Embed Size (px)
Citation preview

UNIVERSIDADE FEDERAL DO PARÁ
CENTRO DE CIÊNCIAS BIOLÓGICAS
PROGRAMA DE PÓS-GRADUAÇÃO
DOUTORADO EM BIOLOGIA DE AGENTES INFECCIOSOS E PARASITÁRIOS
AVALIAÇÃO DO PERFIL IMUNOLÓGICO COM ÊNFASE NA RESPOSTA T
REGULATÓRIA ANTE A INFECÇÃO DO HELICOBACTER PYLORI EM
PACIENTES COM GASTRITE
JAIR FRANCISCO DE SANTANA GRAIM
Belém-Pará
2014

JAIR FRANCISCO DE SANTANA GRAIM
AVALIAÇÃO DO PERFIL IMUNOLÓGICOCOM ÊNFASE NA RESPOSTA T
REGULATÓRIA ANTE A INFECÇÃO DO HELICOBACTER PYLORI EM
PACIENTES COM GASTRITE
Tese apresentada ao Programa de Pós-
Graduação em Biologia de Agentes
Infecciosos e Parasitários do Instituto de
Ciências Biológicas da Universidade Federal
do Pará como requisito parcial para a obtenção
do grau de Doutor em Biologia dos Agentes
Infecciosos e Parasitários.
Orientador: Prof. Dr. Juarez Antonio Simões
Quaresma
Belém – Pará
2014

Graim, Jair Francisco de Santana
Avaliação do perfil imunológico com ênfase na resposta T regulatória
ante a infecção do H. Pylori em pacientes com gastrite, Belém-Pará,
2013, 129p, Tese de Doutorado em Biologia dos Agentes Infecciosos e
Parasitários.
1. Helicobacter pylori 2. Resposta T regulatória 3. Gastrites

1
JAIR FRANCISCO DE SANTANA GRAIM
AVALIAÇÃO DO PERFIL IMUNOLÓGICO COM ÊNFASE NA RESPOSTA T
REGULATÓRIA ANTE A INFECÇÃO DO HELICOBACTER PYLORI EM
PACIENTES COM GASTRITE
Tese apresentada ao Programa de Pós-Graduação em Biologia de Agentes Infecciosos e
Parasitários do Centro de Ciências Biológicas da Universidade Federal do Pará como
requisito parcial para a obtenção do grau de Doutor em Biologia dos Agentes
Infecciosos e Parasitários.
Orientador: Prof. Dr. Juarez Antonio Simões Quaresma
Instituto de Ciências Biológicas - UFPA.
Banca Examinadora: Profa. Dra. Ana Cecília Ribeiro Cruz
Instituto Evandro Chagas - IEC/PA
Prof. Dr. Ismaelino Mauro Nunes Magno
Centro de Estudos Superiores do Pará - CESUPA
Profa. Dra. Helen Thais Fuzii
Universidade Federal do Pará - UFPA
Profa. Dra. Fabiola Elizabeth Villanova
Universidade Federal do Pará - UFPA
Profa. Dra. Luisa Carício Martins
Núcleo de Medicina Tropical - NMT/UFPA
Belém, 24 de junho de 2014

2
“A mente que se abre a uma nova idéia
jamais voltará ao seu tamanho original.”
Albert Einstein

3
À memória de meu pai, por todos os aprendizados e,
em particular por me ensinar “o que é certo e o que é errado”.
de minha avó, Hilda, exemplo de mulher, minha maior incentivadora.
do meu tio, Cecílio, por sempre ter acreditado em mim e por ter sido meu esteio na
hora que mais precisava.

4
AGRADECIMENTOS
A meu Deus por ter me dado o bem mais valioso que um ser humano pode ter, a vida.
À Universidade do Estado do Pará, por ter proporcionado o Programa de Pós-
Graduação Interinstitucional com a Universidade Federal do Pará, contribuindo para
melhor qualificação de seus docentes;
Ao Instituto de Ciências Biológicas da Universidade Federal do Pará, em especial ao
Programa de Pós-Graduação em Biologia dos Agentes Infecciosos e Parasitários
(BAIP), pelo esmero em contribuir para a formação de profissionais que contribuirão
para o engrandecimento de nosso Estado e de toda a Amazônia;
Ao meu orientador, Prof. Dr. Juarez Antônio Simões Quaresma, por ter aceitado o
desafio de orientação deste trabalho, assim como pela sua atenção, paciência,
compreensão, incentivo e apoio que nos foi tão valiosos até mesmo nos momentos mais
difíceis;
Ao Núcleo de Medicina Tropical (NMT/UFPA) por ter permitido a realização deste
trabalho em seus domínios.
Ao Laboratório Sabin que, gentilmente, cedeu o material para a realização desse estudo.
Aos Professores do curso de Pós-graduação a nível de Doutorado em Biologia dos
Agentes Infecciosos e Parasitários do Centro de Ciências Biológicas da Universidade
Federal do Pará.

5
Ao Prof. Dr. Ricardo Ishak que muito acreditou e me incentivou para que eu pudesse
estar realizando e concluindo mais uma das etapas importantes da minha carreira
acadêmica.
A Profa. Dra. Rosana Libonati, pela grande contribuição nas orientações em Estatística.
Ao Hospital Ophir Loyola, meu local de trabalho, pela compreensão permitindo que eu
pudesse me ausentar para realização deste curso de doutorado.
Ao Prof. Doutor Jorge Rodrigues de Souza, pesquisador do Laboratório de
imunopatologia do Núcleo de Medicina Tropical (NMT) da UFPA, pela grande
contribuição nas orientações para melhor desenvolvimento deste trabalho.
A Profa. Dra. Tinara Leila de Souza Aarão, pesquisadora do Laboratório de
imunopatologia do Núcleo de Medicina Tropical (NMT) da UFPA, professora de
imunologia da UEPA, grande colaboradora e incentivadora deste trabalho.
Aos professores da Banca de Qualificação, Profa. Dra. Ana Cecília Ribeiro Cruz, Prof.
Dr. Ismaelino Mauro Nunes Magno, Profa. Dra. Luisa Carício Martins, Profa. Dra.
Hellen Thais Fuzii, a Profa. Dra. Fabiola Elizabeth Villanova.
A Sra. Luciene Dias Cavalcante, Bibliotecária - Coordenadora da Divisão de
Documentação e Biblioteca do Hospital Ophir Loyola, pela sua prestimosa colaboração
na revisão deste documento.
Aos pacientes que participaram desta pesquisa pela importante contribuição para o
desenvolvimento deste estudo.
Aos meus queridos irmãos Rosi e Beto, pelo incentivo e pelo amor que nos mantêm
sempre unidos.
A minha esposa, Nildinha, que com paciência e sabedoria soube contornar e
compreender os momentos mais tensos;

6
Aos meus filhos, Antonio Neto e Priscila, presentes de Deus;
Aos meus pais, Antonio (in memorian) e Rosa, meus maiores incentivadores, meus
espelhos, meus amigos, meus amores, por tudo que me proporcionaram para que eu
pudesse chegar aonde cheguei.

7
SUMÁRIO
LISTA DE TABELAS 10
LISTA DE FIGURAS 11
LISTA DE ABREVIATURAS 13
RESUMO 15
ABSTRACT 16
1 INTRODUÇÃO 17
1.1 O GÊNERO HELICOBACTER 18
1.1.1 A bactéria Helicobacter pylori 19
1.1.1.1 Histórico 19
1.1.1.2 Morfologia da bactéria Helicobacter pylori 21
1.1.1.3 Genoma da bactéria Helicobacter pylori 22
1.1.1.4 Modos de transmissão do H. pylori 25
1.2 EPIDEMIOLOGIA DO H. pylori 26
1.2.1 Prevalência 26
1.3 PATOGÊNESE E FATORES DE VIRULÊNCIA 26
1.3.1 Fatores de colonização 28
1.3.1.1 Motilidade 28
1.3.1.2 Microaerofilismo 30
1.3.1.3 Urease 30
1.3.1.4 HspA e HspB 31
1.3.1.5 Adesinas 31
1.3.2 Fatores de persistência 33
1.3.2.1 Lipopolissacarídeos 33
1.3.2.2 Mimetismo molecular 33
1.3.2.3 Forma cocóide 34
1.3.2.4 Catalase e superóxido dismutase 34
1.3.3 Fatores que induzem a doença 34
1.3.3.1 Proteases e lipases 35

8
1.3.3.2 Citotoxina vacuolizante 35
1.3.3.3 Citotoxina associada ao grupo A (CagA) 38
1.4 ASSOCIAÇÃO ENTRE DOENÇAS GASTROINTESTINAIS E H.
pylori
42
1.5 RESPOSTA IMUNOLÓGICA AO H. pylori 45
1.6 PRINCIPAIS CÉLULAS ENVOLVIDAS NA RESPOSTA IMUNE
PARA H. pylori
47
1.6.1 Macrófagos 47
1.6.2 Células Epiteliais 49
1.6.3 Células Dendríticas 49
1.6.4 Linfócitos 50
1.6.4.1 Células B 50
1.6.4.2 Células T 51
1.7 LINFÓCITO T REGULADOR 52
1.7.1 Fator de Transcrição FOXP3 54
1.7.2 Mecanismos de ação das Tregs 55
1.7.3 Papel das células T reguladoras nas doenças infecciosas 56
1.8 DIAGNÓSTICO DA INFECÇÃO PELO H. PYLORI 57
1.9 OBJETIVOS 61
1.9.1 Objetivo Geral 61
1.9.2 Objetivos Específicos 61
2 MATERIAL E MÉTODOS 62
2.1 POPULAÇÃO DE ESTUDO 62
2.2 CRITÉRIOS DE INCLUSÃO E EXCLUSÃO NO ESTUDO 62
2.2.1 Critérios de inclusão 62
2.2.2 Critérios de exclusão 62
2.3 COLETA E PROCESSAMENTO DAS AMOSTRAS 63
2.3.1
2.3.2
Biópsias
Análise Histopatológica
63
63
2.4 DESCRIÇÃO DAS TÉCNICAS LABORATORIAIS 63
2.4.1 Detecção Histopatológica do H. pylori 63
2.4.2 Detecção das células CD4, CD8, CD20, Neutrófilos e Linfócitos T
regulatórios na biópsia gástrica
63

9
2.5 ANÁLISE ESTATÍSTICA 66
3 RESULTADOS 67
4 DISCUSSÃO 80
5 CONCLUSÃO 89
REFERÊNCIAS BIBLIOGRÁFICAS 90
ANEXOS

10
LISTA DE TABELAS
Tabela 1 - Resultado das amostras estudadas quanto a presença e ausência de
H. pylori.............................................................................................
67
Tabela 2 - Estatística descritiva da variável CD20.............................................
68
Tabela 3 - Estatística descritiva da variável CD4...............................................
70
Tabela 4 - Estatística descritiva da variável CD8..............................................
71
Tabela 5 - Estatística descritiva da variável Neutrófilos...................................
72
Tabela 6 - Estatística descritiva da variável FoxP3...........................................
73
Tabela 7 - Frequência de H. pylori quanto ao número de cruzes, onde 0 =
ausência; 1 = uma cruz; 2 = duas cruzes; 3 = três cruzes .................
74

11
LISTA DE FIGURAS
Figura 1 Microfotografia Eletrônica do H. pylori, demonstrando sua
morfologia curvada e seus flagelos
21
Figura 2 Representação gráfica do primeiro genoma sequenciado do H.
pylori 26695
23
Figura 3 Mosaico do gene vacA adaptado de Atherton et al.
24
Figura 4 Fatores de virulência da bactéria H. pylori
28
Figura 5 Fator de colonização: motilidade e adesão do H. pylori
29
Figura 6 Atividade ureásica do H. pylori
30
Figura 7 Adesão do H. pylori à mucosa gástrica por meio de adesinas
32
Figura 8 Mosaico do gene vacA
36
Figura 9 Representação esquemática dos dois modelos de mecanismo de
ação da citotoxina VacA
38
Figura 10 Ilha de patogenicidade Cag (citotoxin antigen associated)
39
Figura 11 Representação esquemática da ação da citotoxina CagA
41
Figura 12 Representação esquemática da resposta imunológica à infecção
pelo H. pylori
45
Figura 13 Estrutura da proteína FOXP3, com domínio repressor, central
composto por dedos de zinco (ZnF) e zíper de leucina (Zip), e o
domínio forkhead
55
Figura 14 Modelos de mecanismos de ação das Tregs
56
Figura 15 Quantidade de células CD20 encontradas em lâminas H. pylori
positivo e H. pylori negativo
69
Figura 16 Quantidade de células CD4 encontradas nas amostras contendo H.
pylori positivo e H. pylori negativo
70
Figura 17 Quantidade de células CD8 encontradas nas amostras contendo H.
pylori positivo e H. pylori negativo
71
Figura 18 Quantidade de Neutrófilos encontrados na presença de H. pylori
positivo e H. pylori negativo
72

12
Figura 19 Quantidade de células Treg (FoxP3) encontradas nas amostras de
H. pylori positivo e H. pylori negativo
73
Figura 20 Correlação negativa entre a quantidade de células CD4 e H. pylori
75
Figura 21 Correlação negativa entre a quantidade de células CD8 e H. pylori
76
Figura 22 Correlação negativa entre a quantidade de células CD20 e H.
pylori
77
Figura 23 Correlação positiva entre a quantidade de células Treg FoxP3 e H.
pylori
78
Figura 24 Correlação negativa entre a quantidade de neutrófilos e H. pylori
79

13
LISTA DE ABREVIATURAS
AA Aminoácidos
Ag Antígeno
AGS Human gastric adenocarcinoma cells
AlpA Lipoproteina associada à aderência A
AlpB Lipoproteina associada à aderência B
APC Antigen presenting cell
ATPase Enzima de ativação da bomba de prótons
BabA Blood group antigen binding adesin A
BSA Soro Albumina bovina
cagPAI Ilha de patogenicidade cag
cagA Cytotoxin associated gene A
CCR Receptores de quimiocina
CD4 Cluster of diferentation 4
CD8 Cluster of diferentation 8
CTLA Linfócito T citotóxico
DNA Deoxyribonucleic acid
ELISA Enzime-linked immunosorbent assay
h hora
FISH Hibridização in situ com fluorescência
HE Hematoxilina-Eosina
H. pylori Helicobacter pylori
IARC International Agency for Research on Câncer
IFN-γ Interferon gama
IgA Imunoglobulina A
IgG Imunoglobulina G
IgM Imunoglobulina M
IL Interleucina
INCA Instituto Nacional do Câncer
IPEX Immunodeficiency, poliendocrinopathy and enteropathy X-linked syndrome
IS Sequência de inserção
Kb Kilobites
KDa Kilodaltons
KP Lisina-prolina
LPS Lipopolissacarídeos
Mb Megabites
MALT Mucosa associated lymphoid tissue
MAP Mitogen activated protein
min minuto
NAP Proteína ativadora de neutrófilos
NF-kB Factor nuclear kB
NOD Nucleotid binding oligomerization domain containing protein
OiPA Outer inflamatory protein A
OMPs Outer membrane proteins
PCR Polimerase chain reaction
pH Produto hidrogeniônico
RNA Ribonucleic acid
s segundo

14
SabA Sialicacid binding adhesin
SOD Superóxido dimutase
T.A. Temperatura ambiente
TCD4+ Linfócito T auxiliar 4
TCD8+ Linfócito T auxiliar 8
TCR Receptor de células T
Th1 Type 1 helper T cells
Th2 Type 2 helper T cells
TLRs Toll-like receptors
TNF-α Fator de necrose tumoral alfa
Treg Célula T reguladora
vacA Vacuolating cytotoxin gene A
µm Micronmicra

15
RESUMO
A infecção do estômago pelo Helicobacter pylori é a principal causa de gastrite
crônica ativa na metade da população do mundo, muitas vezes evoluindo para formas
mais graves de doenças gastroduodenais. Nos casos em que a resposta imune do
hospedeiro encontra-se ineficiente a bactéria persiste e a inflamação pode continuar por
décadas. A ativação bacteriana das células epiteliais, células dendríticas, monócitos,
macrófagos e neutrófilos é guiada pelas células Thelp tipo 1 de resposta adaptativa,
porém se fazendo de maneira inadequada. A literatura demonstra que muitos tem sido
os caminhos postulados na tentativa de se explicar a falha da resposta imune pelo
hospedeiro, dentre eles destacam-se a apoptose das células epiteliais e macrófagos, a
inadequada função das células efetoras dos macrófagos e células dendríticas, inibição
das células T pelo Vac A e efeito supressor das células Treg. Em muitas doenças,
inclusive as formas crônicas de infecção, a desregulação do sistema imune é o ponto de
partida. No caso do H. pylori, a condição sine qua non da infecção é a presença da
gastrite crônica ativa, caracterizada pelas duas formas de infecção, a forma aguda
(neutrófilos) e a crônica (linfócitos). Foram selecionadas 50 amostras de lâminas de
pacientes que haviam se submetido ao exame de endoscopia digestiva alta e biópsia da
mucosa gástrica, oriundas do Laboratório SABIN, obedecendo os critérios de inclusão e
após ter concordado e assinado o Termo de Consentimento Livre e Esclarecido. A
análise histopatológica e a detecção histopatológica do H. pylori das amostras foram
realizadas no Núcleo de Medicina Tropical da Universidade Federal do Pará. Para
quantificação das interleucinas na mucosa gástrica foram utilizado kits de
imunohistoquímica. Todos os dados foram tabulados e analisados estatisticamente,
aplicando-se o teste t de MANN-WHITNEY e, também, foi aplicado o teste de
correlação de Pearson. Das 50 amostras estudadas, 36 (72%) apresentaram-se infectadas
pelo H. pylori e, 14 (28%) não apresentaram contaminação por essa bactéria. As células
CD20 apresentaram-se em maior quantidade nas amostras que não continham a bactéria,
com média de 8,71, enquanto que as amostras contaminadas pelo H. pylori
apresentaram uma média de 7,27. A média de células CD4 encontradas na amostra em
pacientes contaminados foi de 15,44, enquanto que nas amostras negativas para o H.
pylori a média foi 20,14. A quantidade de células CD8 presentes nas amostras
contaminadas determinou uma média de 12,31, naquelas em que a bactéria não estava
presente foi de 14,43. Avaliando-se a variável neutrófilo, encontrou-se uma média de
11,14 nos casos em que a bactéria estava presente e, 21,79 naqueles onde não foi
encontrada a bactéria. Com relação as células T reguladoras FoxP3, a média encontrada
nas amostras infectadas pelo H. pylori foi de 7,4, já nas amostras negativas para esse
microrganismo a média foi 2,92.
Palavras-chaves: Helicobacter pylori, Resposta T regulatória, Gastrites

16
ABSTRACT
The infection of the stomach by Helicobacter pylori is the major cause of chronic active
gastritis in half the world's population, often progressing to more severe forms of
gastroduodenal diseases. In cases where the host immune response is inefficient by the
bacteria and inflammation may persists for decades. The bacterial activation of
epithelial cells, dendritic cells, monocytes, macrophages and neutrophils is guided by
the type 1 cells Thelp adaptive response, but doing in inapropriate way. The literature
shows that many ways has been postulated in an attempt to explain the failure of the
immune response by the host, among them stand out apoptosis of epithelial cells and
macrophages, the inadequate effector cell function of macrophages and dendritic cells,
inhibition of T cells by Vac A and suppressive effect of Treg cells. In many diseases,
including chronic forms of infection, the deregulation of the immune system is the
starting point. In the case of H. pylori, the sine qua non condition of infection is the
presence of chronic active gastritis, characterized by two forms of infection, the acute
form ( neutrophils ) and chronic ( lymphocytes ) . 50 samples were selected slides from
patients who were submitted to the examination of high digestive endoscopy and biopsy
of the gastric mucosa, resulting from the SABIN Laboratory, following the inclusion
criteria and having agreed and signed the Instrument of Consent . Histopathological
analysis and histopathological detection of H. pylori samples were performed at the
Department of Tropical Medicine of the Federal University of Pará for quantification of
interleukins in the gastric mucosa were used immunohistochemistry kits . All data were
statistically tabulated and analyzed by applying the t MANN-WHITNEY test and also
was applied Pearson correlation test . Of the 50 samples studied , 36 ( 72 % ) were
infected by H. pylori, and 14 ( 28 % ) showed no contamination by this bacteria . The
CD20 cells showed up in greater quantities in the samples that did not contain the
bacteria , with an average of 8.71, while the samples contaminated by H. pylori had an
average of 7.27 . The average CD4 found in patients infected sample was 15.44 , while
the negative samples for H. pylori average was 20.14. The quantity of CD8 cells present
in contaminated samples determined an average of 12.31 , those in which bacteria was
not present was 14.43 . Evaluating the variable neutrophils, was found a mean of 11.14
in cases where the bacteria was present and 21.79 those where the bacteria was not
found. About FoxP3 regulatory T cells, the average found in the samples infected by H.
pylori was 7.4, while in the negative samples for this microorganism, the average was
2.92 .
Keywords : Helicobacter pylori,T Regulatory Response, Gastritis

17
1. INTRODUÇÃO
O Helicobacter pylori (H. pylori) é uma bactéria espiralada gram-negativo, que
coloniza a mucosa gastroduodenal dos seres humanos (Blaser, 1990). A fase inicial da
infecção pelo H. pylori é caracterizada pelo desenvolvimento de uma gastrite aguda,
com aumento transitório de secreção ácida e da resposta inflamatória, sendo precursora
para o desenvolvimento de gastrite crônica ativa com denso infiltrado celular na
mucosa, que com o passar dos anos tende a decrescer (Gisbert et al., 2000). Contudo, o
quadro inflamatório persiste por vários anos e causa sérios danos à mucosa gástrica,
podendo evoluir para doenças gastroduodenais mais graves (Dunn et al., 1997; Gisbert
et al., 2000; Ladeira et al., 2003).
A infecção pelo H. pylori é considerada um dos maiores fatores na patogênese
de várias doenças gastrointestinais, tais como: gastrite crônica, gastrite atrófica, úlceras
pépticas, carcinoma e linfoma gástrico (Longuimire, 1977, Machi et al., 1996, César et
al., 2005; Álvares et al., 2006). Contudo, a maioria dos pacientes infectados permanece
portadores assintomáticos por toda a vida e somente 20% podem evoluir para uma
patologia gastroduodenal mais grave no decorrer da vida (Ladeira et al., 2004;
Thomazini et al., 2006). A grande variabilidade nas manifestações clínicas da infecção
pelo H. pylori está associada a vários fatores, tais como, fatores de virulência
bacteriana, fatores ambientais e fatores genéticos dos hospedeiros, ou a combinação
destes (Coelho et al., 2000, Roder, 2002; Ladeira et al., 2003).
Muitos estudos vêm demonstrando que o H. pylori apresenta uma grande
diversidade genômica, linhagens geneticamente distintas vem sendo descritas em
diferentes regiões, sendo associadas com o desenvolvimento de diferentes doenças
(Peek et al., 1999; Blaser & Berg, 2001; Salama et al., 2001).
Nos últimos anos vem se intensificando a pesquisa de marcadores de
patogenicidade, por técnicas moleculares, na tentativa de detectar cepas bacterianas
associadas ao desenvolvimento de doenças gastroduodenais graves, como úlceras
pépticas e câncer gástrico. Entre estes marcadores, o CagA e os alelos do VacA têm
sido amplamente estudados (González-Valencia et al., 2000; Censini et al., 2001),
estando associados à patogênese das doenças gastroduodenais, fato relevante para sua
identificação (Atherton et al., 1995; Higashi et al., 2004)
Altas taxas de prevalência de doenças gastrointestinais são encontradas no
Brasil, em particular, a região Norte é que apresenta maiores valores, principalmente no

18
que diz respeito ao número de casos de câncer gástrico (Brasil, Ministério da Saúde,
2008)
As estimativas do Instituto Nacional do Câncer (INCA) para 2010 demonstram
uma incidência de 14 casos novos para cada 100 mil homens e de 8 para cada 100 mil
mulheres, variando de acordo com as diversas regiões (Brasil, Ministério da Saúde,
2010).
Apesar de o câncer gástrico vir apresentando uma diminuição da incidência e da
taxa de mortalidade em vários países, até mesmo no Brasil, este tipo de câncer ainda se
constitui em importante problema de saúde pública, especialmente no Estado do Pará
(Araujo et al., 1993; Resende et al., 2006)
A estimativa para incidência de casos novos de câncer de estômago em 2010 no
Estado do Pará, segundo o Instituto Nacional do Câncer, é de 420 casos em homens
(10,82 do total de neoplasias) e 230 casos em mulheres (6,15 do total de neoplasias),
sendo que na capital do Estado, Belém, estima-se destes casos 190 homens e 120
mulheres (Brasil, Ministério da Saúde, 2010).
1.1 O GÊNERO HELICOBACTER
O gênero Helicobacter foi definido por estudos de composição do RNA
ribossômico (Romaniuk, 1987), de seqüenciamento e hibridação do DNA da bactéria
(Goodwin et al, 1989). Juntamente com outros gêneros (Campylobacter, Arcobacter e
Wolinella), constitui a superfamília VI de bactérias gram-negativos (Vandamme et al,
1991).
A morfologia do H. pylori é homogênea, quando observada à microscopia ótica
e eletrônica, apresentando-se com estrutura encurvada ou espiralada, de superfície lisa e
extremidades arredondadas, móvel, não esporulada e microaerófila. Tem a largura
aproximada entre 0,5μm a 0,1μm e 3μm de comprimento, constituindo-se de quatro a
seis flagelos unipolares embainhados e bulbos terminais nas extremidades lisas (Warren
& Marshall, 1984).
Atualmente, o gênero Helicobacter é composto de, no mínimo, 27 espécies com
propriedades comuns, particularmente aquelas relacionadas com a vida no estômago,
podendo-se localizar no fundo e no corpo gástrico, contudo essas bactérias são
encontradas, principalmente, na região antral do estômago (Blaser & Berg, 2001). O H.
pylori pode distribuir-se de maneira focal, segmentar ou difusa ao longo da mucosa

19
gástrica (Warren & Marshall, 1984), localizando-se no interior ou sob a camada de
muco que recobre o epitélio da superfície ou das foveólas, em íntimo contato com a
membrana luminal das células epiteliais que revestem a mucosa gástrica (Ladeira et al,
2003).
Esta bactéria apresenta uma capacidade excepcional de aderência (Thomsen et
al., 1990). Estando adaptada para colonizar somente a mucosa gástrica, sendo que
raramente é observada em áreas de metaplasia intestinal. Fator de grande importância
para o seu papel na patogênese da úlcera péptica duodenal é que no duodeno o H. pylori
coloniza áreas de metaplasia gástrica (Marshall et al., 1985; Queiroz & Mendes, 1993).
O H. pylori apresenta afinidade pelas células mucíparas gástricas, fato este ligado a
composição neutra do muco gástrico, que é diferente dos mucopolissacarídeos ácidos
produzidos pelas células caliciformes da metaplasia intestinal (Queiroz & Mendes,
1993).
1.1.1 A bactéria Helicobacter pylori
1.1.1.1 Histórico
Ao longo de séculos, a presença de um microorganismo na mucosa gástrica foi
tida como improvável, por se acreditar que nesta região não haveria condições
habitáveis, em razão da presença de ácido clorídrico produzido a partir das células
parietais do estômago (Dunn et al., 1997).
Pesquisadores alemães, em 1875, detectaram na mucosa gástrica bactérias
espirais, que não cresceram em culturas, sendo, por este motivo, desprezadas (Blaser,
2005).
O cientista italiano Giulio Bizzozero, em 1892, demonstrou bactérias espiraladas
colonizando o ambiente ácido de estômago de cães. Passados três anos, Salomon
também encontrou espiroquetas no estômago de cães e gatos (Marshall & Warren,
1984).
Krienitz, em 1906, descreveu a presença de bactérias espiraladas em estômago
humano em pacientes portadores de câncer gástrico (Krienitz, 1906). Neste mesmo ano,
Balfour, encontrou microorganismos espiralados em úlceras gástricas de cães e
macacos.

20
Em 1939, Doenges (apud Marshall & Warren, 1999) descreveu também a
presença de microorganismos espiralados, similares ao descrito por Balfour, quando de
autópsias de estômagos de macacos Rhesus e seres humanos.
Palmer, em 1954, ao corar espécimes gástricas com Hematoxilina-eosina (HE),
não identificou tais microorganismos, fato este que desestimulou as pesquisas nesta
área, pois acreditava-se que a presença de bactérias guardava relação com a
contaminação da mucosa do estômago (Palmer, 1954).
Steer e Steer & Colin Jones, em 1975, utilizando microscopia ótica e eletrônica
para estudo de espécimes gástricos de pacientes sem lesão da mucosa gástrica e outros
com úlcera gástrica, demonstraram a presença de bactérias (Steer, 1975; Steer & Colin
Jones, 1975).
Robin Warren, em 1979, redescobriu a bactéria, que, posteriormente, juntamente
com Barry Marshall, utilizando-se de biópsias gástricas de pacientes portadores de
gastrite crônica, retornou com a pesquisa na tentativa de novamente isolar este
microorganismo, sendo que para isso aplicou o método de isolamento a semelhança do
gênero Campilobacter (Marshall & Warren, 1984).
Em 1982, de maneira acidental, Marshall e Warren, aumentando o período de
incubação, conseguiram, pela primeira vez, isolar a bactéria Helicobacter pylori
(Marshall & Warren, 1984; Marshall, 1986; Marshall, 1994). Inicialmente recebeu o
nome de Campilobacter pyloridis, em função de sua localização e similaridade com as
bactérias do gênero Campilobacter, observada na coloração pelo Gram e cultura em
condições microaerófilas (Dunn et al., 1997; Parsonnet et al., 1997). Passado algum
tempo o nome da bactéria passou a se chamar de Campilobacter pylori (Marshall &
Goldwin, 1987).
No ano de 1984, voluntariamente, Marshall e Warren, ingeriram caldo de cultura
da bactéria, correlacionando-a, desta feita, como agente causal da inflamação gástrica
(Marshall, 1986).
Por volta do ano de 1989, após vários estudos taxonômicos envolvendo esta
bactéria, observou-se diferenças em relação ao gênero Campilobacter, reconhecendo-se
um novo gênero, o Helicobacter. Inicialmente, envolvendo duas espécies: H. pylori,
microorganismo da mucosa gástrica humana e Helicobacter mustalae, encontrada na
mucosa gástrica de mustelídeos, popularmente conhecidos como furões (De Groote et
al., 1999)

21
Em 2005, Marshall e Warren, em razão dos estudos realizados com o
Helicobacter pylori, receberam o Prêmio Nobel de Medicina (Mandeville et al, 2009).
1.1.1.2 Morfologia da bactéria Helicobacter pylori
O Helicobacter pylori é uma bactéria gram-negativo, pertencente à Família
Helicobacteriaceae, que apresenta duas formas diferentes: a espiralada e a cocóide. A
forma espiralada (Figura 1) tem as extremidades arredondadas, estrutura curva, que
muitas vezes lembra a letra “S”, aproximadamente tem de 2,5 a 5,0μm de comprimento
e, 0,5 a 1,0μm de espessura. Em geral, possui de 4 a 6 flagelos unipolares e
embainhados e em formato de hélices. Cada flagelo possui, aproximadamente, 30μ de
comprimento e 2,5μm de espessura (Goldwin & Amstrong, 1990).
Figura 1: Microfotografia eletrônica da bactéria H. pylori
Fonte: http://www.bio.davidson.edu/people/sosarafova/Assets/Bio307/vinardone
A forma cocóide apresenta membrana circular, superfície espessa e irregular,
apresentando poucas estruturas intracitoplasmáticas e grânulos, onde se observa em seu
interior um bacilo em forma de “U” circundado pelos flagelos (Saito et al., 2003).
Em condições adversas, tais como aerobiose, pH alcalino, alta temperatura,
incubação prolongada, tratamento com antibióticos, tratamento com inibidores da
bomba de prótons, tratamento com óxido nítrico, ocorre mudança da forma espiralada
para a cocóide. O que faz pensar que a forma cocóide é uma morfologia de resistência,
capaz de suportar as condições adversas do meio, associada à transmissão da bactéria,
sendo capaz de infectar água e alimentos, retornando à forma espiralada quando as
condições do meio ambiente forem propícias (Cole et al., 1997).

