Embed Size (px)
Citation preview

Predicting Protein Structure:Predicting Protein Structure:Comparative ModelingComparative Modeling(homology modeling)(homology modeling)

??
KQFTKCELSQNLYDIDGYGRIALPELICTMFHTSGYDTQAIVENDESTEYGLFQISNALWCKSSQSPQSRNICDITCDKFLDDDITDDIMCAKKILDIKGIDYWIAHKALCTEKLEQWLCEKE
Predicting Protein Structure:Predicting Protein Structure:Comparative ModelingComparative Modeling
(formerly, homology modeling)(formerly, homology modeling)
Use as template & model
8lyz1alc
KVFGRCELAAAMKRHGLDNYRGYSLGNWVCAAKFESNFNTQATNRNTDGSTDYGILQINSRWWCNDGRTPGSRNLCNIPCSALLSSDITASVNCAKKIVSDGNGMNAWVAWRNRCKGTDVQAWIRGCRLShare
Similar Sequence
Homologous

• In an ideal world, we would be able to accurately predict protein structure from the sequence only!
• Because of the myriad possible configurations of a protein chain – This goal can’t reliably be achieved, yet.
• Knowledge based prediction vs. Simulation based on physical forces.
• Here we will only concern ourselves with knowledge-based methods, although we might use simulation in order to optimize our models.
Structure prediction
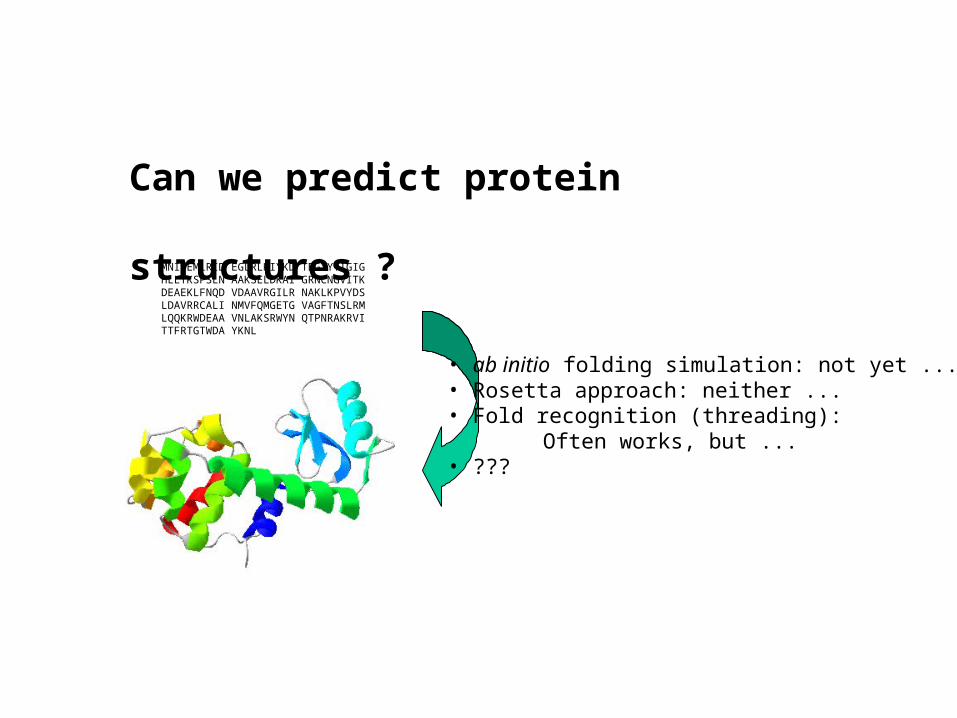
MNIFEMLRID EGLRLKIYKD TEGYYTIGIG HLLTKSPSLN AAKSELDKAI GRNCNGVITKDEAEKLFNQD VDAAVRGILR NAKLKPVYDS LDAVRRCALI NMVFQMGETG VAGFTNSLRMLQQKRWDEAA VNLAKSRWYN QTPNRAKRVI TTFRTGTWDA YKNL
Can we predict protein
structures ?
• ab initio folding simulation: not yet ...• Rosetta approach: neither ...• Fold recognition (threading):
Often works, but ...• ???
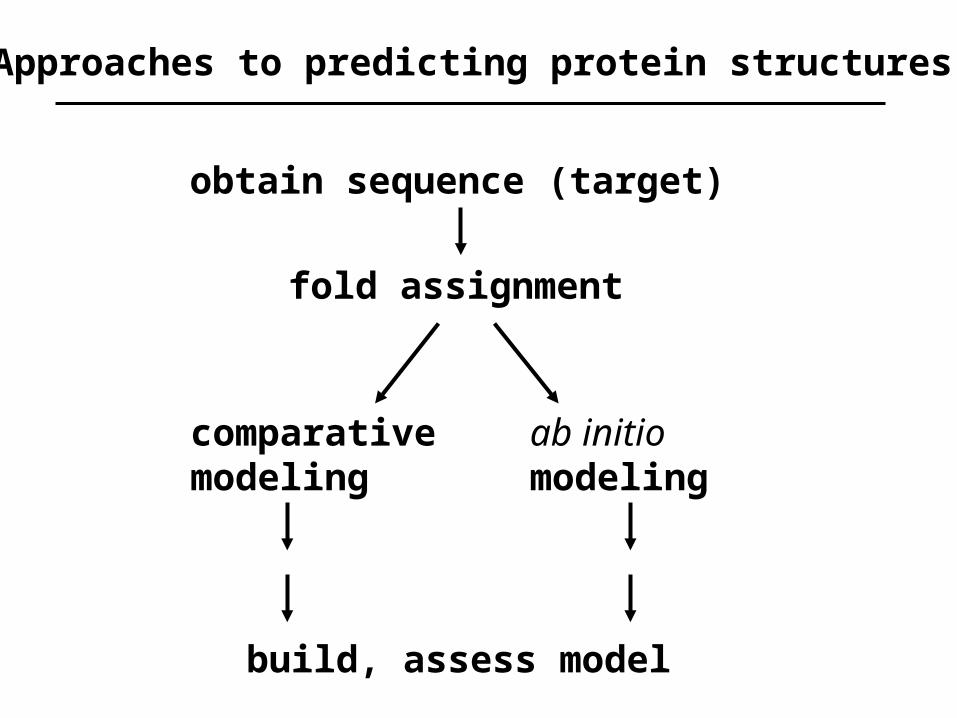
obtain sequence (target)
fold assignment
comparativemodeling
ab initiomodeling
build, assess model
Approaches to predicting protein structures

