Embed Size (px)
Citation preview

THE JOURNAL OB BIOLOGICAL CHEMISTRY Vol. 247, No. 19, Issue of October 10, pp. 6212-6217, 1972
Printed in U.S.A.
The Purification of a Lipoprotein Lipase
from Bovine Skim Milk* (Received for publication, May 19, 1972)
TORBJ~~RN EGELRUD AND THOMAS OLIVECRONA
From. the Department of Chemistry, Section on Physiological Chemistry, University of [ime&, S-90187 Ume&, Sweden
SUMMARY
The purification of a lipase from skim milk is described. The enzyme had the characteristics of a lipoprotein lipase, i.e. its activity against emulsified long chain triglyceride was stimulated more than ZO-fold by addition of suitable amounts of serum to the assay system and the activity was almost completely inhibited by 1 M NaCl. After an initial fractiona- tion of the skim milk, the main purification was obtained by afhnity chromatography on Sepharose 4B with covalently linked heparin. The preparation obtained was purified 5,000- to 7,000-fold. Gel electrophoresis of this preparation in urea or in sodium dodecyl sulfate revealed one major component which stained for protein and for carbohydrate and which comprised more than 80% of the total protein. The apparent minimum molecular weight of this component was 62,000 to 66,000 as determined by electrophoresis in polyacrylamide gels in the presence of sodium dodecyl sulfate.
Lipoprotein lipases (LPL, glycerol ester hydrolase, EC 3.1.1.3) are enzymes which catalyze the liberation of long chain fatty acids from emulsified triglycerides at a maximal rate only if the triglyceride substrate has been previously incubated with a serum lipoprotein component (l-3). There is a lipoprotein lipase in bovine milk, both in the skim milk fraction and in the cream (4). We have previously demonstrated that skim milk lipoprotein lipase binds to agarose gel to which heparin has been covalently linked (5). The reversibility and the relatively high specificity of this binding suggest that it can be exploited for the purification of the enzyme. In this paper we describe a method for obtaining milligram quantities of skim milk lipoprotein lipase purified 5000 to 7000 times. The preparation thus obtained may be 80 to 100% pure.
MATERIALS AND METHODS
Unpasteurized bovine milk was obtained from a local dairy. The preparatory work was started within a few hours of delivery. The rennet preparation used was an extract of calf stomach ob- tained from Kochlight Laboratories, Ltd., Colnbrook, England.
* This work was supported by grants from the Swedish Medical Research Council (B-72-13X-727) and the Medical Faculty, Uni- versity of Ume%.
Commercial grade ammonium sulfate and analytical grade ace- tone and ethyl ether were used. To prepare dialyzed human serum, outdated titrated plasma from the local hospital blood bank was recalcified, the fibrin clot removed, and the serum dialyzed against 0.154 M NaCl. Bovine serum albumin was from Sigma Chemical Company, St. Louis, MO.; heparin and triglyceride emulsion (Intralipid, 10%) for the incubation mix- tures were from Vitrum, Stockholm, Sweden.
The heparin-substituted agarose gel (Sepharose 4B, Pharmacia, Uppsala, Sweden), hereafter called “heparinSepharose” was a kind gift from Dr. Per-Henrik Iverius and was prepared as he has previously described (6).
The assay for lipoprotein lipase in most experiments was based on that described by Robinson (2) and the same as that used by Olivecrona d al. (5) except that the albumin solution was made up in 0.154 M NaCl. Each assay mixture consisted of: 0.5 ml of 1.35 M Tris-HCl, pH 8.1, 1.2 ml of 18.7% bovine serum albumin in 0.154 M NaCl, pH 8.1,0.8 ml of dialyzed human serum, 0.1 ml of triglyceride emulsion (Intralipid, lo%), 0.1 ml of heparin (20 i.u. per ml, made up in water). Enzyme solution and 0.154 M NaCl were added to give a final volume of 4.7 ml. The total amount of NaCl in the assay system was never high enough to cause inhibition of the lipoprotein lipase. The sensi- tivity of the enzyme to NaCl was markedly decreased by the amounts of heparin added (6a). Fifteen-minute preliminary incubation and incubations were performed in a shaking water bath at 37”, 50 strokes per min. Duplicate l-ml aliquots were withdrawn at 0 min and after 60 min incubation, and the free fatty acid contents were determined by extraction and titration (7). A standard amount of palmitic acid was taken through the extraction procedure and titrated at each experiment, making adequate corrections for the fatty acid content of the assay mix- tures possible. One lipoprotein lipase unit is defined as that amount of enzyme that releases 1 peq of fatty acid per hour. In one experiment lipoprotein lipase activity was determined by the method of Schotz et al. (8). Protein was determined by the method of Lowry et al. (9) in a total volume of 1.3 ml (of which the protein solution comprised 0 to 0.6 ml). Bovine serum albumin was used as standard, 10 pg giving an absorption at 750 nm of approximately 0.13. No corrections to obtain true pro- tein concentrations were made. Chromatography profiles were measured with a Uvicord II (LKB produkter AB, Stockholm, Sweden). Gel electrophoresis in 10% polyacrylamide gel con- taining 0.1 y0 sodium dodecyl sulfate before and after reduction with &mercaptoethanol and molecular weight determinations
6212
by guest on April 23, 2020
http://ww
w.jbc.org/
Dow
nloaded from
6213
were performed by the method of Weber and Osborn (10). Gel electrophoresis in 7.5% polyacrylamide gel containing 6.25 M
urea, pH 3.2, was a modification of the method of Panyim and Chalkley (11). After electrophoresis the gels were stained for 20 min in 1% (w/v) Amido black in ethanol-acetic acid-water (400 :75:525). They were then destained in the same ethanol- acetic acid-water mixture with a small amount of an anion ex- changer on the bottom of the tubes. After destaining, the gels were kept in 7.5% (v/v) acetic acid in water. This staining method was chosen because it gave less background than the Coomassie stain commonly used to stain SDS’ gels. Impurities were more easily detected by this method. Carbohydrate staining was as described by Zacharius et al. (12). Spectro- metric scanning of the gels was done using an Acta III Beckman spectrophotometer with a gel scanner GS-2 at 620 nm.
