Embed Size (px)
Citation preview

Myostatin Induces DNA Damage in Skeletal Muscle ofStreptozotocin-induced Type 1 Diabetic Mice*
Received for publication, May 20, 2013, and in revised form, December 22, 2013 Published, JBC Papers in Press, January 14, 2014, DOI 10.1074/jbc.M113.483115
Sandhya Sriram‡, Subha Subramanian‡, Prasanna Kumar Juvvuna§, Craig McFarlane¶, Monica Senna Salerno�,Ravi Kambadur‡¶, and Mridula Sharma§1
From the ‡Division of Molecular Genetics and Cell Biology, School of Biological Sciences, Nanyang Technological University,60 Nanyang Drive, Singapore 637551, the §Department of Biochemistry, YLLSoM, National University of Singapore, 14 MedicalDrive, Centre for Translational Medicine MD6, Level 14, Singapore 117599, the ¶Growth, Development and Metabolism Program,Singapore Institute of Clinical Sciences, Brenner Centre for Molecular Medicine, 30 Medical Drive, Singapore 117609, and�AgResearch, Hamilton 3240, New Zealand
Background: Uncontrolled type 1 diabetes leads to DNA damage and skeletal muscle atrophy.Results: STZ-induced Foxa2 up-regulates Mstn leading to DNA damage via p63/REDD1 pathway in skeletal muscle.Conclusion: Mstn is a target of Foxa2. Blocking Mstn can attenuate DNA damage in the diabetic muscle.Significance: The findings reveal a mechanism of induction of Mstn and DNA damage during diabetes.
One of the features of uncontrolled type 1 diabetes is oxida-tive stress that induces DNA damage and cell death. Skeletalmuscle atrophy is also considerable in type 1 diabetes, however,the signaling mechanisms that induce oxidative stress culminat-ing in muscle atrophy are not fully known. Here, we show that inStreptozotocin-induced diabetic wild type mice, hypo-phosphory-lation of Akt, resulted in activation of Foxa2 transcription factor inthe muscle. Foxa2 transcriptionally up-regulated Myostatin, con-tributing to exaggerated oxidative stress leading to DNA damagevia p63/REDD1 pathway in skeletal muscle of Streptozotocin-treated wild type mice. In Myostatin�/� mice however, Streptozo-tocin treatment did not reduce Akt phosphorylation despitereduced IRS-1 signaling. Moreover, Foxa2 levels remained unal-tered in Myostatin�/� mice, while levels of p63/REDD1 werehigher compared with wild type mice. Consistent with theseresults, relatively less DNA damage and muscle atrophy wasobserved in Myostatin�/� muscle in response to Streptozotocintreatment. Taken together, our results for the first time show therole of Foxa2 in Myostatin regulation in skeletal muscle in diabeticmice. Altogether, these results demonstrate the mechanism bywhich Myostatin contributes to DNA damage in skeletal muscle ofthe diabetic mice that would lead to myofiber degeneration.
Type 1 diabetes is caused by lack of insulin secretion in thebody due to pancreatic � cell damage. The increased levels ofglucose in circulation dysregulate the prooxidant-antioxidantbalance, thus enhancing oxidative stress and reducing antioxi-dant levels. The exaggerated level of reactive oxygen species(ROS)2 increases protein degradation and reduces protein syn-
thesis eventually leading to skeletal muscle wasting. In additionto proteins, DNA is also susceptible to damage by ROS duringtype 1 diabetes. Increased ROS, especially hydroxyl (-OH) andsuperoxide (O2
. ) radicals can damage DNA by addition to dou-ble bonds of DNA bases resulting in increased 8-oxo-dG levels,a mutagenic base by-product used as a marker for oxidativestress. A recent study conducted on type 1 diabetic patients hasshown that 8-OHdG levels and level of carbonylated proteinswere increased during type 1 diabetes (1). Additionally, DNAdamage consists of single and double strand breaks and AP(apurinic/apyrimidinic) sites. The cellular DNA repair machin-ery comprising base excision repair and nucleotide excisionrepair is able to repair the above mentioned lesions (2). How-ever, DNA repair mechanisms are also affected by ROS result-ing in DNA fragmentation and eventually necrosis of cells.Indeed alterations in the DNA repair capacity and index havebeen observed in diabetic patients (3). In recent years, regulatedin development and DNA damage responses (REDD1), a com-ponent of stress response and a developmentally regulatedtranscriptional target of p63 and p53 (4), has been identified tobe involved in oxidative stress-induced DNA damage in skeletalmuscle during chronic hypoxia (5).
Myostatin (Mstn), a growth and differentiation factor, hasbeen associated closely with skeletal muscle wasting. Our labo-ratory has shown recently that Mstn can induce ROS and inturn Anti-oxidant enzymes in skeletal muscle through TNF-�and NADPH oxidase (Nox) in a feed-forward manner (6). Pre-viously it was reported that Mstn expression and level was up-reg-ulated in skeletal muscle of rodents with type 1 diabetes induced byStreptozotocin (STZ) (7, 8, 9). Furthermore, Mstn expression wasattenuated by insulin administration in STZ-induced type 1 dia-betic mice (7). However, the signaling mechanisms by which highglucose levels induce Mstn expression are still unknown.
Since Mstn levels are increased in skeletal muscle duringSTZ-induced type 1 diabetes and Mstn also acts as a pro-oxi-dant, the aim of this study was to investigate how Mstn levelsare up-regulated during STZ-induced diabetes and whetherMstn can cause DNA damage under hyperglycemic conditions.
* This work was supported by the Academic Research Council (Ministry ofEducation, Singapore) and National Research Foundation, Singapore.
1 To whom correspondence should be addressed: Department of Bio-chemistry, YLLSoM, National University of Singapore, 14 Medical Drive,Centre for Translational Medicine MD6, Level 14, Singapore 117599.Tel.: 65-65167102, E-mail: [email protected].
2 The abbreviations used are: ROS, reactive oxygen species; REDD, regulatedin development and DNA damage response; AP, apurinic/apyrimidinic;Mstn, Myostatin; STZ, Streptozotocin; CCM, control conditioned medium.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 289, NO. 9, pp. 5784 –5798, February 28, 2014© 2014 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A.
5784 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 289 • NUMBER 9 • FEBRUARY 28, 2014
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Here, we present evidence that STZ-induced hyperglycemiaup-regulated Mstn via Foxa2 transcription factor, which in turninduced ROS via TNF-� and Nox. The excessive ROS levelscaused by high glucose and elevated Mstn levels led to p63/REDD1 regulated DNA damage. Using in vitro techniques, wedemonstrate that Mstn induced single-strand and double-strand breaks in DNA, eventually leading to DNA fragmenta-tion in muscle cells via p63/REDD1 pathway. Furthermore,inactivation or inhibition of Mstn in skeletal muscle or cellsattenuated DNA damage by regulating the DNA damage/repairmechanisms.
EXPERIMENTAL PROCEDURES
Animals—7-week-old C57Bl/6J male mice (WT) wereobtained from National University of Singapore-Centre forAnimal Resources. Mstn�/� male mice (7-week-old) wereobtained as previously described (6) and maintained at Nan-yang Technological University Animal house. All animals hadfree access to chow diet and water. All experimental procedureswere approved by the Institutional Animal Care and Use Com-mittee, Singapore.
Reagents and Proteins—STZ was purchased from Sigma-Al-drich. Mstn protein containing conditioned medium wasobtained from the Mstn expressing CHO cell line (10); controlconditioned medium is denoted as CCM and conditionedmedium containing Mstn is denoted as CMM in this report. Toantagonize Mstn, we used Ant1, a C-terminal truncation of Mstnprotein which is a dominant negative (Mstn mimetic) protein.Ant1 was produced and purified as previously published (11).
Induction of Type 1 Diabetes (Hyperglycemia) by STZInjection—To generate an acute type 1 diabetes mouse model,WT and Mstn�/� mice were intraperitoneally injected with asingle high-dose (concentration 22.5 mg/ml) of STZ (150mg/kg body weight). Mice injected with an equal volume ofsodium citrate buffer were maintained as control groups. Non-fasting body weight and random blood glucose levels weredetermined on the day of injection and subsequently on Day 1,2, 4, 5, 7, 8, 9, 11, 12, 13, 14, 15, 16, and 18. Mice were fasted for9 –10 h and body weight and fasting blood glucose was mea-sured on Day 3, 6, 10, and 17. WT-Control and -STZ injectedgroups are denoted as WT-C and WT-STZ, respectively, whileMstn�/� groups are denoted as Mstn�/�-C and Mstn�/�-STZin this report. Mice were sacrificed on Day 4, 7, 11, and 18 andhind limb muscles were collected.
