Embed Size (px)
Citation preview

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 26
CHAPTER – II
STABILITY INDICATING LIQUID
CHROMATOGRAPHIC METHODS FOR
THE SIMULTANEOUS
DETERMINATION OF DRUG
PRODUCTS.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 27
2.1. INTRODUCTION:
Stability testing forms an important part of the drug product testing , it provide evidence
on how quality of a drug substance or drug product varies with time under the influence of a
variety of environmental factors such as temperature, humidity, light and enables
recommendation of storage conditions, retest period and shelf life to be established. The two
main aspects of drug products that play an important role in shelf life determination are assay of
active drug and degradants generated during the stability study. There are several stability-
indicating methods has been reported for assays of various drugs in drug products containing
only one active drug substance. Only few stability indicating methods are reported for the assay
of combination drug products containing two or more active drug substances.
The objective of the current study is to develop and validated stability indicating
reversed-phase HPLC method for the simultaneous determination of drug product. First HPLC
method is for simultaneous determination of Levocetirizine dihydrochloride and
Pseudoephedrine sulfate in Tablet dosage forms, second sensitive LC method for simultaneous
determination of Ciclesonide and Formoterol fumarate in dry powder inhaler and third Sensitive
LC method for the Simultaneous determination of Diacerein and Aceclofenac in tablet dosage
form.
Levocetirizine dihydrochloride is [2-[4-[(R)-(4-Chlorophenyl)phenylmethyl]-1-
piperazinyl]ethoxy]-acetic acid dihydrochloride is the pharmacologically active enantiomer of
cetirizine, and is a potent histamine H-1 receptor antagonist1 and Pseudoephedrine sulfate
chemically [(S-(R*,R*))-alpha-(1-(Methylamino)ethyl)benzenemethanol sulfate].
Pseudoephedrine sulfate is official in USP2. Levocetirizine dihydrochloride is official in Indian
Pharmacopoeia3. Levocetirizine Dihydrochloride is having antiallergic properties used Once
daily for the treatment of allergic rhinitis4.
A Fixed dose combination of 180 mg of Pseudoephedrine sulfate and 5 mg of
Levocetirizine dihydrochloride is available commercially as tablets and are widely used for the
symptomatic treatment of allergic rhinitis.
Formoterol fumarate is N-[2-Hydroxy-5-[[(1RS)-1-hydroxy-2-[[(1RS)-2-(4-
methoxyphenyl)-1-methylethyl] amino] ethyl] phenyl] formamide(E)-2-butenedioate is b2-

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 28
agonist with a long duration of action.5 Ciclesonide is (R)-11b,16a,17,21-Tetrahydroxypregna-
1,4-diene-3, 20-dione cyclic 16,17-acetal with cyclohexanecarboxaldehyde 21-isobutyrate is
effective and novel Inhaled corticosteroids, which has very low affinity for the glucocorticoid
receptor in its native form, but very high affinity when transformed to its active metabolite by
esterase in the lung. 6
Bronchodilator medications are central to the symptomatic management of
chronic obstructive pulmonary disease (COPD). Long-acting inhaled bronchodilators are more
convenient. Also, Systemic corticosteroids are beneficial in the management of acute
exacerbations of COPD. 7
As a result, the combination of b2-agonist and corticosteroids has been
a more useful tool in the management of asthma and COPD. Formoterol fumarate is reported in
British Pharmacopoeia8 whereas Ciclesonide is reported in Indian Pharmacopoeia
9. Fixed dose
combinations of 6 mg of Formoterol fumarate (FF) and 200 mg of Ciclesonide (CS) are available
commercially as dry powder inhaler and are widely used for the treatment of COPD.
Diacerein is chemically 1,8-diacetoxy-3-carboxyanthraquinone and is also known as
diacetylrhein. This drug is used in the treatment of osteoarthritis. After absorption, the drug is
metabolized to its active metabolite rhein10
. Diacerein and rhein are anthraquinone compounds
that ameliorate the course of osteoarthritis11-13
. Aceclofenac is chemically (2-[(2,6-
dichlorophenyl)amino] phenyl acetoxyacetic acid)14
. It has analgesic properties and a good
tolerability profile in a variety of painful conditions. A combined fixed dose of 50 mg Diacerein
and 100 mg Aceclofenac is available commercially as tablets and are widely used for the
treatment of osteoarthritis.
2.2 LITERATURE SURVEY:
In this section summarized some of the important analytical methods for the
determination of Levocetirizine dihydrochloride, Pseudoephedrine sulfate, Formoterol fumarate
Ciclesonide, Aceclofenac and Diacerein as a individual component or in combination with
another drug substance.
Feyyaz O., et. al., (2000)15
Feyyaz O et. al., reported Spectrophotometric method for the simultaneous
determination of Pseudoephedrine Sulfate, Dexbrompheniramine Maleate and Loratadine in

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 29
pharmaceutical preparations using derivative spectrophotometry and ratio spectra derivative
spectrophotometry. Two spectrophotometric methods are described for the simultaneous analysis
of pseudoephedrine sulfate-dexbrompheniramine maleate and pseudoephedrine sulfate-loratadine
combinations. The procedures do not require any separation step. Mean recoveries were found to
be >99% in the methods for these compounds in their synthetic mixtures.
Makhija S.N., et. al., (2001)16
Makhija S.N., reported HPTLC method for the simultaneous determination of
pseudoephedrine and cetirizine in pharmaceutical formulations. The solvent system consisted of
ethyl acetate-methanol-ammonia (7:1.5:1, v/v/v). This system was found to give compact spots
for both pseudoephedrine (Rf value of 0.69+/-0.01) and cetirizine (Rf value of 0.38+/-0.01).
Spectrodensitometric scanning-integration was performed at a wavelength of 240 nm.
Nian W., et. al., (2002)17
Nian W., et. al., reported HPLC method for the quantitative determination of
pseudoephedrine sulfate and its related compounds in pharmaceutical preparation. Isocratic
reversed-phase high performance liquid chromatograghic method for the separation of
pseudoephedrine and its related compounds in pharmaceutical formulations is described. The
separation is achieved on a C-18 column (4.6 mm x 25 cm length, 5µ particle size) using a
mobile phase consisting of a mixture of ammonium acetate and methanol. With run time 35
minutes.
Mabrouk M.M., et. al., (2003)18
Mabrouk M.M., et. al., reported Reversed phase liquid chromatographic and first
derivative spectrophotometric methods for determination of antihistaminic drug Loratadine and
nasal decongestant drug Pseudoephedrine sulfate. The HPLC method involves separation of
Loratadine and Pseudoephedrine sulfate on micro-BondaPak C18 column using mixture of
methanol:water:phosphoric acid:ammonium dihydrogen phosphate in the ratio of 220:300:2:3
(V/V/V/W), 60 and 40 acetonitrile as mobile phase flowing at 2 ml/min with ultraviolet detection

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 30
at 247 nm. The spectrophotometric method is based on recording the first derivative spectra for
Loratadine and Pseudoephedrine and at 307, 266 nm, respectively.
Tahraoui A., et. al., (2005)19
Tahraoui A., reported a new HPLC approach for the determination of hydrophilic and
hydrophobic components: the case of Pseudoephedrine sulfate and Loratadine in tablets.he
chromatographic behavior of Pseudoephedrine sulfate and Loratadine on RP C18 and C8
columns were studied in presence and absence of sodium lauryl sulfate (SLS). The effect of
combining two different types of stationary phases (cyano and C18 or C8) on the relative
retention of the two compounds was investigated. In conclusion, it was found that the
combination of a C18 column followed by a standard cyano column provides a stationary phase
that separates both compounds effectively and within a reasonable time.
Culzoni M.J., et. al., (2007)20
Culzoni M.J., reported determination of Loratadine and Pseudoephedrine sulfate in
pharmaceuticals based on non- linear second-order spectrophotometric data generated by a pH-
gradient flow injection technique and artificial neural networks, Loratadine
and Pseudoephedrine sulfate were determined in pharmaceutical samples by using non-linear
second-order data generated by a pH-gradient flow injection analysis system with diode-array
detection. Determination of both analytes was performed on the basis of differences between the
acid-base and spectral features of each drug species. Non-linearities were detected by using both
qualitative and quantitative tools.
Lakshmana P., et. al., (2008)21
Lakshmana P., et. al., reported UV spectrophotometric method for the estimation of
Ambroxol hydrochloride and Levocetirizine dihydrochloride. The method involved solving
simultaneous equations based on measurement of absorbance at two wavelengths 242 nm and
231 nm, the γ max of Ambroxol hydrochloride and Levocetirizine dihydrochloride, respectively.
Beer's law was obeyed in the concentration range 10–50 µg/ml and 8–24 µg/ml for Ambroxol

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 31
hydrochloride and Levocetirizine dihydrochloride respectively. Results of the method were
validated statistically and by recovery studies.
Hadad G. M., et. al., (2009)22
Hadad G. M., et. al., reported development and validation of a stability-indicating RP
HPLC method for the determination of Paracetamol with Dantrolene or/and Cetirizine
and Pseudoephedrine in two pharmaceutical dosage forms. A gradient mobile phase system
consisting of (A) 50 mmol L(-1) sodium dihydrogen phosphate, 5 mmol L(-1) heptane sulfonic
acid sodium salt, pH 4.2 and (B) acetonitrile was used with discovery reversed-phase HS C(18)
analytical column (250 mm x 4.6 mm i.d., 5 µ particle size). Quantitation was achieved with UV
detection at 214 nm, based on peak area.
Kalogria E., et. al., (2010)23
Kalogria E., et. al., reported a porous graphitized carbon column HPLC method for the
quantification of Paracetamol, Pseudoephedrine, and Chlorpheniramine in a pharmaceutical
formulation. Chromatographic separation was achieved isocratically on an RP porous graphitized
carbon analytical column (125 x 2.1 mm , 5µ) using 5.0 mM ammonium acetate-acetonitrile (35
: 65 v/v) mobile phase at a flow rate of 0.50 mL/min. UV spectrophotometric detection at 220
nm was used.
Reddy J. M., et. al., (2011)24
Reddy J. M., et. al., reported RP-HPLC for simultaneous estimation of
Diethylcarbamazine and Levocetirizine in tablet Formulation. UV detector was carried out at 224
nm, using Princeton Sphere-100 C18 (250×4.6 mm. 5 µ) column. The mobile phase used was
20mM potassium dihydrogen orthophosphate buffer (pH: 3.2):acetonitrile (50:50 v/v) with
isocratic flow rate 1 ml/min. The compounds Diethylcarbamazine, Levocetirizine and
Losartan otassium were eluted at 2.12, 4.27 and 5.96 min, respectively. The peaks were eluted
with better resolution.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 32
Joshi S., et. al., (2012)25
Joshi S., et. al., reported quantization of Dextromethorphan and Levocetirizine in
combined dosage form using a novel validated RP-HPLC method the separation of these
compounds was achieved within 10 min on a Phenomenex (USA) C 18 analytical column,
250Χ4.0 mm i.d., using an isocratic mobile phase consisting of potassium dihydrogen phosphate
buffer (pH 2.5) - acetonitrile- tetrahydrofuran (70:25:5 v/v/v). The analysis was performed at a
flow rate of 1.2 ml/min and at a detection wavelength of 232 nm.
Srividya P., et. al., (2013)26
Srividya P., et. al., reported RP-HPLC method for the simultaneous analysis of
Levocetirizine dihydrochloride, Ambroxol hydrochloride, and Montelukast sodium. Analysis
was carried out on C18 reverse phase column (Phenomenox- RP Aqueous) of 250 × 4.6 mm
dimensions and 5 µm particle size with mobile phase containing 15 mm of Ammonium
acetate:Acetonitrile (40:60 v/v) by using isocratic mode and eluents were monitored at 215 nm.
The retention times for Levocetirizine dihydrochloride, Ambroxol hydrochloride, and
Montelukast sodium. are 2.21, 4.46, and 13.35 nm, respectively.
Other than above mentioned methods few more methods are also reported for the
quantification of Levocetirizine dihydrochloride in combination with another drug substances27,28
and Quantification of Levocetirizine dihydrochloride in human plasma29
. Similarly quantification
of Pseudoephedrine by LC–MS–MS 30
in human plasma and simultaneous quantification
Pseudoephedrine with another drug substances by HPLC31
are reported.
Butter J.J., et. al., (1996)32
Butter J.J., et. al., reported HPLC determination of Formoterol RR and SS
enantiomers in Urine. A method is described for the determination of the R,R- and S,S-
enantiomer of the long-acting ß2-adrenoceptor agonist formoterol, The sample clean-up from
urine takes place by liquid liquid extraction followed by solid phase extraction. An AGP-column
combined with electrochemical detection is used for the separation and detection of the
enantiomers.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 33
Campestrini J., et. al., (1997) 33
Campestrini J., et. al., reported high-performance liquid chromatography and
electrochemical detection method for the determination of formoterol in human plasma. . The
compounds were eluted with pH 6 buffer solution-methanol (70:30, v/v) and the eluate was
further diluted with water. An aliquot of the extract solution was injected and analyzed by
HPLC. The extraction, dilution, injection and chromatographic analysis were combined and
automated using the automate (ASPEC) system. The chromatographic separations were achieved
on a 5 microm, Hypersil ODS analytical column (200 mm x 3 mm I.D.), using (pH 6 phosphate
buffer, 0.035 M + 20 mg/l EDTA)-MeOH-CH3CN (70:25:5, v/v/v) as the mobile phase at a
flow-rate of 0.4 ml/min. The analytes were detected with electrochemical detection at an
operating potential of +0.63 V.
Song J.Z., et. al., (1999)34
Song J.Z., et. al., reported capillary electrophoresis assay method for the determination
formoterol in dry syrup. The development of a capillary zone electrophoresis method with head-
column field-amplified sample stacking injection for the determination of formoterol in a low
dosage dry syrup form was described. To obtain the highest sensitivity, the sample solution was
prepared by high content of organic solvent with the presence of a small amount of H+ (60-100
µ) and the capillary inlet end was dipped in water before electroinjection.
Akapo S., et. al., (2003)35
Akapo S., et. al., reported RP-HPLC Assay of Formoterol and its Related Substances in
Formoterol Fumarate Dehydrate Drug Substance. A stability-indicating reversed-phase high
performance liquid chromatographic (HPLC) method has been developed and validated for
the assay offormoterol fumarate and the related substances, namely, formoterol
fumarate desformyl and formoterol fumarate acetamide analogs, in the active pharmaceutical
ingredient. The separation was achieved by isocratic elution using an Alltech Alltima C18 (150 x
4.6 mm) column, a mobile phase consisting of ammonium acetate (50 mM; pH 5.0)-ethanol
(65:35, v/v), a flow rate of 1.0 ml/min and UV detection at 242 nm.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 34
Akapo S. O., et. al., (2004)36
Akapo S. O., et. al., reported gas chromatographic method for analysis of (RS,SR)-
astereoisomeric impurity in formoterol fumarate. The method involves silylation of formoterol
fumarate with N-(trimethylsilyl)imidazole in N,N-dimethylformamide at room temperature in an
autosampler vial to produce trimethylsilyl derivatives of the enantiomers prior to GC analysis.
The optimized silylation and separation conditions, respectively, produced good yield and
resolution of the analytes. The method appears to be convenient and fast, and permits accurate
determination of (RS,SR)-diastereoisomer in formoterol fumarate with adequate precision.
Nave R., et. al., (2005)37
Nave R., et. al., reported Formation of fatty acid conjugates of ciclesonide active
metabolite in the rat lung after 4-week inhalation of ciclesonide. Ciclesonide and des-CIC
concentrations were determined using solid-phase extraction and reverse-phase high-
performance liquid chromatography with tandem mass spectrometry (LC/MS/MS).
Concentrations of fatty acid ester conjugates were indirectly assessed using enzymatic de-
esterification before LC/MS/MS.
Prasad A.V.S.S., et. al., (2006)38
Prasad A.V.S.S., et. al., reported spectrophotometric determination of Formoterol
Fumarate and Budesonide in their combined dosage forms. Methanol is used as diluent for
carring out analysis. The γ max values for formoterol fumarte and budesonide are 217 nm and
252 nm respectively. The results of analysis by this method have been found to be precise and
accurate.
Kakubari I., et. al., (2007)39
Kakubari I., et. al., reported liquid chromatography-electrospray ionization mass
spectrometry in rat plasma using high performance liquid chromatographic separation with
tandem mass spectrometry. Samples were purified using liquid-liquid extraction and separated
on CAPCELL PAK C18 UG120 (2.0 x 150 mm) with a mobile phase consisting of a mixture of
methanol- 50 mM ammonium hydrogen carbonate (1:1 v/v). Detection was performed with a

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 35
TSQ 7000 mass spectrometer using positive ion electrospray ionisation, monitoring the shift
from precursor ions for formoterol at m/z 344.9 to product ions of m/z 121.0.
Mascher H.J., et. al., (2008)40
Mascher H.J., et. al., reported HPLC-MS/MS for the determination of Ciclesonide,
Ciclesonide-M1-Metabolite and Fluticasone Propionate in Human Serum. Serum was mixed
with the internal standards (IS) D11-CIC and D11-CIC-M1 and extracted with diisopropylether.
A gradient with acetonitrile (containing 10 mM of acetic acid and 10% of acetone) was used.
HPLC-MS/MS of the acetic acid adducts of the analytes was performed in negative mode. The
novel aspect of this method is that instead of the dopant being introduced directly into the source
by means of an external HPLC pump, it was added to the mobile phase. This provided
significantly better sensitivity than the usual method of in-source addition of the dopant, and
with no loss in HPLC performance.
Akapo S., et. al., (2009)41
Akapo S., et. al., reported Chiral HPLC analysis of Formoterol Stereoisomers. Analysis
was performed on a Chiral-AGP column (100 x 4-mm, 5-microm) using a variable mixture of
mobile phase A (50-mM sodium phosphate buffer, pH 7.0) and B (10% v/v IPA) at a flow rate of
1.3 ml min(-1), and UV detection at 242 nm. A chiral HPLC method was validated and
successfully applied for the determination of formoterol stereoisomers and their inversion
products in an aqueous matrix stored at 5-70 °C up to 3 weeks.
Dave N. H., et. al., (2010)42
Dave N.H., et. al., reported rapid Spectrophotometric methods for the determination of
two novel steroids Ciclesonide and Fluticasone propionate in bulk and pressurised metered -
dose preparations. The developed methods for determination of both the steroids are direct
spectrophotometric method and first derivative spectrophotometric method. The absorbance of
Ciclesonide was measured at 243 nm for direct spectrophotometric determination. Whereas
Fluticasone was determined by first derivative spectroscopy, the first derivative spectra were
plotted with delta lambda 8 nm and scaling factor 10.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 36
Fei L., et. al., (2011)43
Fei L., et. al., reported Development of ciclesonide dry powder inhalers and the
anti-asthmatic efficacy in guinea pigs, The ciclesonide was measured with a high
per- formance liquid chromatography system with an ultraviolet detector.
The analysis was performed on ODS C18 column (Alltima, 250 mm×4.6 mm, 5µm) at 30 ºC
and the wavelength was set at 242nm.The mobile phase consisted of anhydrous alcohol
and water (61:39 v/v). The flow rate was 1.0 mL/min, and the injection volume was 20 µL.
Srinivasarao K., et. al., (2012)44
Srinivasarao K., et. al., reported HPLC method for determination of Formoterol
Fumarate and Mometasone Furoate in Metered Dose Inhaler. Separation was achieved on a
reversed-phase C18 column (150 mm×4.6 mm i.d., 5 µm) using a mobile phase consisting of
Sodium dihydrogen orthophosphate buffer/acetonitrile (50:50, v/v) at a flow rate of 1.0 mL/min
and UV detection at 220 nm.
Vaghela V., et. al., (2013)45
Vaghela V., et. al. reported optimized method for rapid estimation of ciclesonide in bulk
and its dosage form . The chromatographic method with isocratic elution by utilizing an inertsil
ODS-C18, 250 mm × 4.6 mm, 5 µm column. A mobile phase consisting of solvent [solution
containing Methanol:Water (95:05%, v/v)] endowed at a flow rate of 1.0 mL min−1
. The analyte
was detected and quantified at λmax 242 nm using UV detector.
Other than above mentioned methods few other methods are also reported for the
determination of formatoerol, such as capillary electrophoresis method46
, Spectophotometric
method47
and HPLC method48-49
for determination of formoterol in human serum and the assay of
Formoterol and Budesonide in Symbicort Turbuhaler.
Hinz B., et. al., (2003)50
Hinz B., et. al., reported HPLC method for simultaneous determination of Aceclofenac
and three of its metabolites in human plasma. A liquid–liquid extraction-based reversed-phase
HPLC method with UV detection was validated and applied for the analysis of Aceclofenac and

