Embed Size (px)
Citation preview

Evaluation of Comparative Protein StructureModeling by MODELLER-3Roberto Sanchez and Andrej Sali*The Rockefeller University, New York, New York
ABSTRACT We evaluate homology-de-rived 3D models of dihydrofolate reductase(DFR1), phosphotransferase enzyme IIA do-main (PTE2A3), and mouse/human UBC9 pro-tein (UBC924) which were submitted to thesecond Meeting on the Critical Assessment ofTechniques for Protein Structure Prediction(CASP). The DFR1 and PTE2A3 models, basedon alignments without large errors, wereslightly closer to their corresponding X-raystructures than the closest template struc-tures. By contrast, the UBC924 model wasslightly worse than the best template due to amisalignment of the N-terminal helix.Althoughthe current models appear to be more accuratethan the models submitted to the CASP meet-ing in 1994, the four major types of errors inside chain packing, position, and conformationof aligned segments, position and conforma-tion of inserted segments, and in alignmentstill occur to almost the same degree. Themodest improvement probably originates fromthe careful manual selection of the templatesand editing of the alignment, as well as fromthe iterative realignment and model buildingguided by various model evaluation tech-niques. This iterative approach to comparativemodeling is likely to overcome at least someinitial alignment errors, as demonstrated bythe correct final alignment of the C terminus ofDFR1. Proteins, Suppl. 1:50–58, 1997.r 1998 Wiley-Liss, Inc.
Key words: evaluation; comparative proteinmodeling; Modeller
INTRODUCTION
Protein modelers were challenged for the secondtime to model sequences without available 3D struc-tures and to submit them to the CASP meeting inDecember 1996 (CASP; URL http://PredictionCenter.llnl.gov/). At the same time, the 3D structures werebeing determined by X-ray crystallography and NMRmethods. Because the experimentally determinedstructures were only released at the meeting, it waspossible to test the modeling methods objectively. Asummary of all comparative models submitted toCASP2 can be found elsewhere in this issue (A.C.R.Martin et al.).
We submitted homology-derived models of threeproteins: DFR1, PTE2A3, and UBC924; the subscriptindicates the target sequence number assigned bythe organizers of CASP2. All three structures havebeen determined by X-ray crystallography: DFR1 at2.6 Å resolution and R factor of 18% (U. Pieper andO. Herzberg, in preparation), PTE2A3 at 2.4 Åresolution (K. Huang and O. Herzberg, in prepara-tion) and UBC924 at 2.0 Å resolution and R factor of16% (H. Tong and T. Sixma, in preparation). Thesethree target sequences were chosen because theyhave a relatively low, ,43% sequence identity withtheir templates. In this range of sequence similarity,the largest errors in comparative modeling due tomisalignments begin to appear.1,2 It is important toconcentrate on this range of sequence similaritybecause most of the detectable related sequence–structure pairs are related at less than 40% sequenceidentity level,3 despite earlier indications to thecontrary.4
Our approach to comparative protein structuremodeling is based on satisfaction of spatial re-straints and is implemented in program Modeller.†5
This program can be used in all stages of typicalcomparative modeling: Finding suitable templatestructures in the PDB,6 aligning them with thesequence to be modeled, calculating the 3D model,and evaluating the model. Comparative protein mod-eling was recently reviewed.7,8
†Modeller is available at URL http://guitar.rockefeller.edu:pub/modeller and also as part of Quanta and InsightII (MSI,San Diego, CA. E-mail: [email protected]).
Abbreviations: DFR1, Haloferax volcanii dihydrofolate reduc-tase; PTE2A3, Mycoplasma capricolum phosphotransferaseenzyme IIA domain; UBC924, mouse/human UBC9 protein;NMR, nuclear magnetic resonance; PDB, Brookhaven ProteinData Bank; RMSD, root-mean-square deviation; 3D, three-dimensional; CASP, critical assessment of techniques for pro-tein structure prediction.
Contract grant sponsor: National Institutes of Health; Con-tract grant number: GM 54762; Contract grant sponsor: Na-tional Science Foundation; Contract grant number: BIR-9601845.
*Correspondence to: A. Sali, The Rockefeller University, 1230York Avenue, New York, NY 10021.
E-mail: [email protected] 8 May 1997; Accepted 26 August 1997
PROTEINS: Structure, Function, and Genetics, Suppl. 1:50–58 (1997)
r 1998 WILEY-LISS, INC.