Homology Modelling of Proteins
• Definition: Prediction of three dimensional structure of a target protein from the
amino acid sequence (primary structure) of a homologous (template) protein for which an X-ray or NMR structure is available.
• Why a Model:A Model is desirable when either X-ray crystallography or NMR spectroscopy cannot determine the structure of a protein in time or at all. The built model provides a wealth of information of how the protein functions with information at residue property level. This information can than be used for mutational studies or for drug design.

Homology modeling
= Comparative protein modeling = Knowledge-based modeling
Idea: Extrapolation of the structure for a new (target) sequence from the known 3D-structures of related family members (templates).
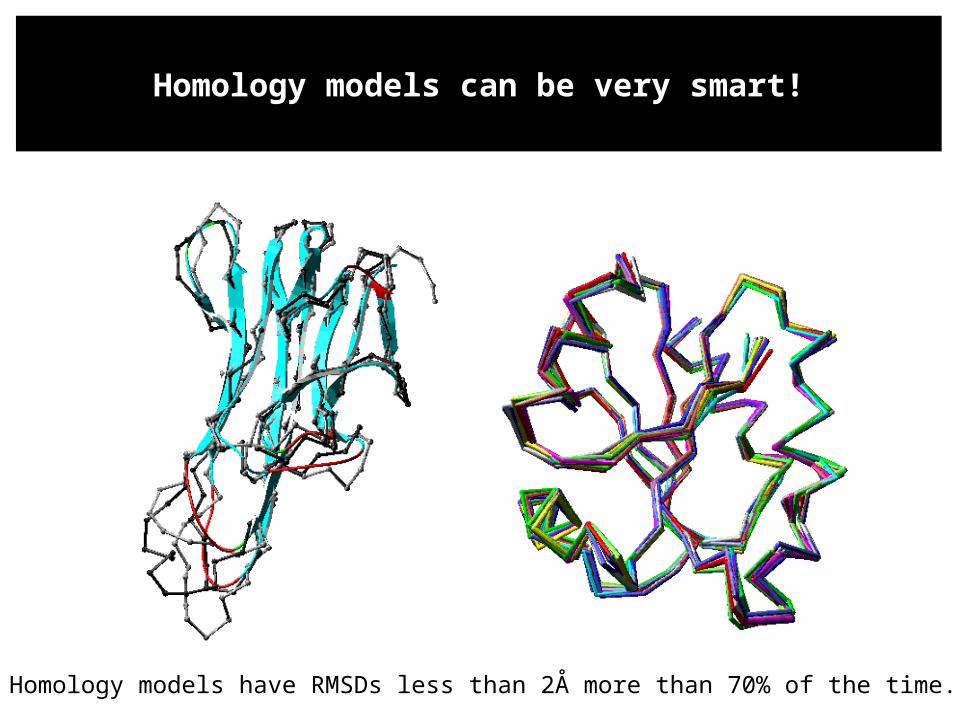
Homology models have RMSDs less than 2Å more than 70% of the time.
Homology models can be very smart!
.
0
20
40
60
80
100
0 50 100 150 200 250
identity
Number of residues aligned
Perc
enta
ge s
equence
identi
ty/s
imila
rity
(B.Rost, Columbia, NewYork)
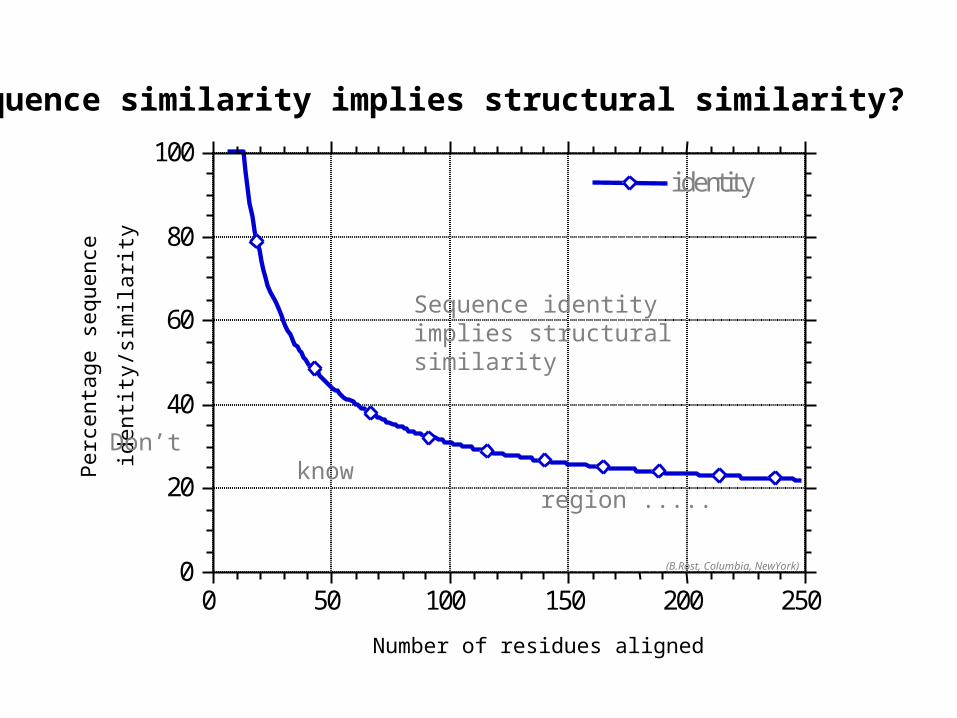
Sequence identity implies structural similarity
Don’t know region .....
Sequence similarity implies structural similarity?