RESULTS
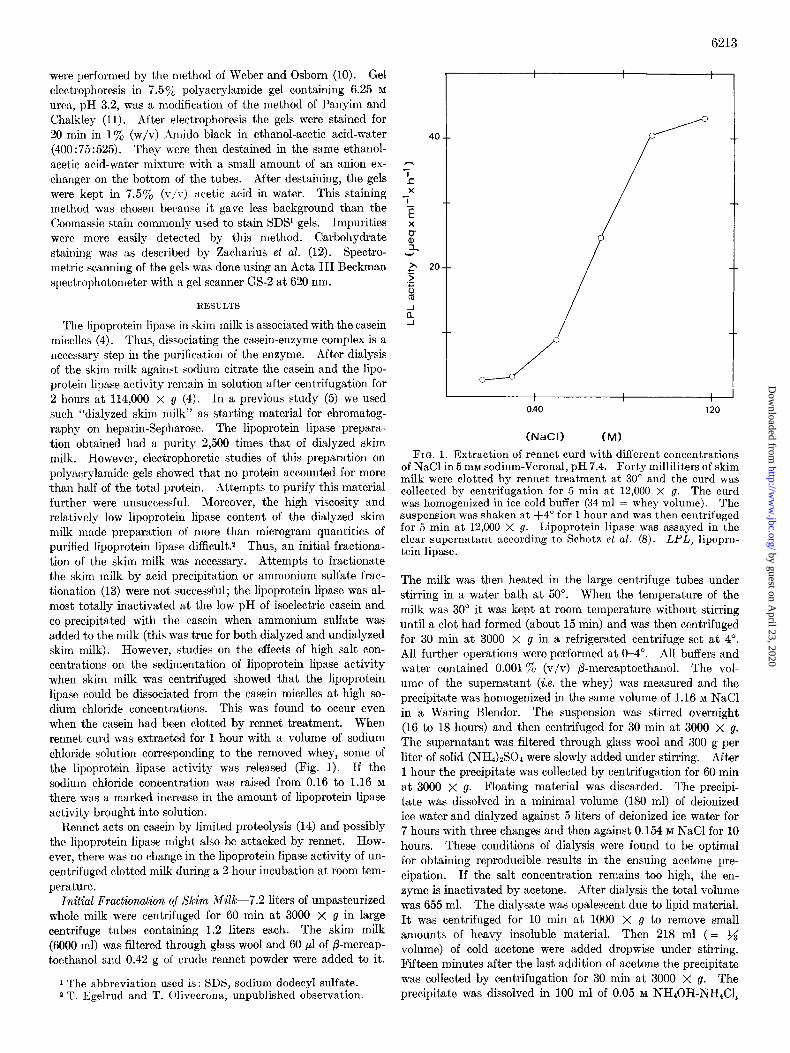
The lipoprotein lipase in skim milk is associated with the casein micelles (4). Thus, dissociating t.he casein-enzyme complex is a necessary step in the purification of the enzyme. After dialysis of the skim milk against sodium citrate the casein and the lipo- protein lipase activity remain in solution after centrifugation for 2 hours at 114,000 x g (4). In a previous study (5) we used such “dialyzed skim milk” as starting material for chromatog- raphy on heparin-Sepharose. The lipoprotein lipase prepara- tion obtained had a purit’y 2,500 times that of dialyzed skim milk. However, electrophoretic studies of this preparation on polyacrylamide gels showed that no protein accounted for more than half of the total protein. Attempts to purify this material further were unsuccessful. Moreover, the high viscosity and relatively low lipoprotein lipase content of the dialyzed skim milk made preparation of more than microgram quantities of purified lipoprotein lipase difficult.* Thus, an initial fractiona- tion of the skim milk was necessary. Attempts to fractionate the skim milk by acid precipitation or ammonium sulfate frac- tionation (13) were not’ successful; the lipoprotein lipase was al- most totally inactivat)ed at the low pH of isoelectric casein and co-precipitated with t#he casein when ammonium sulfate was added to the milk (this was true for both dialyzed and undialyzed skim milk). However, studies on the effects of high salt con- centrations on the sedimentation of lipoprotein lipase activity when skim milk was centrifuged showed that the lipoprotein lipase could be dissociated from the casein micelles at high so- dium chloride concentrations. This was found to occur even when the casein had been clotted by rennet treatment. When renriet curd was extracted for 1 hour with a volume of sodium chloride solution corresponding to the removed whey, some of the lipoprotein lipase activity was released (Fig. 1). I f the sodium chloride concentration was raised from 0.16 to 1.16 M
there was a marked increase in the amount of lipoprotein lipase activity brought into solution.
