Embed Size (px)
Citation preview

IMPORTANT CLINICAL TRIALS IN CARDIOLOGY – 2016-17
1

HEART FAILURE
2

3August 28,2016

METHODS
• In a randomized, controlled trial, 556 patients with symptomatic systolic heart failure (left ventricular ejection fraction, ≤35%) not caused by coronary artery disease were assigned to receive an ICD, and 560 patients were assigned to receive usual clinical care (control group).
• In both groups, 58% of the patients received CRT. The primary outcome of the trial was death from any cause. The secondary outcomes were sudden cardiac death and cardiovascular death.
4

RESULTS
• After a median follow-up period of 67.6 months, the primary outcome had occurred in 120 patients (21.6%) in the ICD group and in 131 patients (23.4%) in the control group (hazard ratio, 0.87; 95% [CI], 0.68 to 1.12; P = 0.28).
• Sudden cardiac death occurred in 24 patients (4.3%) in the ICD group and in 46 patients (8.2%) in the control group (hazard ratio, 0.50; 95% CI, 0.31 to 0.82; P = 0.005).
• Device infection occurred in 27 patients (4.9%) in the ICD group and in 20 patients (3.6%) in the control group (P = 0.29).
5

6The median follow up period was 67.6 months

CONCLUSION
• In this trial, prophylactic ICD implantation in patients with symptomatic systolic heart failure not caused by coronary artery disease was not associated with a significantly lower long-term rate of death from any cause than was usual clinical care.
7

8April 12,2017

METHODS
• In this double-blind trial, investigators randomly assigned 2157 patients with acute heart failure to receive a continuous intravenous infusion of either ularitide at a dose of 15 ng per kilogram of body weight per minute or matching placebo for 48 hours, in addition to accepted therapy.
• Treatment was initiated a median of 6 hours after the initial clinical evaluation.
• The coprimary outcomes were death from cardiovascular causes during a median follow-up of 15 months and a hierarchical composite end point that evaluated the initial 48-hour clinical course.
9

RESULTS
• Death from cardiovascular causes occurred in 236 patients in the ularitide group and 225 patients in the placebo group (21.7% vs. 21.0%; hazard ratio, 1.03; 96% confidence interval, 0.85 to 1.25; P = 0.75).
• In the intention-to-treat analysis, there was no significant between-group difference with respect to the hierarchical composite outcome.
• The ularitide group had greater reductions in systolic blood pressure and in levels of N-terminal pro–brain natriuretic peptide than the placebo group.
• However, changes in cardiac troponin T levels during the infusion did not differ between the two groups in the 55% of patients with paired data. 10

11

12
The mean decrease in SBP in the ularitide group was greater by 6.8 mm Hg at 6 hours and by 3.9 mm Hg at 48 hours than in the placebo
group (P<0.001 for both comparisons); these between-group differences dissipated over a period of 72 to 120 hours.

CONCLUSIONS
• In patients with acute heart failure, ularitide exerted favorable physiological effects (without affecting cardiac troponin levels), but short-term treatment did not affect a clinical composite end point or reduce long-term cardiovascular mortality.
13

14
July 12,2017

METHODS
• To assess the effect of high-dose spironolactone(100mg) and usual care(25mg) on N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels compared with usual care alone.
• This double-blind and placebo (or low-dose)-controlled randomized clinical trial was conducted in 22 US acute care hospitals among patients with AHF who were previously receiving no or low-dose (12.5mg or 25mg daily) spironolactone and had NT-proBNP levels of 1000 pg/mL or more or B-type natriuretic peptide levels of 250 pg/mL or more, regardless of ejection fraction.
.15

RESULTS
• A total of 360 patients were randomized, of whom the median age was 65 years, 129 (36%) were women, 200 (55.5%) were white, 151 (42%) were black, 8 (2%) were Hispanic or Latino, 9 (2.5%) were of other race/ethnicity, and the median left ventricular ejection fraction was 34%.
• Baseline median (interquartile range) NT-proBNP levels were 4601 (2697-9596) pg/mL among the group treated with high-dose spironolactone and 3753 (1968-7633) pg/mL among the group who received usual care.
16

• There was no significant difference in the log NT-proBNP reduction between the 2 groups (−0.55 [95%CI, −0.92 to −0.18] with high-dose spironolactone and −0.49 [95%CI, −0.98 to −0.14] with usual care, P = .57). None of the secondary end point or day-30 all-cause mortality or heart failure hospitalization rate differed between the 2 groups.
• The changes in serum potassium and estimated glomerular filtration rate at 24, 48, 72, and 96 hours were similar between the 2 groups.
17

18

19

CONCLUSIONS
• Adding treatment with high-dose spironolactone to usual care for patients with AHF for 96 hours was well tolerated but did not improve the primary or secondary efficacy end points.
20

INTERVENTIONS
21

22DECEMBER 8,2016

• Investigators randomly assigned 1905 eligible patients with left main coronary artery disease of low or intermediate anatomical complexity to undergo either percutaneous coronary intervention (PCI) with fluoropolymer-based cobalt–chromium everolimus-eluting stents (PCI group, 948 patients) or CABG (CABG group, 957 patients).
• Anatomic complexity was assessed at the sites and defined by a Synergy between Percutaneous Coronary Intervention with Taxus and Cardiac Surgery (SYNTAX) score of 32 or lower.
23

• The primary end point was the rate of a composite of death from any cause, stroke, or myocardial infarction at 3 years, and the trial was powered for noninferiority testing of the primary end point (noninferiority margin, 4.2 percentage points).
• Major secondary end points included the rate of a composite of death from any cause, stroke, or myocardial infarction at 30 days and the rate of a composite of death, stroke, myocardial infarction, or ischemia-driven revascularization at 3 years.
• Event rates were based on Kaplan–Meier estimates in time-to-first-event analyses.
24