Hematoxylin and Eosin Staining—Tibialis anterior musclesections (10 �m) were stained with hematoxylin and eosin asdescribed previously (12). Images were taken at 10� magnifi-cation using a Leica upright microscope, equipped with ImagePro Plus software.
Cell Culture—C2C12 myoblasts (13) were grown in prolifer-ation medium: DMEM with high glucose (4.5 g/liter) (PAA Lab-oratories-Cat. No. G0006, 3050), 10% FBS, and 1% penicillin/streptomycin (P/S), as previously described (6). The hepatocellularcarcinoma cell line, HepG2 was used for ChIP assays. HepG2 cellswere grown in medium containing DMEM with high glucose (4.5g/liter), L-glutamine, and sodium pyruvate (PAA Laboratories-Cat. No. G0006, 3050), 10% FBS, and 1% P/S.
Isolation of Primary Myoblasts—The hind limb skeletal mus-cles from WT and Mstn�/� mice were dissected, and primarymyoblasts were isolated according to the previously establishedprotocol (14, 15).
Treatment of C2C12 and Primary Myoblasts—STZ at twodifferent concentrations (STZ1 � 0.25 mg/ml; STZ2 � 1mg/ml) was used to treat proliferating C2C12 myoblasts andprimary myoblasts isolated from WT and Mstn�/� mice for48 h. The conditioned medium from CHO cells (CMM) wasfound to have Mstn at a concentration of 3.5 ng/ml (as deter-mined by Enzyme Immuno Assay (EIA) (ImmundiagnostikAG)). Two different concentrations of CMM (CMM1–7 ng;CMM2–17.5 ng) were used to treat C2C12 and primary myo-blasts in proliferation medium for 48 h. Ant1 at a concentrationof 1 ng/ml was used to pretreat cells 1 h before STZ or CMMtreatment.
RT-qPCR (Reverse Transcriptase Quantitative PolymeraseChain Reaction)—RNA was isolated from Gastrocnemius mus-cle and liver tissue, and RT-qPCR was performed exactly asdescribed in Sriram et al. (6). The forward and reverse primersused are available upon request.
Protein Isolation—Protein lysates from Gastrocnemius mus-cles and myoblasts were made as previously described (6). Cyto-plasmic and nuclear fractions from Biceps femoris muscleswere isolated as previously described (16). The protein concen-trations were measured by Bradford’s assay (17).
Western Blot Analysis—Western blotting was performed asdescribed previously (6). List of primary and secondary anti-bodies are available upon request.
Electrophoretic Mobility Shift Assay—The Foxa2 binding sitewas identified using the TFSEARCH tool. The oligonucleotidescontaining the Foxa2 binding site on mouse Mstn promoter(5�-TTTTTTCCCTCAAATATTTGTTTTAGTAACAA-3�)were labeled at the 3�-end with Biotin Tetra-ethylene glycol(Sigma-Aldrich). The nuclear extracts from WT-C and WT-STZ Biceps femoris muscle were used for the assay. The elec-trophoretic mobility shift assays were performed using theLightshift Chemiluminescent EMSA kit (Thermo Scientific) aspreviously described (6).
Chromatin Immunoprecipitation (ChIP) Assay—HepG2cells were transfected with 1.7 kb mouse Mstn promoter con-struct (1.7P) (18) using Lipofectamine 2000 according to themanufacturer’s instructions (Invitrogen). ChIP assay was per-formed according to the published protocol (19). The followingset of primers was used for PCR: Foxa2 forward primer5�-GTCAGCTCTTCCTAGTTTTTACTTCTC-3� and Foxa2reverse primer 5�-TCCTTTAAGACTTGGAGTGCTGT-3�.The resulting PCR products were electrophoresed on 1.5% aga-rose gel and stained with ethidium bromide.
Luciferase Assay—C2C12 myoblasts were transfected witheither pGL3-basic and pFLAG-CMV2 empty vectors or 1.7 kbmouse Mstn promoter construct (1.7P) and pFLAG-CMV2 or1.7P and pFLAG-Foxa2, together with the control Renilla lucif-erase vector pRL-TK using Lipofectamine 2000 (Invitrogen),per the manufacturer’s guidelines. Sixteen hours after transfec-tion, the medium was replaced with fresh proliferation mediumand the myoblasts were incubated for a further 24 h. Luciferaseassays were performed using the Dual Luciferase Assay System,
Myostatin-induced DNA Damage in Skeletal Muscle
FEBRUARY 28, 2014 • VOLUME 289 • NUMBER 9 JOURNAL OF BIOLOGICAL CHEMISTRY 5785
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

as per the manufacturer’s protocol (Promega). Relative lucifer-ase activity was measured in triplicate using the FluoroskanAscent Microplate Fluorometer and Luminometer (ThermoFisher Scientific Inc.).
Preparation of Muscle Homogenates for Enzyme Assays—Quadriceps muscle homogenates were made according to apreviously published protocol (6), and total protein concentra-tion was measured by Bradford’s assay (17).
Estimation of Lipid Peroxidation Product—The lipid peroxi-dation product (malonaldehyde) was determined as describedpreviously (20).
Estimation of Superoxide Dismutase and GlutathionePeroxidase—The activity of Superoxide dismutase was esti-mated as described by Sriram et al. (6). Glutathione peroxidaseenzyme assay was performed by the modified method ofRotruck et al. (21) as described previously (6).
Estimation of Reduced Glutathione—Reduced glutathionelevels were determined by the method of Moron et al. (22).
Analysis of Intracellular ROS Production—ROS productionwas assayed in C2C12 myoblasts treated with STZ during pro-liferation as previously described (6) using the fluorescent dye,CM-H2DCFDA (Molecular Probes).
Myoblast Proliferation Assay—Myoblast proliferation assaywas performed as previously described (6) using the methyleneblue photometric end point assay (23).
Immunohistochemistry for REDD1 and OGG1—OCT-em-bedded Tibialis anterior muscles were sectioned and fixed with4% paraformaldehyde in PBS. The immunohistochemistry pro-tocol for REDD1 or OGG1 primary antibodies was followed asper manufacturer’s instructions (Proteintech). Images of thestained sections were taken at 5� magnification using a Leicaupright microscope.
Immunocytochemistry (ICC) for 8-oxo-dG—C2C12 and pri-mary myoblasts were seeded at a density of 15,000 cells/cm2.Next day, proliferation medium with or without STZ1, STZ2,CCM, CMM1, CMM2, and with or without pretreatment withAnt1 was added to the myoblasts and incubated for 48 h. Theimmunostaining for 8-oxo-dG was performed according tomanufacturer’s instructions (Trevigen Inc.). Images (DAPI-blue; 8-oxo-dG-green) were taken at 10� magnification using aLeica upright microscope.
Comet Assay—C2C12 myoblasts were grown and treated asdescribed in the previous section. A single cell suspension wasmade and comet assay was performed as previously described (24).The alkaline lysis method was used to detect the combination ofsingle strand breaks, double strand breaks, and alkali-labile sites inthe DNA, and neutral lysis method was performed to detect onlyDNA double strand breaks. Using propidium iodide (PI), the com-ets were visualized and at least 50 comet images/slide and 3 slides/treatment were examined. Comet image analysis software wasused to quantify various parameters.
Transient Transfection of shRNA to Knockdown Mstn—C2C12 myoblasts were transfected with 4 �g/well of emptyvector control (pGFP-V-RS), scrambled shRNA or Mstn-spe-cific shRNA expression vector (shMstn) (OriGene Technolo-gies, Inc.) using Lipofectamine 2000 (Invitrogen), as perthe manufacturer’s guidelines. Next day, fresh proliferationmedium containing CCM, CMM, STZ, and/or Ant1 was added
for a further 48 h. The myoblasts’ protein lysates were made asdescribed previously (6).
Statistical Analysis—The p value was calculated usingANOVA, and p � 0.05 was considered as significant. Five orseven mice for each treatment were used for various experi-ments. The results are presented as mean � S.E. of three inde-pendent experiments.
