Embed Size (px)
Citation preview

Trithianes as Coinitiators in Benzophenone-InducedPhotopolymerizations
Ewa Andrzejewska,† Gordon L. Hug,*,‡ Maciej Andrzejewski,† andBronislaw Marciniak§
Institute of Chemical Technology and Engineering, Poznan University of Technology, Pl.M.Sklodowskiej -Curie 2, 60-965 Poznan, Poland; Radiation Laboratory, University of Notre Dame,Notre Dame, Indiana 46556; and Faculty of Chemistry, A. Mickiewicz University,Grunwaldzka 6, 60-780 Poznan, Poland
Received September 29, 1998; Revised Manuscript Received January 27, 1999
ABSTRACT: A series of 1,3,5-trithiane derivatives, including R- and â-isomers of the methyl and phenylderivatives, was investigated for use as coinitiators in benzophenone-induced photopolymerizations. Tostudy the mechanism of the photoinduced production of initiating radicals, nanosecond laser flashphotolysis was used. The rate constants for quenching the benzophenone triplet state by various trithianesand the associated quantum yields of ketyl-radical formation were determined. Photopolymerizationswere carried out with a multifunctional methacrylate model monomer, and the progress of the polymeriza-tions was followed by differential scanning calorimetry. The resulting plots showing the progress of therates of polymerization and the conversions of double bonds were compared to the photochemical informa-tion for the various trithianes. It was found that the polymerization efficiency of the coinitiators did notfollow the efficiency of photoinduced formation of the initiating radicals. The reasons for this lack ofcorrelation were discussed in terms of how the hydrogen-donating ability of coinitiators and the reactivityof the radicals formed affected the various stages of the polymerization. Further studies with a cleavage-type photoinitiator in the presence of the trithianes lent supporting evidence for the role that the trithianeradicals play in the polymerization of the benzophenone/trithiane systems investigated in this work.
Introduction
Photopolymerization of multifunctional monomersprovides a simple method for the production of highlycross-linked polymers. Such dense polymer networks areresistant to solvent penetration and show enhancedmechanical and thermal stability. These properties, inconjunction with rapid curing of the formulations, haveled to applications of these materials for the productionof protective coatings (e.g. for optical fibers), dentalfillings, and restorative materials, in microlithography,microelectronics, optical data storage, etc.1,2
Photoinduced radical polymerizations are usuallyperformed in the presence of photoinitiators of the“cleavage” type (e.g., benzoin derivatives) or H-abstrac-tion type (e.g., aromatic ketones). In the second class ofreactions, e.g. with benzophenone (BP), initiation occursmainly through hydrogen abstraction by 3BP from themonomer and/or a coinitiator molecule, such as an ether,alcohol, or amine.1,2 The initiating radical is derivedfrom the hydrogen-donating molecule, whereas the ketylradical that is formed can participate in the terminationprocess.
Our previous studies showed that aliphatic sulfidescan also be efficient coinitiators for BP-induced photo-polymerizations.3,4 A number of works has been devotedto the influence of aliphatic sulfides on the polymeri-zation of multi(meth)acrylates both in the presence ofphotofragmenting initiators4,5 as well as BP.3,5 Theeffect of the sulfide group built into the monomermolecule has also been described.6-10
The activity of sulfides in photopolymerizations canrange from low to high, depending on their hydrogen-donating ability. Moreover, aliphatic sulfides are alsoeasily oxidized in air, leading to a reduction of oxygeninhibition with an activity comparable to that reachedby aliphatic amines.5 There are also additional advan-tages of using aliphatic sulfides as coinitiators: theyincrease the hydrophobicity of the product and improveits thermo-oxidative stability.5
Among the sulfides investigated (linear sulfides,sulfides containing other functionalities, cyclic dithio-acetals) an exceptionally strong accelerating effect onBP-induced photopolymerization was shown by 2,4,6-trimethyl-1,3,5-trithiane (TMT).3 A laser flash photoly-sis study showed that photoreduction of BP by TMT (thecis-cis isomer, R ) CH3 in eq 1) is very efficient andoccurs by electron transfer from the sulfur atom to theBP triplet, followed by proton transfer:11
* Corresponding author. Telephone: (219) 631-4504. Fax: (219)631-8068. E-mail: [email protected].
† Poznan University of Technology.‡ University of Notre Dame.§ A. Mickiewicz University.
2173Macromolecules 1999, 32, 2173-2179
10.1021/ma9815408 CCC: $18.00 © 1999 American Chemical SocietyPublished on Web 03/12/1999
Under the same conditions, simple aliphatic sulfidesdid not reduce BP.12 In this case, the back electrontransfer (kbet) within the radical-ion pair formed wasthe main process. Thus, the main reason for the highactivity of TMT had to be the large quantum yield ofketyl radical formation and an associated formation ofinitiating TMT radicals. This suggestion was confirmedby photopolymerization studies.11 Polymerizations weresensitized by BP alone and by BP in the presence of thetwo simple sulfides, di-n-propyl sulfide and 2,2′-thio-bisethanol, in addition to TMT. Butane-1,4-diol di-methacrylate (BDM) was used as the model monomer.The formation of initiating radicals can occur by theexcited BP abstracting hydrogen from the additives and/or from the monomer itself. Under the conditions used,polymerization occurred only in the presence of TMT.This correlates well with the efficiency of the ketylradical formation in the photoreduction of BP by TMTand simple sulfides. That means also that the formationof initiating radicals by direct hydrogen abstraction fromthe monomer used was not efficient.