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 37
three of its metabolites (4′-hydroxy-Aceclofenac, Diclofenac, 4′-hydroxy-Diclofenac) in human
plasma. The analytes were separated using an Acetonitrile–Phosphate buffer gradient at a flow
rate of 1 mL/min, and UV detection at 282 nm. The retention times for Aceclofenac, Diclofenac,
4′-hydroxy-aceclofenac, 4′-hydroxy-diclofenac and ketoprofen (internal standard) were 69.1,
60.9, 46.9, 28.4 and 21.2 min, respectively.
Chen H., et. al., (2004)51
Chen H., et. al., reported HPLC method for the determination of Aceclofenac in human
plasma by reversed-phase high performance liquid chromatography. Chromatography was
performed on an ODS column with methanol-0.1 mol/L ammonium acetate (pH 6.0) (7:3, v/v) as
the mobile phase. The flow rate was 1.0 mL/min. The UV-Vis detector was set at 275 nm.
Angelo Z., et. al., (2005)52
Angelo Z., et. al. reported separation of Aceclofenac and Diclofenac in human plasma
by free zone capillary electrophoresis using N-methyl-D-glucamine as an effective electrolyte
additive. The effect of increasing concentrations of N-methyl-D-glucamine organic base on
borate run buffer was investigated. A good separation was achieved using a 40 cm × 75 µm
uncoated silica capillary, 300 mmol/l sodium borate buffer, 200 mmol/l N-methyl-D-glucamine,
pH 8.9, in about 3 min. Momin M.Y., et. al., (2006)
53
Momin M.Y., et. al., reported HPLC method for determination of Aceclofenac and
Paracetamol in tablet dosage form. Chromatography was perform on reverse phase C-18 column
(Intersile 4.6 mm×25 cm, 10 µm) using Acetonitrile : 50 mM Sodium dihydrogen
orthophosphate in a ratio of 65:35 (pH adjusted to 3.0 with orthophosphoric acid) as a mobile
phase at a flow rate of 1.5 ml/min and detection at 276 nm. The retention time for Aceclofenac
and Paracetamol was found to be 1.58 and 4.01 min respectively. The method can be used for
estimation of combination of these drugs in tablets.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 38
Han C.Y., et. al., (2007)54
Han C.Y., et. al., reported flow injection chemiluminescence method for sensitive
determination of Nanogram levels of Diacerein in a pharmaceutical formulation. It was based on
the greatly enhancive effect of diacerein on the CL reaction between luminal and hydrogen
peroxide in alkaline medium. The enhanced CL intensity was linear with the concentration of
diacerein over the range 1.0-500 ng/mL. The degradation of diacerein was also investigated
briefly.
Borgmann S. M., et. al., (2008)55
Borgmann S. M., et. al., reported spectrophotometric method for estimation of Diacerhein
in capsules dosage form. The dissolution established conditions were: 900 mL of sodium
phosphate buffer pH 7.0 with 0.75 % of sodium lauryl sulphate as dissolution medium, using a
basket apparatus at a stirring rate of 50 rpm. The drug release was evaluated by UV
spectrophotometric method at 258 nm. The method was validated to meet requirements for a
global regulatory filing.
Ojha A., et. al., (2009)56
Ojha A., et. al., reported HPLC method for determination of Rhein and Aeclofenac in
human plasma. Simple HPLC method with UV detection for simultaneous determination of
rhein and aceclofenac from human plasma samples. Sample preparation was accomplished
through liquid-liquid extraction with ethyl acetate and chromatographic separation was
performed on a reversed-phase ODS column. Mobile phase consisted of a mixture of acetate
buffer and acetonitrile run under gradient at flow rate of 1.0 ml/min. Wavelength was set at 258
nm.
Narade S., et. al., (2010)57
Narade S., reported UV spectrophotometric method for the determination of Diacerein in
capsules. DMF: Distilled water (1:4) used as a diluent and λmax was found to be 258.5 nm. The
solution was filtered through Whatman filter paper No. 41. The absorbance of standard and
sample solutions was measured at 258.5 nm using diluent as blank.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 39
Maheshwari R. et. al., (2011)58
Maheshwari R. reported application of mixed-hydrotropy in titrimetric analysis of
Aceclofenac bulk drug sample, Investigation includes the enhancement of solubility of
Aceclofenac by more than 1155 fold in (20% N,N-dimethyl urea + 20% sodium citrate) solution
as compared to solubility in distilled water, utilizing the concept of mixed-hydrotropy. Mixed
hydrotropic solution was employed to solubilize a poorly water-soluble drug - aceclofenac, in
bulk to carry out titrimetric estimation precluding the use of organic solvents which are toxic,
eco-pollutant and costlier. Statistical data proved the accuracy, reproducibility and the precision
of the proposed method.
Shirwaikar A., et. al., (2012)59
Shirwaikar A., et. al., reported determination of Diacerein in Rabbit Plasma by LC -
Mass Spectroscopy. Chromatography was performed on C18 (4.6mm i.d.×50mm) analytical
column and operated at 40°C. The mobile phase was acetonitrile: 1mM ammonium formate
(70:30, v/v) at a flow-rate of 400µl/min. Detection was made at m/z 283.2/183 for Diacerein and
380/316.4 for internal standard (Celecoxib).
Somashekar P.L., et. al., (2013)60
Somashekar P.L. et .al. reported HPLC method for the quantification of Aceclofenac
Specified Impurity-B (Methyl[2-[(2, 6-dichlorophenyl)amino] phenyl]acetate) in Aceclofenac
Bulk Drug. Early chromatographic work was performed GraceSmart - RP-C18, 250 mm x 4.6
mm, 5 µm columns as stationary phase and various combinations of buffered (pH 4.5-5.0)
organic phases (Acetonitrile and /or Methanol). The flow rate of mobile phase was varied within
0.5-1.5 mL/min. All noted measurements were performed with an injection volume of 100 µL, 1
mL/min flow rate and UV detection at 275 nm using mobile phase- acetonitrile and phosphate
buffer pH-5 (60:40).
Other than above mentioned methods few other methods are also reported for the
determination of Aceclofenac, such as capillary Spectophotometric method61
and HPLC
method62-66
for determination of Aceclofenac in tablet dosage form or in combination with
another drug substance.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 40
2.3. A STABILITY-INDICATING LC METHOD FOR THE SIMULTANEOUS
DETERMINATION OF LEVOCETIRIZINE DIHYDROCHLORIDE AND
PSEUDOEPHEDRINE SULFATE IN TABLET DOSAGE FORMS.
A comprehensive literature survey revealed the lack of suitable stability indicating assay
method for the determination of these two drugs in pharmaceutical dosage forms also Analytical
method for the determination of Levocetirizine dihydrochloride and Pseudoephedrine in tablet
dosage form not official in any pharmacopeia. Hence it is felt essential to develop HPLC
method for the determination of Levocetirizine dihydrochloride and Pseudoephedrine sulfate in
the tablet dosage form.
The mobile phase A consisted of Potassium dihydrogen phosphate Buffer 0.05M and 1-
Ocatne sulphonic acid sodium salt 0.25%, pH adjusted to 3.0 with orthophospheric acid. Mobile
Phase B: Acetonitrile, Gradient elution at flow rate of 1 mL/min and Column temperature at
40◦C. Detector wavelength of 242 nm using a photodiode array detector. The described method
shows excellent linearity over a range of 200–10 µg ml−1 for Levocetirizine dihydrochloride and
7200-360 µg ml−1 for Pseudoephedrine sulfate. The correlation coefficient for Levocetirizine
dihydrochloride and Pseudoephedrine sulfate are 0.9999.
The proposed stability indicating HPLC method for simultaneous determination of
Levocetirizine dihydrochloride and Pseudoephedrine sulfate in tablet dosage form is cost
effective, precise, accurate, linear and suitable for routine analysis and quality control stability
study of drug product.
2.3.1. DRUG PROFILE:
2.3.1.1. Levocetirizine dihydrochloride:
1. Chemical Name: 2-(2-{4-[(R)-(4-chlorophenyl)(phenyl)methyl]piperazin-1-
yl}ethoxy)acetic acid
2. Molecular Formula: C21H25ClN2O3•2HCl.
3. Molecular Weight: 461.82
4. Description: White crystalline powder

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 41
5. Chemical Structure:
6. Solubility: Soluble in water.
7. Melting Point: 215-220 ºC
8. Category: antihistaminic
2.3.1.2. Pseudoephedrine sulfate:
1. Chemical Name: (S-(R*,R*))-alpha-(1-(Methylamino)ethyl)benzenemethanol sulfate
2. Chemical Structure:
3. Molecular Formula: (C10H15NO)2·H2SO4
4. Molecular Weight: 428.54
5. Description: White crystalline powder
6. Solubility: Freely soluble in water
7. Melting Point: 174- 179 C
8. Category: sympathomimetic
2.3.2. EXPERIMENTAL:
2.3.2.1. Working standard:
The working standards were obtained from Dr. Reddys Laboratories Ltd Hyderabad,
India having following batch number and potency.
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 42
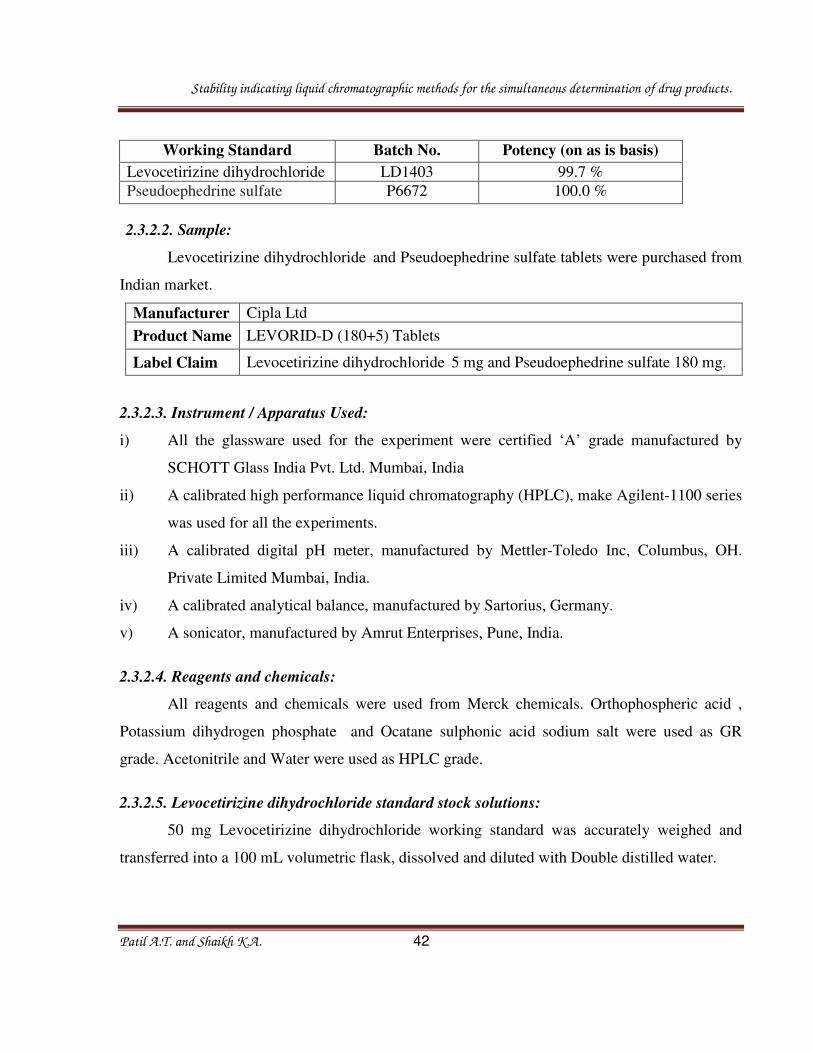
Working Standard Batch No. Potency (on as is basis)
Levocetirizine dihydrochloride LD1403 99.7 %
Pseudoephedrine sulfate P6672 100.0 %
2.3.2.2. Sample:
Levocetirizine dihydrochloride and Pseudoephedrine sulfate tablets were purchased from
Indian market.
Manufacturer Cipla Ltd
Product Name LEVORID-D (180+5) Tablets
Label Claim Levocetirizine dihydrochloride 5 mg and Pseudoephedrine sulfate 180 mg.
2.3.2.3. Instrument / Apparatus Used:
i) All the glassware used for the experiment were certified ‘A’ grade manufactured by
SCHOTT Glass India Pvt. Ltd. Mumbai, India
ii) A calibrated high performance liquid chromatography (HPLC), make Agilent-1100 series
was used for all the experiments.
iii) A calibrated digital pH meter, manufactured by Mettler-Toledo Inc, Columbus, OH.
Private Limited Mumbai, India.
iv) A calibrated analytical balance, manufactured by Sartorius, Germany.
v) A sonicator, manufactured by Amrut Enterprises, Pune, India.
2.3.2.4. Reagents and chemicals:
All reagents and chemicals were used from Merck chemicals. Orthophospheric acid ,
Potassium dihydrogen phosphate and Ocatane sulphonic acid sodium salt were used as GR
grade. Acetonitrile and Water were used as HPLC grade.
2.3.2.5. Levocetirizine dihydrochloride standard stock solutions:
50 mg Levocetirizine dihydrochloride working standard was accurately weighed and
transferred into a 100 mL volumetric flask, dissolved and diluted with Double distilled water.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 43
2.3.2.6. Mixed standard solution:
A 90 mg Pseudoephedrine sulfate working standard was accurately weighed, transferred
into a 25 mL volumetric flask, transferred 5 ml Levocetrizine dihydrochloride Standard Stock
solution, dissolved and diluted with Double distilled water.
2.3.2.7. Preparation of sample solution:
Ten tablets were weighed and finely powdered. A quantity of powder equivalent to one
tablet containing 180 mg of Pseudoephedrine sulfate and 5 mg of Levocetirizine dihydrochloride was Transferred into a 50 mL volumetric flask. To this flask, 10 mL of methanol was added, and
the solution was sonicated for 20 min with intermittent shaking. The solution was cooled to
ambient Temperature, further added 20 mL of double distilled water and the solution was
sonicated for 20 min with intermittent shaking. The solution was cooled to ambient temperature.
Then the volume was made up with double distilled water and centrifuged at 4,000 rpm for 10
min. The Centrifuged solution filtered through a 0.45-um nylon filter.
2.3.2.8. Chromatographic conditions:
Mobile phase A: Potassium dihydrogen phosphate Buffer 0.05M and 1-Ocatne
Sulphonic acid sodium salt 0.25%, pH adjusted to 3.0 with
orthophospheric acid.
Mobile phase B: Acetonitrile
Column: Cosmosil C8, 250 x 4.6 mm, 5 µm
Column oven temperature: 40◦C
Flow: 1.0 mL/min
Wavelength: 242 nm
Injection volume: 20 µL
Runtime: 20 minutes.
Gradient program :
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 44
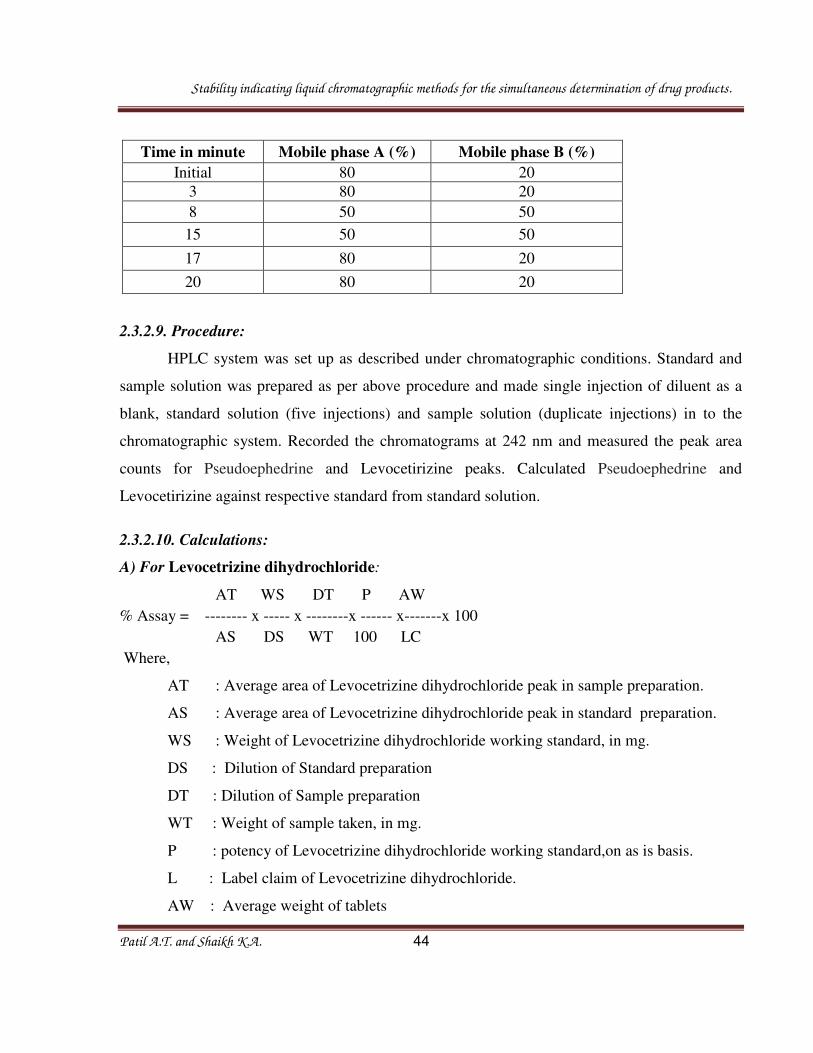
Time in minute Mobile phase A (%) Mobile phase B (%)
Initial 80 20
3 80 20
8 50 50
15 50 50
17 80 20
20 80 20
2.3.2.9. Procedure:
HPLC system was set up as described under chromatographic conditions. Standard and
sample solution was prepared as per above procedure and made single injection of diluent as a
blank, standard solution (five injections) and sample solution (duplicate injections) in to the
chromatographic system. Recorded the chromatograms at 242 nm and measured the peak area
counts for Pseudoephedrine and Levocetirizine peaks. Calculated Pseudoephedrine and
Levocetirizine against respective standard from standard solution.
2.3.2.10. Calculations:
A) For Levocetrizine dihydrochloride:
AT WS DT P AW
% Assay = -------- x ----- x --------x ------ x-------x 100
AS DS WT 100 LC
Where,
AT : Average area of Levocetrizine dihydrochloride peak in sample preparation.
AS : Average area of Levocetrizine dihydrochloride peak in standard preparation.
WS : Weight of Levocetrizine dihydrochloride working standard, in mg.
DS : Dilution of Standard preparation
DT : Dilution of Sample preparation
WT : Weight of sample taken, in mg.
P : potency of Levocetrizine dihydrochloride working standard,on as is basis.
L : Label claim of Levocetrizine dihydrochloride.
AW : Average weight of tablets

