Embed Size (px)
Citation preview

University of Groningen
Neuroprotective signaling mechanisms in the mammalian brainDolga, Amalia Mihaela
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2008
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Dolga, A. M. (2008). Neuroprotective signaling mechanisms in the mammalian brain. s.n.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 21-04-2020
CHAPTER 1
Introduction

2 Introduction
1.1 Neurodegeneration
Neurodegeneration is defined as progressive loss of neuronal structure and func-tion that ultimately leads to neuronal death. Neurodegeneration occurs in variousdiseases affecting the central nervous system. The loss of specific populations of neu-rons related to functional neuronal networks determines the clinical presentation ofthe neurodegenerative disease. For example, degeneration of neurons located in thefrontal lobes and caudate nucleus/striatum of the basal ganglia are associated withHuntington’s disease, although the loss of neuronal function in the substantia nigraand striatum is related to Parkinson’s disease.
Because of the social and financial impact of these diseases to modern westernsocieties, the development of affordable and effective therapies to prevent and protectagainst neurodegenerative diseases is of great interest. However, the prospects ofadequate treatment of brain diseases are still very limited in spite of the impressiveincrease of neuroscience research during the last four decades. Due to the high levelof complexity of brain function and dysfunction, the progress in development of newtreatments and safe drugs is still impeded by insufficient knowledge of the causes andthe mechanisms by which neurons die in neurodegenerative disorders.
Classification of neurodegenerative disorders has been a matter of serious dis-pute for a long time since many disorders overlap with one another in their clini-cal representation and their neuropathological characteristics. Traditionally, diseasesof the brain were categorized based on the main clinical feature or the anatomicaldistribution of the predominant lesion. According to the anatomical regions, neu-rodegenerative disorders include roughly diseases of the cerebral cortex, the basalganglia, the brainstem, the cerebellum or the spinal cord (Dickson, 2003). Withineach group a further classification was made, based on particular clinical features.The cerebral cortex diseases were subdivided into dementing (e.g. Alzheimer’s dis-ease (AD)) and non-dementing illnesses. However, dementia is not specific to ADonly. It can accompany a diversity of conditions besides neurodegenerative disorderssuch as metabolic or infectious brain diseases. More recent classifications tend tobe based not on anatomical dysfunctions but on common molecular defects. Themajor group of molecules implicated in neurodegenerative processes includes amyloid(Alzheimer’s disease (AD), some forms of Creutzfeldt-Jacob disease); tau (Alzheimer’sdisease, Frontotemporal-17 dementia, Parkinson’s disease, Pick’s disease, ProgressiveSupranuclear Palsy), a-synuclein (Parkinson’s disease, Multiple System Atrophy de-mentia, Diffuse Lewy Body dementia and some forms of AD), trinucleotide-repeatsequence (Huntington disease, Spino-Cerebellar Atrophy, Myotonic Dystrophy) andprions (Creutzfeldt-Jacob disease, Fatal Familial Insomnia, Gerstmann-Straussler-Scheinker, Kuru, Scrapie) (Dickson, 2003).
Given that regenerative capacities are rather limited in the adult central nervoussystem neuronal death represents a catastrophic event during neurodegenerative pro-cesses. Neuronal cell death can be roughly divided into necrotic and non-necrotictypes. Necrotic cell death is a fast process characterized by cell swelling that requires

1.1 Neurodegeneration 3
no active contribution of the degenerative cell. In contrast, non-necrotic cell deathis tightly regulated by autonomous processes, requires the engagements of active cel-lular processes and ultimately induces distinctive ultrastructural alterations. Thiscell death type can be further divided into apoptotic and autophagic type (Clarke,1990). The hallmark features of apoptosis include chromatin condensation, nuclearfragmentation, margination and cytoplasmic blebbing (Clarke, 1999). Apoptosis isthe most common and well-investigated form of programmed cell death. It is impli-cated in several neurodegenerative disorders and the main regulators include Bcl-2and caspase families (Korsmeyer, 1999; Wellington and Hayden, 2000).
1.1.1 Alzheimer’s disease
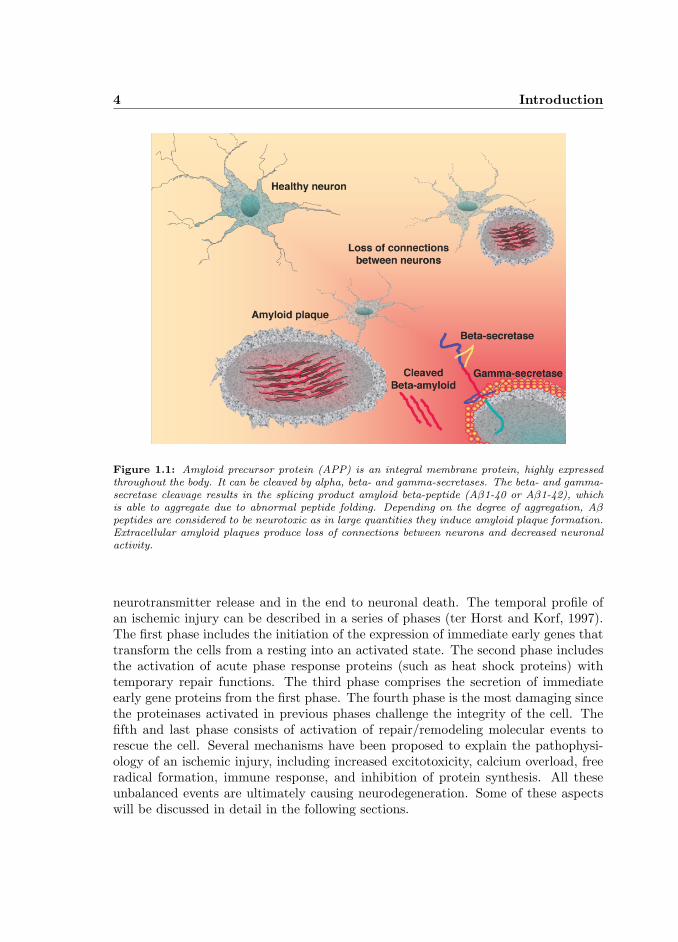
AD represents the most common cause of dementia. Behavioral abnormalities fol-lowed by impairments in language proficiency, sensory perceptions and motor skillsoften occur during the progression of AD. The disease includes sporadic and familialforms with both “early-onset” (less than 65 years of age) and “late-onset” (over 65years of age) forms. The diagnosis is rather difficult since the clinical features of ADoverlap with the symptoms of various other neuropathological conditions. In addi-tion, a definite confirmation of AD is achieved only by morphological and histologicalexamination of the brain at autopsy (Jellinger, 1998). AD pathology is characterizedby an accumulation of “senile plaques” and “neurofibrillary tangles” in brain regionsinvolved in learning and memory processes and degeneration of basal forebrain cellgroups. Senile plaques are extracellular deposits of fibrils and amorphous or diffuseaggregates of amyloid β-peptide (Aβ). Amyloid precursor protein (APP) is an in-tegral membrane protein, highly expressed throughout the body. In AD, APP isabnormally cleaved by several secretases and this results in formation of the insol-uble Aβ peptides (Fig. 1.1). Neurofibrillary tangles are intracellular accumulationsof hyperphosphorylated microtubule-associated protein tau. As far as we currentlyknow, tangle formation is for a large part the final result of amyloid-induced nerve celldegeneration. Another hallmark of AD is the degeneration of synapses and the deathof specific groups of neurons. In particular cholinergic and glutamatergic neurons areaffected but even those producing norepinephirine or serotonin have been observed todegenerate (Dickson, 2003).
1.1.2 Stroke/ischemia
Cerebrovascular accident (CVA) is a clinical definition used to describe the symp-toms of a perturbation in the cerebral blood supply. Decreased or interrupted bloodsupply has several consequences. The major one is the strong reduction in glucoseand oxygen availability in the territory of the affected vascular brain areas, a phe-nomenon designated as cerebral ischemia. Ischemia leads, within seconds or minutes,to a cellular energy crisis, initiation of anaerobic glycolysis, disruption in the activ-ity of cellular pumps, increase in intracellular calcium and extracellular potassium,
4 Introduction
Figure 1.1: Amyloid precursor protein (APP) is an integral membrane protein, highly expressedthroughout the body. It can be cleaved by alpha, beta- and gamma-secretases. The beta- and gamma-secretase cleavage results in the splicing product amyloid beta-peptide (Aβ1-40 or Aβ1-42), whichis able to aggregate due to abnormal peptide folding. Depending on the degree of aggregation, Aβpeptides are considered to be neurotoxic as in large quantities they induce amyloid plaque formation.Extracellular amyloid plaques produce loss of connections between neurons and decreased neuronalactivity.
neurotransmitter release and in the end to neuronal death. The temporal profile ofan ischemic injury can be described in a series of phases (ter Horst and Korf, 1997).The first phase includes the initiation of the expression of immediate early genes thattransform the cells from a resting into an activated state. The second phase includesthe activation of acute phase response proteins (such as heat shock proteins) withtemporary repair functions. The third phase comprises the secretion of immediateearly gene proteins from the first phase. The fourth phase is the most damaging sincethe proteinases activated in previous phases challenge the integrity of the cell. Thefifth and last phase consists of activation of repair/remodeling molecular events torescue the cell. Several mechanisms have been proposed to explain the pathophysi-ology of an ischemic injury, including increased excitotoxicity, calcium overload, freeradical formation, immune response, and inhibition of protein synthesis. All theseunbalanced events are ultimately causing neurodegeneration. Some of these aspectswill be discussed in detail in the following sections.

