Embed Size (px)
Citation preview

SYNTHESIS Journal ofSynthetic OrganicChemistry REPRINT
������������� ��������������
Thieme

REVIEW 1929
Chiral Phosphoric Acids as Versatile Catalysts for Enantioselective TransformationsChiral Phosphoric Acids as Enantioselective CatalystsMasahiro Terada*Department of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan Fax +81(22)7956602; E-mail: [email protected] 25 March 2010; revised 13 April 2010
SYNTHESIS 2010, No. 12, pp 1929–1982xx.xx.2010Advanced online publication: 27.05.2010DOI: 10.1055/s-0029-1218801; Art ID: E26710SS© Georg Thieme Verlag Stuttgart · New York
Abstract: Chiral phosphoric acids derived from axially chiral biar-yls and related chiral Brønsted acids have emerged as an attractiveand widely applicable class of enantioselective organocatalysts fora variety of organic transformations. This review focuses on recentachievements in the development of enantioselective transforma-tions using these axially chiral phosphoric acids and their analoguesas chiral Brønsted acid catalysts. The contents are arranged accord-ing to the specific types of carbon–carbon bond-forming reactions,followed by carbon–heteroatom bond-forming reactions and func-tional group transformations, including reduction and oxidation.Further applications to combined phosphoric acid and metal com-plex catalytic systems and new aspects in the development of chiralBrønsted acid catalysts are also highlighted.
1 Introduction2 Chiral Phosphoric Acids as Enantioselective Brønsted Acid
Catalysts 3 Mannich and Related Reactions4 One-Carbon Homologation Reactions via Activation of
Imines5 Friedel–Crafts and Related Reactions 6 Ene (Type) Reactions7 Cycloaddition Reactions8 Cyclization Reactions9 Transformations via Protonation of Electron-Rich Double
Bonds10 Miscellaneous Carbon–Carbon Bond-Forming Reactions11 Carbon–Heteroatom Bond-Forming Reactions 12 Transfer Hydrogenation Reactions13 Oxidations14 Combined Use of Metal Complex and Phosphoric Acid
Catalysts15 New Aspects in the Development of Chiral Brønsted Acid
Catalysts16 Conclusions
Key words: asymmetric catalysis, diastereoselectivity, electro-philic addition, enantioselectivity, multicomponent reaction
1 Introduction
Over the past decade, enantioselective catalysis by a smallorganic molecule, so-called organocatalysis, has becomea rapidly growing area of research as it offers operationalsimplicity, uses mild reaction conditions, and is environ-mentally benign.1 Among the procedures for organocatal-ysis reported to date, researchers have focused on ‘chiral
Brønsted acid catalysis’, the innovative methods of whichhave afforded enantioenriched products using a catalyticamount of a chiral organic molecule bearing an acidicfunctionality.2 The first example of chiral Brønsted acidcatalysis was reported by Jacobsen and co-workers in theenantioselective Strecker reaction catalyzed by peptide-based thiourea derivatives acting as hydrogen-bond donorcatalysts.3 Their landmark report in 1998 has fueled greatinterest in the enantioselective catalysis by chiral Brønstedacids. Their achievement has clearly indicated that achiral Brønsted acid enables discrimination between theenantiotopic faces of an imine substrate via hydrogenbonds, and has opened up a new avenue in enantioselec-tive catalysis without the use of chiral metal (Lewis acid)catalysts. Thereafter, excellent work on the enantioselec-tive hetero-Diels–Alder reaction was reported by Rawaland co-workers in 2003 using TADDOL (tetraaryl-1,3-di-oxolane-4,5-dimethanol) as the chiral Brønsted acid cata-lyst.4 These milestones have strongly influenced currentstudies on the development of chiral Brønsted acid cata-lysts. However, the acidity of the thiourea and aliphatic al-cohol functionalities is rather weak, with pKa valuesranging from 20 to 28 in DMSO.5,6 In contrast to these ad-vanced studies, an innovative approach to the develop-ment of chiral Brønsted acid catalysts that possessstrongly acidic functionalities was independently accom-plished by Akiyama and co-workers7 and Terada and co-workers8 in 2004. Highly enantioselective transforma-tions using 1,1¢-bi-2-naphthol (BINOL)-derived mono-phosphoric acids 1 as chiral Brønsted acid catalysts weredemonstrated by these two research groups (Figure 1).2a,c,9
Among the chiral Brønsted acids reported hitherto,2 chiralphosphoric acids derived from axially chiral biaryls repre-sent an attractive and widely applicable class of enantiose-lective organocatalysts for a variety of organictransformations. The present review focuses on recentachievements in the development of enantioselectivetransformations using axially chiral phosphoric acid cata-lysts 1 and related chiral Brønsted acids.
Figure 1 BINOL-derived monophosphoric acids 1 as chiralBrønsted acid catalysts
O
O
PO
OH
G
G
(R)-1
O
O
POH
O
G
G
(S)-1
Synthesis 2000, No. X, x–xx ISSN 0039-7881 © Thieme Stuttgart · New York
���
����
����
���
���
��� �
����
����
� �
����
���
����
���
����
����
����
��
�����
���
����
���
���
�����
��
����
Synthesis 2000, No. X, x–xx ISSN 0039-7881 © Thieme Stuttgart · New York
���
����
����
���
���
��� �
����
����
� �
����
���
����
���
����
����
����
��
�����
���
����
���
���
�����
��
����

1930 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
2 Chiral Phosphoric Acids as Enantioselective Brønsted Acid Catalysts
The desirable features of phosphoric acids as chiral Brønstedacid catalysts are summarized as follows (Figure 2):
1) Phosphoric acids are expected to capture electrophiliccomponents through hydrogen-bonding interactions with-out forming loose ion pairs, owing to their relativelystrong yet appropriate acidity.10
2) The phosphoryl oxygen would function as a Brønstedbasic site and hence it is anticipated that it would conveyacid/base dual function even to monofunctional phospho-ric acid catalysts. This catalyst design is conceptually sim-ilar to the mainstream of bifunctional organocatalystdesign reported to date.1,11 However, in a strict sense, thephosphoric acid catalysts should be distinguished frommost bifunctional organocatalysts, in which rather weaklyacidic and basic functionalities are introduced individual-ly to the catalyst molecule.
3) An acidic functionality is available even with the intro-duction of a ring system. It is likely that this ring systemeffectively restricts the conformational flexibility of thechiral backbone.
4) Substituents (G in Figure 2) can be introduced to thering system to provide a chiral environment for enantiose-lective transformations.
It is anticipated that an efficient substrate recognition sitewould be constructed around the activation site, namely,the acidic proton, of the phosphoric acid catalyst, as a re-sult of the acid/base dual function and the steric and elec-tronic influence of the substituents (G).
Figure 2 Phosphoric acids to be developed as chiral Brønsted acidcatalysts
As shown in Figure 1, axially chiral biaryls, especiallyBINOL derivatives, were mainly employed as chiralsources for the construction of the ring structure. BINOLis well known as an axially chiral molecule having C2-symmetry. Its derivatives have been utilized extensivelyas chiral ligands for metal catalysts.12 This C2-symmetryis crucial because it means that the same catalyst moleculeis generated when the acidic proton migrates to the phos-phoryl oxygen. In addition, both enantiomers of BINOLsare commercially available and numerous protocols forthe modification of BINOLs have been reported to date, inwhich a variety of substituents (G) can be introduced tothe 3,3¢-positions of the binaphthyl backbone. These ster-ically but also electronically adjustable substituents (G)can be utilized to create an appropriate chiral environmentfor enantioselective transformations.13
3 Mannich and Related Reactions
Akiyama et al. reported enantioselective catalysis in theMukaiyama type Mannich reaction that used BINOL-derived phosphoric acids 1 as chiral Brønsted acid cata-lysts.7 In the same year, Terada and Uraguchi indepen-dently demonstrated a highly enantioselective directMannich reaction using similar phosphoric acids.8 Asthese enantioselective Mannich reactions were success-fully developed, chiral phosphoric acids have been widelyutilized as efficient enantioselective organocatalysts fornumerous organic transformations. Among the asymmet-ric reactions investigated, the electrophilic activation ofimines by chiral phosphoric acid has been proven to be anattractive and efficient method for the construction ofnitrogen-substituted stereogenic centers in optically ac-tive forms.14
3.1 Mannich Reaction
Enantioselective Mannich reactions are widely utilizedfor the construction of optically active b-amino carbonylcompounds15 that serve as versatile intermediates for thesynthesis of biologically active compounds and drug can-didates. Highly enantioselective Mannich reactions have
Masahiro Terada was bornin 1964 in Tokyo, Japan. Hegraduated in 1986 and re-ceived his PhD in 1993 fromTokyo Institute of Technol-ogy. He was appointed as anAssistant Professor in Pro-fessor Mikami’s Laboratoryat Tokyo Institute of Tech-nology in 1989. He workedas a postdoctoral fellowwith Professor M. D. Shair
at Harvard University in1999–2000 and moved toTohoku University as anAssociate Professor in2001. He has been a Profes-sor of Chemistry at theGraduate School of Science,Tohoku University (Japan)since 2006. He is the recipi-ent of the Incentive Awardin Synthetic Organic Chem-istry, Japan (2003), The
Chemical Society of JapanAward for Creative Work(2008), and the MukaiyamaAward (2010). His currentresearch interests are fo-cused on the development ofnew and useful syntheticmethodologies based on thedesign of novel chiralBrønsted acid and base cata-lysts as well as the utilizationof transition-metal catalysts.
Biographical Sketch
OPO
O
O H
G
G
Brønsted basic site
stereo & electronic effect
Brønsted acidic site
substrate recognition site
dual function by monofunctional catalyst

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1931
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
been established using other types of organocatalysts,such as proline and its derivatives, chiral secondaryamines.16
In 2004, Terada and Uraguchi developed the chiral phos-phoric acid 1 catalyzed enantioselective direct Mannichreaction of imine 2 with acetylacetone (3).8 Chiral phos-phoric acid 1a exhibited extremely high catalytic activityfor the direct Mannich reaction of N-Boc-protected imine2aa with acetylacetone (3) (Scheme 1). The resulting b-amino ketone 4a was obtained in an optically active form(12% ee). The beneficial effects of the diaryl substituentsat the 3,3¢-positions of the binaphthyl backbone are note-worthy in regards to the enantioselectivity. For instance,performing the direct Mannich reaction using 3,3¢-phenyl-substituted phosphoric acid 1b furnished the correspond-ing product in 56% ee. Interestingly, the simple extensionof the aromatic substitution to the para-position improvedthe enantioselectivity dramatically. Use of 1d as the cata-lyst further increased the enantioselectivity to 95% ee,giving the product 4a in nearly quantitative yield.
Scheme 1 Direct Mannich reaction catalyzed by 1
The present catalytic reaction was applicable to ortho-,meta-, and para-substituted N-Boc-protected aromaticimines 2a and the corresponding products were obtainedin excellent chemical yields with high enantioselectivities(Scheme 2). The reaction proceeded smoothly withoutany detrimental effects even on a gram scale and the cata-lyst load could be decreased to 1 mol% while maintaininghigh yield and enantioselectivity. In addition, more than80% of catalyst 1d could be recovered.
In the present direct Mannich reaction, it is consideredthat the dual function of the phosphoric acid moietysmoothly accelerates the reaction (Figure 3). The enolproton of the acetylacetone tautomer and the O-H protonof 1 function as acidic sites, while the nitrogen atom of 2aand the phosphoryl oxygen function as basic sites. In thedirect Mannich reaction of 2a with 3, 1 would enable theformation of a transient structure through a hydrogen-bonding network that connects the acidic and basic siteswith each other (Figure 3a). Thus, phosphoric acid cata-lyst 1 electrophilically activates 2a through the acidic pro-ton, and the Brønsted basic phosphoryl oxygen interactswith the O-H proton of the enol form of 3. Subsequentbond recombination results in the formation of Mannichproduct 4 and the regeneration of catalyst 1 (Figure 3b).More importantly, the reaction proceeds under a chiral en-vironment created by the chiral conjugate base of 1through hydrogen-bonding interactions to furnish optical-ly active products 4.
Figure 3 Assumed mechanism of enantioselective direct Mannichreaction catalyzed by chiral phosphoric acid 1
Akiyama and co-workers independently reported theMukaiyama-type Mannich reaction of ketene silyl acetals5 with imines 2b derived from 2-hydroxyaniline(Scheme 3).7,17 The introduction of 4-nitrophenyl groupsat the 3,3¢-positions of the catalyst binaphthyl backbone,giving 1e, yielded the best results in terms of catalytic ac-tivity and enantioselectivity. Mannich products 6 were ob-tained with high syn-selectivity and the enantioselectivityof the major syn-isomers reached as high as 96% ee. In thepresent Mannich reaction, an ortho-hydroxy functionalityintroduced onto the N-aryl moiety of the imine is essentialto achieve the high stereoselectivities. Yamanaka, Akiyama,and co-workers rationalized the re-facial selectivity of thepresent reaction on the basis of experimental results anddensity functional theory (DFT) calculations,18 in whichthe phosphoric acid catalyst and the imine form a nine-membered cyclic structure (A) through a double hydro-gen-bonding interaction.
The creation of a new structural motif of chiral phosphoricacids is a challenging task to broaden the scope of chiralBrønsted acid catalysis. Akiyama et al. demonstrated that
N
Ph
Boc
H
+
NH
Ph
Boc
O
OCH2Cl2, r.t., 1 h
Phβ-naphthyl
1a: G = H 1b: G =
1c: G = 1d: G =
92%, 12% ee 95%, 56% ee
88%, 90% ee 99%, 95% ee
(R)-1 (2 mol%)
(R)-4a
O
O
O
O
PO
OH
G
G
(R)-1
2aa 3
Scheme 2 Direct Mannich reaction of imines 2a
N
Ar
Boc
H
+
NH
Ar
Boc
O
OCH2Cl2, r.t., 1 h
493–99%, 90–98% ee
X
X = MeO, Me, Br, F
Ar =
(R)-1d (2 mol%)
O
O
2a 3
N
Ar
H
Boc
HOP
O
H
O
O
O
O
hydrogen-bonding network under chiral environment
a) b)
: acidic sites : basic sites : hydrogen bonds
Ar
NBoc
HOO
HOP
OO
O
*
G
G
G
G
formation of product 4via bond recombination
3
2a
4
1 1

1932 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
chiral phosphoric acid 7, derived from TADDOL, func-tioned as an efficient enantioselective catalyst for theMukaiyama-type Mannich reaction of imines 2c withketene silyl acetals 5d (Scheme 4a).19 Akiyama et al. alsodeveloped a phosphoric acid catalyst derived from an ax-ially chiral biphenol,20 which was also applied to theMukaiyama-type Mannich reaction of ketene silyl acetals5 with imines 2b, affording Mannich products in goodyields albeit with a slight decrease in enantioselectivities.Terada et al. also developed phosphorodiamidic acid 9 foruse as an efficient Brønsted acid catalyst in the directMannich reaction of N-acyl imines 2d with acetylacetone(3) (Scheme 4b).21 Although the asymmetric induction ofMannich product 10 is still moderate, phosphorodiamidicacid 9 is a viable structural motif of chiral Brønsted acidcatalysts. Further modification of the chiral diamine back-bone or the substituents on the nitrogen atoms of the cata-lyst could lead to its becoming an efficientenantioselective catalyst.
Gong and co-workers applied chiral phosphoric acid cat-alysts to the three-component direct Mannich reaction torealize a one-pot reaction among aromatic aldehyde 11,aniline (12a), and ketones (Scheme 5).22 The reaction ofcyclic ketones 13 as the nucleophilic component proceed-ed smoothly in the presence of 0.5 mol% 1f or 2 mol%14a, giving desired products 15 in good yields with highenantio- and anti-selectivities. They also extended thecoupling method to the reaction of acyclic ketones, suchas acetone and acetophenone, which gave products ingood yields when the catalyst load was increased to 5mol% and excess amounts of these ketones (10 equiv)were used, although slight decreases in enantioselectivi-ties were observed. Shortly thereafter, Rueping et al. re-ported achiral Brønsted acid assisted chiral Brønsted acid
catalysis in the direct Mannich reaction of acyclic ke-tones.23 The reaction of N-aryl imines with acetophenonewas conducted using a chiral phosphoric acid catalyst incombination with acetic acid as the co-catalyst. The cor-responding products were obtained in acceptable yieldseven when the amount of acetophenone employed was de-creased (2 equiv).
Scheme 5 Three-component direct Mannich reaction
3.2 Nucleophilic Addition of Diazoacetates to Aldimines
a-Diazocarbonyl compounds have an electronicallyunique sp2-carbon to which the diazo group is attached.Thus, these compounds have a partially negative chargeand function as nucleophiles. In the reactions of imines, a-diazocarbonyl compounds are commonly used in aziri-dine formation reactions (aza-Darzens reaction) under
Scheme 3 Mukaiyama-type Mannich reaction of imines 2b with ke-tene silyl acetals 5
N
R H
+
HN
RCO2R2
R1
(R)-1e (10 mol%)
HO
R1
OTMS
OR2
HN
RCO2R2
R1
toluene–78 °C, 24 h
+
G = NO2
HO HO
R1 = Me
R1 = Bn
R1 = Ph3SiO
R2 = Et
R2 = Et
R2 = Me
87 (96% ee)
93 (91% ee)
100 (91% ee)
100% yield
100% yield
79% yield
: 13
: 7
: 0
syn-6 anti-6
(87% E)
(87% E)
(91% E)
N
Ph H
OH
P
O
OO
O H*
A
2b (R = Ph)5
syn-6 anti-6
5a:
5b:
5c:
Scheme 4 Other structural motifs of chiral phosphoric acid catalysts
N
Ar H
+7 (5 mol%)
HO
OTMS
OMe
HN
ArCO2Me
HO
8100%, 89% ee
toluene –78 °C, 30 h
O
P
OO
O
ArAr
ArAr
O
OH
(Ar = 4-F3CC6H4)
a)
b)
N
Ar H
+
NH
Ar
O
O
toluene, r.t., 4 h
(R)-9 (2 mol%)
1042%, 56% ee
N
N
PO
OH
O
O
O2S
O2S Ar
Ar
Ph
O
O
Ph(Ar = 4-MeC6H4)
2c (Ar = Ph)
2d (Ar = Ph)
5d
3
O
Ar H+
NH
Ar
Ph
toluene, 0 °C, 48 h
(R)-1f (0.5 mol%)or (R)-14a (2 mol%)
O
O
PO
OH
Ph
Ph
O
77–92% anti,89–98% anti,
83 to >99%,74–97%,
(R)-1f (R)-14a
H2N
X
O
Xanti-15
+
Cl1f: G =
(R)-14a
X = CH2, O, NBocX = S
80–95% ee83–98% ee
11 12a
13

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1933
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Lewis24 and Brønsted25 acid catalyzed conditions. Mean-while, Terada and co-workers reported an enantioselec-tive direct substitution at the a-position of diazoacetateusing a chiral phosphoric acid catalyst.26 Phosphoric acidcatalyst 1g efficiently promoted the substitution reactionof a-diazoacetates 16 with N-acyl imines 2e to giveMannich-type products 17, which are b-amino esters hav-ing a diazo substituent at the a-position, in optically activeforms (Scheme 6). Interestingly, the electronic propertiesof the acyl protective group of the imine nitrogen pro-foundly affected the enantioselectivity. The para-substit-uents of the N-acyl aromatic moiety had a marked effecton the enantioselectivity, and the introduction of anelectron-donating dimethylamino moiety gave the best re-sults. A series of aromatic imines 2e were applicable tothese substitution reactions. Para-substituted aromaticsshowed excellent enantioselectivities irrespective of theirelectronic properties. Ortho- and meta-substitutions aswell as fused-ring systems were also applicable. The thus-obtained b-amino-a-diazoester products 17 could betransformed into common synthetic intermediates, such asb-amino acid derivatives, via simple reduction or oxida-tion of the diazo moiety.
Scheme 6 a-Substitution reaction of diazoacetate with imines
The phosphoric acid is expected to promote the substitu-tion reaction as a result of its dual function (Figure 4a). In-tracomplex deprotonation from B by the basic phosphoryloxygen would allow the direct substitution of diazoace-tate, giving Mannich-type products without the formationof aziridine products.
Akiyama et al. reported the aziridine formation reaction(aza-Darzens reaction) using a-diazoacetate 16b and p-methoxyphenyl (PMP)-protected imines 18 prepared insitu from glyoxal monohydrate 19 and 4-methoxyaniline(12b) in the presence of magnesium sulfate (Scheme 7).27
Chiral phosphoric acid catalyst 1h gave the correspondingaziridine products 20 with high enantioselectivities andthe cis-isomer was formed exclusively. The PMP protec-tive group would preserve the nucleophilicity of the nitro-gen atom and hence the intramolecular substitution by thenitrogen atom would proceed predominantly via interme-diate C (Figure 4b). This contrasts the reaction of N-ben-zoyl imines where Mannich-type products were obtained
in high yields. The electron-withdrawing property of theN-benzoyl protective group significantly decreased theelectron density of the nitrogen atom, effectively sup-pressing nucleophilic substitution by the nitrogen atom.
Meanwhile, Zhong and co-workers reported the trans-selective aza-Darzens reaction of diazoacetamide 21 withN-Boc aromatic imines 2a (Scheme 8).28 In the presenceof chiral phosphoric acid catalyst 1g, the correspondingaziridine products 22 were obtained with excellent trans-selectivities, in this case, with high enantioselectivities.Although the dramatic changes in the stereochemical out-comes have not been clarified yet, the amide moiety mayfunction as a hydrogen-bond donor site to the phosphoric
Ar =
Ar
N
H
O
4-Me2NC6H4
Ar
NH
t-BuO2C
N2
+ (R)-1g (2 mol%)
X
O
4-Me2NC6H4
toluene, r.t., 24 h
X = F, Ph, Me, MeO
62–74%97% ee
X = 2-F, 2-MeO, 3-F
X
84–89%91–93% ee
O
O
75%95% ee
N2
Ht-BuO2C
G =
16a 2e17
Figure 4 Mechanistic proposal for reactions of diazoacetate withimines
R2O2C
N
N
H
N
H R1
R2O2C
N
N
NH
R1
a) PG =
R2O2C
H
N
R1
N
N
R2O2CR1
N
b) PG =
aziridine
elimination
PG
PG
PG
C
R2O2C
H
R1
N
N
B
H
N
H
OP
O
OO
PG
addition
O P
O
O
OPG H
HOP
O
O
O
O
NMe2
OMe
deprotonation
Mannich-type product
Scheme 7 Enantio- and cis-selective aza-Darzens reaction
HN
PMP
CO2Et
H2N OMe
O
Ar
OH
OH +
O
Ar
(R)-1h (2.5 mol%)
G = (4-t-BuC6H4)3Si
MgSO4
toluene, r.t., 1 h
toluene–30 °C, 23 hO
Ar
N
H
PMP
cis-2091–100%
92–97% ee
N2
H CO2Et
12b
16b
19
18

1934 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
acid catalyst and affect the configuration of the transientassembly composed of ternary constituents.
3.3 Biginelli Reaction
Gong and co-workers reported the enantioselectiveBiginelli reaction via the three-component coupling of al-dehydes 11, (thio)urea 23, and b-keto esters 24(Scheme 9).29 Utilizing the Mannich reaction as the initialstep, the method enabled efficient access to 3,4-dihydro-pyrimidine-2(1H)-one derivatives 2530 that possess a widearray of pharmaceutical properties and hence are consid-ered to be medicinally relevant compounds. Catalyst 14ahaving the H8-binaphthyl backbone most effectively fur-nished the corresponding pyrimidinone derivatives inmoderate to high yields with high enantioselectivities. Awide variety of aldehydes 11 are employable in this reac-tion. Gong and co-workers further optimized the substitu-ent of catalyst 1 in the present Biginelli reaction.31
Triphenylsilyl-substituted catalyst 1i was found to pro-vide higher enantioselectivity than that obtained by 14a inmost cases. In addition, the reaction catalyzed by (R)-1igave product 25 whose enantiofacial selection is oppositethat of catalyst (R)-14a even when using the same axialchirality of the catalysts. They performed transition-stateanalysis by DFT calculations to elucidate the reverse ofthe enantiofacial selection in these catalyses. They alsodemonstrated the synthetic utility of the enantioselectiveBiginelli reaction in the preparation of optically active in-termediate 26 of (S)-L-771688,32 a selective a1a receptorantagonist (Scheme 10).
Gong and co-workers further applied this reaction usingcatalyst 1i to the Biginelli-like condensation, employingenolizable ketones, including not only cyclic ketones 13but also acyclic ketones 27, in place of b-keto esters 24(Scheme 11).31 A broad range of cyclic ketones 13 andacyclic ketones 27 were applicable to the Biginelli-likecondensation with aromatic aldehyde 11 and N-benzyl-thiourea (28), giving the corresponding products 29 and30 with excellent enantioselectivities.
3.4 Vinylogous Mannich Reaction
The asymmetric Mannich reaction is one of the most ubiq-uitous carbon–carbon bond-forming reactions in organicchemistry. The vinylogous extension of this fundamentalcarbon–carbon bond-forming reaction to nucleophiliccomponents, namely, the vinylogous Mannich reaction,
Scheme 8 Enantio- and trans-selective aza-Darzens reaction
HAr
NBoc
+ (R)-1g (5 mol%)
CH2Cl2, r.t., 10 min
trans-2290–97%, 88–96% ee
N2
HN
Ar H
Boc
2aO
HN
PMP
21
G =
O NH
PMP
Scheme 9 Biginelli reaction
+
(R)-14a (10 mol%)or
G = Ph3Si (R)-1i (10 mol%)
+O
R1 H
O
OR2
OX
NH2H2N
CH2Cl225 °C, 6 d
2551–86%, 85–97% ee (R)
40%, 92% ee (R)
HN NH
X
CO2R2
R1
23(X = S or O)
*
11 24
R1 = aromaticR1 = cyclohexyl
(R1 = aromatic or cyclohexyl)
(R)-14a
O
O
PO
OH
Ph
Ph
(R)-1i54–98%, 91–98% ee (S)
54%, 90% ee (S)R1 = aromaticR1 = cyclohexyl
toluene50 °C, 60 h
Scheme 10 Application to asymmetric synthesis of (S)-L-771688precursor
+
(R)-1i (10 mol%)
+
O
HO
OMe
OS
NH2H2N
CH2Cl2, 50 °C, 6 d
25a86%, 91% ee (S)
HN NH
S
CO2Me
Ar
23a11a
24a
F
F
N NH
O
CO2Me
F
F
OMe
NH
O
N
N
(S)-L-771688
HN NH
O
CO2Me
ArOMe
26
ref. 32
Ar = F
F
Scheme 11 Biginelli-like reaction via three-component coupling
+
(R)-1i (10 mol%)
+O
Ar H
S
NHBnH2N
toluene50 °C, 6 d
39–86%90 to >99% ee
HN N
S
Ar
2811X
O
X = CH2, O, NBoc, S
n = 1–3
13
O
R2or
27
X
HN N
S
Ar
R1
R2or
(R1 = aliphatic)(R2 = aliphatic or aromatic)
33–83%,49%,
R2 = aliphaticR2 = aromatic
30 + regio isomer <20%
BnBn
29
R1
92–96% ee 61% ee
n
n

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1935
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
has been little exploited33 despite its potential to provideefficient access to highly functionalized d-amino carbonylcompounds bearing a double bond. 2-Siloxyfuran 31 is anattractive vinylogous nucleophile that has been utilizedextensively in vinylogous variants of fundamental trans-formations, such as the aldol reaction,34 because the reac-tions provide g-substituted butenolides, an importantstructural motif existing in naturally occurring productsand biologically active compounds. Akiyama et al. suc-cessfully developed the enantioselective vinylogousMannich reaction of 2-trimethylsiloxyfuran (31) with al-dimines 2b using chiral phosphoric acid catalyst 1j bear-ing iodine groups at the 6,6¢-positions of the binaphthylbackbone (Scheme 12).35 A series of aldimines, includingaliphatic ones, were utilized in the present reaction and g-butenolide products 32 were obtained in good yields withmoderate to high diastereo- and enantioselectivities.
Scheme 12 Vinylogous Mannich reaction of 2-trimethylsiloxyfuran(31)
Scheme 13 Vinylogous Mannich reaction of silyl dienol ether 33
Schneider and Sickert were the first to demonstrate thecatalytic and enantioselective vinylogous Mannich reac-tion of acyclic silyl dienol ether 33 with aldimines 2f us-
ing chiral phosphoric acid catalyst 1k (Scheme 13).36 Thereaction provided highly valuable d-amino-a,b-unsaturat-ed carboxylic esters 34 in optically active forms withcomplete regioselectivities. Schneider and co-workersalso reported the enantioselective vinylogous Mannichreaction of vinylketene silyl N,O-acetals with aromaticimines.37 The piperidine-derived vinylketene silyl N,O-acetals proved to be more suitable substrates than their di-alkylamine- and pyrrolidine-derived counterparts, fur-nishing the corresponding vinylogous products, d-amino-a,b-unsaturated amides, in good yields with high enantio-selectivities.
3.5 Aza-Petasis–Ferrier Rearrangement
Organocatalysis in the direct Mannich reactions of alde-hydes with aldimines using chiral secondary amine cata-lysts has emerged as a powerful tool to provide b-aminoaldehydes with high diastereo- and enantioselectivities.38
However, one critical drawback inherent to the methodol-ogies reported to date is that aromatic or glyoxylate-derived aldimines are employed as adaptive substrates inmost cases. The enantioselective direct Mannich reactionof aliphatic aldimines has largely been unexploited.38c
Terada and Toda developed an alternative strategy to fur-nish optically active b-amino aldehydes 35 having an ali-phatic substituent (R) at the b-position by combining twocatalytic reactions (Scheme 14).39 The sequence involvesinitial nickel complex catalyzed Z-selective isomerizationof a double bond, followed by chiral phosphoric acid cat-alyzed aza-Petasis–Ferrier rearrangement,40 using readilyavailable hemiaminal allyl ethers 36 as the substrate. Theaza-Petasis–Ferrier rearrangement of hemiaminal vinylethers 37 proceeded via carbon–oxygen bond cleavage ofthe ether moiety by acid catalyst 1l, generating a reactiveiminium intermediate and an enol form of the aldehyde.Subsequent recombination with carbon–carbon bond for-
N
R H
+
HN
R
(R)-1j (5 mol%)
HO
toluene, 0 °C, 18–22 h+
G = i-Pr
HO
R = PhR = 4-F3CC6H4
R = 4-O2NC6H4
R = i-Pr
91 (82% ee)69 (99% ee)97 (96% ee)88 (92% ee)
100% yield95% yield85% yield84% yield
: : :
syn-32
O OTMS
O
O
O
O
PO
OH
G
GI
I
i-Pr
i-Pr
anti-32
9313
12
2b
31
(R)-1jsyn-32anti-32
(R)-1k (5 mol%)
G =
N
R H
PMP
R = PhR = 4-EtC6H4
R = t-Bu
87%, 88% ee88%, 92% ee83%, 82% ee
OTBS
OEt
NH
R
O
OEt
PMP
+
THF–t-BuOH–2-Me-2-BuOH = 1:1:1
–30 °C, 3–48 h2f 33
34
Scheme 14 Double-bond isomerization/aza-Petasis–Ferrier rear-rangement sequence for the preparation of b-amino aldehydes 35
H R
HNBoc
(±)-36
O R
HNBoc
[Ni-H] (cat.)
(R)-1l (2 mol%)
O R
HNBoc
>95% Z
O
HO R
HNBoc
anti syn
+HO R
HNBoc
AcOEt or acetone40 °C, 2–7 h
G = t-Bu
i-Pr
i-Pr
MeOH, 0 °C, 1 h
NaBH4
R = BnR = isobutylR = cyclohexylR = Ph
(94% ee)(97% ee)
(>99% ee)(98% ee)
70% yield55% yield75% yield87% yield
95959593
5557
::::
35
(±)-37
>95% Z(±)-37

1936 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
mation resulted in rearranged products 35, thus providingb-amino aldehydes having not only aliphatic but also aro-matic substituents at the b-position with high anti- andenantioselectivities.
4 One-Carbon Homologation Reactions via Activation of Imines
4.1 Strecker Reaction and Related Reactions
The hydrocyanation of imines, namely, the Strecker reac-tion, offers a practical route to a-amino acids or 1,2-di-amines, which can be transformed from the correspondinga-amino nitrile products through simple hydrolysis or re-duction of the nitrile moiety. The development of the cat-alytic enantioselective Strecker reaction is hence a vitalstep toward the efficient synthesis of these amines in op-tically active forms. A number of methods for the enan-tioselective Strecker reaction have been intensivelyinvestigated using either chiral metal catalysts or organo-catalysts.41 Chiral phosphoric acid catalysts were also suc-cessfully applied to the enantioselective Strecker reaction(Scheme 15). Rueping et al. were able to attain excellentperformance using chiral catalyst 1m, which has stericallydemanding 9-phenanthryl substituents at the 3,3¢-posi-tions of the binaphthyl backbone.42 The method offers ef-ficient access to a broad range of aromatic amino nitriles39 with high enantioselectivities (Scheme 15a). They alsodeveloped the enantioselective Strecker reaction ofketimines 40 catalyzed by the same chiral phosphoric acid1m to give products 41, having a chiral quaternary stereo-genic center, with moderate to high enantioselectivities(Scheme 15b).43,44
Scheme 15 Strecker reaction
In contrast to the Strecker reaction that involves the hy-drocyanation of imines, the cyanation of hydrazone togive synthetically valuable a-hydrazinonitriles is poorlyexploited45 despite its ability to provide efficient access toa-hydrazino acids of therapeutically intriguing molecules.
Tsogoeva and Zamfir developed a highly enantioselectiveStrecker-type reaction of hydrazones 42 using chiralphosphoric acid 1e (Scheme 16).46 Readily prepared andair-stable aliphatic hydrazones 42 are employable for thisreaction, giving synthetically valuable a-hydrazinonitriles44 in moderate to good yields with high enantioselectivi-ties. The thus-obtained a-hydrazinonitriles 44 can bereadily transformed into the corresponding a-hydrazinoacids 45 in a quantitative manner under acid hydrolysis.The authors also proposed that the active catalyst is an insitu generated O-silylated BINOL-phosphate, thus shift-ing the mode of catalysis from a Brønsted acid catalysis toa Lewis acid catalysis.
Scheme 16 Hydrocyanation of hydrazones 42 derived from alipha-tic aldehydes
4.2 Aza-Henry Reaction
The synthesis of b-nitro amines via the addition of nitroal-kanes to imines, or the so-called aza-Henry reaction, is anattractive tool for the creation of carbon–carbon bonds.The products obtained can be readily converted into vici-nal diamines and a-amino acids by simple reduction andthe Nef reaction, respectively, highlighting the several im-portant synthetic applications of these compounds. In thiscontext, enantioselective versions of the aza-Henry reac-tion have been actively investigated using chiral metalcomplexes and organocatalysts.47 Rueping and Antonchickdemonstrated the enantioselective direct aza-Henry reac-tion of a-imino esters 2h with nitroalkanes 46 using chiralphosphoric acid catalysts (Scheme 17).48 Catalyst 14b,with its sterically demanding triphenylsilyl substituents,significantly accelerated the reaction to provide b-nitro-a-amino acid esters 47 in good yields. A variety of nitroal-kanes were applicable to this reaction, giving desiredproducts 47 with high diastereo- and enantioselectivities.
4.3 Imino-Azaenamine Reaction
Formaldehyde dialkylhydrazones, such as azaenamines(nitrogen analogues of enamines), possess a carbon atomthat shows nucleophilic character, and are thus utilized asformyl anion equivalents.49 The reaction enables efficientone-carbon homologation of electrophilic substrates.Rueping et al. utilized this nucleophilic species in a reac-
N
HAr XHN
CNAr X
G =
(R)-1m (10 mol%)
toluene, –40 °C, 2–3 d+
HCN
X = HX = OMe
55–97%,53–87%,
85–99% ee86–96% ee
38
2g
39
N
Ar XHN
**CNAr X
(R)-1m (5 mol%)
toluene, –40 °C, 3 d+
HCN
X = OMeX = Br
81–95%,69–90%,
56–72% ee76–80% ee
38
40
41
a)
b)
N
HR
HN
O
(R)-1e (5 mol%)t-BuOH (20 mol%)
CH2Cl2, –10 °C, 3 d+ TMSCN
26–95%, 71–93% ee
43
42 (R = aliphatic)
44
G = NO2
NO2
HN
CNR
HN
O
NO2
HCl HN
COOHR
NH2⋅HCl
45