Step 1 in Homology Modeling - Fold Identification
Aim: To find a template or templates structures from protein data base
Improved Multiple sequence alignment methods improves sensitivity - remote homologs
PSIBLAST, CLUSTAL
pairwise sequence alignment - finds high homology sequences BLAST
http://www.ncbi.nlm.nih.gov/BLAST/
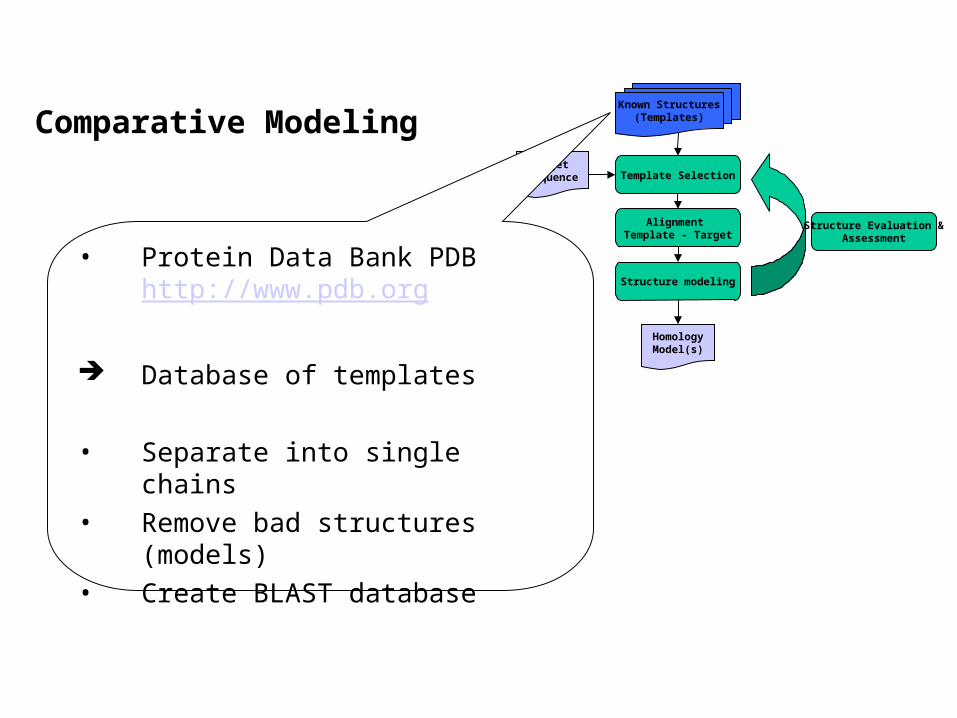
Comparative ModelingKnown Structures
(Templates)
Target Sequence Template Selection
Alignment Template - Target
Structure modeling
Structure Evaluation &Assessment
