Embed Size (px)
Citation preview

This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution4.0 International License.
Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschungin Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung derWissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht:Creative Commons Namensnennung 4.0 Lizenz.
Selbststapelnde Systeme, I 4,4' ,5,5'-Tetramethoxy-2,2'-dithiobiphenyl-Iod (6/7), eine Verbindung mit Radikalkationen und Polyiodidketten Selfstacking Systems, I 4,4',5,5'-Tetramethoxy-2,2'-dithiobiphenyl-Iodine (6/7), a Compound with Radical Cations and Polyiodide Chains
Winfried Hinrichs, Jürgen Kopf, Klaus-Wilhelm Stender und Günter Klar* Institut für Anorganische und Angewandte Chemie der Universität Hamburg, Martin-Luther-King-Platz 6, D-2000 Hamburg 13
Z. Naturforsch. 40b, 39 -44 (1985); eingegangen am 20. September 1984 4,4',5,5'-Tetramethoxy-2,2'-dithiobiphenyl-Iodine (6/7), Molecular and Crystal Structure, Self-stacking Systems, Polyiodide Chains
The title compound can be isolated from concentrated solutions of 4,4',5,5'-tetramethoxy-2,2'-dithiobiphenyl (DTB, 2) and iodine as blue needles with brass-coloured luster. The crystal struc-ture consists of stacks of (partially oxidized) DTB units with alternating orientations and of disordered polyiodide chains in channels between these stacks. The DTB molecule is not planar; the dihedral angle between the two phenyl rings is 25.1°, the dithio bridge is inclined by 41.7° to the longitudinal axis. The bond distances and angles of the disulfide group are in the normal range, the dihedral angles (CArSS'CAr- = 55.6°, CArCArSS' = 41.3°) differ from the ideal values of ca. 90° as a consequence of the cyclic structure.
Die Darstellung von 2,3,7,8-Tetramethoxythian-thren („TMT", 1), einem Dibenzo-l,4-dithiin, aus 1,2-Dimethoxybenzol und Schwefeldichlorid [1] ver-läuft über tieffarbene Oxidationsprodukte von 1 als Zwischenstufen, deren Natur von uns inzwischen aufgeklärt werden konnte [2]. Es sollte nun geprüft werden, ob auch von vergleichbaren Dibenzo-1,2-dithiinen entsprechende Oxidationsprodukte erhal-ten werden können. Dazu wurde das 4,4',5,5'-Tetra-methoxy-2,2'-dithiobiphenyl („DTB", 2) erstmals synthetisiert [3] und sein Verhalten gegenüber Iod näher untersucht.
H,C CH-, s—s
m H-jC CH,
1 (TMT) 2 (DTB)
* Sonderdruckanforderungen an Prof. Dr. W. S. Sheldrick. 0340 - 5087/85/0100 - 0039/$ 01.00/0
Darstellung und Eigenschaften der Titelverbindung
Während 1 mit Iod glatt zu dem wohldefinierten, tiefblauen (TMT)+I3~ oxidiert werden kann, liefert 2 unter den entsprechenden Bedingungen kein streng stöchiometrisch zusammengesetztes Produkt.
Beim Zusammengeben der verdünnten Lösungen von 2 und Iod beobachtet man keine Reaktion. Das Elektronenspektrum der Mischung erweist sich als Überlagerung der Spektren der Ausgangsstoffe, wie aus Abb. 1 zu ersehen ist. Ebensowenig ändert sich das 'H-NMR-Spektrum von 2 (Lösungsmittel: CDC13) bei Zugabe von Iod, wodurch die Bildung von Radikalkationen in der Reaktionslösung ausge-schlossen werden kann; konsequenterweise zeigt diese Lösung dann auch kein ESR-Signal.