RESULTS
STZ Treatment Induced Muscle Atrophy in Mice—STZ treat-ment has been shown to induce hyperglycemia in rodents (25),hence we injected mice with STZ to establish a type 1 diabetesmodel. The results showed maximum induction of ROS, indi-cated by expression of TNF-� (Fig. 1A) and Nox1 (Fig. 1B) andanti-oxidant enzymes (data not shown) on Day 7 in STZ-treated muscles; hence, further experiments were performedusing Day 7 STZ-treated muscles. Furthermore by Day 7, thepercentage loss of Gastrocnemius and Quadriceps muscleweights normalized to body weight were significantly reducedin WT-STZ and Mstn�/�-STZ groups when compared withtheir respective control groups (data not shown).
Histology of Tibialis anterior muscle revealed that musclefiber number was reduced in the WT-STZ and Mstn�/�-STZmuscle as compared with respective controls (Data not shown).The muscle fiber cross-sectional area (CSA) was significantlydecreased in WT-STZ muscle when compared with WT-Cmuscle (Fig. 1C). Specifically, in WT-C muscle, �40% of musclefibers only had an area �2500 �m2; however, in the WT-STZmuscle, �64% were found to have an area �2500 �m2. InMstn�/�-C muscle, �60% of the muscle fibers had an area�2500 �m2, while in the Mstn�/�-STZ muscle, �50% fibershad an area �2500 �m2 (Fig. 1C). These results confirmed thatSTZ treatment led to extensive skeletal muscle atrophy in WTmice and relatively less muscle atrophy in Mstn�/� mice.
Foxa2 Mediated Up-regulation of Mstn in Response to STZ—The expression and levels of Mstn and its downstream targets,p-Smad2/3 ((Fig. 1D (i), (ii), and (iii)) were significantly up-reg-ulated in WT-STZ muscle (lane 2) when compared with WT-Cmuscle (lane 1). To investigate the mechanism involved in STZ-induced Mstn transcription, an in silico analysis was performedon the 1.7 kb upstream sequence of the mouse Mstn gene toidentify various transcription factor binding sites. Thesequence analysis identified a putative Foxa2 binding site (5�-CAAATATTTGTT-3�) within the 1.7 kb sequence of themouse Mstn promoter. Foxa2 has been shown to regulate glu-cose homeostasis and glucose-induced insulin release (26). RT-qPCR and Western blot analysis of Foxa2 indicated that Foxa2mRNA expression and protein level (Fig. 2A (i) and (ii)) wassignificantly up-regulated in WT-STZ muscles (lane 2) andbarely detectable in Mstn�/� muscles even upon STZ treat-ment (lanes 3 and 4). Next, to determine whether high glucoselevels induced by STZ can enhance Foxa2 binding to Mstn pro-moter, electrophoretic mobility shift assay was performed. Asshown in Fig. 2B (i), STZ treatment in WT mice led to increasedFoxa2 binding as indicated by the shifted band (lane 3). Fur-thermore, when nuclear extracts from WT-STZ Biceps femorismuscles were incubated with increasing concentrations ofcompetitor oligos, the disappearance of the shifted band was
Myostatin-induced DNA Damage in Skeletal Muscle
5786 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 289 • NUMBER 9 • FEBRUARY 28, 2014
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

observed (Fig. 2B (ii)- lanes 3 and 4). The specificity of Foxa2binding was confirmed using Foxa2 specific antibody; the resultsshowed a supershift of the Foxa2 specific band (Fig. 2B (ii)-lane 5).ChIP assay further demonstrated enhanced binding of Foxa2 tothe Mstn promoter in HepG2 cells transfected with the Mstn pro-moter (lane 10) when compared with control (lane 9, Fig. 2C).
Myoblasts transfected with 1.7 kb mouse Mstn promoterconstruct showed an �8.0-fold increase in Luciferase activity,when compared with myoblasts transfected with the emptyvectors (Fig. 2D). A further significant increase in Luciferaseactivity was observed in myoblasts transfected with both Foxa2expression vector and 1.7 kb mouse Mstn promoter construct
FIGURE 1. STZ treatment induced skeletal muscle atrophy in mice. Representative graph showing mRNA expression of TNF-� (A) and Nox1 (B) in Gastro-cnemius muscle from C and STZ groups of WT and Mstn�/� mice (Day 7), (*, p � 0.05 and **, p � 0.01 when compared with WT-C muscle, n � 7). C,representative graph showing frequency distribution of Tibialis anterior skeletal muscle fiber cross-sectional area (�m2) in WT-C, WT-STZ, Mstn�/�-C andMstn�/�-STZ mice (n � 7). D, representative graph (i) showing mRNA expression of Mstn and representative Western blot (ii) and densitometric analysis (iii)showing protein levels of Mstn, p-Smad2/3 and Smad2/3 in WT-C and WT-STZ Gastrocnemius muscle (n � 7, *, p � 0.05; **, p � 0.01; ***, p � 0.001; lane 1-WT-C,lane 2-WT-STZ). GAPDH was used as an internal control for equal protein loading on the gel.
Myostatin-induced DNA Damage in Skeletal Muscle
FEBRUARY 28, 2014 • VOLUME 289 • NUMBER 9 JOURNAL OF BIOLOGICAL CHEMISTRY 5787
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Myostatin-induced DNA Damage in Skeletal Muscle
5788 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 289 • NUMBER 9 • FEBRUARY 28, 2014
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

when compared with myoblasts transfected with control vec-tors (Fig. 2D). These data confirm that Foxa2 regulates tran-scription of Mstn gene.
In addition, overexpression of Foxa2 in C2C12 cells led toup-regulated levels of Mstn and p-Smad2/3 and reduced levelsof p-Akt1/2/3 (Figs. 2E (i) and 2F (i)-lane 2). Knockdown ofFoxa2 using Foxa2-siRNA resulted in reduced p-Smad2/3 lev-els, increased p-Akt1/2/3 levels and no change in Mstn levels(lane 2) (Fig. 2, E (ii) and F (ii)). Previously, insulin signaling (27)and Akt phosphorylation (28) has been shown to regulate Foxa2activity. Western blot results showed that the levels of p-Akt1/2/3 and Akt1/2/3 were reduced in WT-STZ muscles (lane 2),when compared with WT-C muscles (lane 1), while the levelswere significantly higher in Mstn�/� muscles (lanes 3 and 4)(Figs. 3A (i) and (ii)). IRS-1 levels were significantly decreased inMstn�/�-STZ mice (lane 4). IGF-1 levels were significantlyreduced in WT-STZ muscles (lane 2) compared with controlswhile the levels were higher in Mstn�/� muscles even uponSTZ treatment (lane 4) (Figs. 3A (i) and (ii)).
Absence of Mstn Abrogated STZ-induced Changes in p63 andREDD1 Signaling—STZ-induced ROS has been implicated inDNA damage, thus we investigated if STZ-induced Mstn cancause DNA damage in skeletal muscle. STZ treatment in WTmice reduced REDD1 and p63 levels and increased OGG1 lev-els, significantly (lane 2, Fig. 3B (i) and (ii)). REDD1 and p63levels were elevated in Mstn�/�-C muscles (lane 3) when com-pared with WT-C muscles (lane 1). Upon STZ treatment inMstn�/� mice (lane 4), no change in REDD1 and OGG1levels was observed when compared with Mstn�/�-C group(lane 3). A decrease in p63 levels was observed in Mstn�/�-STZ muscles (lane 4), which were comparable to WT-Cmuscles (lane 1) (Fig. 3B (i) and (ii)). No significant changewas observed in p53 levels in STZ-treated WT and Mstn�/�
muscles (Fig. 3B (i) and (ii)). Immunohistochemistry analysisof Tibialis anterior muscle revealed that REDD1 staining wasreduced in WT-STZ muscle sections when compared withWT-C sections (Fig. 3C (i)), while the staining was higher inboth Mstn�/�-C and -STZ sections (Fig. 3C (ii)). Only STZ-treated WT muscle showed increased OGG1 staining ascompared with WT-C muscle, Mstn�/�-C and -STZ musclesections (Fig. 3D (i) and (ii)). These results indicated thatSTZ-induced changes in p63/REDD1 signaling are rescuedin the absence of Mstn.