1.2 Glutamate in neurodegeneration 5
1.2 Glutamate in neurodegeneration
The neurodegenerative molecular pathways are poorly understood largely due tothe difficulty in distinguishing primary from secondary events. One important playerin neurodegeneration is glutamate, the major excitatory neurotransmitter in par-ticular in the forebrain regions. In many neurodegenerative disorders like cerebralischemia and AD glutamate is locally released in high quantities and promotes aneffect called excitotoxicity, which ultimately leads to programmed neuronal death(Smith-Swintosky and Mattson, 1994).
1.2.1 Glutamate-induced excitotoxicity
The neurotoxic action of the excitatory amino acid glutamate arises from its ca-pacity to trigger a pathophysiological chain of reactions when it acts continuously onits receptors. L-Glutamate (L-Glu) is the major excitatory neurotransmitter in thebrain being present at approximately two thirds of central synapses (Fonnum, 1984).Glutamate receptors are divided on the basis of their mode of action and pharmaco-logical properties into two major subdivisions: ionotropic channel receptors (iGluR)and metabotropic G-protein coupled receptors (mGluR). iGluRs are characterized bytheir selective affinity for specific agonists: N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and kainic acid (KA). Uponbinding of these ligands to the receptor, the channel opening occurs which allows theinflux of mainly sodium and/or calcium ions into the nerve cell. A differential distribu-tion and a specific pharmacological profile exists for each iGluR subtype (Monaghanet al., 1989).
The excitotoxicity theory asserts that the physiological excitatory transmissioncan be changed from a physiological into a pathological state leading to neuronal de-struction (Whetsell and Shapira, 1993). The drastic increase in L-Glu in the synapticcleft during brain injury could initiate two detrimental processes. These processesdiffer in time-dependency and ionic characteristics. The first process involves acuteswelling of cell bodies and dendrites via the opening of membrane cation channels,causing depolarization. The Na+ influx and passive influx of Cl– ions and H2O pre-cedes the cell volume expansion. Swelling occurs within minutes of L-Glu exposureand is critically dependent on the extracellular concentrations of Na+ and Cl– ions.The second process is marked by delayed neuronal degeneration. In vitro observationssuggest that the neuronal death is closely related with the increase in Ca2+ influx,mainly via NMDA receptors. NMDA receptors exhibit the highest permeability toCa2+ compared to AMPA or KA receptors and posses a superior capacity for induc-ing intracellular Ca2+ influx and thus initiating neurodegenerative processes (Choi,1992).

6 Introduction
1.2.2 NMDA-receptors
NMDA receptors constitute a major class of Glu receptors in the mammaliancentral nervous system (MacDonald et al., 1989). They are localized at the post-synaptic membrane of excitatory synapses on almost all neurons, but are specificallyenriched on pyramidal neurons in the neocortex and hippocampus. Pyramidal neuronsare particularly susceptible to neurodegeneration and are e.g. massively lost in AD(Hynd et al., 2004). Normally speaking NMDA receptors are involved in a widerange of cellular processes, including neuronal differentiation, synaptic plasticity, andlong-term potentiation (LTP) (Carroll and Zukin, 2002). In addition they mediatethe neurotoxic effects of excitatory amino acids in the adult brain under pathologicalconditions of overstimulation (Choi, 1994).
NMDA receptors are activated by glutamate and glycine which makes NMDAreceptors unique among other neurotransmitter receptors. NMDA receptor activityis modulated by several modulators such as divalent cations (Ca2+, Mg2+, Zn2+),redox substances, pH and polyamines.
NMDA receptors are ligand-gated channels and are composed of heteromultimericsubunits: NR1 and NR2 (NR2A-D) (McBain and Mayer, 1994). Although the struc-ture and stoichiometry of the NMDA channel is unknown, in vivo receptors containan obligatory NR1 and one of the NR2 subunits (Dingledine et al., 1999). The typeof NR2 subunit determines agonist affinity, Mg2+, Zn2+ sensitivity, deactivation ki-netics and channel conductance. In AD, NMDA receptors are significantly altered:NR1 and NR2B protein levels are significantly reduced, while the NR2A expression isincreased in the regions involved in learning and memory processes (Mishizen-Eberzet al., 2004). Therefore, subunit composition defines the response of the receptor toglutamate activation, which subsequently affects neuronal function.
1.3 Neuroprotective signaling
1.3.1 TNF-α signaling
Cytokines are defined as small soluble proteins secreted by a cell, which can alterthe behavior or properties of the cell itself or of another cell. Cytokines are involvedin a variety of inflammatory and infectious conditions. They are not expressed con-stitutively but rather transiently after an inducing stimulus. The most potent signalsfor cytokine expression are other cytokines. In the end this led to the concept of acytokine matrix in which they can stimulate or inhibit each other (Zhu and Emer-son, 2002). This concept accounts for the complexity of the cytokine network foundwith any neurodegenerative disorder. Cytokine receptors are constitutively expressedand their activity is modulated by ligand interaction. Cytokine receptors are cleavedby metalloproteinase enzymes to produce soluble cytokine receptors (Williams et al.,1996), which are able to capture soluble cytokines, and hence act as inhibitors bycompeting with membrane-bound receptors.

1.3 Neuroprotective signaling 7
Tumor necrosis factor-alpha (TNF-α), one of the best-characterized cytokines wasdiscovered in the 1970s by Old and colleagues (Carswell et al., 1975; Old, 1985). TNF-α is produced mainly by the monocyte/macrophage lineage, but T lymphocytes, neu-trophils, mast cells, endothelial cells and neurons can express it also under particularcircumstances. TNF-α is highly expressed under physical (UV, X-radiation, heat),chemical or immunological challenges. In vivo, TNF-α is considered to be the mostrapidly secreted pro-inflammatory cytokine (Sorimachi et al., 1999) from preformedstores. It is produced by cleavage of the membrane TNF-α by TNF-α convertingenzyme (TACE/ADAM17) (Cerretti et al., 1999). Membrane TNF-α is a 26-kDa cellsurface transmembrane type II polypeptide. The result of the TACE cleavage consistsof a 17-kDa soluble TNF-α form. TACE cleavage results in a decreased cell surfacemembrane-bound receptor density. Since clustering of TNF receptors is necessary forsignaling, their overall activity is inhibited in this way. Most of the reported TNF-α-mediated biological effects are attributed to the soluble TNF-α form, whereas cellsurface transmembrane TNF-α-mediated physiological effects are less known. The bi-ological effects exerted by transmembrane TNF-α are mediated by direct cell-to-cellinteraction (Probert et al., 1997).
In vivo TNF-α coordinates the cytokine response to injury. If its production isblocked, the expression of other cytokines, such as IL-1 and IL-6 or chemokines isdown-regulated as well (Probert et al., 1996). Under pathophysiological conditionsTNF-α acts as a switch-on molecule for the immune system. Following prolongedexposure to an excess of TNF-α its inflammatory properties are tailored towardsimmunosuppressive properties (Correale and Villa, 2004).
a) TNF-R1 and TNF-R2 signaling
TNF-α binds two distinct cell surface receptors: TNF-R1 and TNF-R2. TNF-Rmolecular pathways, either cooperatively or individually lead to cytotoxicity as wellas differentiation and growth regulatory activities.
TNF-R1 activation can trigger fibroblast growth, and endothelial cell adhesion,while TNF-R2 signaling promotes proliferation of thymocytes and peripheral T cellsand inhibition of early haematopoiesis (MacEwan, 2002). Because of its low affinityto soluble TNF, TNF-R2 was for a long time thought to have an “accessory function”by enhancing TNF-R1 signaling through a ”ligand passing” process by which TNF-αbinds to TNF-R2, dissociates and subsequently binds to TNF-R1.
Both TNF-Rs potentiate NF-κB complex activation. It was reported that neuro-protection is dependent on TNF-R1 expression in kainic acid-induced seizures model(Gary et al., 1998) or on TNF-R2 expression in a retinal-induced ischemic model(Fontaine et al., 2002). TNF-R2-induced neuroprotection was associated with thePKB/Akt pathway, since inhibition of PKB/Akt signaling abolished the neuropro-tective effect (Fontaine et al., 2002). These in vivo studies were paralleled by studiesin cultured cortical neurons where TNF-α induced neuroprotection by activation ofTNF-R2 pathway. Moreover, it was suggested that activation of NF-κB by TNF-R1

