Embed Size (px)
DESCRIPTION
Neurobiology, Medicine,
Citation preview

1141The Journal of Experimental Biology 201, 1141–1152 (1998)Printed in Great Britain © The Company of Biologists Limited 1998JEB1381
2, USA
ADAPTATIONS OF VERTEBRATE NEURONS TO HYPOXIA AND ANOXIA:MAINTAINING CRITICAL Ca 2+ CONCENTRATIONS
PHILIP E. BICKLER1,* AND LESLIE T. BUCK2
1Medical Sciences 261, Department of Anesthesia, University of California at San Francisco, CA 94143-054and 2Department of Zoology, University of Toronto, Canada M5S 3G5
*e-mail: [email protected]
Accepted 28 October 1997; published on WWW 24 March 1998
Down-regulation of ion channel activity (‘channelarrest’), which aids in preserving critical ion gradients inconcert with greatly diminished energy production, is oneimportant strategy by which anoxia-tolerant neurons adaptto O2 shortage. Channel arrest results in the elimination ofaction potentials and neurotransmission and also decreasesthe need for ion transport, which normally requires a largeenergy expenditure. Important targets of this down-regulation may be channels in which activity wouldotherwise result in the toxic increases in intracellular[Ca2+] characteristic of anoxia-sensitive mammalianneurons. In turtles, Na+ channels and the Ca2+-permeableion channel of the N-methyl-D-aspartate (NMDA)-typeglutamate receptor undergo down-regulation during
anoxia. Inactivation of NMDA receptors during hypoxiaoccurs by a variety of mechanisms, including alterations inthe phosphorylation state of ion channel subunits, Ca2+-dependent second messenger activation, changes in Ca2+-dependent polymerization/depolymerization of actin topostsynaptic receptors and activation of other G-protein-coupled receptors. Release of inhibitory neurotransmitters(e.g. γ-aminobutyrate) and neuromodulators (e.g.adenosine) into the brain extracellular fluids may play animportant role in the down-regulation of these and othertypes of ion channels.
Key words: anoxia, hypoxia, neuron, Ca2+, vertebrate, turtle,Chrysemys picta belli.
Summary
lthe
. beoutlyuld
orith
us to
ofan
thebleisear
that
d and
Oxygen deprivation is a stress occasionally encounteredsome degree by members of all vertebrate taxa, but tvertebrate anaerobes, which can tolerate anoxia for prolonperiods without damage to their organ systems, are unusThe champion anaerobes, members of several groups of fiand turtles, can withstand anoxia for periods more than 10times as long as hypoxia-sensitive species such as smammals (Adolph, 1948; Nilsson, 1993; Ultsch and Jacks1982). In some vertebrates, hypoxic stress is largely confito one particular developmental stage or period of life histo(e.g. birth trauma in primates), and in other groups it is a daexperience associated with diving, burrowing or flying extreme altitude.
In principle, neurons in the central nervous system cousurvive prolonged transitions from aerobic to anaerobmetabolism by using the following strategies.
Keeping the brain running. Anaerobic metabolism couldincrease via stimulation of glycolysis to provide ATP at almosthe rate achieved under aerobic conditions. Critical organs (band heart) might adopt this strategy at the expense of others muscle and liver), thus receiving substrates to meet their nefor heightened glycolysis, while other organs go wantinExamples of this strategy are some anoxia-tolerant fishes (Land Nilsson, 1997) which remain active in anoxic water.
Introduction
toruegedual.shes00
mallon,nedryily
at
ldic
train(e.g.edsg.utz
Turning the brain off. Anaerobic metabolism wouldcontinue only at ‘pilot light’ levels, while normal neuronafunctions would assume a dormant posture. An example is brain of Chrysemys picta belli, the western painted turtle,which survives 5 months of anoxia during winter dormancy
Of course, a combination of these strategies might alsoadopted to serve neuronal function early and later into a bof anoxia or dormancy. We hypothesize, however, that onthe second strategy, with its attendant energy savings, wobe a viable strategy for really long bouts of anoxia dormancy. Just as the metabolic strategies for coping woxygen deprivation are varied, hypoxia-tolerant nervosystems no doubt employ a number of different strategiesavoid injury from forced limitation of aerobic metabolism.
In contrast to our knowledge of metabolism, the study how neurons adapt to anaerobic conditions remains in embryonic stage. Several recent reviews concerning adaptations of the vertebrate brain to anoxia are availa(Lutz, 1992; Lutz and Nilsson, 1997; Nilsson, 1993). In threview, we will emphasize pathways and processes that appto contribute to the second of the strategies outlined above, of metabolic arrest coupled to ion channel arrest.
Down-regulation of energy utilization, decreasetransmembrane ion fluxes through membrane ion channels

1142
ns
keyothfor
ty ofld-rgy
xiaobicmeiremelar
ATP
e
aoringasnreonctinaldedls
asmeant int’) innyly
xictsdth
nce ased
P. E. BICKLER AND L. T. BUCK
Table 1.Effects of ion channel inactivation on criticalelements of neuronal signaling
Na+ channel inactivationElevated action potential thresholdDecreased nerve conduction and velocityDecreased Na+ leak, decreased likelihood of Na+ gradient collapseStabilized Na+-gradient-dependent transporters (glucose, Ca2+,
neurotransmitters, H+)
K+ channel inactivationAltered action potential shapeAltered excitability
Glutamate receptor inactivationDecreased excitatory neurotransmission, saving energyStabilized [Ca2+]i
Voltage-gated Ca2+ channel inactivationDecreased excitabilityDecreased neurotransmitter releaseDecreased [Ca2+]i accumulation
Acetylcholine receptor inactivationDecreased excitatory neurotransmissionDecreased Ca2+ influx
decreased neuronal excitability are hallmarks of the responof anoxia-tolerant neurons that apparently follow the secostrategy. These processes allow the maintenance of criticagradients, particularly that of Ca2+, despite drastically reducedrates of substrate utilization and ATP production. Tprocesses that control ion channel activity in these neuronsalmost completely unexplored and offer fertile ground for nexplorations. The processes uncovered will be of valueunderstanding dormancy in a wide variety of organisms awill even lead eventually to improvements in the clinical caof patients with conditions such as strokes, degeneranervous system diseases and chronic pain syndromes; theyalso point to strategies for the suspended animation of orgfor transplantation. Ultimately, such research will lead to tability to create dormancy-like states for space travel ahuman adventures not yet imagined.
Problems for anaerobic neurons: energetics and iongradients
For neurons exhibiting metabolic arrest strategies surviving long-term anoxia, a critical problem is how to coupa decreased metabolic rate with membrane ion channel anpump activities. The maintenance of strongly hyperpolarizmembrane potentials through energetically intensmembrane ion pumping is characteristic of hypoxia-sensitvertebrate neurons, such as those found in ‘typical’ laboratmammals and humans. The absence of aerobic ATP producin anoxia-sensitive neurons causes membrane depolarizaand ion gradient collapse, excitatory neurotransmitter releand lethal increases in [Ca2+]i. Glutamate, the chief excitatoryneurotransmitter in the vertebrate central nervous sys(CNS), is a primary cause of Ca2+ influx and cell death via the‘glutamate cascade’ (Choi, 1995). Hypoxia-tolerant neuroavoid this chain reaction (Hochachka et al. 1996; Lutz andNilsson, 1997). We believe that decreases in the activity of ion channels in hypoxia-tolerant neurons play a key role bin energy conservation and in decreasing the potential injurious translocations of Ca2+ into the intracellularcompartment.
Targets for energy savings: ion transport down-regulation
Hochachka (1986) proposed that a decreased permeabilimembranes to ions would be a hallmark of anoxia- or cotolerant cells. The available evidence shows that eneproduction in hypoxia-tolerant animals during long-term anodecreases substantially and that increased anaermetabolism does not occur (Hochachka, 1986). At the satime, transmembrane ion gradients, which normally reqularge inputs of energy, remain almost unchanged. In sosystems (for example, the anoxic turtle brain), the extracelluconcentrations of key ions, such as Ca2+ and Mg2+, increase bya factor of five or more during prolonged anoxia (Cserr et al.1988), compounding the problems of ionic regulation. substantial amount of energy (up to 70–80 % of total A
sesnd
l ion
he are
ew inndretive willanshend
forle
d ioned
iveiveorytiontionase
tem
turnover) is expended by ion transport pumps, with just onsuch pump, the Na+/K+-ATPase, accounting for at least 50 %of ATP turnover in mammalian cells (Else, 1991; Hochachket al. 1996). The only known mechanism which can account fthese facts is a large decrease in ion translocation – a tightenof the conduits through which large numbers of ions such Ca2+ regularly flow. These conduits are undoubtedly protein iochannels; non-specific leaks through the lipid bilayer aunlikely to account for more than a small percentage of net itranslocation and, furthermore, would probably not be subjeto the rapid down-regulation characteristic of ion transport the anoxia-tolerant cells studied thus far. Such a functionconstriction of ion flow through ion channels could be initiateby a number of processes, all leading to effectively decreasneurotransmission, reduced number of action potentia(‘spiking’), decreased size of slow postsynaptic potentials, well as suppression of miniature endplate potentials, etc. Soeffects of decreased ion translocation through several importclasses of ion channels and receptors are summarizedTable 1. While cessation of action potentials (‘spike arresmay alone account for a significant fraction of the decreasenet ion translocation, one would predict suppression of matypes of ion translocation processes in order to achieve trusubstantial energy savings during prolonged periods of anodormancy. Furthermore, stabilization of some ionic gradien(e.g. that for Ca2+) may be needed to prevent disordereintracellular signaling and induction of programmed cell dea(apoptosis) during dormancy.
