Embed Size (px)
Citation preview

Creating Your IQCP for Microbiology:
Real-Life Examples
1
Susan E. Sharp, Ph.D., ABMM, FAAM Director, Regional Microbiology and Molecular Infectious Diseases
Laboratories Kaiser Permanente, Department of Pathology
Associate Professor, Oregon Health and Sciences University,
Department of Pathology Portland, OR

Objectives:
1. Identify components that should be included in an IQCP
2. Analyze the process of creating an IQCP’s step by step, using IQCP examples
3. Evaluate the impact of IQCP on quality control procedures in the microbiology laboratory
4. Develop IQCPs for your own microbiology test systems
2

IQCP - Components
Risk Assessment (RA) • Review of literature • Review of manufacturer/regulatory information • Review of your verification and historical QC data • Review of your laboratory protocols, training, CA, PT, etc. • Preanalytical; Analytical; Postanalytical phases (mandated by CMS) • Risks associated with specimen, reagents, environment, testing personnel, test
system (5 mandated CMS components)
Quality Control Plan (QCP) • Defines the type, frequency and acceptability of QC
Quality Assessment (QA) • Develop an on going assessment of your QCP
3

THESE ARE ONLY EXAMPLES !
Your plans will differ!! •Use your own literature/manufacturer reviews •Use your own historical QC •Use your own risk criteria (frequency and severity) •Use your own measures to control risks •Use your own monitors
Use your own everything…..
4

Frequency of occurrence: Unlikely (once every 2-3 years)
Occasional (once per year)
Probable (once per month)
Frequent (once a week)
Severity of harm to patient: Negligible (temporary discomfort)
Minor (temporary injury; not requiring medical intervention)
Serious (impairment requiring medical intervention)
Critical (life threatening consequences)
5
Define your own Risk Criteria
This is only an example (adapted from CLSI EP23)

EXAMPLES
Vitek-2 Antimicrobial Susceptibility Testing System
Microscan Microbial Identification System
Alere i Influenza A&B assay
CLSI “Exempt” media
6

EXAMPLE
IQCP for the Vitek-2 AST System
7

Regulatory and Accreditation Requirements: Our IQCP is in compliance with CMS and CAP (ALL COMMON and MICROBIOLOGY) requirements. Copies of these requirements are located in our Compliance Department.
Method verification: The instrument was received and the test system verified in 2012. Subsequent verifications were performed when new drugs were added (1/12/14 and 4/14/15). This documentation is filed in Vitek-2 verification files. All verifications were deemed acceptable by the Laboratory Director.
Training of personnel: Completion of personnel training is documented in Compliance Department office.
Competency Assessment: New employees have CA ~6 months after initial training and annually thereafter. Documentation is filed in Compliance Department office.
Proficiency Testing: All testing personnel will perform PT testing and also review results. Proficiency testing records are filed in supervisor’s office.
IQCP - Vitek-2 Antimicrobial Susceptibility Testing (AST) System

VITEK-2 Manufacturer Information: Package insert (PI) contains system performance data and describes testing principle and procedure, QC recommendations, and limitations. PI is located in the supervisor’s office. Manufacturer alerts and bulletins are located the supervisor’s office. NOTE: No risks were identified when looking at the PI for this system. Both the Limitations and Performance data sections were closely reviewed to identify possible risks associated with the system and none were found.
Reference Information: Scientific publications used during collection of information for RA: 1. Smith et al. 2012. J Laboratory Testing. 52:109 2. Jones and Cartwright. 2015. Microbiology Today. 18:1821 3. CLSI document M07-A10. 2015 NOTE: Upon review of these articles no risk factors for using this test system were identified for our laboratory.

VITEK-2 Historical QC Data Summary of in-house QC data from routine testing of QC strains. Review period from 3-14-14 to 3-13-15. Review of QC records for 1 year of ~ 3500 results demonstrated: • 0.8% occurrence of random QC errors (corrected upon repeat
testing) • 0.02% occurrence (one incident) of potential system QC error that
required corrective action. • This involved an out-of-range QC results with imipenem that was presumed to be due to drug degradation following improper storage of 1 box of panels. • Additional panels from this box were QC’d and found to be out-of-range; panels were discarded, none were used for patient testing.

Vitek-2 Instrument Data: Summary of in-house data from routine instrument maintenance and performance checks: • Review of instrument QC records for the past 12 months (3-14-14
to 3-13-15) included approximately 55 routine checks of instrument A and 55 routine checks of instrument B.
• There was 1 scheduled maintenance performed by the company’s service engineer (8-1-14).
NOTE: None of these reviews showed any performance problems with the Vitek-2 that would impact patient results.

Laboratory Error Data: Summary of corrected reports and physician complaints (documentation located in supervisor’s office):
Review of reporting errors (corrected reports), physician complaints, and delayed reports (> 5 days after specimen collection) for these 12 months showed… 38 corrected reports showed errors were due to one or more of the following:
1. inappropriate antimicrobial agent reported for the species/body site (n=17) 2. incorrect result released due to mixed culture (n=9) 3. incorrect result released due to use of inappropriate MIC interpretation (n=8) 4. failure to perform a susceptibility test when warranted (n=4)
2 formal physician complaints revealed: 1. question of a result for S.aureus isolate - repeat testing = initial results were incorrect 2. delay in reporting results for a CRE due to the need to confirm results
5 AST reports delayed > 5 days were due to: 1. verification of an MDR phenotype needing confirmatory testing (n=4) 2. failure of the operator to “finalize” the report (n=1)
Note: during this review of corrected and delayed reports and physician complaints, none of the errors could have been avoided by any changes in protocol for testing of QC strains including frequency of testing QC strains.