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1937
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
tion of imines 2a under the influence of chiral phosphoricacid 14c (Scheme 18).50 The reaction of pyrrolidine-derived methylenehydrazine 48 with a series of N-Boc ar-omatic imines 2a provided a-amino hydrazones 49 withmoderate to high enantioselectivities. Products 49 havebeen proven to be useful synthetic intermediates that canbe readily transformed into a diverse array of chiral nitro-gen-containing compounds, such as a-amino aldehydes,a-amino nitriles, and 1,2-diamines, without racemization.
Scheme 18 Imino-azaenamine reaction of hydrazone 48
4.4 Addition of Isocyanide to Aldimines
Isocyanides are useful one-carbon sources in organic syn-thesis. The Passerini three-component reaction51 and theUgi four-component reaction52 are representative trans-formations that use isocyanides as one-carbon homologa-tion reagents having a formally divalent carbon center,and the isocyanides can react with electrophiles and nu-cleophiles. Wang, Zhu, and co-workers developed enan-tioselective Ugi-type multicomponent reactions53 thatinvolved the a-addition of isocyanides to imines54 cata-lyzed by chiral phosphoric acids (Scheme 19). Catalyst 1kefficiently accelerated the three-component reaction ofaliphatic aldehydes 11, aniline derivative 12c, and a-iso-cyanoacetamides 50, giving 2-(1-aminoalkyl)-5-amino-
oxazoles 51 in good yields with moderate to highenantioselectivities.
5 Friedel–Crafts and Related Reactions
Enantioselective Friedel–Crafts reactions using metal-based chiral catalysts or chiral organocatalysts have beeninvestigated intensively, and provide atom-economicalmethods for the production of alkylated arenes in opticallyactive forms.55 The chiral phosphoric acid catalyzedFriedel–Crafts reaction was first accomplished via the ac-tivation of imines, but currently, the scope of electrophiliccomponents has been broadened to include (a,b-unsatur-ated) carbonyl compounds, nitroalkenes, and others.
5.1 Friedel–Crafts Reaction via Activation of Aldimines or Hemiaminals
The Friedel–Crafts reaction via the activation of electro-philes functionalized by a nitrogen atom, such as imines,is undoubtedly the most practical approach to introduce anitrogen-substituted side chain onto aromatic compounds.The enantioselective version of the Friedel–Crafts reac-tion of imines with electron-rich aromatic compounds en-ables efficient access to enantioenriched arylmethanamine derivatives. Several excellent approaches tohighly enantioselective Friedel–Crafts reactions havebeen established using chiral phosphoric acid catalysts.
Terada and co-workers successfully demonstrated for thefirst time an enantioselective 1,2-aza-Friedel–Crafts reac-tion of 2-methoxyfuran (52) with N-Boc aldimines 2a us-ing a catalytic amount of chiral phosphoric acid(Scheme 20).56 In the presence of 2 mol% 1n having ster-ically hindered 3,5-dimesitylphenyl substituents, the cor-responding Friedel–Crafts products 53 were obtainedwith excellent enantioselectivities, irrespective of theelectronic properties of the aromatic imines 2a employed.Most notable was that the reaction could be performed inthe presence of as little as 0.5 mol% 1n without any detri-mental effects even on a gram scale (Ar = Ph: 95%, 97%ee).
Scheme 17 Aza-Henry reaction of a-imino esters 2h
O
O
PO
OH
SiPh3
SiPh3
N
HR1O2C
PMP(R)-14b (10 mol%)
benzene, 30 °C, 12–166 h
+R2
NO2
HN
MeO2C
PMP
NO2
R2
anti-47
syn-47+
R2 = MeR2 = Me(CH2)4
R2 = Bn
(92% ee)(92% ee)(88% ee)
61% yield65% yield93% yield
919093
9107
:::
(10 equiv)2h (R1 = Me) 46
O
O
PO
OH
G
G
(R)-14c: G =
N
HAr
Boc
HN
Ar
Boc
(R)-14c (10 mol%)
CHCl3, 0 °C, 16 h+
48–82%, 74–90% ee
NN
NN
H
H
2a
48
49
Scheme 19 Three-component reaction of isocyanides, aldehydes,and anilines
O
HR1
HN
R1
toluene, –20 °C, 16 h
+
51–93%, 56–90% ee
11 (R1 = aliphatic)
51
(R)-1k (20 mol%)
G =
H2N
CF3
12cR2
CN
O
N
X
50 (X = O or CH2)
N
ON
R2
CF3
X
+

1938 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Scheme 20 1,2-Aza-Friedel–Crafts reaction of N-Boc imines 2awith 2-methoxyfuran (52)
The synthetic utility of this transformation is highlightedby the derivatization of the furyl ring to form a g-buteno-lide (Scheme 21). As the g-butenolide architecture is acommon building block in the synthesis of various naturalproducts, Friedel–Crafts reaction product 53 represents anew addition to the synthetic precursors of nitrogen-containing molecules. The aza-Achmatowicz reaction,followed by reductive cyclization of 1,4-dicarbonyl inter-mediate 54 under Luche conditions, produced g-buteno-lide 55 in good yield.
Scheme 21 Synthetic utility of furan-2-yl amine products
Further applications of the chiral phosphoric acid cata-lyzed 1,2-aza-Friedel–Crafts reaction were investigatedby several research groups and the developed methodsyielded a diverse array of optically active aryl methan-amine derivatives with high enantioselectivities. In partic-ular, the 1,2-aza-Friedel–Crafts reaction of indoles wasintensively investigated because the enantioenrichedproducts, namely, 3-indolyl methanamine derivatives, arewidely known as valuable structures of pharmacophoresand are present in thousands of natural products and manymedicinal agents possessing versatile therapeutic ef-fects.57 You and co-workers (Scheme 22a),58 Antilla andco-workers (Scheme 22b),59 and Terada et al.(Scheme 22c)60 almost simultaneously developed thehighly enantioselective Friedel–Crafts reaction of indoleswith aromatic imines. Their approaches differed in thesubstituents (G) of catalysts 1 introduced at the 3,3¢-posi-tions of the binaphthyl backbone and the protective groupof imines 2, and more interestingly, with or without theprotective group at the nitrogen atom of indoles 56.Goodman and Simón reported that, in the reaction of N-to-
syl imine 2i with unprotected indole 56a (Scheme 22a),coordination between the phosphoric acid and both com-ponents, the imine and the indole, was observed by DFTcalculations of the transition states.61 These componentswere brought together under the chiral environment creat-ed by the chiral phosphoric acid catalyst through hydro-gen bonds formed between the OH proton of thephosphoric acid and the nitrogen atom of imine 2i, as wellas the NH proton of indole 56a and the phosphoryl oxy-gen. It is noteworthy that the enantioselectivities were ashigh as that observed in the reaction using unprotected in-doles 56a, even when N-protected indoles 56b and 56cwere employed (Schemes 22b and 22c).
Scheme 22 1,2-Aza-Friedel–Crafts reaction of indoles 56 with aro-matic imines
The reaction of a-imino esters as an electrophilic compo-nent was also reported independently by Hiemstra and co-workers (Scheme 23a)62 and You and co-workers(Scheme 23b).63 In Hiemstra’s approach, indolyl glycineproducts 58a were obtained with opposite absolute con-figurations, depending on the sulfur substituent as well asthe structures of catalysts 1i and 14d, even when chiralphosphoric acids having axial chirality in the same R-configuration were employed.
Ma and co-workers reported an enantioselective synthesisof trifluoromethylated indole derivatives via the three-component coupling reaction among trifluoroacetalde-hyde hemiaminal 59, aniline derivative 12, and indoles56a (Scheme 24).64 Catalyst 1q having bulky 2,4,6-triiso-propylphenyl substituents worked well,9b giving the cor-responding products 60 in excellent yields. In the presentFriedel–Crafts reaction, the aniline derivative exhibited amarked effect on the enantioselectivity: 3,4,5-trimethoxy-aniline (12d) gave products 60 with much higher enantio-selectivities than 4-methoxyaniline (12b). Although theintroduction of a substituent at the 2-position of the indolering resulted in slight decreases in both reactivity andenantioselectivity, a wide range of functional groups, in-cluding electron-donating and electron-withdrawing sub-
Ar H
NBoc
+(R)-1n (2 mol%)
82–96%, 86–97% ee
OOMe
Ar
NH
OOMe
Boc
(CH2Cl)2, –35 °C, 24 h
2a
52
53
1n: G =
Ph
NHBoc
OOMe
Ph
NHBoc
OO
Ph
NHBoc
O OMeO
CeCl3⋅7H2O, NaBH4
MeOH, –78 °C to r.t., 5 h
NBS, NaHCO3
Et2O–H2O, 0 °C, 30 min
53a97% ee
54
5595%, 85% syn, 96% ee
90% (R)-1p (2–10 mol%)
65–91%82–98% ee
G =
Ph
Ph
(CHCl2)2
–40 °C24 h
c)
68–90%89 to >99% ee
N N
NH
Ar
PG1
+Ar H
NPG1
(S)-1o (10 mol%)
G =
toluene–60 °C
15 min to 2 ha)
*
89–99%90–97% ee
G = Ph3Si(S)-1i (5 mol%)
4 Å MSCH2Cl2–30 °C
15 h
b)
R R
PG2 PG2
PG1 = TsPG2 = H
PG1 = PhCO-PG2 = Bn
PG1 = BocPG2 = TBS
1 (cat.)
2
56
2i:56a:
2d:56b:
2a:56c:
57

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1939
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
stituents, were tolerated by the indole ring. The authorsextended this methodology to difluoroacetaldehyde hemi-aminal to demonstrate the scope of this three-componentreaction.
Scheme 24 Friedel–Crafts reaction via three-component couplingof trifluoroacetaldehyde hemiaminal 59, aniline derivative 12d, andindoles 56a
The enantioselective Friedel–Crafts reactions catalyzedby chiral phosphoric acids were further applied to suchelectron-rich aromatic compounds as pyrroles 61(Schemes 25a and 25b)65,66 and 4,7-dihydroindoles 63a(Scheme 25c).67 The reaction of 4,7-dihydroindoles 63ayielded 2-substituted indole derivatives 65 after oxidationof Friedel–Crafts products 64. This approach comple-ments current studies of enantioselective Friedel–Craftsreactions of parent indoles 56, where 3-substituted indole
derivatives 57 were obtained exclusively (seeScheme 22).
Enders et al. demonstrated the one-pot synthesis of enan-tioenriched isoindolines based on a consecutive transfor-mation that involved a chiral Brønsted acid catalyzedFriedel–Crafts reaction followed by a base-catalyzed aza-Michael addition reaction using indole (56aa) and bifunc-tional e-iminoenoates 2l as substrates (Scheme 26).68 Inthis one-pot transformation, the initial Friedel–Crafts re-action catalyzed by chiral BINOL-derived N-triflyl phos-phoramide 66, which was originally developed by
Scheme 23 1,2-Aza-Friedel–Crafts reaction of indoles 56 with a-imino esters
(R)-1i: G = Ph3Si
(R)-14d
NH
MeO2C H
NSRa)
b)
+(R)-1i or (R)-14d (5 mol%)
NH
MeO2C
benezene–toluene (4:1)5 Å MS
R = 2-nitrophenyl
R = Ph3C
86% ee (S)
88% ee (R)
*MS, 0 °C, 24 h
O
O
PO
OH
CHPh2
CHPh2
(R)-14d
56aa
2j
58a
R1O2C H
NPMP
NH
R2
+
G =
(S)-1m (10 mol%)
5 Å MStoluene, –50 °C, 1.5 h N
H
**
NH
EtO2C
PMP
R2
58b85–93%, 51–87% ee
56a
2h (R1 = Et)
CH2Cl23 Å MS
NHRS
NH
56a
60
(S)-1q (10 mol%)
G = i-Pr
i-Pr
i-Pr
NH
NH
F3C
MeO
MeO
OMe
R1
R1
R2
R2
4 Å MSCH2Cl2, r.t., 1–4 d
H2N
OMe
OMe
OMe
12d
OH
F3C OMe
59
+ +
R1 = HR1 = Me, R2 = H
90–98% ee79% ee
83–99%,80%,
Scheme 25 1,2-Aza-Friedel–Crafts reaction of pyrroles 61 and 4,7-dihydroindoles 63a
6480–97%, 86 to >99% ee
NH
Ar
Ts
+
Ar H
NTs
toluene, –40 °C, 5–80 min
c)
62b52–80%, 64–95% ee
NH
Ar
PyO2S
+
Ar H
NSO2 toluene, r.t., 12 h
b)
*
62a66–97%, 42 to >99% ee
NH
Ar
PhOC
+Ar H
N
CHCl3–60 °C, 23–41 h
a)Ph
O
G = Ph3Si(S)-1i (5 mol%)N
Me
R
N
Me
R
HN H
N
*
G = Ph3Si(R)-1i (10 mol%)
N
HN
R
HN
R
G = Ph3Si(S)-1i (10 mol%)
p-benzoquinone
NH
Ar
Ts
HN
R
2d 61a
61b (R = H)
2k
2i 63a
65
R R
Scheme 26 One-pot synthesis of enantioenriched isoindolines 67based on chiral Brønsted acid catalyzed Friedel–Crafts reaction/base-catalyzed aza-Michael addition sequence
+
(R)-66a (10 mol%)
CH2Cl2, r.t., 10 min
NO2(R)-66a: G =
NH
6794%, 90% cis, 90% ee
NTs
H
O
O
PO
NHTf
G
G
(R)-66
NTs
NH
CO2Me
DBU(50 mol%)
15 min
56aa 2lCO2Me
O
O
PO
NHTf
G
G

1940 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Yamamoto and Nakashima,69 followed by the DBU-promoted intramolecular aza-Michael addition, furnishedisoindoline derivatives 67 in good yields with highdiastereo- and enantioselectivities.
Rueping and Nachtsheim reported the nucleophilic substi-tution reaction of g-alkyl-g-hydroxylactams 68 with in-dole (56aa) catalyzed by chiral phosphoramide 66(Scheme 27).70 The reaction proceeds via in situ generat-ed N-acyliminium ions 69 from 68 under the influence of66b. The reaction provides optically active g-lactams 70having a quaternary stereogenic center at the g-position.The method furnishes an efficient approach to the g-ami-no acid derivatives of biologically relevant molecules andcan be extended to other nucleophilic components.
5.2 Pictet–Spengler Reaction
The Pictet–Spengler reaction of tryptamines and phenylethylamines with an aldehyde is a powerful and efficientmethod for the preparation of tetrahydro-b-carbolines andtetrahydroisoquinolines, respectively,71 which are struc-tural motifs of many alkaloids and related naturally occur-ring compounds. The enantioselective variants of thePictet–Spengler reaction are in high demand because ofthe versatile synthetic applicability of the correspondingproducts to biologically active molecules. The reactionconsists of a two-step transformation: an initial dehydra-tive imine formation, followed by an intramolecularFriedel–Crafts reaction. An excess amount of Brønstedacid is usually required to promote the reaction. List andco-workers accomplished the enantioselective Pictet–Spengler reaction by directly reacting substitutedtryptamines 71 with aldehydes 11 in the presence of chiralphosphoric acid catalyst 1q (Scheme 28a).72 The methodprovided efficient access to tetrahydro-b-carboline deriv-atives 72 with high enantioselectivities, although the in-troduction of a gem-disubstituent adjacent to the reactiveimine was required to suppress competing aldol pathways.Hiemstra and co-workers circumvented this intrinsicdrawback, the requirement of a gem-disubstituent, by im-plementing an iminium ion strategy. They used N-sulfe-
nyliminium ions as intermediates, generated from thereaction of N-sulfenyl tryptamines 73 with aldehydes 11,in the Pictet–Spengler reaction and established an effi-cient method for the preparation of tetrahydro-b-carbolinederivatives 74 in optically active forms using chiral phos-phoric acid 1r (Scheme 28b).73 With this approach, notonly aromatic but also aliphatic substituents can be intro-duced to the stereogenic center.
Scheme 28 Pictet–Spengler reaction of tryptamine derivatives withaldehydes
Hiemstra and co-workers applied their iminium ion strat-egy to natural product synthesis and accomplished theasymmetric total synthesis of the tetracyclic indole alka-loid (–)-arboricine (78) using the enantioselective Pictet–Spengler reaction as a key step (Scheme 29).74 The Pictet–Spengler reaction of N-allyl-substituted tryptamine 75with aldehyde 76 catalyzed by phosphoric acid 14b gavethe corresponding product 77 in good yield with highenantioselectivity.
5.3 Friedel–Crafts Reaction via Activation of Nitroalkenes
The enantioselective Friedel–Crafts reaction of indoleswith nitroalkenes has been known for a long time. This re-action offers considerable synthetic utility by enabling ac-cess to versatile precursors for the preparation of indolealkaloids. The enantioselective catalysis of this reactionhas been investigated by using chiral catalysts, includingmetal complexes and organic molecules.75 However, theenantioselectivities were not always good. Akiyama andco-workers developed a highly enantioselective Friedel–Crafts reaction of nitroalkenes 79 with indoles 56a usingchiral phosphoric acid 1i (Scheme 30).76 They pointed out
Scheme 27 Nucleophilic substitution reaction of g-alkyl-g-hydro-xylactams 68 with indole
+(R)-66b (5 mol%)
CH2Cl2, –65 to 20°C, 2–3 dNH
7039~93%, 53~84% ee
O
O
PO
NHTf
SiPh3
SiPh3
56aa 68 (R = aliphatic)
N
NH
∗∗ NO
Ph
R
O
Ph
R
HO NO
Ph
R
69
NH
NH2
7240–98%, 62–94% ee
(S)-1q (20 mol%)
Na2SO4
toluene or CH2Cl2–30 to –10 °C, 3–6 d
a)
RCHO+CO2Et
CO2EtX
NH
NH
CO2Et
CO2EtX
R
b)
(R)-1r (5–10 mol%)
3 Å MS, BHTtoluene, 0 °C, 0.5–24 h
NH
HNS
RCHO+
PhPh Ph
BHT: 3,5-di(tert-butyl)-4-hydroxytoluene
1q: G = i-Pr
i-Pr
i-Pr
71
11(R = aliphatic or aromatic)
73
11(R = aliphatic or aromatic)
NH
NH
R
HCl
PhSH
7477–90%, 30–88% ee
1r: G =
CF3
CF3

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1941
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
the beneficial effects of molecular sieves on both chemi-cal yield and enantioselectivity in the present Friedel–Crafts reaction. Among the molecular sieves tested, theMS 3A exhibited the best results, giving Friedel–Craftsproducts 80 in good yields with high enantioselectivities.The method is applicable not only to aromatic but also toaliphatic nitroalkenes. Friedel–Crafts product 80 can bereadily transformed into tryptamine derivative 81 via asimple reduction of the nitro group, giving melatonin ana-logue 82 and tetrahydro-b-carboline derivative 83 withoutracemization. The reaction of N-methylindole 56d provid-ed a racemic Friedel–Crafts product in low yield andhence, the N-H moiety of indoles was deemed essentialfor accelerating the reaction as well as controlling the ste-reochemical outcome.
You and co-workers employed 4,7-dihydroindoles 63a,instead of indoles 56, in the Friedel–Crafts reaction withnitroalkenes 79 (Scheme 31).77 Catalyst 1g displayed ex-
cellent performance; however, the syringe pump tech-nique was required to achieve high enantioselectivitiesbecause of considerable background reaction. Aromaticnitroalkenes 79 were useful substrates for this transforma-tion. Meanwhile, the use of aliphatic nitroalkenes signifi-cantly decreased the enantioselectivity. Oxidation ofFriedel–Crafts products 84 by p-benzoquinone provided2-substituted indole derivatives 85 in excellent yield with-out racemization. As shown in Scheme 30, this approachcomplements the reaction of parent indoles 56 in which 3-substituted indole derivatives 80 are obtained exclusively.
Scheme 31 Friedel–Crafts reaction of 4,7-dihydroindoles 63a withnitroalkenes 79
Shortly thereafter, You and co-workers further extendedthe substrate scope to include pyrroles 61b in the enantio-selective Friedel–Crafts reaction of nitroalkenes 79, giv-ing products 86 in high yields and enantioselectivities(Scheme 32).78
Scheme 32 Friedel–Crafts reaction of pyrroles 61b with nitro-alkenes 79
5.4 Friedel–Crafts Reaction via Activation of Carbonyl Compounds
A number of enantioselective transformations have beenestablished using chiral phosphoric acids via the activa-tion of imines and related nitrogen-substituted functional-ities. However, the activation of carbonyl compounds,including a,b-unsaturated carbonyl compounds, by chiralphosphoric acids has been limited and hence it is a chal-lenging task to expand the synthetic utility of chiral phos-phoric acid catalysts. Although the Friedel–Crafts
Scheme 29 Synthetic application of the enantioselective Pictet–Spengler reaction
NH
HN
I
+
OH
O
O
(R)-14b (2 mol%)
4 Å MStoluene, r.t., 24 h
O
O
PO
OH
SiPh3
SiPh3
NH
N
I
O
O
7786%, 89% ee
NH
N
O
H
H
78(–)-arboricine
75 76
Scheme 30 Friedel–Crafts reaction of indoles 56 with nitroalkenes79
NH
+(R)-1i (10 mol%)
3 Å MSbenzene–DCE (1:1)
–35 °C, 2–5 d
79
X
56a
RNO2
G = Ph3Si
NH
XR
NO2
80R = aromaticR = aliphatic
63–84%, 90–94% ee57–77%, 88–91% ee
NH
Ph
NH2[H]
81
NH
PhHN
NH
Ph
NTsAc
82
(X = H, R = Ph)
83Ph
+4 Å MS
CH2Cl2–benzene (1:1), r.t.syringe pump addition over 2 h
7963a
RNO2
84
R = aromaticR = cyclohexyl
93–98%, 94%,
85
(S)-1g (0.5 mol%)
G =
NH
X
NH
X
R
88–97% ee24% ee
p-benzoquinone
NH
X
R
NO2 NO2
+4 Å MS
CH2Cl2–benzene (1:1)r.t., 3~24 h
syringe pump addition over 2 h79
61b
RNO2
86
R = aromatic ortho-substituted (2-BrC6H4)R = cyclohexyl
87–94%,81%,<5%
(S)-1g (5 mol%)
G =
81–94% ee11% ee
NH
X
NH
X
R
NO2

1942 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
reaction with a,b-unsaturated carbonyl compounds in-volves fundamental transformations, simple a,b-unsatur-ated ketones are still challenging substrates in the chiralphosphoric acid catalyzed Friedel–Crafts reactions. Onlymoderate enantioselectivities were reported by Zhou, He,and co-workers in the Friedel–Crafts reaction of chalconederivatives 87 with indole (56aa) using H8-BINOL-derived chiral phosphoric acid 14e as catalyst(Scheme 33).79
Scheme 33 Friedel–Crafts reaction of chalcone derivatives 87 withindole
On the other hand, activated b,g-unsaturated a-keto esters89 functioned as efficient electrophiles in the enantio-selective Friedel–Crafts reaction catalyzed by chiral phos-phoramides 66 (Scheme 34). Rueping et al.80 and You andco-workers81 independently reported the enantioselectiveFriedel–Crafts reactions of b,g-unsaturated a-keto esters89 with N-methylindole 56d (Scheme 34a) and N-methyl-4,7-dihydroindole 63b (Scheme 34b), respectively. Theuse of chiral N-triflyl phosphoramide 66, which was de-veloped by Yamamoto and Nakashima69 and is a strongeracid than chiral phosphoric acid, is crucial for the acceler-ation of the reaction. The C3 and C2 functionalization ofthe indole derivatives was accomplished to give 90 and92, respectively, in a highly enantioselective manner.
Rueping et al. also reported interesting regioselectivity(1,2- vs. 1,4-addition) in the Friedel–Crafts reaction ofb,g-unsaturated a-keto esters 89 with N-methylindole 56dby tuning the steric and electronic effects of the substitu-ents introduced at the 3,3¢-positions of phosphoramidecatalysts 66. When a sterically demanding triphenylsilylgroup was introduced at the 3,3¢-positions of the binaph-thyl backbone to give catalyst 66b, 1,4-addition products90 were obtained exclusively (Scheme 34a). In contrast,when aryl groups were introduced at the 3,3¢-positions,the catalytic reaction yielded predominantly bisindolylproduct 93, which was derived from intermediary 1,2-ad-dition product 94 under the acid-catalyzed conditions(Scheme 35).80 The formation of bisindolyl products iswell known in the reactions of aldehydes, ketones, and1,2-diketones with indole derivatives catalyzed by Lewisand Brønsted acids.82 However, most interestingly, thisbisindolyl product 93 exhibited atropisomerism and was
separable as a stable isomer. After optimization of the cat-alyst substituents and the reaction conditions, the authorssucceeded in obtaining bisindolyl product 93 in an atrop-isomeric ratio of 81:19 using catalyst 66d.
Scheme 35 Formation of atropisomeric bisindolyl product 93 via1,2-addition reaction
O
O
PO
OH
G
G
(R)-14e (2 mol%)
CH2Cl2, 25 °C, 24 hNH
+
87
56aa
Ar1
NH
Ar1
8867–89%, 34–56% ee
O
Ar2
O
Ar2
(R)-14e: G = Cl
Scheme 34 Friedel–Crafts reaction of b,g-unsaturated a-keto esters89 with indole derivatives
O
O
PO
NHTf
SiPh3
SiPh3
(R)-66b (5 mol%)
CH2Cl2, –75 °C, 15–24 hNMe
+
89a
56d
Ar
NMe
Ar
9043–88%, 80–92% ee
O
CO2Me
O
CO2Me
(R)-66b
R
NMe
R
89b
Ar
O
CO2Et
63b
+
O
O
PNHTf
O
G
G
NMe
**
Ar O
CO2Et
(S)-66c (5 mol%)
Et2O, –60 °C, 1–30 h
9159–96%, 87–98% ee
R
R
a)
b)
NMe
**
Ph O
CO2Et
92
p-benzoquinone
(S)-66c: G = i-Pr
i-Pr
i-Pr
(Ar = Ph, R = H)
(R)-66d (5 mol%)
toluene, –78 °C+
89aa
Ph93
62% ee
O
CO2Me
O
O
PO
NHTf
G
G
(R)-66d: G =
NMe
56da
NMe
Ph
CO2Me
MeN
NMe
HO
Ph
CO2Me
94 95
(R)-66d
NMe
56da
MeO2C
Ph
– H2O(R)-66d

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1943
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
You and co-workers focused their attention on the forma-tion of intermediary vinylogous iminium ion 95 during thetransformation into bisindolyl products (Scheme 35).They successfully demonstrated the tandem intermolecu-lar/intramolecular Friedel–Crafts reaction of indole 56awith 2-formylbiphenyl derivatives 96 catalyzed by chiralphosphoric acid 1o (Scheme 36).83 In general, the use ofmolecular sieves enhanced the chemical yield and theenantioselectivity, because water is generated during thecourse of the second Friedel–Crafts reaction. Amongthose screened, MS 5A was the best, giving fluorene de-rivatives 97 in good yields with high enantioselectivities.In the reaction of N-methylindole derivative 56d, both thereaction rate and the enantioselectivity (35% ee) deterio-rated considerably and hence the N-H moiety of indoles56a functions not only to accelerate the reaction but alsoto efficiently control the stereoselectivity in the interme-diary benzylidene indolium ion 98.
Scheme 36 Tandem intermolecular/intramolecular Friedel–Craftsreaction of indole 56a with 2-formylbiphenyl derivatives 96
You and co-workers further applied the methodology tothe in situ generation of the benzylidene indolium ionfrom N-tosyl(3-indolyl)methanamine 99 under the influ-ence of chiral phosphoric acid 1s (Scheme 37).84 Themethod provides efficient access to the enantioselectivesynthesis of indole-containing unsymmetrically substitut-ed triarylmethane derivatives 100, albeit with moderateenantioselectivities. In this catalytic system, the authorsobserved an interesting feature: the kinetic resolution ofracemic (3-indolyl)methanamine 99 by chiral phosphoricacid catalyst 1s.
Ma and co-workers successfully developed the enantiose-lective Friedel–Crafts alkylation of indoles 56a with tri-fluoromethyl ketones 101, giving chiral tertiary alcohols102 in good yields without the formation of bisindolylbyproducts (Scheme 38a).85 Further substitution at thequaternary stereogenic center by indole 56a would be ef-ficiently suppressed by the trifluoromethyl group. Cata-lyst 1q is also effective for the relatively less reactive
trifluoromethyl alkyl ketone to afford the desired productin high yield albeit with a slight decrease in enantioselec-tivity. The method is applicable to the reaction of difluo-romethyl- and perfluoroethyl-substituted phenyl ketones,giving the corresponding products with high enantioselec-tivities, and hence provides an efficient route to the asym-metric synthesis of tertiary alcohols bearing a fluorinatedalkyl group in a highly enantioselective manner. Ma andco-workers also reported the enantioselective Friedel–Crafts alkylation of indoles 56a with trifluoroacetoacetate103 (Scheme 38b).86 Optically active trifluoromethylatedtertiary alcohols 104 were also obtained with moderate tohigh enantioselectivities.
Scheme 38 Friedel–Crafts reaction of trifluoromethyl-substitutedketones with indoles 56a
6 Ene (Type) Reactions
6.1 Aza-Ene-type Reaction of Aldimines with Enecarbamates
Kobayashi and co-workers pioneered the use of enamidesor enecarbamates as nucleophiles in enantioselective reac-
(S)-1o (5 mol%)
5 Å MSCCl4, –15 °C to r.t., 0.33–24 h
+
9737–96%, 73–96% ee
HN
56a
G =
HN
OHC
MeO OMe
R3
R2
R1
96
R1
R2
R3
MeOOMe
H
N
R1
R2
H
MeO OMe
R3
98
Scheme 37 Enantioselective synthesis of unsymmetrically substitu-ted triarylmethanes 100
(S)-1s (5 mol%)
5 Å MSbenzene or m-xylene, r.t., 4–52 h
+
NH
CF3
CF3
G =
99 56da
NMe
ArTsHN
NH
**
Ar
NMe
10064–96%, 45–68% ee
R
R
(S)-1q (5 mol%)
CH2Cl2, r.t., 1.5–3 d
NH
56a
NH
102
R1
R1
O
R2 CF3101
R2 = aromaticR2 = Bn
52–99%,86%,
85–98% ee76% ee
i-Pr
i-Pr
i-Pr
(S)-1q (10 mol%)
CH2Cl2, r.t., 3–5 d
O
CF3103
1q: G =
10436–99%, 55–78% ee
O
EtO
R2
CF3HO
NH
R1
**
CF3HO
CO2Et
a)
b)

1944 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
tions with either glyoxylate-derived imines or glyoxylatescatalyzed by chiral copper complexes.87 The reaction us-ing enamides or enecarbamates as nucleophilic compo-nents, namely, the aza-ene-type reaction, with iminesprovides b-amino imines that can be readily transformedinto 1,3-diamine derivatives via nucleophilic addition tothe imine moiety of the corresponding products.
Organocatalysis has been proven to be beneficial in manyrespects. However, one critical drawback inherent to themethodologies reported to date is the inadequate catalyticefficiency. Most organocatalytic reactions are performedat catalyst loads of 10 mol% or more to achieve sufficientchemical yields and avoid loss of enantioselectivity. Toensure high efficiency, one of the greatest challenges inpractical organocatalysis is to decrease the catalystload.3c,88 Terada et al. demonstrated a highly efficient or-ganocatalytic reaction that uses phosphoric acid with asignificantly low catalyst load in the aza-ene-type reaction(Scheme 39).89 The reaction of aromatic imines 2d withenecarbamates 105 could be accomplished in the presenceof 0.1 mol% phosphoric acid catalyst 1g. Both para- andmeta-substitution to aromatic imines, or substitution tofused aromatic and a,b-unsaturated imines, resulted in ex-cellent chemical yields and enantioselectivities, irrespec-tive of the electronic properties of the substituents.Although ortho-substitution reduced the catalytic effi-ciency, giving products 106 in moderate chemical yields,the yields were improved by increasing the catalyst loadto 0.5 mol%. It is noteworthy that the reaction can be per-formed without considerable loss of enantioselectivityeven when the catalyst load is decreased to as little as 0.05mol%. The synthetic applicability of the present highly ef-ficient organocatalysis was demonstrated by the reductionof the imine moiety by Red-Al, giving predominantly theanti-1,3-diamine derivatives 108.
Scheme 39 Enantioselective aza-ene-type reaction under low cata-lyst load
6.2 Cascade Transformation Based on Tandem Aza-Ene-type Reaction
The development of efficient methods to access complexmolecules with multiple stereogenic centers continues tobe a formidable challenge in both academia and industry.One approach is the use of catalytic enantioselective cas-cade reactions90 that have emerged as powerful tools torapidly increase molecular complexity from simple andreadily available starting materials, thus producing enan-tioenriched compounds in a single operation.
Terada et al. successfully applied the aza-ene-type reac-tion to the cascade transformation by taking advantage ofthe formation of imine products (Scheme 40).91 They em-ployed monosubstituted enecarbamates 10987f instead ofthe disubstituted versions and, as a result, piperidine de-rivatives 110 with multiple stereogenic centers were ob-tained with high stereoselectivities. The acid-catalyzedaza-ene-type reaction of initial aldimines 2a with mono-substituted enecarbamates 109a afforded aza-ene-typeproducts of N-acyl aldimines 111 as reactive intermedi-ates and 111 underwent further aza-ene-type reaction togenerate aldimines 112. The intramolecular cyclization ofintermediate 112 was conducted to terminate the tandemaza-ene-type reaction sequence. It is noteworthy that onestereoisomer was formed exclusively from among theeight possible stereoisomers, consisting of four pairs ofenantiomers, under the influence of phosphoric acid cata-lyst 1c. Not only aromatic but also aliphatic aldimines 2acould be used in this cascade reaction. Moreover, the gly-oxylate-derived aldimine could be transformed into thehighly functionalized piperidine derivative with excellentenantioselectivity. This cascade methodology allows rap-id access to piperidine derivatives 110 with multiple ste-
Ar H
NBz
+(R)-1g (0.05–0.5 mol%)
toluene, r.t., 5 h
HN
Ph
CO2Me Ar
NHBz
Ph
NCO2Me
NHBz
Ph
O
Ar
H3O+
G =
Ar
NHBz
Ph
HN
X
X = Me, Br
Ar = X = 4-MeO, 4-Me, 4-F, 4-Cl, 4-Br, 4-NC, 3-Me, 3-Br
X
CO2Me
Ar =
Red-Al
0.1 mol% of (R)-1g: 82–97%, 92–98% ee
0.5 mol% of (R)-1g: 82–84%, 93–96% ee
2d
105
α-naphthyl, β-naphthyl, (E)-PhCH=CH-
108 (Ar = Ph)87% anti
107
106
Scheme 40 One-pot access to piperidine derivatives 110 via tandemaza-ene-type reaction/cyclization cascade
H
HNCbz
H
N
R
Boc
+
NH NH
R
BocCbz
*
N
H
Cbz
*
1st aza-ene
2nd aza-ene
cyclization
(2.1 equiv)
N
R
HN
NH
Cbz
Boc
N
R
HN
NH
Cbz
Boc
+
trans-110(2,4-trans-4,6-trans)
cis-110(2,4-cis-4,6-cis)
(R)-1c (2–5 mol%)
CH2Cl2, 0 °C, 1–5 h
46
2
Cbz
Cbz
112
NH N
R
Boc Cbz
H*111
G =
76 to >99%88–95% trans 98 to >99% ee
70%95% trans
97% ee
84%88% trans
98% ee
68%94% trans
97% ee
2a
109a
(E)-PhCH=CH- MeCO2- cyclohexylR = aromatic