The net effects of the decreases in ion channel conducta(summarized in Table 1) on global neuronal function includestable membrane potential, decreased excitability, decrea
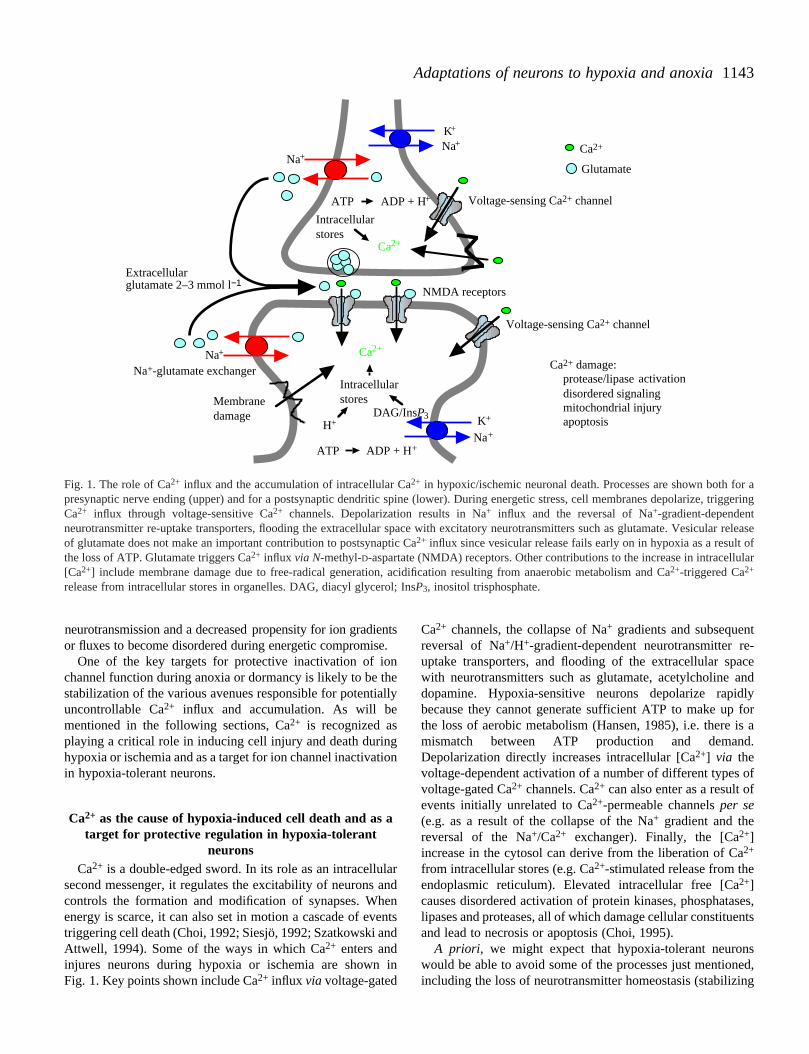
1143Adaptations of neurons to hypoxia and anoxia
-cendlyfors a.
of
ses,nts
sed,g
Na+K+
Na+
K+
Na+
Na+
Voltage-sensing Ca2+ channel
Extracellularglutamate 2–3 mmol l−1
DAG/InsP3H+
ATP ADP + H+
ATP ADP + H+
Voltage-sensing Ca2+ channel
Ca2+
NMDA receptors
Ca2+
Ca2+ damage: protease/lipase activation disordered signaling mitochondrial injury apoptosis
Ca2+
Glutamate
Na+-glutamate exchanger
Membranedamage
Intracellularstores
Intracellularstores
Fig. 1. The role of Ca2+ influx and the accumulation of intracellular Ca2+ in hypoxic/ischemic neuronal death. Processes are shown both for apresynaptic nerve ending (upper) and for a postsynaptic dendritic spine (lower). During energetic stress, cell membranes depolarize, triggeringCa2+ influx through voltage-sensitive Ca2+ channels. Depolarization results in Na+ influx and the reversal of Na+-gradient-dependentneurotransmitter re-uptake transporters, flooding the extracellular space with excitatory neurotransmitters such as glutamate. Vesicular releaseof glutamate does not make an important contribution to postsynaptic Ca2+ influx since vesicular release fails early on in hypoxia as a result ofthe loss of ATP. Glutamate triggers Ca2+ influx via N-methyl-D-aspartate (NMDA) receptors. Other contributions to the increase in intracellular[Ca2+] include membrane damage due to free-radical generation, acidification resulting from anaerobic metabolism and Ca2+-triggered Ca2+
release from intracellular stores in organelles. DAG, diacyl glycerol; InsP3, inositol trisphosphate.
neurotransmission and a decreased propensity for ion grador fluxes to become disordered during energetic compromis
One of the key targets for protective inactivation of iochannel function during anoxia or dormancy is likely to be tstabilization of the various avenues responsible for potentiauncontrollable Ca2+ influx and accumulation. As will bementioned in the following sections, Ca2+ is recognized asplaying a critical role in inducing cell injury and death durinhypoxia or ischemia and as a target for ion channel inactivain hypoxia-tolerant neurons.
Ca2+ as the cause of hypoxia-induced cell death and as atarget for protective regulation in hypoxia-tolerant
neuronsCa2+ is a double-edged sword. In its role as an intracellu
second messenger, it regulates the excitability of neurons controls the formation and modification of synapses. Whenergy is scarce, it can also set in motion a cascade of evtriggering cell death (Choi, 1992; Siesjö, 1992; Szatkowski aAttwell, 1994). Some of the ways in which Ca2+ enters andinjures neurons during hypoxia or ischemia are shownFig. 1. Key points shown include Ca2+ influx via voltage-gated
ientse.nhelly
gtion
larandenentsnd
in
Ca2+ channels, the collapse of Na+ gradients and subsequentreversal of Na+/H+-gradient-dependent neurotransmitter reuptake transporters, and flooding of the extracellular spawith neurotransmitters such as glutamate, acetylcholine adopamine. Hypoxia-sensitive neurons depolarize rapidbecause they cannot generate sufficient ATP to make up the loss of aerobic metabolism (Hansen, 1985), i.e. there imismatch between ATP production and demandDepolarization directly increases intracellular [Ca2+] via thevoltage-dependent activation of a number of different types voltage-gated Ca2+ channels. Ca2+ can also enter as a result ofevents initially unrelated to Ca2+-permeable channels per se(e.g. as a result of the collapse of the Na+ gradient and thereversal of the Na+/Ca2+ exchanger). Finally, the [Ca2+]increase in the cytosol can derive from the liberation of Ca2+
from intracellular stores (e.g. Ca2+-stimulated release from theendoplasmic reticulum). Elevated intracellular free [Ca2+]causes disordered activation of protein kinases, phosphatalipases and proteases, all of which damage cellular constitueand lead to necrosis or apoptosis (Choi, 1995).
A priori, we might expect that hypoxia-tolerant neuronwould be able to avoid some of the processes just mentionincluding the loss of neurotransmitter homeostasis (stabilizin

1144
rengf
a
e
ndd
g inialof
e,
rcee,tialandts
nia.
rs,d,de,lee of
ite7;
xic ins.
thins
re or
P. E. BICKLER AND L. T. BUCK
Na+ gradients), depolarization and Ca2+ channel activation,and inactivation of other receptors and ion channcontributing to Ca2+ influx (e.g. the Ca2+-permeable glutamatereceptors and acetylcholine receptors). We will next revisome of the specifics known about these possibilities.
Neuronal ion channel regulation during anoxiaNa+ channels
Current flowing through Na+ channels constitutes a majocomponent of action potentials (‘spikes’). Spike arrest has bproposed as an important mechanism in the adaptationneurons to hypoxia by Sick, Rosenthal, Lutz and others (Set al. 1993) because it would decrease the energetic costneurotransmission and nerve conduction. The arrest of N+
channels would favor the maintenance of transcellular N+
gradients, which are crucial for preventing cellular swellinthe failure of neurotransmitter re-uptake and Ca2+ influx (Table1).
Several differences in Na+ channel abundance betweeanoxia-sensitive and anoxia-insensitive animals (rats versusturtles) have been found and could be related to phenomenon of spike arrest. The density of Na+ channels inturtles is lower than in mammals (Edwards et al. 1989), insynaptosomes and in several fractions of brain homogen(Xia and Haddad, 1991), and is perhaps related to the tenlower metabolic rate of reptiles compared with mammals (El1991). Na+ channels further decrease in density during anoin turtles. The most dramatic change with anoxia is a 42decrease in density of Na+ channels in cerebellum (PerezPinzon et al. 1992). It is not clear whether this differenc(based solely on the numbers of binding sites in brahomogenates) is alone enough to account for the 100-reduction in metabolic rate in turtle brain, even if it leads complete ‘spike arrest’. Whether or not the open probabilityindividual Na+ channels is regulated during anoxia neestudy, because channel numbers cannot be used to channel activity or channel currents, and Na+ channels, like allchannels studied thus far, are subject to a large numbeallosteric controls involving phosphorylation, etcUnfortunately, to our knowledge, no direct measurementsNa+ channel activity during anoxia in anoxia-tolerant neurohave been made. This is required, since determining numbers of channels through binding studies gives no usinformation on channel activity per se.
Na+ channel cycling from an active membrane-bound forinto one in the cytosol is a form of regulation that could involved in anoxia-induced channel inactivation. Thmovement of Na+ channels from the cell membrane to thcytoplasm and back again has been demonstrated in ssystems. Both protein kinase A and protein kinase phosphorylation modulate the activity of neuronal Na+
channels (Frohnweiser et al. 1995; Murphy et al. 1993), andthese phosphorylation/dephosphorylation events could eitdirectly influence receptor activity (open probability) or labreceptors for internalization or for persistence in an act
els
ew
reen oficks of
aag,
n
the
atefoldse,xia%
-ein
foldto ofdsinfer
r of. ofnstheeful
mbeeeomeC
herelive
role in the membrane. As a variety of other channels aanchored to the cytoskeleton, it is conceivable that, durianoxia, Ca2+-dependent events (e.g. depolymerization ochannels to actin in the cytoskeleton) remove the N+
channels from the membrane or inactivate them in situ. Muchexciting work on these possibilities seems inevitable in thnear future.
