Embed Size (px)
Citation preview

SwissMedical Forum
Offizielles Fortbildungsorgan der FMHOrgane officiel de la FMH pour la formation continueBollettino ufficiale per la formazione della FMHOrgan da perfecziunament uffizial da la FMH www.medicalforum.ch
With extended abstracts from “Swiss Medical Weekly”
10
9. 3
. 201
6
241 S. Hofmann, A. Buser, A. TaegtmeyerGlucose-6-Phosphat-Dehydro genase-Mangel
245 A. Burri, V. Garelli, N. PetitpierreDen Tramlinien auf der Spur
249 C. Burkhardt, C. Neuwirth, M. WeberScreening von Kognitions- und Verhaltensänderungen bei der Amyotrophen Lateralsklerose
236 A. Durovic, S. Tschudin-SutterUpdate zu Clostridium difficile
SMF – FMS Schweizerisches Medizin-Forum – Forum Médical Suisse – Forum Medico Svizzero – Forum Medical Svizzer

Und anderswo …?
A. de Torrenté
235 Schweres heterogenes Emphysem: eine neue Behandlung? Übersichtsartikel
A. Durovic, S. Tschudin-Sutter
236 Update zu Clostridium difficileClostridium difficile ist die häufigste infektiöse Ursache nosokomialer Diarrhoe und gewinnt zunehmend an Bedeutung als Erreger ambulant erworbener Durchfall erkrankungen. C. difficile-Infektionen sind mit hoher Morbidität und Mortalität assoziiert und gehen mit einem breiten Spektrum klinischer Symptome einher.
S. Hofmann, A. Buser, A. Taegtmeyer
241 Glucose-6-Phosphat-Dehydro genase-MangelImmer häufiger kommen Patienten in die Praxis und ins Spital, die einen G6PD-Mangel aufweisen und bei denen es wichtig ist zu wissen, welche Medikamente poten tiell gefährlich sind. In diesem Übersichtsartikel werden der G6PD-Mangel, die Diagnostik und die Medikamente, die zu vermeiden sind, beschrieben.
Was ist Ihre Diagnose?
A. Burri, V. Garelli, N. Petitpierre
245 Den Tramlinien auf der SpurBei einer 73-jährigen Patientin bestand seit drei Monaten ein unstillbarer Husten mit maulvollen Expektorationen. Ferner beschrieb sie eine progrediente Dyspnoe, die sich bei ihrer Vorstellung im Stadium NYHA II befand. Es bestand kein Status febrilis.Die Patientin war uns bereits aufgrund ihrer Vor geschichte mit Energie- und Eiweissmangelernährung, Osteoporose sowie einer seit 30 Jahren bestehenden Colitis ulcerosa bekannt.
Aus der Forschung
C. Burkhardt, C. Neuwirth, M. Weber
249 Screening von Kognitions- und Verhaltensänderungen bei der Amyotrophen LateralskleroseDie bisher gängigen neuropsycho logischen Tests zur Untersuchung fronto-temporaler Funktionen bei ALS wurden nicht eigens für die ALS-Erkrankten entwickelt und weisen dadurch keine An passung an die besonderen körperlichen Einschränkungen dieser Patienten auf. Die Durchführung und Auswertung wird hierdurch erschwert oder gar verunmöglicht. Mit dem ECAS wurde ein schnell und einfach anwendbares klinisches Screening-Tool entwickelt.
INHALTSVERZEICHNIS 233
Redaktion
Prof. Dr. Nicolas Rodondi, Bern (Chefredaktor); Dr. Nadja Pecinska, Basel (Managing editor); Prof. Dr. David Conen, Basel; Prof. Dr. Martin Krause, Münsterlingen; Prof. Dr. Klaus Neftel, Bern; Prof. Dr. Antoine de Torrenté, La Chaux-de-Fonds; Prof. Dr. Gérard Waeber, Lausanne; PD Dr. Maria Monika Wertli, Bern
Beratende Redaktoren
Prof. Dr. Reto Krapf, Luzern; Prof. Dr. Ludwig T. Heuss, Zollikerberg; Dr. Pierre Périat, Basel; Prof. Dr. Rolf A. Streuli, Langenthal
Advisory Board
Dr. Sebastian Carballo, Genève; Dr. Daniel Franzen, Zürich; Dr. Francine Glassey Perrenoud, La Chaux-de-Fonds; Dr. Markus Gnädinger, Steinach; Dr. Matteo Monti, Lausanne; Dr. Sven Streit, Bern; PD Dr. Ryan Tandjung, Zürich

Fallberichte
A. Koster-Rusch, J. Capraro, W. Nagel, T. Clerici, S. J. Stoeckli, F. Forrer, M. Brändle, S. Bilz
253 Bilaterale Halsschwellung bei einer jungen FrauParagangliome des Kopf- und Halsbereichs («Glomus tumoren») sind seltene neuroendokrine Tumoren, die von den parasympathischen Paraganglien ausgehen.
T. Rauer, T. De Zulueta, U. Caspar, B. Wagner, M. Zünd
256 Das histiozytäre SarkomDiese äusserst seltene und häufig aggressive Non-Langerhanszell-Histiozytose erfordert sowohl in der Diagnosestellung als auch in der Therapieplanung eine sehr enge interdisziplinäre Zusammenarbeit.
Extended abstracts from SMW
New articles from the online journal “Swiss Medical Weekly” are presented after page 258.
INHALTSVERZEICHNIS 234
ImpressumSwiss Medical Forum – Schweizerisches Medizin-ForumOffizielles Fortbildungsorgan der FMH und der Schweizerischen Gesellschaft für Innere Medizin
Redaktionsadresse: Ruth Schindler, Redaktionsassistentin SMF, EMH Schweizerischer Ärzteverlag AG, Farnsburgerstrasse 8, 4132 Muttenz, Tel. +41 (0)61 467 85 58, Fax +41 (0)61 467 85 56, [email protected], www.medicalforum.ch
Manuskripteinreichung online: http://www.edmgr.com/smf
Verlag: EMH Schweizerischer Ärzte-verlag AG, Farnsburgerstrasse 8, 4132 Muttenz, Tel. +41 (0)61 467 85 55,Fax +41 (0)61 467 85 56, www.emh.ch
Marketing EMH / Inserate: Dr. phil. II Karin Würz, Leiterin Marketing und Kommunikation, Tel. +41 (0)61 467 85 49, Fax +41 (0)61 467 85 56, [email protected]
Abonnemente FMH-Mitglieder: FMH Verbindung der Schweizer Ärztinnen und Ärzte, Elfenstrasse 18, 3000 Bern 15, Tel. +41 (0)31 359 11 11, Fax +41 (0)31 359 11 12, [email protected] Abonnemente: EMH Schweize-rischer Ärzteverlag AG, Abonnemente, Farnsburgerstrasse 8, 4132 Muttenz, Tel. +41 (0)61 467 85 75, Fax +41 (0)61 467 85 76, [email protected]: zusammen mit der Schweizerischen Ärzte- zeitung 1 Jahr CHF 395.– / Studenten CHF 198.– zzgl. Porto; ohne Schweize-rische Ärzte zeitung 1 Jahr CHF 175.– / Studenten CHF 88.– zzgl. Porto (kürzere Abonnementsdauern: siehe www.medicalforum.ch)
ISSN: Printversion: 1424-3784 / elektronische Ausgabe: 1424-4020Erscheint jeden Mittwoch
© EMH Schweizerischer Ärzteverlag AG(EMH), 2016. Das Swiss Medical Forum ist eine Open- Access-Publika tion von EMH. Entsprechend gewährt EMH allen Nutzern auf der Basis der Creative-Commons-Lizenz «Namensnennung – Nicht kommerziell – Keine Bearbei-tung en 4.0 International» das zeitlich unbeschränkte Recht, das Werk zu ver-viel fältigen, zu verbreiten und öffentlich zugänglich zu machen unter den Bedin-gungen, dass (1) der Name des Autors genannt wird, (2) das Werk nicht für kommerzielle Zwecke verwendet wird und (3) das Werk in keiner Weise bear-beitet oder in anderer Weise verändert wird. Die kommerzielle Nutzung ist nur mit ausdrück licher vorgängiger Erlaub-nis von EMH und auf der Basis einer schriftlichen Vereinbarung zulässig.
Hinweis: Alle in dieser Zeitschrift pu blizierten Angaben wurden mit der grössten Sorgfalt überprüft. Die mit Verfassernamen gezeichneten Ver-öffentlichungen geben in erster Linie die Auffassung der Autoren und nicht zwangsläufig die Meinung der SMF-Redaktion wieder. Die angegebenen Dosierungen, Indikationen und Appli-kationsformen, vor allem von Neuzu-lassungen, sollten in jedem Fall mit den Fachinformationen der verwende-ten Medikamente verglichen werden.
Herstellung: Schwabe AG, Muttenz, www.schwabe.ch
Titelbild: © CDC / James Archer

Und anderswo …?Antoine de Torrenté
Schweres heterogenes Emphysem: eine neue Behandlung?
FragestellungTrotz Optimierung der medikamentösen Be-handlungen sind Patienten mit schwerer COPD nach wie vor stark eingeschränkt. Bei einigen stark selektierten Patienten kann durch die chirurgische Entfernung der am schwersten betroffenen Lungenabschnitte eine Verbesserung der Lungenfunktion, des allgemeinen Gesundheitszustands und sogar der Überlebenszeit erzielt werden. Der an sehr schwachen Patienten vorgenommene Eingriff geht jedoch nach wie vor mit einer Mortalität von ca. 5% einher. Um eine OP zu vermeiden, können mittels Broncho skopie Einweg-Lungenventile eingesetzt werden, die die Überblähung des erkrankten Lungenflü-gels verringern und in manchen Fällen sogar dafür sorgen können, dass dieser kollabiert, was erwünscht ist. Idealerweise sollten die In-terlobien intakt sein, um die gewünschte Ent-lüftung des Lungenflügels zu gewährleisten. Ist diese Methode ungefährlicher als eine OP?
MethodeDie analysierten Patienten wiesen eine FEV1 von <50% des Sollwerts, ein Residualvolu-
men von >150% und eine 6-Minuten-Gehstre-cke von <450 m auf. Alle waren optimal medi-kamentös eingestellt. Im Lungen-CT mussten ein heterogenes Emphysem, ein fast vollstän-dig zerstörter, zu behandelnder Lungenflügel sowie ein intaktes Interlobium nachgewiesen worden sein. Die Studie war randomisiert und verblindet. Eine Gruppe erhielt ein oder mehrere Lungenventile und die Plazebo-gruppe eine Bronchoskopie ohne Lungenven-tilimplantation. Primärer Endpunkt war der prozentuale Unterschied der FEV1 zwischen Studieneinschluss und drei Monaten danach. Zu den sekundären Endpunkten zählten die Belastungsfähigkeit auf dem Fahrradergome-ter, die 6-Minuten-Gehstrecke und die Ver-besserung des Gesundheitszustands (Saint-George-Fragebogen für COPD-Patienten).
Resultate25 Patienten wurden durchschnittlich 3 Lun-genventile eingesetzt, und 25 dienten als Kon-trollgruppe. Aus der umfassenden Daten-sammlung ging hervor, dass die FEV1 drei Mona te nach Studieneinschluss um 8,7% zu-genommen hatte, gegenüber 2,8% in der Kon-trollgruppe, p = 0,032. Zudem war in der Lun-genventilgruppe ein signifikanter Anstieg der Belastungsfähigkeit und der Gehstrecke zu
beobachten. Es traten jedoch zahlreiche Kom-plikationen auf: 2 Pneumonien je Gruppe. In der Verumgruppe mussten 2 Lungenventile entfernt werden und 5 wurden spontan ausge-hustet. Ferner gab es in dieser zwei, in der Kon-trollgruppe hingegen keine Todesfälle.
Probleme und KommentarDie Studie hat gezeigt, dass die Exklusion eines ventilatorisch insuffizienten, gesundheitsge-fährdenden Lungenflügels durch Einweg-Lun-genventile die Lungenfunktion verbessern kann. Hervorzuheben ist der Mut der Patien-ten, die eine Scheinbronchoskopie über sich ergehen liessen. Ob die Lungenventilmethode der Resektion bezüglich Wirksamkeit oder Sicherheit überlegen ist, kann jedoch nicht eindeutig gesagt werden. Bei Betrachtung der FEV1-Zunahme wird deutlich, dass lediglich 5 von 25 Patienten, deren FEV1 um >50% zuge-nommen hat, tatsächlich von der Methode profitiert haben. Alles in allem stellt die Re-duktion des Lungenvolumens bei schwerer COPD eine Option dar. Die Wahl der Behand-lungsmethode hängt wahrscheinlich von der Erfahrung der behandelnden Ärzteteams ab.Davey C, et al. Lancet. 2015 Sep 12;386(9998): 1066–73.
Prävention kolorektaler AdenomeEpidemiologische Studien haben ergeben, dass ein hoher Serum-Vitamin-D-Spiegel und Kalzi-umsupplemente die Risiken für kolorektale Neoplasien verringern. ~2300 Patienten mit Adenomen ohne Restläsionen nach Kolo skopie wurden randomisiert und erhielten entweder 1000 IE Vitamin D sowie 1200 mg Kalzium/d oder keine Therapie. Bei einer Koloskopie 3–5 Jahre später wurden bei 43% der Patienten in beiden Gruppen Adenome entdeckt. Die «Prä-ventivbehandlung» hatte somit keinen Nut-zen. Womit dieses Thema geklärt wäre …Baron JA, et al. N Engl J Med. 2015 Oct 15;373(16):1519–30.
Medikamente und Internet: Chaos!Eine Schätzung der WHO hat ergeben, dass 50% der im Internet verkauften Medikamente nicht den pharmazeutischen Standards ent-sprechen: Zu geringer Wirkstoffgehalt und Kontaminationen verschiedenster Art. Schät-zungsweise 5 Millionen Amerikaner erwerben pro Jahr online Medikamente im Wert von 70–200 Milliarden Dollar. Hauptlieferanten: Indien und China. Dies könnte sowohl für Einzel personen als auch für die Volksgesund-
heit dramatische Folgen haben, vor allem bei Mala riamedikamenten.Clark F, Lancet. 2015 Oct 3;386(10 001):1327–8.
Spironolacton (Aldactone®) und therapierefraktäre Hypertonie300 Patienten mit therapierefraktärer Hyper-tonie (systolischer BD von >140 mm Hg in der Arztpraxis), die bereits eine Dreifachtherapie erhielten, wurde 12 Wochen lang zusätzlich zur gewohnten Behandlung Spironolacton (25–50 mg), Doxazosin (Cardura®), Bisoprolol (Concor®) oder Plazebo gegeben. Spironolac-ton senkte den systolischen BD signifikant um fast 9 mm Hg. Vorsicht vor Hyperkaliämie!Williams B, et al. Lancet. 2015 Sep 18. pii: S0140-6736(15)00257-3.
Harnsäurewert, Gicht und kardiovasku läres RisikoHyperurikämie und Gicht sind mit einem erhöh ten kardiovaskulären Risiko assoziiert. Von ca. 2650 aus einer Kohorte von 40 000 Pa-tienten stammenden Probanden erhielten 55% eine Behandlung mit einem harnsäure-senkenden Medikament, 45% blieben unbe-handelt. Die unbehandelten Patienten wiesen
ein erhöhtes kardiovaskuläres Todesrisiko auf, HR 2,4. Die behandelten Patienten hatten eine HR von 0,5 in Bezug auf die Gesamtsterb-lichkeit. Ist eine Behandlung von Patienten mit erhöhtem Harnsäurewert, jedoch ohne Gicht, ebenfalls ratsam? Wahrscheinlich …Chen JH, et al. J Rheumatol. 2015 Sep;42(9): 1694–701.
Langzeitauswirkungen von Eingriffen bei abdominalem AortenaneurysmaCa. 80 000 >65-jährige Patienten, die entweder eine endovaskuläre Prothese oder einer offe-nen Operation erhalten hatten, wurden gemat-ched. Die postoperative Sterblichkeit betrug bei einem endovask. Eingriff 1,6 gegenüber 5,2%. Die Zahl der nachträglich erforderlichen offenen Operationen sank von 2,2% im Jahr 2001 auf 0,3% im Jahr 2008. Während des 8-jährigen Follow-up trat jedoch bei 5,4% in der endovask. Gruppe gegenüber 1,4% eine Aortenruptur auf. Der Überlebensvorteil der endovask. Gruppe schwand nach 3-jährigem Follow-up und glich sich der Kurve bei offe-nem Eingriff an. Diese Zahlen sind wichtig für die Entscheidungsfindung!Shermerhorn ML, et al. N Engl J Med. 2015;373:328.
UND ANDERSWO …? 235
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):235