Risk Analysis Table
13
EXAMPLE – 1 Risk Analysis

Risk Factor (Possible Sources of Error)
Frequency of
occurrence
Severity of harm to patient
Measures to Control Risk/Relevant Documentation
Preanalytical SPECIMEN (PRIMARY):
Patient identification occasional serious Patient identification, collection, transport, storage requirements and acceptability criteria are defined (SOP.xxx); competency assessment is performed (CA.xxx); remedial actions to control improperly handled specimens are defined (SOP.xxx). All errors in specimen collection and handling are reviewed with involved individuals; frequent errors are addressed with all staff involved with patient specimen collection and handling.
Collection/container/volume occasional minor
Integrity occasional minor
Transport occasional minor
Storage unlikely minor
SPECIMEN (ORGANISM): Clinically relevant occasional minor SOP.xxx describes selecting organisms to test based clinical relevancy. CA performed. Colony age occasional minor Organism growth requirements are defined (SOP.xxx). CA performed. Media type unlikely minor Appropriate media for inoculum is defined (SOP.xxx). CA performed. Pure isolate unlikely serious SOP.xxx describes selecting pure isolates to test. Inoculate purity plate. CA performed. Inoculum suspension occasional minor Turbidity meter for inoculum standardization is defined (SOP.xxx). CA performed.
Analytical TESTING PERSONNEL: Training and Competency Assessment (CA) occasional serious Training and Competency are performed as required (SOP.xxx) with retraining and
remedial action taken as necessary when issues are found. Proficiency Testing (PT) unlikely negligible PT are performed by all testing personnel. Remedial action taken as necessary to
address errors in reporting; all staff to review/sign off on all PT critiques. Staffing unlikely minor Supervisor reviews staffing needs daily and schedules staff accordingly. REAGENTS: Receiving/storage/expirations dates occasional minor
Appropriate handling of reagents is defined (SOP.xxx). Training is documented. CA performed.
Preparation/use occasional minor QC strain storage/preparation occasional negligible ENVIRONMENT: Temperature /airflow/ humidity/ ventilation/ utilities/noise/ vibration
unlikely negligible Environmental conditions are defined and controlled as per SOP.xxx and manufacturer’s PI. Training is documented. CA performed.
TEST SYSTEM: Mechanical/electronic/jam occasional negligible Perform preventive maintenance according to recommended schedule. CA performed. Software/antimicrobial reporting rules (ARR) unlikely serious Software rules will flag most potential errors – results are to be checked by tech daily. Transmission of results to LIS unlikely serious Daily supervisory review of reported results; annual check of test system-LIS interface as
outlined in SOP.xxx. Postanalytical
TEST RESULTS: Results reported within 5 days occasional serious Supervisor reviews delayed results - takes corrective action (SOP.xxx). Transmission of results from LIS to HIS unlikely serious Supervisor reviews incorrect/delayed results… - takes corrective action (SOP.xxx). Review reported results occasional serious Results are checked at multiple steps by techs prior to release (SOP.xxx). Clinician feedback occasional serious Supervisor investigates and adds new risks into QA plan as appropriate.
Risk
Ana
lysis
Tab
le

Risk Factor (Possible Sources of Error)
Frequency of
occurrence
Severity of harm
to patient
Measures to Control Risk/Relevant Documentation
Preanalytical SPECIMEN (PRIMARY):
Patient identification occasional serious Patient identification, collection, transport, storage requirements and acceptability criteria are defined (SOP.xxx); competency assessment is performed (CA.xxx); remedial actions to control improperly handled specimens are defined (SOP.xxx). All errors in specimen collection and handling are reviewed with involved individuals; frequent errors are addressed with all staff involved with patient specimen collection and handling.
Collection/container/volume occasional minor Integrity occasional minor Transport occasional minor Storage unlikely minor
SPECIMEN (ORGANISM):
Clinically relevant occasional minor SOP.xxx describes selecting organisms to test based clinical relevancy. CA performed.
Colony age occasional minor Organism growth requirements are defined (SOP.xxx). CA performed.
Media type unlikely minor Appropriate media for inoculum is defined (SOP.xxx). CA performed.
Pure isolate unlikely serious SOP.xxx describes selecting pure isolates to test. Inoculate purity plate. CA performed.
Inoculum suspension occasional minor Turbidity meter for inoculum standardization is defined (SOP.xxx). CA performed.
Risk
Ana
lysis
Tab
le

Risk Factor (Possible Sources of Error)
Frequency of
occurrence
Severity of harm
to patient
Measures to Control Risk/Relevant Documentation
Preanalytical SPECIMEN (PRIMARY): Patient identification occasiona
l serious Patient identification, collection, transport, storage
requirements and acceptability criteria are defined (SOP.xxx); competency assessment is performed (CA.xxx); remedial actions to control improperly handled specimens are defined (SOP.xxx). All errors in specimen collection and handling are reviewed with involved individuals; frequent errors are addressed with all staff involved with patient specimen collection and handling.
Collection/container/volume occasional
minor
Integrity occasional
minor
Transport occasional
minor
Storage unlikely minor
Analytical TESTING PERSONNEL: Training and Competency Assessment (CA)
occasional serious Training and Competency are performed as required (SOP.xxx) with retraining and remedial action taken as necessary when issues are found.
Proficiency Testing (PT) unlikely negligible PT are performed by all testing personnel. Remedial action taken as necessary to address errors in reporting; all staff to review/sign off on all PT critiques.
Staffing unlikely minor Supervisor reviews staffing needs daily and schedules staff accordingly.
REAGENTS: Receiving/storage/expirations dates
occasional minor Appropriate handling of reagents is defined (SOP.xxx). Training is documented. CA performed. Preparation/use occasional minor
QC strain storage/preparation occasional negligible ENVIRONMENT: Temperature /airflow/ humidity/ ventilation/ utilities/noise/ vibration
unlikely negligible Environmental conditions are defined and controlled as per SOP.xxx and manufacturer’s PI. Training is documented. CA performed.
TEST SYSTEM: Mechanical/electronic/jam occasional negligible Perform preventive maintenance according to
recommended schedule. CA performed. Software/antimicrobial reporting rules (AES)
unlikely serious Software rules will flag most potential errors – results are to be checked by tech daily.
Transmission of results to LIS unlikely serious There is daily supervisory review of reported results. There is annual checks of test system-LIS interface as outlined in SOP.xxx.
Risk
Ana
lysis
Tab
le

Risk Factor (Possible Sources of Error)
Frequency of occurrence
Severity of harm
to patient
Measures to Control Risk/Relevant Documentation
Preanalytical SPECIMEN (PRIMARY): Patient identification occasiona
l serious
Collection/container/volume occasional
minor
Integrity occasional
minor
Transport occasional
minor
Postanalytical
TEST RESULTS:
Results reported within 5 days occasional serious Supervisor reviews delayed results - takes corrective action (SOP.xxx).
Transmission of results from LIS to HIS
unlikely serious Supervisor reviews incorrect/delayed results due to transmission errors - takes corrective action (SOP.xxx).
Review reported results occasional serious Results are checked at multiple steps by techs prior to release (SOP.xxx).
Clinician feedback occasional serious Supervisor investigates and adds new risks into QC plan as appropriate.
Risk
Ana
lysis
Tab
le

Quality Control Plan (QCP) for the Vitek-2 AST System
Based on our Risk Assessment the QCP consists of following the in Quality Control Section of SOP.xxx for Performance of AST. This is summarized below: • Test of appropriate QC strains on each new lot/shipment of panels before or
concurrently with placing these materials into use for patient testing isolates.
• Test of appropriate QC strains on each panel type weekly.
• Test of appropriate QC strains on each panel type after major system maintenance or software upgrades before or concurrently with placing the instrumentation back into service.
• Test of appropriate QC strains against any new antimicrobial agent added to the panel with the 3x5 plan (over 5 days) prior to weekly QC testing of the panel.
• Record and evaluate QC results according to QC acceptability criteria as defined in SOP.xxx. Any out-of-range result is immediately investigated and corrective action performed prior to releasing any patient results.