HomologyModel(s)
• Protein Data Bank PDB http://www.pdb.org
Database of templates
• Separate into single chains• Remove bad structures
(models)• Create BLAST database
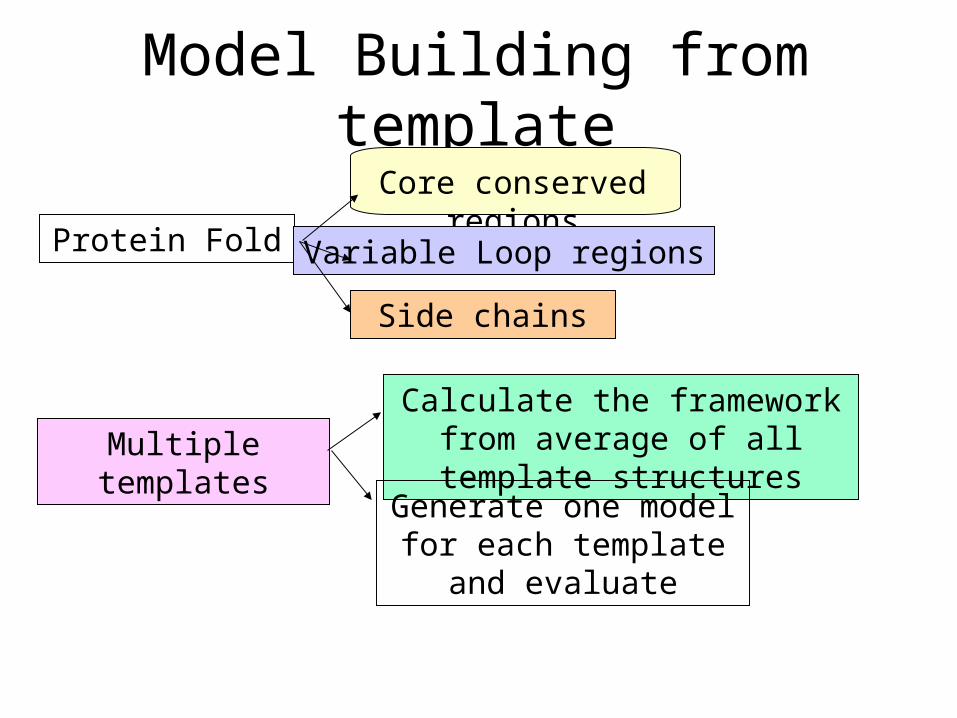
Model Building from template
Multiple templates
Protein Fold
Core conserved regions
Variable Loop regions
Side chains
Calculate the framework from average of all template structures
Generate one model for each template and evaluate
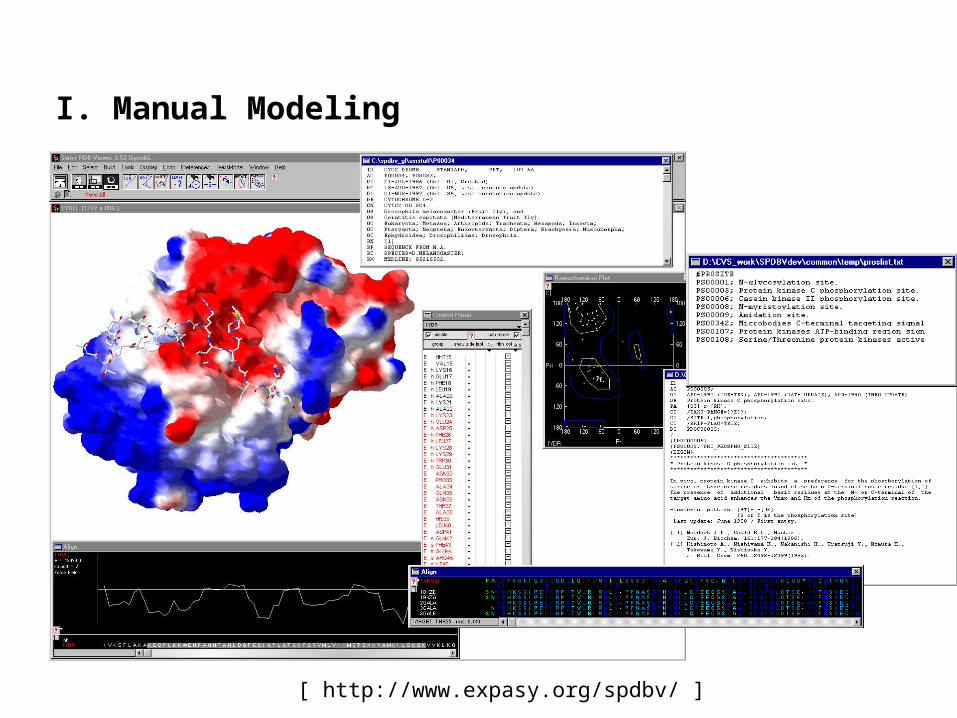
I. Manual Modeling
[ http://www.expasy.org/spdbv/ ]