In this article, we briefly describe the modelingmethod and then concentrate on evaluation of thethree submitted models. In particular, we discussthe question of whether or not the models aregenerally closer to the X-ray structure being modeledthan the template structures.
METHODS
The first step in comparative modeling of the threetarget proteins was identification of potential tem-plate structures. This was followed by several cyclesof template selection, target-template alignment,model building, and model evaluation. The aim ofthe iteration was to minimize the errors in the modelreported by various model evaluation techniques.This iterative process, including careful manualselection of the templates and editing of the align-ments, is the main difference between the currentapproach and that followed two years ago for theCASP1 meeting.2 The final alignments and 3D mod-els are available from the CASP2 Web site at URLhttp://PredictionCenter.llnl.gov/CASP/CM_results/.
Template Selection
Proteins that have known 3D structure and aresimilar to the sequences being modeled had to beidentified. This was achieved by searching a set ofsequences representative of the whole PDB (July 1,1996) [6], using the SEQUENCE_SEARCH com-mand of Modeller.9 The representative set of proteinstructures included 916 chains whose sequence iden-tity was less than 30% to any other chain in the set.The final templates were as follows: For DFR1,4DFR-B (30%, 1.4 Å, 91%), 3DFR (24%, 1.5 Å, 93%),and 8DFR (22%, 1.6 Å, 94%); for PTE2A3 , 1GPR(43%, 1.3 Å , 94%) and 1F3G (36%, 1.1 Å , 94%); andfor UBC924, 1AAK (35%, 1.1 Å, 90%) and 2UCE(30%, 1.2 Å, 90%). The numbers in the parenthesesare the percentage sequence identity, RMSD for Caatoms, and the fraction of the equivalent Ca atoms.These were all obtained from pairwise template-target least-squares superpositions with a 3.5 Åcutoff.
Target-Template Alignment
Initial multiple template-target alignments wereobtained by aligning the target sequences with theprealigned template structures, using the ALIGN2Dcommand of Modeller.9 This command implements aglobal dynamic programming10 algorithm with avariable gap-penalty function that depends on thestructural context of an insertion or a deletion (R.Sanchez and A. Sali, in preparation). The gap pen-alty is constructed such that insertions and deletionsare less preferred within helices and sheets, buriedregions, straight segments, and also between tworesidues that are distant in space. The alignmentsalso depended on a 20 3 20 amino acid residuesubstitution matrix that was derived from 105 struc-
ture–structure alignments.4 The initial calculatedalignments were edited by hand as appropriate (seebelow).
Model Building
The 3D models containing all nonhydrogen atomswere obtained automatically by satisfying restraintson many distances, angles, and dihedral angles.4,5
Spatial restraints were extracted from the align-ment of the target sequence with the templatestructures4,5 and from the Charmm-22 force field.11
The whole model, including backbone, side chains,loops, and insertions, was build in one optimization.Conformation of the regions aligned with the tem-plates was based mostly on the template structures,while the insertions were restrained mostly by thepreferences of the different residue types for thedifferent areas of the Ramachandran plot.
Model Evaluation
The models had to satisfy most restraints used tocalculate them, especially the stereochemical re-straints. These tests were done by the ModellerENERGY command,9 the Procheck program,12 andthe WhatCheck program.13 The most importantevaluation was done by ‘‘energy’’ profiles calculatedby ProsaII, which relies on statistical potentialsinvolving single residues and pairs of residues.14
Additional evaluation was done by ‘‘energy’’ profilescalculated from a new set of statistical potentialsinvolving pairs of atoms.15 Side chain packing waschecked by calculating cavities in the core of aprotein, using the Quanta Protein Health module(MSI, San Diego, CA). If any of the model evaluationtools indicated an error in the model, the model waschanged manually. For example, side chains weremanually repositioned to eliminate a cavity in thecore. Another example is a selection of differenttemplates and editing of the alignment around theregion with a bad ProsaII profile, followed by an-other round of the automated model building.