Quality Assessment: Ongoing Monitoring for QCP Effectiveness QC failures, PT failures, and patient isolate reporting errors will be investigated and addressed as needed in a new/updated risk assessment and the QCP revised as needed. Daily review of patient results looking for reporting errors. Clinician complaints are investigated ASAP. Take corrective action and revise QCP as needed. Monthly review of QC results performed. Take corrective action and revise QCP as needed.
Monthly review of results delayed beyond 5 days. Take corrective action and the QCP will be revised as needed when number of delayed reports exceeds acceptable limit (more than 2%).
Review Proficiency Testing results. Take corrective action and revise QCP as necessary when PT results are not acceptable. Monthly review of all equipment maintenance/monitoring logs. Take corrective action and revise QCP as needed. Perform training and competency assessment. Modify training/CA and revise QCP as needed. Continual participation in this institution’s quality program that addresses specimen handling and labeling. Take corrective action and revise QCP as needed.
This QCP has been reviewed and is approved by the laboratory director.
Signature Date

Example
20
IQCP for the Microscan Microbial Identification System

21
Regulatory and Accreditation Requirements: Our IQCP is in compliance with CMS and CAP requirements. Copies of these requirements are located in our Compliance Department.
Method verification: The instrument was received and test system verification completed in year 2010. Streamlined ID was verified in year 2014 as per CLSI M50 (Briefly, we documented QC
performance for 3 consecutive lot numbers of gram positive and gram negative panels from 3 different shipments that spanned 3 consecutive seasons).
Documentation filed in supervisor’s office. Training of personnel: Completion of training documented in training tracker on-line system.
Competency Assessment: New employees have CA 6 months after initial training and annually thereafter. Documentation filed in supervisor’s office. Proficiency Testing: Rotated among testing personnel; all testing personnel review results. Proficiency testing records filed in supervisor’s office. Quality Control: CLIA ’88 and CAP require testing of QC strains on each new lot/shipment of commercial microbial ID systems to include a positive and a negative reactivity control for each substrate. Alternatively, an IQCP can be developed for streamlined QC of microbial identification.
IQCP - Microscan Identification System / Streamlined QC

22
Test System Information: Manufacturer: • Package insert contains system performance data and describes testing principle and procedure, QC
recommendations (including streamlined QC), and limitations. Package insert (PI) is located in the supervisor’s office.
• Manufacturer’s alerts and bulletins are located the supervisor’s office. • Operator’s manual including troubleshooting guide is located next to the instrument. References used during collection of information for RA: 1 CLSI: Quality Control for Commercial Microbial Identification Systems; Approved Guideline. Document M50-A. CLSI, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA. 2008. 2 CMS letter, Streamlined QC Ref: S&C-09-06. 2008 http://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/SurveyCertificationGenInfo/downloads/SCLetter09-06.pdf Summary of in-house data from streamlined QC testing: Streamlined QC testing was performed according to SOP.xxx and that of CLSI M50. Review of QC records for the past 12 months that contained approximately 200 results demonstrated: • <0.2% occurrence of random QC errors that corrected upon repeat testing (less than 0.5% is considered
acceptable for this IQCP). Summary of in-house data from routine instrument performance checks: Instrument checks were done according to SOP.xxx and manufacturer instructions. Review of instrument QC records for the past 12 months contained approximately 55 routine checks of the Microscan instrument and 1 check following scheduled maintenance performed by the company; none revealed any instrument performance problems that would impact patient results. Summary of corrected reports and physician complaints: Documentation located supervisor’s office. Review of corrected reports for the last 12 months indicated: 10 corrected reports with erroneous identification performed on the Microscan ID System due to mixed
inoculum suspensions. (Note: none of these errors could have been avoided by performing CLIA ’88 microbial identification QC, rather than CLSI streamlined QC).
There were no physician complaints as a result of unsatisfactory microbial identification.

EXAMPLE – 2
Risk Acceptability Table
and
Risk Acceptability Assignment Table
23
Risk Analysis

24
Risk Acceptability Table Risk Factor
(Possible Sources of Error) Frequency of occurrence
Severity of harm to patient Risk Level
Preanalytical Specimen (Primary): Patient identification probable minor Collection/container/volume frequent negligible Integrity frequent negligible Transport frequent negligible Storage probable negligible Specimen (Organism): Clinically relevant occasional negligible Colony age/viability/sampling occasional minor Media type unlikely minor Pure isolate unlikely minor Inoculum suspension preparation occasional minor
Analytical Testing Personnel: Training probable serious Competency probable serious Experience probable serious Proficiency Testing unlikely negligible Staffing occasional minor Reagents: Shipping/receiving/storage occasional minor Expiration dates unlikely minor Preparation/use occasional minor QC strain storage/prep occasional negligible Environment: Temperature/airflow/humidity/ ventilation unlikely negligible Utilities occasional minor Space unlikely negligible Noise/vibration unlikely negligible Test System: Mechanical/electronic stability of instrument/equipment/jam occasional negligible Software unlikely negligible Transmission of results to LIS unlikely serious
Postanalytical Test Results: Results reported within 5 days occasional minor Transmission of results to Electronic Health Record occasional serious Review reported results frequent serious Clinician feedback unlikely serious

Use your own Risk Matrix
25 This is only an example (modified from CLSI EP-23)
Frequency Negligible Minor Serious Critical
Frequent Not Acceptable
Not Acceptable
Not Acceptable
Not Acceptable
Probable Acceptable Not Acceptable
Not Acceptable
Not Acceptable
Occasional Acceptable Acceptable Acceptable Not Acceptable
Unlikely Acceptable Acceptable Acceptable Not Acceptable
Risk Matrix Severity of Harm

26
Risk Factor (Possible Sources of Error)
Frequency of occurrence
Severity of harm to patient Risk Level
Preanalytical Specimen (Primary): Patient identification probable minor Not Acceptable Collection/container/volume frequent negligible Not Acceptable Integrity frequent negligible Not Acceptable Transport frequent negligible Not Acceptable Storage probable negligible Acceptable Specimen (Organism): Clinically relevant occasional negligible Acceptable Colony age/viability/sampling occasional minor Acceptable Media type unlikely minor Acceptable Pure isolate unlikely minor Acceptable Inoculum suspension preparation occasional minor Acceptable
Analytical Testing Personnel: Training probable serious Not Acceptable Competency probable serious Not Acceptable Experience probable serious Not Acceptable Proficiency Testing unlikely negligible Acceptable Staffing occasional minor Acceptable Reagents: Shipping/receiving/storage occasional minor Acceptable Expiration dates unlikely minor Acceptable Preparation/use occasional minor Acceptable QC strain storage/prep occasional negligible Acceptable Environment: Temperature/airflow/humidity/ ventilation unlikely negligible Acceptable Utilities occasional minor Acceptable Space unlikely negligible Acceptable Noise/vibration unlikely negligible Acceptable Test System: Mechanical/electronic stability of instrument/equipment/jam occasional negligible Acceptable Software unlikely negligible Acceptable Transmission of results to LIS unlikely serious Acceptable
Postanalytical Test Results: Results reported within 5 days occasional minor Acceptable Transmission of results to Electronic Health Record occasional serious Acceptable Review reported results probable serious Not Acceptable Clinician feedback unlikely serious Acceptable
Risk Acceptability Table