ÜBERSICHTSARTIKEL 236
Die häufigste infektiöse Ursache nosokomialer Diarrhoe
Update zu Clostridium difficileAna Durovic, Sarah Tschudin-Sutter
Klinik für Infektiologie und Spitalhygiene, Universitätsspital Basel
Clostridium difficile ist die häufigste infektiöse Ursache nosokomialer Diarrhoe und gewinnt zunehmend an Bedeutung als Erreger ambulant erworbener Durchfallerkrankungen. C. difficile-Infektionen sind mit hoher Morbidität und Mortalität assoziiert und gehen mit einem breiten Spektrum klinischer Symptome – von milder Diarrhoe bis hin zu schwerer toxischer Kolitis – einher. Schwere klinische Verlaufsformen und die hohe Rezidivrate stellen besondere Herausforderungen der Therapie dar.
Grundlagen
Einführung/HintergrundEine Clostridium difficile-Infektion (CDI) versursacht akute Diarrhoe und Kolitis. Ihr Erreger wurde erstmals 1935 als Bestandteil der normalen intestinalen Flora bei Neugeborenen entdeckt. Erst 1978 gelang es, dieses Grampositive anaerobe Stäbchen als Verursacher der pseudomembranösen Kolitis zu identifizieren. Seine Fähigkeit zur Sporenbildung ermöglicht diesem Bakterium, selbst bei ungünstigen Umweltbedingungen über Monate auf inerten Oberflächen zu überleben. Die meisten Desinfektionsmittel sowie Antibiotika sind nicht in der Lage, diese Dauerformen zu zerstören, so dass lebensfähige Keime an den Händen des Pflegepersonals oder auf Oberflächen eine potentielle Infektionsquelle im Spital darstellen. Nach der Aufnahme von Sporen gehen diese im Kolon in ihre vegetative, toxinproduzierende Form über und können dort ihren pathogenen Effekt auf den Wirt ausüben. Der wichtigste Risikofaktor für die Entwicklung einer CDI ist eine vorangegangene AntibiotikaExposition, insbesondere mit Cephalosporinen, Chinolonen, Penicillinen und Clindamycin. Diese verändern die Zusammensetzung der normalen intestinalen Darmflora und verschaffen somit C. difficile – das gegenüber einer grossen Anzahl verschiedener Antibiotika resistent ist – einen Überlebensvorteil. Weitere Risiko fak toren sind der Erhalt von Antazida (Protonenpumpeninhibitoren und H2RezeptorAntagonisten), vorangegangene Hospitalisationen, Hospitalisationsdauer, Alter, schwerwiegende Grunderkrankungen, viszeralchirurgische Eingriffe und Sondenernährung.Im Kolon produziert C. difficile vor allem zwei Toxine, Toxin A und B, die eine starke neutrophile Entzün
dungsreaktion verursachen. Diese führt zu Diarrhoe, Erosionen der Darmmukosa sowie der Ausbildung von Pseudomembranen.Die meisten Stämme produzieren gleichzeitig beide Toxine, jedoch reicht bereits eines aus, um eine Diarrhoe oder Kolitis auszulösen. Einige Stämme produzieren zusätzlich das binäre Toxin. Es wird häufiger von «hypervirulenten» Ribotypen produziert und gilt daher als Virulenzfaktor, auch wenn seine Relevanz bisher noch unklar ist. Diejenigen Stämme, die nicht in der Lage sind, Toxine zu produzieren, können den Darm zwar besiedeln, sind aber apathogen. Ana Durovic
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):236–240

ÜBERSICHTSARTIKEL 237
EpidemiologieSeit Beginn des 21. Jahrhunderts hat sich die Epidemiologie der CDI deutlich verändert. Insbesondere in den USA und Kanada wurden zunehmend Epidemien von besonders schwer verlaufenden CDI in vielen Spitälern
beobachtet, die auf einen spezifischen Stamm, den sogenannten hypervirulenten PCRRibotyp 027, zurückgeführt werden konnten. Bei der PCRRibotypisierung handelt es sich um ein molekulares Typisierungsverfahren, das verwendet wird, um die Ausbreitung verschie dener Stämme von C. difficile untersuchen zu können. Der PCRRibotyp 027 breitete sich anschliessend erfolgreich in Grossbritannien und weiteren europäischen Ländern aus. In den letzten Jahren konnte auch eine deutliche Zunahme von ambulant erworbenen CDI dokumentiert werden. Als mögliche Ursachen werden unter anderem kürzere Hospitalisationen und zunehmende Verabreichung von Antibiotikatherapien im ambulanten Bereich diskutiert. Hypervirulente Stämme werden möglicherweise mit CDI bei Patienten mit geringem Risikoprofil, wie postpartalen Frauen und Kindern, in Verbindung gebracht.
KlinikDas klinische Spektrum einer CDI reicht von asymptomatischer Kolonisation bis hin zu schwersten septischen Zustandsbildern mit toxischem Megakolon. Der Schweregrad der Diarrhoe variiert von minimal bis profus. Fieber, abdominelle Schmerzen und Krämpfe sind mögliche Begleitsymptome. Im Blutbild kann oftmals eine Leukozytose dokumentiert werden. Tabelle 1 fasst wichtige Definitionen sowie die Klinik der CDI zusammen.
Diagnostik [1]Der Hauptpfeiler der CDIDiagnostik ist der Nachweis von Toxin A oder B im Stuhl. Als Referenzmethode gilt der Ansatz einer Selektivkultur mit Toxinnachweis in der Zellkultur. Durch den enormen Zeit und Arbeitsaufwand stehen die Resultate meist erst nach 48 Stunden zur Verfügung. Diese Methode wurde daher zugunsten von schnelleren und günstigeren Verfahren weitgehend verlassen. Mittlerweile haben sich die Enzymimmunoassays (EIA) als Alternative etabliert. Mit diesen standardisierten Tests können direkt aus dem Stuhl innerhalb einer Stunde ein ClostridienAntigen (Glutamatdehydrogenase = GDH) und teilweise auch Toxine nachgewiesen werden. Der Nachweis von GDH ist sehr sensibel (Tab. 2); das Enzym wird aber auch von nicht toxinbildenden C. difficileStämmen gebildet und kann daher nicht als alleiniger Test eingesetzt werden. Die Sensitivität des Toxinnachweises mittels EIA ist für eine zuverlässige Diagnostik inakzeptabel tief (Tab. 2), so dass die aktuellen Guidelines ein ZweiStufenModell zur Abklärung empfehlen. Hierbei sind verschiedene Testkombinationen möglich. Eine Option besteht darin, jede Stuhlprobe zunächst auf GDH zu untersuchen und positive Proben zusätzlich auf das Vorhan
Tabelle 2: Sensitivität und Spezifität der verschiedenen diagnostischen Tests zum Nachweis von Clostridium difficile [1].
Sensitivität Spezifität
Nachweis von C. difficile (Toxin-positive und -negative Stämme)
EIA zum Nachweis der GDH 0,71–1,00 0,67–1,00
Nachweis von Toxinen
EIA zum Nachweis der Toxine A und/oder B 0,31–0,99 0,65–1,00
RT-PCR zum Nachweis des Toxin-B-Gens 0,86–1,00 0,94–1,00
Abkürzungen: EIA = Enzymimmunoassay; GDH = Glutamatdehydrogenase; RT-PCR = realtime polymerase chain reaction.
Tabelle 1: Definitionen und Klinik der Clostridium difficile-Infektion (CDI) [2].
Klinik der CDI – Diarrhoe in variabler Ausprägung mit/ohne systemische Reaktion.– Typischerweise im Zusammenhang mit Antibiotikatherapie. – Schwere Verläufe mit pseudomembranöser Kolitis, toxischem
Megakolon, Ileus.
CDI-Episode – CDI-typische Klinik mit Nachweis von freien Toxinen und C. difficile im Stuhl, ohne Hinweise für eine andere Ursache der Diarrhoe.
– Histologischer/koloskopischer Nachweis von pseudomembranöser Kolitis.
CDI-Rezidiv – Erneutes Auftreten einer symptomatischen CDI-Episode innerhalb von 8 Wochen nach Beginn der ersten Episode (vorausgesetzt, dass die Symptome der initialen Episode nach Therapie sistierten).
Milde CDI – Diarrhoe durch C. difficile ohne Symptome einer schweren/ komplizierten Infektion.
Patienten ohne Zeichen einer schweren CDI mit Alter ≥65 Jahre, mit schweren Grundkrankheiten, intensivmedizinischer Behandlung oder Immunsuppression haben ein erhöhtes Risiko, einen schweren Ver-lauf zu erleiden.
Schwere CDI – CDI-Episode begleitet von ≥1 Symptom einer schweren Kolitis oder von einem komplizierten Verlauf der CDI mit einer schweren systemischen Entzündungsreaktion:
Klinik:1. Fieber >38,5 °C, Schüttelfrost2. Hämodynamische Instabilität oder respiratorisches Versagen3. Klinische Zeichen einer Peritonitis oder eines Ileus
Labor:1. Leukozytose >15× 109/l , Linksverschiebung >20% stabkernige
Neutrophile2. Anstieg des Serum-Kreatinins um >50% des Ausgangswertes3. Tiefes Serum-Albumin <30 g/l4. Anstieg des Serum-Laktats ≥5 mmol/l
Koloskopie oder Sigmoskopie:Endoskopischer Nachweis einer pseudomembranösen Kolitis
Bildgebung:Verdickung der Kolonwand, Dilatation der Kolonschlingen, Fett-gewebsimbibierung um das Kolon, Aszites (ohne andere Ursache)
Schwere CDI mitKomplikationen
– Schwere CDI mit schwerer systemischer Reaktion und Schock, die eine intensivmedizinische Behandlung, Kolektomie oder Tod zur Folge hat.
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):236–240

ÜBERSICHTSARTIKEL 238
densein der Toxine A und B zu testen. Immer öfter wird zur Diagnostik auch die RTPCR (realtime poly-merase chain reaction) eingesetzt, die mittlerweile als kommerzielles System erhältlich ist und mit hoher Zuverlässigkeit toxinproduzierende C. difficile nachweist (Tab. 2).Wichtige Voraussetzung zur Interpretation der Laborbefunde ist die klinische Präsentation. C. difficile kann trotz erfolgreicher Therapie während einiger Zeit im Stuhl persistieren, bei einigen Personen ist der Keim sogar Teil der Normalflora. Untersuchungen von Stuhl mit normaler Konsistenz, insbesondere zur Therapiekontrolle, bringen daher kaum Informationsgewinn und sollten aus ökonomischen Überlegungen vermieden werden.
Therapie [2]Die Behandlungsstrategie einer CDI richtet sich nach dem klinischen Schweregrad. Grundsätzlich sollte bei jeder Diagnosestellung die auslösende Antibiotikatherapie kritisch reevaluiert und bei fehlender Indikation zur Fortführung sistiert werden. Volumen und Elektrolyte müssen substituiert und antiperistaltische Medikamente vermieden werden. Des Weiteren sollte die Indikation allfälliger Antazida überprüft werden. Die Standardtherapie leichter Verlaufsformen bleibt weiterhin die orale Gabe von Metronidazol oder Vancomycin (Tab. 3). Zur Behandlung der schweren CDI wird Vanconmycin aufgrund seiner pharmakologischen Eigenschaften dem Metronidazol als überlegen beurteilt. Die gängigen Therapien sind aber mit Rezidivraten von bis zu 20% noch nicht ideal. Nicht zuletzt verzögern die beiden Antibiotika aufgrund ihres Wirkspektrums selbst die Rekonstitution der Mikroflora. Eine Therapie mit Probiotika wird aktuell nicht empfohlen.
PräventionEin rationaler Einsatz von Antibiotika ist die wahrscheinlich wichtigste Massnahme zur Prävention einer C. difficileInfektion. Ergänzend sollen insbesondere bei stationären Patienten Hygienemassnahmen (insbesondere konsequente Händehygiene) den Kontakt mit dem Bakterium verhindern. Bei bekannter CDIInfektion können das Tragen von Handschuhen und Einwegschürzen bei Kontakt mit dem Patienten eine Transmission zusätzlich verhindern. Toiletten sollten mindestens bis zur Normalisierung der Stuhlkonsistenz nicht von anderen Personen mitbenutzt werden. Idealerweise werden diese Patienten im Einzelzimmer gepflegt.Präventive Therapien oder Screening asymptomatischer Träger werden aktuell nicht empfohlen.
Neue Erkenntnisse und aktuelle Entwicklungen
Inzidenz und Prävalenz International werden grosse Anstrengungen unternommen, um die Tragweite von CDI zu erfassen. Die bisher publizierten Zahlen zeigen eine anhaltend hohe Inzidenz an Infektionen weltweit. In einer kürzlich durchgeführten Studie betreffend die Prävalenz nosokomialer Infektionen in den USA wurde C. difficile als häufigster Erreger erfasst [3]. 2008 ergab eine epidemiologische Studie in 34 Ländern Europas eine durchschnittliche Inzidenz von 4,1 pro 10 000 Patiententage. Die Schweiz lag leicht über dem europäischen Durchschnitt (4,8/10 000). Die erhobenen Raten variierten insgesamt stark zwischen den Ländern und den teilnehmenden Spitälern [4].Grosser Schwachpunkt solcher statistischer Auswertungen ist die rasch wechselnde Epidemiologie der CDI. Zudem ist man auf konsequente und korrekte Abklärungen angewiesen, um das wahre Ausmass der Krankheit abschätzen zu können. Die kürzlich publizierte EUCLIDStudie [5] zeigt ein grosses Optimierungspotential: Gemäss Hochrechnung werden europaweit 23% der CDI nicht diagnostiziert – schlicht, weil keine Indikation zur Abklärung gestellt wird. Dieses Problem wird zusätzlich durch den Einsatz ungenügender Labormethoden in einigen Kliniken verschärft. Die rapportierte Infektionsrate der teilnehmenden Länder für die Jahre 2011/12 lag im Schnitt bei 7 Fällen pro 10 000 Patiententage. Die wahre Infektionsrate für denselben Zeitraum dürfte aber aufgrund des hohen Prozentsatzes der nicht diagnostizierten CDI wesentlich höher sein. Vor diesem Hintergrund wird deutlich, dass das wahre Ausmass von C. difficile wahrscheinlich weitgehend unterschätzt wird.
Epidemiologischer ShiftVermutlich ist der Anteil an ambulant erworbener CDI höher als bisher angenommen. Diese ausserhalb des Spitals erworbenen Infektionen betreffen eher jüngere Patienten mit geringerer Exposition gegenüber Anti bio tika oder anderen Risikofaktoren. Neu sind auch Berichte über CDI bei Schwangeren oder Kindern. Die Gründe für diese Verschiebungen sind noch nicht geklärt. Möglich sind neue Reservoirs (Lebensmittel, Tiere) aber auch Selektionsvorteile bestimmter Stämme.
Korrelation von Ribotyp und KlinikVermehrtes Augenmerk auf die Typisierung isolierter C. difficile-Stämme bringt neue Informationen über deren Eigenschaften und klinische Präsentation.
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):236–240