These results prompted us to study other sym-metrically substituted 1,3,5-trithianes as reducing agentsfor excited BP. Trisubstituted trithianes exist as twostereoisomers13 whose activity in the photoreduction ofBP may differ. The aim of this work was to study theprimary photochemical reaction between BP and aseries of 1,3,5-trithiane derivatives and to correlate theiractivity in photoreduction of BP with their efficiency ascoinitiators in BP-induced polymerization.
Experimental SectionBenzophenone (BP) was obtained from Aldrich as the best
available grade and was used without further purification. The1,3,5-trithiane derivatives were synthesized from sodiumthiosulfate or hydrogen sulfide and the corresponding aldehydeor ketone according to refs 14 and 15. They were purified byrepeated crystallization. Tri(ethylene glycol) dimethacrylate(TEGDM) was purchased from Aldrich and 2,2-dimethoxy-2-phenylacetophenone (DMPA) from Ciba-Geigy. Acetonitrile(Fisher, HPLC grade) was used as received.
The nanosecond laser flash photolysis setup has beendescribed generally elsewhere16 and also as specifically con-figured in this work.17 The concentration of benzophenone was2 × 10-3 M in all experiments. The concentrations of trithianeswere in the range 10-5-10-3 M for the quenching experiments.However, the concentrations of these quenchers were 0.01 Min the quantum yield determinations and for the experimentsthat involved recording transient absorption spectra in thetime range following complete quenching (>95%) of the BPtriplet state. Transient absorption spectra and quantum yieldmeasurements were performed using a flow-cell system. Allsolutions were deoxygenated by bubbling high-purity argonthrough them.
The polymerization kinetics was monitored under isother-mal conditions at 40 ( 0.01 °C in a high-purity argonatmosphere (<0.0005% of O2) by a differential scanningcalorimeter (DSC 605 M, Unipan-Termal, Warsaw), equippedwith a lid specially designed for photochemical measurements.The procedure was the same as that given in ref 7. Polymer-ization was initiated by a BP (0.06 M)-trithiane (0.02 or 0.04M) system with radiation (366 nm) from a Hg medium-pressure lamp. From the heat flux measured by the DSC, therate of polymerization (Rp) as a function of irradiation timewas determined. By integration of this curve, the double-bondconversion (p) was also determined as a function of irradiationtime. For computations, the heat of polymerization was takento be 56 kJ/mol per double bond.
Results and DiscussionThe trithianes investigated were 1,3,5-trithiane (TT),
R ) H; R-2,4,6-trimethyl-1,3,5-trithiane (R-TMT), R )
CH3; â-2,4,6-trimethyl-1,3,5-trithiane (â-TMT), R )CH3; R-2,4,6-triphenyl-1,3,5-trithiane (R-TPT), R )C6H5; â-2,4,6-triphenyl-1,3,5-trithiane (â-TPT), R )C6H5; and 2,4,6-trimethyl-2,4,6-triphenyl-1,3,5-trithiane(TMTPT).
TMTPT exists only in the cis-trans form.Benzophenone Photoreduction Study. Quench-
ing of the BP triplet state by trithianes in acetonitrilewas investigated by means of nanosecond laser flashphotolysis. The quenching rate constants, kq, weremeasured by monitoring the decays of the triplet-tripletabsorption of BP at 520 nm. The concentration of BPwas held fixed (2 mM), while the concentrations oftrithianes varied over a range that reduced the lifetimeof the triplet. The kq values were determined by a linearleast-squares fit of kobs vs the quencher concentrationplots by employing the formula
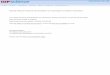
where τT is the BP triplet-state lifetime in the absenceof quencher, Q. An example of this method is illustratedin Figure 1, which shows the quenching of the BP tripletby â-TPT. The resulting rate constants kq for alltrithianes used are listed in Table 1.