Risk Factor
Possible Error
How can identified sources of error be reduced?
Preanalytical 1A: Specimen - Biological Patient/specimen identification Collection/container/ volume Integrity Transport Storage
Improper specimen procurement/ handling/processing
Train to SOP.xxx that addresses patient identification and specimen collection, labeling, transport, storage and remedial actions to control improperly handled specimens. Annually review representative specimen processing errors (N=10 to 15) with all staff involved with patient specimens. During initial training and competency assessment, emphasize: • Proper specimen handling/processing is the most critical part of any test • Failure to inoculate/streak correctly (no isolated colonies) and delayed incubation may result in delayed microbiology reports
1B: Specimen - Organism Clinically relevant
Clinically irrelevant organisms tested
Train to SOP.xxx and during initial training and competency assessment, emphasize: • differentiation of organisms included in normal flora group vs. potential pathogens requiring identification
Colony age Organism non-viable (or beyond maximum age for identification testing as described by the manufacturer
Train to SOP.xxx and during initial training and competency assessment, emphasize: • Time various organisms generally remain viable on various media • Proper age of colony for identification testing
Media Media for inoculum source other than that recommended is used Train to SOP.xxx and during initial training and competency assessment, emphasize: • Appropriate media for inoculum preparation • Species that can be reliably identified by the test system based on manufacturer’s recommendations
Pure isolate
Mixed inoculum
Train to SOP.xxx and during initial training and competency assessment, emphasize: • Proper selection of colonies for inoculum preparation • Hazards of picking from plates not incubated for sufficient time (“young” colonies) or poorly isolated colonies • Potential sources of contamination during testing process • Use of purity plates • Impact of delayed results if retesting needed
Inoculum suspension preparation Over- or under-inoculation Use of nonviable colonies Inoculum suspension sets too long before inoculating the identification panel
Train to SOP.xxx and during initial training and competency assessment, emphasize: • Proper use of turbidity meter for inoculum standardization • Proper inoculum suspension preparation
Analytical 2: Testing Personnel Training Competency Experience Proficiency Testing Staffing
Incompletely trained Unaware of updated protocols
All staff to train and have CA to SOP.xxx; read (and sign off) on PT sample critiques Supervisor annual review of any changes in microbial identification recommendations by accrediting agencies or manufacturer Supervisor to annually review appropriate staffing needs for identification testing and schedule staff accordingly During initial training and competency assessment, emphasize: • Critical nature of correct microbial identifications
3: Reagents Receiving/storage Expiration dates Preparation/use
Incorrect ordering Depleted reagent supply Reagent integrity compromised Use incorrect panel for select organism
Designated staff member(s) assigned to inventory (order/receipt) microbial identification materials to ensure supply is properly maintained and testing materials are handled appropriately on receipt Use color codes on boxes of panels Train to SOP.xxx and during initial training and competency assessment, emphasize standard rules to always: • Take responsibility for reagents/supplies (all staff) • Maintain reagents at proper storage conditions • Check expiration dates • Perform required QC
QC strain storage/prep QC out of control due to improper QC strain maintenance
Train to SOP.xxx and during initial training and competency assessment, emphasize: • Proper maintenance of QC strains (limited number of subcultures) • Potential sources of QC failures • QC troubleshooting • QC frequency
4: Environment Temperature/airflow/humidity/ ventilation Utilities Space Noise/vibration
Results not reported (ancillary equipment failure, e.g., incubator malfunction)
Train to SOP.xxx and during initial training and competency assessment, emphasize standard rules for: • Take responsibility for any possible instrument/ environmental problem (out of the ordinary observation)(all staff) Equipment maintenance Temperature recording (done automatically with continuous monitoring device) Electrical supply
5: Test System Mechanical/electronic/jam
Results not reported (e.g., instrument malfunction and/or aborted test)
Train to SOP.xxx and during initial training and competency assessment, emphasize standard rules for: • Take responsibility for any possible instrument/test system problem (out of the ordinary observation) During initial training and competency assessment, emphasize: • How to avoid jams
Software Erroneous results reported
• Daily supervisor (or supervisor designee) review of reported results • Software flags unusual results requiring supervisor review (e.g. organism identification is not consistent with associated AST
results) During initial training and competency assessment, emphasize: • Results requiring follow up action (e.g., confirmation by repeat testing) • Atypical results requiring consultation with supervisor
Transmission of results to LIS Incorrect transmission of results Delay in transmission of results
• Daily supervisor (or supervisor designee) review of reported results • Annual check of test system- LIS computer interface
Postanalytical 6: Test Results Results reported within 5 days Transmission of results to Electronic Health Record Review reported results Clinician feedback
Results delayed beyond that expected Incorrect transmission of results Delay in transmission of results Erroneous results reported Complaints/suggestions regarding delayed or potential erroneous results
Supervisor maintains summary of incorrect and delayed results and meets with laboratory director monthly to review this summary and remediate issues During initial training and competency assessment, emphasize: timely reporting of both preliminary results and final reports Incorporate suggestions into QA plan, as appropriate
Risk Acceptability Assignment Table
28
Risk Factor
Possible Error
How can identified sources of error be reduced?
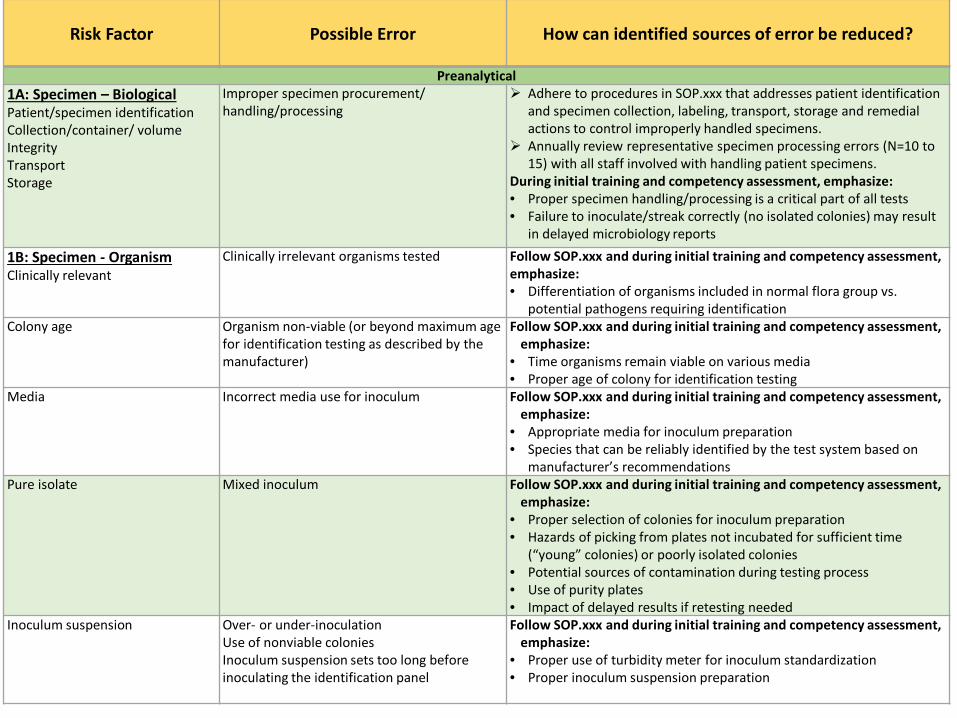
Preanalytical 1A: Specimen – Biological Patient/specimen identification Collection/container/ volume Integrity Transport Storage
Improper specimen procurement/ handling/processing
Adhere to procedures in SOP.xxx that addresses patient identification and specimen collection, labeling, transport, storage and remedial actions to control improperly handled specimens.
Annually review representative specimen processing errors (N=10 to 15) with all staff involved with handling patient specimens.
During initial training and competency assessment, emphasize: • Proper specimen handling/processing is a critical part of all tests • Failure to inoculate/streak correctly (no isolated colonies) may result
in delayed microbiology reports
1B: Specimen - Organism Clinically relevant
Clinically irrelevant organisms tested Follow SOP.xxx and during initial training and competency assessment, emphasize: • Differentiation of organisms included in normal flora group vs.
potential pathogens requiring identification Colony age Organism non-viable (or beyond maximum age
for identification testing as described by the manufacturer)
Follow SOP.xxx and during initial training and competency assessment, emphasize:
• Time organisms remain viable on various media • Proper age of colony for identification testing
Media Incorrect media use for inoculum Follow SOP.xxx and during initial training and competency assessment, emphasize:
• Appropriate media for inoculum preparation • Species that can be reliably identified by the test system based on
manufacturer’s recommendations Pure isolate
Mixed inoculum
Follow SOP.xxx and during initial training and competency assessment, emphasize:
• Proper selection of colonies for inoculum preparation • Hazards of picking from plates not incubated for sufficient time
(“young” colonies) or poorly isolated colonies • Potential sources of contamination during testing process • Use of purity plates • Impact of delayed results if retesting needed
Inoculum suspension Over- or under-inoculation Use of nonviable colonies Inoculum suspension sets too long before inoculating the identification panel
Follow SOP.xxx and during initial training and competency assessment, emphasize:
• Proper use of turbidity meter for inoculum standardization • Proper inoculum suspension preparation

