Embed Size (px)
Citation preview

University of Massachusetts Amherst University of Massachusetts Amherst
ScholarWorks@UMass Amherst ScholarWorks@UMass Amherst
Open Access Dissertations
5-2013
Photo-Reaction of Copolymers with Pendent Benzophenone Photo-Reaction of Copolymers with Pendent Benzophenone
Scott Kenneth Christensen University of Massachusetts Amherst
Follow this and additional works at: https://scholarworks.umass.edu/open_access_dissertations
Part of the Polymer Science Commons
Recommended Citation Recommended Citation Christensen, Scott Kenneth, "Photo-Reaction of Copolymers with Pendent Benzophenone" (2013). Open Access Dissertations. 730. https://doi.org/10.7275/69rm-4g21 https://scholarworks.umass.edu/open_access_dissertations/730
This Open Access Dissertation is brought to you for free and open access by ScholarWorks@UMass Amherst. It has been accepted for inclusion in Open Access Dissertations by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected].

PHOTO-REACTION OF COPOLYMERS WITH PENDENT BENZOPHENONE
A Dissertation Presented
by
SCOTT KENNETH CHRISTENSEN
Submitted to the Graduate School of the
University of Massachusetts Amherst in partial fulfillment
of the requirements for the degree of
DOCTOR OF PHILOSOPHY
May 2013
Polymer Science and Engineering

© Copyright by Scott Kenneth Christensen 2013
All Rights Reserved

PHOTO-REACTION OF COPOLYMERS WITH PENDENT BENZOPHENONE
A Dissertation Presented
by
SCOTT KENNETH CHRISTENSEN
Approved as to style and content by:
___________________________________________
Ryan C. Hayward, Chair
___________________________________________
Kenneth R. Carter, Member
___________________________________________
Dhandapani Venkataraman, Member
____________________________________
David Hoagland, Department Head
Polymer Science and Engineering

To my best friend, my wife Jen

v
ACKNOWLEDGMENTS
I feel that the list of individuals responsible for the completion of this work could
span its entirety, yet I will do my best to be concise. First and foremost, my sincerest
gratitude goes to Professor Ryan Hayward, who I have been lucky enough to work with
these past six years. Ryan I thank you for being patient and encouraging throughout my
time, yet assertive when needed. It has been a privilege to grow as a scientist with your
guidance, and if I had to repeat this process I would once more make the same decision. I
must also extend my thanks to Professors Ken Carter and Dhandapani Venkataraman.
Each of you has played a vital role in the completion of my PhD, and I truly appreciate
the time and effort you have expended at my request. Ken you are one of the reasons I
came to UMass PSE, not only due to your scientific achievements but also because when
I met you during recruitment I noticed the leg lamp in your office (A Christmas Story is a
personal favorite of mine…). DV you have personally affected me greatly as my
collaboration with you and your group have served to give me confidence in my scientific
knowledge and perspective on topics I would otherwise be ignorant to.
I have had the joy of working with many qualified people during my time here,
and it is through involvement with you all that I find myself completing this work. I find
myself reminiscing about simpler times, when the Hayward group consisted of only 7
people and group meeting would be over in an hour every week. Jintao, Ian, Felicia, Le,
and Jungwook each witnessed the growing pains of my earliest attempts and I thank you
for your patience and guidance in getting through them without too many scars. The
group has expanded greatly to this point to include so many individuals who contribute to
my everyday life, from discussing the frustrations of research (looking at you Dayong,

vi
Maria, Cheol, and Nakul), to helping me run experiments (hey Rachel), to enriching my
knowledge by discussing their work (Kyle and Daniel). It has been a learning experience
just understanding how I fit into such a large group of qualified scientists, and I truly
appreciate it. I have also had many opportunities for collaboration, and I would be remiss
not to mention the fantastic people involved. The many members of the Center for
Chemical Innovation allowed me to explore new fields which were both a challenge and
a treat, and solid scientific connections sprang from the open and willing environment
created by them. I must give special thanks to Dipankar Basak, from whom I learned the
most effective ways to work together toward a common goal, and two amazing
undergraduate students, each of whom I worked with for a summer, Kate Arpino and
Anna Melker. Thanks are also due to the PSE faculty who taught me in and out of the
classroom, and to the technical staff members who keep our instruments running and my
blood pressure a little lower: Lou Raboin, Sekar Thirunavukkarasu, Weiguo Hu, Steve
Eyles, and Wim de Jeu.
Focusing on the scientific side of my PhD is only possible because of the superb
administrative staff we have here in our department. Sophia Hsu has made my life
especially easy, as I have never had to deal directly with a Procard audit and not once
missed a pay period, and everything she does is with a friendly and willing smile! The
front office staff has worked with me through arriving at PSE, my time as PSE Club
president, and the completion of my PhD, and Lisa Groth, Maria Farrington, Ann
Brainerd, and Vivien Venskowski are deserving of their prominent placement welcoming
everyone in the door of Conte. Thank you all!

vii
I have been lucky to have an unbelievable support system in place ever since I
began here in Fall 2007, and I apologize in advance for forgetting you (clever, right?).
I’m going to start by thanking my incoming classmates from 2007 – it’s amazing how
you can forge such bonds with people after only months crammed together in a
classroom. I think I could meet any of you in a decade and be right at home. I want to
personally thank Bugra, Eric, Sam, Nick, Ruosty, Felicia, Katrina, Yuri, and my many
other roommates for being there when I needed you to be. Of course I would be ashamed
to forget the Valley Radicals, Spare Parts, and all the folks from Amherst Sandlot soccer
who helped me release tension these past few years.
Finally I must thank my family, who I owe for helping me to become the person I
am today. Mom and Dad you have pushed all the right buttons along the way and given
me opportunities for which I am forever grateful. My brothers Matt and Robb have been
my best friends for longer than I realized, and I thank you for providing me with such
wonderful sisters Ortal and Kate. I am also extremely lucky to have amazing in-laws, as
Rick, Mary Anne, RJ, and Brian have been welcoming from the start and never once
made me feel like I wasn’t family. Last but certainly not least I owe my wife Jen my
sincere gratitude. Over the past decade she has been loving, patient, supportive, and
understanding of my mania and without her I could not reach this point.

viii
ABSTRACT
PHOTO-REACTION OF COPOLYMERS WITH PENDENT BENZOPHENONE
MAY 2013
SCOTT KENNETH CHRISTENSEN, B.S.E., CASE WESTERN RESERVE UNIVERSITY
M.S., UNIVERSITY OF MASSACHUSETTS AMHERST
Ph.D., UNIVERSITY OF MASSACHUSETTS AMHERST
Directed by: Professor Ryan C. Hayward
This dissertation aims to both deepen and broaden our understanding of
copolymers with pendent benzophenone (BP) in relation to both established applications
and novel directions in materials science. Photo-reaction of these BP copolymers is
explored in attempts to achieve three distinct goals: (1) robust and efficiently photo-
crosslinkable solid polymer films, (2) photo-reacted polymer blends with disordered
bicontinuous nanostructures, and (3) photo-patterned hydrogel materials with
environmental UV stability. We begin by investigating the fundamental gelation behavior
of solid polymer films, finding BP copolymers to be particularly effective crosslinkable
materials. Gelation efficiency can be tuned according to comonomer chemistry, as BP
hydrogen abstraction on the main polymer chain increases chain scission, reducing
crosslinking efficiency. This knowledge is then applied in Chapter 3, wherein we discuss
two potential methods for preparing nanostructured polymer blends from these
copolymers, namely spinodal decomposition of a photo-crosslinked polymer blend and
solution-state photografting to create interfacially active species. While each technique
shows promise, the ultimate goal of a disordered bicontinuous morphology will require
further tuning of materials systems and protocols. Finally, chemical deactivation of BP

ix
photo-crosslinker in copolymers for use as photo-patternable and environmentally stable
hydrogel materials is investigated. Reduction of BP by sodium borohydride proves a
feasible route toward deactivating residual photo-crosslinker in patterned hydrogel films.
These results confirm the utility of copolymers with pendent benzophenone photo-
crosslinkers as useful tools for complex material systems.

x
TABLE OF CONTENTS
Page
ACKNOWLEDGMENTS ...................................................................................................v
ABSTRACT ..................................................................................................................... viii
LIST OF TABLES ........................................................................................................... xiii
LIST OF SCHEMES........................................................................................................ xiv
LIST OF FIGURES ...........................................................................................................xv
CHAPTER
1. INTRODUCTION‡ ..........................................................................................................1
1.1 Benzophenone photo-reaction and applications ..........................................2
1.2 References ....................................................................................................8
2. GELATION OF COPOLYMERS WITH PENDENT BENZOPHENONE
PHOTO-CROSSLINKERS† ..................................................................................16
2.1 Abstract ......................................................................................................16
2.2 Motivation ..................................................................................................17
2.3 Experimental ..............................................................................................19
2.4 Results and Discussion ..............................................................................23
2.5 Conclusions ................................................................................................42
2.6 References ..................................................................................................43
3. ROUTES TOWARD NANOSTRUCTURED POLYMER BLENDS BY
BENZOPHENONE PHOTO-REACTION ............................................................49
3.1 Abstract ......................................................................................................49
3.2 Motivation ..................................................................................................50

xi
3.3 Arresting phase separation of LCST blends by BP photo-
crosslinking ................................................................................................51
3.3.1 Experimental ..................................................................................56
3.3.2 Results and Discussion ..................................................................59
3.3.3 Conclusion .....................................................................................63
3.3.4 Critical Blend Future Directions ....................................................63
3.4 Solution-state asymmetric photo-grafting of benzophenone
copolymers .................................................................................................65
3.4.1 Experimental ..................................................................................69
3.4.2 Results and Discussion ..................................................................72
3.4.3 Conclusion .....................................................................................81
3.4.4 Solution-state Photo-Grafting Future Directions ...........................82
3.5 Conclusions ................................................................................................83
3.6 References ..................................................................................................84
4. CHEMICAL DEACTIVATION OF BENZOPHENONE IN PHOTO-
PATTERNED HYDROGELS ...............................................................................94
4.1 Abstract ......................................................................................................94
4.2 Motivation ..................................................................................................95
4.3 Experimental ..............................................................................................97
4.4 Results and discussion .............................................................................101
4.5 Conclusion ...............................................................................................110
4.6 Future directions ......................................................................................110
4.7 References ................................................................................................113

xii
5. CONCLUSIONS..........................................................................................................116
BIBLIOGRAPHY ............................................................................................................119

xiii
LIST OF TABLES
Table Page
2.1: Properties of copolymers studied ................................................................................24
2.2: Results of Charlesby-Pinner Analysis; Number of crosslinks per unit AmBP
converted, ηc; Number of dislinks per crosslink, ηd/ηc (*value based on
SEC analysis of samples); Fraction of crosslinks formed by
benzopinacol formation, F/2ηc. ........................................................................31
2.3: Rate constants for hydrogen abstraction by benzophenone ........................................35

xiv
LIST OF SCHEMES
Scheme Page
1.1: Photo-reaction of benzophenone: Absorbance of ultraviolet irradiation
promotes BP from the ground state (S0) to the n-π* singlet (S1) which
undergoes rapid intersystem crossing to the diradicaloid n-π* triplet
(T1). This diradical then abstracts aliphatic hydrogen from its
environment, generating an aliphatic radical and a BP-centered radical
which then initiate further reaction. ...................................................................3
1.2: Possible triplet states for benzophenone. The n-π* triplet (left) is often
represented as a diradical and is the most reactive of excited state
species in BP. Possible π-π* triplets are shown in the bracket, with the
most common labeled as π-π* and π-π* CT (charge transfer). The CT
state is the least reactive of BP triplets. Schematic after reference 44
. ...............4
2.1: Several possible reaction pathways following hydrogen abstraction by triplet
BP: (a) Routes yielding a net cross-link via recombination of ketyl and
aliphatic- centered radicals; (b) One possible route yielding a net
dislink via β-scission of an aliphatic-centered radical .....................................19
2.2: Copolymer architecture and monomers used for copolymer synthesis (S:
styrene; pMS: p-methylstyrene; MAA: methacrylic acid; MMA:
methyl methacrylate; MA: methyl acrylate; iBMA: isobutyl
methacrylate; iBA: isobutyl acrylate; NiPAm: N-isopropyl
acrylamide); )( mnmxAmBP , where m and n are the mole fractions
of AmBP and comonomer, respectively. .........................................................22

xv
LIST OF FIGURES
Figure Page
1.1: Energy levels of p-aminobenzophenone in isopropanol and cyclohexane. The
more polar isopropanol causes inversion of the n-π* and CT states,
causing p-aminobenzophenone to be inert in this solvent. Solvation in
nonpolar cyclohexane permits reaction due to the n-π* triplet having
the lowest energy. Schematic after reference 38
. ................................................6
2.1: a) AmBP conversion is monitored via UV/Vis absorption of the π -π* peak
(~ 300 nm). Absorbance data have been normalized between 0 and 1
and fit with a single exponential decay (inset: spectra for PMMA (11K
Mw, xAmBP = 0.067)); b) Gel fraction is determined by measuring the
thickness reduction for surface-attached polymer films following
exposure to UV and extraction of the sol component. .....................................25
2.2: Charlesby-Pinner plot of PMMA (103K Mw, xAmBP = 0.033), used to
determine the number of crosslinks, ηc, and dislinks, ηd, per unit BP
converted according to Equation 6. .................................................................30
2.3: A comparison of the gel curves for PMMA copolymerized with AmBP (75K
Mw, xAmBP = 0.051, filled symbols), and doped with an equivalent
molar concentration of BP (55K Mw, open symbols), reveals a roughly
30-fold increase in gelation efficiency for the copolymer. ..............................34
2.4: a) SEC traces for PS (79K Mw, xAmBP = 0.05); b) reveal a nearly constant Mn
and increasing polydispersity with UV dose, indicating a system with
ηd/ηc ≈ 1. ...........................................................................................................38
2.5: SEC traces for PMAA (117K Mw, xAmBP = 0.030), show a decrease in Mn with
UV dose, indicating an overall ηd/ηc > 1. .........................................................40
2.6: Gel curves in both air and nitrogen for select copolymers reveal little
dependence on atmosphere, with the exception of PS which exhibits a
slightly higher ηeff under nitrogen; a) PMMA, 42K Mw, xAmBP = 0.048;
b) PS, 79K Mw, xAmBP = 0.05; c) PNiPAm, 43K Mw, xAmBP = 0.078; d)
PiBMA, 37K Mw, xAmBP = 0.055. .....................................................................41

xvi
3.1: Phase separation mechanisms of a binary polymer mixture. Nucleation and
growth (top) occurs when the homogeneous blend is quenched to a
metastable region of the phase diagram. Isolated particles of
equilibrium composition form and grow, forming a droplet in matrix
morphology. Spinodal decomposition (bottom) occurs when the
homogeneous mixture is quenched to a thermodynamically unstable
state. Concentration fluctuations vary with time, initially maintaining
constant size while increasing in amplitude. At later stages, both
amplitude and wavelength increase. Figure from Bates, F. S. Polymer-
Polymer Phase-Behavior. Science 251, 898–905 (1991). Reprinted
with permission from AAAS.21
........................................................................54
3.2: Sample turbidity data obtained from small angle light scattering. a)
Transmitted intensity versus temperature for a 25/75 PS-BP/PVME
sample exposed to 365 nm UV irradiation 30 s prior to heating at 5
°C/min through the cloud point. Inset images show light scattered prior
to and after clouding occurs. b) Cloud points determined for different
heating rates, showing the rate dependence on observed phase
separation. Reported cloud points have been extrapolated to a heating
rate of 0 °C/min. ...............................................................................................58
3.3: LCST Cloud points measured by SALS for PS/PVME58K Mn (black squares )
and PS-BP (55K Mn, xAmBP = 0.05)/PVME58K (red circles ) showing
little change in phase behavior upon incorporation of benzophenone
monomer. Inset is a representative bright field optical microscope
image of a 1/1 PS-BP/PVME blend after heating from 120 – 200 °C at
3 °K/min. Phase separation is observed. ..........................................................59
3.4: Cloud points measured by SALS for a 30/70 PS-BP (55K, xAmBP =
0.05)/PVME (58K Mn) blend exposed to 365 nm UV irradiation prior
to heating. a) Cloud points increase with increasing exposure energy
until 90 s, after which point the clouding behavior becomes difficult to
discern. b) Data plotted with inverse cloud point on the y-axis,
showing a dependence on radiation similar to that shown by Bauer and
Briber67
. ............................................................................................................61

xvii
3.5: Small angle X-ray scattering analysis of selected 50/50 PS-BP (55K, xAmBP =
0.05)/PVME (58K Mn) blends which have been pre-exposed to 365
nm UV irradiation for different times prior to phase separation at 175
°C. No discernible scattering peaks are observed in any sample from
30 – 240 s irradiation time, indicating a lack of resolvable features on
the 4 – 25 nm length scale. ...............................................................................62
3.6: Experimental technique for reactive blending. i) PS-BP copolymer and either
PEG or POEGMA (each polymer at 40 mg/mL) are dissolved in
benzene, and ii) are injected into a capillary tube, degassed, and
exposed to 365 nm UV for a prescribed irradiation time. After
exposure the solution is cast onto a glass slide and annealed prior to
characterization. ...............................................................................................71
3.7: UV/Visible absorption spectra of small molecule analogs at 45 mM
concentration in benzene. Benzophenone (black) shows the slowest
decay while incorporation of cumene with benzophenone (red) decays
faster and diethyl ether with benzophenone (blue) decays the most
rapidly, showing that benzophenone reacts most rapidly with the ether
species. Decay constants are extracted from single exponential fits. ..............74
3.8: SAXS traces for reactive blends of 1/1 PS-BP /PEG6K Mn exposed to 365 nm
UV for different durations. In the exposure range 65 – 90 minutes
annealed powder samples exhibit scattering peaks, indicating that
nanostructures are indeed present. ...................................................................75
3.9: Reactive blending results for 1/1 blends of PS-BP /PEG; a) Exposure times
required to acquire scattering peaks in SAXS for different molecular
weights of PEG, showing that with increased PEG molecular weight
the exposure time required is diminished. b) Length scales observed by
SAXS for blends with varying PEG molecular weights. .................................76
3.10: TEM image of a 1/1 PS-BP/PEG6K blend which has been exposed to 365
nm UV for 60 minutes. A hierarchical phase separation is observed,
with large domains on the order of 100 nm containing smaller features
on the 10 nm length scale, corresponding well with SAXS results
indicating 11.5 nm features. This sample has been cryo-microtomed
into 70 nm thick sections at -90 °C and stained with RuO4 vapor for 40
minutes prior to imaging. .................................................................................78

xviii
3.11: SEC traces of a 1/1 blend of PS-BP/PEG6K and the corresponding individual
polymers. The black and red curves are the PS-BP copolymer and PEG
homopolymer, respectively, while the green and blue curves represent
refractive index and ultraviolet signal from the polymer blend after 60
minutes of UV exposure. The refractive index detector peak (green
curve) at high retention times in the blend indicates a fraction of PEG
which has decreased molecular weight compared to the homopolymer.
This is indication of main chain scission during irradiation. Refractive
index curves magnified 25X. ...........................................................................79
3.12: SEC traces of a 1/1 blend of PS-BP/POEGMA and the corresponding
individual polymers. a) the black and red curves are the PS-BP and
POEGMA copolymers, respectively, while the green and black curves
represent refractive index and ultraviolet signals from the polymer
blend after 70 minutes of UV exposure. Unlike the PS/PEO blends,
this material clearly shows an increased molecular weight with UV
exposure. Refractive index curves have been magnified 25X. b)
Progression of molecular weight distribution (UV signal) with
exposure time for this blend signal. A peak at higher molecular weight
arises between 50 and 70 minutes of exposure, and disappears by 90
minutes, indicating the formation of insoluble species at high reaction
times. ................................................................................................................80
4.1: Floating hydrogel disc deactivation. i) PNiPAm-AAc-AmBP film is exposed
to UV through a mask and developed. ii) The sacrificial layer is
dissolved in an aqueous 1 mM phosphate buffer with 1 mM NaCl.
Deactivation is achieved through inclusion of solid NaBH4 to a
concentration of 0.15 M. iii) Concentration of NaBH4 is decreased
such that ionic strength of the buffer is at least 30X greater than that of
NaBH4. The samples are permitted to swell at room temperature and
are imaged, yielding discs with area A1. iv) Samples are heated
through the LCST to 60 °C, deswelling the discs. They are then
subjected to UV exposure overnight in the deswelled state. v) The
samples are swelled once more at room temperature, yielding discs
with area A2......................................................................................................99

xix
4.2: Deactivation of AmBP in PNiPAm-AAc-AmBP copolymer as studied by
UV/Vis and SEC. a) The inset shows spectra of polymer solution prior
to and post deactivation with 0.15 M NaBH4 (in 2:1 ethanol:water),
showing the disappearance of AmBP absorbance at 300 nm with a
corresponding rise of photo-product at 250 nm. Monitoring absorbance
at 300 nm during deactivation in we observe >90% conversion within
2500 s in ethanol solution, yet in a 2:1 ethanol:water mixture
deactivation is achieved within 300 s; b) SEC traces of polymer
solutions prior to and after treatment with NaBH4. The curves are
nearly identical, indicating that treatment does not significantly
influence molecular weight distribution of the copolymer. ...........................102
4.3: Swelling curve of PNiPAm-AAc-AmBP hydrogel discs after one exposure.
Areal swelling is calculated as A1/A0, with A0 the area of the initial
photo-mask. The minimal difference in swelling between samples with
and without NaBH4 treatment is indication that the deactivation
process does not disturb the existing hydrogel network. ...............................103
4.4: Final versus initial swelling for PNiPAm-AAc-AmBP hydrogel discs
subjected to the protocol depicted in Figure 4.1. By treating the
hydrogel discs with NaBH4 we are able to recover over 85% of the
initial swelling over the entire first exposure energy range. Conversely,
we see that the sample without NaBH4 treatment was only able to
achieve a fraction of the initial swelling. At high energy the curves
intersect due to conversion of all AmBP photo-crosslinker in the first
UV exposure step. ..........................................................................................104
4.5: Extent of reaction of AmBP photo-crosslinker in PNiPAm hydrogel discs
following each UV exposure. Values are calculated based upon
equilibrium swelling theory of a polyelectrolyte gel in the low salt
limit33
. a) After the first UV exposure, extent of reaction 1p ranges
from 0.59 to 1.0, and results are consistent between samples treated
with NaBH4 and controls; b) After deswelling and subsequent UV
irradiation, extent of reaction 2p for samples that have been treated
with NaBH4 are nearly identical to their 1p values, while controls
exhibit significantly higher values, further evidence that reduction with
NaBH4 prevents crosslinking upon subsequent UV irradiation in these
photo-patterned hydrogels. ............................................................................107

xx
4.6: Fluorescence microscopy images of PNiPAm-AAc-AmBP-RhBMA/PpMS-
AmBP bilayers. The untreated bilayer in both the deswelled (i) and
swelled (ii) states is nearly flat. Importantly, no distinction in swelling
contrast is observed between the halves of this sample. The bilayer that
has been treated with NaBH4 swells from the flat (iii) state to a curled
cylinder (iv) with a distinct difference between low and high swelling
areas, indicating successful deactivation of AmBP. ......................................109

1
CHAPTER 1
INTRODUCTION‡
Photochemistry, the study of reactions which proceed by the absorption of
photons, is a relatively new field when one considers that photo-reactions have been
utilized for millennia. Ancient Egyptians would soak linen in oil and place them in the
sun to dry, creating waterproof and robust materials which were applied to
mummification and boat building. What they did not fully appreciate at the time was that
the lavender oil used contained Syrian asphalt, which would effectively photo-crosslink
with radiation, and thus they were applying photochemistry over 4,000 years ago1,2
. More
recently, the advantages of photo-reactions have become apparent, as evidenced by the
vast attention they have received in the scientific and technological communities. The use
of photo-reactions allow processing of materials after their initial synthesis, and the
availability of low-cost and tunable radiation sources make photo-chemical techniques
exceedingly versatile3–5
. Furthermore, radiation processing permits scaling for high
throughput fabrication and spatial variation of intensity (i.e. lithography), with theoretical
resolution dependent on the wavelength of irradiation2,6–8
. As a result photo-induced
reactions have become essential to modern life, used in coatings, biosensors, inks,
photoresists, adhesives, and many other applications9–12
.
In polymer systems, photo-reaction generally proceeds by one of several
mechanisms including radical formation and cycloaddition. Radicals can be generated by
‡ Portions modified and reprinted (adapted) with permission from Christensen, S.K.,
Chiappelli, M.C., and Hayward, R.C. Macromolecules, 2012, 45 (12), pp 5237–5246.
Copyright 2012 American Chemical Society.

2
either Norrish Type I or Type II photo-initiation. Norrish Type I is achieved by α-
cleavage, whereby photon absorption is followed by homolytic cleavage of a bond alpha
to the excited carbonyl, producing a radical pair (as in acetophenone). Conversely,
Norrish Type II photo-initiation occurs by hydrogen abstraction to generate radicals13
.
Phenyl azides are also extensively used, as they will undergo one of several insertion
reactions after conversion to the highly reactive nitrene species when activated14–16
.
These processes are reasonably nonspecific, meaning that species so formed have the
potential to take part in any number of subsequent reactions and thus no additional
functionalization is necessary. A more selective set of photo-reactive materials are based
on self-cycloaddition, under which two identical unsaturated species dimerize to generate
a crosslink (examples include cinnamate esters, stilbazolium salts, coumarin,
styrylpyridinium, or maleimides)5,7,17–21
.
1.1 Benzophenone photo-reaction and applications
A set of photo-active molecules that have attracted a great deal of attention are
based on benzophenone (BP), and have been widely studied due to the photophore’s
numerous attractive properties. BP has proven to be extremely amenable to
experimentation, as it is more chemically robust than alternative molecules, stable to
ambient illumination, and active under wavelengths which do not damage the majority of
biomolecules10,11,22–24
. The modest molar absorbance of BP also permits photo-reaction
of reasonably thick samples with minimal gradient in illumination intensity3,25
. Coupling
this ease of use with high photochemical activity, broad reactivity, and low cost allows
BP to be applied in a diverse range of applications5,9,13,22,26–28
.

3
A Norrish Type II photo-initiator, BP is active under UV irradiation, where upon
absorption of a photon with wavelength of ~ 250 - 365 nm, ground state BP (S0) is
excited to a singlet state (S1) that rapidly and efficiently undergoes intersystem crossing
to yield the lowest energy reactive triplet13,26,29
(Scheme 1.1). The n-π* triplet (T1), which
can be represented as a diradical, is well known to abstract aliphatic hydrogens, with
especially high reactivity for those located alpha to electron donating heteroatoms (most
commonly nitrogen, sulfur, and oxygen)22,30
, thus converting the triplet BP to a ketyl
radical and also yielding an aliphatic carbon-centered radical. This process is known to
proceed with reasonable efficiency even in the presence of oxygen31–37
, making BP a
robust and broadly useful photo-chemically reactive species.
Scheme 1.1: Photo-reaction of benzophenone: Absorbance of ultraviolet irradiation
promotes BP from the ground state (S0) to the n-π* singlet (S1) which undergoes rapid
intersystem crossing to the diradicaloid n-π* triplet (T1). This diradical then abstracts
aliphatic hydrogen from its environment, generating an aliphatic radical and a BP-
centered radical which then initiate further reaction.

4
Reactivity of benzophenone derivatives can vary broadly depending upon
substituent groups and solvent conditions. In particular, reactivity depends on the lowest
energy triplet state in a given material, where the π-π* charge transfer (CT) state is least
reactive and the n-π* triplet is the most reactive (Scheme 1.2)38–40
. While unsubstituted
BP attains a lowest energy n-π* triplet, substitution with certain functionalities alters the
energy levels of these triplet states, even inverting them to make the unreactive CT state
lower energy26,41
. This inversion is caused by incorporation of an electron donating
species (i.e. amino, dimethylamino, hydroxy, methoxy, etc.) at the meta or para position
of BP, which serves to stabilize the CT state seen in Scheme 1.2, thereby lowering its
energy and making the BP species much less reactive38,42–44
. This effect can be made
more clear if we consider the hydrogen abstraction process in Scheme 1.1 as a reaction
between the electrophilic n-π* diradical and an electron-rich hydrogen donor substrate.
Including an electron donor on the aromatic ring serves to increase the electron density at
the carbonyl oxygen, making this group less electrophilic and thus less reactive. The
Scheme 1.2: Possible triplet states for benzophenone. The n-π* triplet (left) is often
represented as a diradical and is the most reactive of excited state species in BP. Possible
π-π* triplets are shown in the bracket, with the most common labeled as π-π* and π-π*
CT (charge transfer). The CT state is the least reactive of BP triplets. Schematic after
reference 44
.

5
opposite is true of electron withdrawing groups, which generally enhance reactivity of BP
by maintaining the n-π* as the lowest energy triplet13,39,40,44
. A special case is found when
substitution is at the ortho position of BP, which prevents reaction with substrate
materials due to the occurrence of intramolecular hydrogen abstraction26,38,41,44
.
Solvent conditions are also known to affect reactivity of benzophenone
derivatives, as changes in solvent polarity will change the relative energy levels of
substituted BP, sometimes inverting the n-π* and CT states when they are close in
energy. An example is shown in Figure 1.1 for p-aminobenzophenone, where in the more
polar isopropanol the π-π* CT state is the lowest triplet, yet in nonpolar cyclohexane the
n-π* is of lower energy38
. Generally, the energy of an excited state will decrease in polar
solvents if its polarity is higher than that of the ground state. As the π-π* CT state as
shown in Scheme 1.2 is more polar than the n-π* triplet, its stabilization in polar media is
greater, preventing reaction of some BP derivatives in polar solvents38,42–44
. It must be
said, however, that even when the lowest energy triplet is of π-π* (or CT) nature, some
reactivity will likely be observed as thermal fluctuation to an n-π* triplet will occur with
some frequency38,44
.
Due to its generally high reactivity, the scientific community has made extensive
use of BP, particularly in polymer systems where BP acts as both a photo-initiator and a
photo-grafting agent. As the n-π* triplet behaves as a diradical, many vinyl monomers
are susceptible to direct photo-polymerization by BP45,46
. Furthermore, when combined
with alkyl amine as a co-initiator, BP derivatives initiate polymerization of acrylate
monomers with very high efficiency, and as such are commonly used in
photoresists9,47–49
. Numerous polymers have also been successfully photo-grafted to

6
substrates by activation of BP dissolved in solution50,51
or anchored to a
surface11,23,45,52–57
, which allows coupling of radicals generated on polymer chains to
those on the substrate58
. These techniques have improved adhesion of polymer films54–56
,
increased solubility of nanoparticles in a polymer matrix57
, and allowed the photo-
patterning of biomolecule probes10,11,52
. Furthermore, addition of BP as a dopant has also
been used to achieve photo-crosslinking of a variety of polymers in the solid and melt
states3,4,6,25,28,59–61
. Despite their effectiveness in many arenas, the use of small-molecule
BP-species suffers from several intrinsic limitations, namely the possibility for the dopant
to macrophase separate, migrate preferentially to certain regions of the sample, and even
be volatilized at high temperature6,28,47,62–65
.
.
Figure 1.1: Energy levels of p-aminobenzophenone in isopropanol and cyclohexane. The
more polar isopropanol causes inversion of the n-π* and CT states, causing p-
aminobenzophenone to be inert in this solvent. Solvation in nonpolar cyclohexane
permits reaction due to the n-π* triplet having the lowest energy. Schematic after
reference 38
.

7
The use of a one-component material, i.e., a copolymer containing covalently
attached BP moieties, can potentially eliminate these issues. By including the BP
functionality in a macromolecule, phase separation and migration are prevented by
chemical compatibilization with the matrix material, and the vapor pressure of BP is
effectively decreased. What’s more, in several cases the photochemical activity of BP
copolymers has proven higher than their small molecule counterparts9,28,65
. It is thus no
surprise that copolymers containing covalently-bound BP as photo-crosslinkers and
photo-grafting agents have been described in the literature going back over fifty
years6,31,32,62,66–72
.
More recently, however, BP copolymers have gained attention as a powerful route
to prepare hydrogels as surface attached films23,52,54,55,73
, micro-devices8,12,74–76
,
biosensors5,77
, biomedical coatings56
, and patterned sheets78,79
. These versatile materials
have also been used to generate resist layers80
, pressure-sensitive adhesive coatings24,81
,
optical multilayers82
, and even to align liquid crystals83
. Therefore, despite having existed
for over half a century, it is clear that benzophenone copolymers are an increasingly
important tool for expanding the fundamental and practical applications of materials
science.
This dissertation aims to both deepen and broaden our understanding of
copolymers with pendent benzophenone in relation to both established applications and
novel directions in materials science. BP copolymers wherein the photo-crosslinker is
located pendent to the main polymer chain have been synthesized by conventional free
radical polymerization, and other materials are acquired from commercial sources or
facile chemical means in order to limit synthetic demands. Photo-reaction of these BP

8
copolymers is then explored in attempts to achieve three distinct goals: i) robust and
efficiently photo-crosslinkable solid polymer films, ii) photo-reacted polymer blends with
disordered bicontinuous nanostructures, and iii) UV stable photo-patterned hydrogels for
multilayer systems. In Chapter 2 we investigate the fundamental gelation behavior of
solid polymer films by a combination of UV/Visible absorption spectroscopy, gel fraction
measurements, and chromatography. Knowledge gained from this study is then applied in
Chapter 3, wherein we discuss two potential methods for preparing nanostructured
polymer blends from these materials. Finally, Chapter 4 investigates the chemical
deactivation of BP photo-crosslinker in copolymers for use as photo-patternable and
environmentally stable hydrogel materials. The combined results from this report serve to
define the capabilities of BP copolymers in material systems of the future.
1.2 References
1. Decker, C. & Bendaikha, T. Interpenetrating polymer networks. II. Sunlight-
induced polymerization of multifunctional acrylates. J. Appl. Polym. Sci. 70,
2269–2282 (1998).
2. Wondraczek, H., Kotiaho, A., Fardim, P. & Heinze, T. Photoactive
polysaccharides. Carbohydr. Polym. 83, 1048–1061 (2011).
3. Qu, B. J. & Ranby, B. Photocross-linking of Low-Density Polyethylene. 1.
Kinetics and Reaction Parameters. J. Appl. Polym. Sci. 48, 701–709 (1993).
4. Doytcheva, M. et al. Ultraviolet-induced crosslinking of solid poly(ethylene
oxide). J. Appl. Polym. Sci. 64, 2299–2307 (1997).
5. Mateescu, A., Wang, Y., Dostalek, J. & Jonas, U. Thin Hydrogel Films for Optical
Biosensor Applications. Membranes 2, 40–69 (2012).
6. Oster, G., Oster, G. K. & Moroson, H. Ultraviolet Induced Crosslinking and
Grafting of Solid High Polymers. J. Polym. Sci. 34, 671–684 (1959).