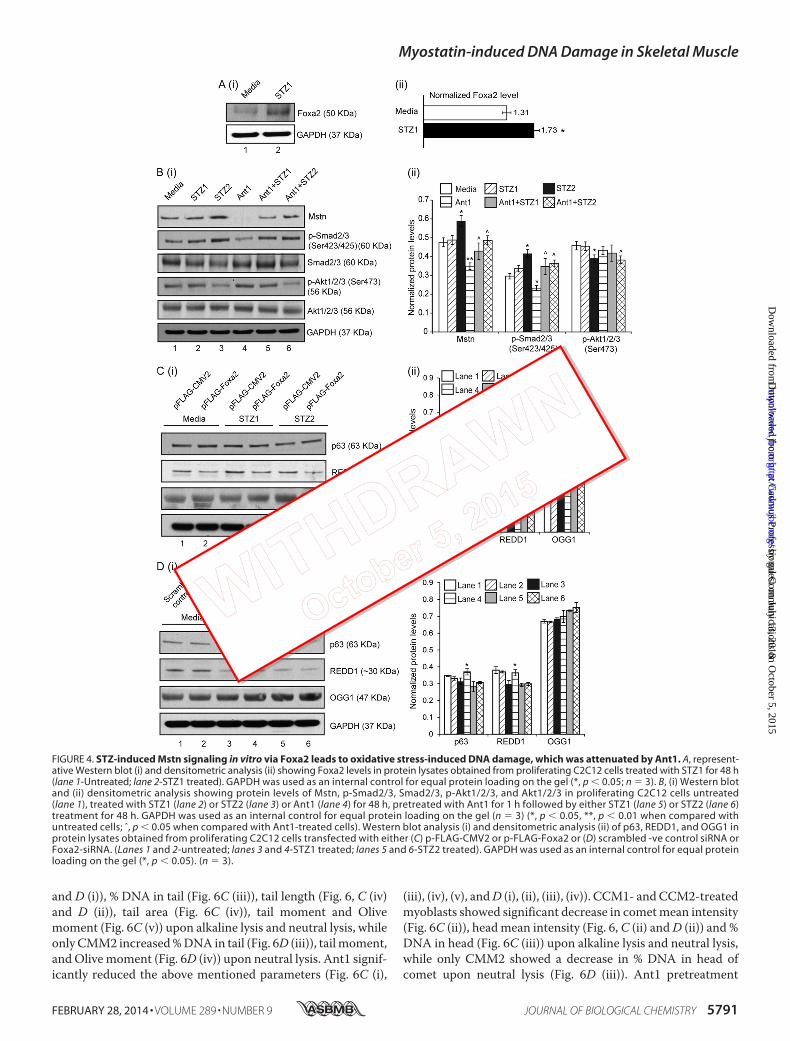
STZ-induced Mstn Signaling in Vitro via Foxa2 Leads toOxidative Stress-induced DNA Damage—STZ (STZ1– 0.25mg/ml) treatment on C2C12 cells resulted in increased ROS
(Fig. 3E) (as previously shown (29)) and Foxa2 levels in proteinlysates (lane 2) (Fig. 4A (i) and (ii)). As shown in Fig. 4B (i) and(ii), STZ treatment (STZ2 - 1 mg/ml) showed increased levels ofMstn (lane 3), while Ant1 (11) treatment alone significantlyinhibited Mstn levels (lane 4). Ant1 treatment along with STZpartially reduced Mstn levels (lanes 5 and 6). Dose-dependentincrease in p-Smad2/3 levels were observed upon STZ treat-ment (lanes 2 and 3), while Ant1 treatment partially rescued theincrease (lanes 4 – 6). p-Akt1/2/3 levels were significantlyreduced upon STZ2 treatment (lane 3) (Fig. 4B (i) and (ii)).
Western blot analysis for p63, REDD1, and OGG1 was also per-formed on protein lysates obtained from STZ-treated C2C12 cellswith gain or loss of Foxa2 expression. As shown in Fig. 4C (i) and(ii), overexpression of Foxa2 reduced the levels of REDD1 in theuntreated (lane 2) and STZ-treated (lanes 4 and 6) cells; whileOGG1 levels were increased upon overexpression of Foxa2 andSTZ2 treatment (lane 6). In agreement with these results, p63 andREDD1 levels were higher in cells transfected with Foxa2-siRNAand treated with STZ1 as compared with control (lanes 3, 4-Fig.4D (i) and (ii)). However, higher concentration of STZ (STZ2)reduced the levels of p63 and REDD1 even in cells transfected withFoxa2-siRNA (lane 6) when compared with Foxa2-siRNA trans-fected and STZ1-treated cells. No significant differences wereobserved in OGG1 levels in Foxa2-siRNA transfected and STZ-treated cells in comparison to control (Fig. 4D).
The effect of STZ or Mstn on DNA damage in C2C12 myoblastswas also evaluated by immunocytochemistry for 8-oxo-dG, a sen-sitive marker of ROS-induced DNA damage. The intensity of8-oxo-dG staining was increased upon STZ1 (Fig. 5A) and CMM1(7 ng) (Fig. 5B) treatment, while Ant1 pre-treatment reduced thestaining intensity. In primary myoblasts isolated from WT andMstn�/� mice, STZ (Fig. 5C) and CMM (Fig. 5D) treatmentincreased DNA damage and Ant1 pre-treatment was able to pro-tect WT myoblasts from DNA damage, even upon treatment withhigher concentrations of STZ (STZ2) or CMM (CMM2–17.5 ng).In Mstn�/� myoblasts, there was no change in the intensity of8-oxo-dG staining upon treatment with two different concentra-tions of STZ (Fig. 5C) or CMM (Fig. 5D). These results indicatedthat STZ and Mstn induced DNA damage in myoblasts that wasattenuated by inhibition of Mstn.
STZ and CMM Cause Single Strand Breaks and DoubleStrand Breaks in DNA in Proliferating Myoblasts—Cometassay was performed to detect single strand breaks and dou-ble strand breaks in DNA. The arrows in Fig. 6A (i) (alkalinelysis) and (ii) (neutral lysis) indicate DNA fragmentation anddamage, as detected by the increased comet tail length and
FIGURE 2. Foxa2 mediated up-regulation of Mstn in response to STZ. A, representative graph (i) showing mRNA expression of Foxa2 and representative Westernblot (ii) showing protein levels of Foxa2 in WT-C, WT-STZ, Mstn�/�-C and Mstn�/�-STZ muscle (**, p � 0.01, ***, p � 0.001 when compared with WT-C muscle, n � 7).B, (i) left panel, representative electrophoretic mobility shift assay gel showing increased Foxa2 binding to the DNA upon STZ treatment as indicated by the shiftedband in lane 3 (lane 1-oligo only, lane 2-WT-C, lane 3-WT-STZ). (ii) Right panel, representative gel showing the disappearance of the shifted band when nuclear extractsof WT-STZ Biceps femoris muscles were incubated with increasing concentrations of competitor oligos (100� and 500�). Supershift of the Foxa2 specific band whenWT-STZ Biceps femoris muscle nuclear extracts were pre-incubated with Foxa2 antibody (lane 1-oligo only, lane 2-WT-STZ, lane 3-with 100� competitor oligo, lane4-with 500� competitor oligo, lane 5-with Foxa2 antibody) (n � 3). C, representative agarose gel image showing the binding of Foxa2 to 1.7 kb murine Mstn promoter(1.7P) (lanes 9 and 10), as assessed by ChIP. The relative amounts of both the control and Mstn promoter in the input were also assessed (lanes 3 and 4). Both no antibody(No Ab) (lanes 5 and 6) and isotype specific IgG (lanes 7 and 8) controls are shown. D, assessment of promoter-luciferase reporter activity, expressed as relativeluminescence units (RLU) in C2C12 myoblasts transfected with either pGL3-basic and pFLAG-CMV2 empty vectors or 1.7 kb mouse Mstn promoter construct (1.7P) andpFLAG-CMV2 or 1.7P and Foxa2 expression vector (pFLAG-Foxa2), together with the control Renilla luciferase vector pRL-TK, (****, p � 0.0001, ˆˆˆˆ, p � 0.0001; n � 3).E, Western blot and (F) densitometric analysis of Foxa2, Mstn, p-Smad2/3, Smad2/3, p-Akt1/2/3, and Akt1/2/3 in protein lysates obtained from proliferating C2C12 cellstransfected with either (i) p-FLAG-CMV2 or p-FLAG-Foxa2 or (ii) scrambled -ve control siRNA or Foxa2-siRNA. GAPDH was used as an internal control for equal proteinloading on the gel (*, p � 0.05; **, p � 0.01; ***, p � 0.001). (n � 3).