Dagegen erhält man beim Eindunsten der Lösun-gen von 2 und Iod in Acetonitril, Toluol oder Chlo-roform ein tiefblaues Produkt, das aus den heißen, konzentrierten Lösungen in Form blauer, messing-glänzender Nadeln auskristallisiert. Nach den Ele-mentaranalysen besitzt die Verbindung eine gewisse Phasenbreite. Ihre Zusammensetzung schwankte zwischen (DTB)Ij 05 und (DTB)Ij 18 und läßt sich am einfachsten mit der im Titel angegebenen Formel er-fassen.

40 W. Hinrichs et al. • 4,4' ,5,5'-Tetramethoxy-2.2'-dithiobiphenyl-Iod (6/7)
Abb. 1. Elektronenspektren von DTB (2) ( ), Iod ( ) und DTB/Iod ( ) in Acetonitril als Lösungs-mittel.
Darstellungsvorschrift: In 50 ml Acetonitril wer-den 500 mg (1,4 mmol) DTB (2) und 535 mg (2,1 mmol) Diiod nacheinander gelöst. Nach kurzem Aufkochen der Lösung und Stehen über Nacht fällt 3 kristallin aus. Ausbeute 350 mg (76,2% d.Th.).
C16H16I1;07O4S2 (472,2) Ber. C 40,70 H 3,42 I 28,76, Gef. C 40,43 H 3,40 I 28,36.
Die Verbindung 3 bildet sich auch, wenn festes DTB (2) bei ca. 80 °C in eine Iodatmosphäre ge-bracht wird. Die Aufnahme von Iod in die Kristalle von 2 erkennt man gut an dem Farbumschlag von gelb nach blau unter gleichzeitiger Ausbildung des metallischen Glanzes. Die Iodaufnahme ist reversi-bel. Durch Tempern bei ca. 120 °C wird das Iod wie-
der abgegeben, wobei unverändertes DTB zurückge-wonnen werden kann.
Im festen Zustand ist 3 paramagnetisch. Das ESR-Spektrum zeigt ein Singulett ohne Feinstruktur mit g = 2,00606, also einem g-Faktor, der für organische Radikale charakteristisch ist. Erste, mehr orientie-rende Spindichtebestimmungen ergaben ca. 1,5 • 1023 Spins/mol, was in etwa einem Oxidations-grad von [(CTB)4]+ entspricht. Als Gegen-Ionen müßten dann in 3 Iodid-Ionen neben Iod oder Poly-iodid-Ionen vorliegen. Im Ramanspektrum der Sub-stanz fehlt die Resonanzlinie der Streckschwingung von L-Molekülen, die bei 215 cm"1 auftritt [4]. Im Spektrum finden sich aber die Absorptionen bei 160 und 110 cm - 1 , die entweder I5~-Einheiten [4] oder verzerrten I3~-Einheiten [5] zugeordnet werden kön-nen und damit das Vorhandensein von Polyiodidket-ten nahelegen.
Strukturbestimmung
Um weitere Aussagen über die Natur der Verbin-dungen 3 zu erhalten, wurde eine Röntgenstruktur-analyse durchgeführt.
Ein Kristall der Größe 0,05x0,15x0,05 cm3 wur-de auf einem Syntex-P21-Vierkreisdiffraktometer unter Verwendung von MoKa-Strahlung (X = 71,069 pm) nach der 0/20-Scan-Methode vermessen. Die Strukturdaten finden sich in Tab. I. Die Um-wandlung der Intensitäten in Strukturamplituden wurde mit den üblichen Lorentz- und Polarisations-korrekturen durchgeführt, wobei wegen des kleinen Absorptionskoeffizienten auf eine Absorptionskor-rektur verzichtet werden konnte. Die Struktur wurde
Tab. I. Strukturdaten für 3 mit Standardabweichungen in Klammern.