9
7. Kuckling, D., Adler, H.-J. P. & Arndt, K.-F. Polymer Gels. 833, 312–325
(American Chemical Society: Washington, DC, 2002).
8. Chiappelli, M. C. & Hayward, R. C. Photonic multilayer sensors from photo-
crosslinkable polymer films. Adv. Mater. 24, 6100–4 (2012).
9. George, B. & Dhamodharan, R. A study of the photopolymerization kinetics of
methyl methacrylate using novel benzophenone initiators. Polym. Int. 50, 897–905
(2001).
10. Marcon, L. et al. Photochemical immobilization of proteins and peptides on
benzophenone-terminated boron-doped diamond surfaces. Langmuir 26, 1075–80
(2010).
11. Martin, T. A. et al. Quantitative photochemical immobilization of biomolecules on
planar and corrugated substrates: a versatile strategy for creating functional
biointerfaces. ACS Appl. Mater. Interfaces 3, 3762–71 (2011).
12. Belardi, J., Schorr, N., Prucker, O. & Rühe, J. Artificial Cilia: Generation of
Magnetic Actuators in Microfluidic Systems. Adv. Funct. Mater. 21, 3314–3320
(2011).
13. Turro, N. et al. Molecular Photochemistry of Alkanones in Solution - Alpha-
cleavage, Hydrogen Abstraction, Cycloaddition, and Sensitization Reactions. Acc.
Chem. Res. 5, 92–101 (1972).
14. Chen, G., Ito, Y. & Imanishi, Y. Micropattern Immobilization of a pH-Sensitive
Polymer. Macromolecules 30, 7001–7003 (1997).
15. Ito, Y., Chen, G., Guan, Y. & Imanishi, Y. Patterned Immobilization of
Thermoresponsive Polymer. Langmuir 13, 2756–2759 (1997).
16. Chen, G., Imanishi, Y. & Ito, Y. Photolithographic Synthesis of Hydrogels.
Macromolecules 31, 4379–4381 (1998).
17. Chujo, Y., Sada, K. & Saegusa, T. Polyoxazoline having a coumarin moiety as a
pendant group. Synthesis and photogelation. Macromolecules 23, 2693–2697
(1990).
18. Shindo, Y., Katagiri, N., Ebisuno, T., Hasegawa, M. & Mitsuda, M. Network
formation and swelling behavior of photosensitive poly(vinyl alcohol) gels
prepared by photogenerated crosslinking. Angew. Makromol. Chem. 240, 231–239
(1996).

10
19. Cockburn, E. S., Davidson, R. S. & Pratt, J. E. The photocrosslinking of
styrylpyridinium salts via a [2 + 2]-cycloaddition reaction. J. Photochem.
Photobiol., A 94, 83–88 (1996).
20. Williams, J. L. R. & Border, D. G. The preparation and properties of photoreactive
polymers. I. 2-(arylvinyl)-N-vinylpyridinium arylsulfonate polymers. Macromol.
Chem. 73, 203–214 (1964).
21. Kuckling, D. & Pareek, P. Bilayer hydrogel assembly. Polymer 49, 1435–1439
(2008).
22. Dorman, G. & Prestwich, G. D. Benzophenone Photophores in Biochemistry.
Biochemistry 33, 5661–5673 (1994).
23. Prucker, O., Naumann, C. A., Rühe, J., Knoll, W. & Frank, C. W. Photochemical
Attachment of Polymer Films to Solid Surfaces via Monolayers of Benzophenone
Derivatives. J. Am. Chem. Soc. 121, 8766–8770 (1999).
24. Scherzer, T., Tauber, A. & Mehnert, R. UV curing of pressure sensitive adhesives
studied by real-time FTIR-ATR spectroscopy. Vib. Spectrosc. 29, 125–131 (2002).
25. Wu, S. K. & Rabek, J. F. Benzophenone-Initiated Photo-Crosslinking of
Polynorbornene in the Solid-State. Polym. Degrad. Stab. 21, 365–376 (1988).
26. Rabek, J. F. Mechanisms of photophysical processes and photochemical reactions
in polymers: Theory and Applications. Energy 272–300 (John Wiley & Sons:
Chichester, 1987).
27. Rabek, J. F. Photosensitized Processes in Polymer Chemistry: A Review.
Photochem. Photobiol. 7, 5–57 (1968).
28. Wu, Q. H. & Qu, B. J. Synthesis of Di(4-hydroxyl benzophenone) sebacate and its
usage as initiator in the photocrosslinking of polyethylene. J. Appl. Polym. Sci. 85,
1581–1586 (2002).
29. Porter, G. & Wilkinson, F. Primary photochemical processes in aromatic
molecules. Part 5- Flash photolysis of benzophenone in solution. Trans. Faraday
Soc. 57, 1686–1691 (1961).
30. Shoute, L. C. T. & Huie, R. E. Reactions of triplet decafluorobenzophenone with
alkenes. A laser flash photolysis study. J. Phys. Chem. A 101, 3467–3471 (1997).
31. Rehmer, G., Boettcher, A. & Portugall, M. (Meth)acrylate copolymer based UV-
crosslinkable materials. US Patent 5,389,699 1–7 (1995).

11
32. Rehmer, G., Boettcher, A. & Portugall, M. Benzophenone derivatives and their
preparation. US Patent 5,264,533 1–7 (1993).
33. Smets, G. J., Elhamouly, S. N. & Oh, T. J. Photochemical-Reactions on Polymers.
Pure Appl. Chem. 56, 439–446 (1984).
34. Lin, A. A., Sastri, V. R., Tesoro, G., Reiser, A. & Eachus, R. On the Cross-Linking
Mechanism of Benzophenone-Containing Polyimides. Macromolecules 21, 1165–
1169 (1988).
35. Tsubakiyama, K., Kuzuba, M., Yoshimura, K., Yamamoto, M. & Nishijima, Y.
Photochemical-Reaction of Polymers Having Arylketone and Tertiary Amine as
Pendant Groups. Polym. J. (Tokyo, Jpn.) 23, 781–788 (1991).
36. Eisele, G., Fouassier, J. P. & Reeb, R. Kinetics of photocrosslinking reactions of a
DCPA/EA matrix in the presence of thiols and acrylates. J. Polym. Sci., Part A:
Polym. Chem. 35, 2333–2345 (1997).
37. Carroll, G. T., Triplett, L. D., Moscatelli, A., Koberstein, J. T. & Turro, N. J.
Photogeneration of gelatinous networks from pre-existing polymers. J. Appl.
Polym. Sci. 122, 168–174 (2011).
38. Porter, G. & Suppan, P. Primary photochemical processes in aromatic molecules.
Part 12 - Excited states of benzophenone derivatives. Trans. Faraday Soc. 61,
1664–1673 (1965).
39. Aspari, P. et al. Photoinduced electron transfer between triethylamine and
aromatic carbonyl compounds: the role of the nature of the lowest triplet state.
Trans. Faraday Soc. 92, 1689 (1996).
40. Li, W., Xue, J., Cheng, S. C., Du, Y. & Phillips, D. L. Influence of the chloro
substituent position on the triplet reactivity of benzophenone derivatives: a time-
resolved resonance Raman and density functional theory study. J. Raman
Spectrosc. 43, 774–780 (2012).
41. Porter, G. & Suppan, P. Primary photochemical processes in aromatic molecules.
Part 14 - Comparative photochemistry of aromatic carbonyl compounds. Trans.
Faraday Soc. 62, 3375 (1966).
42. Ghoneim, N., Monbelli, A., Pilloud, D. & Suppan, P. Photochemical reactivity of
para-aminobenzophenone in polar and non-polar solvents. J. Photochem.
Photobiol., A 94, 145–148 (1996).
43. Singh, A. K., Bhasikuttan, A. C., Palit, D. K. & Mittal, J. P. Excited-state
dynamics and photophysical properties of para-aminobenzophenone. J. Phys.
Chem. A 104, 7002–7009 (2000).

12
44. Bhasikuttan, A. C., Singh, A. K., Palit, D. K., Sapre, A. V. & Mittal, J. P. Laser
Flash Photolysis Studies on the Monohydroxy Derivatives of Benzophenone. J.
Phys. Chem. A 102, 3470–3480 (1998).
45. Rohr, T., Hilder, E. F., Donovan, J. J., Svec, F. & Frechet, J. M. J. Photografting
and the Control of Surface Chemistry in Three-Dimensional Porous Polymer
Monoliths. Macromolecules 36, 1677–1684 (2003).
46. Yang, B. & Yang, W. T. Thermo-sensitive switching membranes regulated by
pore-covering polymer brushes. J. Membr. Sci. 218, 247–255 (2003).
47. Allen, N. S. et al. Photochemistry and photopolymerization activity of novel 4-
alkylamino benzophenone initiators-synthesis, characterization, spectroscopic and
photopolymerization activity. Eur. Polym. J. 26, 1345–1353 (1990).
48. Allen, N. S. et al. Photochemistry of Novel 4-Alkylamino Benzophenone Initiators
- A Conventional Laser Flash-Photolysis and Mass-Spectrometry Study. J.
Photochem. Photobiol., A 54, 367–388 (1990).
49. Fouassier, J. P., Ruhlmann, D., Graff, B., Morletsavary, F. & Wieder, F. Excited-
State Processes in Polymerization Photoinitiators. Prog. Org. Coat. 25, 235–271
(1995).
50. Costamagna, V., Strumia, M., Lopez-Gonzalez, M. & Riande, E. Gas transport in
surface grafted polypropylene films with poly(acrylic acid) chains. J. Polym. Sci.,
Part B: Polym. Phys. 45, 2421–2431 (2007).
51. Li, G., He, G., Zheng, Y., Wang, X. & Wang, H. Surface photografting initiated by
benzophenone in water and mixed solvents containing water and ethanol. J. Appl.
Polym. Sci. 123, 1951–1959 (2012).
52. Naumann, C. A. et al. The Polymer-Supported Phospholipid Bilayer: Tethering as
a New Approach to Substrate−Membrane Stabilization. Biomacromolecules 3, 27–
35 (2002).
53. Griep-Raming, N., Karger, M. & Menzel, H. Using benzophenone-functionalized
phosphonic acid to attach thin polymer films to titanium surfaces. Langmuir 20,
11811–4 (2004).
54. Toomey, R., Freidank, D. & Rühe, J. Swelling Behavior of Thin, Surface-Attached
Polymer Networks. Macromolecules 37, 882–887 (2004).
55. Beines, P. W., Klosterkamp, I., Menges, B., Jonas, U. & Knoll, W. Responsive
thin hydrogel layers from photo-cross-linkable poly(N-isopropylacrylamide)
terpolymers. Langmuir 23, 2231–2238 (2007).

13
56. Schwartz, V. B. et al. Antibacterial Surface Coatings from Zinc Oxide
Nanoparticles Embedded in Poly(N-isopropylacrylamide) Hydrogel Surface
Layers. Adv. Funct. Mater. 22, 2376–2386 (2012).
57. Roth, P. J. & Theato, P. Covalent Attachment of Gold Nanoparticles to Surfaces
and Polymeric Substrates Using UV Light. Macromol. Chem. Phys. 213, 2550–
2556 (2012).
58. Kato, K., Uchida, E., Kang, E.-T., Uyama, Y. & Ikada, Y. Polymer surface with
graft chains. Prog. Polym. Sci. 28, 209–259 (2003).
59. Graves, R. & Pintauro, P. N. Polyphosphazene membranes. II. Solid-state
photocrosslinking of poly[(alkylphenoxy)(phenoxy)phosphazene] films. J. Appl.
Polym. Sci. 68, 827–836 (1998).
60. Choi, J.-E. & Ko, K.-Y. Zeolite H-beta: an Efficient and Recyclable Catalyst for
the Tetrahydropyranylation of Alcohols and Phenols , and the Deprotection of
Tetrahydropyranyl Ethers. Bull. Korean Chem. Soc. 22, 1177–1178 (2001).
61. Qu, B. J. Reaction mechanism of benzophenone-photoinitiated crosslinking of
polyethylene. Chin. J. Polym. Sci. 20, 291–307 (2002).
62. Tocker, S. Cross-linked blended polymers containing polymeric acryloxy
benzophenones. US Patent 3,265,772 1–5 (1966).
63. Zhao, Y. & Dan, Y. Synthesis and characterization of a polymerizable
benzophenone derivative and its application in styrenic polymers as UV-stabilizer.
Eur. Polym. J. 43, 4541–4551 (2007).
64. Zhou, Z., Li, S., Zhang, Y., Liu, M. & Li, W. Promotion of proton conduction in
polymer electrolyte membranes by 1H-1,2,3-triazole. J. Am. Chem. Soc. 127,
10824–5 (2005).
65. Wu, G. et al. UV exposure effects on photoinitiator-grafted styrene-butadiene-
styrene triblock copolymer. J. Appl. Polym. Sci. 120, 2627–2631 (2011).
66. Tocker, S. Organic polymeric articles and preparation thereof from derivatives of
ethylene and unsaturated benzophenones. US Patent 3,214,492 1–8 (1965).
67. Tocker, S. Graft and cross linked copolymers of polar vinylidene monomers with
acryloxyphenones. US Patent 3,315,013 1–7 (1967).
68. Agolini, F. Polymeric phenone photosensitizers and blends thereof with other
polymers. US Patent 3,632,493 1–4 (1972).

14
69. Sumitomo, H., Nobutoki, K. & Susaki, K. Photo-Graft Polymerization of Methyl
Methacrylate onto Styrene-Vinylbenzophenone Copolymer. J. Polym. Sci., Part A-
1: Polym. Chem. 9, 809–812 (1971).
70. Carlini, C., Ciardelli, F., Donati, D. & Gurzoni, F. Polymers containing side-chain
benzophenone chromophores: a new class of highly efficient polymerization
photoinitiators. Polymer 24, 599–606 (1983).
71. Jiang, X., Luo, X. & Yin, J. Polymeric photoinitiators containing in-chain
benzophenone and coinitiators amine: Effect of the structure of coinitiator amine
on photopolymerization. J. Photochem. Photobiol., A 174, 165–170 (2005).
72. Rufs, A. M. et al. Synthesis and photoinitiation activity of macroinitiators
comprising benzophenone derivatives. Polymer 49, 3671–3676 (2008).
73. Buller, J., Laschewsky, A. & Wischerhoff, E. Photoreactive oligoethylene glycol
polymers – versatile compounds for surface modification by thin hydrogel films.
Soft Matter 9, 929–937 (2013).
74. Castellanos, A. et al. Size-Exclusion “Capture and Release” Separations Using
Surface-Patterned Poly(N-isopropylacrylamide) Hydrogels. Langmuir 23, 6391–
6395 (2007).
75. Stoychev, G., Zakharchenko, S., Turcaud, S., Dunlop, J. W. C. & Ionov, L. Shape-
Programmed Folding of Stimuli-Responsive Polymer Bilayers. ACS Nano 6,
3925–3934 (2012).
76. Zakharchenko, S., Puretskiy, N., Stoychev, G., Stamm, M. & Ionov, L.
Temperature controlled encapsulation and release using partially biodegradable
thermo-magneto-sensitive self-rolling tubes. Soft Matter 6, 2633 (2010).
77. Wang, Y., Brunsen, A., Jonas, U., Dostálek, J. & Knoll, W. Prostate specific
antigen biosensor based on long range surface plasmon-enhanced fluorescence
spectroscopy and dextran hydrogel binding matrix. Anal. Chem. 81, 9625–9632
(2009).
78. Kim, J., Hanna, J. A., Byun, M., Santangelo, C. D. & Hayward, R. C. Designing
Responsive Buckled Surfaces by Halftone Gel Lithography. Science 335, 1201–
1205 (2012).
79. Kim, J., Hanna, J. A., Hayward, R. C. & Santangelo, C. D. Thermally responsive
rolling of thin gel strips with discrete variations in swelling. Soft Matter 8, 2375–
2381 (2012).
80. McCartin, P. & Nebe, W. Photoactive plastisols: positive washout image. J.
Imaging Sci. 32, 222–225 (1988).

15
81. Do, H.-S., Park, Y.-J. & Kim, H.-J. Preparation and adhesion performance of UV-
crosslinkable acrylic pressure sensitive adhesives. J. Adhes. Sci. Technol. 20,
1529–1545 (2006).
82. Mönch, W., Dehnert, J., Prucker, O., Rühe, J. & Zappe, H. Tunable Bragg filters
based on polymer swelling. Appl. Opt. 45, 4284–4290 (2006).
83. Cox, J. R., Simpson, J. H. & Swager, T. M. Photoalignment Layers for Liquid
Crystals from the Di-π-methane Rearrangement. J. Am. Chem. Soc. 135, 640–3
(2013).

16
CHAPTER 2
GELATION OF COPOLYMERS WITH PENDENT BENZOPHENONE PHOTO-
CROSSLINKERS†
2.1 Abstract
A series of copolymers containing covalently attached benzophenone (BP) photo-
crosslinkers was synthesized and their UV-induced gelation monitored as a function of
the extent of BP conversion. For poly(methyl methacrylate) copolymers, the
recombination yield between BP- and aliphatic-centered radicals was estimated and
compared to that for dimerization of each species, directly confirming that the high
gelation efficiencies observed for these copolymers arises due to the additional
crosslinking pathways provided by covalently incorporated BP, as compared to doping
with a small-molecule crosslinker. The placement of the hydrogen species most
susceptible to abstraction by triplet benzophenone is found to greatly influence gelation
efficiency, since radical generation on the polymer backbone typically increases the
probability of dislinking events, while hydrogen abstraction pendent to the copolymer
backbone tends to enhance crosslinking. Finally, the presence of atmospheric oxygen
during photo-crosslinking was found to yield only modest changes in the gelation
behavior of these copolymers.
† Reprinted (adapted) with permission from Christensen, S.K., Chiappelli, M.C., and
Hayward, R.C. Macromolecules, 2012, 45 (12), pp 5237–5246. Copyright 2012
American Chemical Society.

17
2.2 Motivation
Benzophenone (BP) has long been used as a dopant to achieve photo-crosslinking
of a variety of polymers in the solid and melt states, including poly(ethylene)1–5
,
poly(ethylene oxide)6, poly(norbornene)
7, and poly(phosphazene)s
8. While this doping
with small-molecule BP-species is a simple route to achieve photo-crosslinking, it suffers
from several intrinsic limitations, including the possibility for small molecule
crosslinkers to macrophase separate, migrate preferentially to certain regions of the
sample, and even be volatilized at high temperature4,9,10
.
The use of a one-component material, i.e., a copolymer containing covalently
attached BP moieties, eliminates issues of phase separation and migration, and can also
significantly enhance gelation efficiency since it provides additional pathways to
crosslink formation. As shown in Scheme 2.1a, following hydrogen abstraction there are
three pathways by which radical recombination can yield crosslinks: (i) coupling of ketyl
and aliphatic radicals, (ii) dimerization of ketyl radicals to form a benzopinacol, and (iii)
dimerization of aliphatic radicals. By contrast, for samples doped with unattached BP
derivatives, only the third mechanism provides a crosslink. Given the possibility for
radicals placed on the polymer backbone to undergo competing reactions leading to chain
scission or “dislinking” (an example of which is shown in Scheme 2.1b), these additional
pathways can in many cases be critical to obtaining reasonable crosslinking yields.
Copolymers containing covalently-bound BP as photo-crosslinkers and photo-
grafting agents have been described in the patent literature going back nearly fifty
years9,11–15
, but have only more recently gained attention in the open scientific literature
as a powerful route to prepare hydrogels as surface attached films16–19
, micro-devices20,21
,

18
biosensors22,23
, biomedical coatings24
, and patterned sheets25,26
, as well as to generate
resist layers27
, pressure-sensitive adhesive coatings28,29
, and optical multilayers30
. Several
efforts have been made to investigate how the photoreaction of these copolymers depends
on BP concentration and mode of substitution29,31–34
, comonomer composition29,33,35,36
,
and reaction temperature28,37,38
. These studies on crosslinking are largely incomplete,
however, as they are complicated by the use of multi-component28,29,39
systems and
exotic monomers33,36
, or substitution effects due to incorporation of BP in the polymer
backbone32,37,40
, making the extension of these results to common material systems
difficult.
As copolymers containing covalently bound BP represent an increasingly
important tool for materials chemists, the current report seeks to better understand the
gelation of these polymers, in particular the efficiency of photo-crosslinking and its
dependence on comonomer chemistry. Here, a series of copolymers containing
acrylamidobenzophenone (AmBP) monomers, and thus pendent BP moieties, were
synthesized and crosslinked using 365 nm UV light, with the conversion of BP monitored
by UV/visible absorption spectroscopy and the gel fractions determined from film
thickness measurements following extraction of sol components. Effective crosslinking
efficiencies, and the number of crosslinks and dislinks per BP unit converted, were
estimated using mean-field and Charlesby-Pinner theories, respectively. Gelation of
select polymers was also examined under a nitrogen environment to quantify the effect of
oxygen on crosslinking.
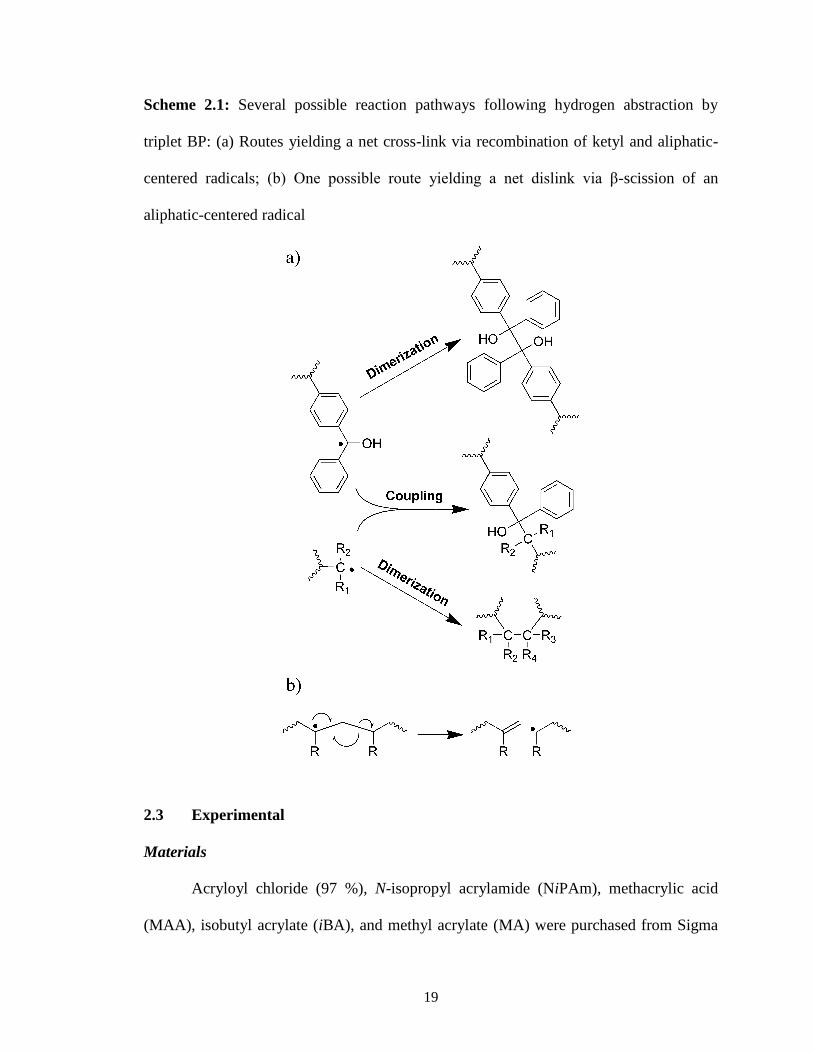
19
Scheme 2.1: Several possible reaction pathways following hydrogen abstraction by
triplet BP: (a) Routes yielding a net cross-link via recombination of ketyl and aliphatic-
centered radicals; (b) One possible route yielding a net dislink via β-scission of an
aliphatic-centered radical
2.3 Experimental
Materials
Acryloyl chloride (97 %), N-isopropyl acrylamide (NiPAm), methacrylic acid
(MAA), isobutyl acrylate (iBA), and methyl acrylate (MA) were purchased from Sigma

20
Aldrich. 4-aminobenzophenone (98 %), triethylamine (99.7 %), p-methyl styrene (pMS)
and isobutyl methacrylate (iBMA) were obtained from Acros Organics. Methyl
methacrylate (MMA) was obtained from Fisher Scientific and styrene (S) from
Malinckrodt-Baker. Inhibitor was removed from monomers by extraction with KOH (aq)
and water, and subsequently drying over anhydrous MgSO4 (Fisher).
Azobisisobutyronitrile (AIBN, Aldrich) was recrystallized from methanol prior to use
while 1,4-dioxane (Aldrich), methanol (Fisher Scientific) and
methacryloxypropyltrichlorosilane (Gelest) were used without further purification.
Synthesis
Acrylamidobenzophenone (AmBP) was synthesized from acryloyl chloride and 4-
aminobenzophenone according to the method of Jia et al41
. Polymers were synthesized by
free radical polymerization under nitrogen at 80 °C in 1,4-dioxane with AIBN as initiator
(for methacrylic acid, a 3:1 methanol:1,4-dioxane by volume mixture was used).
Resulting polymers were then precipitated and dried under vacuum. 1H-NMR was
performed with a Bruker Avance 400 MHz or DPX 300 MHz spectrometer to determine
AmBP content. Molecular weights were determined via size exclusion chromatography
(SEC) with a Polymer Laboratories GPC-50 with a polystyrene column and THF as
eluent (0.5 mL/min). A refractive index detector was used, and molecular weights were
calibrated with poly(methyl methacrylate) standards, or poly(styrene) standards for
styrenic materials. Poly(methacrylic acid) molecular weight was characterized with a
Hewlett Packard Series 1050 aqueous SEC with 0.1M NaNO3 and 3 mM NaN3 as eluent
and a refractive index detector against poly(ethylene glycol) standards.

21
UV Exposure
Polymer film samples (on quartz or silicon substrates) were exposed to 365 nm
UV radiation using a UVGL-58 Handheld UV lamp (UVP; ~ 2 mW/cm2, spectral range
340 - 410 nm). For samples on quartz substrates, a silicon wafer was placed behind the
sample to mimic the reflection of light that occurs for samples on silicon substrates. The
lamp was allowed to equilibrate for at least 1 h prior to use, and power was frequently
measured with an X-Cite XR2100 Power Meter (EXFO) to ensure accuracy of the
reported dose. Exposures under inert atmosphere were performed by placing samples
inside an Instec HCS621V microscope hot stage purged with N2 gas. The sample was
maintained at a temperature of 35 °C, matching that of the surface of the UV source.
Unless otherwise specified, all exposures were performed in open atmospheric conditions
with the sample placed in contact with the window on the UV light source.
AmBP Conversion
Samples for UV/visible absorption spectroscopy (UV/vis) were cast by placing
~ 100 L of a filtered 10 mg/mL polymer solution (PMMA, PiBMA, PiBA, PS, PpMS in
toluene; PMA in chloroform; PNiPAm in ethanol) onto UV-ozone treated 1 x ½ in quartz
plates, yielding films of ~ 1 m thickness, which were subsequently annealed under
vacuum for at least 12 h at 30 K above the glass transition temperature (Tg) of each
material. Conversion of AmBP was monitored as a function of UV dose with a Hitachi
U-3010 spectrophotometer in absorbance mode over a wavelength range of 200 - 500 nm.

22
Sol/Gel Measurements
Silicon wafers were silanized with the adhesion promoter
methacryloxypropyltrichlorosilane and samples were subsequently spin coated from 40
mg/mL solutions (PMMA, PiBMA, PiBA, PS, PpMS in toluene; PMA in chloroform;
PNiPAm in ethanol; PMAA in methanol) at 2000 rpm for 50 s, yielding film thicknesses
of 150 - 400 nm upon annealing at Tg + 30 K. Each sample was exposed to UV for a
prescribed period of time, then immersed in a marginal solvent mixture for at least 5 h
which was sufficient to completely remove the sol fraction (solvent mixtures: v/v
Scheme 2.2: Copolymer architecture and monomers used for copolymer synthesis
(S: styrene; pMS: p-methylstyrene; MAA: methacrylic acid; MMA: methyl methacrylate;
MA: methyl acrylate; iBMA: isobutyl methacrylate; iBA: isobutyl acrylate; NiPAm: N-
isopropyl acrylamide); )( mnmxAmBP , where m and n are the mole fractions of
AmBP and comonomer, respectively.

23
THF/water: PMMA 80/20, PMA 73/27, PiBMA 84/16, PiBA 84/16, PS 78/12; v/v
toluene/hexanes: PpMS 75/25; v/v ethanol/water: PNiPAm 31/69; water: PMAA).
Samples were then annealed for at least 12 h (at Tg + 30 K) prior to thickness
measurement, which was performed with a Filmetrics F20 thin film measurement system
with a tungsten-halogen fiber light source over a wavelength range of 400 - 900 nm. Each
film was sampled four times, and uncertainties reported as plus or minus one standard
deviation.
2.4 Results and Discussion
A series of copolymers containing the photo-crosslinker AmBP and one of eight
other monomers were first prepared, as summarized in Scheme 2. The chemistries
chosen include the commonly-used materials poly(styrene), poly(methyl methacrylate),
and poly(N-isopropyl acrylamide), as well as five other (meth)acrylate or styrenic
monomers that allow for comparisons between materials with varying reactivity, density,
and location of abstractable hydrogens. The copolymers synthesized had molecular
weights (weight average, Mw) ranging from 11,000 to 117,000 g/mol and dispersities (Ð)
between 1.5 and 2.2, as indicated in Table 2.1. The mole fraction of photo-crosslinkers
xAmBP in these copolymers ranged from 0.03 to 0.125, corresponding to an average
number of AmBP units per chain between 3 and 21. For the case of PMMA, four
polymers with different molecular weights and crosslinker contents were prepared to
determine whether these parameters significantly influence our results.
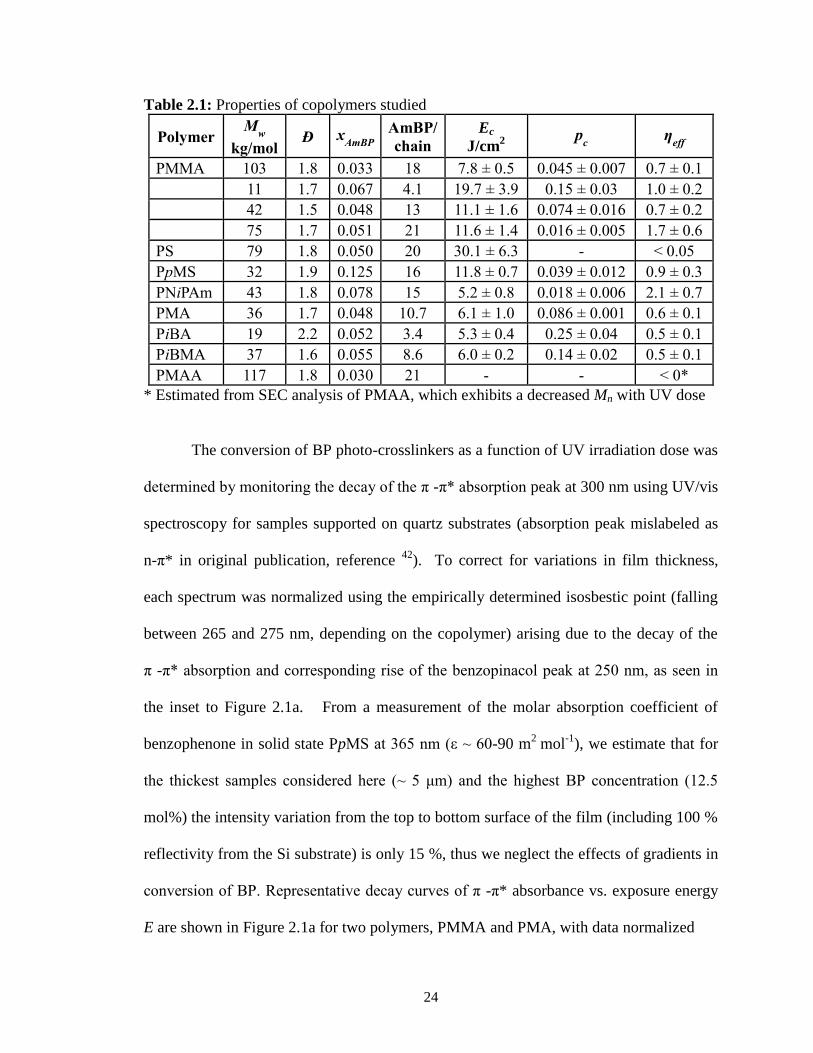
24
Table 2.1: Properties of copolymers studied
Polymer M
w
kg/mol Ð x
AmBP AmBP/
chain Ec
J/cm2
pc η
eff
PMMA 103 1.8 0.033 18 7.8 ± 0.5 0.045 ± 0.007 0.7 ± 0.1 11 1.7 0.067 4.1 19.7 ± 3.9 0.15 ± 0.03 1.0 ± 0.2 42 1.5 0.048 13 11.1 ± 1.6 0.074 ± 0.016 0.7 ± 0.2 75 1.7 0.051 21 11.6 ± 1.4 0.016 ± 0.005 1.7 ± 0.6 PS 79 1.8 0.050 20 30.1 ± 6.3 - < 0.05 PpMS 32 1.9 0.125 16 11.8 ± 0.7 0.039 ± 0.012 0.9 ± 0.3 PNiPAm 43 1.8 0.078 15 5.2 ± 0.8 0.018 ± 0.006 2.1 ± 0.7 PMA 36 1.7 0.048 10.7 6.1 ± 1.0 0.086 ± 0.001 0.6 ± 0.1 PiBA 19 2.2 0.052 3.4 5.3 ± 0.4 0.25 ± 0.04 0.5 ± 0.1 PiBMA 37 1.6 0.055 8.6 6.0 ± 0.2 0.14 ± 0.02 0.5 ± 0.1 PMAA 117 1.8 0.030 21 - - < 0*
* Estimated from SEC analysis of PMAA, which exhibits a decreased Mn with UV dose
The conversion of BP photo-crosslinkers as a function of UV irradiation dose was
determined by monitoring the decay of the π -π* absorption peak at 300 nm using UV/vis
spectroscopy for samples supported on quartz substrates (absorption peak mislabeled as
n-π* in original publication, reference 42
). To correct for variations in film thickness,
each spectrum was normalized using the empirically determined isosbestic point (falling
between 265 and 275 nm, depending on the copolymer) arising due to the decay of the
π -π* absorption and corresponding rise of the benzopinacol peak at 250 nm, as seen in
the inset to Figure 2.1a. From a measurement of the molar absorption coefficient of
benzophenone in solid state PpMS at 365 nm (ε ~ 60-90 m2
mol-1
), we estimate that for
the thickest samples considered here (~ 5 μm) and the highest BP concentration (12.5
mol%) the intensity variation from the top to bottom surface of the film (including 100 %
reflectivity from the Si substrate) is only 15 %, thus we neglect the effects of gradients in
conversion of BP. Representative decay curves of π -π* absorbance vs. exposure energy
E are shown in Figure 2.1a for two polymers, PMMA and PMA, with data normalized