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1945
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
reogenic centers, as key structural elements of naturalproducts.
6.3 Two-Carbon Homologation Reaction
The homologation of a carbon unit is an important andfundamental methodology in the construction of carbonframeworks in synthetic organic chemistry. Much atten-tion has been devoted to the development of the two-carbon homologation reaction using acetaldehyde anionequivalents, as these can be directly utilized in furthertransformations.92 From a synthetic viewpoint, monosub-stituted enecarbamates are attractive as acetaldehyde an-ion equivalents for the two-carbon homologation reactionbecause they are readily available and can provide ald-imine products.87f,93 These aldimines can be directly trans-formed into 1,3-diamine derivatives when the two-carbonhomologation reactions are conducted with imines as thesubstrate.
Although the high reactivity of aldimine products 111 hin-dered the completion of the two-carbon homologation re-action because of overreaction, Terada et al. neatlydemonstrated two-carbon homologation using enecar-bamates 109 and hemiaminal ethers 113, instead of imines2a, as the substrate (Scheme 41).94 In the acid-catalyzedreaction of hemiaminal methyl ethers 113, intermediaryand reactive aldimine 111 was trapped by methanol thatwas generated during the formation of imine 2a. A seriesof aromatic hemiaminal ethers 113a were employable forthis homologation reaction in the presence of phosphoricacid catalyst 1p, giving hemiaminal products 114a withhigh enantioselectivities. The enantioselectivities weredetermined only after the reduction of the hemiaminalmoiety to 1,3-diamine derivatives 115, although the reac-tivity of hemiaminal ethers 113a was considerably depen-dent on the electronic properties of the substituents on thearomatic ring. Aliphatic hemiaminal ethers 113b werealso applicable to the present homologation, but in thiscase, protection of the nitrogen atom of 119 by the moreelectron-withdrawing trichloroethoxycarbonyl (Troc)group was required to suppress the formation of byprod-ucts.
This homologation reaction can be applied to substitutedenecarbamate 116a (Scheme 42a). Either anti- or syn-products 118 were obtained in a highly diastereoselectivemanner from the respective geometric isomers of enecar-bamates 116a, and each of the major diastereomers 118,furnished by the reduction of the hemiaminal moiety of117, exhibited good to high enantioselectivities. The syn-thetic utility of this homologation is highlighted by thesequential transformation of the homologation/Friedel–Crafts reaction in one pot (Scheme 42b). Catalyst 1q alsoaccelerated the Friedel–Crafts reaction of hemiaminalether 114ba with indole (56aa) to afford the desired 1,3-diamine derivatives 119 in good yields and nearly optical-ly pure forms, albeit with only moderate syn-diastereose-lectivities. The method enables facile access to highlyenantioenriched 1,3-diamine derivatives as pharmaceuti-cally and biologically intriguing molecules.
Shortly thereafter, Masson, Zhu, and co-workers devel-oped a multicomponent approach to the two-carbon ho-mologation reaction among aldehydes 11, anilines 12, andenecarbamates 109 in the presence of excess ethanol asthe trapping reagent to intercept initial product 120(Scheme 43).95 The use of aniline derivative 12e, with itselectron-withdrawing nitro group, resulted exclusively in
Scheme 41 Two-carbon homologation of hemiaminal ethers byenecarbamate
1p: G =
Ph
Ph
R1 = aromatic
109a: CO2R2 = Cbz
R1 = aliphatic
109b: CO2R2 = Troc1q: G = i-Pr
i-Pr
i-Pr
115a44–99%, 84–95% ee
115b56–84%, 86–98% ee
HN
OMeR1
Boc
N
HR1
Boc
2a
R1
NH N
H
Boc CO2R2
R1
NH HN
OMe
Boc CO2R2
MeOH
111
(R)-1p or 1q(2 mol%)HN
H
CO2R2
R1
NH HNBoc CO2R3
+
a
115a: R1 = aromatic, R3 = Bn
i-Pr2O ortoluene
r.t. to 50 °C109
109
115b: R1 = aliphatic, R3 = Me115
(a) K-Selectride
(a) 1) KOH, MeOH 2) K-Selectride
113a: R1 = aromatic113b: R1 = aliphatic
114a: R1 = aromatic114b: R1 = aliphatic
Scheme 42 Application of the two-carbon homologation reaction
HN
H
CO2Me
+
Ph
NH HNBoc CO2Me
syn-118
HN
OMePh
Boc(R)-1p (2 mol%)
K-Selectride
CH2Cl2, r.t., 24 h
(E)-116a(Z)-116a
99% yield67% yield
(94% ee)(51% ee)
(3% ee)(88% ee)
295
985
::
Ph
NH HNBoc CO2Me
anti-118
Ph
NH HNBoc CO2Me
OMe
+
Et
NH HN
OMe
Boc Troc
Et
NH HNBoc Troc
(R)-1q (2 mol%)
toluene, r.t., 12 h
NH
indole56aa
40 °C, 24 h
1,3-syn-11970%, 76% syn, >99% ee
HN
H
Troc
+HN
OMeEt
Boc
a)
b)
113aa
113ba 109b
116a117
114ba

1946 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
four-component adducts 121 at the expense of thePovarov products, tetrahydroquinolines 122, which wereformed from the inverse-electron-demand aza-Diels–Alder reaction of N-aryl imines with enecarbamate 109(see Section 7.3).96 In this regard, tetrahydroquinolines122 were the only products obtained in the absence of eth-anol under otherwise identical conditions. Aromatic aswell as aliphatic aldehydes 11 can be used in the presentfour-component reaction, affording hemiaminal ethylethers 121 in good yields with high enantioselectivities.The potential of this multicomponent reaction is furtherdemonstrated using substituted enecarbamate 116b. Thereaction of 4-nitroaniline (12e) and (E)-116b with a rangeof aromatic and aliphatic aldehydes 11 gave anti-products123 with high diastereoselectivities (>95% anti) after thereduction of the hemiaminal moiety of 121.
Scheme 43 Two-carbon homologation via multicomponent ap-proach
6.4 Aza-Ene-type Reaction of Aldehyde with Enecarbamates
Terada et al. accomplished the enantioselective activationof aldehydes using a chiral phosphoric acid catalyst.97
Their aza-ene-type reaction of glyoxylate 124 as a reac-tive aldehyde with enecarbamate 105 afforded the corre-sponding products 125 with excellent enantioselectivities(Scheme 44).87d–g Catalyst 1b efficiently accelerated theaza-ene-type reaction of glyoxylate 124 in the presence ofMS 4A, which was employed as a scavenger of acidic im-purities.98 After hydrolysis of products 125 to b-hydroxyketones 126, excellent enantioselectivities were observedeven when catalyst 1b, bearing unmodified phenyl groups
(G = Ph), was used. The fact that the simple phenyl-substituted catalyst provides excellent enantioselectivityis noteworthy, since in experiments on the activation ofimines, it was found that catalysts 1 required modifiedphenyl substituents, bulky ones in general, to achieve highenantioselectivities. To rationalize the stereochemicaloutcome, Terada and co-workers97 proposed double hy-drogen-bonding interaction D as the complexation mode99
of glyoxylate 124 and phosphoric acids 1 on the basis of ascreening of catalysts 1 having a series of substituted phe-nyl rings instead of simple phenyl groups, and DFT com-putational analysis of the hydrogen-bonding interactionbetween glyoxylate 124 and phosphoric acids 1(Scheme 44). The additional hydrogen bond that existsbetween the formyl hydrogen atom and the phosphoryloxygen atom creates a coplanar orientation between theformyl group and the phosphoric acid subunit,100 andhence the formyl carbon of glyoxylate 124 would be ex-posed to an efficient chiral environment created by thephosphoric acid catalyst.
Scheme 44 Aza-ene-type reaction of glyoxylate 124 with ene-carbamate 105 catalyzed by (R)-1b
The applicability of the aza-ene-type reaction of glyoxy-late 124 with a series of substituted enecarbamates 127demonstrates the stereochemical issue of enantio- anddiastereoselection (Scheme 45). Catalysts 1t exhibited ex-cellent performance in the reaction of (E)-127 to giveanti-products 128 in nearly optically pure forms. How-ever, (Z)-127 retarded the reaction markedly and lowenantioselectivity was observed for major anti-128. Theexclusive formation of anti-products from the E-isomerscould be attributed to the well-defined exo-transitionstate.87d
6.5 Carbonyl-Ene Reaction
The carbonyl-ene reaction is an important carbon–carbonbond-forming reaction that affords synthetically valuablehomoallylic alcohols.101 The enantioselective catalysis ofthe carbonyl-ene reaction has been extensively investigat-
HN
H
Cbz
+O
HR1
(R)-14e (10 mol%)
R2
NaBH3CNp-TsOH
EtOH (17 equiv)CH2Cl2, 0 °C, 24 h
R1
NH HNAr Cbz
R2
11109a: R2 = H116b: R2 = Me
H2N
NO2
12e
R1
NH NAr Cbz
R2
120
R1
NH HN
OEt
Ar Cbz
R2
H
123
121
+
X
NH
R1
HN
R2
Cbz
122 (X = NO2)
O
O
PO
OH
G
G
Cl(R)-14e: G =
EtOH
R1 = aromaticR1 = aliphatic
R2 = HR2 = H
R1 = aromaticR1 = aliphatic
R2 = MeR2 = Me
Ar = NO2
74–86%, 79–96% ee76–97%, 88–90% ee
82 to >99% ee90–97% ee
55–80%, 62–95%,
>95% anti, >95% anti,
OH
EtO2C
NCO2Me
Ph
124
+(R)-1b (5 mol%)
4 Å MS CH2Cl2, r.t., 1 h
O
HEtO2C
12693%, 95% ee
H3O+
Ph
HNCO2Me
OH
EtO2C
O
Ph
G =
O
O
P
G
G
O
OHO
HMeO
O
D
105
125

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1947
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
ed using chiral metal complexes.102 However, no highlyenantioselective organocatalysis of the carbonyl-ene reac-tion was reported103 until the work of Rueping et al.,104
who used chiral phosphoramide 6669 as the strongly acidiccatalyst (Scheme 46). The reaction of trifluoropyruvate129 with a-methyl styrene derivatives 130 in the presenceof chiral phosphoramide 66e gave the corresponding ho-moallylic alcohols 131, having the quaternary stereogeniccenter substituted by a trifluoromethyl group, with highenantioselectivities.
Scheme 46 Carbonyl-ene reaction of trifluoropyruvate 129 with a-methyl styrene derivatives 130
7 Cycloaddition Reactions
7.1 Hetero-Diels–Alder Reaction of Aldimines
The enantioselective hetero-Diels–Alder reaction of sil-oxydienes, such as Danishefsky’s and Brassard’s dienes,with imines provides an efficient route for the preparationof functionalized nitrogen heterocycles in optically activeforms, which can be utilized as synthetic precursors of bi-ologically interesting alkaloid families and aza sugars.Akiyama et al. developed the enantioselective hetero-Diels–Alder reaction of Danishefsky’s diene (132) witharomatic imines 2m using chiral phosphoric acid catalyst1q (Scheme 47a).105 The reaction catalyzed by 1q provid-ed dihydropyridin-4(1H)-one derivatives 133 in goodyields with high enantioselectivities in the presence ofacetic acid as the stoichiometric additive. Akiyama andco-workers also reported an enantioselective hetero-Diels–Alder reaction of Brassard’s diene (134) with imines
2m (Scheme 47b).106 One of the intrinsic problems asso-ciated with Brassard’s diene lies in its instability underacidic conditions. In order to suppress the decompositionof the diene by the phosphoric acid catalysts, they em-ployed pyridinium salts of the catalysts. The pyridiniumsalt of 1g displayed excellent performance for the hetero-Diels–Alder reaction of Brassard’s diene with a broadrange of imines, including aromatic and aliphatic deriva-tives. Uniformly high enantioselectivities and chemicalyields of dihydropyridin-2(1H)-one products 135 werenoted after treatment with an acid.
Scheme 47 Enantioenriched nitrogen heterocycles by hetero-Diels–Alder reaction of siloxydienes
7.2 Direct Cycloaddition Reaction of Aldimines with Cyclohexenone
Gong and co-workers107 and Rueping and Azap108 inde-pendently reported the enantioselective direct cycloaddi-tion reaction of cyclohexenone 136 with aromatic imines2n. Rueping and Azap employed acetic acid as the co-cat-alyst to accelerate the tautomerization of cyclohexenone136, by which a reactive dienol for the enantioselectivecycloaddition was generated in situ (Scheme 48). Theyproposed a stepwise mechanism for the reaction. The ini-tial step is the chiral phosphoric acid 1u catalyzed Mannichreaction of imines 2n with the dienol generated in thepresence of acetic acid, giving intermediate 137. The sub-sequent intramolecular aza-Michael reaction provides thecorresponding cycloaddition products 138, the so-calledisoquinuclidine derivatives, in moderate to good yields.
Scheme 45 Diastereo- and enantioselective aza-ene-type reactionof glyoxylate 124 with enecarbamate 127
124
+(R)-1t (5 mol%)
4 Å MSCH2Cl2, r.t., 2–24 h
O
HEtO2C
syn-128
H3O+
R
HNCO2Me
OH
EtO2C
O
R
G =
127 (R = Ph, Et)
anti-128
t-Bu
OH
EtO2C
O
R+
(E)-127
(Z)-127
73%,
11–74%,
96 to >99% anti,
50–72% anti,
99 to >99% ee for anti-12826–28% ee for anti-12869–88% ee for syn-128
O
O
PO
NHTf
G
G
OMe(R)-66e: G =
129
+
(R)-66e(1 mol%)
o-xylene10 °C, 34 h
O
CF3EtO2C Ar
130EtO2C Ar
HO CF3
13155–96%, 92–97% ee
N
HAr
HO
Me
OMe
OTMS
Me
OH
N
OAr
(R)-1q (5 mol%)
AcOH (1.2 equiv)toluene, –78 °C, 10–35 h 133
72–100%, 76–91% ee
+
a)
R = aromatic R = aliphatic
OTMS
OMe
+
b)
MeO
(R)-1g/pyridine (3 mol%) PhCOOH Ar
N
OMeR
O
HO
MeAr =
mesitylene–40 °C, 24 h
G = i-Pr
i-Pr
i-Pr2m
132
2m
134
135
63–91%, 94–99% ee65–69%, 93–99% ee
O
O
PO
O
G
G
(R)-1g/pyridine
G =HN
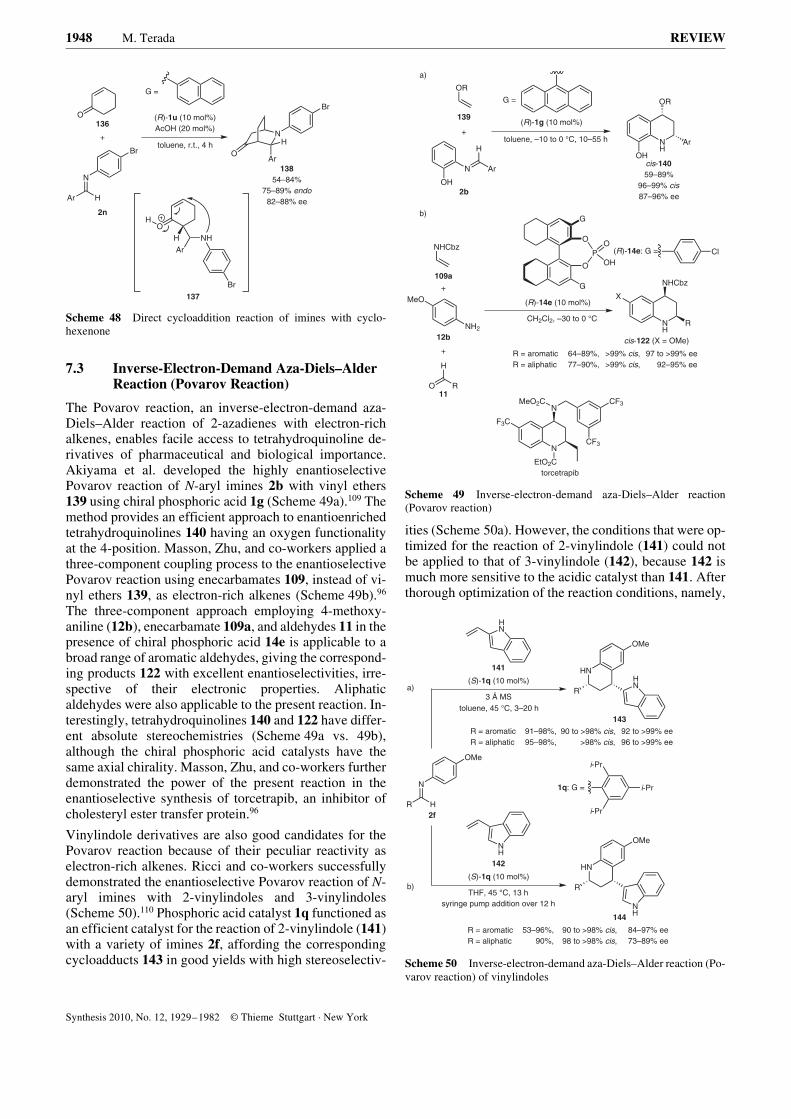
1948 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Scheme 48 Direct cycloaddition reaction of imines with cyclo-hexenone
7.3 Inverse-Electron-Demand Aza-Diels–Alder Reaction (Povarov Reaction)
The Povarov reaction, an inverse-electron-demand aza-Diels–Alder reaction of 2-azadienes with electron-richalkenes, enables facile access to tetrahydroquinoline de-rivatives of pharmaceutical and biological importance.Akiyama et al. developed the highly enantioselectivePovarov reaction of N-aryl imines 2b with vinyl ethers139 using chiral phosphoric acid 1g (Scheme 49a).109 Themethod provides an efficient approach to enantioenrichedtetrahydroquinolines 140 having an oxygen functionalityat the 4-position. Masson, Zhu, and co-workers applied athree-component coupling process to the enantioselectivePovarov reaction using enecarbamates 109, instead of vi-nyl ethers 139, as electron-rich alkenes (Scheme 49b).96
The three-component approach employing 4-methoxy-aniline (12b), enecarbamate 109a, and aldehydes 11 in thepresence of chiral phosphoric acid 14e is applicable to abroad range of aromatic aldehydes, giving the correspond-ing products 122 with excellent enantioselectivities, irre-spective of their electronic properties. Aliphaticaldehydes were also applicable to the present reaction. In-terestingly, tetrahydroquinolines 140 and 122 have differ-ent absolute stereochemistries (Scheme 49a vs. 49b),although the chiral phosphoric acid catalysts have thesame axial chirality. Masson, Zhu, and co-workers furtherdemonstrated the power of the present reaction in theenantioselective synthesis of torcetrapib, an inhibitor ofcholesteryl ester transfer protein.96
Vinylindole derivatives are also good candidates for thePovarov reaction because of their peculiar reactivity aselectron-rich alkenes. Ricci and co-workers successfullydemonstrated the enantioselective Povarov reaction of N-aryl imines with 2-vinylindoles and 3-vinylindoles(Scheme 50).110 Phosphoric acid catalyst 1q functioned asan efficient catalyst for the reaction of 2-vinylindole (141)with a variety of imines 2f, affording the correspondingcycloadducts 143 in good yields with high stereoselectiv-
ities (Scheme 50a). However, the conditions that were op-timized for the reaction of 2-vinylindole (141) could notbe applied to that of 3-vinylindole (142), because 142 ismuch more sensitive to the acidic catalyst than 141. Afterthorough optimization of the reaction conditions, namely,
N
Ar H
Br
O
+ N
OAr
H
Br
13854–84%
75–89% endo82–88% ee
(R)-1u (10 mol%)AcOH (20 mol%)
toluene, r.t., 4 h
G =
OH
Ar
NH
Br
H
137
2n
136
Scheme 49 Inverse-electron-demand aza-Diels–Alder reaction(Povarov reaction)
+toluene, –10 to 0 °C, 10–55 h
(R)-1g (10 mol%)
G =
OH
N Ar
HOH
NH
OR
Ar
cis-14059–89%
96–99% cis87–96% ee
MeO
NH2
O
O
PO
OH
G
G
Cl(R)-14e: G =
a)
b)
O R
H
OR
CH2Cl2, –30 to 0 °C NH
NHCbz
R
X
NHCbz
+
(R)-14e (10 mol%)
R = aromaticR = aliphatic
139
2b
109a
12b
+
11
64–89%,77–90%,
cis-122 (X = OMe)
>99% cis,>99% cis,
97 to >99% ee92–95% ee
N
N
F3C
MeO2C
EtO2C
CF3
CF3
torcetrapib
Scheme 50 Inverse-electron-demand aza-Diels–Alder reaction (Po-varov reaction) of vinylindoles
HN
141
R H
N
OMe
2f
NH
142
1q: G = i-Pr
3 Å MStoluene, 45 °C, 3–20 h
THF, 45 °C, 13 hsyringe pump addition over 12 h
HN
OMe
R
HN
143
HN
OMe
R
NH144
(S)-1q (10 mol%)
(S)-1q (10 mol%)
i-Pr
i-Pr
91–98%,95–98%,
53–96%,90%,
90 to >98% cis,>98% cis,
90 to >98% cis,98 to >98% cis,
92 to >99% ee96 to >99% ee
84–97% ee73–89% ee
R = aromaticR = aliphatic
R = aromaticR = aliphatic
a)
b)

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1949
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
use of a coordinating solvent such as tetrahydrofuran andthe slow addition of 142 over 12 hours, cycloadducts 144were obtained (Scheme 50b) with results that were com-parable to those of 143 using the same catalyst 1q. A va-riety of aryl substituents introduced at the nitrogen atomcan be utilized in this reaction, giving the cycloadducts inhigh yields with excellent stereoselectivities.
7.4 Diels–Alder Reaction of Enone
The enantioselective Diels–Alder reaction of a,b-unsatur-ated carbonyl compounds with dienes is one of the mostwell-studied transformations using chiral metalcatalysts111 and organocatalysts.112 However, chiral phos-phoric acids do not effectively accelerate the Diels–Alderreaction of a,b-unsaturated carbonyl compounds becausethey lack sufficient acidity to activate these dienophiles.Yamamoto and Nakashima overcame this intrinsic draw-back by designing a novel chiral Brønsted acid catalystwith improved acidity (Scheme 51).69 They successfullydeveloped stronger chiral Brønsted acids, N-triflyl phos-phoramides 66, by introducing a strong electron acceptor,the TfN-H group,113 into the phosphoric acid subunit inplace of the phosphoric acid O-H group. N-Triflyl phos-phoramide catalyst 66c exhibited significant activity inthe reaction of a,b-unsaturated ketone 145 with siloxy-dienes 146a, affording enantioenriched cycloadducts 147in excellent yields in most cases, although the yields werequite sensitive to the stability of the siloxydiene 146a, be-cause the protonation of the diene resulted in silylationand thus deactivation of the catalyst. In contrast, parentphosphoric acid 1b (G = Ph) showed no catalytic activityand thus, the introduction of the electron-withdrawingNTf group into the phosphoric acid is quite effective in in-creasing the catalytic activity. The reaction of non-substi-tuted and methyl-substituted siloxydienes at the R groupfurnished olefin regioisomers 148. However, olefin mi-
gration was not observed when sterically demanding sub-stituents, such as benzyl moieties, were employed as theR group.
7.5 Hetero-Diels–Alder Reaction of Aldehydes
The hetero-Diels–Alder reactions of dienes with alde-hydes are an efficient way to provide dihydropyran deriv-atives.114 The development of catalytic enantio- anddiastereoselective variants is an area of considerable im-portance.115 Extensive studies of the use of chiral Lewisacid catalysts to control high levels of stereoselectivityhave been conducted.116 It is noteworthy that vicinal sub-stituents of the dihydropyran are controlled exclusively ina syn-selective manner, where the diene approaches thealdehyde with an endo-orientation, presumably becauseof secondary p-orbital interactions and more importantlyto avoid steric repulsion between the incoming diene andthe sterically demanding Lewis acid catalyst.117 In con-trast, alternative exo-oriented enantioselective processesthat afford anti-isomers have yet to be fully established.118
Terada and co-workers developed an unprecedented anti-and enantioselective hetero-Diels–Alder reaction of sil-oxy- or methoxydienes with glyoxylate 124 using chiralphosphoric acid catalyst 1 (Schemes 52 and 53).119 Cata-lyst 1b, bearing unmodified phenyl groups (G = Ph), ex-hibited excellent performance in the hetero-Diels–Alder
Scheme 51 Diels–Alder reaction of enone 145 with siloxydienes146 catalyzed by N-triflyl phosphoramide 66c
(S)-66c: G = i-Pr
i-Pr
i-Pr
Et
O
145
(S)-66c (5 mol%)
toluene, –78 °C, 3–12 h
147
O
O
PNHTf
O
G
G
OTIPS
Me
R
Et
O
OTIPS
Me
R
Et
O
R
OTIPS
146a (1.5 equiv)
+
148 (R = H or Me)
+
R = Me, Bn, BzOCH2CH2, 4-TBSOC6H4CH2: 95 to >99%, >98% endo, 85–92% eeR = H: 43%, >98% endo, 88% ee
Scheme 52 Hetero-Diels–Alder reaction of siloxydienes 146 withglyoxylate 124 catalyzed by (R)-1b
OTBS
+H
O
EtO
O
(R)-1b (5 mol%)
OTBS
O
EtO2C
4 Å MStoluene, r.t., 24 h
anti-149a
OTBS
O
EtO2C
syn-149a
+
95%, >99% anti, 99% ee for anti
146b(1.5 equiv)
(Z,E)/(E,E) = 90:10
124
G =
POO
O
O
O
O
OH
H
OSi
E
anti-149a
OTBS
Ph
O
EtO2COTBS
O
EtO2C
149b92%
>99% anti 98% ee
149e 90%
>99% anti98% ee
OTBS
R
O
EtO2C
149d (R = CH=CHMe)75%
>95% anti99% ee
149c (R = Ph)92%
>99% anti97% ee

1950 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
reaction of glyoxylate 124 with tert-butyldimethylsilyl-protected siloxydienes 146b, giving the correspondingproduct 149a in high yield with extremely high stereose-lectivity (Scheme 52). It is noteworthy that chiral phos-phoric acid 1b is uniquely efficient in affording anti-cycloadduct 149a as the single diastereomer in nearly op-tically pure form. In order to rationalize this unprecedent-ed anti-selectivity observed in the 1b-catalyzed reaction,they proposed exo-transition state E,120 where the smallphenyl groups at the 3,3¢-positions of 1b allow the dieneto occupy an exo-position, giving the anti-isomer exclu-sively.
The anti- and enantioselective hetero-Diels–Alder reac-tion catalyzed by 1b is applicable to a series of 2-siloxy-dienes, giving the corresponding products 149 in goodyields with excellent stereoselectivities (Scheme 52). Al-though the alkenyl-substituted siloxydiene resulted in adecrease in reactivity, the stereoselectivity was main-tained at a high level. Furthermore, methoxydienes 150were also well tolerated, providing the correspondinganti-dihydropyrans 151 predominantly with excellentenantioselectivities (Scheme 53).
7.6 1,3-Dipolar Cycloaddition Reactions
The 1,3-dipolar cycloaddition reaction of nitrones with vi-nyl ethers provides an efficient route to 1,3-amino alco-hols,121 which can be transformed from the correspondingisoxazolidine products through reductive cleavage of thenitogen–oxygen bond. The development of the catalyticenantioselective 1,3-dipolar cycloaddition reaction ishence a vital step toward the efficient synthesis of theseamino alcohols in optically active forms. Yamamoto andco-workers successfully developed the enantioselective1,3-dipolar cycloaddition reaction of diaryl nitrones 152with ethyl vinyl ether (139a) (Scheme 54).122 They ap-plied acidic NHTf-substituted chiral phosphoramide cata-lysts 66, originally developed by the same researchgroup,69 to the present dipolar cycloaddition reaction.Chiral phosphoramide 66f effectively catalyzed the cy-cloaddition reaction to give the corresponding isoxazoli-dine derivatives 153 in high yields. The introduction of an
electron-withdrawing group to the aromatic ring, Ar2, isnecessary to ensure the high enantioselectivity. In addi-tion, endo-153 was furnished as the major diastereomer inthe present reaction. The predominance of endo-productscontrasted the formation of exo-isomers as major productsunder Lewis acid catalysis.123
Scheme 54 1,3-Dipolar cycloaddition reaction of nitrones 152 withethyl vinyl ether (139a)
The asymmetric 1,3-dipolar cycloaddition reaction ofazomethine ylides with electron-deficient olefins provideschiral pyrrolidine derivatives, an important class of nitro-gen heterocycles,121c,124 as the precursors of biologicallyactive compounds. Gong and co-workers reported theenantioselective catalysis by chiral Brønsted acids in thethree-component 1,3-dipolar cycloaddition reactionamong aldehydes 11, a-amino-1,3-dicarbonyl compounds154, and maleate 155 as an electron-deficient dipolaro-phile (Scheme 55).125 As none of the chiral monophos-phoric acids 1 provided sufficient enantioselectivities,they developed the new bisphosphoric acid 156, derivedfrom linked BINOL, that displayed excellent performancein this cycloaddition reaction in terms of diastereo- andenantioselectivities as well as catalytic efficiency. Abroad range of aldehydes 11, including aromatic and a,b-unsaturated aldehydes, were used in the reaction, giving
Scheme 53 Hetero-Diels–Alder reaction of methoxydienes 150with glyoxylate 124 catalyzed by (R)-1b
OMe
O
EtO2C
OMe
O
EtO2C
R
OMe
O
EtO2C
151a84%
93% anti98% ee
151b (R = H)93%
91% anti99% ee
151c (R = Me)51%
94% anti95% ee
151d75%
95% anti97% ee
R2
R3
OMe(R)-1b
(5 mol%)
R2
R3
OMe
O
EtO2C4 Å MStoluene, r.t., 48 h
anti-151
R1 R1
+124
150
(S)-66f: G =
O
O
PNHTf
O
G
G
i-Pr
i-Pr
Ad
N
H Ar2
OAr1OEt
(S)-66f(Ad = 1-adamantyl)
(5 mol%)
CHCl3–55 to –40 °C, 1–6 h
+
endo-15366 to >99%
87–97% endo70–93% ee
N OAr1
Ar2
OEt
139a152
Scheme 55 1,3-Dipolar cycloaddition reaction via three-componentcoupling
O
HR
CO2Et
CO2EtH2N
CO2MeMeO2C
NH
CO2Et
CO2EtMeO2C
CO2Me
R
O
O PO
HO
O
O
P
OHO
O
(R,R)-156
+ +
(R,R)-156 (10 mol%)
3 Å MSCH2Cl2, r.t., 24–96 h
15767–97%, >99% endo, 84–99% ee
11 154 155

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1951
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
the corresponding pyrrolidine derivatives 157 with excel-lent endo- and enantioselectivities.
Gong and co-workers further developed an enantioselec-tive three-component 1,3-dipolar cycloaddition reactionamong aldehydes 11, a-amino-1,3-dicarbonyl compound154, and a series of electron-deficient dipolarophiles,namely, N-aryl imines 158, 2,3-allenoates 159, and meth-yleneindolinones 160 (Scheme 56). The reaction of N-aryl imines 158 as the dipolarophiles, generated in situfrom aldehydes 11 and 4-tert-butoxyaniline (12f), afford-ed diastereomeric mixtures of cycloadducts 161 with ex-cellent enantioselectivities under the influence ofphosphoric acid 1q (Scheme 56a).126 The diastereoselec-tivities were markedly dependent on the substitution pat-tern of the aldehyde 11. The syn-isomers 161 wereobtained predominantly in the reaction of para- and meta-substituted benzaldehydes, regardless of the electronicproperties of these substituents. In contrast, ortho-substi-tution resulted in a significant decrease in diastereoselec-tivities. Meanwhile, in the reaction of 2,3-allenoates 159as the dipolarophile, bisphosphoric acid catalyst 156 wasthe most effective in terms of enantioselectivity to givecycloadducts 162 as a single diastereomer having an exo-
methylene moiety (Scheme 56b).127 Although aliphaticaldehydes resulted in decreased chemical yields and enan-tioselectivities, excellent enantioselectivities wereachieved using aromatic aldehydes as the 1,3-dipolaro-phile precursors in most cases. The activation of azometh-ine ylides by chiral phosphoric acid catalysts is alsoapplicable to the cycloaddition reaction of methylenein-dolinones 160 (Scheme 56c).128 A broad range of alde-hydes 11 and 160 were well tolerated for the cyclizationreaction in the presence of chiral phosphoric acid 1u, giv-ing spirocyclic adducts 163 in good yields with high enan-tioselectivities. Thus, both aromatic and aliphaticaldehydes 11, as well as 160 with either aromatic or ali-phatic R2 substituents, were adaptive substrates, despitethe formation of regioisomeric byproducts when aliphaticR2 substituents were introduced into 160.
8 Cyclization Reactions
8.1 Electrocyclic Reactions
The Nazarov reaction is classified as an electrocyclic re-action and is one of the most versatile methods for theconstruction of five-membered cyclic enones, which arethe key structural motif of numerous natural products.129
In general, the Nazarov cyclization is accelerated byLewis acid or Brønsted acid catalysts. However, the enan-tioselective Nazarov cyclization has rarely been exploit-ed, even by using chiral metal complexes.130 Rueping etal. developed for the first time an organocatalytic ap-proach to the enantioselective Nazarov cyclization ofdienones 164 using chiral phosphoramide 66 as theBrønsted acid catalyst (Scheme 57).131 The reactivitiesand the enantioselectivities were strongly dependent onthe solvent employed. While no reaction was observed inpolar solvents, such as tetrahydrofuran and acetonitrile,the reaction proceeded smoothly in halogenated and aro-matic solvents to afford diastereomeric mixtures of cy-clized products 165. The best enantioselectivities wereachieved using chiral phosphoramide 66d in chloroform,and a variety of dienones 164 were transformed into cy-clopentenones 165 with moderate to high diastereoselec-tivities. Major product syn-165 can be isomerizedexclusively to anti-165 under basic conditions withoutany loss of enantiomeric purity.
Scheme 56 1,3-Dipolar cycloaddition reaction of 1,3-dipolarophi-les via multicomponent coupling
NHR1
N
R2
O
Ac
O
HR1
CO2Et
CO2EtH2N
N
NH
CO2Et
CO2EtAr
R1
R1
+
(R)-1q (10 mol%)
3 Å MStoluene, –10 °C, 60 h
76–91% syn,
45–88% syn,
11
154
NH
CO2Et
CO2Et
R1
H2N
Ot-Bu
12f
11
syn-161R1 = para- or meta-substituents 73–91%,R1 = ortho-substituents 76–99%,
93–98% ee
85–90% ee
+
N
H
R1Ar
158
R3
R2O2C
•
CO2R2
R3
159
160
162
Ot-Bu
Ar =
(R,R)-156 (10 mol%)
3 Å MStoluene, 25 °C, 36–144 h
N
O
Ac
R2
163
R2 = 9-anthracenylmethyl
R1 = aromaticR1 = aliphatic
75–96% ee17–69% ee
(R)-1u (10 mol%)
3 Å MSCH2Cl2, 25 °C, 24–96 h
67–99%,29–69%,
a)
b)
c)
R3
R3
1q: G = i-Pr
i-Pr
i-Pr
1u: G = R1 = aromatic R1 = aliphatic
81–98% ee82–93% ee
74–97%,59–95%,
in situ
CO2EtCO2Et
Scheme 57 Nazarov cyclization of dienones 164
O
O
PO
NHTf
G
G
O
O
R
Ar164
(R)-66d: G =
(R)-66d (2 mol%)
CHCl3, 0 °C, 1–2 h
O
Ar
R
OO
Ar
R
O
syn-16587–93% ee
anti-16591–98% ee
61–92%, 60–90% syn
+