K+ channels
K+ channels are diverse in function and structure, amany types are still being defined genetically anelectrophysiologically. Increased activity of K+ channelsbrings cell membrane potential closer to the K+ equilibriumpotential, which is hyperpolarized from the normal restinpotential. Hyperpolarization makes neurons less excitablethe sense that they are farther from the action potentthreshold, but the hyperpolarization comes at the cost increased gradients for Na+ and Ca2+, requiring greater tonicenergy expenditure to expel these ions. It is difficult to setherefore, that K+ channel activation would form areasonable strategy for saving energy in an energy-scasituation. K+ channels also have other functions, of courssuch as shaping the upstroke and duration of action potenspikes, because they are active at depolarized potentials will tend to pull the membrane potential back towards iresting level.
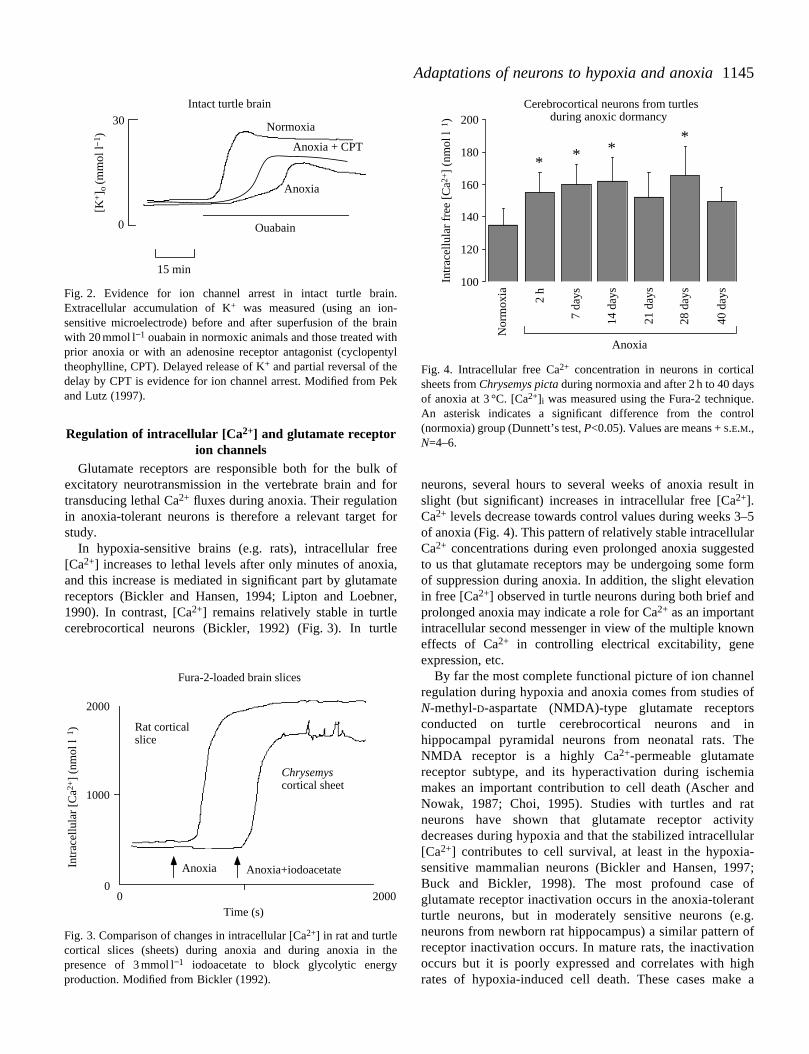
Measurements of K+ channel activity during anoxia havealso only been made indirectly. The efflux of K+ from neuronshas been used as an index of K+ channel activity in intact turtlebrain or isolated cerebellum (Chih et al. 1989). Measurementsof K+ accumulation in extracellular fluid (ECF) have beemade when metabolic production of ATP is blocked by anoxand an inhibitor of glycolysis (usually iodoacetate)Measurements of K+ accumulation in the extracellular fluidmade using this technique depend upon a number of factoincluding the activation of multiple types of ion channels anof course, on rates of ATP depletion. These problems asireduced efflux of K+ has been seen in anoxia-adapted turtcerebellum and cortex and has been interpreted as evidencchannel arrest (Fig. 2). The efflux of K+ is depressed byextracellular adenosine, a ‘retaliatory’ adenylate metabolaccumulated in turtle ECF during anoxia (Pek and Lutz, 199Perez-Pinzon et al. 1993).
One specific group of K+ channels, those sensitive to ATP(KATP channels), is involved in the initial hyperpolarization(i.e. before the cells depolarize and die) seen in anomammalian neurons. These channels could play a rolestabilizing membrane potential in anoxia-tolerant cellHowever, glibenclamide, an antagonist of mammalian KATP
channels, fails to alter K+ release from turtle brain (Jiang et al.1992) or carp brain during anoxia or during depolarization wiouabain (Johansson and Nilsson, 1995). Much work remato be done to define the roles of K+ channels in the adaptationsof neurons to hypoxic conditions, including the use of mospecific tools for measurement (e.g. patch-clamp studieswhole-cell K+ current measurements).
1145Adaptations of neurons to hypoxia and anoxia
in
5rd
rmn
n
Ouabain
Normoxia
Anoxia + CPT
Anoxia
15 min
30
0
[K+] o
(mm
ol l
1 )Intact turtle brain
Fig. 2. Evidence for ion channel arrest in intact turtle brain.Extracellular accumulation of K+ was measured (using an ion-sensitive microelectrode) before and after superfusion of the brainwith 20 mmol l−1 ouabain in normoxic animals and those treated withprior anoxia or with an adenosine receptor antagonist (cyclopentyltheophylline, CPT). Delayed release of K+ and partial reversal of thedelay by CPT is evidence for ion channel arrest. Modified from Pekand Lutz (1997).
100
120
140
160
180
200
Nor
mox
ia
2 h
7 da
ys
14 d
ays
21 d
ays
28 d
ays
40 d
ays
Intr
acel
lula
r fr
ee [
Ca2
+]
(nm
ol l
1 )
Cerebrocortical neurons from turtles during anoxic dormancy
* * **
Anoxia
Fig. 4. Intracellular free Ca2+ concentration in neurons in corticalsheets from Chrysemys pictaduring normoxia and after 2 h to 40 daysof anoxia at 3 °C. [Ca2+]i was measured using the Fura-2 technique.An asterisk indicates a significant difference from the control(normoxia) group (Dunnett’s test, P<0.05). Values are means + S.E.M.,N=4–6.
Regulation of intracellular [Ca2+] and glutamate receptorion channels
Glutamate receptors are responsible both for the bulkexcitatory neurotransmission in the vertebrate brain andtransducing lethal Ca2+ fluxes during anoxia. Their regulationin anoxia-tolerant neurons is therefore a relevant targetstudy.
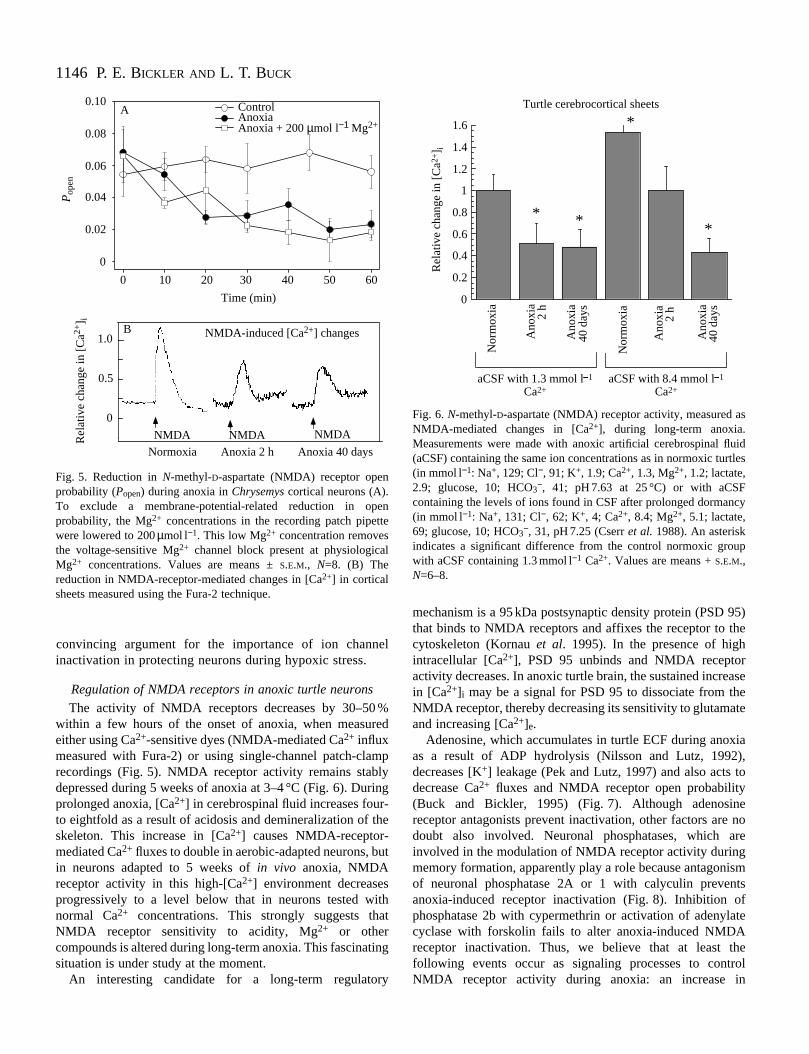
In hypoxia-sensitive brains (e.g. rats), intracellular fr[Ca2+] increases to lethal levels after only minutes of anoxand this increase is mediated in significant part by glutamreceptors (Bickler and Hansen, 1994; Lipton and Loebn1990). In contrast, [Ca2+] remains relatively stable in turtlecerebrocortical neurons (Bickler, 1992) (Fig. 3). In turt
lf
ne
iadtyar
7;fntg.ofnh a
Fig. 3. Comparison of changes in intracellular [Ca2+] in rat and turtlecortical slices (sheets) during anoxia and during anoxia in presence of 3 mmol l−1 iodoacetate to block glycolytic energyproduction. Modified from Bickler (1992).