ÜBERSICHTSARTIKEL 239
GenomVergleiche über mehrere Jahre zeigen eine gros se Diversität und einen regen Wechsel der dominierenden Ribotypen. Inwiefern der Ribotyp mit der Schwere der Infektion korreliert, kann zurzeit nicht eindeutig beantwortet werden. Die bisherigen Berichte über hypervirulente Stämme mit höherer Inzidenz und schlechterem Outcome stammen grösstenteils aus Daten, die während Ausbrüchen gesammelt wurden. Analysen dieser Stämme ohne zeitnahe Häufung konnten bisher keinen sicheren Zusammenhang zwischen Ribotyp, Schwere der Krankheit und Mortalität herstellen. Als hypervirulent gelten hauptsächlich die PCRRibotypen 027 und 078. Der Stamm NAP1/027/B1 (oder PCRRibotyp 027) konnte für einige Epidemien in Kanada und Amerika zu Beginn des Jahrtausends verantwortlich gemacht werden und ist seither mit schwererem Krankheitsbild und höheren Rezidivraten assoziiert. Inzwischen wurde der Stamm weltweit als Ursache von CDI nachgewiesen. Der PCRRibotyp 078 scheint in Europa gehäuft vorzukommen und wird vor allem mit CDI bei jüngeren Patienten, ambulant erworbenen In
fektionen sowie CDI bei Tieren in Verbindung gebracht. Standardisierte Messungen in vitro entkräften zunehmend das bisherige Konzept der Hypervirulenz durch exzessive Toxinproduktion. Auffallend hingegen ist die signifikant grössere Menge an Sporen, was die erfolgreiche Ausbreitung und erhöhte Rezidivraten solcher Stämme erklären könnte.Eine einmalige Gelegenheit zum direkten Vergleich bot sich während eines Ausbruchs mit zeitgleichem Auftreten von zwei verschiedenen Ribotypen. Einer dieser Stämme (PCRRibotyp 027) gilt als hypervirulent, der andere (PCRRibotyp 017) trägt keine Virulenzmarker. Dennoch war die 30TageMortalitätsrate für beide Stämme hoch (017: 23%; 027: 26%). Infektionen mit anderen, nichtepidemischen Stämmen zeigten hingegen eine Mortalität von nur 3%. Die Autoren spekulieren, dass die erhöhte Mortalität nicht durch den Ribotyp an sich zu erklären sei, sondern eher Ausdruck einer schweren Grundkrankheit mit erhöhter Infektanfälligkeit ist. Aufgrund der Widersprüchlichkeit der bestehenden Daten betreffend die Assoziation zwischen verschiedenen Ribotypen und klinischem Schweregrad der Erkrankung empfiehlt die European Society of Clinical Micro biology and Infectious Diseases (ESCMID) aktuell, die Therapie der CDI der Klinik und nicht dem auslösenden Stamm anzupassen [2].
Neue TherapieoptionenFidaxomicin ist seit Mai 2014 in der Schweiz zugelassen. Es gehört zu einer neuen Antibiotikaklasse der Makrozykline. Durch Hemmung der RNAPolymerase wirkt es bakterizid gegen C. difficile mit engem Wirkspektrum und hohen Konzentrationen im Stuhl. In vitro vermag es die Sporen und Toxinbildung von C. difficile zu hemmen. In zwei randomisierten Studien konnte nachgewiesen werden, dass zur Behandlung der CDI Fidaxomicin im Vergleich zu Vancomycin nicht weni ger effektiv war, bei jedoch signifikant tieferer Rezidivrate [2]. Da Fidaxomicin kaum systemisch resorbiert wird, zeichnet sich diese Substanz durch ein günstiges Interaktions und Nebenwirkungsprofil aus. Erbrechen, Übelkeit und Obstipation wurden als Neben wirkungen am häufigsten dokumentiert. Eine gleichzeitige Verabreichung von Ciclosporin, Ketoconazol, Erythromycin, Clarithromycin, Verapamil, Dronedaron und Amiodaron wird nicht empfohlen (www.compendium.ch). Die Kosten von Fidaxomicin sind im Vergleich zu den Standardtherapien höher, weshalb der Einsatz dieser Substanz in der Schweiz bei Patienten mit einer milden CDI zurzeit nicht zu empfehlen, jedoch insbesondere bei Patienten mit Rezidivgefahr zu erwägen ist (Tab. 3). In den europäischen Richtlinien
Tabelle 3: Empfehlungen zur Behandlung einer Clostridium difficile-Infektion (CDI) [2].
Schweregrad Empfohlene Therapie
Milde CDI Antibiotische TherapieMetronidazol 3× 500 mg per os für 10 Tage (Evidenzgrad A-I) oderVancomycin 4× 125 mg per os für 10 Tage (Evidenzgrad B-I) oder
Keine antibiotische Therapie (Evidenzgrad C-II)Voraussetzung: Nicht-epidemische Situation und klarer Zusammenhang mit einer auslösenden antibiotischen Therapie– Stopp der auslösenden Therapie– Engmaschige klinische Verlaufsbeobachtung über 48 Stunden– Beginn einer antibiotischen Therapie bei klinischer Verschlechterung
Schwere CDI Vancomycin 4× 125 mg per os für 10 Tage (Evidenzgrad A-I) oderFidaxomicin 2× 200 mg per os für 10 Tage (Evidenzgrad B-I)
Erstes Rezidiv oder erhöhtes Rezidivrisiko
Vancomycin 4× 125 mg per os für 10 Tage (Evidenzgrad B-I) oderFidaxomicin 2× 200 mg per os für 10 Tage (Evidenzgrad B-I) oderMetronidazol 3× 500 mg per os für 10 Tage (Evidenzgrad C-I)
Mehrfach rezidivierende CDI
Vancomycin 4× 125 mg per os für 10 Tage (Evidenzgrad B-II)gefolgt von einer Pulstherapie/Tapering* oderFidaxomicin 2× 200 mg per os für 10 Tage (Evidenzgrad B-II)
Bei fehlendem Ansprechen nach wiederholter antibiotischer Therapie ist eine Stuhltrans- plantation in Kombination mit einer oralen antibiotischen Therapie empfohlen (Evidenzgrad A-I)
Orale Therapie nicht möglich
Milde CDIMetronidazol 3× 500 mg intravenös für 10 Tage (Evidenzgrad A-II)
Schwere CDIMetronidazol 3× 500 mg intravenös für 10 Tage (Evidenzgrad A-II) kombiniert mit Vancomycin-Einläufen 500 mgin 100 ml NaCl 4×/d oder kombiniert mit Vancomycin 500 mg in 100 ml NaCl 4×/d über die Magensonde für 10 Tage (Evidenzgrad B-III)
* Pulstherapie: Vancomycin 125–500 mg/Tag alle 2–3 Tage für mindestens 3 Wochen.Tapering: graduelle Reduktion der Dosis auf 125 mg/Tag.
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):236–240

ÜBERSICHTSARTIKEL 240
wird Fidaxomicin, mit einem geringeren Evidenzgrad als Metronidazol, wie Vancomycin als Alternative zur Behandlung einer milden CDI empfohlen [2]. Die Verabreichung monoklonaler Antikörper, die gegen die C. difficileToxine A und B gerichtet sind, zusätzlich zur Standardtherapie mit Metronidazol oder Vancomycin, zeigte ebenfalls eine signifikante Reduktion der Rezidivrate – aktuell ist die Evidenz zur Anwendung noch ungenügend [2]. Übertragung von Stuhl gesunder Spender führt zu einer raschen Verbesserung der normalen Darmfunktion. Mit Erfolgsraten von >90% etabliert sich die Stuhltransplantation zunehmend als Therapiealternative bei rezidivierenden CDIEpisoden [2]. Gelingt es, die dafür verantwortlichen Faktoren und Bakterienarten zu identifizieren, würden sich damit immense Ansätze zur gezielteren Therapie und Prophylaxe von CDI und anderen Darmerkrankungen ergeben.
Neues zur PräventionImpfstoffe zur Prävention von CDI werden aktuell erforscht. Grundlage hierfür bilden Studien, die einen Zusammenhang zwischen asymptomatischer Kolonisation mit C. difficile und hohen IgGAntikörpern gegen C. difficileToxin A aufzeigen konnten. Auch haben Patien
ten, die während einer CDI eine Immunantwort gegen Toxin A ausbilden, ein tieferes Rezidivrisiko. Verschiedene Forschungsgruppen entwickeln daher Impfstoffe, die eine anhaltend hohe Konzentration an Antikörpern gegen C. difficile-Toxine im Serum erzielen sollen. Ein alternativer Ansatz besteht in der oralen Verabreichung inaktivierter bakterieller Sporen, die eine mukosale Immunabwehr gegen C. difficile herbeiführen soll [6].
Ausblick
C. difficile ist nicht nur einer der häufigsten Keime der im Spital erworbenen Infektionen, sondern hat sich auch als wichtiger DurchfallErreger im ambulanten Bereich durchgesetzt. Eine der wesentlichen Massnahmen zur Prävention ist die Umsetzung eines restriktiven Antibiotikaeinsatzes in allen Bereichen.Die dynamische Epidemiologie von C. difficile wird uns auch in Zukunft herausfordern. Die Verbreitung verschiedener Ribotypen und deren Assoziationen mit dem klinischen Schweregrad der Erkrankung müssen weiterhin erforscht werden, da das Verständnis solcher Zusammenhänge die Grundlage für neue Therapieund Präventionsempfehlungen bildet.Neue Therapieoptionen, wie Fidaxomicin, monoklonale Antikörper und Stuhltransplantation, sind vielversprechende Optionen zur Reduktion der hohen Rezidivrate der CDI. Zukünftige Behandlungsempfehlungen müssen sich daher mit der Frage auseinandersetzen, welche Patienten am meisten von solch neuen Therapien profitieren können.
Disclosure statementS. TschudinSutter ist ein Mitglied des Fidaxomicin Advisory Board (Astellas) und hat Forschungsfonds von Astellas, Pfizer International Operations und dem Schweizerischen Nationalfonds erhalten.
TitelbildCDC / James Archer.
Literatur1 Crobach MJ, Dekkers OM, Wilcox MH, Kuijper EJ. European Society
of Clinical Microbiology and Infectious Diseases (ESCMID): data review and recommendations for diagnosing Clostridium difficileinfection (CDI). Clin Microbiol Infect. 2009;15:1053–66.
2 Debast SB, Bauer MP, Kuijper EJ, Committee. European Society of Clinical Microbiology and Infectious Diseases: update of the treatment guidance document for Clostridium difficile infection. Clin Microbiol Infect 2014;20 Suppl 2:1–26.
3 Magill SS, Edwards JR, Bamberg W, Beldavs ZG, Dumyati G, Kainer MA, et al. Multistate pointprevalence survey of health careassociated infections. N Engl J Med. 2014;370:1198–208.
4 Bauer MP, Notermans DW, van Benthem BH, Brazier JS, Wilcox MH, Rupnik M, et al. Clostridium difficile infection in Europe: a hospitalbased survey. Lancet. 2011;377:63–73.
5 Davies KA, Longshaw CM, Davis GL, Bouza E, Barbut F, Barna Z, et al. Underdiagnosis of Clostridium difficile across Europe: the European, multicentre, prospective, biannual, pointprevalence study of Clostridium difficile infection in hospitalised patients with diarrhoea (EUCLID). Lancet Infect Dis. 2014;14:1208–19.
6 Ivarsson ME, Leroux J, Castagner B. Investigational new treatments for Clostridium difficile infection. Drug Discov Today 2014.
Korrespondenz: PD Dr. med. Sarah TschudinSutter, MSc FMH Infektiologie und Innere Medizin Klinik für Infektiologie und Spitalhygiene Universitätsspital Basel Petersgraben 4 CH4031 Basel Sarah.Tschudin[at]usb.ch
Das Wichtigste für die Praxis • Clostridium difficile ist einer der wichtigsten Erreger nosokomialer In-
fektionen und eine der Hauptursachen der im Spital erworbenen Diar-
rhoe.
• In den letzten Jahren hat der Anteil ambulant erworbener C. difficile-In-
fektionen (CDI) deutlich zugenommen; Patienten mit geringem Risiko-
profil können auch ohne vorangegangene Antibiotikaexposition betrof-
fen sein.
• Die aktuelle Datenlage zur Korrelation von Ribotyp mit Krankheits-
schwere ist widersprüchlich. Die Therapie sollte sich deshalb weiterhin
nach dem klinischen Schweregrad richten.
• Der wichtigste Risikofaktor für die Entwicklung einer CDI ist eine voran-
gegangene Antibiotikaexposition. Weitere Risikofaktoren sind der Er-
halt von Antazida, Hospitalisation, Alter, schwerwiegende Grunderkran-
kungen, viszeralchirurgische Eingriffe und Sondenernährung.
• Mikrobiologische Untersuchungen sind nur bei symptomatischen Pati-
enten sinnvoll. Der Hauptpfeiler der CDI-Diagnostik ist der Nachweis
von Toxin A oder B im Stuhl. Die Sensitivität des Toxinnachweises mit-
tels Enzymimmunoassay (EIA) ist für eine zuverlässige Diagnostik in-
akzeptabel tief, so dass die aktuellen Guidelines ein Zwei-Stufen-Modell
zur Abklärung empfehlen.
• Die Behandlung wird der Krankheitsschwere angepasst. Neue Therapie-
optionen richten sich hauptsächlich gegen Rezidive.
• Ein rationaler und restriktiver Einsatz von Antibiotika ist die wirksamste
präventive Massnahme. Zusätzliche Hygienemassnahmen helfen, Über-
tragungen im Spital zu verhindern.
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):236–240

ÜBERSICHTSARTIKEL 241
Ein Enzymdefekt mit unterschiedlichen Folgen
Glucose-6-Phosphat-Dehydro-genase-MangelSarah Hofmanna, b, Andreas Buserc, Anne Taegtmeyera
a Klinik für Klinische Pharmakologie & Toxikologie, Universitätsspital Baselb Klinik für Innere Medizin, Universitätsspital Baselc Blutspendezentrum beider Basel & Klinik für Hämatologie, Universitätsspital Basel
Immer häufiger kommen Patienten in die Praxis und ins Spital, die einen G6PD-Mangel aufweisen und bei denen es wichtig ist zu wissen, welche Medikamente poten tiell gefährlich sind. In diesem Übersichtsartikel werden der G6PD-Mangel, die Diagnostik und die Medikamente, die zu vermeiden sind, beschrieben.
Einführung
Glucose-6-Phosphat-Dehydrogenase-Mangel (G6PD-Mangel) ist der häufigste angeborene Enzymdefekt welt-weit. Circa 400 Millionen Menschen sind davon be-troffen mit unterschiedlicher klinischer Manifestation. Durch die Globalisierung und das Migrations- und Rei-severhalten kann auch in der Schweiz immer häufiger ein G6PD-Mangel beobachtet werden.
Geschichte und Epidemiologie
Erstmals wurde eine hämolytische Anämie auf Prima-quin – das damals einzige Malariamedikament – 1926 von Cordes beschrieben [1]. Es dauerte weitere 20 Jahre, bis man während des 2. Weltkriegs, des Koreakriegs und Indochinakriegs aufgrund der Massenanwendung von Primaquin an Armeeangehörigen von regelmässi-gem Auftreten einer Hämolyse unter Primaquin berich-tete, und diese als «primaquin-sensitive syndrome» be-zeichnet wurde [2]. Erst Mitte der 50er Jahre wurde der Enzymdefekt und der Pathomechanismus durch Car-son und sein Team entdeckt [3]. Mittlerweile sind circa 400 Enzymvarianten des G6PD-Enzyms und mehr als 180 Mutationen des Gens bekannt. Die WHO-Klassifi-kation beruht auf der Enzymaktivität. Dabei liegt in der WHO-Klasse I ein schwerer Enzymmangel (<10% normale Enzymaktivität) vor, was sich als chronische hämolytische Anämie präsentiert. Bei Klasse II liegt ebenfalls ein schwerer Enzymdefekt vor, jedoch mit klinisch intermittierender hämolytischer Anämie. Ein mäs siger Enzymdefekt (10–60% normale Enzymaktivi-tät) liegt bei Klasse III vor, wobei es dabei meist im Rah-men einer Infektion oder Medikamentenexposition zu
einer hämolytischen Anämie kommt. Die Klassen IV und V sind klinisch nicht relevant und weisen eine nahe zu normale Enzymaktivität bzw. eine gesteigerte Aktivität auf.Der Enzymdefekt wird X-chromosomal-rezessiv vererbt. In Subsahara-Afrika ist die Variante G6PD A am häu-figsten zu finden. 10–15% der Bevölkerung aus West- und Zentralafrika sind davon betroffen; eine ähnliche Prävalenz findet sich bei Afroamerikanern. Bei Kauka-siern liegt vorwiegend die G6PD-Mediterranean-Variante vor; eine Häufung ist in den Küstenregionen von Sar-dinien und Griechenland (20–35%) sowie im Mittleren Sarah Hofmann
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):241–244

ÜBERSICHTSARTIKEL 242
Osten (kurdische Juden 60–70%) zu finden. Auch in Südostasien gibt es ein unterschiedliches Vorkommen (Abb. 1) [4]. Aufgrund der hohen Prävalenz in Regionen, in denen Malaria endemisch vorkommt (bzw. vorge-kommen ist), wird postuliert, dass der G6PD-Mangel ei-nen Selektionsvorteil gegen die Infektion mit Plasmo-dium falciparum aufweist. Verschiedene Autoren konnten diesen Zusammenhang zeigen [5–7]. Der Me-chanismus, der für die Hemmung des Parasitenwachs-tums respektive erhöhte Phago zytose der infizierten Erythrozyten mit dem G6PD-Mangel verantwortlich ist, ist nach wie vor unbekannt.
Pathomechanismus
Die Glucose-6-Phosphat-Dehydrogenase schützt über den Pentosephosphatweg die Zelle vor einem oxidati-ven Schaden (Abb. 2). Da die Erythrozyten keinen ande-ren Weg als den Pentosephosphatweg zur Gewinnung von NADPH kennen, kommt es bei einem Mangel des
Enzyms durch Oxidantien zu Schädigungen an Zellbe-standteilen und zur Hämolyse. Die Glucose-6-Phos-phat-Dehydrogenase katalysiert den ersten Schritt, sie oxidiert Glucose-6-Phosphat zu 6-Phos phogluconat und reduziert Nicotinamidadenindinu cleotid phos-phat (NADP) zu NADPH. NADPH reduziert Glutathion-disulfid (GSSG) zu Glutathion (GSH). Re duziertes Gluta-thion ist das wichtigste Antioxidans und hat die Fähigkeit, das durch Oxidantien generierte Wasser-stoffperoxid (H2O2) zu Wasser zu reduzieren und somit unschädlich zu machen.Liegt ein G6PD-Mangel vor, so kommt es zur Kumula-tion von Oxidantien und Schädigung der Erythrozyten. Verschiedene Faktoren beeinflussen den Schweregrad der Hämolyse:1. Alter der Erythrozyten: Das normal funktionierende
G6PD-Enzym hat eine Halbwertszeit von 62 Tagen. Junge Erythrozyten weisen eine noch nahezu nor-male G6PD-Aktivität auf. Patienten mit einer G6PD-A-Variante beispielsweise haben normalerweise eine milde Hämolyse (WHO-Klasse III), die sich auf ältere Erythrozyten beschränkt, da bei ihnen die Halb-wertszeit von G6PD auf 13 Tage reduziert ist. Die G6PD Mediterranean-Variante ist instabiler, mit einer G6PD-Halbwertszeit von Stunden und entsprechend stärker ausgeprägter Hämolyse (WHO-Klasse II) [8].
2. Dosis des Medikaments: Ist ein Medikament der Aus-löser für den oxidativen Stress, so ist die Dosis mit-verantwortlich für das Ausmass der Hämolyse. Bei-spielsweise kann die Einnahme von 45 mg Primaquin zu einer schweren Hämolyse führen, während unter einer Therapie mit 15 mg die Hämolyse klinisch kaum relevant sein kann [9].
3. Genetik: Es gibt 187 bekannte Mutationen im G6PD-Gen und mindestens 35 der mutierten Allele sind poly morph. Auch das X-chromosomal-rezessive Ver-erbungsmuster führt zu unterschiedlich ausgepräg-tem Schweregrad, je nachdem, ob ein homozygoter oder ein heterozygoter Enzymmangel bei der Frau bzw. beim Mann vorliegt [10].
Klinik
Verschiedene Auslöser können zu einem oxidativen Stress führen: Infektionen, Chemikalien, Medikamente und Nahrungsmittel (z.B. Favabohnen). In der Regel kommt es nach 5–72 Stunden zur Hämolyse mit Ikterus, Müdigkeit, Blässe und Hämaturie. Zusätzlich können auch Bauch-/Rücken- und/oder Kopfschmerzen sowie Nausea und Fieber auftreten. Diese Symptome können unterschiedlich stark ausgeprägt sein, das Spektrum reicht von leichteren hämolytischen Schüben bis hin zu transfusionsbedürftigen hämolytischen Krisen. Eine
Abbildung 1: Verteilung des G6PD-Mangels. Die Prävalenz in der Schweiz beträgt <0,5%.
Quelle: [21]. Nachdruck mit freundlicher Genehmigung von Elsevier.
<0–5%0,5–2,9%3–6,9%7–9,9%10–11,9%15–26%
Abbildung 2: Pentosephosphatweg. Elimination von Oxidantien (H2O2 zu H2O) in den
Erythrozyten.
NADP = Nicotinamidadenindinukleotidphosphat, NADPH = reduziertes Nicotinamidaden-
indinukleotidphosphat, GSH = reduziertes Glutathion, GSSG = oxidiertes Glutathion.
Glucose-6-Phosphat NADP GSH H2O2
H2OGSSGNADPH6-Phosphogluconat
Glucose-6-Phosphat-Dehydrogenase
Oxidans
GSSG- Reduktase
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):241–244