20-Bereich [°] 48 Zahl der unabhängigen Reflexe 3777 Reflexe mit |F0| > 3CJ(F0) 2463 Kristallsystem monoklin Raumgruppe C2/c a [pm] 2312,3(10) b [pm] 698,5(2) c [pm] 2555,8(10) ß [°] 123,52(3) Z 8 V [106 pm3] 3441.6 pber [g/cm3] 2,78 /?, = 1.\A F /2 F0 0.062 R 2 = V 2 w | j F p / 2 w | F 0 | 2 ° - 0 5 2
w 1 = ( / ( F . )

41 W. Hinrichs et al. • 4,4',5,5'-Tetramethoxy-2.2'-dithiobiphenyl-Iod (6/7)
mit Hilfe von Direktmethoden (MULTAN 78 [7]) und Differenz-Fourier-Rechnungen (SHELX 76 [8]) gelöst. Die Direktmethoden lieferten ein fast voll-ständiges Bild der DTB-Einheit. Die Bestimmung der Iodlagen gestaltete sich schwieriger, weil schon aus Drehkristallaufnahmen zu ersehen war, daß ent-lang der £>-Achse eine Fehlordnungsperiode von 310 pm vorhanden war, die für Polyiodidketten ty-pisch ist [9]. Die Art der Fehlordnung wurde mit Hilfe von Elektronendichtediagrammen aus Diffe-renz-Fourier-Rechnungen mit der vorgegebenen DTB-Einheit bestimmt. Danach besetzen die Iod-atome für x/a = 0,25 und z/c = 0,0 entlang der b-Achse beliebige Lagen, da die Elektronendichte in diesen Kanälen nirgendwo Null wird, sondern allen-falls auf ca. 65% des Maximalwerts absinkt. Für die nachfolgenden kombinierten Differenz-Fourier- und LSQ-Rechnungen wurden 3 Iodlagen (entsprechend den Elektronendichtemaxima) mit variablen fraktio-nellen Koordinaten für xla und z/c auf der £>-Achse fixiert und abwechselnd mit freien Besetzungs- bzw. Temperaturfaktoren in einer Reihe von LSQ-Rech-nungen bis zur Konvergenz verfeinert. Der beson-dere Zustand der Iodatome wird aus den Werten der Tab. III deutlich. Die Lagen der Wasserstoffatome wurden in abschließenden Differenz-Fourier-Rech-
nungen lokalisiert und dann mit starren Bindungsab-ständen von 96 pm berechnet. Die schweren Atome der DTB-Einheit wurden wie üblich ohne Beschrän-kung mit freien Variablen für Orts- und Temperatur-parameter verfeinert. Die endgültigen Atomkoordi-naten finden sich in Tab. II*, das Numerierungs-schema für die DTB-Einheit ergibt sich aus Abb. 2.
S2 — S1 / \
C27 C23 — C22 C12—C13 C17 \ / \ / \ / 02U—C2L C21 —C11 O t t — O t t
\ / \ / C25—C26 C16—C15 / \
025 015 \ / C28 C18
Abb. 2. Numerierungsschema für die DTB-Einheit von 3.
* Die Lagen der Wasserstoffatome, die thermischen Para-meter und die Strukturfaktoren können beim Fachinfor-mationszentrum Energie, Physik, Mathematik, D-7513 Eggenstein-Leopoldshafen 2, unter Angabe der Hinter-legungsnummer CSD 51052, der Autoren und des Zeit-schriftenzitats angefordert werden.
Tab. II. Atomkoordinaten für 3 mit Standardabweichungen in Klammern.