Anoxia+iodoacetateAnoxia
Rat corticalslice
Chrysemyscortical sheet
2000
00 2000
Time (s)
Intr
acel
lula
r [C
a2+]
(nm
ol l
1 )
Fura-2-loaded brain slices
1000
of for
for
eeia,ateer,
le
neurons, several hours to several weeks of anoxia result slight (but significant) increases in intracellular free [Ca2+].Ca2+ levels decrease towards control values during weeks 3–of anoxia (Fig. 4). This pattern of relatively stable intracellulaCa2+ concentrations during even prolonged anoxia suggesteto us that glutamate receptors may be undergoing some foof suppression during anoxia. In addition, the slight elevatioin free [Ca2+] observed in turtle neurons during both brief andprolonged anoxia may indicate a role for Ca2+ as an importantintracellular second messenger in view of the multiple knoweffects of Ca2+ in controlling electrical excitability, geneexpression, etc.
By far the most complete functional picture of ion channeregulation during hypoxia and anoxia comes from studies oN-methyl-D-aspartate (NMDA)-type glutamate receptorsconducted on turtle cerebrocortical neurons and ihippocampal pyramidal neurons from neonatal rats. ThNMDA receptor is a highly Ca2+-permeable glutamatereceptor subtype, and its hyperactivation during ischemmakes an important contribution to cell death (Ascher anNowak, 1987; Choi, 1995). Studies with turtles and raneurons have shown that glutamate receptor activitdecreases during hypoxia and that the stabilized intracellul[Ca2+] contributes to cell survival, at least in the hypoxia-sensitive mammalian neurons (Bickler and Hansen, 199Buck and Bickler, 1998). The most profound case oglutamate receptor inactivation occurs in the anoxia-toleraturtle neurons, but in moderately sensitive neurons (e.neurons from newborn rat hippocampus) a similar pattern receptor inactivation occurs. In mature rats, the inactivatiooccurs but it is poorly expressed and correlates with higrates of hypoxia-induced cell death. These cases make
the
1146
5)e
seee
a,o
nore
ms
te
eol
P. E. BICKLER AND L. T. BUCK
Normoxia Anoxia 2 h Anoxia 40 days
1.0
0.5
0
NMDARel
ativ
e ch
ange
in [
Ca2+
] i
NMDA-induced [Ca2+] changesB
NMDA NMDA
ControlAnoxia Anoxia + 200 µmol l−1 Mg2+
A
Time (min)
0 10 20 30 40 50 60
Pop
en
0
0.02
0.04
0.06
0.08
0.10
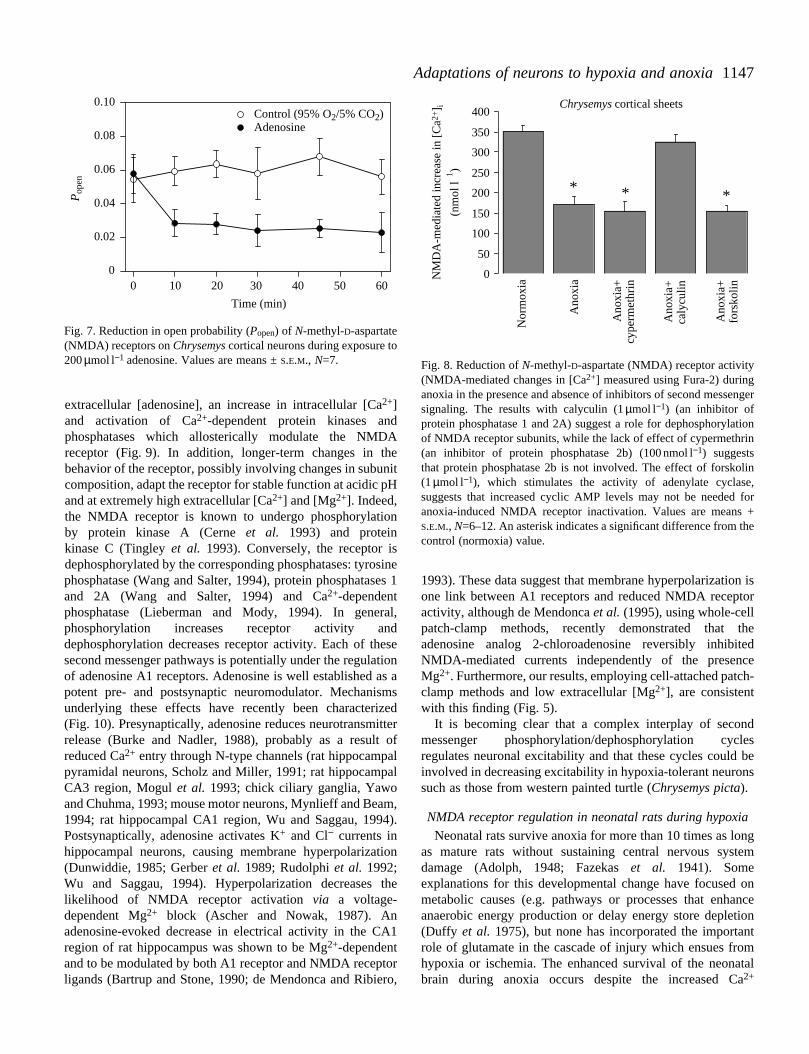
Fig. 5. Reduction in N-methyl-D-aspartate (NMDA) receptor openprobability (Popen) during anoxia in Chrysemyscortical neurons (A).To exclude a membrane-potential-related reduction in openprobability, the Mg2+ concentrations in the recording patch pipettewere lowered to 200µmol l−1. This low Mg2+ concentration removesthe voltage-sensitive Mg2+ channel block present at physiologicalMg2+ concentrations. Values are means ±S.E.M., N=8. (B) Thereduction in NMDA-receptor-mediated changes in [Ca2+] in corticalsheets measured using the Fura-2 technique.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
Nor
mox
ia
Ano
xia
2 h
Ano
xia
40 d
ays
Nor
mox
ia
Ano
xia
2 h
Ano
xia
40 d
ays
Rel
ativ
e ch
ange
in [
Ca2
+] i
Turtle cerebrocortical sheets
*
* * *
aCSF with 1.3 mmol l 1
Ca2+aCSF with 8.4 mmol l 1
Ca2+
Fig. 6. N-methyl-D-aspartate (NMDA) receptor activity, measured asNMDA-mediated changes in [Ca2+], during long-term anoxia.Measurements were made with anoxic artificial cerebrospinal fluid(aCSF) containing the same ion concentrations as in normoxic turtles(in mmol l−1: Na+, 129; Cl−, 91; K+, 1.9; Ca2+, 1.3, Mg2+, 1.2; lactate,2.9; glucose, 10; HCO3−, 41; pH 7.63 at 25 °C) or with aCSFcontaining the levels of ions found in CSF after prolonged dormancy(in mmol l−1: Na+, 131; Cl−, 62; K+, 4; Ca2+, 8.4; Mg2+, 5.1; lactate,69; glucose, 10; HCO3−, 31, pH 7.25 (Cserr et al.1988). An asteriskindicates a significant difference from the control normoxic groupwith aCSF containing 1.3 mmol l−1 Ca2+. Values are means + S.E.M.,N=6–8.
convincing argument for the importance of ion channinactivation in protecting neurons during hypoxic stress.
Regulation of NMDA receptors in anoxic turtle neurons
The activity of NMDA receptors decreases by 30–50within a few hours of the onset of anoxia, when measueither using Ca2+-sensitive dyes (NMDA-mediated Ca2+ influxmeasured with Fura-2) or using single-channel patch-clarecordings (Fig. 5). NMDA receptor activity remains stabdepressed during 5 weeks of anoxia at 3–4 °C (Fig. 6). Durprolonged anoxia, [Ca2+] in cerebrospinal fluid increases fourto eightfold as a result of acidosis and demineralization of skeleton. This increase in [Ca2+] causes NMDA-receptor-mediated Ca2+ fluxes to double in aerobic-adapted neurons, bin neurons adapted to 5 weeks of in vivo anoxia, NMDAreceptor activity in this high-[Ca2+] environment decreasesprogressively to a level below that in neurons tested wnormal Ca2+ concentrations. This strongly suggests thNMDA receptor sensitivity to acidity, Mg2+ or othercompounds is altered during long-term anoxia. This fascinasituation is under study at the moment.
An interesting candidate for a long-term regulato
el
%red
mplying-the
ut
ithat
ting
ry
mechanism is a 95 kDa postsynaptic density protein (PSD 9that binds to NMDA receptors and affixes the receptor to thcytoskeleton (Kornau et al. 1995). In the presence of highintracellular [Ca2+], PSD 95 unbinds and NMDA receptoractivity decreases. In anoxic turtle brain, the sustained increain [Ca2+]i may be a signal for PSD 95 to dissociate from thNMDA receptor, thereby decreasing its sensitivity to glutamatand increasing [Ca2+]e.