22
Estudos demonstram que existem três modos de colonização da bactéria na
mucosa gástrica: aderidas à superfície das células do epitélio gástrico, livres na secreção
mucosa (sem contato com as células gástricas) e intercelular quando se encontram entre
as células epiteliais (Goldwin & Amstrong, 1990).
1.1.1.3 Genoma da bactéria Helicobacter pylori
Para se chegar ao completo seqüenciamento do genoma do H. pylori, foram
realizadas diversas análises a fim de se demonstrar a estrutura genômica e a diversidade
desta bactéria. O H. pylori foi a primeira espécie bacteriana a ter seu genoma
seqüenciado e comparado de duas cepas isoladas independentemente (Tomb et al.,
1997).
O tamanho do genoma do H. pylori é variável, entre 1.6 a 1.73 Mb. Estudos
demonstram que em aproximadamente 40% dos H. pylori isolados contém plasmídeos
que variam de tamanho, entre 1.5 a 23.2, entretanto não foram encontrados neles genes
que codificam fatores de virulência (Tomb et al., 1997).
A primeira cepa seqüenciada foi a 26695, isolada de um paciente que
apresentava gastrite, no Reino Unido (Tomb et al.,1997). No ano de 1998, o genoma da
linhagem J99 foi isolado nos Estados Unidos a partir de um paciente com úlcera
duodenal (Hancock et al., 1998).
Observa-se diferenças na organização, quando se alinha os dois genomas, onde
regiões estão invertidas ou translocadas para outra região e, nessas situações, também, é
possível observar a presença de elementos de inserção ou repetitivos (Tomb et al., 1997;
Alm et al., 1999).
O genoma de cepa “26695” consiste em aproximadamente 1,7 milhão de pares
de bases, com aproximadamente 1620 genes , enquanto que o genoma de cepa “J99”
consiste também de aproximadamente 1,7 milhão de pares de bases, com 1490 genes
(Figura 2). Quando comparadas as duas linhagens seqüenciadas, observa-se que a
organização do genoma e do proteoma de ambas as cepas são similares, onde somente
7% da J99 e 26965 são únicos para cada uma das cepas (gene 89 e 117,
respectivamente). Estudos demonstram que a maioria desses genes cepa específicos
estão localizados em uma região única e variável do genoma denominada zona de
plasticidade (Marshall et al., 1998; Alm et al., 1999). Observa-se também grandes

23
diferenças genéticas nas duas cepas seqüenciadas, chegando até a 6% de diferença nos
nucleotídeos (Pylori Gene Web Server).
Figura 2: Representação gráfica do genoma seqüenciado do H. pylori J99.
Fonte: http://www.helicobacterspain.com/imagenes/rggJ99.htm
Estudos a respeito do genoma do H. pylori demonstram que vários mecanismos
podem contribuir para a diversidade genética desta bactéria. Dentre eles destacam-se os
rearranjos intragênicos que podem ocorrer na bactéria, tais como deleções, duplicações,
inversões e translocações de partes do genoma. Observa-se que muitos desses rearranjos
podem ocorrer como resultado da presença de DNA repetitivo. O DNA do H. pylori
possui um grande número de sequências de DNA repetitivo, bem como sequências de
inserção (IS605) (Alm et al., 1999; Björkholm & Salama, 2003).
Garcia-Vallvé afirma que o H. pylori é competente para transformação natural e,
aparentemente, recombina com eficiência o DNA ganho por transformação ao genoma,
levando a uma diversidade considerável (Garcia-Vallvé et al., 2002).
Estudos apontam que as variações genômicas das cepas podem ser responsáveis
pela codificação de diferentes fatores de virulência, sendo capazes de determinar
diversos tipos de lesão no hospedeiro. Por isso, o estudo do genoma está direcionado
para a compreensão da patogênese e o conhecimento da habilidade deste organismo
para causar doença (Ladeira et al., 2003).
No banco de dados do genoma, há 62 genes da categoria “patogênese”. As cepas
sequenciadas têm Cag (ilha de patogenicidade) longas de aproximadamente 40 Kb
(sucessão de genes responsáveis pela patogênese), com mais de 40 genes. O gene cagA
(citotoxin antigen associated) é considerado marcador de ilha de patogenicidade cag

24
(cag-PAI), e está fortemente associado ao risco para desenvolvimento do câncer gástrico
(Ladeira et al., 2003). Tal ilha de patogenicidade está normalmente ausente em cepas de
H. pylori isoladas de humanos que são portadores assintomáticos (Telford et al., 1994).
O gene vacA encontra-se presente em todas as cepas do H. pylori e compreende
duas partes variáveis, “s” e “m” (Figura 3). A região s (codifica sinal peptídico)
encontra-se localizada no final da cadeia 5’ e tem dois alelos, s1 ou s2, sendo que para o
alelo s1 ainda existem três subtipos: s1a, s1b e s1c. Já a região média (m) tem os alelos
m1 ou m2. A combinação em mosaico dos alelos da região s com os alelos da região m
determina a produção de citotoxinas, responsáveis pelo grau de virulência do H. pylori
(Ladeira et al., 2003). Assim, as cepas portadoras do genótipo vacA s1/m1 produzem
grande quantidade de toxina, enquanto as cepas s1/m2, pouca ou nenhuma toxina. As
cepas vacA do tipo s1a parecem ser mais patogênicas que as s1b e s1c ou s2, estando
mais relacionadas com úlcera péptica. As cepas do tipo m1 estão associadas a um maior
risco de danos para as células epiteliais do que as do tipo m2 (Ladeira et al., 2003).
Figura 3: Mosaico do gene vacA adaptado de Atherton et al.
Fonte: Ladeira et al., 2003, p. 339.
Estudos têm demonstrado que o H. pylori possui uma grande diversidade
genômica, assim como linhagens geneticamente distintas vem sendo descritas em
diferentes regiões, estando relacionadas com o desenvolvimento de diversas doenças
(Van Doorn et al., 1999; Martins et al., 2005; Wen et al., 2007).
Membros de uma determinada população são infectados por cepas de H. pylori
que, caracteristicamente, infectam aquela população, assim como em toda a população

25
humana. Com isso pesquisadores se permitem estudar padrões de migração humana
utilizando-se de cepas do H. pylori. A exemplo disto poderia ser estabelecido que o H.
pylori em índios amazônicos tem sua origem no leste asiático em vez de européia, o que
faz crer que os primeiros imigrantes chegaram aqui há pelo menos 11.000 anos (Wen et
al., 2007).
1.1.1.4 Modos de Transmissão do H. pylori
Apesar de cerca de 50% da população mundial estar contaminada pelo
Helicobacter pylori, os mecanismos de transmissão constituem-se, ainda, em motivos
de muita controvérsia. Têm sido atribuídas as vias oral-oral e fecal-oral como sendo as
principais formas de transmissão, muito embora as taxas reais não tenham sido
estabelecidas até o momento (Marschall, 2000).
O homem é o principal hospedeiro do Helicobacter pylori, enquanto que o
estômago é considerado o seu reservatório (Dunn et al., 1997).
A água contaminada por matéria fecal constitui importante fonte de infecção
(Klein et al, 1991). Já se conseguiu isolar a bactéria das fezes de indivíduos colonizados
(Kelly et al., 1994). Mais recentemente foi relatado que a bactéria Helicobacter pylori
pode ser transmitida sexualmente por via oral-anal (Eslick, 2001).
Outro fator importante diz respeito à aglomeração familial como causa da
transmissão da bactéria Helicobacter pylori, que pode ser observado nos estudos em que
se obteve sorologia positiva em mais de 80% de irmãos colonizados com esta bactéria
(Drumm et al, 1990; Rodrigues et al., 2005). Corroborando este fato, foi constatada
uma maior incidência de H. pylori em crianças com pais infectados, em relação às
crianças cujos pais não eram portadores deste microorganismo (Malaty et al, 1991;
Gold et al., 2001).
Esta bactéria já foi isolada em culturas realizadas com material proveniente de
vômitos, fezes diarréicas e salivas (Perry et al., 2006).
Além das causas ambientais que contribuem para a transmissão do Helicobacter
pylori, há estudos que indicam que fatores do hospedeiro exercem importante papel nas
taxas de infecção e nas conseqüências patológicas induzidas pelo microorganismo
(Atherton, 1997; Ladeira et al, 2003).

26
1.2 EPIDEMIOLOGIA DO H. PYLORI
1.2.1 Prevalência
O Helicobacter pylori apresenta distribuição cosmopolita, sendo encontrada em
habitantes dos cinco continentes (Go, 2002). Cerca da metade da população do globo
terrestre apresenta gastrite induzida pelo H. pylori (Blaser & Berg, 2001). A prevalência
da infecção do H. pylori varia em conformidade com alguns fatores, tais como idade,
nível sócio-econômico e a raça. Estudos sorológicos demonstram que a prevalência de
infecção por Helicobacter pylori aumenta com a idade e é maior nos países em
desenvolvimento (Logan & Walker, 2001). Na França, a soropositividade em indivíduos
menores de 18 anos é de 70%, enquanto na Argélia e na Costa do Marfim, está em torno
de 62% e 64%, respectivamente (Ladeira et al, 2003).
Estudos demonstram que a infecção pelo Helicobacter pylori, em países
desenvolvidos, ocorre após os três ou cinco anos de idade; já em países em
desenvolvimento, observa-se que em crianças com menos de um ano de idade já podem
estar contaminadas (Mégraud et al., 1989; Carvalho et al, 1991). No Brasil, na cidade
de Belo Horizonte, foi desenvolvido um estudo com indivíduos entre sete meses e 16
anos de idade, onde pode ser observado que o indivíduo mais jovem infectado tinha 3
anos e que a positividade de infecção pela bactéria aumentava com a idade, atingindo
82% dos indivíduos maiores de 12 anos e com baixo nível sócio-econômico (Carvalho
et al, 1991).
1.3 PATOGÊNESE E FATORES DE VIRULÊNCIA
A infecção pelo Helicobacter pylori é considerada um dos mais importantes
fatores na patogênese de um amplo espectro de patologias gástricas, tais como gastrite
crônica, gastrite atrófica, úlceras pépticas, carcinoma e linfoma gástrico (César et al,
2005; Álvares et al, 2006). Entretanto, a maioria dos pacientes infectados pela bactéria
permanecem portadores assintomáticos por toda a vida, sendo que somente 20% desses
pacientes podem evoluir para uma patologia gastroduodenal mais grave no decorrer da
vida (Ladeira et al, 2004; Thomazini et al, 2006). O resultado clínico da infecção pelo
H. pylori é determinado pela complexa interação entre fatores do hospedeiro e da

27
bactéria. Enquanto que os fatores do hospedeiro permanecem desconhecidos, a
identificação dos da bactéria avança continuamente (Roder, 2002; Ladeira et al, 2003).
Sabe-se que a resistência ao ácido clorídrico é de vital importância na
patogênese do Helicobacter pylori, uma vez que, sem este atributo biológico, a bactéria
não teria condições de colonizar a mucosa do estômago (Ladeira et al, 2003).
Na fase precoce de colonização, o Helicobacter pylori necessita atravessar a
camada de muco que protege o epitélio gástrico. Esta camada é formada por um gel
viscoelástico que confere proteção química e mecânica ao revestimento epitelial,
inclusive contra bactérias (Jenks & Kusters, 2000). Contudo, lipases e proteases
sintetizadas pelo Helicobacter pylori degradam a camada de muco, o que facilita a
progressão da bactéria (Wallace et al., 1991; Hwang et al., 2002). Além disso, o
Helicobacter pylori move-se facilmente devido à morfologia em espiral e aos flagelos e,
com isso, atravessa a camada de muco, fazendo com que estabeleça íntimo contato com
as células epiteliais de revestimento (Jenks & Kusters, 2000). Outras enzimas,
sintetizadas pela bactéria, tais como superóxido dismutase, catalase e arginase,
conferem proteção contra a atividade lítica de macrófagos e neutrófilos, impedindo uma
resposta eficaz do hospedeiro (Hazell et al., 1991).
Muito embora a mucosa gástrica esteja bem protegida contra infecções
bacterianas, o H. pylori encontra-se altamente adaptado a este meio, através do
desenvolvimento de mecanismos próprios que garantem a sua sobrevivência. Vários são
os fatores envolvidos nesse processo, podendo ser divididos em três classes (Figura 4):
fatores de colonização; fatores de persistência e fatores que induzem doença (Moran,
1996).

28
Figura 4 – Fatores de virulência da bactéria H. pylori (Moran, 1996)
1.3.1 Fatores de colonização
Tais fatores permitem ao patógeno se estabelecer no hospedeiro.
Aproximadamente, 20% da bactéria H. pylori encontram-se aderidas ao nível da
superfície do muco das células epiteliais gástricas. Espécimes gástricas quando
observadas a luz da microscopia eletrônicas encontram-se aderidas ao nível das junções
intercelulares, algumas vezes no fundo dos espaços celulares, com mudança do
citoesqueleto no local de adesão e, também, internalizadas nas células epiteliais,
sugerindo que a adesão envolve interações moleculares especializadas com a mucosa
gástrica (Chen et al., 2001, Necchi et al., 2007).
Para que o H. pylori possa sobreviver no ambiente gástrico esta capacidade de
colonizar a superfície do epitélio gástrico é de fundamental importância, sendo que
vários fatores de colonização encontram-se envolvidos, dentre eles destacam-se como
principais:
1.3.1.1 Motilidade
A forma espiralada da bactéria, associada à presença de quatro ou seis flagelos
unipolares faz com que a bactéria H. pylori rapidamente se movimente na luz do

29
estômago (onde o pH é muito baixo), atravessando o lago mucoso, que é um dos
mecanismos de defesa da mucosa gástrica (Figura 5), para uma área onde o pH
encontre-se próximo do neutro, favorecendo seu crescimento. Tal motilidade constitui-
se em um mecanismo imprescindível para que a bactéria continue colonizando o
estômago, ainda que mecanismos de defesa do hospedeiro, como a acidez e
peristaltismo gástrico, possam atuar, permitindo a aproximação da bactéria às células
epiteliais gástricas (Aguilar et al., 2001). Os flagelos do H. pylori são compostos por
sub unidades de flagelina e cobertos com uma dupla camada de fosfolipídeos, que tem
como finalidade protegê-los contra a acidez gástrica (Fox & Wang, 2002).
Figura 5 - Fator de colonização: motilidade e adesão do H. pylor.
Fonte: h.pylori.info/.

30
1.3.1.2 Microaerofilismo
A bactéria H. pylori é microaerofílica, ou seja, necessita de pequenas
quantidades de oxigênio para sobreviver (Olson & Maier, 2002).
1.3.1.3 Urease
Dada a sua potente atividade ureásica o H. pylori é capaz de sobreviver no meio
ácido do estômago (Figura 6). A enzima urease, que é uma proteína de alto peso
molecular (500 a 600KDa), atua promovendo a hidrólise da uréia [CO(NH2)2], que está
presente em condições fisiológicas no suco gástrico, convertendo a uréia em dióxido de
carbono (CO2) e amônia (NH3). As moléculas de amônia reagem com a água formando
íons de amônio (NH4+) e bicarbonato (HCO3
-), que são capazes de neutralizar o meio ao
redor da bactéria, atingindo um pH em torno de 6 (Aguilar et al.,2001). A amônia atua
como receptor de íons H+
, gerando pH neutro no interior da bactéria, o que confere ao
Helicobacter pylori resistência à acidez gástrica (Weeks & Sachs, 2001). Assim, a
bactéria fica protegida dos efeitos deletérios do pH ácido do estômago. A urease
compreende 6% do total de proteínas sintetizadas pela bactéria, o que representa grande
investimento energético motivado pela sua ação essencial como fator de colonização
(Jenks & Kusters, 2000).
Figura 6 - Atividade ureásica do H. pylori. Fonte: h.pylori.info/.

31
A maior parte da urease sintetizada pela bactéria situa-se em seu citoplasma. A
produção de amônia depende da entrada de uréia na bactéria, que é controlada por uma
proteína de membrana sensível ao pH. Esta proteína é codificada por um gene da
família urease, conhecido como urel (Walsh, 2000; Weeks, 2000; Weeks & Saches,
2001). Cepas de Helicobacter pylori com deleção de urel não sobrevivem em pH ácido
(Ladeira et al, 2003). A entrada de uréia na bactéria é acelerada em pH 5 e diminuída
em pH 7 (Weeks, 2000). A entrada de uréia é altamente específica, não sendo
facilmente saturada, e independe de temperatura e energia (Weeks & Sachs, 2001).
Portanto o Helicobacter pylori possui um mecanismo que permite a liberação do
substrato uréia sobre a urease em condições que é necessária a alcalinização local do
meio ambiente (Ladeira et al, 2003). A proteína urel atua como portão de um canal, que
também permite o refluxo de urease, aumentando o pH periplasmático e do
microambiente próximo, prevenindo acúmulo tóxico de uréia dentro da bactéria (Walsh,
2000; Weeks & Sachs, 2001).
A proteção do H. pylori pela urease ainda confere uma apoptose das células
gástricas in vitro e a inibição da somatostatina gástrica liberada em animais, o que traria
como conseqüências alterações na fisiologia da digestão como um todo (Aguilar et al.,
2001).
Desta forma, o H. pylori fica protegido dos efeitos adversos do pH ácido do
estômago, o que lhe confere acesso à camada protetora de muco. Somado a este fato, a
amônia ainda tem atividade citotóxica, aumentando a permeabilidade da célula epitelial
para prótons e, possivelmente, participe como mediador da resposta imunológica local,
uma vez que é potente ativador de monócito in vitro (Gobert et al., 2002).
1.3.1.4 HspA e HspB
O H. pylori possui as proteínas de choque térmico (Hsp) homólogas as de
humanos, HspA e HspB, cuja a expressão destas proteínas acredita-se aumentar a
atividade da urease e influenciem na habilidade da bactéria tolerar as condições
extremas do estômago (Dunn & Blaser, 1997).
1.3.1.5 Adesinas
Para garantir a sua colonização, o H. pylori necessita aderir-se firmemente as
células gástricas, para tanto lança mão de um conjunto de substâncias que facilitarão

32
essa adesão, as adesinas (Figura 7). Presentes na superfície da bactéria, as adesinas são
estruturas as quais reconhecem especificamente determinados receptores da mucosa
gástrica, que são estruturas glicoconjugadas, ligando-se e iniciando a colonização
bacteriana (Blaser, 1992; Dunn et al., 1997; Covacci et al., 1999).
Figura 7 - Adesão do H. pylori à mucosa gástrica por meio de adesinas.
Fonte: h.pylori.info/.
Estudos demonstram que o mecanismo de adesão da bactéria ao epitélio
gástrico ainda não está totalmente elucidado, estes estudos fazem referência que a
adesina BabA (blood-group antigen-binding adhesin A) do H. pylori utiliza como
receptores os antígenos fucolizados de grupo sanguíneos Lewis b (Leb) e H tipo 1 (H-1),
expressos na superfície epitelial da mucosa gástrica (Covacci et al., 1999; Yamaoka et
al., 2008). Parece que a aderência promovida pela BabA tem um papel crítico no que
diz respeito a transferência de fatores de virulência bacterianos, que são responsáveis
em promover lesão na mucosa do estômago, quer seja de maneira direta ou em
decorrência da resposta inflamatória e/ou auto-imunidade (Ilver et al., 1998). Além do
que, bactérias que se mantêm aderidas firmemente estão menos vulneráveis à acidez
gástrica e, também, não são eliminadas através dos movimentos peristálticos (Silveira et
al., 2005).
O estudo realizado por Ilver et al. (1998) demonstraram que a maior freqüência
de úlcera péptica encontra-se naqueles indivíduos que apresentam grupo sanguíneo O,

33
que equivale ao antígeno de Lewis b, em razão da carga bacteriana ser maior nesses
indivíduos (Ilver et al., 1998).
Em estudos mais recentes observa-se que a análise da sequência genômica das
cepas 26695, J99 e HPAG1 H. pylori, mostra a presença de cinco famílias maiores de
proteínas da membrana externa, que corresponde a 4% do genoma bacteriano. Dentre
estas, temos as adesinas, que se ligam a carboidratos modificados nas glicoproteínas das
células dos hospedeiros, como a adesina ligada ao antígeno do grupo sanguíneo (BabA),
adesina ligada ao ácido siálico (SabA), lipoproteínas associada à aderência (AlpA e
AlpB), proteína inflamatória da membrana externa (OipA) (Yamaoka, 2008; Matteo et
al., 2010).
1.3.2 Fatores de persistência
A literatura demonstra que a bactéria H. pylori permanece no estômago por um
período longo de tempo, muitas vezes permanecendo por toda a vida do indivíduo
infectado. Uma vez que a infecção pelo H. pylori é crônica, é capaz de ativar a resposta
imunológica no hospedeiro, determinando um processo inflamatório crônico. Dentre os
mecanismos de persistência, destacam-se:
1.3.2.1 Lipopolissacarídeos (LPS)
Também denominados endotoxinas, os LPS são glicolipídios tóxicos
fosforilados, presentes na superfície das bactérias gram-negativos, atuando como
principal antígeno de superfície (Berstad et al., 2001).
1.3.2.2 Mimetismo molecular
O mimetismo de antígenos é considerado um dos mais importantes mecanismos
de evasão do sistema imunológico do hospedeiro (Aguilar et al., 2001; Aguiar et al.,
2002). A estrutura da cadeia O-específica do LPS em diferentes tipos de H. pylori pode
mimetizar antígenos da estrutura do grupo sanguíneo Lewis (Lea, Le
b, Le
x, Le
y) que
estão presentes na mucosa do estômago normal de seres humanos e a expressão desses
antígenos Lewis na superfície da bactéria pode camuflar a bactéria e contribuir para a
sobrevivência da mesma no estômago (Aguilar et al., 2001; Berstad et al., 2001; Aguiar

34
et al., 2002). O papel do mimetismo, portanto, é permitir o escape da bactéria aos
mecanismos de reconhecimento do sistema imune, o que contribui para a cronicidade da
infecção (Dunn et al., 1997; GO & Crowe, 2000). Segundo Wirth et al.(1997), a
bactéria H. pylori não expressaria antígenos Lewis apenas, mas também a expressão
fenotípica Le do hospedeiro, o que sugere uma adaptação da bactéria ao hospedeiro.
1.3.2.3 Forma cocóide
Em determinadas condições, que envolva estresse ambiental, a bactéria H. pylori
modifica-se, convertendo a sua forma espiral para a forma cocóide, considerada uma
forma de resistência, originada da forma bacilar, que se adapta transitoriamente às
condições impróprias do ambiente, garantindo ao microorganismo sua sobrevivência ao
meio até que esta possa chegar ao estômago do hospedeiro onde irá replicar-se (Costa et
al., 1999).
1.3.2.4 Catalase e superóxido dismutase
As enzimas catalase e superóxido dismutase (SOD) extracelular são produzidas
em quantidades satisfatórias e são considerados fatores de resistência da bactéria aos
mecanismos líticos oxidativos dos fagócitos polimorfonucleares, o que causa a morte
desses últimos e determina lesão epitelial aguda, sendo importantes fatores de
sobrevivência da bactéria frente à mucosa gástrica inflamada (Ladeira et al., 2003).
1.3.3 Fatores que induzem a doença
Além dos fatores de colonização e persistência, a bactéria H. pylori possui
fatores de virulência que são responsáveis pela produção de substâncias tóxicas que
atuam agredindo as células epiteliais gástricas para obtenção de nutrientes e de melhor
fixação na mucosa gástrica, entretanto tais fatores determinam, normalmente, sérios
danos ao hospedeiro (Yakoob et al., 2009). Dentre esses fatores, destacam-se:

35
1.3.3.1 Proteases e lipases
Enzimas sintetizadas pela bactéria H. pylori, atuam diminuindo o efeito protetor
da camada de muco do epitélio gástrico, facilitando a progressão da bactéria, levando à
penetração do ácido na superfície das células epiteliais, causando sérios danos à mucosa
gástrica (Vinall et al., 2002).
1.3.3.2 Citotoxina vacuolizante (VacA)
Considerada a maior toxina secretada pelo H. pylori sua ação citotóxica
encontra-se produzida em mais de 50% dos casos isolados de H. pylori, estando
relacionada epidemiologicamente com a destruição dos tecidos e úlceras pépticas
(Aguilar et al., 2001; Muller et al., 2002).
Na literatura encontram-se vários estudos demonstrando diversos mecanismos
da atividade tóxica da VacA contra as células do hospedeiro (Aguilar et al., 2001;
Ashour et al., 2002, Muller et al., 2002; Ando et al., 2006). Acredita-se que esta toxina
induz alterações no trânsito intracelular vesicular em células eucarióticas, o que leva a
formação de grandes vacúolos contendo sinais de recentes lisossomos e endossomos.
Tal mecanismo interfere na apresentação de antígeno ao sistema imunológico por
prejudicar o processo e apresentação do antígeno pela célula apresentadora de antígeno
(Aguilar et al., 2001; Muller et al., 2002; Gebert et al., 2003, Ando et al., 2006).
Cerca de 50% a 60% dos diferentes tipos da bactéria H. pylori produzem a
citotoxina vacuolizante VacA, muito embora o gene da VacA esteja sempre presente
em todas as cepas bacterianas isoladas (Ando et al., 2006).
Apesar do gene da citotoxina vacuolizante (VacA) estar presente em todos os
tipos de H. pylori, o mesmo apresenta variações estruturais entre os alelos vacA,
particularmente em duas regiões. Essas variações alélicas se concentram na região N-
terminal (s1a, s1b, s1c e s2) e próximo à região do meio (m1 e m2), que descreve as
diferenças na produção da citotoxina ou na atividade vacuolizante (Figura 8). Assim,
alelos VacA estão relacionados com incremento de úlceras pépticas. As regiões s e m
têm sido associadas a diferentes manifestações clínicas; assim, o alelo s1a/m1 é
associado com maior destruição do epitélio gástrico e úlcera duodenal (Ladeira et al.,
2003; Ando et al., 2006).

36
Figura 8 – Mosaico do gene vacA. Fonte: Adaptado de Athernon et al., 1997
Estudos mostram ainda que s1a/m1 tem maior virulência, enquanto vacAs2 são
raramente associados com doença ulcerosa péptica, assim como são raras em
determinadas populações. Com base nessas informações, os genótipos vacA não podem
ser usados de maneira segura no sentido de discernir o risco de desenvolvimento de
doença gastroduodenal no hospedeiro. Esses estudos ainda demonstram que o alelo s1c
é fenotipicamente diferente de s1a e s1b (Aguilar et al. 2001; Yamaoka et al., 2002;
Ladeira et al., 2003; Ando et al., 2006).
O gene vacA codifica uma protoxina de aproximadamente 140 kDa de massa.
Tal prototoxina é processada antes de ser secretada, sendo retirada da região
aminoterminal a porção do peptídeo sinal e, posteriormente, da região carboxiterminal,
o fragmento transmembrânico, que é separado da toxina no momento da secreção,
resultando em uma toxina com peso de aproximadamente 88 kDa. A proteína
processada pode ser secretada ou permanecer na superfície da bactéria (Cover et al.,
1994; Telford et al., 1994).
A proteína secretada forma uma estrutura monomérica que é composta na região
aminoterminal pelo domínio P37 (34kDa) e na porção carboxiterminal pelo domínio
P58 (54kDa), esses dois domínios estão interligados por uma região de loop, sensível a
ação de proteases (Telford et al., 1994; Cover et al., 1994).
As estruturas monoméricas quando expostas a um pH ácido (<5,5) ou básico
(>8,0) unem-se formando uma estrutura oligomérica, que pode ser composta por 6 a 7
estruturas monoméricas e possui baixa atividade citotóxica. Mas em condições

37
favoráveis a estrutura oligomérica se dissocia e os monômeros exercem alto poder
citotóxico (Mcclain et al., 2000).
A ligação da VacA com a superfície da célula gástrica é o primeiro passo para a
intoxicação celular. Estudos de Padilla et al. (2000) demonstraram que a VacA possui a
capacidade de ligar-se a diferentes tipos de células e tem múltiplos sítios de ligação na
superfície das células.
A porção P37 da proteína VacA é considerada importante na internalização da
toxina e no desenvolvimento do processo de intoxicação celular e a porção P58 é
responsável pela ligação do monômero ao receptor celular (Mcclain et al., 2000).
Alguns estudos demonstram que a citotoxina VacA é capaz de alterar a junção
célula-célula e modificar a resistência elétrica trans-epitelial celular, criando uma rota
paracelular (célula → bactéria) de permeabilidade de pequenas moléculas orgânicas e
íons, tal como, Fe3+ e Ni2
+, que são essenciais para o crescimento da bactéria.
Entretanto, o mecanismo que causa esse processo ainda não foi identificado (Papini et
al., 1998; Montecucco et al., 2001).
Experimentos de Tombola et al.(1999) têm demonstrado que a inserção da
toxina VacA na membrana plasmática leva a formação de poros ânions seletivos,
importantes a sobrevida da bactéria na célula.
Reyrat et al. (2000) propõem dois diferentes modelos de mecanismos de ação da
VacA. O primeiro modelo propõe que o domínio P58 media a ligação à célula alvo, e a
porção P37 seria injetada no citosol da célula alvo, onde enzimaticamente alteraria alvos
citoplasmáticos específicos levando a formação de vacúolos e a apoptose. Entretanto,
ainda não se conseguiu demonstrar alguma atividade enzimática da porção P37, nem tão
pouco foram descritos possíveis alvos citoplasmáticos (Galmiche et al., 2000; Reyrat et
al., 2000; Papini et al., 2001).
O segundo modelo descreve que tanto a porção P37, quanto a porção P58 estão
envolvidos na capacidade de inserir-se na membrana plasmática da célula alvo e na
formação de canais condutores de íons. Sendo que a porção P58 seria responsável pela
interação com a membrana plasmática e, também, pela inserção do P37 na porção
transmembrana. O domínio P37 é capaz de suportar a ação dos lisossomos, importante
na manutenção dos canais, uma vez que este é endocitado e transportado para o
citoplasma dentro de um endossoma (Tombola et al., 1999). No endossoma, a toxina
VacA aumenta a permeabilidade dos ânions deste compartimento, determinando a
ativação da bomba de prótons ATPase, levando à introdução do Hidrogênio no
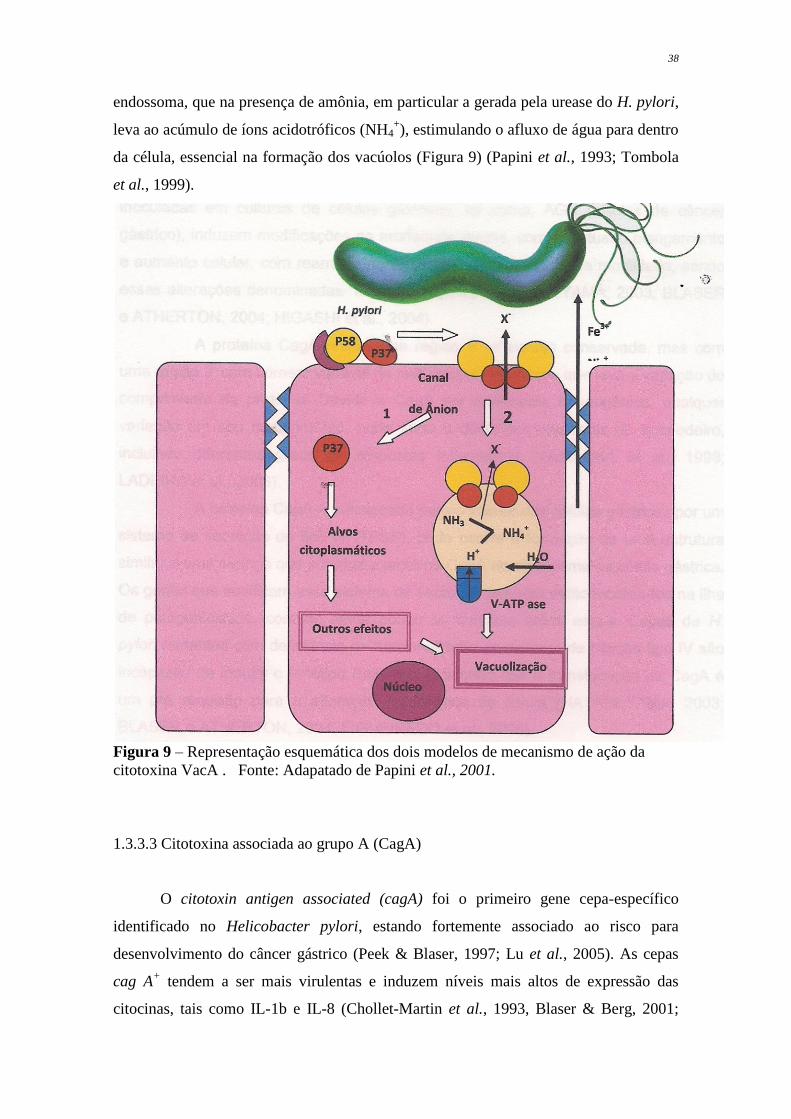
38
endossoma, que na presença de amônia, em particular a gerada pela urease do H. pylori,
leva ao acúmulo de íons acidotróficos (NH4+), estimulando o afluxo de água para dentro
da célula, essencial na formação dos vacúolos (Figura 9) (Papini et al., 1993; Tombola
et al., 1999).
Figura 9 – Representação esquemática dos dois modelos de mecanismo de ação da
citotoxina VacA . Fonte: Adapatado de Papini et al., 2001.
1.3.3.3 Citotoxina associada ao grupo A (CagA)
O citotoxin antigen associated (cagA) foi o primeiro gene cepa-específico
identificado no Helicobacter pylori, estando fortemente associado ao risco para
desenvolvimento do câncer gástrico (Peek & Blaser, 1997; Lu et al., 2005). As cepas
cag A+ tendem a ser mais virulentas e induzem níveis mais altos de expressão das
citocinas, tais como IL-1b e IL-8 (Chollet-Martin et al., 1993, Blaser & Berg, 2001;