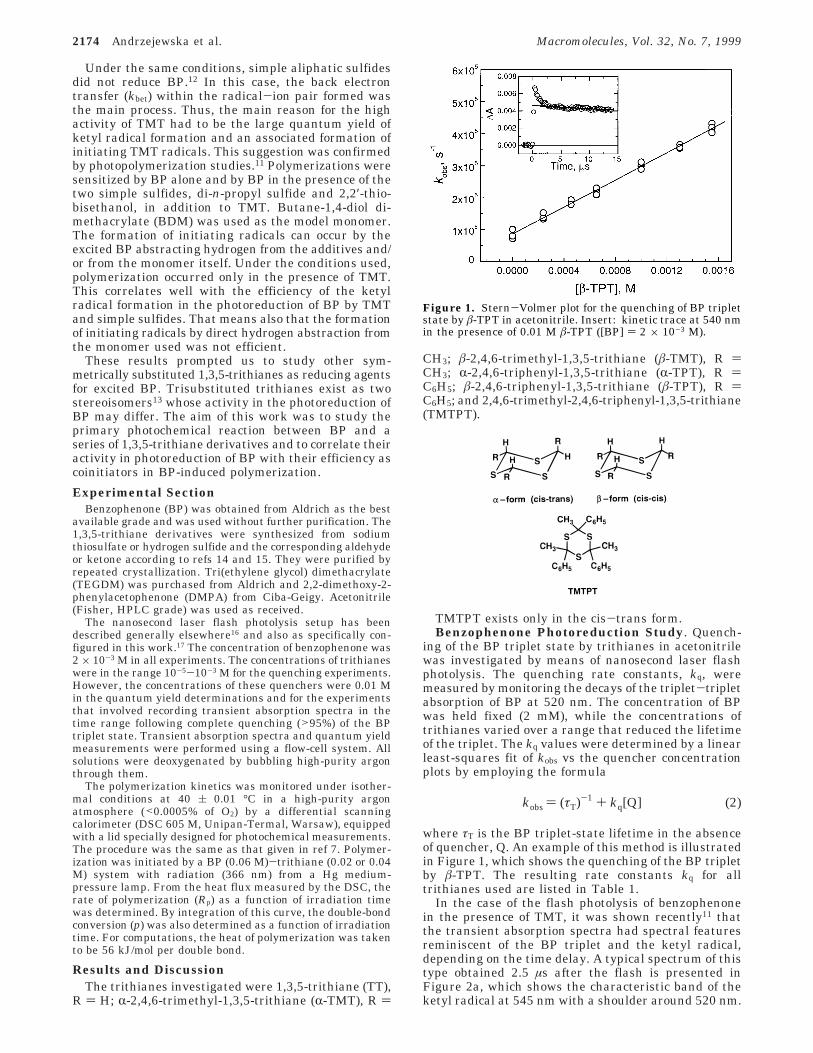
In the case of the flash photolysis of benzophenonein the presence of TMT, it was shown recently11 thatthe transient absorption spectra had spectral featuresreminiscent of the BP triplet and the ketyl radical,depending on the time delay. A typical spectrum of thistype obtained 2.5 µs after the flash is presented inFigure 2a, which shows the characteristic band of theketyl radical at 545 nm with a shoulder around 520 nm.
Figure 1. Stern-Volmer plot for the quenching of BP tripletstate by â-TPT in acetonitrile. Insert: kinetic trace at 540 nmin the presence of 0.01 M â-TPT ([BP] ) 2 × 10-3 M).
kobs ) (τT)-1 + kq[Q] (2)
2174 Andrzejewska et al. Macromolecules, Vol. 32, No. 7, 1999
A reference spectrum of the ketyl radical is also shownin Figure 2a; it is taken from ref 11 and normalized tothe maximum of the BPH• absorption.
The presence of an equivalent amount of R-alkylthioalkyl radical (see reaction 1) was not observed forTMT and TT in the spectral range studied. However,the use of phenyl substituents in the trithiane ring(TPT) led to the formation of R-phenyl thioalkyl radicalsthat can absorb in the spectral region investigated. Thisis shown in Figure 2b for the â-TPT isomer, where thedisplayed spectral range has been truncated from theoriginal data to illustrate how the quantum yields weredetermined from the spectrum. When the differencebetween the observed spectrum (open circles) and theketyl radical was taken over the range 310-620 nm,there was a sharp maximum of ∆A ≈ 0.03 in the rangeof 310-320 nm. The strong absorption around 310-320nm is reminiscent of the benzyl radical18 and lends somesupport to the suggestion that the excess absorptionbetween 300 and 500 nm is that of an R-phenyl thioalkylradical.
The quantum yields of ketyl radical formation weredetermined for concentrations of trithianes large enoughto quench 95% of the BP triplets. To measure thequantum yields, the relative actinometry method19 wasused. An acetonitrile solution of BP with no quencherwas used as an actinometer. The experiments were
performed in matched optical cells having identicalabsorption at 337 nm due to the presence of benzophe-none. The quantum yields were determined using thefollowing equation:
where ∆Ap is the ketyl radical’s absorption change at545 nm extrapolated to the end of the BP triplet decayand ∆AT is the absorption change in the actinometerimmediately after the flash due to the BP triplet (at 520nm) under conditions of no quenching. εp ) 3400 M-1
cm-1 and εT ) 6500 M-1 cm-1 are the correspondingmolar absorption coefficients of the ketyl radical andthe BP triplet state, respectively.20 Using this procedure,the quantum yields of ketyl radical formation for thereduction of BP by various trithianes were determined,and they are summarized in Table 1.