25
Figure 2.1: a) AmBP conversion is monitored via UV/Vis absorption of the π -π* peak
(~ 300 nm). Absorbance data have been normalized between 0 and 1 and fit with a single
exponential decay (inset: spectra for PMMA (11K Mw, xAmBP = 0.067)); b) Gel fraction is
determined by measuring the thickness reduction for surface-attached polymer films
following exposure to UV and extraction of the sol component.
between zero and one (after subtracting the constant absorbance at infinite energy K) for
clarity. In all cases, the data are well described by a single exponential decay

26
KeAA cEE
0 , with a characteristic dose for conversion of BP units Ec that is
related to the extent of conversion p by
cEE
ep
1 . (2.1)
As summarized in Table 2.1, values of Ec ranged from ~ 5 - 30 J/cm2
for the samples
considered here.
To approximate the yield of the photoproduct benzopinacol relative to
consumption of AmBP, the peak maxima were monitored as a function of UV dose. An
exponential background was subtracted from each spectrum, as a significant background
due to absorbance from the comonomers is present at low wavelengths. Combining the
initial and final absorbances of AmBP ( 0
AmBPA and f
AmBPA , respectively) and benzopinacol
( 0
bpA and f
bpA , respectively) in these copolymer systems allows the fraction of AmBP
converted to benzopinacol F to be calculated as
)(1
)(2
0
0
f
AmBPAmBP
AmBP
bp
f
bp
bp
AA
AA
F
(2.2)
where εAmBP (540 m2/mol) and εbp (1180 m
2/mol) are the peak molar absorption
coefficients of AmBP and benzopinacol, respectively, measured in methanol. Values of F
range from 0.15 to 0.58, indicating significant formation of benzopinacol through the
combination of AmBP-centered radicals.
For each copolymer, a corresponding set of films were prepared on silicon
substrates treated with an adhesion promoter to allow measurement of the gel fraction.
These films were exposed to UV and subsequently developed by immersion in a solvent

27
mixture of moderate quality to extract the sol fraction without extensively swelling the
gelled material. The gel fraction for each sample was calculated as the ratio of the (dry)
film thickness after solvent extraction to that of the initial film prior to UV exposure.
Figure 2.1b shows data on gel fraction vs. irradiation energy for the same PMMA and
PMA polymers as in Figure 2.1a. Each curve shows a clear gel point, as evidenced in the
jump from zero to finite gel fraction at a particular dose. Using Equation 1 to relate the
energy at the gel point to an extent of reaction, we convert this dose to a critical extent of
reaction pc for each sample. Experimental values are reported as the average of the last
point with zero gel fraction and the first point with non-zero gel fraction for each
copolymer. As summarized in Table 2.1, these values ranged from 0.016 to 0.25 for the
samples studied, with the exceptions of PS and PMAA, which showed zero gel fraction
even at full conversion (p = 1).
Mean-Field Model
A crude estimate of the efficiency of BP photo-crosslinkers is obtained by
comparison with the mean-field bond percolation model of gelation for lattice sites with
functionality f. For the case of a polymer with a weight-average43,44
degree of
polymerization N, if each monomer is capable of forming a crosslink and N >> 1, then
the mean field prediction for the critical extent of reaction is:
Nfp fm
c
1
1
1
, (2.3)
i.e., gelation occurs at an average of one crosslink per chain45,46
. For the copolymers
considered here, only a fraction xAmBP of the monomers are capable of forming crosslinks,
thus we modify Equation 3 to yield

28
AmBP
fm
cxN
p
1, (2.4)
as the mean field prediction for gelation47
. An overall efficiency of conversion of BP
units into crosslinks can then be calculated from the experimentally measured gel point pc
by
AmBPcc
fm
ceff
xNpp
p
1
, (2.5)
with values for each copolymer listed in Table 2.1. Importantly, the use of PS or PMMA-
equivalent molecular weights to determine N introduces a modest systematic error into
this analysis which varies depending on the copolymer being tested. However, we do not
correct for this since Mark-Houwink parameters are not readily available for all the
copolymers studied.
Charlesby-Pinner Analysis
While mean-field theory provides a simple and useful metric for crosslinking
efficiency, it suffers from the well-known limitation that it ignores cyclization, and also
cannot distinguish the influence of dislinking reactions from inefficient crosslinking.
While the former effect is difficult to correct for and will generally lead to an
underestimate (~5%) of crosslink formation from experimental pc values48
, dislinking
reactions can be taken into account by a Charlesby-Pinner analysis49
. While the original
theory is developed in terms of the radiation dose, this treatment cannot be directly used
as AmBP reactivity varies with different co-monomers. Further, since our experiments
reach BP conversions near unity, we rewrite the Charlesby-Pinner equation as:
pxNSS
AmBPcc
d
1
22
1
(2.6)

29
where S is the sol fraction, and c and d represent the number of crosslinks and dislinks
per unit of BP converted, respectively. Due to the presence of significant uncertainties in
our measured values of both p and S, we calculated effective error bars in S incorporating
both components for purposes of curve fitting. Figure 2.2 shows representative data for a
PMMA copolymer plotted according to the form of Equation 6. At low values of 1/p the
data are in good agreement with a linear relationship, allowing the determination of c
and d from the slope and intercept of the uncertainty-weighted least squares best-fit line.
Fits were also performed over the entire range of 1/p, and while this leads to changes in
the values of c and d determined, the qualitative trends reported remain intact. The
observed deviations from linearity at high p1 are presumably caused by the assumption
of a random molecular weight distribution ( 2nw NN ) in Charlesby-Pinner theory. As
the prepared copolymers have initially non-random molecular weight distributions, it is
only upon random crosslinking and dislinking (at low 1/p) that samples approach this
polydispersity value.
Crosslinking Efficiency by Mean-Field and Charlesby-Pinner Analyses
The values of effective crosslinking efficiency ηeff determined from mean-field
analysis, as summarized in Table 2.1, are at least 0.5 for all copolymer samples, with the
exception of PS and PMAA, which did not gel even at full AmBP conversion (ηeff <
0.05). Although this approach has significant limitations due to the inherent
simplifications in the mean field model, it suggests that pendent AmBP units are highly
efficient crosslinkers in all but these two cases. Only one polymer, PNiPAm, gave a
value of ηeff (2.1 ± 0.7) that with reasonable confidence can be said to exceed unity. As
we are not aware of reactions that would yield multiple crosslinks per AmBP converted,
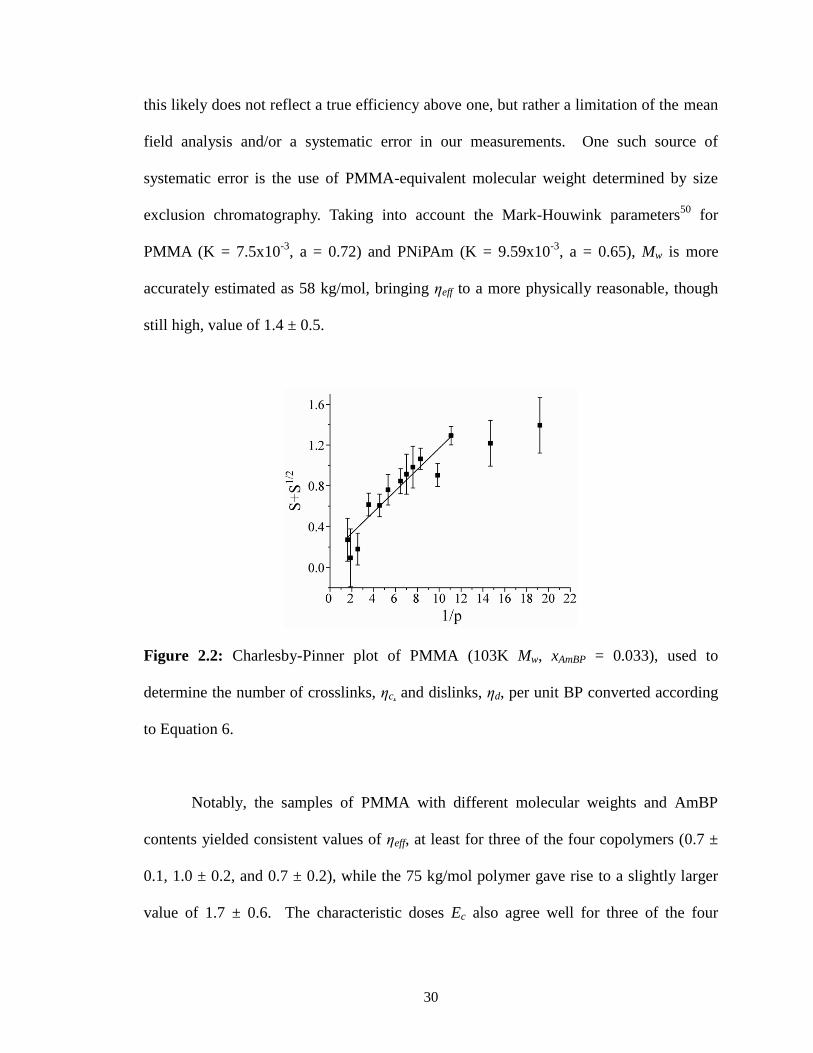
30
this likely does not reflect a true efficiency above one, but rather a limitation of the mean
field analysis and/or a systematic error in our measurements. One such source of
systematic error is the use of PMMA-equivalent molecular weight determined by size
exclusion chromatography. Taking into account the Mark-Houwink parameters50
for
PMMA (K = 7.5x10-3
, a = 0.72) and PNiPAm (K = 9.59x10-3
, a = 0.65), Mw is more
accurately estimated as 58 kg/mol, bringing ηeff to a more physically reasonable, though
still high, value of 1.4 ± 0.5.
Figure 2.2: Charlesby-Pinner plot of PMMA (103K Mw, xAmBP = 0.033), used to
determine the number of crosslinks, ηc, and dislinks, ηd, per unit BP converted according
to Equation 6.
Notably, the samples of PMMA with different molecular weights and AmBP
contents yielded consistent values of ηeff, at least for three of the four copolymers (0.7 ±
0.1, 1.0 ± 0.2, and 0.7 ± 0.2), while the 75 kg/mol polymer gave rise to a slightly larger
value of 1.7 ± 0.6. The characteristic doses Ec also agree well for three of the four

31
PMMA copolymers, though the 75 kg/mol sample shows a value roughly twice as large
as the others. We note that differences in xAmBP and “blockiness” of the random
copolymers (i.e. due to differences in the degree of conversion of monomer used during
polymerization) could influence the ability of AmBP to locally aggregate, and therefore
lead to real differences in the photo-chemical activity of AmBP in copolymers of
otherwise identical chemistry. However, the reasonably good agreement obtained for
these PMMA samples provides an indication that the trends we report here indeed arise
due to chemistry of the predominant comonomer.
Table 2.2: Results of Charlesby-Pinner Analysis; Number of crosslinks per unit AmBP
converted, ηc; Number of dislinks per crosslink, ηd/ηc (*value based on SEC analysis of
samples); Fraction of crosslinks formed by benzopinacol formation, F/2ηc.
Polymer Mw
(kg/mol) c cd cF 2
PMMA 103 0.53±0.11 0.25±0.19 0.20±0.05
11 0.62±0.12 -0.18±0.14 0.12±0.02
42 0.50±0.13 -0.04±0.14 0.16±0.04
75 1.08±0.14 0.56±0.1 0.07±0.02
PS 79 - ~1* -
PpMS 32 0.85±0.06 0.61±0.09 0.34±0.02
PNiPAm 43 1.06±0.15 0.33± 0.1 0.14±0.03
PMA 36 0.43±0.06 0.22±0.08 0.21±0.12
PiBA 19 0.37±0.07 -0.50±0.3 0.33±0.06
PiBMA 37 0.35±0.05 0.01±0.2 0.51±0.08
PMAA 117 - >1* -
The results of Charlesby-Pinner analysis, summarized in Table 2.2, provide
information on both crosslinking and dislinking reactions. Importantly, these values are
dominated by the gelation behavior at high conversion, whereas the mean-field analysis

32
considers only the gel point, thus they provide a complementary picture of the activity of
AmBP in these copolymers. The number of crosslinks per unit AmBP converted c,
excluding the values for PS and PMAA, all lie between 0.25 and 1, and are in general
agreement with the efficiencies determined by mean-field analysis. Dislinking reactions,
expressed in terms of the ratio d/c, and again excluding PS and PMAA, range from
approximately 0 to 0.6, thus they appreciably reduce the net efficiency of crosslink
formation for some copolymers. Only one sample (PiBA) yielded a value of d/c that
was negative with reasonable confidence (-0.50 ± 0.3), which is an unphysical result that
again must reflect limitations in the model and/or systematic errors. As in the mean-field
analysis, three of the four PMMA copolymer samples show very good agreement, with ηc
values of 0.5 – 0.6, and d/c close to zero. Surprisingly, however, the 75 kg/mol PMMA
sample shows larger values of ηc = 1.08 ± 0.14 and d/c = 0.56 ± 0.1. The reason for this
discrepancy is not clear, but it does indicate that sample-to-sample variations can be
significant.
As a Charlesby-Pinner analysis yields the number of crosslinks formed per AmBP
converted ηc, a direct comparison can be made with the fraction of benzophenone
converted to the dimer benzopinacol, F, defined in Equation 2. Assuming that each
benzopinacol formed yields one elastically active crosslink (i.e., ignoring loops), an
estimate for the maximum fraction of crosslinks due to benzopinacol formation is
obtained from the quantity cF 2 . The factor of two is introduced since each AmBP
converted to benzopinacol yields half of a crosslink. Results are seen in Table 2.2, and
fall between 0.07 and 0.51, indicating a significant fraction of crosslinks formed due to
AmBP-centered radical recombination in these copolymers. Interestingly, PMMA

33
copolymers exhibit a relatively low overall fraction, ranging from 0.07 to 0.20, again
with three of the four copolymers falling close to one another (0.20 ± 0.05, 0.12 ± 0.02,
and 0.16 ± 0.04). The lower value of cF 2 for 75K Mw PMMA (0.07 ± 0.02) could
potentially be associated with a lower incidence of intramolecular AmBP recombination,
again suggesting a different compositional gradient compared to the other three PMMA
copolymers. Significantly higher values of cF 2 (0.33 – 0.51) are seen for PpMS, PiBA
and PiBMA, indicating a higher fraction of crosslinks due to benzopinacol formation.
Pendent vs. Doped Benzophenone
We next performed a direct comparison between a PMMA copolymer containing
AmBP (xAmBP = 0.051, Mw = 75 kg/mol), and a similar molecular weight PMMA
homopolymer (Mw = 55 kg/mol, Ð = 1.7) doped with an equivalent molar concentration
of free benzophenone. As shown in Figure 2.3, while the doped sample does gel, it
requires a conversion of approximately 0.6 to do so, in comparison to only 0.016 ± 0.005
for the copolymer. Despite variations seen among PMMA copolymers, this result
represents a significant decrease of crosslinking efficiency in the doped system, with a
roughly 30-fold lower value of eff. While we cannot rule out the possibility that BP
aggregation reduces the measured efficiency in the doped case, the samples were
similarly transparent, at least excluding the possibility of large-scale phase separation.
This result, combined with values of cF 2 between 0.07 and 0.20 for PMMA, provides
a clear picture of the contribution of each crosslink-forming photoproduct to the overall
gelation behavior (Scheme 2.1a). In polymer doped with BP, the only route for crosslink
formation is recombination of aliphatic radicals, and thus the 30-fold difference in eff
indicates that ~ 3 % of the total crosslinks in the copolymer are formed through aliphatic-

34
aliphatic radical recombination. Considering that only 7 – 20 % of the crosslinks formed
in the copolymer are due to benzopinacol formation, recombination of BP- and aliphatic-
centered radicals is revealed to be the dominant mechanism, accounting for 77 – 90 % of
crosslinks formed in PMMA copolymers.
Figure 2.3: A comparison of the gel curves for PMMA copolymerized with AmBP (75K
Mw, xAmBP = 0.051, filled symbols), and doped with an equivalent molar concentration of
BP (55K Mw, open symbols), reveals a roughly 30-fold increase in gelation efficiency for
the copolymer.
Reactivity of Hydrogen to BP Abstraction
As previously mentioned, the selected copolymers were chosen to provide
variations in reactivity, density, and location of abstractable hydrogens, and thus of the
radicals resulting from reaction with triplet BP. Numerous studies have detailed the
sensitivity of hydrogen abstraction by BP to chemical environment, and for reference,
rate constants from several sources are reported in Table 2.3. Photoinitiation studies have

35
indicated that aliphatic hydrogens alpha to certain heteroatoms (nitrogen, sulfur, and
oxygen in particular) are especially susceptible to BP abstraction, thus the reason that
amines are often used as coinitiators with BP. This increased reactivity is generally
attributed to charge transfer interactions between the BP triplet and electrons in the
heteroatom prior to abstraction. Of these heteroatoms, nitrogen-containing materials have
been found to be the most reactive compared to materials containing oxygen or
sulfur51–54
. Aliphatic sites lacking heteroatoms at the alpha position are less susceptible to
hydrogen abstraction by BP, with reactivity decreasing as the stability of the resulting
radical decreases55,56
. As radical stability generally increases with substitution, rate
constants for hydrogen abstraction increase in the order of primary (1°), secondary
Table 2.3: Rate constants for hydrogen abstraction by benzophenone
Species k [s-1
] k [M-1
s-1
] Relative to
primary Ref
Primarya 1.70x10
3 1 57
Secondarya 6.8x10
4 40 57
Tertiarya 5.1x10
5 300 57
Secondary
cyclohexyla
6.0x104 35 57
Primary benzylica 1.3x10
5 74 57
Benzene C-Ha 2.7x10
1 0.016 58
Tertiary benzylicd 1.9-5.0x10
5 59
t-C4H9NH2b 1.1x10
8 60
sec-C4H9NH2b 3.0x10
8 60
n-(C3H7)2NHc 3.4x10
9 60
C6H12e 3.3x10
6 3.6x10
5 55
CH3OHe 3.8x10
6 1.5x10
5 55
C2H5OHe 12.0x10
6 7.0x10
5 55
(CH3)2CHOHe 23.0x10
6 1.8x10
6 55
a: 25 °C in acetonitrile, rates per R-H hydrogen bond broken; b: 25 °C in acetonitrile; c:
25 °C in benzene; d: 25 °C in acetonitrile, estimated from ratio of primary benzylic to
tertiary benzylic photoproducts; e: values originally obtained at 20 °C in pure solvent
(s-1
), divided by molarity of pure solution to obtain M-1
s-1

36
cyclohexyl, secondary (2°), primary benzylic, and tertiary (3°). Aromatic hydrogen is by
far the least reactive, with the rate of hydrogen abstraction two orders of magnitude lower
than that of 1° hydrogen57
. While we note that amides and esters may have reduced
reactivity compared to amines or alcohols, we still expect these functional groups to
enhance susceptibility to abstraction relative to aliphatic sites lacking heteroatoms at the
alpha position. Therefore, by considering the results from mean field and Charlesby-
Pinner analyses in light of the reactivity of the different hydrogen species in each
material, we can rationalize the effect of comonomer chemistry on gelation behavior.
(Meth)acrylate copolymer gelation
We first compare acrylate- and methacrylate-based copolymers, which contain a
range of hydrogens with varying reactivity and location. Poly(methyl methacrylate)
(PMMA) contains 1° and 2° hydrogens, with three 1° located alpha to oxygen, and
exhibits a characteristic dose for AmBP conversion Ec between 7.8 and 19.7 J/cm2. The
presence of 3° hydrogens as in poly(methyl acrylate) (PMA), poly(isobutyl methacrylate)
(PiBMA), or poly(isobutyl acrylate) (PiBA) yields a corresponding decrease in Ec (5.3 –
6.1 J/cm2), consistent with the greater susceptibility of 3° hydrogen species to abstraction
by BP. Despite this lower reactivity for PMMA copolymers, however, ηeff is as high or
higher for PMMA than any other acrylate-based copolymer. For PMA, which contains 3°
hydrogens on the polymer main chain, a higher ηd/ηc value is found, indicating an
increased occurrence of dislinking in PMA than the corresponding (similar N*xAmBP) 42K
PMMA. This clearly suggests that hydrogen abstraction, and thus radical placement, on
the 3° position of the polymer main chain is unfavorable for gelation of these
copolymers. The increase in dislinking is likely caused by β-scission, as previously

37
suggested by Torikai et al (Scheme 2.1b)61
. For PiBA, in contrast to PMA, a negligible
occurrence of dislinking was observed, suggesting that the additional 3° hydrogen
pendent to the main chain in PiBA greatly decreases the probability of hydrogen
abstraction from the backbone. A comparison of PiBA and PiBMA shows the values of
Ec, ηeff, ηc, and ηd/ηc to be nearly identical for these copolymers, further suggesting that the
reactivity of 3° hydrogens located pendent to the polymer chain are enhanced compared
to those on the backbone, presumably due to steric effects. Together, these results on
acrylate and methacrylate copolymers suggest that hydrogen abstraction, and thus radical
placement, pendent to the polymer backbone is conducive to gelation, since radicals
placed on the main-chain have an increased probability of undergoing dislinking.
Styrenic Copolymer Gelation
We next consider the complementary case of styrenic copolymers. Poly(styrene)
(PS) contains a majority of relatively unreactive aromatic hydrogens, with two 2° and one
3°-benzylic hydrogen on the polymer backbone. The high Ec for PS (30.1 ± 6.3 J/cm2,
nearly three times larger than any other material) despite the presence of what should be a
quite reactive 3°-benzylic position suggests a steric effect here as well. While quenching
of AmBP triplet by the styrenic polymer62
could also contribute to lower reactivity, we
note that the incorporation of a 1°-benzylic position pendent to the polymer chain as in
poly(p-methyl styrene) (PpMS) lowers Ec considerably (11.8 ± 0.7 J/cm2). Together with
the aforementioned results for PiBA and PiBMA, these findings paint a consistent picture
that steric effects lead to generally reduced reactivity of backbone hydrogens. In addition
to this reduced reactivity, PS exhibits a very low ηeff (< 0.05), not gelling under
atmospheric conditions even at full conversion. To determine whether any crosslinking or
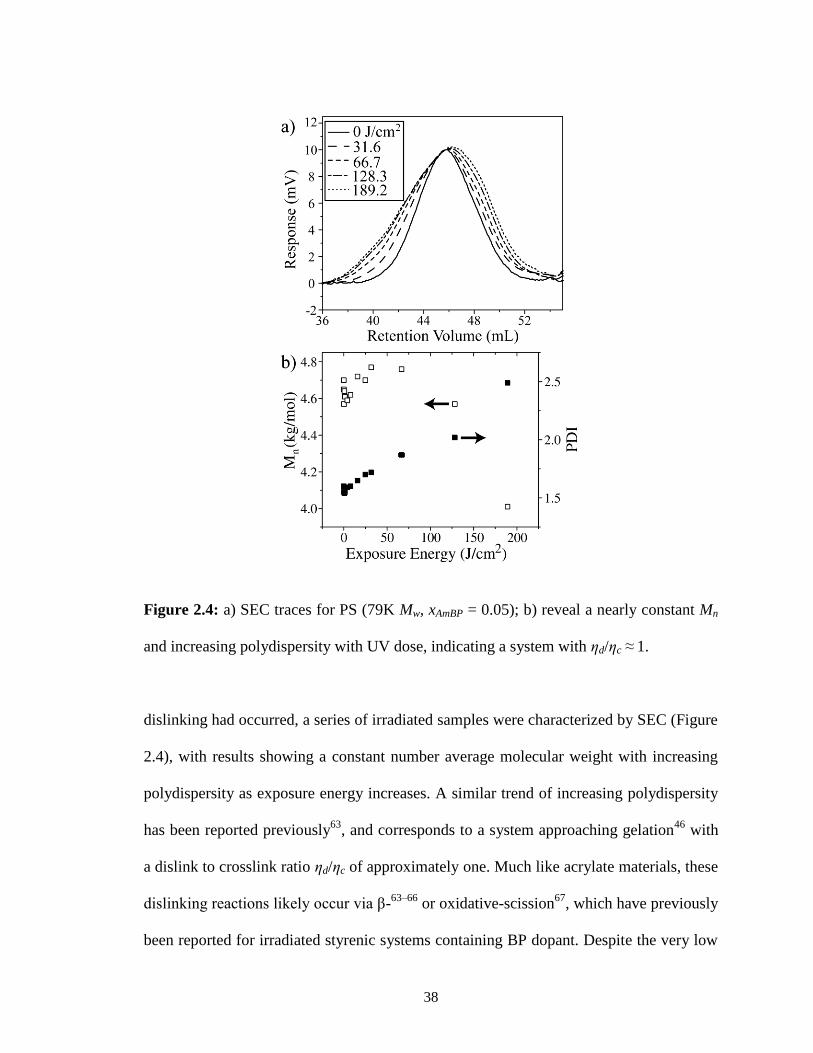
38
Figure 2.4: a) SEC traces for PS (79K Mw, xAmBP = 0.05); b) reveal a nearly constant Mn
and increasing polydispersity with UV dose, indicating a system with ηd/ηc ≈ 1.
dislinking had occurred, a series of irradiated samples were characterized by SEC (Figure
2.4), with results showing a constant number average molecular weight with increasing
polydispersity as exposure energy increases. A similar trend of increasing polydispersity
has been reported previously63
, and corresponds to a system approaching gelation46
with
a dislink to crosslink ratio ηd/ηc of approximately one. Much like acrylate materials, these
dislinking reactions likely occur via β-63–66
or oxidative-scission67
, which have previously
been reported for irradiated styrenic systems containing BP dopant. Despite the very low

39
effective crosslinking efficiency for PS, when compared to the results of a study of PS
doped with free BP65
, which yielded ηd/ηc ≈ 7.7 - 25, this provides further evidence that
polymers with covalently-attached BP provide superior crosslinking efficiency.
As PS yields a nearly equal rate of dislinking and crosslinking reactions, a second
styrenic copolymer (PpMS), was studied, which incorporates three additional 1°-benzylic
hydrogens pendent to the polymer main chain. The inclusion of these additional reactive
positions serves to increase ηeff to nearly one while decreasing ηd/ηc to 0.61 ± 0.09. This
result is similar to our findings for the acrylate copolymers, i.e. that shifting hydrogen
abstraction away from the polymer backbone to pendent 1°-benzylic hydrogens enhances
crosslinking, thus providing an effective means to gel even polymers that are susceptible
to degradation.
Gelation of poly(N-isopropyl acrylamide) and poly(methacrylic acid)
To further extend our understanding of these systems, two additional copolymers
were investigated, with each presenting a high degree of asymmetry between the lability
of hydrogens located pendent to the polymer main chain compared to those attached
directly on the backbone. The first of these materials is poly(N-isopropyl acrylamide)
(PNiPAm), which contains 3° hydrogens alpha to nitrogen and pendent to the polymer
backbone. As this position is anticipated to be highly reactive, the vast majority of
hydrogen abstraction should occur on the side chains. Consistent with this expectation, a
previous study has shown that BP-containing copolymers with pendent aliphatic amine
are extremely susceptible to photocrosslinking36
. Our results show PNiPAm to exhibit
among the lowest Ec of any material studied, confirming the high reactivity of these
hydrogens. Further, PNiPAm yields a high ηeff (1.4 ± 0.5, with the molecular weight

40
correction) and ηc (1.06 ± 0.15). While a reasonably high ηd/ηc (0.33 ± 0.1) is also
observed in this sample, these results reinforce that hydrogen abstraction pendent to the
main polymer chain is conducive to gelation.
The second material chosen is poly(methacrylic acid) (PMAA), whose most
reactive hydrogen (2°) is located on the polymer main-chain. As our copolymer systems
have, to this point, indicated an increased occurrence of dislinking with hydrogen
abstraction from the polymer backbone, this material should be inefficient in gelation.
This notion is confirmed as PMAA, much like PS, does not gel in air. SEC analysis of
this material (Figure 2.5) shows a decreasing molecular weight with increasing irradiation
energy, indicating ηd/ηc to be greater than one. While conversion of AmBP was not
monitored for this sample, the nearly constant molecular weight distributions seen for the
highest doses indicate that p in these samples has approached unity. This result further
confirms that hydrogen abstraction from the polymer backbone favors dislinking,
hindering gelation.
Figure 2.5: SEC traces for PMAA (117K Mw, xAmBP = 0.030), show a decrease in Mn with
UV dose, indicating an overall ηd/ηc > 1.

41
Effect of oxygen on copolymer gelation
Since an attractive feature of BP is that it retains high photochemical activity even
in air, an open atmosphere was used to this point in the study. However, to determine
whether oxygen does affect gelation of these copolymers, several samples were studied
under a nitrogen atmosphere (Figure 2.6, Table 2.4). With the exception of PS, only
minor variations in gelation behavior are observed, with PMMA, PNiPAm, and PiBMA
exhibiting ηeff values nearly identical to atmospheric conditions, in agreement with prior
results for dimethylaminostyrene-containing terpolymers68
. Small changes to the gel
curves for the latter three polymers indicate only a slight susceptibility of these materials
Figure 2.6: Gel curves in both air and nitrogen for select copolymers reveal little
dependence on atmosphere, with the exception of PS which exhibits a slightly higher ηeff
under nitrogen; a) PMMA, 42K Mw, xAmBP = 0.048; b) PS, 79K Mw, xAmBP = 0.05; c)
PNiPAm, 43K Mw, xAmBP = 0.078; d) PiBMA, 37K Mw, xAmBP = 0.055.

42
to oxygen quenching of triplet AmBP and oxidative scission. We note that the
characteristic time for oxygen diffusion69
across a 10 μm PMMA film at 35 °C is roughly
30 s, whereas our experimental Ec values indicate deactivation of AmBP on a time scale
about 80 times longer. Therefore, we conclude that insensitivity to oxygen in these solid-
state copolymers is not simply a matter of mass transport limitations, in agreement with
previous reports37,40
. Conversely, PS shows an increase in gelation efficiency from < 0.05
in air to 0.08 ± 0.02 under nitrogen, showing that oxygen plays a slight role in preventing
crosslinking in this case. These results agree with those for BP-doped polymer films,
wherein increased peroxy radical concentration in PS causes an increase in chain
scission67,70
. As PS in an open atmosphere yields a dislink to crosslink ratio ηd/ηc of
roughly one, preventing oxidative scission allows the material to gel, albeit with very
poor efficiency.
2.5 Conclusions
Copolymers containing pendent BP have been prepared, and their gelation
behavior examined via UV/Vis and sol-gel measurements. Compared to doping with BP,
covalent attachment of BP photo-crosslinker provides two additional routes by which
radical recombination can result in crosslinking, thus greatly increasing gelation
efficiency. The location of highly abstractable hydrogens is found to be the dominant
factor, with hydrogen abstraction on the main polymer chain tending to favor dislinking,
while abstraction from side chains promotes gelation. Finally, oxygen was found to exert
little influence on gelation of most copolymers, with the exception of PS, which exhibits
a slightly higher efficiency under a nitrogen atmosphere. The insights into crosslinking

43
efficiency provided by this study should help to guide the choice of appropriate
copolymer chemistries for fabrication of robust photo-crosslinkable materials.
Acknowledgements
We are grateful to Jungwook Kim for synthesis of the PNiPAm copolymer. This
work was funded by the US Army Research Office through grant W911NF-11-1-0080
and the Defense Threat Reduction Agency through grant HDTRA1-10-1-0099, and made
use of facilities supported by the National Science Foundation MRSEC on Polymers at
UMass (DMR-0820506).
2.6 References
1. Oster, G., Oster, G. K. & Moroson, H. Ultraviolet Induced Crosslinking and
Grafting of Solid High Polymers. J. Polym. Sci. 34, 671–684 (1959).
2. Qu, B. J. & Ranby, B. Photocross-linking of Low-Density Polyethylene. 1.
Kinetics and Reaction Parameters. J. Appl. Polym. Sci. 48, 701–709 (1993).
3. Qu, B. J. Reaction mechanism of benzophenone-photoinitiated crosslinking of
polyethylene. Chin. J. Polym. Sci. 20, 291–307 (2002).
4. Wu, Q. H. & Qu, B. J. Synthesis of Di(4-hydroxyl benzophenone) sebacate and its
usage as initiator in the photocrosslinking of polyethylene. J. Appl. Polym. Sci. 85,
1581–1586 (2002).
5. Wu, Q. & Qu, B. Photoinitiating characteristics of benzophenone derivatives as
new initiators in the photocrosslinking of polyethylene. Polym. Eng. Sci. 41, 1220–
1226 (2001).
6. Doytcheva, M. et al. Ultraviolet-induced crosslinking of solid poly(ethylene
oxide). J. Appl. Polym. Sci. 64, 2299–2307 (1997).
7. Wu, S. K. & Rabek, J. F. Benzophenone-Initiated Photo-Crosslinking of
Polynorbornene in the Solid-State. Polym. Degrad. Stab. 21, 365–376 (1988).

44
8. Graves, R. & Pintauro, P. N. Polyphosphazene membranes. II. Solid-state
photocrosslinking of poly[(alkylphenoxy)(phenoxy)phosphazene] films. J. Appl.
Polym. Sci. 68, 827–836 (1998).
9. Tocker, S. Cross-linked blended polymers containing polymeric acryloxy
benzophenones. US Patent 3,265,772 1–5 (1966).
10. Zhao, Y. & Dan, Y. Synthesis and characterization of a polymerizable
benzophenone derivative and its application in styrenic polymers as UV-stabilizer.
Eur. Polym. J. 43, 4541–4551 (2007).
11. Tocker, S. Organic polymeric articles and preparation thereof from derivatives of
ethylene and unsaturated benzophenones. US Patent 3,214,492 1–8 (1965).
12. Tocker, S. Graft and cross linked copolymers of polar vinylidene monomers with
acryloxyphenones. US Patent 3,315,013 1–7 (1967).
13. Agolini, F. Polymeric phenone photosensitizers and blends thereof with other
polymers. US Patent 3,632,493 1–4 (1972).
14. Rehmer, G., Boettcher, A. & Portugall, M. Benzophenone derivatives and their
preparation. US Patent 5,264,533 1–7 (1993).
15. Rehmer, G., Boettcher, A. & Portugall, M. (Meth)acrylate copolymer based UV-
crosslinkable materials. US Patent 5,389,699 1–7 (1995).
16. Prucker, O., Naumann, C. A., Rühe, J., Knoll, W. & Frank, C. W. Photochemical
Attachment of Polymer Films to Solid Surfaces via Monolayers of Benzophenone
Derivatives. J. Am. Chem. Soc. 121, 8766–8770 (1999).
17. Naumann, C. A. et al. The Polymer-Supported Phospholipid Bilayer: Tethering as
a New Approach to Substrate−Membrane Stabilization. Biomacromolecules 3, 27–
35 (2002).
18. Toomey, R., Freidank, D. & Rühe, J. Swelling Behavior of Thin, Surface-Attached
Polymer Networks. Macromolecules 37, 882–887 (2004).
19. Beines, P. W., Klosterkamp, I., Menges, B., Jonas, U. & Knoll, W. Responsive
thin hydrogel layers from photo-cross-linkable poly(N-isopropylacrylamide)
terpolymers. Langmuir 23, 2231–2238 (2007).
20. Belardi, J., Schorr, N., Prucker, O. & Rühe, J. Artificial Cilia: Generation of
Magnetic Actuators in Microfluidic Systems. Adv. Funct. Mater. 21, 3314–3320
(2011).