Myostatin-induced DNA Damage in Skeletal Muscle
FEBRUARY 28, 2014 • VOLUME 289 • NUMBER 9 JOURNAL OF BIOLOGICAL CHEMISTRY 5789
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

height, in C2C12 myoblasts caused by STZ. Pretreatmentwith Ant1 reduced the comet formation to the level similarto untreated myoblasts (quantitation data not shown).
Similarly, CMM1 and CMM2 induced single strand breaks inDNA, detected by the alkaline lysis method (Fig. 6B (i)). Only
higher concentrations of CMM (CMM2) caused double strandbreaks in DNA, as detected by neutral lysis of cells (Fig. 6B (ii)).Pretreatment with Ant1 reduced the comets formed by CMM1or CMM2 treatment. CMM1 and CMM2 treatment signifi-cantly increased comet length and comet height (Fig. 6, C (i)
FIGURE 3. Absence of Mstn abrogated STZ-induced changes in p63 and REDD1 signaling. A, Western blot analysis (i) and densitometric analysis (ii) ofp-Akt1/2/3, Akt1/2/3, IRS-1 and IGF-1 in WT-C (lane 1), WT-STZ (lane 2), Mstn�/�-C (lane 3) and Mstn�/�-STZ (lane 4) Gastrocnemius muscle protein lysates. *, p �0.05, **, p � 0.01 when compared with WT-C; ˆˆ, p � 0.01 when compared with Mstn�/�-C. GAPDH was used as an internal control for equal protein loading onthe gel (n � 7). B, Western blot analysis (i) and densitometric analysis (ii) of REDD1, OGG1, p53, and p63 protein levels in WT-C (lane 1), WT-STZ (lane 2),Mstn�/�-C (lane 3), and Mstn�/�-STZ (lane 4) Gastrocnemius muscle. GAPDH was used as an internal control for equal protein loading on the gel (n � 5). *, p �0.05, ***, p � 0.001 when compared with WT-C. Immunohistochemistry for REDD1 (C) and OGG1 (D) was performed on cryosections of Tibialis anterior musclesfrom WT-C and WT-STZ (i) and Mstn�/�-C and Mstn�/�-STZ (ii) mice. The fluorescence was viewed under a Leica upright microscope and images were takenat 5� magnification. Increased or decreased fluorescence (green) indicates changes in expression of REDD1 (C) or OGG1 (D); scale bar represents 100 �m (n �3). E, ROS production was measured in proliferating C2C12 myoblasts treated with STZ1 for 48 h in Permanox chamber slides using the CM-H2DCFDAfluorescent probe. The fluorescence was viewed under a Leica upright microscope and images were taken at 10� magnification. Increased fluorescence(green) intensity is directly proportional to increased ROS production; scale bar represents 100 �m (n � 2).
Myostatin-induced DNA Damage in Skeletal Muscle
5790 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 289 • NUMBER 9 • FEBRUARY 28, 2014
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from
and D (i)), % DNA in tail (Fig. 6C (iii)), tail length (Fig. 6, C (iv)and D (ii)), tail area (Fig. 6C (iv)), tail moment and Olivemoment (Fig. 6C (v)) upon alkaline lysis and neutral lysis, whileonly CMM2 increased % DNA in tail (Fig. 6D (iii)), tail moment,and Olive moment (Fig. 6D (iv)) upon neutral lysis. Ant1 signif-icantly reduced the above mentioned parameters (Fig. 6C (i),
(iii), (iv), (v), and D (i), (ii), (iii), (iv)). CCM1- and CCM2-treatedmyoblasts showed significant decrease in comet mean intensity(Fig. 6C (ii)), head mean intensity (Fig. 6, C (ii) and D (ii)) and %DNA in head (Fig. 6C (iii)) upon alkaline lysis and neutral lysis,while only CMM2 showed a decrease in % DNA in head ofcomet upon neutral lysis (Fig. 6D (iii)). Ant1 pretreatment
FIGURE 4. STZ-induced Mstn signaling in vitro via Foxa2 leads to oxidative stress-induced DNA damage, which was attenuated by Ant1. A, represent-ative Western blot (i) and densitometric analysis (ii) showing Foxa2 levels in protein lysates obtained from proliferating C2C12 cells treated with STZ1 for 48 h(lane 1-Untreated; lane 2-STZ1 treated). GAPDH was used as an internal control for equal protein loading on the gel (*, p � 0.05; n � 3). B, (i) Western blotand (ii) densitometric analysis showing protein levels of Mstn, p-Smad2/3, Smad2/3, p-Akt1/2/3, and Akt1/2/3 in proliferating C2C12 cells untreated(lane 1), treated with STZ1 (lane 2) or STZ2 (lane 3) or Ant1 (lane 4) for 48 h, pretreated with Ant1 for 1 h followed by either STZ1 (lane 5) or STZ2 (lane 6)treatment for 48 h. GAPDH was used as an internal control for equal protein loading on the gel (n � 3) (*, p � 0.05, **, p � 0.01 when compared withuntreated cells; ˆ, p � 0.05 when compared with Ant1-treated cells). Western blot analysis (i) and densitometric analysis (ii) of p63, REDD1, and OGG1 inprotein lysates obtained from proliferating C2C12 cells transfected with either (C) p-FLAG-CMV2 or p-FLAG-Foxa2 or (D) scrambled -ve control siRNA orFoxa2-siRNA. (Lanes 1 and 2-untreated; lanes 3 and 4-STZ1 treated; lanes 5 and 6-STZ2 treated). GAPDH was used as an internal control for equal proteinloading on the gel (*, p � 0.05). (n � 3).
Myostatin-induced DNA Damage in Skeletal Muscle
FEBRUARY 28, 2014 • VOLUME 289 • NUMBER 9 JOURNAL OF BIOLOGICAL CHEMISTRY 5791
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

brought the levels of these parameters back to levels similar toCCM-treated myoblasts (Fig. 6, C and D).
STZ Treatment Altered Genes Involved in DNA Repair inVivo—In muscles, Western blot analyses for XRCC1, a proteininvolved in repair of single strand breaks in DNA, and HistoneH2A.X, involved in DNA double strand break repair were per-formed. The results indicated that XRCC1 protein levels weresignificantly reduced in WT-STZ muscles (lane 2), while therewas no change observed in Mstn�/� muscles (lanes 3 and 4),when compared with WT-C muscles (lane 1) (Fig. 7A (i) and(ii)). The level of phosphorylated Histone H2A.X was increasedand total Histone H2A.X was correspondingly decreased inWT-STZ muscles (lane 2) when compared with WT-C muscles(lane 1) (Fig. 7A (i) and (ii)). Mstn�/�-C and Mstn�/�-STZmuscles (lanes 3 and 4, respectively) showed higher levels ofphosphorylated and total Histone H2A.X when compared withWT-C muscles (lane 1) (Fig. 7A (i) and (ii)).
STZ and CMM Alter p63/REDD1 Signaling and DNA RepairGenes in Proliferating Primary Myoblasts—The effect of STZand Mstn on p63 and REDD1 levels and DNA repair genes wasalso analyzed in STZ- (data not shown) or Mstn-treated (Fig. 7B(i) and (ii)) WT and Mstn�/� primary myoblasts. The resultsshow that p63 levels were decreased in CMM1- and CMM2-treated WT myoblasts in a dose-dependent manner (lanes 2and 3), while only CMM2 treatment reduced p63 level inMstn�/� myoblasts (lane 9) (Fig. 7B (i) and (ii)). CMM1 andCMM2 treatment increased OGG1 protein levels (lanes 2 and3), while Ant1 pretreatment rescued this increase (lanes 4 – 6),when compared with CCM-treated WT primary myoblasts(lane 1). In Mstn�/� myoblasts, the increase in OGG1 levels onCMM treatment (lanes 8 and 9) was lesser than CMM-treatedWT myoblasts. CMM2 treatment on WT myoblasts reducedREDD1 and p-Histone H2A.X levels (lane 3), while Ant1 pre-treatment rescued the levels (lanes 4 – 6). CMM2 treatment on
FIGURE 5. STZ-induced DNA damage in proliferating myoblasts is attenuated by antagonizing Mstn. A, immunocytochemistry was performed for8-oxo-dG on proliferating C2C12 myoblasts treated for 48 h with or without STZ1 and with or without pre-treatment of Ant1. The fluorescence for 8-oxo-dG(green) and DAPI (blue) was viewed under a Leica upright microscope and images were taken at 10� magnification. Increased fluorescence is directlyproportional to increased DNA damage; scale bar represents 100 �m (n � 3). B, 8-oxo-dG immunostaining in proliferating C2C12 myoblasts treated for 48 hwith CCM, CMM1, Ant1CCM or Ant1CMM1. Images were taken as mentioned above in A; scale bar represents 100 �m (n � 3). Immunostaining for 8-oxo-dGwas performed on WT and Mstn�/� primary myoblasts treated for 48 h during proliferation with STZ1, STZ2, Ant1 alone, Ant1STZ1, or Ant1STZ2 (C) or withCCM, CMM1, CMM2, Ant1CCM, Ant1CMM1, or Ant1CMM2 (D). Representative images showing an increase or decrease in 8-oxo-dG staining. Images weretaken as mentioned above in A; scale bar represents 100 �m (n � 3).