The quenching rate constants, which reflect theability of trithianes to form radical-ion pairs with BP,are on the same order and are very close in value. Theonly exception is the hexasubstituted trithiane. The lowvalue of kq for TMTPT suggests that the attack of thecarbonyl group of the excited BP on the lone electronpairs of the sulfur atoms in the trithiane ring is notlikely to occur, probably due to steric hindrance (threebulky substituent groups on each side of the ring). Thatmeans also that methyl hydrogens in this compound arenot sufficiently labile to be directly abstracted by excitedBP. The steric factors can also influence kq values ofthe remaining trithianes, since â-isomers always showhigher quenching rate constants than R-isomers (seeTable 1).
As can be seen in Table 1, the quantum yields of ketylradical formation are rather high and decrease in thefollowing order:
The quantum yields of ketyl radical formation are thehighest for the â-isomers that have three bulky sub-stituent groups on one side of the trithiane ring. Becausethe first step of the reaction is the attack of the BPtriplet on the lone electron pairs of the sulfur atoms,we may conclude that this attack is determined by sterichindrance. This is of special importance in the case ofphenyl substituents, where the change in configurationfrom the cis-cis to the cis-trans isomer drasticallyreduces radical formation.
The next step in the photoreduction is proton transferwithin the radical-ion pair (kH in eq 1). It is determinedby the acidity of the hydrogens of the trithiane ring. Theacidity is higher for phenyl-containing derivatives;therefore, radical formation is more effective for â-TPTthan for â-TMT. Another factor that plays a role indetermining acidity is the overlap of the sulfur orbitalsand the C-H bond that is being broken. Such overlapis better for axial hydrogens. This gives an additionalfactor favoring radical formation in â-isomers, becausethey have all their hydrogen atoms axial to the trithianering. However, radical formation is also affected by thestability of the radical being formed. In the case of theunsubstituted trithiane, whose radical is not stabilizedby substituents, its quantum yield is the lowest.
Photopolymerization Study. Photopolymerizationstudies were undertaken to compare the efficiency ofradical formation by the trithiane-BP system with the
Figure 2. Transient absorption spectrum following quenchingof the BP triplet state by â-TMT (0.01 M) (Figure 2a) and byâ-TPT (0.01 M) (Figure 2b) in acetonitrile taken 2.5 µs afterthe flash and the reference spectrum of BPH• radical inacetonitrile (shown as b b b).
Table 1. Rate Constants for Quenching of the BP Triplet(kq) and Quantum Yields of Ketyl Radical Formation(ΦBPH•) during Photoreduction of BP by Trithianes in
Acetonitrile
compound kq (M-1 s-1) ΦBPH•
TT (2.5 ( 0.1) × 108 0.28â-TMT (4.2 ( 0.4) × 108 0.62R-TMT (3.4 ( 0.2) × 108 0.53â -TPT (2.1 ( 0.1) × 108 0.81R -TPT (1.6 ( 0.1) × 108 0.32TPTMT e5 × 106
Φ ) ∆ApεT/(∆ATεp) (3)
â-TPT (0.81) > â-TMT (0.62) > R-TMT (0.53) >R-TPT (0.32) > TT (0.28)
Macromolecules, Vol. 32, No. 7, 1999 Trithianes as Coinitiators 2175
ability of trithianes to accelerate the polymerization. Asa model monomer, tri(ethylene glycol) dimethacrylate(TEGDM) was chosen. This monomer was chosen be-
cause the BP-induced polymerization of TEGDM occursmuch faster and efficiently than the polymerization ofBDM, which we used previously.11 This enables a moreprecise study of differences in the activities of thevarious trithianes. The polymerization was carried outin an inert atmosphere, to avoid the complicatinginfluence of atmospheric oxygen. The unsubstitutedtrithiane was not checked as the coinitiator due to itsvery poor solubility.