Rennet acts on casein by limited proteolysis (14) and possibly the lipoprotein lipase might also be attacked by rennet. How- ever, there was no change in the lipoprotein lipase activity of un- centrifuged clotted milk during a 2-hour incubation at room tem- perature.
Initial Fractionation of Skim Mills-7.2 liters of unpasteurized whole milk were centrifuged for 60 min at 3000 x g in large centrifuge tubes containing 1.2 liters each. The skim milk (6000 ml) was filtered t,hrough glass wool and 60 ~1 of /3-mercap- toethanol and 0.42 g of crude rennet powder were added to it.
1 The abbreviation used is: SDS, sodium dodecyl sulfate. 2 T. Egelrud and T. Olivecrona, unpublished observation
I -c t / t X I / I
0.40 1.20
(NaCI) (Ml
FIG. 1. Extraction of rennet curd with different concentrations of NaCl in 5 mM sodium-Veronal, pH 7.4. Forty milliliters of skim milk were clotted by rennet treatment at 30” and the curd was collected by centrifugation for 5 min at 12,000 X g. The curd was homogenized in ice cold buffer (34 ml = whey volume). The suspension was shaken at +4” for 1 hour and was then centrifuged for 5 min at 12,000 X g. Lipoprotein lipase was assayed in the clear supernatant according to Schotx et al. (8). LPL, lipopro- tein lipase.
The milk was then heated in the large centrifuge tubes under stirring in a water bath at 50”. When the temperature of the milk was 30” it was kept at room temperature without stirring until a clot had formed (about 15 min) and was then centrifuged for 30 min at 3000 x g in a refrigerated centrifuge set at 4”. All further operations were performed at O-4”. All buffers and water contained 0.001% (v/v) fl-mercaptoethanol. The vol- ume of the supernatant (i.e. the whey) was measured and the precipitate was homogenized in the same volume of 1.16 M NaCl in a Waring Blendor. The suspension was stirred overnight (16 to 18 hours) and then centrifuged for 30 mm at 3000 x g. The supernatant was filtered through glass wool and 300 g per liter of solid (NH&S04 were slowly added under stirring. After 1 hour the precipitate was collected by centrifugation for 60 min at 3000 x g. Floating material was discarded. The precipi- tate was dissolved in a minimal volume (180 ml) of deionized ice water and dialyzed against 5 liters of deionized ice water for 7 hours with three changes and then against 0.154 M NaCl for 10 hours. These conditions of dialysis were found to be optimal for obtaining reproducible results in the ensuing acetone pre- cipation. If the salt concentration remains too high, the en- zyme is inactivated by acetone. After dialysis the total volume was 655 ml. The dialysate was opalescent due to lipid material. It was centrifuged for 10 min at 1000 X g to remove small amounts of heavy insoluble material. Then 218 ml (= 35 volume) of cold acetone were added dropwise under stirring. Fifteen minutes after the last addition of acetone the precipitate was collected by centrifugation for 30 min at 3000 X g. The precipitate was dissolved in 100 ml of 0.05 M NHIOH-NH&I,
by guest on April 23, 2020
http://ww
w.jbc.org/
Dow
nloaded from

6214
pH 8.5, and reprecipitated by addition of 1500 ml of acetone. The precipitate was collected on a Btichner funnel and washed with 500 ml of ethyl ether. The acetone-ether powder could be stored at -20” for several weeks without loss of lipoprotein lipase activity. From this powder lipoprotein lipase can be brought into solution by extraction with 5 mM Verona1 buffer, pH 7.4, containing NaCI. NaCl concentrations from 0.16 to 0.50 M were equally effective.
The initial fractionation of skim milk is summarized in Table I. The sum of the lipoprotein lipase activity of the whey, the rennet curd after extraction, and the rennet curd extract ac- counts for only 65% of the lipoprotein lipase activity of the skim milk. However, this may be an underestimate for two reasons. Heparin was present in the assay of lipoprotein lipase activity (see “Material and Methods”). The skim milk lipo- protein lipase may be stimulated by heparin to different degrees at different stages of purification (6a). In one experiment similar to that summarized in Table I, the lipoprotein lipase activity was determined with no heparin in the assay system. The yield of lipoprotein lipase activity of the whey and the rennet curd ex- tract was estimated to be 56% of the total lipoprotein lipase activity in the skim milk used as starting material, whereas it was only estimated to be 42% when heparin was present in the assay system. Moreover, the lipoprotein lipase activity re- maining in the rennet curd may be underestimated due to in- complete homogenization (see legend to Table I). Thus, the total amount of lipoprotein lipase inactivated during the prep- aration and extraction of the rennet curd may be less than 35% of the initial activity. The losses in the further processing of the rennet extract are much less. The preparation yielded 130 ml of enzyme solution containing about 14 mg of protein per ml and with a total lipoprotein lipase content of 76,000 units. Its specific activity was 10 times higher than that of the skim milk
and the yield of lipoprotein lipase activity was 9.5%. In other preparations the yields were 10 to 15s.” At all steps of the preparation the lipolytic activity was stimulated 20-fold or more by serum in the assay system (Table I).