Myostatin-induced DNA Damage in Skeletal Muscle
5792 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 289 • NUMBER 9 • FEBRUARY 28, 2014
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Myostatin-induced DNA Damage in Skeletal Muscle
FEBRUARY 28, 2014 • VOLUME 289 • NUMBER 9 JOURNAL OF BIOLOGICAL CHEMISTRY 5793
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Mstn�/� myoblasts (lane 9) increased p-Histone H2A.X andtotal H2A.X levels (Fig. 7B (i) and (ii)).
Knockdown of Mstn by shRNA Partially Attenuates STZ- andCMM-induced DNA Damage—C2C12 myoblasts were trans-fected with shMstn and treated with STZ (data not shown) orCMM (Fig. 8A (i) and (ii)). Results showed that indeed Mstnlevels were significantly reduced in shMstn transfected myo-blasts (data not shown). As shown in Fig. 8A (i) and (ii), p63levels were reduced upon CMM treatment in a dose-dependentmanner (lanes 1–9). REDD1 levels were reduced in CMM-treated empty vector (lanes 2 and 3) and scrambled shRNA(lanes 5 and 6) transfected myoblasts while only CMM2 showeda decrease in REDD1 levels in shMstn-transfected myoblasts(lane 9) (Fig. 8A (i) and (ii)). CMM1 treatment decreased thelevels of p-Histone H2A.X (lanes 1–9), while CMM2 treatmentincreased the levels of p-Histone H2A.X and total H2A.X inshMstn-transfected myoblasts (lanes 7–9) (Fig. 8A (i) and (ii)).
DISCUSSION
Type 1 diabetes is characterized by lack of insulin, hypergly-cemia and oxidative stress. In this report, we elucidate themechanisms by which STZ treatment leads to an increase inMstn expression through activation of Foxa2 transcription fac-tor. Enhanced Mstn contributes to ROS that causes DNA dam-age mediated by p63/REDD1 in the muscle of diabetic mice.
Over the recent years, various studies have shown that Mstnexpression is increased in STZ treated mice (7–9), however themechanism behind the increase in Mstn levels was not identi-fied. Since, our results presented here showed an increase inMstn RNA and protein levels, we surmised that Mstn could beregulated at the transcriptional level during type 1 diabetes.Indeed, in silico analysis of Mstn 5�-upregulatory sequencesrevealed the presence of a binding site for Foxa2, a transcriptionfactor regulated by insulin (30). Previous studies demonstratedthat in the absence of insulin, Foxa2 activity and level wasenhanced in the liver. Foxa2 has been reported to regulate genesimplicated in glucose (26) and lipid metabolism in the liver (28).Our results indicate that in the muscle from STZ-treated WTmice, the Foxa2 expression and protein was up-regulated (Fig.2A (i) and (ii), respectively). Further confirmation of increasedFoxa2 levels and activity came from the electrophoretic mobil-ity shift assay and Mstn promoter-reporter assays. The ChIPresults clearly indicated the binding of endogenous Foxa2 onthe Mstn promoter transfected in HepG2 cells. Taken together,these results for the first time demonstrate the activation ofFoxa2-induced Mstn expression. Furthermore, in vitro experi-ments of overexpression and knockdown of Foxa2 confirm thatFoxa2 mediated the up-regulation of Mstn and its downstreamsignaling.
Previously it has been reported that insulin signaling inhibitsFoxa2 activity by phosphorylation mediated by Akt (28, 30).Accordingly, Akt phosphorylation was down regulated uponSTZ treatment in WT mice (Fig. 3A (i) and (ii), lanes 1 and 2)leading to higher Foxa2 and Mstn levels. These findings notonly indicate that one of the transcriptional targets of Foxa2 isMstn but also extend the role of Foxa2 in skeletal muscle ofdiabetic mice. However, despite developing overt diabetes, thelevels of Foxa2 were significantly lower in Mstn�/� muscle evenafter treatment with STZ, which we propose could be due toenhanced Akt signaling in these mice (Fig. 3A (i) and (ii), lanes3 and 4). Furthermore, Foxa2 has been reported to auto regulateits expression through a positive feedback loop (31). Such anautoregulatory mechanism would help maintain increased lev-els of Foxa2 in STZ-treated WT mice and lower levels inMstn�/� mice.
In line with earlier findings (32, 33, 34), increased ROS wasobserved (and thus Anti-oxidant enzymes) in skeletal musclesof WT diabetic mice via TNF-�, Nox1 (Fig. 1, A and B) and lipidperoxidation (data not shown). However, our results revealedno change in these parameters in Mstn�/�-STZ mice (Fig. 1, Aand B) indicating that in the absence of Mstn these miceresisted the induction of ROS by STZ. ROS are potent mole-cules that are toxic to the cells; if uncontrolled, lead to proteindegradation as well as DNA damage. It is evident that STZ-induced ROS can lead to DNA damage (35, 36). In this commu-nication, we show for the first time that significantly higherlevels of Mstn-induced ROS led to DNA damage and inhibitedDNA repair systems in the skeletal muscle. Earlier, it has beensuggested that STZ also has a direct effect on myoblasts mani-fested by an up-regulation in ROS and inhibition of myoblastproliferation (29). Regardless of direct or indirect effect, wenoticed not only an increase in ROS but also Foxa2 in STZ-treated myoblasts thus recapitulating in vivo effects of STZ onmuscle.
Earlier, Andican and Bursac reported oxidative DNA damageduring STZ-induced type 1 diabetes with a concomitantincrease in oxidized DNA bases like 8-oxo-dG in the liver ofSTZ treated diabetic rats (37). Furthermore, 8-oxo-dG contentwas found to be higher in the skeletal muscle of diabeticpatients (38). Similarly, occurrence of higher levels of OGG1has been reported in pancreas of diabetic patients that was cor-related with DNA damage (39). Consistently, our results dem-onstrate that both STZ and Mstn can cause an increase in8-oxo-dG and OGG1 indicative of DNA damage and repairrespectively and inhibition of Mstn partially reversed thesechanges.
FIGURE 6. STZ or CMM treatment leads to single strand breaks and double strand breaks in DNA in proliferating myoblasts. A, comet assay wasperformed by alkaline lysis method (i) and neutral lysis method (ii) on C2C12 myoblasts treated with STZ1, Ant1, or Ant1STZ1 for 48 h during proliferation.Representative images showing the head and tail of comets detected by PI stain during various treatments. DNA fragmentation is observed by the formationof tail in the comet as indicated by arrows. Comet images were taken using a Leica upright microscope (n � 3). B, comet assay was performed by alkaline lysismethod (i) and neutral lysis method (ii) on C2C12 myoblasts treated with CCM, CMM1, CMM2, Ant1CCM, Ant1CMM1, or Ant1CMM2 for 48 h duringproliferation. Representative images showing the head and tail of comets detected by PI stain during various treatments. DNA fragmentation is observed bythe formation of tail in the comet as indicated by arrows. Comet images were taken using a Leica upright microscope. Quantitative analysis of variousparameters of comet assay following alkaline lysis (C) and neutral lysis (D); comet length and comet height (C i) and (D i), comet mean intensity (C ii), head meanintensity (C ii) and (D ii), % DNA in head and % DNA in tail (C iii) and (D iii), tail length (C iv) and (D ii), tail area (C iv), tail moment and olive moment (C v) and (Div). *, p � 0.05, **, p � 0.01, ***, p � 0.001, ****, p � 0.0001 when compared with CCM-treated myoblasts; ˆ, p � 0.05, ˆˆ, p � 0.01, ˆˆˆ, p � 0.001, ˆˆˆˆ, p � 0.0001when compared with CMM1- or CMM2-treated myoblasts (n � 3).