8 Introduction
and TNF-R2 displays differential temporal kinetics: while TNF-R1 signaling led to atransient NF-κB activity, TNF-R2 signaling resulted in a persistent NF-κB activation,which turned out to be crucial for neuroprotection (Marchetti et al., 2004). In addi-tion, the TNF-R2 gene contains the consensus elements for transcription factors, suchas nuclear factor-kappa B (NF-κB) in the 5’-flanking region suggesting receptor-selfpromotion (Rasmussen et al., 2001; Santee and Owen-Schaub, 1996).
b) TNF-α in pathology and therapy
Several reports showed the involvement of TNF-α in neurodegenerative illnesses(Probert et al., 1996; Ghezzi and Mennini, 2001; Sriram and O’Callaghan, 2007; Perryet al., 2001). It has been suggested that in CNS disorders, in which apoptosis isan underlying process for neuronal death such as AD, Parkinson’s disease, retinitispigmentosa, cerebellar degeneration and ischemic injury, TNF-α is a major player.
TNF-α signaling in AD. A number of studies have shown that TNF-α is up-regulated in AD (Perry et al., 2001). Furthermore, TNF promoter polymorphismsare associated with AD genes (Ma et al., 2004). It was reported that the chromosome1p and chromosome 12p regions are involved in late-onset AD and these two regionsharbor the TNF-R1 and TNF-R2 genes. However, only TNF-R2 exon 6 polymorphismwas found to be linked to late-onset AD in families with no apolipoprotein E-epsilon4 (ApoE-e4) genotype (Perry et al., 2001).
In addition, it has been demonstrated that overexpression of TNF-R1 promotesAβ-induced neuronal death (Li et al., 2004). Cross-breeding transgenic APP23 mice,which are able to develop Aβ plaques, with TNF-R1-/- mice (APP23/TNF-R1-/-) re-sulted in a strong decrease of Aβ plaques compared with APP23 mice. APP23/TNF-R1-/- mice have lower expression of BACE1 and show increased learning abilitiescompared to APP23 mice. The authors suggested that TNF-R1 is connected to ab-normal Aβ processing which could lead to Aβ plaque formation, neuronal damageand learning deficits (He et al., 2007). Thus, anti-TNF-R1-based therapies might bean efficient therapeutic target in treating AD (Rosenberg, 2005).
TNF signaling in ischemia. TNF-α expression in the cerebrospinal fluid (CSF)and in postmortem brain tissue correlates with the extent of ischemic injury in humanssuffering from ischemic stroke (Zaremba et al., 2001). Similarly, TNF-α expressionwas increased both at the protein and mRNA level in an experimental focal ischemiamodel in rats (Botchkina et al., 1997). Additional evidence for the role of TNF-αsignaling in models of focal cerebral ischemia was previously reported using micedouble-deficient for both TNF receptors (Bruce et al., 1996). In these studies TNF-R1 ameliorated hippocampal damage (Gary et al., 1998). In contrast, the depletionof TNF-R2 signaling increased the cellular degeneration in retinal ischemia (Fontaineet al., 2002). These two differential effects of TNF-R1 suggest a distinct signaling withrespect to cellular composition in the brain regions affected by the ischemic injury.

1.3 Neuroprotective signaling 9
Furthermore, middle cerebral artery occlusion/focal cerebral ischemia induces a dra-matic up-regulation of TNF receptors, with TNF-R1 appearing within 6 hr, followedby the appearance of TNF-R2 at 24 hr after the onset of ischemia (Botchkina et al.,1997). TNF-R1 can induce neuroprotection in some cell systems by increasing Fas-associated death domain-like interleukin-1-beta-converting enzyme-inhibitory protein(FLIP(L) (Taoufik et al., 2007) and NF-κB activation. TNF-R2 promotes neuronalsurvival by activation of PKB/Akt and NF-κB activation (Fontaine et al., 2002). TheTNF-R2 neuroprotective effect persisted for 8 days following the retinal ischemic in-jury (Fontaine et al., 2002), which suggests that TNF-R2 may be a promising newapproach in preventing irreversible neuronal loss by ischemic insults.
1.3.2 PKB/Akt signaling
Protein kinase B (PKB/Akt) belongs to the serine/threonine protein kinase familycalled AGC protein kinases (EC 2.7.11.1). PKB/Akt research started back in 1977,when Staal and co-workers identified a transforming leukemia virus in mice developingspontaneous lymphoma. This virus, termed Akt8, induced tumor formation confirm-ing the oncogenic potential of the Akt gene (Staal et al., 1977). Together with thisdiscovery, the PKB/Akt molecular homology was identified as significantly related toprotein kinase A (PKA) and C (PKC) using a PCR screening approach (Coffer andWoodgett, 1991).
The first PKB/Akt transgenic mouse model was generated in 2000 (Jones et al.,2000). Overall, results from transgenic PKB/Akt mice demonstrate that PKB/Aktis an important modulator of cellular growth and cell survival and it controls thedevelopment and progression of various tumors.
PKB/Akt isoforms. Hitherto three isoforms of PKB/Akt have been identified anddescribed in mice and humans (Brazil and Hemmings, 2001). Characterization andanalyses of PKB/Akt isoform mutants provided new insight into the function of thethree PKB/Akt proteins. PKBα/Akt1 is expressed in all organs and tissues and playse.g. an essential role in the modulation of fetal growth. In the brain PKBα/Akt1was shown to mediate neuroprotection in adult mice against ischemia-induced injury(Miao et al., 2005) by increasing endothelial nitric oxide synthase (eNOS) expressionvia phosphatidylinositol 3 (PI3)-kinase pathways (Hashiguchi et al., 2004). These datawere confirmed in PKBα/Akt1 ko mice. These mice display diminished PKB/Aktphosphorylation and a reduction in eNOS (Yang et al., 2003), which could lead toenhanced neuronal degeneration. PKBβ/Akt2 is predominantly found in fat tissue,liver and skeletal muscle suggesting an involvement in glucose metabolism (Altomareet al., 1998). The role of PKBβ/Akt2 in the central nervous system is not known yet.PKBγ/Akt3 is found in the brain, testis, lung, mammary gland and fat (Yang et al.,2005). In contrast to PKBα/Akt1 or PKBβ/Akt2 mutant mice, PKBγ/Akt3 mutantmice display normal glucose metabolism and no growth retardation. However, theirbrain size is dramatically reduced by about 25% with a significant decrease in both

10 Introduction
cell size and cell number (Tschopp et al., 2005). Characterization of the isoforms ofPKB/Akt in double or triple knockout mice also provided more information on thespecific function of the PKB/Akt isoforms. These studies concluded that the Akt1gene is more important than Akt3 for embryo survival but that both are required forembryonic development (Yang et al., 2005; Tschopp et al., 2005).
PKB/Akt activation. PKB/Akt is activated by several cytokines, growth fac-tors or neurotransmitters. PKB/Akt activation can be mediated via PI3-kinase(Datta et al., 1999). This pathway includes membrane phospholipids, especiallyphosphatidylinositol 3,4,5-triphosphate [PtIns(3,4,5)P3] that recruits PKB/Akt tothe plasma membrane where it becomes phosphorylated. In the case of PKBα/Akt1,activation is reached when threonine 308 (Thr308) and serine 473 (Ser473) are phos-phorylated by 3-phosphoinositide-dependent protein kinase 1 (PDK1) and a still tobe identified Ser473 kinase (Yang et al., 2003).
PKB/Akt regulation. So far several mechanisms of PKB/Akt regulation havebeen proposed. One hypothesis is that carboxyl-terminal modulator protein (CTMP)keeps PKB/Akt in an unphosphorylated and inactive state by physical protein-proteininteractions (Maira et al., 2001). Another known negative regulator is protein phos-phatase and tensin homologue deleted on chromosome 10 (PTEN) that inactivatesPKB/Akt in a PI3-kinase dependent manner. PTEN knockout mice develop a broadrange of tumors. Furthermore, they display atypical social interactions, exaggeratedresponses to stressful sensory stimuli in paradigms designed to assess anxiety andlearning. Their brains are enlarged in the regions in which the PTEN gene wasdeleted and this effect is associated with hypertrophy of the cell bodies and with ab-normal growth of neuronal processes (Kwon et al., 2006). Similar pathophysiologicalconditions were described for PKB/Akt overexpression in mice (Yang et al., 2003). Inaddition, heat shock proteins (Hsp) also bind and regulate PKB/Akt activity. Hsp arestress proteins that regulate protein stabilization and protect cells from several stresscircumstances. It has been reported that overexpression of Hsp27 in neurons protectsagainst excitotoxicity and may act as an inhibitor of neurodegeneration (Wagstaffet al., 1999). To date, Hsp27 and Hsp90 proteins have been reported to specificallybind and activate PKB/Akt leading to an inhibition of apoptosis (Rane et al., 2003).
PKB/Akt is interacting and regulated as well by scaffold proteins, such as scaffoldproteins in the stress-mediated MAP-kinase (SAPK) signaling: JNK interacting pro-tein 1 (JIP1) a scaffold protein for the c-jun amino-terminal kinase (JNK) pathwayin neuronal cells and plenty of Src homology 3 (POSH), a scaffold protein for themixed-lineage kinase (MLK)-JNK pathway (Kim et al., 2002; Figueroa et al., 2003).It has been suggested that PKB/Akt suppresses the JNK-dependent death mecha-nism not only upstream but also downstream of MLKs, thus leading to a decrease inthe neuronal susceptibility to degeneration (Xu et al., 2001).