39
Hull et al., 2001). Pacientes infectados por cepas que expressam cagA têm
probabilidade três vezes maior de desenvolver câncer gástrico do que aqueles infectados
por cepas cagA- (Parsonnet et al, 1997). O gene cagA é considerado marcador da ilha de
patogenicidade cag (cag-PAI), que possui de 35 a 40 Kb, comporta 31 genes (Blaser &
Berg, 2001) e é encontrada em cerca de 60% das cepas ocidentais. A ilha cag-PAI é um
componente do genoma do Helicobacter pylori que contém genes homólogos aos de
outras bactérias que codificam componentes do sistema de secreção do tipo IV, que atua
como agulha e serve para injetar moléculas efetoras da bactéria na célula hospedeira,
permitindo que a bactéria module vias do metabolismo celular da célula hospedeira
(Figura 10), incluindo a expressão de proto-oncogenes (Covacci et al, 1999; Ladeira et
al, 2003).
Figura 10 - Ilha de patogenicidade Cag (citotoxin antigen associated).
Fonte: Suerbaum & Michetti, 2002, p. 1117.
Componentes do Helicobacter pylori induzem alterações na fosforilação de
tirosinas, nas vias de sinalização e de transdução da célula hospedeira, resultando em
rearranjos do citoesqueleto e alterações morfológicas que estimulam a célula a se
espalhar e se alongar de maneira idêntica à produzida pelo fator de crescimento de

40
hepatócitos (Segal et al, 1996; Asahi et al, 2000; Ondebreit et al, 2000; Stein et al,
2000). Além disso, fatores da bactéria podem induzir a desfosforilação de proteínas
normalmente fosforiladas (Atherton, 2000).
A proteína cagA do Helicobacter pylori atua como um antígeno altamente
imunogênico. A estrutura do gene revela uma região 5’ altamente conservada, mas com
uma região 3’ com número variável de seqüências repetitivas, o que leva à variação do
comprimento da proteína. Como a proteína CagA é fortemente imunogênica, qualquer
variação em seu comprimento pode levar a diferentes respostas do hospedeiro, incluindo
diferentes graus de resposta inflamatória (Ladeira et al, 2003).
Estudos da variabilidade da região 3’ do gene cagA para investigar as diferenças
nesta região estariam relacionadas a diferentes processos patológicos, demonstraram
que 86% de uma das variantes eram provenientes de pacientes com câncer gástrico e
concluíram que outras diferenças na região 3’ do gene cagA podem estar relacionadas a
diferentes processos patológicos (Yamaoka et al, 1998).
A função da citotoxina CagA ainda não está totalmente elucidada, entretanto,
alguns experimentos têm demonstrado que cepas cagA positivas quando inoculadas em
cultura de células gástricas, como por exemplo, AGS (células do câncer gástrico),
induzem alterações na morfologia destas células, com acentuado alongamento e
aumento celular, determinando rearranjo no citoesqueleto e aumento da motilidade,
alterações essas denominadas de “fenótipo Beija-flor” (Hatakeyama, 2003; Blaser &
Atherton, 2004; Higashi et al., 2004).
Assim, o CagA é um marcador de cepas virulentas do H. pylori, associadas mais
comumente, com úlcera péptica e câncer gástrico, e sua detecção tem se mostrado
clinicamente relevante, ao menos na maioria dos países do Ocidente (Kuipers et al,
1995; Higashi et al, 2002; Higashi et al, 2004).
Experimentos demonstram que a proteína CagA é translocada para dentro das
células do estômago através de um sistema de secreção do tipo IV (TFSS), onde ocorre
a formação de uma estrutura similar a uma seringa que introduz a proteína CagA no
citoplasma da célula gástrica. Embora pouco se conheça a respeito, sabe-se que os genes
que codificam esse sistema de secreção também estão localizados na ilha de
patogenicidade. Esses experimentos revelam que cepas do H. pylori mutantes com
deficiência na funcionalidade do sistema de injeção tipo IV são incapazes de induzir o
fenótipo Beija-flor, o que faz crer que a translocação da CagA é um pré requisito para

41
alteração morfológica da célula (Hatakeyama, 2003; Blaser & Atherton, 2004;
Figueiredo et al., 2005).
A molécula CagA apresenta sítios de fosforilação de tirosina, reconhecidos pelos
domínios SHP2, presentes nas moléculas sinalizadoras, o que faz com que essas
moléculas se liguem aos receptores tirosinoquinases. Tais receptores podem ativar as
proteínas Ras assim como a cascata de Map-quinase (proteinoquinases ativadas por
mitógenos), determinando sinalização ao núcleo da célula e alterando o citoesqueleto da
mesma, estimulando a proliferação e diferenciação celular (Hatakeyama, 2003; Blaser
& Atherton, 2004; Higashi et al., 2004). Contudo, experimentos de Higashi et al.
(2004), demonstraram que a citotoxina CagA pode, através das proteínas sinalizadoras,
enviar sinalização diretamente ao núcleo da célula, independentemente da ativação de
proteínas Ras, conforme se observa na Figura 11.
O prolongado estímulo das proteínas sinalizadoras pela toxina CagA, determina
uma sinalização inadequada estimulando a apoptose celular. Somado a este fato, uma
replicação celular aumentada determina uma ocorrência maior de mutações, e o
acúmulo dessas mutações pode condicionar a uma expressão de oncogenes, alterando o
controle celular e, conseqüentemente, o desenvolvimento de neoplasias (Higashi et al.,
2002; Hatakeyama, 2003; Blaser & Atherton, 2004).
Figura 11 - Representação esquemática da ação da citotoxina CagA.
Fonte: Adaptado de Higashi et al., 2004.

42
1.4 ASSOCIAÇÃO ENTRE DOENÇAS GASTROINTESTINAS E H. PYLORI
A fase inicial da infecção pelo H. pylori é caracterizada pelo desenvolvimento de
uma gastrite aguda, com aumento transitório de secreção ácida e da resposta
inflamatória, sendo precursora para o desenvolvimento da gastrite crônica ativa com
denso infiltrado celular na mucosa, que com o passar dos anos tende a decrescer
(Gisbert et al., 2000). Entretanto, o quadro inflamatório perdura por vários anos
causando sérios danos à mucosa gástrica, podendo evoluir para patologias mais graves
(Blaser, 1990; Dunn et al., 1997; Suerbaun & Michetti, 2002; Ladeira et al., 2003).
Estudos de auto-inoculação, relatados por Marshall, são a maior evidência de que a
infecção pelo H. pylori está associada ao desenvolvimento de gastrite crônica (Warren
&Marshall, 1984).
Predominantemente, a infecção pelo H. pylori acomete a região do antro
gástrico, o que estimula as células G a produzirem gastrina e, conseqüentemente, eleva a
produção de ácido clorídrico. Essa elevação da quantidade de ácido no estômago faz
com que haja um maior afluxo de ácido para o duodeno, determinando uma agressão ao
tecido intestinal, que num mecanismo de auto-defesa sofre metaplasia para tecido
gástrico, contribuindo para colonização do duodeno pelo H. pylori. O grande fluxo de
ácido para o duodeno e a presença da colonização deste tecido pelo H. pylori. Levam ao
desenvolvimento da úlcera duodenal (Suerbaun & Michetti, 2002; Calam & Baron,
2001).
Ocorrendo a gastrite na região do corpo gástrico, observa-se uma diminuição na
produção de ácido clorídrico, em razão da inibição das células enterocromomatóficas,
produtoras de histaminas e células parietais. É comum nestes casos a observação de
destruição das glândulas gástricas (gastrite atrófica) assim como da diminuição da
camada de muco, ambiente este favorável ao desenvolvimento de úlcera gástrica e/ou
câncer gástrico (Suerbaun & Michetti, 2002).
Marshall e Warren observaram que a infecção pelo H. pylori estava associada
com o desenvolvimento de úlcera duodenal, e esta observação também foi rapidamente
confirmada e aplicada para os casos de úlcera gástrica (Warren & Marshall, 1984). Em
1994, a conferência do National Institutes of Health concluiu que o H. pylori era a
maior causa de doença ulcerosa péptica, recomendando que a associação entre úlcera
péptica e aquela bactéria deveria ser tratada através da erradicação dessa bactéria
(Marshall, 1994; Dunn et al., 1997, Sokol & Hoffenberg, 1997, Silva et al., 2004).

43
Estudos têm demonstrado que a infecção pelo H. pylori pode causar uma
elevação na produção de gastrina, determinando um quadro de hipergastrenemia, e que
com a erradicação da bactéria com antibióticos, a secreção da gastrina retornaria a
níveis normais (Silva et al., 2004). Entretanto, os mecanismos pelos quais o H. pylori
mantém as células G em estados hiperfuncionantes ainda não são conhecidos (Blaser,
1990; Chehter, 1999). Pacientes infectados pelo H. pylori possuem uma diminuição no
número de células G da mucosa gástrica, sofrendo sensível aumento após a erradicação
da bactéria. A maioria dos portadores de úlcera duodenal possuem esvaziamento
gástrico mais acelerado que o normal, fazendo com que haja um contato mais intenso da
mucosa do duodeno com a secreção ácida (Moran, 1996). Este contato prolongado da
mucosa do duodeno com a excessiva quantidade de secreção ácida do estômago pode
determinar o aparecimento de metaplasia gástrica no tecido duodenal. Nos pacientes
infectados pelo H. pylori ocorre comumente duodenite, pois as áreas de metaplasia
gástrica do duodeno encontram-se constantemente colonizadas pela bactéria. A presença
do H. pylori no duodeno, somada à ação contínua da secreção cloridropéptica do
estômago, podem ser os determinantes pela perpetuação da úlcera (Figueiredo et al,
2005; Suerbaun & Michetti, 2002).
Geralmente, a úlcera gástrica é única, localizando-se normalmente ao nível da
pequena curvatura no antro gástrico, próximo a incisura angularis, em região adjacente a
mucosa secretora de ácido (Martins et al., 2002). A úlcera gástrica encontra-se
relacionada à deficiência dos fatores de defesa da mucosa do estômago, que não são
capazes de proteger o epitélio contra os efeitos corrosivos do ácido e da pepsina
(Martins et al., 2006). Entretanto, ao que parece, o maior responsável pela diminuição
da resistência da mucosa ao ataque do ácido é a inflamação crônica (Censini et al.,
2001). A responsabilidade pela diminuição da defesa da mucosa gástrica está
relacionada com a associação entre o H. pylori e gastrite (Censini et al., 2001; Martins
et al., 2002). A infecção pelo H. pylori leva a um processo inflamatório agudo ou
crônico da mucosa gástrica (Blaser, 1990; Warren & Marshall, 1984; Martins et al.,
2002). Desta maneira as úlceras gástricas ocorrem tanto na presença de gastrite crônica
superficial como atrófica, desenvolvendo-se no limite do processo inflamatório (Blaser,
1990; Martins et al., 2002).
Estudos demonstram que o H. pylori produz várias proteínas que parecem
mediar ou facilitar seus efeitos deletérios sobre a mucosa gástrica (Martins et al., 2002).
A associação da citotoxina CagA com aumento da inflamação gástrica e

44
conseqüentemente o aparecimento de quadros clínicos mais graves, tem sido bastante
ressaltada atualmente (Martins et al., 2002). As cepas virulentas (CagA+) induzem forte
processo inflamatório, com denso infiltrado de neutrófilos, que causam graves danos à
mucosa gástrica (Lin et al., 2000; Queiroz et al., 2000, Martins et al., 2002).
Estudos sugerem que a soroprevalência para o antígeno CagA do H. pylori em
adultos com úlcera gástrica está aumentada, existindo uma associação entre a presença
do CagA e o risco aumentado de desenvolver úlcera péptica e câncer gástrico (Williams
& Pounder, 1999, Lin et al., 2000; Martins et al., 2002).
Estudos sorológicos demonstram que pacientes com úlcera péptica e tumor
gástrico são mais freqüentemente infectados por cepas virulentas, em torno de 60% a
100%, o que leva a crer que o gene CagA pode ser considerado um marcador genotípico
e fenotípico para linhagens virulentas (Lin et al., 2000; Queiroz et al., 2000; Censini et
al., 2001; Covacci et al., 1999; Williams & Pounder, 1999).
Inúmeros estudos, mais recentemente, apontam evidências de que exista uma
relação entre H. pylori e câncer gástrico (Palli, 1997; Butov et al., 1998; Wang et al.,
1998; Kim et al., 1997; Rangel et al., 2003; Perez-Perez et al., 2005).
A International Agency for Research on Câncer, World Health Organization
(IARC), no ano de 1994, baseando-se no resultado de nove estudos retrospectivos de
casos-controle e quatro estudos coorte, demonstrou uma associação causal entre
infecção por H. pylori e o câncer gástrico (Tanida et al., 1997; Rangel et al., 2003).
Em alguns estudos, há relato de que a gastrite atrófica decorrente de vários anos
de infecção pelo H. pylori funcionaria como lesão precursora da metaplasia intestinal,
para o desenvolvimento da neoplasia gástrica maligna (Huang et al., 1998; Debongnie
et al., 1997; Shibata et al., 1997, Rangel et al., 2003).
Estudos de sorologia para o H. pylori em portadores de neoplasia maligna do
estômago indicam que um maior risco de desenvolvimento da neoplasia estaria
relacionada à infecção pelas cepas CagA desta bactéria (Torres & Pérez, 1998, Rangel
et al., 2003)
O reconhecimento do papel do H. pylori na associação com o câncer gástrico
proporcionou um grande avanço do conhecimento científico, ajudando a elucidar
inúmeros aspectos fisiopatológicos (El-Omar et al., 2000; Uemura et al., 2001).
Contudo, é sabido que alguns aspectos dessa associação ainda permanecem bastante
obscuros, sendo necessário que se prossiga com investigações no que diz respeito a real
45
virulência do H. pylori, a genética do hospedeiro, assim como fatores ambientais e
epidemiológicos.
1.5. RESPOSTA IMUNOLÓGICA AO H. pylori
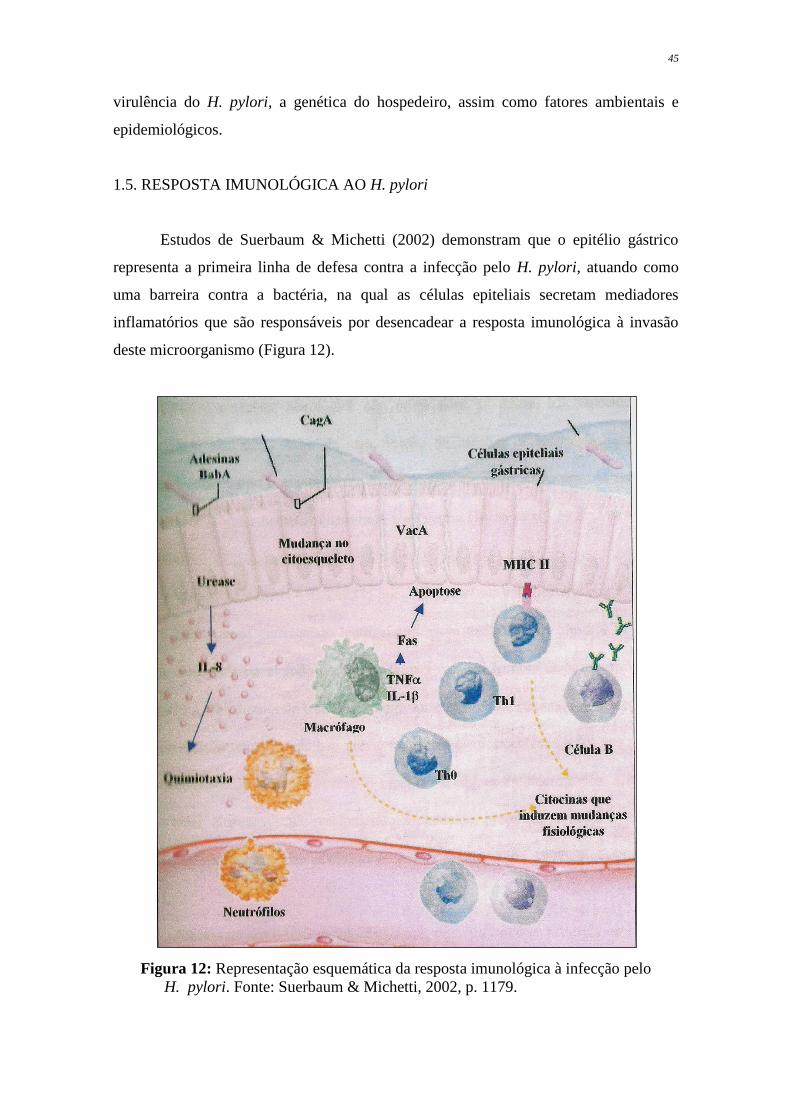
Estudos de Suerbaum & Michetti (2002) demonstram que o epitélio gástrico
representa a primeira linha de defesa contra a infecção pelo H. pylori, atuando como
uma barreira contra a bactéria, na qual as células epiteliais secretam mediadores
inflamatórios que são responsáveis por desencadear a resposta imunológica à invasão
deste microorganismo (Figura 12).
Figura 12: Representação esquemática da resposta imunológica à infecção pelo
H. pylori. Fonte: Suerbaum & Michetti, 2002, p. 1179.

46
Depois que há o contato com os antígenos bacterianos, as células epiteliais
gástricas secretam citocinas pró-inflamatórias, como a IL-8, que é um potente mediador
inflamatório, que por mecanismos quimiotáxicos recrutam e ativam neutrófilos que
migram para o sítio da infecção (Baggiolini et al., 1994, D'elios et al, 1997; Suerbaum
& Michetti, 2002; Sarvestani et al., 2006; D’elios & Andersen, 2007).
Quando cronicamente ativados, os neutrófilos apresentam propriedades que
contribuem para aumentar a lesão tecidual, uma vez que além de atuarem na fagocitose
e digestão das bactérias liberam enzimas intracelulares e produtos reativos, sendo este
evento um importante mecanismo patogênico, entretanto incapaz de debelar a infecção
em razão da produção bacteriana de catalase e superóxido de desmutase, que protegem a
bactéria (Rautemaa et al., 2001).
A contínua exposição ao microorganismo resulta na ativação da resposta
imunológica específica. Esta resposta se dá pela estimulação dos linfócitos T auxiliares
(TCD4+), direcionando a resposta imunológica tanto para o perfil Th1, quanto Th2.
Entretanto, a resposta celular Th1 é predominante, onde se pode observar que a maioria
dos clones das células CD4+
, específicas contra o H. pylori, secretam interferon γ (IFN-
γ) como resposta à estimulação antigênica, indicando uma resposta com fenótipo Th1
(Suerbaum & Michetti, 2002).
A resposta Th1 leva não somente a produção de IFN-γ, como também de IL-12
(interleucina 12), IL-18 (interleucina 18), IL-17 (interleucina 17) e o fator de necrose
tumoral (TNF-α), que ocorre ao nível da região do antro gástrico (Shibata et al., 1999).
Tudo isso pode ser explicado pela estimulação de neutrófilos, monócitos e células
dendríticas pelo gene da proteína ativadora de neutrófilos (HP-NAP). Em pacientes
infectados há uma proliferação importante de diferentes antígenos H. pylori, incluindo
HP-NAP, que determina a produção de níveis elevados de IFN-γ e TNF-α, para
antígenos gástricos específicos das células Th, resultando numa potente atividade
citotóxica com polarização da resposta Th1. Outros fatores também podem contribuir
para a resposta Th1, além do HP-NAP (Amedei et al., 2006).
A resposta humoral também pode ser observada na infecção pelo H. pylori.
Observam-se anticorpos, predominantemente, do tipo IgG, atuando contra vários
antígenos do H. pylori, entretanto estes não conferem proteção, nem evitam uma nova
infecção (Suerbaum & Michetti, 2002).
De um modo geral, a resposta imunológica que não consegue eliminar a bactéria
resulta do predomínio de células Th1, uma vez que as Th2 estão praticamente ausentes

47
durante a infecção pelo H. pylori. Além do que, clones de células T, específicos para H.
pylori, isolados de pacientes com úlcera péptica, são mais freqüentemente do tipo Th1
do que clones isolados de pacientes apresentando somente gastrite crônica (D’elios et
al., 1997).
Com base nesses estudos, Israel & Peek (2001) levantam a hipótese de que uma
resposta aberrante do hospedeiro (Th1) para um organismo que deveria induzir
respostas imunológicas secretórias (Th2) pode influenciar e perpetuar a inflamação
gástrica.
Estudos de Wang et al. (2007) demonstram que os pacientes infectados pelo H.
pylori CagA+, que apresentam imunidade celular mediada por células Th1, estão
associados com estágios precoces da carcinogênese gástrica, enquanto que, a imunidade
humoral mediada por células Th2, estão relacionados com os estágios avançados da
doença. Nos casos em que pacientes apresentam cepas H. pylori CagA negativo e
naqueles com ausência de bactéria, não há tendência de polarização Th1/Th2. Há um
predomínio da imunidade local sobre a sistêmica. A polarização da resposta
imunológica mediada pelas células Th está associada com a cronicidade e evolução das
patologias gástricas, especialmente o câncer gástrico (Wang et al., 2007).
1.6 PRINCIPAIS CÉLULAS ENVOLVIDAS NA RESPOSTA IMUNOLÓGICA PARA
HELICOBACTER PYLORI
1.6.1 Macrófagos
São fagócitos mononucleares resultantes da maturação de monócitos. Apresenta
diferentes denominações especiais dependendo do tecido onde esteja localizado.
Responsáveis por realizar várias funções importantes nas imunidades inata e adquirida,
tais como ingestão e morte de microorganismo, ingestão de células mortas do
hospedeiro, secretam citocinas, ativam linfócitos T e promovem a angiogênese e síntese
do colágeno (Mai et al., 1991, Abbas et al, 2011).
Dependendo do tipo de estímulo para a ativação celular, os macrófagos podem
adquirir distintas propriedades funcionais. Subpopulações de células T se constituem no
exemplo mais evidente de resposta de macrófagos a diferentes citocinas, em que algumas
destas citocinas, num processo chamado de ativação clássica, ativam macrófagos para se
tornarem eficientes na eliminação de microorganismos. Outras, num processo chamado

48
de ativação alternativa, ativam macrófagos que promovem o remodelamento e o reparo
teciduais. Os macrófagos ainda, por ativação de estímulos externos, como os
microorganismos, podem assumir diferentes formas. Em razão de sua semelhança com
células epiteliais da pele, alguns macrófagos são chamados de células epitelióides.
Quando ativados, podem sofrer fusão para formar células gigantes multinucleadas (Mai
et al., 1991, Abbas et al., 2011).
De um modo geral, os macrófagos respondem aos microrganismos tão
rapidamente quanto os neutrófilos, entretanto sobrevivem por mais tempo nos sítios de
inflamação. Sua diferença em relação aos neutrófilos é que os macrófagos são células em
fase terminal de diferenciação. Portanto, macrófagos são células efetoras dominantes que
atuam nos estágios mais tardios da resposta imunológica inata, ou seja, alguns dias após
a infecção ter se instalada (Mai et al., 1991, Abbas et al., 2011).
Os macrófagos são considerados como peças essenciais ante a resposta inata ao
H. pylori , através das células epiteliais, no momento em que esta bactéria entra em
contato direto com a superfície da mucosa gástrica. Os monócitos e macrófagos são
importantes coordenadores de resposta imune frente a agentes patogênicos e, no caso do
H. pylori, são prováveis ativadores, juntamente com as DCs, de produção de fatores de
imunidade adaptativa, tais como a IL-12, que estimulam as células Th1 resultando numa
produção de citocinas como o intérferon gama (IFN-γ). Estudos recentes demonstraram
que a proteína de ativação de neutrófilos (NAP) do H. pylori contribui para a polarização
do Th1 através da estimulação da secreção de IL-12 e IL-23 de neutrófilos e manócitos.
A produção de IL-12 na mucosa gástrica está associada ao desenvolvimento de úlcera
péptica quando associada a infecção por cepas de H.pylori cagA-positiva, muito
provavelmente em razão da estimulação da resposta Th1(Grimm & Doc, 1996).
Os macrófagos também estão envolvidos na amplificação da resposta
inflamatória produzidas pelas citocinas, tais como as IL-1, TNF-α e IL-6. A ativação da
IL-6 tem sido associada a ativação de TLR4, MAP quinase e eventos de sinalização de
NF-kB.
Os macrófagos também são células efetoras. Algo que tem sido bastante
explorado é a geração de NO derivado da indução da enzima NO sintaxe (iNOS, NOS2),
na qual tem sido demonstrado ser regulada pelo H. pylori em macrófagos tanto in vitro,
quanto in vivo. Estudos demonstram que o H. pylori pode ser morto por macrófagos,
mesmo quando essas bactérias são fisicamente separadas a partir destas células efetoras

49
por um suporte de filtro e que esta defesa antimicrobiana é NO dependente (Bussiere et
al., 2005).
1.6.2 Células Epiteliais
A interação entre H. pylori e células epiteliais tem sido intensamente avaliada
(Peek & Blaser, 2002; Panchal et al., 2003; Naumann & Crabtree, 2004; Peek &
Crabtree, 2006; Crabtree & Naumann, 2006). Entretanto, apesar do contexto de toda essa
revisão, torna-se importante enfatizar o envolvimento das células epiteliais gástricas na
falha da resposta imune. Provavelmente, o H. pylori tira proveito do elevado nível de
tolerância oral no intestino para sobreviver no estômago. Exemplo disso é a falta de
indução da TLR5 pelo H. pylori comparadas com outros agentes patogênicos gástricos
(Gewirtz et al., 2004).
1.6.3 Células Dendríticas
"As células dendríticas são as APC mais importantes para a ativação de células T
virgens, e desempenham grande papel na resposta imune inata às infecções e na ligação
entre imunidade inata e adquirida" (Nish et al., 2003, Abbas et al., 2011).
As células dendríticas (DCs) são de grande interesse no estudo da imunologia da
infecção causada pela bactéria Helicobacter pylori, em razão de representar o elo
fundamental entre a resposta imune inata e a adaptativa. Identificadas como
respondedores primários aos estímulos causados pela bactéria e como células
apresentadoras de antígenos (Pulendran et al., 2001).
Alguns experimentos in vitro têm demonstrado que as células dendríticas
penetram a camada epitelial da mucosa gástrica e, a barreira epitelial do intestino in vivo,
englobando a bactéria diretamente (Rescigno, et al., 2001, Niess et al, 2005, Chieppa et
al., 2006). Entretanto, isto ainda não está bem comprovado no que diz respeito a
infecção por H. pylori, mas a interrupção do complexo apical juncional do epitélio pelo
H. pylori poderia facilitar a interação tanto luminal, quanto subepitelial das células
dendríticas com o H. pylori e os antígenos produzidos pela bactéria (Amieva et al.,
2003). Trabalhos demonstraram que células dendríticas derivadas de células
mononucleares de sangue periférico de seres humanos, produziram IL-12 e IL-10

50
quando estas eram contaminadas por H. pylori (Guiney et al., 2003, Kranzer et al.,
2004).
Determinante importante na orquestração da resposta imune, talvez possa ser o
potencial antigênico do H. pylori de proteína de membrana externa (Voland et al,, 2003).
Recentemente, tem-se demonstrado que as DCs interagem com H. pylori pela
ligação de estruturas de hidrato de carbono de glicoconjugados para Dc-específico
(Appelmelk et al., 2003).
Experimentos demonstraram que em ratos infectados por H. pylori, as DCs são
rapidamente recrutadas para o estômago, levando, em média, em torno de seis horas após
a inoculação, porém desaparecem em torno do quinto dia, não se sabendo se este
desaparecimento está ligado a migração das células CD11-positivas para os nódulos
linfáticos, ou se ocorre apoptose dessas células, ou ainda, uma falha de sustentação dessa
resposta, por vez necessitando de melhores investigações (Kao et al, 2006).
Trabalhos mais recentes demonstram que as DCs quando estimuladas pelo H.
pylori durante 48 horas, exibem uma capacidade nitidamente atenuada para induzir a
produção de IF-γ mediante cultura com células T naive comparadas com as DCs
pulsadas com H. pylori em apenas 8 horas, havendo perda semelhante de resposta para
ligação com CD40, sugerindo que a exposição crônica de DCs ao H. pylori resulta na
perda de capacidade de induzir uma resposta Th1 que poderia estar contribuindo para a
persistência da infecção ( Mitchell et al, 2007).
1.6.4 Linfócitos
1.6.4.1Células B
Apesar de terem sido bem menos estudadas que as células T, há uma grande
evidência de que as células B contribuam para a imunopatogênese da bactéria H. pylori
(Akhiani et al., 2004).
Akhiani e colaboradores realizando estudo experimental com ratos deficientes em
células B (µMT), infectados com H. pylori, comparando estes com ratos de linhagem
selvagem, observaram que não havia diferença na colonização após 2 semanas de
infecção, porém em dois casos houve uma redução por volta da oitava e décima sexta
semana respectivamente, tendo sido associados com o aumento da inflamação gástrica e
infiltração de células CD4+ T (Akhiani et al., 2004). Embora já se tenha descrito

51
resposta IgG e IgA tanto no soro, quanto na mucosa gástrica podendo estar envolvida na
proteção imunológica, estudos aposteriores dão conta de que há uma resposta negativa
de anticorpos IgA, sugerindo que a resposta mediada por anticorpos de células B pode
ser contraproducente. O maior foco de investigação que tem sido relatado, está voltado
para o estudo da associação do desenvolvimento de tumores de células linfóides da
mucosa gástrica. Um grupo de cientistas demonstraram que as células esplênicas de ratos
expostas ao H. pylori estavam protegidas contra apoptose espontâneas e sofreram
proliferação em resposta baixa, e não alta, da multiplicação da infecção, e estas células
respondedoras eram derivadas da população de células B (Bussiere et al., 2006).
Experimentos demonstraram que a infecção crônica da mucosa gástrica pelo H.
pylori encontrava-se protegida por células B esplênicas derivadas de apoptose, indicando
que há uma ativação/sobrevivência fenotípica de célula B, podendo estar relacionado
com o tumor de células linfóides da mucosa gástrica (Bussiere et al., 2006).
Sabe-se que além de produzir anticorpos específicos para antígenos, as células B
também demonstraram produzir anticorpos autorreativos que podem ser patogênicos
(Crabtree et al., 1991, Yamanishi et al., 2006).
1.6.4.2 Células T
A partir do momento em que houve a descoberta do H. pylori como um patógeno
humano, muito se tem investigado a respeito do papel das células T. Os primeiros
estudos dão conta de que os linfócitos gástricos de pacientes infectados pelo H. pylori
tem um incremento de células T produtoras de IFN-γ, com base na resposta da citocina
Th1 e que clones de células T H. pylori-específicos derivados da mucosa gástrica
também possuem um perfil de resposta Th1 em pacientes com doença ulcerosa péptica
(Karttunen et al.,1995, D'Elios et al.,1997, Bamford et al., 1998). As células T da
mucosa gástrica infectada por H. pylori produzem níveis abundantes de citocinas Th1,
IFN-γ e IL-2 e, níveis baixos de citocinas Th2, IL-4 e IL-5 (Karttunen et al., 1995,
Bamford et al., 1998). Trabalhos adicionais demonstraram a importância da ativação da
produção de IL-12 a partir de monócitos, macrófagos ou DCs na indução da resposta
imune por linfócitos Th1 (Haeberle et al., 1997, Meyer et al, 2003, Guiney et al., 2003,
Hafsi et al., 2004), assim como o papel das células epiteliais gástricas como células
apresentadoras de antígenos na ativação de células CD4+ Th (Ye et al., 1997). Estudos
com ratos tem demonstrado que a resposta Th1 está associada com o aumento da

52
gastrite, isto porque IFN-γ/- de ratos demonstra diminuição da gastrite (Akhiani et al.,
2002) e a combinação severa de ratos portadores de doença que determinam
imunodeficiência com deficiência de células T e B com infecção por H. pylori
necessitam da transferência adaptativa de células CD4+ T por gastrites (Eaton et al.,
2001).
Trabalhos tem demonstrado a implicação das células Treg na patogênese da
infecção pelo H, pylori (Lundgren et al., 2003, Lundgren et al., 2005).
1.7 - LINFÓCITO T REGULADOR
Durante a década de 1980 foram realizadas vários tipos de experimentos com
diversos tipos de hibridomas, onde foi identificada um subtipo de células T que tem a
capacidade de suprimir as respostas imunes frente a antígenos específicos. Essa
capacidade supressora foi observada tanto in vitro como in vivo, acometendo qualquer
indivíduo simplesmente transferindo estas células, que passaram a ser denominadas de
células supressoras (Hernández, 2009).
Contudo, as tentativas iniciais para definir populações de células supressoras e
seus mecanismos de ação não foram bem sucedidas, tendo sido os trabalhos
abandonados por mais de duas décadas (Abbas, 2011).
Passados mais de 20 anos, com a utilização de melhores abordagens com intuito
de definir, purificar e analisar populações de linfócitos T que inibem respostas
imunológicas, fizeram com que o interesse e as pesquisas ressurgissem de maneira
impressionante (Abbas, 2011).
As células T reguladoras foram descritas pela primeira vez em 1995 por
Sakaguchi e colaboradores, como uma célula reguladora da resposta imune e da
ativação celular (Belkaid & Rouse, 2005).
Os linfócitos T reguladores representam uma subpopulação de linfócitos T de
células TCD4+, caracterizados pela expressão da molécula CD25+ e do fator nuclear
FOXP3, cuja a função é suprimir as respostas imunológicas e manter a autotolerância
(Abbas, 2011; Melo & Carvalho, 2009).
Os linfócitos T reguladores são componentes da tolerância imunológica, e são
moduladores essenciais na resposta imune à patógenos, alérgenos, células cancerígenas
e antígenos próprios (Melo & Carvalho, 2009).