1952 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Rueping and Ieawsuwan further applied the enantioselec-tive Nazarov cyclization to the preparation of a-substitut-ed cyclopentenones 167, lacking a b-substituent, usingdienones 166 (Scheme 58).132 In this transformation, thestereo-determining step is the Brønsted acid catalyzedenantioselective protonation of intermediately generatedenol 168.130c H8-BINOL-derived phosphoramide 66g ex-hibited the best performance in this case, giving cyclopen-tenones 167 with fairly good enantioselectivities.
Scheme 58 Nazarov cyclization of dienones 166
In contrast to the Nazarov reaction, which is a 4p-electro-cyclization, List and Müller reported the enantioselectivecatalysis of 6p-electrocyclization using benzylidene-acetone-derived phenylhydrazone 169 as a 6p-electronsystem for the first time (Scheme 59).133,134 Chiral phos-phoric acid 1g exhibited promising catalytic activity andenantioselectivity in the 6p-electrocyclization of 169, giv-ing the cyclized products, pyrazolines 170, which are in-teresting synthetic targets in terms of broad potentbiological activities, in excellent yields with high enantio-selectivities. The electrocyclic reaction is well suited forhydrazones bearing either an electron-withdrawing orelectron-donating substituent at the aromatic ring of theenone moiety. To undergo the cyclization, carbon–nitro-gen double-bond isomerization of hydrazone (E)-169 isrequired and this E/Z isomerization is also catalyzed byphosphoric acid 1g. After adopting the cyclization-reac-tive Z-s-cis-conformation, protonated intermediate F un-dergoes 6p-electrocyclization in the chiral environmentcreated by a chiral counteranion of the phosphoric acid,affording optically active 3-pyrazolines 171. Subsequentisomerization of the double bond yields thermodynami-cally more stable 2-pyrazolines 170. They also developeda direct method for the enantioselective 6p-electrocycliza-tion from enones and hydrazines without the isolation ofhydrazones 169. In this direct method, the prior formationof hydrazones 169 using MS 4A was necessary in the ab-sence of catalyst 1g, because the overall reaction proceed-ed sluggishly when enones and phenylhydrazine reactedin the presence of the catalyst.
Scheme 59 6p-Electrocyclic reaction of phenylhydrazones 169 lea-ding to pyrazolines 170
8.2 Multicomponent Cyclization Reactions
Multicomponent reactions, in which three or more reac-tants are combined in a single process to deliver new mol-ecules containing substantial elements of all reactants,have received much attention with respect to synthetic ef-ficiency and an increase in molecular diversity from thereactants.135 Gong and co-workers developed an efficientthree-component cyclization that involved a,b-unsaturat-ed aldehydes 172, aniline derivatives 12, and b-keto esters24 in the presence of chiral phosphoric acid catalyst 14c,and 1,4-dihydropyridine derivatives 173 were obtained inthe optically active forms (Scheme 60).136 Both the reac-tion efficiency and the enantioselectivity displayed signif-icant dependence upon the substituents of these reactants.Among the aniline derivatives 12 tested, 3-methoxy-aniline (12g) gave the highest enantioselectivities albeitwith accompanying decrease in chemical yields. In mostcases, the reaction of ethyl or isopropyl b-keto esters 24provided cyclized products 173 with higher enantioselec-tivities than the reaction of their methyl or allyl counter-parts. a,b-Unsaturated aldehydes 172 with electron-withdrawing substituents, such as a nitro group or halogenatom, at the aromatic ring exhibited high enantioselectiv-ities compared with the parent cinnamaldehyde and enalsbearing an electron-donating group on the aromatic ring.The application of enantioenriched 1,4-dihydropyridineshas been reported in the synthesis of alkaloids and a vari-ety of biologically active molecules,137 in particular, mol-ecules that show activity against calcium channels,138 andoptically pure 4-substituted 1,4-dihydropyridine deriva-tives have been utilized as chiral models of NAD(P)H.139
The present method enables efficient access to these dihy-dropyridine derivatives in a highly enantioselective man-ner.
The Hantzsch synthesis is one of the most efficient meth-ods for the preparation of 1,4-dihydropyridine deriva-tives. Gestwicki and Evans reported the highly
O
O
PO
NHTf
G
G
O
O
R
166
(R)-66g: G =
(R)-66g (5 mol%)
CHCl3, –10 °C, 20 h
O
R
O
16744–93%, 67–78% ee
O
R
OH
168
169
1g: G =
(S)-1g (10 mol%)
C6H5Cl30 °C, 75–96 h
17085–99%, 76–96% ee
NNH
N N
R1
R2
R2
R1
N
F
Ar1
Ar2
N
O P
O
O
OH
H
(Z)-s-cis conformation
171
N NH
R1
R2

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1953
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
enantioselective Hantzsch four-component coupling reac-tion of dimedone (174), ethyl acetoacetate (24b), aromaticaldehydes 11, and ammonium acetate (175) using chiralphosphoric acid 1v (Scheme 61).140 The use of excessdimedone (174) was required to avoid the formation ofbyproducts, namely, symmetrical 1,4-dihydropyridines,and the desired four-component coupling products 176were afforded in good yields with high enantioselectivi-ties.
Scheme 61 1,4-Dihydropyridine synthesis via Hantzsch four-component coupling reaction
Azlactones 177 possess multiple reactive sites.141 For ex-ample, the acidic nature at the C4 position allows for theformation of an enol tautomer that functions as a nucleo-philic site and reacts with a variety of electrophiles, whilethe electrophilic nature of the carbonyl allows for nucleo-philic attack to furnish a ring-opening product. Hence, therich reactivity of the azlactone scaffold enables a large va-riety of transformations, making azlactones very useful asversatile reactants in the synthesis of biologically interest-ing molecules. Gong and co-workers developed an enan-tioselective cyclization reaction via three-componentcoupling by taking advantage of the multiple reactive sitesof azlactones 177a (Scheme 62).142 Chiral phosphoricacid 1i efficiently catalyzed the three-component cou-pling among azlactones 177a, aniline derivatives 12, anda,b-unsaturated aldehydes 172, affording cyclized prod-ucts 178 as a single diastereomer with high enantioselec-tivity, although aliphatic enals failed to give the desired
products. The use of MS 4A was crucial to obtain desiredproducts 178 in good yields. When the reaction was con-ducted in the absence of molecular sieves, considerableamounts of byproducts 179 were formed via direct nu-cleophilic ring opening of azlactones 177a by 12.
Scheme 62 Cyclization reaction via three-component couplingamong azlactones 177a, aniline derivatives 12, and a,b-unsaturatedaldehydes 172
Gong and co-workers also accomplished the enantioselec-tive synthesis of benzo[a]quinolizidine derivatives 180using aryl ethylamine derivative 181, instead of anilinederivatives 12 (Scheme 63).142 Aryl ethylamine derivative181 participated in the smooth cyclization reaction ofazlactone 177aa with various cinnamaldehydes 172 in thepresence of 1i, affording cyclized products 182. Subse-quent treatment of 182 with boron trifluoride–diethylether complex resulted in the clean formation of ben-zo[a]quinolizidine derivatives 180 via the Pictet–Spengler-type cyclization in good overall yield with ex-cellent diastereo- and enantioselectivities.
Scheme 63 Cyclization reaction via three-component couplingamong azlactones 177aa, aryl ethylamine derivative 181, and a,b-un-saturated aldehydes 172
8.3 Intramolecular Aldol Reaction (Robinson-type Annulation)
The Robinson annulation reaction is one of the most fun-damental methods for providing cyclohexenone structuralmotifs and has been applied to a number of natural productsyntheses. The enantioselective catalysis of the Robinson
Scheme 60 1,4-Dihydropyridine synthesis via three-componentcoupling
(S)-14c (20 mol%)
C6H5CN, 50 °C, 24 h
17340–93%, 81–98% ee
O
O
POH
O
G
G
(S)-14c: G =
OH
Ar1
Ar2NH2
172
12
24
O O
OR
N
Ar1
Ar2
CO2R+
+
MeCN, r.t., 5 h
17669–94%, 87 to >99% ee
O
HAr1124b
O O
OEt
NH
**
Ar
CO2Et
++
(S)-1v (10 mol%)
G =
O
O174
(1.5 equiv)
O
NH4OAc
175
+
4 Å MSCHCl3, 0–20 °C, 72 h
17862–90%, 62–96% ee
++
G = Ph3Si(R)-1i (15–20 mol%)
O
N
O
Ph
R
Ar1
O
HAr2NH2
12 172177a
N
Ar1
HN
R
Ar2
O
Ph
O
OAr2HN
RHN Ph
O
179
4 Å MSCHCl3, 0 °C, 72 h
18065–75%, 90–97% ee
+
+
(R)-1i (20 mol%)
O
N
O
Ph
G = Ph3Si
181
172
177aa
N
Ar1
NHCOPh
O
CO2Et
CO2Et
N
Ar1
NHCOPh
O
CO2Et
CO2Et
R
R
NH2
CO2Et
CO2EtAr
182
BF3⋅OEt2
CH2Cl2, –15 °C

1954 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
annulation reaction is recognized as a prominent transfor-mation in organocatalysis research.143 The key step in theenantioselective Robinson annulation reaction is the ini-tial enantioselective Michael addition to enones using thechiral catalysts. Akiyama et al. have developed a novelstrategy for the enantioselective Robinson annulation re-action, which has two enantioselective processes. Theycombined a chiral phosphoric acid catalyzed enantiose-lective Michael addition of a-alkyl-b-keto esters 183 tomethyl vinyl ketone (184) with a chiral phosphoric acidcatalyzed kinetic resolution in the intramolecular aldol re-action followed by dehydration (Scheme 64).144 Enan-tiomers 185 that were delivered predominantly by theenantioselective Michael addition were transformed pref-erentially by the kinetic resolution to give the correspond-ing Robinson-type annulation products 186 with excellentenantioselectivities.
Scheme 64 Robinson-type annulation reaction via two enantiosel-ective processes
Akiyama and co-workers further developed the chiralphosphoric acid catalyzed desymmetrization of meso-1,3-diones 187 to yield optically active cyclohexenone deriv-atives 188 based on consecutive processes that includedthe enantioselective intramolecular aldol reaction as thekey step (Scheme 65).145 The method provides an efficientapproach to cyclohexenone derivatives 188 in a highlyenantioselective manner via the desymmetrization of s-symmetric triketones 187 followed by dehydration in aone-pot operation.
9 Transformations via Protonation of Electron-Rich Double Bonds
Brønsted acid activation of an electron-rich double bondis a fundamental method to generate reactive carbocationspecies. Brønsted acid catalyst molecules 1 undoubtedlypossess relatively strong acidity and hence can be utilized
as protonating agents of electron-rich double bonds togenerate cationic species for further elaboration. The ap-proach opens a new avenue for utilization of chiral phos-phoric acids towards advanced enantioselective catalysis.
9.1 Carbon–Carbon Bond Formation via Proto-nation of Enamides or Enecarbamates
Enantioselective Friedel–Crafts reactions have been in-vestigated intensively, using metal-based chiral catalystsor chiral organocatalysts.55 These enantioselective cataly-ses have been accomplished via the activation of electron-deficient multiple bonds, such as C=O, C=NR, and C=C–X (X: electron-withdrawing group). The acid-catalyzedFriedel–Crafts reactions of arenes with electron-rich alk-enes are practical and atom-economical methods for theproduction of alkylated arenes and have been applied tonumerous industrial processes. However, there had beenno reports of the enantioselective catalysis of the Friedel–Crafts reaction initiated by the activation of electron-richmultiple bonds, even using chiral metal catalysts, until2007. Terada and Sorimachi successfully developed ahighly enantioselective Friedel–Crafts reaction initiatedby the activation of electron-rich multiple bonds using achiral Brønsted acid catalyst for the first time. Chiralphosphoric acid 1q exhibited excellent performance forthe activation, enabling the catalytic reaction of indoles56a with enecarbamates 189 as electron-rich alkenes(Scheme 66).146 Uniformly high enantioselectivities andchemical yields were achieved in the reaction of indole(56aa) with enecarbamates 189 bearing either a linear ora branched alkyl group as well as an aromatic substituent.In addition, the enantioselectivities were maintained at anequally high level for a wide variety of indole derivatives56a, irrespective of their electronic properties. Thepresent approach provides efficient access to enantioen-riched 3-indolyl methanamines 190 with a variety of ali-phatic substituents and effectively complements previousmethods that afforded aromatic-group-substituted 3-in-dolyl methanamines 57 via the activation of aromaticimines (see Scheme 22).
The present Friedel–Crafts reaction proceeds through thein situ generation of aliphatic imines 191 that are deliv-ered via the protonation of enecarbamates 189 by thephosphoric acid catalyst (Figure 5). Phosphoric acid func-tions as an efficient catalyst for the dual transformation
O
O
PO
OH
G
G
CF3(R)-14f: G =
+
(R)-14f(2–10 mol%)
m-xylene20–50 °C, 24–48 h184
18654–74%, 96–99% ee
ZCO2Me
O
R
183(Z = CH2 or O)
F3C
O
18579–99%, 70–83% ee
ZR
CO2Me
O
ZR
O O
CO2Me
(R)-1q(10 mol%)
toluenereflux, 48 h
G = i-Pr
i-Pr
i-Pr
Scheme 65 Desymmetrization of s-symmetric triketones 187 lea-ding to enantioenriched cyclohexenones 188
18872–94%, 87–94% ee
187
R
OO O
R
(R)-1q(10 mol%)
n-hexane or toluene70–90 °C, 48–96 h
G = i-Pr
i-Pr
i-Pr
OO

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1955
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
that involves the in situ generation of imine 191 and theenantioselective carbon–carbon bond formation with in-doles 56a. This protocol offers the distinct advantage ofgenerating, in situ, unstable aliphatic imines 191 fromstorable and thus easily handled enecarbamates 189, andhence is applicable to other organic transformations. Infact, Terada et al. applied this method to an enantioselec-tive direct Mannich reaction (Scheme 67).147 The methodprovides an efficient pathway to produce b-alkyl-b-ami-nocarbonyl derivatives 192 in optically active forms.
Figure 5 Mechanistic considerations of the Friedel–Crafts reactionof enecarbamates 189 with indole 56a
Scheme 67 Direct Mannich reaction via activation of enecarba-mates 189
Shortly thereafter, Zhou and co-workers independentlyreported the enantioselective Friedel–Crafts reaction ofindoles 56a with aryl-substituted enamides 193 catalyzedby chiral phosphoric acid catalyst 1q (Scheme 68).148 The
yield of Friedel–Crafts products 194 was remarkably im-proved by the addition of MS 4A that prevented the hy-drolysis of enamides 193. It is noteworthy that in thiscatalytic Friedel–Crafts reaction, a quaternary stereogeniccenter bearing a nitrogen atom can be constructed in ahighly enantioselective manner.
Scheme 68 Formation of quaternary stereogenic center bearing anitrogen atom in the Friedel–Crafts reaction
Tsogoeva and co-workers reported the enantioselectiveformation of a quaternary stereogenic center bearing a ni-trogen atom through the homo-coupling reaction of en-amides 193 (Scheme 69).149 Enamides 193 were isomerizedin situ to ketimines 195 under the influence of phosphoricacid catalyst 1e, as depicted in Scheme 69. Electrophilicketimines 195 were generated under equilibrium condi-tions and their homo-coupling reaction with nucleophilicenamides 193 proceeded via the aza-ene-type mechanismto give the corresponding imine products 196.87,89 Result-ant imines 196 were readily isomerized to the enamineform under acidic conditions, giving enamide products197 with high enantioselectivities. Hydrolysis of the en-amide moiety yielded optically active b-amino ketoneshaving a quaternary carbon atom, which are potentiallyuseful as synthetic building blocks.
Scheme 69 Homo-coupling reaction of enamides 193
9.2 Enantioselective Protonation of Silyl Enol Ethers
The enantioselective protonation of prochiral enol deriva-tives is an attractive route for the preparation of opticallyactive a-substituted carbonyl compounds and has beenstudied extensively for the enantioselective catalysis us-ing chiral metal complexes or a metal complex (Lewis
Scheme 66 Friedel–Crafts reaction via activation of enecarbamates189 by phosphoric acid catalyst
G = i-Pr
i-Pr
i-Pr
+(R)-1q (2 mol%)
NH
NH
NHBoc
MeCN, –20 to 50 °C, 12–48 h
HN
H
NH
NHBoc
X
NH
NHBoc
Br
R1 = H, R2 = H, Me, n-Bu, i-Pr, Ph
R1 = Me, R2 = Me
X = MeO, Me, Br, CO2Me 84–91%, 90–93% ee
78%, 96% ee
R1
Boc
R1R2
R2
56aa
189
190a
190b 190c
63–98%, 90–94% ee
69%, 94% ee
1q
N
H
BocH
O P
O
OO
R1
R2
N
H
Boc HO
P
O
OO
H
R2
R1
56a
aliphatic imines 191 generated via protonation of enecarbamates 189
189 190
G = i-Pr
i-Pr
i-Pr
HN
HR
Boc
+
R
NH
O
O
(R)-1q (5 mol%)
THF, 50 °C, 24 hO
O
Boc
R = Me, n-Bu, i-Bu, cyclohexyl, Bn 49–73%, 49–89% ee
189
3
192
+
NH
Ar4 Å MS
toluene, 0–25 °C, 6–48 h
HN
Ar
NH
R
95–99%, 86–97% ee
R
HN Me
Ar: except ortho-substituents
AcG = i-Pr
i-Pr
i-Pr(S)-1q (10 mol%)
56a
194
193
Ac
193
HN
Ar
Ac(R)-1e (10 mol%)
toluene, r.t., 72 h
N
Ar
NO2G =
Ar
AcHN Ar
NHAc*
Ar
AcHN Ar
NAc*
2
aza-ene
isomerization isomerization
Ac
195 196
193
19779–83%, 88 to >99% ee

1956 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
acid) assisted chiral Brønsted acid system in the presenceof achiral proton sources.150 Yamamoto and Cheon report-ed a highly enantioselective protonation of cyclic silylenol ethers 198 using a metal-free chiral Brønsted acid forthe first time (Scheme 70).88i They developed a new fam-ily of chiral phosphoramide catalysts 199 and 200 in orderto achieve higher acidity to promote the enantioselectiveprotonation.
Scheme 70 Protonation of cyclic silyl enol ethers 198
It is noteworthy that almost no reaction was observedwhen the parent chiral phosphoric acid 1q and its thiolanalogue 201151 were employed. Meanwhile, the intro-duction of an NHTf group into the phosphoryl moiety,i.e., phosphoramide catalyst 66c, is crucial to enhance thecatalytic activity, leading to the quantitative formation ofenantioenriched product 202. Further substitution of theoxygen with sulfur 199a or selenium 200 in the phos-phoramide catalyst improved enantioselectivity as well asreactivity. The enantioselectivities and the reactivitieswere also strongly dependent on the achiral proton sourc-es and higher enantioselectivities could be achieved withsterically less hindered phenol or carboxylic acid deriva-tives. Among the catalytic systems tested, 199b coupledwith phenol (203) as the achiral proton source displayedthe best result in terms of catalytic activity and enantiose-lectivity. Although moderate enantioselectivities were ob-served in the protonation of cyclic ketones having analiphatic substituent at the a-position, the method is wellsuited for cyclic ketones bearing either an electron-withdrawing or an electron-donating aromatic substituent.More importantly, the catalyst load can be reduced to aslittle as 0.05 mol% without any significant loss of enantio-selectivity.88
9.3 Aldol-type Reaction via Protonation of Vinyl Ethers
The activation of vinyl ethers by a Brønsted acid catalystis an extensively utilized and fundamental method in syn-thetic organic chemistry, and is employed in the protec-tion of alcohols and the formation of carbon–carbonbonds, among other processes. In this activation mode, the
use of chiral phosphoric acids gives rise to ion pairs of achiral conjugate base and an oxocarbenium ion via theprotonation of vinyl ether 139. Terada et al. developedchiral conjugate base controlled enantioselectivetransformations152 involving the oxocarbenium ion as thereactive intermediate.153 They applied the intermediaryoxocarbenium154 to a direct aldol-type reaction of azlac-tones 177b141 via their oxazole tautomers 177b¢(Scheme 71). The method enables efficient access to bio-logically and pharmaceutically intriguing b-hydroxy-a-amino acid derivatives having a quaternary stereogeniccenter at the a-carbon atom.
Scheme 71 Aldol-type reaction of azlactones 177b via protonationof vinyl ethers 139
The electronic manipulation of Ar1 substituents intro-duced at the C2 position of azlactone 177 had a significantimpact not only on the reactivity but also on the stereo-chemical outcome in terms of both enantio- and diastereo-selectivities. When electron-donating methoxy sub-stituents were introduced to the 3,5-positions of the phe-nyl ring, giving 177b, b-alkoxy-a-amino acid derivatives205 were obtained with excellent enantio- and diastereo-selectivities after opening the azlactone ring of initial al-dol products 204. An investigation of the substituenteffect of vinyl ethers 139 showed that the sterically de-manding tert-butyl ether is important to achieve high dia-stereoselectivity. Vinyl ethers 139 with an alkyl groupsubstitution at the terminal position were also applicable,
PhOH (203) (1.1 equiv)toluene, r.t., 6–40 h
>99%,>99%,
(S)-199b (1–5 mol%)
202
G =
O
O
PY
X
G
G
i-Pr
i-Pr
i-Pr
66c:199a:200:1q:
201:
X = OX = SX = SeX = OX = O
199b:G =
i-Pr
i-Pr
t-Bu
Y = NHTfY = NHTfY = NHTfY = OHY = SH
X = S
OTMS
R
O
R
198(n = 1 or 2) R = aromatic
R = aliphatic72–90% ee58–68% ee
Y = NHTf
n n
G =
i-Pr
i-Pr
i-Pr
N
O
O
Ar2
Ar1
N
O**
O
Ar1
**
R1O
R2
Ar2
MeONa
MeOH
syn-205
anti-205+
O
Ar2 NH
OAr1
R1
OR1
R2 +CH2Cl2, 0 °C to r.t., 12–48 h
(R)-1q (5 mol%)
N
O
OH
Ar2
Ar1
R2
Ar1 = 3,5-(MeO)2C6H3
139177b'
177b
204
OR1
Ot-Bu
R2Ar2 = Ph Ar2 = Ph
99%98% syn98% ee
94%82% syn96% ee
62%80% syn93% ee
99%96% syn97% ee
66%88% syn94% ee
R1 = R2 =t-Bu n-Bu Bn Me n-Pr
Ot-Bu
Ar2 = X
X = 4-MeO, 4-Me, 4-CF3, 4-Br, 3-Me X = 2-F
45%70% syn37% ee
67–99%94–98% syn91–97% ee
OMe
O

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1957
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
affording b-alkoxy-a-amino acid derivatives 205 withhigh enantioselectivities. Azlactones 177b having a seriesof aromatic groups (Ar2) revealed uniformly high enantio-and diastereoselectivities for para- and meta-substitutedaromatic rings (Ar2), irrespective of their electronic prop-erties. However, the ortho substitution led to a marked re-duction of selectivity and chemical yield. Terada et al.proposed that the interaction, namely, C–H···O hydrogen-bond formation, between the chiral conjugate base and theoxocarbenium ion would allow the reaction to proceed un-der a chiral environment regulated by the chiral conjugatebase,153 and hence high stereoselectivities were achievedin this transformation.
9.4 Semi-Pinacol Rearrangement via Protona-tion of Vinyl Ethers
The semi-pinacol rearrangement is one of the most funda-mental skeletal rearrangements of carbon frameworks andis considered to be a variant of the pinacol rearrangement,where a carbon–oxygen double bond is formed at C1 withconcomitant migration of a group from C1 to adjacent C2,with or without loss of a leaving group, other than the hy-droxy group, from C2.155 Tu and co-workers accom-plished the enantioselective synthesis of spiroethers thatfeature two fused rings joined by a single chiral oxo qua-ternary carbon center, on the basis of the semi-pinacol re-arrangement (Scheme 72).156 Although a highly strainedcyclobutanol unit was necessary to promote the rearrange-ment, allylic alcohol derivatives 206 were rearranged tospiroethers 207 via protonation of the vinyl ether by chiralphosphoric acid catalyst 1q and subsequent ring expan-sion of the cyclobutane skeleton. The addition of MS 5Awas necessary for the reproducibility of the enantioselec-tivity, affording the corresponding products 207 in goodyields with high enantioselectivities. The authors also at-tempted the in situ generation of phosphoric acid 1q, us-ing the corresponding silver phosphate 208 of 1q, becauseof the lability of 206 under acidic conditions. This proce-dure, which involves a silver–proton exchange betweenalcohol 206 and silver phosphate 208, provides relatively
mild and weakly acidic conditions, effectively realizingthe semi-pinacol rearrangement of 206.
9.5 Hydroamination via Protonation of Non-activated Alkenes
The intramolecular addition of amines to unsaturated car-bon–carbon multiple bonds is one of the most direct meth-ods for the preparation of nitrogen heterocycles. A varietyof methodologies for the intramolecular hydroaminationsof alkynes and allenes have been reported so far.157 In con-trast, the hydroamination of electronically nonactivatedalkenes remains a challenging topic. The enantioselectivehydroamination of nonactivated alkenes has relied thusfar entirely on the use of chiral metal complexes.Ackermann and Althammer reported the enantioselectiveintramolecular hydroamination of nonactivated alkeneshaving a basic amine terminus under metal-free condi-tions (Scheme 73).158 Chiral phosphoric acid 1r enabledthe hydroamination of aminoalkene 209, giving the corre-sponding pyrrolidine derivatives 210 in optically activeforms, although the enantioselectivity was not sufficientlyhigh.
Scheme 73 Intramolecular hydroamination of nonactivated alkene209
10 Miscellaneous Carbon–Carbon Bond-Forming Reactions
10.1 Aza-Cope Rearrangement
Sigmatropic rearrangements are one of the most funda-mental methods for the construction of carbon frame-works and have widespread application in the synthesis ofbiologically relevant molecules. A variety of enantiose-lective sigmatropic rearrangements have been establishedusing chiral metal catalysts or organocatalysts.159 Enantio-selective versions of the aza-Cope rearrangement provideefficient routes to optically active homoallylic amines,which are useful precursors for the synthesis of naturalproducts. However, this valuable transformation was notsuccessfully developed until 2008. Rueping and Anton-chick reported for the first time a highly enantioselectiveaza-Cope rearrangement that uses chiral phosphoric acidcatalysts (Scheme 74).160 The reaction of aromatic alde-hydes 11 with achiral homoallylic amine 211 proceedssmoothly via the in situ generation of imines 212 in thepresence of catalyst 14g to afford rearranged products213, homoallylic amine derivatives, in good yields withhigh enantioselectivities. The method furnishes a practical
Scheme 72 Semi-pinacol rearrangement of allylic alcohols 206 viaprotonation of vinyl ether
OOH
R
R
G =
i-Pr
i-Pr
i-Pr
(R)-1q (10 mol%)or (R)-208 (10 mol%) OR
R
O5 Å MS
CCl4, 0 °C to r.t., 2 h
206(n = 1, 2)
20751–98%, 74–98% ee
O
O
PO
OAg
G
G
(R)-208: G =
i-Pr
i-Pr
i-Pr
n n
NH
Ph
Ph
Bn
G =
(R)-1r (20 mol%)
1,4-dioxane, 130 °C, 20 h
CF3
CF3
N
Ph
PhBn
21072%, 17% ee209

1958 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
route to the catalytic enantioselective aminoallylation ofaromatic aldehydes on the basis of a condensation–rear-rangement sequence. Free primary homoallylic amines214 were obtained by treating rearranged products 213with hydroxylamine hydrochloride (215).
Scheme 74 Aza-Cope rearrangement of in situ generated imine 212
10.2 Radical Reaction
Stereoselective radical reactions have received much at-tention due to the uniqueness of the radical processes andhence, the enantioselective catalysis of radical reactionshas been approached in various ways.161 In particular, be-cause it is favorable for catalytic processes, reductivealkylation has been investigated extensively in enantiose-lective radical reactions, where the addition of alkyl radi-cals to multiple bonds, followed by trapping of theresulting radicals with a hydrogen atom source, leads toreduced products. Kim and Lee developed an enantiose-lective radical reaction of imines 2o catalyzed by chiralphosphoramide 66c on the basis of the reductive alkyla-tion process using tris(trimethylsilyl)silane (217) as thehydrogen source and triethylborane as the initiator(Scheme 75).162 The enantioselective reductive alkylationof imines 2o by ethyl iodide (216a) gave the correspond-ing products 218a in good yields with relatively highenantioselectivities. Meanwhile, in the reaction of isopro-pyl iodide (216b) and tert-butyl iodide (216c), desiredproducts 218b and 218c were obtained in modest yieldsbecause of the considerable amount of byproduct 218afurnished by the direct addition of an ethyl radical, gener-ated from triethylborane, to imines 2o.
10.3 Alkylation of Enamides via Activation of Alcohols
The catalytic enantioselective a-alkylation of carbonylderivatives is a highly valuable carbon–carbon bond-
forming strategy in organic synthesis. However, this im-portant methodology has remained a longstanding issue inasymmetric catalysis and hence various approaches havebeen investigated. Among those approaches, Petrini,Melchiorre, and co-workers and Cozzi et al. accomplishedefficient methods for the enantioselective a-alkylation ofsimple aldehydes with stable and unsymmetrically substi-tuted diarylcarbocations using chiral secondary aminecatalysts.163 Shortly thereafter, Gong and co-workers re-ported that H8-BINOL-derived phosphoric acid 14a en-abled the highly enantioselective a-alkylation ofenamides 219 with indolyl alcohols 220 to give b-aryl-b-indolylpropanones 221 in good yields (Scheme 76).164
The reaction proceeds via stabilized cation 222, a vinylo-gous iminium intermediate, generated in situ from indolylalcohols 219 under the influence of acid catalyst 14a.
(R)-14g: G =
O
HAr
H2N
Ph Ph
+(R)-14g (10 mol%)
3 Å MSt-BuOMe, 50 °C, 48 h
Ar
N Ph
Ph
O
O
PO
OH
G
G
N
Ph Ph
Ar
N
Ar
Ph
Ph
aza-Cope rearrangement
21352–87%, 80–94% ee
Ar
NH2(R)-14g
11
211
214
212 213
H2NOH⋅HCl215
Scheme 75 Reductive alkylation of imines 2o catalyzed by chiralphosphoramide 66c
O
O
PO
NHTf
G
G
(R)-66c: G =
i-Pr
i-Pr
i-Pr
N
HAr
+(R)-66c (30 mol%)
BEt3, O2
THF, –40 °C, 24 h2o
R I
Ph
Ar ** R
HN
Ph
216
+
218a20–35%, 4–85% ee
Ar ** Et
HN
Ph
+
HSi(SiMe3)3
217
R = Et (218a): 56–77%, 73–84% ee
R = i-Pr (218b), t-Bu (218c): 31–56%, 74–80% ee
a: R = Etb: R = i-Prc: R = t-Bu
byproducts obtained in the reaction of 216b and 216c
Scheme 76 a-Alkylation of enamides 219 with indolyl alcohols 220
+(R)-14a (10 mol%)
CH2Cl2, –30 °C, 34–68 h
22169–96%, 88–96% ee
NH
NH
HO
Ar2
HN
Ar1
Bz
219220
**
Ar2O
Ar1
O
O
PO
OH
Ph
Ph
NH222
Ar2
R
R
R

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1959
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
11 Carbon–Heteroatom Bond-Forming Reac-tions
11.1 Hydrophosphonylation (Kabachnik–Fields Reaction)
The Kabachnik–Fields reaction, which involves the hy-drophosphonylation of phosphites with imines generatedin situ from carbonyl compounds and amines, is an attrac-tive method for the preparation of a-amino phosphonates.Optically active a-amino phosphonic acids and theirphosphonate esters are an interesting class of compoundsdue to their potent biological activities as non-proteino-genic analogues of a-amino acids. Therefore, consider-able attention has been paid to their enantioselectivesynthesis by the hydrophosphonylation of preformedimines, using either metal-based catalysts or organocata-lysts.165
Akiyama et al. reported that chiral phosphoric acid 1rfunctioned as an efficient catalyst in the addition reactionof diisopropyl phosphite (223a) with N-PMP-protectedaldimines 2f (Scheme 77).166 Aromatic and a,b-unsaturat-ed imines could be used in the reaction, giving a-aminophosphonate esters 224 in high yields. The use of sterical-ly demanding dialkyl phosphites 223 and aldimines de-rived from cinnamaldehyde derivatives is essential toachieve high enantioselectivities. The same researchgroup and Yamanaka and Hirata conducted DFT compu-tational analysis of the hydrophosphonylation reaction.167
Chiral phosphoric acid 1r plays a significant role in theactivation of both imine 2f and phosphite 223 based on itsdual functional catalysis.
Scheme 77 Hydrophosphonylation of imines
List and co-workers reported an excellent approach to theenantioselective synthesis of b-branched a-amino phos-phonates 225, which involved the dynamic kinetic resolu-tion strategy (Scheme 78)168 that they had previouslyapplied to the enantioselective reductive amination of ra-cemic a-branched aldehydes 226 (see Scheme 91).169
They successfully accomplished the highly stereoselec-tive direct three-component Kabachnik–Fields reaction ofone equivalent each of racemic aldehyde 226, 4-methoxy-aniline (12b), and di(3-pentyl)phosphite (223b) in thepresence of the newly developed chiral phosphoric acid1w by combining dynamic kinetic resolution with the par-allel creation of an additional stereogenic center. The hy-drophosphonylation proceeds through a tautomerization
between imines 227 and enamines 228 with concomitantracemization at the a-position. Dynamic kinetic resolu-tion occurs under the influence of catalyst (S)-1w, where-by the hydrophosphonylation of (R)-227 proceeds fasterthan that of (S)-227 to give b-branched a-amino phospho-nates 225 with high diastereo- and enantioselectivities, es-pecially for aldehydes 226 bearing a secondary alkylgroup at the a-position.
Scheme 78 Stereoselective synthesis of b-branched a-amino phos-phonates 225 by Kabachnik–Fields reaction
11.2 Formation of (Hemi)Aminals
Aminals, compounds having two amino groups bound tothe same carbon atom, are represented in many medicinalagents that have versatile therapeutic actions, such as pro-teinase inhibitors and neurotensins. Antilla and co-work-ers developed an enantioselective synthesis of protectedaminals 229, which involved the addition reaction of ni-trogen nucleophiles 230 with N-Boc aromatic imines 2acatalyzed by chiral phosphoric acids (Scheme 79).170 Inthis novel enantioselective transformation, they employeda new type of axially chiral phosphoric acid 231, derivedfrom VAPOL (vaulted 3,3¢-biphenanthrol), originally de-veloped by Antilla’s research group. Indeed, 231 exhibit-ed excellent catalytic activity and enantioselectivity in theaddition to N-Boc aromatic imines 2a. Enantioenrichedaminal products 229a were obtained in the reaction of sul-fonamides 230a (Scheme 79a) and were stable on storage;neither decomposition nor racemization was observed insolution over several days. The same research group re-ported the enantioselective addition reaction of phthal-imide or its derivatives 230b with N-Boc aromatic imines2a, affording the corresponding aminals 229b with excel-lent enantioselectivities (Scheme 79b).171
(R)-1r (10 mol%)
m-xylene, r.t., 24–171 h
72–84%, 52–77% ee76–97%, 81–90% ee
G =
CF3
CF3
N
HR
PMP
HP
O
Oi-Pr
Oi-Pr+
R = aromatic R = alkenyl
HN
P(Oi-Pr)2R
PMP
O2f 223a224
1w: G =
i-Pr
i-Pr
(S)-1w (10 mol%)
5 Å MScyclohexane50 °C, 168 h
61–86%,
63–84%,
HP
OO
+
R = secondary alkyl (cyclopentyl, i-Pr, cyclohexyl)
R = primary alkyl (Me, Et)
HN
P
PMP
O
O
HR
ArNH2
OMe
R
Ar
O
Et Et
EtEt
O
O Et
Et
Et Et
+
76–94% ee
2–84% ee
87–97% anti,
60–67% anti,
223b
225
(±)-22612b
Ar
RH
NPMP
Ar
RH
HNPMP
Ar
RH
NPMP
fasthydrophosphonylation
slow hydrophosphonylation
(S)-227 (R)-227228(racemization)