1.3 Neuroprotective signaling 11
PKB/Akt substrates. PKB/Akt isoforms are able to phosphorylate several sub-strates providing a variety of cellular responses. Glycogen synthase kinase-3 (GSK-3)was the first identified PKB/Akt substrate (Burgering and Coffer, 1995). Impor-tantly, PKB/Akt phosphorylates the Bcl-2/Bcl-X antagonist (BAD). BAD binds theproteins Bcl-2 and Bcl-X at the mitochondrial membrane. Upon phosphorylation byPKB/Akt, BAD translocates from the mitochondrial membrane and Bcl-2 is released.Bcl-2 is then translocated from the mitochondria to the nucleus where it promotesanti-apoptotic activities (Datta et al., 1999). Bcl-2 proteins exhibit neuroprotectivefunctions against various excitotoxic challenges, such as glutamate or amyloid betapeptides. Another substrate which is activated and phosphorylated by PKB/Akt ina PI3-kinase-dependent manner (Fulton et al., 1999) is eNOS. eNOS activation hasbeen shown to have neuroprotective function in various neurodegenerative conditions(Endres et al., 2004).
Other PKB/Akt phosphorylation targets essential for neuroprotection are severaltranscription factors, including cyclic AMP (cAMP)-response element binding protein(CREB) and NF-κB (Kane et al., 1999). Upon phosphorylation CREB possesses ahigher affinity for its co-activator, resulting in transcriptional activation. The detailedmechanism of how CREB and PKB/Akt both lead to cellular survival still remainsto be elucidated. Activation of NF-κB is dependent on the IkB kinase (IKK) com-plex. PKB/Akt is a direct regulator of IKK activity in a PI3-kinase-dependent man-ner. PKB/Akt-mediated NF-κB activation contributes to the suppression of apopto-sis since NF-κB activity initiates the transcription of several anti-apoptotic proteins(Kane et al., 1999; Lawlor and Alessi, 2001).
1.3.3 NF-κB signaling
NF-κB is a transcription factor involved in the development and progression ofseveral diseases such as autoimmune disease, cancer and neurodegenerative disorders.Depending on the cell type and its regulators NF-κB complex could induces thetranscription of pro- and anti-apoptotic genes. It was first identified in B lymphocytesas an activator of immunoglobulin κ light chain transcription (Sen and Baltimore,1987). The NF-κB complex consists of an inactive form of DNA-binding dimers andinhibitory subunits. In neurons, the most common subunits expressed are p50, p65(RelA) and the inhibitory subunit IκB, which is composed of IκBα and IκBβ (Mattsonand Meffert, 2006).
NF-κB activation. The NF-κB complex is activated in neurons by several moleculessuch as TNF-α, Fas ligands, glutamate, nerve growth factor (NGF), cell adhesionmolecules and a secreted form of amyloid precursor protein (APP) or even by synaptictransmission between neurons (Mattson et al., 2000). These stimuli activate variouscellular signaling pathways such as the protein kinase C (PKC), mitogen-activatedprotein (MAP) kinase kinase kinase-1 (MEKK1) and also the PKB/Akt signalingpathway, which all have the potential to phosphorylate IKK. This kinase consists of

12 Introduction
two catalytic subunits (IKKα and IKKβ) and a regulatory subunit (IKKγ). IKKphosphorylates the inhibitory subunit IκB and induces the dissociation of IκB fromthe NF-κB complex. Upon dissociation of IkappaB from the NF-κB complex, thep50-p65 dimer of the NF-κB complex translocates from the cytosol to the nucleuswhere it binds to NF-κB responsive genes (Mattson and Meffert, 2006).
NF-κB substrates. The NF-κB complex induces the expression of genes involvedin the regulation of several cellular processes, including cellular survival (Bcl-2, in-hibitor of apoptosis proteins (IAP), TNF-α or TNF-R2 genes); immune response(TNF-α, interleukins (IL)-2, IL-6 genes); ion homeostasis (subunits of NMDA recep-tors and small conductance Ca2+ activated potassium (SK) channels). Furthermore,NF-κB can promote the transcription of the inhibitory subunit IκB and in this wayregulates its own activity in a negative feedback loop (Mattson and Meffert, 2006).
The NF-κB complex is activated in neurons and glia cells in response to acute orchronic neurodegenerative processes. In several neurodegenerative models for trau-matic brain injury, stroke, epilepsy or AD an increase in NF-κB activity was observed.These data were paralleled by in vitro studies which showed an activation of NF-κB inresponse to glutamate-induced excitotoxicity, metabolic insults or glucose deprivation.In neuronal cells sustained activation of NF-κB was shown to induce neuroprotection(Marchetti et al., 2004; Mattson et al., 2000). This beneficial outcome was in part theresult of PI3-kinase-PKB/Akt pathway activation, increase in mitochondrial antiox-idant Cu/Zn-SOD, Mn-SOD and the induction of anti-apoptotic proteins Bcl-2 andBcl-x. Furthermore, NF-κB cellular survival mechanisms underlie cytokine-inducedneuroprotective pathways, including transforming growth factor-beta1 (TGF-beta1)and TNF-α (Zhu et al., 2004).
NF-κB in relation to neurodegenerative conditions. Several reports showedthat in AD NF-κB activity is increased in cells associated with neurodegenerative pro-cesses such as neurons and astrocytes located in close proximity to amyloid-β plaques(Lezoualc’h and Behl, 1998; Collister and Albensi, 2005). Furthermore, NF-κB ac-tivity is increased in cholinergic neurons in the basal forebrain and in the superiortemporal lobe gyrus of AD patients (Lukiw and Bazan, 1998; Boissiere et al., 1997).These brain regions are known to be susceptible to neurodegeneration. In culturedneurons amyloid-β and a secreted form of APP induce an up-regulation of NF-κBactivity. This NF-κB activation was reported to be protective against amyloid-βtoxicity in cultured neurons (Barger et al., 1995). Interestingly, in early-stage ADpathology NF-κB activity is strongly increased while in later stages NF-κB activitydecreases tremendously.

1.4 Neuroprotective agents 13
1.4 Neuroprotective agents
1.4.1 Statins
Statins, 3-hydroxy-3-methylglutaryl co-enzyme A (HMG-CoA) reductase inhibi-tors, are widely used as medication for lowering cholesterol levels. Today nine phar-maceutical compounds are available: lovastatin, mevastatin, simvastatin, pravastatin,fluvastatin, atorvastatin, rosuvastatin, pitavastatin and cerivastatin. They are classi-fied in relation to their inhibitory properties for either the purified HMG-CoA reduc-tase enzyme or for cellular cholesterol biosynthesis (McTaggart et al., 2001).
Statin signaling. Besides their acknowledged role on the regulation of cardiovas-cular functions, statins are able to exert neuroprotective effects, mainly attributedto anti-inflammatory effects (Leung et al., 2003), stimulation of eNOS (Harris et al.,2004; Hernandez-Perera et al., 1998) and inhibition of inducible nitric oxide synthase(iNOS) (Vaughan and Delanty, 1999). In general, statins inhibit mevalonate synthesisand prevent the production of several isoprenoids, including farnesylpyrophosphateand geranylgeranylpyrophosphate that modulate small G proteins (GTPases). Thus,statins suppress the activation of GTPases, such as Rho, Ras and Rac by preventingtheir isoprenylation and thus their translocation from the inactive GDP-bound formslocated in the cytoplasm to the active GTP-bound forms in the plasma membrane(Maltese, 1990; Seabra, 1998). Inhibition of Rho proteins by statins prevents down-regulation of eNOS expression and activity under hypoxia conditions, leading to thestabilization of eNOS mRNA (Laufs et al., 2000). Furthermore, Rho isoprenylationinhibits neuronal outgrowth in hippocampal neuronal cultures (Pooler et al., 2006).
Although our understanding of statin-mediated cellular pathways increased tremen-dously in the last decade, statin-induced neuroprotective mechanisms are still elu-sive. In vitro studies using primary neuronal cultures described merely three possiblestatin-mediated neuroprotective mechanisms. Although acute treatment with statinsdid not rescue neurons from cellular death and did not prevent the rise in [Ca2+]caused by NMDA treatment in their system, Zacco and colleagues managed to showthe neuroprotective potential of various chronic statin treatments against NMDA ex-citotoxicity. In this case the neuroprotective effect of statins was not attributed todirect regulation of NMDA receptor-mediated events but rather to depletion of thecellular pool of cholesterol (Zacco et al., 2003). In an oxygen and glucose depriva-tion (OGD)/reoxygenation-evoked neuronal death model simvastatin treatment wasreported to induce neuroprotection (Lim et al., 2006). Simvastatin mediated this ben-eficial effect by reducing the production and toxicity of 4-hydroxy-2E-nonenal (HNE),a major cytotoxic end product of lipid peroxidation. The statin-induced neuroprotec-tive mechanism was not ascribed to NMDA or AMPA receptor-mediated events butrather to modulation of NF-κB activity, since HNE directly inhibits NF-κB basal ac-tivity (Lim et al., 2006). It was concluded that simvastatin treatment suppresses HNEcytotoxicity by restoring the NF-κB activity in the OGD model leading to increased