53
A sinalização para o desenvolvimento das Tregs naturais que ocorre durante a
timopoiese normal ainda é desconhecida, segundo Piccirilo (2008), entretanto Abbas
(2005) acredita que essas células possam ser geradas mediante reconhecimento de
antígenos próprios no timo, através de receptores de alta afinidade (TCR).
Em experimentos realizados com ratos observou-se que a IL-2 tem papel
importante no desenvolvimento das células T reguladoras. Tais experimentos
demonstraram que a deficiência tanto da citocina quanto do receptor, determinaram
graves defeitos de Tregs. Fato este também observado em pacientes com deficiência
congênita da molécula CD25 (Abbas, 2005; Piccirillo, 2008).
São descritos, atualmente, dois tipos principais de Tregs: naturais e adaptativas.
As chamadas Tregs naturais expressam continuamente o receptor de nos corpúsculos de
Hassal no timo como uma subpopulação de células T principalmente distintas e
maduras, representando 5 a 10% das células TCD4+ do sangue periférico (Yagi et al.,
2004; Sakaguchi, 2005; Melo & Carvalho, 2009). Ainda pode-se citar, como
marcadores dessas células, além da expressão constitutiva do fator de transição FOXP3,
o receptor da família TNF induzida por glucocorticóide (GITR), o antígeno 4 associado
ao linfócito T citotóxico (CTLA-4) e elevado nível da cadeia alfa do receptor de IL-2
(CD25) (Cruvinel et al., 2008; Borges et al., 2010).
Outros receptores de superfície descritos nas Tregs são CD27, Fas, CD62L; e os
receptores de quimiocina CCR6, CCR7, CCR8 e CD103, o que permite a migração das
Tregs até o local da inflamação (Melo & Carvalho, 2009).
As células T reguladoras adaptativas são produzidas na periferia após uma
variedade de estímulos antigênicos ou em condições tolerogênicas (Bacchetta et al.,
2007; Taams et al., 2006). Exercem sua função com a liberação de citocinas inibitórias
como IL-10 e TGF-β (Jonuleit & Schimitt, 2003; Sakaguchi, 2006). Vários tipos de
Tregs adaptativas têm sido descritos, como TR1, que produzem IL-10, cuja a função
supressora pode ser bem observada nos trabalhos relativos as doenças alérgicas,
autoimunes e transplante alogênico (Bocchetta et al., 2007; Melo & Carvalho, 2009).
Ainda são citadas na literatura outras Tregs adaptativas tais como TR3 (produtoras de
TGF-β), células T CD4-CD8-, natural killer, CD8+ supressora e gama-delta (Tang &
Bluestone, 2008; Melo & Carvalho, 2009).

54
1.7.1 Fator de Transcrição FOXP3
A partir do trabalho de Brunkow et al. é que a importância do FOXP3 nas
células T reguladoras ficaram bem definidas, quando identificaram a mutação do tipo
frameship do gene FOXP3 (Torgerson & Ochs, 2007). Esta mutação é responsável pelo
fenótipo de camundongos scurfy, uma linhagem mutante recessiva, ligada ao X que
apresenta distúrbios auto-imunes graves com depleção completa de Tregs e óbito
precoce (Melo & Carvalho, 2009).
A deleção funcional do FOXP3 em seres humanos tem sido descrita em
pacientes com a síndrome IPEX (Immunodeficiency, Poliendocrinopathy and
enteropathy X-linked syndrome), caracterizada clinicamente por múltiplas doenças
autoimunes, incluindo diarréia, eczema, diabetes com destruição das glândulas
endócrinas, insulinite e tireoidite, com predileção para meninos em torno dos dois anos
de idade, culminando com o óbito precoce (Gambineri et al., 2003; Campbell & Ziegler,
2007; Torgerson & Ochs, 2007; Melo & Carvalho, 2009).
Localizado no braço curto do cromossomo X, o FOXP3 está constituído de 11
exons e codifica uma proteína de 431 aminoácidos, denominada também de FOXP3.
Predominantemente, encontra-se expresso nas células do timo, baço e linfonodos e,
particularmente, nas células T CD4+CD25+ (Yagi et al., 2004; Torgerson & Oches,
2007).
A proteína FOXP3 é um fator de transcrição, cuja função é exercida sobre
regiões reguladoras específicas dentro do DNA através de um fator ligante altamente
preservado denominado forkhead/winged-helix, aumentando ou suprimindo a
transcrição de genes específicos (Abbas, 2005; Torgerson & Ochs, 2007; Melo &
Carvalho, 2009). Exerce função efetora e facilitadora sobre genes de proteínas chaves
na ativação celular, incluindo a IL-2 e o GM-CSF (Torgerson & Ochs, 2007; Melo &
Carvalho, 2009).
Membro da subfamília P das proteínas FOX (forkhead box), o fator FOXP3 é
composto por três domínios (repressor, central e ligante de DNA ou forkhead (Coffer &
Burgering, 2004; Campbell & Ziegler, 2007; Melo & Carvalho, 2009). (Figura 13). A
maioria das mutações que afetam o gene que levam a substituição de um aminoácido
pelo outro); entretanto, é sabido que ocorrem vários outros tipos de mutações (deleções
ou substituições) e que podem afetar outros domínios (Torgerson & Ochs, 2007; Melo
& Carvalho, 2009).
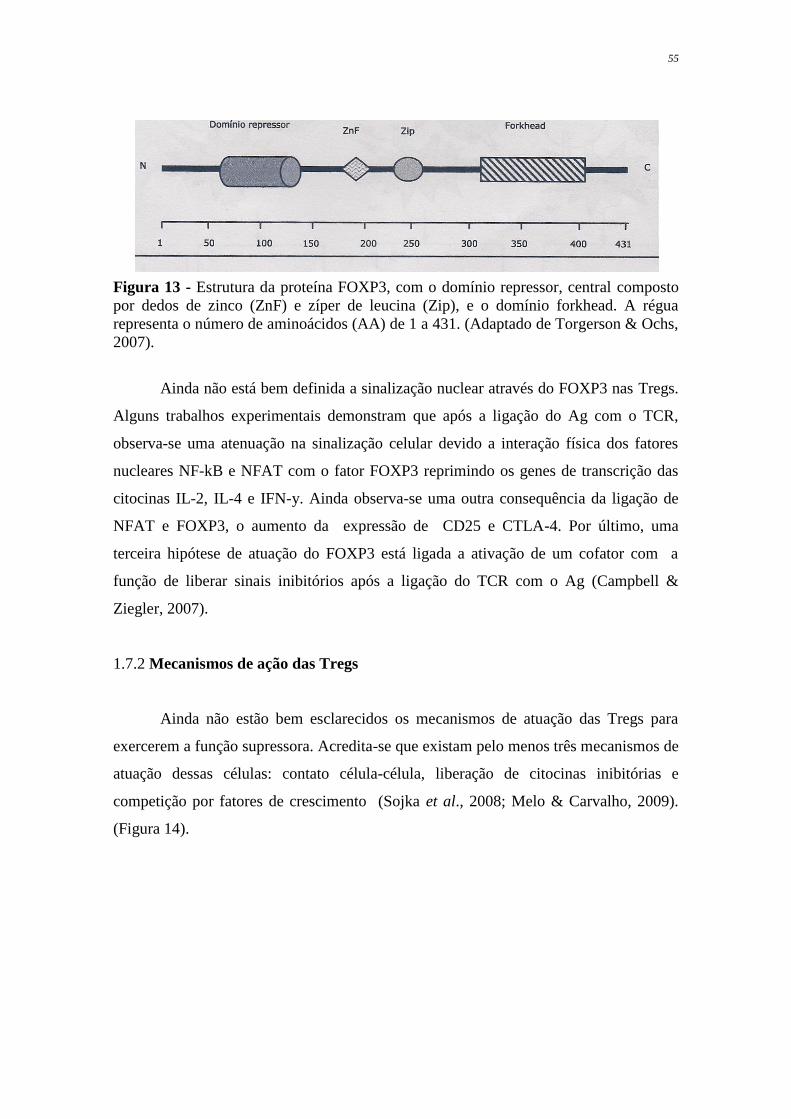
55
Figura 13 - Estrutura da proteína FOXP3, com o domínio repressor, central composto
por dedos de zinco (ZnF) e zíper de leucina (Zip), e o domínio forkhead. A régua
representa o número de aminoácidos (AA) de 1 a 431. (Adaptado de Torgerson & Ochs,
2007).
Ainda não está bem definida a sinalização nuclear através do FOXP3 nas Tregs.
Alguns trabalhos experimentais demonstram que após a ligação do Ag com o TCR,
observa-se uma atenuação na sinalização celular devido a interação física dos fatores
nucleares NF-kB e NFAT com o fator FOXP3 reprimindo os genes de transcrição das
citocinas IL-2, IL-4 e IFN-y. Ainda observa-se uma outra consequência da ligação de
NFAT e FOXP3, o aumento da expressão de CD25 e CTLA-4. Por último, uma
terceira hipótese de atuação do FOXP3 está ligada a ativação de um cofator com a
função de liberar sinais inibitórios após a ligação do TCR com o Ag (Campbell &
Ziegler, 2007).
1.7.2 Mecanismos de ação das Tregs
Ainda não estão bem esclarecidos os mecanismos de atuação das Tregs para
exercerem a função supressora. Acredita-se que existam pelo menos três mecanismos de
atuação dessas células: contato célula-célula, liberação de citocinas inibitórias e
competição por fatores de crescimento (Sojka et al., 2008; Melo & Carvalho, 2009).
(Figura 14).

56
Figura 14 - Modelos de mecanismos de ação das Tregs. (a) Contato célula-célula -
Supressão da célula-alvo com liberação de fatores de supressão incluindo o monofosfato
de adenosina cíclica (AMPc), citocinas supressivas como o TGF-β, citólise direta ou
sinalização negativa através da molécula CTLA-4; (b) Fatores de supressão solúveis
como citocinas IL-10, TGF-β e IL-35 ou secreção de fatores supressivos pelas APC
como adenosina; (c) Competição - Competição por citocinas que sinalizam através de
receptores da cadeia γ comum (IL-2, IL-4 e IL-7). APC: célula apresentadora de
antígeno (Adaptado de Sojka et al., 2008).
1.7.3 Papel das células T reguladoras nas doenças infecciosas
Diversos mecanismos são acionados para o controle das respostas imunológicas
a infecção e as Tregs participam ativamente no controle dessas respostas. Alguns
trabalhados sobre a infecção causado pela bactéria Helicobacter pylori em seres
humanos tem sido demonstrado o aumento da frequência de células T reguladoras tanto
no estômago quanto no duodeno desses indivíduos infectados por essa bactéria. Outros
experimentos demonstram que em pacientes com hepatite pelo vírus B (HBC) e C
(HCV), também há um aumento de Tregs em sangue periférico (Taams et al., 2006)
Diferente do que ocorre tanto na infecção pelo Helicobacter pylori quanto na
produzida pelos vírus B e C da hepatite, nos pacientes contaminados pelo vírus HIV, a
progressão da doença está diretamente proporcional a um estado de hiperativação
imune, levando a uma redução no número e na função das Tregs (Eggema et al.2005;
Ndhlovu et al., 2008).

57
1.8 DIAGNÓSTICO DA INFECÇÃO PELO H. PYLORI
Atualmente encontram-se disponíveis vários métodos de diagnóstico para a
infecção pelo H. pylori, estando divididos em métodos endoscópicos e não-
endoscópicos. O método endoscópico utiliza-se da esofagogastroduodenoscopia para
realização da biópsia gástrica e assim proceder a coleta do material através de biópsia
gástrica, onde, a partir do qual será realizada a pesquisa através ou do estudo
histopatológico, ou do teste da urease, ou da cultura, ou PCR (Hammar et al., 1992;
Logan & Walker, 2001; Bittencourt et al., 2006).
Para a realização do método histopatológico procede-se a realização do exame
endoscópico, retira-se de um, dois ou mais fragmentos da mucosa gástrica,
preferencialmente do antro gástrico, posteriormente encaminhando este material a
patologia que procederá a identificação do microorganismo através dos diversos
métodos de coloração histológica, sendo que os mais utilizados são o Giemsa, Warthin-
Starry, Steiner e carbolfucsina. Este método, além da identificação da bactéria, permite
também avaliar o tipo e a intensidade da inflamação da mucosa gástrica. Contudo, não é
um método isento de falhas, pois uma amostra inadequada pode comprometer o
resultado do exame (Bittencourt et al., 2006).
A cultura da bactéria é considerada como o método mais específico para o
diagnóstico da infecção por este microorganismo, pois permite não só a identificação do
H. pylori, como, também, proporciona o estudo da amostra quanto à presença de fatores
de virulência, podendo ser usado para determinar a suscetibilidade aos antimicrobianos,
visando uma terapêutica mais adequada (Scholte et al., 2002; César et al., 2005).
Dado seu crescimento lento é muito difícil cultivar in vitro a bactéria H. pylori.
Entretanto, vários meios de cultura podem ser empregados no sentido de identificar a
bactéria. Vários fatores podem influenciar o crescimento da bactéria H. pylori em meio
de cultura como o número de biópsias, o próprio meio empregado, a duração e a
temperatura de transporte e, até mesmo, o método de cultivo (Tonelli & Freire, 2000;
Bittencourt et al., 2006). Alguns medicamentos também podem influenciar a detecção
do H. pylori, como por exemplo, o omeprazol, antimicrobianos, sais de bismuto e
benzocainas. Outro dado importante reside no fato de que alguns autores afirmem que
resíduos de glutaraldeído, utilizado no processo de desinfecção das pinças de biópsias,
possam estar presentes nestas por ocasião da realização da biópsia e, com isso, afetar a
viabilidade do microorganismo (Tonelli & Freire, 2000).

58
Outro método diagnóstico bastante empregado atualmente para identificação da
bactéria H. pylori é o teste da urease, baseado na potente atividade ureásica do
microorganismo. O teste da urease é altamente específico e sensível e, também, o mais
empregado no diagnóstico endoscópico. Como regra, biopsia um fragmento do antro ou
um fragmento do antro e outro do corpo gástrico que, após a coleta, são introduzidos de
imediato em um pequeno tubo contendo uréia e um indicador de pH (vermelho fenol).
Observa-se que a urease hidrolisa a uréia em amônia e dióxido de carbono,
determinando, conseqüentemente, um aumento do pH e mudança de coloração do meio,
passando da cor amarela para a rosa. O teste será considerado positivo quando esta
mudança de coloração possa ocorrer num espaço de tempo menor que 24 horas
(Ornellas et al., 2000).
Embora largamente utilizado na prática clínica, o teste da urease tamponado não
fornece informações a respeito da intensidade da inflamação (Chu et al., 1997).
A genotipagem dos marcadores de virulência do H. pylori e determinação de
suscetibilidade da bactéria aos microorganismos, tanto em amostras isoladas quanto em
fragmentos de biópsia, são as técnicas de Biologia Molecular empregadas para o
diagnóstico da infecção (Bittencourt et al., 2006).
A Reação em Cadeia da Polimerase (PCR) apresenta altíssima sensibilidade e
especificidade, podendo ser feita diretamente da biópsia gástrica ou duodenal, do suco
gástrico, da placa dentária, da saliva, da cultura e até mesmo das fezes (Siqueira et al.,
2007). Em função da sua alta sensibilidade, a PCR é bastante utilizada em estudos
epidemiológicos ligados à identificação de reservatórios ambientais, assim como em
trabalhos de determinação do modo de transmissão desta bactéria (Lehours et al., 2003).
Com sensibilidade semelhante a da PCR, a reação de hibridização in situ com
fluorescência (FISH) é outro método de pesquisa do DNA da bactéria em tecido
(Lehours et al., 2003).
Para a identificação de fatores de virulência do H. pylori, como cagA e vacA,
diversos métodos de biologia molecular podem ser empregados, como PCR, PCR
seguida de hibridação (LIPA – line probe assay), PCR em tempo real rtPCR (Van doorn
et al., 1998; Queiroz et al., 2000; Boonjakuakul et al, 2004). Levando-se em
consideração as variações regionais na seqüência dos genes que codificam esses fatores,
os iniciadores das reações devem ser testados para as diferentes populações (Queiroz et
al., 2006).

59
Também chamados de indiretos, os métodos não endoscópicos têm sua
indicação para detecção do H. pylori em pacientes que não necessitam exame direto da
mucosa gástrica, dificuldades na obtenção de biópsias (úlceras com sangramento e
terapia anticoagulante), avaliação da eficácia da erradicação do microorganismo e
estudos epidemiológicos (Hirschl & Makristathis, 2007). Compreendem a pesquisa de
anticorpos anti-H. pylori em amostras de soro, urina e saliva, a pesquisa de antígenos de
H. pylori nas fezes e o teste respiratório através da ingestão de solução de uréia marcada
com carbono-13 e carbono-14 (Bittencourt et al., 2006; Siqueira et al., 2007).
De grande importância nos estudos epidemiológicos, os testes sorológicos se
baseiam na detecção de anticorpos específicos: IgG, IgM ou IgA, contra o H. pylori
(Bittencourt et al., 2006).
Uma resposta imunológica celular e humoral, que resulta na produção de
anticorpos anti-H. pylori das classes IgM, IgA e IgG é induzida no hospedeiro através
da infecção pela bactéria H. pylori. Os anticorpos específicos da classe IgA e IgG
atingem níveis detectáveis, aproximadamente, 3 semanas a 3 meses depois do início da
infecção aguda e podem ser observados até cerca de 2 anos após a erradicação do H.
pylori. Em razão da infecção provocada pela bactéria ser restrita à mucosa, em
determinados casos, a estimulação antigênica pode ser lenta e, em poucas semanas ou
meses após nova infecção, resultados falsos-negativos podem ocorrer (Bittencourt et al.,
2006).
Em razão de sua rapidez, simplicidade, baixo custo, reprodutibilidade, com
inúmeros kits disponíveis no mercado, o ensaio imunoenzimático (ELISA), dentre os
vários métodos disponíveis, é o mais utilizado. Contudo, podem apresentar resultados
falsos-negativos em crianças, idosos e pessoas imunodeprimidas, que não desenvolvem
reação imunológica contra a infecção (Gilger et al., 2002).
A pesquisa da bactéria nas fezes é realizada através de método imunoenzimático
empregando anticorpos mono ou policlonais (Bittencourt et al., 2006).
Recentemente foram lançados no mercado testes imunocromatográficos para
detecção de antígenos nas fezes, de execução fácil, que permitem a obtenção de
resultados em poucos minutos, com sensibilidade e especificidade semelhantes àquelas
observadas com os testes imunoenzimáticos (Kato et al., 2004; Antos et al, 2005).
Para a realização do teste respiratório administra-se uma solução de uréia
marcada com carbono-13 e carbono-14; a urease bacteriana converte a uréia em amônia
e bicabornato, que é absorvido e convertido em CO2 nos pulmões; o paciente expira em

60
um balão ou outro recipiente próprio onde a presença de carbono marcado pode ser
detectada por cintilação ou espectrografia (Tonelli & Freire, 2000). Este teste apresenta
mais de 90% de sensibilidade e é altamente específico. Também pode ser utilizado em
estudos epidemiológicos, sendo de preferência para o controle da erradicação da
infecção pelo H. pylori, por não ser um teste invasivo (Siqueira et al., 2007).
Recomenda-se a utilização de, pelo menos, dois testes para o diagnóstico mais
acurado da infecção pelo H. pylori (Bittencourt et al., 2006).

61
1.9 OBJETIVOS
1.9.1Objetivo Geral
Determinar o padrão de resposta imunológica ao H. pylori em amostras de
biópsias gástricas de pacientes com gastrite com ênfase na resposta T regulatória.
1.9.2 Objetivos específicos
Quantificar no tecido gástrico a presença de células CD4, CD8, Células B,
Neutrófilos e Linfócitos T regulatórios.
Correlacionar a presença de infecção pelo H. pylori entre os pacientes com o
perfil fenotípico do infiltrado inflamatório e os achados histopatológicos.

62
2. MATERIAL E MÉTODOS
2.1 POPULAÇÃO DE ESTUDO
O estudo realizado caracterizou-se como estudo transversal analítico, constituído
de cinquenta amostras de biópsias gástricas, provenientes do arquivo de anatomia
patológica de um Laboratório de Análises Clínicas e Patologia Privado de Belém-PA, de
pacientes com diagnóstico clínico (endoscópico) de gastrite.
Previamente, todos os sujeitos pesquisados foram informados sobre a pesquisa, de
maneira acessível. Após o esclarecimento da importância deste estudo, foi solicitada sua
permissão, através da assinatura do Termo de Consentimento Livre e Esclarecido (Anexo
1), conforme rege a Resolução 196/96, do Conselho Nacional de Saúde, sobre aspectos
éticos envolvendo a pesquisa com seres humanos, autorizando suas participações nesta
pesquisa, possibilitando a coleta de material biológico para pesquisa.
2.2 CRITÉRIOS DE INCLUSÃO E EXCLUSÃO NO ESTUDO
2.2.1 Critérios de inclusão
Indivíduos com idade igual ou superior a 18 anos;
Não fizeram uso de antagonistas de receptores H2, inibidores de bomba de
prótons; ou drogas antimicrobianas, há pelo menos 60 dias anteriores à obtenção do
material;
Não apresentarem deficiência de natureza mental;
Assinarem o Termo de Esclarecimento e Consentimento Livre.
2.2.2 Critérios de exclusão
Menores de idade;
Fizeram uso de antagonistas de receptores H2, inibidores de bomba de prótons;
ou drogas antimicrobianas, a pelo menos 60 dias anteriores à obtenção do
material;
Portadores de deficiência mental de qualquer natureza;

63
Portadores de outras patologias gástricas que não gastrite;
Portadores de doenças infecto contagiosas;
Portadores de doenças consuptivas;
Não assinarem o Termo de Esclarecimento e Consentimento Livre.
2.3 COLETA E PROCESSAMENTO DAS AMOSTRAS
2.3.1. Biópsias: A biópsia gástrica foi coletada durante o exame de endoscopia digestiva
alta, utilizando-se uma pinça gástrica. Sendo retiradas duas amostras da região antral
para extração do DNA, que foram acondicionadas em tubo eppendorf estéril e
congeladas para posterior análises. E biópsias gástricas da região antral e da área de
lesão da mucosa, quando houvesse, para análise histopatológica.
2.3.2 Análise Histopatológica
A análise histopatológica e a detecção histopatológica do H. pylori das amostras
foram realizadas no Núcleo de Medicina Tropical da Universidade Federal do Pará.
As biópsias chegaram ao laboratório, fixadas em solução de 10% de formalina em
tampão fosfato, onde foram processadas, submetidas a banhos seqüenciais de álcoois
70%, 80%, 90%, 100% e 100% uma hora em cada, e incluídas em parafina.
Posteriormente, foram feitos cortes histológicos seqüenciais de aproximadamente 4 mm
de espessura, dispostos em lâminas e corado pela Hematoxilina-Eosina (HE), para a
análise histopatológica.
2.4 DESCRIÇÃO DAS TÉCNICAS LABORATORIAIS
2.4.1 Detecção Histopatológica do H. pylori
Para a detecção do H. pylori foi utilizado o método Gram modificado e
microscopia óptica convencional, identificando a bactéria mediante suas características
morfológicas, forma curva e espiralada, e coloração azul intensa.
2.4.2 Detecção das células CD4, CD8, linfócitos B, Neutrófilos e linfócitos T
regulatórios na biópsia gástrica.

64
Para quantificação das interleucinas na mucosa gástrica foram utilizado kits de
imunohistoquímica, que obedeceu às seguintes etapas:
1- Desparafinização das lâminas em xilol I (20 minutos), xilol II (10 minutos) em
temperatura ambiente;
2- Hidratação das lâminas em etanol absoluto I (5 minutos), etanol absoluto II (2
minutos), etanol 95% (2 minutos), etanol 80% (2 minutos), etanol 70% (2minutos);
3- Lavar os cortes com água corrente (5 minutos), água destilada (5 minutos) e PBS (5
minutos). OBS: PBS pH 7,4;
4- Bloqueio de pigmento formólico, onde os cortes foram imersos em solução de
hidróxido de amônia 25% por 30 minutos em temperatura ambiente.
- 250 ml de Hidróxido de amônia
- 750 ml de Álcool 95% / 1000ml
5- Lavar os cortes em água corrente (5 minutos), água destilada (5 minutos) e PBS (5
minutos)
6- Bloqueio da peroxidase endógena com 3 incubações de 15 minutos cada em peróxido
de hidrogênio 3% em câmara escura.
- 85,7 ml de Peróxido de hidrogênio
- 914,3 ml de Água destilada / 1000ml
7- Lavagem em água corrente (5 minutos), água destilada (5 minutos), PBS (5 minutos)
Para a recuperação antigênica os cortes foram imersos em tampão de ácido acético
diaminotetraetileno (Tris/EDTA) pH 9,0 já aquecido em banho-maria a 95ºC por 20 a 25
minutos. A Seguir, lavou-se em água corrente (5 minutos), água destilada (5 minutos),
PBS (5 minutos).
Para bloqueio das proteínas inespecíficas foi realizada a imersão dos cortes em
solução de leite molico 10% por 30 minutos.
O passo seguinte será a incubação das lâminas com anticorpo primário diluídos em
Soro Albumina Bovina (BSA) a 1% nas concentrações pré-estabelecidas por 12 horas a
4ºC. Pinga-se 100µl do anticorpo diluído.
Para análise das citocinas as lâminas foram colocadas em solução de saponina por
10 minutos. Posteriormente, fez-se duas lavagens em PBS (5 minutos); incubou-se com
o anticorpo secundário; pingou-se sobre as lâminas a biotina , deixou-se a temperatura de
37ºC e aguardou-se por 30 minutos; realizou-se mais duas lavagens com PBS (5
minutos).

65
Para análise das citocinas, as lâminas foram colocadas em solução de saponina por
10 minutos. Posteriormente, fez-se duas lavagens de PBS (5 minutos); incubou-se com
anticorpo secundários; pingou-se sobre as lâminas a biotina, deixou-se a temperatura de
37°C e aguardou-se por 30 minutos, realizou-se mais duas lavagens com PBS (5
minutos).
Após esse tempo pingou-se sobre as lâminas a estreptavidina, a 37ºC por 30
minutos.
Mais duas lavagens em PBS, por cinco minutos foram realizadas.
Revelou-se as lâminas em solução substrato (DAB), por 2 a 3 minutos.
Lavou-se as lâminas em água corrente por cinco minutos.
O passo seguinte foi contracorar as lâminas em hematoxicilina de Mayers por 1
minuto.
Lavou-se em água corrente até que a mesma ficasse clara.
Desidratou-se em concentrações crescentes de álcool (70%, 95%, 100%, 100%) por
dois minutos cada.
Deixou-se secar em T.A. over night ou em estufa por 5 a 10 minutos a temperatura
de 37ºC.
Finalmente, montou-se entre lamínula e cola apropriada.

66
2.5 ANÁLISE ESTATÍSTICA
Após a análise descritiva, os resultados foram apresentados sob a forma de
tabelas e gráficos.
Na análise das variáveis quantitativas, foram utilizadas medidas de tendência
central (média, mediana) e medidas de variabilidade (máximo, mínimo e desvio-
padrão).
Para verificar se havia diferença significativa entre as médias foi utilizado Teste
t de Mann-Whitney.
Para verificar a existência de correlação entre as variáveis foi adotado o Teste de
Correlação de Pearson.
Para aplicação dos testes foi utilizado o programa de computação estatística
BIOESTAT 5.0 (Ayres et al., 2003).
A interpretação dos testes foi realizada de acordo com a convenção científica,
isto é, o resultado foi chamado de estatisticamente significante se o valor de p era menor
que 0,05 (p>0,05). Sugerindo a não rejeição da hipótese de nulidade de que as variáveis
envolvidas estão de alguma forma associada.