Analytical 2: Testing Personnel Training Competency Experience Proficiency Testing Staffing
Incomplete training Unaware of updated protocols
All staff to train and have CA to SOP.xxx; read (and sign off) on PT sample critiques Supervisor annual review of any changes in microbial identification recommendations
by accrediting agencies or manufacturer Supervisor to annually review appropriate staffing needs for identification testing and
schedule staff accordingly During initial training and competency assessment, emphasize: • Critical nature of correct microbial identifications
3: Reagents Receiving/storage Expiration dates Preparation/use
Failure to solve problems occurring with test reagents Incorrect ordering Depleted reagent supply Reagent integrity compromised Use incorrect panel for select organism
Designated staff member(s) assigned to inventory (order/receipt) microbial identification materials to ensure supply is properly maintained and testing materials are handled appropriately on receipt
Use color codes on boxes of panels Train to SOP.xxx and during initial training and competency assessment, emphasize: • Taking responsibility for reagents/supplies (all staff) • Maintaining reagents at proper storage conditions • Checking expiration dates • Performing required QC
QC strain storage/prep QC out of control due to improper QC strain maintenance
Train to SOP.xxx and during initial training and competency assessment, emphasize: • Proper maintenance of QC strains (limited number of subcultures) • Potential sources of QC failures • QC troubleshooting • QC frequency
4: Environment Temperature/airflow/humidity/ ventilation Utilities Space Noise/vibration
Results not reported (ancillary equipment failure, e.g., incubator malfunction)
Instrument installed following manufacturer’s suggestions Equipment maintenance SOP.xxx followed after training Temperature recording (done automatically w/continuous monitoring device) Electrical supply – checked annually by engineering department Space – N/A (sufficient space for testing and instrumentation) During initial training and competency assessment, emphasize: • Taking responsibility for any possible instrument/ environmental problem (out of the
ordinary observation)(all staff)
5: Test System Mechanical/electronic/jam
Results not reported (e.g., instrument malfunction and/or aborted test)
Train to preventive maintenance according to SOP.xxx and recommended schedule During initial training and competency assessment, emphasize: • Take responsibility for any possible instrument/test system problem (out of the
ordinary observation) • How to avoid jams when loading instrument
Software Erroneous results reported
Daily supervisor (or supervisor designee) review of reported results Software flags unusual results requiring supervisor review (e.g. organism identification
is not consistent with associated AST results) During initial training and competency assessment, emphasize: • Results requiring follow up action (e.g., confirmation by repeat testing) • Atypical results requiring consultation with supervisor
Transmission of results to LIS Incorrect transmission of results
Delay in transmission of results
Daily supervisor (or supervisor designee) review of reported results
Annual check of test system- LIS computer interface
Risk Factor Possible Error How can identified sources of error be reduced?

30
Postanalytical 6: Test Results Incorrect or delayed results Transmission of results to Electronic Health Record Review reported results Clinician feedback
Results delayed beyond that expected Incorrect transmission of results; delay in transmission of results Erroneous results reported Complaints/suggestions regarding delayed or potential erroneous results
Supervisor maintains summary of incorrect and delayed results and meets with laboratory director monthly to review this summary and remediate issues
Incorporate clinician suggestions into QA plan, as appropriate.
During initial training and competency assessment, emphasize: • timely reporting of both preliminary
results and final report
Risk Factor Possible Error How can identified sources of error be reduced?

31
QCP for Streamlined QC of Microscan Identification System
Based on our risk assessment the QCP consists of following the instructions that are provided in SOP.xxx (Microscan ID System).
Testing of appropriate QC strains for streamlined QC on each new lot/shipment of panels before or concurrently with placing these materials into use for patient testing.
Testing of appropriate QC strains on each panel type after major system maintenance or software upgrade before or concurrently with placing the instrument back into service.
QC acceptability criteria is defined in SOP.xxx (Quality Control for Microscan ID). Any abnormal result is immediately investigated.