14 Introduction
neuronal survival. Johnson-Anuna and colleagues reported simvastatin-mediated neu-roprotective effects against Aβ toxicity via increased Bcl-2 mRNA and protein levelsin primary cortical neurons (Johnson-Anuna et al., 2007).
Several in vivo studies have shown statin-mediated neuroprotective effects againstvarious neurodegeneration-like events. This beneficial neuroprotective effect was main-ly attributed to increased eNOS expression (Kurosaki et al., 2004), Hsp cooperation,Bcl2 up-regulation, NF-κB and PKB/Akt pathways (Lu et al., 2007). In a model forbrain injury, statins (simvastatin and atorvastatin) were shown to increase neurogen-esis in neurogenic brain areas such as the hippocampal dentate gyrus and to reduceneuronal loss in the non-neurogenic brain areas such as the cornu ammonis 3 (CA3)(Lu et al., 2007). In a different study simvastatin enhanced neuronal survival in axo-tomized retinal ganglion cells after optic nerve injury via the overexpression of Hsp27and activation of PKB/Akt pathways (Kretz et al., 2006). Furthermore, statin treat-ment increases the expression level of 26 genes (as observed in a microarray study)in particular genes related to apoptosis (c-myc, Bcl2) (Johnson-Anuna et al., 2005)and PKB/Akt phosphorylation (Li et al., 2006). These microarray studies were par-alleled by in vivo chronic simvastatin administration to investigate whether statinswould provide neuroprotection in brain cells after a challenge with the Bcl-2 inhibitorHA 14-1 or the NO donor sodium nitroprusside (SNP). Bcl-2 levels were significantlyincreased in brains of simvastatin-treated and these brains were less vulnerable tomitochondrial dysfunction or caspase activation (Franke et al., 2007).
1.4.2 SK channels
SK channels are voltage insensitive channels and belong to the KCNN family(potassium intermediate/small conductance calcium-activated channel, subfamily N).In the mammalian brain, SK channels are the product of three paralogous genes,namely KCNN1/KCa2.1/SKCa1 (SK1), KCNN2/KCa2.2/SKCa2 (SK2) and KCNN3/KCa2.3/SKCa3/hKCa3 (SK3). Recently, a new KCNN4/ KCa3.1/SK4/IK1 channelwas discovered being highly expressed in rat microglia (reviewed by Stocker (2004)).
SK1 and SK2 channel subunits show extensive colocalization. They are mainlyfound in the entorhinal cortex, the subiculum, in pyramidal cortical neurons, theCA1 - CA3 region from the hippocampus and in the thalamus. SK3 channel subunitshave a complementary distribution, and are mainly located in the brain stem and inmonominergic neurons (Sailer et al., 2002).
Functional SK channels are composed of heteromeric subunits with constitutivelybound calmodulin (CaM). The channels are activated in response to low (< 1µM)intracellular Ca2+ that binds to CaM. It has been shown that SK channels contributeto basal synaptic integration at least in four different ways. In cortical and hippocam-pal pyramidal central neurons, SK channel activity dampens membrane excitability,contributes to dendritic response and regulates the synaptic integration. In certainneuronal cell types, SK channels are localized to dendritic spines where they inter-act with NMDA receptors. There SK channel activity suppresses the amplitude of

1.5 Outline of this thesis 15
evoked synaptic potentials by inhibiting NMDA receptor-dependent activation (Ngo-Anh et al., 2005; Faber et al., 2005). In tonically firing cells, including midbraindopamine neurons SK channels affect pacemaking by decreasing interspike intervalsduring a burst of action potentials as well as the length of the burst. In high firing-frequency cells, including cerebellar Purkinje neurons, SK channel activity is requiredfor enduring basal rapid firing and SK channels act as modulators for firing pattern.In auditory hair cells SK channels are able to convert excitatory neurotransmittersignal into inhibitory signals (reviewed by Bond et al. (2005)). Overall, it can be con-cluded that SK channels are important regulators of synaptic integration and neuronalexcitability in such a way that an increase in SK channel activity normally leads todecreased excitability of the cells (reviewed by Stocker (2004)).
Hitherto, there is only limited information on the role of SK channels in aging andneurodegenerative disorders. Changes in neuronal excitability are currently thoughtto underlie learning and memory processes. Aging is often characterized by a decreasein neuronal excitability and cognitive deficits. Since SK channel activity leads toa reduction of neuronal excitability it was hypothesized that increased SK channelexpression and activity can in part account for the cognitive impairments found inaged individuals. In agreement with this hypothesis SK3 channel expression wasfound to be up-regulated in the hippocampus of aged mice. This elevation in SK3channel expression was shown to result in a strong decrease in neuronal excitabilityand was paralleled by impaired memory and learning abilities (Blank et al., 2003).Since SK channels can regulate neuronal excitability a role in neurodegeneration andneuroprotection may be expected. Indeed overexpression of SK2 channels in in vitrocultured hippocampal neurons induced neuroprotection against kainite and glutamateexcitotoxicity. Furthermore, in vivo overexpression of SK2 channels in dentate gyrusdiminished a kainate-induced CA3 lesion (Lee et al., 2003). Thus, SK channels mayprove to be important drug targets in the development of new effective therapeuticstrategies against degenerative events.
1.5 Outline of this thesis
Neurodegenerative processes lead to neuronal loss which may ultimately resultin functional impairments or even death. Thus, the understanding of the molecularbasis of neuroprotective mechanisms for developing effective therapies to prevent andtreat neurodegenerative diseases is of paramount importance. Hitherto, a number ofstudies showed that TNF-α is able to promote protection against glutamate-inducedexcitotoxicity in cortical neurons. Until now, the molecular mechanisms underlyingthese beneficial effects of TNF-α are largely unknown. An initial study showed thatTNF-α increases neuronal survival by mobilizing the TNF-R2 pathway. TNF-R2-associated neuroprotective effects are mediated by the activation of PKB/Akt andsubsequent sustained NF-κB activation.
In the first part of this thesis (Chapters 2, 3 and 4) the TNF-α-mediated pro-

16 Introduction
tective mechanisms in cortical neurons are investigated in more detail. In Chapter2 we studied the kinetics of the neuroprotective effect of TNF-α treatment. SincePTEN is a counter-regulator of PKB/Akt signaling we investigated the time courseof PKB/Akt and PTEN expression during TNF-α-treatment in wild-type, TNF-R1-/-
and TNF-R2-/- neurons. In particular, we checked whether PTEN activation precedesPKB/Akt activation during long-term TNF-α treatment. Finally, we identified whichPKB/Akt isoform is important in TNF-α-induced neuroprotective pathways.
To gain further insight into the regulation of PKB/Akt we studied the phosphory-lation of PKB/Akt by two cAMP effector systems, PKA and Epac in cortical neuronsin Chapter 3. PKA activation was shown to dephosphorylate PKB/Akt whereas Epacactivation lead to increased PKB/Akt phosphorylation. Since both PKA and Epacwere found to be complexed to the A-kinase anchoring protein AKAP150, we hy-pothesized that AKAP150 acts as a coordinator of PKA/Epac signaling to modulatePKB/Akt phosphorylation.
To date, only limited data is available on potential neuroprotective downstreamtargets of TNF-α induced NF-κB activation. SK channels are important regula-tors of neuronal excitability and were recently identified as a downstream target forNF-κB-mediated promoter regulation. Therefore, we investigated whether increasedSK channel expression is part of the mechanism of TNF-α-induced cellular survival(Chapter 4).
Chapter 5 describes the neuroprotective effect of lovastatin pretreatment on glu-tamate-induced neuronal death in neurons. Interestingly, in primary human vascularendothelial cells (HUVEC) lovastatin treatment was shown to increase the expressionof TNF-R2 proteins, without affecting TNF-R1 expression levels. We investigatedwhether lovastatin can also induce an increase in TNF-R2 expression levels in corticalneurons and whether activation of TNF-R2 signaling might be the mechanism thatunderlies the neuroprotective effect exerted by lovastatin. Moreover, we investigatedthe effect of lovastatin on PKB/Akt and NF-κB phosphorylation.
In Chapter 6 we wanted to confirm the observed in vitro neuroprotective effect oflovastatin in an in vivo mouse model. The damage of cholinergic neurons and theircortical target areas from the magnocellular nucleus basalis (MNB) that occurs duringaging or AD could be mimicked using an NMDA-induced lesions into MNB model. Inthis in vivo model we found that lovastatin has a neuroprotective effect on NMDA-induced excitotoxic lesions. In addition, we investigated whether this neuroprotectiveeffect of lovastatin administration was dependent on PKB/Akt activation. Since thedegeneration of cortical cholinergic fibers is associated with a cognitive decline in ADwe studied the effects of lovastatin treatment on learning and memory abilities usingdifferent behavior paradigms.
It is known that glutamatergic and cholinergic systems interact functionally at thelevel of the cholinergic basal forebrain. The N-methyl-D-aspartate receptor (NMDA-R) is a multiprotein complex composed of NR1, NR2 and/or NR3 subunits. Thesubunit composition of NMDA-R of cholinergic cells in the nucleus basalis has not yet