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 45
B) For Pseudoephedrine sulfate:
AT1 WS1 DT1 P1 AW1
% Assay = -------- x ----- x --------x ------ x-------x 100
AS1 DS1 WT1 100 LC1
Where,
AT1 : Average area counts of Pseudoephedrine sulfate peak in sample preparation.
AS1 : Average area counts of Pseudoephedrine sulfate peak in standard preparation.
WS1 : Weight Pseudoephedrine sulfate working standard, in mg.
DS1 : Dilution of Standard preparation
DT1 : Dilution of Sample preparation
WT1 : Weight of sample taken, in mg.
P1 : Percentage potency of Pseudoephedrine sulfate working standard, on as is basis.
LC1 : Label claim of Pseudoephedrine sulfate in mg per tablet.
AW1 : Average weight of tablets
2.3.3. RESULTS AND DISCUSSION:
2.3.3.1. Optimization of the chromatographic conditions:
The main criteria for development of a successful HPLC method for determination of
Levocetirizine dihydrochloride and Pseudoephedrine sulfate in tablet was the method should
be able to determine assay of both drugs in single run and should be accurate, reproducible,
robust, stability indicating, free of interference from degradation products, and straight forward
enough for routine use in the quality control laboratory67-75
. In order to optimize the LC
separation of Pseudoephedrine sulfate and Levocetirizine dihydrochloride initially, the retention
behavior of both the components was studied in the pH range of 2.5–6.8, using mobile phases of
buffer (pH 2.5–6.8) and acetonitrile, methanol as organic modifier. were found that
Pseudoephedrine sulfate eluted in void volume and more retention time Levocetirizine
dihydrochloride. Hence, it was decided to work by adding ion pairing reagent(1-Ocatne
sulphonic acid sodium salt) in the mobile phase.
To ensure that Pseudoephedrine sulfate gives better retention, resolution between
Levocetirizine dihydrochloride and Pseudoephedrine sulfate not less than 5 and the method was
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 46
as fast as possible, a gradient run was optimized using buffer pH 3.0 and acetonitrile, finally The
mobile phase A consisted of Potassium dihydrogen phosphate Buffer 0.05M and 1-Ocatne
sulphonic acid sodium salt 0.25%, pH adjusted to 3.0 with orthophospheric acid. Mobile Phase
B: Acetonitrile at Gradient elution at flow rate of 1 mL/min and Column temperature at 40◦C,
and detector wavelength of 242 nm using a photodiode array detector was selected as an
appropriate chromatographic conditions, which gave good resolution and acceptable peak
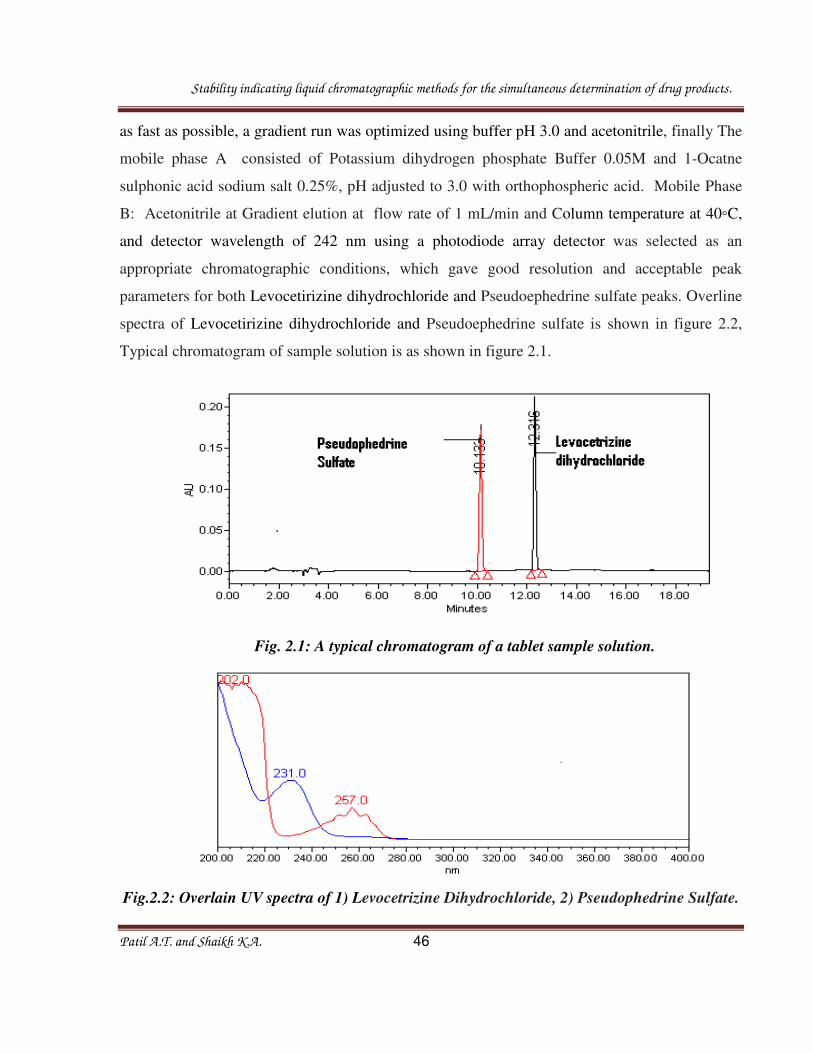
parameters for both Levocetirizine dihydrochloride and Pseudoephedrine sulfate peaks. Overline
spectra of Levocetirizine dihydrochloride and Pseudoephedrine sulfate is shown in figure 2.2,
Typical chromatogram of sample solution is as shown in figure 2.1.
Fig. 2.1: A typical chromatogram of a tablet sample solution.
Fig.2.2: Overlain UV spectra of 1) Levocetrizine Dihydrochloride, 2) Pseudophedrine Sulfate.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 47
2.3.3.2. Procedure for forced degradation study of drug product:
Forced degradation studies were performed to demonstrate the selectivity and stability
indicating capability of the proposed method. The powdered samples of tablets were exposed to
acidic, alkaline, oxidizing and thermal degradation conditions. The stress conditions engaged for
degradation studies as per ICH recommendation
2.3.3.2.1. Acid degradation:
A quantity of powder equivalent to one tablet containing 180 mg of Pseudoephedrine
sulfate and 5 mg of Levocetirizine dihydrochloride was transferred into a 50 mL volumetric
flask. To this flask, 10 mL of methanol was added, and the solution was sonicated for 20min
with intermittent shaking. then 5 mL0 .1 N HCl added and the mixture kept at 60 ◦C for 45 min
in a water bath. The solution was allowed to attend ambient temperature, then it was neutralized
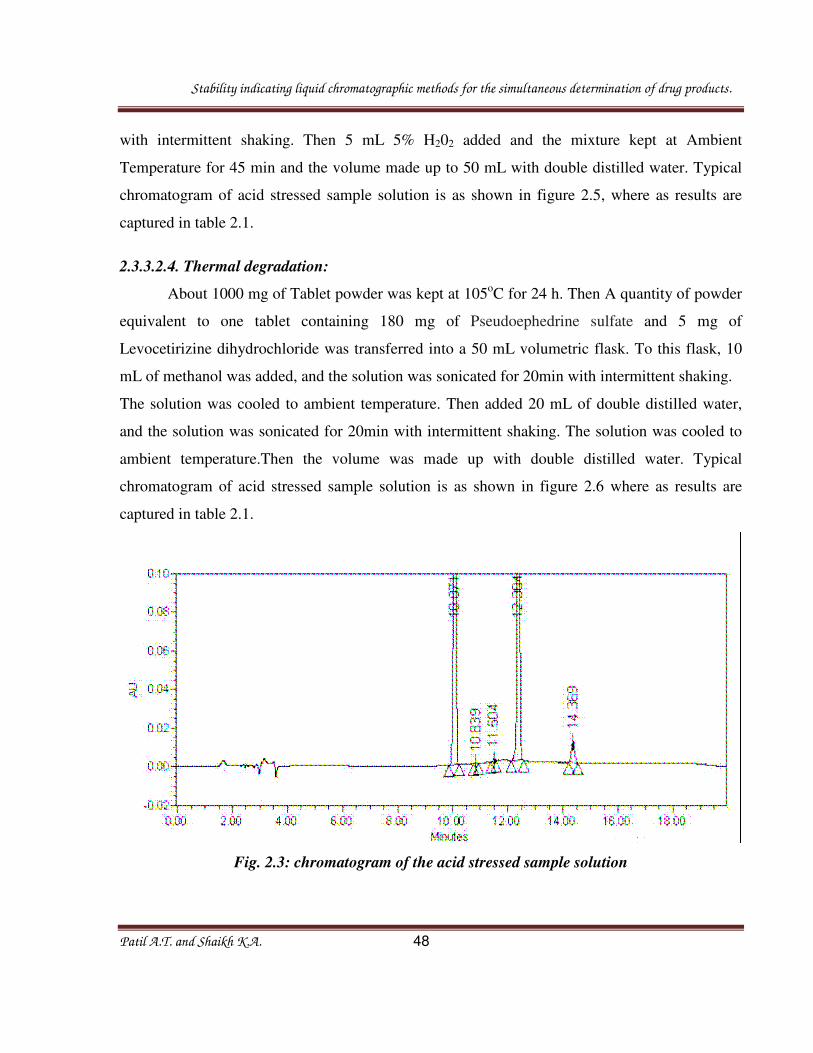
with 0.1 N NaOH and the volume made up to 50 mL with double distilled water. Typical
chromatogram of acid stressed sample solution is as shown in figure 2.3, where as results are
captured in table 2.1.
2.3.3.2.2. Base degradation:
A quantity of powder equivalent to one tablet containing 180 mg of Pseudoephedrine
sulfate and 5 mg of Levocetirizine dihydrochloride was transferred into a 50 mL volumetric
flask. To this flask, 10 mL of methanol was added, and the solution was sonicated for 20min
with intermittent shaking.then 5 mL 1 N NaOH added and the mixture kept at 60 ◦C for 45 min
in a water bath. The solution was allowed to attend ambient temperature, then it was neutralized
with 1 N HCL and the volume made up to 50 mL with double distilled water. Typical
chromatogram of acid stressed sample solution is as shown in figure 2.4, where as results are
captured in table 2.1.
2.3.3.2.3. Oxidative degradation:
A quantity of powder equivalent to one tablet containing 180 mg of Pseudoephedrine
sulfate and 5 mg of Levocetirizine dihydrochloride was transferred into a 50 mL volumetric
flask. To this flask, 10 mL of methanol were added, and the solution was sonicated for 20min
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 48
with intermittent shaking. Then 5 mL 5% H202 added and the mixture kept at Ambient
Temperature for 45 min and the volume made up to 50 mL with double distilled water. Typical
chromatogram of acid stressed sample solution is as shown in figure 2.5, where as results are
captured in table 2.1.
2.3.3.2.4. Thermal degradation:
About 1000 mg of Tablet powder was kept at 105oC for 24 h. Then A quantity of powder
equivalent to one tablet containing 180 mg of Pseudoephedrine sulfate and 5 mg of
Levocetirizine dihydrochloride was transferred into a 50 mL volumetric flask. To this flask, 10
mL of methanol was added, and the solution was sonicated for 20min with intermittent shaking.
The solution was cooled to ambient temperature. Then added 20 mL of double distilled water,
and the solution was sonicated for 20min with intermittent shaking. The solution was cooled to
ambient temperature.Then the volume was made up with double distilled water. Typical
chromatogram of acid stressed sample solution is as shown in figure 2.6 where as results are
captured in table 2.1.
Fig. 2.3: chromatogram of the acid stressed sample solution
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 49
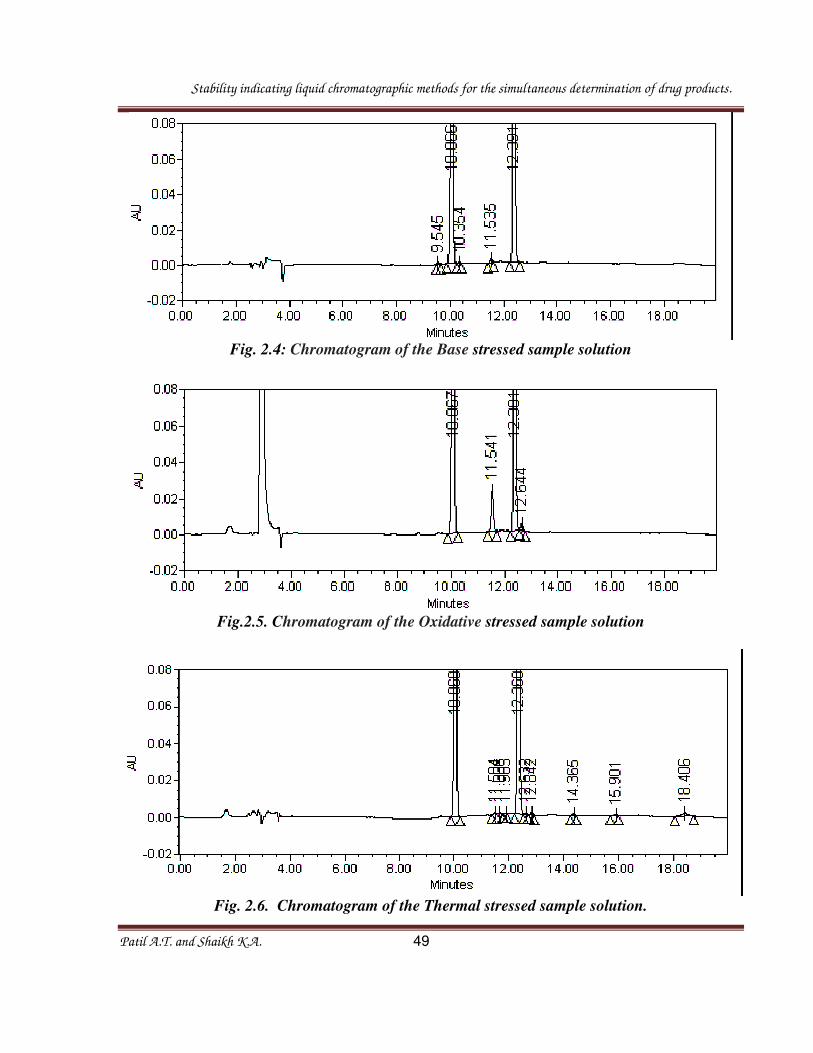
Fig. 2.4: Chromatogram of the Base stressed sample solution
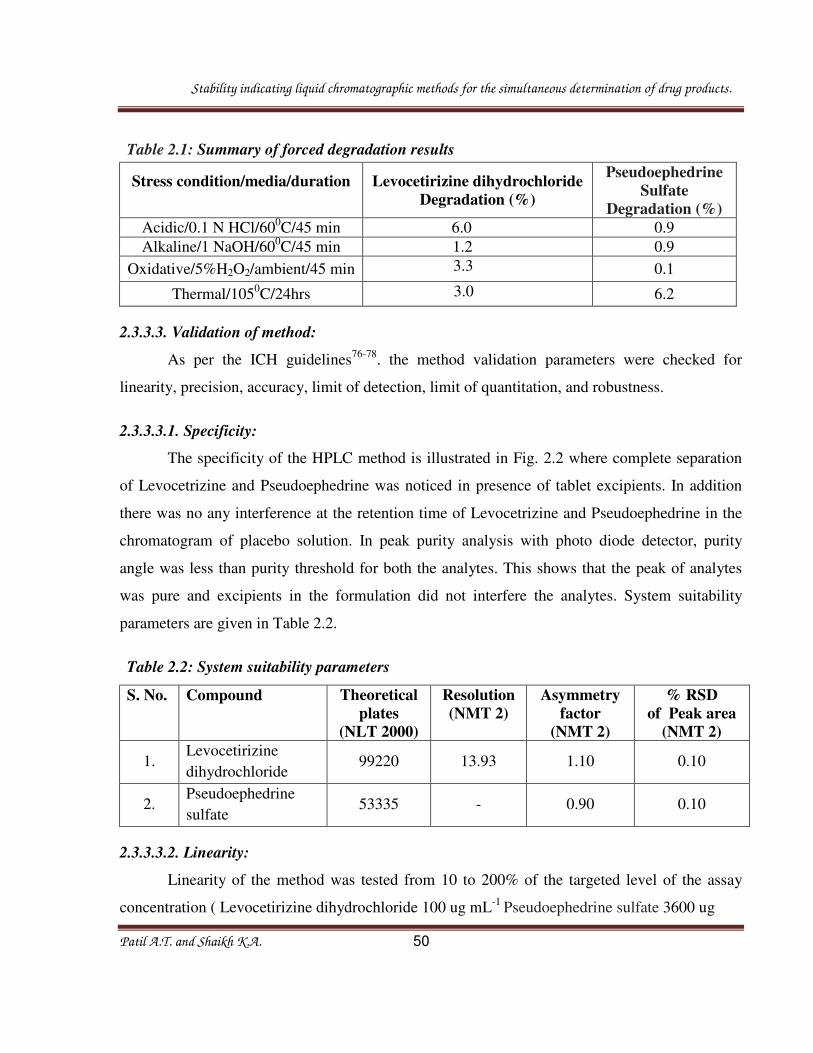
Fig.2.5. Chromatogram of the Oxidative stressed sample solution
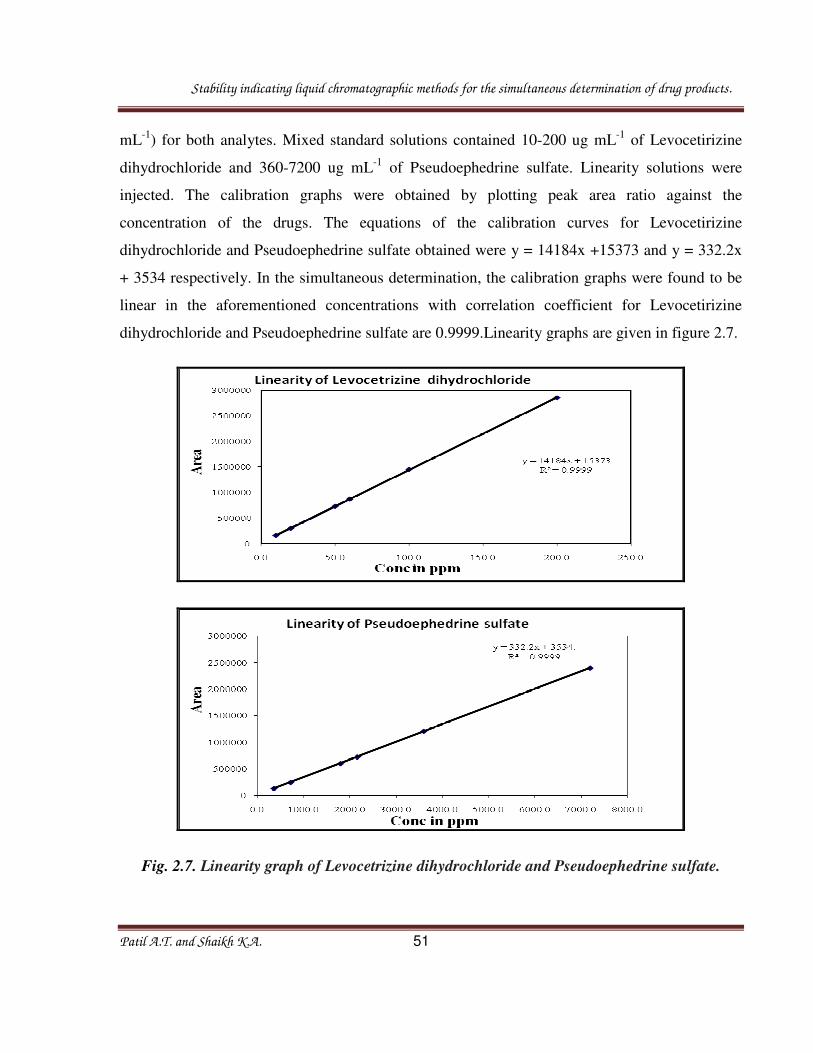
Fig. 2.6. Chromatogram of the Thermal stressed sample solution.
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 50
Table 2.1: Summary of forced degradation results
Stress condition/media/duration
Levocetirizine dihydrochloride
Degradation (%)
Pseudoephedrine
Sulfate
Degradation (%)
Acidic/0.1 N HCl/600C/45 min 6.0 0.9
Alkaline/1 NaOH/600C/45 min 1.2 0.9
Oxidative/5%H2O2/ambient/45 min 3.3 0.1
Thermal/1050C/24hrs 3.0 6.2
2.3.3.3. Validation of method:
As per the ICH guidelines76-78
. the method validation parameters were checked for
linearity, precision, accuracy, limit of detection, limit of quantitation, and robustness.
2.3.3.3.1. Specificity:
The specificity of the HPLC method is illustrated in Fig. 2.2 where complete separation
of Levocetrizine and Pseudoephedrine was noticed in presence of tablet excipients. In addition
there was no any interference at the retention time of Levocetrizine and Pseudoephedrine in the
chromatogram of placebo solution. In peak purity analysis with photo diode detector, purity
angle was less than purity threshold for both the analytes. This shows that the peak of analytes
was pure and excipients in the formulation did not interfere the analytes. System suitability
parameters are given in Table 2.2.
Table 2.2: System suitability parameters
S. No. Compound Theoretical
plates
(NLT 2000)
Resolution
(NMT 2)
Asymmetry
factor
(NMT 2)
% RSD
of Peak area
(NMT 2)
1. Levocetirizine
dihydrochloride 99220 13.93 1.10 0.10
2. Pseudoephedrine
sulfate 53335 - 0.90 0.10
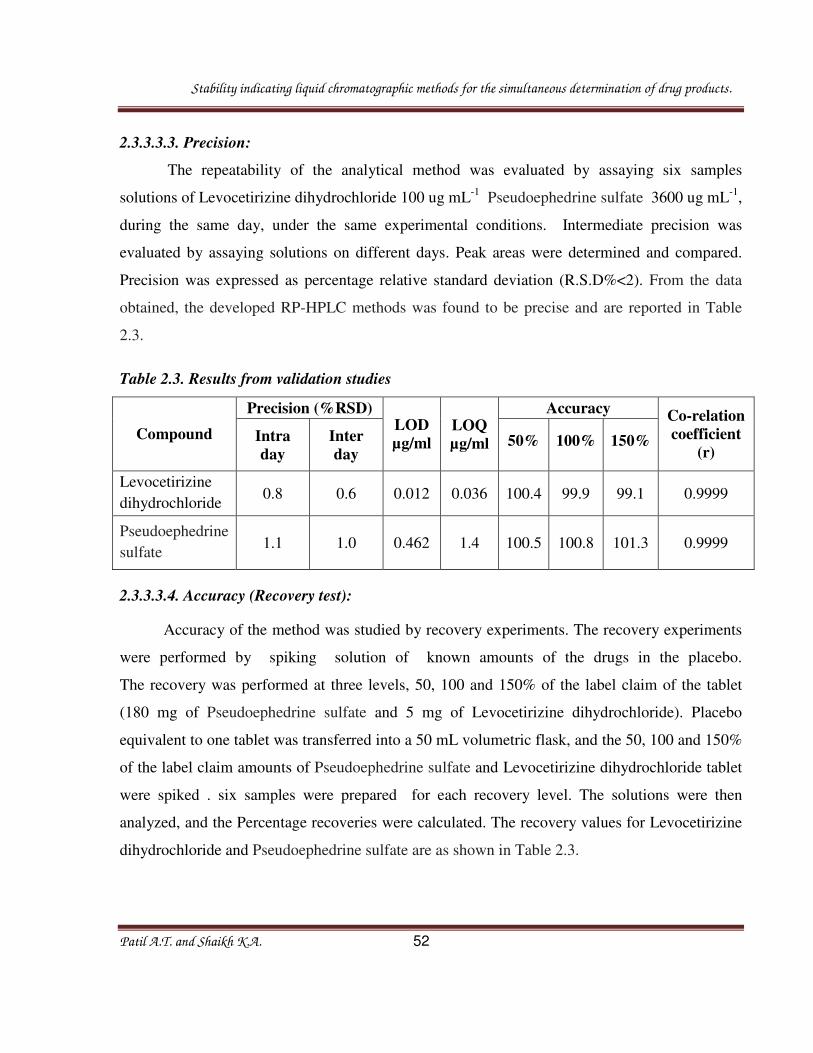
2.3.3.3.2. Linearity:
Linearity of the method was tested from 10 to 200% of the targeted level of the assay
concentration ( Levocetirizine dihydrochloride 100 ug mL-1
Pseudoephedrine sulfate 3600 ug
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 51
mL-1
) for both analytes. Mixed standard solutions contained 10-200 ug mL-1
of Levocetirizine
dihydrochloride and 360-7200 ug mL-1
of Pseudoephedrine sulfate. Linearity solutions were
injected. The calibration graphs were obtained by plotting peak area ratio against the
concentration of the drugs. The equations of the calibration curves for Levocetirizine
dihydrochloride and Pseudoephedrine sulfate obtained were y = 14184x +15373 and y = 332.2x
+ 3534 respectively. In the simultaneous determination, the calibration graphs were found to be
linear in the aforementioned concentrations with correlation coefficient for Levocetirizine
dihydrochloride and Pseudoephedrine sulfate are 0.9999.Linearity graphs are given in figure 2.7.
Fig. 2.7. Linearity graph of Levocetrizine dihydrochloride and Pseudoephedrine sulfate.
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 52
2.3.3.3.3. Precision:
The repeatability of the analytical method was evaluated by assaying six samples
solutions of Levocetirizine dihydrochloride 100 ug mL-1
Pseudoephedrine sulfate 3600 ug mL-1
,
during the same day, under the same experimental conditions. Intermediate precision was
evaluated by assaying solutions on different days. Peak areas were determined and compared.
Precision was expressed as percentage relative standard deviation (R.S.D%<2). From the data
obtained, the developed RP-HPLC methods was found to be precise and are reported in Table
2.3.
Table 2.3. Results from validation studies
Precision (%RSD) Accuracy
Compound Intra
day
Inter
day
LOD
µg/ml
LOQ
µg/ml
50% 100% 150%
Co-relation
coefficient
(r)
Levocetirizine
dihydrochloride 0.8 0.6 0.012 0.036 100.4 99.9 99.1 0.9999
Pseudoephedrine
sulfate 1.1 1.0 0.462 1.4 100.5 100.8 101.3 0.9999
2.3.3.3.4. Accuracy,(Recovery,test):
Accuracy of the method was studied by recovery experiments. The recovery experiments
were performed by spiking solution of known amounts of the drugs in the placebo.
The recovery was performed at three levels, 50, 100 and 150% of the label claim of the tablet
(180 mg of Pseudoephedrine sulfate and 5 mg of Levocetirizine dihydrochloride). Placebo
equivalent to one tablet was transferred into a 50 mL volumetric flask, and the 50, 100 and 150%
of the label claim amounts of Pseudoephedrine sulfate and Levocetirizine dihydrochloride tablet
were spiked . six samples were prepared for each recovery level. The solutions were then
analyzed, and the Percentage recoveries were calculated. The recovery values for Levocetirizine
dihydrochloride and Pseudoephedrine sulfate are as shown in Table 2.3.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 53
2.3.3.3.5. LOD and LOQ:
The LOD and LOQ for Levocetirizine dihydrochloride and pseudoephedrine sulfate were
determined at a signal to-noise ratio of 3:1 and 10:1, respectively by injecting a series of dilute
solutions with known concentrations. The LOD and LOQ are as shown in Table 2.3.
2.3.3.3.6. Robustness:
The robustness of a method is the ability to remain unaffected by small changes in
parameters. The evaluation of robustness should be considered during the development phase
and depends on the type of procedure under study. It should show the reliability of an analysis
with respect to deliberate variations in method parameters. If measurements are susceptible to
variations in analytical conditions, the analytical conditions should be suitably controlled or a
precautionary statement should be included in the procedure. One consequence of the evaluation
of robustness should be that a series of system suitability parameters is established to ensure that
the validity of the analytical procedure is maintained whenever used. In the case of liquid
chromatography, typical variations are influence of variations of pH in a mobile phase; influence
of variations in mobile phase composition; Column temperature; mobile phase flow rate.
System suitability testing is an integral part of many analytical procedures hence to
determine robustness of the method, experimental conditions were purposely altered and
chromatographic resolution between Levocetirizine dihydrochloride and Pseudoephedrine sulfate
were evaluated. The flow rate of the mobile phase was 1.0 mLmin-1
. To study the effect of flow
rate on the resolution of Levocetirizine dihydrochloride and Pseudoephedrine sulfate, it was
changed to 0.2 units from 1.0 mL min-1
to1.2 mL min-1
and 0.8 mL min-1
. The effect of column
temperature on the resolution was studied at 45 and 35 0C instead of 40
0C and The effect of pH
Variation of Mobile phase A( Buffer) on the resolution was studied at pH 3.2 and pH 2.8 instead
of pH 3.0. Robustness results are as shown in Table 2.4.
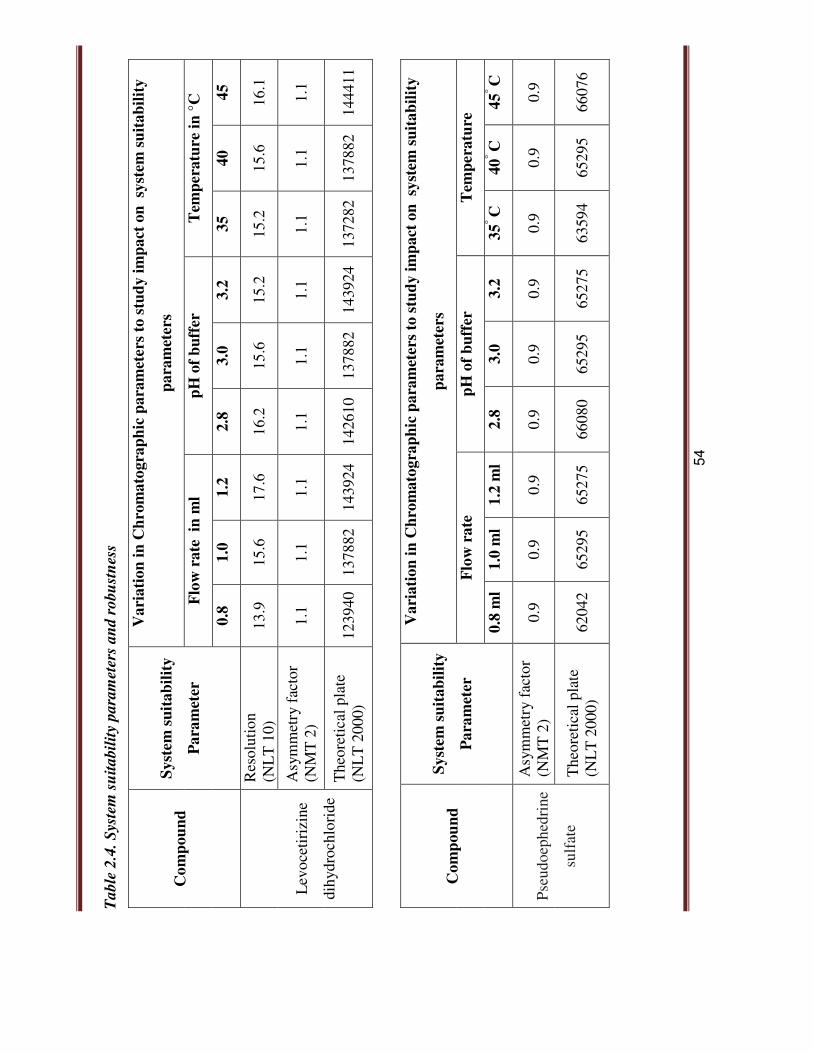
54
Table
2.4
. S
yste
m s
uit
abil
ity
para
met
ers
an
d r
obu
stn
ess
Vari
ati
on
in
Ch
rom
ato
gra
ph
ic p
ara
met
ers
to s
tud
y i
mp
act
on
sy
stem
su
itab
ilit
y
para
met
ers
Flo
w r
ate
in
ml
pH
of
bu
ffer
T
emp
era
ture
in
°C
C
om
pou
nd
S
yst
em
su
itab
ilit
y
Para
met
er
0.8
1.0
1.2
2.8
3.0
3.2
35
40
45
Res
olu
tion
(NL
T 1
0)
13.9
15.6
17.6
16.2
15.6
15.2
15.2
15.6
16.1
Asy
mm
etry
fac
tor
(NM
T 2
) 1.1
1.1
1.1
1.1
1.1
1.1
1.1
1.1
1.1
Lev
oce
tiri
zine
dih
ydro
chlo
ride
Theo
reti
cal
pla
te
(NL
T 2
000)
123940
137882
143924
142610
137882
143924
137282
137882
144411
Vari
ati
on
in
Ch
rom
ato
gra
ph
ic p
ara
met
ers
to s
tud
y i
mp
act
on
sy
stem
su
itab
ilit
y
para
met
ers
Flo
w r
ate
pH
of
bu
ffer
T
emp
era
ture
C
om
pou
nd
S
yst
em
su
itab
ilit
y
Para
met
er
0.8
ml
1.0
ml
1.2
ml
2.8
3.0
3.2
35
° C
40
° C
45
° C
Asy
mm
etry
fac
tor
(NM
T 2
) 0.9
0.9
0.9
0.9
0.9
0.9
0.9
0.9
0.9
P
seudoep
hed
rine
sulf
ate
Theo
reti
cal
pla
te
(NL
T 2
000)
62042
65295
65275
66080
65295
65275
63594
65295
66076