45
21. Castellanos, A. et al. Size-Exclusion “Capture and Release” Separations Using
Surface-Patterned Poly(N-isopropylacrylamide) Hydrogels. Langmuir 23, 6391–
6395 (2007).
22. Mateescu, A., Wang, Y., Dostalek, J. & Jonas, U. Thin Hydrogel Films for Optical
Biosensor Applications. Membranes 2, 40–69 (2012).
23. Wang, Y., Brunsen, A., Jonas, U., Dostálek, J. & Knoll, W. Prostate specific
antigen biosensor based on long range surface plasmon-enhanced fluorescence
spectroscopy and dextran hydrogel binding matrix. Anal. Chem. 81, 9625–9632
(2009).
24. Schwartz, V. B. et al. Antibacterial Surface Coatings from Zinc Oxide
Nanoparticles Embedded in Poly(N-isopropylacrylamide) Hydrogel Surface
Layers. Adv. Funct. Mater. 22, 2376–2386 (2012).
25. Kim, J., Hanna, J. A., Byun, M., Santangelo, C. D. & Hayward, R. C. Designing
Responsive Buckled Surfaces by Halftone Gel Lithography. Science 335, 1201–
1205 (2012).
26. Kim, J., Hanna, J. A., Hayward, R. C. & Santangelo, C. D. Thermally responsive
rolling of thin gel strips with discrete variations in swelling. Soft Matter 8, 2375–
2381 (2012).
27. McCartin, P. & Nebe, W. Photoactive plastisols: positive washout image. J.
Imaging Sci. 32, 222–225 (1988).
28. Scherzer, T., Tauber, A. & Mehnert, R. UV curing of pressure sensitive adhesives
studied by real-time FTIR-ATR spectroscopy. Vib. Spectrosc. 29, 125–131 (2002).
29. Do, H.-S., Park, Y.-J. & Kim, H.-J. Preparation and adhesion performance of UV-
crosslinkable acrylic pressure sensitive adhesives. J. Adhes. Sci. Technol. 20,
1529–1545 (2006).
30. Mönch, W., Dehnert, J., Prucker, O., Rühe, J. & Zappe, H. Tunable Bragg filters
based on polymer swelling. Appl. Opt. 45, 4284–4290 (2006).
31. Jiang, X., Luo, X. & Yin, J. Polymeric photoinitiators containing in-chain
benzophenone and coinitiators amine: Effect of the structure of coinitiator amine
on photopolymerization. J. Photochem. Photobiol., A 174, 165–170 (2005).
32. Wang, J. Z., Nayak, B. R., Creed, D., Hoyle, C. E. & Mathias, L. J.
Photocrosslinking of poly(ethylene terephthalate) copolymers containing
photoreactive comonomers. Polymer 46, 6897–6909 (2005).

46
33. Suyama, K., Miyamoto, Y., Matsuoka, T., Wada, S. & Tsunooka, M. Photo-
initiated thermal crosslinking of copolymers bearing pendant base generating
groups. Polym. Adv. Technol. 11, 589–596 (2000).
34. Wu, G. et al. UV exposure effects on photoinitiator-grafted styrene-butadiene-
styrene triblock copolymer. J. Appl. Polym. Sci. 120, 2627–2631 (2011).
35. Carlini, C. & Gurzoni, F. Optically active polymers containing side-chain
benzophenone chromophores. Polymer 24, 101–106 (1983).
36. Tsubakiyama, K., Kuzuba, M., Yoshimura, K., Yamamoto, M. & Nishijima, Y.
Photochemical-Reaction of Polymers Having Arylketone and Tertiary Amine as
Pendant Groups. Polym. J. (Tokyo, Jpn.) 23, 781–788 (1991).
37. Higuchi, H., Yamashita, T., Horie, K. & Mita, I. Photo-Cross-Linking Reaction of
Benzophenone-Containing Polyimide and Its Model Compounds. Chem. Mater. 3,
188–194 (1991).
38. Sumitomo, H., Nobutoki, K. & Susaki, K. Photo-Graft Polymerization of Methyl
Methacrylate onto Styrene-Vinylbenzophenone Copolymer. J. Polym. Sci., Part A-
1: Polym. Chem. 9, 809–812 (1971).
39. Ikeda, R. M., Rogers, F. F., Tocker, S. & Gelin, B. R. Photocrosslinking of
polyethylene using polyethylene-acryloxybenzophenone - a physical study. ACS
Symp. Ser. 76–89 (1976).
40. Lin, A. A., Sastri, V. R., Tesoro, G., Reiser, A. & Eachus, R. On the Cross-Linking
Mechanism of Benzophenone-Containing Polyimides. Macromolecules 21, 1165–
1169 (1988).
41. Jia, J., Sarker, M., Steinmetz, M. G., Shukla, R. & Rathore, R. Photochemical
Elimination of Leaving Groups from Zwitterionic Intermediates Generated via
Electrocyclic Ring Closure of alpha, beta-Unsaturated Anilides. J. Org. Chem. 73,
8867–8879 (2008).
42. Christensen, S. K., Chiappelli, M. C. & Hayward, R. C. Gelation of Copolymers
with Pendent Benzophenone Photo-Cross-Linkers. Macromolecules 45, 5237–
5246 (2012).
43. Charlesby, A. Gel formation and molecular weight distribution in long-chain
polymers. Proc. R. Soc. London, Ser. A 222, 542–557 (1954).
44. Flory, P. J. Molecular size distribution in three dimensional polymers. II.
Trifunctional branching units. J. Am. Chem. Soc. 63, 3091–3096 (1941).

47
45. Flory, P. J. Constitution of Three-dimensional Polymers and the Theory of
Gelation. J. Phys. Chem. 46, 132–140 (1942).
46. Rubinstein, M. & Colby, R. H. Polymer Physics. (Oxford University Press, Inc:
New York, 2003).
47. Stockmayer, W. H. Theory of molecular size distribution and gel formation in
branched polymers II. General cross linking. J. Chem. Phys. 12, 125–131 (1944).
48. Stockmayer, W. H. Theory of Molecular Size Distribution and Gel Formation in
Branched-Chain Polymers. J. Chem. Phys. 11, 45–55 (1943).
49. Charlesby, A. & Pinner, S. H. Analysis of the Solubility Behaviour of Irradiated
Polyethylene and Other Polymers. Proc. R. Soc. London, Ser. A 249, 367–386
(1959).
50. Kurata, M. & Tsunashima, Y. Viscosity-Molecular Weight Relationships and
Unperturbed Dimensions of Linear Chain Molecules. Polymer Handbook VII/1–
VII/83 (1999).
51. Cohen, S. G., Parola, A. & Parsons, G. H. Photoreduction by Amines. Chem. Rev.
73, 141–161 (1973).
52. Cohen, S. G. & Cohen, J. I. Photoreduction of aminobenzophenones in nonpolar
media. Effects of tertiary amines. J. Phys. Chem. 72, 3782–3793 (1968).
53. Cohen, S. G. & Chao, H. M. Photoreduction of aromatic ketones by amines.
Studies of quantum yields and mechanism. J. Am. Chem. Soc. 90, 165–173 (1968).
54. Griller, D., Howard, J. A., Marriott, P. R. & Scaiano, J. C. Absolute rate constants
for the reactions of tert-butoxyl, tert-butylperoxyl, and benzophenone triplet with
amines: the importance of a stereoelectronic effect. J. Am. Chem. Soc. 103, 619–
623 (1981).
55. Topp, M. R. Activation-controlled hydrogen abstraction by benzophenone triplet.
Chem. Phys. Lett. 32, 144–149 (1975).
56. Wagner, P. & Park, B. Photoinduced hydrogen atom abstraction by carbonyl
compounds. Org. Photochem. 11, 227–366 (1991).
57. Giering, L., Berger, M. & Steel, C. Rate Studies of Aromatic Triplet Carbonyls
with Hydrocarbons. J. Am. Chem. Soc. 96, 953–958 (1974).
58. Dedinas, J. Photoreduction of benzophenone in benzene. I. Mechanism of
secondary reactions. J. Phys. Chem. 75, 181–186 (1971).

48
59. Wagner, P. J., Truman, R. J., Puchalski, A. E. & Wake, R. Extent of charge
transfer in the photoreduction of phenyl ketones by alkylbenzenes. J. Am. Chem.
Soc. 108, 7727–7738 (1986).
60. Inbar, S., Linschitz, H. & Cohen, S. G. Nanosecond Flash Studies of Reduction of
Benzophenone by Aliphatic Amines. Quantum Yields and Kinetic Isotope Effects.
J. Am. Chem. Soc. 103, 1048–1054 (1981).
61. Torikai, A., Sekigawa, Y. & Fueki, K. Photo-degradation of blends of polystyrene
and poly (methyl methacrylate). Polym. Degrad. Stab. 21, 43–54 (1988).
62. Horie, K., Tsukamoto, M., Morishita, K. & Mita, I. Photochemistry in Polymer
Solids. 5. Decay of Benzophenone Phosphorescence in Polystyrene and in
Polycarbonate. Polym. J. (Tokyo, Jpn.) 17, 517–524 (1985).
63. Mita, I., Hisano, T., Horie, K. & Okamoto, A. Photoinitiated Thermal-Degradation
of Polymers. 1. Elementary Processes of Degradation of Polystyrene.
Macromolecules 21, 3003–3010 (1988).
64. Ikeda, T., Kawaguchi, K., Yamaoka, H. & Okamura, S. Photoinduced and
Radiation-Induced Degradation of Vinyl-Polymers in Solution. 2. Photosensitized
Degradation of Poly-Alpha-Methylstyrene by Benzophenone. Macromolecules 11,
735–739 (1978).
65. Mita, I., Takagi, T., Horie, K. & Shindo, Y. Photosensitized Degradation of
Polystyrene by Benzophenone in Benzene Solution. Macromolecules 17, 2256–
2260 (1984).
66. Kaczmarek, H., Kaminska, A., Swiatek, M. & Sanyal, S. Photoinitiated
degradation of polystyrene in the presence of low-molecular organic compounds.
Eur. Polym. J. 36, 1167–1173 (2000).
67. Torikai, A., Takeuchi, T. & Fueki, K. Photodegradation of Polystyrene and
Polystyrene Containing Benzophenone. Polym. Photochem. 3, 307–320 (1983).
68. Smets, G. J., Elhamouly, S. N. & Oh, T. J. Photochemical-Reactions on Polymers.
Pure Appl. Chem. 56, 439–446 (1984).
69. Kaptan, H. Y. ESR study on the effect of temperature on the diffusion of oxygen
into PMMA and PVAC polymers. J. Appl. Polym. Sci. 71, 1203–1207 (1999).
70. Lin, C. S., Liu, W. L., Chiu, Y. S. & Ho, S. Y. Benzophenone-Sensitized
Photodegradation of Polystyrene Films under Atmospheric Conditions. Polym.
Degrad. Stab. 38, 125–130 (1992).

49
CHAPTER 3
ROUTES TOWARD NANOSTRUCTURED POLYMER BLENDS BY
BENZOPHENONE PHOTO-REACTION
3.1 Abstract
Photo-reaction of copolymers containing pendent benzophenones (BP) was
utilized in two approaches in attempts to prepare highly interconnected three-dimensional
nanostructured polymer blends. Arresting of spinodal decomposition by crosslinking was
first investigated by UV exposure in a poly(styrene-AmBP)/poly(vinyl methyl ether)
critical blend prior to phase separation. Photo-crosslinking elevated the cloud point of
blends due to A-B reaction which constrains phase separation. Morphological
characterization of this system was impossible, however, as the electron density contrast
between the two polymers is insufficient for SAXS. In the second technique we prepared
nanostructured polymer blends by UV-initiated solution-state photo-grafting of
poly(styrene-AmBP) to PEG-containing homopolymer. Nanoscale features were
generated upon UV exposure as evidenced by SAXS, yet clear signatures of bicontinuous
morphologies remain elusive. Refinement of each method is discussed in an effort to
provide successful realization of photochemically prepared bicontinuous polymer blends
in the future.

50
3.2 Motivation
Nanostructured polymer materials have garnered immense interest from
technological perspectives due to their potential to enhance material properties. For
instance, polymer blends with nanoscale domains have been shown to improve
thermomechanical properties, optical transparency, and toughness compared to
conventional blends1, often attributed to the high surface area of such nanoscale features
2.
This improvement of properties represents an opportunity to vastly increase performance
in a wide range of applications, from proton exchange3 and lithium ion
4 membranes to
peptide biosensors5. In fact, proton exchange membranes (PEMs) with nanoscale
domains have previously been found to exhibit conductivity orders of magnitude higher
than their homogeneous counterparts due to the connectivity of ionic domains promoted
by nanoscale phase separation6,7
. Furthermore, nanometer domain size in PEMs increases
selectivity and can even promote retention of water in hydrated films due to capillary
forces8, further improving function.
While nanoscale domains are important, equally important is how these domains
are arranged in space. For many applications (i.e. sound damping, gas transport, and
impact resistant materials) it is desirable for blends to demonstrate properties of both
components in a single volume, and as such a bicontinuous morphology is preferred. This
morphology is composed of two chemically distinct material networks that percolate in
three dimensions with high connectivity. Because each material occupies the entire three-
dimensional space, each component gives its maximum contribution to the blend,
improving physical properties (modulus, strain at break, toughness, and impact
strength)9,10
. Furthermore, because of the high interconnectivity of each domain, these

51
materials have found use in ion transport3,6,11–13
, charge dissipation10,14
, photovoltaic
technology15
, and templates for porous membranes which can be used for filtration or
controlled drug release16,17
.
Current routes toward generation of bicontinuous morphologies generally rely on
kinetic trapping during melt mixing or spinodal decomposition, or the use of block
copolymers18
. Spinodal decomposition and block copolymers are discussed in Sections
3.3 and 3.4, respectively. By incorporating compatibilizer into a polymer melt, it is
possible to achieve kinetically trapped cocontinuous morphologies, yet these features are
micron scale and will only ripen with time2,9,14,19
. In this chapter we discuss new
photochemical methods for the preparation of nanoscale bicontinuous morphologies
using copolymers with pendent benzophenones. We believe that these materials offer
several advantages to traditional approaches for the creation of three-dimensional
interconnected domains, and we explore two potential methods toward their creation.
First we describe attempts to arrest the phase separation of a lower critical polymer blend
by photo-crosslinking, and in Section 3.4 we discuss attempts to create nanostructures by
UV-initiated solution-state photo-grafting. While these approaches garner limited
success, significant refinement of the materials systems and processes would be
necessary to achieve the desired bicontinuous morphologies.
3.3 Arresting phase separation of LCST blends by BP photo-crosslinking
Polymer blends are known to develop nanoscale bicontinuous morphologies
during phase separation20,21
, and as such represent one potential route toward
cocontinuity for copolymers containing pendent benzophenones. For phase separation to

52
occur, polymers must first be miscible, meaning that at some condition there must exist a
negative free energy of mixing, mixG , defined as
mixmixmix STHG (3.1)
where mixH and mixS are the enthalpy and entropy of mixing, respectively. Adjusting
the free energy equation to split the combinatorial ( combS ) and noncombinatorial
entropies of mixing allows us to rewrite Equation 3.1 as
comb
E
mixmix STGG (3.2)
where E
mixG represents the excess free energy of mixing, or a combination of the
noncombinatorial entropy and enthalpy of mixing. This excess free energy of mixing
E
mixG encompasses three distinct effects: (1) interactions between the two polymers
( 0 mixH favors miscibility), (2) free volume effects caused by differences in thermal
expansion coefficient (a difference of >4% between the two polymers is generally
sufficient to yield phase separation22,23
), and (3) size effects from differences in
segmental size (usually minimal in polymers, favors phase separation)24
. Because combS
is small due to the long chain length of polymers, miscibility in a polymer blend requires
some favorable interaction between the two component materials. Therefore within some
temperature range the favorable interactions between component polymers cause
diminished free volume effects and enhanced size effects, rendering the blend
homogeneous22
. These favorable interactions and size effects generally diminish with
temperature while free volume effects are enhanced, and thus the blend will phase
separate above a given temperature, yielding a lower critical solution temperature (LCST)
blend. If upon cooling the size effects overcome the interaction term phase separation

53
may also occur, resulting in an upper critical solution temperature (UCST) blend24
. While
both phenomenon are thermodynamically possible for any polymer blend with favorable
segmental interactions, crystallization or glass transition temperatures often occur above
the UCST (see e.g. Briber25
or Isayeva26
), and consequently LCST blends are much more
common than UCST blends. There have been numerous experimentally realized LCST
blends27–42
, UCST blends38,43–51
, and even some systems exhibiting both UCST and
LCST behavior52–56
.
Quenching of these critical blends into the two-phase range induces phase
separation of the component polymers, which occurs by one of two mechanisms:
nucleation and growth or spinodal decomposition (Figure 3.1). If the blend is quenched
from a homogeneous to a metastable region, nucleation and growth will occur, whereby
isolated particles of equilibrium composition appear and grow, forming droplets of one
polymer in a matrix of the other57
. If instead the miscible blend crosses into the unstable
region, spinodal decomposition will occur whereby concentration fluctuations initially
vary with time, maintaining a constant size while increasing in amplitude58,59
. This
spinodal morphology is characterized by an almost uniform, yet random and highly
interconnected cocontinuous structure36
. At later stages of spinodal decomposition, both
amplitude and wavelength of this cocontinuous morphology increase with time until
finally interconnectivity is lost57
. Thus while it is possible to achieve a bicontinuous
morphology via spinodal decomposition of critical blends, in the absence of additional
stimuli the architectures will ripen to macroscopic length scales, resulting in fully phase
separated morphologies.

54
Figure 3.1: Phase separation mechanisms of a binary polymer mixture. Nucleation and
growth (top) occurs when the homogeneous blend is quenched to a metastable region of
the phase diagram. Isolated particles of equilibrium composition form and grow, forming
a droplet in matrix morphology. Spinodal decomposition (bottom) occurs when the
homogeneous mixture is quenched to a thermodynamically unstable state. Concentration
fluctuations vary with time, initially maintaining constant size while increasing in
amplitude. At later stages, both amplitude and wavelength increase. Figure from Bates, F.
S. Polymer-Polymer Phase-Behavior. Science 251, 898–905 (1991). Reprinted with
permission from AAAS.21
As a consequence of macrophase separation in these materials, many studies have
attempted to kinetically trap the bicontinuous spinodal morphology. The incorporation of
particles into polymer systems can effectively stem the phase separation process, as
evidenced by Fillipone60,61
, Li62
, and Gubbels63,64
, wherein nanoparticles preferentially
occupy one material and prevent the phases from coalescing. “Bijels” have also been
experimentally realized, wherein microparticles exist at the interface between two phases,

55
thus stabilizing the bicontinuous morphology, albeit on the micron scale65,66
.
Nanoparticle-loaded blends, however, require specific interactions be imparted into the
nanoparticles, and suffer limitations due to difficulty of achieving an initially
homogeneous distribution of particles, and the changing properties of said materials
through the incorporation of a high filler volume fraction.
A second method of trapping these structures involves the photo-crosslinking of
blends prior to phase separation. First theoretically described by deGennes in 1979 and
expanded upon by Briber in 1988, it was believed that by crosslinking the polymers,
concentration fluctuations can be constrained, thus limiting the wavelength of phase
separation to a finite size67–70
. Briber extended this theory to a poly(styrene)/poly(vinyl
methyl ether) (PS/PVME) blend, which upon exposure to gamma radiation increased the
single phase regime and exhibited scattering by features on a 60 nm scale upon phase
separation67
, in qualitative agreement with the theory. In years since, a number of studies
by Tran–Cong Miyata have investigated blends consisting of anthracene-functionalized-
PS and PVME, which upon UV exposure will undergo anthracene dimerization to yield a
crosslinked PS phase 20,71–80
. While in some cases this system has resulted in crosslinked
spinodal morphologies20,71,72,77
, the A-A self-crosslinking inherent to this photochemistry
consistently shifts the LCST phase boundary down due to increased PS molecular weight,
decreasing the window of miscibility and often inducing phase separation without any
thermal treatment20,73,75,76,78,80–83
. The use of A-A (anthracene) crosslinking in these
studies has therefore limited the usable range of the LCST blend, while crosslinking in
gamma irradiated blends is poorly defined and structure in these materials has never been
studied in detail.

56
Copolymers functionalized with benzophenone thus represent a versatile system
for the study of photo-crosslinkable critical polymer blends, and in this section we
investigate the possibility of achieving a bicontinuous morphology by photo-crosslinking
of a benzophenone-containing polystyrene (PS-BP)/PVME LCST blend prior to phase
separation. Benzophenone offers the capability of tuning crosslinking in the blend to be
preferentially A-A, B-B, or A-B depending on the asymmetry of hydrogen abstraction in
the chosen polymers and the material in which the photo-crosslinker resides. As
PS/PVME undergoes a well-described phase separation at a LCST falling above the glass
transition and below the decomposition temperature, this is the blend system of
choice84,85
. To promote A-B crosslinking, acrylamidobenzophenone (AmBP) will be
copolymerized into PS thus expanding the miscibility window and allowing phase
separation to be better controlled. Phase behavior of these blends is investigated by a
combination of scattering techniques, and the effect of UV irradiation is described.
Morphological characterization is incomplete, however, as this particular blend system
does not yield strong enough X-ray scattering for our purposes.
3.3.1 Experimental
Materials and Sample Preparation
Poly(styrene) (PS; Mn = 16,000 g/mol) and poly(styrene-AmBP) copolymer (PS-
BP; Mn = 55,000 g/mol, xAmBP = 0.05) were synthesized as in Chapter 2, while poly(vinyl
methyl ether) (PVME) with a molecular weight of 58,000 g/mol was purchased directly
from Polysciences, Inc. Water was removed from PVME by rotary evaporation and the
dried polymers were subsequently dissolved in toluene. Blend solutions were prepared at

57
20 mg/mL and filtered through a 0.45 μm PTFE syringe filter prior to drop casting onto
untreated glass cover slips. After evaporation at ambient conditions, the solid polymer
films were annealed under vacuum at 100 °C for at least 10 hours and cooled slowly,
forming clear blend films with thicknesses of 5 – 10 μm. UV exposures were performed
in the same way as in Chapter 2.
Small Angle Light Scattering
Small angle light scattering (SALS) was performed on an in-house setup
operating with a 632 nm HeNe laser. The beam passes through two pinholes prior to a hot
stage containing the sample of interest. Behind the sample plane is a 2-dimensional image
plate coupled to a CCD camera and a photodiode which captures the intensity of the
direct beam (turbidity measurement). Samples were placed in the hot stage at room
temperature and heated through the cloud point under a constant nitrogen purge, as
shown in Figure 3.2a. At low temperatures the sample is transparent, causing minimal
scattering of the incident beam and thus a high transmitted intensity. At high
temperatures the sample phase separates, becoming opaque and scattering the direct beam
away from the photodiode, causing a substantial decrease in transmission. The intercept
of the initial slope and the slope after phase separation defines the cloud point in these
experiments. It is important to note that the measured cloud point will depend on the
heating rate, and thus for each experiment several heating rates were utilized, with a
linear fit extrapolated to 0 °K/min to yield the reported cloud point for each blend
composition or exposure time (Figure 3.2b).

58
Small Angle X-Ray Scattering
X-Ray scattering was done using an in-house setup from Molecular Metrology Inc.
(presently sold as Rigaku S-Max3000). It uses a 30 W microsource (Bede) with a 30 x 30
μm2 spot size matched to a Maxflux® optical system (Osmic) leading to a low-
divergence beam of monochromatic CuKα radiation (wavelength λ = 0.1542 nm). After
passing beam defining and guard pinholes, the beam of about 0.4 mm diameter enters the
sample chamber. The whole system is evacuated, and the actual scattering angles are
calibrated using the accurately known reflections from silver behenate.
Figure 3.2: Sample turbidity data obtained from small angle light scattering. a)
Transmitted intensity versus temperature for a 25/75 PS-BP/PVME sample exposed to
365 nm UV irradiation 30 s prior to heating at 5 °C/min through the cloud point. Inset
images show light scattered prior to and after clouding occurs. b) Cloud points
determined for different heating rates, showing the rate dependence on observed phase
separation. Reported cloud points have been extrapolated to a heating rate of 0 °C/min.

59
3.3.2 Results and Discussion
Blend films were prepared as described and subjected to SALS experiments at
several heating rates (5, 10, and 30 °C/min) to determine cloud points extrapolated to 0
°K/min. Blends ranging from a polystyrene mass fraction (ΦPS) of 0.1 to 0.9 were first
investigated to define the phase behavior of our materials. As shown in Figure 3.3 (black
squares), a blend of poly(styrene) homopolymer with PVME yields a LCST blend with a
critical composition between ΦPS = 0.3 – 0.5 at 155 °C. This temperature is higher than
Figure 3.3: LCST Cloud points measured by SALS for PS/PVME58K Mn (black squares )
and PS-BP (55K Mn, xAmBP = 0.05)/PVME58K (red circles ) showing little change in
phase behavior upon incorporation of benzophenone monomer. Inset is a representative
bright field optical microscope image of a 1/1 PS-BP/PVME blend after heating from 120
– 200 °C at 3 °K/min. Phase separation is observed.
most reported in the literature, but it must be noted that the molecular weights
investigated in this study were significantly lower than in previous

60
publications20,38,68,74,80,82
. Replacing the poly(styrene) with a corresponding PS-BP is
shown to have little effect upon phase behavior, indicating that incorporation of
benzophenone in the poly(styrene) phase preserves the LCST nature of the blend (Figure
3.3, red circles). A slight lowering (Tc ≈ 150 °C) and shift (ΦPS-BP = 0.3 – 0.4) of the
critical point toward low PS-BP compositions is observed, however, and likely reflects an
increase in molecular weight of the PS-BP relative to neat poly(styrene). This molecular
weight change decreases the entropy of mixing and therefore results in a less miscible
system. The inset in Figure 3.3 shows a bright field optical microscope image of a
symmetric PS-BP/PVME blend film which has been heated to 200 °C at 3 °K/min. It is
clear from this image that phase separation has occurred in the sample, which exhibits
features characteristic of a nucleation and growth mechanism, which is to be expected at
an off-critical ΦPS-BP = 0.5.
After determining the phase diagram of the PS-BP/PVME blend system, blends
located near the critical point (ΦPS-BP = 0.3) were exposed to 365 nm UV irradiation for
various times to determine its effect on the cloud point of the system. As seen in Figure
3.4a, UV exposure up to 90 s raises the cloud point by 70 °C, after which point the data
varies significantly up to 240 s. Samples irradiated up to 90 s are consistent with previous
theoretical and experimental work, with the finding that the inverse critical temperature is
proportional to radiation dose (Figure 3.4b)67,70
, however at longer exposure times
clouding behavior is either absent or poorly defined. As the majority of crosslinking in
this system should occur between PS-BP and PVME (A-B crosslinking due to the high
reactivity of the methyl hydrogen alpha to the ether of PVME, which should minimize
chain scission in this polymer system by promoting abstraction on the side groups), the

61
size of the one-phase envelope is greater due to constraints on phase separation of the two
chemically unique polymers67,68
. This constraint causes the unpredictable clouding
behavior observed above 90 s, as the samples have gelled and thus cannot phase separate
on the length scale observed here (samples exposed for >300 s never yield a measurable
cloud point). Below 30 s, the deviation from linearity is likely due to breakdown of the
theoretical treatment afforded by deGennes near the sol-gel transition70
. Phase behavior
of samples irradiated between 15 and 90 s are strikingly similar to those seen by Briber
and Bauer, indicating a strong likelihood that these PS-BP/PVME blends will exhibit
phase separated morphologies with a characteristic length scale67
.
Figure 3.4: Cloud points measured by SALS for a 30/70 PS-BP (55K, xAmBP =
0.05)/PVME (58K Mn) blend exposed to 365 nm UV irradiation prior to heating. a)
Cloud points increase with increasing exposure energy until 90 s, after which point the
clouding behavior becomes difficult to discern. b) Data plotted with inverse cloud point
on the y-axis, showing a dependence on radiation similar to that shown by Bauer and
Briber67
.

62
In an effort to quantify any nanoscale morphology present in these materials,
ΦPS-BP = 0.5 blends were cast onto Kapton 300HN polyimide films and subjected to
small-angle X-ray scattering. Selected results are shown in Figure 3.5 for samples
irradiated for several exposure times, and unfortunately it is impossible to define any
scattering features from these results. Weak scattering intensity across the entire range of
UV exposures (30 - 240 s) provides no quantifiable data, and without some indication of
nanoscale ordering further characterization was not performed. It is important to note that
while these X-ray results do not definitively eliminate the possibility of nanoscale
features in these samples, it does bring to light some significant experimental
considerations when performing these tests. The first is that the electron density contrast
Figure 3.5: Small angle X-ray scattering analysis of selected 50/50 PS-BP (55K, xAmBP =
0.05)/PVME (58K Mn) blends which have been pre-exposed to 365 nm UV irradiation
for different times prior to phase separation at 175 °C. No discernible scattering peaks are
observed in any sample from 30 – 240 s irradiation time, indicating a lack of resolvable
features on the 4 – 25 nm length scale.

63
in this PS-BP/PVME blend system may in fact be too low to adequately scatter X-rays
within any accessible experimental time frame, as suggested by Tran-Cong et al81
. If this
is true, it would be impossible to obtain scattering results similar to those obtained by
Briber and Bauer without resorting to selective deuteration, as in their work67
. It may,
however, be possible to obtain evidence of phase separated morphologies via electron
microscopy or AFM, yet these techniques are very time consuming and would likely
require investigation of many samples.
3.3.3 Conclusion
Photo-crosslinking of the LCST blend poly(styrene-AmBP)/poly(vinyl methyl
ether) was investigated by a combination of light and X-ray scattering. Incorporation of
benzophenone preserves the LCST nature of the blend, and exposure with 365 nm UV
causes an increase in phase separation temperature as defined by the cloud point. This
elevation is caused by A-B crosslinking in the blend, which constrains phase separation
of the two components, even preventing it altogether at high UV doses (>300 s). While
results are consistent with phase separation, morphological characterization has proved
difficult as the electron density contrast between the two polymers is insufficient for
SAXS.
3.3.4 Critical Blend Future Directions
While this series of experiments was unsuccessful in demonstrating a
bicontinuous blend morphology, there are numerous routes that could be taken in future
experiments to achieve this goal. Several paths are possible if one does not wish to

64
change the material system significantly (PS/PVME with benzophenone photo-
crosslinker), each focusing on simplifying characterization of the materials. First and
foremost, deuteration of one component (i.e. deuterated PS, as in Briber and Bauer’s
work67
) would allow for neutron scattering of the UV-exposed polymer blends and thus
more effective morphological characterization. Since neutron scattering provides
characterization on the nano- and micro-scale, this would allow investigation of samples
fabricated with current methods, determining if spinodal morphologies have been
arrested and perhaps completing the project. It should be noted, however, that deuteration
is known to influence phase behavior of PS/PVME blends, raising the spinodal
temperature by 40 °C in one investigation86
. It would thus be necessary to repeat all
experiments in this study. Second, if sample size were increased characterization with
AFM could be performed. AFM is chosen over TEM due to the extensive sample
preparation required for TEM. Since AFM is a surface characterization technique, this
would require a uniform film where neither the PS-BP nor the PVME have a preference
for the substrate; otherwise it is possible that only one of the component polymers would
be present at the surface. If high throughput characterization is possible, the PS-
BP/PVME LCST blend system could yet result in the desired bicontinuous morphologies
on the nanoscale.
While PS-BP/PVME remains a suitable material system for these model
experiments, there are numerous other critical polymer blend systems that may be
utilized (see introduction). Rather than select one specific polymer blend, requirements
that should yield the most promising results will be discussed. Of primary importance is
accessibility of the temperature at which phase separation will occur. While most

65
polymer blends will exhibit some critical temperature, this transition is often impeded by
the glass transition, crystallization, or degradation of one or both polymers. Crosslinking
of the polymer blend will also alter the temperature at which phase separation occurs, and
this effect will vary depending upon the polymers and crosslinker selected. A-B
crosslinking is desired as this will increase the area of the miscible regime while A-A
crosslinking of a glassy polymer will generally shrink this window by increasing Tg and
decreasing TLCST82
. A-B crosslinking is further beneficial because A-A crosslinking can
require extremely high crosslink density to achieve nanostructured morphologies (for
example, 36 crosslinks per chain were required to create nanostructures in a PS-
anthracene/PVME blend20
, whereas 5 crosslinks per chain led to micron scale features in
the same material system72
). By chemically immobilizing the two component polymers
together as in A-B crosslinking, fewer junctions should be required to achieve
nanostructured materials. A final suggestion resulting directly from this work is that
characterization of materials should be fully considered prior to material selection, as
without scattering methods it is very difficult to demonstrate the desired goals.
3.4 Solution-state asymmetric photo-grafting of benzophenone copolymers
A second means to achieving nanoscale three-dimensional interconnected
polymer networks involves the blending of two homopolymers with corresponding A-B
diblock copolymer. Analogous to oil/water mixtures with surfactant87,88
, these polymeric
systems are often referred to as bicontinuous microemulsions (BμEs), wherein the
diblock copolymer acts as a polymer surfactant, stabilizing the interface between A and B
domains. These blends have been shown to form highly connected three-dimensional

66
morphologies in many polymer systems, and as such have received much attention in
recent years10,89–93
. This section investigates the possibility of creating
thermodynamically stable three-dimensional interconnected polymer networks by the
creation of interfacially active graft copolymers through solution-state photo-reaction of
copolymers with pendent benzophenone.
As the basis for BμEs, any discussion must begin with block copolymers. These
materials have been extensively studied and well characterized, with several
morphologies accessible by varying volume fraction and molecular weight of the
component polymers with a given χ interaction parameter94
. Bicontinuous morphologies
are usually found within phase diagrams of block copolymers, yet the range over which
they are accessible is very small, requiring a precise balance of experimental variables
and chemical incompatibility (i.e. gyroid morphology generally forms only with modest
segregation strenths91
). BμEs represent an attractive alternative route toward these three-
dimensional morphologies via dilution of the pure block copolymer with the
corresponding homopolymer. It has been shown that with equal volume fractions of
homopolymer, only 10 - 20% diblock copolymer is necessary for BμE generation in the
blend9,90,95
, which significantly increases synthetic throughput. A symmetric volume
fraction of components is necessary as it has been found that swelling of lamellar
domains with homopolymer causes fluctuation in the lamellar morphology, generating
this interconnected network morphology89,93
.
As defined above, BμEs utilize block copolymers with low polydispersity,
materials which generally require time consuming and costly synthetic routes. The
benzophenone copolymers in Chapter 2 are synthesized via conventional free radical