References 17
been investigated. By means of choline acetyl-transferase and NR2B or NR2C doublestaining, we investigated whether mice express both the NR2C and NR2B subunits innucleus basalis cholinergic cells (Chapter 7). Since we found increased acetyl choline(ACh) in the frontal cortex and amygdala, we assessed behavioral habituation to novelenvironments and objects as well as object recognition in NR2C-2B subunit exchangemice.
Overall this thesis provides more insight into TNF-α mediated neuroprotective sig-naling. We investigated both upstream and downstream targets of TNF-α pathways.In addition, we provide more information about PKB/Akt regulation, lovastatin-induced neuroprotective mechanisms.
References
Altomare, D. A., Lyons, G. E., Mitsuuchi, Y., Cheng, J. Q., and Testa, J. R. (1998).Akt2 mRNA is highly expressed in embryonic brown fat and the AKT2 kinase isactivated by insulin. Oncogene, 16:2407–2411.
Barger, S. W., Horster, D., Furukawa, K., Goodman, Y., Krieglstein, J., and Matt-son, M. P. (1995). Tumor necrosis factors alpha and beta protect neurons againstamyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding fac-tor and attenuation of peroxide and Ca2+ accumulation. Proc. Natl. Acad. Sci.U.S.A., 92:9328–9332.
Blank, T., Nijholt, I., Kye, M., Radulovic, J., and Spiess, J. (2003). Small-conductance, Ca2+-activated K+ channel SK3 generates age-related memory andLTP deficits. Nat. Neurosci., 6:911–912.
Boissiere, F., Hunot, S., Faucheux, B., Duyckaerts, C., Hauw, J. J., Agid, Y., andHirsch, E. C. (1997). Nuclear translocation of NF-kappaB in cholinergic neurons ofpatients with Alzheimer’s disease. Neuroreport, 8:2849–2852.
Bond, C. T., Maylie, J., and Adelman, J. P. (2005). SK channels in excitability,pacemaking and synaptic integration. Curr. Opin. Neurobiol., 15:305–311.
Botchkina, G. I., Meistrell, M. E., Botchkina, I. L., and Tracey, K. J. (1997). Expres-sion of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebralischemia. Mol. Med., 3:765–781.
Brazil, D. P. and Hemmings, B. A. (2001). Ten years of protein kinase b signalling:a hard akt to follow. Trends Biochem. Sci., 26(11):657–664.
Bruce, A. J., Boling, W., Kindy, M. S., Peschon, J., Kraemer, P. J., Carpenter, M. K.,Holtsberg, F. W., and Mattson, M. P. (1996). Altered neuronal and microglialresponses to excitotoxic and ischemic brain injury in mice lacking TNF receptors.Nat. Med., 2:788–794.

18 Introduction
Burgering, B. M. and Coffer, P. J. (1995). Protein kinase B (c-Akt) inphosphatidylinositol-3-OH kinase signal transduction. Nature, 376:599–602.
Carroll, R. C. and Zukin, R. S. (2002). NMDA-receptor trafficking and targeting:implications for synaptic transmission and plasticity. Trends Neurosci., 25:571–577.
Carswell, E. A., Old, L. J., Kassel, R. L., Green, S., Fiore, N., and Williamson, B.(1975). An endotoxin-induced serum factor that causes necrosis of tumors. Proc.Natl. Acad. Sci. U.S.A., 72:3666–3670.
Cerretti, D. P., Poindexter, K., Castner, B. J., Means, G., Copeland, N. G., Gilbert,D. J., Jenkins, N. A., Black, R. A., and Nelson, N. (1999). Characterization ofthe cDNA and gene for mouse tumour necrosis factor alpha converting enzyme(TACE/ADAM17) and its location to mouse chromosome 12 and human chromo-some 2p25. Cytokine, 11:541–551.
Choi, D. W. (1992). Excitotoxic cell death. J. Neurobiol., 23:1261–1276.
Choi, D. W. (1994). Glutamate receptors and the induction of excitotoxic neuronaldeath. Prog. Brain Res., 100:47–51.
Clarke, P. G. H. (1990). Developmental cell death: morphological diversity and mul-tiple mechanisms. Anat. Embryol., 181(3):195–213.
Clarke, P. G. H. (1999). Cell death and diseases of the nervous system, chapterApoptosis versus necrosis. How valid a dichotomy for neurons?, pages 3–28. HumanaPress.
Coffer, P. J. and Woodgett, J. R. (1991). Molecular cloning and characterisation ofa novel putative protein-serine kinase related to the cAMP-dependent and proteinkinase C families. Eur. J. Biochem., 201:475–481.
Collister, K. A. and Albensi, B. C. (2005). Potential therapeutic targets in the NF-kappaB pathway for Alzheimer’s disease. Drug News Perspect., 18:623–629.
Correale, J. and Villa, A. (2004). The neuroprotective role of inflammation in nervoussystem injuries. J. Neurol., 251:1304–1316.
Datta, S. R., Brunet, A., and Greenberg, M. E. (1999). Cellular survival: a play inthree Akts. Genes Dev., 13:2905–2927.
Dickson, D. (2003). Neurodegeneration: The Molecular Pathology of Dementia andMovement Disorders, chapter Apoptosis and neurodegeneration, pages 2–17. JohnWiley and Sons.
Dingledine, R., Borges, K., Bowie, D., and Traynelis, S. F. (1999). The glutamatereceptor ion channels. Pharmacol. Rev., 51:7–61.

References 19
Endres, M., Laufs, U., Liao, J. K., and Moskowitz, M. A. (2004). Targeting eNOSfor stroke protection. Trends Neurosci., 27:283–289.
Faber, E. S., Delaney, A. J., and Sah, P. (2005). SK channels regulate excitatorysynaptic transmission and plasticity in the lateral amygdala. Nat. Neurosci., 8:635–641.
Figueroa, C., Tarras, S., Taylor, J., and Vojtek, A. B. (2003). Akt2 negativelyregulates assembly of the POSH-MLK-JNK signaling complex. J. Biol. Chem.,278:47922–47927.
Fonnum, F. (1984). Glutamate: a neurotransmitter in mammalian brain. J. Neu-rochem., 42(1):1–11.
Fontaine, V., Mohand-Said, S., Hanoteau, N., Fuchs, C., Pfizenmaier, K., and Eisel,U. (2002). Neurodegenerative and neuroprotective effects of tumor Necrosis factor(TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2.J. Neurosci., 22:RC216.
Franke, C., Nuldner, M., Abdel-Kader, R., Johnson-Anuna, L. N., Gibson Wood, W.,Muller, W. E., and Eckert, G. P. (2007). Bcl-2 upregulation and neuroprotection inguinea pig brain following chronic simvastatin treatment. Neurobiol. Dis., 25:438–445.
Fulton, D., Gratton, J. P., McCabe, T. J., Fontana, J., Fujio, Y., Walsh, K., Franke,T. F., Papapetropoulos, A., and Sessa, W. C. (1999). Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature, 399:597–601.
Gary, D. S., Bruce-Keller, A. J., Kindy, M. S., and Mattson, M. P. (1998). Ischemicand excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosisfactor receptor. J. Cereb. Blood Flow Metab., 18:1283–1287.
Ghezzi, P. and Mennini, T. (2001). Tumor necrosis factor and motoneuronal degen-eration: an open problem. Neuroimmunomodulation, 9:178–182.
Harris, M. B., Blackstone, M. A., Sood, S. G., Li, C., Goolsby, J. M., Venema, V. J.,Kemp, B. E., and Venema, R. C. (2004). Acute activation and phosphorylationof endothelial nitric oxide synthase by HMG-CoA reductase inhibitors. Am. J.Physiol. Heart Circ. Physiol., 287:H560–566.
Hashiguchi, A., Yano, S., Morioka, M., Hamada, J., Ushio, Y., Takeuchi, Y., andFukunaga, K. (2004). Up-regulation of endothelial nitric oxide synthase via phos-phatidylinositol 3-kinase pathway contributes to ischemic tolerance in the CA1subfield of gerbil hippocampus. J. Cereb. Blood Flow Metab., 24:271–279.