ÜBERSICHTSARTIKEL 243
spontane klinische Besserung ist innerhalb von 5–10 Ta-gen zu erwarten. Je nach Enzymvariante kann aber auch eine chronische hämolytische Anämie vorliegen. Eine zusätzliche Hämoglobinopathie, wie beispielsweise eine Thalassämie oder Sichezellanämie, kann den klinischen Verlauf ebenfalls beeinflussen. In seltenen Fällen kann ein Ikterus bereits wenige Tage nach der Geburt beobachtet werden. Das klinische Bild unterscheidet sich von der typischen Rhesus-Inkom-patibilität: Der Ikterus beim G6PD-Mangel ist selten bereits bei Geburt sichtbar, sondern manifestiert sich typischerweise zwischen dem 2. und 3. Tag. Zudem ist der Ikterus im Vergleich zur Schwere der Anämie beim G6PD-Mangel deutlich stärker ausgeprägt als bei der Rhesus-Inkompatibilität [11]. Daten aus den USA zeigten, dass 21% aller Kernikterusfälle mit einem G6PD- Mangel assoziiert waren [12]. Dies wirft die Frage auf, ob eine Testung auf G6PD-Mangel in das Neugeborenen-Screening-Programm aufgenommen werden soll. In der Schweiz gehört ein G6PD-Test nicht zum Neugeborenen-Screening, es sei denn, es liegt ein Ikterus vor.
Diagnostik
Der G6PD-Mangel ist primär eine klinische Diagnose (Abb. 3). Eine genaue Anamnese gibt Hinweise darauf, ob ein oxidativer Stress vor der Manifestation der Hä-molyse stattgefunden hat oder ob eine andere Ursache für eine hämolytische Anämie wahrscheinlich ist (z.B. mechanische Aortenklappe, Aufenthalt in Tropenge-biet). Ein Differentialblutbild mit Bilirubin (direkt und
indirekt), Laktatdehydrogenase (erhöht) und Haptoglo-bin (erniedrigt) sind wegweisend und können den Ver-dacht bestätigen. Zum Ausschluss einer immunhämo-lytischen Anämie ist ein negativer direkter Coombs-Test nützlich. Zur Bestätigung des G6PD-Mangels zeigt sich zum einen im Blutausstrich eine typische Konstellation (Heinz-Körperchen, Bite-Zellen), zum anderen kann die G6PD-Aktivität laborchemisch gemessen werden. Eine normale Aktivität zum Zeitpunkt der Hämolyse schliesst jedoch einen G6PD-Mangel nicht aus, da es aufgrund der Rekrutierung von jungen, Enzym-akti-ven Erythrozyten zu einer falsch-normalen Enzymak-tivität kommen kann. Eine Wiederholung der Messung wäre in diesem Fall nach drei Monaten empfohlen, weil dann ein Gleichgewicht zwischen älteren und jüngeren Erythrozyten besteht.
Therapie und Prävention
Bei Auftreten einer hämolytischen Anämie aufgrund eines G6PD-Mangels sollte natürlich nach Möglichkeit das auslösende Medikament abgesetzt werden. Je nach Schweregrad ist in seltenen Fällen eine Bluttransfusion notwendig. Innerhalb von 5–10 Tagen kommt es zur Erholung und im Verlauf zur Normalisierung des Blut-bildes. Eine spezifische Therapie gibt es nicht.Wichtig ist vor allem, den Patienten über das Krank-heitsbild aufzuklären und zukünftig auslösende Fakto-ren zu meiden. Bis heute gibt es keinen Konsens über die auslösenden Medikamente, und aufgrund des gene-tischen Polymorphismus ist eine genaue Aussage kaum
Abbildung 3: Vorschlag zum Diagnostik-Algorithmus bei Verdacht auf Hämolyse bei G6PD-Mangel.
Anamnese:Mechanische Herzklappe, chemische/toxische Agentien, Transfusionen, Tropenaufenthalte, vorangehende Infekte, persönliche und Familienanamnese, Medikamente
Suche nach Ätiologie
Blutausstrich(Fragmentozyten?
Bite-cells?Mikrosphärozyten)
BestätigungQuantitative Bestimmung der Glucose-6-Phosphat- Dehydrogenase-Aktivität
–
BestätigunghämolytischeAnämie
Basisdiagnostik– Differentialblutbild, inkl. Retikulozyten– Bilirubin (direkt, indirekt)– LDH , Haptoglobin – Direkter Coombs-Test
+ Immunhämo lytische Anämie möglich
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):241–244

ÜBERSICHTSARTIKEL 244
möglich, welcher Patient auf welches Medikament wie stark reagiert. Ein Zusammenhang zwischen der che-mischen Struktur des Arzneimittels und dem Pathome-chanismus des Enzymmangels hat sich bisher nicht gezeigt; deshalb ist es auch nicht möglich, aufgrund dieser Kenntnisse eine Voraussage machen zu können. Youngster [13] hat 2010 eine evidence-based review ver-öffentlicht, in der er eine umfangreiche Literatur-recherche durchgeführt und ausgewertet hat. Dabei hat
Tabelle 1: Medikamente, die bei Patienten mit einem G6PD-Mangel vermieden werden sollten (nach Youngster und Luzzatto) [10, 13].
Kategorie Vorhersehbare Hämolyse («unsafe»)
Mögliche Hämolyse
Antimalaria Primaquin Dapson
Chloroquin (Nivaquine®) Chinin (Limptar®)
Analgetika Phenazopyridin Acetylsalicylsäure (Aspirin®)Paracetamol (Dafalgan®, Panadol®)
Antibiotika Nitrofurantoin (Uvamin®)
Sulfasalazin (Salazopyrin®)Sulfadiazin (Flammazine®, Ialugen plus®)Cotrimoxazol(Trimethoprim/Sulfmethoxazol, Bactrim®, Nopil®)*Chinolon (Ciprofloxacin, Norfloxacin, Levofloxacin)*
Andere Rasburicase (Fasturtec®)MethylenblauToluidinblau
IsoniazidAscorbinsäure (Vitamin C)Glibenclamid (Daonil®)Vitamin K (Konakion®)Isosorbiddinitrat (Isoket®)Succimer (Succicaptal®)
Chemikalien Anilinfarbstoff (Teerfarbstoffe)Naphthalen (Mottenkugeln)HennaFavabohnen
* Gemäss Luzzatto [10] als «unsafe» bewertet.
Das Wichtigste für die Praxis• Aufgrund der Migrationszunahme ist damit zu rechnen, dass mehr Per-
sonen mit Glucose-6-Phosphat-Dehydrogenase-(G6PD-)Mangel in der
Schweiz medizinische Versorgung in Anspruch nehmen werden.
• Patienten mit G6PD-Mangel können sich mit einer hämolytischen Krise
präsentieren, wofür die Kenntnis der Diagnostik und Behandlung in dieser
Notfallsituation wichtig ist.
• Für die Betreuung dieser Patienten in der Praxis braucht es eine Kenntnis
der zu vermeidenden Medikamente.
Korrespondenz: Sarah Hofmann Klinik für Innere Medizin Universitätsspital Basel CH-4031 Basel sarah.hofmann[at]usb.ch
sich gezeigt, dass die Datenlage zu vielen Medikamen-ten, von denen befürchtet wurde, sie könnten bei Pati-enten mit G6PD-Mangel eine Hämolyse auslösen, sehr spärlich ist. Beispielsweise wurde in einigen Fallberich-ten eine Hämolyse unter Paracetamol zwar dokumen-tiert, aber in den meisten Fällen lag eine klare Überdo-sierung vor, oder aber die Patienten hatten hohes Fieber, weshalb der auslösende Faktor nicht sicher zu bestimmen war [14–18]. Zwei Studien an Kindern mit G6PD-Mangel konnten keine Hämolyse nach Gabe von Paracetamol nachweisen [19, 20]. In Anbetracht des sehr häufigen und weit verbreiteten Einsatzes von Para cetamol und dieser spärlichen Fallberichte kann davon ausgegangen werden, dass man dieses Medi-kament bei Patienten mit einem G6PD-Mangel in nor-malen Dosierungen problemlos verabreichen kann. Die Tabelle 1 zeigt auf, welche Medikamente gemäss Luz-zatto und Youngster [10, 13] wiederholt zur Hämolyse bei betroffenen Patienten geführt haben und deshalb als «unsafe» deklariert werden. In der Schweiz sind da-von lediglich Nitrofurantoin (Uvamin®, Furadantin®) und Rasburicase (Fasturtec®) sowie Methylenblau und Toluidinblau erhältlich. Eine Bestimmung der G6PD-Aktivität, bevor eine The-rapie mit Dapson oder Primaquin begonnen wird, ist nur bei entsprechender persönlicher und Familien-anamnese oder klinischem Verdacht notwendig. Für Methylenblau und Rasburicase, die in Notfallsituatio-nen verabreicht werden, reicht oftmals die Zeit nicht für eine Abklärung im Voraus. Des Weiteren pflegt die «Associazione Italiana Favismo – Deficit di G6PD» (G6PD Deficiency favism association) eine Website (http://g6pd.org) inklusive Listen mit «sicheren» und «unsicheren» Medikamenten sowie In-formationen für Patienten, Angehörige und Gesund-heitspersonal.
Disclosure statementDie Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
TitelbildWikimedia Commons.
LiteraturDie vollständige nummerierte Literaturliste finden Sie als Anhang des Online-Artikels unter www.medicalforum.ch.
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):241–244

LITERATUR / RÉFÉRENCES Online-Appendix
Literatur / Références
1. Cordes W. Experiences with plasmochin in malaria. Med Depart 15th Annual Report. Boston United Fruit Co; 1926:66-71
2. Dern RJ, Beutler E, Alving AS. The hemolytic effect of primaquine. V. Primaquine sensitivity as a manifestation of a multiple drug sensitivity. J Lab Clin Med. 1955 Jan;45(1):30-9.
3. Alving AS, Carson PE, Flanagan CL, Ickes CE. Enzymatic deficiency in primaquine-sensitive erythrocytes. Science. 1956 Sep 14;124(3220):484-5
4. Nkhoma ET1, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis. 2009 May-Jun;42(3):267-78.
5. Ruwende C, Khoo SC, Snow RW, Yates SN, Kwiatkowski D, Gupta S, Warn P, Allsopp CE, Gilbert SC, Peschu N, et al. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. 1995 Jul 20;376(6537):246-9.
6. Luzzatto L, Usanga FA, Reddy S. Glucose-6-phosphate dehydrogenase deficient red cells: resistance to infection by malarial parasites. Science. 1969 May 16;164(3881):839-42.
7. Cappadoro M, Giribaldi G, O'Brien E, Turrini F, Mannu F, Ulliers D, Simula G, Luzzatto L, Arese P. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood. 1998 Oct 1;92(7):2527-34
8. Piomelli S, Corash LM, Davenport DD, Miraglia J, Amorosi EL. In vivo lability of glucose-6-phosphate dehydrogenase in GdA- and GdMediterranean deficiency. J Clin Invest. 1968 Apr;47(4):940-8.
9. Kellermeyer RW, Tarlov AR, Brewer GJ, Carson PE, Alving AS. Hemolytic effect of therapeutic drugs. Clinical considerations of the primaquine-type hemolysis. JAMA. 1962 May 5;180:388-94.
10. Luzzatto L, Seneca E. G6PD deficiency: a classic example of pharmacogenetics with on-going clinical implications. Br J Haematol. 2014 Feb;164(4):469-80.
11. Kaplan M, Hammerman C, Glucose-6-phosphate dehydrogenase deficiency: a hidden risk for kernicterus.
Semin Perinatol. 2004;28(5):356. 12. L Johnson, V K Bhutani, K Karp, E M Sivieri and S M
Shapiro Clinical report from the pilot USA Kernicterus Registry (1992 to 2004) Journal of Perinatology (2009) 29, S25–S45).
13. Youngster I, Arcavi L, Schechmaster R, Akayzen Y, Popliski H, Shimonov J, Beig S, Berkovitch M. Medications and glucose-6-phosphate dehydrogenase deficiency: an evidence-based review. Drug Saf. 2010 Sep 1;33(9):713-26.
14. Sklar GE, Hemolysis as a potential complication of acetaminophen overdose in a patient with glucose-6-phosphate dehydrogenase deficiency , Pharmacotherapy. 2002 May;22(5):656-8.
15. Wright RO, Perry HE, Woolf AD, Shannon MW. Hemolysis after acetaminophen overdose in a patient with glucose-6-phosphate dehydrogenase deficiency. J Toxicol Clin Toxicol. 1996;34(6):731-4.
16. Phillpotts S, Tash E, Sen S. Glucose-6-phosphate dehydrogenase deficiency: an unusual cause of acute jaundice after paracetamol overdose. Eur J Haematol. 2014 Mar 29
17. Oliver M, Coton T, Badens C, Dehan C, Lena-Russo D, Moalic JL. Homozygous G6PD deficiency and propacetamol induced hemolysis. Haematologica. 2001 Sep;86(9):987-8
18. Ruha AM, Seldem B. Hemolytic anemia after acetaminophen overdose in patient with glucose-6-phosphate dehydrogenase deficiency. Am J Med. 2001 Feb 15;110(3):240-1.
19. Cottafava F, Nieri S, Franzone G, Sanguinetti M, Bertolazzi L, Ravera G. [Double-blind controlled comparison of placebo and paracetamol in patients with G-6-PD deficiency]. Pediatr Med Chir. 1990 Nov-Dec;12(6):631-7. Italian.
20. Najafi N, Van de Velde A, Poelaert J. Potential risks of hemolysis after short-term administration of analgesics in children with glucose-6-phosphate dehydrogenase deficiency. J Pediatr. 2011 Dec;159(6):1023-8.
21. Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008 Jan 5;371(9606):64-74. Review.
SWISS MEDICAL FORUM