RESULTS AND DISCUSSION
Although the DFR1, UBC924, and PTE2A3 modelshave good stereochemistry, they have errors in fourother categories: Distortions or shifts of a region thatis aligned correctly with the templates (e.g., loops,helices, strands); errors in side chain packing; distor-tions or shifts of a region that does not have anequivalent segment in any of the templates (e.g.,inserted loops); and distortions or shifts of a regionthat is aligned incorrectly with the templates (e.g.,loops and larger segments with low sequence iden-tity to the templates). Examples of these errors aredescribed in the following sections. We also discussthe lessons learned from this experiment with re-spect to automated template mimicking in differentregions of a model; the cycle of template selection,alignment, model building, and model evaluation;
51EVALUATION OF COMPARATIVE PROTEIN MODELING

and the relative overall similarity of a model and thetemplates to the target X-ray structure.
Stereochemistry of the Models
The stereochemical features of the models, such asthose evaluated by the Procheck12 and WhatCheck13
programs, are comparable to those in the highresolution X-ray structures. These features includebond lengths, angles, improper dihedral angles, posi-tion of residues in the Ramachandran plot, peptidebonds planarity, Ca tetrahedral distortion, non-bonded interactions, hydrogen bond energies, andcloseness of side chain dihedral angles to idealvalues. It is not surprising that the models arestereochemically correct since they were calculatedpartly by optimizing the stereochemical features asencoded in the Charmm-22 force field.11
Errors in Side Chain Packing
The side chain rotamers were predicted surpris-ingly inaccurately. For example, the percentage of x1angles for DFR1, PTE2A3, and UBC924 predictedwithin 30° of the target values was 42%, 48%, and65%, respectively. Since at least the UBC924 X-raystructure has been refined at a high resolution of 2 Åand an R factor of 16%, the low prediction accuracymust reflect significant problems with our side chainmodeling procedure in this range of backbone andside chain similarities. However, the mistakes madewere not trivial because the models followed theirtemplates for conserved and similar side chains,because the model rotamers were not distorted, andbecause the cavities in the models were not largerthan those in the X-ray structures. It is not clearwhat kind of improvements are needed beyond aself-evident need for a more accurate energy functionand perhaps a better optimizer.
The difficulty of the side chain modeling problemin this range of sequence similarity is illustrated bythe fact that the template and target X-ray struc-tures have different rotamers for up to 45% of theconserved residues. For example, DFR1 has 125residues with at least one side chain dihedral angle,29 of which are conserved in one of the templates(PDB code 3DFR), but 12 of these occur in differentrotamer states.Asystematic analysis of this phenom-enon, based on highly refined structures, would beuseful. If the target and template X-ray structuresare accurate and the finding proves to be general,this indicates that the side chains should be modeledon the basis of more general physical principles16–19
rather than by mimicking the templates,20,21 espe-cially when the backbones of the target and thetemplate have an RMSD larger than 2 Å. An addi-tional complication for the evaluation of side chainmodels is that for the two targets refined at a lowresolution of 2.6 Å (DFR1) and 2.4 Å (PTE2A3), it isnot clear that all the differences between the models
and the X-ray structures are due to the mistakes inthe modeling procedure.22
Distortions or Shifts in Correctly AlignedRegions: Template Mimicking in DifferentRegions of a Model
For all three models, at least two template struc-tures were used. Thus, it was possible to determinehow frequently the automated model building se-lected the best template for a given segment wherethe templates shared different degrees of structuralsimilarity with the target structure. The ability topick locally optimal templates is important becauseit allows the model to be overall closer to the correctstructure than any of the individual templates.
The distances of the positions of the Ca atoms ofthe model and the templates from the equivalentatoms in the superposed target X-ray structure areshown for DFR1 and UBC924 in Figure 1. For thecorrectly aligned regions, the model always followsone of the templates. When two templates differ in agiven correctly aligned region, the model generallyfollows the template that is structurally closer to theexperimental structure: Six such segments of atleast three residues with distances between thetemplates of at least 1 Å occur in the DFR1 andUBC924 models. For the correctly aligned regions,there are no examples of the model following asuboptimal template. As a consequence, the model isgenerally closer overall to the experimental struc-ture than any of the templates (see also Fig. 4).However, for a given region, model building does notresult in a model that is better than the besttemplate in that region (Fig. 1).