32
Quality Assessment for ongoing monitoring for QCP Effectiveness (Performed by supervisor) Reasons for QC failures, PT failures, and patient reporting errors will be examined and addressed as needed in a new/updated risk assessment: 1) Has a new risk factor been identified? 2) Does this change the frequency of risk? 3) Does the risk factor change the potential severity of harm to patient? Revision of the QCP will be done if necessary based on new risk assessment.
Daily review of patient results for reporting errors and clinician complaints. Take corrective action and revise QCP as needed. Monthly review of QC results by supervisor or section head. Take corrective action and revise QCP when unexpected QC failures indicate modification is needed.
Monthly review of time from specimen collection to microbial ID result to determine incidence of reports delayed beyond 5 days. Take corrective action and revise QCP when number of delayed reports exceeds acceptable limit (defined in SOP.xxx). Regular review of Proficiency Testing results. Take corrective action and revise QCP if necessary when PT results are not acceptable. Monthly review of all equipment maintenance/monitoring logs and take corrective action and revise QCP as needed.
Regular training and competency assessment and modify training/CA and revise QCP as needed.
This QCP has been reviewed and is approved by the laboratory director (as named on the CLIA license).
Signature: Date:

Example
33
IQCP for the Alere i Influenza A&B assay

IQCP - Alere i Influenza A&B assay Package insert (PI) contains system performance data and describes testing principle and procedure, QC recommendations, and limitations.
• This PI is located in the microbiology laboratory’s PI files.
• No specific limitations were noted in the PI that would affect the use of this test for our patients; no risks were identified upon review of the PI.
• The manufacturer recommends to test positive and negative
external controls (supplied in the kit) once with each new shipment. • An internal control is included in each test designed to control for
sample inhibition, amplification and assay reagent function. This control must be VALID in order to generate a patient result.
Manufacturer alerts and bulletins are located with the microbiology laboratory’s PI files.
• No alerts/bulletins have been released for this product.
34

Literature • Copies of the following pertinent published studies are located
along with the initial verification testing documentation performed on this system (located in the microbiology supervisor’s office files).
• J Clin Virol. 2014 Sep;61(1):81-6. doi: 10.1016/j.jcv.2014.06.001. Epub 2014 Jun 7. Multicenter clinical evaluation of the novel Alere™ i Influenza A&B isothermal nucleic acid amplification test. Bell J, Bonner A Cohen DM, Birkhahn R, Yogev R, Triner W, Cohen J, Palavecino E, Selvarangan R.
• J Clin Microbiol. 2014 Nov; 52(11): 3992–3995. doi: 10.1128/JCM.01639-14 Evaluation of the Alere i Influenza A&B Nucleic Acid Amplification Test by Use of Respiratory Specimens Collected in Viral Transport Medium. J. Bell J and Selvarangan R.
• No additional risks were identified from this literature for performing this testing
in our laboratory with both adult and pediatric populations when received in VTM*
• *This assay is considered moderately complex when used with nasal or NP swabs in VTM.
35

CAP IQCP Accreditation Requirements
This IQCP is in compliance with all CAP IQCP Checklist Requirements contained in the ALL Common and the Microbiology checklists.
36

Historical QC data - Summary Review of Verification Data performed from 8/1/14-11/30/14 showed no failures of positive and negative external controls, or internal controls in 30 days of testing (verification testing on file in supervisor’s office).
Review of data for 6 months of our last influenza season (12/1/14 - 5/30/15) showed:
• Lot and shipment QC (6 lot/shipment checks with positive and negative controls)
• Instrument maintenance (performed once during the time period w/positive and negative controls)
• Software upgrades – no software upgrades were performed during this time period
• Upon monthly QC (6 monthly QC cycles with positive and negative controls)
0% failures of external QC were noted (12 positive/12 negative). No external QC failures were noted after instrument maintenance (1 positive/
1 negative). No failures of internal QC were noted each day patient samples were tested
(165 days).
37

1 Specimen
2 Operator 4
Environment
3 Reagents
5 Test System
Identify Potential Hazards
Incorrect Test Results Released
-Verification -Maintenance -QC failures
-Training -Competency Assessment -Proficiency testing -Staffing levels
-Identification -Collection -Transport -Storage -Volume
-Receiving/Storage -Expirations date -QC materials
-Temperature -Amplicon free
Pre-analytical Analytical Post-analytical
RISK ASSESSMENT: Identification of Potential Failures - Alere i Influenza A&B Assay
38
6 Test Results
-Review of released results -Clinician feedback -Release from LIS to HIS

RISK FREQUENCY OF
OCCURRENCE
SEVERITY OF
HARM
MEASURES TO CONTROL RISK SUPPORTING DOCUMENTATION
PRE-ANALYTICAL - SPECIMEN
Identification Occasional Critical Specimen identification criteria are defined. Competency assessment (CA) performed. SOP.xxxx; CA.xxxx
Collection Occasional Minor Collection criteria are defined. CA performed. SOP.xxxx; CA.xxxx
Transport Occasional Minor Transport criteria are defined. CA performed. SOP.xxxx; CA.xxxx
Storage Occasional Minor Storage criteria are defined. CA performed. SOP.xxxx; CA.xxxx
Volume Occasional Minor Rejection criteria are defined. CA performed. SOP.xxxx; CA.xxxx
ANALYTICAL - OPERATOR
Training Occasional Critical All testing personnel have had appropriate training before performing assay. TR.xxxx
CA Unlikely Critical All personnel have appropriate competency assessment performed on assay. CA.xxxx
PT Occasional Negligible All PT failures with this assay are addressed with corrective action. SOP.xxxx
Staffing Occasional Minor Adequate staffing to support this test and TAT is available on all shifts. SOP.xxxx
ANALYTICAL - REAGENTS
Recev’g/Storage Occasional Critical Reagents for the assay are stored according to manufacturer’s instructions. SOP.xxxx
Exp. dates Occasional Minor All reagents used in the are used within expiration dates. SOP.xxxx
QC Occasional Minor All QC results for the assay are within parameters prior to releasing pt results. All specimen-specific QC parameters are controlled within the system; if there is a failure, the assay will not deliver a result; result will be "invalid” - see PI.
SOP.xxxx manufacturer’s PI
ANALYTICAL - ENVIRONMENT
Temperature Unlikely Minor Appropriate environmental conditions are maintained in the laboratory. SOP.xxxx
Amplicon free Unlikely Minor Clean work benches with bleach; use gloves for testing; procedures for reducing cross-contamination are used. Also, the test cartridges for the Alere i Influenza A&B assay are self-contained – material cannot escape unless the cartridge is damaged. Proper discarding and decontamination protocols are followed.
SOP.xxxx manufacturer’s PI
ANALYTICAL - TEST SYSTEM
Electric Unlikely Negligible Appropriate utilities are employed in the laboratory to serve the Alere i instrumentation. SOP.xxxx
Maintenance Unlikely Minor Criteria are defined to address Alere FLU instrument maintenance. SOP.xxxx; QCF.xxxx
QC failures Occasional Negligible Criteria are defined to address QC failures (external and internal) associated with the Alere i instrument & Influenza A&B assay. SOP.xxxx; QCF.xxxx
POSTANALYTICAL - TEST RESULT
Review results Occasional Critical Criteria are defined to address review of released results and investigate any errors detected related to this assay. SOP.xxxx
Phys feedback Occasional Critical Criteria are defined to address clinician feedback and investigate concerns with this assay. SOP.xxxx
LIS to HIS Occasional Negligible Criteria are defined for periodic review of results transfer related to this assay. SOP.xxxx
Risk Assessment Table; Alere i Influenza A&B assay