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 55
2.3.4. CONCLUSION:
The developed simple LC method for assay determination of Levocetirizine
dihydrochloride and Pseudoephedrine sulfate is linear, precise, accurate and specific. The
method was validated to the requirements of ICH and the results were satisfactory. The
developed stability-indicating analytical method can be used for the routine analysis of
production samples, where sample load is higher and high throughput is essential for faster
delivery of results. Overall, the method provides a high throughput solution for determination of
Levocetirizine dihydrochloride and Pseudoephedrine sulfate in tablet dosage form with excellent
selectivity, precision, and accuracy.
2.4. SENSITIVE LC METHOD FOR SIMULTANEOUS DETERMINATION OF
CICLESONIDE AND FORMOTEROL FUMARATE IN DRY POWDER INHALER.
To the best of our knowledge, a stability indicating RP-HPLC method for the
simultaneous determination of Formoterol fumarate and Ciclesonide in dry powder inhaler is not
available in any pharmacopoeia. Hence, it was felt essential to develop and validate a sensitive,
accurate, and stability indicating RP-HPLC method for the simultaneous determination of
Formoterol fumarate and Ciclesonide in dry powder inhaler.
The chromatographic separation was achieved on Hypersil BDS C8 250x4.6 mm, 5 um
column using a mobile phase consisting of 0.1% orthophospheric acid and acetonitrile in the
ratio of 35:65 (v/v), at a flow rate of 2.0 mLmin-1
. The column compartment temperature was set
at 40°C. The typical HPLC chromatograms was extracted at 214 nm using photodiode array
detector (PDA). The described method shows excellent linearity over a range of 3.6 µg mL-1
to
1.1 ng mL-1
for formoterol fumarate and 120 µg mL-1
to 34 ng mL-1
for ciclesonide. The
correlation coefficient for formoterol fumarate and ciclesonide was 0.9998. The limit of
detection and limit of quantification for formoterol fumarate was 0.33 ng mL-1
, 1.1 ng mL-1
and
for ciclesonide 10.2 ng mL-1
, 34 ng mL-1
, respectively. The proposed method was found to be
very sensitive and accurate for the determination of Formoterol Fumarate and ciclesonide in dry
powder inhaler.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 56
2.4.1. DRUG PROFILE:
2.4.1.1. Formetrol fumarate:
1. Chemical Name: N-[2-Hydroxy-5-[[(1RS)-1-hydroxy-2-[[(1RS)-2-(4-methoxyphenyl)-1-
methylethyl] amino] ethyl] phenyl] formamide(E)-2-butenedioate
2. Chemical Structure:
3. Molecular Formula: (C19H24N2O4)2•C4H4O4•2H2O.
4. Molecular weight: 840.92
5. Description: white to yellowish crystalline powder
6.Solubility: Freely soluble in glacial acetic acid, soluble in methanol, sparingly soluble in
ethanol and isopropanol, slightly soluble in water, and practically insoluble in acetone, ethyl
acetate, and diethyl ether.
7. Melting Point: 138-140°C
8. Category: beta2-adrenergic bronchodilator.
2.4.1.2. Ciclesonide:
1. Chemical Name: (R)-11b,16a,17,21-Tetrahydroxypregna-1,4-diene-3, 20-dione cyclic 16,17-
acetal with cyclohexanecarboxaldehyde 21-isobutyrate
2. Chemical Structure:
3. Molecular Formula: C32H44O7

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 57
4. Molecular weight: 540.688
5. Description: white to yellow-white powder
6. Solubility: practically insoluble in water and freely soluble in ethanol and Acetone.
7. Melting Point: 207 ºC
8. Category: non-halogenated glucocorticoid
2.4.2. EXPERIMENTAL:
2.4.2.1. Working standard:
The working standards were obtained from Indian market with following batch number
and potency.
Working Standard Batch No. Potency (on as is basis)
Formetrol fumarate FF3121 94.5 % w/w
Ciclesonide CN4517 98.9 % w/w
2.4.2.2. Sample:
Formetrol fumarate and Ciclesonide capsules were purchased from Indian market.
Manufacturer Cipla Ltd
Product Name Simply One Rotacaps
Label Claim Formoterol fumarate 6mcg, Ciclesonide 200 mcg capsules
2.4.2.3. Instrument / Apparatus Used:
i) All the glassware used for the experiment were certified ‘A’ grade manufactured by
SCHOTT Glass India Pvt. Ltd. Mumbai, India
ii) A calibrated high performance liquid chromatography (HPLC), make Agilent-1100 series
was used for all the experiments.
iii) A calibrated digital pH meter, manufactured by Mettler-Toledo Inc, Columbus, OH.
Private Limited Mumbai, India.
iv) A calibrated analytical balance, manufactured by Sartorius, Germany.
v) A sonicator, manufactured by Amrut Enterprises, Pune, India.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 58
2.4.2.4.. Reagent, chemicals and diluent:
All reagents and chemicals were used from Merck chemicals. Orthophospheric acid and
Potassium dihydrogen phosphate were used as GR grade. Acetonitrile and Water were used as
HPLC grade. Purified water and Acetonitrile in the ratio of 50:50 v/v were used as diluent for
the experiment.
2.4.2.5. Preparation of standard stock solution A :
Accurately weighed 24mg working standard of Formoterol fumarate is transfer to a
100ml volumetric flask, 70 ml diluent is added and sonicate to dissolve it completely. Make up
the volume to 100 ml with diluent and mix.
2.4.2.6. Preparation of standard stock solution B :
Accurately weighed 80.0 mg working standard of Ciclesonide is transfer to a 100 mL
volumetric flask, 70 mL diluent is added and sonicate to dissolve it completely. Make up the
volume to 100 mL with diluent and mix.
2.4.2.7. Preparation of standard solution:
Transferred 2.0 ml of the standard stock solution A and 10.0 ml of the standard stock
solution B into a 100ml volumetric flask and make up the volume with diluent and mix.
2.4.2.8. Preparation of Sample solution:
The average fill weight of 20 capsules was determined. Accurately weighed sample
powder was transferred in the amount of 120 mg of FF and 4000 mg of CS in to a 50mL
volumetric flask, to which was added 20mL of diluent and sonicated for about 20 min to
dissolve; then, diluent was added to obtain a volume of 50 mL.
2.4.2.9. Chromatographic conditions:
Buffer solution : 0.1% orthophospheric acid
Mobile Phase : Mixture of buffer and acetonitrile in a ratio of (35:65)v/v.
Column: Hypersil BDS C8, 250x4.6 mm, 5 um

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 59
Column oven temperature: 40◦C
Flow: 2.0 mL/min
Wavelength: 214 nm
Injection volume: 200 µL
Runtime: 10 minutes
2.4.2.10. Procedure:
HPLC system was set up as described under chromatographic conditions. Standard and
sample solution was prepared as per above procedure and made single injection of diluent as a
blank, standard solution (five injections) and sample solution (duplicate injections) in to the
chromatographic system.Recorded the chromatograms at 214 nm for Formoterol fumarate and
Ciclesonide peaks. Calculated % assay of Formoterol fumarate and Ciclesonide against
respective standard from standard solution.
2.4.2.11. Calculations:
A) For Formoterol fumarate (% Assay ) :
AT WS DT P AW
% Assay = -------- x ----- x -----x ------ x-------x 100
AS DS WT 100 LC
Where,
AT :Average area of Formoterol fumarate peak in sample preparation.
AS :Average area of Formoterol fumarate peak in standard preparation.
WS :Weight of Formoterol fumarate working standard, in mg.
DS :Dilution of Standard preparation
DT : Dilution of Sample preparation
WT :Weight of sample taken, in mg.
P :Percentage potency of Formoterol fumarate working standard,
on as is basis.
L : Label claim of Formoterol fumarate.
AW : Average weight of tablets

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 60
B) For Ciclesonide:
AT1 WS1 DT1 P1 AW1
% Assay = -------- x ----- x --------x ------ x-------x 100
AS1 DS1 WT1 100 LC1
Where,
AT1 : Average area counts of Ciclesonide peak in sample preparation
AS1 : Average area counts of Ciclesonide peak in standard preparation
WS1 : Weight Ciclesonide working standard, in mg
DS1 : Dilution of Standard preparation
DT1 : Dilution of Sample preparation
WT1 : Weight of sample taken, in mg
P1 : Percentage potency of Ciclesonide working standard, on as is basis
LC1 : Label claim of Ciclesonide in mg per tablet
AW1 : Average weight of tablets
2.4.3. RESULTS AND DISCUSSION:
2.4.3.1. Optimization of the chromatographic conditions:
The main objective of development of a HPLC method for determination of FF and CS in
dry powder inhaler was that the method should be able to determine assays of both drugs in
single run with run time <10 min. As dose strength is at mcg level, method should be more
sensitive, stability indicating, free of interference from degradation products, and straight
forward enough for routine use in the quality control laboratory25-33
.
During optimization of chromatographic conditions, different mobile phase compositions,
different HPLC columns, organic modifiers such as acetonitrile and methanol, and flow rate
were tried to achieve acceptable system suitability parameters, as well as good separation
between FF and CS. Optimum wavelength selected was 214nm because of higher sensitivity of
FF at this wavelength and also good absorption shown by CS at this wavelength (Figure 2.8).