These observations are a direct consequence of theform of the homology-derived distance restraints.4,5
The restraints are expressed as probability densityfunctions. When several templates are aligned witha given segment in the target sequence, a restrainton an inter- or intrasegment distance has a multimo-dal shape with the peaks corresponding to the equiva-lent distances in the templates, not to the averagedistance. The heights and the widths of the peaksare determined by the overall and local sequencesimilarities between the templates and the targetsequence, such that the model is most likely toresemble the template with the most similar se-quence. This means that the model is generallycloser to one or the other template by construction.In order to allow for the modeling of distortions orshifts relative to the template structures, a scoringfunction that guides the model in the correct direc-tion from the template to the target structure isnecessary. A combination of homology-derived re-straints with atom based statistical potentials15,23–25
is perhaps one way of achieving this aim.
52 R. SANCHEZ AND A. SALI
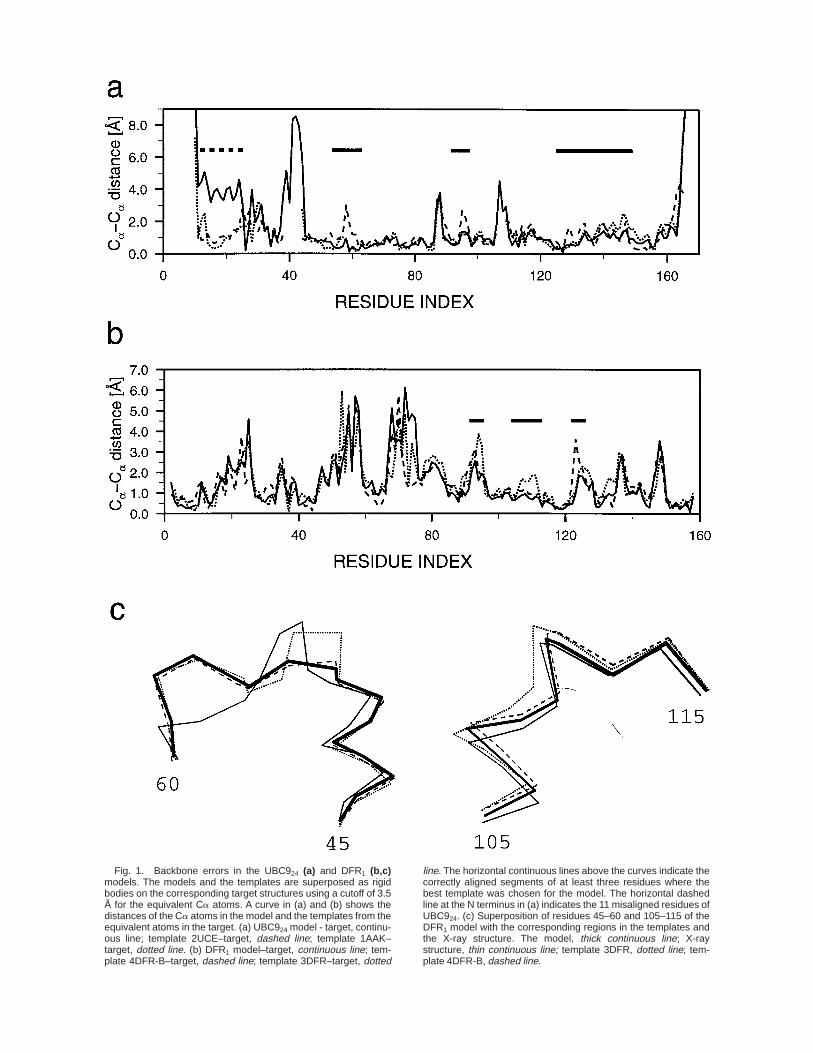
Fig. 1. Backbone errors in the UBC924 (a) and DFR1 (b,c)models. The models and the templates are superposed as rigidbodies on the corresponding target structures using a cutoff of 3.5Å for the equivalent Ca atoms. A curve in (a) and (b) shows thedistances of the Ca atoms in the model and the templates from theequivalent atoms in the target. (a) UBC924 model - target, continu-ous line; template 2UCE–target, dashed line; template 1AAK–target, dotted line. (b) DFR1 model–target, continuous line; tem-plate 4DFR-B–target, dashed line; template 3DFR–target, dotted
line. The horizontal continuous lines above the curves indicate thecorrectly aligned segments of at least three residues where thebest template was chosen for the model. The horizontal dashedline at the N terminus in (a) indicates the 11 misaligned residues ofUBC924. (c) Superposition of residues 45–60 and 105–115 of theDFR1 model with the corresponding regions in the templates andthe X-ray structure. The model, thick continuous line; X-raystructure, thin continuous line; template 3DFR, dotted line; tem-plate 4DFR-B, dashed line.