WAS IST IHRE DIAGNOSE? 245
Produktiver Husten und Gewichtsverlust
Den Tramlinien auf der SpurAmélie Burria, Valentina Garellib, Nicolas Petitpierreb
a Service de Médecine interne, Centre Hospitalier Universitaire Vaudois CHUV, Lausanneb Service de Pneumologie, Centre Hospitalier Universitaire Vaudois CHUV, Lausanne
Fallbeschreibung
Bei einer 73-jährigen Patientin bestand seit drei Mona-ten ein unstillbarer Husten mit maulvollem Auswurf, die sowohl tagsüber als auch nachts auftraten, jedoch im Liegen stärker waren. Ferner beschrieb sie eine pro-grediente Dyspnoe, die sich bei ihrer Vorstellung im Sta-dium NYHA II (Klassifikation der New York Heart Associ-ation) befand. Es bestand kein Status febrilis.Bei der Patientin war eine Mangelernährung, Osteopo-rose sowie eine seit 30 Jahren bestehenden Colitis ulce-rosa (CU) bekannt, die bei der Vorstellung im Spital me-dikamentös kontrolliert war. Im Jahr 2011 hatte sie eine Karotisdissektion mit transitorischer ischä mischer At-tacke erlitten und war im Jahr 1975 Opfer eines Ver-kehrsunfalls mit schwerer Gehirnerschüt terung und neurologischen Folgeschäden (Gang stö rungen, Tre-mor) geworden. Als Standardmedikation erhielt sie Clopidogrel, Simvastatin, Levetiracetam, Mesalazin, Thiamin und Alprazolam.Bei der klinischen Untersuchung wies die Patientin einen Body Mass Index von 18 kg/m2 auf. Ihre Sauer-stoffsättigung unter Raumluft betrug 95%. Die Lungen-auskultation ergab einige feine Rasselgeräusche und diffuses Giemen.
1) Welche der folgenden Untersuchungen gehört nicht
zur Erstuntersuchung?
a) Röntgen-Thorax
b) Sputumkultur
c) Lungenfunktionstest
d) Bronchoskopie
Die nichtrauchende Patientin litt an chronischem Hus-ten. Der erste Schritt bestand darin, nach einer medi-kamentösen Ursache, insbesondere einer Behandlung mit einem Angiotensin-Converting-Enzyme-(ACE-)Hemmer zu suchen. Es konnten keine klinischen Hin-weise für ein Postnasal-Drip-Syndrom oder einen gastro ösophagealen Reflux gefunden werden. Eine Ex-juvantibus-Behandlung mit einem Protonenpumpen-hemmer bewirkte keine Besserung der Symptomatik. Ausser dem Husten deutete nichts auf Asthma hin. Es wurde ein Röntgen-Thorax angefertigt, das ein interstitielles und alveoläres Infiltrat rechts basal mit positivem Bronchopneumogramm zeigte (Abb. 1).
Der Spirometriebefund war unauffällig und ohne ob-struktives Syndrom, jedoch musste die Patientin wäh-rend der Durchführung häufig husten. Aufgrund der Bronchorrhoe wurde eine Sputumkultur angeordnet, die eine Kontamination mit oropharynge-aler Flora ergab.Eine Bronchoskopie gehört nicht zur Erstuntersuchung bei chronischem Husten.Aufgrund der Symptomatik und der radiologisch nach-gewiesenen Infiltrate wurde der Verdacht auf eine ambulant erworbene Pneumonie geäussert und eine Cefuroxim-Behandlung (10 Tage 250 mg 2×/täglich) be-gonnen.
Abbildung 1: Interstitielles und alveoläres Infiltrat rechts
basal mit positivem Aerobronchogramm (Pfeil).
Abbildung 2: Kleine alveoläre Infiltrate rechts basal
mit «Tramlinien» (Pfeile).
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):245–248
WAS IST IHRE DIAGNOSE? 246
Drei Wochen später berichtete die Patientin, dass sich ihr Husten gebessert habe, sie aber weiterhin, insbe-sondere nachts, unter Hustenanfällen leide. Erneut wurde eine Thorax-Röntgenaufnahme angefertigt. Diese zeigte neue Verdichtungen und «Tramlinien» (Abb. 2), was den Verdacht auf Bronchiektasen nahe-legte. Infolgedessen wurde ein Thorax-CT angefertigt, welches das Vorhandensein von Bronchiektasen mit Infiltraten im rechten Mittel- und Unterlappen bestä-tigte (Abb. 3).
2) Welche der folgenden Untersuchungen gehört nicht
zur Erstuntersuchung bei Bronchiektasen?
a) Tb-Spot-TB-Test und Mykobakterienkultur
b) Serum-Immunglobulin-Bestimmung (IgG)
c) Bestimmung von Gesamt-IgE und Aspergillus-Präzipitinen
d) Suche nach einer Mutation im CFTR-Gen (Cystic fibrosis
transmembrane conductance regulator)
Als Bronchiektase wird eine irreversible Ausweitung eines Bronchus bezeichnet. Die Diagnosestellung er-folgt anhand des Nachweises eines grösseren Durch-messers des Bronchuslumens im Vergleich zum beglei-tenden Blutgefäss mittels hochauflösendem Thorax-CT. Welche Laboruntersuchungen angeordnet werden, ist von der klinischen Präsentation abhängig [1].Aufgrund der diffusen Bronchiektasen vermuteten wir bei der Patientin folgende Differentialdiagnosen: primärer oder sekundärer Immundefekt (primäre Hy-po gammaglobulinämie), humanes Immundefi zi enz -Virus (HIV), Folgen einer infektiösen Atemwegserkran-
kung, atypische Mykobakterieninfektion, Assoziation mit einer systemischen Erkrankung (rheumatoide Poly arthritis, Sjögren-Syndrom usw.) oder mit einer chronisch-entzündlichen Darmerkrankung (CED).Eine allergische bronchopulmonale Aspergillose er-schien aufgrund des anamnestisch nicht bekannten Asthmas zwar unwahrscheinlich, wurde aber dennoch gesucht.Eine genetische Erkrankung (Mukoviszidose, Primäre Ciliäre Dyskinesie) ist in dieser Altersgruppe und ohne HNO- bzw. gastrointestinale Symptomatik selten.Die durchgeführten Laboruntersuchungen umfassten einen IgG- und IgG-Subklassen-Test, einen Tb-Spot-TB-Test, einen HIV-Test, eine Bestimmung des Gesamt-IgE sowie des spezifischen IgE auf Aspergillus fumigatus, einen Test auf Aspergillus-Präzipitine sowie Tests auf Autoimmunerkrankungen und mikrobiologische Ana-lysen.Alle Untersuchungen, einschliesslich der Sputumkul-turen (klassische Bakterien, Mykobakterien und Pilze), waren unauffällig.Angesichts der trotz Antibiotikatherapie persistieren-den Symptome und Lungeninfiltrate wurde eine Bron-choskopie durchgeführt, die eine stark entzündete Luftröhren- und Bronchialschleimhaut mit grossen Mengen mukopurulenten Sekrets zeigte (Abb. 4).Die Zellverteilung in der bronchoalveolären Lavage (BAL) ergab eine Prädominanz von Neutrophilen (33%). In der BAL-Kultur wurde eine oropharyngeale Flora (103 Keime/Milliliter) und im Sekret der bronchialen Absaugung ein Serratia marcescens-Keim festgestellt.
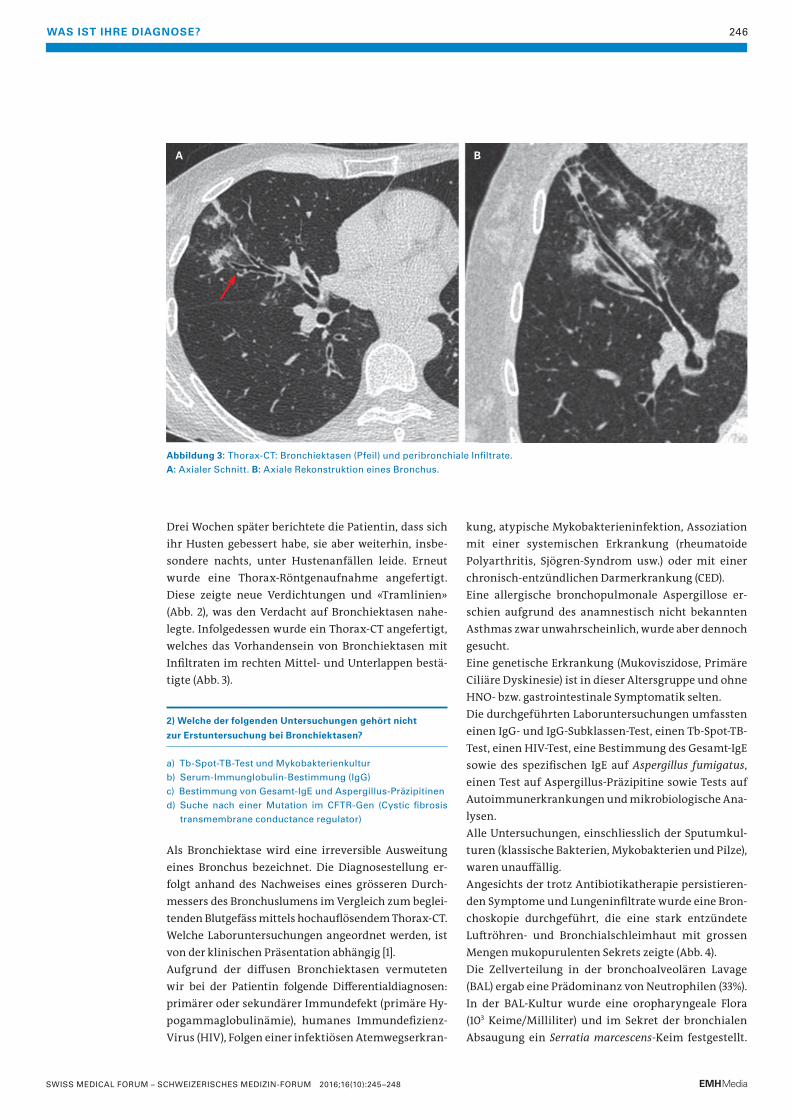
Abbildung 3: Thorax-CT: Bronchiektasen (Pfeil) und peribronchiale Infiltrate.
A: Axialer Schnitt. B: Axiale Rekonstruktion eines Bronchus.
A B
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):245–248
WAS IST IHRE DIAGNOSE? 247
Aufgrund dessen wurde eine dreiwöchige Behand-lung mit Sulfamethoxazol/Trimethoprim versucht. Zusätzlich wurde eine Mykobakterienkultur angelegt, die auch diesmal negativ ausfiel.Zwei Monate später war der Husten zurückgegangen, jedoch nicht vollständig abgeklungen. Die Lungen-auskultation und der Lungenfunktionstest waren un-verändert.Einige Tage später erlitt die Patientin eine erste Epi-sode von Hämoptysen (5–6 Esslöffel). Ausserdem klagte sie über Gewichtsverlust ohne damit verbundene Stuhlgangprobleme.Das aufgrund der Hämoptysen angefertigte Notfall-Thorax-CT zeigte eine Progression der Bronchiektasen, assoziiert mit zahlreichen alveolären Infiltraten (Abb. 5).Alles in allem hatte eine rasche Progression der Bron-chiektasen stattgefunden, das makroskopische Erschei-nungsbild zeigte eine chronische Entzündung, und die übliche ätiologische Abklärung war unauffällig.
3) Welche Diagnose würden Sie als Erstes vermuten?
a) Eine Lungenentzündung mit atypischen Mykobakterien
b) Eine Mukoviszidose
c) Bronchiektasien infolge der Colitis ulcerosa
d) Eine allergische bronchopulmonale Aspergillose
Eine derart rasche Progression von Bronchiektasen ist absolut ungewöhnlich. Nach dem hinreichenden Aus-schluss einer infektiös, autoimmun oder genetisch be-dingten Ätiologie stellten wir die Diagnose Bronchiek-tasen infolge der CU. Bronchitiden aufgrund einer CED trotz fehlender Anzeichen einer Krankheitsaktivität im Gastrointestinaltrakt sind ein bekanntes Phäno-men [2].
4) Welche Therapie schlagen Sie vor?
a) Systemische Kortikoidtherapie, anschliessend inhalative
Kortikosteroide
b) Ausschliesslich inhalative Kortikoide
c) Mesalazin
d) Infliximab
Wir begannen eine Kortikoidtherapie mit Prednison (30 mg/Tag) sowie zusätzlicher Osteoporose- und Pneumonieprophylaxe gegen Pneumocystis jiroveci.Zwei Monate später zeigte sich ein guter Verlauf mit vollständig abgeklungenem Husten und Gewichts-zunahme. Infolgedessen konnte das Prednison ausge-schlichen werden.Gleichzeitig wurde mit einer inhalativen Kortikoid-therapie (Budesonid 400 μg 2×/Tag) begonnen, in der Hoffnung, auf diese Weise das Ausschleichen der syste-mischen Kortikoidtherapie zu erleichtern.
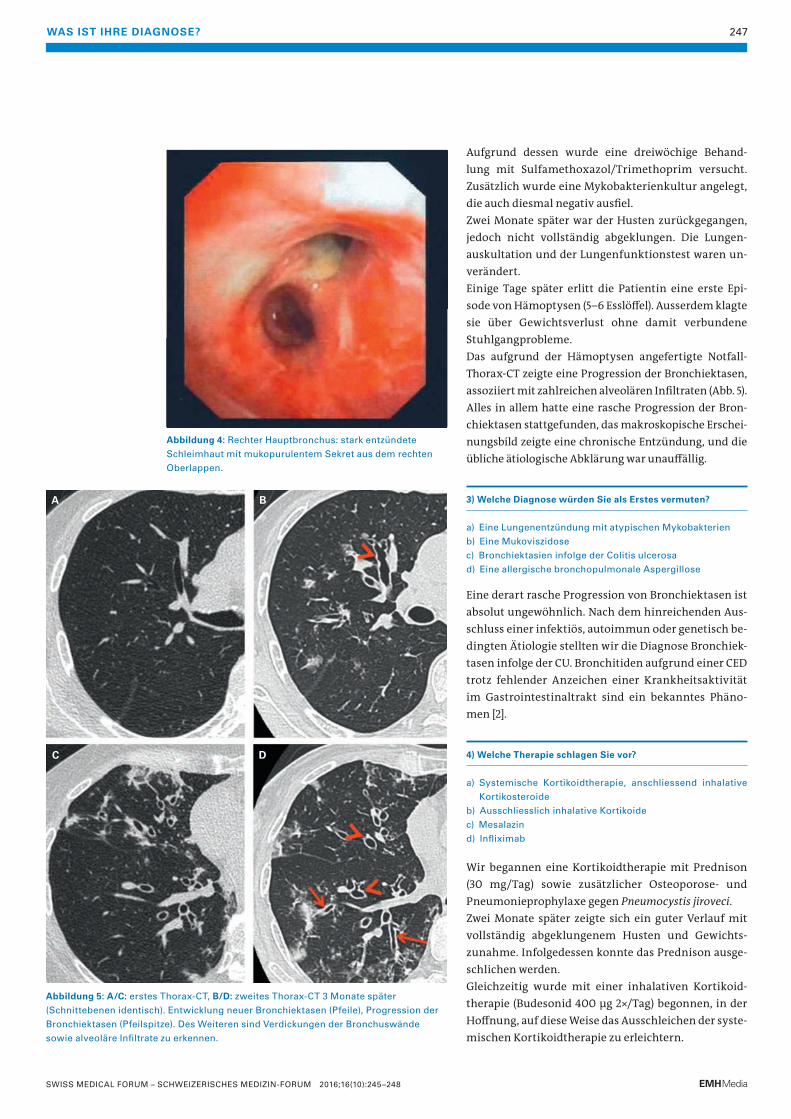
Abbildung 4: Rechter Hauptbronchus: stark entzündete
Schleimhaut mit mukopurulentem Sekret aus dem rechten
Oberlappen.
Abbildung 5: A/C: erstes Thorax-CT, B/D: zweites Thorax-CT 3 Monate später
(Schnitt ebenen identisch). Entwicklung neuer Bronchiektasen (Pfeile), Progression der
Bronchiektasen (Pfeilspitze). Des Weiteren sind Verdickungen der Bronchuswände
sowie alveoläre Infiltrate zu erkennen.
A
C
B
D
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):245–248

WAS IST IHRE DIAGNOSE? 248
Diskussion
Chronisch-entzündliche Darmerkrankungen (CED) kön-nen zahlreiche extraintestinale Manifestationen auf-weisen, haupt sächlich an Gelenken, Augen und Haut.Obgleich eine hohe Prävalenz subklinischer Atem-wegsbeteiligungen zu bestehen scheint [3, 4], treten manifeste Atemwegsbeschwerden nur selten auf. Die Läsionen und klinischen Manifestationen sind äusserst vielfältig und können das gesamte respiratorische Sys-tem betreffen: die Atemwege, die Luftröhre bis hin zu den Bronchiolen, das Lungenparenchym, die Pleura und das Lungengefässsystem. Atemwegsentzündungen gehören zu den häufigsten Beschwerden und können sich zu Bronchiektasen, schweren Bronchialstenosen oder Bronchiolitiden entwickeln. Bei Colitis ulcerosa kommt es häufiger zu bronchialen Symptomen als bei Morbus Crohn.Neben den Beschwerden, die direkt durch die Entzün-dung entstehen, können auch Nebenwirkungen der medikamentösen Therapie wie opportunistische Infek-tionen oder eine interstitielle Pneumonie vorliegen.80–85% der Patienten entwickeln nach einer CED-Dia-gnose respiratorische Symptome. Die Atemwegsbe-schwerden können als Erstmanifestation der Erkran-kung, jedoch auch erst Jahre nach der Diagnosestellung und selbst bei Patienten in Remission vorkommen. Auch das Auftreten von Atemwegsbeschwerden nach einer Kolektomie wurde häufig beschrieben.
Die Pathophysiologie ist nicht vollständig geklärt, hängt jedoch wahrscheinlich mit dem gemeinsamen embryonalen Ursprung des respiratorischen und Ko-lon-Epithels (Schleimzellen und submukose Drüsen) zusammen.Die Behandlung mit Kortikosteroiden stellt hier, ent-gegen anderen Formen von Bronchiektasen, den Gold-standard dar. Meist erfolgt die Therapie systemisch und anschliessend in hochdosierter inhalativer Form.Bei leichteren Manifestationen kann die ausschliessli-che Verabreichung inhalativer Kortikoide ausreichend sein.Zusätzlich zur antiinflammatorischen Behandlung sind die üblichen Therapiemassnahmen bei Bronchi-ektasen erforderlich: Physiotherapie zur Förderung der Bronchialdrainage, eventuell unterstützt durch die Inhalation von NaCl, Grippeschutz- und Pneumo-kokkenimpfungen sowie die früh- und langzeitige Be-handlung von Bronchialinfekten. Azithromycin, das häufig bei Bronchiektasen anderen Ursprungs einge-setzt wird, wurde in Bezug auf die oben genannte Po-pulation nicht spezifisch untersucht. In manchen Fäl-len wird es bei wiederholten Exazerbationen als Ex-juvantibus-Therapie verordnet.Die Prognose ist im Allgemeinen gut, es gibt jedoch Kortikoid-resistente Lungenerkrankungen. In diesem Fall sind die therapeutischen Optionen beschränkt. Es wurden Bronchiallavagen mit Methylprednisolon be-schrieben. Klassische immunsuppressive CED-Thera-pien (Azathioprin, Mercaptopurin und Methotrexat) scheinen mässig wirksam zu sein.
Disclosure statementDie Autoren haben keine finanziellen oder persönlichen Verbin-dungen im Zusammenhang mit diesem Beitrag erklärt.
Literatur1 Espinosa V, Rochat T. Bronchiectasies chez l’adulte: recherche
d’une étiologie. Rev Med Suisse. 2013;9(407):2155–9.2 Camus Ph, Colby TV. Bronchiectasis associated with inflammatory
bowel disease., in ERS monograph. Bronchiectasis, Floto RA et Haworth CS Editors. 2011, European Respiratory Society: Plymouth. p. 163–77.
3 Herrlinger KR, Noftz MK, Dalhoff K, Ludwig D, Stange EF, Fellermann K. Alterations in pulmonary function in inflamm-atory bowel disease are frequent and persist during remission. Am J Gastroenterol. 2002;97(2):377–81.
4 Desai D, Patil S, Udwadia Z, Maheshwari S, Abraham P, Joshi Al. Pulmonary manifestations in inflammatory bowel disease: a prospective study. Indian J Gastroenterol. 2011;30(5):225–8.
Das Wichtigste für die Praxis
Bei Patienten mit CED sollte man neben den üblichen extraintestinalen Ma-
nifestationen auch an Komplikationen im Lungenbereich, insbesondere an
tracheobronchiale Beschwerden, denken. Diese können auch bei inaktiver
Erkrankung oder nach einer Kolektomie auftreten. Dies kann das Erkennen
eines Zusammenhangs zwischen beiden Erkrankungen in einigen Fälle n
schwierig gestalten. Die Diagnose von Bronchiektasen infolge einer CED
ist wichtig, da diese Erkrankung im Allgemeinen sehr gut auf eine
Kortikoid therapie anspricht.
Antworten:
Frage 1: d. Frage 2: d. Frage 3: c. Frage 4: a.
Korrespondenz: Dr. med. Amélie Burri Service de Médecine Interne CHUV CH-1011 Lausanne amelie.burri[at]chuv.ch
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):245–248