67
4. RESULTADOS
4.1 AMOSTRA
A população deste estudo foi constituída por 50 amostras de indivíduos
portadores de gastrite, em que foram realizadas biópsias gástricas durante exame de
endoscopia digestiva alta, com posterior análise das lâminas oriundas do arquivo de
patologia do Laboratório Sabin.
4.2 DETECÇÃO HISTOPATOLÓGICA DO H.pylori
Através da utilização do método Gram modificado e microscopia óptica
convencional, foi possível detectar a presença ou ausência da bactéria no material
estudado. Deste modo, o H. pylori foi encontrado em 72% (36) da amostra estudada,
enquanto que 28% (14) desta não apresentava a bactéria (Tabela 1).
Tabela 1 - Resultado das amostras estudadas quanto a presença e ausência de H. pylori.
H. pylori Frequência Porcentagem Porcentagem acumulada
não 14 28.0% 28.0%
sim 36 72.0% 100.0%
Total 50 100.0% 100.0%
sim = H. pylori presente; não = H. pylori ausente
Int. Conf. 95%
não 16.2% 42.5%
sim 57.5% 83.8%
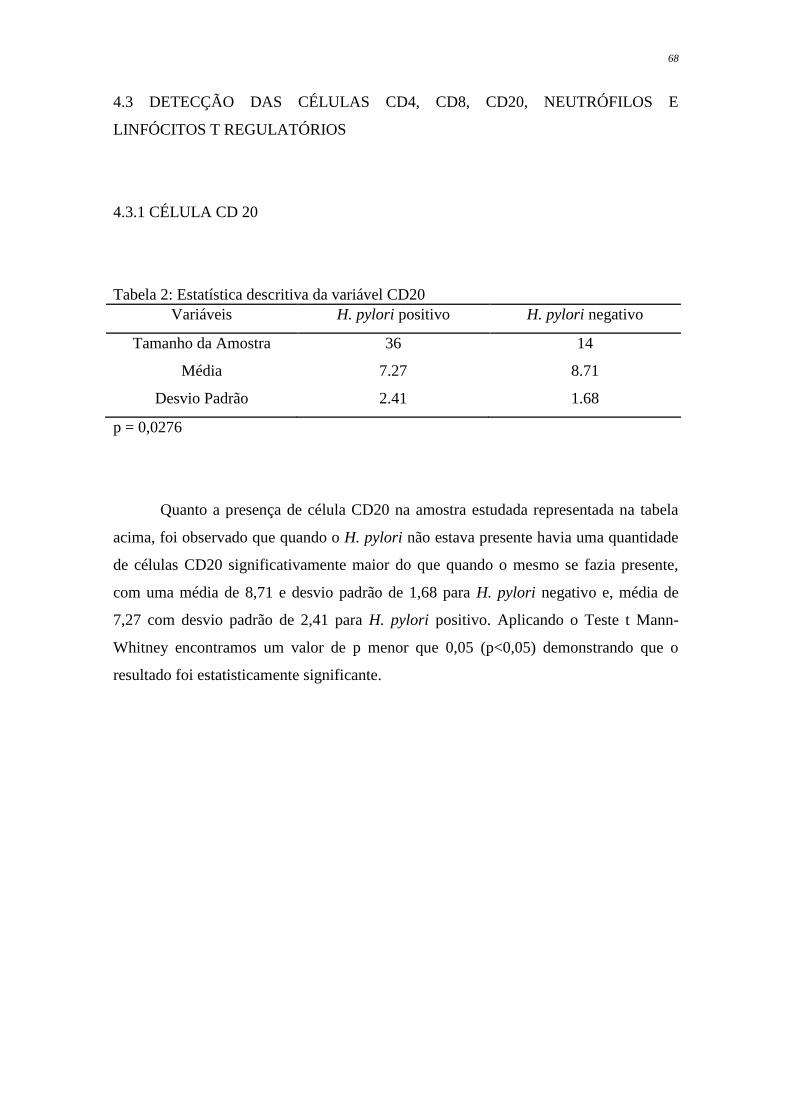
68
4.3 DETECÇÃO DAS CÉLULAS CD4, CD8, CD20, NEUTRÓFILOS E
LINFÓCITOS T REGULATÓRIOS
4.3.1 CÉLULA CD 20
Tabela 2: Estatística descritiva da variável CD20
Variáveis H. pylori positivo H. pylori negativo
Tamanho da Amostra 36 14
Média 7.27 8.71
Desvio Padrão 2.41 1.68
p = 0,0276
Quanto a presença de célula CD20 na amostra estudada representada na tabela
acima, foi observado que quando o H. pylori não estava presente havia uma quantidade
de células CD20 significativamente maior do que quando o mesmo se fazia presente,
com uma média de 8,71 e desvio padrão de 1,68 para H. pylori negativo e, média de
7,27 com desvio padrão de 2,41 para H. pylori positivo. Aplicando o Teste t Mann-
Whitney encontramos um valor de p menor que 0,05 (p<0,05) demonstrando que o
resultado foi estatisticamente significante.
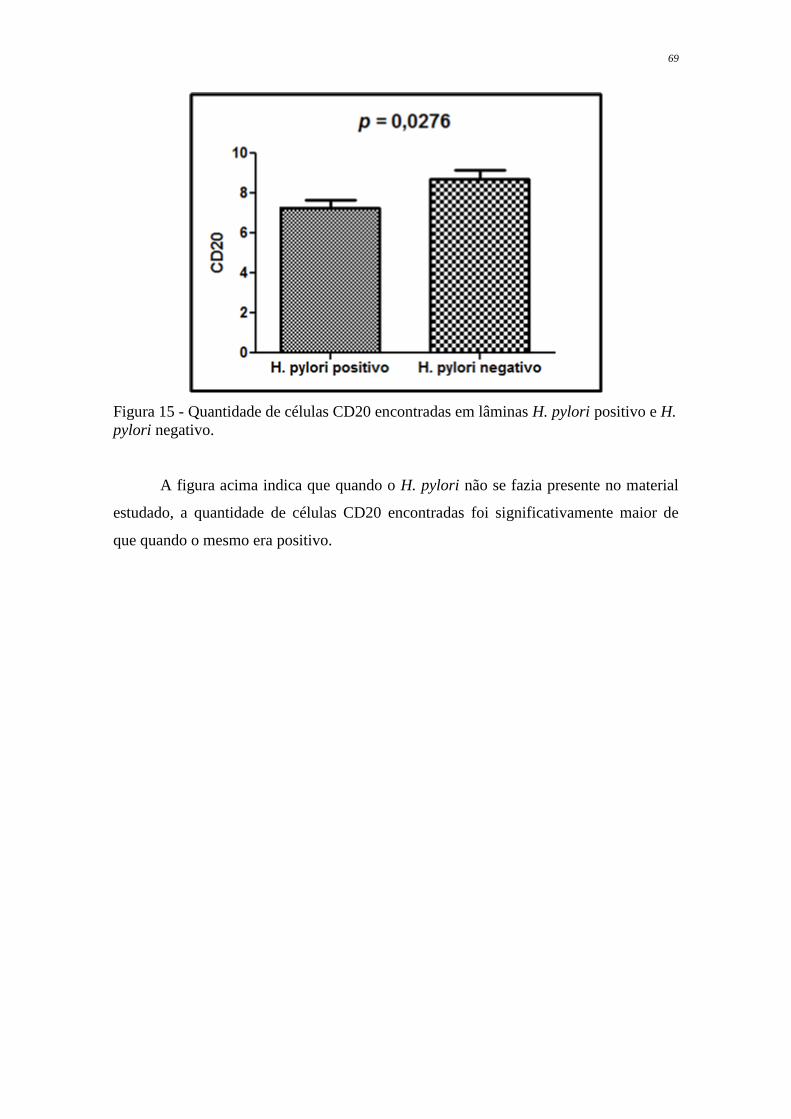
69
Figura 15 - Quantidade de células CD20 encontradas em lâminas H. pylori positivo e H.
pylori negativo.
A figura acima indica que quando o H. pylori não se fazia presente no material
estudado, a quantidade de células CD20 encontradas foi significativamente maior de
que quando o mesmo era positivo.
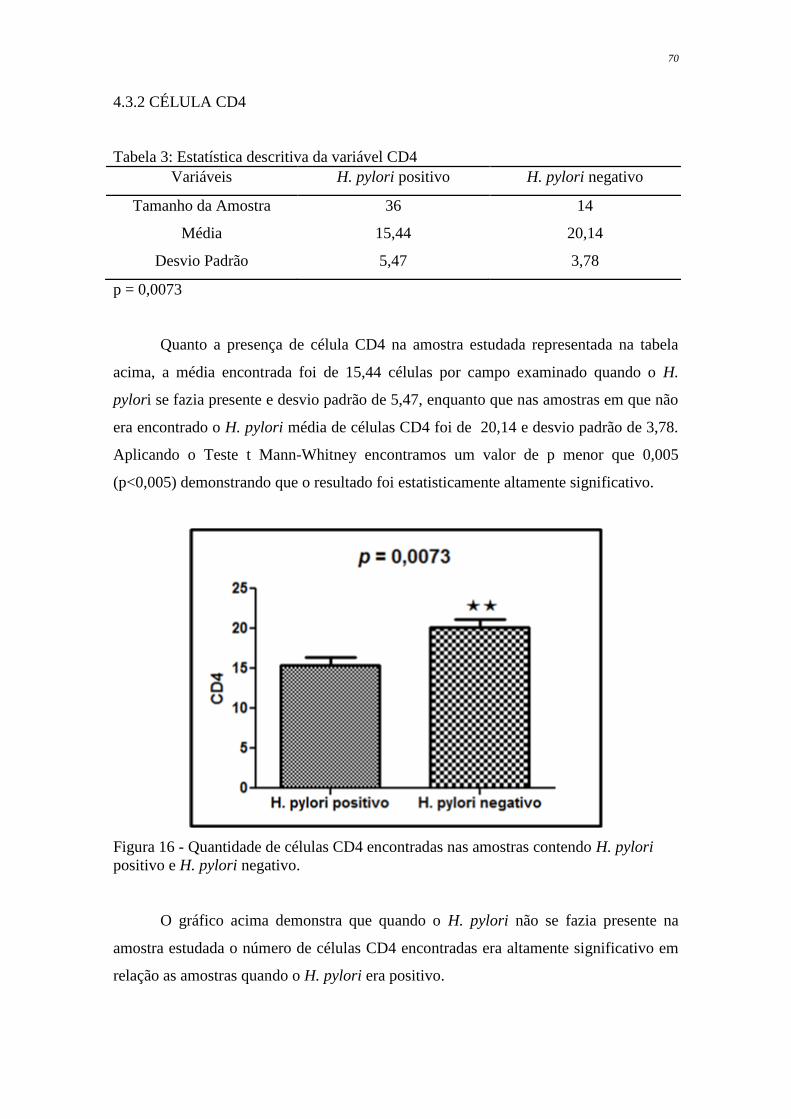
70
4.3.2 CÉLULA CD4
Tabela 3: Estatística descritiva da variável CD4
Variáveis H. pylori positivo H. pylori negativo
Tamanho da Amostra 36 14
Média 15,44 20,14
Desvio Padrão 5,47 3,78
p = 0,0073
Quanto a presença de célula CD4 na amostra estudada representada na tabela
acima, a média encontrada foi de 15,44 células por campo examinado quando o H.
pylori se fazia presente e desvio padrão de 5,47, enquanto que nas amostras em que não
era encontrado o H. pylori média de células CD4 foi de 20,14 e desvio padrão de 3,78.
Aplicando o Teste t Mann-Whitney encontramos um valor de p menor que 0,005
(p<0,005) demonstrando que o resultado foi estatisticamente altamente significativo.
Figura 16 - Quantidade de células CD4 encontradas nas amostras contendo H. pylori
positivo e H. pylori negativo.
O gráfico acima demonstra que quando o H. pylori não se fazia presente na
amostra estudada o número de células CD4 encontradas era altamente significativo em
relação as amostras quando o H. pylori era positivo.
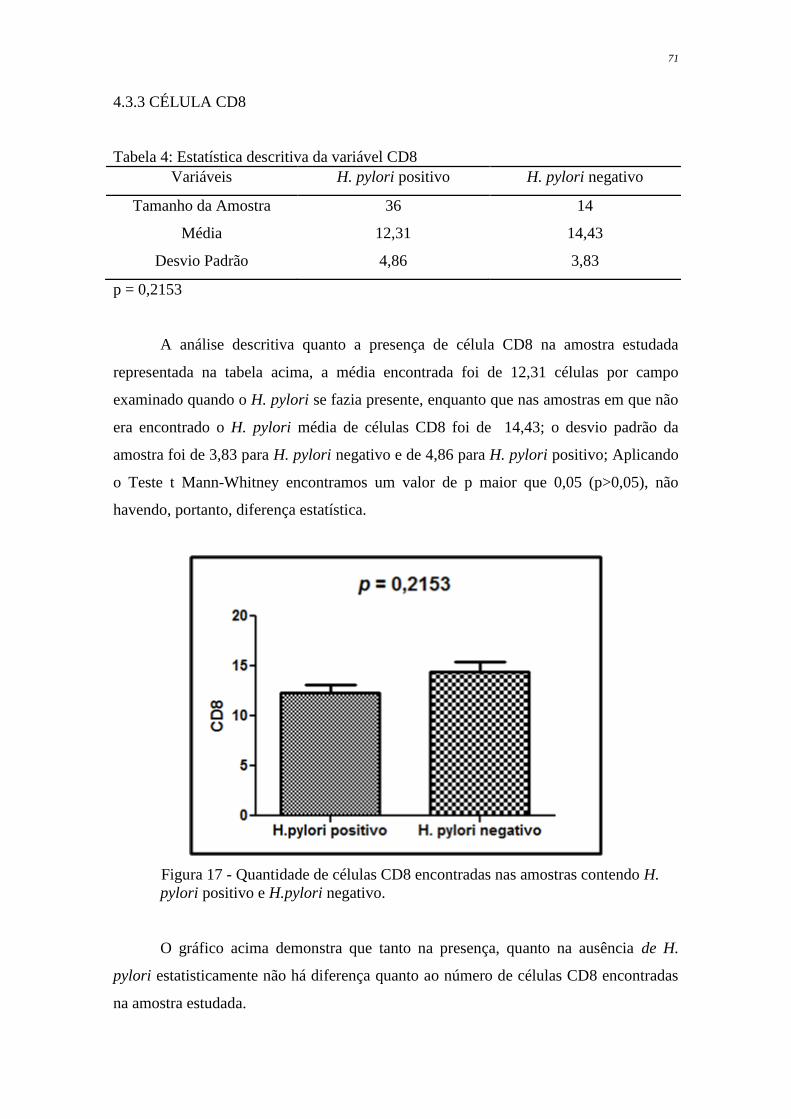
71
4.3.3 CÉLULA CD8
Tabela 4: Estatística descritiva da variável CD8
Variáveis H. pylori positivo H. pylori negativo
Tamanho da Amostra 36 14
Média 12,31 14,43
Desvio Padrão 4,86 3,83
p = 0,2153
A análise descritiva quanto a presença de célula CD8 na amostra estudada
representada na tabela acima, a média encontrada foi de 12,31 células por campo
examinado quando o H. pylori se fazia presente, enquanto que nas amostras em que não
era encontrado o H. pylori média de células CD8 foi de 14,43; o desvio padrão da
amostra foi de 3,83 para H. pylori negativo e de 4,86 para H. pylori positivo; Aplicando
o Teste t Mann-Whitney encontramos um valor de p maior que 0,05 (p>0,05), não
havendo, portanto, diferença estatística.
Figura 17 - Quantidade de células CD8 encontradas nas amostras contendo H.
pylori positivo e H.pylori negativo.
O gráfico acima demonstra que tanto na presença, quanto na ausência de H.
pylori estatisticamente não há diferença quanto ao número de células CD8 encontradas
na amostra estudada.
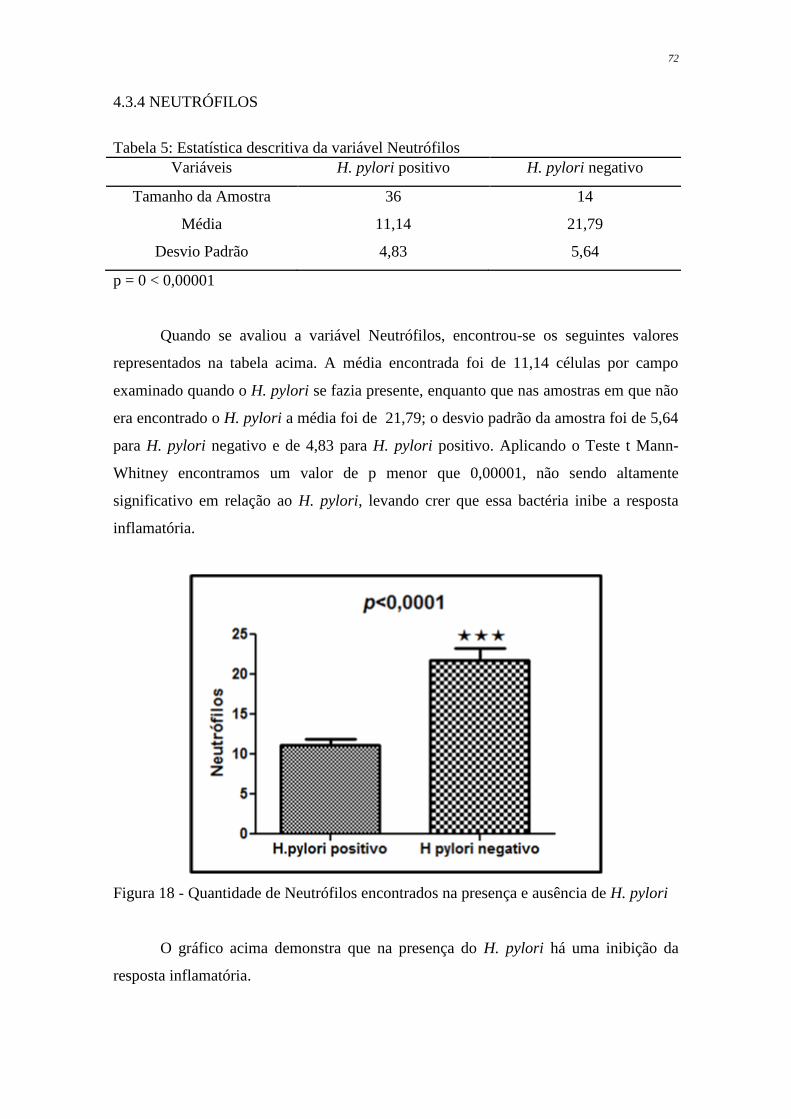
72
4.3.4 NEUTRÓFILOS
Tabela 5: Estatística descritiva da variável Neutrófilos
Variáveis H. pylori positivo H. pylori negativo
Tamanho da Amostra 36 14
Média 11,14 21,79
Desvio Padrão 4,83 5,64
p = 0 < 0,00001
Quando se avaliou a variável Neutrófilos, encontrou-se os seguintes valores
representados na tabela acima. A média encontrada foi de 11,14 células por campo
examinado quando o H. pylori se fazia presente, enquanto que nas amostras em que não
era encontrado o H. pylori a média foi de 21,79; o desvio padrão da amostra foi de 5,64
para H. pylori negativo e de 4,83 para H. pylori positivo. Aplicando o Teste t Mann-
Whitney encontramos um valor de p menor que 0,00001, não sendo altamente
significativo em relação ao H. pylori, levando crer que essa bactéria inibe a resposta
inflamatória.
Figura 18 - Quantidade de Neutrófilos encontrados na presença e ausência de H. pylori
O gráfico acima demonstra que na presença do H. pylori há uma inibição da
resposta inflamatória.
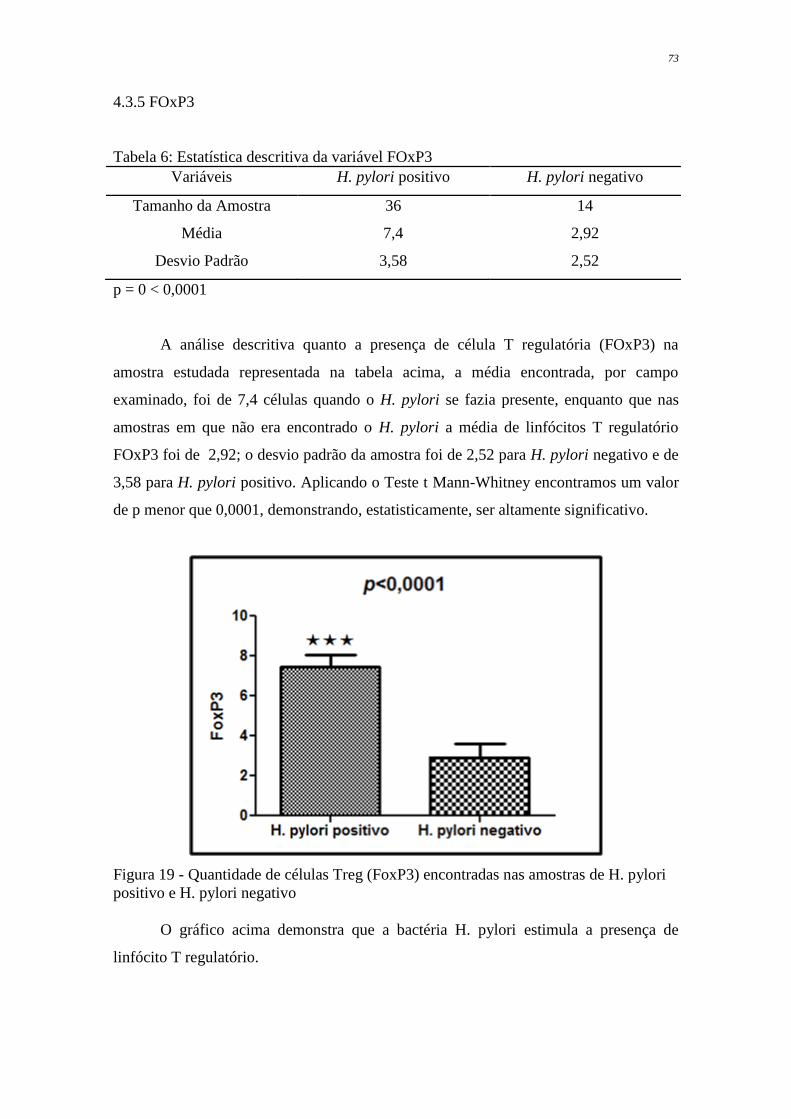
73
4.3.5 FOxP3
Tabela 6: Estatística descritiva da variável FOxP3
Variáveis H. pylori positivo H. pylori negativo
Tamanho da Amostra 36 14
Média 7,4 2,92
Desvio Padrão 3,58 2,52
p = 0 < 0,0001
A análise descritiva quanto a presença de célula T regulatória (FOxP3) na
amostra estudada representada na tabela acima, a média encontrada, por campo
examinado, foi de 7,4 células quando o H. pylori se fazia presente, enquanto que nas
amostras em que não era encontrado o H. pylori a média de linfócitos T regulatório
FOxP3 foi de 2,92; o desvio padrão da amostra foi de 2,52 para H. pylori negativo e de
3,58 para H. pylori positivo. Aplicando o Teste t Mann-Whitney encontramos um valor
de p menor que 0,0001, demonstrando, estatisticamente, ser altamente significativo.
Figura 19 - Quantidade de células Treg (FoxP3) encontradas nas amostras de H. pylori
positivo e H. pylori negativo
O gráfico acima demonstra que a bactéria H. pylori estimula a presença de
linfócito T regulatório.
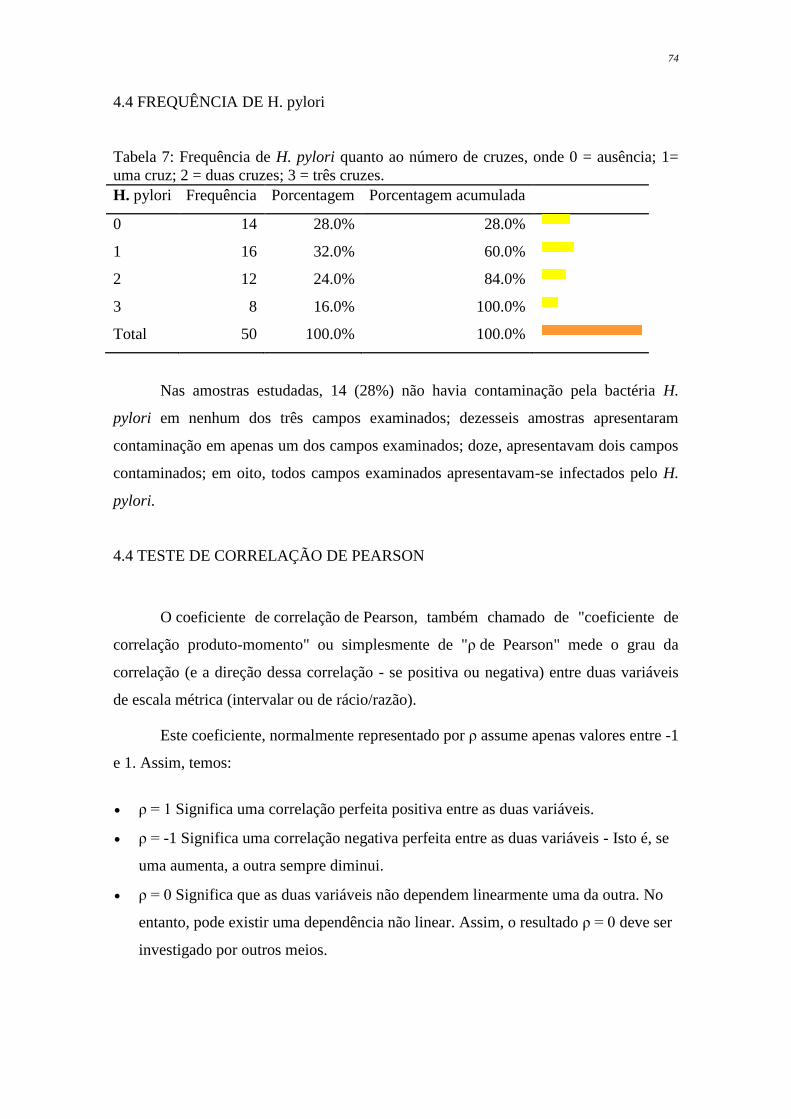
74
4.4 FREQUÊNCIA DE H. pylori
Tabela 7: Frequência de H. pylori quanto ao número de cruzes, onde 0 = ausência; 1=
uma cruz; 2 = duas cruzes; 3 = três cruzes.
H. pylori Frequência Porcentagem Porcentagem acumulada
0 14 28.0% 28.0%
1 16 32.0% 60.0%
2 12 24.0% 84.0%
3 8 16.0% 100.0%
Total 50 100.0% 100.0%
Nas amostras estudadas, 14 (28%) não havia contaminação pela bactéria H.
pylori em nenhum dos três campos examinados; dezesseis amostras apresentaram
contaminação em apenas um dos campos examinados; doze, apresentavam dois campos
contaminados; em oito, todos campos examinados apresentavam-se infectados pelo H.
pylori.
4.4 TESTE DE CORRELAÇÃO DE PEARSON
O coeficiente de correlação de Pearson, também chamado de "coeficiente de
correlação produto-momento" ou simplesmente de "ρ de Pearson" mede o grau da
correlação (e a direção dessa correlação - se positiva ou negativa) entre duas variáveis
de escala métrica (intervalar ou de rácio/razão).
Este coeficiente, normalmente representado por ρ assume apenas valores entre -1
e 1. Assim, temos:
ρ = 1 Significa uma correlação perfeita positiva entre as duas variáveis.
ρ = -1 Significa uma correlação negativa perfeita entre as duas variáveis - Isto é, se
uma aumenta, a outra sempre diminui.
ρ = 0 Significa que as duas variáveis não dependem linearmente uma da outra. No
entanto, pode existir uma dependência não linear. Assim, o resultado ρ = 0 deve ser
investigado por outros meios.
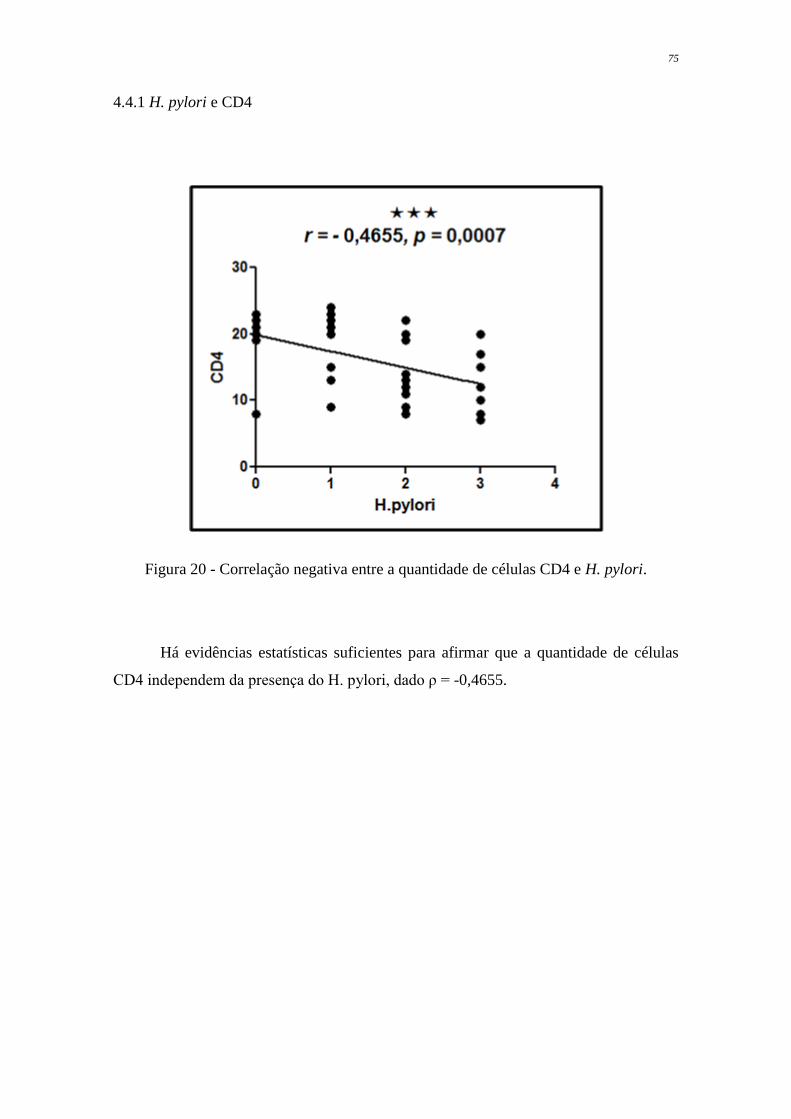
75
4.4.1 H. pylori e CD4
Figura 20 - Correlação negativa entre a quantidade de células CD4 e H. pylori.
Há evidências estatísticas suficientes para afirmar que a quantidade de células
CD4 independem da presença do H. pylori, dado ρ = -0,4655.
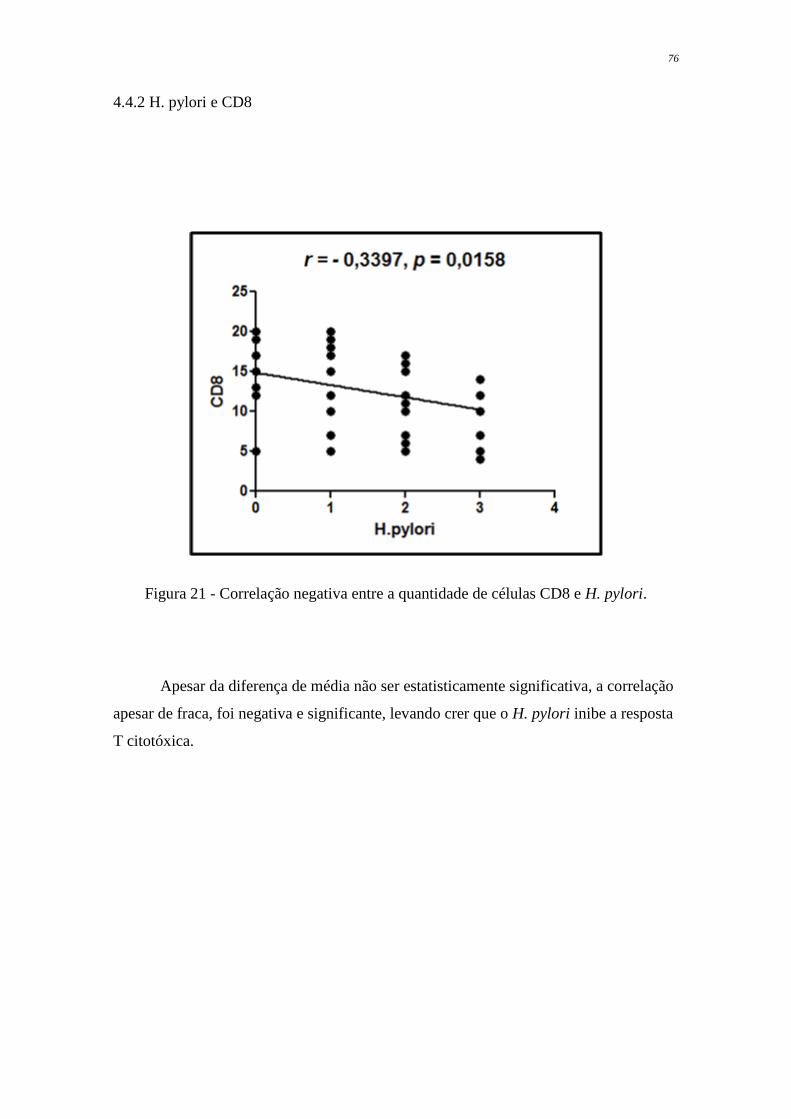
76
4.4.2 H. pylori e CD8
Figura 21 - Correlação negativa entre a quantidade de células CD8 e H. pylori.
Apesar da diferença de média não ser estatisticamente significativa, a correlação
apesar de fraca, foi negativa e significante, levando crer que o H. pylori inibe a resposta
T citotóxica.
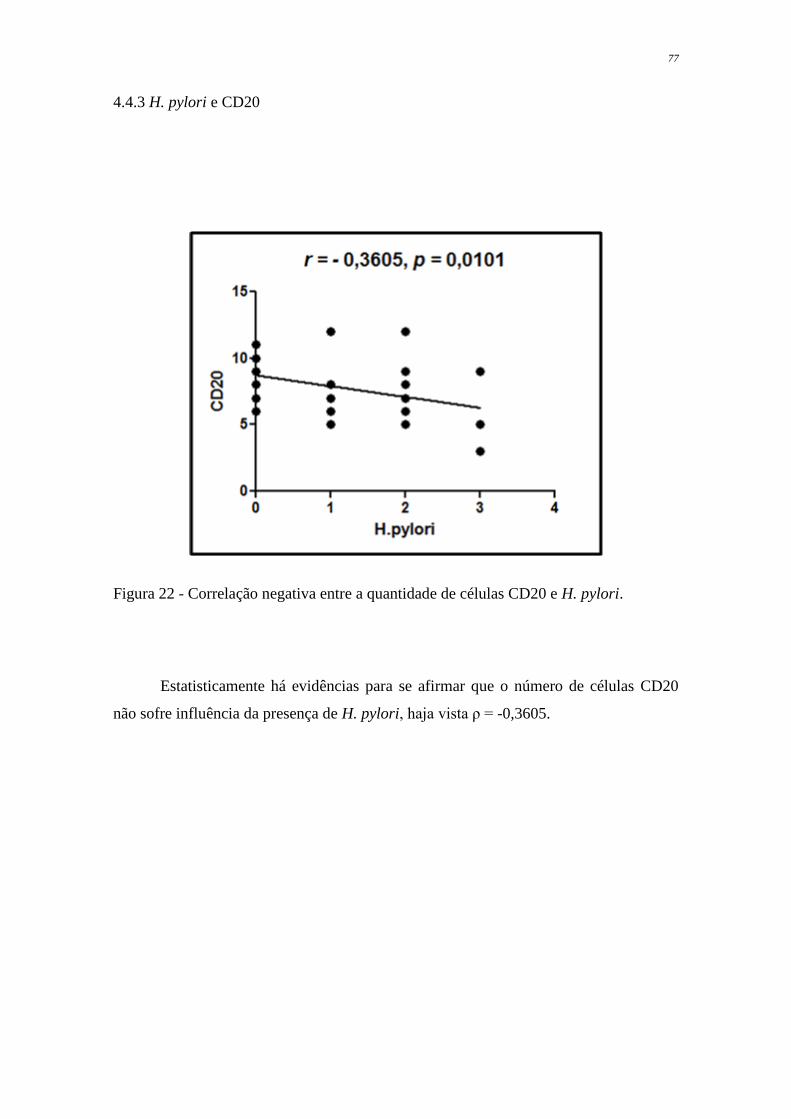
77
4.4.3 H. pylori e CD20
Figura 22 - Correlação negativa entre a quantidade de células CD20 e H. pylori.
Estatisticamente há evidências para se afirmar que o número de células CD20
não sofre influência da presença de H. pylori, haja vista ρ = -0,3605.
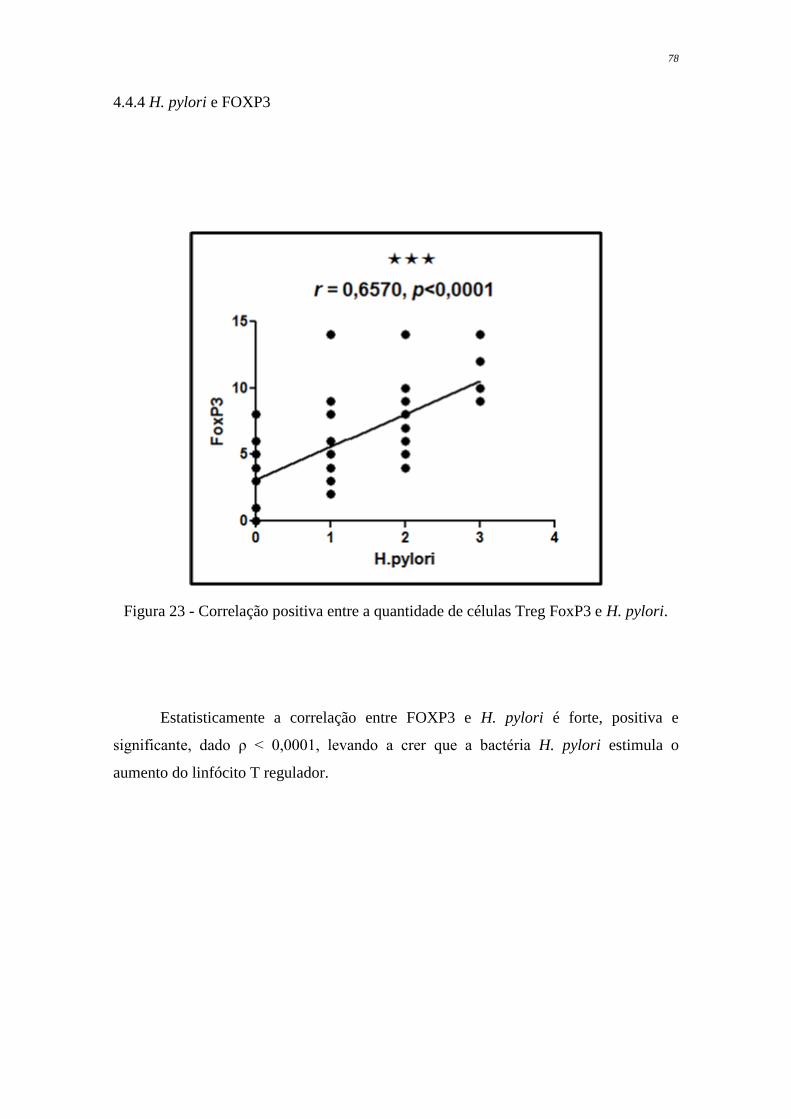
78
4.4.4 H. pylori e FOXP3
Figura 23 - Correlação positiva entre a quantidade de células Treg FoxP3 e H. pylori.
Estatisticamente a correlação entre FOXP3 e H. pylori é forte, positiva e
significante, dado ρ < 0,0001, levando a crer que a bactéria H. pylori estimula o
aumento do linfócito T regulador.
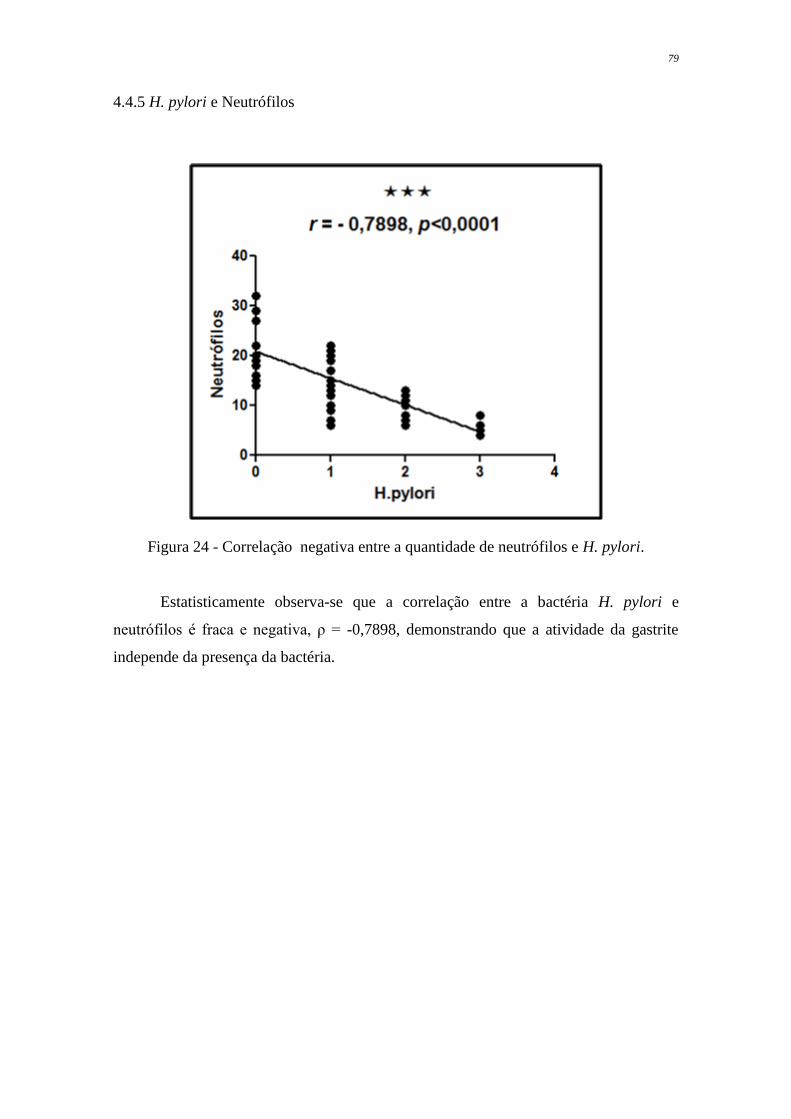
79
4.4.5 H. pylori e Neutrófilos
Figura 24 - Correlação negativa entre a quantidade de neutrófilos e H. pylori.
Estatisticamente observa-se que a correlação entre a bactéria H. pylori e
neutrófilos é fraca e negativa, ρ = -0,7898, demonstrando que a atividade da gastrite
independe da presença da bactéria.

