P., Ramos, A. and Rodrlguez, A. (1991) J. Chromatogr.
563, 37-52. [33] Pinto, R.M., Costas, M.J., Fernfindez, A., Canales, J., Garcia-
Agfindez, J.A. and Cameselle, J.C. (1991) FEBS Lett. 287, 85-88. [34] Wierzchowski, J., Sierakowska, H. and Shugar, D. (1985) Biochim.
Biophys. Acta 828, 109-115. [35] Bradford, M.M. (1976) Anal. Biochem. 72, 248-254. [36] Pintor, J. and Miras-Portugal, M.T. (1993) Drug. Dev. Res. 28,
259-262. [37] Ramos, A., Pintor, J., Miras-Portugal, M.T. and Rotll~in, P. (1995)
Anal. Biochem. 228, 74-81. [38] Lazewska, D., Starzynska, E. and Guranowski, A. (1993) Protein
Expr. Purif. 4, 45-51. [39] Jakubowski, H. and Guranowski, A. (1983) J. Biol. Chem. 258,
9982-9989. [40] Prescott, M., Milne, D.A. and McLennan, A.G. (1989) Biochem. J.,
259, 831-838. [41] Robinson, A.K., De la Pefia, C.E. and Barnes L.D. (1993) Biochim.
Biophys. Acta 1161, 139-148. [42] Costas, M.J., Montero, M.J., Cameselle, J.C., Sillero, M.A.G. and
Sillero, A. (1984) Int. J. Biochem. 16, 757-762. [43] Prescott, M., Thorne, N.M.H., Milne, A.D. and McLennan, A.
(1992) Int. J. Biochem. 24, 565-571. [44] Guranowski, A., Brown, P., Ashton, A. and Blackburn G.M. (1994)
Biochemistry 33, 235-240. [45] Vallejo, C.G. and Le6n, P. (1989) Int. J. Biochem. 21, 1223-1228. [46] Guranowski, A., Sillero, M.A.G. and Sillero, A. (1990) FEBS Lett.
271,215-218. [47] Warner, A.H., Beers, P.C. and Huang, F.L. (1974) Can. J. Biochem.
52, 241-251. [48] Liu, J.J. and McLennan, A.G. (1994) J. Biol. Chem. 269, 11787-
11794.





