Errors in Loops
There were only two insertions in the three mod-els, both of them in UBC924 (Fig. 2). The longestinsertion was only five residues long (residues 40–44), and the second insertion was two residues long(residues 108–109). When the whole model wassuperposed on the X-ray structure, the RMSD be-tween the backbones for the five-residue loop was 6.7Å; when the backbones of only the two loops weresuperposed locally, the RMSD was 1.7 Å. Thus, boththe orientation and conformation of the predictedloop were incorrect. The large difference between thetwo numbers shows that the positioning of the looprelative to the rest of the protein can be a veryimportant contributor to the total error even in thecase of relatively short loops. The alignment in theneighborhood of the loop was correct, except perhaps
for the alignment of residue 39, which probablyshould not have been aligned with any residue in thetemplates (Fig. 2). The RMSD for the backbones ofthe three residues preceding (37–39) and the threeresidues following the loop (45–47) was 2.3 Å and 1.5Å for the global and local superposition, respectively.The average backbone isotropic temperature factorsfor the five- and two-residue insertions were 24.4 Å2
and 22.2 Å2, respectively, compared to the slightly
Fig. 2. Errors in the two UBC924 loop models. The loopscorresponding to the two insertions in the UBC924 model (continu-ous thick line) are shown superposed with the target X-raystructure (continuous thin line), the templates 1AAK (dotted line),and 2UCE (dashed line). The numbers indicate the beginning andending residues of each segment in UBC924. The correspondingregions of the modeling alignment are shown below each set of thestructures.
Fig. 3. Alignment problems and solutions. (a) Alignment of theN-terminal region of UBC924. The alignment used for modelbuilding (modeling) and the correct alignment derived from thesuperposition of the experimental structures of the templates andthe target (3D) are shown. The ProsaII energy profiles for themodel (continuous line) and the target X-ray structure (dashedline) are shown below the alignment. Note the lower energy of theX-ray structure in the misaligned region. (b) The correct andalternative alignments for the C-terminal region of DFR1. TheProsaII energy profiles for the corresponding 3D models areshown below the alignment. The model based on the correct DFR1
alignment, continuous line; the model based on the alternativealignment, dashed line. Note the positive energy for the alternativemodel in the C-terminal region. (c) Superposition of the C-terminalregion of the correct (continuous line) and alternative model ofDFR1 (dashed line) with the X-ray structure (thin line).
54 R. SANCHEZ AND A. SALI

Figure 4 (legend on following page).
55EVALUATION OF COMPARATIVE PROTEIN MODELING

lower average of 16.4 Å2 for the backbone of thewhole protein. Thus, if the loops are not in contactwith other protein molecules in the crystal, it islikely that the differences between the insertions inthe crystal structure and the model reflect errors inthe model.
Distortions or Shifts in Incorrectly AlignedRegions: The Cycle of Alignment, ModelBuilding, and Model Evaluation
In the three models, there was only one secondarystructure segment that was misaligned, the N-terminal helix of UBC924. In addition, there werethree, zero, and one gaps in the modeling alignmentsfor DFR1, UBC924, and PTE2A3, respectively, whereone or a few residues were misaligned.
In the UBC924 model, the N-terminal segment of11 residues was misaligned by one position, whichresulted in large errors in the model (Fig. 1a). Thismisalignment was unexpected because the correctalignment corresponded to a significantly lower se-quence similarity between the target and the tem-plate (Fig. 3a). For example, the number of matchesbetween hydrophobic residues is decreased and thenumber of matches between hydrophobic and polarresidues is increased when the incorrect alignmentis corrected. The misalignment was not detected bythe ProsaII profile of the model (Fig. 3a). However,the comparison of the profiles for the X-ray structureand the model shows that the X-ray structure has alower ProsaII score in that region (Fig. 3a). Thissuggests that the search for the alignment with thelowest ProsaII profiles of the implied model couldconceivably result in the correct alignment and thusa significantly better model in this case.
Another interesting observation is that the overallsequence identity between the target sequence andthe more similar of the two templates dropped from39% to 35% for the correct alignment. This makesthe point that optimizing only sequence similarity isnot always best in comparative modeling.