67
polymerization and are thus polydisperse, so we must now consider this effect on
morphology generation. Fortunately, this question has previously been raised and
polydispersity has been found to actually stabilize nanostructured morphology in block
copolymer systems (both di-93,96–98
and tri-blocks99,100
). This stabilization is attributed to
a reduced entropic cost of space filling by the polydisperse component, reducing packing
frustration and causing the polydisperse block to occupy a smaller volume than
expected96,97,99,101
. It has thus been found that a polydisperse minority phase is preferred.
Polydispersity also favors curved interfaces and larger domain spacing than a
corresponding monodisperse block copolymer, and as such reduces long range order97,102
.
While this may appear detrimental, decreased long range order is actually beneficial for
our purposes, as a disordered three dimensional BμE morphology is desired. In fact,
polydispersity may expand the range of polymer blends for which nanophase separation
occurs99,103
. It has even been theorized that polydisperse block copolymers will form
more flexible interfaces and suppress attractive copolymer interactions, allowing
formation of BμEs from highly immiscible polymers. Polydispersity is therefore a
potentially useful tool for the generation of highly interconnected morphologies.
While BμEs represent an effective route to highly interconnected three-
dimensional network morphologies, significant reduction of domain size is possible by
the formation of copolymers in-situ, which are known to stabilize interfaces more
effectively than premade block copolymers104
. This is commonly achieved by reactive
blending, whereby compatibilization is accomplished by thermal coupling reactions at the
interface between polymers105
. This process has the added benefit of eliminating the
transport limitation of block copolymer dispersion to the interface in the melt state10
. A

68
number of studies have created nanostructured polymer blends by this
technique1,16,106–109
, yet the inspiration for this section is derived from the method
developed by Pernot et al110
. This process consists of the polymerization of two separate
materials: poly(ethylene) with maleic anhydride groups placed randomly along the
polymer and polyimide end-functionalized with complementary amine. Extrusion of the
materials at high temperatures causes reaction of the functional groups and in-situ
formation of graft copolymers with various architectures. These interfacially active
species each have a preferred curvature defined by the relative volume fraction of each
component, and when combined with unreacted homopolymer, stable cocontinuous
morphologies are developed. The stability of these morphologies are attributed to several
factors: (1) the homogeneous stabilization afforded by randomly distributed functional
groups109
, (2) the relative difficulty of removing graft copolymers from the interface108
,
and (3) to the improved reduction of interfacial tension by graft copolymers than by
corresponding diblock copolymers104,111
. While this method is successful at preparing
bicontinuous morphologies, it still suffers from the necessity of functionalizing both
polymers with complementary moieties and the potentially harsh processing conditions
required.
While the previous section focused on the potential to arrest spinodal
decomposition in a polymer blend, this section instead discusses the possibility of
creating thermodynamically stable nanoscale bicontinuous morphologies by the
formation of graft copolymers through solution-state photo-reaction of copolymers with
pendent benzophenone. These materials offer an improvement in that they represent
polydisperse materials functionalized randomly with a photo-crosslinking group, much

69
like the poly(ethylene) in Pernot et al’s work. Unlike this work, however, BP offers the
added advantages in that existing hydrogen in the component polymers can be exploited
in place of additional chemical modification, and milder processing conditions can be
achieved through the use of photochemistry. The selected material systems consist of PS-
BP and PEG-containing homopolymer (both PEG and poly(oligoethylene glycol
methacrylate) (POEGMA)). After exposure in benzene solution the morphology of these
materials is interrogated by X-ray scattering and TEM, finding that nanoscale features
can be generated with this technique. Unfortunately, bicontinuous morphologies remain
elusive due to significant chain scission in PEG and unresolvable morphology in
POEGMA.
3.4.1 Experimental
Materials and Sample Preparation
Poly(styrene-co-acrylamidobenzophenone) (PS-BP) and poly(oligoethylene
glycol methacrylate) (POEGMA) were synthesized as discussed in Chapter 2. PS-BP
exhibited Mn = 55,000 g/mol (Ð = 1.3) and xAmBP = 0.05. POEGMA was prepared from
poly(ethylene glycol) methyl ether methacrylate (Aldrich, Average Mn = 475 g/mol) and
di(ethylene glycol) methyl ether methacrylate (Aldrich) in 1,4-dioxane yielding liquid
polymer with Mn = 13,400 g/mol (Ð = 1.69) and 8.6 mol% monomer with PEG side
chains. Poly(ethylene glycol) (PEG) with molecular weights ranging from Mn = 2,050 to
35,000 g/mol were purchased from Aldrich and used as received. Benzophenone (>99%)
was purchased from Sigma Aldrich, benzene was purchased from TCI America, while

70
cumene (99.9%) and extra dry ether (99.5%) over molecular sieves were purchased from
Acros Organics. All reagents were used without further purification.
Solution-state reactive blending was performed by preparing concentrated
solutions of PS-BP and PEG in benzene and mixing in an appropriate ratio such that both
PS-BP and PEG were present at 40 mg/mL (Figure 3.6). This solution was then filtered
through a 0.45 μm PTFE syringe filter and injected to a capillary tube fitted with a
needle. The capillary tube is then sealed with a Luer-lok three-way stopcock attached to
the needle and degassed by three freeze-pump-thaw cycles with liquid nitrogen followed
by a dry nitrogen purge. Samples were then exposed to 365 nm ultraviolet irradiation
(same source as Chapter 2, ~2 mW/cm2) at a distance of 1 cm from the lamp surface for a
given time. After exposure, capillary tubes were broken and the contents cast onto glass
slides, removing solvent. These samples were then annealed under vacuum at 130 °C for
at least 4 hours prior to characterization.
UV/Visible Absorption Spectroscopy
Benzophenone, cumene, and diethyl ether solutions were prepared in benzene and
diluted to a final 43 mM concentration of each species prior to injection into 2 mm path
length quartz cuvettes fitted with ground PTFE stoppers to limit evaporation of solvent.
Three samples (benzophenone alone, benzophenone with cumene, benzophenone with
ether) were exposed to UV (1.58 mW/cm2 at plane of exposure) for different times and
the benzophenone π-π* peak maximum at 336 nm was monitored as a function of UV
exposure time with a Hitachi U-3010 spectrophotometer in absorbance mode over a
wavelength range of 200 - 500 nm. Decays were then normalized to the same maximum
absorbance at 0 s.

71
Figure 3.6: Experimental technique for reactive blending. i) PS-BP copolymer and either
PEG or POEGMA (each polymer at 40 mg/mL) are dissolved in benzene, and ii) are
injected into a capillary tube, degassed, and exposed to 365 nm UV for a prescribed
irradiation time. After exposure the solution is cast onto a glass slide and annealed prior
to characterization.
Morphological Characterization
Annealed samples were embedded in epoxy and cured at room temperature prior
to preparing 70 nm sections with a Leica Ultracryomicrotome at -90 °C. Grids were then
treated with ruthenium tetroxide vapor for 40 min to provide sample contrast.
Transmission electron microscopy was performed using a JEOL 2000FX 200 kV
transmission electron microscope with a lanthanum hexaboride filament in bright field
mode. Small angle X-ray scattering was performed on powder samples with the same
setup as Section 3.3.
Size Exclusion Chromatography
Molecular weights were analyzed via size exclusion chromatography (SEC) with
a Polymer Laboratories GPC-50 with a polystyrene column and THF as eluent (0.5

72
mL/min). Refractive index and UV detectors were simultaneously used, and molecular
weights were calibrated with poly(styrene) standards. Refractive index signals have been
magnified by 25X in reported data.
3.4.2 Results and Discussion
Solution state asymmetric photo-grafting between PS-BP and five different PEG
homopolymers and a single POEGMA homopolymer were investigated by a combination
of scattering, microscopy, and chromatography techniques. Blends were exposed to 365
nm UV for between 10 and 100 min prior to casting and annealing, in most cases yielding
solid films which could then be either redissolved for SEC or scraped off as powder for
TEM and SAXS. In several cases, particularly at high NPEG samples gelled in capillary
tubes and could not be further analyzed. Benzophenone-containing poly(styrene) and
PEG-containing polymers were chosen because of the anticipated requirement for
successful preparation of nanostructures, namely that the preference for hydrogen
abstraction by benzophenone (BP) should be highly asymmetric to promote A-B grafting
and prepare interfacially active species. As discussed in Chapter 2, PS-BP exhibits the
highest characteristic dose for BP conversion, and as such represents the polymer that is
least likely to undergo self (A-A) reaction. Combining PS-BP with PEG-containing
homopolymers, each of which contain a large number of aliphatic hydrogen located alpha
to oxygen (highly reactive hydrogen species for BP abstraction112
), we should thus
promote A-B grafting from the PS-BP to the homopolymer. We must also consider
results from Chapter 2, which indicate that abstraction of hydrogen from PEG will

73
potentially result in chain scission. It is hoped that this will prove to be inconsequential
for our system, yet we investigate this effect in detail later in this section.
To ensure this desired reactivity, small molecule analogues for the polymers of
interest were first investigated by UV/Visible absorption spectroscopy. Cumene and
diethyl ether were selected for investigation, as each molecule should behave in a similar
fashion as a single repeat unit of the corresponding polymer. Based on reactivity of
functional groups to hydrogen abstraction by BP, cumene (containing a single tertiary
benzylic hydrogen with much less reactive primary and aromatic hydrogen) should be
close in reactivity to a styrene repeat unit while diethyl ether (with four hydrogen alpha to
an oxygen molecule with less reactive primary hydrogen) is analogous to an ethylene
glycol. Solutions were prepared in benzene, which should be relatively inert to hydrogen
abstraction by BP, and decay of the π-π* peak absorbance of BP was monitored as a
function of UV exposure time, as shown in Figure 3.7. Clearly evident from these results
is that conversion of BP is slowest with benzene as a hydrogen abstraction substrate,
increases with the addition of cumene, and is still more rapid in the presence of diethyl
ether. This result is consistent with a previous report stating that hydrogen abstraction
from benzene by BP is minimal113
. Considering that the abstraction rate for cumene has
earlier been shown to be equal to that of oligostyrenes in acetonitrile, and since
oligostyrenes were shown to quench BP triplet three times faster than cumene, it is
reasonable to believe that PS-BP will not readily react with itself114
. Therefore, it is likely
that reaction of PEG with PS-BP will be much faster than reaction of PS-BP with itself,
creating A-B grafted species and making this system a candidate for formation of blends
with nanoscale morphology.
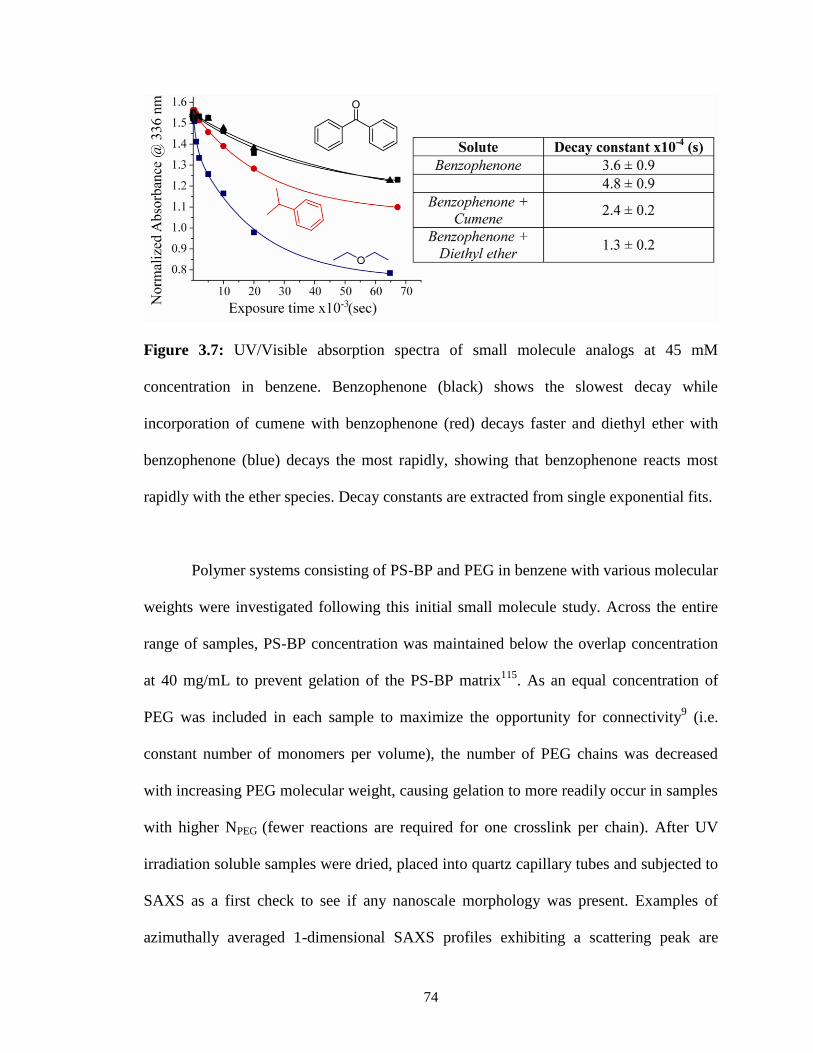
74
Figure 3.7: UV/Visible absorption spectra of small molecule analogs at 45 mM
concentration in benzene. Benzophenone (black) shows the slowest decay while
incorporation of cumene with benzophenone (red) decays faster and diethyl ether with
benzophenone (blue) decays the most rapidly, showing that benzophenone reacts most
rapidly with the ether species. Decay constants are extracted from single exponential fits.
Polymer systems consisting of PS-BP and PEG in benzene with various molecular
weights were investigated following this initial small molecule study. Across the entire
range of samples, PS-BP concentration was maintained below the overlap concentration
at 40 mg/mL to prevent gelation of the PS-BP matrix115
. As an equal concentration of
PEG was included in each sample to maximize the opportunity for connectivity9 (i.e.
constant number of monomers per volume), the number of PEG chains was decreased
with increasing PEG molecular weight, causing gelation to more readily occur in samples
with higher NPEG (fewer reactions are required for one crosslink per chain). After UV
irradiation soluble samples were dried, placed into quartz capillary tubes and subjected to
SAXS as a first check to see if any nanoscale morphology was present. Examples of
azimuthally averaged 1-dimensional SAXS profiles exhibiting a scattering peak are

75
shown in Figure 3.8, each with a very poorly defined scattering peak located at d = 13 nm
for this particular PEG (Mn = 6,000 g/mol). Samples which exhibited any resolvable
scattering peak were considered to have nanoscale domains, with results plotted in Figure
3.9. As is plainly seen in Figure 3.9, the exposure time required to achieve nanoscale
features decreases while the length scale of these features increases with increasing NPEG.
The exposure time dependence is caused by a more rapid decrease in the amount of
ungrafted materials as PEG molecular weight increases. The observed increase of domain
spacing in Figure 3.9b is also expected, as the volume fraction of PEG in graft
Figure 3.8: SAXS traces for reactive blends of 1/1 PS-BP /PEG6K Mn exposed to 365 nm
UV for different durations. In the exposure range 65 – 90 minutes annealed powder
samples exhibit scattering peaks, indicating that nanostructures are indeed present.
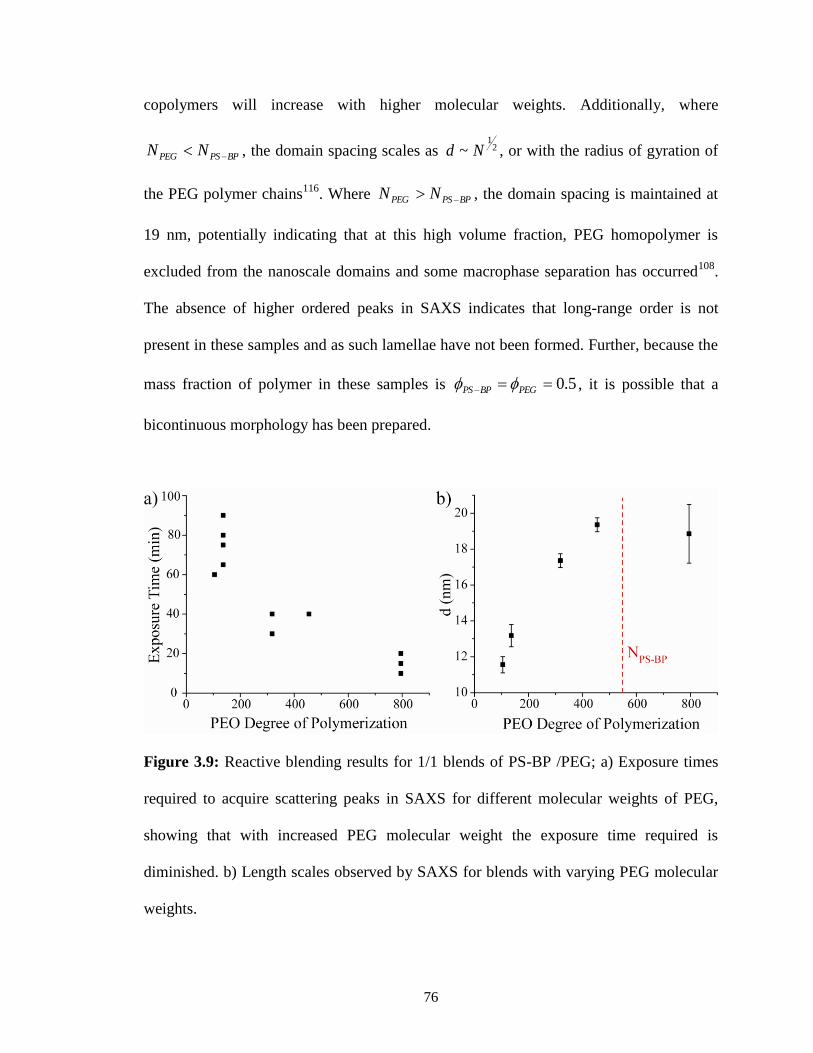
76
copolymers will increase with higher molecular weights. Additionally, where
BPPSPEG NN , the domain spacing scales as 21
~ Nd , or with the radius of gyration of
the PEG polymer chains116
. Where BPPSPEG NN , the domain spacing is maintained at
19 nm, potentially indicating that at this high volume fraction, PEG homopolymer is
excluded from the nanoscale domains and some macrophase separation has occurred108
.
The absence of higher ordered peaks in SAXS indicates that long-range order is not
present in these samples and as such lamellae have not been formed. Further, because the
mass fraction of polymer in these samples is 5.0 PEGBPPS , it is possible that a
bicontinuous morphology has been prepared.
Figure 3.9: Reactive blending results for 1/1 blends of PS-BP /PEG; a) Exposure times
required to acquire scattering peaks in SAXS for different molecular weights of PEG,
showing that with increased PEG molecular weight the exposure time required is
diminished. b) Length scales observed by SAXS for blends with varying PEG molecular
weights.

77
To further interrogate the morphology of these photo-grafted PS-BP/PEG blends,
transmission electron microscopy was utilized, with a representative image of a blend
with 000,6, PEGnM g/mol shown in Figure 3.10. It is clear from this image that there
are two length scales of phase separation in this material, with structure on both the 10
nm and 100 nm length scales. The shorter of the two corresponds well with X-ray
scattering results which showed features at 5.11~d nm, however the resulting
morphology is certainly not bicontinuous, as the features are not present uniformly across
the sample.
To better understand this photo-grafting process, we next examined the
progression of molecular weight in this blend system using a combination of refractive
index (RI) and ultraviolet (UV) detection in size exclusion chromatography. Because
PEG will not provide signal from the UV detector, combining the two detectors allows us
to distinguish between polymers containing poly(styrene) and those without it. Figure
3.11 shows RI SEC traces for pure PS-BP (black) and 6,000 g/mol PEG (red), with both
RI and UV traces for a blend exposed to 365 nm UV for 60 minutes (in green and blue,
respectively). Evidence of graft copolymer formation is present via the slight increase in
molecular weight observed at low retention time in both RI and UV signals of the blend,
however there is also evidence of chain scission in this material system. Examining the
blend traces at high retention times, we see a significant signal from the RI detector
without a corresponding signal from the UV detector. This indicates that the fraction of
sample at this molecular weight does not contain poly(styrene), and thus must be PEG.
Comparing the position of this feature to the neat PEG homopolymer, one sees clearly
that PEG molecular weight is decreased by exposure of the blend to UV. This can be

78
Figure 3.10: TEM image of a 1/1 PS-BP/PEG6K blend which has been exposed to 365
nm UV for 60 minutes. A hierarchical phase separation is observed, with large domains
on the order of 100 nm containing smaller features on the 10 nm length scale,
corresponding well with SAXS results indicating 11.5 nm features. This sample has been
cryo-microtomed into 70 nm thick sections at -90 °C and stained with RuO4 vapor for 40
minutes prior to imaging.
explained by considering the results of Chapter 2, which stated that abstraction of
hydrogen from the main polymer chain will often lead to a dislinking event. As PEG
contains only main-chain hydrogen species, it comes as no surprise that a significant

79
fraction of photo-reaction in this blend system is main chain PEG scission. For effective
solution state photo-grafting with BP copolymers one should encourage hydrogen
abstraction on a side group of the polymer.
Figure 3.11: SEC traces of a 1/1 blend of PS-BP/PEG6K and the corresponding individual
polymers. The black and red curves are the PS-BP copolymer and PEG homopolymer,
respectively, while the green and blue curves represent refractive index and ultraviolet
signal from the polymer blend after 60 minutes of UV exposure. The refractive index
detector peak (green curve) at high retention times in the blend indicates a fraction of
PEG which has decreased molecular weight compared to the homopolymer. This is
indication of main chain scission during irradiation. Refractive index curves magnified
25X.
With this in mind we then alter the blend system to promote hydrogen abstraction
on the homopolymer side chain by incorporating PEG functional groups into the side
chains of a methacrylate based polymer, which we call poly(oligoethylene glycol
methacrylate) or POEGMA. Performing the same SEC analysis with a blend system of

80
PS-BP and POEGMA exposed to UV for 70 min, we see in Figure 3.12a that the
molecular weight of the blends exhibits a peak at lower retention time than the
corresponding homopolymers, indicating the presence of higher molecular weight
species. Because the RI and UV traces of the blend agree well with one another, it can be
concluded that these polymers contain both PS-BP and POEGMA, and thus graft
copolymers have been successfully prepared.
Figure 3.12: SEC traces of a 1/1 blend of PS-BP/POEGMA and the corresponding
individual polymers. a) the black and red curves are the PS-BP and POEGMA
copolymers, respectively, while the green and black curves represent refractive index and
ultraviolet signals from the polymer blend after 70 min of UV exposure. Unlike the
PS/PEO blends, this material clearly shows an increased molecular weight with UV
exposure. Refractive index curves have been magnified 25X. b) Progression of molecular
weight distribution (UV signal) with exposure time for this blend signal. A peak at higher
molecular weight arises between 50 and 70 min of exposure, and disappears by 90 min,
indicating the formation of insoluble species at high reaction times.

81
More importantly, there is no significant signal at high retention time, and therefore the
POEGMA appears stable to hydrogen abstraction by PS-BP. Figure 3.12b shows the
progression of blend molecular weight monitored by UV detector from 50 to 90 min of
UV exposure. Slight variations in signal intensity of the primary peaks centered at 1200 s
are likely due to variation of solution concentration. The high molecular weight graft
peak is present in all samples, yet nearly disappears after 90 min of exposure. We believe
this to be due to further grafting of the polymers which creates species which are either
insoluble or filtered from the solution prior to sampling. SAXS investigation of these
blend systems did not yield any scattering, however, indicating that these blends do not
phase separate on the nanometer scale.
3.4.3 Conclusion
Graft copolymers have been formed in blends of PS-BP and either PEG and
POEGMA by a solution state photo-grafting process. While nanostructured materials
were observed by both SAXS and TEM in PEG blends, bicontinuous morphologies were
not achieved. SEC analysis revealed a significant amount of PEG chain scission due to
hydrogen abstraction by BP on the main polymer chain, an issue which appears to have
been remedied by POEGMA, whose abstractable hydrogen are on the side groups of the
polymer. This was encouraging for the PS-BP/POEGMA blends, however they
unfortunately did not exhibit any phase separation on the length scale probed by SAXS.

82
3.4.4 Solution-state Photo-Grafting Future Directions
While it is possible that increased reaction time may potentially increase the
number of grafted species formed in the PS-BP/PEG system, significant refinement of
reaction conditions would be required to overcome the chain scission and achieve the
desired morphology, with no guarantee of success. Much like in Section 3.3., there are
several different paths which could ultimately achieve the desired goal of a nanoscale
bicontinuous morphology by solution state photochemical reaction. The first possibility
involves utilizing a BP copolymer but altering the material system to give the greatest
possible chance of success. The same requirements must be met, namely that A-B
grafting is necessary and reactivity of BP with its parent polymer must be minimized,
therefore BP copolymers with very few (or no) abstractable hydrogen should be used.
Candidate materials could be poly(α-methyl styrene-BP), poly(methacrylonitrile-BP), or
more synthetically challenging copolymers which only contain aromatic or primary
hydrogen (i.e. BP copolymer prepared from α,β,β-trimethylstyrene). The corresponding
homopolymer should have an adequate number of abstractable hydrogens on the side
chains to encourage grafting and prevent scission of the main chain. Due to the
persistence of secondary hydrogen on the backbone of the majority of common polymers,
it is necessary that either tertiary hydrogen or hydrogen on carbon alpha to nitrogen or
oxygen be present on the side chains. Poly(N-isopropyl methacrylamide) or a more exotic
material containing alkylamine or alkanol groups could potentially yield the desired
results. Whenever deciding on a material system, care must be taken to ensure adequate
contrast for the characterization method of choice, preferably a high-throughput
scattering technique.

83
It may be of value to change the photochemistry used in this photo-grafting
process. Without a hydrogen abstracting or alpha-cleavage photo-reactive agent,
however, it will be necessary to functionalize both component materials of the blend,
eliminating the most attractive feature of the current technique – synthetic simplicity. If
one were to utilize photochemistries which undergo self cycloaddition (i.e. anthracene or
cinnamate esters), grafting would not occur and nanoscale domains are very unlikely to
be formed. Functionalizing both materials with complementary groups would make the
photo-grafting process much cleaner, as the reaction would be better defined than in the
case of BP copolymers. As an alternative photochemistry, UV initiated thiol-ene could be
an attractive option. This reaction is known to proceed quickly and with appropriate
concentration of polymer and functional group the desired bicontinuity could be
achieved.
3.5 Conclusions
The ability of benzophenone (BP) copolymers to form nanostructured
bicontinuous polymer blends was investigated, with limited success. Two approaches
were used in this study, and while nanostructured blends were realized, the ultimate goal
was not achieved. This is attributed to several factors, mainly generation of radicals on
the polymer backbone which induces chain scission, and the inability to characterize the
blends with a high throughput technique. Upon consideration of the above results,
copolymers containing pendent BP photo-crosslinkers are candidates for the formation of
nanoscale bicontinuous morphology, yet significant refinement for each method is
necessary. Benzophenone as a UV-active agent is certainly capable of providing A-B

84
crosslinking and grafting in a critical polymer blend and solution, respectively, but a
significant limitation exists: in order to prepare the desired blend morphology significant
synthetic challenges must first be overcome. Unfortunately this negates a very attractive
feature of the as-synthesized BP copolymers, in that they are very simple to prepare via
conventional free radical chemistry. If more complex chemistry is to be undertaken there
is no reasonable need to use benzophenone as the UV active chromophore, and much
more convenient photochemistries can (and probably should) be used.
3.6 References
1. Bhardwaj, R. & Mohanty, A. K. Modification of brittle polylactide by novel
hyperbranched polymer-based nanostructures. Biomacromolecules 8, 2476–84
(2007).
2. Li, Y., Iwakura, Y., Zhao, L. & Shimizu, H. Nanostructured Poly(vinylidene
fluoride) Materials by Melt Blending with Several Percent of Acrylic Rubber.
Macromolecules 41, 3120–3124 (2008).
3. Choi, J., Lee, K. M., Wycisk, R., Pintauro, P. N. & Mather, P. T. Nanofiber
Network Ion-Exchange Membranes. Macromolecules 41, 4569–4572 (2008).
4. Nam, K. T. et al. Virus-enabled synthesis and assembly of nanowires for lithium
ion battery electrodes. Science 312, 885–888 (2006).
5. Yemini, M., Reches, M., Gazit, E. & Rishpon, J. Peptide nanotube-modified
electrodes for enzyme-biosensor applications. Anal. Chem. 77, 5155–5159 (2005).
6. Chen, L., Hallinan, D. T., Elabd, Y. A. & Hillmyer, M. A. Highly Selective
Polymer Electrolyte Membranes from Reactive Block Polymers. Macromolecules
42, 6075–6085 (2009).
7. Chen, Y. B. et al. Enhancement of anhydrous proton transport by supramolecular
nanochannels in comb polymers. Nature Chem. 2, 503–508
8. Park, M. J. et al. Increased water retention in polymer electrolyte membranes at
elevated temperatures assisted by capillary condensation. Nano Lett. 7, 3547–52
(2007).

85
9. Potschke, P. & Paul, D. R. Formation of Co-continuous Structures in Melt-Mixed
Immiscible Polymer Blends. J. Macromol. Sci., Polym. Rev. 43, 87 (2003).
10. Fredrickson, G. H. & Bates, F. S. Design of bicontinuous polymeric
microemulsions. J. Polym. Sci., Part B: Polym. Phys. 35, 2775–2786 (1997).
11. Pitet, L. M., Amendt, M. A. & Hillmyer, M. A. Nanoporous linear polyethylene
from a block polymer precursor. J. Am. Chem. Soc. 132, 8230–1 (2010).
12. Green, M. D., Choi, J.-H., Winey, K. I. & Long, T. E. Synthesis of Imidazolium-
Containing ABA Triblock Copolymers: Role of Charge Placement, Charge
Density, and Ionic Liquid Incorporation. Macromolecules 45, 4749–4757 (2012).
13. Wong, D. T., Mullin, S. A., Battaglia, V. S. & Balsara, N. P. Relationship between
morphology and conductivity of block-copolymer based battery separators. J.
Membr. Sci. 394-395, 175–183 (2012).
14. Galloway, J. A. & Macosko, C. W. Comparison of methods for the detection of
cocontinuity in poly(ethylene oxide)/polystyrene blends. Polym. Eng. Sci. 44, 714–
727 (2004).
15. Register, R. A. Continuity through dispersity. Nature 483, 167–168 (2012).
16. Shi, H. C. et al. A facile route for preparing stable co-continuous morphology of
LLDPE/PA6 blends with low PA6 content. Polymer 51, 4958–4968
17. Seo, M., Amendt, M. A. & Hillmyer, M. A. Cross-Linked Nanoporous Materials
from Reactive and Multifunctional Block Polymers. Macromolecules 44, 9310–
9318 (2011).
18. Ruzette, A. V & Leibler, L. Block copolymers in tomorrow’s plastics. Nat. Mater.
4, 19–31 (2005).
19. Zhang, C.-L. et al. Efficiency of graft copolymers at stabilizing co-continuous
polymer blends during quiescent annealing. Polymer 49, 3462–3469 (2008).
20. Tamai, T., Imagawa, A. & Tran-Cong, Q. Semi-Interpenetrating Polymer
Networks Prepared by in situ Photo-Crosslinking of Miscible Polymer Blends.
Macromolecules 27, 7486–7489 (1994).
21. Bates, F. S. Polymer-Polymer Phase-Behavior. Science 251, 898–905 (1991).
22. McMaster, L. P. Aspects of Polymer-Polymer Thermodynamics. Macromolecules
6, 760–773 (1973).

86
23. Koningsveld, R., Kleintjens, L. A. & Schoffeleers, H. M. Thermodynamic aspects
of polymer compatibility. Pure Appl. Chem. 39, 1–32 (1974).
24. Kammer, H. The phase behavior of polymer blends–Effects of thermodynamics
and rheology. Acta Polym. 42, 571–576 (1991).
25. Briber, R. M. & Khoury, F. The phase diagram and morphology of blends of
poly(vinylidene fluoride) and poly(ethyl acrylate). Polymer 28, 38–46 (1987).
26. Isayeva, I., Kyu, T. & St. John Manley, R. Phase transitions, structure evolution,
and mechanical properties of blends of two crystalline polymers: poly (vinylidene
fluoride) and poly (butylene adipate). Polymer 39, 4599–4608 (1998).
27. Kim, C. K. & Paul, D. R. Effects of Polycarbonate Molecular-Structure on the
Miscibility with Other Polymers. Macromolecules 25, 3097–3105 (1992).
28. Hu, H., Shangguan, Y. & Zuo, M. Investigation on LCST behavior of a new
amorphous/crystalline polymer blend: Poly (n-methyl methacrylimide)/poly
(vinylidene fluoride). J. Polym. Sci., Part B: Polym. Phys. 46, 1923–1931 (2008).
29. Paul, D. R., Bernstein, R. E., Wahrmund, D. C. & Barlow, J. W. Polymer Blends
Containing Poly(vinylidene fluoride). Part IV: Thermodynamic Interpretations.
Polym. Eng. Sci. 18, 1225–1234 (1978).
30. Kim, C. & Paul, D. Miscibility of poly(methyl methacrylate) blends with halogen-
containing polycarbonates and copolymers. Polymer 33, 4929–4940 (1992).
31. Gan, P. & Paul, D. Phase behaviour of blends of homopolymers with α-
methylstyrene/acrylonitrile copolymer. Polymer 35, 1487–1502 (1994).
32. Kim, C. K. & Paul, D. R. Interaction Parameters for Blends Containing
Polycarbonates. 1. Tetramethyl Bisphenol-a Polycarbonate Polystyrene. Polymer
33, 1630–1639 (1992).
33. Sasaki, H., Kanti Bala, P., Yoshida, H. & Ito, E. Miscibility of PVDF/PMMA
blends examined by crystallization dynamics. Polymer 36, 4805–4810 (1995).
34. Chen, W. J., David, D. J., MacKnight, W. J. & Karasz, F. E. Miscibility and phase
behavior in blends of poly(vinyl butyral) and poly(methyl methacrylate).
Macromolecules 34, 4277–4284 (2001).
35. Penning, J. P. & St. John Manley, R. Miscible Blends of Two Crystalline
Polymers. 1. Phase Behavior and Miscibility in Blends of Poly(vinylidene
fluoride) and Poly(1,4-butylene adipate). Macromolecules 29, 77–83 (1996).