RISK FREQUENCY OF
OCCURRENCE
SEVERITY OF
HARM
MEASURES TO CONTROL RISK SUPPORTING DOCUMENTATION
PREANALYTICAL - SPECIMEN
Identification Occasional Critical Specimen identification criteria are defined. Competency assessment (CA) performed.
SOP.xxxx; CA.xxxx
Collection Occasional Minor Collection criteria are defined. CA performed. SOP.xxxx; CA.xxxx
Transport Occasional Minor Transport criteria are defined. CA performed. SOP.xxxx; CA.xxxx
Storage Occasional Minor Storage criteria are defined. CA performed. SOP.xxxx; CA.xxxx
Volume Occasional Minor Rejection criteria are defined. CA performed. SOP.xxxx; CA.xxxx
Risk Assessment Table; Alere i Influenza A&B assay

41
ANALYTICAL - OPERATOR
Training Occasional Critical All testing personnel have had appropriate training before performing assay. TR.xxxx
CA Unlikely Critical All personnel have appropriate competency assessment performed on assay. CA.xxxx
PT Occasional Negligible All PT failures with this assay are addressed with corrective action. SOP.xxxx
Staffing Occasional Minor Adequate staffing to support this test and TAT is available on all shifts. SOP.xxxx
ANALYTICAL - REAGENTS
Receiving /Storage
Occasional Critical Reagents for the assay are stored according to manufacturer’s instructions. SOP.xxxx
Exp. dates Occasional Minor All reagents used in the are used within expiration dates. SOP.xxxx
QC Occasional Minor All QC results for the assay are within parameters prior to releasing patient results. All specimen-specific QC parameters are controlled within the system; if there is a failure, the assay will not deliver a result; result will be "invalid” - see PI.
SOP.xxxx manufacturer’s
PI
ANALYTICAL - ENVIRONMENT
Temperature Unlikely Minor Appropriate environmental conditions are maintained in the laboratory. SOP.xxxx
Amplicon free
Unlikely Minor Clean work benches with bleach; use gloves for testing; procedures for reducing cross-contamination are used. Also, the test cartridges for the Alere i Influenza A&B assay are self-contained – material cannot escape unless the cartridge is damaged. Proper discarding and decontamination protocols are followed.
SOP.xxxx manufacturer’s
PI
ANALYTICAL - TEST SYSTEM
Electric Unlikely Negligible Appropriate utilities are employed in the laboratory to serve the Alere i instrumentation.
SOP.xxxx
Maintenance Unlikely Minor Criteria are defined to address Alere i instrument maintenance. SOP.xxxx; QCF.xxxx
QC failures Occasional Negligible Criteria are defined to address QC failures (external and internal) associated with the Alere i Influenza A&B assay.
SOP.xxxx; QCF.xxxx
RISK FREQUENCY OF
OCCURRENCE
SEVERITY OF
HARM
MEASURES TO CONTROL RISK SUPPORTING DOCUMENTA-
TION

RISK FREQUENCY OF
OCCURRENCE
SEVERITY OF HARM
MEASURES TO CONTROL RISK SUPPORTING DOCUMENTATION
POSTANALYTICAL - TEST RESULT
Review results
Occasional Critical Criteria are defined to address review of released results and investigate any errors detected related to this assay.
SOP.xxxx
Physician feedback
Occasional Critical Criteria are defined to address clinician feedback and investigate concerns with this assay.
SOP.xxxx
LIS to HIS Occasional Negligible Criteria are defined for periodic review of results transfer related to this assay.
SOP.xxxx
Risk Assessment Table; Alere i Influenza A&B assay

IQCP for Alere i FLU assay
Based on our Risk Assessment: QC for the Alere i Influenza A&B assay will consist of following instructions in SOP.1111 and recording results on the appropriate QC form.
• QC will include: • External QC (positive and negative QC swabs) performed per lot/shipment.* • External QC (positive and negative QC) performed once each 31 days.* • The Internal Control must be acceptable and documented each day patient samples
are tested. (* CAP requires external QC to be performed at least every 31 days and for each new lot/ shipment.)
Further QC details and acceptability criteria are defined in SOP.1111.
This IQCP/QCP has been reviewed and is approved by the CLIA laboratory director:
_______________________(director signature) ______(date)
43

• Specimen collection/acceptability guidelines reviewed annually • See SOP.xxxx
• Staff Training updated annually • See SOP.xxxx
• Competency assessment performed semi-annually and annually as required • See SOP.xxxx
• Proficiency Testing results reviewed and mediated ASAP as required • See SOP.xxxx
• QC/Instrument Function checks reviewed and mediated ASAP as required • See SOP.xxxx, SOP.xxxx. QC-FORM.xxxx, QC-FORM.xxxx
• Unexpected Errors investigation/remediation performed ASAP • See SOP.xxxx
• Incorrect result investigation/remediation performed ASAP • See SOP.xxxx
• Complaint investigation/remediation performed ASAP • See SOP.xxxx
44
Quality Assessment (QA): Our monitoring process will include
Pre-analytical Analytical Post-analytical
For errors/ failures/ concerns a reassessment of risk will be performed.
• The reason for failure will be identified and related to a new/updated risk.
• Additional control measures will be implemented if necessary as determined by the new risk assessment.

Example
45
IQCP for CLSI Exempt Media

List of CLSI-exempt media (CLSI M22-A3): Blood culture media Blood agar MacConkey agar PEA agar CNA agar Mannitol salt agar Blood/SXT agar CIN agar Brucella agar
46
Middlebrook agar Lowenstein-Jensen agar IMA Saboaraud’s dextrose agar LIM broth Selenite broth Thioglycolate broth TSI agar Urease agar
IQCP – Remel commercially prepared CLSI-exempt media

Information Review
47
Regulatory Requirements CMS CAP accreditation requirements Be in compliance w/manufacturer instructions/recommendations
CMS

Information Review
48
Regulatory Requirements CMS CAP accreditation requirements Be in compliance w/ the manufacturer instructions/recommendations
CMS guidelines: CMS’s FAQ for IQCP, revised April 2015, Question 42 – states in part: “For example, laboratory documentation showing visual quality checks of media are acceptable in-house data. The laboratory may also include manufacturer’s quality certificates as part of the information considered in its risk assessment.” Reference http://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Downloads/FAQs-IQCP.pdf