In the DFR1 model, it was obviously difficult toalign the last 13 residues, corresponding to the laststrand of the last b hairpin (Fig. 3). Two plausiblealternative alignments were generated manually bytaking into account local sequence similarity, second-ary structure predictions for DFR1,26–28 and the
structures of the template proteins. The alignmentswere evaluated by comparing the ProsaII profiles ofthe models based on those alignments (Fig. 3b). Oneof the models had a positive profile, and the other onehad a negative profile at the C terminus. A compari-son of the two models with the X-ray structureshowed that the model with the negative profile wasindeed correctly aligned with the template (Fig. 3c).
As illustrated above, alignment errors are a majorsource of large errors in comparative models. Weattempted to overcome this limitation by iteratingthrough several cycles of careful manual templateselection and alignment, followed by automatedmodel building and model evaluation. This processwas guided by a reduction in the errors predicted bya number of model evaluation techniques, mostimportantly the ‘‘energy’’ profiles calculated by theProsaII program and a program of Melo and Feyt-mans.15 Despite our limited experience, we believethat evaluation of an alignment at the level of theimplied model is likely to overcome a significantfraction of initial alignment errors, especially whenbetter potential functions for model evaluation be-come available and when the iterative procedure isautomated so that a larger number of alternativealignments can be explored.29
Overall Accuracy of the Models: RelativeOverall Similarity of a Model and theTemplates to the Target X-ray Structure
We now wish to answer the question of whetherthe predicted structures are a better model of theexperimental structures than the templates used inthe calculation of the models. In other words, howmuch closer is a comparative model of the targetsequence to the target X-ray structure than theclosest template structure?
Although a single RMSD value is useful for measur-ing a difference between two relatively similar struc-tures, RMSD depends on the number of equivalentatom pairs that are compared, which in turn de-pends on the maximal allowed distance between twoequivalent atoms. This makes a single RMSD valueinconvenient for comparing differences between pairsof different proteins. One solution to this problem isto define a similarity curve for a pairwise structure-structure comparison by plotting RMSD as a func-tion of the number of equivalent atoms. The similar-ity curve is obtained by calculating RMSD at differentcutoff values for equivalencing intermolecular pairsof Ca atoms and plotting the resulting RMSD valuesagainst the number of equivalent positions obtainedat each cutoff. Two similarity curves, instead of twosingle RMSD numbers, can then be inspected for acomparison of two protein–protein matches.
The similarity curves for the three pairwise com-parisons of the DFR1 model and the two templateswith the target structure are plotted in Figure 4a.The curves show that over a large range of the
Fig. 4. Similarity curves for the DFR1 (a, b, c) and UBC924 (d,e) models and templates. See the Methods section for thedefinition of the similarity curves. (a and d) The optimal superposi-tion of the templates and the X-ray structure was used to define theequivalent residues. (b, c, and e) The modeling alignment wasused to define the equivalences between the templates and thetarget. (a), (b), (d), and (e), only the Ca atoms are used to calculateRMSD. (c) All atoms are used to calculate RMSD. Model–target,thick continuous line; template 4DFR-B–target and template 2UCE–target, dashed line; template–target and template 1AAK–target,dotted line; template 8DFR–target, thin continuous line.
56 R. SANCHEZ AND A. SALI

number of equivalent atoms, the model is slightlycloser to the experimental structure (lower RMSDvalue) than either of the two templates. In otherwords, at a fixed number of atoms compared, themodel atoms have a lower RMSD from the X-raystructure than the template atoms; conversely, at afixed RMSD, the model has more atoms equivalent tothe X-ray structure than either of the templates.However, the differences are small, ,10% over mostof the similarity range.
Errors in the positioning of three gaps in the DFR1
modeling alignment contributed to the similaritycurve for the model-target comparison, but not to thetemplate-target similarity curves in Figure 4a, whichwere obtained from the superposition of the crystallo-graphic structures. In order to eliminate the contri-bution of the alignment errors and evaluate themodel building procedure on its own, the similaritycurves were recalculated using the modeling align-ment for comparison of the templates with the targetstructure (Fig. 4b). Since the template-target com-parisons now include the alignment errors, the tem-plates are less similar to the target X-ray structurethan in Figure 4a. However, the difference in howrepresentative of the target structure are the modeland the templates is still small, on the order of 10%of RMSD.