RESULTS
• At 3 years, a primary end-point event had occurred in 15.4% of the patients in the PCI group and in 14.7% of the patients in the CABG group (difference, 0.7 percentage points; upper 97.5% confidence limit, 4.0 percentage points; P = 0.02 for noninferiority; hazard ratio, 1.00; 95% confidence interval, 0.79 to 1.26; P = 0.98 for superiority).
25

• The secondary end-point event of death, stroke, or myocardial infarction at 30 days occurred in 4.9% of the patients in the PCI group and in 7.9% in the CABG group (P<0.001 for noninferiority, P = 0.008 for superiority).
• The secondary end-point event of death, stroke, myocardial infarction, or ischemia-driven revascularization at 3 years occurred in 23.1% of the patients in the PCI group and in 19.1% in the CABG group (P = 0.01 for noninferiority, P = 0.10 for superiority).
26

27

CONCLUSIONS
• In patients with left main coronary artery disease and low or intermediate SYNTAX scores by site assessment, PCI with everolimus-eluting stents was noninferior to CABG with respect to the rate of the composite end point of death, stroke, or myocardial infarction at 3 years.
28

29OCTOBER 31,2016

METHODS
• In this prospective, randomised, open-label, non-inferiority trial, patients with left main coronary artery disease were enrolled in 36 centres in northern Europe and randomised 1:1 to treatment with PCI or CABG.
• Eligible patients had stable angina pectoris, unstable angina pectoris, or non-ST-elevation myocardial infarction.
• Exclusion criteria were ST-elevation myocardial infarction within 24 h, being considered too high risk for CABG or PCI, or expected survival of less than 1 year.
30

• The primary endpoint was major adverse cardiac or cerebrovascular events (MACCE), a composite of all-cause mortality, non-procedural myocardial infarction, any repeat coronary revascularisation, and stroke.
• Non-inferiority of PCI to CABG required the lower end of the 95% CI not to exceed a hazard ratio (HR) of 1·35 after up to 5 years of follow-up.
31

RESULTS
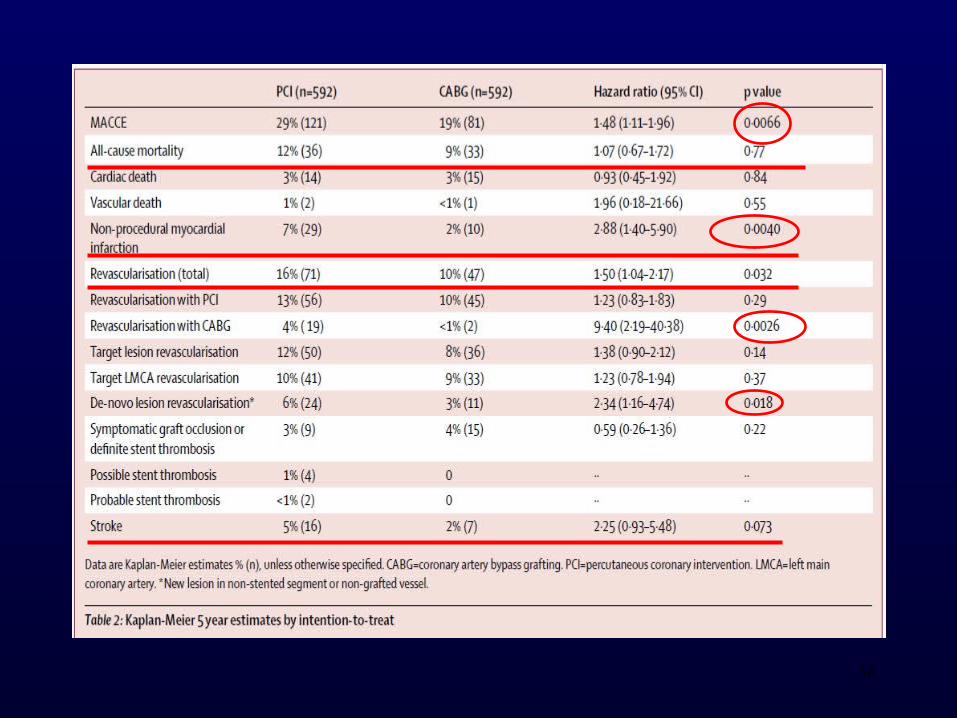
• Between Dec 9, 2008, and Jan 21, 2015, 1201 patients were randomly assigned, 598 to PCI and 603 to CABG, and 592 in each group entered analysis by intention to treat.
• Kaplan-Meier 5 year estimates of MACCE were 29% for PCI (121 events) and 19% for CABG (81 events), HR 1·48 (95% CI 1·11–1·96), exceeding the limit for non-inferiority, and CABG was significantly better than PCI (p=0·0066). As-treated estimates were 28% versus 19% (1·55, 1·18–2·04, p=0·0015).
32

• Comparing PCI with CABG, 5 year estimates were 12% versus 9% (1·07, 0·67–1·72, p=0·77) for all-cause mortality, 7% versus 2% (2·88, 1·40–5·90, p=0·0040) for non-procedural myocardial infarction, 16% versus 10% (1·50, 1·04–2·17, p=0·032) for any revascularisation, and 5% versus 2% (2·25, 0·93–5·48, p=0·073) for stroke.
33
34

CONCLUSIONS
• The findings of this study suggest that CABG might be better than PCI for treatment of left main stem coronary artery disease.
35

36AUGUST 5 , 2015

METHODS
• Investigators undertook an open-label, randomised controlled trial at two university hospitals in Denmark.
• Patients presenting with STEMI who had one or more clinically significant coronary stenosis in addition to the lesion in the infarct-related artery were included.
• After successful percutaneous coronary intervention (PCI) of the infarct-related artery, patients were randomly allocated (in a 1:1 ratio) either no further invasive treatment or complete FFR-guided revascularisation before discharge.
37

• Randomisation was done electronically via a web-based system in permuted blocks of varying size by the clinician who did the primary PCI. All patients received best medical treatment.
• The primary endpoint was a composite of all-cause mortality, non-fatal reinfarction, and ischaemia-driven revascularisation of lesions in non-infarct-related arteries and was assessed when the last enrolled patient had been followed up for 1 year. Analysis was on an intention-to-treat basis.
38