Myostatin-induced DNA Damage in Skeletal Muscle
5794 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 289 • NUMBER 9 • FEBRUARY 28, 2014
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Myostatin-induced DNA Damage in Skeletal Muscle
FEBRUARY 28, 2014 • VOLUME 289 • NUMBER 9 JOURNAL OF BIOLOGICAL CHEMISTRY 5795
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

REDD1 is known to regulate cellular ROS through p63 andhence plays an important role in stress response and modula-tion of growth factors (4). Here, our results have establishedMstn-mediated down-regulation of REDD1 and p63 levels viaROS production. However, recently, Hulmi et al. reported anincrease in p63 and REDD1 levels in STZ-treated mice at week1 of STZ treatment plausibly due to higher dose of STZ (180mg/kg) and/or the type of muscles used in the study (8). Ourresults indicated that the basal levels of REDD1 and p63 werehigher in Mstn�/� mice probably in part due to higher levels ofAMP-activated protein kinase (AMPK) in these mice (40)
because AMPK was shown to stimulate REDD1 in vitro (41).Another reason could be due to the elevated IGF-1 signaling inMstn�/� mice, since IGF-1 treatment was reported to up-reg-ulate the protein levels of REDD1 in muscle (42). Nevertheless,deficiency of p63 is associated with inefficient DNA repair (43)as well as with reduced transcription of REDD1 (4). Consis-tently, in vitro experiments confirmed Foxa2-mediated up-reg-ulation of Mstn upon STZ treatment leading to DNA damagevia p63/REDD1 signaling (Fig. 4, C and D).
At the cellular level, both 8-oxo-dG staining and comet assayshowed that both concentrations of CMM were able to induceDNA single strand breaks, while only a higher concentration ofCMM (CMM2) was able to induce double strand breaks inDNA. Furthermore, XRCC1, involved in single strand breakrepair, was decreased in WT-STZ muscle (Fig. 7A (i) and (ii))indicating that in the diabetic condition due to enhanced ROS,an increase in DNA single strand break would occur and therepair would be impaired due to lower levels of the repairenzyme. XRCC1 is a critical enzyme involved in various stagesof DNA repair via its interactions with other repair proteins. Anincrease in ROS is known to down-regulate XRCC1 that resultsin inefficient single strand break repair and build-up of repairintermediate products (44). In the Mstn�/� mice, the levels ofXRCC1 remain unchanged even after STZ treatment suggest-ing that single strand break repair mechanism was not compro-mised in these muscles.
Histone H2A.X, involved in double strand break repair, wasactivated upon STZ treatment in WT mice thus indicating theresponse of the cell survival system against the DNA damagedue to increased oxidative stress in these mice. The basal levelsof both H2A.X and p-H2A.X in Mstn�/� muscles were higherthan WT muscles, which remained unaltered even after STZtreatment. It is difficult to predict the significance of this resulthowever a recent report by Turinetto et al. suggests a role ofp-H2A.X in self-renewal of mouse embryonic and induced plu-ripotent stem cells that is independent of DNA damageresponse function (45). Overall, our findings suggest that dele-tion/inhibition of Mstn abrogates DNA damage in the diabeticmuscle/cells.
Our results showed that STZ administration in Mstn�/�
mice resulted in high glucose levels and greater decrease inbody weights initially when compared with the WT mice (datanot shown). This anomaly could be due to higher gluconeogen-esis in these mice. In fact, the expression of genes involved ingluconeogenesis like PEPCK and G6P was elevated in theMstn�/� mice relative to the WT mice (data not shown), whichis in agreement with Wang et al. (46). As demonstrated previ-ously, Mstn�/� mice have reduced amount of adipose tissue(47) indicating that muscle would be the predominant source toprovide precursors for the enhanced gluconeogenesis following
FIGURE 7. STZ treatment altered genes involved in DNA repair in vivo; changes in p63/REDD1 signaling and DNA repair genes occur in proliferatingprimary myoblasts upon CMM treatment. A, Western blot analysis (i) and densitometric analysis (ii) of XRCC1, p-Histone H2A.X and H2A.X in WT-C (lane 1),WT-STZ (lane 2), Mstn�/�-C (lane 3), and Mstn�/�-STZ (lane 4) Gastrocnemius muscle. GAPDH was used as loading control on the gel (n � 5). **, p � 0.01 whencompared with WT-C. B, representative Western blot (i) and densitometric analysis (ii) showing protein levels of p63, REDD1, OGG1, p-Histone H2A.X, and H2A.Xin 48 h proliferating WT (lanes 1– 6) and Mstn�/� (lanes 7–9) primary myoblasts; lanes 1 and 7-CCM-treated, lanes 2 and 8-CMM1-treated, lanes 3 and 9-CMM2-treated, lane 4-Ant1-treated, pretreated for 1 h with Ant1 followed by CMM1 (lane 5) or CMM2 (lane 6) treatment. GAPDH was used as an internal control forequal protein loading on the gel (*, p � 0.05, **, p � 0.01 when compared with WT-CCM-treated cells; ˆ, p � 0.05 when compared with Mstn�/�-CCM-treatedcells) (n � 3).
FIGURE 8. Knockdown of Mstn by shRNA partially attenuated CMM-in-duced DNA damage. A, representative Western blot (i) and densitometricanalysis (ii) showing p63, REDD1, p-Histone H2A.X and H2A.X protein levels in48 h proliferating C2C12 myoblasts transfected with empty vector control(pGFP-V-RS) (lanes 1–3), scrambled shRNA (lanes 4 – 6), or shMstn vector (lanes7–9) and treated with two different concentrations of CMM; lanes 1, 4, 7-CCM-treated, lanes 2, 5, 8-CMM1-treated, lanes 3, 6, 9-CMM2-treated. GAPDH wasused as internal loading control on the gel (*, p � 0.05, **, p � 0.01 whencompared with empty vector control transfected-CCM-treated cells; ˆ, p �0.05 when compared with shMstn-transfected-CCM-treated cells) (n � 3).
Myostatin-induced DNA Damage in Skeletal Muscle
5796 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 289 • NUMBER 9 • FEBRUARY 28, 2014
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

STZ treatment, thus accounting for the increased muscle lossobserved in Mstn�/� mice. However, even though Mstn�/�
mice exhibit higher glucose levels upon STZ treatment, appre-ciable DNA damage was not observed in these mice. The inac-tivation/inhibition of Mstn was not able to improve the primarydefect of type 1 diabetes but was able to rescue skeletal musclefrom oxidative stress-induced DNA damage to a certain extent.
In summary, our results illustrate the mechanism of Mstnregulation in type 1 diabetes and Mstn-mediated DNA damagein skeletal muscle. Our findings also indicate that inhibition ofMstn could be an effective preventive measure to mitigate DNAdamage in diabetic muscle.
Acknowledgments—We thank Prof. Se-Jin Lee (The Johns HopkinsUniversity) for providing Mstn�/� heterozygous mice. We are gratefulto Dr. Xu Lin (Fujian Medical University, PR China) for providing uswith the Foxa2 expression vector (pFLAG-Foxa2). We are also thank-ful to Kelvin Tan Suan Liang, Isuru W. Wijesoma, and KottaiswamyAmuthavalli for help.