87
36. Nishi, T., Wang, T. T. & Kwei, T. K. Thermally Induced Phase Separation
Behavior of Compatible Polymer Mixtures. Macromolecules 8, 227–234 (1975).
37. Ishino, S., Nakanishi, H., Norisuye, T., Tran-Cong-Miyata, Q. & Awatsuji, Y.
Designing a polymer blend with phase separation tunable by visible light for
computer-assisted irradiation experiments. Macromol. Rapid Commun. 27, 758–
762 (2006).
38. Kim, J. K., Lee, H. H., Son, H. W. & Han, C. D. Phase Behavior and Rheology of
Polystyrene/Poly(α-methylstyrene) and Polystyrene/Poly(vinyl methyl ether)
Blend Systems. Macromolecules 31, 8566–8578 (1998).
39. Barlow, J. W. & Paul, D. R. Polymer alloys. Ann. Rev. Mater. Sci. 1981. 11, 299–
319 (1981).
40. Li, S. H. & Woo, E. M. Weak interaction, marginal miscibility, and ring-band
spherulites in blends of poly (vinylidene fluoride) with polyesters. J. Appl. Polym.
Sci. 107, 766–777 (2008).
41. Wahrmund, D. C., Barlow, J. W., Paul, D. R. & Bernstein, R. E. Polymer Blends
Containing Polyvinylidene Fluoride. Part I: Poly(Alkyl Acrylates). Polym. Eng.
Sci. 18, 677–682 (1978).
42. Bernstein, R. E., Cruz, C. A., Paul, D. R. & Barlow, J. W. LCST Behavior in
Polymer Blends. Macromolecules 10, 681–686 (1977).
43. Ougizawa, T. & Walsh, D. J. Upper Critical Solution Temperature Behavior in
Polystyrene Poly(Methyl Methacrylate) Mixture. Polym. J. (Tokyo, Jpn.) 25,
1315–1318 (1993).
44. Kressler, J., Higashida, N., Shimomai, K., Inoue, T. & Ougizawa, T. Temperature-
Dependence of the Interaction Parameter between Polystyrene and Poly(Methyl
Methacrylate). Macromolecules 27, 2448–2453 (1994).
45. Lee, J. S., Prabu, A. A. & Kim, K. J. UCST-Type Phase Separation and
Crystallization Behavior in Poly(vinylidene fluoride)/Poly(methyl methacrylate)
Blends under an External Electric Field. Macromolecules 42, 5660–5669 (2009).
46. Tomura, H., Saito, H. & Inoue, T. Light scattering analysis of upper critical
solution temperature behavior in a poly(vinylidene fluoride)/poly(methyl
methacrylate) blend. Macromolecules 25, 1611–1614 (1992).
47. Li, Y., Stein, M. & Jungnickel, B.-J. Competition between crystallization and
phase separation in polymer blends I. Diffusion controlled supermolecular
structures and phase morphologies in poly (e-caprolactone)/polystyrene blends.
Colloid Polym. Sci. 269, 772–780 (1991).

88
48. Li, S. H. & Woo, E. M. Effects of Chain Configuration on UCST Behavior in
Blends of Poly(L-lactic acid) with Tactic Poly(methyl methacrylate)s. J. Polym.
Sci., Part B: Polym. Phys. 46, 2355–2369 (2008).
49. Li, S. H. & Woo, E. M. Immiscibility-miscibility phase transitions in blends of
poly(L-lactide) with poly(methyl methacrylate). Polym. Int. 57, 1242–1251 (2008).
50. Sato, T., Endo, M., Shiomi, T. & Imai, K. UCST behaviour for high-molecular-
weight polymer blends and estimation of segmental chi parameters from their
miscibility. Polymer 37, 2131–2136 (1996).
51. Li, S. H. & Woo, E. M. Immiscibility with upper-critical solution temperature
phase diagrams for poly(methyl methacrylate)/polyesters blends. Colloid Polym.
Sci. 286, 253–265 (2008).
52. Li, H., Yang, Y., Fujitsuka, R., Ougizawa, T. & Inoue, T. Studies on miscibility in
polycarbonate/poly(styrene-co-acrylonitrile) blends by ellipsometry. Polymer 40,
927–933 (1999).
53. Kim, J. K., Jang, J., Lee, D. H. & Ryu, D. Y. Phase behavior of polystyrene and
poly(n-pentyl methacrylate) blend with small amounts of symmetric polystyrene-
block-poly(n-pentyl methacrylate) copolymers. Macromolecules 37, 8599–8605
(2004).
54. Ryu, D. Y. et al. Phase behavior of polystyrene and poly(n-pentyl methacrylate)
blend. Macromolecules 35, 8676–8680 (2002).
55. Ahn, H., Naidu, S., Ryu, D. Y. & Cho, J. Phase Behavior of a Weakly Interacting
Polystyrene and Poly(n-hexyl methacrylate) System: Evidence for the Coexistence
of UCST and LCST. Macromol. Rapid Commun. 30, 469–474 (2009).
56. Browarzik, D. Calculation of the phase behavior of polystyrene plus poly(n-pentyl
methacrylate) blends. J. Macromol. Sci., Part A: Pure Appl. Chem. A42, 1339–
1353 (2005).
57. Inoue, T. Reaction-induced phase decomposition in polymer blends. Prog. Polym.
Sci. 20, 119–153 (1995).
58. Cahn, J. W. Phase separation by spinodal decomposition in isotropic systems. J.
Chem. Phys. 42, 93 (1965).
59. Cahn, J. W. & Hilliard, J. E. Free Energy of a Nonuniform System. I. Interfacial
Free Energy. J. Chem. Phys. 28, 258 (1958).

89
60. Filippone, G., Dintcheva, N. T., Acierno, D. & La Mantia, F. P. The role of
organoclay in promoting co-continuous morphology in high-density
poly(ethylene)/poly(amide) 6 blends. Polymer 49, 1312–1322 (2008).
61. Filippone, G., Romeo, G. & Acierno, D. Role of Interface Rheology in Altering
the Onset of Co-Continuity in Nanoparticle-Filled Polymer Blends. Macromol.
Mater. Eng. 296, 658–665 (2011).
62. Li, L. et al. Kinetically Trapped Co-continuous Polymer Morphologies through
Intraphase Gelation of Nanoparticles. Nano Lett. 11, 1997–2003
63. Gubbels, F. et al. Selective Localization of Carbon-Black in Immiscible Polymer
Blends - a Useful Tool to Design Electrical Conductive Composites.
Macromolecules 27, 1972–1974 (1994).
64. Gubbels, F. et al. Design of Electrical Conductive Composites - Key Role of the
Morphology on the Electrical-Properties of Carbon-Black Filled Polymer Blends.
Macromolecules 28, 1559–1566 (1995).
65. Chung, H., Ohno, K., Fukuda, T. & Composto, R. J. Self-regulated structures in
nanocomposites by directed nanoparticle assembly. Nano Lett. 5, 1878–1882
(2005).
66. Herzig, E., White, K., Schofield, A., Poon, W. & Clegg, P. Bicontinuous
emulsions stabilized solely by colloidal particles. Nat. Mater. 6, 966–971 (2007).
67. Briber, R. M. & Bauer, B. J. Effect of Cross-Links on the Phase-Separation
Behavior of a Miscible Polymer Blend. Macromolecules 21, 3296–3303 (1988).
68. Bauer, B. J., Briber, R. M. & Han, C. C. Small-Angle Neutron-Scattering Studies
of Compatible Blends of Linear Polyvinyl Methyl-Ether) and Cross-Linked
Deuterated Polystyrene. Macromolecules 22, 940–948 (1989).
69. Derouiche, A., Bettachy, A., Benhamou, M. & Daoud, M. Kinetics of microphase
separation in crosslinked polymer blends. Macromolecules 25, 7188–7191 (1992).
70. DeGennes, P. G. Effect of cross-links on a mixture of polymers. J. Phys. Lett. 40,
L–69 (1979).
71. Trancong, Q., Nagaki, T., Nakagawa, T., Yano, O. & Soen, T. Immobilization of
Transient Structures in Polystyrene Poly(2-Chlorostyrene) Blends Undergoing
Phase-Separation by Using Photo-Cross-Linking. Macromolecules 22, 2720–2723
(1989).

90
72. Imagawa, A. & Tran-Cong, Q. Structure-Property Relationship of Polymer Blends
with Co-Continuous Structures Prepared by Photo-Cross-Linking.
Macromolecules 28, 8388–8394 (1995).
73. Tran-Cong, Q. & Harada, A. Reaction-induced ordering phenomena in binary
polymer mixtures. Phys. Rev. Lett. 76, 1162–1165 (1996).
74. Kawazoe, N., Imagawa, A., Tamai, T. & Tran-Cong, Q. Photocrosslinking kinetics
of polymer blends undergoing spinodal decomposition process. J. Appl. Polym.
Sci. 67, 885–893 (1998).
75. Tran-Cong, Q., Kawai, J. & Endoh, K. Modes selection in polymer mixtures
undergoing phase separation by photochemical reactions. Chaos 9, 298–307
(1999).
76. Tran-Cong-Miyata, Q. Phase separation of polymer mixtures induced by
polarization-selective chemical reactions: Spatial symmetry breaking and mode
selection. Macromol. Symp. 160, 91–97 (2000).
77. Tran-Cong-Miyata, Q., Nishigami, S., Ito, T., Komatsu, S. & Norisuye, T.
Controlling the morphology of polymer blends using periodic irradiation. Nat.
Mater. 3, 448–451 (2004).
78. Nakanishi, T. L., Namikawa, N., Norisuye, T. & Tran-Cong-Miyata, Q.
Autocatalytic phase separation and graded co-continuous morphology generated
by photocuring. Soft Matter 2, 149–156 (2006).
79. Trinh, X. A. et al. Effects of elastic deformation on phase separation of a polymer
blend driven by a reversible photo-cross-linking reaction. Macromolecules 40,
5566–5574 (2007).
80. El Mabrouk, K. & Bousmina, M. Effect of molecular weight and shear on phase
diagram of PS/PVME blend. Rheol. Acta 45, 959–969 (2006).
81. Tran-Cong, Q., Kawakubo, R. & Sakurai, S. Effects of chemical labelling on the
phase behaviour of poly (2-chlorostyrene)/poly (vinyl methyl ether) blends.
Polymer 35, 1236–1241 (1994).
82. Kataoka, K., Harada, A., Tamai, T. & Tran-Cong, Q. Phase behavior and
photocrosslinking kinetics of semi-interpenetrating polymer networks prepared
from miscible polymer blends. J. Polym. Sci., Part B: Polym. Phys. 36, 455–462
(1998).
83. Nakanishi, H., Satoh, M., Norisuye, T. & Tran-Cong-Miyata, Q. Phase Separation
of Interpenetrating Polymer Networks Synthesized by Using an Autocatalytic
Reaction. Macromolecules 39, 9456–9466 (2006).

91
84. Okada, M. Experimental study of thermal fluctuation in spinodal decomposition of
a binary polymer mixture. J. Chem. Phys. 85, 5317–5327 (1986).
85. Russell, T. P., Hadziioannou, G. & Warburton, W. Phase separation in low
molecular weight polymer mixtures. Macromolecules 18, 78–83 (1985).
86. Yang, H., Shibayama, M., Stein, R. S., Shimizu, N. & Hashimoto, T. Deuteration
effects on the miscibility and phase separation kinetics of polymer blends.
Macromolecules 19, 1667–1674 (1986).
87. Clausse, M., Peyrelasse, J., Heil, J., Boned, C. & Lagourette, B. Bicontinuous
Structure Zones in Micro-Emulsions. Nature 293, 636–638 (1981).
88. Komura, S. Mesoscale structures in microemulsions. J. Phys.: Condens. Matter 19,
(2007).
89. Bates, F. S. et al. Polymeric bicontinuous microemulsions. Phys. Rev. Lett. 79,
849–852 (1997).
90. Hillmyer, M. A., Maurer, W. W., Lodge, T. P., Bates, F. S. & Almdal, K. Model
bicontinuous microemulsions in ternary homopolymer block copolymer blends. J.
Phys. Chem. B 103, 4814–4824 (1999).
91. Washburn, N. R., Lodge, T. P. & Bates, F. S. Ternary polymer blends as model
surfactant systems. J. Phys. Chem. B 104, 6987–6997 (2000).
92. Pu, G. W., Luo, Y. W., Lou, Q. C. & Li, B. G. Co-Continuous Polymeric
Nanostructures via Simple Melt Mixing of PS/PMMA. Macromol. Rapid
Commun. 30, 133–137 (2009).
93. Ellison, C. J. et al. Bicontinuous Polymeric Microemulsions from Polydisperse
Diblock Copolymers. J. Phys. Chem. B 113, 3726–3737 (2009).
94. Bates, F. S. & Fredrickson, G. H. Block copolymers - Designer soft materials.
Physics Today 52, 32–38 (1999).
95. Zhou, N., Lodge, T. P. & Bates, F. S. Influence of conformational asymmetry on
the phase behavior of ternary homopolymer/block copolymer blends around the
bicontinuous microemulsion channel. J. Phys. Chem. B 110, 3979–89 (2006).
96. Lynd, N. A. & Hillmyer, M. A. Effects of polydispersity on the order-disorder
transition in block copolymer melts. Macromolecules 40, 8050–8055 (2007).
97. Matsen, M. W. Polydispersity-induced macrophase separation in diblock
copolymer melts. Phys. Rev. Lett. 99, (2007).

92
98. Lynd, N. A., Meuler, A. J. & Hillmyer, M. A. Polydispersity and block copolymer
self-assembly. Prog. Polym. Sci. 33, 875–893 (2008).
99. Widin, J. M., Schmitt, A. K., Im, K., Schmitt, A. L. & Mahanthappa, M. K.
Polydispersity-Induced Stabilization of a Disordered Bicontinuous Morphology in
ABA Triblock Copolymers. Macromolecules 43, 7913–7915 (2010).
100. Widin, J. M., Schmitt, A. K., Schmitt, A. L., Im, K. & Mahanthappa, M. K.
Unexpected consequences of block polydispersity on the self-assembly of ABA
triblock copolymers. J. Am. Chem. Soc. 134, 3834–44 (2012).
101. Schmitt, A. L. & Mahanthappa, M. K. Polydispersity-driven shift in the lamellar
mesophase composition window of PEO-PB-PEO triblock copolymers. Soft
Matter 8, 2294 (2012).
102. Cooke, D. M. & Shi, A. C. Effects of polydispersity on phase behavior of diblock
copolymers. Macromolecules 39, 6661–6671 (2006).
103. Thompson, R. B. & Matsen, M. W. Improving polymeric microemulsions with
block copolymer polydispersity. Phys. Rev. Lett. 85, 670–673 (2000).
104. Jeon, H. K., Zhang, J. B. & Macosko, C. W. Premade vs. reactively formed
compatibilizers for PMMA/PS melt blends. Polymer 46, 12422–12429 (2005).
105. Macosko, C. W., Jeon, H. K. & Hoye, T. R. Reactions at polymer–polymer
interfaces for blend compatibilization. Prog. Polym. Sci. 30, 939–947 (2005).
106. Koulic, C. & Jérôme, R. Nanostructured Polyamide by Reactive Blending. 1.
Effect of the Reactive Diblock Composition. Macromolecules 37, 3459–3469
(2004).
107. Yin, B., Zhao, Y., Pan, M. M. & Yang, M. B. Morphology and thermal properties
of a PC/PE blend with reactive compatibilization. Polym. Adv. Technol. 18, 439–
445 (2007).
108. Gani, L., Tencé-Girault, S., Milléquant, M., Bizet, S. & Leibler, L. Co-continuous
Nanostructured Blend by Reactive Blending: Incorporation of High Molecular
Weight Polymers. Macromol. Chem. Phys. 211, 736–743 (2010).
109. Manna, S., Mandal, A. & Nandi, A. K. Fabrication of Nanostructured Poly(3-
thiophene methyl acetate) within Poly(vinylidene fluoride) Matrix: New Physical
and Conducting Properties. J. Phys. Chem. B 114, 2342–2352 (2010).
110. Pernot, H., Baumert, M., Court, F. & Leibler, L. Design and properties of co-
continuous nanostructured polymers by reactive blending. Nat. Mater. 1, 54–58
(2002).

93
111. Sundararaj, U. & Macosko, C. W. Drop Breakup and Coalescence in Polymer
Blends: The Effects of Concentration and Compatibilization. Macromolecules 28,
2647–2657 (1995).
112. Topp, M. R. Activation-controlled hydrogen abstraction by benzophenone triplet.
Chem. Phys. Lett. 32, 144–149 (1975).
113. Dedinas, J. Photoreduction of benzophenone in benzene. I. Mechanism of
secondary reactions. J. Phys. Chem. 75, 181–186 (1971).
114. Horie, K. et al. Rates of Quenching and Hydrogen Abstraction of Benzophenone
Triplet with Cumene and Oligostyrenes. Polym. J. (Tokyo, Jpn.) 16, 887–893
(1984).
115. Hadgraft, J., Hyde, A. J. & Richards, R. W. Diffusion of Polystyrene in
Poly(Methyl Methacrylate)+Benzene Solutions Measured by Photon-Correlation
Spectroscopy. Trans. Faraday Soc. 75, 1495–1505 (1979).
116. Rubinstein, M. & Colby, R. H. Polymer Physics. (Oxford University Press, Inc:
New York, 2003).

94
CHAPTER 4
CHEMICAL DEACTIVATION OF BENZOPHENONE IN PHOTO-
PATTERNED HYDROGELS
4.1 Abstract
Benzophenone copolymers offer an attractive route to patterned hydrogels,
however residual unreacted photo-crosslinker present in these systems causes these
materials to be unstable to further UV radiation. To remedy this issue, deactivation of
residual benzophenone photo-crosslinker with sodium borohydride was monitored by
UV/Vis and hydrogel disc swelling. Nearly complete deactivation was seen within five
minutes in a mixture of ethanol and water, and treated hydrogels recovered on average
98% of initial swelling after full conversion of AmBP. These results indicate that UV-
stable hydrogels can be prepared from benzophenone based photo-crosslinkable hydrogel
materials by reduction with NaBH4. This opens up the possibility of preparing
independently photo-patterned multilayers, and as proof-of-concept a bilayer was
prepared from photo-crosslinkable poly(N-isopropylacrylamide) and poly(p-
methylstyrene), demonstrating promise for complex photo-patterned hydrogel systems.

95
4.2 Motivation
Hydrogels have been intensely studied for many years, due in large part to their
biocompatible, non-fouling, and non-cytotoxic nature. The high porosity present in these
aqueous gel networks allows for high loading of functional material, making hydrogels a
versatile basis for various applications1–5
. Furthermore, many hydrogels are known to be
stimulus responsive, changing material properties with variation of temperature, pH,
pressure, humidity, or electromagnetic stimulus1–3,6–9
, and as such hold great potential for
actuating and sensing devices, piezoelectric current generation10
, microfluidic devices9,
and the mimicking of muscle tissue11
, among many other applications12
.
While the majority of stimulus-responsive hydrogels rely upon critical phase
behavior or changes in osmotic pressure, hydrogel properties can also be tuned by
changing the density of crosslinks in the polymer network. This serves to shift the
equilibrium swelling, and is experimentally realized by either changing the fraction of
multifunctional monomer during hydrogel formation or by some post-synthetic method,
such as chemical exposure, thermal treatment, or photo-processing4. Of these options,
photo-reaction is the most attractive, as it is an accessible, scalable, and low cost
technique which allows fine tuning of crosslink density in hydrogels6,13–16
. An added
benefit of post-synthetic photo-crosslinking is that it allows for simple fabrication of thin
hydrogel films by either spin coating or drop casting prior to gelation of the network.
This reduction of thickness to the micron-scale greatly increases response time, as
swelling and deswelling are typically dominated by poroelastic relaxation, yielding
diffusive scaling6.

96
Post-synthetic photo-crosslinking has therefore been utilized in many hydrogel
studies by preparing linear copolymers containing photo-reactive monomers. The most
commonly used reagents are activated by ultraviolet radiation, and crosslink by
cycloaddition 6,17–19
, hydrogen abstraction1,3,5,7,14–16,20–22
, or reaction of phenyl azide23–25
.
Spatial variation of UV dose in these hydrogel materials has proven capable of producing
significant swelling variations in a single sample9,14,15
, yet in the absence of full
conversion these materials are susceptible to further reaction by additional radiation. In
the case of three-dimensionally morphing hydrogels, this excess reaction would
homogenize crosslinking in the sample, eliminating function by the removal of
differential swelling14,15
. It is therefore attractive to deactivate residual crosslinker that
remains after photo-patterning in order to obtain environmentally robust patterned
hydrogels onto which additional layers can be independently photo-reacted without
concern for the initial film.
In this work we investigate the possibility of preparing stable photo-patterned
hydrogel materials by selective deactivation of residual crosslinker. Poly(N-
isopropylacrylamide)-based hydrogel materials with copolymerized
acrylamidobenzophenone (AmBP) photo-crosslinker are studied, as their material
properties and UV crosslinking are well known. To selectively deactivate AmBP, we
utilize sodium borohydride, a mild and selective reducing agent for ketones and
aldehydes26–28
. Deactivation is monitored by UV/Visible absorption spectroscopy and
swelling changes of hydrogel discs. Finally, as a proof-of-concept that this technique can
be applied to multilayer fabrication methods, a bilayer photo-patterned polymer film is

97
fabricated from deactivated poly(N-isopropylacrylamide-AmBP) hydrogel and glassy
poly(p-methylstyrene-AmBP).
4.3 Experimental
Materials and sample preparation
Benzophenone copolymers were synthesized as in Chapter 2 and previous
publications14,15,29
. In brief, N-isopropylacrylamide (NiPAm), acrylic acid (AAc),
acrylamidobenzophenone (AmBP), and rhodamine B methacrylate (RhBMA) were
copolymerized via conventional free radical polymerization with AIBN initiator in
1,4-dioxane. After reaction overnight at 75 °C, copolymers were recovered by
precipitation in diethyl ether followed by drying under vacuum, yielding two copolymers
with NiPAm-AAc-AmBP-RhBMA molar compositions of 91.6 - 3.6 - 4.8 - 0 and
86.7 - 6.4 - 6.6 - 0.3, respectively. Poly(p-methylstyrene-AmBP) was prepared similarly,
and recovered by precipitation in cold methanol. Reactants were purchased from Sigma
Aldrich, with the exceptions of AmBP and RhBMA, which were synthesized according
to published protocols15
.
Silicon wafers were cleaned by sonication and subjected to UV-ozone treatment
for 30 minutes prior to coating with a Ca2+
-ion crosslinked poly(acrylic acid) sacrificial
layer (Sigma Aldrich, 000,30wM g/mol)30
. 100 μL of 1 wt% PNiPAm copolymer
solution was then drop cast onto coated substrates and solvent removed at 60 °C
overnight in a closed glass container to yield solid polymer films. These films were then
photo-crosslinked through a photo-mask with a Zeiss Axiovert 200 epi-fluorescence
microscope-mounted UV source (365 nm) for a prescribed radiation dose, as monitored

98
by an X-Cite XR2100 Power Meter (EXFO). Uncrosslinked material was then developed
in a 2:1 ethanol:water mixture, yielding surface-attached PNiPAm gels. Immersion of the
wafer in an aqueous 1 mM phosphate buffer containing 1 mM NaCl (hereafter
“phosphate buffer”) dissolved the poly(acrylic acid) sacrificial layer, permitting the
crosslinked gel material to release from the surface and swell.
UV/Visible monitoring of AmBP conversion
To ensure the ability of NaBH4 to convert AmBP, solid NaBH4 (Fisher Scientific)
was first added to a quartz cuvette followed by a 2 mg/mL solution of PNiPAm-AAc-BP
copolymer in either ethanol or a mixture of 2:1 ethanol:water. Conversion of AmBP was
monitored as a function of time by observing the absorbance of AmBP at 300 nm with a
Hitachi U-3010 spectrophotometer in absorbance mode. Spectral profiles were also
obtained over a wavelength range of 200 - 500 nm both prior to and after reaction.
Chemical deactivation of AmBP in hydrogel discs
After development of PNiPAm-AAc-AmBP discs with 400 μm diameter,
deactivation of AmBP was performed by adding solid NaBH4 to a poly(styrene) petri dish
prior to the addition of 250 μL of phosphate buffer (Figure 4.1, step ii). Samples were
then immersed, releasing the discs from the substrate, and allowed to react for 90 min. At
this time the solution was diluted with fresh phosphate buffer such that the ionic strength
of NaBH4 was less than 3% of phosphate ionic strength. Samples were then allowed to
swell at room temperature in the phosphate buffer at least 90 min and imaged on an
Axiovert 200 epi-fluorescence microscope in DIC mode (Figure 4.1, step iii). Subsequent
heating of the samples to 60 °C crosses the LCST of PNiPAm, expelling the aqueous
phase and causing the hydrogel discs to deswell. Samples were irradiated overnight (>8

99
h) with a low power UVGL-58 Handheld UV lamp operating at 365 nm (UVP; ~ 2
mW/cm2, spectral range 340 - 410 nm), reacting any remaining AmBP in the hydrogel
(Figure 4.1, step iv). Finally the hydrogel discs were cooled to room temperature and
swelled for at least 90 min prior to imaging their final dimensions in DIC mode (Figure
4.1, step v). Data points reported are for one exposure which yields anywhere from 5 - 9
hydrogel discs for imaging, and errors are reported as one standard deviation.
Figure 4.1: Floating hydrogel disc deactivation. i) PNiPAm-AAc-AmBP film is exposed
to UV through a mask and developed. ii) The sacrificial layer is dissolved in an aqueous
1 mM phosphate buffer with 1 mM NaCl. Deactivation is achieved through inclusion of
solid NaBH4 to a concentration of 0.15 M. iii) Concentration of NaBH4 is decreased such
that ionic strength of the buffer is at least 30X greater than that of NaBH4. The samples
are permitted to swell at room temperature and are imaged, yielding discs with area A1.
iv) Samples are heated through the LCST to 60 °C, deswelling the discs. They are then
subjected to UV exposure overnight in the deswelled state. v) The samples are swelled
once more at room temperature, yielding discs with area A2.

100
Preparation of multilayer film
PNiPAm-AAc-AmBP-RhBMA films were cast on a 1 cm2
wafer as described
above and photo-patterned with a series of two masks, yielding an exposed 800 x 100 μm
rectangle. One half (400 x 100 μm) of the rectangle was subjected to a UV dose of 100
J/cm2 while the other was given 10 J/cm
2. After development in 2:1 ethanol:water,
deactivation was performed by immersion in a series of solutions: (1) 1 M CaCl2 in 2:1
ethanol water, (2) 1 M CaCl2 + 0.15 M NaBH4 in 2:1 ethanol:water, and (3) 2:1
ethanol:water with no salt present. Calcium chloride solution served to stabilize the
underlying sacrificial layer to dissolution by sodium borohydride, and rinsing with clean
solvent prevented the deposition of salts on the sample. The sample was immersed in
each solution for 15 seconds followed by a 30 second drying step in between to prevent
excessive swelling of the sample. This process (3 immersions with 3 drying steps) was
repeated five times for a total of 75 seconds immersed in the NaBH4 deactivation
solution. After drying any remaining solvent, a poly(p-methylstyrene-AmBP) thin film
was spin-cast onto the sample (40 mg/mL in chloroform, 2000 rpm, 1000 ramp, 60 s). A
slightly larger rectangular area (900 x 200 μm) was aligned to the previously developed
fluorescent film and exposed to 100 J/cm2 UV dose and developed in a 1:0.65
toluene:hexanes developer, creating a rectangular bilayer consisting of PNiPAm-AAc-
AmBP-RhBMA and PpMS-AmBP. Immersing this sample in the phosphate buffer
permitted release from the substrate and subsequent fluorescence imaging.

101
4.4 Results and discussion
Chemical reduction has been utilized in an attempt to prepare photo-patterned
hydrogels which are stable to subsequent UV irradiation. To achieve this goal sodium
borohydride has been chosen as reducing agent, as it is well known to selectively reduce
ketones and aldehydes, including benzophenone. Since our copolymer system is a
complex combination of different functionalities (amide, carboxylic acid, etc.) and
benzophenone is a reasonably stable biaryl ketone with extended conjugation, we first
must confirm the selectivity and efficacy of NaBH4 in benzophenone reduction in our
copolymers. Primary investigation was made via UV/Visible absorption spectroscopy.
Solid NaBH4 was added to the quartz cuvette prior to injection of polymer solution to
limit borate formation26
(and thus neutralization of the reducing species) prior to
interaction with AmBP. Monitoring the decay of the π-π* transition of AmBP (300 nm)
as shown in Figure 4.2a, we observe a pronounced decrease in absorbance, indicating
effective reduction of the photo-crosslinker. In ethanol, this process nears completion
(>90 % absorbance decrease) after 2,500 s, while a mixture of 2:1 ethanol:water requires
only 300 s to achieve a similar conversion. This increased rate of reaction is due to
inclusion of water, which is more protic than ethanol and thus promotes NaBH4 reduction
by acting as a more effective hydrogen donor31
. The inset to Figure 4.2a shows UV
spectra of the copolymer solution in 2:1 ethanol:water both before and after reaction, and
it is clearly seen that the AmBP absorbance at 300 nm has disappeared after reaction with
a corresponding increase of a different species at 250 nm, likely the diphenyl methanol
product32
. This chemical reduction is not accompanied by any molecular weight
variation, as evidenced by the nearly identical SEC traces of both NaBH4-treated and

102
-untreated samples in Figure 4.2b. Reduction with NaBH4 therefore does not significantly
degrade the polymer chain, and when combined with the lack of any additional
absorbance peaks in the post-reaction UV spectrum it appears that NaBH4 is a selective
and effective reducing agent for AmBP in our acrylamide-based copolymer systems. We
note that while UV spectra would be insensitive to reduction of amide or carboxylic acid
in our materials, reaction of these functionalities with NaBH4 is rare26–28
.
Figure 4.2: Deactivation of AmBP in PNiPAm-AAc-AmBP copolymer as studied by
UV/Vis and SEC. a) The inset shows spectra of polymer solution prior to and post
deactivation with 0.15 M NaBH4 (in 2:1 ethanol:water), showing the disappearance of
AmBP absorbance at 300 nm with a corresponding rise of photo-product at 250 nm.
Monitoring absorbance at 300 nm during deactivation in we observe >90% conversion
within 2500 s in ethanol solution, yet in a 2:1 ethanol:water mixture deactivation is
achieved within 300 s; b) SEC traces of polymer solutions prior to and after treatment
with NaBH4. The curves are nearly identical, indicating that treatment does not
significantly influence molecular weight distribution of the copolymer.

103
PNiPAm-AAc-AmBP hydrogel discs were then prepared by photo-crosslinking
400 μm diameter discs with different UV doses, as described above. After development
these samples were released into phosphate buffer, and their swelling monitored after at
least 90 min of immersion. In samples that were treated with NaBH4, swelling was
observed at least 90 min after dilution with fresh phosphate buffer. Results for discs that
were UV exposed to a dose between 3 and 100 J/cm2 are found in Figure 4.3, with areal
swelling defined as the swelled area (A1) divided by the initial mask area (A0). Swelling
ratios between 2.5 and 6 are obtained for these samples, with little difference between
samples treated with NaBH4 and those without. The consistency of the two swelling
Figure 4.3: Swelling curve of PNiPAm-AAc-AmBP hydrogel discs after one exposure.
Areal swelling is calculated as A1/A0, with A0 the area of the initial photo-mask. The
minimal difference in swelling between samples with and without NaBH4 treatment is
indication that the deactivation process does not disturb the existing hydrogel network.

104
curves, when combined with the clean UV and SEC experiments performed above,
seemingly indicate that NaBH4 treatment does not have any adverse effect on the
PNiPam hydrogel network, at least insofar as it influences the equilibrium swelling
behavior of the material. We note that while it is possible to prepare hydrogels at 1 and 2
J/cm2 UV dose, these samples no longer retain the shape of the photo-mask and are
poorly defined, likely indicating that these doses are close to the gel point for the system.
Figure 4.4: Final versus initial swelling for PNiPAm-AAc-AmBP hydrogel discs
subjected to the protocol depicted in Figure 4.1. By treating the hydrogel discs with
NaBH4 we are able to recover over 85% of the initial swelling over the entire first
exposure energy range. Conversely, we see that the sample without NaBH4 treatment was
only able to achieve a fraction of the initial swelling. At high energy the curves intersect
due to conversion of all AmBP photo-crosslinker in the first UV exposure step.

105
After the initial swelling, hydrogel discs are then heated (deswelled) and exposed
to UV radiation overnight to react any remaining AmBP in the copolymer. Cooling the
discs to room temperature and reswelling for at least 90 min, we are then able to measure
the equilibrium swelling area A2 and compare to the initial swelling, with A2/A1 reported
in Figure 4.4. This quantity represents the fraction of initial swelling recovered after UV
exposure, and should be a good indicator of whether AmBP has been deactivated. It is
clear that samples which have not been treated with NaBH4 recover significantly less
swelling than those which have been treated. In fact, over 50 individual discs which have
been swelled in the presence of NaBH4 recover an average of 98% of their initial
swelling across the entire range of UV doses investigated, with a minimum average
recovery of 92% for any given initial UV dose. Conversely, untreated samples can
recover as little as 25% at low initial UV dose. The gap between treated and untreated
samples narrows with increasing first exposure dose because at high energy nearly all the
AmBP has been reacted in the initial crosslinking step, limiting crosslink formation
during the second exposure step. At lower than 3 J/cm2, significant sample to sample
variation was observed, again likely due to the fact that at this low dose the sample is
very near its gel point. This is strong evidence that sodium borohydride effectively
reduces AmBP photo-crosslinker to prepare UV-stable hydrogel films.
Estimation of extent of reaction after UV irradiation
To estimate the extent of AmBP reaction in these discs, we now consider the
theory for equilibrium swelling of a polyelectrolyte gel, as discussed by Rubinstein and
Colby33
. In this work, the equilibrium swelling of a polyelectrolyte gel without salt in the
preparation state scales as

106
KNQ (4.1)
where Q is the equilibrium volumetric swelling ratio, K encompasses material constants,
N is the degree of polymerization between crosslinks, and α an exponent which depends
upon the salt concentration of the swelling solution ( 23 and 53 in the low and high
salt limits, respectively). Our materials fall near the low salt limit as the concentration of
charged monomer is significantly higher than that of salt in the swelling medium. For our
AmBP copolymer gels, the degree of polymerization between crosslinks can be
calculated as
pxN
AmBP
1 (4.2)
with AmBPx the molar fraction of AmBP in the copolymer and p the extent of reaction of
the photo-crosslinker. By combining equations (4.1) and (4.2) we determine that in our
copolymer gels the extent of reaction can be estimated by the equilibrium swelling as
32
32
1
iAmBP
i
QKxp . (4.3)
The material constant K is calculated by considering that after 50 J/cm2 the material is
fully crosslinked ( 1p ), and the average areal swelling ratio for higher energies (Q for
50 – 100 J/cm2) is 2.64 ± 0.09. We then calculate the extents of reaction achieved after
the first ( 1p , Figure 4.1 step iii) and second ( 2p , Figure 4.1 step v) UV exposures,
reporting these results in Figure 4.5.
As observed in Figure 4.5a, extents of reaction after the first UV exposure range
from 0.58 to 1.0 as energy increases, with little difference between samples that have
been treated with NaBH4 and those that have not. Following the second UV exposure

107
step, extents of reaction 2p remain nearly identical for NaBH4 treated samples, while
values increase substantially for the control discs (Figure 4.5b). Extents of reaction for all
control discs are estimated as greater than one, which likely represents limitations of both
the experimental procedure and the theoretical model. At low first exposure energies, the
values of 2p are especially high, which we believe is due to the hydrogel discs having
Figure 4.5: Extent of reaction of AmBP photo-crosslinker in PNiPAm hydrogel discs
following each UV exposure. Values are calculated based upon equilibrium swelling
theory of a polyelectrolyte gel in the low salt limit33
. a) After the first UV exposure,
extent of reaction 1p ranges from 0.59 to 1.0, and results are consistent between samples
treated with NaBH4 and controls; b) After deswelling and subsequent UV irradiation,
extent of reaction 2p for samples that have been treated with NaBH4 are nearly identical
to their 1p values, while controls exhibit significantly higher values, further evidence that
reduction with NaBH4 prevents crosslinking upon subsequent UV irradiation in these
photo-patterned hydrogels.