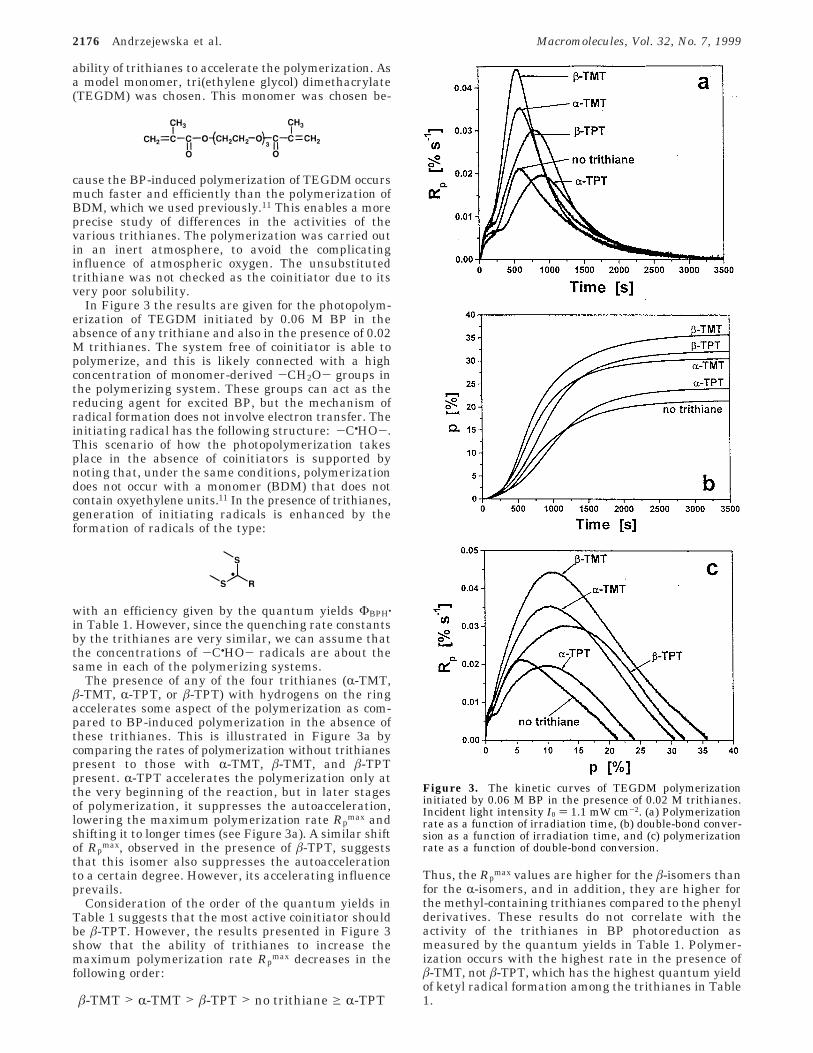
In Figure 3 the results are given for the photopolym-erization of TEGDM initiated by 0.06 M BP in theabsence of any trithiane and also in the presence of 0.02M trithianes. The system free of coinitiator is able topolymerize, and this is likely connected with a highconcentration of monomer-derived -CH2O- groups inthe polymerizing system. These groups can act as thereducing agent for excited BP, but the mechanism ofradical formation does not involve electron transfer. Theinitiating radical has the following structure: -C•HO-.This scenario of how the photopolymerization takesplace in the absence of coinitiators is supported bynoting that, under the same conditions, polymerizationdoes not occur with a monomer (BDM) that does notcontain oxyethylene units.11 In the presence of trithianes,generation of initiating radicals is enhanced by theformation of radicals of the type:
with an efficiency given by the quantum yields ΦBPH•
in Table 1. However, since the quenching rate constantsby the trithianes are very similar, we can assume thatthe concentrations of -C•HO- radicals are about thesame in each of the polymerizing systems.
The presence of any of the four trithianes (R-TMT,â-TMT, R-TPT, or â-TPT) with hydrogens on the ringaccelerates some aspect of the polymerization as com-pared to BP-induced polymerization in the absence ofthese trithianes. This is illustrated in Figure 3a bycomparing the rates of polymerization without trithianespresent to those with R-TMT, â-TMT, and â-TPTpresent. R-TPT accelerates the polymerization only atthe very beginning of the reaction, but in later stagesof polymerization, it suppresses the autoacceleration,lowering the maximum polymerization rate Rp
max andshifting it to longer times (see Figure 3a). A similar shiftof Rp
max, observed in the presence of â-TPT, suggeststhat this isomer also suppresses the autoaccelerationto a certain degree. However, its accelerating influenceprevails.
Consideration of the order of the quantum yields inTable 1 suggests that the most active coinitiator shouldbe â-TPT. However, the results presented in Figure 3show that the ability of trithianes to increase themaximum polymerization rate Rp
max decreases in thefollowing order:
Thus, the Rpmax values are higher for the â-isomers than
for the R-isomers, and in addition, they are higher forthe methyl-containing trithianes compared to the phenylderivatives. These results do not correlate with theactivity of the trithianes in BP photoreduction asmeasured by the quantum yields in Table 1. Polymer-ization occurs with the highest rate in the presence ofâ-TMT, not â-TPT, which has the highest quantum yieldof ketyl radical formation among the trithianes in Table1.
Figure 3. The kinetic curves of TEGDM polymerizationinitiated by 0.06 M BP in the presence of 0.02 M trithianes.Incident light intensity I0 ) 1.1 mW cm-2. (a) Polymerizationrate as a function of irradiation time, (b) double-bond conver-sion as a function of irradiation time, and (c) polymerizationrate as a function of double-bond conversion.