80
4. DISCUSSÃO
Na grande maioria dos indivíduos infectados pela bactéria Helicobacter pylori,
apresentam uma inflamação que é restrita à mucosa da região antral do estômago.
Porém, em outros indivíduos, observa-se que há um comprometimento, também, ao
nível do corpo gástrico, determinando um quadro de pangastrite, podendo, por sua vez,
evoluir para vários graus de atrofia, tendo como consequência a diminuição da produção
de ácido clorídrico, levando a presumir que tais eventos sejam precursores do câncer
gástrico (Ladeira et al., 2003).
Apesar do alerta, o que se tem observado é que apenas 1% desses indivíduos que
foram infectados pelo H. pylori desenvolverão a neoplasia maligna do estômago. Este
fato pode estar relacionado com as diferenças na expressão de produtos bacterianos
específicos, a diferentes respostas do hospedeiro, ou a diferentes interações entre a
bactéria e o hospedeiro (Peek et al., 2002; Peek et al., 2006).
O Helicobacter pylori ao colonizar a mucosa do estômago promove uma
destruição da barreira de proteção da mesma, fazendo com que o ácido gástrico agrida a
própria mucosa, levando à inflamação, caracterizando a gastrite (Ladeira et al., 2003).
Nos últimos anos, uma variedade de testes tem sido usados no diagnóstico do H.
pylori. A escolha de qual teste a ser utilizado vai depender do número de casos, dos
dados clínicos informados, da localização precisa e dos custos e benefícios que
envolvem toda metodologia a ser empregada (Dunn et al, 1997).
A detecção histopatológica do Helicobacter pylori em amostras de biópsia
gástrica é considerada padrão ouro (El Zirraity et al., 1996).
Este exame é feito após a endoscopia, com a retirada de fragmento, no qual
utiliza-se uma variedade de colorações para detectar a bactéria, sendo necessário várias
horas ou mesmo dois dias para se obter o resultado. O microorganismo pode ser
identificado por diversas colorações histológicas, como Giemsa, hematoxilina e eosina
Warthin-starry, steiner, carbofulcsina. Nesse experimento foi adotado a coloração pela
hematoxilina-eosina para identificação da bactéria H. pylori.
Nesta pesquisa, utilizamos como método de escolha a endoscopia digestiva alta
associada a biópsia da mucosa do antro e corpo do estômago, a semelhança de inúmeros
trabalhos encontrados na literatura médica como de Dunn et al. (1997), Shomer et al.
(1998).

81
Pudemos observar, a semelhança do que foi observado por Dunn et al. (1997),
que o grau de inflamação da mucosa gástrica varia muito, indo desde uma leve
inflamação da lâmina própria, mantendo-se intacta a arquitetura celular, até a uma
inflamação intensa com formação de microabcessos e reação atípica do epitélio.
Neste estudo foi adotado o Sistema Sidney modificado para quantificar a
presença do Helicobacter pylori. Assim, quando em nenhum dos campos examinados
era encontrada a bactéria foi denominado de ausente; quando apenas 1 dos campos
examinados estava contaminado, denominou-se leve; quando dois dos campos
examinados estavam contaminados, chamamos de moderado; quando os três campos
examinados apresentavam-se contaminados pelo H. pylori, foi chamado de intenso.
Observa-se que na literatura, na grande maioria dos trabalhos, é adotado o método
descrito acima, como podemos observar em trabalhos de Gold et al.(2001), Ohyaguchi
et al. (2005), Shomer et al. (1998), Pereira et al. (2001).
Dos cinquenta pacientes estudados, catorze não apresentaram contaminação pela
bactéria e, trinta e seis estavam contaminados, sendo que 16 na forma leve, treze na
forma moderada e, sete na forma intensa. Corroborando com nossos achados,
encontramos que no trabalho de Gold et al. (2001) demonstraram resultados
semelhantes quando analisando histologicamente 33 casos de pacientes que foram
submetidos a endoscopia digestiva alta com biópsia da mucosa antral e corpo gástrico.
Outro importante trabalho é o de Ohyaguchi et al. (2005), que ao realizar estudo em
população japonesa, adotou o método de biópsia semelhante aquele realizado em nosso
experimento, onde guarda semelhança com o resultado desta pesquisa, apenas adotando
um quantitativo maior de casos. Shomer et al. (1998), em estudo experimental,
observaram, também, que houve uma variação histológica de moderada a intensa
quando a gastrite estava associada a presença do Helicobacter pylori.
Nesse experimento foi adotada a coloração pela hematoxilina-eosina para
identificação da bactéria H. pylori. Nas condições deste experimento, ocorreu 100% de
aproveitamento de todo material usado no desenvolvimento desta pesquisa.
A desregulação do sistema imune em muitas doenças é uma marca registrada.
No que diz respeito a infecção pelo H. pylori, a condição sine qua non é a gastrite, tanto
na forma crônica (linfócitos), quanto na forma aguda (neutrófilos) de inflamação. Nas
últimas décadas, estudos tem demonstrado descobertas importantes no que diz respeito
a patogenia da infecção pelo H. pylori, identificando os principais fatores de virulência

82
que, por sua vez, contribuem para os variados resultados clínicos (Wilson & Crabtree,
2007).
Assim, entender se a resposta de um tipo de célula em particular, representa
meramente uma resposta inata, ou adaptativa, é difícil, e o reconhecimento de que as
células, tais como células B podem responder diretamente ao H. pylori, ou através da
via de interação das células T ativadas, demonstra a complexidade da resposta imune
(Mai et al., 1992; Gobert et al., 2002; Panchal et al., 2003).
O tecido contaminado pelo H. pylori caracteriza-se pela presença de infiltrado de
polimorfonucleares (PMN), principalmente neutrófilos, que invadem o epitélio gástrico,
determinando uma série de mudanças neste epitélio, tais como depleção mucínica,
vacuolização citoplasmática e uma grande desorganização das glândulas mucosas, que
com o tratamento antimicrobiano esses desarranjos tendem a desaparecer. Entretanto,
uma boa quantidade de PMN persistem por muitos meses (Dunn et al., 1997).
Nesse estudo foi utilizado como parâmetro para avaliar a atividade da gastrite a
contagem de neutrófilos. Os resultados encontrados demonstraram que quando o
paciente, portador de gastrite, apresentava contaminação pelo H. pylori a quantidade de
neutrófilos era menor que na amostra de pacientes não contaminados pela bactéria,
levando a crer que essa inibe a resposta inflamatória.
Estudos realizados por Kandulski et al. (2010) demonstraram resultados
semelhentes ao desta pesquisa, chegando a conclusão semelhante.
Entretanto, os estudos de Karttunen et al. (1995), Israel & Peek (2001),
Rautemaa et al. (2001), Kondo et al. (2004), Algood et al. (2006) e Harris et al. (2008)
demonstraram resultados diferentes desta pesquisa, ou seja, quando havia a
contaminação pela bactéria o número de neutrófilos era maior que aqueles não
contaminados, podendo estar relacionado com outras variáveis não enquadradas nesta
pesquisa, levando-se em conta que esse resultado vai de encontro com o esperado, haja
vista que os neutrófilos medeiam as fases iniciais das reações inflamatórias. Não
obstante, acreditamos que tal fato pode estar relacionado com a intensidade da gastrite,
compartilhando o pensamento de alguns autores, sendo provável que a intensidade e a
forma das respostas inflamatória e imune, associadas a virulência da cepa, possam ser
responsáveis pelas diversas manifestações de doenças gastroduodenais.
Por outro lado, Yuceyar et al.(2002) não relataram nenhuma diferença
significativa entre os pacientes H. pylori infectados e indivíduos não infectados no que
diz respeito ao perfil de linfócitos periféricos, alegando que a infecção pelo H. pylori

83
induz inflamação crônica e local na mucosa gástrica, podendo não influenciar na
contagem absoluta de glóbulos.
Contudo, quando correlacionamos a presença de H. pylori e neutrófilos,
observamos que esta correlação é fraca e negativa, fazendo-nos pensar que a atividade
da gastrite independe da presença da bactéria.
Embora as respostas inflamatórias e imune possam agravar as lesões
degenerativas do epitélio, desencadeadas pela bactéria, são incapazes de eliminá-la.
É provável que a intensidade e a forma das respostas inflamatórias e imune,
associadas a virulência da cepa, possam estabelecer doenças gastroduodenais diferentes.
O linfócito T auxiliar (CD4+) tem a função de coordenar a defesa imunológica
contra vírus, bactérias e fungos, principalmente através da produção e liberação de
substâncias chamadas citocinas (Abbas et al., 2011).
Estudos demonstram que a gastrite crônica ativa está associada ao acúmulo de
células CD4+ na lâmina própria do estômago (Shomer et al., 1998; Scholte et al., 2002;
Israel & Peek, 2001).
Nesta pesquisa foi observado que quando não havia contaminação pelo H.
pylori, o número de células CD4+ era altamente significativo em relação as amostras
contendo a bactéria, demonstrando que a quantidade de células CD4+ independem da
presença da bactéria Helicobacter pylori, haja vista que todas as reações imunológicas,
fenotipicamente, são dependentes das células TCD4+. Por outro lado, nas condições
deste estudo, podemos concluir que a quantidade de células CD4+ não guarda relação
com grau evolutivo da gastrite, nem tampouco com a presença ou ausência da bactéria
H. pylori, mas sim com a patologia em si.
Em estudo envolvendo variadas doenças gastrointestinais desenvolvido por
Chen et al. (2013), demonstraram que as células CD4+ foram observadas em maior
quantidade nos indivíduos infectados pelo H. pylori. Entretanto, não podemos afirmar
que tais resultados divergem dos encontrados neste experimento, haja vista que em
nosso estudo foram envolvidos apenas indivíduos portadores de gastrite.
Estudos desenvolvidos por Beswick et al. (2009), demonstraram que as células T
CD4+ são suprimidas anti a infecção pelo H. pylori.
Contudo, Chen et al. (2013) demonstraram que as células T CD4+ estão
associadas com a redução do risco do desenvolvimento de doenças gástricas mais
graves.

84
Wu et al. (2011) concluiram que pacientes com gastrite crônica intensa
apresentam uma quantidade de células CD4+ mais elevada em comparação com aqueles
que apresentavam gastrite crônica leve e moderada, o que em nosso experimento foi
indiferente.
Quando aplicamos a correlação de Pearson em nosso experimento pudemos
observar que há evidências estatísticas suficientes para afirmar que a quantidade de
células CD4 independem da presença da bactéria Helicobacter pylori.
Com base nos resultados encontrados na literatura, comparados com os deste
experimento, entendemos que esta pesquisa não se afasta daquilo que foi concluído por
outros autores apesar de ter demonstrado resultados diferentes, pois entendemos que há
uma correlação entre essas células e a patogênese causada pela infecção do H. pylori.
Outrossim, entendemos também que em número de casos maiores, talvez pudéssemos
ter encontrado resultados semelhantes àqueles encontrados na literatura, assim como
poderia estar relacionado ao tempo de exposição ao microorganismo.
O linfócito T citotóxico é um importante leucócito que ataca células que se
tornam anormais, geralmente tumorais ou infectadas por vírus e bactérias. Os linfócitos
T citotóxicos (CD8+) são importantes para a saúde, principalmente por inibirem a
instalação e proliferação de cânceres e outros males atrelados ao incorreto
funcionamento de células do próprio organismo (Cocchi et al., 1995).
Segundo Abbas et al. (2011), a ativação completa das células TCD8+ virgens e a
diferenciação em CTL funcionais e células de memória podem exigir a participação das
células TCD4+ auxiliares.
Tummala et al. (2004), descreve em seu trabalho a relação entre o aumento de
células CD8+, assim como CD4+ em pacientes portadores de gastrite crônica.
A infecção pelo H. pylori estimula o epitélio gástrico a secretar uma variedade
de citocinas que determinam uma reação inflamatória no interior da mucosa gástrica
(Tummala et al., 2004).
Em nosso estudo pudemos observar que a quantidade de células CD8+
encontradas tanto nas amostras contaminadas pelo H. pylori, quanto as que não
continham a bactéria foram aproximadamente iguais, tanto que não houve diferença
estatística.
Resultado semelhante ao da nossa pesquisa pudemos observar no trabalho
desenvolvido por Choileain & Redmond (2006).

85
Ruiza et al. (2012) observaram em seu experimento que o número de células
CD8+ não eram estatísticamente significantes quando havia contaminação do estômago
de ratos pelo H. pylori.
Quando, em nosso experimento, aplicamos a correlação de Pearson entre a
CD8+ e presença do H. pylori, observa-se que esta correlação é fraca e negativa, porém
significante, levando a crer que a bactéria H. pylori inibe a resposta T citotóxica.
Em seres humanos a imunomarcação da molécula CD20 vem sendo usada para
avaliar qualitativamente a inflamação crônica (Lins et al., 2008).
A presença de um grande número de linfócitos B e células plasmáticas na
mucosa gástrica é a evidência de uma resposta humoral ante uma infecção crônica por
H. pylori (Tummala et al., 2004).
Este marcador de linfócitos B é positivo na membrana plasmática das células
atípicas do linfoma, definindo-o como de linhagem ou imunofenótipo B. Mesmo em
algumas células em mitose ou em apoptose é possível observar marcação.
Esta pesquisa demonstrou uma ligeiro predomínio de células CD20 nas amostras
em que não havia contaminação pelo H. pylori, entretanto, estatisticamente, foi
significante, demonstrando que as células CD20 tem papel importante na resposta
humoral ante a infecção crônica pelo H. pylori.
É escassa a literatura envolvendo gastrite, infectada ou não por H. pylori e
contagem de células CD20 (linfócitos B).
Porém, em experimento desenvolvido por Tummala et al. (2004), demonstraram
a presença das células CD20 na mucosa gástrica de portadores de gastrite crônica
infectados por H. pylori, entretanto carece de estudo comparativo com aqueles não
contaminados pela bactéria a semelhança de nosso experimento.
Quando correlacionamos células CD20 e H. pylori, observamos que
estatisticamente estas células não sofrem qualquer tipo de influência na presença da
bactéria.
As células Treg apresentam um papel importante na tolerância e controle
periférico da resposta imunológica, agindo sobre diversos tipos celulares, entre eles
Células TCD4+, TCD8+, células B, natural killers e células dendríticas. Não se sabe,
contudo, qual o(s) mecanismo(s) utilizado, e se este é o mesmo para cada tipo celular
específico (Rudensky & Campbell, 2006).
Esta supressão e imunorregulação exercida pelas células Treg constituem uma
forma de controlar a resposta inflamatória excessiva e danosa ao organismo, haja vista

86
que a inexistência deste controle pode determinar a hiper-reatividade do sistema imune
levando ao surgimento de doenças de natureza autoimune e alérgicas. Ao mesmo tempo,
se estas células exercem sua função de maneira acentuada, o excesso de controle levará
a falta de uma resposta inflamatória eficaz e vigorosa, necessária para controlar
infecções e neoplasias (Borrego et al., 2007; Cruvinel et al., 2008; Ferreira et al., 2010).
Assim, pode-se afirmar que a ausência de células Treg no decorrer da instalação
de uma doença infecciosa promoveria resistência à infecção, bem como sua presença
pode traduzir maior susceptibilidade e supressão da resposta imunológica. (Belkaid &
Rouse, 2005; Belkaid, 2007).
O interesse em se estudar células T regulatórias se deve à função chave dessa
população celular na manutenção dos mecanismos de autotelerância e regulação da
resposta imune (Sakaguchi et al., 1995).
Apenas como exemplo, muito embora não guarde relação com esta pesquisa,
Hori et al. (2003) demonstraram que o fator de transcrição FoxP3 é predominantemente
expresso nas Tregs tímicas e periféricas. Células CD4+CD25-transfectadas com FoxP3,
são capazes de suprimir a proliferação de outras células T e o desenvolvimento de
doença auto-imune e doença inflamatória in vivo e (Hori et al., 2003; Fontenot et al.,
2003).
Tudo leva crer que FoxP3 seja um gene de grande importância no
desenvolvimento e função das Tregs no ser humano (Hori et al., 2003; Sakaguchi,
2004). Fato este, que nos levou a nos interessarmos em pesquisar FoxP3.
A principal função das Tregs é regular negativamente as respostas imunes. Elas
são capazes de inibir várias células, tanto relacionadas a imunidade inata quanto a
adaptativa (Thornton et al., 2004).
Analisando artigos da literatura pudemos observar que há inúmeras evidências
sugerindo a relação das Tregs com o desencadeamento de doenças que efetivamente
determina quebra da autotolerância. Outrossi, o inadequado funcionamento das Tregs
poderá estar relacionado à indução, intensidade e forma de evolução de diversas
patologias que acometem os seres humanos.
Estudos demonstram que a função das Tregs nesse contexto é bastante
complexa. No caso das doenças crônicas, sua função supressora pode ser desfavorável
para a eliminação de alguns patógenos. Em contrapartida, suas características
regulatórias são fundamentais para evitar que o sistema imunitário elabore uma resposta

87
contra bactérias comensais, como por exemplo, as bactérias que colonizam a mucosa
gastrointestinal, ressalte-se o H. pylori.
Ultimamente, tem sido sugerida a influência da participação das Tregs em
doenças causadas por Helicobacter hepaticus, Helicobacter pylori e Cytomegalovirus
entre outras.
Nas condições deste experimento, ao ser quantificada a presença de linfócitos T
reguladores (Treg) em 50 pacientes portadores de gastrite, através da detecção
imunohistoquímica do fator transcricional Foxp3+, constatou-se que a presença de
Foxp3+ é maior naqueles pacientes que apresentavam gastrite com H. pylori, o que nos
faz crer que a bactéria H. pylori estimula o aumento do linfócito T regulador, podendo
restringir uma resposta inflamatória eficiente, o que culminaria em formas mais graves
da doença.
Fernández et al. (2008) estudando a ativação do sistema de células T regulatórias
por H. pylori, demonstrou que a infecção de células da mucosa gástrica na presença de
H. pylori provoca uma subexpressão do receptor de cadeia alfa (CD25) nos linfócitos T
CD4+ migrados para o foco inflamatório. Estes linfócitos também expressam anergia de
outras células T, o que poderia favorecer a persistência bacteriana e a cronificação da
infecção. O mesmo pode ser observado através dos resultados desta pesquisa quando
encontramos valores bem expressivos de FoxP3 nos indivíduos com gastrite positiva
para H. pylori em relação aos casos de gastrite sem a presença da bactéria.
Kao et al (2006), demonstraram que a infecção pelo H. pylori altera o equilíbrio
das DCs em direção a uma resposta T regulatória, suprimindo a indução eficaz do TH17
H. pylori específico. Nesse estudo pudemos observar que a infecção produzida pelo H.
pylori ao nível da mucosa gástrica determina um aumento da resposta celular gástrica T
reguladora.
Um estudo experimental, envolvendo ratos, desenvolvido por Rad et al (2006),
identificou um grande número de linfócitos FoxP3 em biópsias de estômagos de ratos
H. pylori positivos, concluindo que estas células participam da resposta imune para H.
pylori.
Em outro estudo experimental com ratos, Raghavan et al. (2003) demostraram
que quando ratos encontravam-se infectados por H. pylori haveria uma ausência de
células T reg, estando associada com aumento da patologia gástrica.

88
Lundgren et al. (2003) concluiram em seu experimento que células T reg
específicas circulam para suprimir as resposta de memória de células T em pessoas
infectadas pelo H. pylori.
Harris et al. (2008), estudando 115 pacientes entre crianças e adultos, residentes
em Santiago do Chile, concluíram que as patologias gástricas são bem menos agressivas
em crianças quando infectadas pelo H. pylori e apresentam resposta T reguladora
aumentada, quando comparadas com adultos infectados, sugerindo que Treg regula a
resposta inflamatória induzida por H. pylori em crianças.
A correlação de Pearson entre H. pylori e FOXP3 é forte, positiva e significante,
o que nos leva a crer que a bactéria atua estimulando o aumento do linfócito T
regulador.

89
5. CONCLUSÕES
Nas condições desta pesquisa podemos afirmar:
72% das amostras estavam contaminadas pelo H. pylori, destes o maior grupo
encontrava-se na faixa de leve a moderada (77,77%).
A quantidade de células CD4 foi bem maior nas amostras que não estavam
contaminadas pelo H. pylori. Portanto, pode-se afirmar que a quantidade de células CD4
independe da presença do H. pylori.
É possivel afirmar que a bactéria H. pylori inibe a resposta T citotóxica.
Os linfócitos B não sofrem influência na presença do H. pylori.
A atividade da gastrite não depende da presença do H. pylori. Por outro lado,
pode-se afirmar que a bactéria inibe a resposta inflamatória.
A quantidade de células T regulatórias FOXP3 encontradas nas amostras
contaminadas pelo H. pylori nos leva a crer que a bactéria estimula o aumento de
linfócito T regulatório, podendo está relacionado com a supressão de uma resposta
inflamatória eficaz, culminando com formas mais agressivas de gastrite.
Por fim, apesar do desenvolvimento de inúmeros trabalhos envolvendo a
bactéria Helicobacter pylori, entendemos que careça de mais estudos com propostas de
acompanhamento e esclarecimento quanto a função exata das células Treg, bem como
suas implicações funcionais, relacionados a patologia inflamatória gástrica, no intuito de
esclarecer os mecanismos relacionados ao adoecimento e resistência à doença, para
então buscar terapias eficazes que atuarão nos mecanismos da resposta imunológica

90
REFERÊNCIAS BIBLIOGRÁFICAS
ABBAS, A.K., LICHTMAN, A.H. Cellular and molecular immunology, 5th
ed.
Philadelphia: Saunders 225-243, 2005.
ABBAS, A.K., LICHTMAN, A.H., PILLAI, S. Imunologia Celular e Molecular.
Editora Elsevier, Rio de Janeiro, 2011.592p.
AGUIAR, D.C.F., CORVELO, T.C., ARAÚJO, M., CRUZ, E.M., DAIBES, S.,
ASSUMPÇÃO, M.B. Expressão dos antígenos ABH e Lewis na gastrite
crônica e alterações pré-neoplásicas da mucosa gástrica. Arquivos de
Gastroenterologia 39: 222-232, 2002.
AGUILAR, G. R., AYALA, G., FIERROS-ZÁRATE, G. Helicobacter pylori:
Recent advances in the study of its pathogenicity and prevention. Salud
Pública de México 43 (3): 237-247, 2001.
AKHIANI, A.A., PAPPO, J., KABOK, Z., SCHON, K., GAO, W., FRANZEN,
L.E., LYCKE, N. Protection against Helicobacter pylori infection following
immunization is IL-12-dependent and mediated by Th1 cells. J Immunol 169:
6977-6984, 2002.
AKHIANI, A.A., SCHON, K., FRANZEN, L.E., PAPPO, J., LYCKE, N.,
Helicobacter pylori-especific antibodies impair the development of gastritis,
facilitate bacterial colonization, and counteract resistance against infection. J
Immunol 172: 5024-5033, 2004.
ALGOOD, H.M., COVER, T.L. Helicobacter pylori persistence: an overwiew of
interactionsbetween H. pylori and host immune defense. Clin. Microbiol.
Rev. 597-613, 2006.
ALM, R. A., LING, L. S., MOIR, D. T., KING, B. L., BROWN, E. D., DOIG, P.
C., SMITH, D. R., NOONAN, B., GUILD, B. C., DEJONGE, B. L.,
CARMEL, G., TUMMINO, P. J., CARUSO, A., URIA-NICKELSEN, M.,
MILLS, D. M., IVES, C., GIBSON, R., MERBER, G. D., MILLS, S. D.,
JIANG, Q., TAYLOR, D. E., VOVIS, G. S., TRUST, T. J. Genomic-sequence
of two unrelated isolates of the human gastric pathogen Helicobacter pylori.
Nature 397 (6715): 176-180, 1999.

91
ÁLVARES, M.M.D., MARINO, M., OLIVEIRA, C.A., MENDES, C.C., COSTA,
A.C.F., GUERRA, J., QUEIROZ, D.M.M., NOGUEIRA, A.M.M.F.
Características da gastrite crônica associada a Helicobacter pylori: aspectos
topográficos, doenças associadas e correlação com o status cagA. Jornal
Brasileiro de Patologia e Medicina Laboratorial 42(1): 51-59, 2006.
AMEDEI, A., CAPPON, A., CODOLO, G. The neutrophil-activating protein of
H. pylori promotes Th1 immune responses. Journal of Clinical Investigation
116: 1092-1101, 2006.
AMIEVA, M.R., VOGELMANN, R., COVACCI, A., TOMPKUNS, L.S.,
NELSON, W.J. FALKOW, S. Disruption of the epithelial apical-junctional
complex by Helicobacter pylori CagA. Science 300: 1430-1434, 2003.
ANDO, T., GOTO, Y., MAEDA, O., WATANABE, O., ISHIGURO, K., GOTO,
H. Causal role of Helicobacter pylori infection in gastric cancer. World
Journal of Gastroenterology 12 (2): 181-186, 2006.
ANTOS, D., CRONE, J., KONSTANTOPOULOS, N., KOLETZKO, S.
Evaluation of a novel rapid one-step immunochromatographic assay for
detection of monoclonal Helicobacter pylori antigen in stool samples from
children. Journal Clinical Microbiology 43 (6): 2598-2601, 2005.
APPELMELK, B.J., VAN DIE, I., VAN VILET, S.J., VANDENBROUCKE-
GRAULS, C.M., GEIJTENBEEK, T.B., VAN KOOYK, Y. Cutting edge:
carbohydrate profiling identifies new pathogens that interact with dendritic
cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J Immunol
170: 1635-1639, 2003.
ARAUJO, M. T. F., ALVES JUNIOR, J. M., ARAUJO, R., MAKINO, E.,
KHAWAGE, S. Frequency of Helicobacter pylori in gastric cancer patients in
Belem, Pará, Brasil. Arquivo Brasileiro de Cirurgia Digestiva 8 (4): 92-95,
1993.
ASAHI, M., AZUMA, T., ITO, S., ITO, Y., SUTO, H., NAGAI,Y.,
TSUBOKAWA, M., TOHYAMA, Y., MAEDA, S., OMATA, M., SUZUKY,
T., SASAKAWA, T. Helicobacter pylori cagA protein can be tyrosine
phosphorylated in gastric epithelial cells. Journal Experimental Medicine
191 (4): 593-602, 2000.

92
ASHOUR, A.A.R., GUSMÃO, V.R., MAGALHÃES, P.P., COLLARES, G.B.,
MENDES, E.N., QUEIROZ, D.M.M., ROCHA, G.A., ROCHA,A.M.C.,
CARVALHO,A.S.T. Associação entre cagA e alelos do vacA de Helicobacter
pylori e úlcera duodenal em crianças no Brasil. Jornal Brasileiro de
Patologia e Medicina Laboratorial 38(2): 79-85, 2002.
ATHERTON JC, CAO P, PEEK RM JR, TUMMURU MK, BLASER MJ,
COVER TL. Mosaicism in vacuolations alleles of Helicobacter pylori. The
Journal of Biological Chemistry 270 (30):17771-77, 1995.
ATHERTON, J. C. CagA: a role at last. Gut 47 (3): 330-331, 2000.
ATHERTON, J. C. The clinical relevance of strain types of Helicobacter pylori.
Gut 40: 701-703, 1997.
AYRES, M; AYRES, M.J; AYRES, D.L; SANTOS, A.S. Bioestat 5.0:
aplicações estatísticas nas áreas das ciências biológicas e médicas. Belém,
Sociedade Civil Mamirauá MCT - CNPq, 2003. 364p.
BACCHETTA, R., GAMBINERI, E., RONCAROLO, M.G. Role of regulatory T-
cells and FOXP3 in human diseases. J Allergy Clin Immunol 120 (2): 227-
235, quiz 36-37, 2007.
BAGGIOLINI, M., DEWALD, B., MOSER, B. Interleukin-8 and related
chemoatractic cytokines - CXC and CC chemokines. Adv Immunol 55: 97-
179, 1994.
BAMFORD, K.B., FAN, X., CROWE, S.E., LEARY, J.F., GOURLEY, W.K.,
LUTHRA, G.K., BROOKS, E.G., GRAHAM, D.Y., REYES, V.E., ERNST,
P.B. Lymphocytes in the human gastric mucosa during Helicobacter pylori
have a T helper 1 effector phenotype. Gastroenterology 114: 482-492, 1998.
BELKAID, Y. Regulatory T cells and infection: a dangerous necessity. Nature
Reviews Immunology 7 (11) 875–888, 2007.
BELKAID, Y.; ROUSE, B.T. Natural regulatory T cells in infectious disease.
Nature Immunology. n.6, p. 353 – 360, 2005.
BERSTAD, A. E., HOGASEN, K., BUKHOLM, G., MORAN, A. P.,
BRANDTZAEG, P. Complement activation directy induced by Helicobacter
pylori. Gastroenterology 120 (5): 1108-1116, 2001.