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 61
2.4.3.2. Effect of the organic modifier:
0.1% orthophosphoric acid was used as the buffer for the mobile phase preparation.
Different combinations of buffer and acetonitrile within the range of 10:90–90:10, as well as
buffer and methanol within the range of 10:90–90:10 were tested. It was observed that methanol
in combination with a buffer leads to more retention of CS peak and asymmetric peak shape. An
increase in the organic modifier volume in the mobile phase produces a reduced retention time of
CS. The mobile phase combination of buffer: Acetonitirle (35:65) gives a symmetric peak shape,
an acceptable tailing factor, and a shorter run time up to 10 min.
2.4.3.3. Effect of stationary phase and pH of mobile phase:
Reversed-phase columns are silica based bonded phases, and C18-type bonded phase is
most frequently used. On an Inertsil ODS 3 V 250mmx 4.6 mm, 5 mm column FF was eluted
with a solvent front peak, as the basic polar compound was protonated in acidic pH and eluted
more quickly. Polar compounds were eluted slowly on C8 column as it is less hydrophobic than
C18 and by use of base deactivated silica column (Hypersil BDS C8 250x4.6 mm, 5 um). Also,
by keeping the pH of the mobile phase on the acidic side, the silanol interaction with basic
analyte was minimized and peak symmetry and sensitivity were improved. The flow rate of the
mobile phase was changed to obtain the expected retention time of the analyte. An increase in
the flow rate led to reduced retention time of the analyte. Typical chromatogram of formoterol
fumarate and Ciclesonide tablet is as show in figure 2.9, where as system suitability results are
reported in table 2.5.
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 62
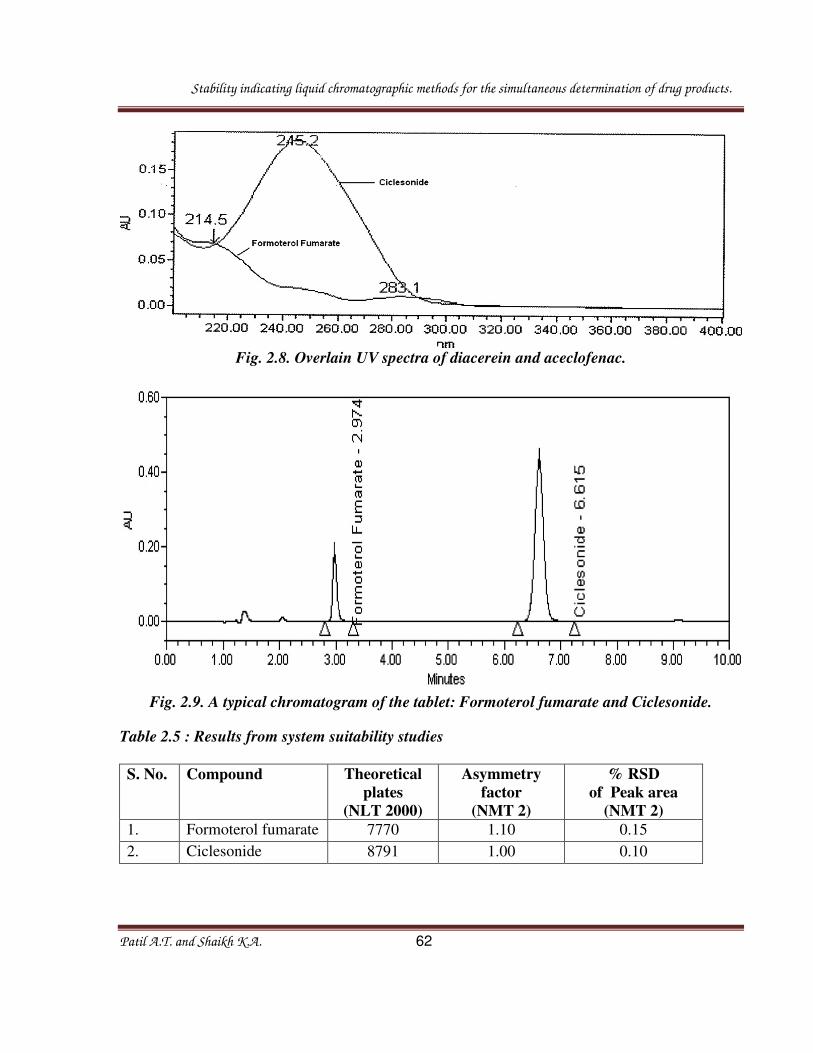
Fig. 2.8. Overlain UV spectra of diacerein and aceclofenac.
Fig. 2.9. A typical chromatogram of the tablet: Formoterol fumarate and Ciclesonide.
Table 2.5 : Results from system suitability studies
S. No. Compound Theoretical
plates
(NLT 2000)
Asymmetry
factor
(NMT 2)
% RSD
of Peak area
(NMT 2)
1. Formoterol fumarate 7770 1.10 0.15
2. Ciclesonide 8791 1.00 0.10
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 63
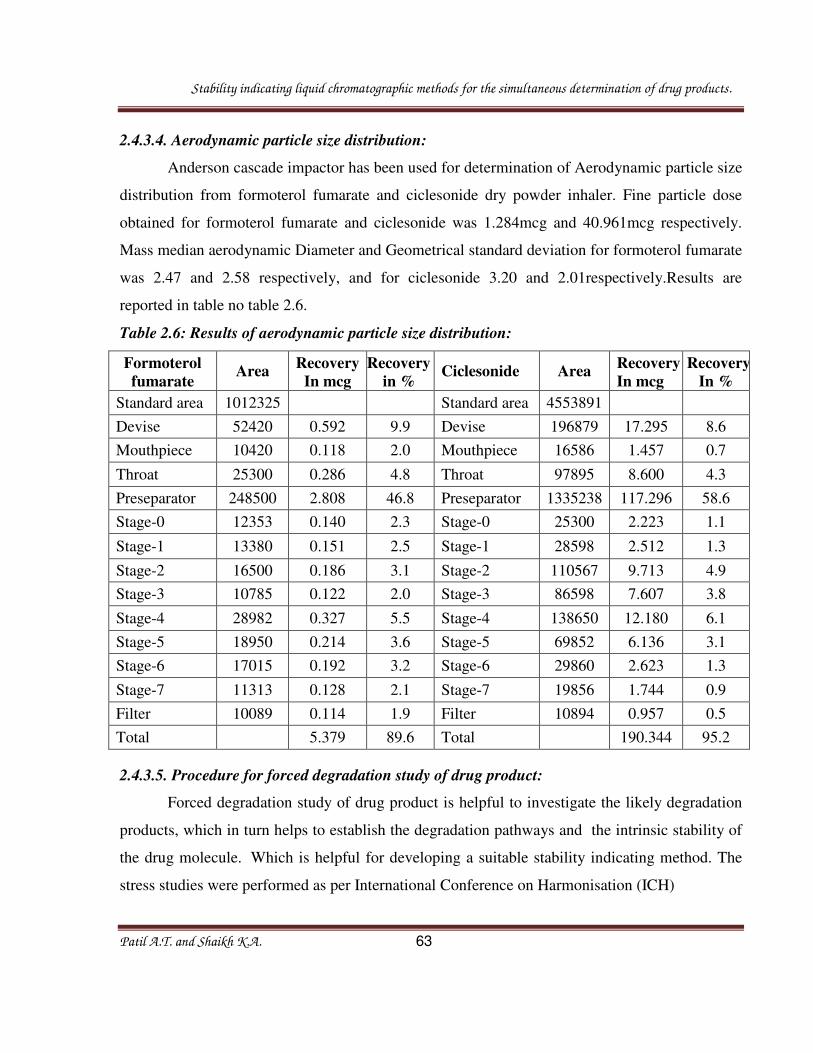
2.4.3.4. Aerodynamic particle size distribution:
Anderson cascade impactor has been used for determination of Aerodynamic particle size
distribution from formoterol fumarate and ciclesonide dry powder inhaler. Fine particle dose
obtained for formoterol fumarate and ciclesonide was 1.284mcg and 40.961mcg respectively.
Mass median aerodynamic Diameter and Geometrical standard deviation for formoterol fumarate
was 2.47 and 2.58 respectively, and for ciclesonide 3.20 and 2.01respectively.Results are
reported in table no table 2.6.
Table 2.6: Results of aerodynamic particle size distribution:
Formoterol
fumarate Area
Recovery
In mcg
Recovery
in % Ciclesonide Area
Recovery
In mcg
Recovery in
In %
Standard area 1012325 Standard area 4553891
Devise 52420 0.592 9.9 Devise 196879 17.295 8.6
Mouthpiece 10420 0.118 2.0 Mouthpiece 16586 1.457 0.7
Throat 25300 0.286 4.8 Throat 97895 8.600 4.3
Preseparator 248500 2.808 46.8 Preseparator 1335238 117.296 58.6
Stage-0 12353 0.140 2.3 Stage-0 25300 2.223 1.1
Stage-1 13380 0.151 2.5 Stage-1 28598 2.512 1.3
Stage-2 16500 0.186 3.1 Stage-2 110567 9.713 4.9
Stage-3 10785 0.122 2.0 Stage-3 86598 7.607 3.8
Stage-4 28982 0.327 5.5 Stage-4 138650 12.180 6.1
Stage-5 18950 0.214 3.6 Stage-5 69852 6.136 3.1
Stage-6 17015 0.192 3.2 Stage-6 29860 2.623 1.3
Stage-7 11313 0.128 2.1 Stage-7 19856 1.744 0.9
Filter 10089 0.114 1.9 Filter 10894 0.957 0.5
Total 5.379 89.6 Total 190.344 95.2
2.4.3.5. Procedure for forced degradation study of drug product:
Forced degradation study of drug product is helpful to investigate the likely degradation
products, which in turn helps to establish the degradation pathways and the intrinsic stability of
the drug molecule. Which is helpful for developing a suitable stability indicating method. The
stress studies were performed as per International Conference on Harmonisation (ICH)
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 64
recommendation. Following stressed conditions has been carried out in order to establish
degradation pathway of drug product.
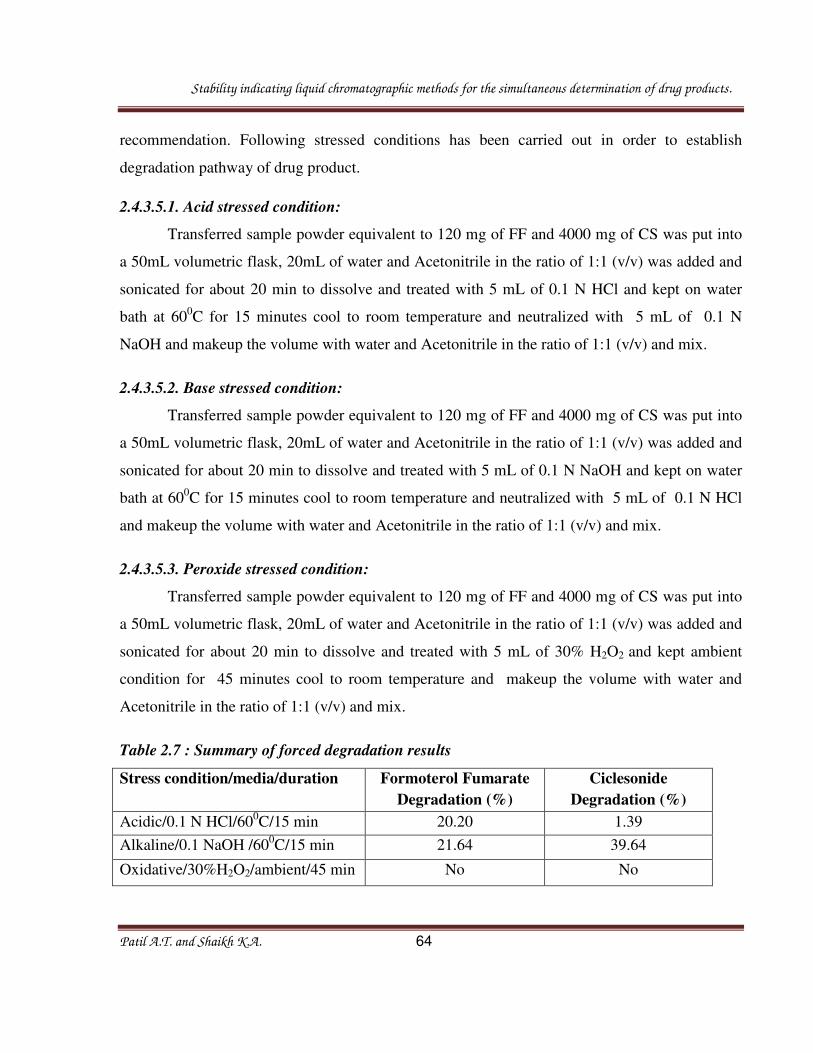
2.4.3.5.1. Acid stressed condition:
Transferred sample powder equivalent to 120 mg of FF and 4000 mg of CS was put into
a 50mL volumetric flask, 20mL of water and Acetonitrile in the ratio of 1:1 (v/v) was added and
sonicated for about 20 min to dissolve and treated with 5 mL of 0.1 N HCl and kept on water
bath at 600C for 15 minutes cool to room temperature and neutralized with 5 mL of 0.1 N
NaOH and makeup the volume with water and Acetonitrile in the ratio of 1:1 (v/v) and mix.
2.4.3.5.2. Base stressed condition:
Transferred sample powder equivalent to 120 mg of FF and 4000 mg of CS was put into
a 50mL volumetric flask, 20mL of water and Acetonitrile in the ratio of 1:1 (v/v) was added and
sonicated for about 20 min to dissolve and treated with 5 mL of 0.1 N NaOH and kept on water
bath at 600C for 15 minutes cool to room temperature and neutralized with 5 mL of 0.1 N HCl
and makeup the volume with water and Acetonitrile in the ratio of 1:1 (v/v) and mix.
2.4.3.5.3. Peroxide stressed condition:
Transferred sample powder equivalent to 120 mg of FF and 4000 mg of CS was put into
a 50mL volumetric flask, 20mL of water and Acetonitrile in the ratio of 1:1 (v/v) was added and
sonicated for about 20 min to dissolve and treated with 5 mL of 30% H2O2 and kept ambient
condition for 45 minutes cool to room temperature and makeup the volume with water and
Acetonitrile in the ratio of 1:1 (v/v) and mix.
Table 2.7 : Summary of forced degradation results
Stress condition/media/duration
Formoterol Fumarate
Degradation (%)
Ciclesonide
Degradation (%)
Acidic/0.1 N HCl/600C/15 min 20.20 1.39
Alkaline/0.1 NaOH /600C/15 min 21.64 39.64
Oxidative/30%H2O2/ambient/45 min No No
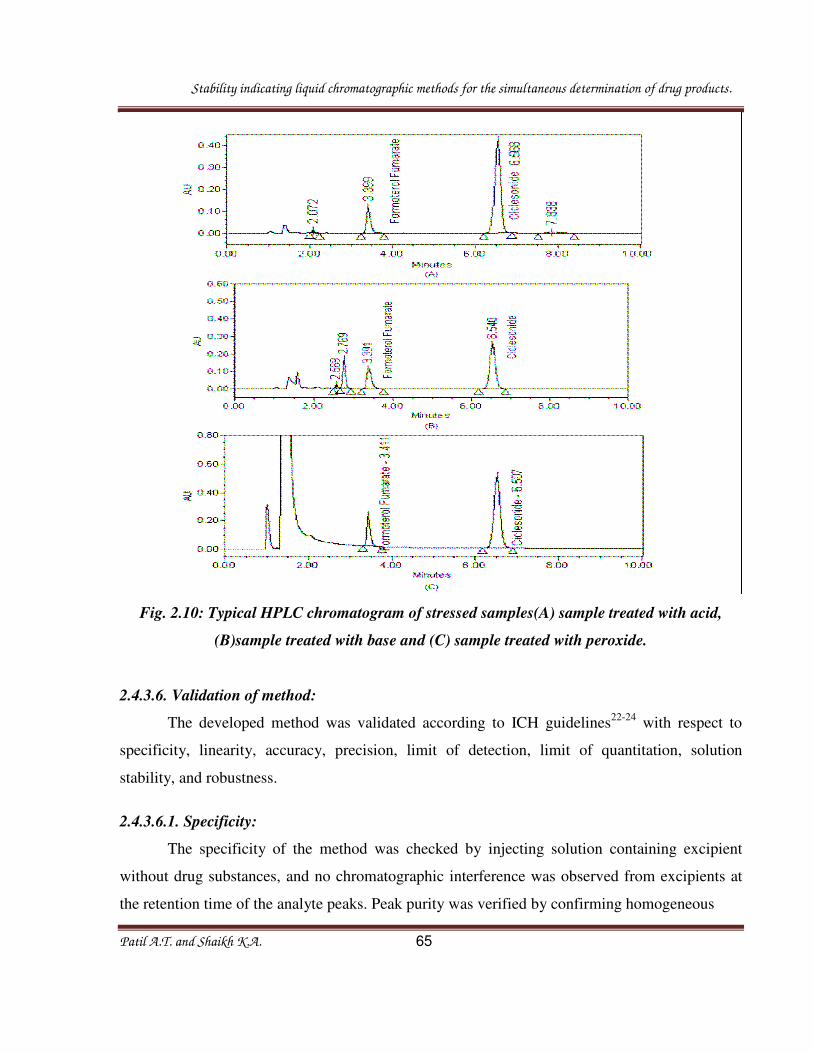
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 65
Fig. 2.10: Typical HPLC chromatogram of stressed samples(A) sample treated with acid,
(B)sample treated with base and (C) sample treated with peroxide.
2.4.3.6. Validation of method:
The developed method was validated according to ICH guidelines22-24
with respect to
specificity, linearity, accuracy, precision, limit of detection, limit of quantitation, solution
stability, and robustness.
2.4.3.6.1. Specificity:
The specificity of the method was checked by injecting solution containing excipient
without drug substances, and no chromatographic interference was observed from excipients at
the retention time of the analyte peaks. Peak purity was verified by confirming homogeneous
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 66
spectral data for FF and CS. The specificity of the method was also checked by performing the
stressed study and no interference from degradation products at the retention time of FF and CS
was found.
2.4.3.6.2. Precision:
The repeatability of the analytical method was evaluated by analyzing six test samples
solutions of Formoterol FumarateþCiclesonide dry powder for Inhaler, during the same day,
under the same experimental conditions. Intermediate precision was evaluated by analyzing six
test solutions on different days. The percentage assay for each component were determined and
compared. Precision was expressed as percentage relative standard deviation of percentage
assay, which was found to be well within the limit (<2). Hence, the method was found to be
precise.
2.4.3.6.3. Accuracy:
Accuracy was evaluated by the simultaneous determination of analytes in solution
prepared by standard addition method. The experiment was carried out by adding known amount
of each component corresponding to three concentration levels of 50%, 100%, and 150% of
target analyte concentration in placebo solution. The samples were prepared in triplicate at each
level. The solutions were then analyzed as per the proposed method and the quantification of
added analyte (% weight/weight) was carried out by using an external standard of corresponding
main drug prepared at the analytical concentration. The recovery values for Formoterol Fumarate
and Ciclesonide are as shown in Table 2.8.
Table 2.8: Recovery data
Formoterol Fumarate Ciclesoinde Recovery levels
Recovery (%) RSD (%) Recovery (%) RSD (%)
50 99.4 0.43 99.5 0.31
100 99.1 0.37 99.7 0.24
150 99.8 0.54 100.3 0.48
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 67
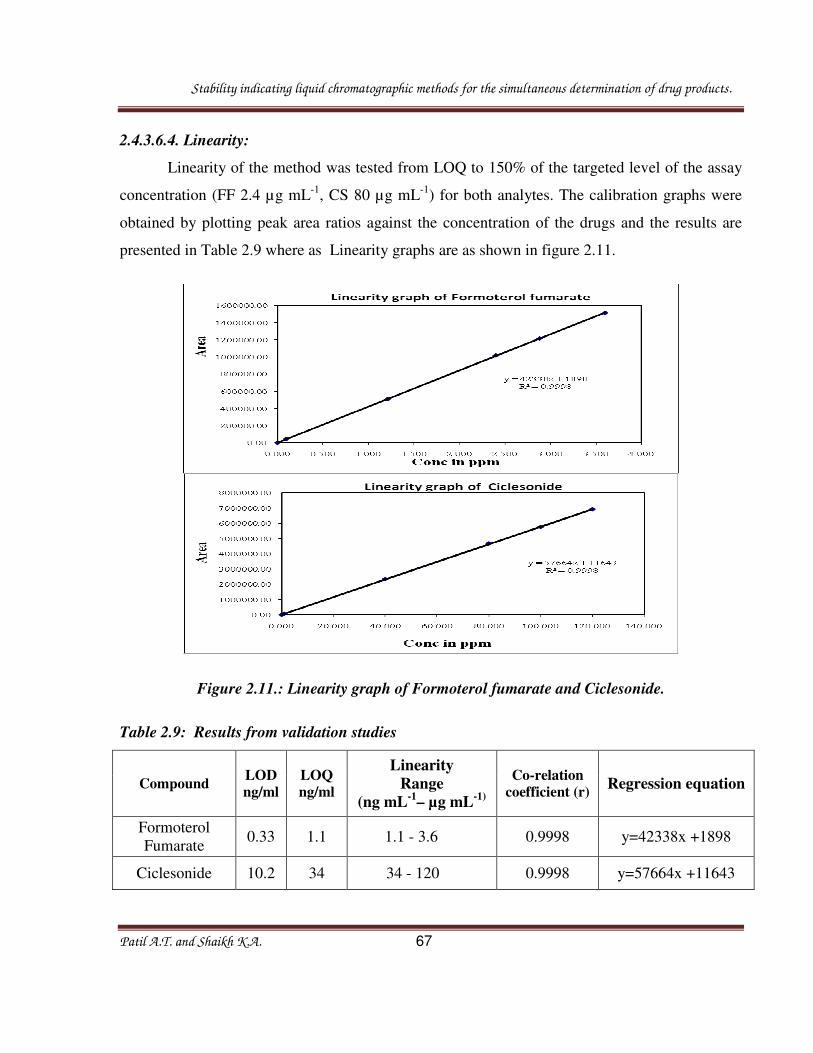
2.4.3.6.4. Linearity:
Linearity of the method was tested from LOQ to 150% of the targeted level of the assay
concentration (FF 2.4 µg mL-1
, CS 80 µg mL-1
) for both analytes. The calibration graphs were
obtained by plotting peak area ratios against the concentration of the drugs and the results are
presented in Table 2.9 where as Linearity graphs are as shown in figure 2.11.
Figure 2.11.: Linearity graph of Formoterol fumarate and Ciclesonide.
Table 2.9: Results from validation studies
Compound LOD
ng/ml
LOQ
ng/ml
Linearity
Range
(ng mL-1
– µg mL-1)
Co-relation
coefficient (r) Regression equation
Formoterol
Fumarate 0.33 1.1 1.1 - 3.6 0.9998 y=42338x +1898
Ciclesonide 10.2 34 34 - 120 0.9998 y=57664x +11643