â-TMT > R-TMT > â-TPT > no trithiane g R-TPT
2176 Andrzejewska et al. Macromolecules, Vol. 32, No. 7, 1999
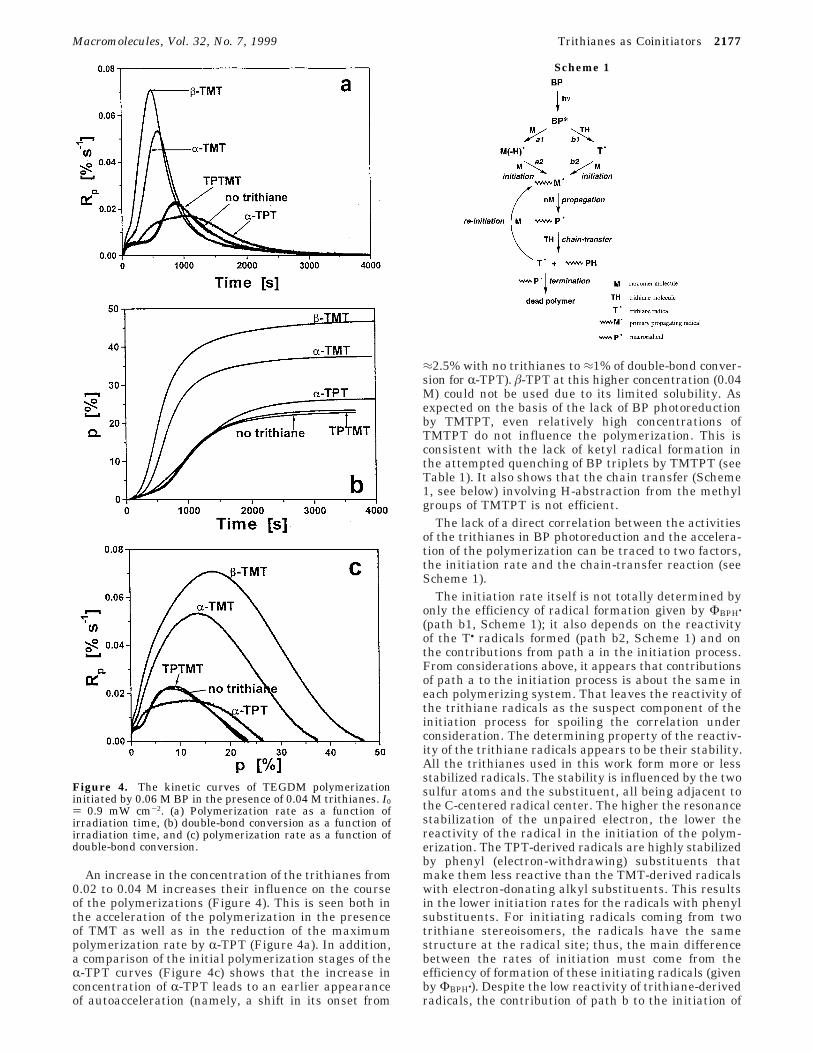
An increase in the concentration of the trithianes from0.02 to 0.04 M increases their influence on the courseof the polymerizations (Figure 4). This is seen both inthe acceleration of the polymerization in the presenceof TMT as well as in the reduction of the maximumpolymerization rate by R-TPT (Figure 4a). In addition,a comparison of the initial polymerization stages of theR-TPT curves (Figure 4c) shows that the increase inconcentration of R-TPT leads to an earlier appearanceof autoacceleration (namely, a shift in its onset from
≈2.5% with no trithianes to ≈1% of double-bond conver-sion for R-TPT). â-TPT at this higher concentration (0.04M) could not be used due to its limited solubility. Asexpected on the basis of the lack of BP photoreductionby TMTPT, even relatively high concentrations ofTMTPT do not influence the polymerization. This isconsistent with the lack of ketyl radical formation inthe attempted quenching of BP triplets by TMTPT (seeTable 1). It also shows that the chain transfer (Scheme1, see below) involving H-abstraction from the methylgroups of TMTPT is not efficient.
The lack of a direct correlation between the activitiesof the trithianes in BP photoreduction and the accelera-tion of the polymerization can be traced to two factors,the initiation rate and the chain-transfer reaction (seeScheme 1).
The initiation rate itself is not totally determined byonly the efficiency of radical formation given by ΦBPH•
(path b1, Scheme 1); it also depends on the reactivityof the T• radicals formed (path b2, Scheme 1) and onthe contributions from path a in the initiation process.From considerations above, it appears that contributionsof path a to the initiation process is about the same ineach polymerizing system. That leaves the reactivity ofthe trithiane radicals as the suspect component of theinitiation process for spoiling the correlation underconsideration. The determining property of the reactiv-ity of the trithiane radicals appears to be their stability.All the trithianes used in this work form more or lessstabilized radicals. The stability is influenced by the twosulfur atoms and the substituent, all being adjacent tothe C-centered radical center. The higher the resonancestabilization of the unpaired electron, the lower thereactivity of the radical in the initiation of the polym-erization. The TPT-derived radicals are highly stabilizedby phenyl (electron-withdrawing) substituents thatmake them less reactive than the TMT-derived radicalswith electron-donating alkyl substituents. This resultsin the lower initiation rates for the radicals with phenylsubstituents. For initiating radicals coming from twotrithiane stereoisomers, the radicals have the samestructure at the radical site; thus, the main differencebetween the rates of initiation must come from theefficiency of formation of these initiating radicals (givenby ΦBPH•). Despite the low reactivity of trithiane-derivedradicals, the contribution of path b to the initiation of
Figure 4. The kinetic curves of TEGDM polymerizationinitiated by 0.06 M BP in the presence of 0.04 M trithianes. I0) 0.9 mW cm-2. (a) Polymerization rate as a function ofirradiation time, (b) double-bond conversion as a function ofirradiation time, and (c) polymerization rate as a function ofdouble-bond conversion.