108
some sol fraction which is extracted prior to the deswelled exposure. What is clear,
however, is that the observed difference in 2p between samples treated with and without
NaBH4 indicates that NaBH4 is an effective agent for deactivation of AmBP photo-
crosslinker, and may potentially enable the preparation of complex UV-stable hydrogel
materials composed of multiple, independently photo-patterned layers.
Finally, as proof-of-concept that NaBH4 chemical reduction can be integrated to a
multistory fabrication procedure, we prepared a multilayer strip containing two photo-
crosslinkable copolymers with the same AmBP photo-crosslinker. For this experiment,
fluorescent PNiPAm-AAc-AmBP-RhBMA was first photo-patterned, yielding a 800 x
100 μm rectangle with two distinct UV doses (each half subjected to either 100 or 10
J/cm2) within the uncrosslinked polymer film matrix. This substrate-bound film was then
subjected to repeated 15 s immersions in solutions prepared with 2:1 ethanol:water to
sequentially stabilize the sacrificial layer, deactivate AmBP, and remove residual salt
from the sample surface. A 30 s dry was included between each step to limit swelling of
the film. After this process, development was completed in a neat 2:1 ethanol:water
mixture to yield a rectangular hydrogel feature on the surface. It should be noted that
despite our efforts the portion of the hydrogel film with a lower crosslinking dose (10
J/cm2) underwent extensive swelling during development, as seen in Figure 4.6i and iii.
Poly(p-methylstyrene-AmBP) was then spin cast onto the wafer and a larger rectangular
photo-mask (900 x 200 μm) aligned to the fluorescent PNiPAm feature for an exposure
of 100 J/cm2 over the entire region. A larger mask was required in this step in order to
restrict swelling over the entire hydrogel surface. If photo-crosslinker in the underlying
film were not deactivated, this final UV exposure would fully crosslink the entire

109
PNiPAm film, removing any swelling contrast and yielding a bilayer with homogeneous
properties over its area. This is exactly what is seen in Figure 4.6i-ii, where upon swelling
very little change is noticed from 55 °C to 30 °C, indicating that photo-crosslinker in the
PNiPAm layer has fully crosslinked over the entire sample. If, however, the photo-
crosslinker has been effectively converted to a nonreactive product the swelling contrast
will remain in the hydrogel layer and the bilayer will demonstrate different properties in
each half of the rectangle. This is precisely what is shown in Figure 4.6iii-iv, as upon
cooling from 55 °C to 30 °C, one half of the strip curls significantly more than the other.
Figure 4.6: Fluorescence microscopy images of PNiPAm-AAc-AmBP-RhBMA/PpMS-
AmBP bilayers. The untreated bilayer in both the deswelled (i) and swelled (ii) states is
nearly flat. Importantly, no distinction in swelling contrast is observed between the halves
of this sample. The bilayer that has been treated with NaBH4 swells from the flat (iii)
state to a curled cylinder (iv) with a distinct difference between low and high swelling
areas, indicating successful deactivation of AmBP.

110
Shown in Figure 4.6v, one observes that the right side of the sample is nearly a complete
cylinder while the left side is mostly flat, indicating that a significant swelling contrast
remains in the sample. This is proof that chemical reduction of AmBP in the PNiPAm
hydrogel materials effectively prevents additional photo-chemical reaction due to
imparted UV, making these materials environmentally stable and permitting the
fabrication of multilayer materials with the same photo-chemistry.
4.5 Conclusion
Deactivation of benzophenone photo-crosslinker by reduction with sodium
borohydride was monitored by UV/Vis and hydrogel disc swelling. Nearly complete
deactivation was seen within five minutes in a mixture of ethanol and water, and treated
hydrogel recovered at least 85% of its initial swelling after full conversion of AmBP.
These results indicate that UV-stable hydrogels can be prepared by NaBH4 reduction of
residual unreacted AmBP groups in benzophenone based photo-crosslinkable hydrogel
materials. Finally, as proof-of-concept that this technique can be integrated to a
multilayer fabrication procedure, a bilayer was prepared from photo-crosslinkable
poly(N-isopropylacrylamide) and poly(p-methylstyrene), demonstrating promise for
complex photo-patterned hydrogel systems.
4.6 Future directions
While treatment with NaBH4 was moderately successful, this is certainly not an
ideal method for deactivation of benzophenone in these materials. First, it appears that
the NaBH4 deactivation process for photo-patterned hydrogels imparts some added stress

111
to the samples. This has been determined by first photo-patterning a hydrogel sample
which can be actuated from a target shape to a flat state by swelling and deswelling
through the LCST. Treatment with NaBH4 as in Figure 4.1 proved to affect this actuation,
as samples would not completely flatten upon deswelling. Additionally, the protocols
used to prepare multilayer materials are far from ideal, as swelling of a layer during
deactivation affects deposition and alignment of the next layer, and the sacrificial layer is
compromised by the presence of Na+ and the formation of diborane and hydrogen gases.
It is certainly a viable option for AmBP deactivation, yet practical use requires
modification of the sacrificial layer (preferably not ionically crosslinked) and
optimization of solvent conditions. NaBH4 is, however, still the best candidate for
chemically reducing AmBP as other reagents are likely too aggressive and would damage
the polymer network in addition to reducing benzophenone (treatment with KOH and
sec-butanol did just this).
If, however, one is willing to allow some amount of crosslinking to occur during
the deactivation process, incorporation of sacrificial hydrogen abstraction substrates may
be an attractive alternative to chemical reduction. This process consists of swelling the
crosslinked gel with a small molecule during exposure which will preferentially react
with the n-π* triplet state of AmBP. This converts residual AmBP, prevents formation of
an elastically active crosslink, and instead grafts a small molecule species to the gel
network. The table of hydrogen abstraction rates provided in Chapter 2 thus gives
guidance for selection of candidate materials for this process. Since PNiPAm contains
tertiary hydrogen alpha to an amide, we must find functional groups that will be more
reactive than this species, namely either secondary or primary hydrogen on the alpha

112
carbon to a nitrogen atom (the order of reactivity of hydrogen alpha to amine is reversed
from aliphatic groups: 1° > 2° > 3°). Small molecule amines are therefore attractive
candidate materials, and given the appropriate swelling of the hydrogel network could be
effective at benzophenone deactivation. In fact, preliminary experiments with
triethylamine show promise for AmBP deactivation. This process may not be completely
efficient, however, and it must be considered that amines with multiple hydrogen at the
alpha positions could lead to crosslinking in their own right by reaction with more than
one photo-crosslinker (assumedly concentration of these species would be high enough to
prevent crosslink formation).
Because this process would likely not be 100% efficient, it is also worth
considering the possibility of incorporating functionality to the small molecule that would
increase swelling in aqueous media. For instance, grafting of polar groups (i.e. carboxylic
or sulfonic acids) during benzophenone deactivation would serve to increase swelling of
the hydrogel, perhaps offsetting whatever extra crosslinks form. Amino acids are
especially attractive candidates for such reactions as they are water soluble and contain
both the polar group and candidate hydrogen for abstraction by benzophenone. One of the
simplest molecules to begin with is alanine, which has a single hydrogen alpha to amine,
which would prevent crosslink formation by multiple reaction. This hydrogen is also
located alpha to the acid group, however, and may not undergo the charge transfer
necessary for rapid abstraction. Instead, β-alanine may be more electronically favorable
for abstraction due to an ethyl spacer between amine and acid. The use of amino acids
would permit a systematic study of their influence in deactivation and also provide

113
evidence for the ability to tether different functionalities to previously crosslinked
hydrogels.
4.7 References
1. Toomey, R., Freidank, D. & Rühe, J. Swelling Behavior of Thin, Surface-Attached
Polymer Networks. Macromolecules 37, 882–887 (2004).
2. Orlov, Y., Xu, X. & Maurer, G. Equilibrium swelling of N-isopropyl acrylamide
based ionic hydrogels in aqueous solutions of organic solvents: Comparison of
experiment with theory. Fluid Phase Equilib. 249, 6–16 (2006).
3. Beines, P. W., Klosterkamp, I., Menges, B., Jonas, U. & Knoll, W. Responsive
thin hydrogel layers from photo-cross-linkable poly(N-isopropylacrylamide)
terpolymers. Langmuir 23, 2231–2238 (2007).
4. Mateescu, A., Wang, Y., Dostalek, J. & Jonas, U. Thin Hydrogel Films for Optical
Biosensor Applications. Membranes 2, 40–69 (2012).
5. Schwartz, V. B. et al. Antibacterial Surface Coatings from Zinc Oxide
Nanoparticles Embedded in Poly(N-isopropylacrylamide) Hydrogel Surface
Layers. Adv. Funct. Mater. 22, 2376–2386 (2012).
6. Kuckling, D., Adler, H.-J. P. & Arndt, K.-F. Polymer Gels. 833, 312–325
(American Chemical Society: Washington, DC, 2002).
7. Castellanos, A. et al. Size-Exclusion “Capture and Release” Separations Using
Surface-Patterned Poly(N-isopropylacrylamide) Hydrogels. Langmuir 23, 6391–
6395 (2007).
8. Saeed, A., Georget, D. M. R. & Mayes, A. G. Synthesis, characterisation and
solution thermal behaviour of a family of poly (N-isopropyl acrylamide-co-N-
hydroxymethyl acrylamide) copolymers. React. Funct. Polym. 70, 230–237
(2010).
9. Liu, D., Bastiaansen, C. W. M., Den Toonder, J. M. J. & Broer, D. J. Single-
composition three-dimensionally morphing hydrogels. Soft Matter 9, 588 (2013).
10. Ma, M., Guo, L., Anderson, D. G. & Langer, R. Bio-inspired polymer composite
actuator and generator driven by water gradients. Science 339, 186–9 (2013).

114
11. De Rossi, D. & Chiarelli, P. Biomimetic Macromolecular Actuators. Macro-ion
Characterization 548, 517–530 (1993).
12. Meng, H. A Brief Review of Stimulus-active Polymers Responsive to Thermal,
Light, Magnetic, Electric, and Water/Solvent Stimuli. J. Intel. Mat. Syst. Str. 21,
859–885 (2010).
13. Harmon, M. E., Kuckling, D., Pareek, P. & Frank, C. W. Photo-Cross-Linkable
PNIPAAm Copolymers. 4. Effects of Copolymerization and Cross-Linking on the
Volume-Phase Transition in Constrained Hydrogel Layers. Langmuir 19, 10947–
10956 (2003).
14. Kim, J., Hanna, J. A., Hayward, R. C. & Santangelo, C. D. Thermally responsive
rolling of thin gel strips with discrete variations in swelling. Soft Matter 8, 2375–
2381 (2012).
15. Kim, J., Hanna, J. A., Byun, M., Santangelo, C. D. & Hayward, R. C. Designing
Responsive Buckled Surfaces by Halftone Gel Lithography. Science 335, 1201–
1205 (2012).
16. Buller, J., Laschewsky, A. & Wischerhoff, E. Photoreactive oligoethylene glycol
polymers – versatile compounds for surface modification by thin hydrogel films.
Soft Matter 9, 929–937 (2013).
17. Kuckling, D. & Pareek, P. Bilayer hydrogel assembly. Polymer 49, 1435–1439
(2008).
18. Williams, J. L. R. & Border, D. G. The preparation and properties of photoreactive
polymers. I. 2-(arylvinyl)-N-vinylpyridinium arylsulfonate polymers. Macromol.
Chem. 73, 203–214 (1964).
19. Shindo, Y., Katagiri, N., Ebisuno, T., Hasegawa, M. & Mitsuda, M. Network
formation and swelling behavior of photosensitive poly(vinyl alcohol) gels
prepared by photogenerated crosslinking. Angew. Makromol. Chem. 240, 231–239
(1996).
20. Zakharchenko, S., Puretskiy, N., Stoychev, G., Stamm, M. & Ionov, L.
Temperature controlled encapsulation and release using partially biodegradable
thermo-magneto-sensitive self-rolling tubes. Soft Matter 6, 2633 (2010).
21. Chiappelli, M. C. & Hayward, R. C. Photonic multilayer sensors from photo-
crosslinkable polymer films. Adv. Mater. 24, 6100–4 (2012).
22. Belardi, J., Schorr, N., Prucker, O. & Rühe, J. Artificial Cilia: Generation of
Magnetic Actuators in Microfluidic Systems. Adv. Funct. Mater. 21, 3314–3320
(2011).

115
23. Ito, Y., Chen, G., Guan, Y. & Imanishi, Y. Patterned Immobilization of
Thermoresponsive Polymer. Langmuir 13, 2756–2759 (1997).
24. Chen, G., Imanishi, Y. & Ito, Y. Photolithographic Synthesis of Hydrogels.
Macromolecules 31, 4379–4381 (1998).
25. Chen, G., Ito, Y. & Imanishi, Y. Micropattern Immobilization of a pH-Sensitive
Polymer. Macromolecules 30, 7001–7003 (1997).
26. Oxidizing and Reducing Agents. Handbook of Reagents for Organic Synthesis
394–400 (1999).
27. Hutchins, R. O. et al. Nucleophilic borohydride: selective reductive displacement
of halides, sulfonate esters, tertiary amines, and N,N-disulfonimides with
borohydride reagents in polar aprotic solvents. J. Org. Chem. 43, 2259–2267
(1978).
28. Kikugawa, Y., Ikegami, S. & Yamada, S.-I. Chemistry of Diborane and Sodium
Borohydride. VI. The Reaction of Amides with Sodium Borohydride. Chem.
Pharm. Bull. 17, 98–104 (1969).
29. Christensen, S. K., Chiappelli, M. C. & Hayward, R. C. Gelation of Copolymers
with Pendent Benzophenone Photo-Cross-Linkers. Macromolecules 45, 5237–
5246 (2012).
30. Linder, V., Gates, B. D., Ryan, D., Parviz, B. A. & Whitesides, G. M. Water-
soluble sacrificial layers for surface micromachining. Small 1, 730–6 (2005).
31. Ward, D. E. & Rhee, C. K. Chemoselective reductions with sodium borohydride.
Can. J. Chem. 67, 1206–1211 (1989).
32. Lang, L. Absorption Spectra in the Ultraviolet and Visible Region. 73 (Academic
Press: New York, 1967).
33. Rubinstein, M., Colby, R. H., Dobrynin, A. V & Joanny, J.-F. Elastic Modulus and
Equilibrium Swelling of Polyelectrolyte Gels. Macromolecules 29, 398–406
(1996).

116
CHAPTER 5
CONCLUSIONS
The goal of this dissertation was to both deepen and broaden our understanding of
copolymers with pendent benzophenone (BP) in relation to both established applications
and novel directions in materials science. Photo-reaction of these BP copolymers was
explored in attempts to achieve three distinct goals: (1) robust and efficiently photo-
crosslinkable solid polymer films, (2) photo-reacted polymer blends with disordered
bicontinuous nanostructures, and (3) photo-patterned hydrogel materials with
environmental UV stability. The work described herein makes it clear that copolymers
with pendent BP are a useful and versatile tool in materials science, however limitations
exist which restrict their efficacy in certain applications.
As described, copolymers with pendent BP are very synthetically accessible,
requiring only the preparation of a BP monomer and subsequent free radical
polymerization, yielding photo-reactive materials which abstract hydrogen from a
multitude of substrates. These represent the greatest benefits of BP copolymers: synthetic
ease and versatile reactivity, requiring no additional functionalization of complementary
materials for effective reaction. Photo-reaction proceeds via abstraction of hydrogen
followed by radical generation, which then initiates further reaction in multiple ways.
This nonspecific reactivity of BP is what largely serves to define practical and
impractical applications for copolymers with pendent BP, and this thesis demonstrates
both their advantages and limitations. In Chapters 2 and 4, wherein highly crosslinkable
films and photo-patterned hydrogels were prepared with BP copolymers, the reaction is

117
allowed to be somewhat undefined, and as long as an elastically active crosslink is
formed after radical generation, BP copolymers are an ideal tool. In Chapter 3, wherein
we attempt to crosslink or graft BP copolymer to a corresponding homopolymer, the
reaction must be more controlled and as such BP copolymers are not ideal tools.
In the case of photo-crosslinkable polymeric materials, copolymers with pendent
BP are a robust and attractive system. We have shown that their crosslinking is much
more efficient than a doped material, and as long as a sufficiently high concentration of
BP exists in the copolymers, gelation will proceed so long as hydrogen abstraction is not
limited solely to the polymer backbone. This work provides a much improved design
criteria for selection of copolymer chemistries, and this knowledge will facilitate future
studies. The one potential downside of using AmBP photochemistry for this application
lies in its relatively low reactivity to UV. If a rapidly photo-crosslinkable polymer film is
desired one may turn either to a more highly reactive substituted BP or an alternative
photochemistry such as a phenyl azide. With higher reactivity comes an entirely different
set of problems, however, and the materials will be significantly more sensitive to
ambient illumination making them challenging from a practical aspect.
The relatively low reactivity of AmBP to UV illumination has proven to be an
advantage for photo-patterning of hydrogel materials, as it allows facile spatial variation
in crosslinking and thus swelling, and BP copolymers have proven once more to be an
appealing material system for the preparation of complex hydrogels. Multilayer hydrogel
films are possible through treatment of a patterned hydrogel film with NaBH4, yet
modifications must be made to the procedure to prevent extensive swelling of the film
prior to deposition of the next layer. This is not a downfall of copolymers with pendent

118
BP, but rather a reflection of solvent conditions currently used, and alternative routes to
deactivation of BP photo-crosslinker are outlined in Chapter 4.
Copolymers with pendent BP are not ideal for every application, however, as
evidenced by our failed attempts to obtain bicontinuous nanostructures in Chapter 3. It
may yet be possible to obtain the desired results from critical polymer blends with BP
copolymers, yet it is clear that these materials are not ideal for solution-state photo-
grafting. Their failure lies in the aforementioned unspecific reaction of BP, which is
generally considered as an advantage yet in this case precludes the realization of our goal.
Because photo-reactive molecules which undergo self-cycloaddition will only react with
themselves, in order to achieve exclusively A-B grafting in these systems,
functionalization of both materials is necessary. This would result in a much more
specific photo-reaction, and an obvious candidate photochemistry is found in thiol-ene
reaction, which could promote graft copolymer formation much more efficiently than a
radical generating agent such as BP.
Copolymers with pendent BP photo-crosslinkers have therefore proved to be
useful tools for the achievement of complex materials systems. This work has expanded
knowledge of the benefits and downfalls of their use, providing guidance for materials
selection in future studies. Even though limitations do exist, they are far outweighed by
advantages, and thus BP copolymers remain powerful and extremely useful
photochemical tools for technological applications which will be increasingly used in the
future.

119
BIBLIOGRAPHY
Agolini, F. Polymeric phenone photosensitizers and blends thereof with other polymers.
US Patent 3,632,493 1–4 (1972).
Ahn, H., Naidu, S., Ryu, D. Y. & Cho, J. Phase Behavior of a Weakly Interacting
Polystyrene and Poly(n-hexyl methacrylate) System: Evidence for the Coexistence
of UCST and LCST. Macromol. Rapid Commun. 30, 469–474 (2009).
Allen, N. S. et al. Photochemistry and photopolymerization activity of novel 4-
alkylamino benzophenone initiators-synthesis, characterization, spectroscopic and
photopolymerization activity. Eur. Polym. J. 26, 1345–1353 (1990).
Allen, N. S. et al. Photochemistry of Novel 4-Alkylamino Benzophenone Initiators - A
Conventional Laser Flash-Photolysis and Mass-Spectrometry Study. J. Photochem.
Photobiol., A 54, 367–388 (1990).
Aspari, P. et al. Photoinduced electron transfer between triethylamine and aromatic
carbonyl compounds: the role of the nature of the lowest triplet state. Trans. Faraday
Soc. 92, 1689 (1996).
Barlow, J. W. & Paul, D. R. Polymer alloys. Ann. Rev. Mater. Sci. 1981. 11, 299–319
(1981).
Bates, F. S. Polymer-Polymer Phase-Behavior. Science 251, 898–905 (1991).
Bates, F. S. et al. Polymeric bicontinuous microemulsions. Phys. Rev. Lett. 79, 849–852
(1997).
Bates, F. S. & Fredrickson, G. H. Block copolymers - Designer soft materials. Physics
Today 52, 32–38 (1999).
Bauer, B. J., Briber, R. M. & Han, C. C. Small-Angle Neutron-Scattering Studies of
Compatible Blends of Linear Polyvinyl Methyl-Ether) and Cross-Linked Deuterated
Polystyrene. Macromolecules 22, 940–948 (1989).
Beines, P. W., Klosterkamp, I., Menges, B., Jonas, U. & Knoll, W. Responsive thin
hydrogel layers from photo-cross-linkable poly(N-isopropylacrylamide) terpolymers.
Langmuir 23, 2231–2238 (2007).

120
Belardi, J., Schorr, N., Prucker, O. & Rühe, J. Artificial Cilia: Generation of Magnetic
Actuators in Microfluidic Systems. Adv. Funct. Mater. 21, 3314–3320 (2011).
Bernstein, R. E., Cruz, C. A., Paul, D. R. & Barlow, J. W. LCST Behavior in Polymer
Blends. Macromolecules 10, 681–686 (1977).
Bhardwaj, R. & Mohanty, A. K. Modification of brittle polylactide by novel
hyperbranched polymer-based nanostructures. Biomacromolecules 8, 2476–84
(2007).
Bhasikuttan, A. C., Singh, A. K., Palit, D. K., Sapre, A. V. & Mittal, J. P. Laser Flash
Photolysis Studies on the Monohydroxy Derivatives of Benzophenone. J. Phys.
Chem. A 102, 3470–3480 (1998).
Briber, R. M. & Khoury, F. The phase diagram and morphology of blends of
poly(vinylidene fluoride) and poly(ethyl acrylate). Polymer 28, 38–46 (1987).
Briber, R. M. & Bauer, B. J. Effect of Cross-Links on the Phase-Separation Behavior of a
Miscible Polymer Blend. Macromolecules 21, 3296–3303 (1988).
Browarzik, D. Calculation of the phase behavior of polystyrene plus poly(n-pentyl
methacrylate) blends. J. Macromol. Sci., Part A: Pure Appl. Chem. A42, 1339–1353
(2005).
Buller, J., Laschewsky, A. & Wischerhoff, E. Photoreactive oligoethylene glycol
polymers – versatile compounds for surface modification by thin hydrogel films. Soft
Matter 9, 929–937 (2013).
Cahn, J. W. & Hilliard, J. E. Free Energy of a Nonuniform System. I. Interfacial Free
Energy. J. Chem. Phys. 28, 258 (1958).
Cahn, J. W. Phase separation by spinodal decomposition in isotropic systems. J. Chem.
Phys. 42, 93 (1965).
Carlini, C., Ciardelli, F., Donati, D. & Gurzoni, F. Polymers containing side-chain
benzophenone chromophores: a new class of highly efficient polymerization
photoinitiators. Polymer 24, 599–606 (1983).

121
Carlini, C. & Gurzoni, F. Optically active polymers containing side-chain benzophenone
chromophores. Polymer 24, 101–106 (1983).
Carroll, G. T., Triplett, L. D., Moscatelli, A., Koberstein, J. T. & Turro, N. J.
Photogeneration of gelatinous networks from pre-existing polymers. J. Appl. Polym.
Sci. 122, 168–174 (2011).
Castellanos, A. et al. Size-Exclusion “Capture and Release” Separations Using Surface-
Patterned Poly(N-isopropylacrylamide) Hydrogels. Langmuir 23, 6391–6395 (2007).
Charlesby, A. Gel formation and molecular weight distribution in long-chain polymers.
Proc. R. Soc. London, Ser. A 222, 542–557 (1954).
Charlesby, A. & Pinner, S. H. Analysis of the Solubility Behaviour of Irradiated
Polyethylene and Other Polymers. Proc. R. Soc. London, Ser. A 249, 367–386
(1959).
Chen, G., Ito, Y. & Imanishi, Y. Micropattern Immobilization of a pH-Sensitive Polymer.
Macromolecules 30, 7001–7003 (1997).
Chen, G., Imanishi, Y. & Ito, Y. Photolithographic Synthesis of Hydrogels.
Macromolecules 31, 4379–4381 (1998).
Chen, L., Hallinan, D. T., Elabd, Y. A. & Hillmyer, M. A. Highly Selective Polymer
Electrolyte Membranes from Reactive Block Polymers. Macromolecules 42, 6075–
6085 (2009).
Chen, W. J., David, D. J., MacKnight, W. J. & Karasz, F. E. Miscibility and phase
behavior in blends of poly(vinyl butyral) and poly(methyl methacrylate).
Macromolecules 34, 4277–4284 (2001).
Chen, Y. B. et al. Enhancement of anhydrous proton transport by supramolecular
nanochannels in comb polymers. Nature Chem. 2, 503–508.
Chiappelli, M. C. & Hayward, R. C. Photonic multilayer sensors from photo-
crosslinkable polymer films. Adv. Mater. 24, 6100–4 (2012).
Choi, J.-E. & Ko, K.-Y. Zeolite H-beta: an Efficient and Recyclable Catalyst for the
Tetrahydropyranylation of Alcohols and Phenols , and the Deprotection of
Tetrahydropyranyl Ethers. Bull. Korean Chem. Soc. 22, 1177–1178 (2001).

122
Choi, J., Lee, K. M., Wycisk, R., Pintauro, P. N. & Mather, P. T. Nanofiber Network Ion-
Exchange Membranes. Macromolecules 41, 4569–4572 (2008).
Christensen, S. K., Chiappelli, M. C. & Hayward, R. C. Gelation of Copolymers with
Pendent Benzophenone Photo-Cross-Linkers. Macromolecules 45, 5237–5246
(2012).
Chujo, Y., Sada, K. & Saegusa, T. Polyoxazoline having a coumarin moiety as a pendant
group. Synthesis and photogelation. Macromolecules 23, 2693–2697 (1990).
Chung, H., Ohno, K., Fukuda, T. & Composto, R. J. Self-regulated structures in
nanocomposites by directed nanoparticle assembly. Nano Lett. 5, 1878–1882 (2005).
Clausse, M., Peyrelasse, J., Heil, J., Boned, C. & Lagourette, B. Bicontinuous Structure
Zones in Micro-Emulsions. Nature 293, 636–638 (1981).
Cockburn, E. S., Davidson, R. S. & Pratt, J. E. The photocrosslinking of styrylpyridinium
salts via a [2 + 2]-cycloaddition reaction. J. Photochem. Photobiol., A 94, 83–88
(1996).
Cohen, S. G. & Chao, H. M. Photoreduction of aromatic ketones by amines. Studies of
quantum yields and mechanism. J. Am. Chem. Soc. 90, 165–173 (1968).
Cohen, S. G. & Cohen, J. I. Photoreduction of aminobenzophenones in nonpolar media.
Effects of tertiary amines. J. Phys. Chem. 72, 3782–3793 (1968).
Cohen, S. G., Parola, A. & Parsons, G. H. Photoreduction by Amines. Chem. Rev. 73,
141–161 (1973).
Cooke, D. M. & Shi, A. C. Effects of polydispersity on phase behavior of diblock
copolymers. Macromolecules 39, 6661–6671 (2006).
Costamagna, V., Strumia, M., Lopez-Gonzalez, M. & Riande, E. Gas transport in surface
grafted polypropylene films with poly(acrylic acid) chains. J. Polym. Sci., Part B:
Polym. Phys. 45, 2421–2431 (2007).
Cox, J. R., Simpson, J. H. & Swager, T. M. Photoalignment Layers for Liquid Crystals
from the Di-π-methane Rearrangement. J. Am. Chem. Soc. 135, 640–3 (2013).
Decker, C. & Bendaikha, T. Interpenetrating polymer networks. II. Sunlight-induced
polymerization of multifunctional acrylates. J. Appl. Polym. Sci. 70, 2269–2282
(1998).

123
Dedinas, J. Photoreduction of benzophenone in benzene. I. Mechanism of secondary
reactions. J. Phys. Chem. 75, 181–186 (1971).
DeGennes, P. G. Effect of cross-links on a mixture of polymers. J. Phys. Lett. 40, L–69
(1979).
De Rossi, D. & Chiarelli, P. Biomimetic Macromolecular Actuators. Macro-ion
Characterization 548, 517–530 (1993).
Derouiche, A., Bettachy, A., Benhamou, M. & Daoud, M. Kinetics of microphase
separation in crosslinked polymer blends. Macromolecules 25, 7188–7191 (1992).
Do, H.-S., Park, Y.-J. & Kim, H.-J. Preparation and adhesion performance of UV-
crosslinkable acrylic pressure sensitive adhesives. J. Adhes. Sci. Technol. 20, 1529–
1545 (2006).
Dorman, G. & Prestwich, G. D. Benzophenone Photophores in Biochemistry.
Biochemistry 33, 5661–5673 (1994).
Doytcheva, M. et al. Ultraviolet-induced crosslinking of solid poly(ethylene oxide). J.
Appl. Polym. Sci. 64, 2299–2307 (1997).
Eisele, G., Fouassier, J. P. & Reeb, R. Kinetics of photocrosslinking reactions of a
DCPA/EA matrix in the presence of thiols and acrylates. J. Polym. Sci., Part A:
Polym. Chem. 35, 2333–2345 (1997).
Ellison, C. J. et al. Bicontinuous Polymeric Microemulsions from Polydisperse Diblock
Copolymers. J. Phys. Chem. B 113, 3726–3737 (2009).
El Mabrouk, K. & Bousmina, M. Effect of molecular weight and shear on phase diagram
of PS/PVME blend. Rheol. Acta 45, 959–969 (2006).
Filippone, G., Dintcheva, N. T., Acierno, D. & La Mantia, F. P. The role of organoclay in
promoting co-continuous morphology in high-density poly(ethylene)/poly(amide)
6 blends. Polymer 49, 1312–1322 (2008).
Filippone, G., Romeo, G. & Acierno, D. Role of Interface Rheology in Altering the Onset
of Co-Continuity in Nanoparticle-Filled Polymer Blends. Macromol. Mater. Eng.
296, 658–665 (2011).

124
Flory, P. J. Molecular size distribution in three dimensional polymers. II. Trifunctional
branching units. J. Am. Chem. Soc. 63, 3091–3096 (1941).
Flory, P. J. Constitution of Three-dimensional Polymers and the Theory of Gelation. J.
Phys. Chem. 46, 132–140 (1942).
Fouassier, J. P., Ruhlmann, D., Graff, B., Morletsavary, F. & Wieder, F. Excited-State
Processes in Polymerization Photoinitiators. Prog. Org. Coat. 25, 235–271 (1995).
Fredrickson, G. H. & Bates, F. S. Design of bicontinuous polymeric microemulsions. J.
Polym. Sci., Part B: Polym. Phys. 35, 2775–2786 (1997).
Galloway, J. A. & Macosko, C. W. Comparison of methods for the detection of
cocontinuity in poly(ethylene oxide)/polystyrene blends. Polym. Eng. Sci. 44, 714–
727 (2004).
Gan, P. & Paul, D. Phase behaviour of blends of homopolymers with α-
methylstyrene/acrylonitrile copolymer. Polymer 35, 1487–1502 (1994).
Gani, L., Tencé-Girault, S., Milléquant, M., Bizet, S. & Leibler, L. Co-continuous
Nanostructured Blend by Reactive Blending: Incorporation of High Molecular
Weight Polymers. Macromol. Chem. Phys. 211, 736–743 (2010).
George, B. & Dhamodharan, R. A study of the photopolymerization kinetics of methyl
methacrylate using novel benzophenone initiators. Polym. Int. 50, 897–905 (2001).
Ghoneim, N., Monbelli, A., Pilloud, D. & Suppan, P. Photochemical reactivity of para-
aminobenzophenone in polar and non-polar solvents. J. Photochem. Photobiol., A
94, 145–148 (1996).
Giering, L., Berger, M. & Steel, C. Rate Studies of Aromatic Triplet Carbonyls with
Hydrocarbons. J. Am. Chem. Soc. 96, 953–958 (1974).
Graves, R. & Pintauro, P. N. Polyphosphazene membranes. II. Solid-state
photocrosslinking of poly[(alkylphenoxy)(phenoxy)phosphazene] films. J. Appl.
Polym. Sci. 68, 827–836 (1998).
Green, M. D., Choi, J.-H., Winey, K. I. & Long, T. E. Synthesis of Imidazolium-
Containing ABA Triblock Copolymers: Role of Charge Placement, Charge Density,
and Ionic Liquid Incorporation. Macromolecules 45, 4749–4757 (2012).

125
Griep-Raming, N., Karger, M. & Menzel, H. Using benzophenone-functionalized
phosphonic acid to attach thin polymer films to titanium surfaces. Langmuir 20,
11811–4 (2004).
Griller, D., Howard, J. A., Marriott, P. R. & Scaiano, J. C. Absolute rate constants for the
reactions of tert-butoxyl, tert-butylperoxyl, and benzophenone triplet with amines:
the importance of a stereoelectronic effect. J. Am. Chem. Soc. 103, 619–623
(1981).
Gubbels, F. et al. Selective Localization of Carbon-Black in Immiscible Polymer Blends
- a Useful Tool to Design Electrical Conductive Composites. Macromolecules 27,
1972–1974 (1994).
Gubbels, F. et al. Design of Electrical Conductive Composites - Key Role of the
Morphology on the Electrical-Properties of Carbon-Black Filled Polymer Blends.
Macromolecules 28, 1559–1566 (1995).
Hadgraft, J., Hyde, A. J. & Richards, R. W. Diffusion of Polystyrene in Poly(Methyl
Methacrylate)+Benzene Solutions Measured by Photon-Correlation Spectroscopy.
Trans. Faraday Soc. 75, 1495–1505 (1979).
Harmon, M. E., Kuckling, D., Pareek, P. & Frank, C. W. Photo-Cross-Linkable
PNIPAAm Copolymers. 4. Effects of Copolymerization and Cross-Linking on the
Volume-Phase Transition in Constrained Hydrogel Layers. Langmuir 19, 10947–
10956 (2003).
Herzig, E., White, K., Schofield, A., Poon, W. & Clegg, P. Bicontinuous emulsions
stabilized solely by colloidal particles. Nat. Mater. 6, 966–971 (2007).
Higuchi, H., Yamashita, T., Horie, K. & Mita, I. Photo-Cross-Linking Reaction of
Benzophenone-Containing Polyimide and Its Model Compounds. Chem. Mater. 3,
188–194 (1991).
Hillmyer, M. A., Maurer, W. W., Lodge, T. P., Bates, F. S. & Almdal, K. Model
bicontinuous microemulsions in ternary homopolymer block copolymer blends. J.
Phys. Chem. B 103, 4814–4824 (1999).
Horie, K. et al. Rates of Quenching and Hydrogen Abstraction of Benzophenone Triplet
with Cumene and Oligostyrenes. Polym. J. (Tokyo, Jpn.) 16, 887–893 (1984).
Horie, K., Tsukamoto, M., Morishita, K. & Mita, I. Photochemistry in Polymer Solids. 5.
Decay of Benzophenone Phosphorescence in Polystyrene and in Polycarbonate.
Polym. J. (Tokyo, Jpn.) 17, 517–524 (1985).