• averaging core template backbone atoms
(weighted by local sequence similarity with the target sequence)
• Leave non-conserved regions (loops) for later ….
a) Build conserved core framework
II. Template based fragment assembly
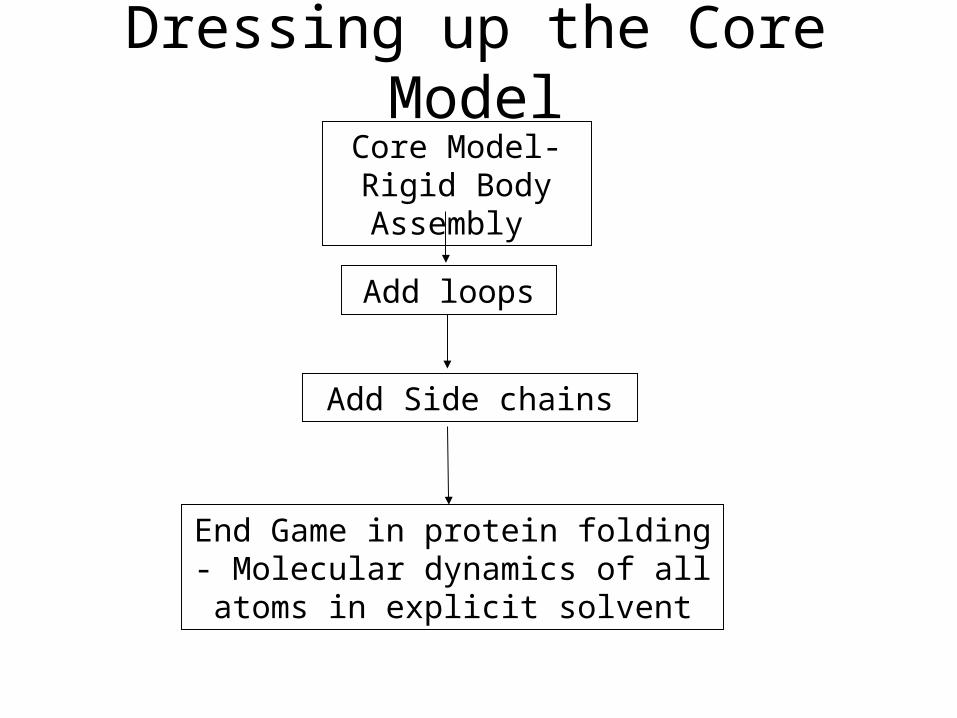
Dressing up the Core ModelCore Model-Rigid Body Assembly
Add Side chains
Add loops
End Game in protein folding - Molecular dynamics of all atoms in
explicit solvent
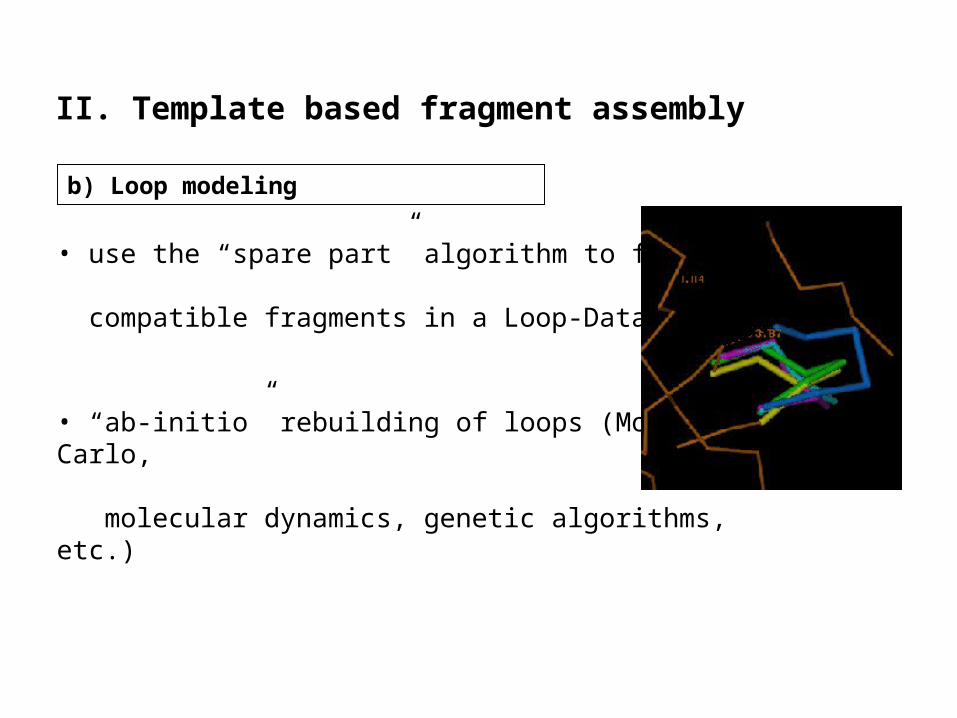
• use the “spare part” algorithm to find
compatible fragments in a Loop-Database
• “ab-initio” rebuilding of loops (Monte Carlo,
molecular dynamics, genetic algorithms, etc.)
b) Loop modeling
II. Template based fragment assembly