A major difficulty in the purification of lipoprotein lipase is the limited stability of the enzyme. In unfractionated skim milk at 4” the enzyme activity decreased by about 40% in 3 days. Addition of 0.001% (v/v) p- mercaptoethanol to the skim milk stabilized the enzyme activity. The enzyme in an extract of the acetone-ether powder lost only 15% of its activity in 3 days and was not significantly stabilized by P-mercaptoethanol. I f the salt concentration in the extract was increased, the rate of inactivation also increased and this could not be prevented by &mercaptoethanol. At 20” the stability of the enzyme was much less than at 4”.
Heparin-Xepharose Chromatography-Two thousand milli- grams of the acetone-ether powder prepared as described above were dissolved in 100 ml of 0.5 M NaCl in 5 mM sodium-Veronal, pH 7.4. After 1 hour it was centrifuged for 60 min at 37,000 x
g. The clear solution (99 ml, 670 lipoprotein lipase units per ml, 15 mg of protein per ml) was passed through a column of heparin- Sepharose (5 x 1 cm) equilibrated with the same buffer. The column was washed with 0.16 M NaCl in 5 mM sodium-Veronal, pH 7.4. The effluent up to this point (112 ml) contained 15.5% of the lipoprotein lipase activity applied. Elution was then performed with a linear salt gradient: 0.16 to 1.5 M NaCl in 5 mM sodium-Veronal, pH 7.4, in a total volume of 200 ml. The use of different ionic strengths and the choice of the gradient was a compromise. The enzyme solution was applied to the gel in 0.5 M NaCl to prevent adsorption of large amounts of proteins other than lipoprotein lipase. The total yield of purified lipo- protein lipase was higher when the gradient was between 0.16
TABLE I
Initial fractionation of skim milk
step ?otal LPL unit9
Skim milk, Whey . Extract of rennet curd. Rennet curd after extractiond. Dissolved pellet after (NH&SOd. : 1 : : Supernatant after (NH.&S04e. Dissolved pellet after % volume of acetone,
acetone and ether/............ Supernatant after s volume of acetone pre-
cipitated with 9 volumes of acetone, washed with ether and redissolvedg.
??d
6,000 4,780 4,520
655 4,870
130
pep jatty acids released/hr
796,000 64,500
206,000 248,000 175,000
2,200
75,900
1,254 87,400
a LPL, lipoprotein lipase. b Serum was replaced by 0.8 ml of 57, bovine serum albumin in
0.154 M NaCl. c Not determined. d The remaining curd was weighed and 5.00 g were homogenized
in a total volume of 25 ml of 0.154 M NaCl, and the lipoprotein li- pase activity was determined on an aliquot of the homogenate.
e An aliquot was dialyzed free of ammonium sulfate against water and then against 0.154 M NaCI.
f The dried powder was extracted with 0.5 M NaCl in 5 mrvf so-
dium-Verona1 pH 7.4 (20 mg per ml) for 1 hour and the suspension was centrifuged at 37,000 X g for 30 min. Lipoprotein lipase was determined on the clear supernatant.
0 The volume of the supernatant was measured. Remaining enzyme and other proteins were precipitated from an aliquot by addition of 9 volumes of acetone. The precipitate was collected on a Biichner funnel and washed with 5 volumes of ethyl ether. The lipoprotein lipase activity of this preparation was measured as under f.
; of maxima .PL-activity
with no erum in the assay mix-
ture”
I
_-
Yield of LPL-activity
% 100
8.1 25.9 31.2 22.0
0.3
9.5
11.0
Total protein pecific activit] Purification
mg
195,600 48,300 24,400
e
22,800 3,900
1,780
15,000
LPL units/?ng protein
4.1 1.3 8.4 c
7.7 0.9
43
5.8
fold
1
2
2
10
by guest on April 23, 2020
http://ww
w.jbc.org/
Dow
nloaded from
and 1.5 M NaCl than when it was between 0.5 and 1.5 M NaCI. The purification obtained was approximately the same.