Adenosine, which accumulates in turtle ECF during anoxias a result of ADP hydrolysis (Nilsson and Lutz, 1992)decreases [K+] leakage (Pek and Lutz, 1997) and also acts tdecrease Ca2+ fluxes and NMDA receptor open probability(Buck and Bickler, 1995) (Fig. 7). Although adenosinereceptor antagonists prevent inactivation, other factors are doubt also involved. Neuronal phosphatases, which ainvolved in the modulation of NMDA receptor activity duringmemory formation, apparently play a role because antagonisof neuronal phosphatase 2A or 1 with calyculin preventanoxia-induced receptor inactivation (Fig. 8). Inhibition ofphosphatase 2b with cypermethrin or activation of adenylacyclase with forskolin fails to alter anoxia-induced NMDAreceptor inactivation. Thus, we believe that at least thfollowing events occur as signaling processes to contrNMDA receptor activity during anoxia: an increase in
1147Adaptations of neurons to hypoxia and anoxia
dAe
nitpH
n
n isor
heedeh-
desbes
ngem
onnce
ionntmtal
Time (min)
0 10 20 30 40 50 60
Pop
en
0
0.02
0.04
0.06
0.08
0.10Control (95% O2/5% CO2)Adenosine
Fig. 7. Reduction in open probability (Popen) of N-methyl-D-aspartate(NMDA) receptors on Chrysemyscortical neurons during exposure to200µmol l−1 adenosine. Values are means ±S.E.M., N=7.
0
50
100
150
200
250
300
350
400
Nor
mox
ia
Ano
xia
Ano
xia+
cype
rmet
hrin
Ano
xia+
caly
culin
Ano
xia+
fors
kolin
Chrysemys cortical sheets
*
*
*
NM
DA
-med
iate
d in
crea
se in
[C
a2+] i
(nm
ol l
1 )
Fig. 8. Reduction of N-methyl-D-aspartate (NMDA) receptor activity(NMDA-mediated changes in [Ca2+] measured using Fura-2) duringanoxia in the presence and absence of inhibitors of second messengersignaling. The results with calyculin (1µmol l−1) (an inhibitor ofprotein phosphatase 1 and 2A) suggest a role for dephosphorylationof NMDA receptor subunits, while the lack of effect of cypermethrin(an inhibitor of protein phosphatase 2b) (100 nmol l−1) suggeststhat protein phosphatase 2b is not involved. The effect of forskolin(1µmol l−1), which stimulates the activity of adenylate cyclase,suggests that increased cyclic AMP levels may not be needed foranoxia-induced NMDA receptor inactivation. Values are means +S.E.M., N=6–12. An asterisk indicates a significant difference from thecontrol (normoxia) value.
extracellular [adenosine], an increase in intracellular [Ca2+]and activation of Ca2+-dependent protein kinases anphosphatases which allosterically modulate the NMDreceptor (Fig. 9). In addition, longer-term changes in thbehavior of the receptor, possibly involving changes in subucomposition, adapt the receptor for stable function at acidic and at extremely high extracellular [Ca2+] and [Mg2+]. Indeed,the NMDA receptor is known to undergo phosphorylatioby protein kinase A (Cerne et al. 1993) and proteinkinase C (Tingley et al. 1993). Conversely, the receptor isdephosphorylated by the corresponding phosphatases: tyrophosphatase (Wang and Salter, 1994), protein phosphatasand 2A (Wang and Salter, 1994) and Ca2+-dependentphosphatase (Lieberman and Mody, 1994). In generphosphorylation increases receptor activity andephosphorylation decreases receptor activity. Each of thsecond messenger pathways is potentially under the regulaof adenosine A1 receptors. Adenosine is well established apotent pre- and postsynaptic neuromodulator. Mechanisunderlying these effects have recently been characteri(Fig. 10). Presynaptically, adenosine reduces neurotransmrelease (Burke and Nadler, 1988), probably as a resultreduced Ca2+ entry through N-type channels (rat hippocamppyramidal neurons, Scholz and Miller, 1991; rat hippocampCA3 region, Mogul et al. 1993; chick ciliary ganglia, Yawoand Chuhma, 1993; mouse motor neurons, Mynlieff and Bea1994; rat hippocampal CA1 region, Wu and Saggau, 199Postsynaptically, adenosine activates K+ and Cl− currents inhippocampal neurons, causing membrane hyperpolarizat(Dunwiddie, 1985; Gerber et al. 1989; Rudolphi et al. 1992;Wu and Saggau, 1994). Hyperpolarization decreases likelihood of NMDA receptor activation via a voltage-dependent Mg2+ block (Ascher and Nowak, 1987). Anadenosine-evoked decrease in electrical activity in the Cregion of rat hippocampus was shown to be Mg2+-dependentand to be modulated by both A1 receptor and NMDA recepligands (Bartrup and Stone, 1990; de Mendonca and Ribie
sinees 1
al,desetions amszeditter ofalal
m,4).
ion
the
A1
torro,
1993). These data suggest that membrane hyperpolarizatioone link between A1 receptors and reduced NMDA receptactivity, although de Mendonca et al. (1995), using whole-cellpatch-clamp methods, recently demonstrated that tadenosine analog 2-chloroadenosine reversibly inhibitNMDA-mediated currents independently of the presencMg2+. Furthermore, our results, employing cell-attached patcclamp methods and low extracellular [Mg2+], are consistentwith this finding (Fig. 5).
It is becoming clear that a complex interplay of seconmessenger phosphorylation/dephosphorylation cyclregulates neuronal excitability and that these cycles could involved in decreasing excitability in hypoxia-tolerant neuronsuch as those from western painted turtle (Chrysemys picta).
NMDA receptor regulation in neonatal rats during hypoxia
Neonatal rats survive anoxia for more than 10 times as loas mature rats without sustaining central nervous systdamage (Adolph, 1948; Fazekas et al. 1941). Someexplanations for this developmental change have focused metabolic causes (e.g. pathways or processes that enhaanaerobic energy production or delay energy store deplet(Duffy et al. 1975), but none has incorporated the importarole of glutamate in the cascade of injury which ensues frohypoxia or ischemia. The enhanced survival of the neonabrain during anoxia occurs despite the increased Ca2+
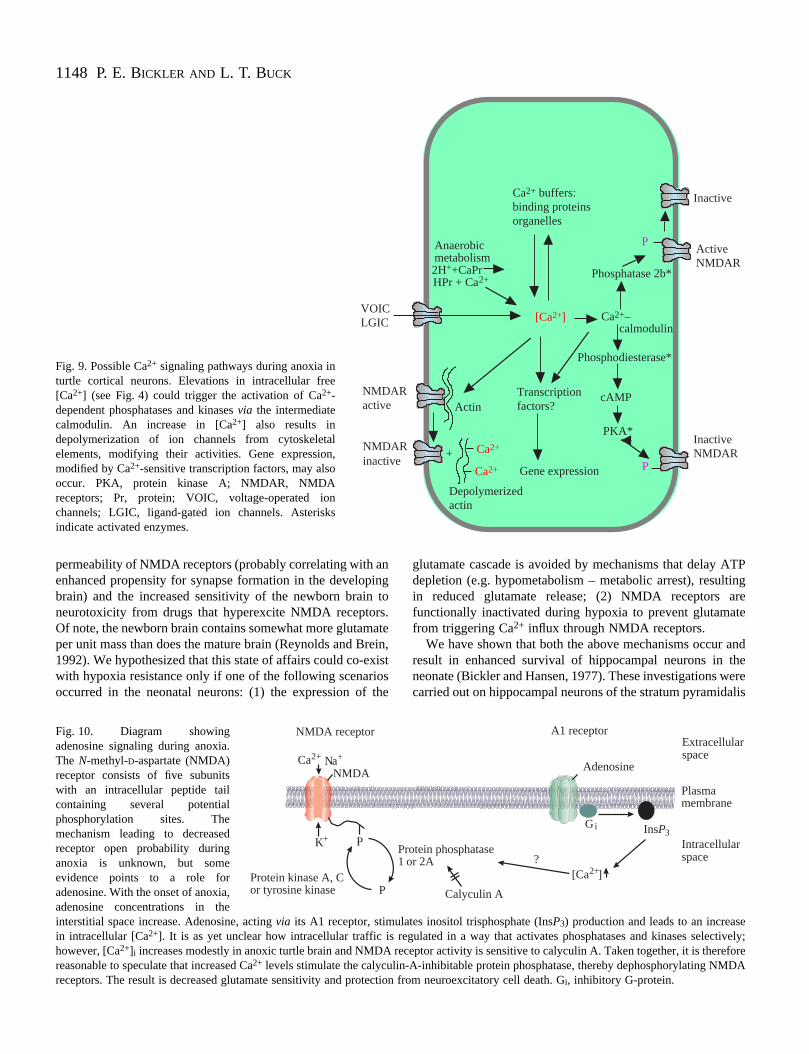
1148
TPngrete
andheerelis
P. E. BICKLER AND L. T. BUCK
[Ca2+]
+
NMDARactive
NMDARinactive
Actin
Depolymerizedactin
Anaerobicmetabolism2H+ +CaPr HPr + Ca2+
VOICLGIC
Transcriptionfactors?