Atom j = 1 J = 2 x/a y/b z/c xla y/b z/c
Sj 0,7893(1) -0,148(1) 0,1573(1) 0,7094(1) 0,047(1) 0,1178(1) C j l 0,7878(3) -0,053(1) 0,2616(3) 0,7112(3) -0,054(1) 0,2234(3) Cj 2 0,8270(3) -0,087(1) 0,2364(3) 0,6716(3) -0,019(1) 0,1590(3) Cj3 0,8997(3) -0,096(1) 0,2750(3) 0,5993(3) -0,022(1) 0,1251(3) Cj4 0,9334(3) -0,064(1) 0,3383(3) 0,5654(3) -0,060(1) 0,1541(3) Cj5 0,8954(3) -0,022(1) 0,3645(3) 0,6043(3) -0,092(1) 0,2194(3) Cj6 0,8236(3) -0,012(1) 0,3263(3) 0,6759(3) -0,092(1) 0,2523(1) Cj7 1,0451(3) -0,102(1) 0,3547(4) 0,4537(3) -0,057(1) 0,0580(3) Cj 8 0,8967(3) 0,041(1) 0,4572(3) 0,6040(3) -0,137(1) 0,3116(3) Oj4 1,0036(2) -0,070(1) 0,3799(2) 0,4956(2) -0,071(1) 0,1254(2) Oj5 0,9331(2) 0,006(1) 0,4277(2) 0,5669(2) -0,122(1) 0,2448(2) I I 0,2416(7) 0,285(0) -0,0032(8) 12 0,2478(2) 0,460(0) -0,0095(2) 13 0,2589(2) 0,660(0) 0,0036(2)
Tab. III. Temperatur- und Besetzungsfaktoren für die Iodlagen in 3. Der anisotrope Temperaturfaktor ist definiert als exp[ -2 ; r 2 (U n h 2 a 2 + \J22k2b2 + U 3 3 / V + 2 U 2 3 k lb c + 2 U n h l a c + 2 U n h k a b)].
Atom U„ U22 U33 U23 U13 U12 Besetzungs-faktor
11 0,055(5) 0,493(21) 0,060(4) -0,099(7) 0,034(4) -0,055(5) 0,299(5) 12 0,040(2) 0,432(14) 0,063(2) 0,015(5) 0,026(2) 0,024(5) 0,384(9) 13 0,048(2) 0,445(20) 0,059(2) 0,034(4) 0,025(2) -0,025(3) 0,453(5)
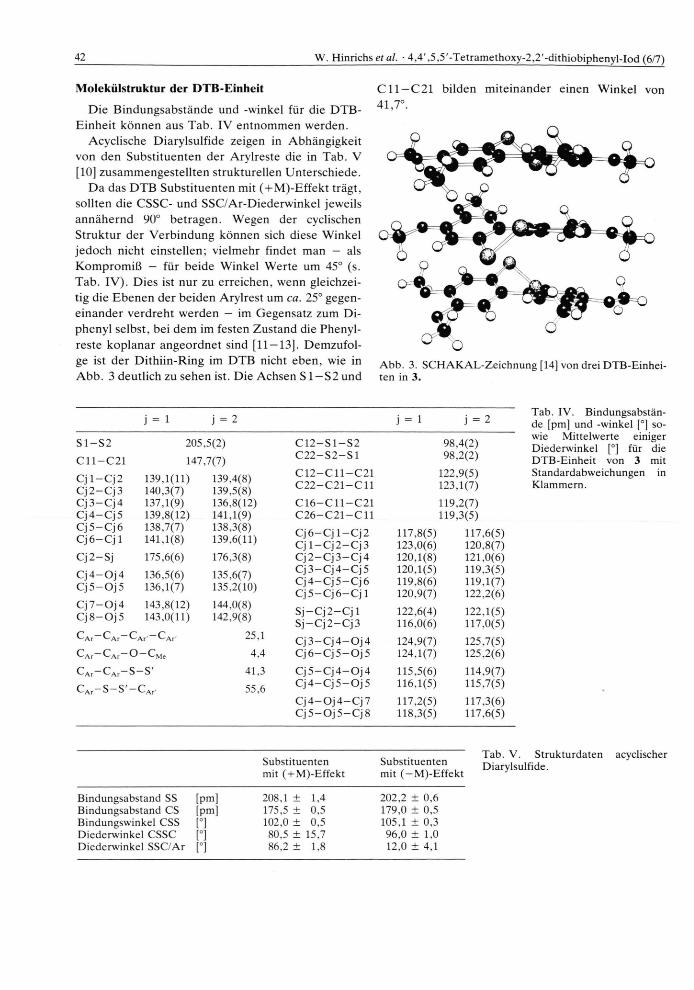
42 W. Hinrichs et al. • 4,4',5,5'-Tetramethoxy-2.2'-dithiobiphenyl-Iod (6/7)
Molekülstruktur der DTB-Einheit
Die Bindungsabs tände und -winkel für die DTB-Einhei t können aus Tab . IV en tnommen werden .