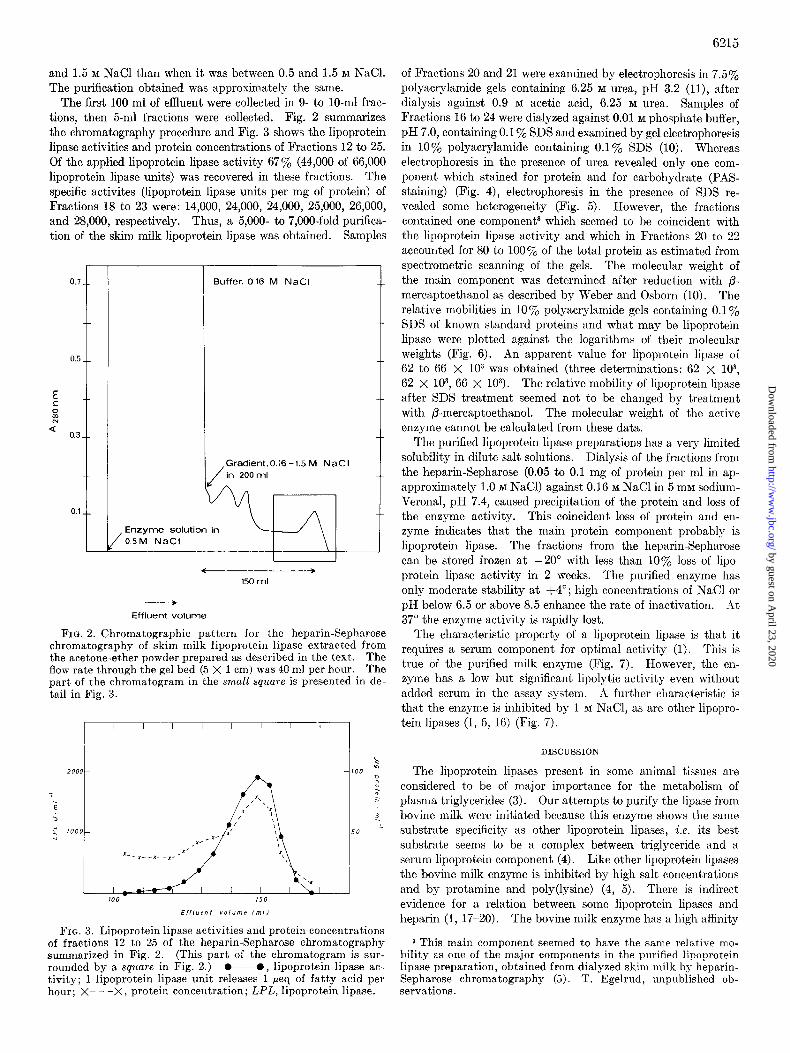
The first 100 ml of effluent were collected in 9- to lo-ml frac- tions, then 5-ml fractions were collected. Fig. 2 summarizes the chromatography procedure and Fig. 3 shows the lipoprotein lipase activities and protein concentrations of Fractions 12 to 25. Of the applied lipoprotein lipase activity 67% (44,000 of 66,000 lipoprotein lipase units) was recovered in these fractions. The specific activites (lipoprotein lipase units per mg of protein) of Fractions 18 to 23 were: 14,000, 24,000, 24,000, 25,000, 26,000, and 28,000, respectively. Thus, a 5,000- to 7,000-fold purifica- tion of the skim milk lipoprotein lipase was obtained. Samples
Buffer. 0 16 M NaCl
Gradient.O.;G-1.5M NaCl /in 200 ml
- Effluent volume
FIG. 2. Chromatographic pattern for the heparin-Sepharose chromatography of skim milk lipoprotein lipase extracted from the acetone-ether powder prepared as described in the text. The flow rate through the gel bed (5 X 1 cm) was 40 ml per hour. The part of the chromatogram in the small square is presented in de- tail in Fig. 3.
/-I I I I I
’ 1
FIG. 3. Lipoprotein lipase activities and protein concentrations of fractions 12 to 25 of the heparin-Sepharose chromatography summarized in Fig. 2. (This part of the chromatogram is sur- rounded by a square in Fig. 2.) O-0, lipoprotein lipase ac- tivity; 1 lipoprotein lipase unit releases 1 peq of fatty acid per hour; X- - -X, protein concentration; LPL, lipoprotein lipase.
6215
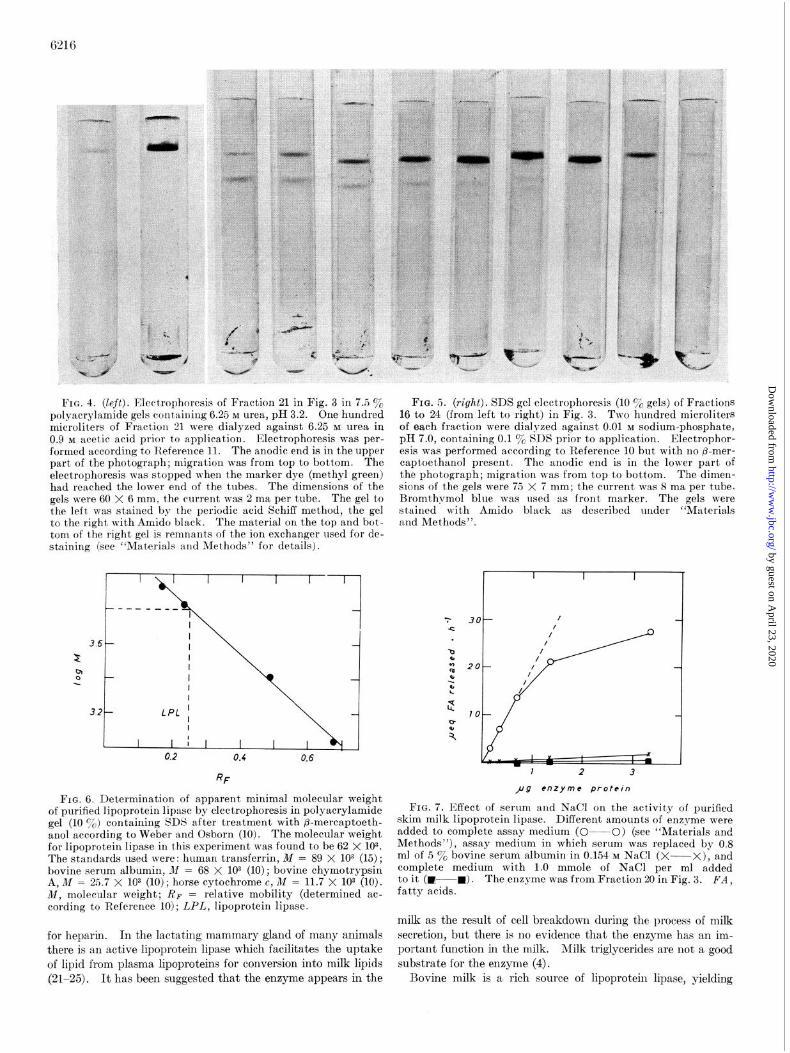
of Fractions 20 and 21 were examined by electrophoresis in 7.5~~ polyacrylamide gels containing 6.25 M urea, pH 3.2 (II), after dialysis against 0.9 M acetic acid, 6.25 M urea. Samples of Fractions 16 to 24 were dialyzed against 0.01 M phosphate buffer, pH 7.0, containing 0.1 y0 SDS and examined by gel electrophoresis in 10% polyacrylamide containing 0.1 y0 SDS (10). Whereas electrophoresis in the presence of urea revealed only one com- ponent which stained for protein and for carbohydrate (PAS- staining) (Fig. 4), electrophoresis in the presence of SDS re- vealed some heterogeneity (Fig. 5). However, the fractions contained one componenta which seemed to be coincident with the lipoprotein lipase activity and which in Fractions 20 to 22 accounted for 80 to 100% of the total protein as estimated from spectrometric scanning of the gels. The molecular weight of the main component was determined after reduction with p- mercaptoethanol as described by Weber and Osborn (10). The relative mobilities in 10% polyacrylamide gels containing 0.1% SDS of known standard proteins and what may be lipoprotein lipase were plotted against the logarithms of their molecular weights (Fig. 6). An apparent value for lipoprotein lipase of 62 to 66 x lo3 was obtained (three determinations: 62 x 103, 62 x lOa, 66 x 103). The relative mobility of lipoprotein lipase after SDS treatment seemed not to be changed by treatment with /3-mercaptoethanol. The molecular weight of the active enzyme cannot be calculated from these data.