Loop BuildersMini protein folding problem-
3 to 10 residues longer in membrane proteins
Ab Initio methods - generates various
random conformations of loops and score
Compare the loop sequence string to
DB and get hits and evaluate.
Some Homology modeling methods
have less number of loops to be added
because of extensive multiple
sequence alignment of profiles
Loops result from
substitutions, insertions and deletions in
the same family

Using database of loops which appear in known structures. The loops could be catagorised by their length or sequence
Ab initio methods - without any prior knowledge. This is done by empirical scoring functions that check large number of conformations and evaluates each of them.
Construction of loops might be done by:

c) Side Chain placement
Find the most probable side chain
conformation, using
• homologues structures • back-bone dependent rotamer libraries
• energetic and packing criteria
II. Template based fragment assembly

• modeling will produce unfavorable contacts and bonds
idealization of local bond and angle geometry
• extensive energy minimization will move coordinates away
keep it to a minimum
• SwissModel is using GROMOS 96 force field for a steepest descent
d) Energy minimization
II. Template based fragment assembly
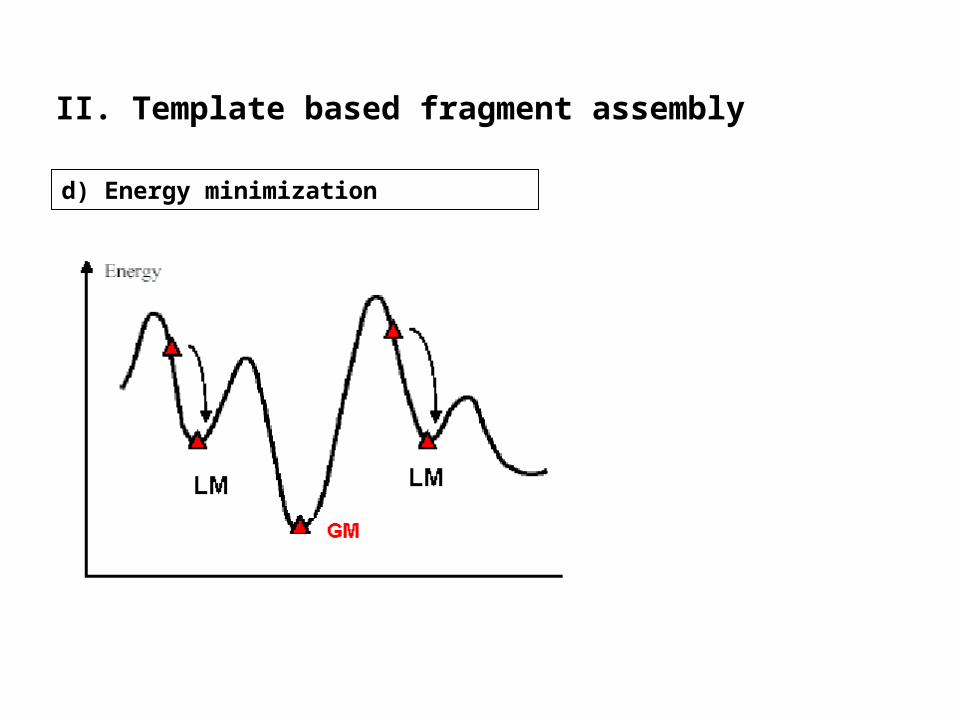
d) Energy minimization
II. Template based fragment assembly

Homology Modeling Programs
Modeller (http://guitar.rockefeller.edu/modeller)
Swiss-Model (http://www.expasy.ch/swissmod)
Whatif (http://www.cmbi.kun.nl/whatif)

Swiss-Model• Method:
Knowledge-based approach.
• Requirements:At least one known 3D-structure of a related protein. Good quality sequence alignements.
• Procedures:Superposition of related 3D-structures. Generation of a multiple a alignement.Generation of a framework for the new sequence. Rebuild lacking loops. Complete and correct backbone. Correct and rebuild side chains. Verify model structure quality and check packing. Refine structure by energy minimisation and molecular dynamics.

Model Confidence Factors
The Model B-factors are determined as follows:
• The number of template structures used for model building.
• The deviation of the model from the template structures.
• The Distance trap value used for framework building.
The Model B-factor is computed as:
85.0 * (1/ # selected template str.) * (Distance trap / 2.5)
and
99.9 for all atoms added during loop and side-chain building