Information Review
49
Regulatory Requirements CMS
CAP accreditation requirements Be in compliance w/ the manufacturer instructions/recommendations
Per CAP - Commercially prepared CLSI ‘exempt’ media may have an IQCP. Reference: CAP MIC.21240 Revised 7/28/15; Media QC-Purchased

50

http://www.remel.com/IFUs/TM93.pdf Technical Manual of Microbiological Media
51

Information Review
52
Regulatory Requirements Regulatory requirement (e.g.; CMS) CAP accreditation requirements Be in compliance w/ manufacturer instructions/recommendations Manufacturer information:
– Remel’s “Certificates of Quality” certify that specific lot numbers of exempt media have met all performance and QC criteria for the product. See link: http://www.remel.com/IFUs/TM93.pdf (--> what we just looked at)
– No additional risks were identified from review of the Remel’s IFU (Instructions For Use), alerts or bulletins associated with these media products.
• IFU for these media can be found at the below link: http://www.remel.com/Support/SearchDocument.aspx
• All alerts and bulletins pertaining to these media can be found in the microbiology laboratory’s media QC files.
– Remel has no recommendation for end-user QC of CLSI exempt media. See link: http://www.remel.com/IFUs/TM93.pdf (--> what we just looked at)

Summary of Historical In-house data for CLSI-exempt Remel media
Media quality data were reviewed for 12 months (4/1/14 - 3/31/15). When evaluating media we undergo a visual inspection looking for:*
change in expected color of media cracked or damaged plates
agar detached from the plates excessive bubbles or rough surfaces
frozen or melted agar excessive moisture or dehydration
unequal filling of plates obvious contamination
insufficient agar in the plates presence of precipitates
hemolysis of blood containing media
In addition: Media is checked for contamination immediately before inoculation with specimens. When reviewing cultured specimens, microbiologists look for abnormal growth patterns
that may suggest plate contamination.
When using the above parameters: 0.2% occurrence of unacceptable quality was noted. These plates were not used for
patient care but were discarded. This was not associated with any one particular media type. (We consider anything less than 0.5% acceptable*).
< 0.05% of the time when using media was plate contamination suspected upon review of culture results; this was taken into account when resulting cultures after review with supervisor.
53 * reference: NCCLS/CLSI document M22-A3

RISK
FREQUENCY OF
OCCURRENCE
SEVERITY OF
HARM
MEASURES TO CONTROL RISK
SUPPORTING DOCUMENTATION
PREANALYTICAL - SPECIMEN
Identification Occasional Critical Patient & specimen identification acceptability criteria are defined. Competency assessment (CA) performed. SOP.xxx; CA.xxx
Collection/ Container
Occasional Minor Collection criteria are defined. CA performed. SOP.xxx; CA.xxx
Transport Occasional Minor Transport criteria are defined. CA performed. SOP.xxx; CA.xxx
Storage Occasional Minor Storage criteria are defined. CA performed. SOP.xxx; CA.xxx
Volume Occasional Minor Rejection criteria are defined. CA performed. SOP.xxx; CA.xxx
ANALYTICAL – OPERATOR
Training Occasional Critical All testing personnel have had training in appropriate utilization of media and media quality parameters. TR.xxx
CA Occasional Critical All personnel have appropriate CA performed regarding appropriate utilization of media and media quality parameters. CA.xxx
Proficiency Testing
Occasional Negligible All PT failures are addressed with corrective action; media quality is always investigated as deemed necessary. SOP.xxx
Staffing Occasional Minor Adequate staffing to support appropriate evaluation of media upon arrival and prior to use. SOP.xxx
ANALYTICAL – REAGENTS
Receiving/Storage Occasional Minor Media are received and stored according to manufacturer’s instructions. SOP.xxx; manufacturer’s PI
Expirations dates Occasional Minor All media are used within expiration dates; no expired media are ever used. SOP.xxx
Visual Inspections Unlikely Negligible Training and procedures are provided for appropriate visual inspection of media upon receipt. Visual quality checks are documented. CA performed.
SOP.xxx; CA.xxx; QC-Form.xxx;
ANALYTICAL – ENVIRONMENT
Temperature/ airflow/ humidity
Unlikely Negligible Appropriate environmental conditions are maintained in the laboratory for proper storage of media as specified by manufacturer.
SOP.xxx; manufacturer’s PI
Utilities Unlikely Negligible Appropriate utilities are maintained in the laboratory for proper storage of media as specified by manufacturer. SOP.xxx; manufacturer’s PI
ANALYTICAL - TEST SYSTEM
Contamination Unlikely Negligible Training and procedures are provided to check for contamination prior to plating patient specimens. Documentation is recorded on appropriate QC forms. CA performed.
SOP.xxx; QC-Form.xxx; CA.xxx
Organism growth Unlikely Negligible Training and procedures are provided to check for inconsistencies in organism growth on all media types. Suspect cultures are reviewed with the supervisor prior to release. CA performed.
SOP.xxx; CA.xxx
POSTANALYTICAL - TEST RESULT
Review results Unlikely Negligible There is review of all released results. Appropriate investigation for all reporting errors is undertaken including media quality review as necessary.
SOP.xxx
Clinician feedback Unlikely Critical Appropriate investigations are undertaken for all clinician feedback, issues, complaints, etc., including media quality review as necessary.
SOP.xxx
RISK ASSESSMENT TABLE

Quality Control Plan (QCP) Upon receipt of exempt media visual inspection will be performed as
outlined in the CLSI-M22 and SOP.xxx. Failed media will be brought to the attention of the supervisor and addressed immediately.
Media will be checked for contamination immediately before inoculation with patient specimens. Contaminated media will be brought to the attention of the supervisor and addressed immediately.
Suspected media contamination when reviewing cultures will be brought to the attention of the supervisor and addressed immediately.
Remel QC alerts and bulletins will be reviewed and acted on appropriately as necessary.
QC Acceptability Criteria is defined in SOP.xxx
55

Our post-implementation quality assessment monitoring process will include the following:
For issues identified from the above review of documents, procedures, PT, CA, QC, complaints, and other errors/concerns, a reassessment of risk will be performed. The reason for failure will be identified and investigated, and additional control measures will be implemented as necessary in the QCP.
56
Media receipt and storage guidelines are reviewed and updated annually as necessary.
Staff training documents are reviewed and updated annually. Competency assessment performed semi-annually and annually as required,
updated as necessary. Proficiency testing results are reviewed and corrective action taken. Media quality notifications are reviewed and corrective action taken. Unexpected errors are investigated and corrective action taken. Incorrect laboratory results are investigated and corrective action taken. Complaints are investigated timely and corrective action taken.

Remel CLSI-exempt media
This IQCP/QCP has been reviewed and is approved by the CLIA laboratory director. ______________________________ _______ (CLIA Laboratory Director signature) (date)
57

58
http://clinmicro.asm.org/iqcp ASM/CAP/CLSI collaborative effort for IQCP on AST, Exempt Media, Microbial Identification Systems





