When side chain atoms were included in thecalculation of the similarity curves, the DFR1 modelbecame an even better representation of the targetstructure relative to the templates (Fig. 4c). Forexample, the model had approximately 95% of itsatoms superposed with an RMSD from the targetstructure of 2 Å, while the closest template only had78% of the atoms at that level of similarity (Fig. 4c).This was expected because the templates do notshare all the side chain atoms with the targetstructure while the model does.
In contrast to DFR1, the UBC924 model is worsethan the best template because of the alignmenterrors, primarily the shift for one position of theN-terminal 11 residues (Fig. 4d). The PTE2A3 modelis as close to the target structure as the best tem-plate (data not shown).
All comparative modeling methods start with analignment of the target sequence with the templatestructures, followed by model building that is de-coupled from the alignment procedure. Therefore,when evaluating comparative modeling methods, itis important for method developers to distinguishbetween errors due to misalignments and errors dueto the model building procedure. This distinction isalso important for the method users because themodeling alignment, not the correct alignment, wouldbe used to extract information from the templatestructure in the absence of any model building.When the modeling alignment is used to compareboth the model and the templates with the targetstructure, all three models are a better representa-
tion of the experimental structure than the tem-plates used in their derivation (Fig. 4b,e; data notshown for PTE2A3). This is especially true when theside chain as well as backbone atoms are compared(e.g., Fig. 4c). These comparisons suggest that it isbetter to use a comparative model of the target thanhomologous structures, unless only coarse predic-tions are made.
CONCLUSIONS
The modest improvement in our models relative toCASP1 probably originates from the careful manualselection of the templates and editing of the align-ment, as well as from the iterative re alignment andmodel building. This suggests directions for futuredevelopment of the algorithms that will, it is hoped,result in larger increases in the model accuracy.8,29–32
ACKNOWLEDGMENTS
We thank crystallographers U. Pieper, O. Herz-berg, K. Huang, H. Tong and T. Sixma for providingthe structures before their release to the PDB, and F.Melo and E. Feytmans for evaluating our modelswith their evaluation program. R.S. is a HowardHughes Medical Institute predoctoral fellow. A.S. isa Sinsheimer Scholar.
REFERENCES1. Delbaere, L.T.J., Brayer, G.D., James, M.N.G. Comparison
of the predicted model of ff-lytic protease with the x-raystructure. Nature 279:165–168, 1979.
2. Sali, A., Potterton, L., Yuan, F., van Vlijmen, H., Karplus,M. Evaluation of comparative protein modeling by MOD-ELLER. Proteins 23:318–326, 1995.
3. Rost, B. Protein structures sustain evolutionary drift.Folding Design 2:S19–S24, 1997.
4. Sali, A. and Overington, J. Derivation of rules for compara-tive protein modeling from a database of protein structurealignments. Protein Sci. 3:1582–1596, 1994.
5. Sali, A., Blundell, T.L. Comparative protein modelling bysatisfaction of spatial restraints. J. Mol. Biol. 234:779–815,1993.
6. Abola, E.E., Bernstein, F.C., Bryant, S.H., Koetzle, T.,Weng, J. Protein data bank. In: ‘‘Crystallographic Data-bases: Information, Content, Software Systems, ScientificApplications.’’ Allen, F.H., Bergerhoff, G., Sievers, R., eds.Bonn: Data Commission of the International Union ofCrystallography, 1987:107–132.
7. Johnson, M.S., Srinivasan, N., Sowdhamini, R., Blundell,T.L. Knowledge-based protein modelling. CRC Crit. Rev.Biochem. Mol. Biol. 29:1–68, 1994.
8. Sanchez, R., Sali, A. Advances in comparative protein–structure modeling. Curr. Opin. Str. Biol. 7:206–214, 1997.
9. Sanchez, R., Sali, A. Modeller, A Protein Structure Model-ing Program, Release 3. URL http://guitar.rockefeller.edu/,1995.
10. Needleman, S.B., Wunsch, C.D. A general method appli-cable to the search for similarities in the amino acidsequence of two proteins. J. Mol. Biol. 48:443–453, 1970.
11. Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J.,Swaminathan, S., Karplus, M. CHARMM: A program formacromolecular energy minimization and dynamics calcu-lations. J. Comp. Chem. 4:187–217, 1983.