RESULTS
• From March, 2011, to February, 2014, investigators enrolled 627 patients to the trial; 313 were allocated no further invasive treatment after primary PCI of the infarct-related artery only and 314 were assigned complete revascularisation guided by FFR values.
• Median follow-up was 27 months (range 12–44 months). • Events comprising the primary endpoint were recorded in
68 (22%) patients who had PCI of the infarct-related artery only and in 40 (13%) patients who had complete revascularisation (hazard ratio 0∙56, 95% CI 0∙38–0∙83; p=0∙004).
39

40

CONCLUSIONS
• In patients with STEMI and multivessel disease, complete revascularisation guided by FFR measurements significantly reduces the risk of future events compared with no further invasive intervention after primary PCI.
• This effect is driven by significantly fewer repeat revascularisations, because all-cause mortality and non-fatal reinfarction did not differ between groups.
• Thus, to avoid repeat revascularisation, patients can safely have all their lesions treated during the index admission. Future studies should clarify whether complete revascularisation should be done acutely during the index procedure or at later time and whether it has an effect on hard endpoints.
41

42MARCH 18, 2017

METHODS
• Investigators randomly assigned 885 patients with STEMI and multivessel disease who had undergone primary PCI of an infarct-related coronary artery in a 1:2 ratio to undergo complete revascularization of non–infarct-related coronary arteries guided by fractional flow reserve (FFR) (295 patients) or to undergo no revascularization of non– infarct-related coronary arteries (590 patients).
• The FFR procedure was performed in both groups, but in the latter group, both the patients and their cardiologist were unaware of the findings on FFR.
43

• The primary end point was a composite of death from any cause, nonfatal myocardial infarction, revascularization, and cerebrovascular events at 12 months.
• Clinically indicated elective revascularizations performed within 45 days after primary PCI were not counted as events in the group receiving PCI for an infarct-related coronary artery only.
44

RESULTS
• The primary outcome occurred in 23 patients in the complete-revascularization group and in 121 patients in the infarct-artery-only group that did not receive complete revascularization, a finding that translates to 8 and 21 events per 100 patients, respectively (hazard ratio, 0.35; 95% confidence interval [CI], 0.22 to 0.55; P<0.001).
45

• Death occurred in 4 patients in the complete-revascularization group and in 10 patients in the infarct-artery-only group (1.4% vs. 1.7%) (hazard ratio, 0.80; 95% CI, 0.25 to 2.56), myocardial infarction in 7 and 28 patients, respectively (2.4% vs. 4.7%) (hazard ratio, 0.50; 95% CI, 0.22 to 1.13), revascularization in 18 and 103 patients (6.1% vs.17.5%) (hazard ratio, 0.32; 95% CI, 0.20 to 0.54), and cerebrovascular events in 0 and 4 patients (0 vs. 0.7%).
• An FFR-related serious adverse event occurred in 2 patients (both in the group receiving infarct-related treatment only).
46

RESULTS
• In patients with STEMI and multivessel disease who underwent primary PCI of an infarct-related artery, the addition of FFR-guided complete revascularization of non–infarct-related arteries in the acute setting resulted in a risk of a composite cardiovascular outcome that was lower than the risk among those who were treated for the infarct-related artery only.
• This finding was mainly supported by a reduction in subsequent revascularizations.
47

48

CONCLUSIONS
• In patients with STEMI and multivessel disease who underwent primary PCI of an infarct-related artery, the addition of FFR-guided complete revascularization of non– infarct-related arteries in the acute setting resulted in a risk of a composite cardiovascular outcome that was lower than the risk among those who were treated for the infarct-related artery only.
• This finding was mainly supported by a reduction in subsequent revascularizations.
49

50MARCH 29 ,2017

METHODS
• Investigators randomly assigned 1845 patients undergoing PCI to receive either a bioresorbable vascular scaffold (924 patients) or a metallic stent (921 patients).
• The primary end point was target-vessel failure (a composite of cardiac death, target-vessel myocardial infarction, or target-vessel revascularization).
• The data and safety monitoring board recommended early reporting of the study results because of safety concerns. This report provides descriptive information on end-point events.
51

RESULTS
• The median follow-up was 707 days. • Target-vessel failure occurred in 105 patients in the
scaffold group and in 94 patients in the stent group (2-year cumulative event rates, 11.7% and 10.7%, respectively; hazard ratio, 1.12; 95% confidence interval [CI], 0.85 to 1.48; P = 0.43); event rates were based on Kaplan–Meier estimates in time-to-event analyses.
52

a. Cardiac death occurred in 18 patients in the scaffold group and in 23 patients in the stent group (2-year cumulative event rates, 2.0% and 2.7%, respectively),
b. Target-vessel myocardial infarction occurred in 48 patients in the scaffold group and in 30 patients in the stent group (2-year cumulative event rates, 5.5% and 3.2%),
c. Target-vessel revascularization occurred in 76 patients in the scaffold group and in 65 patients in the stent group (2-year cumulative event rates, 8.7% and 7.5%).
53

• Definite or probable device thrombosis occurred in 31 patients in the scaffold group as compared with 8 patients in the stent group (2-year cumulative event rates, 3.5%vs. 0.9%; hazard ratio, 3.87; 95% CI, 1.78 to 8.42; P<0.001).
54

55

CONCLUSIONS
• In this preliminary report of a trial involving patients undergoing PCI, there was no significant difference in the rate of target-vessel failure between the patients who received a bioresorbable scaffold and the patients who received a metallic stent.
• The bioresorbable scaffold was associated with a higher incidence of device thrombosis than the metallic stent through 2 years of follow-up.
56