1960 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Scheme 79 Preparation of aminals via addition of nitrogen nucleo-philes to imines
Cyclic aminals are relatively common structural elementsof diverse pharmaceutical compounds. However, no effi-cient method was available to prepare these compoundsuntil 2008. The research groups of List172 and Rueping173
independently reported a highly enantioselective directsynthesis of cyclic aminals 232 from aldehydes 11 and o-aminobenzamide derivatives 233 using chiral phosphoricacids (Scheme 80). The catalytic reaction involves theimine formation/intramolecular amine addition sequenceusing 11 and 233. In List’s approach, aliphatic aldehydeswere mainly employed as the substrate and the corre-sponding dihydroquinazolinones 232 were furnished withexcellent enantioselectivities when primary alkyl-substi-tuted aldehydes and phosphoric acid catalyst 1w wereused (Scheme 80a). They also applied the enantioselec-tive method to sulfonamide analogues instead of benz-amides 233. The method provides a practical and efficientroute to pharmaceutically relevant compounds, namely,the benzo(thia)diazine class of cyclic aminals. In fact,they synthesized several benzo(thia)diazines, includingaquamox, thiabutazide, and penflutizide, with high enan-tioselectivities using their modified methodology. Mean-while, Rueping et al. focused on the preparation ofaromatic-substituted cyclic aminals 232 using aromaticaldehydes and 233a as the substrates (Scheme 80b). Phos-phoric acid 1g was the best catalyst in their case.
Antilla and co-workers further extended their methodolo-gy to the addition of an oxygen nucleophile, instead of anitrogen nucleophile (see Scheme 79), to imines(Scheme 81).174 They successfully developed the catalyticenantioselective addition reaction of alcohols 234 as oxy-gen nucleophiles with N-benzoyl imines 2d using chiral
phosphoric acid 1g. The method provides straightforwardaccess to chiral hemiaminal ethers 235 with high enantio-selectivities.
11.3 Nucleophilic Ring Opening of Aziridines and Related Reactions
The ring opening of aziridines with nitrogen nucleophilesprovides an efficient route to a vicinal diamine structuralmoiety175 that exists in numerous biologically active com-pounds and medicinal agents, as well as chiral ligands ofmetal complexes. In this context, considerable effort hasbeen devoted to their enantioselective synthesis. One of
N
HAr
Boc HN
NAr
Boc
(S)-231 (5–20 mol%)
229a80–99%, 73–99% ee
Ph
Ph
O
O
POH
O
H2N SO2R2
PG+
R2 = aromatic or Me
230
HN
PG
R1R1
HN
O
O
R2HN
O
O
229b82–94%, 89–99% ee
ora) b)
*
(S)-231 (5 mol%), r.t., 24 h
Et2O
(S)-231 (5–20 mol%), r.t., 1–20 h
HN
NAr
Boc
SO2R2
H
*
HN
NAr
BocO
O R2
2a 229
(230a)
230b
Scheme 80 Direct transformation of o-aminobenzamides 233 withaldehydes into cyclic aminals 232
(R)-1g (10 mol%)
3 Å MSCHCl3, r.t., 24–48 h
23273–93%, 80–92% ee
NH2
O
NH2
+
O
H R
NH
NH
O
R
233a
11(R = aromatic)
b)
(S)-1w (10 mol%)
5 Å MS toluene, –45 °C, 24 h
NH2
O
NH2X
+
O
H R
a)
233
11
aquamox(R)-231: 78%, 61% ee
thiabutazide(S)-1x: 81%, 91% ee
penflutizide(S)-1x: 74%, 90% ee
NH
NH
O
H2NO2S
Cl NH
NHSH2NO2S
Cl
O O
NH
NHSH2NO2S
F3C
O O
1x: G = Ad
i-Pr
i-Pr
Ad = 1-adamantyl
G =
R = aromatic
79–96%, 67–72%,
R = primary alkylR = Ph or i-Pr
89–98% ee26–50% ee
NH
NH
O
RX
232
G =
i-Pr
i-Pr
4
Scheme 81 Preparation of hemiaminal ethers 235 via addition of al-cohols 234 to imines 2d
(R)-1g (5 mol%)
EtOAc, r.t., 24 h+
N
R1 H
G =
76–99%, 81–84%,
62%,
R2 = primary alkyl R2 = secondary alkyl R2 = Me
83–95% ee81–88% ee
65% ee
Ph
O
HN
R1 OR2
Ph
O
R1 = aromatic
R1 = Et
2d 235234
R2OH

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1961
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
the most direct methods for the preparation of enantio-enriched vicinal diamines is the enantioselective desym-metrization of meso-aziridines 236 with nitrogennucleophiles.176 Antilla and co-workers developed a high-ly enantioselective synthesis of vicinal diamines based onthe desymmetrization strategy using chiral phosphoricacid 231 and azidotrimethylsilylane (237) as the nitrogennucleophile (Scheme 82).177 The 3,5-bis(trifluorometh-yl)benzoyl substituent introduced on the nitrogen atom ofaziridines, giving 236a, optimized both yield and enantio-selectivity. A series of meso-aziridines 236a derived from(hetero)cyclic and acyclic alkanes having aliphatic or aro-matic substituents were applicable to the enantioselectiveaziridine ring opening reaction, giving the correspondingproducts 238 in optically active forms. They proposed thatthe silylated phosphate generated in situ by the reaction ofchiral phosphoric acid with azidotrimethylsilylane (237)is a catalytically active species.
Scheme 82 Desymmetrization of meso-aziridines 236a with azide
Antilla and co-workers further applied their method to theenantioselective ring opening of meso-aziridines 236 by aseries of thiols 239, instead of azide, as the nucleophile(Scheme 83).178 This expansion of the substrate scope en-abled the asymmetric synthesis of optically active b-ami-no thioether derivatives 240. Phosphoric acid 231 alsofunctioned as an efficient catalyst for the ring opening bythiols 239 in terms of catalytic activity and enantioselec-tivity. Interestingly, a silyl group is not required in thisring opening of aziridines 236, which is in contrast to thereaction with azide as the nucleophile (Scheme 82). Theintroduction of 3,5-dinitrobenzoyl to the nitrogen atom ofaziridine, giving 236b, is highly desirable in achieving thehigh enantioselectivities. 3,5-Bis(trifluoromethyl)ben-zoyl, which was employed for the ring opening by azide(Scheme 82),177 and 4-nitrobenzoyl protective groups dis-played much lower enantioselectivities. Arylthiols aresuitable for the reaction. Thus, a wide variety of electron-withdrawing and electron-donating substituents can be in-troduced to the ortho-, meta-, and para-positions of thearomatic ring. Meanwhile, heteroaryl- and alkylthiolsproved to be relatively poor reaction partners, giving the
corresponding products 240 with low to moderate enantio-selectivities. Della Sala and Lattanzi independently re-ported the highly enantioselective ring opening of meso-aziridine 236 by (phenylthio)trimethylsilane (241; notshown), instead of thiols 239, using the same catalyst231.179 A range of meso-aziridines 236b having a 3,5-dinitrobenzoyl group at the nitrogen atom could be usedfor this catalytic system, affording phenyl thioethers ingood yields with high enantioselectivities. They also pro-posed that silylated phosphate generated in situ from cat-alyst 231 with silylated nucleophile 241 would be acatalytically active species, following Antilla’s mechanis-tic proposal in the ring opening by azidotrimethylsilylane(237).177
Scheme 83 Desymmetrization of meso-aziridines 236b with thiols239
Toste and co-workers successfully demonstrated an enan-tioselective synthesis of b-alkoxy amines from racemic b-chloro tertiary amines 242 and alcohols 243 as an oxygennucleophile under the influence of an equimolar amountof silver(I) salt and a catalytic amount of chiral phospho-ric acid 1q (Scheme 84).180 The methodology is based onthe chiral counteranion mediated enantioselective trans-formation.152 The reaction proceeds via the asymmetricring opening of intermediary meso-aziridinium ions 244that were generated from the ring closure of b-chloro ter-tiary amines 242 by silver(I) salts. Subsequent nucleo-philic ring opening of meso-aziridinium ions 244 byalcohols 243 provided the corresponding b-alkoxy amines245. Secondary, tertiary, and relatively hindered primaryalcohols could be used in the reaction, giving b-alkoxyamines 245 with high enantioselectivities. The efficientdesymmetrization of meso-aziridinium ion 244 wasachieved under the influence of the chiral counteranionderived from phosphoric acid 1q, as the ion pair G wasformed between cationic 244 and the counteranion of 1q.They pointed out that the resulting catalytic process canbe regarded as chiral anion phase-transfer catalysis inanalogy to the established chiral cation alternative.181
Toste and co-workers further extended their chemistry tothe sulfur analogue of the ring opening reaction, namely,the desymmetrization of meso-episulfonium ions 246(Scheme 85).180a However, in this application, the silver–
(S)-231 (10 mol%)
64–97%, 91–95% ee
DCE, r.t., 21–91 h N3
HN
COAr
O
CF3
CF3N
n = 1–3
TMSN3+
R
R
X
R = Ph95%, 83% ee
84%, 92% ee
X = O49%, 87% ee
90%, 70% ee
R = Me88%, 86% ee
X = NCbz94%, 71% ee
Ar = 3,5-(CF3)2C6H3236a
237238
N3 N3
N3
N3
N3
HN
HN
HN
HN
HN
COAr COAr
COAr
COAr
COAr
n
(S)-231 (10 mol%)
Et2O, r.t., 20 hSR1
HN
COAr
O
NO2
NO2N R1SH+
R2
R2
R2 = n-Pr94%, 87% ee
99%, 95% ee 95%, 96% eeR2 = Me97%, 95% ee
236b
239
240Ar = 3,5-(O2N)2C6H3
R1 = aromaticR1 = heteroaromaticR1 = aliphatic
70–99%,71–78%,15–86%,
74 to >99% ee55–82% ee18–62% ee
HN
SPh
HN
HN
SPhSPh
COAr COArCOAr

1962 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
halogen abstraction method for the generation of the cat-ionic intermediate could not be utilized, because the sul-fide would likely bind silver(I). Therefore, they employedtrichloroacetimidate as the leaving group. The trichloro-acetimidate moiety is directly activated by chiral phos-phoric acid 1q, and this is followed by ring closure withconcomitant loss of trichloroacetamide from substrates247 generating ion pair H of meso-episulfonium ions 246and the chiral counteranion. The interception of interme-diate 246 with an oxygen nucleophile, such as alcohols243, affords chiral sulfide products 248 in excellent yieldswith good enantioselectivities.
Scheme 85 Desymmetrization of meso-episulfonium ions 246 withalcohols 243
12 Transfer Hydrogenation Reactions
12.1 Transfer Hydrogenation of Acyclic and Cyclic Imines
The enantioselective reduction of ketimines is a straight-forward approach to synthesizing optically active amines.Although the catalytic enantioselective hydrogenationsand the transfer hydrogenations of ketones and olefinshave been intensively investigated using chiral metalcomplexes, the corresponding enantioselective reductionsof imines are less advanced and hence the development ofefficient methods for these transformations is still one ofthe greatest challenges in the practical synthesis of chiralamines. Most catalytic enantioselective hydrogenations ofketimines have been conducted using common hydrogensources, such as high-pressure hydrogen gases, di- or tri-alkylsilanes, and ammonium formate, under the influenceof chiral transition-metal complexes.182 Recently, List andco-workers and MacMillan and co-workers independentlyreported the organocatalytic transfer hydrogenations ofa,b-unsaturated aldehydes using Hantzsch esters 249(Figure 6), which are dihydropyridine derivatives, as abiomimetic hydrogen source.183 Following these excellentreports, Hantzsch esters gained popularity as an efficienthydrogen source for the enantioselective reduction of or-ganic compounds, including imines.184
Figure 6 Hantzsch esters 249
In 2005, Rueping et al. reported that chiral phosphoricacid 1r functions as an efficient catalyst for the enantiose-lective reduction of ketimines 250 (Scheme 86a-1).185 Avariety of aryl methyl ketimines 250 were reduced to thecorresponding amines 251 in optically active forms usingHantzsch ester 249a as the hydrogenation transfer re-agent.186 In the same year, List and co-workers indepen-dently reported the highly enantioselective transferhydrogenation reaction of ketimines 250 using Hantzschester 249a by thorough optimization of the substituents(G) that were introduced onto the phosphoric acid catalyst(Scheme 86a-2).187 Almost simultaneously, MacMillanand co-workers successfully developed the enantioselec-tive reductive amination reaction using Hantzsch ester249a via simple fragment coupling between ketones 252and aniline derivatives 12b (Scheme 86b).188 Acid cata-lyst 1i, with its sterically demanding triphenylsilyl groups,was optimal for the enantioselectivity. Although the reac-tion required a methyl ketone subunit, this operationallysimple method is applicable to not only aryl but also alkylmethyl ketones and provides a highly efficient route tostructurally diverse chiral amines. In their conscientiousstudies on the reductive aminations, MacMillan and co-
Scheme 84 Desymmetrization of meso-aziridinium ions 244 withalcohols 243
+
245d70%, 99% ee
245b87%, 96% ee
245a81%, 92% ee
N
Ph
Ph
Cl
(±)-242
(S)-1q (15 mol%)
G = i-Pr
i-Pr
i-Pr
Ag2CO3 (0.6 equiv)4 Å MS
toluene, 50 °C, 24–36 h
N
Ph
Ph
O
245c56%, 92% ee
N
Ph
Ph
O
N
Ph
Ph
O
N
O
NO2
HO
OMe
243a
R = R = t-Bu
Ph
Ph
NR
R
244
O P
O
O
O
1qG
R
OMe
+
90–98%, 87–92% ee
SR1
Ph
Ph
O
(±)-247
(S)-1q (15 mol%)
G = i-Pr
i-Pr
i-Pr
toluene, r.t., 12 hSR1
Ph
Ph
OR2
243
Ph
Ph
SR1
246
HN CCl3
248
(R1 = Me or Bn)
O P
O
O
O
1qH
(R2 = primary or secondary alkyl)
R2OH
NH
R3
R1O2C CO2R2
249
a: R1, R2 = Et, R3 = Meb: R1 = Me, R2 = t-Bu, R3 = Mec: R1, R2 = allyl, R3 = Med: R1, R2 = t-Bu, R3 = Mee: R1, R2 = Me, R3 = i-Pr
H H

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1963
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
workers also reported a couple of examples using cyclicimines as the substrate.
Shortly thereafter, a detailed investigation of the transferhydrogenation of cyclic imines was conducted byRueping et al. (Scheme 87a).88g Benzoxazines 253a un-derwent the transfer hydrogenation in good yields withextremely high enantioselectivities even with a remark-ably low catalyst load of 1m (0.1 mol%). Benzothiazines253b (Scheme 87a) and benzoxazinones 254a(Scheme 87b) were also hydrogenated by Hantzsch ester249a in the presence of the same catalyst 1m (1 mol%) togive the corresponding products, 255b and 256a, with ex-cellent enantioselectivities. They further extended thetransfer hydrogenation to quinoxalines 253c and quinox-alinones 254b using 1g (10 mol%) as the catalyst, givingrise to 2-tetrahydroquinoxalines 255c and 3-dihydroqui-noxalinones 256b, respectively, with high enantioselec-tivities (Scheme 87c).189
Soon after these initial reports, the groups of Antilla190
and You191 independently applied the chiral phosphoricacid catalysis approach to the enantioselective hydrogena-tion of a-imino esters 257. The method provides an alter-native route to the enantioselective synthesis of a-aminoesters 258. Antilla and co-workers employed VAPOL-derived phosphoric acid 231,177 originally developed byhis research group (Scheme 88), whereas 1g was used in
You’s studies. In both cases, excellent enantioselectivitieswere achieved. You and co-workers further applied themethod to the enantioselective reduction of a-imino esters259 having an alkynyl substituent at the a-position(Scheme 89).192 Both alkyne and imine moieties were re-duced under transfer hydrogenation conditions with anexcess amount of Hantzsch ester 249a (2.2 equiv) to giveb,g-unsaturated a-amino esters 260. Although the chemi-cal yields were moderate, high enantio- and trans-selec-tivities were achieved using the same chiral phosphoricacid catalyst 1g.
Scheme 88 Transfer hydrogenation of a-imino esters 257
Scheme 86 Transfer hydrogenation of acyclic ketimines 250 usingHantzsch ester 249 as the biomimetic hydrogen source
1 (cat.)249aN
R
PMP
(R)-1r: G =
CF3
CF3
R = aromaticbenzene, 60 °C, 16 h46–91%, 70–84% ee (R)
HN
R
PMP
*
(S)-1q: G =
R = aromatic or isopropyltoluene, 35 °C, 42–71 h80–98%, 80–93% ee (S)
a)
b)
+O
R
HN
R
PMP
G = Ph3Si(R)-1i (10 mol%)
249a
5 Å MSbenzene, 40–50 °C
24–96 h
a-1) a-2)
(R = aromatic or aliphatic)
(10 mol%) (1 mol%)
H2N
OMe
i-Pr
i-Pr
i-Pr
250 251
252 12b 25149–92%
83–96% ee
HNPMP
Ph
HNPMP
70%, 88% ee75%, 85% ee90%, 93% ee
HN
Ph
TsN
F
HNPMP
BzO
72%, 81% ee 82%, 97% eeO
HN
O
Scheme 87 Transfer hydrogenation of cyclic imines, 253 and 254
(R)-1m249a
CHCl3, r.t., 12 h
G =
N
X
Ar NH
X
Ar
a)
b)
N
O
Ar NH
O
Ar
O
255a: X = O 0.1 mol% of 1m92–95%, 98 to >99% ee
1 mol% of 1m50–87%, 93 to >99% ee
255b: X = S
O(R)-1m (1 mol%)
249a
CHCl3, r.t. to 60 °C, 15–24 h
255253
254a
a: X = Ob: X = S
256a55–92%, 90 to >99% ee
(R)-1g (10 mol%)249a
N
HN
Ar NH
HN
Ar
c)
255c: Z = H, H CHCl3, 35 °C, 24 h73–98%, 92–98% ee
THF, 50 °C, 24 h42–75%, 92–98% ee
256b: Z = O
253254
c: Z = H, Hb: Z = O
Z Z
G =
c: Z = H, Hb: Z = O
255256
XX
N
CO2EtR
PMPHN
CO2EtR
PMP(S)-231 (5 mol%)249a
toluene, 50 °C, 18–22 h
25885–98%, 94–99% ee
Ph
Ph
O
O
POH
O
(S)-231
257(R = aromatic or
aliphatic)

1964 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Scheme 89 Transfer hydrogenation of a-imino esters 259 having analkynyl substituent at the a-position
As shown in Schemes 86, 88, and 89, the reductive ami-nation of ketones and the hydrogenation of ketimines cat-alyzed by chiral phosphoric acids were accomplished withhigh enantioselectivities. However, in these reductions,aryl derivatives, such as PMP, were primarily employedas the nitrogen protective group. The removal of theseprotective groups usually required somewhat harsh reac-tion conditions. Antilla and Li circumvented this draw-back by using enamides 193 as the substrates(Scheme 90).193 The reduction of enamides 193 proceed-ed via tautomerization to intermediary imines to giveamine products 261 having an N-acetyl protective groupthat can be readily removed under standard conditions.Acetic acid was employed as the co-catalyst, which al-lowed for the reduction of the catalyst load of 1g to 1mol% while maintaining the high enantioselectivity. Ace-tic acid accelerated the tautomerization of the enamide tothe imine, while it was inactive in the subsequent reduc-tion and only chiral phosphoric acid 1g functioned as theactive catalyst for the successive transfer hydrogenationof the imine.
Scheme 90 Transfer hydrogenation of enamides 193
List and co-workers developed an excellent approach tosynthesize b-branched amines in optically active forms bycombining the enamine and reductive amination process-es (Scheme 91).169 The reductive amination of unsymmet-rically a,a-disubstituted aldehydes 226 and anilinederivatives 12a–c proceeded through a tautomerizationbetween imine 227 and enamine 228. Dynamic kineticresolution occurred under the influence of catalyst (R)-1q,whereby the hydrogenation of (R)-227 proceeded morerapidly than that of (S)-227 to give b-branched amines 262with moderate to high enantioselectivities (seeScheme 78).
Gong and co-workers reported a facial synthesis of chiral1,3-diamine derivatives 263 bearing a quaternary stereo-
genic center via the transfer hydrogenation of 2,4-diaryl-2,3-dihydrobenzodiazepine derivatives 264 on the basisof dynamic kinetic resolution (Scheme 92).194 The trans-fer hydrogenation of the S-enantiomer among racemic264 proceeds rapidly in the presence of (R)-14a to afford(2S,4R)-syn-263 predominantly along with (2S,4S)-anti-263. Meanwhile, the opposite enantiomer, (R)-264, un-dergoes slow transfer hydrogenation and the remaining(R)-264 racemizes via intermediate 265 through the retro-Mannich and Mannich sequences.
Scheme 92 Enantioselective synthesis of 1,3-diamine derivatives263 through dynamic kinetic resolution
N
CO2Et
PMPHN
CO2Et
PMP(S)-1g (1 mol%)249a (2.2 equiv)
Et2O, r.t., 24 h
26027–64%, 83–96% ee
Ar
Ar
G =
259
HN
Ar
AcHN
Ar
Ac(S)-1g (1 mol%), AcOH (10 mol%)249a
toluene, 50 °C, 14–44 h261
43–99%, 71–95% eeexcept o-substituted aryl
G =
193
Scheme 91 Enantioselective synthesis of b-branched amines 262through dynamic kinetic resolution
(R)-1q (5 mol%)249b
5 Å MS, benzene, 6 °C, 72 h
46–96%, 39–81%,
R2
R1
H
O
H2NAr+
R2
R1
HNAr
(R1 = aromatic, aliphatic, CF3)(R2 = Me, Et)
88–98% ee40–80% ee
(Ar = Ph, PMP, 4-CF3C6H4)
R1 = aromaticR1 = aliphatic, CF3
R2
R1
H
NAr
R2
R1
H
HNAr
R2
R1
H
NAr
(racemization)
fast reduction slow reduction
(R)-227 (S)-227
G = i-Pr
i-Pr
i-Pr
(±)-226
12a–c
262
228
(R)-14a (10 mol%)249c
Na2SO4
CHCl3, –10 °C, 5.5 d
(racemization)fast reduction slow reduction265
(R)-264(S)-264Mannich
retro-Mannich
N NH
ArAr
HN N
Ar Ar
HN NH
ArAr
HN NH
ArArO
O
PO
OH
Ph
Ph
(R)-14a
(±)-264
syn-263 anti-263
+
Mannich
retro-Mannich
63–97%syn/anti = 2:1 to 8:1
63–86% ee for syn (2S,4R)75–94% ee for anti (2S,4S)

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1965
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
In organocatalytic hydrogenations, the hydrogen sourceswere, until recently, limited to Hantzsch esters 249, whichare the most well used cofactors in biochemical hydroge-nation reactions.184 Akiyama and Zhu utilized benzothia-zoline derivative 266,195 which functioned as an efficientantioxidant with potent reducing ability,196 as a novel hy-drogen source, instead of Hantzsch esters 249(Scheme 93). Aromatic methyl ketimines 250 havingelectron-donating and electron-withdrawing substituentswere well tolerated in this hydrogenation system and evenaliphatic ketimine underwent the reduction while main-taining excellent enantioselectivity. It is noteworthy thatthe present method, using 266 as a novel hydrogen source,provides the corresponding products 251 with higherenantioselectivities than those obtained in previous re-ports (see Scheme 86)185,187,188 in which Hantzsch ester249a was employed. The distinct advantage of this proto-col is that benzothiazoline 266 can be generated in situfrom 2-naphthalenecarbaldehyde and 2-aminothiophenol.Thus, the three-component reaction between 250 and thein situ generated 266 becomes operative without consid-erable loss of enantioselectivity and chemical yield.
Scheme 93 Transfer hydrogenation using benzothiazoline derivati-ve 266 as a novel hydrogen source
12.2 Tandem Transfer Hydrogenation of Quino-lines and Pyridine Derivatives
The asymmetric hydrogenation of nitrogen-substitutedheteroaromatic compounds provides a straightforwardand attractive route to enantioenriched nitrogen heterocy-cles. In contrast, the enantioselective hydrogenation ofthese heteroaromatics using chiral transition-metal cata-lysts has not been fully established and successful exam-ples of these transformations are limited.197 The use ofHantzsch esters 249 as the hydrogen source was proven tobe beneficial for the asymmetric hydrogenation of theseheteroaromatics by Rueping et al. The enantioselective re-duction of quinolines 267 to tetrahydroquinolines 268 us-ing Hantzsch ester 249a proceeded smoothly in thepresence of chiral phosphoric acid catalyst 1m(Scheme 94).198 The method is applicable to a variety of2-aryl- and 2-alkyl-substituted quinolines 267 and pro-vides direct access to tetrahydroquinoline alkaloids 268 ina highly enantioselective manner. They proposed that the
reaction proceeds via a tandem hydrogenation cascade.The first step of the cascade generates enamines 269 viathe 1,4-hydride addition to quinolines 267. Under acidicconditions, the subsequent isomerization of 269 providesimines 270, which are delivered to the second step of thecascade. The 1,2-hydride addition to imines 270 with gen-eration of the stereogenic center provides enantioenrichedtetrahydroquinoline products 268.
Scheme 94 Transfer hydrogenation of 2-substituted quinolines 267to tetrahydroquinolines 268
Du and co-workers reported the enantioselective hydroge-nation of quinolines using a new type of axially chiralphosphoric acid catalyst 271 having a bis-BINOL scaffold(Scheme 95).88h Newly developed catalyst 271 showedhigher efficiency than the parent mono-BINOL-derivedphosphoric acid catalysts. The enantioselectivities ofalkyl and aryl 2-substituted quinolines 267 were uniform-ly high and the corresponding tetrahydroquinolines 268were obtained quantitatively even when the catalyst loadwas reduced to as little as 0.2 mol%. 2,3-Disubstitutedquinolines were also hydrogenated with high diastereo-and enantioselectivities.
Scheme 95 Transfer hydrogenation of quinolines 267 using newlydeveloped chiral phosphoric acid catalyst 271
(R)-1q (2 mol%)266
84–97%,80%,
95–98% ee98% ee
N
R
PMPHN
R
PMP
G = i-Pr
i-Pr
i-Pr
250 251
S
HN
266
R = aromaticR = cyclohexyl
mesitylene, 50 °C, 25–30 h
(R)-1m (1–5 mol%)249a (2.4 equiv)
benzene, 60 °C, 12–60 h
G =
N R NH
R
54–93%,88–95%,
R = aromatic:R = aliphatic:
(R = aromatic or aliphatic)91 to >99% ee
87–91% ee
NH
R N R
acid-catalyzedisomerization
1,4-hydrideaddition 1,2-hydride
addition
269 270
267268
N R1 NH
R1
(R1 = aromatic or aliphatic)267
268
(R,R)-271 (0.2 mol%)249d (2.4 equiv)
Et2O, 35 °C, 20 h
>99%,
>99%,
R1 = aromatic except ortho-substituentR1 = aliphatic
94–98% ee
88–95% ee
(R,R)-271
R2 =
O
OO
OP
O OHR2 R2

1966 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Rueping and Antonchick successively applied the cascadetransfer hydrogenation strategy to the enantioselective re-duction of pyridine derivatives (Scheme 96a).199 Catalyst1g functioned efficiently in the hydrogenation of pyridinederivatives 272 to furnish the corresponding products 273,hexahydroquinolinones, with high enantioselectivities.The method allows access to enantioenriched nitrogenheterocycles as useful precursors of various natural prod-ucts. Metallinos et al. reported the enantioselective reduc-tion of 2-substituted and 2,9-disubstituted 1,10-phenanthrolines 274 (Scheme 96b).200 Although consider-able amounts of meso-isomers were formed in the hydro-genation of 2,9-disubstituted 1,10-phenanthrolines 274b,dl-octahydrophenanthrolines 275b were obtained with ex-cellent enantioselectivities.
Scheme 96 Transfer hydrogenation of pyridine and 1,10-phenan-throline derivatives
Rueping et al. further extended the method to the hydro-genation of 3-substituted quinolines 276 (Scheme 97).201
Here, the key step is the enantioselective protonation ofintermediary enamine 277. The mechanisms of the stereo-determining step are entirely different from those of theasymmetric hydrogenation of 2-substituted quinolines267, where the 1,2-hydride addition step is the key to de-termining the stereochemical outcome (see Scheme 94).After thorough optimization of the phosphoric acid cata-lyst, 14b was found to be the best, giving 3-substituted tet-rahydroquinolines 279 with good enantioselectivities.
12.3 Transfer Hydrogenation of a,b-Unsaturated Carbonyl Compounds
Most organic transformations proceed via ionic interme-diates or transition states and hence are influenced by thecounterion, particularly in organic solvents, because ionpairs are ineffectively solvated in these media. Althoughefficient asymmetric catalytic transformations involvinganionic intermediates have been realized using chiral cat-ionic catalysts, such as phase-transfer catalysts,181 an anal-ogous version of inverse polarity was not accomplisheduntil recently. In this context, List and Mayer introducedthe concept of ‘asymmetric counteranion directed cataly-sis (ACDC)’ as a useful strategy in enantioselective orga-nocatalytic transformations152 and applied their ACDCconcept to a metal-free biomimetic transfer hydrogena-tion of a,b-unsaturated aldehydes 280,183 in which a bina-ry catalytic system of an achiral amine and a chiralphosphoric acid was employed (Scheme 98).152 Theyfound that the morpholine salt of 1q, prepared readily bysimple mixing, was a widely applicable and highly enan-tioselective catalyst for the transfer hydrogenation of a,b-unsaturated aldehydes 280. The catalytic salt of morpho-line and 1q generates the ion pair, I, of achiral iminiumcation and chiral phosphate anion, where the chiral phos-phate anion could induce the asymmetry in the process.a,b-Unsaturated aldehydes having aromatic substituentsat the b-position were reduced to their corresponding sat-urated aldehydes 281 in good yields with excellent enan-tioselectivities. The method is also applicable to a,b-unsaturated aldehydes with less sterically hindered ali-phatic substituents, the transfer hydrogenations of whichcould not be achieved by previous methods using chiralamine base catalysts in a highly enantioselective man-ner.183,184
List and Martin further applied their salt catalyst to theenantioselective hydrogenation of a,b-unsaturated ke-tones 282 (Scheme 99).202 Although, in the reduction of282, the morpholine/chiral phosphoric acid salt systemfailed to provide the corresponding saturated ketones 283in satisfactory yields and enantioselectivities, ammoniumphosphate, derived from (R)-1q and primary amine (S)-valine tert-butyl ester, proved to be effective for thepresent transfer hydrogenation. It is noteworthy that thesalt derived from the opposite enantiomer of 1q, namely,(S)-1q, and (S)-valine tert-butyl ester displayed a consid-
(R)-1g (5 mol%)249a (4 equiv)
benzene50 °C, 30–36 hN N
H
27366–84%, 87–92% ee
R
O O
R
G =a)
b)
N
N
R1
R2
(R)-1m (2 mol%)249a (6 equiv)
G =
benzene60 °C, 24 h
NH
NH
R1
R2
54%, 78–83% ee
72–88%,
275a: R1 = Me, n-Bu; R2 = H
275b: R1 = R2 = Me, n-Bu
dl/meso = 3:2, 99% ee for dl-isomer
272
274 275
Scheme 97 Transfer hydrogenation of 3-substituted quinolines totetrahydroquinolines
(R)-14b (5 mol%)249c (2.4 equiv)
benzene60 °C, 22–48 h
enantioselectiveprotonation
O
O
PO
OH
SiPh3
SiPh3
N NH
NH
N
1,4-hydrideaddition 1,2-hydride
addition
277278
276279
30–84%, 77–86% ee
Ar Ar
Ar Ar