AUS DER FORSCHUNG 249
Validierungsstudie einer Version des ECAS im Schweizer Hochdeutsch
Screening von Kognitions- undVerhaltensänderungen beider Amyotrophen LateralskleroseChristian Burkhardt, Christoph Neuwirth, Markus Weber
Muskelzentrum/ALS Clinic, Kantonsspital St. Gallen
Hintergrund
Die Amyotrophe Lateralsklerose (ALS) ist die häufigsteMotoneuronerkrankung beim Erwachsenen. Sie führt durch Degeneration des ersten und zweiten motori-schen Neurons zu fortschreitenden Lähmungen. In denletzten beiden Dekaden änderte sich die Wahrnehmungder Erkrankung radikal. Neben Paresen und Spastiksind kognitive Defizite und Verhaltensänderungen als häufige Symptome dieser Multisystem-Erkrankung akzeptiert, wenn auch gegenwärtig noch nicht in den Diagnosekriterien verankert. Auf Basis von geneti-schen (z.B. c9orf72-Expansion) und histomorphologi-schen (z.B. mit TDP-43-positiven Einschlüssen im ZNS) Gemeinsamkeiten konnte die Brücke zwischen der ALSund der fronto-temporalen Demenz (FTD) geschlossen werden [1]. Beide Erkrankungen bilden somit ein Kon-tinuum mit variabler phänotypischer Präsentation.Frontotemporale Störungen der ALS decken ein breites klinisches Spektrum von Symptomen ab. Einige Pa-tienten weisen lediglich in kognitiven TeilbereichenDefizite (kognitive Beeinträchtigungen, ALS-ci) auf. Ebenso werden isolierte Verhaltensänderungen (Ver-haltensstörungen, ALS-bi) beobachtet, und einige Pa-tienten präsentieren ein Mischbild aus beiden Berei-chen. Nur eine Minderheit der ALS-Patienten (5–15%)erfüllt die Kriterien einer «ausgewachsenen» ALS-FTD. Für ALS-ci schwanken die Prozentangaben von 15–60%[2]. Die Defizite bei der ALS-ci liegen meist in der Wort-flüssigkeit (fluency), Beeinträchtigung von exekutivenFunktionen, eingeschränkten Fähigkeiten zur Planung und Bearbeitung von Problemstellungen, Aufmerk-samkeits- und Konzentrationsstörungen sowie dem Multitasking. Bei der ALS-bi reicht das Spektrum vongeringfügigen, kaum wahrnehmbaren Verhaltens- änderungen mit wenig Einfluss auf das tägliche Leben bis hin zu Verhaltensauffälligkeiten einer frontalenDemenz. Hier sind sowohl Minussymptome wie Apa-thie, Interessenverlust, Verlust an Empathie und sozial-
emotionalen Beziehungen als auch Plussymptome wie Reizbarkeit und eine unzureichende Impulskontrolle zu nennen. Faktoren, welche die grosse Heterogenitätder Symptompräsentation dieses Kontinuums der ALS-FTD in der Literatur erklären, liegen zum einen in denunterschiedlichen Erkrankungsstadien der Patienten,der hohen genetischen Variabilität sowie der sehr unterschiedlichen diagnostischen Testbatterien, die zur Detektion der fronto-temporalen Defizite ein-gesetzt werden. Die bisher gängigen neuropsycho-logischen Tests zur Untersuchung fronto-temporalerFunktionen bei ALS wurden nicht eigens für dieALS-Erkrankten entwickelt und weisen dadurch keineAnpassung an die besonderen, körperlichen Ein-schränkungen dieser Patienten, vor allem in spätenKrankheitsstadien, auf. Die Durchführung und Aus-wertung wird hierdurch erschwert oder gar verun-möglicht. Oftmals werden mit diesen Tests nur einzelne kognitive Teilbereiche geprüft, was bei der Heterogeni-tät der Patienten zu einer unzureichenden Empfindlich-keit des Screeningtools führen kann. Fronto-tempo-rale Dysfunktionen können in nahezu allen Bereichendes täglichen Lebens des Patienten bei Interaktion mit Angehörigen und medizinischem Personal sowie beiEntscheidungsprozessen Relevanz zeigen und die Lebensqualität von Patienten und Angehörigen beein-trächtigen. Zudem stellt eine FTD einen Risikofaktorfür ein verkürztes Überleben dar. So ist zum Beispieldie Adaptation an die fortschreitenden körperlichen Einschränkungen sowie die Fähigkeit zu Problem-lösungen vermindert. Patienten stossen rasch an die Grenzen ihrer Ressourcen bei der Anwendung von Kom-munikations-, Ernährungs- und Beatmungshilfen. Be-reits das Wissen und Verständnis der Angehörigen und Pflegenden um die kognitiven Defizite als Teil der Er-krankung entlastet und hilft, den Alltag besser zu be-wältigen. Mit dem ECAS (Edinburgh Cognitive andBehavioural ALS Screen) wurde kürzlich von Abrahamset al. [3] ein schnell und einfach anwendbares klinisches Screening-Tool zur Aufdeckung von fronto-tempora-Christian Burkhardt
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):249–252

AUS DER FORSCHUNG 250
len Auffälligkeiten bei ALS-Patienten entwickelt, dasauch in fortgeschrittenen Krankheitsstadien mit motorischen Einschränkungen anwendbar ist. Seine Sensitivität und Spezifität ist vergleichbar mit einerdetaillierten neuropsychologischen Testung.
Zielsetzung und Hypothese
Das Ziel der vorliegenden Studie war es, eine Versiondes ECAS im Schweizer Hochdeutsch für den klinischen Gebrauch zu validieren. Es handelt sich hierbei um eine Übersetzung ins schweizerische Deutsch. Es liegtauch eine hochdeutsche Version des ECAS vor, die sichetwas von dieser unterscheidet. Die Studienergebnissesind Teil einer bereits publizierten Untersuchung einer multinationalen Kooperation zur Harmonisierungund Standardisierung von Biomarkern bei ALS [4].
Methodik
Die Studie wurde bei Einverständnis der Teilnehmernach Prüfung der Ethikkommision des Kantons St.Gallenin Einklang mit den ethischen Standards der Helsinki- Deklaration von 1964 durchgeführt. Die englische Versiondes ECAS wurde durch ein zertifiziertes Übersetzungs-büro ins Schweizer Hochdeutsch übersetzt. Zur Prüfung der Stimmigkeit der Übersetzung erfolgte eine Rück-übersetzung durch eine zweisprachige Linguistin insEnglische und ein Vergleich zur Ursprungsversion. Der ECAS (Abb. 1) beinhaltet neben einem ALS-spezi-fischen Abschnitt (exekutive Funktionen, Wortflüssig-keit, Sprache und soziale Kognition) einen nicht-ALS-spezifischen Abschnitt (Gedächtnis, visuell-räumliches Verständnis) zur Abgrenzung von anderen Demenz-formen. Zusätzlich beinhaltet der ECAS die Befragung von engen Bezugspersonen bezüglich Verhaltensände-rungen in fünf charakteristischen Kompetenzberei-chen (Enthemmung, Apathie, Empathie, Stereotypien,Hyperoralität) und für psychotische Symptome [3]. Der ECAS kann entweder in einer schriftlichen oder münd-lichen Version erfolgen, um den Einfluss von motori-schen Handicaps auf das Testergebnis zu minimieren. Bei 40 Patienten mit Diagnose einer ALS gemäss den revidierten El-Escorial-Kriterien wurde der ECAS ange-wandt. Diese Ergebnisse wurden mit jenen von 49 ge-sunden Kontrollpersonen zur Festlegung der Grenz-werte (zwei Standardabweichungen unterhalb desMittelwerts der Kontrollen) verglichen. Wir definier-ten vier Untergruppen für die Cut-off-Werte in Bezug auf Alter und Ausbildung, da beide Parameter einen grossen Einfluss auf Leistungen in kognitiven Tests haben (Tab. 1). Vierunddreissig enge Bezugspersonen wurden hinsichtlich Verhaltensänderungen der Pa-tienten befragt. Die Untersuchung erfolgte durch einenNeurologen oder eine nicht-neuropsychologisch aus-gebildete Study Nurse. Die Testdauer betrug im Durch-schnitt 25 Minuten.
Ergebnisse
Die Basischarakteristika der Patienten und Kontrollensind in Tabelle 2 aufgeführt. ALS-Patienten wiesen vorallem in den Bereichen der exekutiven Funktionenund der Sprache Einschränkungen gegenüber gesun-den Kontrollpersonen auf. Auffälligkeiten der Wort-flüssigkeit waren seltener nachweisbar. Jedoch zeigten einige ALS-Patienten auch in nicht-ALS-spezifischen Bereichen wie Gedächtnis Veränderungen im Ver-gleich zu den Kontrollprobanden. Insgesamt konntenbei 7/40 Patienten in einem oder mehreren Bereichenkognitive Defizite nachgewiesen werden. Weitaus häu-figer, bei 17/34 Patienten, wurden durch die Angehöri-
Abbildung 1: Beispielseite des Edinburgher kognitiven ALS-Testverfahrens (ECAS).
Nachdruck mit freundlicher Genehmigung von Professor Sharon Abrahams,
Chair of Neuropsychology & Consultant Clinical Neuropsychologist, Edinburgh.
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):249–252
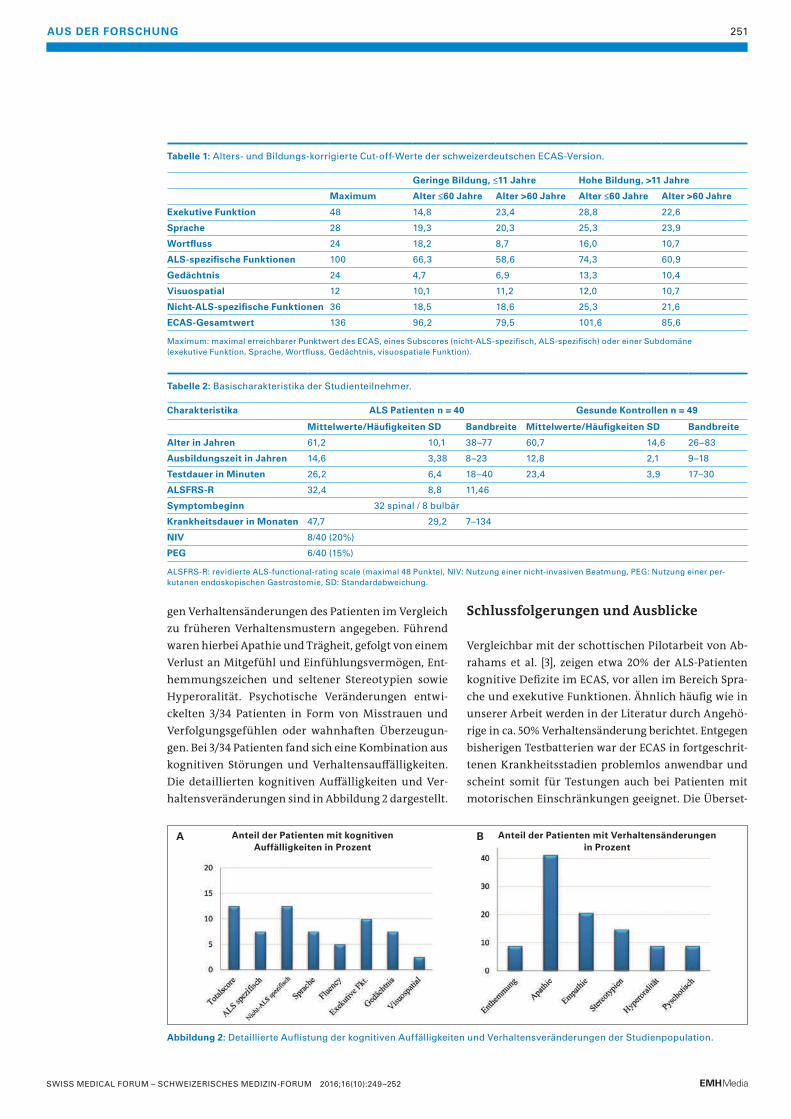
AUS DER FORSCHUNG 251
gen Verhaltensänderungen des Patienten im Vergleichzu früheren Verhaltensmustern angegeben. Führend waren hierbei Apathie und Trägheit, gefolgt von einem Verlust an Mitgefühl und Einfühlungsvermögen, Ent-hemmungszeichen und seltener Stereotypien sowie Hyperoralität. Psychotische Veränderungen entwi-ckelten 3/34 Patienten in Form von Misstrauen und Verfolgungsgefühlen oder wahnhaften Überzeugun-gen. Bei 3/34 Patienten fand sich eine Kombination auskognitiven Störungen und Verhaltensauffälligkeiten. Die detaillierten kognitiven Auffälligkeiten und Ver-haltensveränderungen sind in Abbildung 2 dargestellt.
Schlussfolgerungen und Ausblicke
Vergleichbar mit der schottischen Pilotarbeit von Ab-rahams et al. [3], zeigen etwa 20% der ALS-Patienten kognitive Defizite im ECAS, vor allen im Bereich Spra-che und exekutive Funktionen. Ähnlich häufig wie inunserer Arbeit werden in der Literatur durch Angehö-rige in ca. 50% Verhaltensänderung berichtet. Entgegenbisherigen Testbatterien war der ECAS in fortgeschrit-tenen Krankheitsstadien problemlos anwendbar undscheint somit für Testungen auch bei Patienten mit motorischen Einschränkungen geeignet. Die Überset-
Tabelle 1: Alters- und Bildungs-korrigierte Cut-off-Werte der schweizerdeutschen ECAS-Version.
Geringe Bildung, ≤11 Jahre Hohe Bildung, >11 Jahre
Maximum Alter ≤60 Jahre Alter >60 Jahre Alter ≤60 Jahre Alter >60 Jahre
Exekutive Funktion 48 14,8 23,4 28,8 22,6
Sprache 28 19,3 20,3 25,3 23,9
Wortfluss 24 18,2 8,7 16,0 10,7
ALS-spezifische Funktionen 100 66,3 58,6 74,3 60,9
Gedächtnis 24 4,7 6,9 13,3 10,4
Visuospatial 12 10,1 11,2 12,0 10,7
Nicht-ALS-spezifische Funktionen 36 18,5 18,6 25,3 21,6
ECAS-Gesamtwert 136 96,2 79,5 101,6 85,6
Maximum: maximal erreichbarer Punktwert des ECAS, eines Subscores (nicht-ALS-spezifisch, ALS-spezifisch) oder einer Subdomäne (exekutive Funktion, Sprache, Wortfluss, Gedächtnis, visuospatiale Funktion).
Tabelle 2: Basischarakteristika der Studienteilnehmer.
Charakteristika ALS Patienten n = 40 Gesunde Kontrollen n = 49
Mittelwerte/Häufigkeiten SD Bandbreite Mittelwerte/Häufigkeiten SD Bandbreite
Alter in Jahren 61,2 10,1 38–77 60,7 14,6 26–83
Ausbildungszeit in Jahren 14,6 3,38 8–23 12,8 2,1 9–18
Testdauer in Minuten 26,2 6,4 18–40 23,4 3,9 17–30
ALSFRS-R 32,4 8,8 11,46
Symptombeginn 32 spinal / 8 bulbär
Krankheitsdauer in Monaten 47,7 29,2 7–134
NIV 8/40 (20%)
PEG 6/40 (15%)
ALSFRS-R: revidierte ALS-functional-rating scale (maximal 48 Punkte), NIV: Nutzung einer nicht-invasiven Beatmung, PEG: Nutzung einer per-kutanen endoskopischen Gastrostomie, SD: Standardabweichung.
Abbildung 2: Detaillierte Auflistung der kognitiven Auffälligkeiten und Verhaltensveränderungen der Studienpopulation.
A BAnteil der Patienten mit kognitivenAuffälligkeiten in Prozent
Anteil der Patienten mit Verhaltensänderungenin Prozent
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):249–252