57AUGUST 30, ,2016

METHODS
• Investigators randomly assigned 9013 patients who had stable or unstable coronary artery disease to undergo percutaneous coronary intervention (PCI) with the implantation of either contemporary drug-eluting stents or bare-metal stents.
• In the group receiving drug-eluting stents, 96% of the patients received either everolimus- or zotarolimus-eluting stents.
• The primary outcome was a composite of death from any cause and nonfatal spontaneous myocardial infarction after a median of 5 years of follow-up.
• Secondary outcomes included repeat revascularization, stent thrombosis ,and quality of life.
58

RESULTS
• At 6 years, the rates of the primary outcome were 16.6% in the group receiving drug-eluting stents and 17.1% in the group receiving bare-metal stents (hazard ratio,0.98; [CI], 0.88 to 1.09; P = 0.66).
• There were no significant between-group differences in the components of the primary outcome.
• The 6-year rates of any repeat revascularization were 16.5% in the group receiving drug-eluting stents and 19.8% in the group receiving bare-metal stents (P<0.001); the rates of definite stent thrombosis were 0.8% and 1.2%, respectively (P = 0.0498).
• Quality-of-life measures did not differ significantly between the two groups.
59

60

• In patients undergoing PCI, there were no significant differences between those receiving drug-eluting stents and those receiving bare-metal stents in the composite outcome of death from any cause and nonfatal spontaneous myocardial infarction.
• Rates of repeat revascularization were lower in the group receiving drug eluting stents.
61

62MARCH 18,2017

METHODS
• Investigators conducted a multicenter, randomized, controlled, open-label clinical trial using the Swedish Coronary Angiography and Angioplasty Registry for enrollment.
• A total of 2037 participants with stable angina or an acute coronary syndrome who had an indication for physiologically guided assessment of coronary-artery stenosis were randomly assigned to undergo revascularization guided by either iFR or FFR.
• The primary end point was the rate of a composite of death from any cause, nonfatal myocardial infarction, or unplanned revascularization within 12 months after the procedure.
63

RESULTS
• A primary end-point event occurred in 68 of 1012 patients (6.7%) in the iFR group and in 61 of 1007 (6.1%) in the FFR group (difference in event rates, 0.7 percentage points; [CI], −1.5 to 2.8%; P = 0.007 for noninferiority; hazard ratio, 1.12; 95% CI, 0.79 to 1.58; P = 0.53); the upper limit of the 95% confidence interval for the difference in event rates fell within the prespecified noninferiority margin of 3.2 percentage points.
64

• The results were similar among major subgroups.• The rates of myocardial infarction, target-lesion
revascularization, restenosis, and stent thrombosis did not differ significantly between the two groups.
• A significantly higher proportion of patients in the FFR group than in the iFR group reported chest discomfort during the procedure.
65

66

CONCLUSIONS
• Among patients with stable angina or an acute coronary syndrome, an iFR-guided revascularization strategy was noninferior to an FFR-guided revascularization strategy with respect to the rate of major adverse cardiac events at 12 months.
67

68
MARCH 17,2017

METHODS
• Investigators evaluated the clinical outcomes in intermediate-risk patients with severe, symptomatic aortic stenosis in a randomized trial comparing TAVR (performed with the use of a self-expanding prosthesis) with surgical aortic-valve replacement.
• The primary end point was a composite of death from any cause or disabling stroke at 24 months in patients undergoing attempted aortic-valve replacement.
• Investigators used Bayesian analytical methods (with a margin of 0.07) to evaluate the noninferiority of TAVR as compared with surgical valve replacement.
69

RESULTS
• A total of 1746 patients underwent randomization at 87 centers. Of these patients, 1660 underwent an attempted TAVR or surgical procedure.
• The mean(±SD) age of the patients was 79.8±6.2 years, and all were at intermediate risk for surgery (Society of Thoracic Surgeons Predicted Risk of Mortality, 4.5±1.6%).
• At 24 months, the estimated incidence of the primary end point was 12.6% in the TAVR group and 14.0% in the surgery group (95% credible interval [Bayesian analysis] for difference, −5.2 to 2.3%; posterior probability of noninferiority, >0.999).
70

• Surgery was associated with higher rates of acute kidney injury, atrial fibrillation, and transfusion requirements, whereas TAVR had higher rates of residual aortic regurgitation and need for pacemaker implantation.
• TAVR resulted in lower mean gradients and larger aortic-valve areas than surgery.
• Structural valve deterioration at 24 months did not occur in either group.
71

72

CONCLUSION
• TAVR was a noninferior alternative to surgery in patients with severe aortic stenosis at intermediate surgical risk, with a different pattern of adverse events associated with each procedure.
73

ORAL ANTICOAGULANTS
74

75MARCH 18,2017

METHODS
• In this randomized, double-blind, phase 3 study, investigators assigned 3396 patients with venous thromboembolism to receive either once-daily rivaroxaban (at doses of 20 mg or 10 mg) or 100 mg of aspirin.
• All the study patients had completed 6 to 12 months of anticoagulation therapy and were in equipoise regarding the need for continued anticoagulation. Study drugs were administered for up to 12 months.
• The primary efficacy outcome was symptomatic recurrent fatal or nonfatal venous thromboembolism, and the principal safety outcome was major bleeding.
76

RESULTS
• A total of 3365 patients were included in the intention-to-treat analyses (median treatment duration, 351 days).
• The primary efficacy outcome occurred in 17 of 1107 patients (1.5%) receiving 20 mg of rivaroxaban and in 13 of 1127 patients (1.2%) receiving 10 mg of rivaroxaban, as compared with 50 of 1131 patients (4.4%) receiving aspirin (hazard ratio for 20 mg of rivaroxaban vs. aspirin, 0.34; [CI], 0.20 to 0.59; hazard ratio for 10 mg of rivaroxaban vs. aspirin, 0.26; 95% CI, 0.14 to 0.47; P<0.001 for both comparisons).
77