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1967
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
erable decrease in yields and enantioselectivities. Notonly cyclic but also acyclic ketones 282 were reduced ef-ficiently in the presence of (R)-1q and (S)-valine tert-bu-tyl ester, giving saturated products 283 in good yields andenantioselectivities.
12.4 Application of Transfer Hydrogenation to Cascade Reaction
Cascade transformations involving different organocata-lytic processes have emerged as a powerful and efficientmethod for the rapid construction of complex moleculesstarting from simple materials in one pot.90 In particular,organocatalyses by amines or their salts were intensivelyapplied to cascade transformations, including enamine oriminium ion, as the reactive intermediate.203 List andZhou successfully combined chiral phosphoric acid catal-ysis with these amine catalyses based on enamine and imi-nium ion mechanisms (Scheme 100).204 They developed ahighly enantioselective synthesis of 3-substituted (hete-ro)cyclohexylamines 284 from 2,6-diketones 285 via athree-step organocatalysis,205 namely, (i) intramolecularaldol condensation via enamine 286 formation to give cy-clized product 287; (ii) 1,4-hydride addition under theiminium ion mechanism to give saturated imine 288; and(iii) 1,2-hydride addition cascade catalyzed by chiralphosphoric acid and accelerated by aniline substrate 12bor 12h. Acid catalyst 1q exhibited excellent performance
for the cascade transformation, giving the corresponding3-substituted (hetero)cyclohexylamines 284 with highenantio- and cis-selectivities. The reductive amination of3-substituted cyclohexanones, intermediate 288 of thepresent cascade, generally provides the correspondingtrans-isomer under standard reductive amination condi-tions. In contrast, in the present cascade transformation,cis-284 was obtained predominantly.
Scheme 100 Intramolecular aldol condensation/tandem transfer hy-drogenation cascade
Rueping and Antonchick developed an organocatalyticmultiple-cascade sequence using enamines 289 and a,b-unsaturated ketones 290 as substrates to obtain enantioen-riched nitrogen heterocycles 291 (Scheme 101).206 Themultiple-cascade sequence comprises Michael addition,geometrical isomerization, cyclization, dehydration,isomerization, and 1,2-hydride addition, in which eachsingle step is efficiently catalyzed by chiral phosphoricacid catalyst 1g. The method enables direct, rapid, and ef-ficient access to a diverse set of tetrahydropyridine andazadecalinone derivatives 291 with excellent enantio-selectivities.
13 Oxidations
13.1 Epoxidation
Catalytic asymmetric epoxidation occupies a privilegedposition in organic synthesis owing to the fundamentalimportance of enantioenriched epoxides as key syntheticintermediates. A number of efficient protocols for theenantioselective epoxidation using chiral metalcatalysts207 and organocatalysts208 have been reported todate. The Julia–Colonna epoxidation is one of the most ef-ficient synthetic methods for the enantioselective func-tionalization of a,b-unsaturated ketones using hydrogenperoxide under basic conditions.209 Since its discovery in
Scheme 98 Asymmetric counteranion directed organocatalytictransfer hydrogenation of a,b-unsaturated aldehydes 280
(R)-1q/morpholine (20 mol%)249e
1,4-dioxane, 50 °C, 24 hTHF, r.t., 24–96 h
280 281
O
O
PO
O
G
G
NH2
O
G = i-Pr
i-Pr
i-Pr
R
CHO
R
CHO
R = aromaticR = aliphatic
63–90%,71–77%,
96 to >98% ee90–92% ee
O P
O
O
O
1q
IR
N
H
O
Scheme 99 Transfer hydrogenation of a,b-unsaturated ketones 282
(5 mol%)249a
n-Bu2O, 60 °C, 48 h
282 283
(R)-1q
R1, R2 = cyclicR1, R2 = acyclic
68 to >99%,81 to >99%,
94–98% ee70–84% ee
H3N CO2t-Bu
i-PrO
R3R2R1
O
R3R2R1
G = i-Pr
i-Pr
i-Pr
+
(R)-1q (10 mol%)249a (2.2 equiv)
5 Å MScyclohexane, 50 °C, 72 h
(R = 2-naphthyl or aliphatic)(X = CH2, O, S)
35–89%67–99% cis, 82–96% ee
OX
O
R
X
HN
R
Ar
H2NAr
12b:12h:
OX
NHAr
R
X
NAr
RX
NAr
R
1,4-hydrideaddition
1,2-hydrideaddition
intramolecularaldol
condensation
enamineformation
285
286 287 288
Ar = 4-MeOC6H4
Ar = 4-EtOC6H4
12284

1968 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
1980, the enantioselective epoxidation of a,b-unsaturatedcarbonyl compounds has emerged as an efficient route forpreparing highly functionalized synthetic intermedi-ates.210
List et al. applied their ACDC concept152 to the develop-ment of the enantioselective epoxidation of a,b-unsaturat-ed aldehydes 292 using an ammonium phosphate saltcatalytic system (Scheme 102).211 They identified a newcatalytic salt that was derived from dibenzylamine deriv-ative 293 and phosphoric acid 1q, and functioned as an ef-ficient enantioselective catalyst for 1,2-disubstituted enals292a having aromatic substituents at the b-position. Thecorresponding epoxides 294a, with excellent trans- andenantioselectivities, were obtained in the presence of tert-butyl hydroperoxide (TBHP) as the oxidant, although anenal having an aliphatic substituent at the b-position ex-hibited a slight decrease in stereoselectivity. Notably, thissalt-catalyzed reaction is well suited for trisubstitutedenals 292b to afford trisubstituted epoxides 294b withhigh enantioselectivities, because these enals 292b havepreviously been elusive substrates for all types of enantio-selective epoxidation.
13.2 Baeyer–Villiger Oxidation
The Baeyer–Villiger reaction represents one of the mostfundamental and widely applied reactions in organic syn-thesis.212 However, the enantioselective Baeyer–Villigerreaction is largely unexplored in comparison with othercatalytic enantioselective transformations. In addition,there are only a few cases where aqueous hydrogen perox-ide is utilized as the terminal oxidant in the enantioselec-tive Baeyer–Villiger reactions, despite the distinctadvantage that its reduction product is water. Ding and co-workers successfully developed a highly enantioselectiveBaeyer–Villiger reaction of cyclobutanone derivatives295 using chiral phosphoric acid catalyst 14h in combina-tion with aqueous hydrogen peroxide as the oxidant(Scheme 103).213 A variety of 3-aryl- and alkyl-substitut-ed cyclobutanones 295 can be oxidized by 10 mol% 14hwith hydrogen peroxide. High enantioselectivities wereachieved in the reaction of cyclobutanones having aro-matic substituents. It is noteworthy that the value (R = Ph:88% ee) represents the highest enantioselectivity obtainedin the asymmetric Baeyer–Villiger oxidation of 295a(R = Ph) with chemical catalysts, although the oxidationof 295 having aliphatic substituents gave the correspond-ing g-butyrolactones 296 with modest enantioselectivi-ties. Ding, Li, and co-workers also elucidated themechanism of the Baeyer–Villiger oxidation on the basisof experimental and theoretical studies.213b The phospho-ric acid catalyst participates in both the carbonyl additionand the subsequent rearrangement steps in a synergisticmanner through hydrogen-bonding interactions. The cata-lyst simultaneously enhances the electrophilicity of thecarbonyl carbon via protonation and the nucleophilicity ofthe hydrogen peroxide through Brønsted basic site in theaddition step followed by the rearrangement step, inwhich the dissociation of the hydroxy group from theCriegee intermediate is also accelerated by the catalyst.
Scheme 101 Multiple-cascade sequence leading to tetrahydropyri-dine and azadecalinone derivatives 291
O R2
EWG
NH2R1
+
NH
EWG
R1 R2
(R)-1g (5 mol%)249a (1.1 equiv)
3 Å MSCHCl3 or benzene
50 °C, 12–18 h
G =
R1R2
OEWG
H2N
NH
EWG
R1 R2
Michaeladdition
cyclization&
dehydration
geometricalisomerization
289 290291
NH2
R2
OEWG
R1
N
EWG
R1 R2
1,2-hydrideaddition
isomerization
NH
NC
Ar NH
MeO2C
Ar
55–56%, 97–99% ee77–89%, 96–97% ee
R2 = aryl or n-pentyl47–78%, 89–99% ee
NH
Ar NH
R2
O O
42–54%, 97–99% ee
Scheme 102 Stereoselective epoxidation using ammonium phos-phate salt catalyst
(R)-1q/293 (10 mol%)TBHP
292a trans-294a
O
O
PO
OH
G
G
(R)-1q: G = i-Pr
i-Pr
i-Pr
ROHC
ROHC
R = aromaticR = n-hexyl
63–90%,67%,
84–96% ee70% ee
1,4-dioxane, 35 °C, 72 h
HN
CF3
CF3
CF3
F3C293
O
(R)-1q/293 (10 mol%)TBHP
292b 294bR
OHCR
OHC
75–85%, 90–94% ee
t-BuOMe, 0 °C, 24 h
OR R
(R = aliphatic)
98 to >99% trans,94% trans,

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1969
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Scheme 103 Baeyer–Villiger reaction of cyclobutanone derivatives295
13.3 a-Hydroxylation of 1,3-Dicarbonyl Com-pounds
The a-hydroxylation of 1,3-dicarbonyl compounds hasbeen well utilized, affording a-hydroxy carbonyl com-pounds that serve as key structural elements in a numberof natural products and pharmaceuticals. In this regard,considerable effort has been devoted to the establishmentof efficient methods for the preparation of a-hydroxy 1,3-dicarbonyl compounds in optically active forms. Howev-er, the development of catalytic enantioselective a-hy-droxylations of 1,3-dicarbonyl compounds has facedlimited success.214 Zhong and co-workers developed ahighly enantioselective a-hydroxylation of 1,3-dicarbonylcompounds 297 using chiral phosphoric acid catalyst 1i(Scheme 104).215 In general, a-hydroxylation reactionsare accomplished by using oxaziridines and peroxides asoxygen sources. Zhong and co-workers, however, em-ployed 4-chloronitrosobenzene (298) as the oxygensource.216 Chiral phosphoric acid catalyst 1i activates thenitroso compound via protonation of the basic nitrogenatom of the nitroso moiety and promotes bond formationbetween the oxygen atom and the a-carbon atom of 1,3-dicarbonyl compounds 297. Subsequent nitrogen–oxygenbond cleavage affords a-hydroxylation products 299 inoptically active forms. b-Keto esters 297a bearing a five-membered ring were well tolerated in the a-hydroxyla-tion, giving the corresponding products 299a with excel-lent enantioselectivities, irrespective of the steric demandof the ester moiety. Although in the reaction of 1,3-dike-tones 297b, a slight decrease in enantioselectivities wasobserved, the a-hydroxylation of 1,3-diketones wasachieved in high yields with good enantioselectivities forthe first time.
14 Combined Use of Metal Complex and Phos-phoric Acid Catalysts
In the past decade, tremendous progress has been made inthe field of catalysis using small organic molecules,namely, organocatalysis.1 Meanwhile, catalysis by transi-tion-metal complexes has continued to be applied to abroad range of organic transformations and it occupies aprivileged position in synthetic organic chemistry. Muchof the research on catalysis has centered on the use of met-al complexes to activate a variety of chemical bonds. Inrecent years, armed with the idea of taking advantage ofboth of these catalytic processes, researchers have com-bined metal complexes and organic molecules to con-struct binary catalytic systems,217 and this has attractedmuch attention as it has the potential to realize unprece-dented transformations. Recently, several excellent ap-proaches were established with the chiral phosphoric acid/metal complex binary catalytic system in catalytic enanti-oselective transformations. There are two types of combi-nation in the binary catalytic system. One is that eachreactant is activated simultaneously by one type of cata-lyst; thus, the metal complex activates nucleophiles, whilethe phosphoric acid activates electrophiles in a coopera-tive manner. The other is the consecutive transformationusing a binary catalytic system, that is, relay catalysis fora multi-step sequence in which each catalyst promotes onetype of reaction in a one-pot sequential manner.
14.1 Cooperative Catalysis by Metal Complex and Phosphoric Acid
Krische and Komanduri developed a highly enantioselec-tive reductive coupling reaction of 1,3-enynes with het-eroaromatic aldehydes and ketones catalyzed by a chiralrhodium complex under hydrogenation conditions.218 Inthe course of their studies, they found that a Brønsted acidco-catalyst accelerated the rate of the reaction significant-ly. In order to elucidate the role of the Brønsted acid co-catalyst, they attempted to use a combination of achiralrhodium complex 300 with chiral phosphoric acid 1q inthe reductive coupling between 2-pyridinecarboxalde-hyde (301) and 1,3-enyne 302 (Scheme 105). The product303 was obtained in an optically active form, although theenantioselectivity obtained in this combination was lowerthan that observed in the parent combination of the chiralrhodium complex and the achiral Brønsted acid co-cata-lyst. This result indicates that the rhodium complex andthe Brønsted acid functioned as the cooperative catalyst.Thus, 1q protonates 2-pyridinecarboxaldehyde (301) togive intermediate J in advance of the carbon–carbonbond-forming step, hence accelerating the coupling reac-tion because of the lowered LUMO of aldehyde 301. Fur-thermore, the enantioenrichment of product 303 suggeststhat the chiral counteranion of 1q is incorporated into thetransient assembly of the coupling reaction through hy-drogen-bonding interactions. The authors disclosed thatthe protonation of 301 by 1q is responsible for the asym-metric induction and not the ion pairing to the rhodium
O
O
PO
OH
G
G
(R)-14h: G =
+
(R)-14h(1–10 mol%)
CHCl3–40 °C, 18–80 h295
R O aq H2O2
296
OO
R
R = aromaticR = aliphatic
91–99%,99%,
82–93% ee55–58% ee
Scheme 104 a-Hydroxylation of 1,3-dicarbonyl compounds 297 by4-chloronitrosobenzene (298)
O
R1
O
X
(R)-1i(1 mol%)
benzene, 4 °C, 22 h
G = Ph3Si
O
R1
O
XOH
R1 = OR2
R1 = Me73–85%,65–75%,
92 to >98% ee80–86% ee
Cl NO
+
297a:b:
298
299
R1 = OR2
R1 = Me
299a: 299b:

1970 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
complex, on the basis of these results coupled with theirprevious studies.219
Rueping et al. developed an excellent cooperative processthat comprised an enantioselective Brønsted acid and sil-ver complex catalyzed alkynylation (Scheme 106).220 Theenantioselective alkynylation under binary catalytic con-ditions was accomplished in the reaction of a-imino ester2h with a series of aryl-substituted terminal alkynes 304,in which the a-imino ester and the alkyne were activatedby phosphoric acid 1m and the silver complex, respective-ly. a-Amino acid products 305 having an alkynyl substit-uent were obtained in good yields with highenantioselectivities. Although, from a mechanistic view-point, an exchange of the metal counteranion from acetateto chiral phosphate, leading to the formation of a chiralsilver complex, cannot be ruled out, both catalysts are re-quired for the reaction to proceed, and, more importantly,the reaction that proceeded under the influence of a chiralsilver complex and an achiral phosphoric acid catalyst re-sulted in racemic products 305. As shown in reactive in-termediate K, the simultaneous activation of both a-iminoester 2h and alkyne 304 by cooperative organic and me-tallic catalyses is crucial in furnishing a-amino ester prod-ucts 305 in optically active forms.
Hu, Gong, and co-workers developed a system that in-volved cooperative catalysis by chiral phosphoric acid 1mand achiral rhodium complex 306 (Scheme 107).221 Theyapplied the binary catalytic system to a three-componentcoupling reaction among a-diazoesters 307, primary alco-hols, and aldimines 2. The steric bulk of the primary alco-hol had a significant effect on both diastereo- andenantioselectivities. The sterically demanding 9-an-thracenemethanol (308) was the best component to giveb-amino-a-alkoxy esters 309 with excellent stereoselec-tivities under cooperative catalysis by phosphoric acid 1mand rhodium complex 306. Hu and co-workers further ap-plied this cooperative catalysis approach to a four-compo-
nent coupling reaction among a-diazoesters, primaryalcohols, aromatic aldehydes, and aniline derivatives.222
The enantioselective reduction of imines provides directaccess to chiral amines, one of the most important func-tionalities in fine chemical and pharmaceutical com-pounds. As shown in Schemes 86–97, the organocatalyticapproach has been established to provide an efficientroute to the enantioselective transfer hydrogenation ofketimines using Hantzsch ester 249 as the hydrogensource. However, in the hydrogenation of ketimines usingchiral metal catalysts, this transformation remains chal-lenging, particularly in the case of acyclic ketimines 250.Xiao and co-workers successfully developed a binary cat-alytic system for the enantioselective hydrogenation ofacyclic ketimines 250, where diamine-ligated iridiumcomplex 310, in combination with chiral phosphoric acid1q, enabled the highly enantioselective hydrogenation of250, affording the corresponding amines 251 in goodyields with excellent enantioselectivities (Scheme 108).223
Notably, the preformed iridium complex 311, preparedfrom 310 and 1q, functioned as an efficient catalyst with-
Scheme 105 Rhodium complex/chiral phosphoric acid cooperativecatalysis for reductive coupling between heteroaromatic aldehyde301 and 1,3-enyne 302
(R)-1q (4 mol%)
H2 (1 atm), DCE40 °C, 3 h
+
Ph
O
HN
OH
N
Ph
301
302
G = i-Pr
i-Pr
i-Pr
Rh(cod)2OTf/BIPHEP300
(4 mol% each)
30356%, 82% ee
OP
O
O
O
1q
J
O
HN
H
Scheme 106 Silver complex/chiral phosphoric acid cooperative ca-talysis for enantioselective alkynylation of a-imino ester 2h
G =N
HR1O2C
PMP
R2
(R)-1m (10 mol%)AgOAc (5 mol%)
toluene, 30 °C, 10–12 h+
HN
MeO2C
PMP
R2305
60–93%, 86–92% ee
N
HMeO2C
PMP
P
O
O
O O
R2
[Ag]
+
2h (R1 = Me)
304 (R2 = aromatic)
H
K
Scheme 107 Rhodium complex/chiral phosphoric acid cooperativecatalysis for three-component coupling reaction
+N
Ar2HCO2R
N2Ar1 Ar3
OH
+
307(Ar1 = aromatic)(R = Me, Et)
2f:2p:
Ar2 = aromatic, Ar3 = PMPAr2 = aromatic, Ar3 = Ph
G =
(R)-1m (2 mol%)
4 Å MSCH2Cl2, –20 °C, 4 h
82–98%, >99% dr, 84 to >99% ee
HN
Ar2Ar1
O CO2R
Ar3
Ar4
308
309(Ar4 = 9-anthryl)
Rh2(OAc)4 (2 mol%)306

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1971
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
out any detrimental effect on the enantioselectivity. Moreinterestingly, the addition of 1q to preformed 311 marked-ly enhanced the catalytic activity with higher conversionbeing observed at 1 mol% each of 311 and 1q. Xiao andco-workers further applied the present cooperative cataly-sis to the enantioselective reductive amination reaction ofketones with aniline derivatives under hydrogenation con-ditions.224
Scheme 108 Iridium complex/chiral phosphoric acid cooperativecatalysis for enantioselective hydrogenation of ketimines 250
Xiao and co-workers established the present cooperativecatalysis by taking into consideration Norton’s mechanis-tic studies on ruthenium-catalyzed ionic hydrogenation ofiminium cations where imines can be reduced through anionic pathway,225 while it is generally believed that themetal-catalyzed hydrogenation proceeds via the coordina-tion between the nitrogen atom of imine and the metalcenter. Hydrolytically hydrogen-activating catalyst 312 isassociated with chiral counteranion 1q–, which influencesthe enantio-discrimination by ion pairing with resultingiminium cation 313 (Scheme 109). In fact, the enantiose-lectivity observed was significantly affected by the chiralphosphoric acids employed.
As shown in Schemes 106– 108, one type of catalyst acti-vates each reactant simultaneously: the phosphoric acidactivates imines as the electrophilic component while themetal complex activates nucleophiles in a cooperativemanner.
List and Mukherjee developed the enantioselective a-ally-lation of a-branched aldehydes 314 using cooperative ca-talysis (Scheme 110).226 The transformation of 314 withallylamine derivatives 315 was mediated in a highly enan-tioselective manner under the influence of chiral phospho-ric acid 1q with achiral palladium complex 316,Pd(PPh3)4. The reaction likely proceeded through reactiveintermediate L, including a p-allyl palladium complex, anenamine, and chiral phosphate counteranion 1q–, to affordthe corresponding chiral aldehydes 317 bearing a quater-nary stereogenic center with high enantioselectivities. Inthis transformation, the chiral phosphate counteranion ac-tivates the nucleophilic enamine and this activation modeis in contrast to the previous cooperative catalyses wherethe phosphoric acid activates electrophilic imines (seeSchemes 106–108). The method enables efficient accessto the enantioselective construction of all-carbon quater-nary stereogenic centers, which represents an importantyet considerable challenge in organic synthesis.227 Thesame authors also demonstrated a concise synthesis of(+)-cuparene by taking advantage of this method for pre-paring 317a as the key building block (Scheme 111).228
Scheme 110 Palladium complex/chiral phosphoric acid cooperativecatalysis for a-allylation of a-branched aldehydes 314
Scheme 111 Synthetic application of palladium complex/chiralphosphoric acid cooperative catalysis
NH
IrN
S
Ar
O OPh
Ph
G = i-Pr
i-Pr
i-Pr
310 (1 mol%) / (R)-1q (6 mol%)or
311 (1 mol%) / (R)-1q (1 mol%)N
MeR
PMPHN
MeR
PMP
*250 251
H2 (20 atm)toluene, 20 °C, 15–20 h
92–96%,88–91%
60%,
R = aromaticR = aliphaticR = Ph
84–98% ee92–95% ee
97% ee
NH2
IrN
S
Ar
O OPh
Ph
310
310 (1 mol%) / (R)-1q (6 mol%):
311 (1 mol%) / (R)-1q (1 mol%):
X
311 (X = 1q )Ar = 2,4,6-(i-Pr)3C6H2
Scheme 109 Cooperative catalysis by iridium complex/chiral phos-phoric acid binary system
L2Ir
L1
* 1q + H2
L2Ir
L1
*
250 O
P OO
O
N
MeR
H PMP
313312
1q
Cp*
Cp*
H311
(R)-1q (1.5 mol%)
5 Å MSt-BuOMe, 40–60 °C, 8–72 h
G = i-Pr
i-Pr
i-Pr
Pd(PPh3)4 (3 mol%)316
R1
CHO
NH
R2
Ph
Ph
314
315
R2
R1
CHO
+2 M HCl
Et2Or.t., 30 min
71–89%,40–82%,
65%,
R1 = aromaticR1 = PhR1 = cyclohexyl
317
94–97% ee82–92% ee
70% ee
PO
OO
L
R1
NH
O
R3
H
PdR2
R2 = HR2 = Me, PhR2 = H
CHO
317a
Rh(dppe)2Cl(1 mol%)
p-xylene, 160 °C, 12 h30%
p-tol
O
p-tol
(ref. 228)1 step
(+)-cuparene

1972 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
14.2 Relay Catalysis by Metal Complex and Phos-phoric Acid
As shown in Schemes 105– 108 and 110, each reactantwas activated by one type of catalyst simultaneously.Terada and Sorimachi reported an unprecedented consec-utive transformation using a binary catalytic system, thatis, relay catalysis for the tandem isomerization/carbon–carbon bond-formation sequence promoted by a binarycatalyst consisting of ruthenium hydride complex 318 andBrønsted acid 319 (Scheme 112).229 The reaction of N-Boc-protected allylamine 320 with 2-methoxyfuran (52)proceeded smoothly to give product 321, with a nitrogenfunctionality at the stereogenic center, in good yield. Con-trol experiments revealed that both catalysts, rutheniumcomplex 318 and Brønsted acid 319, are indispensable tothe sequential processes. Although a racemic acid catalystwas employed in this case, an enantioselective versionwould also be applicable, considering recent progress inenantioselective Friedel–Crafts reactions using chiralphosphoric acid catalysts (see Sections 5.1 and 9.1).
Scheme 112 Relay catalysis by ruthenium complex/Brønsted acidbinary system
The present sequential transformation involves a three-step relay catalysis where: (i) the isomerization of N-allyl-carbamate 320 to enecarbamate 189a is catalyzed by theruthenium hydride complex 318; (ii) the subsequentisomerization of 189a to intermediary imine 191a is re-layed by acid catalyst 319; and (iii) the catalytic sequenceis terminated by a carbon–carbon bond-forming reactionof 191a with electron-rich aromatic compounds 52 as anucleophilic component under the influence of acid cata-lyst 319. The advantage of this relay catalysis is that themethod enables the generation of reactive imines 191from readily available allylcarbamates in a one-pot reac-tion via tandem isomerization. Terada and Sorimachi alsoapplied the present relay catalysis to the reaction of a 1,3-
dicarbonyl compound, instead of 2-methoxyfuran (52), asthe nucleophilic component (see Section 9.1). The corre-sponding Mannich product was obtained in an acceptableyield.
The enantioselective version of the relay transformationby organic and metallic catalyses was successfully dem-onstrated by Gong and co-workers (Scheme 113).230 Theyaccomplished the direct transformation of o-propargyl-aniline derivatives 322 into tetrahydroquinolines 268 in ahighly enantioselective manner through the hydroamina-tion of alkynes/isomerization/enantioselective transferhydrogenation (see Section 12.2) sequence under the relaycatalysis of an achiral gold complex 323/chiral phospho-ric acid 1m binary system. A control experiment usingchiral gold phosphate, prepared from the silver salt of 1mand Ph3PAuCl, showed that the reaction was incompleteand the enantioselectivity was lower than that obtained inthe binary catalytic system. Therefore, it can be excludedthat the chiral gold phosphate, even if it were generated insitu, functions as the dominant catalytic species at the laststep, namely, the enantioselective transfer hydrogenationof 270. Instead, chiral phosphoric acid 1m plays the dom-inant role in the actual enantioselective catalyst.
Scheme 113 Enantioselective relay catalysis by gold complex/chir-al phosphoric acid binary system for tetrahydroquinoline synthesis
Meanwhile, Che and Liu reported an intermolecular ver-sion of Gong’s relay catalysis, that is, the hydroaminationof alkynes/isomerization/enantioselective transfer hydro-genation sequence (Scheme 114).231 The intermolecularrelay catalysis was accomplished via a three-componentreaction among terminal alkynes 304, aniline derivatives12, and Hantzsch ester 249a in the binary catalytic systemcomposed of gold complex 324 and chiral phosphoric acid1q. This relay catalysis exhibited a very broad substratescope toward diversely substituted chiral amines 325 ingood yields with high enantioselectivities.
O
O
PO
OH
Ph
Ph
1st isomerization[RuH] 318
HN
Boc
2nd isomerizationBrønsted acid 319
NBoc
C–C bond formationBrønsted acid 319
HN
Boc
+toluene, 40 °C, 12 h
(±)-319 (5 mol%) HN
Boc
O
OMe
O
OMe
189a 191a
69%320
52
321
RuClH(CO)(PPh3)3 (1 mol%)318
52
gold-catalyzedhydroamination
acid-catalyzedisomerization
acid-catalyzedenantioselective
transfer hydrogenation
toluene, r.t., 16–27 h
(R)-1m (15 mol%)
NH2
R2R1
NH
R2
R1
G =
NH
EtO2C CO2Et
26882 to >99%, 88 to >99% ee
NH
R2
R1N
R2
R1
249a
322
249a
+
(Ph3P)AuMe (5 mol%)323
269 270

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1973
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Scheme 114 Relay catalysis by gold complex/chiral phosphoricacid binary system for amine synthesis
Shortly thereafter, Dixon and co-workers reported a bina-ry catalytic system composed of gold complex 326 andchiral phosphoric acid 1i for a one-pot sequential transfor-mation of alkynoic acids 327 and tryptamines 328(Scheme 115).232 The initial step of this sequential trans-formation is the gold-catalyzed cycloisomerization of327, affording substituted furanones 329. In a previouspublication, it was pointed out that this initial cyclo-isomerization is not promoted by Brønsted acid cata-lysts.233 Subsequent steps were conducted by addingtryptamines 328 and phosphoric acid catalyst 1i in a one-pot procedure; nucleophilic ring opening of furanone de-rivatives 329 by tryptamines 328 led to acyclic ketoamideintermediate 330, and this was followed by dehydrativerecyclization to afford enamides 331. Resulting enamides331 generated N-acyl iminium ions 332 as the reactive in-termediate under the influence of chiral phosphoric acidcatalyst 1i and, following Pictet–Spengler-type ring clo-
sure, polycyclic tetrahydro-b-carboline derivatives 333were formed in good yields with high enantioselectivities.
You and co-workers combined olefin cross-metathesiswith intramolecular Friedel–Crafts reaction in a one-potsequence using ruthenium complex 334 and chiral phos-phoric acid 1m (Scheme 116).234 This sequential transfor-mation involves two distinct catalytic cycles: the firstcycle is the olefin cross-metathesis between aryl vinyl ke-tones 335 and allyl indolylmethyl ethers 336a by rutheni-um complex 334, giving rise to enones 338, and thesubsequent catalytic cycle involves the intramolecularFriedel–Crafts reaction of 338 under the influence ofchiral phosphoric acid 1m. This binary catalytic processindeed worked well in the reaction of 335 with 336a to af-ford the corresponding cyclized products 337a in goodyields with high enantioselectivities. However, this meth-od affords products 337a with slightly lower enantioselec-tivities than that observed in the chiral phosphoric acidcatalyzed intramolecular Friedel–Crafts reaction of 338a(e.g., 337aa: R1 = Me, R2 = H, Ar = Ph, 94% ee vs. 96%ee), presumably owing to the generation of Lewis acidicruthenium complex 339 during the course of the first cat-alytic cycle.235 Thus, the activation of enones 338 byachiral ruthenium complex 339 leads to undesired racem-ic product 337a, although this achiral catalytic process isnot a dominant cycle because of little loss of enantioselec-tivity during the sequential catalysis. The method is alsoapplicable to the construction of the tetrahydro-b-carbo-line skeleton: the reaction of nitrogen analogue 336b withphenyl vinyl ketone (335a) provided cyclized product
R
+
HN
R
70–98%,54–98%,
12
304
H2NX
X(t-Bu)2(o-biphenyl)PAuOTf (1–2 mol%)324
(S)-1q (5–10 mol%)
5 Å MSbenzene, 40–60 °C, 40–120 h
G = i-Pr
i-Pr
i-Pr
R = aromaticR = aliphatic
325
83–96% ee83–95% ee
+
249a
Scheme 115 Relay catalysis by gold complex/chiral phosphoricacid binary system for polycyclic tetrahydro-b-carboline synthesis
33377–96%, 83–95% ee
O
NH
O
R329
N
R
O
X
NH
NH2
X
328
NH
NH
X
O
R
O
330 331
HO2C
R
327(R = aliphatic)
NH
X
NO
R
NH
N
R
O
X
332
(PhP3)AuCl (0.5 mol%)326
AgOTf (0.5 mol%)toluene, r.t., 1 h
then 328 and (R)-1i (G = Ph3Si) (10 mol%)
80 °C, 24 hthen 110 °C, 24 h
gold-catalyzedcycloisomerization
1i
acid-catalyzedenantioselectivePictet–Spengler-
type reaction
Scheme 116 Relay catalysis by ruthenium complex/chiral phospho-ric acid binary system in olefin cross-metathesis/intramolecularFriedel–Crafts reaction sequence
G =
(S)-1m (5 mol%)334 (5 mol%)
+toluene
40–60 °C, 40–120 h
N
X
R1
R2
N
X
R1
R2
N
R1
R2
X
Ar
O
O
Ar
336
338
a: X = Ob: X = NBoc
Ar
O
335
a: X = Ob: X = NBoc
337
N N
Ru
Mes Mes
O SO2NMe2
Cl
Cl
334
43–97%,90%,
80–94% ee83% ee
337a:337b:
N N
Ru
Mes Mes
Cl
Cl
339
Mes =
a: X = Ob: X = NBoc

1974 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
337b (R1 = Me, R2 = H, Ar = Ph) in good yield, albeitwith a slight decrease in enantioselectivity.
15 New Aspects in the Development of Chiral Brønsted Acid Catalysts
Since the development of chiral phosphoric acids andtheir analogues as enantioselective catalysts, continuouseffort has been made to cultivate novel structural motifs ofchiral Brønsted acid catalysts having acidic functional-ities other than phosphoric acid, and several efficientchiral acid catalysts have been reported to date. This sec-tion reviews recent progress in the development of chiralBrønsted acid catalysts, especially those bearing stronglyacidic functionalities.
15.1 Chiral Brønsted Acid Catalysts Other Than Phosphoric Acid Catalysts
One of the key principles in the design of chiral Brønstedacid catalysts is the choice of a catalyst having an appro-priate acidity. Although an important class of moderateBrønsted acids, carboxylic acids, is often utilized as oneof the most fundamental acid catalysts in a variety of or-ganic transformations, these carboxylic acids have rarelybeen employed in enantioselective catalyses,236 presum-ably because of the poor catalytic activity of the carboxy-lic acid and the difficulty of constructing an efficientchiral environment around it. Maruoka and Hashimotoovercame these intrinsic problems by introducing two car-boxylic acid moieties in the catalyst molecule. They de-veloped novel chiral Brønsted acid catalysts 340, whichconsist of two carboxylic acids and an axially chiral bi-naphthyl moiety (Scheme 117).237 Dicarboxylic acid 340afunctioned as an efficient enantioselective catalyst in theMannich-type reaction of N-Boc imines 2a with diazo-acetate 16a, affording the corresponding products 341with high enantioselectivities. They also applied dicar-boxylic acid catalysts 340 to the enantioselective aziridi-nation of diazoacetamides with N-Boc imines238 and theenantioselective imino azaenamine reaction of arylalde-hyde N,N-dialkylhydrazones with N-Boc imines.239
Scheme 117 Mannich-type reaction of N-Boc imines 2a with diazo-acetate 16a, catalyzed by a novel chiral Brønsted acid having two car-boxylic acids
Combined acid and base salts, such as pyridinium p-tolu-enesulfonate (PPTS), are one of the most utilized acid cat-alysts in organic synthesis. These acid–base salts haveseveral advantages over single-molecule catalysts interms of flexibility in the design of their dynamic com-plexes. Ishihara and co-workers focused their attention onthe superior properties of these acid–base combined or-ganic salts and developed novel chiral Brønsted acids de-rived from binaphthyldisulfonic acid 342 and pyridinederivatives 343 (Scheme 118).240 The salt catalyst gener-ated in situ from 342a (G = H)241 and 2,6-diphenylpyri-dine (343a) exhibited excellent performance in the directMannich reaction of N-Cbz imines 2q with acetylacetone(3), affording the corresponding products 344 with highenantioselectivities. The distinct advantage of the salt cat-alyst is that both the Brønsted acidity and the bulk can bereadily controlled by achiral pyridine derivatives 343without substituents at the 3,3¢-positions of the binaphthylbackbone (G = H). In fact, catalytic activities and enantio-selectivities were markedly dependent on the achiral pyri-dine 343 employed. The use of pyridine derivative 343b,with its sterically bulky tert-butyl substituents at the 2,6-positions, resulted in a considerable decrease in chemicalyield with slightly lower enantioselectivity (76% ee). Theimportance of the salt formation with the pyridine deriva-tive is obvious, because a significant decrease in enantio-selectivity (to only 17% ee) was observed in the absenceof pyridine derivatives, despite furnishing 344a in goodyield.
Scheme 118 Mannich reaction of N-Cbz imines 2q with acetylace-tone (3) catalyzed by chiral disulfonic acid/pyridine derivative com-bined organic salts
Strong chiral Brønsted acids have great potential for high-performance enantioselective catalysis in general and areparticularly promising for the activation of important butless basic substrate classes, such as aldehydes. List andco-workers reported that a disulfonimide is a promisingacid catalyst for the activation of simple aldehydes(Scheme 119).242 They identified that the newly devel-oped chiral cyclic disulfonimide 345, with a binaphthyl
Ar
N
H
Boc
Ar
HN
CO2t-Bu
N2
+
(R)-340a(5 mol%)
Boc
4 Å MSCH2Cl2
0 °C, 5–72 h
N2
H CO2t-Bu
340a: G =
16a2a
G
G
(R)-340
COOH
COOH
34138–89%, 85–96% ee
t-Bu
Ar
N
H
Cbz
SO3H
SO3H
2q
+
NH
Ar
Cbz
O
O
G = H(R)-342a (1 mol%)
343a (2 mol%)
34491–99%, 89–98% ee
O
O
(R)-342a: G = H
3
NR R
343a: R = Phb: R = t-Bu
MgSO4
CH2Cl2, 0 °C, 30 min
(R)-342a (5 mol%)/343a (10 mol%)(R)-342a (5 mol%)/343b (10 mol%)(R)-342a (5 mol%)/ –
74%32%81%
92% ee76% ee17% ee
G
G
acid–base combined salt catalysts
yield of344a (Ar = Ph)