Ca2+
Ca2+ Gene expression
Ca2+– calmodulin
Phosphodiesterase*
cAMP
PKA*InactiveNMDAR
P
Phosphatase 2b*
PActiveNMDAR
InactiveCa2+ buffers:binding proteinsorganelles
Fig. 9. Possible Ca2+ signaling pathways during anoxia inturtle cortical neurons. Elevations in intracellular free[Ca2+] (see Fig. 4) could trigger the activation of Ca2+-dependent phosphatases and kinases via the intermediatecalmodulin. An increase in [Ca2+] also results indepolymerization of ion channels from cytoskeletalelements, modifying their activities. Gene expression,modified by Ca2+-sensitive transcription factors, may alsooccur. PKA, protein kinase A; NMDAR, NMDAreceptors; Pr, protein; VOIC, voltage-operated ionchannels; LGIC, ligand-gated ion channels. Asterisksindicate activated enzymes.
permeability of NMDA receptors (probably correlating with aenhanced propensity for synapse formation in the developbrain) and the increased sensitivity of the newborn brainneurotoxicity from drugs that hyperexcite NMDA receptorOf note, the newborn brain contains somewhat more glutamper unit mass than does the mature brain (Reynolds and B1992). We hypothesized that this state of affairs could co-ewith hypoxia resistance only if one of the following scenarioccurred in the neonatal neurons: (1) the expression of
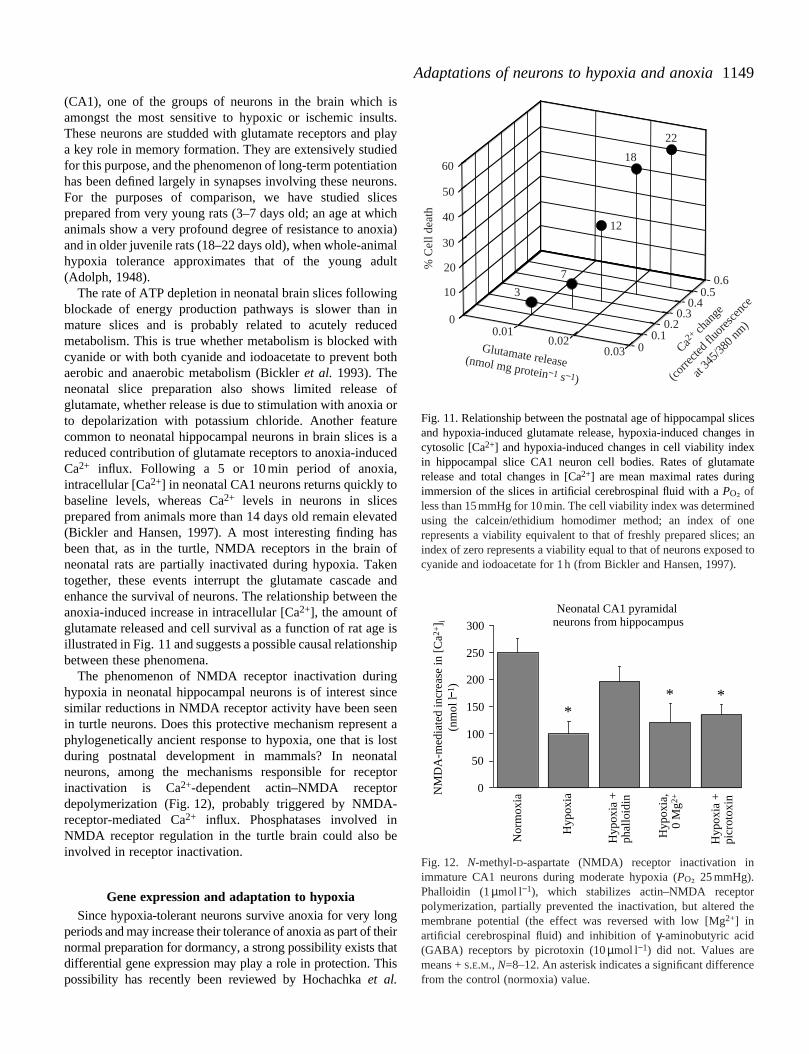
Fig. 10. Diagram showingadenosine signaling during anoxia.The N-methyl-D-aspartate (NMDA)receptor consists of five subunitswith an intracellular peptide tailcontaining several potentialphosphorylation sites. Themechanism leading to decreasedreceptor open probability duringanoxia is unknown, but someevidence points to a role foradenosine. With the onset of anoxia,adenosine concentrations in theinterstitial space increase. Adenosine, acting via its A1 receptor, stimin intracellular [Ca2+]. It is as yet unclear how intracellular traffic however, [Ca2+]i increases modestly in anoxic turtle brain and NMDreasonable to speculate that increased Ca2+ levels stimulate the calycureceptors. The result is decreased glutamate sensitivity and prot
NMDA receptor
NMDA
K P
Ca2+
+
Na+
PProtein kinase A, Cor tyrosine kinase
P1
ning tos.ate
rein,xistos the
glutamate cascade is avoided by mechanisms that delay Adepletion (e.g. hypometabolism – metabolic arrest), resultiin reduced glutamate release; (2) NMDA receptors afunctionally inactivated during hypoxia to prevent glutamafrom triggering Ca2+ influx through NMDA receptors.
We have shown that both the above mechanisms occur result in enhanced survival of hippocampal neurons in tneonate (Bickler and Hansen, 1977). These investigations wcarried out on hippocampal neurons of the stratum pyramida
ulates inositol trisphosphate (InsP3) production and leads to an increaseis regulated in a way that activates phosphatases and kinases selectively;A receptor activity is sensitive to calyculin A. Taken together, it is thereforelin-A-inhibitable protein phosphatase, thereby dephosphorylating NMDA
ection from neuroexcitatory cell death. Gi, inhibitory G-protein.
A1 receptor
Adenosine
rotein phosphataseor 2A
Calyculin A
Plasmamembrane
Gi InsP3
[Ca ]2+?
Extracellularspace
Intracellularspace
1149Adaptations of neurons to hypoxia and anoxia
islts.play
iedtionons.cesichxia)al
ult
g incedithoth
of orreis aced,o
tedasofen
and the
e isship
gnceennt alosttalptorr-
e
ngtheirhathis
7777
1111 2222
11118888
22222222
3333
% C
ell d
eath
Glutamate release(nmol mg protein−1 s−1)
Ca2+ ch
ange
(corre
cted f
luores
cenc
e
at 34
5/380
nm)
60
50
40
30
20
10
00.01
0.020.03 0
0.10.2
0.30.4
0.50.6
22
18
7
3
12
Fig. 11. Relationship between the postnatal age of hippocampal slicesand hypoxia-induced glutamate release, hypoxia-induced changes incytosolic [Ca2+] and hypoxia-induced changes in cell viability indexin hippocampal slice CA1 neuron cell bodies. Rates of glutamaterelease and total changes in [Ca2+] are mean maximal rates duringimmersion of the slices in artificial cerebrospinal fluid with a PO∑ ofless than 15mmHg for 10min. The cell viability index was determinedusing the calcein/ethidium homodimer method; an index of onerepresents a viability equivalent to that of freshly prepared slices; anindex of zero represents a viability equal to that of neurons exposed tocyanide and iodoacetate for 1h (from Bickler and Hansen, 1997).
0
50
100
150
200
250
300
Nor
mox
ia
Hyp
oxia
Hyp
oxia
+ph
allo
idin
Hyp
oxia
,0
Mg2
+
Hyp
oxia
+pi
crot
oxin
NM
DA
-med
iate
d in
crea
se in
[C
a2+] i
(nm
ol l
1 )
Neonatal CA1 pyramidalneurons from hippocampus
** *
Fig. 12. N-methyl-D-aspartate (NMDA) receptor inactivation inimmature CA1 neurons during moderate hypoxia (PO∑ 25 mmHg).Phalloidin (1µmol l−1), which stabilizes actin–NMDA receptorpolymerization, partially prevented the inactivation, but altered themembrane potential (the effect was reversed with low [Mg2+] inartificial cerebrospinal fluid) and inhibition of γ-aminobutyric acid(GABA) receptors by picrotoxin (10µmol l−1) did not. Values aremeans + S.E.M., N=8–12. An asterisk indicates a significant differencefrom the control (normoxia) value.
(CA1), one of the groups of neurons in the brain whichamongst the most sensitive to hypoxic or ischemic insuThese neurons are studded with glutamate receptors and a key role in memory formation. They are extensively studfor this purpose, and the phenomenon of long-term potentiahas been defined largely in synapses involving these neurFor the purposes of comparison, we have studied sliprepared from very young rats (3–7 days old; an age at whanimals show a very profound degree of resistance to anoand in older juvenile rats (18–22 days old), when whole-animhypoxia tolerance approximates that of the young ad(Adolph, 1948).
The rate of ATP depletion in neonatal brain slices followinblockade of energy production pathways is slower thanmature slices and is probably related to acutely redumetabolism. This is true whether metabolism is blocked wcyanide or with both cyanide and iodoacetate to prevent baerobic and anaerobic metabolism (Bickleret al. 1993). Theneonatal slice preparation also shows limited releaseglutamate, whether release is due to stimulation with anoxiato depolarization with potassium chloride. Another featucommon to neonatal hippocampal neurons in brain slices reduced contribution of glutamate receptors to anoxia-induCa2+ influx. Following a 5 or 10 min period of anoxiaintracellular [Ca2+] in neonatal CA1 neurons returns quickly tbaseline levels, whereas Ca2+ levels in neurons in slicesprepared from animals more than 14 days old remain eleva(Bickler and Hansen, 1997). A most interesting finding hbeen that, as in the turtle, NMDA receptors in the brain neonatal rats are partially inactivated during hypoxia. Taktogether, these events interrupt the glutamate cascadeenhance the survival of neurons. The relationship betweenanoxia-induced increase in intracellular [Ca2+], the amount ofglutamate released and cell survival as a function of rat agillustrated in Fig. 11 and suggests a possible causal relationbetween these phenomena.
The phenomenon of NMDA receptor inactivation durinhypoxia in neonatal hippocampal neurons is of interest sisimilar reductions in NMDA receptor activity have been sein turtle neurons. Does this protective mechanism represephylogenetically ancient response to hypoxia, one that is during postnatal development in mammals? In neonaneurons, among the mechanisms responsible for receinactivation is Ca2+-dependent actin–NMDA receptodepolymerization (Fig. 12), probably triggered by NMDAreceptor-mediated Ca2+ influx. Phosphatases involved inNMDA receptor regulation in the turtle brain could also binvolved in receptor inactivation.