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 68
2.4.3.6.5. Limit of Quantification (LOQ) and Detection (LOD:
The limit of detection (LOD) was estimated as three times the signal to noise ratio and
the limit of quantification (LOQ) was estimated as ten times the signal to noise ratio. LOD and
LOQ were achieved by injecting a series of dilute solutions of FF and CS. The precision of the
developed method for FF and CS was checked by analyzing six test solutions prepared at LOQ
and LOD level and determined the percentage relative standard deviation of peak area. The LOD
and LOQ values are as shown in Table 2.9.
2.4.3.6.6. Stability in Analytical Solution:
Standard solution and sample solution were found to be stable for about five days at room
temperature. Similarity factor between freshly prepared standard and the fifth day standard
solution was found to be 1. Percentage assay was checked for initial sample preparation, and on
the fifth day at room temperature against the freshly prepared standard, the percentage difference
between the fifth day sample and initial sample assay was less than 1.
2.4.3.6.7. Robustness:
The robustness of a method is the ability to remain unaffected by small changes in
parameters. To determine robustness of the method, experimental conditions were deliberately
altered and percentage assay was checked for FF and CS. The altered parameters were change in
flow rate (+0.2mLmin_1), column compartment temperature (+5°C), and absolute organic
composition (+10%). The percentage assay for both the components was found to be well within
the limits.
2.4.4. CONCLUSION:
The proposed HPLC method is specific, accurate, and precise for simultaneous
determination of FF and CS from its pharmaceutical dosages form. The method was validated as
per ICH guidelines and the results showed an the stability-indicating property of the method and,
hence, the method is suitable for routine analysis and quality control of pharmaceutical
preparation containing FF and CS as active pharmaceutical ingredients.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 69
2.5. SENSITIVE LC METHOD FOR THE SIMULTANEOUS DETERMINATION OF
DIACEREIN AND ACECLOFENAC IN TABLET DOSAGE FORM.
A comprehensive literature survey revealed the lack of suitable stability indicating assay
method for the determination of Aceclofenac and Diacerein in tablet dosage form also this
method is not official in any pharmacopeia. Hence, it was felt essential to develop and validate a
sensitive, accurate and stability indicating RP-HPLC method for the simultaneous determination
of Aceclofenac and Diacerein in tablet dosage form. the chromatographic separation was
achieved on Kromasil C-18, 150 × 4.6 mm, 3.5-µm analytical column using mobile phase double
distilled water (pH 2.7 with glacial acetic acid)-acetonitrile (45:55 v/v). Detector was set at 256
nm. The described method shows excellent linearity over a range of 75–2.5 µg/mL for diacerein
and 150–5 µg/mL for aceclofenac. The correlation coefficient for diacerein is 0.9999 and for
aceclofenac is 0.9997. The proposed method was found to be suitable for simultaneous
determination and stability study of diacerein and aceclofenac in tablet dosage form.
2.5.1. DRUG PROFILE:
2.5.1.1. Diacerein:
1. Chemical Name: 1,8-diacetoxy-3-carboxyanthraquinone
2. Chemical Structure:
3. Molecular Formula: C19H12O8
4. Molecular Weight: 368.294
5. Description: Orange-yellow needle crystalline powder
6. Solubility: Soluble in dimethyl sulfoxide, dimethyl acetamide; difficult to dissolve in water,
methanol, chloroform.
7. Melting Point: 217-218°C
{8. Category: anti-inflammatory agent prescribed for osteoarthritis

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 70
2.5.1.2. Aceclofenac:
1. Chemical Name: 5-chloro-1-[1-[3-(2-oxo-2,3-dihydro-1H-benzimidazol-1-yl) propyl]--4-yl]-
1,3-dihydro-2H-benzimidazol-2-one
2. Chemical Structure:
3. Molecular Formula: C16 H13Cl2NO4
4. Molecular Weight: 354.185
5. Description: white or off-white crystalline powder
6. Solubility: Practically insoluble in water, freely soluble in acetone, soluble in alcohol.
7. Melting Point:149-153 ºC
8. Category: Antipyretic and Analgesics
2.5.2. EXPERIMENTAL:
2.5.2.1. Working standard:
The working standards were obtained from Glenmark pharmaceuticals Ltd., India having
following batch number and potency.
Working Standard Batch Number Potency (on as is basis)
Diacerein DAC2145 99.00 % w/w
Aceclofenac ACE3412 99.12 % w/w
2.5.2.2. Sample:
Tablets were purchased from Indian market.
Manufacturer Glenmark pharmaceuticals
Product Name Dycerin-A
Label Claim Diacerein 50 mg and Aceclofenac 100 mg per tablet

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 71
2.5.2.3. Instrument / Apparatus Used:
i) All the glassware used for the experiment were certified ‘A’ grade manufactured by
SCHOTT Glass India Pvt. Ltd. Mumbai, India
ii) A calibrated high performance liquid chromatography (HPLC), make Agilent-1100 was
used for all the experiments.
iii) A calibrated digital pH meter, manufactured by Mettler-Toledo Inc, Columbus, OH.
Private Limited Mumbai, India.
iv) A calibrated analytical balance, manufactured by Sartorius, Germany.
v) A sonicator, manufactured by Amrut Enterprises, Pune, India.
2.5.2.4. Reagents and chemicals:
All used reagents and chemicals from Merck fine chemicals. Glacial acetic acid GR
grade, Acetonitrile HPLC grade, Dimethyl acetamide GR grade, Water HPLC grade from Milli-
Q water purification system, was used through the experiment.
2.5.2.5. Preparation of standard stock solution :
Accurately weighed 25mg working standard of Diacerein and 50mg working standard of
Aceclofeanc and transferred it into a 100ml volumetric flask, 10 mL Dimethyl acetamide added
and sonicate to dissolve it completely. 70 ml of mobile phase added and sonicate for 5 minutes.
Made up the volume with mobile phase and mix.
2.5.2.6. Preparation of standard solution:
Transferred 5.0 ml of the Standard stock Solution into a 50ml volumetric flask and made
up the volume with mobile phase.
2.5.2.7. Preparation of Sample solution:
Ten tablets were weighed and finely powdered. A quantity of powder equivalent to one
tablet containing 100 mg of Aceclofenac and 50 mg of Diacerein was Transferred into a 200 mL
volumetric flask. To this flask 20 mL of Dimethyl acetamide were added and the solution was
sonicated for 20min with intermittent shaking. The solution was cooled to ambient temperature.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 72
Then added 70 mL of mobile phase and sonicate for 5 minutes and volume was made up with
mobile phase. The Centrifuged solution filter through 0.45µ nylon filter further dilute 5ml
solution was transferred into a 50 mL volumetric flask and diluted to volume mobile phase.
2.5.2.8. Chromatographic conditions:
Buffer solution: Double distilled water pH adjusted 2.7 with glacial acetic acid.
Mobile Phase : Prepared a mixture of buffer and acetonitrile in a ratio of (45:55) v/v.
Column: Kromasil C18, 150 x 4.6 mm, 3.5 µm
Column oven temperature: 25◦C
Flow: 1.0 mL/min
Wavelength: 256 nm
Injection volume: 10 µL
Runtime: 10 minutes.
2.5.2.9. Procedure:
HPLC system was set up as described under chromatographic conditions. Standard and
sample solution was prepared as per above procedure and made single injection of diluent as a
blank, standard solution (five injections) and sample solution (duplicate injections) in to the
chromatographic system. Recorded the chromatograms at 256 nm for Diacerein and Aceclofenac
peaks. Calculated % assay of Diacerein and Aceclofenac against respective standard from
standard solution.
2.5.2.10. Calculation:
A) For Diacerein:
AT WS DT P AW
% Assay = -------- x ----- x -----x ------ x-------x 100
AS DS WT 100 LC
Where,
AT : Average area of Diacerein peak in sample preparation
AS : Average area of Diacerein peak in standard preparation
WS : Weight of Diacerein working standard, in mg