Acyclische Diarylsulfide zeigen in Abhängigkei t von den Subst i tuenten der Arylreste die in Tab . V [10] zusammengeste l l ten strukturellen Unterschiede .
D a das D T B Subst i tuenten mit ( + M)-Ef fek t trägt , sollten die CSSC- und SSC/Ar-Diederwinkel jeweils annähe rnd 90° be t ragen . Wegen der cyclischen St ruktur der Verb indung können sich diese Winkel jedoch nicht einstellen; vielmehr findet man — als K o m p r o m i ß — für beide Winkel Wer te um 45° (s. Tab . IV) . Dies ist nur zu erreichen, wenn gleichzei-tig die E b e n e n der beiden Arylrest um ca. 25° gegen-e inander verdreht werden — im Gegensatz zum Di-phenyl selbst, bei dem im festen Zustand die Phenyl-reste koplanar angeordne t sind [11 — 13]. Demzufol -ge ist der Dithi in-Ring im D T B nicht eben , wie in A b b . 3 deutlich zu sehen ist. Die Achsen S1 —S2 und
C l l —C21 bilden mi te inander einen Winkel von 41,7°.
Abb. 3. SCHAKAL-Zeichnung [14] von drei DTB-Einhei-ten in 3.
j = 1 j - 2 j = 1
S I - S2 205,5(2) C 1 2 - S 1 - S 2 98,4(2) C H — C21 147,7(7) C 2 2 - S 2 - S 1 98,2(2)
c j i -Cj2-
" C j 2 - C j 3
139,1(11) 140,3(7)
139,4(8) 139,5(8)
C 1 2 - C 1 1 - C 2 1 C 2 2 - C 2 1 - C 1 1
122,9(5) 123,1(7)
Cj3-- C j 4 137,1(9) 136,8(12) C 1 6 - C 1 1 - C 2 1 119,2(7) Cj4-- C j 5 139,8(12) 141,1(9) C 2 6 - C 2 1 - C 1 1 119,3(5) Cj5-Cj6-
- C j 6 - c j i
138,7(7) 141,1(8)
138,3(8) 139,6(11) Cj6—Cj 1 —Cj2
Cj 1 —Cj2-Cj3 117,8(5) 123,0(6)
117,6(5) 120,8(7)
Cj2-- s j 175,6(6) 176,3(8) Cj 2 — Cj 3 — C j 4 120,1(8) 121,0(6) Cj4-Cj5-
- O j 4 - O j 5
136,5(6) 136,1(7)
135,6(7) 135,2(10)
Cj3—Cj4—Cj5 Cj4—Cj5 —Cj6 Cj5 —Cj6—Cj 1
120,1(5) 119,8(6) 120,9(7)
119,3(5) 119,1(7) 122,2(6)
Cj7-Cj8-
- O j 4 - O j 5
143,8(12) 143,0(11)
144,0(8) 142,9(8) Sj—Cj2—Cj 1
S j - C j 2 - C j 3 122,6(4) 116,0(6)
122,1(5) 117,0(5)
CAI-" ~ C A R ~ C A R ' - C A R ' 25,1 Cj3 —Cj4—Oj4 124,9(7) 125,7(5) CAR" ~CA R— o - c M e 4,4 C j 6 - C j 5 - O j 5 124,1(7) 125,2(6)
CAI-" ~ cAr— S - S ' 41,3 Cj5 —Cj4—Oj4 115,5(6) 114,9(7)
C^AR" - S - S ' — CAR' 55,6 Cj4—Cj5 —Oj5
Cj4—Oj4—Cj7 Cj 5 — O j 5 — C j 8
116,1(5)
117,2(5) 118,3(5)
115,7(5)
117,3(6) 117,6(5)
Tab. IV. Bindungsabstän-de [pm] und -winkel [°] so-wie Mittelwerte einiger Diederwinkel [°] für die DTB-Einheit von 3 mit Standardabweichungen in Klammern.