The purified lipoprotein lipase preparations has a very limited solubility in dilute salt solutions. Dialysis of the fractions from the heparin-Sepharose (0.05 to 0.1 mg of protein per ml in ap- approximately 1.0 M NaCl) against 0.16 M NaCl in 5 mM sodium- Veronal, pH 7.4, caused precipitation of the protein and loss of the enzyme activity. This coincident loss of protein and en- zyme indicates that the main protein component probably is lipoprotein lipase. The fractions from the heparin-Sepharose can be stored frozen at -20” with less than 10% loss of lipo- protein lipase activity in 2 weeks. The purified enzyme has only moderate stability at +4”; high concentrations of NaCl or pH below 6.5 or above 8.5 enhance the rate of inactivation. At 37” the enzyme activity is rapidly lost.
The characteristic property of a lipoprotein lipase is that it requires a serum component for optimal activity (1). This is true of the purified milk enzyme (Fig. 7). However, the en- zyme has a low but significant lipolytic activity even without added serum in the assay system. A further charact.eristic is that the enzyme is inhibited by 1 M NaCl, as are other lipopro- tein lipases (1, 5, 16) (Fig. 7).
DISCUSSION
The lipoprotein lipases present in some animal tissues are considered to be of major importance for the metabolism of plasma triglycerides (3). Our attempts to purify the lipase from bovine milk were initiated because this enzyme shows the same substrate specificity as other lipoprotein lipases, i.e. its best substrate seems to be a complex between triglyceride and a serum lipoprotein component (4). Like other lipoprotein lipases the bovine milk enzyme is inhibited by high salt concentrations and by protamine and poly(lysine) (4, 5). There is indirect evidence for a relation between some lipoprotein lipases and heparin (1, 17-20). The bovine milk enzyme has a high affinity
3 This main component seemed to have the same relative mo- bility as one of the major components in the purified lipoprotein lipase preparation, obtained from dialyzed skim milk by heparin- Sepharose chromatography (5). T. Egelrud, unpublished ob- servations.
by guest on April 23, 2020
http://ww
w.jbc.org/
Dow
nloaded from
6216
FIG. 4. (left). Elcct,rophorcsis of Fraction 21 in Fig. 3 in 7.5 y0 polyacrylamide gels cont’aining 6.25 M urea, pH 3.2. One hundred microliters of Fract,ion 21 were dialyzed against 6.25 M urea in 0.9 M acet,ic acid prior to qplication. Electrophoresis was per- formed according to R.eference 11. The anodic end is in the upper part of the photograph; migration was from top to bot’tom. The electrophoresis was stopped when the marker dye (methyl green) had reached the lower end of the tubes. The dimensions of the gels were 60 X 6 mm, t’he current was 2 ma per tube. The gel to the left was stained by the periodic acid Schiff method, the gel to the right with Amido b&k. The material on the top and bot- tom of the right gel is remnants of the ion exchanger used for de- staining (see “Materials and Methods” for details).