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 73
DS : Dilution of Standard preparation
DT : Dilution of Sample preparation
WT : Weight of sample taken, in mg
P : Percentage potency of Diacerein working standard, on as is basis.
L : Label claim of Diacerein
AW : Average weight of tablets
B) For Aceclofenac:
AT1 WS1 DT1 P1 AW1
% Assay= -------- x ----- x --------x ------ x-------x 100
AS1 DS1 WT1 100 LC1
Where
AT1 : Average area counts of Aceclofenac peak in sample preparation
AS1 : Average area counts of Aceclofenac peak in standard preparation
WS1 : Weight Aceclofenac working standard, in mg
DS1 : Dilution of Standard preparation
DT1 : Dilution of Sample preparation
WT1 : Weight of sample taken, in mg
P1 : Percentage potency of Aceclofenac working standard, on as is basis
LC1 : Label claim of Aceclofenac in mg per tablet
AW1 : Average weight of tablets
2.5.3. RESULTS AND DISCUSSION:
2.5.3.1. Optimization of the chromatographic conditions:
The main criteria for development of a successful HPLC method for determination of
Diacerein and Aceclofenac in Tablet were the method should be able to determine assay of
both drugs in single run and should be accurate, reproducible, robust, stability indicating, free of
interference from degradation products, and straight forward enough for routine use in the
quality control laboratory67-75
.
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 74
Our objective of the chromatographic method development was to achieve a peak tailing
factor <2, Run time up to 10 min, along with a resolution between Diacerein and Aceclofenac
>5. In order to optimize the LC separation of Aceclofenac and Diacerein initially the retention
behavior of both the components was studied in the pH range of 2.5–6.8, using mobile phases of
buffer (pH 2.5–6.8) and acetonitrile, methanol as organic modifier.
To ensure resolution between Diacerein and Aceclofenac not less than 5, the method
was as fast as possible, a Isocratic run was optimized by using the mobile phase consisted of
buffer–acetonitrile (45:55 v/v). The buffer used in mobile phase contains double distilled water
pH adjusted 2.7 with glacial acetic acid. Isocratic elution at flow rate of 1 mL/min and Column
temperature at 25◦C. Detector wavelength Kept 256 nm using a photodiode array detector. This
was selected as an appropriate chromatographic conditions, which gave good resolution,
acceptable peak parameters for both Diacerein and Aceclofenac. The analytes of this
combination had adequate retentions, peak shape, less tailing, more resolution and the
chromatographic analysis time was 10 min. In optimized conditions Diacerein, Aceclofenac and
their degradants were well separated. Overlain UV spectra of diacerein and aceclofenac is shown
in figure 2.12, typical chromatogram of tablet is shown in figure 2.13, system suitability results
are shown in table 2.10.
Table 2.10: Results of system suitability studies.
S. No. Compound Theoretical
plates
(NLT 2000)
Resolution
(NMT 2)
Asymmetry
factor
(NMT 2)
% RSD
of Peak area
(NMT 2)
1. Diacerein 9687 25.53 1.2 0.10
2. Aceclofenac 17795 - 1.2 0.10
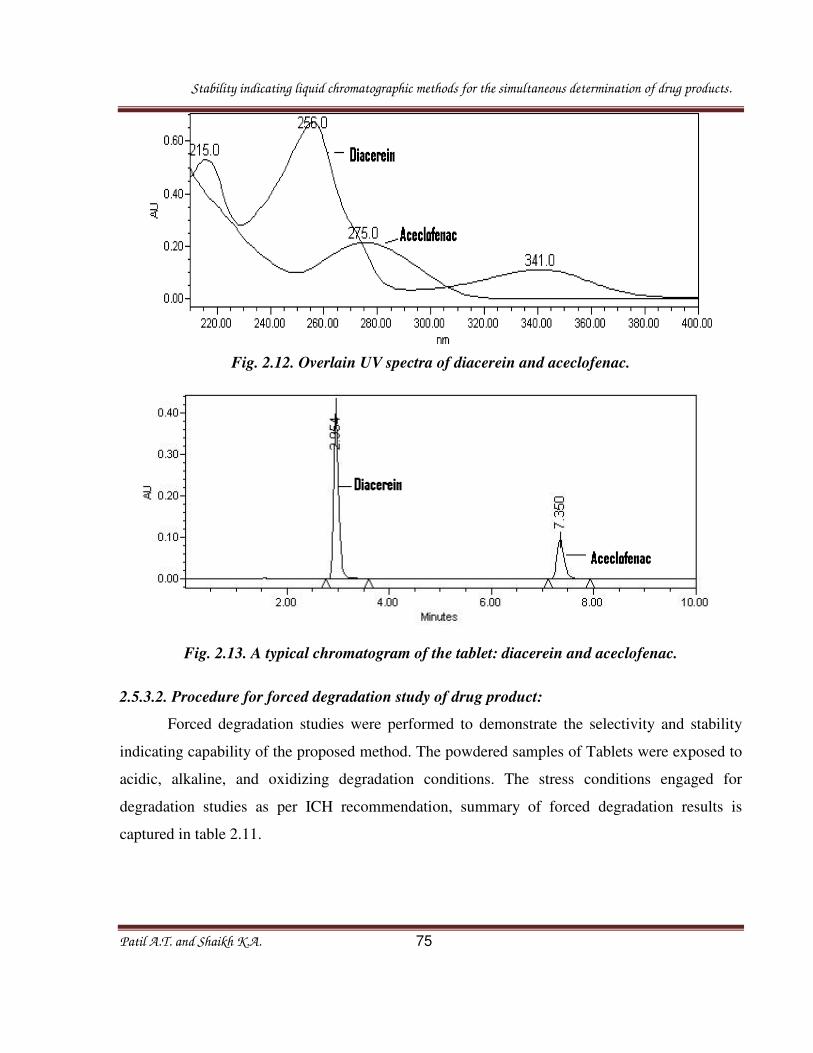
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 75
Fig. 2.12. Overlain UV spectra of diacerein and aceclofenac.
Fig. 2.13. A typical chromatogram of the tablet: diacerein and aceclofenac.
2.5.3.2. Procedure for forced degradation study of drug product:
Forced degradation studies were performed to demonstrate the selectivity and stability
indicating capability of the proposed method. The powdered samples of Tablets were exposed to
acidic, alkaline, and oxidizing degradation conditions. The stress conditions engaged for
degradation studies as per ICH recommendation, summary of forced degradation results is
captured in table 2.11.
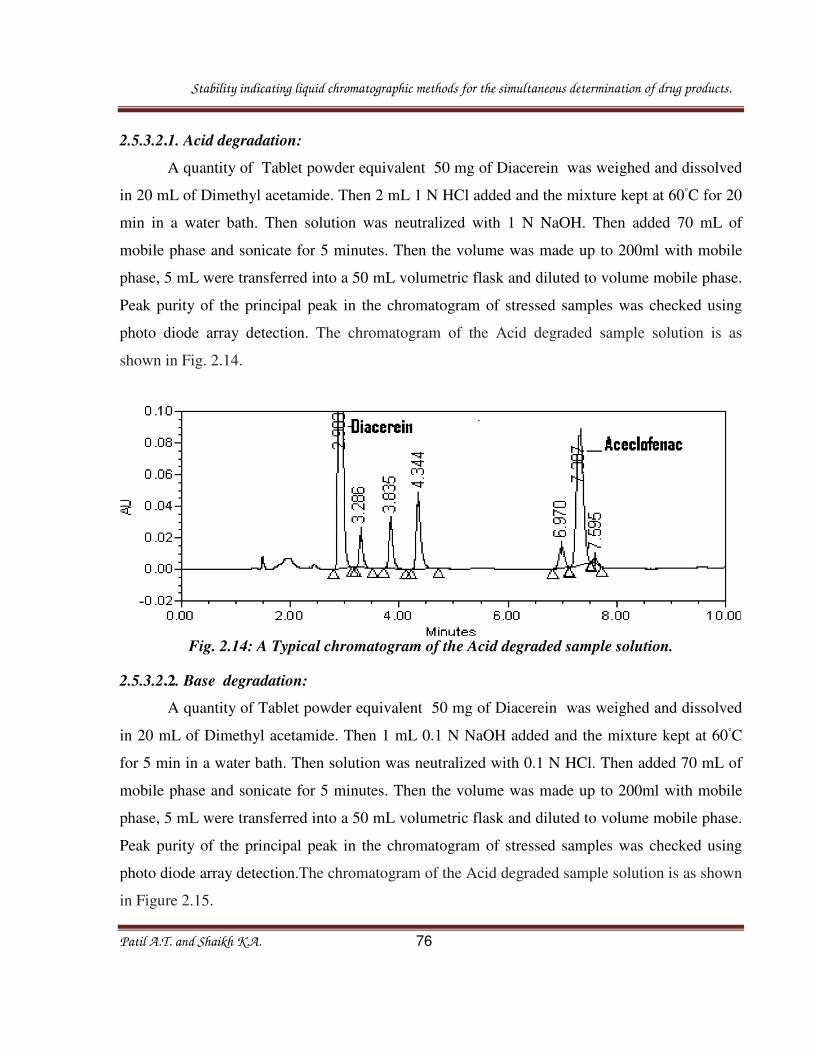
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 76
2.5.3.2.1. Acid degradation:
A quantity of Tablet powder equivalent 50 mg of Diacerein was weighed and dissolved
in 20 mL of Dimethyl acetamide. Then 2 mL 1 N HCl added and the mixture kept at 60◦C for 20
min in a water bath. Then solution was neutralized with 1 N NaOH. Then added 70 mL of
mobile phase and sonicate for 5 minutes. Then the volume was made up to 200ml with mobile
phase, 5 mL were transferred into a 50 mL volumetric flask and diluted to volume mobile phase.
Peak purity of the principal peak in the chromatogram of stressed samples was checked using
photo diode array detection. The chromatogram of the Acid degraded sample solution is as
shown in Fig. 2.14.
Fig. 2.14: A Typical chromatogram of the Acid degraded sample solution.
2.5.3.2.2. Base degradation:
A quantity of Tablet powder equivalent 50 mg of Diacerein was weighed and dissolved
in 20 mL of Dimethyl acetamide. Then 1 mL 0.1 N NaOH added and the mixture kept at 60◦C
for 5 min in a water bath. Then solution was neutralized with 0.1 N HCl. Then added 70 mL of
mobile phase and sonicate for 5 minutes. Then the volume was made up to 200ml with mobile
phase, 5 mL were transferred into a 50 mL volumetric flask and diluted to volume mobile phase.
Peak purity of the principal peak in the chromatogram of stressed samples was checked using
photo diode array detection.The chromatogram of the Acid degraded sample solution is as shown
in Figure 2.15.
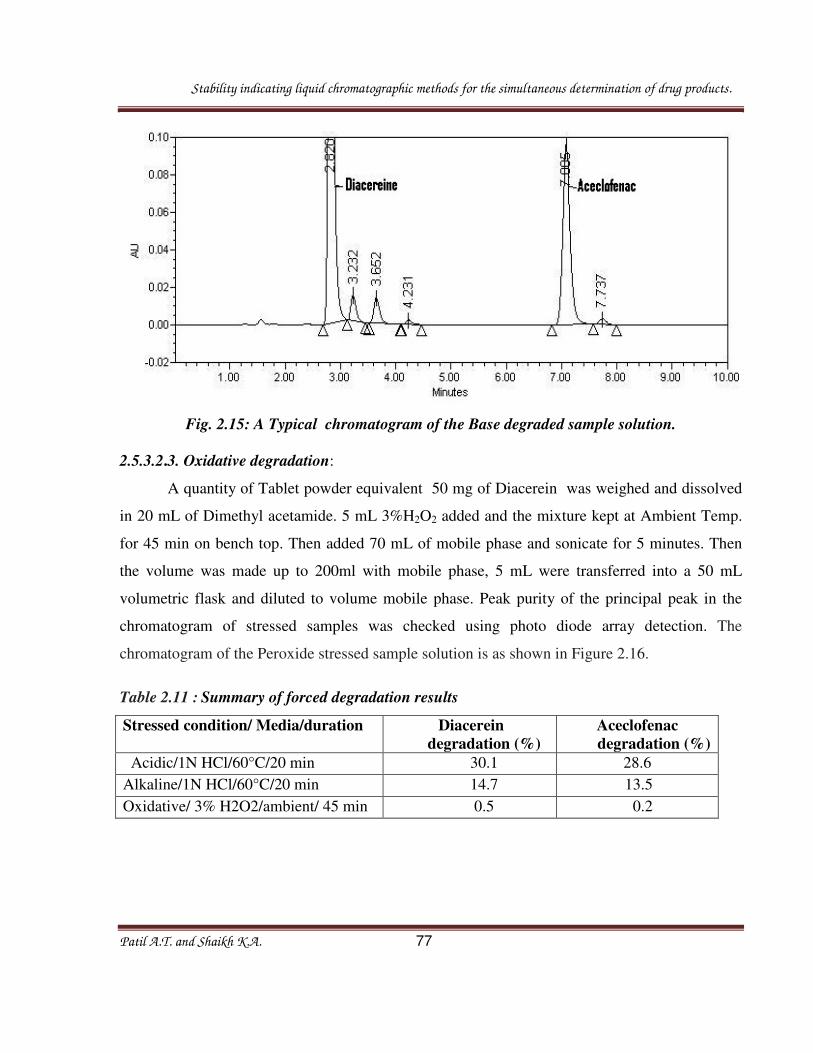
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 77
Fig. 2.15: A Typical chromatogram of the Base degraded sample solution.
2.5.3.2.3. Oxidative,degradation:
A quantity of Tablet powder equivalent 50 mg of Diacerein was weighed and dissolved
in 20 mL of Dimethyl acetamide. 5 mL 3%H2O2 added and the mixture kept at Ambient Temp.
for 45 min on bench top. Then added 70 mL of mobile phase and sonicate for 5 minutes. Then
the volume was made up to 200ml with mobile phase, 5 mL were transferred into a 50 mL
volumetric flask and diluted to volume mobile phase. Peak purity of the principal peak in the
chromatogram of stressed samples was checked using photo diode array detection. The
chromatogram of the Peroxide stressed sample solution is as shown in Figure 2.16.
Table 2.11 : Summary of forced degradation results
Stressed condition/ Media/duration Diacerein
degradation (%)
Aceclofenac
degradation (%)
Acidic/1N HCl/60°C/20 min 30.1 28.6
Alkaline/1N HCl/60°C/20 min 14.7 13.5
Oxidative/ 3% H2O2/ambient/ 45 min 0.5 0.2
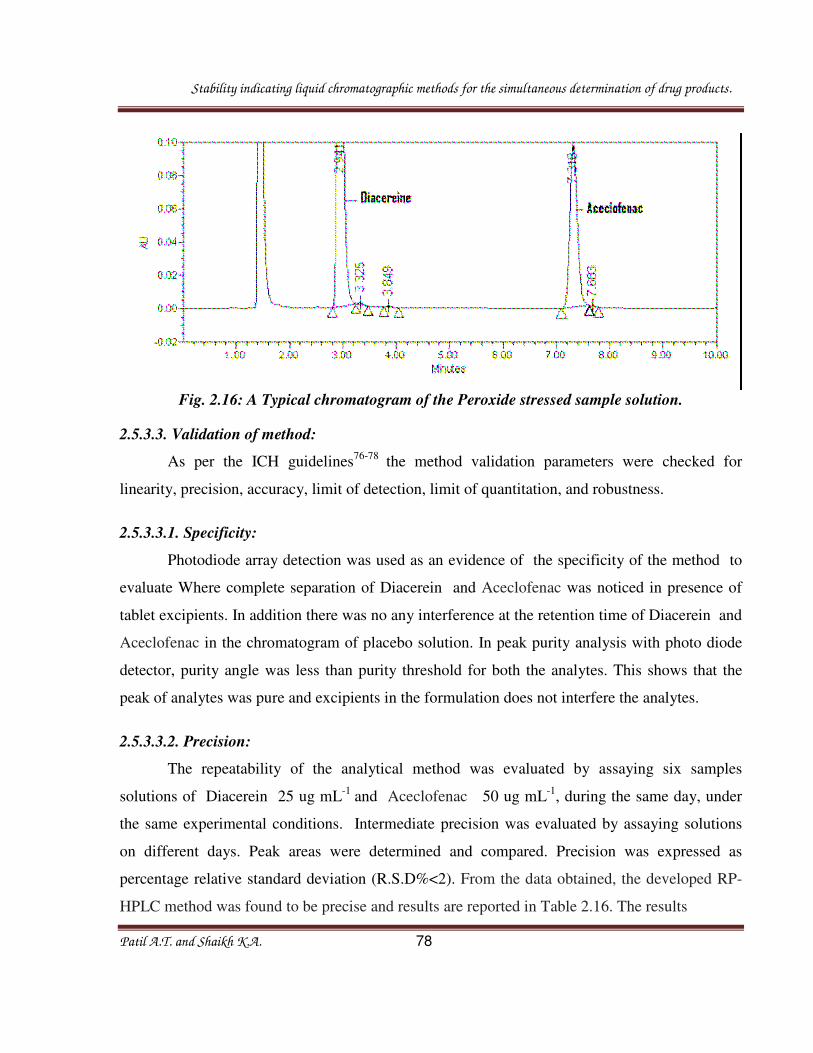
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 78
Fig. 2.16: A Typical chromatogram of the Peroxide stressed sample solution.
2.5.3.3. Validation of method:
As per the ICH guidelines76-78
the method validation parameters were checked for
linearity, precision, accuracy, limit of detection, limit of quantitation, and robustness.
2.5.3.3.1. Specificity:
Photodiode array detection was used as an evidence of the specificity of the method to
evaluate Where complete separation of Diacerein and Aceclofenac was noticed in presence of
tablet excipients. In addition there was no any interference at the retention time of Diacerein and
Aceclofenac in the chromatogram of placebo solution. In peak purity analysis with photo diode
detector, purity angle was less than purity threshold for both the analytes. This shows that the
peak of analytes was pure and excipients in the formulation does not interfere the analytes.
2.5.3.3.2..Precision:
The repeatability of the analytical method was evaluated by assaying six samples
solutions of Diacerein 25 ug mL-1
and Aceclofenac 50 ug mL-1
, during the same day, under
the same experimental conditions. Intermediate precision was evaluated by assaying solutions
on different days. Peak areas were determined and compared. Precision was expressed as
percentage relative standard deviation (R.S.D%<2). From the data obtained, the developed RP-
HPLC method was found to be precise and results are reported in Table 2.16. The results
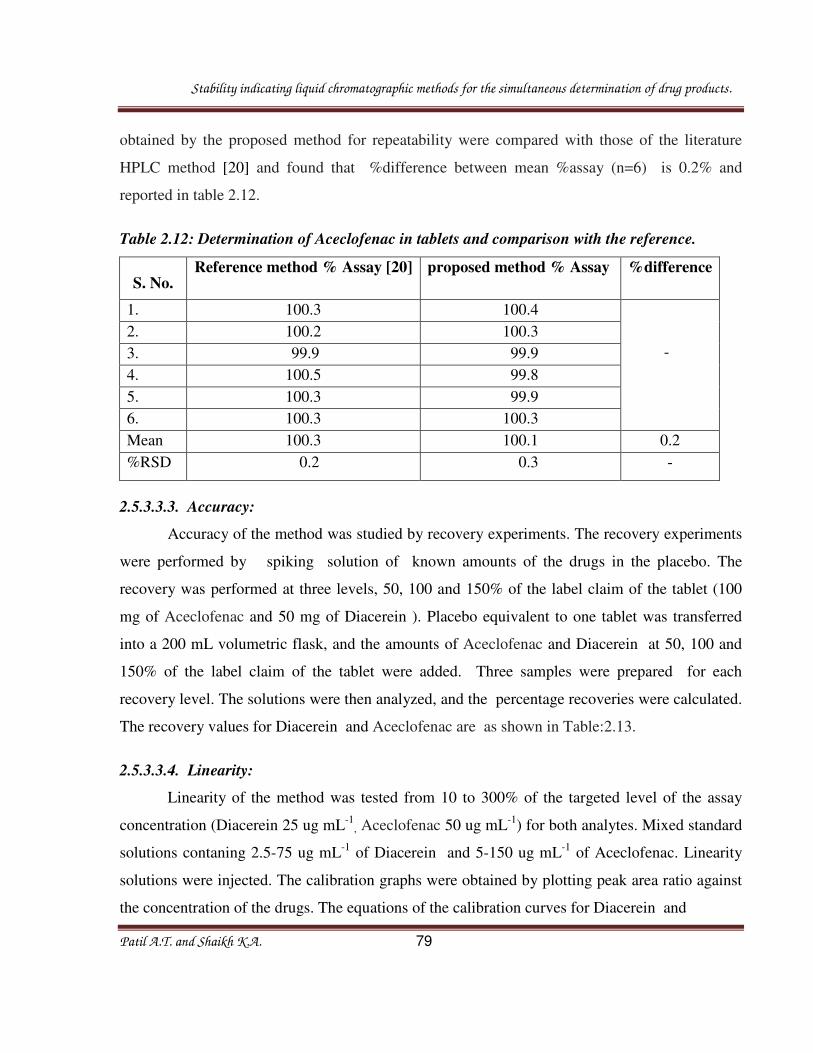
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 79
obtained by the proposed method for repeatability were compared with those of the literature
HPLC method [20] and found that %difference between mean %assay (n=6) is 0.2% and
reported in table 2.12.
Table 2.12: Determination of Aceclofenac in tablets and comparison with the reference.
2.5.3.3.3. Accuracy:
Accuracy of the method was studied by recovery experiments. The recovery experiments
were performed by spiking solution of known amounts of the drugs in the placebo. The
recovery was performed at three levels, 50, 100 and 150% of the label claim of the tablet (100
mg of Aceclofenac and 50 mg of Diacerein ). Placebo equivalent to one tablet was transferred
into a 200 mL volumetric flask, and the amounts of Aceclofenac and Diacerein at 50, 100 and
150% of the label claim of the tablet were added. Three samples were prepared for each
recovery level. The solutions were then analyzed, and the percentage recoveries were calculated.
The recovery values for Diacerein and Aceclofenac are as shown in Table:2.13.
2.5.3.3.4. Linearity:
Linearity of the method was tested from 10 to 300% of the targeted level of the assay
concentration (Diacerein 25 ug mL-1
, Aceclofenac 50 ug mL-1
) for both analytes. Mixed standard
solutions contaning 2.5-75 ug mL-1
of Diacerein and 5-150 ug mL-1
of Aceclofenac. Linearity
solutions were injected. The calibration graphs were obtained by plotting peak area ratio against
the concentration of the drugs. The equations of the calibration curves for Diacerein and
S. No. Reference method % Assay [20] proposed method % Assay
%difference
1. 100.3 100.4
2. 100.2 100.3
3. 99.9 99.9
4. 100.5 99.8
5. 100.3 99.9
6. 100.3 100.3
-
Mean 100.3 100.1 0.2
%RSD 0.2 0.3 -
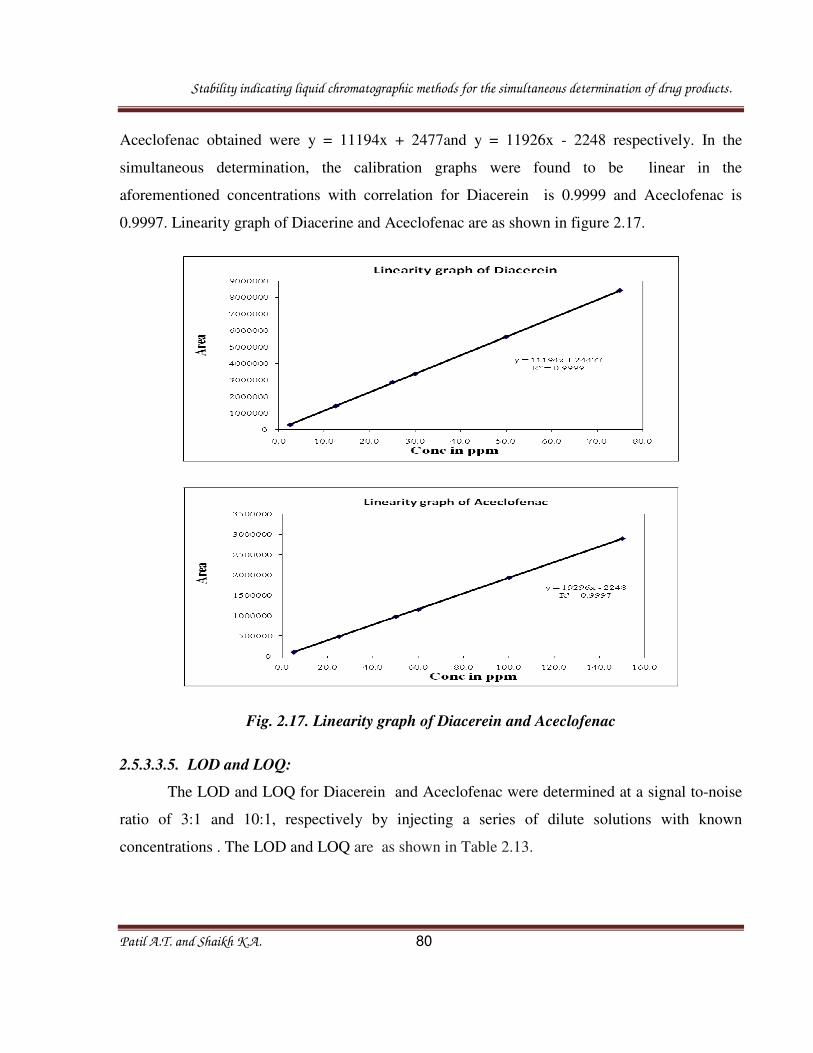
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 80
Aceclofenac obtained were y = 11194x + 2477and y = 11926x - 2248 respectively. In the
simultaneous determination, the calibration graphs were found to be linear in the
aforementioned concentrations with correlation for Diacerein is 0.9999 and Aceclofenac is
0.9997. Linearity graph of Diacerine and Aceclofenac are as shown in figure 2.17.
Fig. 2.17. Linearity graph of Diacerein and Aceclofenac
2.5.3.3.5. LOD and LOQ:
The LOD and LOQ for Diacerein and Aceclofenac were determined at a signal to-noise
ratio of 3:1 and 10:1, respectively by injecting a series of dilute solutions with known
concentrations . The LOD and LOQ are as shown in Table 2.13.
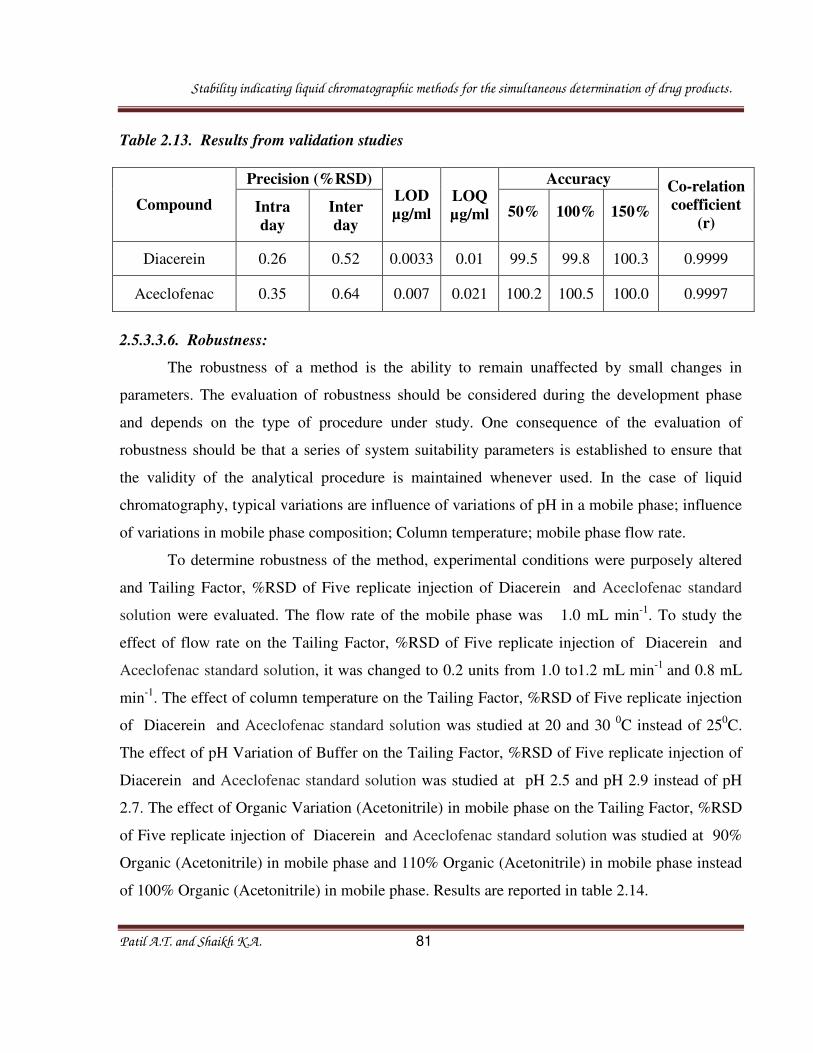
Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 81
Table 2.13. Results from validation studies
Precision (%RSD) Accuracy
Compound Intra
day
Inter
day
LOD
µg/ml
LOQ
µg/ml
50% 100% 150%
Co-relation
coefficient
(r)
Diacerein 0.26 0.52 0.0033 0.01 99.5 99.8 100.3 0.9999
Aceclofenac 0.35 0.64 0.007 0.021 100.2 100.5 100.0 0.9997
2.5.3.3.6. Robustness:
The robustness of a method is the ability to remain unaffected by small changes in
parameters. The evaluation of robustness should be considered during the development phase
and depends on the type of procedure under study. One consequence of the evaluation of
robustness should be that a series of system suitability parameters is established to ensure that
the validity of the analytical procedure is maintained whenever used. In the case of liquid
chromatography, typical variations are influence of variations of pH in a mobile phase; influence
of variations in mobile phase composition; Column temperature; mobile phase flow rate.
To determine robustness of the method, experimental conditions were purposely altered
and Tailing Factor, %RSD of Five replicate injection of Diacerein and Aceclofenac standard
solution were evaluated. The flow rate of the mobile phase was 1.0 mL min-1
. To study the
effect of flow rate on the Tailing Factor, %RSD of Five replicate injection of Diacerein and
Aceclofenac standard solution, it was changed to 0.2 units from 1.0 to1.2 mL min-1
and 0.8 mL
min-1
. The effect of column temperature on the Tailing Factor, %RSD of Five replicate injection
of Diacerein and Aceclofenac standard solution was studied at 20 and 30 0C instead of 25
0C.
The effect of pH Variation of Buffer on the Tailing Factor, %RSD of Five replicate injection of
Diacerein and Aceclofenac standard solution was studied at pH 2.5 and pH 2.9 instead of pH
2.7. The effect of Organic Variation (Acetonitrile) in mobile phase on the Tailing Factor, %RSD
of Five replicate injection of Diacerein and Aceclofenac standard solution was studied at 90%
Organic (Acetonitrile) in mobile phase and 110% Organic (Acetonitrile) in mobile phase instead
of 100% Organic (Acetonitrile) in mobile phase. Results are reported in table 2.14.
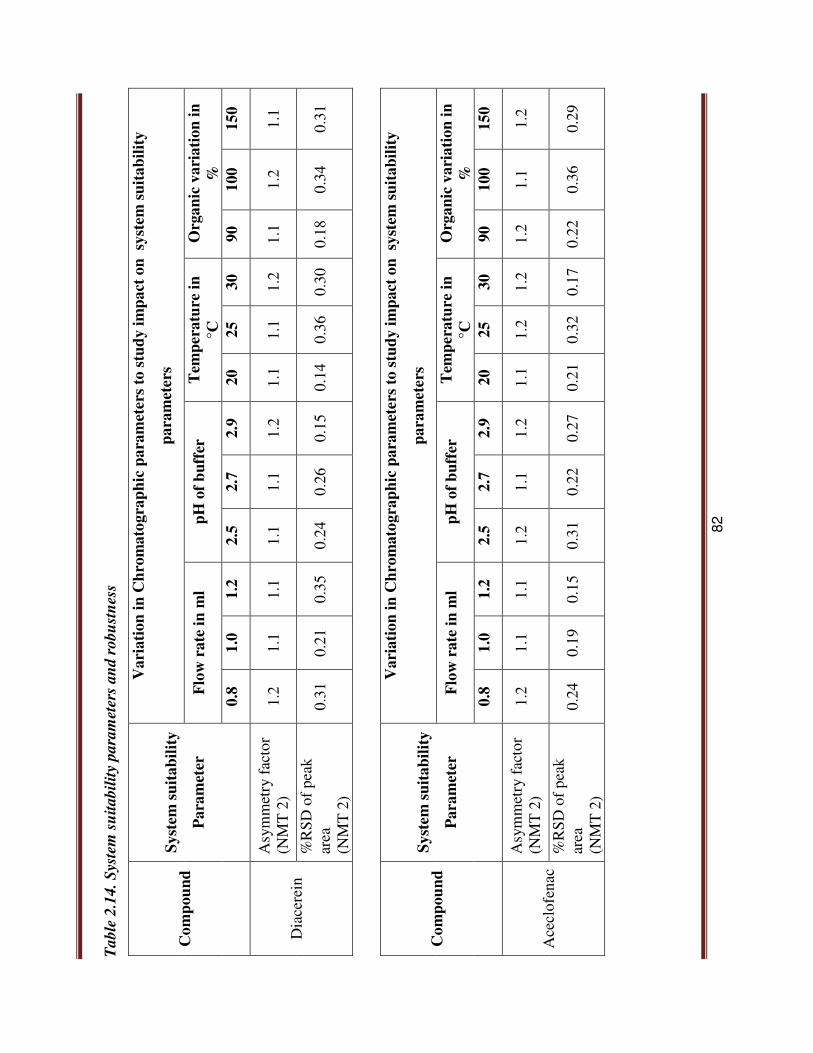
82
Table
2.1
4.
Sys
tem
su
itabil
ity
para
met
ers
an
d r
obu
stn
ess
Vari
ati
on
in
Ch
rom
ato
gra
ph
ic p
ara
met
ers
to s
tud
y i
mp
act
on
sy
stem
su
itab
ilit
y
para
met
ers
Flo
w r
ate
in
ml
pH
of
bu
ffer
T
emp
era
ture
in
°C
Org
an
ic v
ari
ati
on
in
%
Co
mp
ou
nd
S
yst
em
su
itab
ilit
y
Para
met
er
0.8
1.0
1.2
2.5
2.7
2.9
20
25
30
90
100
150
Asy
mm
etry
fac
tor
(NM
T 2
) 1.2
1.1
1.1
1.1
1.1
1.2
1.1
1.1
1.2
1.1
1.2
1.1
Dia
cere
in
%
RS
D o
f pea
k
area
(NM
T 2
)
0.3
1
0.2
1
0.3
5
0.2
4
0.2
6
0.1
5
0.1
4
0.3
6
0.3
0
0.1
8
0.3
4
0.3
1
Vari
ati
on
in
Ch
rom
ato
gra
ph
ic p
ara
met
ers
to s
tud
y i
mp
act
on
sy
stem
su
itab
ilit
y
para
met
ers
Flo
w r
ate
in
ml
pH
of
bu
ffer
T
emp
era
ture
in
°C
Org
an
ic v
ari
ati
on
in
%
Co
mp
ou
nd
S
yst
em
su
itab
ilit
y
Para
met
er
0.8
1.0
1.2
2.5
2.7
2.9
20
25
30
90
100
150
Asy
mm
etry
fac
tor
(NM
T 2
) 1.2
1.1
1.1
1.2
1.1
1.2
1.1
1.2
1.2
1.2
1.1
1.2
Ace
clofe
nac
%
RS
D o
f pea
k
area
(NM
T 2
)
0.2
4
0.1
9
0.1
5
0.3
1
0.2
2
0.2
7
0.2
1
0.3
2
0.1
7
0.2
2
0.3
6
0.2
9