Scheme 1
Macromolecules, Vol. 32, No. 7, 1999 Trithianes as Coinitiators 2177
the polymerization is still significant due to much moreefficient quenching of the BP triplet by trithianes thanby ethers (kq ∼ 106 M-1 s-1).21
The second factor spoiling the correlation is the chain-transfer reaction in which active coinitiators that pos-sess labile hydrogen atoms can be involved. The overalleffect of this reaction is to increase the mobility of theradical population. This overall increase in the mobilityof the radicals enhances bimolecular termination, whichleads to lower reaction rates and lower conversions. Formultifunctional monomers, chain transfer also reducesthe cross-link density (see below). Moreover, chaintransfer transforms a part of the highly reactive propa-gating radicals into less reactive initiating radicals T•
having the same structure as radicals formed in thephotochemical process (path b1). This additionally low-ers the polymerization rate due to slow reinitiation.
From this scenario it can be seen that the influenceof trithianes on BP-induced polymerizations is a com-bination of two opposing processes: acceleration of theinitiation by their relatively efficient formation asinitiating radicals and interference with efficient po-lymerization through their participation in the chain-transfer reaction.
The effect of the chain-transfer reaction can beisolated from that of initiation by addition of trithianesto a polymerizing system initiated by a “cleavage-type”photoinitiator. The results for TEGDM polymerizationinitiated by DMPA in the presence of various trithianesare given in Figure 5. Because of the very high efficiencyof this photoinitiator, the molar ratio of DMPA:trithianehad to be as high as 1:10 to observe any slowing-downeffect from the chain-transfer reaction involving thetrithianes. From Figure 5 it can be seen that â-TMTlowers the polymeriztion rate only slightly. The largesteffect is observed for R-TPT. This trend in slowing downthe polymerization correlates very well with the activityfound for these same trithianes as coinitiators. Thatmeans that the polymerization rate is determinedmainly by the efficiency of the chain-transfer reaction.
The question why R-isomers lower the polymerizationrate more than â-isomers arises. For instance, thereactivity of the radicals formed from both TPT isomersshould be the same because of the conjugation of theunpaired spin with the phenyl group and the resultingplanarity of the radical. The most likely explanationseems to be that there is a larger production of trithianeradicals in the chain-transfer reaction from the R-isomer(two axial, one equatorial hydrogen) compared to theâ-isomer (three axial hydrogens). This effect is likely aconsequence of the presence of the equatorial hydrogenatoms in the R-isomer being more readily abstractableby the growing macroradical than are the axial hydro-gen atoms. For comparison, equatorial hydrogen atomsof trithianes are exchanged faster with deuterium atomsthan are axial hydrogen atoms in the presence of strongbases.22
The time-conversion and conversion-polymerizationrate curves (Figures 3b,c and 4b,c) show that all thetrithianes, even R-TPT, increase the final conversion ofdouble bonds (pf) compared to BP-induced polymeriza-tions in the absence of any trithianes. In general, as therate of polymerization increases, the final double-bondconversion also increases, and the maximum rate ofpolymerization is shifted to higher conversions. This isa direct consequence of the coupling of volume relax-ation with the polymerization kinetics.23 At faster rates
of polymerization, the macroscopic rate of volumeshrinkage is much slower than the rate at which doublebonds are consumed. If the system relaxes much slowerthan the rate of polymerization (which is often the casefor photopolymerization of multifunctional monomers),an excess free volume is present in the system. Thisexcess free volume results in greater mobility of thereacting species, which leads to a higher final double-bond conversion and a shift in the peak maximum ofthe rate to higher conversions.
Figure 5. The kinetic curves of TEGDM polymerizationinitiated by 0.002 M DMPA in the presence of 0.02 Mtrithianes. I0 ) 1.1 mW cm-2. (a) Polymerization rate as afunction of irradiation time, (b) double-bond conversion as afunction of irradiation time, and (c) polymerization rate as afunction of double-bond conversion.
2178 Andrzejewska et al. Macromolecules, Vol. 32, No. 7, 1999

This picture is complicated by the occurrence of thechain-transfer reaction. As indicated earlier, trithianesintensify the chain-transfer process, which is moreefficient in the presence of phenyl derivatives than othertrithianes. Thus, the reduction of the cross-link densityby TPT is greater. Decreasing cross-linking increasesthe free volume of the system at a given conversion andincreases the mobility of the polymerizing system. Theautoacceleration becomes less pronounced, resulting inlower Rp
max, which appears at somewhat higher conver-sion. As a consequence, the onset of autodeceleration(caused by dense network formation and vitrificationeffects that limit diffusion of reacting species) is shiftedalso to higher conversions. This enables the system toreach a higher pf level. This may explain why in twocases, â-TPT vs R-TMT (Figure 3) and R-TPT vs notrithiane (Figures 3 and 4), somewhat higher conver-sions are reached at lower maximum polymerizationrates.