• Rates of major bleeding were 0.5% in the group receiving 20 mg of rivaroxaban, 0.4% in the group receiving 10 mg of rivaroxaban, and 0.3% in the aspirin group; the rates of clinically relevant nonmajor bleeding were 2.7%, 2.0%, and 1.8%, respectively.
• The incidence of adverse events was similar in all three groups.
78

79

CONCLUSIONS
• Among patients with venous thromboembolism in equipoise for continued anticoagulation, the risk of a recurrent event was significantly lower with rivaroxaban at either a treatment dose (20 mg) or a prophylactic dose (10 mg) than with aspirin, without a significant increase in bleeding rates.
80

PIONEER AF PCI
81NOVEMBER 14,2016

METHODS
• Investigators randomly assigned 2124 participants with nonvalvular atrial fibrillation who had undergone PCI with stenting to receive, in a 1:1:1 ratio, low-dose rivaroxaban (15 mg once daily) plus a P2Y12 inhibitor for 12 months (group 1), very-low-dose rivaroxaban(2.5 mg twice daily) plus DAPT for 1, 6, or 12 months (group 2), or standard therapy with a dose-adjusted vitamin K antagonist (once daily) plus DAPT for 1, 6, or 12 months (group 3).
• The primary safety outcome was clinically significant bleeding (a composite of major bleeding or minor bleeding according to Thrombolysis in Myocardial Infarction [TIMI] criteria or bleeding requiring medical attention).
82

RESULTS
• The rates of clinically significant bleeding were lower in the two groups receiving rivaroxaban than in the group receiving standard therapy (16.8% in group 1, 18.0% in group 2, and 26.7% in group 3; hazard ratio for group 1 vs. group 3, 0.59; 95% [CI], 0.47 to 0.76; P<0.001; hazard ratio for group 2 vs. group3, 0.63; 95% CI, 0.50 to 0.80; P<0.001).
• The rates of death from cardiovascular causes, myocardial infarction, or stroke were similar in the three groups (Kaplan–Meier estimates, 6.5% in group 1, 5.6% in group 2, and 6.0% in group 3; P values for all comparisons were nonsignificant).
83

84

CONCLUSIONS
• In participants with atrial fibrillation undergoing PCI with placement of stents, the administration of either low-dose rivaroxaban plus a P2Y12 inhibitor for 12 months or very-low-dose rivaroxaban plus DAPT for 1, 6, or 12 months was associated with a lower rate of clinically significant bleeding than was standard therapy with a vitamin K antagonist plus DAPT for 1, 6, or 12 months.
• The three groups had similar efficacy rates, although the observed broad confidence intervals diminish the surety of any conclusions regarding efficacy.
85

86NOVEMBER13,2016

METHODS
• In this double-blind, event-driven trial, investigators randomly assigned 13,885 patients with symptomatic peripheral artery disease to receive monotherapy with ticagrelor (90 mg twice daily) or clopidogrel (75 mg once daily).
• Patients were eligible if they had an ankle–brachial index (ABI) of 0.80 or less or had undergone previous revascularization of the lower limbs.
• The primary efficacy end point was a composite of adjudicated cardiovascular death, myocardial infarction, or ischemic stroke. The primary safety end point was major bleeding. The median follow-up was 30 months.
87

RESULTS• The median age of the patients was 66 years, and 72% were
men; 43% were enrolled on the basis of the ABI and 57% on the basis of previous revascularization.
• The mean baseline ABI in all patients was 0.71, 76.6% of the patients had claudication, and 4.6% had critical limb ischemia.
• The primary efficacy end point occurred in 751 of 6930 patients (10.8%) receiving ticagrelor and in 740 of 6955 (10.6%) receiving clopidogrel (hazard ratio, 1.02; 95%[CI], 0.92 to 1.13; P = 0.65).
• In each group, acute limb ischemia occurred in 1.7% of the patients (hazard ratio, 1.03; 95% CI, 0.79 to 1.33; P = 0.85) and major bleeding in 1.6% (hazard ratio, 1.10; 95% CI, 0.84 to 1.43; P = 0.49). 88

89

CONCLUSIONS
• In patients with symptomatic peripheral artery disease, ticagrelor was not shown to be superior to clopidogrel for the reduction of cardiovascular events.
• Major bleeding occurred at similar rates among the patients in the two trial groups.
90

STATINS
91

92MARCH 17,2017

METHODS
• Investigators conducted a randomized, double-blind, placebo-controlled trial involving 27,564 patients with atherosclerotic cardiovascular disease and LDL cholesterol levels of 70 mg per deciliter (1.8 mmol per liter) or higher who were receiving statin therapy.
• Patients were randomly assigned to receive evolocumab (either 140 mg every 2 weeks or 420 mg monthly) or matching placebo as subcutaneous injections.
93

• The primary efficacy end point was the composite of cardiovascular death, myocardial infarction, stroke, hospitalization for unstable angina, or coronary revascularization.
• The key secondary efficacy end point was the composite of cardiovascular death, myocardial infarction, or stroke.
• The median duration of follow-up was 2.2 years.
94

RESULTS
• At 48 weeks, the least-squares mean percentage reduction in LDL cholesterol levels with evolocumab, as compared with placebo, was 59%, from a median baseline value of 92 mg per deciliter (2.4 mmol per liter) to 30 mg per deciliter (0.78 mmol per liter) (P<0.001).
• Relative to placebo, evolocumab treatment significantly reduced the risk of the primary end point (1344 patients [9.8%] vs. 1563 patients [11.3%]; hazard ratio, 0.85; 95% confidence interval [CI], 0.79 to 0.92; P<0.001) and the key secondary end point (816 [5.9%] vs. 1013 [7.4%]; hazard ratio, 0.80; 95% CI, 0.73 to 0.88; P<0.001).
95

• The results were consistent across key subgroups, including the subgroup of patients in the lowest quartile for baseline LDL cholesterol levels (median, 74 mg per deciliter [1.9 mmol per liter]).
• There was no significant difference between the study groups with regard to adverse events (including new-onset diabetes and neurocognitive events), with the exception of injection-site reactions, which were more common with evolocumab (2.1% vs. 1.6%).
96