126
Hu, H., Shangguan, Y. & Zuo, M. Investigation on LCST behavior of a new
amorphous/crystalline polymer blend: Poly (n-methyl methacrylimide)/poly
(vinylidene fluoride). J. Polym. Sci., Part B: Polym. Phys. 46, 1923–1931 (2008).
Hutchins, R. O. et al. Nucleophilic borohydride: selective reductive displacement of
halides, sulfonate esters, tertiary amines, and N,N-disulfonimides with borohydride
reagents in polar aprotic solvents. J. Org. Chem. 43, 2259–2267 (1978).
Ikeda, R. M., Rogers, F. F., Tocker, S. & Gelin, B. R. Photocrosslinking of polyethylene
using polyethylene-acryloxybenzophenone - a physical study. ACS Symp. Ser. 76–
89 (1976).
Ikeda, T., Kawaguchi, K., Yamaoka, H. & Okamura, S. Photoinduced and Radiation-
Induced Degradation of Vinyl-Polymers in Solution. 2. Photosensitized
Degradation of Poly-Alpha-Methylstyrene by Benzophenone. Macromolecules 11,
735–739 (1978).
Imagawa, A. & Tran-Cong, Q. Structure-Property Relationship of Polymer Blends with
Co-Continuous Structures Prepared by Photo-Cross-Linking. Macromolecules 28,
8388–8394 (1995).
Inbar, S., Linschitz, H. & Cohen, S. G. Nanosecond Flash Studies of Reduction of
Benzophenone by Aliphatic Amines. Quantum Yields and Kinetic Isotope Effects.
J. Am. Chem. Soc. 103, 1048–1054 (1981).
Inoue, T. Reaction-induced phase decomposition in polymer blends. Prog. Polym. Sci.
20, 119–153 (1995).
Isayeva, I., Kyu, T. & St. John Manley, R. Phase transitions, structure evolution, and
mechanical properties of blends of two crystalline polymers: poly (vinylidene
fluoride) and poly (butylene adipate). Polymer 39, 4599–4608 (1998).
Ishino, S., Nakanishi, H., Norisuye, T., Tran-Cong-Miyata, Q. & Awatsuji, Y. Designing
a polymer blend with phase separation tunable by visible light for computer-assisted
irradiation experiments. Macromol. Rapid Commun. 27, 758–762 (2006).
Ito, Y., Chen, G., Guan, Y. & Imanishi, Y. Patterned Immobilization of
Thermoresponsive Polymer. Langmuir 13, 2756–2759 (1997).
Jeon, H. K., Zhang, J. B. & Macosko, C. W. Premade vs. reactively formed
compatibilizers for PMMA/PS melt blends. Polymer 46, 12422–12429 (2005).

127
Jia, J., Sarker, M., Steinmetz, M. G., Shukla, R. & Rathore, R. Photochemical
Elimination of Leaving Groups from Zwitterionic Intermediates Generated via
Electrocyclic Ring Closure of alpha, beta-Unsaturated Anilides. J. Org. Chem. 73,
8867–8879 (2008).
Jiang, X., Luo, X. & Yin, J. Polymeric photoinitiators containing in-chain benzophenone
and coinitiators amine: Effect of the structure of coinitiator amine on
photopolymerization. J. Photochem. Photobiol., A 174, 165–170 (2005).
Kaczmarek, H., Kaminska, A., Swiatek, M. & Sanyal, S. Photoinitiated degradation of
polystyrene in the presence of low-molecular organic compounds. Eur. Polym. J.
36, 1167–1173 (2000).
Kammer, H. The phase behavior of polymer blends–Effects of thermodynamics and
rheology. Acta Polym. 42, 571–576 (1991).
Kaptan, H. Y. ESR study on the effect of temperature on the diffusion of oxygen into
PMMA and PVAC polymers. J. Appl. Polym. Sci. 71, 1203–1207 (1999).
Kataoka, K., Harada, A., Tamai, T. & Tran-Cong, Q. Phase behavior and
photocrosslinking kinetics of semi-interpenetrating polymer networks prepared from
miscible polymer blends. J. Polym. Sci., Part B: Polym. Phys. 36, 455–462 (1998).
Kato, K., Uchida, E., Kang, E.-T., Uyama, Y. & Ikada, Y. Polymer surface with graft
chains. Prog. Polym. Sci. 28, 209–259 (2003).
Kawazoe, N., Imagawa, A., Tamai, T. & Tran-Cong, Q. Photocrosslinking kinetics of
polymer blends undergoing spinodal decomposition process. J. Appl. Polym. Sci. 67,
885–893 (1998).
Kikugawa, Y., Ikegami, S. & Yamada, S.-I. Chemistry of Diborane and Sodium
Borohydride. VI. The Reaction of Amides with Sodium Borohydride. Chem. Pharm.
Bull. 17, 98–104 (1969).
Kim, C. & Paul, D. Miscibility of poly(methyl methacrylate) blends with halogen-
containing polycarbonates and copolymers. Polymer 33, 4929–4940 (1992).
Kim, C. K. & Paul, D. R. Effects of Polycarbonate Molecular-Structure on the Miscibility
with Other Polymers. Macromolecules 25, 3097–3105 (1992).

128
Kim, C. K. & Paul, D. R. Interaction Parameters for Blends Containing Polycarbonates.
1. Tetramethyl Bisphenol-a Polycarbonate Polystyrene. Polymer 33, 1630–1639
(1992).
Kim, J. K., Lee, H. H., Son, H. W. & Han, C. D. Phase Behavior and Rheology of
Polystyrene/Poly(α-methylstyrene) and Polystyrene/Poly(vinyl methyl ether) Blend
Systems. Macromolecules 31, 8566–8578 (1998).
Kim, J. K., Jang, J., Lee, D. H. & Ryu, D. Y. Phase behavior of polystyrene and poly(n-
pentyl methacrylate) blend with small amounts of symmetric polystyrene-block-
poly(n-pentyl methacrylate) copolymers. Macromolecules 37, 8599–8605 (2004).
Kim, J., Hanna, J. A., Byun, M., Santangelo, C. D. & Hayward, R. C. Designing
Responsive Buckled Surfaces by Halftone Gel Lithography. Science 335, 1201–1205
(2012).
Kim, J., Hanna, J. A., Hayward, R. C. & Santangelo, C. D. Thermally responsive rolling
of thin gel strips with discrete variations in swelling. Soft Matter 8, 2375–2381
(2012).
Komura, S. Mesoscale structures in microemulsions. J. Phys.: Condens. Matter 19,
(2007).
Koningsveld, R., Kleintjens, L. A. & Schoffeleers, H. M. Thermodynamic aspects of
polymer compatibility. Pure Appl. Chem. 39, 1–32 (1974).
Koulic, C. & Jérôme, R. Nanostructured Polyamide by Reactive Blending. 1. Effect of
the Reactive Diblock Composition. Macromolecules 37, 3459–3469 (2004).
Kressler, J., Higashida, N., Shimomai, K., Inoue, T. & Ougizawa, T. Temperature-
Dependence of the Interaction Parameter between Polystyrene and Poly(Methyl
Methacrylate). Macromolecules 27, 2448–2453 (1994).
Kuckling, D., Adler, H.-J. P. & Arndt, K.-F. Polymer Gels. 833, 312–325 (American
Chemical Society: Washington, DC, 2002).
Kuckling, D. & Pareek, P. Bilayer hydrogel assembly. Polymer 49, 1435–1439 (2008).

129
Kurata, M. & Tsunashima, Y. Viscosity-Molecular Weight Relationships and
Unperturbed Dimensions of Linear Chain Molecules. Polymer Handbook VII/1–
VII/83 (1999).
Lang, L. Absorption Spectra in the Ultraviolet and Visible Region. 73 (Academic Press:
New York, 1967).
Lee, J. S., Prabu, A. A. & Kim, K. J. UCST-Type Phase Separation and Crystallization
Behavior in Poly(vinylidene fluoride)/Poly(methyl methacrylate) Blends under an
External Electric Field. Macromolecules 42, 5660–5669 (2009).
Li, G., He, G., Zheng, Y., Wang, X. & Wang, H. Surface photografting initiated by
benzophenone in water and mixed solvents containing water and ethanol. J. Appl.
Polym. Sci. 123, 1951–1959 (2012).
Li, H., Yang, Y., Fujitsuka, R., Ougizawa, T. & Inoue, T. Studies on miscibility in
polycarbonate/poly(styrene-co-acrylonitrile) blends by ellipsometry. Polymer 40,
927–933 (1999).
Li, L. et al. Kinetically Trapped Co-continuous Polymer Morphologies through
Intraphase Gelation of Nanoparticles. Nano Lett. 11, 1997–2003.
Li, S. H. & Woo, E. M. Weak interaction, marginal miscibility, and ring-band spherulites
in blends of poly (vinylidene fluoride) with polyesters. J. Appl. Polym. Sci. 107,
766–777 (2008).
Li, S. H. & Woo, E. M. Effects of Chain Configuration on UCST Behavior in Blends of
Poly(L-lactic acid) with Tactic Poly(methyl methacrylate)s. J. Polym. Sci., Part B:
Polym. Phys. 46, 2355–2369 (2008).
Li, S. H. & Woo, E. M. Immiscibility-miscibility phase transitions in blends of poly(L-
lactide) with poly(methyl methacrylate). Polym. Int. 57, 1242–1251 (2008).
Li, S. H. & Woo, E. M. Immiscibility with upper-critical solution temperature phase
diagrams for poly(methyl methacrylate)/polyesters blends. Colloid Polym. Sci. 286,
253–265 (2008).
Li, W., Xue, J., Cheng, S. C., Du, Y. & Phillips, D. L. Influence of the chloro substituent
position on the triplet reactivity of benzophenone derivatives: a time-resolved
resonance Raman and density functional theory study. J. Raman Spectrosc. 43, 774–
780 (2012).

130
Li, Y., Stein, M. & Jungnickel, B.-J. Competition between crystallization and phase
separation in polymer blends I. Diffusion controlled supermolecular structures and
phase morphologies in poly (e-caprolactone)/polystyrene blends. Colloid Polym. Sci.
269, 772–780 (1991).
Li, Y., Iwakura, Y., Zhao, L. & Shimizu, H. Nanostructured Poly(vinylidene fluoride)
Materials by Melt Blending with Several Percent of Acrylic Rubber.
Macromolecules 41, 3120–3124 (2008).
Lin, A. A., Sastri, V. R., Tesoro, G., Reiser, A. & Eachus, R. On the Cross-Linking
Mechanism of Benzophenone-Containing Polyimides. Macromolecules 21, 1165–
1169 (1988).
Lin, C. S., Liu, W. L., Chiu, Y. S. & Ho, S. Y. Benzophenone-Sensitized
Photodegradation of Polystyrene Films under Atmospheric Conditions. Polym.
Degrad. Stab. 38, 125–130 (1992).
Linder, V., Gates, B. D., Ryan, D., Parviz, B. A. & Whitesides, G. M. Water-soluble
sacrificial layers for surface micromachining. Small 1, 730–6 (2005).
Liu, D., Bastiaansen, C. W. M., Den Toonder, J. M. J. & Broer, D. J. Single-composition
three-dimensionally morphing hydrogels. Soft Matter 9, 588 (2013).
Lynd, N. A. & Hillmyer, M. A. Effects of polydispersity on the order-disorder transition
in block copolymer melts. Macromolecules 40, 8050–8055 (2007).
Lynd, N. A., Meuler, A. J. & Hillmyer, M. A. Polydispersity and block copolymer self-
assembly. Prog. Polym. Sci. 33, 875–893 (2008).
Ma, M., Guo, L., Anderson, D. G. & Langer, R. Bio-inspired polymer composite actuator
and generator driven by water gradients. Science 339, 186–9 (2013).
Macosko, C. W., Jeon, H. K. & Hoye, T. R. Reactions at polymer–polymer interfaces for
blend compatibilization. Prog. Polym. Sci. 30, 939–947 (2005).
Manna, S., Mandal, A. & Nandi, A. K. Fabrication of Nanostructured Poly(3-thiophene
methyl acetate) within Poly(vinylidene fluoride) Matrix: New Physical and
Conducting Properties. J. Phys. Chem. B 114, 2342–2352 (2010).

131
Marcon, L. et al. Photochemical immobilization of proteins and peptides on
benzophenone-terminated boron-doped diamond surfaces. Langmuir 26, 1075–80
(2010).
Martin, T. A. et al. Quantitative photochemical immobilization of biomolecules on planar
and corrugated substrates: a versatile strategy for creating functional biointerfaces.
ACS Appl. Mater. Interfaces 3, 3762–71 (2011).
Mateescu, A., Wang, Y., Dostalek, J. & Jonas, U. Thin Hydrogel Films for Optical
Biosensor Applications. Membranes 2, 40–69 (2012).
Matsen, M. W. Polydispersity-induced macrophase separation in diblock copolymer
melts. Phys. Rev. Lett. 99, (2007).
McCartin, P. & Nebe, W. Photoactive plastisols: positive washout image. J. Imaging Sci.
32, 222–225 (1988).
McMaster, L. P. Aspects of Polymer-Polymer Thermodynamics. Macromolecules 6,
760–773 (1973).
Meng, H. A Brief Review of Stimulus-active Polymers Responsive to Thermal, Light,
Magnetic, Electric, and Water/Solvent Stimuli. J. Intel. Mat. Syst. Str. 21, 859–885
(2010).
Mita, I., Takagi, T., Horie, K. & Shindo, Y. Photosensitized Degradation of Polystyrene
by Benzophenone in Benzene Solution. Macromolecules 17, 2256–2260 (1984).
Mita, I., Hisano, T., Horie, K. & Okamoto, A. Photoinitiated Thermal-Degradation of
Polymers. 1. Elementary Processes of Degradation of Polystyrene.
Macromolecules 21, 3003–3010 (1988).
Mönch, W., Dehnert, J., Prucker, O., Rühe, J. & Zappe, H. Tunable Bragg filters based
on polymer swelling. Appl. Opt. 45, 4284–4290 (2006).
Nakanishi, H., Satoh, M., Norisuye, T. & Tran-Cong-Miyata, Q. Phase Separation of
Interpenetrating Polymer Networks Synthesized by Using an Autocatalytic
Reaction. Macromolecules 39, 9456–9466 (2006).
Nakanishi, T. L., Namikawa, N., Norisuye, T. & Tran-Cong-Miyata, Q. Autocatalytic
phase separation and graded co-continuous morphology generated by photocuring.
Soft Matter 2, 149–156 (2006).

132
Nam, K. T. et al. Virus-enabled synthesis and assembly of nanowires for lithium ion
battery electrodes. Science 312, 885–888 (2006).
Naumann, C. A. et al. The Polymer-Supported Phospholipid Bilayer: Tethering as a New
Approach to Substrate−Membrane Stabilization. Biomacromolecules 3, 27–35
(2002).
Nishi, T., Wang, T. T. & Kwei, T. K. Thermally Induced Phase Separation Behavior of
Compatible Polymer Mixtures. Macromolecules 8, 227–234 (1975).
Okada, M. Experimental study of thermal fluctuation in spinodal decomposition of a
binary polymer mixture. J. Chem. Phys. 85, 5317–5327 (1986).
Orlov, Y., Xu, X. & Maurer, G. Equilibrium swelling of N-isopropyl acrylamide based
ionic hydrogels in aqueous solutions of organic solvents: Comparison of experiment
with theory. Fluid Phase Equilib. 249, 6–16 (2006).
Oster, G., Oster, G. K. & Moroson, H. Ultraviolet Induced Crosslinking and Grafting of
Solid High Polymers. J. Polym. Sci. 34, 671–684 (1959).
Ougizawa, T. & Walsh, D. J. Upper Critical Solution Temperature Behavior in
Polystyrene Poly(Methyl Methacrylate) Mixture. Polym. J. (Tokyo, Jpn.) 25, 1315–
1318 (1993).
Oxidizing and Reducing Agents. Handbook of Reagents for Organic Synthesis 394–400
(1999).
Paul, D. R., Bernstein, R. E., Wahrmund, D. C. & Barlow, J. W. Polymer Blends
Containing Poly(vinylidene fluoride). Part IV: Thermodynamic Interpretations.
Polym. Eng. Sci. 18, 1225–1234 (1978).
Park, M. J. et al. Increased water retention in polymer electrolyte membranes at elevated
temperatures assisted by capillary condensation. Nano Lett. 7, 3547–52 (2007).
Penning, J. P. & St. John Manley, R. Miscible Blends of Two Crystalline Polymers. 1.
Phase Behavior and Miscibility in Blends of Poly(vinylidene fluoride) and Poly(1,4-
butylene adipate). Macromolecules 29, 77–83 (1996).

133
Pernot, H., Baumert, M., Court, F. & Leibler, L. Design and properties of co-continuous
nanostructured polymers by reactive blending. Nat. Mater. 1, 54–58 (2002).
Pitet, L. M., Amendt, M. A. & Hillmyer, M. A. Nanoporous linear polyethylene from a
block polymer precursor. J. Am. Chem. Soc. 132, 8230–1 (2010).
Porter, G. & Wilkinson, F. Primary photochemical processes in aromatic molecules. Part
5- Flash photolysis of benzophenone in solution. Trans. Faraday Soc. 57, 1686–
1691 (1961).
Porter, G. & Suppan, P. Primary photochemical processes in aromatic molecules. Part 12
- Excited states of benzophenone derivatives. Trans. Faraday Soc. 61, 1664–1673
(1965).
Porter, G. & Suppan, P. Primary photochemical processes in aromatic molecules. Part 14
- Comparative photochemistry of aromatic carbonyl compounds. Trans. Faraday
Soc. 62, 3375 (1966).
Potschke, P. & Paul, D. R. Formation of Co-continuous Structures in Melt-Mixed
Immiscible Polymer Blends. J. Macromol. Sci., Polym. Rev. 43, 87 (2003).
Prucker, O., Naumann, C. A., Rühe, J., Knoll, W. & Frank, C. W. Photochemical
Attachment of Polymer Films to Solid Surfaces via Monolayers of Benzophenone
Derivatives. J. Am. Chem. Soc. 121, 8766–8770 (1999).
Pu, G. W., Luo, Y. W., Lou, Q. C. & Li, B. G. Co-Continuous Polymeric Nanostructures
via Simple Melt Mixing of PS/PMMA. Macromol. Rapid Commun. 30, 133–137
(2009).
Qu, B. J. & Ranby, B. Photocross-linking of Low-Density Polyethylene. 1. Kinetics and
Reaction Parameters. J. Appl. Polym. Sci. 48, 701–709 (1993).
Qu, B. J. Reaction mechanism of benzophenone-photoinitiated crosslinking of
polyethylene. Chin. J. Polym. Sci. 20, 291–307 (2002).
Rabek, J. F. Photosensitized Processes in Polymer Chemistry: A Review. Photochem.
Photobiol. 7, 5–57 (1968).

134
Rabek, J. F. Mechanisms of photophysical processes and photochemical reactions in
polymers: Theory and Applications. Energy 272–300 (John Wiley & Sons:
Chichester, 1987).
Register, R. A. Continuity through dispersity. Nature 483, 167–168 (2012).
Rehmer, G., Boettcher, A. & Portugall, M. Benzophenone derivatives and their
preparation. US Patent 5,264,533 1–7 (1993).
Rehmer, G., Boettcher, A. & Portugall, M. (Meth)acrylate copolymer based UV-
crosslinkable materials. US Patent 5,389,699 1–7 (1995).
Rohr, T., Hilder, E. F., Donovan, J. J., Svec, F. & Frechet, J. M. J. Photografting and the
Control of Surface Chemistry in Three-Dimensional Porous Polymer Monoliths.
Macromolecules 36, 1677–1684 (2003).
Roth, P. J. & Theato, P. Covalent Attachment of Gold Nanoparticles to Surfaces and
Polymeric Substrates Using UV Light. Macromol. Chem. Phys. 213, 2550–2556
(2012).
Rubinstein, M., Colby, R. H., Dobrynin, A. V & Joanny, J.-F. Elastic Modulus and
Equilibrium Swelling of Polyelectrolyte Gels. Macromolecules 29, 398–406
(1996).
Rubinstein, M. & Colby, R. H. Polymer Physics. (Oxford University Press, Inc: New
York, 2003).
Rufs, A. M. et al. Synthesis and photoinitiation activity of macroinitiators comprising
benzophenone derivatives. Polymer 49, 3671–3676 (2008).
Russell, T. P., Hadziioannou, G. & Warburton, W. Phase separation in low molecular
weight polymer mixtures. Macromolecules 18, 78–83 (1985).
Ruzette, A. V & Leibler, L. Block copolymers in tomorrow’s plastics. Nat. Mater. 4, 19–
31 (2005).
Ryu, D. Y. et al. Phase behavior of polystyrene and poly(n-pentyl methacrylate) blend.
Macromolecules 35, 8676–8680 (2002).

135
Saeed, A., Georget, D. M. R. & Mayes, A. G. Synthesis, characterisation and solution
thermal behaviour of a family of poly (N-isopropyl acrylamide-co-N-hydroxymethyl
acrylamide) copolymers. React. Funct. Polym. 70, 230–237 (2010).
Sasaki, H., Kanti Bala, P., Yoshida, H. & Ito, E. Miscibility of PVDF/PMMA blends
examined by crystallization dynamics. Polymer 36, 4805–4810 (1995).
Sato, T., Endo, M., Shiomi, T. & Imai, K. UCST behaviour for high-molecular-weight
polymer blends and estimation of segmental chi parameters from their miscibility.
Polymer 37, 2131–2136 (1996).
Scherzer, T., Tauber, A. & Mehnert, R. UV curing of pressure sensitive adhesives studied
by real-time FTIR-ATR spectroscopy. Vib. Spectrosc. 29, 125–131 (2002).
Schmitt, A. L. & Mahanthappa, M. K. Polydispersity-driven shift in the lamellar
mesophase composition window of PEO-PB-PEO triblock copolymers. Soft Matter
8, 2294 (2012).
Schwartz, V. B. et al. Antibacterial Surface Coatings from Zinc Oxide Nanoparticles
Embedded in Poly(N-isopropylacrylamide) Hydrogel Surface Layers. Adv. Funct.
Mater. 22, 2376–2386 (2012).
Seo, M., Amendt, M. A. & Hillmyer, M. A. Cross-Linked Nanoporous Materials from
Reactive and Multifunctional Block Polymers. Macromolecules 44, 9310–9318
(2011).
Shi, H. C. et al. A facile route for preparing stable co-continuous morphology of
LLDPE/PA6 blends with low PA6 content. Polymer 51, 4958–4968.
Shindo, Y., Katagiri, N., Ebisuno, T., Hasegawa, M. & Mitsuda, M. Network formation
and swelling behavior of photosensitive poly(vinyl alcohol) gels prepared by
photogenerated crosslinking. Angew. Makromol. Chem. 240, 231–239 (1996).
Shoute, L. C. T. & Huie, R. E. Reactions of triplet decafluorobenzophenone with alkenes.
A laser flash photolysis study. J. Phys. Chem. A 101, 3467–3471 (1997).
Singh, A. K., Bhasikuttan, A. C., Palit, D. K. & Mittal, J. P. Excited-state dynamics and
photophysical properties of para-aminobenzophenone. J. Phys. Chem. A 104, 7002–
7009 (2000).

136
Smets, G. J., Elhamouly, S. N. & Oh, T. J. Photochemical-Reactions on Polymers. Pure
Appl. Chem. 56, 439–446 (1984).
Stockmayer, W. H. Theory of Molecular Size Distribution and Gel Formation in
Branched-Chain Polymers. J. Chem. Phys. 11, 45–55 (1943).
Stockmayer, W. H. Theory of molecular size distribution and gel formation in branched
polymers II. General cross linking. J. Chem. Phys. 12, 125–131 (1944).
Stoychev, G., Zakharchenko, S., Turcaud, S., Dunlop, J. W. C. & Ionov, L. Shape-
Programmed Folding of Stimuli-Responsive Polymer Bilayers. ACS Nano 6, 3925–
3934 (2012).
Sumitomo, H., Nobutoki, K. & Susaki, K. Photo-Graft Polymerization of Methyl
Methacrylate onto Styrene-Vinylbenzophenone Copolymer. J. Polym. Sci., Part A-1:
Polym. Chem. 9, 809–812 (1971).
Sundararaj, U. & Macosko, C. W. Drop Breakup and Coalescence in Polymer Blends:
The Effects of Concentration and Compatibilization. Macromolecules 28, 2647–
2657 (1995).
Suyama, K., Miyamoto, Y., Matsuoka, T., Wada, S. & Tsunooka, M. Photo-initiated
thermal crosslinking of copolymers bearing pendant base generating groups.
Polym. Adv. Technol. 11, 589–596 (2000).
Tamai, T., Imagawa, A. & Tran-Cong, Q. Semi-Interpenetrating Polymer Networks
Prepared by in situ Photo-Crosslinking of Miscible Polymer Blends.
Macromolecules 27, 7486–7489 (1994).
Thompson, R. B. & Matsen, M. W. Improving polymeric microemulsions with block
copolymer polydispersity. Phys. Rev. Lett. 85, 670–673 (2000).
Tocker, S. Organic polymeric articles and preparation thereof from derivatives of
ethylene and unsaturated benzophenones. US Patent 3,214,492 1–8 (1965).
Tocker, S. Cross-linked blended polymers containing polymeric acryloxy
benzophenones. US Patent 3,265,772 1–5 (1966).
Tocker, S. Graft and cross linked copolymers of polar vinylidene monomers with
acryloxyphenones. US Patent 3,315,013 1–7 (1967).

137
Tomura, H., Saito, H. & Inoue, T. Light scattering analysis of upper critical solution
temperature behavior in a poly(vinylidene fluoride)/poly(methyl methacrylate)
blend. Macromolecules 25, 1611–1614 (1992).
Toomey, R., Freidank, D. & Rühe, J. Swelling Behavior of Thin, Surface-Attached
Polymer Networks. Macromolecules 37, 882–887 (2004).
Topp, M. R. Activation-controlled hydrogen abstraction by benzophenone triplet. Chem.
Phys. Lett. 32, 144–149 (1975).
Torikai, A., Takeuchi, T. & Fueki, K. Photodegradation of Polystyrene and Polystyrene
Containing Benzophenone. Polym. Photochem. 3, 307–320 (1983).
Torikai, A., Sekigawa, Y. & Fueki, K. Photo-degradation of blends of polystyrene and
poly (methyl methacrylate). Polym. Degrad. Stab. 21, 43–54 (1988).
Tran-Cong, Q., Nagaki, T., Nakagawa, T., Yano, O. & Soen, T. Immobilization of
Transient Structures in Polystyrene Poly(2-Chlorostyrene) Blends Undergoing
Phase-Separation by Using Photo-Cross-Linking. Macromolecules 22, 2720–2723
(1989).
Tran-Cong, Q., Kawakubo, R. & Sakurai, S. Effects of chemical labelling on the phase
behaviour of poly (2-chlorostyrene)/poly (vinyl methyl ether) blends. Polymer 35,
1236–1241 (1994).
Tran-Cong, Q. & Harada, A. Reaction-induced ordering phenomena in binary polymer
mixtures. Phys. Rev. Lett. 76, 1162–1165 (1996).
Tran-Cong, Q., Kawai, J. & Endoh, K. Modes selection in polymer mixtures undergoing
phase separation by photochemical reactions. Chaos 9, 298–307 (1999).
Tran-Cong-Miyata, Q. Phase separation of polymer mixtures induced by polarization-
selective chemical reactions: Spatial symmetry breaking and mode selection.
Macromol. Symp. 160, 91–97 (2000).
Tran-Cong-Miyata, Q., Nishigami, S., Ito, T., Komatsu, S. & Norisuye, T. Controlling
the morphology of polymer blends using periodic irradiation. Nat. Mater. 3, 448–451
(2004).

138
Trinh, X. A. et al. Effects of elastic deformation on phase separation of a polymer blend
driven by a reversible photo-cross-linking reaction. Macromolecules 40, 5566–5574
(2007).
Tsubakiyama, K., Kuzuba, M., Yoshimura, K., Yamamoto, M. & Nishijima, Y.
Photochemical-Reaction of Polymers Having Arylketone and Tertiary Amine as
Pendant Groups. Polym. J. (Tokyo, Jpn.) 23, 781–788 (1991).
Turro, N. et al. Molecular Photochemistry of Alkanones in Solution - Alpha-cleavage,
Hydrogen Abstraction, Cycloaddition, and Sensitization Reactions. Acc. Chem. Res.
5, 92–101 (1972).
Wagner, P. J., Truman, R. J., Puchalski, A. E. & Wake, R. Extent of charge transfer in the
photoreduction of phenyl ketones by alkylbenzenes. J. Am. Chem. Soc. 108, 7727–
7738 (1986).
Wagner, P. & Park, B. Photoinduced hydrogen atom abstraction by carbonyl compounds.
Org. Photochem. 11, 227–366 (1991).
Wahrmund, D. C., Barlow, J. W., Paul, D. R. & Bernstein, R. E. Polymer Blends
Containing Polyvinylidene Fluoride. Part I: Poly(Alkyl Acrylates). Polym. Eng.
Sci. 18, 677–682 (1978).
Wang, J. Z., Nayak, B. R., Creed, D., Hoyle, C. E. & Mathias, L. J. Photocrosslinking of
poly(ethylene terephthalate) copolymers containing photoreactive comonomers.
Polymer 46, 6897–6909 (2005).
Wang, Y., Brunsen, A., Jonas, U., Dostálek, J. & Knoll, W. Prostate specific antigen
biosensor based on long range surface plasmon-enhanced fluorescence spectroscopy
and dextran hydrogel binding matrix. Anal. Chem. 81, 9625–9632 (2009).
Ward, D. E. & Rhee, C. K. Chemoselective reductions with sodium borohydride. Can. J.
Chem. 67, 1206–1211 (1989).
Washburn, N. R., Lodge, T. P. & Bates, F. S. Ternary polymer blends as model surfactant
systems. J. Phys. Chem. B 104, 6987–6997 (2000).
Widin, J. M., Schmitt, A. K., Im, K., Schmitt, A. L. & Mahanthappa, M. K.
Polydispersity-Induced Stabilization of a Disordered Bicontinuous Morphology in
ABA Triblock Copolymers. Macromolecules 43, 7913–7915 (2010).

139
Widin, J. M., Schmitt, A. K., Schmitt, A. L., Im, K. & Mahanthappa, M. K. Unexpected
consequences of block polydispersity on the self-assembly of ABA triblock
copolymers. J. Am. Chem. Soc. 134, 3834–44 (2012).
Williams, J. L. R. & Border, D. G. The preparation and properties of photoreactive
polymers. I. 2-(arylvinyl)-N-vinylpyridinium arylsulfonate polymers. Macromol.
Chem. 73, 203–214 (1964).
Wondraczek, H., Kotiaho, A., Fardim, P. & Heinze, T. Photoactive polysaccharides.
Carbohydr. Polym. 83, 1048–1061 (2011).
Wong, D. T., Mullin, S. A., Battaglia, V. S. & Balsara, N. P. Relationship between
morphology and conductivity of block-copolymer based battery separators. J.
Membr. Sci. 394-395, 175–183 (2012).
Wu, G. et al. UV exposure effects on photoinitiator-grafted styrene-butadiene-styrene
triblock copolymer. J. Appl. Polym. Sci. 120, 2627–2631 (2011).
Wu, Q. & Qu, B. Photoinitiating characteristics of benzophenone derivatives as new
initiators in the photocrosslinking of polyethylene. Polym. Eng. Sci. 41, 1220–
1226 (2001).
Wu, Q. H. & Qu, B. J. Synthesis of Di(4-hydroxyl benzophenone) sebacate and its usage
as initiator in the photocrosslinking of polyethylene. J. Appl. Polym. Sci. 85, 1581–
1586 (2002).
Wu, S. K. & Rabek, J. F. Benzophenone-Initiated Photo-Crosslinking of Polynorbornene
in the Solid-State. Polym. Degrad. Stab. 21, 365–376 (1988).
Yang, B. & Yang, W. T. Thermo-sensitive switching membranes regulated by pore-
covering polymer brushes. J. Membr. Sci. 218, 247–255 (2003).
Yang, H., Shibayama, M., Stein, R. S., Shimizu, N. & Hashimoto, T. Deuteration effects
on the miscibility and phase separation kinetics of polymer blends. Macromolecules
19, 1667–1674 (1986).
Yemini, M., Reches, M., Gazit, E. & Rishpon, J. Peptide nanotube-modified electrodes
for enzyme-biosensor applications. Anal. Chem. 77, 5155–5159 (2005).

140
Yin, B., Zhao, Y., Pan, M. M. & Yang, M. B. Morphology and thermal properties of a
PC/PE blend with reactive compatibilization. Polym. Adv. Technol. 18, 439–445
(2007).
Zakharchenko, S., Puretskiy, N., Stoychev, G., Stamm, M. & Ionov, L. Temperature
controlled encapsulation and release using partially biodegradable thermo-magneto-
sensitive self-rolling tubes. Soft Matter 6, 2633 (2010).
Zhang, C.-L. et al. Efficiency of graft copolymers at stabilizing co-continuous polymer
blends during quiescent annealing. Polymer 49, 3462–3469 (2008).
Zhao, Y. & Dan, Y. Synthesis and characterization of a polymerizable benzophenone
derivative and its application in styrenic polymers as UV-stabilizer. Eur. Polym. J.
43, 4541–4551 (2007).
Zhou, N., Lodge, T. P. & Bates, F. S. Influence of conformational asymmetry on the
phase behavior of ternary homopolymer/block copolymer blends around the
bicontinuous microemulsion channel. J. Phys. Chem. B 110, 3979–89 (2006).
Zhou, Z., Li, S., Zhang, Y., Liu, M. & Li, W. Promotion of proton conduction in polymer
electrolyte membranes by 1H-1,2,3-triazole. J. Am. Chem. Soc. 127, 10824–5
(2005).


![Static and Dynamic Density Functional Theory and ...called copolymers. Here we consider the class of copolymers called \block copolymers" [7] while there are many kinds of copolymers](https://img.pdfslide.us/doc/110x75/5eccfbf97d791301bb64d299/static-and-dynamic-density-functional-theory-and-called-copolymers-here-we.jpg)









![1991 the - Library and Archives Canada · 2004-09-21 · benzylprolyl)amino]benzophenone 2-[N-(N-benzy l proly l)amino]benzophenone benzyloxycarbonyl chemical ionization mass spedroscopy](https://img.pdfslide.us/doc/110x75/5f70f230c3335011d7349ea0/1991-the-library-and-archives-2004-09-21-benzylprolylaminobenzophenone-2-n-n-benzy.jpg)
