93
BESWICK, E.J., PINCHUK, I.V., SCHIMITT, D., REYES, V.E. Helicobacter
pylori induces gastric epithelial cell production of TGF-β that suppresses
CD4+ T cells. The Journal of Immunology 182: 9,7, 2009.
BITTENCOURT, P. F. S., ROCHA, G. A., PENNA, F. J., QUEIROZ, D. M.
Úlcera péptica gastroduodenal e infecção pelo Helicobacter pylori na criança e
adolescente. Journal of Pediatric 82 (5): 325-334, 2006.
BJÖRKHOLM, B. & SALAMA, N. R. Genomics of Helicobacter 2003.
Helicobacter 8 (1): 1-7, 2003.
BLASER, M. J. & ATHERTON, J. C. Helicobacter pylori persistence: biology
and disease. The Journal of Clinical Investigation 113 (3): 321-333, 2004.
BLASER, M. J. An endangered species in the stomach. Scientific American 292
(2): 38-45, 2005.
BLASER, M. J. Hypotheses on the pathogenesis and natural history of
Helicobacter pylori-induced inflammation. Gastroenterology 102 (2): 720-
727, 1992.
BLASER, M.J. Helicobacter pylori and the pathogenesis of gastroduodenal
inflammation. The Journal of Infectious Disease 161 (4): 626-633, 1990.
BLASER, M.J..& BERG, D.E. Helicobacter pylori genetic diversity and risk of
human disease. Journa of Clinical Investigation 107 (7): 767-73, 2001.
BOONJAKUAKUL, J. K., SYVANEN, M., SURYAPRASAD, A., BOWLUS, C.
L., SOLNICK, J. V. Transcription profile of the Helicobacter pylori in the
human stomach reflects its physiology in vivo. The Journal of Infectious
Diseases 190 (5): 946-956, 2004.
BORGES, I.K., LIMA, J.L., MILANEZ, P.A.O., BERNARDES, S.S., FELIPE, I.
COSTA, I.C., SARIDAKIS, H.O., WATANABE, M.A.E. Participação de
células T regulatórias (Tregs) na imunopatogênese da infecção pelo vírus da
imunodeficiência humana 1 (HIV-1). Semina; Ciências Biológicas e da
Saúde, Londrina 31 (2): 169-178, 2010.
BORREGO, L.M.; ROSA, S.; ALGUERÓ, C.; TRINDADE, H.; PINTO, J.R.
Células reguladoras. Revista Portuguesa de Pneumologia 13 (3), 2007.

94
BRASIL, MINISTÉRIO DA SAÚDE. Instituto nacional do câncer. Estimativas
da incidência por câncer no Brasil para o ano de 2008. Rio de Janeiro:
INCA, 2008.
BRASIL, MINISTÉRIO DA SAÚDE. Instituto nacional do câncer. Estimativas
da incidência por câncer no Brasil para o ano de 2010. Rio de Janeiro:
INCA, 2010.
BUSSIERE, F.I., CHATURVEDI, R., ASIM, M., HOEK, K.L., CHENG, Y.,
GAINOR, J., SCHOLZ, A., KHAN, W.N., WILSON, K.T. Low multiplicity
of infection of Helicobacter pylori suppresses apoptosis of B lymphocites.
Cancer Res 66: 6834-6842, 2006.
BUSSIERE, F.I., CHATURVEDI, R., CHENG, Y., GOBERT, A.P., ASIM, M.,
BLUMBERG, D.R. et al. Spermine causes loss of innate immune response
to Helicobacter pylori by inhibition of inducible nitric-oxide synthase
translation. J Biol Chem 280: 2409–2412, 2005.
BUTOV, I., SADCHIKOV, V. D., KHUSNUTDINOV, S. H. M.,
KHARCHENKO, A. V. Helicobacter pylori and stomach cancer. Lik Sprava
220 (4): 69-71, 1998.
CALAM, J. & BARON, J. H. ABC of the upper gastrointestinal tract:
pathophysiology of duodenal and gastric ulcer and gastric câncer. British
Medical Journal 323: 980-982, 2001.
CAMPBEL, D.J., ZIEGLER, S.F. FOXP3 modifies the phenotypic and functional
properties of regulatory T cells. Nat Rev Immunol 7 (4): 305-310, 2007.
CARVALHO, A.S.T, QUEIROZ, D.M., MENDES, E.N., ROCHA, G.A.,
PENNA, F.J.. Diagnosis and distribution of Helicobacter pylori in the gastric
mucosa of symptomatic children. Brazilian Journal of Medical and
Biological Research 24 (2): 163-166, 1991.
CENSINI, S; STEIN, M; COVACCI, A. Cellular responses induced after contact
with Helicobacter pylori. Current Opinion in Microbiology 4: 41-48. 2001.
CÉSAR, A.C.G.C; CURY, P.M.; PAYÃO, S.L.M.; LIBERATORE, P.R.; SILVA,
A.E.Comparação dos diagnósticos histológico e molecular do Helicobacter
pylori em lesões benignas e adenocarcinomas gástricos. Jornal Brasileiro de
Microbiologia 36 (1): 12-16, 2005.

95
CHEHTER, L. Úlcera péptica gastroduodenal. In: _ Sinopse de
Gastroenterologia. São Paulo: UNIFESP, 1999. p. 14-18.
CHEN, L., YANG, W-C, HE, J-L, LI, N-Y, HU, J., HE, Y-F, YU, S., ZHAO, Z.,
LUO, F., ZHANG, J-Y, LI, H-B, ZENG, M., LU,D-S, LI, B-S, GUO, H.,
YANG, S-M, GUO, G., MAO, X-H, CHEN, W., WU,C., ZOU, Q-M. A
Dominant CD4+ T-cell Response to Helicobacter pylori Reduces Risk for
Gastric Disease in Humans. Gastroenterology 144 (3): 591-600, 2013.
CHEN, W., SHU, D., CHADWICK, V.S. Helicobacter pylori infection:
mechanism of colonization and functional dyspepsia reduced colonization of
gastric mucosa by Helicobacter pylori in mice deficient in interleukin-10. J
Gastroenterol Hepatol 16: 377-383, 2001.
CHIEPPA, M., RESCIGNO, M., HUANG, A.Y., GERMAIN, R.N. Dynamic
imaging of dendritic cell extension into the small bowel lumen in response to
epithelial cell TLR engagement. J Exp Med 203: 2841-2852, 2006.
CHOILEAIN, N.N., REDMOND, H.P. Regulatory T-cells and autoimmunity. J
Surg Res 130 (1): 124-135, 2006.
CHOLLET-MARTIN, S., MONTRAVERS, P., GIBERT, C. et al. High levels of
interleukin-8 in the blood and alveolar spaces ofpatients with pneumonia and
adult respiratory distress syndrome. Infect Immun 61: 4553-4559, 1993.
CHU, K. M., POON, R., TUEN, H. H., LAW, S. Y. K., BRANICKI, F. J.,
WONG, J. A. Prospective comparison of locally made rapid urease test and
histology for the diagnosis of Helicobacter pylori infection. GastrointestInal
Endoscopy 46 (6): 503-506, 1997.
COCCHI, F., DEVICO, A.L., GARZINO-DEMO, A., ARYA, S.K., GALLO,
R.C., LUSSO, P. Identification of RANTES, MIP-1α, and MIP-1β as the
major HIV-suppressive factors produced by CD8+ T cells. Science 270: 1811-
1815, 1995.
COELHO, L.G.V., LÉON-BARÚA, R., QUIGLEY, E. M. M. and representatives
of the Latin-American National Gastroenterological Societies affiliated with
the Inter-American Association of Gastroenterology (AI-GE). Latin-American
Consensus Conference on Helicobacter pylori infection. American Journal
Gastroenterology 95: 2688-2691, 2000.

96
COFFER, P.J., BURGERING, B.M. Forkhead-box transcriptions factors and their
role in the immune system. Nat Rev Immunol 4 (11): 889-899, 2004.
COLE, S. P., CIRILLO, D. KAGNOFF, M. F. GUINEY, D. G. ECKMANN, L.
Coccoid and spiral Helicobacter pylori differ in their abilities to adhere to
gastric epithelial cells and induce interleukin-8 secretion. Infection and
Immunity 65 (2): 843-846, 1997.
COSTA, K., BACHER, G., ALLMAIER, G., BELLO, M. G. D., EGSTRAND, L.,
FALK, P., PEDRO, M. A., PORTILLO, F. G. The morphological transition of
Helicobacter pylori cells from spiral to coccoide is preceded by substantial
modification of the cell wall. Journal of Bacteriology 181 (12): 3710-3715,
1999.
COVACCI, A, TELFORD, L.J, GIUDICE, D.G, PARSONNET, J., RAPPUOLI,
R. Helicobacter pylori virulence and genetic geography. Science 284: 1328-
1333, 1999.
COVER, T. L., TUMMURU, M. K., CAO, P., THOMPSON, S. A., BLASER, M.
J. Divergence of genetic dequences for the vacuolating cytotoxin among
Helicobacter pylori strains. J. Gastroenterol 267 (15): 1056-1073, 1994.
CRABTREE, J.E., TAYLOR, J.D., WYATT, J.L., HEATLEY, R.V.,
SHALLCROSS, T.M., TOMPKINS, D.S., RATHBONE, B.J. Mucosal IgA
recognition of Helicobacter pylori 120kDa protein, peptic ulceration, and
gastric pathology. Lancet 338: 332-335, 1991.
CRABTREE, J.E., NAUMANN, M. Epithelial cell signalling in Helicobacter
pylori infection. Curr Signal Transduction Ther 1: 53-65, 2006.
CRUVINEL, W.M.; MESQUITA JR.,D.; ARAÚJO, J.A.P.; SALMAZI, K.C.;
KÁLLAS, E.G.; ANDRADE, L.E.C. Células T regulatórias naturais (Treg) em
doenças reumáticas. Revista Brasileira de Reumatologia 48 (6): 342-355,
2008.
D’ELIOS, M. M., ANDERSEN, L. P. Helicobacter pylori inflammation,
immunity, and vaccines. Helicobacter 12 (1): 15-19, 2007.

97
D’ELIOS, M. M., MANGHETTI, M., ALMERIGOGNA, F., AMEDEI, A.,
COSTA, F., BURRONI, D., BALDARI, C. T., ROMAGAGNI, S.,
TELFORD, J. L., DEL PRETE, G. Different cytokine profile and antigen-
especificity repertoire in Helicobacter pylori-specific T cells clones from the
antrum of chronic gastritis patients with or without peptic ulcer. European
Journal of Immunology 27 (7): 1751-1755, 1997.
DE GROOTE, D., VAN DOORN, L. J., DUCATELLE, R., VERSCHUUREN,
A., HAESEBROUCK, F., QUIT, W. G. JALAVA, K. VANDAMME, P.
Candidatus Helicobacter suis’, a gastric from pigs, and its phylogenetic
relatedness to other gastropirilla. International Journal Systematic and
Evolutionary Microbiology 49 (4): 1769-1777, 1999.
DEBONGNIE, J. C., BURETTE, A., GLUPEZYNSKI, Y., PREZ, C. D.,
KONINCK, X., DONNAY, M. Helicobacter pylori and gastric cancer. An
endoscopic series. Acta Gastroenterologica Belgica, 60 (3): 189-191, 1997.
DRUMM, B., PEREZ-PEREZ, G.I., BLASER, M.J., SHERMAN, P.M.
Intrafamilial clustering of Helicobacter pylori infection. New England
Journal of Medicine 22: 359, 1990.
DUNN, B. E., COHEN, H., BLASER, M.J. Helicobacter pylori. Clinical
Microbiology Reviews 10: 720-41, 1997.
EGGEMA, M.P., BARUGAHARE, B., JONES, N., OKELLO, M., MUTALYA,
S., KITYO, C., Depletion of regulatory T cells in HIV infection is associated
with immune activation. J Immunol 174 (7): 4407-4414, 2005.
EATON, K.A., MEFFORD, M., THEVENOT, T. The role of T cell subsets and
cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J
Immunol 166: 7456-7461, 2001.
EL-OMAR, E.M., CARRINGTON, M., CHOW, W.H., et al. Interleukin-1
polymorphisms associated with increased risk of gastric cancer. Nature 404:
398- 402, 2000.
EL-ZIRRAITY, H.M.T., GRAHAM, D.Y., AL-ASSI, T.M., MALATY, H.,
KARTTUNEN, T.J., GRAHAM F, D., HUBERMAN, R.M., GENTA, R.M.
Interobserver variation in the histopatological assessment of Helicobacter
pylori gastritis. Human Pathology 27: 35-41, 1996.
ESLICK, G. Sexual transmission of Helicobacter pylori via oral-anal intercourse.
International Journal Study of Aids 13 (1): 7-11, 2001.

98
FERNÁNDEZ, M.C., MARTINEZ, S.B., CID, T.P. Oxidative stress
by Helicobacter pyloricauses apoptosis through mitochondrial pathway in
gastric epithelial cells. Apoptosis 13 (10): 1267-1280, 2008.
FERREIRA. M.C.; DE OLIVEIRA, R.T.; DA SILVA, R.M.; BLOTTA, M.H.;
MAMONI, R.L.; Involvement of regulatory T cells in the immunossupression
characteristic of patients with paracoccidioidomycosis. Infecction and
Immunity 78: 4392 – 4401, 2010.
FIGUEIREDO, C., MACHADO, J. C., YAMAOKA, Y. Pathogenesis of
Helicobacter pylori infection. Helicobacter 10 (1): 14-20, 2005.
FONTENOT, J.D., GAVIN, M.A., RUDENSKY, A.Y. FoxP3 programs the
development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4
(4): 330-336, 2003.
FOX, J. G. & WANG, T. C. Helicobacter pylori infection: pathogenesis. Current
Opinion in Gastroenterology 18: 15-25, 2002.
GALMICHE, A., RASSOW, J., DOYE, A., CAGNOL, S., CHAMBARD, J. C.,
CONTAMIN, S., DE THILLOT, V., JUST, I., RICCI, V., SOLCIA, E., VAN
OBBERGHEN, E., BOQUET, P. The N-terminal 34 kDa fragment of
Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces
cytochrome c release. The EMBO Journal 19 (23): 6361-6370, 2000.
GAMBINERI, E., TORGERSO, T.R., OCHS, H.D. Immune dysregulation,
polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a
syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical
regulator of T-cell homeostasis. Curr Opin Rheumatol 15 (4): 430-435,
2003.
GARCIA-VALLVÉ, S., JANSEN, P. J., OUZOUNIS, C. A. Genetic variation
between Helicobacter pylori strains: gene acquisition or loss? Trends in
Microbiology, 10 (10): 445-447, 2002.
GEBERT, B., FISCHER, W., WEISS, E., et al. Helicobacter pylori vacuolating
cytotoxin inhibits T lymphocyte activation. Science 301: 1099-1102, 2003.
GEWIRTZ, A.T., YU, Y., KRISHNA, U.S., ISRAEL, D.A., LYONS, S.L., PEEK,
R.M. Jr. Helicobacter pylori flagellin evades toll-like receptor 5-mediated
innate immunity. J Infect Dis 189: 1914-1920, 2004.

99
GILGER, M. A., TOLIA, V., JOHNSON, A., RABINOWITZ, S., JIBALY, R.,
ELITSUR, Y., CHONG, S., ROSENBERG, A., GOLD, B., ROSENTHAL, P.,
ELKAYAM, O., MARCHILDON, P., PEACOCK, J. The use of an oral fluid
immunoglobulin G ELISA for the detection of Helicobacter pylori infection in
children. Helicobacter 7: 105-110, 2002.
GISBERT, J.P., BLANCO, M., CRUZADO, A.I., PAJARES, J.M. Helicobacter
pylori infection, gastric metaplasia in the duodenum and the relationship with
ulcer recurrence. Eur J Gastroenterol Hepatol 12 (12): 1295-1298, 2000.
GO, M. F. & CROWE, S. E. Virulence and pathogenicity of Helicobacter pylori.
Gastroenterology Clinical North American 29 (3): 545-549, 2000.
GO, M. F. Review article: natural history and epidemiology of Helicobacter pylori
infection. Alimentary Pharmacology & Therapeutics 16 (1): 3-15, 2002.
GOBERT, A. P., MERSEY, B. D., CHENG, Y., BLUMBERG, D. R., NEWTON,
J. C., WILSON, K. T. Cutting edge: urease release by Helicobacter pylori
stimulates macrophage inducible nitric oxide synthase. Journal of
Immunology 168 (12): 6002-6006, 2002.
GOLD, B.D., DOORN, L-J, GUARNER, J., OWENS, M., PIERCE-SMITH, D.,
SONG, Q., HUTWAGNER, L., SHERMAN, P.M., MOLA, O.L., CZINN, S.
Genotypic, clinical, and demographic characterisitics of children infected with
Helicobacter pylori. Journal of Clinical Microbiology 39 (4): 1348-1352,
2001.
GOLDWIN, C. S. & AMSTRONG, J. A. Microbilogical aspects of Helicobacter
pylori (Campylobacter pylori). European Journal of Clinical Microbiology
& Infectious Disease 9 (2): 1-13, 1990.
GONZÁLEZ-VALENCIA, G., ATHERTON, J.C., MUÑOZ, O., DEHESA, M.,
MADRAZO-DE LA GARZA, A., TORRES, J. Helicobacter pylori vacA and
cagA genotypes in Mexican Adults and children. The Journal Infectious
Diseases 182: 1450-1454, 2000.
GOODWIN, C.S. et al. Campylobacter pylori become Helicobacter pylori. Int. J.
Bacteriol. 39: 353-405, 1989.

100
GRIMM, M.C., DOC, W.F. Chemokines in inflammatory bowel disease mucosa:
expression of RANTES, macrophage inflammatory protein (MIP)- 1α, MIP-
1β, and γinterferon-inducible protein-10 by macrophages, lymphocytes,
endothelial cells, and granulomas. Inflamm Bowel Dis 2: 88-96, 1996.
GUINEY, D.G., HASEGAWA, P., COLE, S.P. Helicobacter pylori preferentially
induces interleukin 12 (IL-12) rather than IL-6 or IL-10 in human dendritic
cells. Infect Immun 71: 4163-4166, 2003.
HAEBERLE, H.A., KUBIN, M., BAMFORD, K.B., GAROFALO, R.,
GRAHAM, D.Y., EL-ZAATARI, F., KARTTUUNEN, R., CROWE, S.E.,
REYES, V.E., ERNST, P.B. Differential stimulation of interleukin-12 (IL-12)
and IL-10 by live and killed Helicobacter pylori in vitro and association of IL-
12 production with gamma interferon-producing T cells in the human gastric
mucosa. Infect Immun 65: 4229-4235, 1997.
HAFSI, N., VOLAND, P., SCHWENDY, S., RAD, R., REINDL, W.,
GERHARD, M., PRINZ, C. Human dendritic cells respond to Helicobacter
pylori, promoting NK cell and TH1-effector responses in vitro. J Immunol
73: 1249-1257, 2004.
HAMMAR M, TYSZKIEWICZ T, WADSTROM T, O'TOOLE PW. Rapid
detection of Helicobacter pylori in gastric biopsy material by polymerase
chain reaction. Journal of Clinical Microbiology 30 (1):54-8, 1992.
HANCOCK, R. E., ALM, R., BINA, J., TRUST, T. Helicobacter pylori: a
surprisingly conserved bacterium. Nature Biotechnology 16 (3): 216-217,
1998.
HARRIS, R.P., WRIGHT, S.W., SERRANO, C., RIERA, F., DUARTE, I.,
TORRES, J., PEÑA, A., ROLLÁN, A., VIVIANI, P., GÜIRALDES, E.,
SCHIMITZ, J.M., LORENZ, R.G., NOVAK.L., SMYTHIES, L.E., SMITH,
P.D. Helicobacter pylori gastritis in children is associated with a regulatory T
cell response. Gastroenterology 134 (2): 491-499, 2008.
HATAKAYEMA, M. Helicobacter pylori CagA- a potential bacterial oncoprotein
that functionally mimics the mammalian Gab family of adaptor proteins.
Microbes and Infection 5: 143-150, 2003.
HAZELL, S. L., EVANS, D. J., GRAHAM, D. Y. Helicobacter pylori. Journal of
General Microbiology 137 (1): 57-61, 1991.

101
HERNÁNDEZ, A.S. Células colaboradoras (TH1, TH2, TH17) y reguladoras
(Treg, TH3, NKT) en la artritis reumatoide. Reumatologia Clinica 5 (1): 1-5,
2009.
HIGASHI H, NAKAYA A, TSUTSUMI R, YOKOYAMA K, FUJII Y,
ISHIKAWA S, HIGUCHI M, TAKAHASHI A, KURASHIMA Y,
TEISHIKATA Y, TANAKA S, AZUMA T, HATAKEYAMA M.
Helicobacter pylori CagA induces Ras-independent morphogenetic response
through SHP-2 recruitment and activation. Journal of Biological Chemistry
23 (17): 17205-16, 2004.
HIGASHI, H., TSUTSUMI, R., FUJITA, A., YAMAZAKI, S., ASAKA, M.,
AZUMA, T., HATAKEYAMA, M. Biological activity of the Helicobacter
pylori virulence factors CagA is determined by variation in the tyrosine
phosphorylation sites. Proceeds of the National Academy of Sciences of the
United States of America, 99 (22): 14428-14433, 2002.
HIRSCHL, A. M. & MAKRISTATHIS, A. Methods to detect Helicobacter pylori:
from culture to molecular biology. Helicobacter 12 (2): 6-11, 2007.
HORI, S., NOMURA, T., SAKAGUCHI, S. Control of regulatory T cell
development by the transcription factor Foxp3. Science 299 (5609):1057-61,
2003.
HUANG, J., SRIDHAR, S., CHEN, Y., HUNT, R. H. Meta-analisis of the
relantionship between Helicobacter pylori seropositivity and gastric cancer,
Gastroenterology 114 (4): 1169-1179, 1998.
HWANG, I.R., KODAMA, T., KIKUCHI, S. Effect of interleukin 1
polymorphisms on gastric mucosal interleukin 1 beta production in
Helicobacter pylori infection. Gastroenterology 123: 1793-803, 2002.
ILVER, D., ARNQVIST, A., OGREN, J., FRICK, I. M., KERSULYTE, D.,
INCECIK, E. T., BERG, D. E., COVACCI, A. ENGSTRAND, L., BORÉN, T.
Helicobacter pylori adhesion binding fucosylated histo-blood antigens
revealed by retagging. Science 279: 373-377, 1998.
ISRAEL, D. A. & PEEK, R. M. Review article: pathogenesis of Helicobacter
pylori-induced gastric inflammation. Alimentary Pharmacology &
Therapeutics 15 (9): 1271-1290, 2001.

102
JENKS, P. J. & KUSTERS, J. G. Pathogenesis and virulence factors of
Helicobacter pylori. Current Opinion Gastroenterology 16 (1): S11-S18,
2000.
JONULEIT, H., SCHIMITT, E. The regulatory T cell family: distinct subsets and
their interrelations. J Immunol 171 (12): 6323-6327, 2003.
KANDULSKI, A., MALFERTHEINER, P., WEX, T. Role of regulatory T-cells
in H. pylori- induced gastritis and gastric cancer. Anticancer Res 30 (4):
1093-1103, 2010.
KAO, J.Y., RATHINAVELU, S., EATON, K.A., BAI, L., ZAVROS, Y.,
TAKAMI, M., PIERZCHALA, A., MERCHANT, J.L. Helicobacter pylori-
secreted factors inhibit dendritic cell IL-12 secretion: a mechanism of
ineffective host defense. Am J Physiol Gastrointest Liver Physiol 291: G73-
G81, 2006.
KARTTUNEN, R., KARTTUNEN, T., EKRE, H.P., McDONALD, T.T.
Interferon gamma and interleukin 4 secreting cells in the gastric antrum in
Helicobacter pylori positive and negative gastritis. Gut 36: 341-345, 1995.
KATO, S., OZAWA, K., OKUDA, M., NAKAYAMA, Y., YOSHIMURA, N.,
KONNO, M., MINOURA, T., IINUMA, K. Japan pediatric Helicobacter
study group. Multicenter comparison of rapid lateral flow stool antigen
immunoassay and stool antigen enzyme immunoassay for the diagnosis of
Helicobacter pylori infection in children. Helicobacter 9: 669-73, 2004.
KELLY, S. M., PITCHER, M. C. L., FARMERY, S. M. Isolation of Helicobacter
pylori from feces of patients with dyspepsia in the United Kingdom.
Gastroenterology 107 (6): 1671-1674, 1994.
KIM, H. Y., KIM, Y. B., PARK, C. K., YOO, J. Y., GRAHAM, D. Y. Co-existing
gastric cancer and duodenal ulcer disease: role of Helicobacter pylori
infection. Helicobacter 2 (4):205-209, 1997.
KLEIN, P. D., GILMAR, R. H., LEON-BARUA, R., DIAZ, F., SMITH, E.,
GRAHAM, D. Y. The epidemiology of Helicobacter pylori in peruvian
children between 6 and 30 months of age. Gastroenterology 89: 12-16, 1991.
KONDO, Y., JOH, T., SASAKI, M., et al. Helicobacter pylori eradication
decreases blood neutrophil and monocyte counts. alimentary and
Therapeutics, Supplement 20 (1): 74-79, 2004.

103
KRANZER, K., ECKHARDT, A., AIGNER, M., KNOLL, G., DEML, L.,
SPETH, C., LEHN, N., REHLI, M., SCHNEIDER-BRACHERT, W.
Induction of maturation and cytokine release of human dendritic cells respond
to Helicobacter pylori. Infect Immun 72: 4416-4423, 2004.
KRIENITZ, W. Ueber das aufrenten von spirochaeten verschiedener form im
mageninhalt bei carcinoma ventriculi. Deutsche Medizinische
Wochenschrift 32: 872, 1906.
KUIPERS, E.J., PEREZ-PEREZ, G.I., MEUWISSEN, S.G., BLASER, M.J.
Helicobacter pylori and atrophic gastritis. Importance of the cagA status.
Journal National of Cancer Institute 87: 1777-1780, 1995.
LADEIRA, M.S.P., RODRIGUES, M.A., SALVADORI, D.M., NETO,P.P.,
ACHILLES, P., LERCO, M.M., RODRIGUES, P.A., GONÇALVES Jr, I.,
QUEIROZ, D.M., FREIRE-MAIA, D.V. Relationships between cagA, vacA,
and ice genotypes of Helicobacter pylori and DNA damage in the gastric
mucosa. Environmental and Molecular Mutagenesis 44: 91-98, 2004.
LADEIRA, M.S.P., SALVADORI, D.M.F., RODRIGUES, M.A.M. Biopatologia
do Helicobacter pylori. Jornal Brasileiro de Patologia e Medicina
Laboratorial 39(4): 335-342, 2003.
LEHOURS, P., RUSKONE-FOURMESTRAUX, A. LAVERGENE, A.
CANTET, A., MÉGRAUD, F. Wich test to use to detect Helicobacter pylori
infection in patients with low-grade gastric mucosa-associated lymphoid tissue
lymphoma? American Journal of Gastroenterology 98: 291-295, 2003.
LIN, C. W., WU, S. C., LEE, S. C., CHENG, K. S. Genetic analysis and clinical
evaluation of vacuolating cytotoxin gene A and cytotoxin-associated gene A in
Taiwanese Helicobacter pylori isolates from peptic ulcer patients.
Scandinavian Journal of Infectious Diseases 32: 51-57, 2000.
LINS, R.D.A.U. et al. Immunohistochemical evaluation of the inflammatory
response in periodontal disease. Braz. Dent J. 19 (1): 9-14, 2008.
LOGAN, R. P. H. & WALKER, M. M. Epidemiology and diagnosis of H. pylori
infection. British Medical Journal 323 (20): 919-922, 2001.
LONGUIMIRE, W.P. Carcinoma do estômago. In: Tratado de cirurgia.
Sabiston, D.C. Editora Interamericana Ltda., Rio de Janeiro, 1977.

104
LU, W., PAN, K., ZHANG, L., LIN, D., MIAO, X., YOU, W. Genetic
polymorphisms of interleukin (IL)-1B, IL-1RN, IL-8, IL-10 and tumor
necrosis factor α and a risk of gastric cancer in a Chinese population.
Carcinogenesis 26 (3): 631-636, 2005.
LUNDGREN, A., STROMBER, E., SJOLING, A., LINDHOLM, C.,
ENARSSON, K., EDEBO, A., JOHNSSON, E., SURI-PAYER, E.,
LARSSON, P., RUDIN, A., SVENNERHOLM, A.M., LUNDIN, B.S.
Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in
Helicobacter pylori-infected patients. Infect Immun 73: 523-531, 2005.
LUNDGREN, A., SURI-PAYER, E., ENARSSON, K., SVENERHOLM, A.M.,
LUNDING, B.S. hELICOBACTER PYLORI-specefic CD4+ CD25+ high
regulatory T cells suppress memory T-cell responses to H. pylori in infected
individuals. Infect Immun 71: 1755-1762, 2003.
MAI, U.E., PEREZ-PEREZ, G.I., ALLEN, J.B., WAHL S.M., BLASER, M.J.,
SMITH, P.D. Surface proteins from Helicobacter pylori exhibit chemotactic
activity for human leukocytes and are present in gastric mucosa. J Exp Med
175: 517-525, 1992.
MAI, U.E., PEREZ-PEREZ, G.I., WAHL, L.M., WAHL, S.M., BLASER, M.J.,
SMITH, P.D. Soluble surface proteins from Helicobacter pylori activate
monocytes/macrophages by lipolysaccharide-independent mechanism. J Clin
Invest 87: 894-900, 1991.
MALATY, H.M., GRAHAM, D.Y., KLEIN, P.D., EVANS, D.G., ADAM, E.,
EVANS, D.J. Transmission of Helicobacter pylori infection. Studies in
families of healthy individuals. Scandinavian Journal of Gastroenterology,
26 (9): 927-932, 1991.
MANDEVILLE, K.L., KRABSHUIS, J., LADEP, N.G., MULDER, C.J.J.,
QUIGLEY, E.M.M., KHAN, S.A. Gastroenterology in developing
countries :Issues and advances. World journal of Gastroenterology 15 (23):
2839-2854, 2009.
MARSHALL, B. J. & GOLDWIN, C. S. Revisied nomenclature of C. pyloridis.
International Journal Systematic and Evolutionary Microbiology 37: 68,
1987.
MARSHALL, B. J. & WARREN, J. R. Unidentified curved bacilli in the patients
with gastritis and peptic ulceration. Lancet 16 (1): 1311-1315, 1984.

105
MARSHALL, B. J. Campylobacter pylori and gastritis. The journal of Infectious
Disease 153 (4): 650-657, 1986.
MARSHALL, B.J. Helicobacter pylori in the year 2000. Helicobacter pylori
Foundation, 2000. Disponível em:
Http://www.helicobacter.com/hpy2k/frMain.htm.
MARSHALL, B.J., McGECHIE, D.B., ROGERS, P.A., GLANCY, R.J. Pyloric
Campylobacter infection and gastroduodenal disease. The Medical Journal
of Australia 142 (8): 439-444, 1985.
MARSHALL, B. J. Helicobacter pylori. American journal of Gastroenterology
89: 116-128, 1994.
MARSHALL, D. G., DUNDON, W. G., BEESLY, S. M., SMYTH, C. J.
Helicobacter pylori – a conundrum of genetic diversity. Microbiology 144:
2925-2939, 1998.
MARTINS, L. C., CORVELO, T. C. O., OTI, H. T., LOIOLA, R. S. P., AGUIAR,
D. C. F., BERILE, K. A. S., AMARAL, R. K. C., BARBOSA, H. P. M.,
FECURY, A. A., SOUZA, J. T. ABH and Lewis antigen distributions in
blood, saliva and gastric mucosa and H. pylori infection in gastric ulcer
patients. World Journal of Gastroenterology 12 (7): 1120-1124, 2006.
MARTINS, L.C., CORVELO, T.C., DEMACHKI, S., ARAUJO, M.T.,
ASSUMPCAO, M.B., VILAR, S.C., FREITAS, F.B., BARBOSA, H.P.,
FECURY, A.A., DO AMARAL, R.K., DOS SANTOS, S.E. Clinical and
pathological importance of vacA allele heterogeneity and cagA status in peptic
ulcer disease in patients from North Brazil. Memórias do Instituto Oswaldo
Cruz 100(8): 875-81, 2005.
MARTINS, L.C., CORVELO, T.C.O., OTI, H.T., BARILE, K.A.S.
Soroprevalência de anticorpos contra o antígeno cagA do Helicobacter pylori
em pacientes com úlcera gástrica na região Norte do Brasil. Revista da
Sociedade Brasileira de Medicina Tropical 35(4):307-310 , 2002.
MATTEO, M. J., ARMITANO, R. I., GRANADOS, G., WONAGA, A. D.,
SANCHEZ, C., OLMOS, M., CATALANO, M. Helicobacter pylori oipA,
vacA and dupA genetic diversity in individual hosts. Journal of Medical
Microbiology 59 (89), 2010.