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 83
2.5.4. CONCLUSION:
The developed simple LC method for assay determination of Diacerein and Aceclofenac
is linear, precise, accurate and specific. The method was validated to the requirements of ICH
and the results were satisfactory. The developed stability-indicating analytical method can be
used for the routine analysis of production samples, where sample load is higher and high
throughput is essential for faster delivery of results. Overall, the method provides a high
throughput solution for determination of Diacerein and Aceclofenac.
2.6. REFERENCES:
[1] Levocetirizine: a new selective H-1 receptor antagonist for use in allergic disorders,
Day J.H., Ellis A.K., Rafeiro E., Drugs Today (2004)40, 415–421.
[2] U.S. Pharmacopoeia USP31 NF26, VOL 3, 129.
[3] Indian Pharmacopoeia (2007)2, 291.
[4] Once daily Levocetirizine for the treatment of allergic rhinitis and chronic idiopathic,
Urticaria N.E., Calogiuri G.F., Di L.E., Cardinale F., Macchia L., Ferrannini A.,
Vacca1 A., J. Asth. Allergy (2009)2, 17–23.
[5] Formoterol Used as Needed-Clinical Effectiveness, Selroos O., Respir. Med. (2001)95,
17–20.
[6] Ciclesonide has Minimal Oropharyngeal Side Effects in the Treatment of Patients with
Moderate-to-Severe Asthma, Bernstein J.A., Noonan M.J., Rim C., Fish J., Kundu S.,
Williams J., Banerji D.D., Hamedani P., J. Allergy Clin. Immunol. (2004)113-2, 113.
[7] Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive
Pulmonary Disease, Pauwels R.A., Buist A.S., Calverley P.A., Jenkins C.R., Hurd S.
S., Am. J. Respir. Crit. Care. Med. (2001)163, 1256–1276.
[8] British Pharmacopoeia (2009)1, 913.
[9] Indian Pharmacopoeia (2007)2, 931.
[10] An active metabolite of Diacerein, suppresses the interleukin-1alpha-induced
proteoglycan degradation in cultured rabbit articular chondrocytes, Tamura T., Ohmori
K.R., Jpn. J. Pharmacol. (2001)85, 101–104.
[11] New Symptomatic Slow Acting Drug for Osteoarthritis : Diacerein, Mahajan A., Singh

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 84
K., Tandon V.R., Kumar S., Kumar H., A JK Science (2006)8, 173-175.
[12] Diacerhein and Rhein Prevent Interleukin-1β-Induced Nuclear Factor-κB Activation by
Inhibiting the Degradation of Inhibitor κB-α, Alexandrina F.M., Caramona M.M.,
Carvalho A.P., Lopes M.C., Pharma. & Toxi. (2002)91, 22–28.
[13] Bioequivalence pharmacokinetic evaluation of two branded formulations of aceclofenac
100 mg: a single-dose, randomized, open-label, two-period crossover comparison in
healthy Korean adult volunteers, Rhim S.Y., Park J.H., Park Y.S., Lee M.H., Shaw L.
M., and Kang J.S., Clil Therap. (2008)30, 633-640.
[14] Simultaneous HPLC–UV determination of Rhein and Aeclofenac in human plasma,
Ojha A., Rathod R., Padh H., J. Chrom. B (2009)877, 1145- 1148.
[15] Simultaneous determination of Pseudoephedrine Sulfate, Dexbrompheniramine Maleate
and Loratadine in pharmaceutical preparations using derivative spectrophotometry and
ratio spectra derivative spectrophotometry, Feyyaz O., Yücesoy C., Dermi S., Kartal M.,
Kokdil G.,Talanta (2000)51, 269-279.
[16] Stability indicating HPTLC method for the simultaneous determination of
pseudoephedrine and cetirizine in pharmaceutical formulations. Makhija S.N., Vavia
P.R., J Pharm Biomed Anal. (2001) 25, 663-667.
[17] Quantitative and structural determination of pseudoephedrine sulfate and its related
compounds in pharmaceutical preparations using high-performance liquid
chromatography, Nian W., Wenqing F., Elizabeth L., Guodong C., Jayashree P.,
Tze-Ming C., and Birendra P., J. Pharm. Biomed. Anal. (2002)30, 1143-1155.
[18] Simultaneous determination of Loratadine and Pseudoephedrine sulfate in pharmaceutical
Formulation by RP-LC and derivative spectrophotometry, Mabrouk M.M., El-Fatatry H.
M., Hammad S., Abdel A., Wahbi M., J. Pharm. Biomed. Anal. (2003)33, 597-604.
[19] A new HPLC approach for the determination of hydrophilic and hydrophobic
components: the case of pseudoephedrine sulfate and loratadine in tablets. Abu L.
A., Hamdan I.I., Tahraoui A., Drug Dev Ind Pharm (2005)31, 577-588.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 85
[20] Determination of loratadine and pseudoephedrine sulfate in pharmaceuticals based on
non- linear second-order spectrophotometric data generated by a pH-gradient flow
injection technique and artificial neural networks, Culzoni M.J., Goicoechea H.C., Anal
Bioanal Chem., (2007)389, 2217-2225.
[21] Simultaneous UV spectrophotometric estimation of ambroxol hydrochloride and
Levocetirizine dihydrochloride, Lakshmana P., Shirwaikar A.A., Annie S., Kumar C.D.,
Aravind K., Indian J. pharm. Sci. (2008)70-2, 236-238.
[22] Development and validation of a stability-indicating RP-HPLC method for the
determination of paracetamol with dantrolene or/and cetirizine and pseudoephedrine in
two pharmaceutical dosage forms. Hadad G. M., Emara S., Mahmoud W.M.,
Talanta (2009)79, 1360-1367.
[23] A porous graphitized carbon column HPLC method for the quantification of
paracetamol, pseudoephedrine, and chlorpheniramine in a pharmaceutical formulation.
Kalogria E., Koupparis M., Panderi I., J AOAC Int. (2010) 93, 1093-1101.
[24] A Sensitive RP-HPLC Method for Simultaneous Estimation of Diethylcarbamazine and
Levocetirizine in Tablet Formulation, Reddy J. M., Jeyaprakash M. R., Madhuri
K., Meyyanathan S. N., Elango K., Indian J Pharm Sci. (2011) 73, 320–323.
[25] Quantization of Dextromethorphan and Levocetirizine in combined dosage form using a
novel validated RP-HPLC method, Joshi S., Bhatia C., Bal C.S., Indian J Pharma. sci.,
(2012)74, 83-86.
[26] Simultaneous analysis of Levocetirizine dihydrochloride, Ambroxol hydrochloride, and
Montelukast sodium by RP-HPLC - PDA method, Srividya P., Buchi N. N., Tejaswini
M., Sravanthi D., J. Liq. Chrom. Rel. Techno. (2013) 36, 2871-2881.
[27] Simultaneous Quantification of Cefpirome and Cetirizine or Levocetirizine in
Pharmaceutical Formulations and Human Plasma by RP- HPLC, Arayne M.S., Sultana
N., and Nawaz M., J of Anal. Chem. (2008)63-9, 881–887.
[28] Simultaneous Analysis of Ambroxol HCl with Cetirizine HCl and of Ambroxol HCl with
levo-Cetirizine Dihydrochloride in Solid Dosage Forms by RP-HPLC, Birajdar A.S.,

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 86
Meyyanathan S.N., Raja R.B., Krishanaveni N., and Suresh B., Acta Chromatog. (2008)
3, 411–421.
[29] Determination of levocetirizine in human plasma by liquid chromatography–electrospray
tandem mass spectrometry: Application to a bioequivalence study, Morita M. R., Berton
D., Boldin R., Barros A.P., Meurer E. C., Amarante A.R., Campos D.R., Calafatti S.A.,
Pereira R., Abib J.E., and Pedrazolli J., J. Chrom. B (2008)862, 132-139.
[30] LC–MS–MS Assay for Simultaneous Quantification of Fexofenadine and
Pseudoephedrine in Human Plasma, Vijaya D., Bharathi K.R., Banda J., Gajjela R., Indu
B., Naidu A., Mullangi R., Chromatographia (2008)67, 461-466.
[31] Development and validation of a HPLC method for the simultaneous determination of
Bromhexine, Chlorpheniramine, Paracetamol, and Pseudoephedrine in their combined
cold medicine formulations, Silvana E. V., Teodoro S. K., J. Liq. Chrom. Rel. Techno.
(2013) 36, 2829-2843.
[32] Determination by HPLC with Electrochemical Detection of Formoterol RR and SS
Enantiomers in Urine, Butter J.J., Van den Berg B.T.J., Portier E.J.G., Kaiser G., Boxtel
C.J.V., J. Liq. Chromatogr. R. T. (1996)19, 993–1005.
[33] Automated and sensitive method for the determination of formoterol in human plasma by
high-performance liquid chromatography and electrochemical detection. Campestrini J.,
Lecaillon J.B., Godbillon J., J Chromatogr B Biomed Sci Appl. (1997) 704, 221-229.
[34] Assay for the determination of low dosage form of formoterol dry syrup by capillary
electrophoresis with head-column field-amplified sample stacking. Song J.Z., Chen
J., Tian S.J., Sun Z.P., J Pharm Biomed Anal. (1999) 21, 569-576.
[35] Validation of a RP-HPLC Method for the Assay of Formoterol and Its Related Substances
in Formoterol Fumarate Dehydrate Drug Substance, Akapo S., Asif M., J. Pharm.
Biomed. Anal. (2003)33, 935–945.
[36] Optimization and validation of a gas chromatographic method for analysis of (RS,SR)-
diastereoisomeric impurity in formoterol fumarate. Akapo S.O., Wegner M., Mamangun
A., McCrea C., Asif M., Dussex J.C., J Chromatogr A. (2004) 1045, 211-216.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 87
[37] Formation of fatty acid conjugates of ciclesonide active metabolite in the rat lung after 4-
week inhalation of ciclesonide. Nave R., Meyer W., Fuhst R., Zech K.. Pulm Pharmacol
Ther. (2005)18 390-396.
[38] Simutaneous Spectrometric Determination of Formoterol Fumarate and Budesonide in
Their Combined Dosage Forms, Prasad A.V.S.S., Indian J. Chem. Technol. (2006)13,
81–83.
[39] Determination of formoterol in rat plasma by liquid chromatography-electrospray
ionisation mass spectrometry, Kakubari I., Dejima H., Miura K., Koga Y., Mizu
H., Takayasu T., Yamauchi H., Takayama S., Takayama K., Pharmazie (2007) 62
62,94-95.
[40] Sensitive Simultaneous Determination of Ciclesonide, Ciclesonide-M1-Metabolite and
Fluticasone Propionate in Human Serum by HPLC-MS/MS with, Mascher H.J., Zech K.,
Mascher D. J., APPI. J. Chrom. B (2008)869, 84–92.
[41] Chiral HPLC Analysis of Formoterol Stereoisomers and Thermodynamic Study of Their
Interconversion in Aqueous Pharmaceutical Formulations, Akapo S., McCrea C., Gupta
J., Roach M., Skinner W., J. Pharm. Biomed. Anal. (2009)49, 632–637.
[42] Rapid Spectrophotometric methods for the determination of two novel steroids in bulk
and pressurised metered - dose preparations, Dave N. H., Makwana A. G., Int.J.Pharm.&
Health Sci.(2010)1, 68-76.
[43] Development of ciclesonide dry powder inhalers and the anti-asthmatic efficacy in
guinea pigs, Fei L., Wang G.L., Zhang Y., et. al., J Chin Pharma Sci. (2011)20, 473-
482.
[44] Validated Method Development for Estimation of Formoterol Fumarate and
Mometasone Furoate in Metered Dose Inhalation Form by High Performance Liquid
Chromatography, Srinivasarao K., Gorule V.,et. al. J Anal Bioanal Techniques (2012)7,
1-4.
[45] Optimized method for rapid estimation of ciclesonide in bulk and its dosage form
(rotacap) by RP-HPLC, Vaghela V., J Liq Chrom Rel Tech. (2013)36, 1678-1685.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 88
[46] Assay for the Determination of Low Dosage Form of Formoterol Dry Syrup by
Capillary Electrophoresis with Head-Column Field-Amplified Sample, Song J.Z., Chen
J., Tian S.J., Sun Z.P., J. Pharm. Biomed. Anal. (1999)21, 569–576.
[47] Spectrophotometric Determination of Formoterol Fumarate in Rotacap Formulation, Pai
P.N.S., Sandeep S.K., Sekhar A.B., IJPS, (2003)65, 649–650.
[48] Ultra-Sensitive Determination of Fomoterol in Human Serum by HPLC and
Electrospray Tandem Mass Spectrometry, Mascher D.G., Zech K., Nave R.,
Kubesch K.M.H.J., J. Chrom. B (2006 )830, 25–34.
[49] High Performance Liquid Chromatography Assay Method for Simultaneous Quantitation
of Formoterol and Budesonide in Symbicort Turbuhaler, Assi K.H., Tarsin W., Chrystyn
H., J. Pharm. Biomed. Anal. (2006)41, 325–328.
[50] Simultaneous determination of Aceclofenac and three of its metabolites in human plasma
by high- performance liquid chromatography, Hinz B., Auge D., Rau T., Rietbrock S.,
Brune K., Werner U., Bio. Chromate. (2003)17, 268-275.
[51] Determination of aceclofenac in human plasma by reversed-phase high performance liquid
chromatography. Jin Y., Chen H., Gu S., Zeng F., Se Pu. (2004)22, 252-254.
[52] Separation of aceclofenac and diclofenac in human plasma by free zone capillary
electrophoresis using N-methyl-D-glucamine as an effective electrolyte additive, Angelo
Z., Ciriaco C., Salvatore S., Emanuela P., Paolo E., Luca D., Euro j Pharma Sci. (2005)
24,375-380.
[53] Reverse phase HPLC method for determination of aceclofenac and paracetamol in tablet
dosage form, Momin M.Y., Yeole P.G., Puranik M.P., Wadher S.J., Ind. J. Pharma.
Sci. (2006)68-3, 387-389.
[54] Sensitive Determination of Nanogram Levels of Diacerein in a Pharmaceutical
Formulation by Flow Injection Chemiluminescence Analysis, Han-C. Y., Xiao F. Y., Hua
L., J Chin Chemi Soc (2007) 54, 949-956.
[55] Development and Validation of a Dissolution Method with Spectrophotometric Analysis

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 89
for Diacerhein Capsules, Borgmann S. M., parcianello L., Marcela Z., Bajerski L.,
Simone G., Sci. Pharma. (2008)76, 541–554.
[56] Simultaneous HPLC–UV determination of Rhein and Aeclofenac in human plasma,
Ojha A., Rathod R., Padh H., J. Chrom. B (2009)877, 1145- 1148.
[57] Development and validation of UV Spectrophotometric method for the determination of
Diacerein in capsules, Narade S., Patil S., Surve S., Shete D., Pore Y., Dig J Nano
Biostru. (2010)5, 113-118.
[58] Application of mixed-hydrotropy in titrimetric analysis of Aceclofenac bulk drug sample,
Maheshwari R. K., Jawalker S., Jahan F., Patel S., Mehtani D., Bulletin of Pharma Res.
(2011)1, 44-46.
[59] Determination of Diacerein in Rabbit Plasma by Liquid Chromatography - Mass
Spectroscopy and Its Application to Bioavailability Study, Shirwaikar A., Sarala A. D.,
Premalatha K., Sreejith K. R., Int J Res Pharma Biom Sci. (2012) 3, 1738-1744.
[60] A Quantitative and Qualitative High Performance Liquid Chromatographic etermination
of Aceclofenac Specified Impurity-B (Methyl[2-[(2, 6-dichlorophenyl)amino]
phenyl]acetate) in Aceclofenac Bulk Drug, Somashekar P.L., Sanjay Pai., Gopalkrishna
R., Chem Sci Trans., (2013)1, 1-9.
[61] Simple spectrophotometric methods for estimation of aceclofenac from bulk and
formulations, Bose A., Dash P.P., Sahoo M.K., Pharma. Methd. (2010) 1, 57-60.
[62] A simple and sensitive stability-indicating RP-HPLC assay method for the determination
of Aceclofenac, Bhinge J.R., Kumar R.V., Sinha V.R., J chroma sci (2008)46, 440-444.
[63] Single RP-HPLC method for the quantification of Aceclofenac, Paracetamol and
Chlorozoxazone in formulation, Reddy H.K.G., Reddy H.B., Maram R.K., Reddy UM.,
Int. J Sci. Inno. and Disco. (2012) 2, 471-490.
[64] Validated LC method for simultaneous analysis of tramadol hydrochloride and
aceclofenac in a commercial tablet, Kachhadia P.K., Doshi A.S., Ram V.R., Joshi H.S.,
Chromatographia (2008)68, 997–1001.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 90
[65] Reverse Phase HPLC Method for Determination of Aceclofenac and Paracetamol in
Tablet Dosage Form, Godse V.P., Deodhar M.N., Bhosale A.V., Sonawane R.A., Sakpal
P.S., Borkar D.D., and Bafana Y.S., A. J. Res. Chem (2009)2-1, 37-40.
[66] Simultaneous Estimation of Paracetamol, Chlorzoxazone and Aceclofenac in
pharmaceutical formulation by HPLC Method, Karthikeyan V., Yuvaraj V., Nema R.
K., Int. J. ChemTech Res. (2009)1, 457-460.
[67] A Review – Importance of RP-HPLC in Analytical Method Development, Prathap B.,
Akalanka D., Srinivasa G.H., Johnson P. & Arthanariswaran P., int. j. no. tre. in Pharma.
Sci. (2013)3-1,15-23.
[68] On the Reproducibility of Column Performance in Liquid Chromatography and the Role
of the Packing Density, Stanley B.J., Foster C.R., Guiochon G., J. Chrom. A. (1997)
761, 41-51.
[69] Liquid phase separation methods: HPLC, FFF, electrophoresis, Kirkland J.J.,
McCormick R.M., Chromatographia (1987)24-1, 58-76.
[70] On the Reproducibility of Column Performance in Liquid Chromatography and the Role
of the Packing Density, Stanley B.J., Foster C.R., Guiochon G., J. Chrom. A. (1997)
761, 41-51.
[71] Ghost peaks in reversed-phase gradient HPLC: a review and update, Stuart W., J. Chrom.
A (2004)1052, 1-2, 1-11.
[72] Implementation of a rapid and automated high performance liquid chromatography
method development strategy for pharmaceutical drug candidates, Elizabeth F.H., Patrick
L., Sergey G., J. Chrom. A (2006)1107, 1-2, 79-87.
[73] Development of some stationary phases for reversed-phase HPLC, Kirkland J.J.,
J. Chrom. A (2004)1060, 1-2, 9-21.
[74] Simultaneous variation of temperature and gradient steepness for reversed-phase high-
performance liquid chromatography method development, Dolan J.W., Snyder L.R.,
Saunders D.L., Heukelem L.V., J. Chrom. A (1998)803, 1-2, 33–50.

Stability indicating liquid chromatographic methods for the simultaneous determination of drug products.
Patil A.T. and Shaikh K.A. 91
[75] HPLC method development in early phase pharmaceutical development, Henrik T.R.,
Kelly A.S., Sheetal G., Sep. sci. Techn. (2007)8, 353-371.
[76] Validation of analytical procedures: text and Methodology. In: ICH Harmonized
Tripartite Guidelines Q2 (R1). November 2005.
[77] Understanding and Implementing Efficient Analytical Methods Development and
Validation, Jay B., Kevin J., Pierre B., Anal.Chem.Test. (2003)1, 6-13.
[78] Validation of HPLC methods in pharmaceutical analysis, Ashley S. L., Sep. sci. Techn.
(2005)6, 191-192.















![Stability Indicating RP-UPLC Method for Assay of Emtricitabine … › pdf › AJAC_2015092415002585.pdf · 2015-09-24 · forms [9]-[12] and biological fluids [13]. Several liquid](https://img.pdfslide.us/doc/110x75/5f0d7c3d7e708231d43a977c/stability-indicating-rp-uplc-method-for-assay-of-emtricitabine-a-pdf-a-ajac.jpg)