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1975
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
backbone, functioned as a powerful enantioselective cata-lyst for the Mukaiyama aldol reaction of aldehydes 11with ketene silyl acetals 346.
Scheme 119 Mukaiyama aldol reaction catalyzed by chiral disul-fonimide 345
Their basic idea in the design of chiral disulfonimide cat-alysts originates from the resemblance in relative reactiv-ity between triflic acid (TfOH) and triflylimide (Tf2NH),which are both powerful Brønsted acid catalysts: TfOH(pKa –5.9 in water) is much more Brønsted acidic thanTf2NH (pKa 1.7 in water); however, the relationship is re-versed if one regards the Lewis acidity of the correspond-ing silylated species, with that of TMSNTf2 being muchstronger than that of TMSOTf.243 Furthermore, the C2-symmetric binaphthyl disulfonimide topology is of partic-ular interest as it differs from that of the correspondingpseudo-C2-symmetric phosphoric acids, and hence thecorresponding silylated species may serve as promisingchiral Lewis acid catalysts. Indeed, a comparison of phos-phoric acid 1r and phosphoramide 66h with disulfonicacid 342b and disulfonimide 345 revealed that disulfon-imide catalyst 345 was not only far more active than thealternative catalysts, 1r, 66h, and 342b, but also providedproduct 347a with high enantioselectivity in the Mukaiyamaaldol reaction of 11b with 346a (Scheme 120). List andco-workers concluded that the Lewis acid mechanismseems to be more plausible than the Brønsted acid cataly-sis in this Mukaiyama aldol reaction.242 Thus, sulfonimidecatalyst 345 is first silylated by ketene acetal 346, afford-ing N-silyl disulfonimide 348 that is responsible for theactive enantioselective catalyst through the O-silylationof aldehyde 11. They also pointed out that N-silyl disul-fonimide 348 represents a powerful catalyst for enantiose-lective silicon catalysis and a number of reactionscatalyzed by TMSNTf2, and similar catalysts should beaccessible in an enantioselective fashion.
15.2 Chiral Conjugate Acid Catalysts Derived from Chiral Bases
Most of the chiral Brønsted acid catalysts developed canbe classified as nonionic compounds.2 In contrast, the hy-drogen-bonding donor capability of a cationic organicmolecule with a less coordinatable anion, such as tetra-arylborate, has been well investigated. This type of chiral
Brønsted acid is generated as the conjugate acid of a chiralorganic Brønsted base. However, the development of cat-ionic Brønsted acid catalysts for enantioselective transfor-mations has been largely unexploited in comparison withthat of nonionic chiral Brønsted acid catalysts.244 Jacobsenand Uyeda successfully developed a cationic chiralBrønsted acid catalyst that was furnished from a chiralguanidine base (Scheme 121).245 Thus, chiral guanidini-um ion 349 having a fluorinated tetraarylborate as thecounteranion functioned as an efficient enantioselectivecatalyst for the Claisen rearrangement of ester-substitutedallyl vinyl ethers 350. Notably, the enantioselectivity isstrongly dependent on the substrate employed; introduc-tion of the ester substituent on the allyl vinyl ethers is in-dispensable for achieving high enantioselectivity,although the guanidinium catalyst induces rate enhance-ments in the rearrangement of a variety of substituted allylvinyl ethers. Substrates having an alkyl group (Me or Et)at the vinyl terminus afforded rearranged products 351with high enantioselectivities. Although the introductionof substituents at the allyl terminus resulted in a slight de-crease in enantioselectivities, not only anti-isomers butalso products having a quaternary stereogenic center wereobtained with high diastereoselectivities.
Meanwhile, Ooi and co-workers designed and synthe-sized novel cationic chiral Brønsted acid catalyst 352(Scheme 122).246 Their strategy for the molecular designwas to employ a chiral P-spiro tetraaminophosphoniumcation framework as the primary structure, which is aunique structural motif introduced by their researchgroup,247 where the core structure of HN-P+-NH interactswith electrophiles through two hydrogen bonds that seemto regulate the relative location between the electrophile
70–98%,46–95%,
R1
O
H
SO2
SO2
11
+
OSiR43
R1CO2R3
(R)-345(0.01–2 mol%)
347346(R2 = Me or H)
Et2O, –78 °C, 12–24 hR2
R2
OR3
OSiR43
G
G
NH (R)-345: G =
CF3
CF3
R2 R2
R1 = aromaticR1 = aliphatic
72–94% ee50–94% ee
Scheme 120 Mukaiyama aldol reaction catalyzed by various chiralacid catalysts
<2%,<2%,<2%,
>99%,
β-Nap
O
H
SO2
SO2
11b
+
OSiMe3
β-NapCO2Me
catalyst(2 mol%)
347a346a
Et2O, r.t., 12 hOMe
OSiMe3
G
G
NSiMe3 (R)-348: G =
CF3
CF3
---
80% ee
G
G
SO3H
SO3H
O
O
PO
NHTf
G
G
(R)-1r
G =
CF3
CF3
(R)-66h
(R)-342b
catalyst(R)-1r(R)-66h(R)-342b(R)-345
O
O
PO
OH
G
G

1976 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
and the chiral catalyst. In addition, they introduced a bi-naphthyl-derived axially chiral diamine as a readily acces-sible and modifiable chiral source, instead of chiralaliphatic diamines,247 to the phosphorus center by takinginto consideration the enhancement of the acidity of NHprotons. A newly developed arylaminophosphonium cat-ion (R,S)-352, with the [7.7]-spirocyclic core, exhibitedexcellent catalytic activity in the conjugate addition ofaniline derivative 12i to nitroalkenes 79, affording prod-ucts 353 in good yields. The enantioselectivity is marked-ly dependent on the axial chiralities of the diaminesintroduced; switching the unsubstituted binaphthyl unit tothe opposite enantiomer, which gives rise to homochiralarylaminophosphonium cation (R,R)-354a, results in aconsiderable decrease in the enantioselectivity [(R,S)-352a: 83% ee vs. (R,R)-354a: 61% ee]. Heterochiral cata-lyst 352b, with its 3,4,5-trifluorophenyl groups, facilitat-ed the reaction, affording 353a (R = Ph) in nearlyquantitative yield with the highest enantioselectivity. Abroad range of nitroalkenes 79, including aromatic, het-eroaromatic, and aliphatic ones, can be transformed intothe corresponding products 353 in high yields with highenantioselectivities in the presence of catalyst 352b.
16 Conclusions
This review focused on recent advances in the chemistryof chiral Brønsted acid catalysis using BINOL-derivedphosphoric acids of general type 1 for enantioselectivetransformations. In initial studies, chiral phosphoric acidswere utilized for the activation of imines in most cases. Asintroduced in this review, it has been convincingly dem-onstrated that tremendous progress has been made in thedevelopment of chiral phosphoric acid catalysis and thebroadening of the scope of functionalities that can be ac-tivated by chiral phosphoric acid catalysts and their ana-
logues. Numerous bond-forming reactions, includingcarbon–carbon, carbon–heteroatom, and carbon–hydro-gen bond-forming reactions, have been successfully es-tablished in a highly enantioselective manner by usingthese acid catalysts. However, the enantioselective ver-sions of many reactions remain to be developed usingchiral phosphoric acids or other types of chiral Brønstedacids. Further elaboration of novel chiral Brønsted acidsderived not only from other types of chiral backbones,such as diols, diamines, and amino alcohols, but also fromstronger acid functionalities could lead to the discovery ofeven more selective and efficient methods for a wide va-riety of enantioselective organic transformations.
Note Added in Proof
After acceptance of this review article, Ishihara and co-workers re-ported the direct Mannich reaction of N-Boc imine 2aa with ace-tylacetone (3) using chiral phosphoric acids and their metal saltderivatives, where calcium salt of chiral phosphoric acid (R)-1d(G = 4-b-naphthylphenyl) functioned as an efficient catalyst toyield the corresponding product (R)-4a with high enantioselectivi-ty.248 The result obtained with the calcium salt of (R)-1d is compa-rable to that using (R)-1d which was purified by silica gel (the resultshown in Scheme 1). However, an HCl wash of the silica gel puri-fied (R)-1d resulted in the formation of 4a with low and oppositeenantioselectivity. They also reported that chiral phosphoric acid(R)-1g (G = 9-anthryl) washed with HCl exhibited good perfor-mance in the direct Mannich reaction, giving rise to (S)-4a with highenantioselectivity under optimal reaction conditions.
Scheme 121 Claisen rearrangement catalyzed by chiral guanidini-um ion 349
hexanes22–40 °C, 5–14 d
349(20 mol%)
NH
NH
NH2
NNPh Ph
BArF
BArF B
CF3
CF3 4
=
MeO2C
O
R3
R1
349
350
MeO2C
O
R1
R4
R2
R2 R3
R4
351
R1
Me or EtEtMeEtEt
R2
Hn-Pr or Ph
HMe
Ph or alkyl chain
R3
HHH
MeMe
R4
HH
MeHH
92% ee81–85% ee
96% ee81% ee
82–84% ee
80–86%,91–92%,
73%,89%,
73–89%,
anti/syn = >19:<1 (R2 ≠ R3)
Scheme 122 Conjugate addition of aniline derivatives 12i to nitro-alkenes 79 catalyzed by arylaminophosphonium cation
87%,86%,98%,
89–99%,93–98%,
+
83% ee61% ee94% ee
91–97% ee86–87% ee
352b: G =
F
F
N
N
G
G
N
N
P
H H
H HF
(R,S)-352
(R) (S)
N
N
Ph
Ph
N
N
P
H H
H H
(R,R)-354a
(R) (R)
352a: G = Ph
(R,S)-352a:(R,R)-354a:(R,S)-352b:
RNO2
NH2
MeO
OMe
12i
79
catalyst(2 mol%)
toluene or i-Pr2O–15 °C to r.t., 0.5–24 h
RNO2
NH
OMeMeO
353
R = Ph (at 0 °C)R = Ph (at 0 °C)R = Ph (at 0 °C)R = aromaticR = aliphatic
BArF
BArF

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1977
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Acknowledgment
This work was supported by a Grant-in-Aid for Scientific Researchfrom the Ministry of Education, Culture, Sports, and Science andTechnology, Japan.
References
(1) For recent reviews, see: (a) Berkessel, A.; Gröger, H. Asymmetric Organocatalysis – From Biomimetic Concepts to Powerful Methods for Asymmetric Synthesis; Wiley-VCH: Weinheim, 2005. (b) Taylor, M. S.; Jacobsen, E. N. Angew. Chem. Int. Ed. 2006, 45, 1520. (c) Enantioselective Organocatalysis: Reactions and Experimental Procedures; Dalko, P. I., Ed.; Wiley: New York, 2007.
(2) For recent reviews, see: (a) Akiyama, T.; Itoh, J.; Fuchibe, K. Adv. Synth. Catal. 2006, 348, 999. (b) Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713. (c) Akiyama, T. Chem. Rev. 2007, 107, 5744. (d) Yu, X.; Wang, W. Chem. Asian J. 2008, 3, 516. (e) Kampen, D.; Reisinger, C. M.; List, B. Top. Curr. Chem. 2010, 291, 395.
(3) (a) Sigman, M. S.; Jacobsen, E. N. J. Am. Chem. Soc. 1998, 120, 4901. (b) Sigman, M. S.; Vachal, P.; Jacobsen, E. N. Angew. Chem. Int. Ed. 2000, 39, 1279. (c) Vachal, P.; Jacobsen, E. N. J. Am. Chem. Soc. 2002, 124, 10012.
(4) (a) Huang, Y.; Unni, A. K.; Thadani, A. N.; Rawal, V. H. Nature 2003, 424, 146. See also: (b) McDougal, N. T.; Schaus, S. E. J. Am. Chem. Soc. 2003, 125, 12094. (c) Nugent, B. M.; Yoder, R. A.; Johnston, J. N. J. Am. Chem. Soc. 2004, 126, 3418.
(5) For thiourea and urea, see: Bordwell, F. G.; Algrim, D. J.; Harrelson, J. A. Jr. J. Am. Chem. Soc. 1988, 110, 5903.
(6) For aliphatic alcohols, see: (a) Bordwell, F. G.; McCallum, R. J.; Olmstead, W. N. J. Org. Chem. 1984, 49, 1424. (b) Olmstead, W. N.; Margolin, Z.; Bordwell, F. G. J. Org. Chem. 1980, 45, 3295.
(7) Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Angew. Chem. Int. Ed. 2004, 43, 1566.
(8) Uraguchi, D.; Terada, M. J. Am. Chem. Soc. 2004, 126, 5356.
(9) For reviews, see: (a) Connon, S. J. Angew. Chem. Int. Ed. 2006, 45, 3909. (b) Adair, G.; Mukherjee, S.; List, B. Aldrichimica Acta 2008, 41, 31. (c) Terada, M. Chem. Commun. 2008, 4097. (d) Terada, M. Bull. Chem. Soc. Jpn. 2010, 83, 101.
(10) Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley and Sons: New York, 2000, 133.
(11) For a review on asymmetric bifunctional catalysts, see: (a) Shibasaki, M.; Kanai, M.; Funabashi, K. Chem. Commun. 2002, 1989. (b) Shibasaki, M.; Kanai, M. In New Frontiers in Asymmetric Catalysis; Mikami, K.; Lautens, M., Eds.; John Wiley & Sons: Hoboken, 2007, 383. (c) Shibasaki, M.; Kanai, M.; Matsunaga, S.; Kumagai, N. Acc. Chem. Res. 2009, 42, 1117.
(12) For reviews, see: (a) Pu, L. Chem. Rev. 1998, 98, 2405. (b) Kočovský, P.; Vyskočil, S.; Smrčina, M. Chem. Rev. 2003, 103, 3213. (c) Brunel, J. M. Chem. Rev. 2005, 105, 857.
(13) For binaphthol-derived monophosphoric acids as a chiral ligand for transition or rare earth metal catalysts (published before 2004), see: (a) Alper, H.; Hamel, N. J. Am. Chem. Soc. 1990, 112, 2803. (b) McCarthy, N.; McKervey, M. A.; Ye, T.; McCann, M.; Murphy, E.; Doyle, M. P. Tetrahedron Lett. 1992, 33, 5983. (c) Pirrung, M. C.; Zhang, J. Tetrahedron Lett. 1992, 33, 5987. (d) Inanaga, J.;
Sugimoto, Y.; Hanamoto, T. New J. Chem. 1995, 19, 707. (e) Furuno, H.; Hanamoto, T.; Sugimoto, Y.; Inanaga, J. Org. Lett. 2000, 2, 49. (f) Jin, X. L.; Sugihara, H.; Daikai, K.; Tateishi, H.; Jin, Y. Z.; Furuno, H.; Inanaga, J. Tetrahedron 2002, 58, 8321. (g) Furuno, H.; Hayano, T.; Kambara, T.; Sugimoto, Y.; Hanamoto, T.; Tanaka, Y.; Jin, Y. Z.; Kagawa, T.; Inanaga, J. Tetrahedron 2003, 59, 10509. (h) Merlic, C. A.; Zhechman, A. L. Synthesis 2003, 1137.
(14) For a review, see: Terada, M.; Momiyama, N. In Chiral Amine Synthesis. Methods, Developments and Applications; Nugent, T. C., Ed.; Wiley-VCH: Weinheim, 2010, 75.
(15) For general reviews, see: (a) Arend, M.; Westermann, B.; Risch, N. Angew. Chem. Int. Ed. 1998, 37, 1044. (b) Bur, S. K.; Martin, S. F. Tetrahedron 2001, 57, 3221. (c) Córdova, A. Acc. Chem. Res. 2004, 37, 102.
(16) For recent reviews, see: (a) Ting, A.; Schaus, S. E. Eur. J. Org. Chem. 2007, 5797. (b) Verkade, J. M. M.; van Hemert, L. J. C.; Quaedflieg, P. J. L. M.; Rutjes, F. P. J. T. Chem. Soc. Rev. 2008, 37, 29.
(17) Itoh, J.; Fuchibe, K.; Akiyama, T. Synthesis 2008, 1319.(18) Yamanaka, M.; Itoh, J.; Fuchibe, K.; Akiyama, T. J. Am.
Chem. Soc. 2007, 129, 6756.(19) Akiyama, T.; Saitoh, Y.; Morita, H.; Fuchibe, K. Adv. Synth.
Catal. 2005, 347, 1523.(20) (a) Akiyama, T.; Katoh, T.; Mori, K.; Kanno, K. Synlett
2009, 1664. See also: (b) Biaggi, C.; Benaglia, M.; Annunziata, R.; Rossi, S. Chirality 2010, 22, 369.
(21) Terada, M.; Sorimachi, K.; Uraguchi, D. Synlett 2006, 133.(22) Guo, Q.-X.; Liu, H.; Guo, C.; Luo, S.-W.; Gu, Y.; Gong,
L.-Z. J. Am. Chem. Soc. 2007, 129, 3790.(23) Rueping, M.; Sugiono, E.; Schoepke, F. R. Synlett 2007,
1441.(24) (a) Antilla, J. C.; Wulff, W. D. Angew. Chem. Int. Ed. 2000,
39, 4518. (b) Redlich, M.; Hossain, M. M. Tetrahedron Lett. 2004, 45, 8987; and references cited therein.
(25) Williams, A. L.; Johnston, J. N. J. Am. Chem. Soc. 2004, 126, 1612; and references cited therein.
(26) Uraguchi, D.; Sorimachi, K.; Terada, M. J. Am. Chem. Soc. 2005, 127, 9360.
(27) Akiyama, T.; Suzuki, T.; Mori, K. Org. Lett. 2009, 11, 2445.(28) Zeng, X.; Zeng, X.; Xu, Z.; Lu, M.; Zhong, G. Org. Lett.
2009, 11, 3036.(29) Chen, X.-H.; Xu, X.-Y.; Liu, H.; Cun, L.-F.; Gong, L.-Z.
J. Am. Chem. Soc. 2006, 128, 14802.(30) Gong, L.-Z.; Chen, X.-H.; Xu, X.-Y. Chem. Eur. J. 2007, 13,
8920.(31) Li, N.; Chen, X.-H.; Song, J.; Luo, S.-W.; Fan, W.; Gong,
L.-Z. J. Am. Chem. Soc. 2009, 131, 15301.(32) Goss, J. M.; Schaus, S. E. J. Org. Chem. 2008, 73, 7651.(33) (a) Carswell, E. L.; Snapper, M. L.; Hoveyda, A. H. Angew.
Chem. Int. Ed. 2006, 45, 7230. (b) Mandai, H.; Mandai, K.; Snapper, M. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2008, 130, 17961. (c) Yamaguchi, A.; Matsunaga, S.; Shibasaki, M. Org. Lett. 2008, 10, 2319.
(34) For a review, see: Denmark, S. E.; Heemetra, J. R. Jr.; Beutner, G. L. Angew. Chem. Int. Ed. 2005, 44, 4682.
(35) Akiyama, T.; Honma, Y.; Itoh, J.; Fuchibe, K. Adv. Synth. Catal. 2008, 350, 399.
(36) Sickert, M.; Schneider, C. Angew. Chem. Int. Ed. 2008, 47, 3631.
(37) Giera, D. S.; Sickert, M.; Schneider, C. Org. Lett. 2008, 10, 4259.
(38) (a) Yang, J. W.; Stadler, M.; List, B. Angew. Chem. Int. Ed. 2007, 46, 609. (b) Zhang, H.; Mitsumori, S.; Utsumi, N.; Imai, M.; Garcia-Delgado, N.; Mifsud, M.; Albertshofer, K.;

1978 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Cheong, P. H.-Y.; Houk, K. N.; Tanaka, F.; Barbas, C. F. III J. Am. Chem. Soc. 2008, 130, 875. (c) Kano, T.; Yamaguchi, Y.; Maruoka, K. Angew. Chem. Int. Ed. 2009, 48, 1838; and references cited therein.
(39) Terada, M.; Toda, Y. J. Am. Chem. Soc. 2009, 131, 6354.(40) (a) Frauenrath, H.; Arenz, T.; Raabe, G.; Zorn, M. Angew.
Chem., Int. Ed. Engl. 1993, 32, 83. (b) Arenz, T.; Frauenrath, H.; Raabe, G.; Zorn, M. Liebigs Ann. Chem. 1994, 931. (c) Tayama, E.; Otoyama, S.; Isaka, W. Chem. Commun. 2008, 4216.
(41) For reviews on the enantioselective Strecker reaction, see: (a) Gröger, H. Chem. Rev. 2003, 103, 2795. (b) Shibasaki, M.; Kanai, M.; Mita, T. Org. React. 2008, 70, 1. (c) Merino, P.; Marques-Lopez, E.; Tejero, T.; Herrera, R. P. Tetrahedron 2009, 65, 1219.
(42) Rueping, M.; Sugiono, E.; Azap, C. Angew. Chem. Int. Ed. 2006, 45, 2617.
(43) Rueping, M.; Sugiono, E.; Moreth, S. A. Adv. Synth. Catal. 2007, 349, 759.
(44) For theoretical studies on the enantioselective Strecker reaction catalyzed by chiral phosphoric acids, see: Simón, L.; Goodman, J. M. J. Am. Chem. Soc. 2009, 131, 4070.
(45) For the enantioselective hydrocyanation of hydrazones catalyzed by chiral metal complexes, see: (a) Keith, J. M.; Jacobsen, E. N. Org. Lett. 2004, 6, 153. (b) Ding, H.; Friestad, G. K. Heterocycles 2006, 70, 185.
(46) Zamfir, A.; Tsogoeva, S. B. Org. Lett. 2010, 12, 188.(47) For a review, see: Westermann, B. Angew. Chem. Int. Ed.
2003, 42, 151.(48) Rueping, M.; Antonchick, A. P. Org. Lett. 2008, 10, 1731.(49) (a) Lassaletta, J.-M.; Fernández, R. Tetrahedron Lett. 1992,
33, 3691. (b) Lassaletta, J.-M.; Fernández, R.; Martín-Zamora, E.; Díez, E. J. Am. Chem. Soc. 1996, 118, 7002.
(50) Rueping, M.; Sugiono, E.; Theissman, T.; Kuenkel, A.; Köckritz, A.; Pews-Davtyan, A.; Nemati, N.; Beller, M. Org. Lett. 2007, 9, 1065.
(51) For a review, see: Banfi, L.; Riva, R. Org. React. 2005, 65, 1.(52) Dömling, A.; Ugi, I. Angew. Chem. Int. Ed. 2000, 39, 3168.(53) Yue, T.; Wang, M.-X.; Wang, D.-X.; Masson, G.; Zhu, J.
Angew. Chem. Int. Ed. 2009, 48, 6717.(54) For the phosphinic acid catalyzed Ugi-type reaction, see:
Pan, S. C.; List, B. Angew. Chem. Int. Ed. 2008, 47, 3622.(55) For reviews, see: (a) Jørgensen, K. A. Synthesis 2003, 1117.
(b) Bandini, M.; Melloni, A.; Umani-Ronchi, A. Angew. Chem. Int. Ed. 2004, 43, 550. (c) Poulsen, T. B.; Jørgensen, K. A. Chem. Rev. 2008, 108, 2903. (d) You, S.-L.; Cai, Q.; Zeng, M. Chem. Soc. Rev. 2009, 38, 2190.
(56) Uraguchi, D.; Sorimachi, K.; Terada, M. J. Am. Chem. Soc. 2004, 126, 11804.
(57) For reviews, see: (a) Joule, J. A. Science of Synthesis, Vol. 10; Georg Thieme Verlag: Stuttgart, 2001, 361. (b) Aygun, A.; Pindur, U. Curr. Med. Chem. 2003, 10, 1113. (c) Abele, E.; Abele, R.; Dzenitis, O.; Lukevics, E. Chem. Heterocycl. Compd. (Engl. Transl.) 2003, 39, 825. (d) Gribble, G. W. Pure Appl. Chem. 2003, 75, 1417. (e) Sings, H.; Singh, S. Alkaloids 2003, 60, 51. (f) Somei, M.; Yamada, F. Nat. Prod. Rep. 2004, 21, 278. (g) Somei, M.; Yamada, F. Nat. Prod. Rep. 2005, 22, 73.
(58) Kang, Q.; Zhao, Z.-A.; You, S.-L. J. Am. Chem. Soc. 2007, 129, 1484.
(59) Rowland, G. B.; Rowland, E. B.; Liang, Y.; Perman, J. A.; Antilla, J. C. Org. Lett. 2007, 9, 2609.
(60) Terada, M.; Yokoyama, S.; Sorimachi, K.; Uraguchi, D. Adv. Synth. Catal. 2007, 349, 1863.
(61) Simón, L.; Goodman, J. M. J. Org. Chem. 2010, 75, 589.
(62) Wanner, M. J.; Hauwert, P.; Schoemaker, H. E.; de Gelder, R.; van Maarseveen, J. H.; Hiemstra, H. Eur. J. Org. Chem. 2008, 180.
(63) Kang, Q.; Zhao, Z.-A.; You, S.-L. Tetrahedron 2009, 65, 1603.
(64) Zhang, G.-W.; Wang, L.; Nie, J.; Ma, J.-A. Adv. Synth. Catal. 2008, 350, 1457.
(65) Li, G.; Rowland, G. B.; Rowland, E. B.; Antilla, J. C. Org. Lett. 2007, 9, 4065.
(66) Nakamura, S.; Sakurai, Y.; Kakashima, H.; Shibata, N.; Toru, T. Synlett 2009, 1639.
(67) Kang, Q.; Zheng, X.-J.; You, S.-L. Chem. Eur. J. 2008, 14, 3539.
(68) Enders, D.; Narine, A. A.; Toulgoat, F.; Bisschops, T. Angew. Chem. Int. Ed. 2008, 47, 5661.
(69) Nakashima, D.; Yamamoto, H. J. Am. Chem. Soc. 2006, 128, 9626.
(70) Rueping, M.; Nachtsheim, B. J. Synlett 2010, 119.(71) For reviews, see: (a) Cox, E. D.; Cook, J. M. Chem. Rev.
1995, 95, 1797. (b) Chrzanowska, M.; Rozwadowska, M. D. Chem. Rev. 2004, 104, 3341.
(72) Seayad, J.; Seayad, A. M.; List, B. J. Am. Chem. Soc. 2006, 128, 1086.
(73) (a) Wanner, M. J.; van der Haas, R. N. S.; de Cuba, K.; van Maarseveen, J. H.; Hiemstra, H. Angew. Chem. Int. Ed. 2007, 46, 7485. See also: (b) Sewgobind, N. V.; Wanner, M. J.; Ingemann, S.; de Gelder, R.; van Maarseveen, J. H.; Hiemstra, H. J. Org. Chem. 2008, 73, 6405.
(74) Wanner, M. J.; Boots, R. N. A.; Eradus, B.; de Gelder, R.; van Maarseveen, J. H.; Hiemstra, H. Org. Lett. 2009, 11, 2579.
(75) For the Friedel–Crafts reaction catalyzed by chiral metal complexes, see: (a) Bandini, M.; Garelli, A.; Rovinetti, M.; Tommasi, M.; Umani-Ronchi, A. Chirality 2005, 17, 522. (b) Zhuang, W.; Hazell, R. G.; Jørgensen, K. A. Org. Biomol. Chem. 2005, 3, 2566. (c) Jia, Y.-X.; Zhu, S.-F.; Yang, Y.; Zhou, Q.-L. J. Org. Chem. 2006, 71, 75. (d) Lu, S.-F.; Du, D.-M.; Xu, J. Org. Lett. 2006, 8, 2115. (e) Singh, P. K.; Bisai, A.; Singh, V. K. Tetrahedron Lett. 2007, 48, 1127. For the Friedel–Crafts reaction catalyzed by chiral organic molecules, see: (f) Herrera, R. P.; Sgarzani, V.; Bernardi, L.; Ricci, A. Angew. Chem. Int. Ed. 2005, 44, 6576. (g) Fleming, E. M.; McCabe, T.; Connon, S. J. Tetrahedron Lett. 2006, 47, 7037. (h) Ganesh, M.; Seidel, D. J. Am. Chem. Soc. 2008, 130, 16464.
(76) Itoh, J.; Fuchibe, K.; Akiyama, T. Angew. Chem. Int. Ed. 2008, 47, 4016.
(77) (a) Sheng, Y.-F.; Li, G.-Q.; Kang, Q.; Zhang, A.-J.; You, S.-L. Chem. Eur. J. 2009, 15, 3351. (b) Zheng, C.; Sheng, Y.-F.; Li, Y.-X.; You, S.-L. Tetrahedron 2010, 66, 2875.
(78) Sheng, Y.-F.; Gu, Q.; Zhang, A.-J.; You, S.-L. J. Org. Chem. 2009, 74, 6899.
(79) Tang, H.-Y.; Lu, A.-D.; Zhou, Z.-H.; Zhao, G.-F.; He, L.-N.; Tang, C.-C. Eur. J. Org. Chem. 2008, 1406.
(80) Rueping, M.; Nachtsheim, B. J.; Moreth, S. A.; Bolte, M. Angew. Chem. Int. Ed. 2008, 47, 593.
(81) Zeng, M.; Kang, Q.; He, Q.-L.; You, S.-L. Adv. Synth. Catal. 2008, 350, 2169.
(82) For selected examples, see: (a) Yadav, J. S.; Reddy, B. V. S.; Sunitha, S. Adv. Synth. Catal. 2003, 345, 349. (b) Gibbs, T. J. K.; Tomkinson, N. C. O. Org. Biomol. Chem. 2005, 3, 4043. (c) Gu, D. G.; Ji, S. J.; Jiang, Z. Q.; Zhou, M. F.; Loh, T. P. Synlett 2005, 959. (d) Ma, S. M.; Yu, S. C. Org. Lett. 2005, 7, 5063. (e) Nair, V.; Abhilash, K. G.; Vidya, N. Org. Lett. 2005, 7, 5857. (f) Li, H. M.; Wang, Y. Q.; Deng, L. Org. Lett. 2006, 8, 4063. (g) Shirakawa, S.; Kobayashi, S. Org. Lett. 2006, 8, 4939.

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1979
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
(83) Sun, F.-L.; Zeng, M.; Gu, Q.; You, S.-L. Chem. Eur. J. 2009, 15, 8709.
(84) Sun, F.-L.; Zheng, X.-J.; Gu, Q.; He, Q.-L.; You, S.-L. Eur. J. Org. Chem. 2010, 47.
(85) Nie, J.; Zhang, G.-W.; Wang, L.; Fu, A.; Zheng, Y.; Ma, J.-A. Chem. Commun. 2009, 2356.
(86) Nie, J.; Zhang, G.-W.; Wang, L.; Zheng, D.-H.; Zheng, Y.; Ma, J.-A. Eur. J. Org. Chem. 2009, 3145.
(87) For a review, see: (a) Matsubara, R.; Kobayashi, S. Acc. Chem. Res. 2008, 41, 292 . For aza-ene type reactions of a-imino esters, see: (b) Matsubara, R.; Nakamura, Y.; Kobayashi, S. Angew. Chem. Int. Ed. 2004, 43, 1679. (c) Kiyohara, H.; Matsubara, R.; Kobayashi, S. Org. Lett. 2006, 8, 5333. For aza-ene type reactions of glyoxylates, see: (d) Matsubara, R.; Nakamura, Y.; Kobayashi, S. Angew. Chem. Int. Ed. 2004, 43, 3258. (e) Matsubara, R.; Vital, P.; Nakamura, Y.; Kiyohara, H.; Kobayashi, S. Tetrahedron 2004, 60, 9769. (f) Matsubara, R.; Kawai, N.; Kobayashi, S. Angew. Chem. Int. Ed. 2006, 45, 3814. (g) Matsubara, R.; Doko, T.; Uetake, R.; Kobayashi, S. Angew. Chem. Int. Ed. 2007, 46, 3047.
(88) For some excellent studies of low loading of organocatalyst (less than 0.5 mol%), see: (a) Su, J. T.; Vachal, P.; Jacobsen, E. N. Adv. Synth. Catal. 2001, 343, 197. (b) Saaby, S.; Bella, M.; Jørgensen, A. K. J. Am. Chem. Soc. 2004, 126, 8120. (c) Shirakawa, S.; Yamamoto, K.; Kitamura, M.; Ooi, T.; Maruoka, K. Angew. Chem. Int. Ed. 2005, 44, 625. (d) Kitamura, M.; Shirakawa, S.; Maruoka, K. Angew. Chem. Int. Ed. 2005, 44, 1549. (e) Terada, M.; Ube, H.; Yaguchi, Y. J. Am. Chem. Soc. 2006, 128, 1454. (f) Terada, M.; Nakano, M.; Ube, H. J. Am. Chem. Soc. 2006, 128, 16044. (g) Rueping, M.; Antonchick, A. P.; Theissmann, T. Angew. Chem. Int. Ed. 2006, 45, 6751. (h) Guo, Q.-S.; Du, D.-M.; Xu, J. Angew. Chem. Int. Ed. 2008, 47, 759. (i) Cheon, C. H.; Yamamoto, H. J. Am. Chem. Soc. 2008, 130, 9246. (j) Liu, X.; Sun, B.; Deng, L. Synlett 2009, 1685.
(89) Terada, M.; Machioka, K.; Sorimachi, K. Angew. Chem. Int. Ed. 2006, 45, 2254.
(90) For recent reviews, see: (a) Enders, D.; Grondal, C.; Hüttl, M. R. M. Angew. Chem. Int. Ed. 2007, 46, 1570. (b) Yu, X.; Wang, W. Org. Biomol. Chem. 2008, 6, 2037.
(91) Terada, M.; Machioka, K.; Sorimachi, K. J. Am. Chem. Soc. 2007, 129, 10336.
(92) For recent selected examples, see: (a) Denmark, S. E.; Bui, T. J. Org. Chem. 2005, 70, 10190. (b) Boxer, M. B.; Yamamoto, H. Nature Protocols 2006, 1, 2434. (c) Boxer, M. B.; Yamamoto, H. J. Am. Chem. Soc. 2006, 128, 48. (d) Boxer, M. B.; Yamamoto, H. J. Am. Chem. Soc. 2007, 129, 2762. (e) Hayashi, Y.; Itoh, T.; Aratake, S.; Ishikawa, H. Angew. Chem. Int. Ed. 2008, 47, 2082. (f) Yang, J. W.; Chandler, C.; Stadler, M.; Kampen, D.; List, B. Nature 2008, 452, 453. (g) Hayashi, Y.; Itoh, T.; Ohkubo, M.; Ishikawa, H. Angew. Chem. Int. Ed. 2008, 47, 4722.
(93) (a) Gaulon, C.; Gizecki, P.; Dhal, R.; Dujardin, G. Synlett 2002, 952. (b) Prashad, M.; Lu, Y.; Repič, O. J. Org. Chem. 2004, 69, 584.
(94) Terada, M.; Machioka, K.; Sorimachi, K. Angew. Chem. Int. Ed. 2009, 48, 2553.
(95) Dagousset, G.; Drouet, F.; Masson, G.; Zhu, J. Org. Lett. 2009, 11, 5546.
(96) Liu, H.; Dagousset, G.; Masson, G.; Retailleau, P.; Zhu, J. J. Am. Chem. Soc. 2009, 131, 4598.
(97) Terada, M.; Soga, K.; Momiyama, N. Angew. Chem. Int. Ed. 2008, 47, 4122.
(98) Terada, M.; Ikehara, T.; Ube, H. J. Am. Chem. Soc. 2007, 129, 14112; and references cited therein.
(99) For X-ray crystal structure analysis of the double hydrogen bonding interaction between carboxylic acid and dimethylformamide, see: (a) Czugler, M.; Stezowski, J. J.; Weber, E. J. Chem. Soc., Chem. Commun. 1983, 154. (b) Csöregh, I.; Sjögren, A.; Czugler, M.; Cserzö, M.; Weber, E. J. Chem. Soc., Perkin Trans. 2 1986, 507.
(100) For hydrogen bonding in complexes of Lewis acids with formyl C–H hydrogen, see: Corey, E. J.; Lee, T. Chem. Commun. 2001, 1321.
(101) For reviews, see: (a) Snider, B. B. In Comprehensive Organic Synthesis, Vol. 2; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, 1991, 527. (b) Mikami, K.; Shimizu, M. Chem. Rev. 1992, 92, 1021. (c) Clarke, M.; France, M. B. Tetrahedron 2008, 64, 9003.
(102) (a) Mikami, K.; Terada, M.; Narisawa, S.; Nakai, T. Synlett 1992, 255. (b) Berrisford, D. J.; Bolm, C. Angew. Chem. Int. Ed. 1995, 34, 1717. (c) Mikami, K.; Terada, M. In Comprehensive Asymmetric Catalysis, Vol. 3; Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H., Eds.; Springer: Berlin, 1999, 1143.
(103) Clarke, M. W.; Jones, C. E. S.; France, M. B. Beilstein J. Org. Chem. 2007, 3, 24.
(104) Rueping, M.; Theissmann, T.; Kuenkel, A.; Koenigs, R. M. Angew. Chem. Int. Ed. 2008, 47, 6798.
(105) Akiyama, T.; Tamura, Y.; Itoh, J.; Morita, H.; Fuchibe, K. Synlett 2006, 141.
(106) Itoh, J.; Fuchibe, K.; Akiyama, T. Angew. Chem. Int. Ed. 2006, 45, 4796.
(107) Liu, H.; Cun, L.-F.; Mi, A.-Q.; Jiang, Y.-Z.; Gong, L.-Z. Org. Lett. 2006, 8, 6023.
(108) Rueping, M.; Azap, C. Angew. Chem. Int. Ed. 2006, 45, 7832.
(109) Akiyama, T.; Morita, H.; Fuchibe, K. J. Am. Chem. Soc. 2006, 128, 13070.
(110) Bergonzini, G.; Gramigna, L.; Mazzanti, A.; Fochi, M.; Bernardi, L.; Ricci, A. Chem. Commun. 2010, 46, 327.
(111) For reviews, see: (a) Narasaka, K. Synthesis 1991, 1. (b) Kagan, H. B.; Riant, O. Chem. Rev. 1992, 92, 1007. (c) Evans, D. A.; Johnson, J. S. In Comprehensive Asymmetric Catalysis, Vol. 3; Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H., Eds.; Springer Verlag: Berlin, 1999, 1177. (d) Hayashi, Y. In Cycloaddition Reactions in Organic Synthesis; Kobayashi, S.; Jørgensen, K. A., Eds.; Wiley-VCH: Weinheim, 2002, 5. (e) Corey, E. J. Angew. Chem. Int. Ed. 2002, 41, 1650.
(112) For a recent review, see: Merino, P.; Marqués-López, E.; Tejero, T.; Herrera, R. P. Synthesis 2010, 1.
(113) Yagupolskii, L. M.; Petric, V. N.; Kondratenko, N. V.; Sooväli, L.; Kaljurand, I.; Leito, I.; Koppel, I. A. J. Chem. Soc., Perkin Trans. 2 2002, 1950.
(114) (a) Ooi, T.; Maruoka, K. In Comprehensive Asymmetric Catalysis, Vol. 3; Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H., Eds.; Springer-Verlag: Berlin, 1999, 1237. (b) Jørgensen, K. A. In Cycloaddition Reactions in Organic Synthesis; Kobayashi, S.; Jørgensen, K. A., Eds.; Wiley-VCH: Weinheim, 2002, 151.
(115) For representative recent reviews of asymmetric hetero-Diels–Alder reactions, see: (a) Jørgensen, K. A. Angew. Chem. Int. Ed. 2000, 39, 3558. (b) Jørgensen, K. A. Eur. J. Org. Chem. 2004, 2093. (c) Lin, L.; Liu, X.; Feng, X. Synlett 2007, 2147. (d) Pellisser, H. Tetrahedron 2009, 65, 2839.
(116) For representative examples of catalytic enantioselective hetero-Diels–Alder reactions of aldehydes with siloxy- or methoxydienes, see: (a) Mikami, K.; Motoyama, Y.; Terada, M. J. Am. Chem. Soc. 1994, 116, 2812. (b) Dossetter, A. G.; Jamison, T. F.; Jacobsen, E. N. Angew. Chem. Int. Ed. 1999,