ConclusionsThe 1,3,5-trithiane derivatives are active coinitiators
in benzophenone-induced photopolymerizations. Amongthe compounds investigated, the most efficient coini-tiator is the trimethyl derivative: â-TMT isomer (cis-cis), whose reactivity significantly exceeds that of theR-isomer (cis-trans). Generally, the activity of trithianesin accelerating the BP-induced polymerization is aresultant of the efficiency of initiating radical formation(photochemical process, path b1 in Scheme 1), thereactivity of initiating radicals (path b2 in Scheme 1),and the influence of processes resulting from the chaintransfer to coinitiators (Scheme 1). The photochemicalprocess depends on electron transfer from the sulfuratoms to the benzophenone triplet and proton transferwithin the radical-ion pair. This step of initiating radicalformation is governed by steric factors and the acidityof the trithiane-ring protons. The subsequent polymer-ization steps depend on the resonance stabilizationenergy of the various trithiane radicals (determiningtheir reactivity) and on the hydrogen-donating abilityof the various trithiane derivatives (in the chain transferto coinitiator).
Acknowledgment. The work described herein wassupported by the Committee of Scientific ResearchPoland (E.A. and M.A. DS-32/282/98) and the Office ofBasic Energy Sciences of the U.S. Department ofEnergy. This paper is Document No. NDRL 4099 fromthe Notre Dame Radiation Laboratory.
References and Notes
(1) Radiation Curing in Polymer Science and Technology; Fouass-ier, J. P., Rabek, J. F., Eds.; Plenum Press: New York, 1992-1993; Vols. I-IV.
(2) Dietliker, K. In Chemistry and Technology of UV and EBFormulation for Coatings, Inks and Paints; Oldrig, P. K. T.,Ed.; SITA Technology: London, 1991; Vol. 3, p 327.
(3) Andrzejewska, E. J. Polym. Sci.: A. Polym. Chem. 1992, 30,485.
(4) Andrzejewska, E.; Andrzejewski, M. Polimery (Warsaw) 1994,39, 22.
(5) Andrzejewska, E. Polymer 1993, 34, 3899.(6) Andrzejewska, E.; Andrzejewski, M. J. Polym. Sci. A: Polym.
Chem. 1993, 31, 2365.(7) Andrzejewska, E. Polymer 1996, 37, 1039.(8) Andrzejewska, E. Polymer 1996, 37, 1047.(9) Andrzejewska, E. Polym. Bull. 1996, 37, 199.
(10) Andrzejewska, E.; Bogacki, M. Macromol. Chem. Phys. 1997,198, 1649.
(11) Marciniak, B.; Andrzejewska, E.; Hug, G. L. J. Photochem.Photobiol. A: Chem. 1998, 112, 21.
(12) Bobrowski, K.; Marciniak, B.; Hug, G. L. J. Photochem.Photobiol. A: Chem. 1994, 81, 159.
(13) Campaigne, E. Chem. Rev. 1946, 39, 1.(14) El-Hewehi, Z.; Hempel D. J. Prakt. Chem. 1963, 21, 286.(15) Douglas, J. B.; Hydro, W. R. J. Am. Chem. Soc. 1951, 73,
3507.(16) Nagarajan, V.; Fessenden, R. W. J Phys. Chem. 1985, 89,
2330.(17) Hug, G. L.; Marciniak, B.; Bobrowski, K. J. Phys. Chem. 1996,
100, 13914.(18) Porter, G.; Wilkinson, F. Trans. Faraday Soc. 1961, 57, 1686.(19) Carmichael, I.; Hug, G. L. J. Phys. Chem. Ref. Data 1986,
15, 1.(20) Baral-Tosh, S.; Chattopadhyay, S. K.; Das, P. K. J Phys.
Chem. 1984, 88, 1404.(21) Guttenplan, J. B.; Cohen, S. G. J. Am. Chem. Soc. 1972, 94,
4040.(22) Andrzejewska, E.; Zuk, A.; Andrzejewski, M. Tetrahedron
Lett. 1977, 22.(23) Kloosterboer, J. G. Adv. Polym. Sci.1988, 84, 1.
MA9815408
Macromolecules, Vol. 32, No. 7, 1999 Trithianes as Coinitiators 2179
![Toxicological Studies on Benzophenone-Type UV Filters: … · 2020-07-03 · BH Benzhydrol BP Benzophenone BP-d10 Benzophenone-d10 [B] + Base peak ion CDFBS Charcoal dextran-treated](https://img.pdfslide.us/doc/110x75/5fa674776daa1d425832f9d7/toxicological-studies-on-benzophenone-type-uv-filters-2020-07-03-bh-benzhydrol.jpg)