I I\ I
P I b 2 I
I I
32- LPL 1 I
RF FIG. 6. Determination of apparent minimal molecular weight
of purified lipoprotein lipase by electrophoresis in polyacrylamide gel (10 o/00) containing SDS after treatment with P-mercaptoeth- anol according to Weber and Osborn (10). The molecular weight for lipoprotein lipase in this experiment was found to be 62 X 103. The standards used were: human transferrin, M = 89 X lo3 (15); bovine serum albumin, M = 68 X lo3 (10); bovine chymotrypsin A, M = 2.5.7 X lo3 (10); horse cytochrome c, M = 11.7 X lo3 (10). 1M, molecular weight; RF = relative mobility (determined ac- cording to Reference 10); LPL, lipoprotein lipase.
for heparin. In the lactating mammary gland of many animals there is an active lipoprotein lipase which facilitates the uptake of lipid from plasma lipoproteins for conversion into milk lipids (21-25). It has been suggested that the enzyme appears in the
FIG. 5. (right). SDS gel electrophoresis (10 y0 gels) of Fractions 16 to 24 (from left to right) in Fig. 3. Two hundred microliters of each fraction were dialyzed against 0.01 M sodium-phosphate, pH 7.0, containing 0.1 y0 SDS prior to application. Electrophor- esis was performed according to Reference 10 but with no p-mer- captoethanol present. The anodic end is in the lower part of the photograph; migration was from top to bottom. The dimen- sions of the gels were 75 X 7 mm; the current was 8 ma per tube. Bromthpmol blue was used as front marker. The gels were stained with Amido black as described under “Materials and Methods”.
I I I
I 2 3
Yg enzyme protein
FIG. 7. Effect of serum and NaCl on the activity of purified skim milk lipoprotein lipase. Different amounts of enzyme were added to complete assay medium (O---O) (see “Materials and Methods”), assay medium in which serum was replaced by 0.8 ml of 5 y0 bovine serum albumin in 0.154 M NaCl (X--X), and complete medium with 1.0 mmole of NaCl per ml added to it (W-R). The enzyme was from Fraction 20 in Fig. 3. FA, fatty acids.
milk as the result of cell breakdown during the process of milk secretion, but there is no evidence that the enzyme has an im- portant function in the milk. Milk triglycerides are not a good substrate for the enzyme (4).
Bovine milk is a rich source of lipoprotein lipase, yielding
by guest on April 23, 2020
http://ww
w.jbc.org/
Dow
nloaded from

6217
twice as much enzyme activity per unit volume as post-heparin rat plasma. Chicken adipose tissue, which is one of the richest tissue sources of the enzyme contains only >ioo to >iO as much activity per unit weight as does milk.4 It has not yet been possible to purify a lipoprotein lipase by conventional methods of protein separation, mainly due to the very limited stability of the enzyme and its tendency to aggregate. Fielding has purified the lipoprotein lipases of rat and of human post-heparin plasma utilizing the formation of an enzyme-substrate complex which could be isolated (26, 27). This method has been successfully adapted in several other laboratories (28-30) but does not work well with the bovine milk enzyme.5 Instead we have utilized another unusual property of the enzyme to purify it, i.e. the formation of a reversible complex between the enzyme and in- solubilized heparin. By this method we have obtained a prep- aration which appears to be more than 80% pure by polyacryla- mide gel electrophoresis. Unfortunately, due to the limited solubility and the low stability of the enzyme preparation we have not yet been able to obtain rigorous proof that the main component in our preparation really is lipoprotein lipase. For the same reasons, a more thorough characterization of the enzyme molecule has not been possible.
at some site near the capillary lumen by interaction with heparin or a heparin-like substance (3). Our previous studies have shown that the bovine milk lipoprotein lipase does bind very strongly to heparin (5). For optimal activity the enzyme re- quires the presence of a serum lipoprotein component in the assay system (4, 5). However, the role of this component has not yet been defined. It is our hope that the present pro- cedure for obtaining a highly purified lipoprotein lipase will aid in the further exploration of the unusual properties of this physiologically important enzyme.
Acknowledgment-The expert technical Berith Lid& is gratefully acknowledged.
REFERENCES 1. KORN, E. D. (1955) J. Biol. Chem. 216, 2. ROBINSON, D. S. (1963) in Advances in Lipid Research
(PAOLETTI, R., AND KRITCHEVSKY, D., eds) Vol. 1, p. 133, Academic Press, New York
3. ROBINSON, D. S. (1970) in Comprehensive Biochemistry (FLOR- KIN, M., AND STOTZ, E. H., eds), Vol. 18, p. 51, Elsevier, New York
Fox and Tarassuk have previously purified a lipase about 500 times from bovine milk (31). This enzyme, which is usually called the milk lipase, interacts with the casein micelles in a way that closely resembles that of the lipoprotein lipase. That is, it binds to the casein micelles but can be liberated by 0.7 M
NaCl (32). Actually, the initial steps in the procedure of FOX and Tarassuk are quite similar to those in our procedure. They measured the activity of the milk lipase with milk lipids as sub- strate. The characteristic property of a lipoprotein lipase is that it requires a serum component for optimal activity (1). However, even our purest preparations have a low but significant activity against long chain triglyceride even when no serum is added to the assay system. Under the conditions of our assay there is no doubt that the serum-stimulated lipase is the main enzyme measured in crude skim milk (Table I). It is possible that the lipoprotein lipase we have purified is the same enzyme as the so-called milk lipase, but this question requires further study.