20 Introduction
He, P., Zhong, Z., Lindholm, K., Berning, L., Lee, W., Lemere, C., Staufenbiel, M., Li,R., and Shen, Y. (2007). Deletion of tumor necrosis factor death receptor inhibitsamyloid beta generation and prevents learning and memory deficits in Alzheimer’smice. J. Cell Biol., 178:829–841.
Hernandez-Perera, O., Perez-Sala, D., Navarro-Antolın, J., Sanchez-Pascuala, R.,Hernandez, G., Dıaz, C., and Lamas, S. (1998). Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the ex-pression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelialcells. J. Clin. Invest., 101:2711–2719.
Hynd, M. R., Scott, H. L., and Dodd, P. R. (2004). Glutamate-mediated excitotoxicityand neurodegeneration in Alzheimer’s disease. Neurochem. Int., 45:583–595.
Jellinger, K. A. (1998). The neuropathological diagnosis of alzheimer disease. J.Neural. Transm. Suppl., 53:97–118.
Johnson-Anuna, L. N., Eckert, G. P., Franke, C., Igbavboa, U., Muller, W. E.,and Wood, W. G. (2007). Simvastatin protects neurons from cytotoxicity by up-regulating bcl-2 mrna and protein. J. Neurochem., 101(1):77–86.
Johnson-Anuna, L. N., Eckert, G. P., Keller, J. H., Igbavboa, U., Franke, C., Fechner,T., Schubert-Zsilavecz, M., Karas, M., Muller, W. E., and Wood, W. G. (2005).Chronic administration of statins alters multiple gene expression patterns in mousecerebral cortex. J. Pharmacol. Exp. Ther., 312:786–793.
Jones, R. G., Parsons, M., Bonnard, M., Chan, V. S., Yeh, W. C., Woodgett, J. R.,and Ohashi, P. S. (2000). Protein kinase B regulates T lymphocyte survival, nuclearfactor kappaB activation, and Bcl-X(L) levels in vivo. J. Exp. Med., 191:1721–1734.
Kane, L. P., Shapiro, V. S., Stokoe, D., and Weiss, A. (1999). Induction of NF-kappaBby the Akt/PKB kinase. Curr. Biol., 9:601–604.
Kim, A. H., Yano, H., Cho, H., Meyer, D., Monks, B., Margolis, B., Birnbaum, M. J.,and Chao, M. V. (2002). Akt1 regulates a jnk scaffold during excitotoxic apoptosis.Neuron, 35(4):697–709.
Korsmeyer, S. J. (1999). Bcl-2 gene family and the regulation of programmed celldeath. Cancer Res., 59:1693s–1700s.
Kretz, A., Schmeer, C., Tausch, S., and Isenmann, S. (2006). Simvastatin promotesheat shock protein 27 expression and Akt activation in the rat retina and protectsaxotomized retinal ganglion cells in vivo. Neurobiol. Dis., 21:421–430.
Kurosaki, R., Muramatsu, Y., Kato, H., and Araki, T. (2004). Protective effectof pitavastatin, a 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductaseinhibitor, on ischemia-induced neuronal damage. Neurol. Res., 26:684–691.

References 21
Kwon, C. H., Luikart, B. W., Powell, C. M., Zhou, J., Matheny, S. A., Zhang, W.,Li, Y., Baker, S. J., and Parada, L. F. (2006). Pten regulates neuronal arborizationand social interaction in mice. Neuron, 50:377–388.
Laufs, U., Endres, M., Stagliano, N., Amin-Hanjani, S., Chui, D. S., Yang, S. X.,Simoncini, T., Yamada, M., Rabkin, E., Allen, P. G., Huang, P. L., Bohm, M.,Schoen, F. J., Moskowitz, M. A., and Liao, J. K. (2000). Neuroprotection mediatedby changes in the endothelial actin cytoskeleton. J. Clin. Invest., 106(1):15–24.
Lawlor, M. A. and Alessi, D. R. (2001). PKB/Akt: a key mediator of cell proliferation,survival and insulin responses? J. Cell. Sci., 114:2903–2910.
Lee, A. L., Dumas, T. C., Tarapore, P. E., Webster, B. R., Ho, D. Y., Kaufer, D.,and Sapolsky, R. M. (2003). Potassium channel gene therapy can prevent neurondeath resulting from necrotic and apoptotic insults. J. Neurochem., 86:1079–1088.
Leung, B. P., Sattar, N., Crilly, A., Prach, M., McCarey, D. W., Payne, H., Madhok,R., Campbell, C., Gracie, J. A., Liew, F. Y., and McInnes, I. B. (2003). A novel anti-inflammatory role for simvastatin in inflammatory arthritis. J. Immunol., 170:1524–1530.
Lezoualc’h, F. and Behl, C. (1998). Transcription factor NF-kappaB: friend or foe ofneurons? Mol. Psychiatry, 3:15–20.
Li, L., Cao, D., Kim, H., Lester, R., and Fukuchi, K. (2006). Simvastatin enhanceslearning and memory independent of amyloid load in mice. Ann. Neurol., 60:729–739.
Li, R., Yang, L., Lindholm, K., Konishi, Y., Yue, X., Hampel, H., Zhang, D., andShen, Y. (2004). Tumor necrosis factor death receptor signaling cascade is requiredfor amyloid-beta protein-induced neuron death. J. Neurosci., 24:1760–1771.
Lim, J. H., Lee, J. C., Lee, Y. H., Choi, I. Y., Oh, Y. K., Kim, H. S.,Park, J. S., and Kim, W. K. (2006). Simvastatin prevents oxygen and glucosedeprivation/reoxygenation-induced death of cortical neurons by reducing the pro-duction and toxicity of 4-hydroxy-2E-nonenal. J. Neurochem., 97:140–150.
Lu, D., Qu, C., Goussev, A., Jiang, H., Lu, C., Schallert, T., Mahmood, A., Chen, J.,Li, Y., and Chopp, M. (2007). Statins increase neurogenesis in the dentate gyrus,reduce delayed neuronal death in the hippocampal CA3 region, and improve spatiallearning in rat after traumatic brain injury. J. Neurotrauma, 24:1132–1146.
Lukiw, W. J. and Bazan, N. G. (1998). Strong nuclear factor-kappaB-DNA bindingparallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer’sdisease superior temporal lobe neocortex. J. Neurosci. Res., 53:583–592.

22 Introduction
Ma, S. L., Tang, N. L., Lam, L. C., and Chiu, H. F. (2004). Association between tumornecrosis factor-alpha promoter polymorphism and Alzheimer’s disease. Neurology,62:307–309.
MacDonald, J. F., Mody, I., Salter, M. W., Pennefather, P., and Schneiderman, J. H.(1989). The regulation of NMDA receptors in the central nervous system. Prog.Neuropsychopharmacol. Biol. Psychiatry, 13:481–488.
MacEwan, D. J. (2002). TNF receptor subtype signalling: differences and cellularconsequences. Cell. Signal., 14:477–492.
Maira, S. M., Galetic, I., Brazil, D. P., Kaech, S., Ingley, E., Thelen, M., and Hem-mings, B. A. (2001). Carboxyl-terminal modulator protein (CTMP), a negativeregulator of PKB/Akt and v-Akt at the plasma membrane. Science, 294:374–380.
Maltese, W. A. (1990). Posttranslational modification of proteins by isoprenoids inmammalian cells. FASEB J., 4:3319–3328.
Marchetti, L., Klein, M., Schlett, K., Pfizenmaier, K., and Eisel, U. L. (2004). Tumornecrosis factor (TNF)-mediated neuroprotection against glutamate-induced excito-toxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role ofa TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa Bpathway. J. Biol. Chem., 279:32869–32881.
Mattson, M. P., Culmsee, C., Yu, Z., and Camandola, S. (2000). Roles of nuclearfactor kappaB in neuronal survival and plasticity. J. Neurochem., 74:443–456.
Mattson, M. P. and Meffert, M. K. (2006). Roles for NF-kappaB in nerve cell survival,plasticity, and disease. Cell Death Differ., 13:852–860.
McBain, C. J. and Mayer, M. L. (1994). N-methyl-D-aspartic acid receptor structureand function. Physiol. Rev., 74:723–760.
McTaggart, F., Buckett, L., Davidson, R., Holdgate, G., McCormick, A., Schneck,D., Smith, G., and Warwick, M. (2001). Preclinical and clinical pharmacologyof rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor.Am J Cardiol, 87(5A):28B–32B.
Miao, B., Yin, X. H., Pei, D. S., Zhang, Q. G., and Zhang, G. Y. (2005). Neu-roprotective effects of preconditioning ischemia on ischemic brain injury throughdown-regulating activation of JNK1/2 via N-methyl-D-aspartate receptor-mediatedAkt1 activation. J. Biol. Chem., 280:21693–21699.
Mishizen-Eberz, A. J., Rissman, R. A., Carter, T. L., Ikonomovic, M. D., Wolfe,B. B., and Armstrong, D. M. (2004). Biochemical and molecular studies of NMDAreceptor subunits NR1/2A/2B in hippocampal subregions throughout progressionof Alzheimer’s disease pathology. Neurobiol. Dis., 15:80–92.