AUS DER FORSCHUNG 252
zung des ECAS ins Schweizer Hochdeutsch stellt damit ein schnelles und einfaches Werkzeug zur Aufdeckung von fronto-temporalen Dysfunktionen bei ALS dar. BeiNachweis fronto-temporaler Defizite sollten entspre-chende patientenorientierte, massgeschneiderte Pro-gramme in einem multidisziplinären Setting entwi-ckelt werden, um den Patienten und ihren AngehörigenHilfen anbieten zu können. Insbesondere benötigendiese eine Unterstützung bei therapeutischen Ent-scheidungsprozessen, da die daraus resultierenden Konsequenzen vom Patienten nicht immer eingeord-net werden können (z.B. Auswahl aus einer limitierten Anzahl von Alternativen, Meiden impulsiver Entschei-dungen und ausführliche Information über Konse-quenzen). Eine revidierte ECAS-Version mit entspre-chenden Cut-off-Werten ist unter http://ecas.network verfügbar.
VerdankungenWir bedanken uns bei den beteiligten Study Nurses: Bea Goldman undUrsula Schneider. Wir möchten uns bei unseren ALS-Patienten undihren Bezugspersonen sowie den gesunden Kontrollprobanden fürdie Teilnahme an der Studie bedanken. Zudem Bedanken wir uns für
die Unterstützung von Sharon Abrahams, Thomas Bak und Dorothee Lulé bei der Ausarbeitung der schweizerdeutschen Übersetzung undbei Maren Carbon für die Hilfe bei der Rückübersetzung. Diese Studie ist Teil eines EU-geförderten Joint Programme – NeurodegenerativeDisease Research (JPND SNF-Nr. SNF 31ND30_141622 (SOPHIA)) undwird unterstützt durch den Schweizerischen Nationalfonds zur Förde-rung der wissenschaftlichen Forschung.
Disclosure statementThis work was supported by the Swiss ALS Foundation and the EU Joint Programme – Neurodegenerative Disease Research (JPND) projects (grant number SNF 31ND30_141622 (SOPHIA)). The authors report no disclosures with the submitted manuscript.
Literatur1 Ng AS, Rademakers R, Miller BL. Frontotemporal dementia: a
bridge between dementia and neuromuscular disease. Ann N YAcad Sci; 1338;71–93.
2 Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology; 65;586–90.
3 Abrahams S, Newton J, Niven E, Foley J, Bak TH. Screening for cog-nition and behaviour changes in ALS. Amyotroph Lateral SclerFrontotemporal Degener; 15;9–14.
4 Lule D, Burkhardt C, Abdulla S, Bohm S, Kollewe K, Uttner I, et al.The Edinburgh Cognitive and Behavioural Amyotrophic LateralSclerosis Screen: A cross-sectional comparison of establishedscreening tools in a German-Swiss population. Amyotroph LateralScler Frontotemporal Degener; 16;16–23.
Korrespondenz: Christian BurkhardtMuskelzentrum/ALS Clinic Kantonsspital St.GallenGreithstrasse 20 CH-9007 St.Gallenchristian.burkhardt[at] kssg.ch
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):249–252

FALLBERICHTE 253
Was drückt da auf die Hirnnerven?
Bilaterale Halsschwellung bei einer jungen FrauAnita Koster-Ruscha, Joël Capraroa, Wolfgang Nagelb, Thomas Clericib, Sandro J. Stoecklic, Flavio Forrerd, Michael Brändlea, Stefan Bilza
a Klinik für Endokrinologie/Diabetologie, Kantonsspital St. Gallenb Klinik für Chirurgie, Kantonsspital St. Gallenc HNO-Klinik, Kantonsspital St. Gallend Klinik für Radiologie und Nuklearmedizin, Kantonsspital St. Gallen
Fallbericht
AnamneseDie 32-jährige Patientin berichtete über rezidivierende Infekte im Bereich des oberen Respirationstrakts in den letzten eineinhalb Jahren mit intermittierender Heiserkeit. Zudem beklagte sie ein progredientes Druck-gefühl und eine zunehmende sichtbare Schwellung am Hals rechtsbetont. Tachykardien oder erhöhte Blut-druckwerte lagen keine vor. Die Familienanamnese war bezüglich Tumorerkrankungen bland.
StatusDie Patientin präsentierte sich in gutem Allgemeinzu-stand mit normalen Vitalparametern. Zervikal beidseits kaudal des Kieferwinkels fanden sich pulsierende, wenig verschiebliche, indolente Raumforderungen. Hirnner-venausfälle bestanden keine. Der Endolarynx zeigte sich mit symmetrisch beweglichen Stimmlippen ebenso unauffällig wie der weitere internistische Status.
BefundeSonographisch wurden angulär beidseits hypoecho-gene, inhomogene, unregelmässig begrenzte Raum-forderungen dargestellt. Im MRI (Abb. 1) bestätigte sich der Verdacht auf bilaterale Glomus-caroticum-Tumo-ren. Das folgende 18F-FDOPA-PET (Abb. 2) zeigte eine inten sive Radionuklidspeicherung im Bereich der Läsi-onen, fand aber keine Hinweise auf weitere Paragang-liome, Metastasen oder ein Phäochromozytom. Die freien Plasmametanephrine einschliesslich des Me-tho xytyramins waren normwertig. Auch bei ana-mnestisch fehlenden Hinweisen für eine hereditäre Erkran kung wurde eine Mutationsanalyse des SDHB-, SDHC- und SDHD-Gens durchgeführt, die eine Keim-bahnmutation des SDHD-Gens zeigte.
DiagnoseBilaterale Glomus-caroticum-Tumoren aufgrund ei-ner Keimbahnmutation im Succinat-Dehydrogenase-(SDHD)-Gen.
Therapie und VerlaufAufgrund der Beschwerden der Patientin, der fortge-schrittenen Ausdehnung und des jungen Alters wurde die Indikation zur Operation gestellt. Im Wissen um die mögliche Morbidität der Operation wegen der Tu-morausdehnung erfolgte primär die einseitige Resek-tion der dominanten Seite. Im Situs fand sich ein die Karotisgabel umgebender und bis an die Schädelbasis reichender Tumor mit vollständiger Ummauerung des N. vagus, N. glossopharyngeus, N. hypo glossus und N. accessorius. Dieser konnte von den Ge fäs sen, die nicht infiltriert waren, problemlos abpräpa riert wer-den, die entsprechenden Nerven wurden intraoperativ erhalten – dennoch manifestierten sich postoperativ partielle oder vollständige Ausfälle derselben.Die histologische Untersuchung zeigte ein Para-gangliom mit einem Proliferationsindex (Mib-1) von 5% sowie einer Positivität für Somatostatinrezeptoren (Subtyp 2, SSTR2). Insbesondere aufgrund der Vaguspa-rese mit der entsprechenden Schluckstörung im Sinne von Aspirationen und der Heiserkeit aufgrund der
Anita Koster-Rusch
Abbildung 1: T1-gewichtete MR-Sequenz nach Gadolinium-
Gabe: Nachweis zweier irregulär begrenzter, intensiv
Kontrastmittel-aufnehmender zervikaler Raumforderungen.
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):253–255
FALLBERICHTE 254
Stimmlippenlähmung wurde von einer Resektion des linksseitigen Tumors abgesehen, da bei einer bilatera-len Stimmlippenlähmung die Notwendigkeit einer Tra cheo tomie drohte. 3 und 6 Monate postoperativ durchgeführte MRI-Untersuchungen erbrachten keine Anhaltspunkte für einen rechtsseitigen Rest- oder Rezi divtumor und eine stabile Tumorgrösse links. Im Hinblick auf eine allfällige Peptidrezeptor-Radio-nuklidtherapie (PRRT) mit einem radioaktiv markier-ten Somatostatinrezeptorliganden (z.B. 177Lu-DOTA-TOC) wurde ergänzend ein 68Ga-DOTATATE-PET-CT durchgeführt, in dem der verbleibende linksseitige Tu-mor intensiv SSTR-positiv war, bei wiederum fehlen-den Hinweisen auf weitere Tumormanifestationen (Abb. 3). Durch intensive Physiotherapie und Logopä-die konnte eine partielle Verbesserung der Symptome der Hirnnervenausfälle erreicht werden. Aktuell sind Verlaufskontrollen und bei Tumorprogredienz eine DOTATOC- oder perkutane Radiotherapie geplant. Auf-grund des autosomal-dominanten Erbgangs wurde eine Fami lien abklärung eingeleitet.
Diskussion
Hals- und Kopf-Paragangliome (head and neck para-gangliomas, HNPGL) sind seltene Tumoren (Inzidenz 1:30 000–100 000), die von den chromaffinen Zellen der parasympathischen Ganglien entlang des N. glos-so pharyngeus und N. vagus ausgehen. Am häufigsten werden Glomus-caroticum-Tumoren beschrieben
(60%), gefolgt von Tumoren des Glomus jugulare, G. tympanicum und G. vagale. Thorakoabdominale Paragang liome hingegen entstehen in der Regel aus sympa thischen Ganglien [1, 2]. Oft werden die HNPGL inzidentell entdeckt, bei der Abklärung eines familiä-ren Paragangliomsyndroms oder aber aufgrund loka-ler Sym ptome als Folge der Kompression oder Infiltra-tion der umgebenden Strukturen. Die bei unserem Kasus beschriebenen rezidivierenden Infekte der obe-ren Luftwege sind als unabhängig von der schliesslich gestellten Diagnose eines HNPGL zu interpretieren. Pa-resen der Hirnnerven VII, IX, X, XI und XII mit den ent-sprechenden Symptomen der Gesichtslähmung oder von Schluck-, Stimm- und Sprechstörungen sind eher selten und treten nur bei sehr fortgeschrittenen HN-PGL auf, insbesondere solchen des Glomus jugulare. Eine Katecholaminsekretion aus parasympathischen Paragangliomen und hiermit assoziierte Symptome sind ungewöhnlich (<5%), biochemisch kann im Plasma aber bei ca. 30% eine Erhöhung des Dopaminmetabo-liten Methoxytyramin gefunden werden [3]. Die Be-stimmung der Plasmametanephrine im Rahmen der Abklärung ist wichtig und kann hinweisend für ein zusätzliches Phäochromozytom oder thorakoabdomi-nales Paragangliom sein. Die Diagnose eines Glomus-caroticum-Tumors kann mit der Duplexsonographie gestellt werden [4]. Während die lokoregionäre Tumor-ausdehnung in der Folge am besten durch eine MRI-Untersuchung, gegebenenfalls einschliesslich einer MR-Angiographie, erfasst wird, sind funktionelle Bild-
Abbildung 2: 18F-FDOPA-PET-CT: Intensive Radionuklidspei-
cherung in den beiden zervikalen Raumforderungen ohne
Nachweis weiterer Paragangliome oder Phäochromozytome.
Abbildung 3: 68Ga-DOTATATE-PET-CT: Nachweis einer deutli-
chen Somatostatinrezeptorpositivität im verbleibenden links-
seitigen Glomus-caroticum-Tumor.
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):253–255

FALLBERICHTE 255
gebungsverfahren (18F-FDOPA-PET-CT oder 68Ga-DOTA-TATE-PET-CT, Sensitivität >95%) sensitiver, um weitere Tumormanifestationen zu erfassen, und im nächsten Schritt empfohlen [5]. Ca. 35% der HNPGL werden im Rahmen eines hereditären Paragangliomsyndroms als Folge einer Keimbahnmutation in einem der für den Succinat-Dehydrogenase-(SDHx-)Komplex der mito-chondrialen Atmungskette kodierenden Gene beob-achtet. Am häufigsten sind Mutationen des SDHD-Gens (>50%) gefolgt von SDHB- (20–35%) und SDHC- (15%) Mutationen. Die Penetranz der Erkrankung ist hoch, Menschen mit einer SDHD-Mutation haben ein 75%- Risiko, im Verlauf des Lebens ein HNPGL zu entwickeln. Die Vererbung ist autosomal dominant, wobei bei der SDHD-Mutation die Krankheit nur auftritt, wenn die Mutation vom Vater vererbt wird (sog. maternal imprin-ting). Eine genetische (SDHB-, -C- und -D-Mutationen, evtl. SDHA und -AF2) und gegebenenfalls Familien-abklärung ist somit wünschenswert, de-novo-Muta-tionen gelten als Rarität. SDHx-Mutationen führen zu einer Akkumulation von Succinat, der Bildung von reaktiven Sauerstoffspezies und letztlich einer Akti-vierung des HIFα(hypoxia-inducible-factor α)-Signal-weges, der die Tumorentstehung auf mehreren Ebenen begünstigt [1]. Die immunhistochemische Untersu-chung der Expression der verschiedenen SDH-Proteine in chirurgisch entfernten Paragangliomen erlaubt ebenso Rückschlüsse auf eine zugrundeliegende SDHx-Mutation und kann, so diese Abklärung noch nicht erfolgt ist, für die genetische Untersuchung weg-weisend sein [2]. Maligne Paragangliome – diese Dia-gnose kann nur durch das Auftreten von Metastasen gestellt werden – sind bei einer SDHD-Mutation selten (<5%), werden hingegen bei SDHB-assoziierten Tumo-ren in bis zu 30% beobachtet [1]. Mit SDHx-Mutationen können Nierenzellkarzinome (SDHB ca. 14%, SDHD ca.
8%), Gastrointestinale Stromatumoren (GIST) und sel-ten Hypophysenadenome assoziiert sein [3].Prinzipiell können HNPGL bei Beschwerdefreiheit mit-tels MRI beobachtet werden. Dies gilt aufgrund der Operationsmorbidität vor allem für fortgeschrittene temporale Paragangliome, Glomus-vagale-Tumoren und fortgeschrittene Glomus-caroticum-Tumoren. Hier bei spielen auch das Alter und allfällige Komorbi-ditäten des Patienten eine wichtige Rolle. Therapeu-tisch kommen eine Operation, Radiotherapie und Pep-tidrezeptor-Radionuklidtherapie (PRRT) in Betracht. Kleinere HNPGL bei jüngeren Patienten sollen primär operiert werden, wobei bei bilateralen Tumoren ein zweizeitiges Vorgehen angezeigt ist. Bei Malignitäts-verdacht, vor allem bei einer nachgewiesenen SDHB-Mutation, soll ebenso eine Operation angestrebt wer-den. Insbesondere bei älteren Patienten mit ausgedehnten Tumoren hat sich die primäre Bestrah-lung etabliert (konventionelle Radiotherapie oder Ra-diochirurgie). Die PRRT ist besonders für multiple oder maligne Paragang liome ein vielversprechendes Ver-fahren. Für jeden Patienten muss in Abhängigkeit vom Lokalbefund, allfälliger weiterer Paragangliome, des Malignitätsverdachtes sowie der genetischen Konstel-lation ein individuelles Therapiekonzept erstellt wer-den. Auch nach der initialen Therapie sind regelmä-ssige Kontrollen nötig, da noch über Jahre Rezidive, metachrome Paragangliome oder Metastasen auftre-ten können. Eine genetische Beratung ist obligat. Bei im Rahmen eines genetischen Screenings entdeckten asymptomatischen Trägern einer SDHx-Mutation soll jährlich eine klinische und biochemische Untersu-chung sowie alle zwei bis drei Jahre eine MRI-Bildge-bung (Schädelbasis bis Becken) erfolgen [1].
Disclosure statementDie Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Literatur1 Taïeb D, Kaliski A, Boedeker CC, Martucci V, Fojo T, Adler JR, et al.
Current approaches and recent developments in the management of head and neck paragangliomas. Endocr Rev. 2014 Oct;35(5):795–819.
2 Favier J, Amar L, Gimenez-Roqueplo A-P. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015 Feb;11(2):101–11.
3 Benn DE, Robinson BG, Clifton-Bligh RJ. 15 YEARS OF PARAGANGLI-OMA: Clinical manifestations of paraganglioma syndromes types 1–5. Endocr Relat Cancer. 2015 Aug;22(4):T91–103.
4 Stoeckli SJ, Schuknecht B, Alkadhi H, Fisch U. Evaluation of para-gangliomas presenting as a cervical mass on color-coded Doppler sonography. The Laryngoscope. 2002 Jan;112(1):143–6.
5 Castinetti F, Kroiss A, Kumar R, Pacak K, Taieb D. 15 YEARS OF PARAGANGLIOMA: Imaging and imaging-based treatment of pheochromocytoma and paraganglioma. Endocr Relat Cancer. 2015 Aug;22(4):T135–45.
Schlussfolgerung für die PraxisHNPGL sind seltene neuroendokrine Tumoren. Die Diagnose wird durch
anatomische (MRI) und funktionelle (18F-FDOPA- oder 68Ga-DOTATATE-
PET-CT) bildgebende Verfahren bestätigt. Das Behandlungskonzept wird
von einem interdisziplinären Team unter Berücksichtigung der Klinik, Be-
funde aus der Bildgebung und des Resultats der genetischen Abklärung –
in 35% liegt ein familiäres Paragangliomsyndrom als Folge einer SDHx-
Mutation vor – festgelegt. Hierbei kommen eine «wait-and-scan»-Strategie,
eine Operation, eine Radiotherapie oder eine Radionuklidtherapie in Frage.
Bei Nachweis einer SDHx-Mutation ist ein Familienscreening anzustreben.
Korrespondenz: Anita Koster-Rusch Klinik für Allgemeine Innere Medizin und Hausarztmedizin Departement Innere Medizin Kantonsspital St. Gallen Rorschacherstrasse 95 CH-9007 St. Gallen Anita.Koster-Rusch[at] kssg.ch
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):253–255