4. KORN, E. D. (1962) J. Lipid Res. 3, 246 5. OLIVECRONA, T., EGPLRUD, T., IVERIUS, P-H., AND LINDAHL,
U. (1971) Biochem. Biophys. Res. Commun. 43, 524 6. IVERIUS, P-H. (1971) Biochem. J. 134, 677-683 6a. IVERIUS, P-H., LIR’D:~HL, U., E~ELRUD, T., AND OLIVI~~RON~,
T. (1972) J. Riol. Chem. 247, in press 7. DOLE, V. P. (1956) J. C/in. Invest. 36, 150 8. SCHOTZ, M. C., GARFINKEL, A. S., HUEBOTTER, R. J., AND
STEWART, J. E. (1970) J. Lipid Res. 11. 68 9. LOWRY, 0. H., ROSEBROUGH, N. J., FARR,‘A. L. AND RANDALL,
R.. J. (1951) J. Biol. Chem. 193. 265-275 10. WERWR, K., .~ND OSHORN, M. (1969) i. Biol. Chem. 244, 4406-
4412 11.
12.
13.
14.
The yield in our purification is 5 to 10% of the enzyme activity in the starting material. Part of the losses seemed to be due to inactivation during the preparation and extraction of the rennet curd and during the heparin-Sepharose chromatography. In both of these steps the enzyme is exposed to high salt concentra- tions. It has previously been shown that rat tissue lipoprotein lipases are inactivated more rapidly at higher salt concentra- tions. This is also true of bovine milk lipoprotein lipase. Field- ing has shown that the rat tissue enzymes can be stabilized by salts of unsaturated long chain fatty acids (33). This is also true of the lipase in bovine skim milk.5 However, this stabiliza- tion cannot be utilized to increase the yield of active enzyme, since the fatty acid solutions flocculate when the salt concentra- tion is increased. We have not yet been able to find a method of purifying the enzyme without the use of high salt concentrations in some of the steps. Since we do not know how the inactivation of the enzyme occurs, we do not know whether the purified material contains some inactive enzyme in addition to the ac- tive one.
15.
16.
17.
18. 19.
20. 21. 22. 23. 24.
25.
26. 27. 28.
29. 30.
It has been suggested that the lipoprotein lipases are bound 31. 32.
4 T. Egelrud, unpublished observations. 5 T. Olivecrona. unoublished observations. 33.
assistance of Miss
PANYIM, S., .IND CHBLKLEY, R. (1969) Arch. Biochem. Bio- phys. 130, 337-346
ZACHARIUS, R. M., Z~,L, T. E., MORRISON, J. H., AND WOOD- LOCK, J. J. (1969) Ana/. B&hem. 30. 148
MCKENZIE, H: A.’ (1967) in Advance; in Protein Chemistry (ANFINSEN, C. B., JR., ANSON, M. L., EDSALL, J. T., -4%~
RICHARDS, M., eds), Vol. 22, p. 55, Academic Press, New York
ERNSTROM, C. A. AND TITTSLER, li. I’. (1965) in Fundamentals of Dairy Chemistry (Wsm, B. H., AND JOHNSON, A. H., eds) p. 590, The AVI Publishing Co., Inc., Westport, Corm.
PUTNAM, F. W. (1965) in Il’he Proteins (NEURATH, H., ed) 2nd Ed, Vol. 3, p. 172, Academic Press, New York
KORN, E. D., AND QUIGLEY, T. W., JR. (1957) J. Biol. Chem. 226. 833-839
OLIV&RONA, T., AND LINDAHL, U. (1969) A& Chem. &and. 23, 3587
KORN, E. D. (1957) J. Biol. Chem. 226, 827 PATTEN, R. L., AND HOLLENHERG, J. (1969) J. Lipid Res. 10,
374 WHAYNE, T. F., AND FELTS, J. M. (1970) Circ. Res. 27, 941 MCBRIDE, 0. W., AND KORN, E. 1). (1963) J. Lipid Res. 4, 17 ROBINSON, D. S. (1963) J. Lipid Res. 4, 21 MCBRIDE, 0. W., AND KORN, E. D. (1964) J. Lipid Res. 6, 453 ASKEW, E. W., EMERY, R. S., AND THOMAS, J. W. (1970) J.
Dairy Sci. 63, 1415 O~wau, S., .IND RORINSON, I>. S. (196X) Bioch,em. J. 106, 677-
682 FIELDING, C. J. (1969) Biochim. Biophys. Acta 178, 499-507 FIELDING, C. J. (1970) Biochim. Biophys. Acta 206, 109-117 NILSSON-EHLE, P., BELFRAGE, P., AND BORGST~~~~M, B. (1971)
Biochim. Biophys. Acta 248, 114-120 YASU~K~, S., AND FUJII, S. (1971) J. Biochem. 70, 407 GANE:SAN, D., AND BRADFORD, R. H. (1971) Biochem. Biophys.
Res. Commun. 43, 544 Fox, P. F., .~ND TARASSUK, N. P. (1968) J. Dnirly SC?. 61, 826 DO~NI~Y, W. K., AND ANDRX~S, P. (19G6) Wiochem. J. 101, 651-
660 FIELDING, C. J. (1968) Biochim. Biophys. Acta 169, 94-102
by guest on April 23, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Torbjörn Egelrud and Thomas OlivecronaThe Purification of a Lipoprotein Lipase from Bovine Skim Milk
1972, 247:6212-6217.J. Biol. Chem.
http://www.jbc.org/content/247/19/6212Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/247/19/6212.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on April 23, 2020
http://ww
w.jbc.org/
Dow
nloaded from