References 23
Monaghan, D. T., Bridges, R. J., and Cotman, C. W. (1989). The excitatory aminoacid receptors: their classes, pharmacology, and distinct properties in the functionof the central nervous system. Annu. Rev. Pharmacol. Toxicol., 29:365–402.
Ngo-Anh, T. J., Bloodgood, B. L., Lin, M., Sabatini, B. L., Maylie, J., and Adelman,J. P. (2005). SK channels and NMDA receptors form a Ca2+-mediated feedbackloop in dendritic spines. Nat. Neurosci., 8:642–649.
Old, L. J. (1985). Tumor necrosis factor (TNF). Science, 230:630–632.
Perry, R. T., Collins, J. S., Wiener, H., Acton, R., and Go, R. C. (2001). The role ofTNF and its receptors in Alzheimer’s disease. Neurobiol. Aging, 22:873–883.
Pooler, A. M., Xi, S. C., and Wurtman, R. J. (2006). The 3-hydroxy-3-methylglutarylco-enzyme A reductase inhibitor pravastatin enhances neurite outgrowth in hip-pocampal neurons. J. Neurochem., 97:716–723.
Probert, L., Akassoglou, K., Alexopoulou, L., Douni, E., Haralambous, S., Hill,S., Kassiotis, G., Kontoyiannis, D., Pasparakis, M., Plows, D., and Kollias, G.(1996). Dissection of the pathologies induced by transmembrane and wild-typetumor necrosis factor in transgenic mice. J. Leukoc. Biol., 59:518–525.
Probert, L., Akassoglou, K., Kassiotis, G., Pasparakis, M., Alexopoulou, L., and Kol-lias, G. (1997). TNF-alpha transgenic and knockout models of CNS inflammationand degeneration. J. Neuroimmunol., 72:137–141.
Rane, M. J., Pan, Y., Singh, S., Powell, D. W., Wu, R., Cummins, T., Chen, Q.,McLeish, K. R., and Klein, J. B. (2003). Heat shock protein 27 controls apoptosisby regulating Akt activation. J. Biol. Chem., 278:27828–27835.
Rasmussen, L. M., Hansen, P. R., Nabipour, M. T., Olesen, P., Kristiansen, M. T.,and Ledet, T. (2001). Diverse effects of inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase on the expression of VCAM-1 and E-selectin in endothelial cells.Biochem. J., 360:363–370.
Rosenberg, P. B. (2005). Clinical aspects of inflammation in Alzheimer’s disease. IntRev Psychiatry, 17:503–514.
Sailer, C. A., Hu, H., Kaufmann, W. A., Trieb, M., Schwarzer, C., Storm, J. F., andKnaus, H. G. (2002). Regional differences in distribution and functional expres-sion of small-conductance Ca2+-activated K+ channels in rat brain. J. Neurosci.,22:9698–9707.
Santee, S. M. and Owen-Schaub, L. B. (1996). Human tumor necrosis factor receptorp75/80 (CD120b) gene structure and promoter characterization. J. Biol. Chem.,271:21151–21159.

24 Introduction
Seabra, M. C. (1998). Membrane association and targeting of prenylated Ras-likeGTPases. Cell. Signal., 10:167–172.
Sen, R. and Baltimore, D. (1987). In vitro transcription of immunoglobulin genesin a B-cell extract: effects of enhancer and promoter sequences. Mol. Cell. Biol.,7:1989–1994.
Smith-Swintosky, V. L. and Mattson, M. P. (1994). Glutamate, beta-amyloid precur-sor proteins, and calcium mediated neurofibrillary degeneration. J. Neural. Transm.Suppl., 44:29–45.
Sorimachi, K., Akimoto, K., Hattori, Y., Ieiri, T., and Niwa, A. (1999). Secretion ofTNF-alpha, IL-8 and nitric oxide by macrophages activated with polyanions, andinvolvement of interferon-gamma in the regulation of cytokine secretion. Cytokine,11:571–578.
Sriram, K. and O’Callaghan, J. P. (2007). Divergent roles for tumor necrosis factor-alpha in the brain. J Neuroimmune Pharmacol, 2:140–153.
Staal, S. P., Hartley, J. W., and Rowe, W. P. (1977). Isolation of transforming murineleukemia viruses from mice with a high incidence of spontaneous lymphoma. Proc.Natl. Acad. Sci. U.S.A., 74:3065–3067.
Stocker, M. (2004). Ca2+-activated K+ channels: molecular determinants and func-tion of the SK family. Nat. Rev. Neurosci., 5:758–770.
Taoufik, E., Valable, S., Muller, G. J., Roberts, M. L., Divoux, D., Tinel, A., Voulgari-Kokota, A., Tseveleki, V., Altruda, F., Lassmann, H., Petit, E., and Probert, L.(2007). FLIP(L) protects neurons against in vivo ischemia and in vitro glucosedeprivation-induced cell death. J. Neurosci., 27:6633–6646.
ter Horst, G. J. and Korf, J. (1997). Clinical Pharmacology of Cerebral Ischemia.Humana Press.
Tschopp, O., Yang, Z. Z., Brodbeck, D., Dummler, B. A., Hemmings-Mieszczak, M.,Watanabe, T., Michaelis, T., Frahm, J., and Hemmings, B. A. (2005). Essential roleof protein kinase B gamma (PKB gamma/Akt3) in postnatal brain developmentbut not in glucose homeostasis. Development, 132:2943–2954.
Vaughan, C. J. and Delanty, N. (1999). Neuroprotective properties of statins incerebral ischemia and stroke. Stroke, 30:1969–1973.
Wagstaff, M. J., Collaco-Moraes, Y., Smith, J., de Belleroche, J. S., Coffin, R. S.,and Latchman, D. S. (1999). Protection of neuronal cells from apoptosis by Hsp27delivered with a herpes simplex virus-based vector. J. Biol. Chem., 274:5061–5069.

References 25
Wellington, C. L. and Hayden, M. R. (2000). Caspases and neurodegeneration: onthe cutting edge of new therapeutic approaches. Clin. Genet., 57(1):1–10.
Whetsell, W. O. J. and Shapira, N. A. (1993). Neuroexcitation, excitotoxicity andhuman neurological disease. Lab. Invest., 68(4):372–387.
Williams, L. M., Gibbons, D. L., Gearing, A., Maini, R. N., Feldmann, M., and Bren-nan, F. M. (1996). Paradoxical effects of a synthetic metalloproteinase inhibitorthat blocks both p55 and p75 TNF receptor shedding and TNF alpha processingin RA synovial membrane cell cultures. J. Clin. Invest., 97:2833–2841.
Xu, Z., Maroney, A. C., Dobrzanski, P., Kukekov, N. V., and Greene, L. A. (2001).The mlk family mediates c-jun n-terminal kinase activation in neuronal apoptosis.Mol. Cell. Biol., 21(14):4713–4724.
Yang, Z. Z., Tschopp, O., Di-Poı, N., Bruder, E., Baudry, A., Dummler, B., Wahli,W., and Hemmings, B. A. (2005). Dosage-dependent effects of Akt1/protein kinaseBalpha (PKBalpha) and Akt3/PKBgamma on thymus, skin, and cardiovascularand nervous system development in mice. Mol. Cell. Biol., 25:10407–10418.
Yang, Z. Z., Tschopp, O., Hemmings-Mieszczak, M., Feng, J., Brodbeck, D., Perentes,E., and Hemmings, B. A. (2003). Protein kinase B alpha/Akt1 regulates placentaldevelopment and fetal growth. J. Biol. Chem., 278:32124–32131.
Zacco, A., Togo, J., Spence, K., Ellis, A., Lloyd, D., Furlong, S., and Piser, T. (2003).3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors protect cortical neu-rons from excitotoxicity. J. Neurosci., 23:11104–11111.
Zaremba, J., Skrobanski, P., and Losy, J. (2001). Tumour necrosis factor-alpha isincreased in the cerebrospinal fluid and serum of ischaemic stroke patients and cor-relates with the volume of evolving brain infarct. Biomed. Pharmacother., 55:258–263.
Zhu, J. and Emerson, S. G. (2002). Hematopoietic cytokines, transcription factorsand lineage commitment. Oncogene, 21:3295–3313.
Zhu, Y., Culmsee, C., Klumpp, S., and Krieglstein, J. (2004). Neuroprotec-tion by transforming growth factor-beta1 involves activation of nuclear factor-kappaB through phosphatidylinositol-3-OH kinase/Akt and mitogen-activated pro-tein kinase-extracellular-signal regulated kinase1,2 signaling pathways. Neuro-science, 123:897–906.






