FALLBERICHTE 256
Eine Herausforderung für alle Beteiligten
Das histiozytäre SarkomThomas Rauera, Teresa De Zuluetab, Uwe Casparc, Béatrice Wagnerd, Michael Zünda
a Chirurgische Klinik, Zuger Kantonsspital; b Onko-Zentrum, Medizinische Klinik, Zuger Kantonsspital; c Radiologie, Zuger Kantonsspital;d Pathologisches Institut, Luzerner Kantonsspital
Fallbericht
Ein 56-jähriger, HIV-1-positiver Patient (CDC-Stadium A3) stellte sich mit einer dreiwöchigen Anamnese ei-ner indolenten, grössenprogredienten Schwellung imBereich der linken Brust vor. Klinisch wurde die Verdachtsdiagnose eines Lipoms gestellt. Die bildgebende Diagnostik mittels Sonogra-phie und thorakoabdominaler Computertomographie(Abb. 1) zeigte einen solitären, 3,5 × 2 × 4 cm grossen, gutvaskularisierten Tumor im Bereich der linken Pektora-lismuskulatur. Die Stanzbiopsie ergab den dringenden Verdacht auf das Vorliegen eines histiozytären Sar-koms. Im Rahmen einer interdisziplinären Fallbesprechungwurde wegen des unifokalen Tumorbefalls die Indika-tion zur primären radikalen Resektion gestellt. Esfolgte die En-bloc-Resektion des Tumors mit Subkutis und dem grössten Teil des Musculus pectoralis majorbis auf die Thoraxwand unter Mitnahme des Musculus pectoralis minor unterhalb des Tumors. Hierdurch wurde eine R0-Resektion erreicht. Die histologischeAufarbeitung des Resektats (Abb. 2) bestätigte die Diag-nose eines histiozytären Sarkoms mit Expression der histiozytären Marker Faktor XIIIa und CD 68; typi-scherweise fehlte eine Expression der follikulär-dend-ritischen, Langerhanszell- und myeloischen Marker CD 21, CD 1a und MPO sowie spezifischer B- und T-Zellmar-ker, Epithelmarker und Marker der muskulären Diffe-renzierung. Bei komplikationslosem Verlauf konnte der Patient amdritten postoperativen Tag nach Hause entlassen wer-den. Eine adjuvante Strahlentherapie, die im Rahmeneiner interdisziplinären Tumorkonferenz empfohlen wurde, lehnte der Patient ab.In der Nachsorgekontrolle nach 4 Monaten zeigte sichsowohl in der thorakalen Computertomographie alsauch im anschliessendem F-18 FDG-PET/CT (Abb. 3) einsolitärer axillärer Lymphknotenbefall links (3×3×4 cm) ohne Hinweis auf ein Lokalrezidiv oder weitere nodale/extranodale Manifestationen. In der daraufhin durch-geführten therapeutisch/diagnostischen Axilladissek-tion wurden neben dem befallenen Lymphknoten wei-tere 12 tumorfreie Lymphknoten entfernt.
Postoperativ schloss sich eine Bestrahlung der gesamten linken Axilla Level 1 mit einer Gesamtdosis von 40 Gy (20×2 Gy) analog einer Lymphombehandlung an. In denNachsorgekontrollen nach 4 und 8 Monaten zeigte sich in der zerviko-thorako-abdominalen Computertomo-graphie (Abb. 4) erfreulicherweise kein Hinweis auf einLokalrezidiv oder neue nodale/extranodale Manifesta-tionen.
Diskussion
Das histiozytäre Sarkom ist kein Sarkom, sondern einemaligne Erkrankung hämatopoetischen Ursprungs aus-gehend von den reifen Gewebehistiozyten [1, 2]. Es han-delt sich um eine sehr seltene und häufig aggressive Non-Langerhanszell-Histiozytose noch nicht vollstän-dig geklärter Pathogenese, die weniger als 1% aller Non-Hodgkin-Lymphome darstellt [1]. Bisher sind knapp über 40 Fälle in der Literatur beschrieben worden [2]. Das histiozytäre Sarkom kann ohne eindeutige Ge-schlechterbevorzugung (männlich/weiblich 1,5 : 1) in je-
Thomas Rauer
Abbildung 1: Thorakoabdominales CT, sagittaler Strahlen-
gang: Tumor in der linken Pektoralismuskulatur (Pfeil).
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):256–258
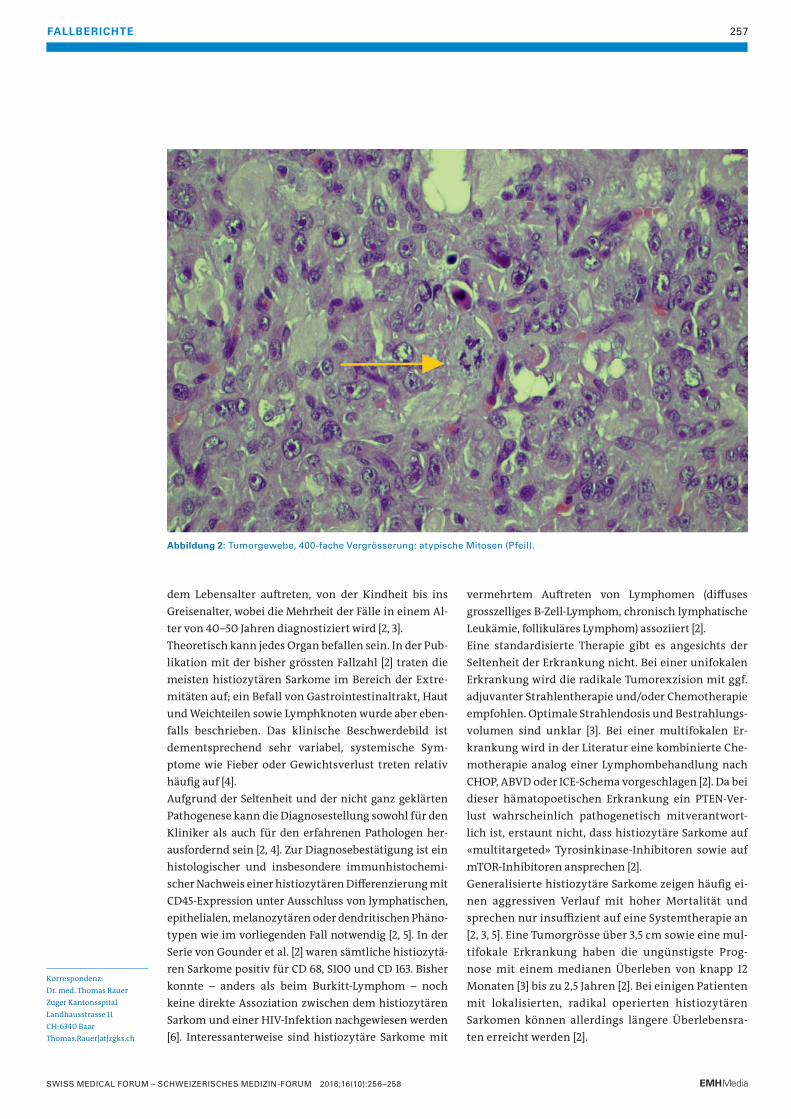
FALLBERICHTE 257
dem Lebensalter auftreten, von der Kindheit bis ins Greisenalter, wobei die Mehrheit der Fälle in einem Al-ter von 40–50 Jahren diagnostiziert wird [2, 3].Theoretisch kann jedes Organ befallen sein. In der Pub-likation mit der bisher grössten Fallzahl [2] traten die meisten histiozytären Sarkome im Bereich der Extre-mitäten auf; ein Befall von Gastrointestinaltrakt, Haut und Weichteilen sowie Lymphknoten wurde aber eben-falls beschrieben. Das klinische Beschwerdebild ist dementsprechend sehr variabel, systemische Sym-ptome wie Fieber oder Gewichtsverlust treten relativhäufig auf [4]. Aufgrund der Seltenheit und der nicht ganz geklärtenPathogenese kann die Diagnosestellung sowohl für denKliniker als auch für den erfahrenen Pathologen her-ausfordernd sein [2, 4]. Zur Diagnosebestätigung ist einhistologischer und insbesondere immunhistochemi-scher Nachweis einer histiozytären Differenzierung mit CD45-Expression unter Ausschluss von lymphatischen, epithelialen, melanozytären oder dendritischen Phäno-typen wie im vorliegenden Fall notwendig [2, 5]. In der Serie von Gounder et al. [2] waren sämtliche histiozytä-ren Sarkome positiv für CD 68, S100 und CD 163. Bisherkonnte – anders als beim Burkitt-Lymphom – nochkeine direkte Assoziation zwischen dem histiozytären Sarkom und einer HIV-Infektion nachgewiesen werden [6]. Interessanterweise sind histiozytäre Sarkome mit
vermehrtem Auftreten von Lymphomen (diffusesgrosszelliges B-Zell-Lymphom, chronisch lymphatische Leukämie, follikuläres Lymphom) assoziiert [2]. Eine standardisierte Therapie gibt es angesichts derSeltenheit der Erkrankung nicht. Bei einer unifokalenErkrankung wird die radikale Tumorexzision mit ggf. adjuvanter Strahlentherapie und/oder Chemotherapie empfohlen. Optimale Strahlendosis und Bestrahlungs-volumen sind unklar [3]. Bei einer multifokalen Er-krankung wird in der Literatur eine kombinierte Che-motherapie analog einer Lymphombehandlung nachCHOP, ABVD oder ICE-Schema vorgeschlagen [2]. Da beidieser hämatopoetischen Erkrankung ein PTEN-Ver-lust wahrscheinlich pathogenetisch mitverantwort-lich ist, erstaunt nicht, dass histiozytäre Sarkome auf«multitargeted» Tyrosinkinase-Inhibitoren sowie aufmTOR-Inhibitoren ansprechen [2].Generalisierte histiozytäre Sarkome zeigen häufig ei-nen aggressiven Verlauf mit hoher Mortalität undsprechen nur insuffizient auf eine Systemtherapie an[2, 3, 5]. Eine Tumorgrösse über 3,5 cm sowie eine mul-tifokale Erkrankung haben die ungünstigste Prog-nose mit einem medianen Überleben von knapp 12Monaten [3] bis zu 2,5 Jahren [2]. Bei einigen Patienten mit lokalisierten, radikal operierten histiozytärenSarkomen können allerdings längere Überlebensra-ten erreicht werden [2].
Abbildung 2: Tumorgewebe, 400-fache Vergrösserung: atypische Mitosen (Pfeil).
Korrespondenz: Dr. med. Thomas RauerZuger Kantonsspital Landhausstrasse 11 CH-6340 Baar Thomas.Rauer[at]zgks.ch
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):256–258
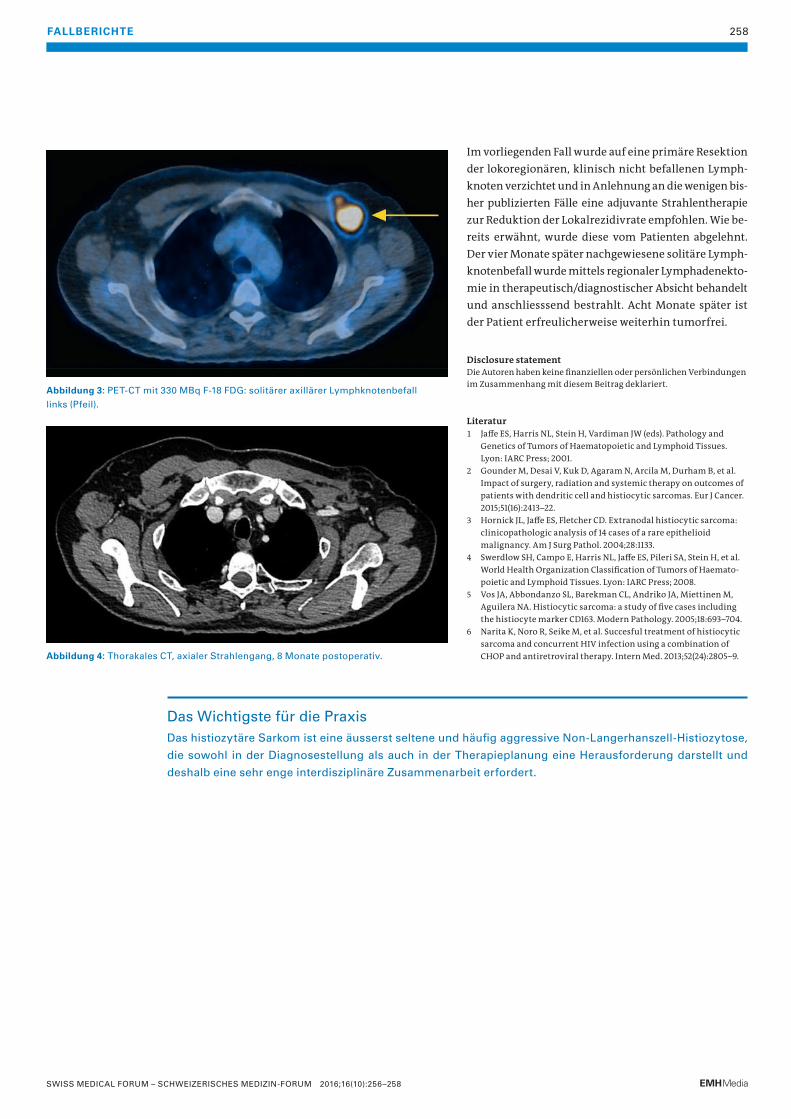
FALLBERICHTE 258
Im vorliegenden Fall wurde auf eine primäre Resektion der lokoregionären, klinisch nicht befallenen Lymph-knoten verzichtet und in Anlehnung an die wenigen bis-her publizierten Fälle eine adjuvante Strahlentherapie zur Reduktion der Lokalrezidivrate empfohlen. Wie be-reits erwähnt, wurde diese vom Patienten abgelehnt.Der vier Monate später nachgewiesene solitäre Lymph-knotenbefall wurde mittels regionaler Lymphadenekto-mie in therapeutisch/diagnostischer Absicht behandeltund anschliesssend bestrahlt. Acht Monate später ist der Patient erfreulicherweise weiterhin tumorfrei.
Disclosure statementDie Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Literatur1 Jaffe ES, Harris NL, Stein H, Vardiman JW (eds). Pathology and
Genetics of Tumors of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2001.
2 Gounder M, Desai V, Kuk D, Agaram N, Arcila M, Durham B, et al.Impact of surgery, radiation and systemic therapy on outcomes of patients with dendritic cell and histiocytic sarcomas. Eur J Cancer. 2015;51(16):2413–22.
3 Hornick JL, Jaffe ES, Fletcher CD. Extranodal histiocytic sarcoma: clinicopathologic analysis of 14 cases of a rare epithelioidmalignancy. Am J Surg Pathol. 2004;28:1133.
4 Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al.World Health Organization Classification of Tumors of Haemato-poietic and Lymphoid Tissues. Lyon: IARC Press; 2008.
5 Vos JA, Abbondanzo SL, Barekman CL, Andriko JA, Miettinen M, Aguilera NA. Histiocytic sarcoma: a study of five cases includingthe histiocyte marker CD163. Modern Pathology. 2005;18:693–704.
6 Narita K, Noro R, Seike M, et al. Succesful treatment of histiocyticsarcoma and concurrent HIV infection using a combination of CHOP and antiretroviral therapy. Intern Med. 2013;52(24):2805–9.
Abbildung 3: PET-CT mit 330 MBq F-18 FDG: solitärer axillärer Lymphknotenbefall
links (Pfeil).
Abbildung 4: Thorakales CT, axialer Strahlengang, 8 Monate postoperativ.
Das Wichtigste für die PraxisDas histiozytäre Sarkom ist eine äusserst seltene und häufig aggressive Non-Langerhanszell-Histiozytose,
die sowohl in der Diagnosestellung als auch in der Therapieplanung eine Herausforderung darstellt und
deshalb eine sehr enge interdisziplinäre Zusammenarbeit erfordert.
SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(10):256–258





