97

CONCLUSIONS
• In this trial, inhibition of PCSK9 with evolocumab on a background of statin therapy lowered LDL cholesterol levels to a median of 30 mg per deciliter (0.78 mmol per liter) and reduced the risk of cardiovascular events.
• These findings show that patients with atherosclerotic cardiovascular disease benefit from lowering of LDL cholesterol levels below current targets.
98

ORION 1
99
MARCH 17,2017

METHODS
• Investigators conducted a phase 2, multicenter, double-blind, placebo-controlled, multiple ascending-dose trial of inclisiran administered as a subcutaneous injection in patients at high risk for cardiovascular disease who had elevated LDL cholesterol levels.
• Patients were randomly assigned to receive a single dose of placebo or 200, 300,or 500 mg of inclisiran or two doses (at days 1 and 90) of placebo or 100, 200, or 300 mg of inclisiran.
100

• The primary end point was the change from baseline in LDL cholesterol level at 180 days.
• Safety data were available through day 210, and data on LDL cholesterol and proprotein convertase subtilisin–kexin type 9 (PCSK9) levels were available through day 240.
101

RESULTS
• A total of 501 patients underwent randomization. Patients who received inclisiran had dose-dependent reductions in PCSK9 and LDL cholesterol levels.
• At day 180, the least-squares mean reductions in LDL cholesterol levels were 27.9 to 41.9% after a single dose of inclisiran and 35.5 to 52.6% after two doses (P<0.001 for all comparisons vs. placebo).
• The two-dose 300-mg inclisiran regimen produced the greatest reduction in LDL cholesterol levels: 48% of the patients who received the regimen had an LDL cholesterol level below 50 mg per deciliter (1.3 mmol per liter) at day 180.
102

• At day 240, PCSK9 and LDL cholesterol levels remained significantly lower than at baseline in association with all inclisiran regimens.
• Serious adverse events occurred in 11% of the patients who received inclisiran and in 8% of the patients who received placebo.
• Injection-site reactions occurred in 5% of the patients who received injections of inclisiran.
103

104

CONCLUSIONS
• In this trial, inclisiran was found to lower PCSK9 and LDL cholesterol levels among patients at high cardiovascular risk who had elevated LDL cholesterol levels
105

106MAY 18, 2017

METHODS• In a multicenter, randomized, double-blind, placebo-controlled
phase 3 trial, investigators enrolled 12,092 patients who had at least one of the following conditions: an acute coronary syndrome within the previous 30 to 365 days, cerebrovascular atherosclerotic disease, peripheral vascular arterial disease, or diabetes mellitus with coronary artery disease.
• Patients were randomly assigned to receive either evacetrapib at a dose of 130 mg or matching placebo, administered daily, in addition to standard medical therapy.
• The primary efficacy end point was the first occurrence of any component of the composite of death from cardiovascular causes, myocardial infarction, stroke, coronary revascularization, or hospitalization for unstable angina.
107

RESULTS
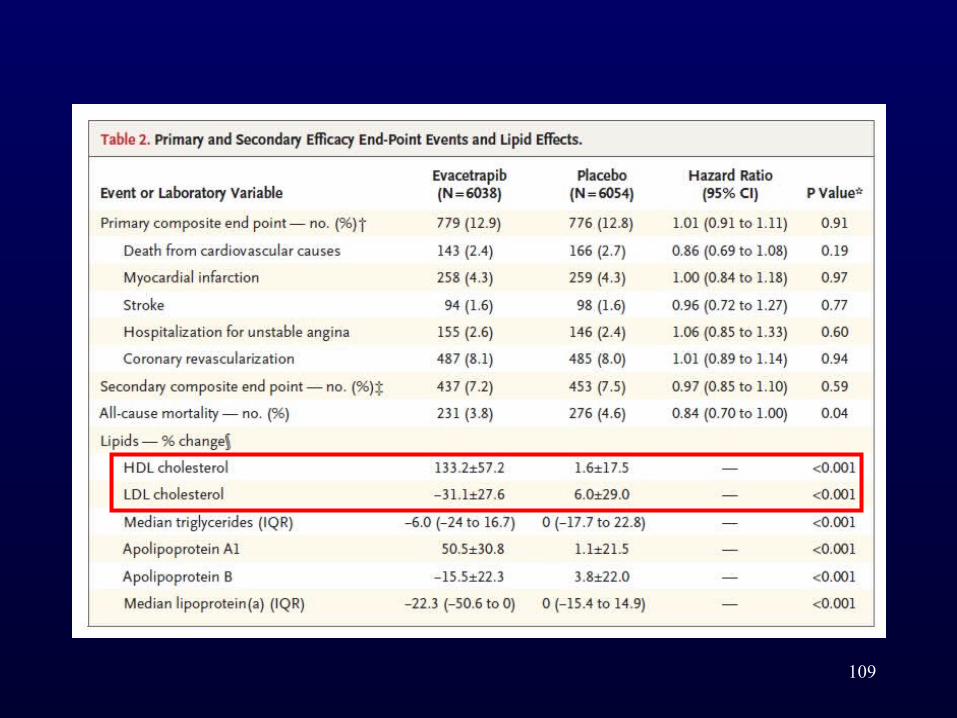
• At 3 months, a 31.1% decrease in the mean LDL cholesterol level was observed with evacetrapib versus a 6.0% increase with placebo, and a 133.2% increase in the mean HDL cholesterol level was seen with evacetrapib versus a 1.6% increase with placebo.
• After 1363 of the planned 1670 primary end-point events had occurred, the data and safety monitoring board recommended that the trial be terminated early because of a lack of efficacy.
• After a median of 26 months of evacetrapib or placebo, a primary end-point event occurred in 12.9% of the patients in the evacetrapib group and in 12.8% of those in the placebo group (hazard ratio, 1.01; 95% CI, 0.91 to 1.11; P = 0.91).
108
109