Verifying the Model
• PROCHECK
• WHAT IF
• PROSA II
• VERIFY 3D, Profile3D

Errors in Models !!!
• Incorrect template selection
• Incorrect alignments
• Errors in positioning of sidechains and loops
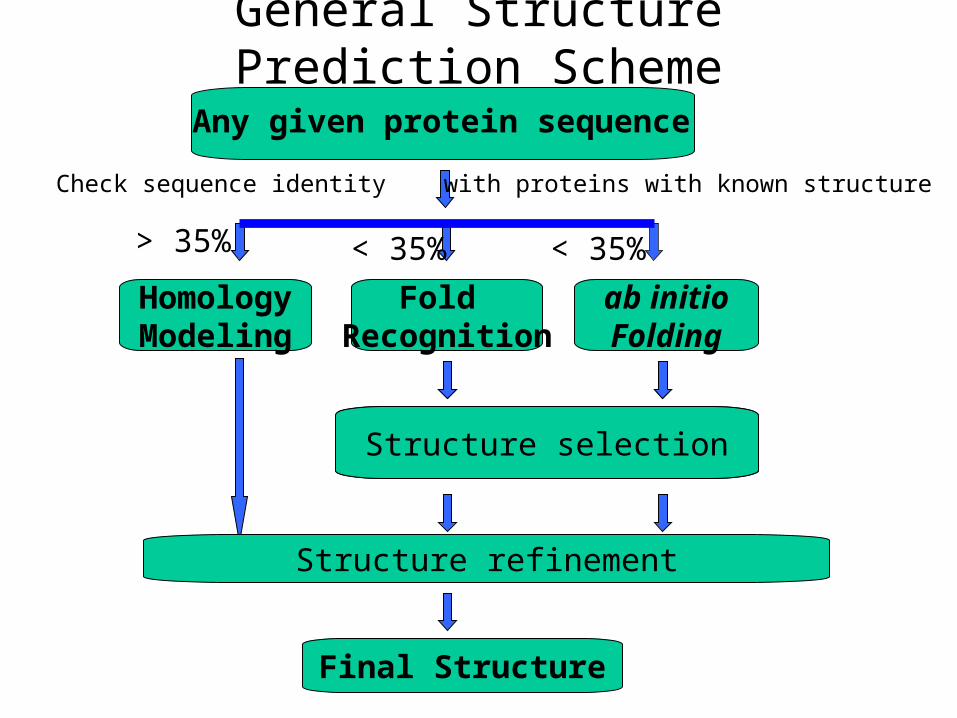
General Structure Prediction Scheme
Any given protein sequence
Structure selection
Check sequence identity with proteins with known structure
HomologyModeling
> 35%
Fold Recognition
ab initioFolding
< 35%< 35%
Structure refinement
Final Structure
Structure selection
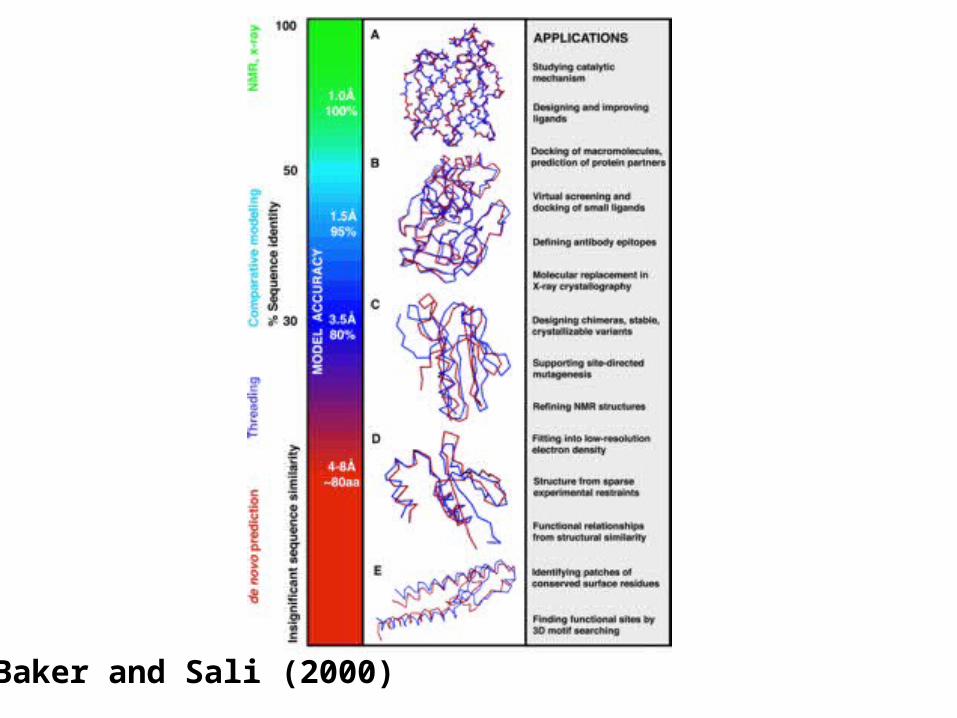
Baker and Sali (2000)

EVA
Evaluation of Automatic protein structure prediction
[ Burkhard Rost, Andrej Sali, http://maple.bioc.columbia.edu/eva/ ]
CASPCommunity Wide Experiment on the Critical Assessment of Techniques for Protein Structure Predictionhttp://PredictionCenter.llnl.gov/casp5/
3D - Crunch
Very Large Scale Protein Modelling Project
http://www.expasy.org/swissmod/SM_LikelyPrecision.html
Model Accuracy Evaluation

Several web pages for homology modeling
COMPOSER – felix.bioccam.ac.uksoft-base.html
MODELLER – guitar.rockefeller.edu/modeller/modeller.html
WHAT IF – www.sander.embl-heidelberg.de/whatif/
SWISS-MODEL – www.expasy.ch/SWISS-MODEL.html



![HOMOLOGY MODELING OF ALPHA- SYNUCLEIN (SNCA ... MODELING...Homology modeling [4] is currently the most accurate computational method [3] to generate reliable structural models and](https://img.pdfslide.us/doc/110x75/60fffa7f52e6de6bf34d31e5/homology-modeling-of-alpha-synuclein-snca-modeling-homology-modeling-4.jpg)

