106
MCCLAIN, M. S., SCHRAW, W., RICCI, V., BOQUET, P., COVER, T. L. Acid
activation of Helicobacter pylori vacuolating cytotoxin (VacA) results in toxin
internalization by eukaryotic cells. Molecular Microbiology 37 (2): 433-442,
2000.
MÉGRAUD, F., BRASSENS-RABBE, M. P., DENIS, F., BELBOURI, A., HOA,
D. Q. Seroepidemiology of Campylobacter pylori infection in various
populations. Journal Clinical Microbiology 27: 1870-1873, 1989.
MELO, K.M., CARVALHO, B.T.C. Células T regulatórias: mecanismos de ação e
função nas doenças humanas. Rev. bras. alerg. imunopatol. 32 (5): 184-188,
2009.
MEYER, F., RAMANUJAM, K.S., GOBERT, A.P., JAMES, S.P., WILSON,
K.T. Cutting edge: cyclooxigenase-2 activation suppresses Th1 polarization in
response to Helicobacter pylori. J Immunol 171: 3913-1917, 2003.
MITCHELL, P., GERMAIN, C., FIORI, P.L., KHAMRI, W., FOSTER, G.R.,
GHOSH, S., LECHLER, R.I., BAMFORD, K.B., LOMBARDI, G. Chronic
exposure to Helicobacter pylori impairs dendritic cell function and inhibits
Th1development. Infect Immun 75: 810-819, 2007.
MONTECUCCO, C., DE BERNARD, M., PAPINI, E., ZORATTI, M.
Helicobacter pylori vacuolating cytotoxin: cell intoxication and anion-specific
channel activity. Currents Topics in Microbiology and Immunology 257:
113-129, 2001.
MORAN, A. P. Pathogenic properties of Helicobacter pylori. Scandinavian
Journal of Gastroenterology 215 (31): 22-31, 1996.
MULLER, I., MEDINA-SELBY, A., PALACIOS, J. L., MARTINEZ, P.,
OPAZO, P., BRUCE, E., MANCILLA, M., VALENZUELA, P.,
YUDELEVICH, A., VENEGAS, A. Cloning and comparison of ten gene
sequences of a Chilean H. pylori strain with other H. pylori strains revealed
higher variability for VacA and CagA virulence factors. Biological Research
35 (1): 67-84, 2002.
NAUMANN, M., CRABTREE, J.E. Helicobacter pylori-induced epithelial cell
signalling in gastric carcinogenesis. Trends Microbiol 12: 29-36, 2004.

107
NDHLOVU, L.C., LOO,C.P., SPOTTS, G., NIXON, D.F., HECHT, F.M. FOXP3
expressing CD127Io CD4+ T cells inversely correlate with CD38+ T cell
activation levels in primary HIV-1 infection. J Leukoc Biol 83 (2): 254-262,
2008.
NECCHI, V., CANDUSSO, M. E., TAVA, F., LUINETTI, O., VENTURA, U.,
FIOCCA, R., RICCI, V., SOLCIA, E. Intracellular, intercellular, and stromal
invasion of gastric mucosa preneoplastic lesions, and cancer by Helicobacter
pylori. Gastroenterology 132: 1009-1023, 2007.
NIESS, J.H., BRAND, S., GU, X., LANDSMAN, L., JUNG, S., McCORMICK,
B.A., VYAS, J.M., BOES, M., PLOEGH, H.L., FOX, J,G, LITTMAN, D.R.,
REINECKER, H.C. CX3CR1-mediated dendritic cell access to the intestinal
lumen and bacterial clearance. Science 307: 254-248, 2005.
NISH, T., OKASAKI, K., KAWASAKI, K., et al. Involvment of myeloid
dendritic cells in the development of gastric secondary lymphoid follicles in
Helicobacter pylori-infected neonatally thymectomized BALB/c mice. Infect
Immunol 71: 2153-2162, 2003.
OHYAUCHI, M., IMATANI, A., YANECHI, M., ASANO, N., MIURA, A.,
LIJIMA, K., KOIKE, T., SEKINE, H., OHARA, S., SHIMOSEGAWA, T.
The polymorphism interleukin 8-251 A/T influences the susceptibility of
Helicobacter pylori related gastric diseases in the Japanese population. Gut
54: 330-335, 2005.
OLSON, J. W. & MAIER, R. J. Molecular hydrogen as an energy source for
Helicobacter pylori. Science 298.5599: 1788-1790, 2002.
ONDEBREIT, S. et al. Translocation of Helicobacter pylori cagA into gastric
epithelial cells by type IV secretion. Science 287: 1497-500, 2000.
ORNELLAS, L. C., CURY, M. S., LIMA, V. M., FERRARI JR, A. P. Avaliação
do teste rápido da urease conservado em geladeira. Arquivo de
Gastroenterologia 37: 155-157, 2000.
PADILLA, P. I., WADA, A., YAHIRO, K., KIMURA, M., NIIDOME, T.,
AOYAGI, H., KUMATORI, A., ANAMI, M., HAYASHI, T., FUJISAWA, J.,
SAITO, H., MOSS, J., HIRAYAMA, T. Morphologic differentiation of HL-60
cells is associated with appearance of RPTPbeta and induction of Helicobacter
pylori VacA sensitivity. Journal of Biological Chemistry 275 (20): 15200-
15206, 2000.

108
PALLI, D. Gastric cancer and Helicobacter pylori: a critical evaluation of the
epidemiological evidence. Helicobacter 2 (1): 550-555, 1997.
PALMER, E. D. Investigation of the gastric mucosa spirochetes of the human.
Gastroenterology 27: 218-220, 1954.
PANCHAL, P.C., FORMAN, J.S., BLUMBERG, D.R., WILSON, K.T.
Helicobacter pylori infection: pathogenesis. Curr Opin Gastroenterol 19: 4-
10, 2003.
PAPINI, E., DE BERNARD, M., BUGNOLI, M., MILIA, E., RAPPUOLI, R.,
MONTECUCCO, C. Cell vacuolization induced by Helicobacter pylori
inhibition by bafilomycins A1, B1, C1 and D. FEMS Microbiology Letters
113 (2): 155-159, 1993.
PAPINI, E., ZORATTI, M., COVER, T.L. In search of Helicobacter pylori VacA
mechanism of action. Toxicon 39: 1757-1767, 2001.
PAPINI, E., SATIN, B., NORAIS, N. Selective increase of the permeability of
polarized epithelial cell monolayers by Helicobacter pylori vacuolating toxin.
The Journal of Clinical Investigation 102: 813-820, 1998.
PARSONNET, J., COVACCI, A., PEREZ-PEREZ, G. Risk for gastric cancer in
people with cagA positive or cagA negative Helicobacter pylori infection. Gut
40: 297-301, 1997.
PEEK JR., R.M., BLASER, M.J. Pathophysiology of Helicobacter pylori-induced
gastritis and peptic ulcer disease. Am J Med 102: 200-205, 1997.
PEEK JR., R.M. et al. Helicobacter pylori strain-specific genotypes and
modulation of the gastric epithelial cell cycle. Cancer Research 59: 6124-31,
1999.
PEEK JR., R.M., BLASER, M.J. Helicobacter pylori and gastrointestinal tract
adenocarcinomas. Nat Rev Cancer 2: 28-37, 2002.
PEEK JR., R.M., CREBTREE, J.E. Helicobacter infection and gastric neoplasia. J
Pathol 208: 233-248, 2006
PEREZ-PEREZ GI, GARZA-GONZALEZ E, PORTAL C, ET AL. Role of
cytokine polymorphisms in the risk of distal gastric cancer development.
Cancer Epidemiology, Biomarkers and Prevention 14 (8):1869-73, 2005.

109
PERRY, S., DE LA LUZ SANCHEZ, M., YANG, S. HAGGERTY, T. D.,
HURST, P., PEREZ-PEREZ, G. PARSONNET, J. Gastroenteritis and
transmission of Helicobacter pylori infection in households. Emergency
Infectious Diseases 48: 1701-1708, 2006.
PICCIRILLO, C.A. Células T reguladoras na saúde e na doença. Citocinas 43 (3):
395-401, 2008.
PULENDRAN, B., PALUCKA, K., BANCHEREAU, J. Sensing pathogens and
tuning immune responses. Science 293: 253-256, 2001.
QUEIROZ, D. M. M., & MENDES, E. N. H. Helicobacter pylori e outros
microorganismos espiralados gástricos: aspectos microbiológicos. In: Tópicos
em Gastroenterologia. Castro, L.P., Rocha, P.R.S., Coelho, L.G.V. Editora
Medsi, Rio de Janeiro, 1993. p. 235-248.
QUEIROZ, M. M. D., MENDES, N. E., CARVALHO, S. T. A., ROCHA, A. G.,
OLIVEIRA, R. M. A., SOARES, F. T., SANTOS, A., CABRAL, A. D. M. M.,
NOGUEIRA, F. M. M. A. Factors associated with Helicobacter pylori
infection by a CagA-positive strain in children. The Journal of Infectious
Diseases, 181: 626-630, 2000.
RAD, R., BRENNER, L., BAUER, S., SCHWENDY, S., LAYLAND, L.,
COSTA, C.F., REINDI, W., DOSSUMBEKOVA, A., FRIEDRICH, M.,
SAUR, D., WAGNER, H., SCHIMID, R.M., FRINZ, C. CD25/FoxP3 T cells
Regulate Gastric inflammation and Helicobacter pylori colonization in vivo.
131 (2): 525-537, 2006.
RAGHAVAN, S., FREDRIKSSON, M., SAVERHOLM, A.M. et al. Absence of
CD4+CD25+ regulatory T cells is associated with a loss of regulation leading
to increased pathology in Helicobacter pylori-infected mice. Clin Exp
Immunol 132: 393–400, 2003.
RANGEL, M. F., AMORIM, W. P., AMORIM, M. F. D., AMORIM, P. D.,
NÓBREGA, L. P. S. Avaliação da prevalência da infecção por Helicobacter
pylori em pacientes portadores de câncer gástrico. Revista do Colégio
Brasileiro de Cirurgia 30 (1): 34-32, 2003.
RAUTEMAA, R., RAUTELIN, H., PUOLAKKAINEN, P., KOKKOLA, A.,
KARKKAINEN, P., MERI, S. Survival of Helicobacter pylori from
complement lysis by binding of GPI – anchored protectin (CD59).
Gastroenterology 120 (2): 470-479, 2001.

110
RESCIGNO, M., URBANO, M., VALZASINA, B., FRANCOLINE, M.,
ROTTA, G., BONASIO, R., GRANUCCI, F., KRAEHENBUHI, J.P.,
RICCIARDI-CASTAGNOLI, P. Dendritic cells express tight junction proteins
and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol 189:
361-367, 2001.
RESENDE, A. L. S., MATTOS, I. E., KOIFMAN, S. Mortalidade por câncer
gástrico no Estado do Pará, 1980-1997. Arquivos de Gastroenterologia 43
(3): 247-252, 2006.
REYRAT, J. M., RAPPUOLI, R., TELFORD, J. L. A structural overview of the
Helicobacter citotoxin. International Journal of Medical Microbiology 290:
375-379, 2000.
RODER, D.M. The epidemiology of gastric cancer. Gastric cancer 5: 5-11, 2002.
RODRIGUES, N.R., QUEIROZ, D.M.M., RODRIGUES, R.T., ROCHA, A.M.C.,
BRAGA, M.B.N., BRAGA, L.L.B.C. Helicobacter pylori infection in adults
from a poor urban community in northeasten Brazil: demographic, lifestyle
and environmental factors. Brazilian Journal of Infectuous Disease 9 (5):
405-410, 2005.
ROMANIUK, P.J., ZOLTOWSKA, B., TRUST, T.J., LANE, D.J., OLSEN, G.J.,
PACE, N.R., STAHL, D.A. Campylobacter pylori, the spiral bacterium
associated with human gastritis, is not a true Campylobacter sp. Journal Of
Bactriology 169 (5): 2137-2141, 1987.
RUDENSKY, A.Y.; CAMPBELL, D.J. In vivo sites and cellular mechanisms of T
reg cell-mediated supression. The Journal of Experimental Medicine 203
(3): 489 – 492, 2006.
RUZAI, V.E., SACHEDEVA, M., ZHANGA, S., WENA, S., MOSSA, S.F.
Isolating immunophenotiping and ex vivo stimulation of CD4+ and CD8+
gastric lymphocytes during murine Helicobacter pylori infection. J. Immunol
Methods 384 (1-2): 157-163, 2012.
SAITO, N., KONISHI, K., SATO, F., KATO, M., TAKEDA, H., SUGIYAMA,
T., ASAKA, M. Plural transformation-process from spiral to cocoid
Helicobacter pylori and its viability. The Journal of Infectious Disease 46:
49-55, 2003.

111
SAKAGUCHI, S., SAKAGUCHI, N., ASANO, M., ITOH, M. Immunologic
tolerance maintained by activated T cells expressing IL-2 receptor â-chains
(CD25): breakdow of a single mechanism of selftolerance causes various
autoimmune diseases. J Immunol 155: 1151-1164, 1995.
SAKAGUCHI, S. Naturally arising CD4+ regulatory T cells for immunologic
selftolerance andnegative control of immune responses. Annu. Rev.
Immunol. 22: 531-562, 2004.
SAKAGUCHI, S. Naturally arising Foxp3-expressing CD25+ CD4+ regulatory T
cells in immunological tolerance to self and non-self. Nature Immunology 6
(4): 345-352, 2005.
SAKAGUCHI, S. Regulatory T cells. Springer Semin Immunopatol 28 (1): 1-2,
2006.
SALAMA, N.R., OTTO, G., TOMPKINS, L., FALKOW, S. Vacuolating
cytotoxin of Helicobacter pylori plays a role during colonization in a mouse
of infection. Infection and Immunity 69(2): 730-6, 2001.
SARVESTANI, E.K., BAZARGANI, A., MASOUDIAN, M., LANKARANI, K.,
TAGHAVI, A., SABERIFIROOZI, M. Association of H. pylori and vacA
genotypes and IL-8 gene polymorphisms with clinical outcome of infection in
Iranian patients with gastrointestinal diseases. World Journal of
Gastroenterology 12(32): 5205-5210, 2006.
SCHOLTE, G.H.A., VAN DOOR, L.J., CATS, A., BLOEMENA, E.,
LINDERMAN, J., QUINT, W.G.V., MEUWISSEN, S.G.M., KUIPERS, E.J.
Genottyping of helicobacter pylori in paraffin-embedded gastric biopsy
specimens: relation to histological parameters and effects of therapy.
American Journal of Gastrenterology 97, 1687-1695, 2002.
SEGAL, E. D., FALKOW, S., TOMPKINS, L. S. Helicobacter pylori attachment
to gastric cells induces cytoskeletal rearrangements and tyrosine
phosphorilation of host cell proteins. Proceeds of the National Academy of
Sciences of the United States of America 93 (3): 1259-1264, 1996.
SHIBATA, J., GOTO, H., ARISAWA, T., et al. Regulation of tumour necrosis
factor (TNF) induced apoptosis by soluble TNF receptors in Helicobacter
pylori infection. Gut 45: 24-31, 1999.

112
SHIBATA, T., IMOTO, I., GABAZZA, E. C. Detection of Helicobacter pylori in
biopsy of patients with gastric carcinoma. Biomedicine & Pharmacotherapy
51 (1): 22-28, 1997.
SHOMER, N.H., DANGLER, C.A., MARINI, R.P., FOX, J.G. Helicobacter
bilis/Helicobacter rodentium co-infection associated with diarrhea in a colony
of scid mice. Comparative Medicine 48 (5): 455-459, 1998.
SILVA, L. B. L., GONÇALVES, T. M., ALENCAR, J. S., NUNES, O. S.,
VASCONCELLOS, A. C. A., SANTANA. Atenção farmacêutica a pacientes
com gastrite Helicobacter pylori positivo. Infarma 16 (7-8): 70-73, 2004.
SILVEIRA, J. A., SOUZA, L. E. O., FILHO, J. E. S., SOUZA, L. B. S.,
SANTANA, W. J., COUTINHO, H. D. M. Fatores de virulência e
características epidemiológicas do Helicobacter pylori. Revista Médica Ana
Costa 10 (2): 28-31, 2005.
SIQUEIRA, J. S., LIMA, P. S. S., BARRETO, A. S., QUINTANS JUNIOR, L. J.
General aspects of Helicobacter pylori infections-review. RBAC 39 (1): 9-13,
2007.
SOJKA, D.K., HUANG, Y.H., FOWELL, D.J. Mechanisms of regulatory T-cell
supression - a diverser arsenal for a moving target. Immunology 124 (1): 13-
22, 2008.
SOKOL, R.J. & HOFFENBERG, E.J. Antioxidantes na doença gastrointestinal
pediátrica. Clin. Ped. Am. Norte 8: 457-72, 1997.
STEER, H. W. & COLIN-JONES, D. G. Mucosal changes in gastric ulceration
and their response to carbenoxolone sodium. Gut 16: 590-597, 1975.
STEER, H. W. Ultrastructure of cell migration trough the gastric epithelium and
its relantionship to bacteria. Journal of Clinical Pathology 28: 639-646,
1975.
STEIN, M., RAPPUOLI, R., COVACCI, A. Tyrosine phosphorilation of the
Helicobacter pylori cagA antigen after cag-driven host cell translocation.
Proc. Natl. Acad. Sci. USA 97: 1263-1268, 2000.
SUERBAUM, S. & MICHETTI, P. Helicobacter pylori infection. New England
Journal of Medicine 347 (15): 1175-1186, 2002.

113
TAAMS, L.S., PALMER, D.B., AKBAR, A.N., ROBINSON, D.S., BROWN, Z.,
HAWRYLOWICS, C.M., Regulatory T cells in human disease and their
potential for therapeutic manipulation. J Immunol 118 (1): 1-9, 2006.
TANG, Q., BLUESTONE, J.A. The FoxP3+ regulatory T cell: a jack of all trades,
master of regulation. Nat Immunol 9 (3): 239-244, 2008.
TANIDA, N., SAKAGAMI, T., SAWADA, Y., SHIMOYAMA, T. Critical
review of the WHO/IARC report regarding carcinogenicity of Helicobacter
pylori. Nippon Rinsho, Japanese Journal of Clinical Medicine 55 (4): 995-
1002, 1997.
TELFORD, J. L., GHIARA, P., DELL-URCO, M., COMANDUCCI, M.,
BURRONI, D., BUGNOLI, M., TECCE, M. F., CENSINI, S. COVACCI, A.,
XIANG, Z., PAPINI, E., MONTECUCCO, C., PARENTE, L., RAPPUOLI,
R. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key
role in gastric disease. The Journal of Experimental Medicine 179: 1653-
1658, 1994.
THOMAZINI, M.T., PINHEIRO, N.A., PARDINI, M.I., NARESSE, L.E.,
RODRIGUES, M.A.M. Infecção por Helicobacter pylori e câncer gástrico:
freqüência de cepas patogênicas cagA e vacA em pacientes com câncer
gástrico. Jornal Brasileiro de Patologia e Medicina Laboratorial 42(1): 25-
30, 2006.
THOMSEN, L. L., GAVI, B. J., TASMAN-JONES, C. Relation of Helicobacter
pylori to human gastritis of the antrum. Gut 32 (1): 230-236, 1990.
THORNTON, A.M., DONOVAN, E.E., PICCIRILO, C.A., SHEVACH, E.M.
Cuttong edge: IL-2 is critically required for the in vitro activation of
CD4+CD25+ T cell suppressor function. J Immunol 172: 6519-6523, 2004.
TOMB, J. F., WHITE, O., KERLAVAGE, A. R., CLAYTON, R. A., SUTTON,
G. G., FLEISCHMANN, R. D., KETCHUM, K. A., KLENK, H. P., GILL, S.,
DOUGHERTY, T. Y., QUACKENBUSH, J., ZHOU, L. KIRKNESS, E. F.,
PETERSON, S., LOFTUS, B., RICHARDSON, D., DODSON, R., KHALAK,
H. G., GLODEK, A., MCHENNEY, K., FITZEGERALD, L. M., LEE, N.,
ADAMS, M. D., VENTER, J. C. The complete genome sequence of the
gastric pathogen Helicobacter pylori. Nature 7 (388): 539-547, 1997.

114
TOMBOLA, F., CARLESSO, C., SZABO, I., DE BERNARD, M., REYRAT, J.
M., TELFORD, J. L., RAPPUOLI, R., MONTECUCCO, C., PAPINI, E.,
ZORATTI, M. Helicobacter pylori vacuolating toxin forms anion-selective
channels in planar lipid bilayers: possible implications for the mechanism of
cellular vacuolation. Biophysical Journal 76 (3): 1401-1409, 1999.
TONELLI, E. & FREIRE, L. M. S. Doenças infecciosas na infância e
adolescência. Editora Medsi, São Paulo, 2000. p. 656-657.
TORGERSON, T.R., OCHS, H.D. Regulatory T cells primary immunodeficiency
diseases. Curr Opin Allergy Clin Immunol 7 (6): 515-521, 2007.
TORRES, J. & PÉREZ, G. I. Infection with CagA Helicobacter pylori strains as a
possible predictor of risk in the development of gastric adenocarcinoma in
Mexico. International Journal of Cancer 78 (3): 298-300, 1998.
TUMMALA, S., KEATES, S., KELLY, C.P. Update on the Immunologic Basis of
Helicobacter pylori Gastritis. Current opinion in gastroenterology 20 (6):
592-597, 2004.
UEMURA, N., OKAMOTO, S., YAMAMOTO, S. Helicobacter pylori infection
and the development of gastric cancer. New England Journal of Medicine
345: 784-789, 2001.
VAN DOORN, L. J., FIGUEIREDO, C. MÉGRAUD, F. PENA, S. MIDOO, P.,
QUEIROZ, D. M. M., CARNEIRO, F., VANDERBORGHT, B., PEGADO,
M. G. F., SANNA, R., BOER, W., SCHNEEBERGER, P. M., CORREA, P.,
NG, E. K., ATHERTON, J., BLASER, M. J., QUINT, W. G. V. Geographic
distribution of vacA allelic types of Helicobacter pylori. Gastroenterology
116: 823-830, 1999.
VAN DOORN, L. J., FIGUIREDO, C., ROSSAU, R., JANNES, G., VAN
ASBROEK, M., SOUSA, J. C., CARNEIRO, F., QUINT, W. G. Typing of
Helicobacter pylori vacA gene and detectionof cagA gene by PCR and reverse
hybridization. Journal Clinical Microbiology 36: 1271-1276, 1998.
VANDAMME, P., FALSEN, E., ROSSAU, R., HOSTE, B., SERGES, P.,
TYTGAT, R., DE LEY, J. Revision of Campylobacter, Helicobacter, and
wolinella taxonomy: emendation of generic descriptions and proposal of
Arcobacter gen. nov. International Journal of Systematic Bacteriology 41
(1): 88-103, 1991.

115
VINALL, L. E., KING, M., NOVELLI, M., GREEN, C. A., DANIELS, G.,
HILKENS, J., SARNER, M., SWALLOW, D. M. Altered expression and
allelic association of the hypervariable membrane muci MUC1 in
Helicobacter pylori gastritis. Gastroenterology 123 (1): 41-49, 2002.
VOLAND, P., HAFSI, N., ZEITNER, M., LAFORSCH, S., WAGNER, H.,
PRINZ, C. Antigenic properties OF HpaA and Omp18, two outer membrane
proteins of Helicobacter pylori. Infect Immun 71: 3837-3843, 2003.
WALLACE, J.L, CUCALA, M., MUGRIDGE, K., PARENTE, L. Secretagogue-
specific effects of interleukin-1 on gastric acid secretion. The American
Journal of Physiology 261 (4 pt 1): 559-564, 1991.
WALSH, J. H. A pH sensitive channel regulates urea across to Helicobacter
pylori urease. Gastroenterology 118: 249-250, 2000.
WANG, J. T., CHANG, C. S., LEE, C. Z., YANG, J. C., LIN, J. T., WANG, T. H.
Antibody to Helicobacter pylori species specific antigen in patients with
adenocarcinoma of the stomach. Biochemical and Biophysical Research
Communications 244 (2): 360-263, 1998.
WANG, S. K., ZHU, H. F., HE, B. S., ZHANG, Z. Y., CHEN, Z. T., WANG, Z.
Z., WU, G. L. CagA+ H. Pylori infection is associated with polarization of T
helper cell immune responses in gastric carcinogenese. World Journal of
Gastroenterology 13 (21): 2923-2931, 2007.
WARREN, J. R. & MARSHALL, B. J. Unidentified curved bacilli in the stomach
of patients with gastritis and peptic ulceration. Lancet 16 1(8390): 1311-1315,
1984.
WEEKS, D. L. & SACHES, G. Sites of pH regulation of the urea channel of
Helicobacter pylori. Molecular Microbiology 40 (6): 1249-1289, 2001.
WEEKS, D. L. H+-gated urea channel: the link between Helicobacter pylori
urease and gastric colonization. Science 287: 482-485, 2000.
WEN, S., VELIN, D., FELLEY, C. P., DU, L., MICHETTI, P., PAN-
HAMMARSTRÖM, Q. Expression of Helicobacter pylori virulence factors
and the associated inflammatory gene expression profiles in the human gastric
mucosa. Infection and Immunity 20 (11): 5118-5126, 2007.

116
WILLIAMS, P. M., POUNDER, M. A. Helicobacter pylori: From the benign to
the malignant. The American Journal of Gastroenterology 94 (II): 115-165,
1999.
WILSON, K.T. & CRABTREE, J.E. Immunology of Helicobacter pylori: insights
into the failure of the immune response and perspectives on vaccine studies.
Gastroenterology 133: 288-308, 2007.
WIRTH, H. P., YANG, M., PEEK, R., THAM, K. T., BLASER, M. J.
Helicobacter pylori Lewis expression in related to the host Lewis phenotype.
Gastroenterology 113: 1091-1098, 1997.
WU, Y-Y, CHEN, J-H, KAO, J-T, LIU, K-C, LAI, C-H, WANG, Y-M, HSICH,
C-T, TZEN, J.T.C., HSU, P.N. Expression of CD25high
regulatory T cells and
PD-1 in gastric infiltrating CD4+ T lymphocytes in patients with Helicobacter
pylori infection. Clinical and Vaccine Immunology, 1198-1201, 2011.
YAGI, H. et al. Crucial role of FOXP3 in the development and function of human
CD25+ CD4+ regulatory T cells. International immunology 16 (11): 1643-
1656, 2004.
YAKOOB, J., ABID, S., ABBAS, Z., JAFRI,W., AHMAD, Z., AHMED, R.,
ISLAM, M. Distribuition of Helicobacter pylori virulence markers in patients
with gastroduodenal diseases in Pakistan. BMC Gastroenterology, 9 (87): 1-
7, 2009.
YAMANISHI, S., LIZUMI, T., WATANABE, E., SHIMIZU, M., KAMIYA, S.,
NAGATA, K., KUMAGAI, Y., FUKUNAGA, Y., TAKAHASHI, H.
Implications for induction of autoimmunity via activation of B-1 cells by
Helicobacter pylori urease. Infect Immun 113: 248-256, 2006.
YAMAOKA, Y. Roles of Helicobacter pylori in gastroduodenal pathogenesis.
World Journal Gastroenterology 14 (27): 4265-4272, 2008.
YAMAOKA, Y., KATO, M., ASAKA, M. Geographic differences in gastric
cancer incidence can be explained by differences between Helicobacter pylori
strains. Internal medicine 47: 1077-1083, 2008.
YAMAOKA, Y., KIKUCH, S., EL-ZIMAITY, H. M., GUTIERREZ, O., OSATO,
M. S., GRAHAM, D. Y. Importance of Helicobacter pylori oipA in clinical
presentation, gastric inflammation, and mucosal interleukin 8 production.
Gastroenterology 123 (2): 414-424, 2002.

117
YAMAOKA, Y., KODAMA, T., KASHIMA, K., GRAHAM, D. Y.,
SEPULVEDA, A. R. Variants of the 3’ region of the cagA gene in
Helicobacter pylori isolates from patients with different H. pylori-associated
diseases. Journal of Clinical Microbiology 36 (8): 2258-2263, 1998.
YE, G., BARRERA, C., FAN, X., GOURLEY, W.K., CROWE, S.E., ERNST,
P.B., REYES, V.E. Expression of B7-1 and B7-2 costimulatory molecules by
human gastric epithelial cells: potential role in CD4+ T cell activation during
Helicobacter pylori infection. J Clin Invest 99: 1628-1636, 1997.
YUCEYAR, H., SARUC, M., KOKULUDAG, A., TERZIOGLU, E., GOK SEL,
G., ISISAG, A. The systemic cellular immune response in the Helicobacter
pylori-assiciated duodenal ulcer and chronic antral gastritis. Hepato-
Gastroenterology 49 (46): 1177-1179, 2002.

ANEXO 1 - TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO
UNIVERSIDADE FEDERAL DO PARÁ
CENTRO DE CIÊNCIAS BIOLOÓGICAS
DEPARTAMENTO DE PATOLOGIA
TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO
Título da pesquisa: Determinação do perfil imunológico com ênfase na resposta T
regulatória ante a infecção pelos diferentes tipos de cepas do H. pylori em pacientes
com gastropatia inflamatória.
Pesquisadores responsáveis: Jair Francisco de Santana Graim e Juarez Antonio
Simões Quaresma.
Justificativa, Objetivos e Procedimentos da Pesquisa: Helicobacter pylori (H. pylori)
é uma bactéria que causa inflamação persistente na mucosa do estômago e do intestino,
com diferentes lesões em humanos, tais como gastrite crônica, úlcera péptica e câncer
de estômago.
Esta pesquisa possui como principal objetivo a identificação de fatores que podem ser
usados como marcadores de risco para essa doença, possibilitando desse modo,
diagnóstico e tratamento mais eficazes. Para tanto é necessário realizar biópsias do
estômago. Com essa finalidade prestamos os seguintes esclarecimentos:
Esta pesquisa não oferece nenhum risco adicional ao paciente que fará a endoscopia
com biópsias do estômago para estudo de cepas virulentas do H. pylori, já que a
endoscopia será realizada por um profissional HABILITADO E EXPERIMENTADO
EM ENDOSCOPIA DIGESTIVA.
Portanto, para o paciente, os riscos de participar da pesquisa será tão somente aqueles a
que ele já está exposto em virtude do procedimento de endoscopia com biópsia visando
um maior conhecimento sobre a infecção causada por esta bactéria em nossa em nosso
Estado.
O material coletado para a pesquisa será usado exclusivamente para este fim, assim
como o questionário e, após o término da pesquisa, serão descartadas de acordo com as
normas de biossegurança.
Ao paciente, o risco de constrangimento pela participação na pesquisa em decorrência
da divulgação de informações será minimizado pelo sigilo absoluto da identidade do
mesmo por parte dos pesquisadores, se for necessário, autoridades de saúde poderão ser
informados para tomar medidas que beneficiem o participante da pesquisa ou outras
pessoas. Aos que se sentirem insatisfeitos ou incomodados é garantido o direito de
abandonar a pesquisa a qualquer momento, sem que isso interfira com o tratamento do

paciente. Aos que permanecerem até o final, é garantido o direito de saber os resultados
parciais e finais da pesquisa.
Os exames realizados pela pesquisa serão gratuitos, não necessitando nenhum custo por
parte do participante para sua realização.
Solicitamos assim, a sua autorização para efetuarmos os referidos exames e realizarmos
uma entrevista, sendo que a mesma é confidencial; para desenvolvermos o estudo em
questão.
Métodos alternativos existentes: Neste trabalho não há prática de outros métodos,
restringindo-se apenas aos descritos acima para a realização do estudo.
Forma de acompanhamento e assistência: Quando necessário, o voluntário receberá
toda assistência médica aos agravos decorrentes das atividades da pesquisa. Basta
procurar os pesquisadores Jair Graim, pelo fone (91) 9146-3281 e Juarez Quaresma,
pelo fone (91) 8125-8200 e também no endereço Av. Magalhães Barata nº 992, no
Hospital Ophir Loyola.
Ressarcimento de pesquisa e indenizações: Não há qualquer tipo de despesa por parte
do paciente e nem sequer qualquer forma de indenização pelo mesmo pela sua
participação neste estudo.
___________________________________________
Dr. Juarez Quaresma
CONSENTIMENTO PÓS-INFORMADO: Declaro que li as informações acima
sobre a pesquisa, e que me sinto perfeitamente esclarecido sobre o conteúdo do mesmo,
assim como seus benefícios. Declaro ainda que por minha livre vontade, aceito
participar da pesquisa cooperando com a coleta de material para exame.
Belém, _____/_____/_____
____________________________________
ASSINATURA DO PACIENTE
Em caso de dúvidas com respeito aos aspectos éticos deste estudo, você poderá
consultar o CEP do Núcleo de Medicina Tropical-NMT/Universidade Federal do
Pará-UFPA
Endereço: Av. Generalíssimo Deodoro, 92
Bairro: Umarizal, Município: Belém, UF: PA
CEP: 66.055-240
Telefone: (91)3201-6857
E-mail: [email protected]

ANEXO 2 – TERMO DE ACEITE DO NÚCLEO DE MEDICINA TROPICAL
DA UNIVERDIDADE FEDERAL DO PARÁ
























![Francisco de Assis e Francisco de Roma [release]](https://img.pdfslide.us/doc/110x75/568ca7bb1a28ab186d968a02/francisco-de-assis-e-francisco-de-roma-release.jpg)