12. Laskowski, R.A., McArthur, M.W., Moss, D.S., Thornton,J.M. PROCHECK: A program to check the stereochemicalquality of protein structures. J. Appl. Crystallogr. 26:283–291, 1993.
57EVALUATION OF COMPARATIVE PROTEIN MODELING

13. Hooft, R., Vriend, G., Sander, C., Abola, E. Errors in proteinstructures. Nature 381:272, 1996.
14. Sippl, M.J. Recognition of errors in three-dimensionalstructures of proteins. Proteins 17:355–362, 1993.
15. Melo, F., Feytmans, E. Novel knowledge-based mean forcepotential at atomic level. J. Mol. Biol. 267:207–222, 1997.
16. Koehl, P., Delarue, M. Application of a self-consistent meanfield theory to predict protein side-chains conformation andestimate their conformational entropy. J. Mol. Biol. 239:249–275, 1994.
17. Lee, C. Predicting protein mutant energetics by self consis-tent ensemble optimisation. J. Mol. Biol. 236:918–939,1994.
18. Maeyer, M.D., Desmet, J., Lasters, I. All in one: A highlydetailed rotamer library improves both accuracy and speedin the modelling of side chains by dead-end elimination.Folding Design 2:53–66, 1997.
19. Chung, S.Y., Subbiah, S. A structural explanation for thetwilight zone of protein sequence homology. Structure4:1123–1127, 1996.
20. Sutcliffe, M.J., Hayes, F.R.F., Blundell, T.L. Knowledgebased modeling of homologous proteins, Part II: Rules forthe conformation of substituted side-chains. Protein Eng.1:385– 392, 1987.
21. Summers, N.L., Karplus, M. Construction of side-chains inhomology modelling: Application to the C-terminal lobe ofrhizopuspepsin. J. Mol. Biol. 210:785–811, 1989.
22. Bower, M.J., Cohen, F.E., Dunbrack, R.L. Prediction ofprotein side-chain rotamers from a backbone-dependentrotamer library: A new homology modeling tool. J. Mol.Biol. 267:1268–1282, 1997.
23. Miyazawa, S., Jernigan, R.L. Estimation of effective inter-residue contact energies from protein crystal structures:Quasi-chemical approximation. Macromolecules 18:534–552, 1985.
24. Sippl, M.J. Calculation of conformational ensembles frompotentials of mean force: An approach to the knowledge-based prediction of local structures in globular proteins. J.Mol. Biol. 213:859–883, 1990.
25. DeBolt, S.E., Skolnick, J. Evaluation of atomic level meanforce potentials via inverse folding and inverse refinementof protein structures: Atomic burial position and pairwisenonbonded interactions. Protein Eng. 9:937–955, 1996.
26. Rost, B., Sander, C. Prediction of protein structure atbetter than 70% accuracy. J. Mol. Biol. 232:584–599, 1993.
27. Biou, V., Gibrat, J.-F., Levin, J., Garnier, J. Secondarystructure prediction: Combination of three different meth-ods. Protein Eng. 2:185–191, 1988.
28. Chandonia, J.M., Karplus, M. The importance of largerdata sets for protein secondary structure prediction withneural networks. Protein Sci. 5:768–774, 1996.
29. Guenther, B., Onrust, R., Sali, A., O’Donnell, M., Kuriyan,J. Crystal structure of the d8 subunit of the clamp-loadercomplex of E. coli DNA polymerase III. Cell 91:335–345,1997.
30. Sali, A. Modelling mutations and homologous proteins.Curr. Opin. Biotech. 6:437–451, 1995.
31. Sali, A. Protein modeling by satisfaction of spatial re-straints. Mol. Med. Today 1:270–277, 1995.
32. Sanchez, R., Sali, A. Comparative protein modeling as anoptimization problem. J. Mol. Struct. (Theochem) 398:489–496, 1997.
58 R. SANCHEZ AND A. SALI








![Comparative Modeling.ppt [Read-Only]cbsu.tc.cornell.edu/Workshop/2002_cd/lectures/Comparative_Modeling.pdfinteractions of a protein with other molecules are ... •Protein-protein](https://img.pdfslide.us/doc/110x75/5f4faad1f5f0f738b7283867/comparative-read-onlycbsutccornelleduworkshop2002cdlecturescomparativemodelingpdf.jpg)




