CONCLUSIONS
• Although the cholesteryl ester transfer protein inhibitor evacetrapib had favorable effects on established lipid biomarkers, treatment with evacetrapib did not result in a lower rate of cardiovascular events than placebo among patients with high-risk vascular disease.
110

HYPERTENSION
111

APRIL 2,2016112

METHODS
• 12,705 participants in 21 countries who did not have cardiovascular disease and were at intermediate risk were randomly assigned to receive candesartan at a dose of 16 mg per day plus hydrochlorothiazide at a dose of 12.5 mg per day or placebo.
• The first coprimary outcome was the composite of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke, and the second coprimary outcome additionally included revascularization, heart failure, and resuscitated cardiac arrest.
• The median follow-up was 5.6 years.113

RESULTS
• The mean blood pressure of the participants at baseline was 138.1/81.9 mm Hg; the decrease in blood pressure was 6.0/3.0 mm Hg greater in the active-treatment group than in the placebo group.
• The first coprimary outcome occurred in 260 participants (4.1%) in the active-treatment group and in 279 (4.4%) in the placebo group (p=0.40);
114

• The second coprimary outcome occurred in 312 participants (4.9%) and 328 participants (5.2%), respectively (p=0.51).
• In one of the three prespecified hypothesis-based subgroups, participants in the subgroup for the upper third of systolic blood pressure (>143.5 mm Hg) who were in the active-treatment group had significantly lower rates of the first and second coprimary outcomes than those in the placebo group; effects were neutral in the middle and lower thirds (P=0.02 and P=0.009, respectively, for trend in the two outcomes).
115

CONCLUSION
• Therapy with candesartan at a dose of 16 mg per day plus hydrochlorothiazide at a dose of 12.5 mg per day was not associated with a lower rate of major cardiovascular events than placebo among persons at intermediate risk who did not have cardiovascular disease.
116

April 2, 2016
117

METHODS
• 12,705 participants in 21 countries who did not have cardiovascular disease and were at intermediate risk were randomly assigned to receive rosuvastatin at a dose of 10 mg per day or placebo.
• The first coprimary outcome was the composite of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke, and the second coprimary outcome additionally included revascularization, heart failure, and resuscitated cardiac arrest.
• The median follow-up was 5.6 years.
118

RESULTS
• The overall mean LDL level was 26.5% lower in the rosuvastatin group than in the placebo group.
• The first coprimary outcome occurred in 235 participants (3.7%) in the rosuvastatin group and in 304 participants (4.8%) in the placebo group (p=0.002).
• The results for the second coprimary outcome were consistent with the results for the first (occurring in 277 participants [4.4%] in the rosuvastatin group and in 363 participants [5.7%] in the placebo group; P<0.001).
119

• The results were also consistent in subgroups defined according to cardiovascular risk at baseline, lipid level, C-reactive protein level, blood pressure, and race or ethnic group.
• In the rosuvastatin group, there was no excess of diabetes or cancers, but there was an excess of cataract surgery (in 3.8% of the participants, vs. 3.1% in the placebo group; P=0.02) and muscle symptoms (in 5.8% of the participants, vs. 4.7% in the placebo group; P=0.005).
120

CONCLUSIONS
• Treatment with rosuvastatin at a dose of 10 mg per day resulted in a significantly lower risk of cardiovascular events than placebo in an intermediate-risk, ethnically diverse population without cardiovascular disease.
121

April 2, 2016
122

METHODS
• 12,705 participants at intermediate risk who did not have cardiovascular disease were randomly assigned to rosuvastatin (10 mg per day) or placebo and to candesartan (16 mg per day) plus hydrochlorothiazide (12.5 mg per day) or placebo. In the analyses reported here, the 3180 participants assigned to combined therapy (with rosuvastatin and the two antihypertensive agents) with the 3168 participants assigned to dual placebo were compared.
• The first coprimary outcome was the composite of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke, and the second coprimary outcome additionally included revascularization, heart failure, and resuscitated cardiac arrest.
• The median follow-up was 5.6 years. 123

RESULTS• The decrease in the LDL cholesterol level was 33.7 mg per
deciliter (0.87 mmol per liter) greater in the combined-therapy group than in the dual-placebo group, and the decrease in systolic blood pressure was 6.2 mm Hg greater with combined therapy than with dual placebo.
• The first coprimary outcome occurred in 113 participants (3.6%) in the combined-therapy group and in 157 (5.0%) in the dual-placebo group (p=0.005).
• The second coprimary outcome occurred in 136 participants (4.3%) and 187 participants (5.9%), respectively (p=0.003).
• Muscle weakness and dizziness were more common in the combined-therapy group than in the dual-placebo group, but the overall rate of discontinuation of the trial regimen was similar in the two groups. 124

CONCLUSION
• The combination of rosuvastatin (10 mg per day), candesartan (16 mg per day), and hydrochlorothiazide (12.5 mg per day) was associated with a significantly lower rate of cardiovascular events than dual placebo among persons at intermediate risk who did not have cardiovascular disease.
125

ARRHYTHMIAS
126

MARCH 19, 2017127

METHODS• In this randomized, open-label, multicenter, controlled trial
with blinded adjudicated end-point assessments, investigators randomly assigned patients scheduled for catheter ablation of paroxysmal or persistent atrial fibrillation to receive either dabigatran (150 mg twice daily) or warfarin (target international normalized ratio, 2.0 to 3.0).
• Ablation was performed after 4 to 8 weeks of uninterrupted anticoagulation, which was continued during and for 8 weeks after ablation.
• The primary end point was the incidence of major bleeding events during and up to 8 weeks after ablation; secondary end points included thromboembolic and other bleeding events.
128