Gene expression and adaptation to hypoxiaSince hypoxia-tolerant neurons survive anoxia for very lo
periods and may increase their tolerance of anoxia as part of normal preparation for dormancy, a strong possibility exists tdifferential gene expression may play a role in protection. Tpossibility has recently been reviewed by Hochachka et al.
1150
anteasre
anteaseer.ies
ost
ingons in,
ings,
elndthe
are
P. E. BICKLER AND L. T. BUCK
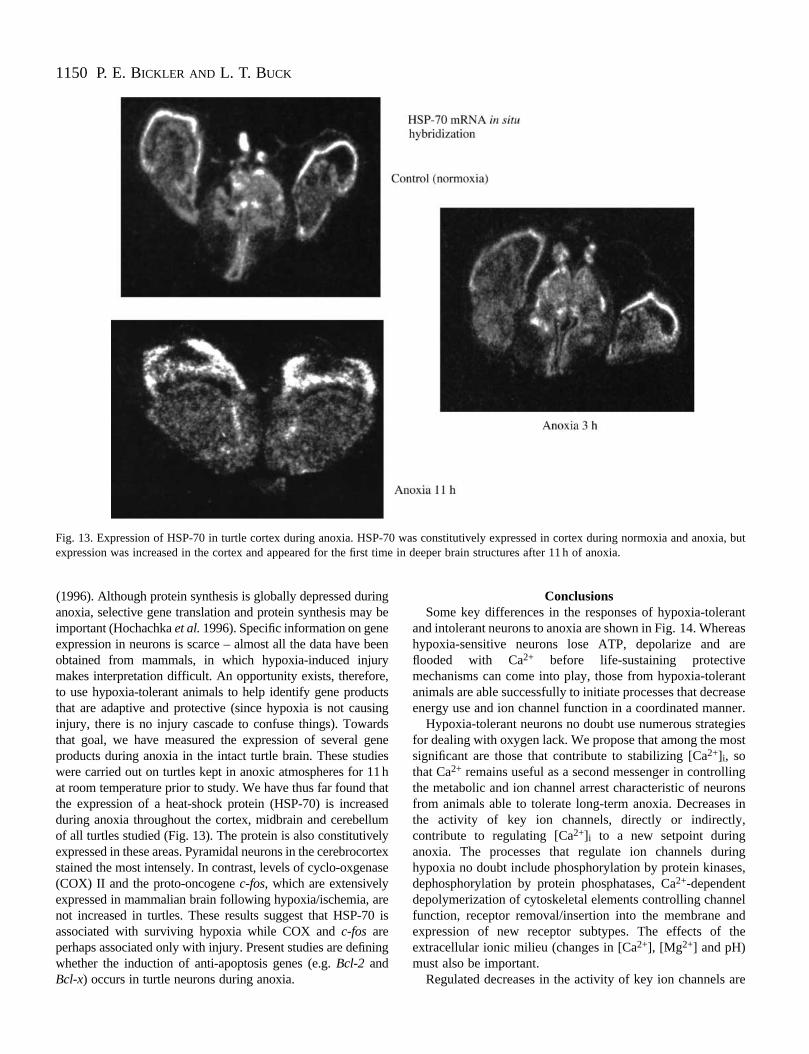
Fig. 13. Expression of HSP-70 in turtle cortex during anoxia. HSP-70 was constitutively expressed in cortex during normoxia and anoxia, butexpression was increased in the cortex and appeared for the first time in deeper brain structures after 11 h of anoxia.
(1996). Although protein synthesis is globally depressed duranoxia, selective gene translation and protein synthesis maimportant (Hochachka et al.1996). Specific information on geneexpression in neurons is scarce – almost all the data have obtained from mammals, in which hypoxia-induced injurmakes interpretation difficult. An opportunity exists, thereforto use hypoxia-tolerant animals to help identify gene produthat are adaptive and protective (since hypoxia is not causinjury, there is no injury cascade to confuse things). Towathat goal, we have measured the expression of several gproducts during anoxia in the intact turtle brain. These studwere carried out on turtles kept in anoxic atmospheres for 1at room temperature prior to study. We have thus far found tthe expression of a heat-shock protein (HSP-70) is increaduring anoxia throughout the cortex, midbrain and cerebelluof all turtles studied (Fig. 13). The protein is also constitutiveexpressed in these areas. Pyramidal neurons in the cerebrocstained the most intensely. In contrast, levels of cyclo-oxgen(COX) II and the proto-oncogene c-fos, which are extensivelyexpressed in mammalian brain following hypoxia/ischemia, anot increased in turtles. These results suggest that HSP-7associated with surviving hypoxia while COX and c-fos areperhaps associated only with injury. Present studies are definwhether the induction of anti-apoptosis genes (e.g. Bcl-2 andBcl-x) occurs in turtle neurons during anoxia.
ingy be
beenye,ctsing
rdseneies1hhatsedmlyortexase
re0 is
ing
ConclusionsSome key differences in the responses of hypoxia-toler
and intolerant neurons to anoxia are shown in Fig. 14. Wherhypoxia-sensitive neurons lose ATP, depolarize and aflooded with Ca2+ before life-sustaining protectivemechanisms can come into play, those from hypoxia-toleranimals are able successfully to initiate processes that decrenergy use and ion channel function in a coordinated mann
Hypoxia-tolerant neurons no doubt use numerous strategfor dealing with oxygen lack. We propose that among the msignificant are those that contribute to stabilizing [Ca2+]i, sothat Ca2+ remains useful as a second messenger in controllthe metabolic and ion channel arrest characteristic of neurfrom animals able to tolerate long-term anoxia. Decreasesthe activity of key ion channels, directly or indirectlycontribute to regulating [Ca2+]i to a new setpoint duringanoxia. The processes that regulate ion channels durhypoxia no doubt include phosphorylation by protein kinasedephosphorylation by protein phosphatases, Ca2+-dependentdepolymerization of cytoskeletal elements controlling channfunction, receptor removal/insertion into the membrane aexpression of new receptor subtypes. The effects of extracellular ionic milieu (changes in [Ca2+], [Mg2+] and pH)must also be important.
Regulated decreases in the activity of key ion channels

1151Adaptations of neurons to hypoxia and anoxia
ndpal
nt
cts
by
c
g
Oxygen present
No oxygen
Na , Ca2+
Na , Ca2+
Na, Ca2+
Ca2+
Ca2+
Ca2+
Ca2+
K+Na , Ca2+
K+
K+
K+
−70 mmol l−1−70 mV
−70 mV
2nd messengers
2nd messengers
ADP
ADP
ADP
ADP
ATP
ATP
gluc
gluc
gluc
gluc
CO2
Turtle
Mammal
CO2
Ion leaks
Ion leaks
Ion leaks
Ion leaks
Ion pumpsIon pumps
Ion pumpsIon pumps
++
+
0 mV
Lactate
ATPLactate
ATP
Fig. 14. Summary of the differencesbetween hypoxia-tolerant (turtle) andhypoxia-sensitive (mammalian)neurons. Relative concentrations ofions, substrates and rates of reactionsare indicated by the size of thearrows and lettering. In mammals,ion pumping and ATP turnover ratesduring normoxia are greater than inturtles and ion pumping rates are notdecreased during anoxia, depletingATP, depolarizing the membrane andincreasing intracellular [Ca2+].Second messengers are not effectivein preventing catastrophic ATP loss,[Ca2+] elevation and cellswelling/membrane damage. Incontrast, lower rates of ATPdepletion allow turtle neurons toactivate a cascade of secondmessengers to inactivate ion pumps,reduce energy turnover and maintainrelatively stable ATP and Ca2+
levels. gluc, glucose. Membranepotentials are also given.
common to hypoxia-tolerant neurons from both mammals areptiles, and thus these processes may represent fundamcellular adaptations to stresses that may decrease enavailability. The key differences between hypoxia-sensitimammalian and hypoxia-tolerant reptilian neurons may notthe mechanism, but the time required: the lower metabolic of hypoxia-tolerant neurons allows for the expression ocoordinated suite of processes before catastrophic enfailure occurs.
ReferencesADOLPH, E. (1948). Tolerance to cold and anoxia in infant rats. Am.
J. Physiol. 155, 366–377.ASCHER, P. AND NOWAK, L. (1987). Electrophysiological studies o
NMDA receptors. Trends neurol. Sci.10, 284–288.BARTRUP, J. T. AND STONE, T. W. (1990). Activation of NMDA
receptor-coupled channels suppresses the inhibitory actionadenosine on hippocampal slices. Brain Res. 530, 330–334.
BICKLER, P. E. (1992). Cerebral anoxia tolerance in turtles: regulatof intracellular calcium and pH. Am. J. Physiol. 263,R1298–R1302.
BICKLER, P. E., GALLEGO, S. M. AND HANSEN, B. M. (1993).Developmental changes in intracellular calcium regulation in cerebral cortex during hypoxia. J. cereb. Blood Flow Metab.13,811–819.
BICKLER, P. E. AND HANSEN, B. M. (1994). Causes of calciumaccumulation in rat cortical brain slices during hypoxia a
ndentalergyve beratef aergy
f
of
ion
rat
nd
ischemia: role of ion channels and membrane damage. Brain Res.665, 269–276.
BICKLER, P. AND HANSEN, B. (1997). Hypoxia-tolerant neonatal CA1neurons: Relationship of survival to evoked glutamate release aglutamate receptor-mediated calcium changes in hippocamslices. Dev. Brain Res. (in press).
BUCK, L. AND BICKLER, P. (1995). Role of adenosine in NMDAreceptor modulation in the cerebral cortex of an anoxia-toleraturtle (Chrysemys picta belli). J. exp. Biol.198, 1621–1628.
BUCK, L. AND BICKLER, P. (1998). Adenosine and anoxia reduce N-methyl-D-aspartate receptor open probability in turtlecerebrocortex. J. exp. Biol.201, 289–297.