1980 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
38, 2398. (c) Quitschalle, M.; Christmann, M.; Bhatt, U.; Kalesse, M. Tetrahedron Lett. 2001, 42, 1263.
(117) (a) Danishefsky, S. J.; Larson, E.; Askin, D.; Kato, N. J. Am. Chem. Soc. 1985, 107, 1246. (b) Jurczak, J.; Golebiowski, A.; Rahm, A. Tetrahedron Lett. 1986, 27, 853. (c) McCarrick, M. A.; Wu, Y. D.; Houk, K. N. J. Org. Chem. 1993, 58, 3330. (d) Zhang, X.; Du, H.; Wang, Z.; Wu, Y. D.; Ding, K. J. Org. Chem. 2006, 71, 2862.
(118) Only one example of the catalytic asymmetric anti-selective hetero-Diels–Alder reaction has been reported using Danishefsky’s dienes. See: Yamashita, Y.; Saito, S.; Ishitani, H.; Kobayashi, S. J. Am. Chem. Soc. 2003, 125, 3793.
(119) Momiyama, N.; Tabuse, H.; Terada, M. J. Am. Chem. Soc. 2009, 131, 12882.
(120) The double hydrogen bonding model of the TS structure has been proposed in the aza-ene type reaction of glyoxylate 124 with enecarbamate 105, see ref. 97.
(121) For reviews, see: (a) Gothelf, K. V.; Jørgensen, K. A. Chem. Commun. 2000, 1449. (b) Kanemasa, S. Synlett 2002, 1371. (c) Nair, V.; Suja, T. D. Tetrahedron 2007, 63, 12247.
(122) Jiao, P.; Nakashima, D.; Yamamoto, H. Angew. Chem. Int. Ed. 2008, 47, 2411.
(123) For an example, see: Simonsen, K. B.; Bayón, P.; Hazell, R. G.; Gothelf, K. V.; Jørgensen, K. A. J. Am. Chem. Soc. 1999, 121, 3845.
(124) For reviews, see: (a) Gothelf, K. V.; Jørgensen, K. A. Chem. Rev. 1998, 98, 863. (b) Nájera, C.; Sansano, J. M. Angew. Chem. Int. Ed. 2005, 44, 6272. (c) Husinec, S.; Savic, V. Tetrahedron: Asymmetry 2005, 16, 2047. (d) Pandey, G.; Banerjee, P.; Gadre, S. R. Chem. Rev. 2006, 106, 4484. (e) Bonin, M.; Chauveau, A.; Micouin, L. Synlett 2006, 2349. (f) Pellisier, H. Tetrahedron 2007, 63, 3235. (g) Stanley, L. M.; Sibi, M. P. Chem. Rev. 2008, 108, 2887.
(125) Chen, X.-H.; Zhang, W.-Q.; Gong, L.-Z. J. Am. Chem. Soc. 2008, 130, 5652.
(126) Liu, W.-J.; Chen, X.-H.; Gong, L.-Z. Org. Lett. 2008, 10, 5357.
(127) Yu, J.; Chen, X.-H.; Song, J.; Chen, W.-J.; Gong, L.-Z. Org. Lett. 2009, 11, 4946.
(128) Chen, X.-H.; Wei, Q.; Luo, S.-W.; Xiao, H.; Gong, L.-Z. J. Am. Chem. Soc. 2009, 131, 13819.
(129) (a) Denmark, S. E. In Comprehensive Organic Synthesis, Vol. 5; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, 1991, 51. (b) Habermas, K. L.; Denmark, S. E.; Jones, T. K. Org. React. 1994, 45, 1. (c) Pellissier, H. Tetrahedron 2005, 61, 6479. (d) Frontier, A. J.; Collison, C. Tetrahedron 2005, 61, 7577. (e) Tius, M. A. Eur. J. Org. Chem. 2005, 2193.
(130) (a) Liang, G.; Gradl, S. N.; Trauner, D. Org. Lett. 2003, 5, 4931. (b) Aggarwal, V. K.; Belfield, A. J. Org. Lett. 2003, 5, 5075. (c) Liang, G.; Trauner, D. J. Am. Chem. Soc. 2004, 126, 9544. (d) Walz, I.; Togni, A. Chem. Commun. 2008, 4315. (e) Yaji, K.; Shindo, M. Synlett 2009, 2524.
(131) Rueping, M.; Ieawsuwan, W.; Antonchick, A. P.; Nachtsheim, B. J. Angew. Chem. Int. Ed. 2007, 46, 2097.
(132) Rueping, M.; Ieawsuwan, W. Adv. Synth. Catal. 2009, 351, 78.
(133) Muller, S.; List, B. Angew. Chem. Int. Ed. 2009, 48, 9975.(134) For the chiral phosphoric acid catalyzed consecutive
condensation/6p-electrocyclization of 4-hydroxycoumarin with crotonaldehyde to give a chromenone derivative with low enantioselectivity, see: Moreau, J.; Hubert, C.; Batany, J.; Toupet, L.; Roisnel, T.; Hurvois, J.-P.; Renaud, J.-L. J. Org. Chem. 2009, 74, 8963.
(135) For selected reviews, see: (a) Burke, M. D.; Schreiber, S. L. Angew. Chem. Int. Ed. 2004, 43, 46. (b) Multicomponent
Reactions; Zhu, J.; Bienaymé, H., Eds.; Wiley-VCH: Weinheim, 2005. (c) Ramon, D. J.; Yus, M. Angew. Chem. Int. Ed. 2005, 44, 1602. (d) Guillena, G.; Ramon, D. J.; Yus, M. Tetrahedron: Asymmetry 2007, 18, 693. (e) Toure, B. B.; Hall, D. G. Chem. Rev. 2009, 109, 4439.
(136) (a) Jiang, J.; Yu, J.; Sun, X.-X.; Rao, Q.-Q.; Gong, L.-Z. Angew. Chem. Int. Ed. 2008, 47, 2458. See also: (b) Moreau, J.; Duboc, A.; Hubert, C.; Hurvois, J.-P.; Renaud, J.-L. Tetrahedron Lett. 2007, 48, 8647.
(137) For reviews, see: (a) Stout, D. M.; Meyers, A. I. Chem. Rev. 1982, 82, 223. (b) Lavilla, R. J. Chem. Soc., Perkin Trans. 1 2002, 1411.
(138) (a) Janis, R. A.; Silver, P. J.; Triggle, D. J. Adv. Drug Res. 1987, 16, 309. (b) Bossert, F.; Vater, W. Med. Res. Rev. 1989, 9, 291.
(139) Murakami, Y.; Kikuchi, J.-i.; Hisaeda, Y. Chem. Rev. 1996, 96, 721.
(140) Evans, C. G.; Gestwicki, J. E. Org. Lett. 2009, 11, 2957.(141) For reviews, see: (a) Fisk, J. S.; Mosey, R. A.; Tepe, J. J.
Chem. Soc. Rev. 2007, 36, 1432. (b) Hewlett, N. M.; Hupp, C. D.; Tepe, J. J. Synthesis 2009, 2825.
(142) Jiang, J.; Qing, J.; Gong, L.-Z. Chem. Eur. J. 2009, 15, 7031.(143) For proline-catalyzed reactions, see: (a) Eder, U.; Sauer, G.;
Wiechert, R. Angew. Chem., Int. Ed. Engl. 1971, 10, 496. (b) Hajos, Z. G.; Parrish, D. R. J. Org. Chem. 1974, 39, 1615. (c) Bui, T.; Barbas, C. F. III Tetrahedron Lett. 2000, 41, 6951. (d) Cheong, P. H.-Y.; Houk, K. N.; Warrier, J. S.; Hanessian, S. Adv. Synth. Catal. 2004, 346, 1111. (e) Inomata, K.; Barragué, M.; Paquette, L. A. J. Org. Chem. 2005, 70, 533. (f) Clemente, F. R.; Houk, K. N. J. Am. Chem. Soc. 2005, 127, 11294. (g) Lacoste, E.; Vaique, E.; Berlande, M.; Pianet, I.; Vincent, J.-M.; Landais, Y. Eur. J. Org. Chem. 2007, 167.
(144) Akiyama, T.; Katoh, T.; Mori, K. Angew. Chem. Int. Ed. 2009, 48, 4226.
(145) Mori, K.; Katoh, T.; Suzuki, T.; Noji, T.; Yamanaka, M.; Akiyama, T. Angew. Chem. Int. Ed. 2009, 48, 9652.
(146) Terada, M.; Sorimachi, K. J. Am. Chem. Soc. 2007, 129, 292.(147) (a) Terada, M.; Tanaka, H.; Sorimachi, K. Synlett 2008,
1661. See also: (b) Kobayashi, S.; Gustafsson, T.; Shimizu, Y.; Kiyohara, H.; Matsubara, R. Org. Lett. 2006, 6, 4923.
(148) Jia, Y.-X.; Zhong, J.; Zhu, S.-F.; Zhang, C.-M.; Zhou, Q.-L. Angew. Chem. Int. Ed. 2007, 46, 5565.
(149) Baudequin, C.; Zamfir, A.; Tsogoeva, S. B. Chem. Commun. 2008, 4637.
(150) For a review of enantioselective protonation, see: Duhamel, L.; Duhamel, P.; Plaquevent, J. C. Tetrahedron: Asymmetry 2004, 15, 3635.
(151) For the development of BINOL-derived phosphorodithioic acids, see: Pousse, G.; Devineau, A.; Dalla, V.; Humphreys, L.; Lasne, M.-C.; Rouden, J.; Blanchet, J. Tetrahedron 2009, 65, 10617.
(152) Mayer, S.; List, B. Angew. Chem. Int. Ed. 2006, 45, 4193; and references cited therein.
(153) Terada, M.; Tanaka, H.; Sorimachi, K. J. Am. Chem. Soc. 2009, 131, 3430.
(154) Reisman, S. E.; Doyle, A. G.; Jacobsen, E. N. J. Am. Chem. Soc. 2008, 130, 7198.
(155) For reviews on the semi-pinacol rearrangement, see: (a) Rickborn, B. In Comprehensive Organic Synthesis, Vol. 3; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, 1991, 733. (b) Snape, T. Chem. Soc. Rev. 2007, 36, 1823.
(156) Zhang, Q.-W.; Fan, C.-A.; Zhang, H.-J.; Tu, Y.-Q.; Zhao, Y.-M.; Gu, P.; Chen, Z.-M. Angew. Chem. Int. Ed. 2009, 48, 8572.

REVIEW Chiral Phosphoric Acids as Enantioselective Catalysts 1981
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
(157) For recent reviews, see: (a) Widenhoefer, R. A. Chem. Eur. J. 2008, 14, 5382. (b) Mueller, T. E.; Hultzsch, K. C.; Yus, M.; Foubelo, F.; Tada, M. Chem. Rev. 2008, 108, 3795. (c) Severin, R.; Doye, S. Chem. Soc. Rev. 2007, 36, 1407. (d) Aillaud, I.; Collin, J.; Hannedouche, J.; Schulz, E. Dalton Trans. 2007, 5105.
(158) Ackermann, L.; Althammer, A. Synlett 2008, 995.(159) For reviews, see: (a) The Claisen Rearrangement;
Hiersemann, M.; Nubbemeyer, U., Eds.; Wiley-VCH: Weinheim, 2007. (b) Martin Castro, A. M. Chem. Rev. 2004, 104, 2939.
(160) Rueping, M.; Antonchick, A. P. Angew. Chem. Int. Ed. 2008, 47, 10090.
(161) For reviews, see: (a) Radicals in Organic Synthesis, Vols. 1 and 2; Renaud, P.; Sibi, M. P., Eds.; Wiley-VCH: Weinheim, 2001. (b) Sibi, M. P.; Manyem, S.; Zimmerman, J. Chem. Rev. 2003, 103, 3263. (c) Bar, G.; Parsons, A. F. Chem. Soc. Rev. 2003, 32, 251. (d) Zimmerman, J.; Sibi, M. P. Top. Curr. Chem. 2006, 263, 107.
(162) Lee, S.; Kim, S. Tetrahedron Lett. 2009, 50, 3345.(163) (a) Shaikh, R. R.; Mazzanti, A.; Petrini, M.; Bartoli, G.;
Melchiorre, P. Angew. Chem. Int. Ed. 2008, 47, 8707. (b) Cozzi, P. G.; Benfatti, F.; Zoli, L. Angew. Chem. Int. Ed. 2009, 48, 1313. See also: (c) Melchiorre, P. Angew. Chem. Int. Ed. 2009, 48, 1360.
(164) Guo, Q.-X.; Peng, Y.-G.; Zhang, J.-W.; Song, L.; Feng, Z.; Gong, L.-Z. Org. Lett. 2009, 11, 4620.
(165) For a review of enantioselective synthesis of a-amino phosphonate derivatives, see: Ma, J.-A. Chem. Soc. Rev. 2006, 35, 630.
(166) Akiyama, T.; Morita, H.; Itoh, J.; Fuchibe, K. Org. Lett. 2005, 7, 2583.
(167) For computational analysis of transition states, see: (a) Akiyama, T.; Morita, H.; Bachu, P.; Mori, K.; Yamanaka, M.; Hirata, T. Tetrahedron 2009, 65, 4950. (b) Yamanaka, M.; Hirata, T. J. Org. Chem. 2009, 74, 3266.
(168) Cheng, X.; Goddard, R.; Buth, G.; List, B. Angew. Chem. Int. Ed. 2008, 47, 5079.
(169) Hoffmann, S.; Nicoletti, M.; List, B. J. Am. Chem. Soc. 2006, 128, 13074.
(170) Rowland, G. B.; Zhang, H.; Rowland, E. B.; Chennamadhavuni, S.; Wang, Y.; Antilla, J. C. J. Am. Chem. Soc. 2005, 127, 15696.
(171) Liang, Y.; Rowland, E. B.; Rowland, G. B.; Perman, J. A.; Antilla, J. C. Chem. Commun. 2007, 4477.
(172) Cheng, X.; Vellalath, S.; Goddard, R.; List, B. J. Am. Chem. Soc. 2008, 130, 15786.
(173) Rueping, M.; Antonchick, A. P.; Sugiono, E.; Grenader, K. Angew. Chem. Int. Ed. 2009, 48, 908.
(174) Li, G.; Fronczek, F. R.; Antilla, J. C. J. Am. Chem. Soc. 2008, 130, 12216.
(175) For a review, see: Yudin, A. K. Aziridines and Epoxides in Organic Synthesis; Wiley-VCH: Weinheim, 2006.
(176) For a review, see: Schneider, C. Angew. Chem. Int. Ed. 2009, 48, 2082.
(177) Rowland, E. B.; Rowland, G. B.; Rivera-Otero, E.; Antilla, J. C. J. Am. Chem. Soc. 2007, 129, 12084.
(178) Larson, S. E.; Baso, J. C.; Li, G.; Antilla, J. C. Org. Lett. 2009, 11, 5186.
(179) Della Sala, G.; Lattanzi, A. Org. Lett. 2009, 11, 3330.(180) (a) Hamilton, G. L.; Kanai, T.; Toste, F. D. J. Am. Chem.
Soc. 2008, 130, 14984. For related chiral counteranion strategies for enantioselective transformations, see: (b) Hamilton, G. L.; Kang, E. J.; Mba, M.; Toste, F. D. Science 2007, 317, 496. (c) LaLonde, R. L.; Wang, Z. J.; Mba, M.; Lackner, A. D.; Toste, F. D. Angew. Chem. Int. Ed. 2010, 49, 598.
(181) For recent reviews, see: (a) Ooi, T.; Maruoka, K. Angew. Chem. Int. Ed. 2007, 46, 4222. (b) Hashimoto, T.; Maruoka, K. Chem. Rev. 2007, 107, 5656. (c) Asymmetric Phase Transfer Catalysis; Maruoka, K., Ed.; Wiley-VCH: Weinheim, 2008.
(182) For reviews, see: (a) Blaser, H.-U.; Malan, C.; Pugin, B.; Spindler, F.; Steiner, H.; Studer, M. Adv. Synth. Catal. 2003, 345, 103. (b) Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029. (c) Riant, O.; Mostefaï, N.; Courmarcel, J. Synthesis 2004, 2943.
(183) (a) Yang, J. W.; Hechavarria Foneca, M. T.; Vignola, N.; List, B. Angew. Chem. Int. Ed. 2005, 44, 110. (b) Ouellet, S. G.; Tuttle, J. B.; MacMillan, D. W. C. J. Am. Chem. Soc. 2005, 127, 32. See also: (c) Yang, J. W.; Hechavarria Foneca, M. T.; List, B. Angew. Chem. Int. Ed. 2004, 43, 6660.
(184) For reviews, see: (a) Ouellet, S. G.; Walji, A. M.; Macmillan, D. W. C. Acc. Chem. Res. 2007, 40, 1327. (b) You, S.-L. Chem. Asian J. 2007, 2, 820. (c) Connon, S. J. Org. Biomol. Chem. 2007, 5, 3407. (d) Rueping, M.; Sugiono, E.; Schoepke, F. R. Synlett 2010, 852.
(185) Rueping, M.; Sugiono, E.; Azap, C.; Theissmann, T.; Bolte, M. Org. Lett. 2005, 7, 3781.
(186) For theoretical studies, see: (a) Simón, L.; Goodman, J. M. J. Am. Chem. Soc. 2008, 130, 8741. (b) Marcelli, T.; Hammar, P.; Himo, F. Chem. Eur. J. 2008, 14, 8562.
(187) Hoffmann, S.; Seayad, A. M.; List, B. Angew. Chem. Int. Ed. 2005, 44, 7424.
(188) Storer, R. I.; Carrera, D. E.; Ni, Y.; MacMillan, D. W. C. J. Am. Chem. Soc. 2006, 128, 84.
(189) Rueping, M.; Tato, F.; Schoepke, F. R. Chem. Eur. J. 2010, 16, 2688.
(190) Li, G.; Liang, Y.; Antilla, J. C. J. Am. Chem. Soc. 2007, 129, 5830.
(191) Kang, Q.; Zhao, Z.-A.; You, S.-L. Adv. Synth. Catal. 2007, 349, 1657.
(192) Kang, Q.; Zhao, Z.-A.; You, S.-L. Org. Lett. 2008, 10, 2031.(193) Li, G.; Antilla, J. C. Org. Lett. 2009, 11, 1075.(194) Han, Z.-Y.; Xiao, H.; Gong, L.-Z. Bioorg. Med. Chem. Lett.
2009, 19, 3729.(195) Zhu, C.; Akiyama, T. Org. Lett. 2009, 11, 4180.(196) (a) Chikashita, H.; Miyazaki, M.; Itoh, K. Synthesis 1984,
308. (b) Chikashita, H.; Miyazaki, M.; Itoh, K. J. Chem. Soc., Perkin Trans. 1 1987, 699.
(197) Glorius, F. Org. Biomol. Chem. 2005, 3, 4171.(198) Rueping, M.; Antonchick, A. P.; Theissmann, T. Angew.
Chem. Int. Ed. 2006, 45, 3683.(199) Rueping, M.; Antonchick, A. P. Angew. Chem. Int. Ed. 2007,
46, 4562.(200) Metallinos, C.; Barrett, F. B.; Xu, S. Synlett 2008, 720.(201) Rueping, M.; Theissmann, T.; Raja, S.; Bats, J. W. Adv.
Synth. Catal. 2008, 350, 1001.(202) (a) Martin, N. J. A.; List, B. J. Am. Chem. Soc. 2006, 128,
13368. See also: (b) Tuttle, J. B.; Ouellet, S. G.; MacMillan, D. W. C. J. Am. Chem. Soc. 2006, 128, 12662.
(203) For reviews, see: (a) List, B. Chem. Commun. 2006, 819. (b) Palomo, C.; Mielgo, A. Angew. Chem. Int. Ed. 2006, 45, 7876. (c) Walji, A. M.; MacMillan, D. W. C. Synlett 2007, 1477.
(204) Zhou, J.; List, B. J. Am. Chem. Soc. 2007, 129, 7498.(205) Schrader, W.; Handayani, P. P.; Zhou, J.; List, B. Angew.
Chem. Int. Ed. 2009, 48, 1463.(206) Rueping, M.; Antonchick, A. P. Angew. Chem. Int. Ed. 2008,
47, 5836.(207) For selected reviews on metal-catalyzed asymmetric
epoxidations, see: (a) Katsuki, T. In Comprehensive Asymmetric Catalysis, Vol. 2; Jacobsen, E. N.; Pfaltz, A.;

1982 M. Terada REVIEW
Synthesis 2010, No. 12, 1929–1982 © Thieme Stuttgart · New York
Yamamoto, H., Eds.; Springer-Verlag: Berlin, 1999, 621. (b) Jacobsen, E. N.; Wu, M. H. In Comprehensive Asymmetric Catalysis, Vol. 2; Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H., Eds.; Springer-Verlag: Berlin, 1999, 649. (c) McGarrigle, E.; Gilheany, D. G. Chem. Rev. 2005, 105, 1563. (d) Xia, Q.-H.; Ge, H.-Q.; Ye, C.-P.; Liu, Z.-M.; Su, K.-X. Chem. Rev. 2005, 105, 1603.
(208) For selected reviews on organocatalytic asymmetric epoxidations, see: (a) Shi, Y. Acc. Chem. Res. 2004, 37, 488. (b) Yang, D. Acc. Chem. Res. 2004, 37, 497. (c) Armstrong, A. Angew. Chem. Int. Ed. 2004, 43, 1460. (d) Dalko, P. I.; Moisan, L. Angew. Chem. Int. Ed. 2004, 43, 5138. (e) Berkessel, A.; Gröger, H. Asymmetric Organocatalysis–From Biomimetic Concepts to Powerful Methods for Asymmetric Synthesis; Wiley-VCH: Weinheim, 2005, 277.
(209) (a) Juliá, S.; Masana, J.; Vega, J. C. Angew. Chem., Int. Ed. Engl. 1980, 19, 929. (b) Juliá, S.; Guixer, J.; Masana, J.; Rocas, J.; Colonna, S.; Annunziata, R.; Molinari, H. J. Chem. Soc., Perkin Trans. 1 1982, 1317.
(210) For a review on the asymmetric epoxidation of a,b-unsaturated carbonyl compounds, see: Porter, M. J.; Skidmore, J. Chem. Commun. 2000, 1215.
(211) Wang, X.; List, B. Angew. Chem. Int. Ed. 2008, 47, 1119.(212) For a review, see: ten Brink, G.-J.; Arends, I. W. C. E.;
Sheldon, R. A. Chem. Rev. 2004, 104, 4105.(213) (a) Xu, S.; Wang, Z.; Zhang, X.; Zhang, X.; Ding, K. Angew.
Chem. Int. Ed. 2008, 47, 2840. (b) Xu, S.; Wang, Z.; Li, Y.; Zhang, X.; Wang, H.; Ding, K. Chem. Eur. J. 2010, 16, 3021.
(214) For Lewis acid catalyzed enantioselective a-hydroxylation of b-keto esters, see: (a) Bonaccorsi, C.; Althaus, M.; Becker, C.; Togni, A.; Mezzetti, A. Pure Appl. Chem. 2006, 78, 391. (b) Ishimaru, T.; Shibata, N.; Nagai, J.; Nakamura, S.; Toru, T.; Kanemasa, S. J. Am. Chem. Soc. 2006, 128, 16488. For organocatalysis, see: (c) Acocella, M. R.; Mancheño, O. G.; Bella, M.; Jørgensen, K. A. J. Org. Chem. 2004, 69, 8165.
(215) Lu, M.; Zhu, D.; Lu, Y.; Zeng, X.; Tan, B.; Xu, Z.; Zhong, G. J. Am. Chem. Soc. 2009, 131, 4562.
(216) For reviews, see: (a) Merino, P.; Tejero, T. Angew. Chem. Int. Ed. 2004, 43, 2995. (b) Yamamoto, H.; Momiyama, N. Chem. Commun. 2005, 3514.
(217) For reviews, see: (a) Shao, Z.; Zhang, H. Chem. Soc. Rev. 2009, 38, 2745. (b) Hashmi, A. S. K.; Hubbert, C. Angew. Chem. Int. Ed. 2010, 49, 1010. (c) de Armas, P.; Tejedor, D.; García-Tellado, F. Angew. Chem. Int. Ed. 2010, 49, 1013. (d) Zhou, J. Chem. Asian J. 2010, 5, 422.
(218) Komanduri, V.; Krische, M. J. J. Am. Chem. Soc. 2006, 128, 16448.
(219) (a) Kong, J.-R.; Ngai, M.-Y.; Krische, M. J. J. Am. Chem. Soc. 2006, 128, 718. (b) Ngai, M.-Y.; Kong, J.-R.; Krische, M. J. J. Org. Chem. 2007, 72, 1063.
(220) Rueping, M.; Antonchick, A. P.; Brinkmann, C. Angew. Chem. Int. Ed. 2007, 46, 6903.
(221) Hu, W.; Xu, X.; Zhou, J.; Liu, W.-J.; Huang, H.; Hu, J.; Yang, L.; Gong, L.-Z. J. Am. Chem. Soc. 2008, 130, 7782.
(222) Xu, X.; Zhou, J.; Yang, L.; Hu, W. Chem. Commun. 2008, 6564.
(223) Li, C.; Wang, C.; Villa-Marcos, B.; Xiao, J. J. Am. Chem. Soc. 2008, 130, 14450.
(224) Li, C.; Villa-Marcos, B.; Xiao, J. J. Am. Chem. Soc. 2009, 131, 6967.
(225) Guan, H.; Iimura, M.; Magee, M. P.; Norton, J. R. J. Am. Chem. Soc. 2005, 127, 7805.
(226) Mukherjee, S.; List, B. J. Am. Chem. Soc. 2007, 129, 11336.(227) For reviews, see: (a) Christoffers, J.; Baro, A. Adv. Synth.
Catal. 2005, 347, 1473. (b) Trost, B. M.; Jiang, C. Synthesis 2006, 369. (c) Cozzi, P. G.; Hilgraf, R.; Zimmermann, N. Eur. J. Org. Chem. 2007, 5969. For a review of an organocatalytic approach, see: (d) Bella, M.; Gasperi, T. Synthesis 2009, 1583.
(228) Reetz, M. T.; Westermann, J.; Kyung, S.-H. Chem. Ber. 1985, 118, 1050.
(229) Sorimachi, K.; Terada, M. J. Am. Chem. Soc. 2008, 130, 14452.
(230) Han, Z.-Y.; Xiao, H.; Chen, X.-H.; Gong, L.-Z. J. Am. Chem. Soc. 2009, 131, 9182.
(231) Liu, X.-Y.; Che, C.-M. Org. Lett. 2009, 11, 4204.(232) Muratore, M. E.; Holloway, C. A.; Pilling, A. W.; Storer, R.
I.; Trevitt, G.; Dixon, D. J. J. Am. Chem. Soc. 2009, 131, 10796.
(233) Genin, E.; Toullec, P. Y.; Antoniotti, S.; Brancour, C.; Genêt, J.-P.; Michelet, V. J. Am. Chem. Soc. 2006, 128, 3112.
(234) Cai, Q.; Zhao, Z.-A.; You, S.-L. Angew. Chem. Int. Ed. 2009, 48, 7428.
(235) Chen, J.-R.; Li, C.-F.; An, X.-L.; Zhang, J.-J.; Zhu, X.-Y.; Xiao, W.-J. Angew. Chem. Int. Ed. 2008, 47, 2489.
(236) Momiyama, N.; Yamamoto, H. J. Am. Chem. Soc. 2005, 127, 1080.
(237) (a) Hashimoto, T.; Maruoka, K. J. Am. Chem. Soc. 2007, 129, 10054. (b) Hashimoto, T.; Maruoka, K. Synthesis 2008, 3703.
(238) Hashimoto, T.; Uchiyama, N.; Maruoka, K. J. Am. Chem. Soc. 2008, 130, 14380.
(239) Hashimoto, T.; Hirose, M.; Maruoka, K. J. Am. Chem. Soc. 2008, 130, 7556.
(240) Hatano, M.; Maki, T.; Moriyama, K.; Arinobe, M.; Ishihara, K. J. Am. Chem. Soc. 2008, 130, 16858.
(241) List and co-workers employed 342a as a chiral Brønsted acid catalyst for the first time; however, no asymmetric induction was obtained. See: (a) Pan, S. C.; List, B. Chem. Asian J. 2008, 3, 430. (b) Kampen, D.; Ladépêche, A.; Claßen, G.; List, B. Adv. Synth. Catal. 2008, 350, 962.
(242) García-García, P.; Lay, F.; García-García, P.; Rabalakos, C.; List, B. Angew. Chem. Int. Ed. 2009, 48, 4363.
(243) Mathieu, B.; Ghosez, L. Tetrahedron Lett. 1997, 38, 5497.(244) (a) Schuster, T.; Bauch, M.; Dürner, G.; Göbel, M. W. Org.
Lett. 2000, 2, 179. (b) Schuster, T.; Kurz, M.; Göbel, M. W. J. Org. Chem. 2000, 65, 1697. (c) Tsogoeva, S. B.; Dürner, G.; Bolte, M.; Göbel, M. W. Eur. J. Org. Chem. 2003, 1661. (d) Akalay, D.; Dürner, G.; Bats, J. W.; Bolte, M.; Göbel, M. W. J. Org. Chem. 2007, 72, 5618. For an example of the stoichiometric use of a cationic chiral Brønsted acid, see: (e) Jang, D. O.; Kim, S. Y. J. Am. Chem. Soc. 2008, 130, 16152.
(245) (a) Uyeda, C.; Jacobsen, E. N. J. Am. Chem. Soc. 2008, 130, 9228. See also: (b) Xu, H.; Zuend, S. J.; Woll, M. G.; Tao, Y.; Jacobsen, E. N. Science 2010, 327, 986.
(246) Uraguchi, D.; Nakashima, D.; Ooi, T. J. Am. Chem. Soc. 2009, 131, 7242.
(247) (a) Uraguchi, D.; Sakaki, S.; Ooi, T. J. Am. Chem. Soc. 2007, 129, 12392. (b) Uraguchi, D.; Ueki, Y.; Ooi, T. J. Am. Chem. Soc. 2008, 130, 14088. (c) Uraguchi, D.; Ito, T.; Ooi, T. J. Am. Chem. Soc. 2009, 131, 3836. (d) Uraguchi, D.; Ueki, Y.; Ooi, T. Science 2009, 326, 120.
(248) Hatano, M.; Moriyama, K.; Maki, T.; Ishihara, K. Angew. Chem. Int. Ed. 2010, 49, 3823.





![四)€¦ · Web view第一章 化學反應 [1-1 認識化學反應] 自然與生活科技 (四) 第一章 化學反應 [1-1 認識化學反應] 第一章 化學反應 [1-1 認識化學反應]](https://img.pdfslide.us/doc/110x75/602b7f91aab14d629e313d77/-web-view-cc-oe-1-1-eeoe-ececc.jpg)