RESULTS• The trial enrolled 704 patients across 104 sites; 635 patients
underwent ablation.• Baseline characteristics were balanced between treatment
groups.
• The incidence of major bleeding events during and up to 8 weeks after ablation was lower with dabigatran than with warfarin (5 patients [1.6%] vs. 22 patients [6.9%]; absolute risk difference, −5.3 percentage points; 95% confidence interval, −8.4 to −2.2; P<0.001).
• Dabigatran was associated with fewer periprocedural pericardial tamponades and groin hematomas than warfarin.
• The two treatment groups had a similar incidence of minor bleeding events. One thromboembolic event occurred in the warfarin group. 129

130

CONCLUSIONS
• In patients undergoing ablation for atrial fibrillation, anticoagulation with uninterrupted dabigatran was associated with fewer bleeding complications than uninterrupted warfarin.
131

13228 JUNE 2016

METHODS AND RESULTS
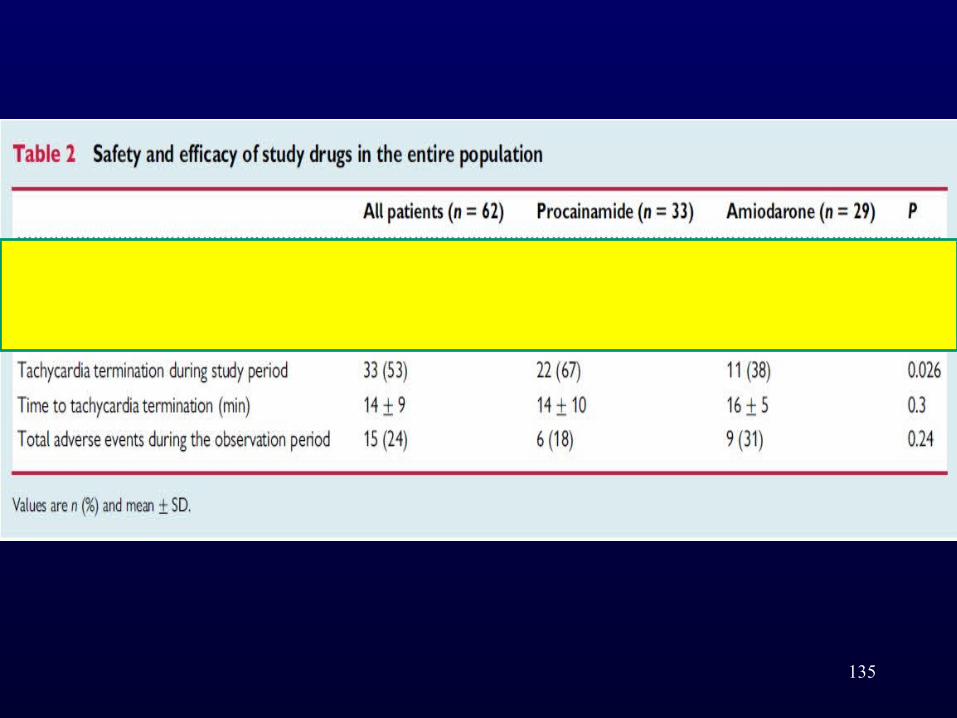
• Patients were randomly assigned to receive intravenous procainamide (10 mg/kg/20 min)or amiodarone (5mg/kg/20min).
• The primary endpoint was the incidence of major predefined cardiac adverse events within 40 min after infusion initiation.
• Of 74 patients included, 62 could be analysed.• The primary endpoint occurred in 3 of 33 (9%)
procainamide and 12 of 29 (41%) amiodarone patients (odd ratio, OR ¼ 0.1; 95% confidence interval, CI 0.03–0.6; P ¼ 0.006).
133

• Tachycardia terminated within 40 min in 22 (67%) procainamide and 11 (38%) amiodarone patients (OR ¼ 3.3; 95% CI 1.2–9.3; P ¼ 0.026).
• In the following 24 h, adverse events occurred in 18% procainamide and 31% amiodarone patients (OR: 0.49; 95% CI: 0.15–1.61; P: 0.24).
• Among 49 patients with structural heart disease, the primary endpoint was less common in procainamide patients (3 [11%] vs. 10 [43%]; OR: 0.17; 95% CI: 0.04–0.73, P ¼ 0.017)
134
135

136

CONCLUSIONS
• This study compares for the first time in a randomized design intravenous procainamide and amiodarone for the treatment of the acute episode of sustained monomorphic well-tolerated (probably) ventricular tachycardia.
• Procainamide therapy was associated with less major cardiac adverse events and a higher proportion of tachycardia termination within 40 min.
137

138

METHODS
• 9361 persons with a systolic blood pressure of 130 mm Hg or higher and an increased cardiovascular risk, but without diabetes, were randomly assigned to a SBP target of less than 120 mm Hg (intensive treatment) or a target of less than 140 mm Hg (standard treatment).
• The primary composite outcome was myocardial infarction, other acute coronary syndromes, stroke, heart failure, or death from cardiovascular causes.
139

RESULTS At 1 year, the mean SBP was 121.4 mm Hg in the intensive
treatment group and 136.2 mm Hg in the standard-treatment group.
The intervention was stopped early after a median follow-up of 3.26 years owing to a significantly lower rate of the primary composite outcome in the intensive-treatment group than in the standard-treatment group (1.65% per year vs. 2.19% per year; P<0.001).
All-cause mortality was also significantly lower in the intensive treatment group (P = 0.003).
Rates of serious adverse events of hypotension, syncope, electrolyte abnormalities, and acute kidney injury or failure, but not of injurious falls, were higher in the intensive treatment group than in the standard-treatment group. 140

141

• Rates of serious adverse events of hypotension, syncope, electrolyte abnormalities, and acute kidney injury or failure, but not of injurious falls, were higher in the intensive treatment group than in the standard-treatment group.
142

CONCLUSION
• Among patients at high risk for cardiovascular events but without diabetes, targeting a systolic blood pressure of less than 120 mm Hg, as compared with less than 140 mm Hg, resulted in lower rates of fatal and nonfatal major cardiovascular events and death from any cause, although significantly higher rates of some adverse events were observed in the intensive-treatment group
143





