BURKE, S. P. AND NADLER, J. V. (1988). Regulation of glutamate andaspartate release from slices of the hippocampal CA1 area: effeof adenosine and baclofen. J. Neurochem.51, 1541–1551.
CERNE, R., RUSIN, K. I. AND RANDIC, M. (1993). Enhancement of theN-methyl-D-aspartate response in spinal dorsal horn neurons cyclic AMP-dependent protein kinase. Neurosci. Lett.161, 124–128.
CHIH, C., ROSENTHAL, M. AND SICK, T. (1989). Ion leakage is reducedduring anoxia in turtle brain: a potential survival strategy. Am. J.Physiol.257, R1562–R1564.
CHOI, D. W. (1992). Excitotoxic cell death.J. Neurobiol. 23,1261–1276.
CHOI, D. W. (1995). Calcium: still center stage in hypoxic–ischemineuronal death. Trends Neurosci.18, 58–60.
CSERR, H., DEPASQUALE, M. AND JACKSON, D. (1988). Brain andcerebrospinal fluid composition after long-term anoxia in divinturtles. Am. J. Physiol.255, R338–R343.
DE MENDONCA, A. AND RIBIERO, J. A. (1993). Adenosine inhibits

1152
in
ns.
e:In
t),
n
in
ea
t
ic
t),
al
te
.
lt)
P. E. BICKLER AND L. T. BUCK
NMDA receptor-mediated excitatory postsynaptic potential in thippocampus. Brain Res.606, 351–356.
DE MENDONCA, A., SEBASTIAO, A. M. AND RIBIERO, J. A. (1995).Inhibition of NMDA-mediated currents in isolated rat hippocampneurones by adenosine A1 receptor activation. Neuroreport 6,1097–1100.
DUFFY, T. E., KOHLE, S. J. AND VANNUCCI, R. C. (1975). Carbohydrateand energy metabolism in perinatal rat brain: relation to survivaanoxia. J. Neurochem.24, 271–276.
DUNWIDDIE, T. V. (1985). The physiological role of adenosine in thcentral nervous system. Int. Rev. Biochem.27, 63–139.
EDWARDS, R., LUTZ, P. AND BADEN, D. (1989). Relationship betweenenergy expenditure and ion channel function in the rat and tubrain. Am. J. Physiol.255, R1345–R1359.
ELSE, P. (1991). Oxygen consumption and sodium pumthermogenesis in a developing mammal. Am. J. Physiol.261,R1575–R1578.
FAZEKAS, J., ALEXANDER, F. AND HIMWICH, H. (1941). Tolerance ofthe newborn to anoxia. Am. J. Physiol.134, 281–287.
FROHNWEISER, B., WEIGL, L. AND SCHREIBMAYER, W. (1995).Modulation of cardiac sodium channel isoform by cyclic-AMPdependent protein kinase does not depend on phosphorylatioserine-1504 in the cytosolic loop interconnecting transmembradomain-III and domain-IV. Pflügers arch. 430, 751–753.
GERBER, U., GREENE, R. W., HAAS, H. L. AND STEVENS, D. R. (1989).Characterization of inhibition mediated by adenosine in thippocampus of the rat in vitro. J. Physiol., Lond.417, 567–578.
HANSEN, A. (1985). Extracellular potassium concentration in juvenand adult brain cortex during anoxia. Acta physiol. scand.99,412–428.
HOCHACHKA, P. (1986). Defense strategies against hypoxia ahypothermia. Science231, 234–241.
HOCHACHKA, P., BUCK, L., DOLL, C. AND LAND, S. (1996). Unifyingtheory of hypoxia tolerance: Molecular/metabolic defense arescue mechanisms for surviving oxygen lack. Proc. natn. Acad.Sci. U.S.A.93, 9493–9498.
JIANG, C., XIA, Y. AND HADDAD, G. (1992). Role of ATP-sensitive K+
channels during anoxia: major differences between rat (newband adult) and turtle neurons. J. Physiol., Lond.448, 599–612.
JOHANSSON, D. AND NILSSON, G. (1995). Roles of energy status, KATP
channels and channel arrest in fish brain K+ gradient dissipationduring anoxia. J. exp. Biol.198, 2575–2580.
KORNAU, H. C., SCHENKER, L. T., KENNEDY, M. B. AND SEEBURG, P. H.(1995). Domain interaction between NMDA receptor subunits athe postsynaptic density protein PSD-95. Science269, 1737–1740.
LIEBERMAN, D. N. AND MODY, I. (1994). Regulation of NMDAreceptor function by endogenous Ca2+-dependent phosphateNature369, 235–239.
LIPTON, P. AND LOEBNER, D. (1990). Mechanisms of intracellularcalcium accumulation in the CA1 region of rat hippocampus duranoxia in vivo. Stroke21, 60–64.
LUTZ, P. (1992). Mechanisms for anoxic survival in the vertebrabrain. A. Rev. Physiol.54, 601–618.
LUTZ, P. AND NILSSON, G. (1997). Contrasting strategies for anoxbrain survival – glycolysis up or down. J. exp. Biol. 200, 411–419.
MOGUL, D. J., ADAMS, M. E. AND FOX, A. P. (1993). Differentialactivation of adenosine receptors decreases N-type but potentP-type current in hippocampal CA3 neurons. Neuron10, 327–334.
MURPHY, B., ROSSIE, S., DEJONGH, K. AND CATTERALL, W. (1993).Identification of the sites of selective phosphorylation adephosphorylation of the rat brain Na+ channel alpha subunit by
he
al
l in
e
rtle
p
-n ofne
he
ile
nd
nd
orn
nd
.
ing
te
ic
iates
nd
cyclic AMP dependent protein kinase and phosphoprotephosphatases. J. biol. Chem.268, 27355–27362.
MYNLIEFF, M. AND BEAM, K. G. (1994). Adenosine acting at an A1receptor decreases N-type calcium current in mouse motoneuroJ. Neurosci.14, 3628–3634.
NILSSON, G. (1993). Neurotransmitters and anoxia resistancComparative physiological and evolutionary perspectives. Surviving Hypoxia: Mechanisms of Control and Adaptation(ed. P.Hochachka, P. Lutz, T. Sick, M. Rosenthal and G. van den Thillarpp. 401–413. Boca Raton, FL: CRC Press.
NILSSON, G. AND LUTZ, P. (1992). Adenosine release in anoxic turtlebrain as a mechanism for anoxic survival. J. exp. Biol.162, 345–351.
PEK, M. AND LUTZ, P. (1997). Role for adenosine in channel arrest ithe anoxic turtle brain. J. exp. Biol.200, 1913–1917.
PEREZ-PINZON, M., LUTZ, P., SICK, T. AND ROSENTHAL, M. (1993).Adenosine, a ‘retaliatory’ metabolite, promotes anoxia tolerance turtle brain. J. cereb. Blood Flow Metab.13, 728–732.
PEREZ-PINZON, M., ROSENTHAL, M., SICK, T., LUTZ, P., PABLO, J. AND
MASH, D. (1992). Down-regulation of sodium channels duringanoxia: a putative survival strategy of turtle brain. Am. J. Physiol.262, R712–R715.
REYNOLDS, J. AND BREIN, J. (1992). Ontogeny of glutamate andgamma-aminobutyric acid release in the hippocampus of the guinpig. J. dev. Physiol.18, 243–252.
RUDOLPHI, K. A., SCHUBERT, P., PARKINSON, F. E. AND FREDHOLM, B.B. (1992). Adenosine and brain ischemia. Cerebrovasc. BrainMetab. Rev.4, 346–369.
SCHOLZ, K. P. AND MILLER, R. J. (1991). Analysis of adenosine actionson Ca2+ currents and synaptic transmission in cultured rahippocampal pyramidal neurons. J. Physiol., Lond.435, 373–393.
SICK, T., PEREZ-PINZON, M., LUTZ, P. AND ROSENTHAL, M. (1993).Maintaining coupled metabolism and membrane function in anoxbrain: A comparison between the turtle and the rat. In SurvivingHypoxia: Mechanisms of Control and Adaptation(ed. P.Hochachka, P. Lutz, T. Sick, M. Rosenthal and G. van den Thillarpp. 351–364. Boca Raton, FL: CRC Press.
SIESJÖ, B. K. (1992). Pathophysiology and treatment of focal cerebrischemia. II. Mechanisms of damage and treatment. J. Neurosurg.77, 337–354.
SZATKOWSKI, M. AND ATTWELL, D. (1994). Triggering and executionof neuronal death in brain ischemia: two phases of glutamarelease by different mechanisms. Trends Neurosci. 17, 359–365.
TINGLEY, W., ROCHE, K. W., THOMPSON, A. K. AND HUGANIR, R. L.(1993). Regulation of NMDA receptor phosphorylation byalternative splicing of the C-terminal domain. Nature364, 70–73.
ULTSCH, G. AND JACKSON, D. (1982). Long-term submergence at 3 °Cof the turtle Chrysemys picta belli, in normoxic and severelyhypoxic water. I. Survival, gas exchange and acid–base statusJ.exp. Biol.96, 11–28.
WANG, Y. T. AND SALTER, M. W. (1994). Regulation of NMDAreceptors by tyrosine kinases and phosphatases. Nature 369,233–235.
WU, L. AND SAGGAU, P. (1994). Adenosine inhibits evoked synaptictransmission primarily by reducing presynaptic calcium influx inarea CA1 of hippocampus. Neuron12, 1139–1148.
XIA, Y. AND HADDAD, G. (1991). Major differences in CNSsulfonylurea receptor distribution between the rat (newborn, aduand turtle. J. comp. Neurol.314, 278–289.
YAWO, H. AND CHUHMA, N. (1993). Preferential inhibition of omega-conotoxin sensitive presynaptic Ca2+ channels by adenosineautoreceptors. Nature365, 256–258.





