Substituenten mit ( + M)-Effekt
Substituenten mit ( -M)-Effekt
Bindungsabstand SS [pm] 208,1 ± 1,4 202,2 ± 0,6 Bindungsabstand CS [pm] 175,5 ± 0,5 179,0 ± 0,5 Bindungswinkel CSS [°] 102,0 ± 0,5 105,1 ± 0,3 Diederwinkel CSSC [°] 80.5 ± 15.7 96,0 ± 1.0 Diederwinkel SSC/Ar [°] 86,2 ± 1.8 12,0 ± 4,1
Tab. V. Strukturdaten acyclischer Diarylsulfide.

W. Hinrichs et al. • 4,4 ,,5,5'-Tetramethoxy-2,2'-dithiobiphenyl-Iod (6/7) 43
Im DTB weist also die ArSSAr-Gruppierung nicht die ideale Konformation auf; daher können sich die mesomeren Effekte der Substituenten gemäß 2 a und 2b nicht voll auswirken. Auch wenn die Abweichun-
'S-S> 'S-S^
2a V / <0
/ 0/ —
2b
gen nicht immer sehr signifikant sind, ist dies zu er-kennen an dem im Vergleich zum Normalfall (Tab. V) kleineren CSS-Bindungswinkel und an dem kürzeren SS- bzw. längeren CS-Bindungsabstand. Andererseits ist im Einklang mit 2 b der Abstand zwischen den beiden Arylresten gegenüber dem Bi-phenyl [11-13] verkürzt (147,7 bzw. 150,6 pm). Da-bei ist zu beachten, daß durch die partielle Oxidation der DTB-Einheit in 3 die Mesomerie begünstigt wird.
Im übrigen liegen die CC-Abstände und CCC-Winkel in den Arylresten im üblichen Bereich. Auch
die Anordnung der beiden jeweils orthoständigen Methoxy-Substituenten jedes Ringes (transartig, ko-planar zum Ring) entspricht völlig den Befunden bei den oligomeren 4,5-Dimethoxy-l,2-phenylenchalko-geniden [15] und ist dort schon ausführlich disku-tiert. Geometrie und Bindungsabstände sprechen ebenfalls für die oben formulierten mesomeren Wechselwirkungen.
Kristallstruktur von 3
Die Kristallstruktur von 3 ist in Abb. 4 dargestellt. Im Kristall bilden die DTB-Einheiten in ^-Rich-
tung parallele Stapel, während die Polyiodidketten in Kanälen zwischen diesen Stapeln untergebracht sind.
Die DTB-Stapel zeigen eine Schichtenfolge AB A B . . . , da direkt aufeinanderfolgende Moleküle um jeweils 180° um die Längsachse (C11-C21) ge-dreht sind. Bei dieser Anordnung stören sich die aus den Ringebenen herausragenden Schwefelatome ge-genseitig nicht. Gleichzeitig wird dadurch erreicht, daß übereinanderliegende Ringe parallel zueinander ausgerichtet sind. Da die beiden Arylringe der DTB-
Abb. 4. Elementarzelle von 3 (Projektion auf die a/c-Ebene).

44 W. Hinrichs et al. • 4,4',5,5'-Tetramethoxy-2.2'-dithiobiphenyl-Iod (6/7)
Einheit nicht koplaner sind, haben die nebeneinan-der verlaufenden Arylstapel eines DTB-Stapels ent-gegengesetzte Anstellwinkel zur Stapelachse (vgl. Abb. 3). Der Abstand zwischen den Arylresten liegt mit 349 pm in einem Bereich, der auch bei anderen Stapelstrukturen mit Polyiodidketten gefunden wird, wie z.B. bei (TTF)I0 j i (TTF = Tetrathiafulvalen) mit 356 pm [16] und bei (TTT)I1 5 (TTT = Tetrathio-tetracen) mit 332 pm [17].
Zwar können aus den drei hauptsächlichen Iod-lagen in den Kanälen konventionelle Iod/Iod-Ab-stände wie im Diiod-Molekül oder im Triiodid-Ion berechnet werden, doch lassen sich diese Spezies nicht exakt lokalisieren. Tatsächlich ist aufgrund der Fehlordnung der Iod-Atome nur eine quasizwei-dimensionale Lösung der Struktur möglich, so daß eine weitergehende Interpretation der Natur der
Iodketten aus dem vorliegenden Datensatz heraus nicht sinnvoll erscheint.
Der Ladungstransport bei der Ausbildung der par-tiell oxidierten DTB-Stapel und der Polyiodid-Ket-ten erfolgt wohl nicht wie bei den a-Cyclodextrin/ Iod-Verbindungen über Iod/Wasserstoff-Brücken [18], sondern eher über Iod/Schwefel-Wechselwir-kungen. Dies ergibt aus einem Vergleich der Abstän-de zwischen den Iodkanälen und den nächstliegen-den Wasserstoff- bzw. Schwefelatome der DTB-Ein-heiten der Stapel (dIH = 320 pm, dIS = 350 pm) einerseits und den van-der-Waals-Abständen [19, 20] (I—H = 335 bzw. 318 pm und I - S = 400 bzw. 378 pm) andererseits.
Wir danken der Stiftung Volkswagenwerk für fi-nanzielle Unterstützung.
[1] T. Weiß und G. Klar, Liebigs Ann. Chem. 1978, 785. [2] W. Hinrichs und G. Klar, in Vorbereitung. [3] K.-W. Stender und G. Klar, in Vorbereitung. [4] A. G. Maki und R. Vorneris, Spectrochim. Acta,
Part A, 23, 867 (1967). [5] M. Cowie, A. Gleizes, G. W. Grynkewich, D. W.
Kaiina, M. S. McClure, R. P. Scaringe, R. T. Teitel-baum, S. L. Ruby, J. A. Ibers, C. R. Kannewurf und T. J. Marks, J. Am. Chem. Soc. 101, 2921 (1979).
[6] E. Mulazzi, I. Pollini, L. Piceri und R. Tubino, Phys. Rev. B 24, 3555 (1981).
[7] P. Main. S. E. Hull, L. Lessinger, G. Germain, J. Declerq und M. Woolfson, MULTAN 78: System of Computer Programs for the Automatic Solution of Crystal Structures from X-Ray Diffraction Data, York 1978.
[8] G. Sheldrick SHELX 76: Programs for Crystal Struc-ture Determination, Cambridge 1976.
[9] M. Kertesz und F. Vonderviszt, J. Am. Chem. Soc. 104, 5889 (1982).
[10] J. Kopf, K. von Deuten, B. Nakhdjavan und G. Klar, Z. Naturforsch. 34b, 48 (1979).
[11] J. Trotter, Acta Crystallogr. 14, 1135 (1961). [12] A. Hargreaves und S. H. Rizvi, Acta Crystallogr. 15,
365 (1962). [13] G.-P. Charbonneau und Y. Delugeard, Acta Crystal-
logr. B 32, 1420 (1976). [14] E. Keller, SCHAKAL: Molekülmodell-Plotpro-
gramm, Universität Freiburg 1980. [15] W. Hinrichs, H.-J. Riedel und G. Klar, J. Chem. Res.
(S), 1982, 334, J. Chem. Res. (M), 1982, 3501. [16] J. J. Daly und F. Sanz, Acta Crystallogr. B 31, 620
(1975). [17] L. I. Buravov, G. I. Zvereva, V. F. Kaminskii. L. P.
Rosenberg. M. L. Khidekel, R. P. Shibaeva, I. F. Shchegolev und E. B. Yagubskii, J. Chem. Soc. Chem. Commun. 1976, 720.
[18] W. Saenger, Angew. Chem. 92, 343 (1980). [19] L. Pauling, Die Natur der chemischen Bindung,
2. Aufl., S. 245, Verlag Chemie, Weinheim 1964. [20] A. Bondi, J. Phys. Chem. 68, 441 (1964).





