REFERENCES1. Su, H., Velly, A. M., Salah, M. H., Benarroch, M., Trifiro, M., Schipper,
H. M., and Gornitsky, M. (2012) Altered redox homeostasis in humandiabetes saliva. J. Oral Path. Med. 41, 235–241
2. Clancy, S. (2008) DNA damage & repair: mechanisms for maintainingDNA integrity. Nature Education 1, 103
3. Pácal, L., Varvarovská, J., Rusavý, Z., Lacigová, S., Stetina, R., Racek, J.,Pomahacová, R., Tanhäuserová, V., and Kanková, K. (2011) Parameters ofoxidative stress, DNA damage and DNA repair in type 1 and type 2 dia-betes mellitus. Arch. Physiol. Biochem. 117, 222–230
4. Ellisen, L. W., Ramsayer, K. D., Johannessen, C. M., Yang, A., Beppu, H.,Minda, K., Oliner, J. D., McKeon, F., and Haber, D. A. (2002) REDD1, aDevelopmentally Regulated Transcriptional Target of p63 and p53, Linksp63 to Regulation of Reactive Oxygen Species. Mol. Cell 10, 995–1005
5. Favier, F. B., Costes, F., Defour, A., Bonnefoy, R., Lefai, E., Baugé, S., Pein-nequin, A., Benoit, H., and Freyssenet, D. (2010) Downregulation of Akt/mammalian target of rapamycin pathway in skeletal muscle is associatedwith increased REDD1 expression in response to chronic hypoxia. Am. J.Physiol. 298, R1659 –R1666
6. Sriram, S., Subramanian, S., Sathiakumar, D., Venkatesh, R., Salerno,M. S., McFarlane, C. D., Kambadur, R., and Sharma, M. (2011) Modulationof reactive oxygen species in skeletal muscle by myostatin is mediatedthrough NF-�B. Aging Cell 10, 931–948
7. Chen, Y., Cao, L., Ye, J., and Zhu, D. (2009) Upregulation of myostatin geneexpression in streptozotocin-induced type 1 diabetes mice is attenuatedby insulin. Biochem. Biophys. Res. Commun. 388, 112–116
8. Hulmi, J. J., Silvennoinen, M., Lehti, M., Kivelä, R., and Kainulainen, H.(2012) Altered REDD1, myostatin, and Akt/mTOR/FoxO/MAPK signal-ing in streptozotocin-induced diabetic muscle atrophy. Am. J. Physiol.302, E307–E315
9. Dutra, D. B., Bueno, P. G., Silva, R. N., Nakahara, N. H., Selistre-Araújo,H. S., Nonaka, K. O., and Leal, A. M. O. (2012) Expression of myostatin,myostatin receptors and follistatin in diabetic rats submitted to exercise.Clin. Exp. Pharmacol. Physiol. 39, 417– 422
10. Zimmers, T. A., Davies, M. V., Koniaris, L. G., Haynes, P., Esquela, A. F.,Tomkinson, K. N., McPherron, A. C., Wolfman, N. M., and Lee, S. J. (2002)Induction of Cachexia in Mice by Systemically Administered Myostatin.Science 296, 1486 –1488
11. Siriett, V., Salerno, M. S., Berry, C., Nicholas, G., Bower, R., Kambadur, R.,and Sharma, M. (2007) Antagonism of Myostatin Enhances Muscle Re-generation During Sarcopenia. Mol. Ther. 15, 1463–1470
12. Ge, X., Vajjala, A., McFarlane, C., Wahli, W., Sharma, M., and Kambadur,R. (2012) Lack of Smad3 signaling leads to impaired skeletal muscle regen-eration. Am. J. Physiol. 303, E90 –E102
13. Yaffe, D., and Saxel, O. R. A. (1977) Serial passaging and differentiation ofmyogenic cells isolated from dystrophic mouse muscle. Nature 270,725–727
14. Partridge, T. A. (1997) Tissue culture of Skeletal Muscle. Methods Mol.Biol. 75, 131–144
15. McCroskery, S., Thomas, M., Maxwell, L., Sharma, M., and Kambadur, R.(2003) Myostatin negatively regulates satellite cell activation and self-re-newal. J. Cell Biol. 162, 1135–1147
16. Ye, J., Cippitelli, M., Dorman, L., Ortaldo, J., and Young, H. (1996) Thenuclear factor YY1 suppresses the human gamma interferon promoterthrough two mechanisms: inhibition of AP1 binding and activation of asilencer element. Mol. Cell Biol. 16, 4744 – 4753
17. Bradford, M. M. (1976) A rapid and sensitive method for the quantitationof microgram quantities of protein utilizing the principle of protein-dyebinding. Anal. Biochem. 72, 248 –254
18. Salerno, M. S., Thomas, M., Forbes, D., Watson, T., Kambadur, R., andSharma, M. (2004) Molecular analysis of fiber type-specific expression ofmurine myostatin promoter. Am. J. Physiol. 287, C1031–C1040
19. Wu, Y.-l., Peng, X.-e., Wang, D., Chen, W.-n., and Lin, X. (2012). Humanliver fatty acid binding protein (hFABP1) gene is regulated by liver-en-riched transcription factors HNF3� and C/EBP�. Biochimie 94, 384 –392
20. Ohkawa, H., Ohishi, N., Yagi, K. (1979) Assay for lipid peroxides in animaltissues by thiobarbituric acid reaction. Anal. Biochem. 95, 351–358
21. Rotruck, J. T., Pope, A. L., Ganther, H. E., Swanson, A. B., Hafeman, D. G.,and Hoekstra, W. G. (1973) Selenium: Biochemical Role as a Componentof Glutathione Peroxidase. Science 179, 588 –590
22. Moron, M. S., Depierre, J. W., and Mannervik, B. (1979) Levels of gluta-thione, glutathione reductase and glutathione S-transferase activities inrat lung and liver. Biochim. Biophys. Acta 582, 67–78
23. Oliver, M., Harrison, N., Bishop, J., Cole, P., and Laurent, G. (1989) A rapidand convenient assay for counting cells cultured in microwell plates: ap-plication for assessment of growth factors. J. Cell Sci. 92, 513–518
24. Olive, P. L., and Banath, J. P. (2006) The comet assay: a method to measureDNA damage in individual cells. Nat. Protocols 1, 23–29
25. West, E., Simon, O. R., and Morrison, E. Y. (1996) Streptozotocin alterspancreatic beta-cell responsiveness to glucose within six hours of injec-tion into rats. West Indian Med. J. 45, 60 – 62
26. Wang, H., Gauthier, B. R., Hagenfeldt-Johansson, K. A., Iezzi, M., andWollheim, C. B. (2002) Foxa2 (HNF3�) Controls Multiple Genes Impli-cated in Metabolism-Secretion Coupling of Glucose-induced Insulin Re-lease. J. Biol. Chem. 277, 17564 –17570
27. Puigserver, P., and Rodgers, J. T. (2006) Foxa2, a novel transciptional reg-ulator of insulin sensitivity. Nat. Med. 12, 38 –39
28. Wolfrum, C., Asilmaz, E., Luca, E., Friedman, J. M., and Stoffel, M. (2004)Foxa2 regulates lipid metabolism and ketogenesis in the liver during fast-ing and in diabetes. Nature 432, 1027–1032
29. Johnston, A. P. W., Campbell, J. E., Found, J. G., Riddell, M. C., and Hawke,T. J. (2007) Streptozotocin induces G2 arrest in skeletal muscle myoblastsand impairs muscle growth in vivo. Am. J. Physiol. 292, C1033–C1040
30. Wolfrum, C., Besser, D., Luca, E., and Stoffel, M. (2003) Insulin regulatesthe activity of forkhead transcription factor HNF-3�/Foxa-2 by Akt-me-diated phosphorylation and nuclear/cyotsolic localization. Proc. Natl.Acad. Sci. U.S.A. 100, 11624 –11629
31. Bochkis, I. M., Schug, J., Rubins, N. E., Chopra, A. R., O’Malley B. W., andKaestner, K. H. (2009) Foxa2-dependent hepatic gene regulatory networksdepend on physiological state. Physiol. Genomics 38, 186 –195
32. Aragno, M., Mastrocola, R., Catalano, M. G., Brignardello, E., Danni, O.,and Boccuzzi, G. (2004) Oxidative Stress Impairs Skeletal Muscle Repairin Diabetic Rats. Diabetes 53, 1082–1088
33. Mastrocola, R., Reffo, P., Penna, F., Tomasinelli, C. E., Boccuzzi, G., Bac-cino, F. M., Aragno, M., and Costelli, P. (2008) Muscle wasting in diabeticand in tumor-bearing rats: Role of oxidative stress. Free Rad. Biol. Med. 44,584 –593
34. Luo, M., Guan, X., Luczak, E. D., Lang, D., Kutschke, W., Gao, Z., Yang, J.,Glynn, P., Sossalla, S., Swaminathan, P. D., Weiss, R. M., Yang, B., Rokita,A. G., Maier, L. S., Efimov, I. R., Hund, T. J., and Anderson, M. E. (2013)Diabetes increases mortality after myocardial infarction by oxidizingCaMKII. J. Clin. Investig. 123, 1262–1274
Myostatin-induced DNA Damage in Skeletal Muscle
FEBRUARY 28, 2014 • VOLUME 289 • NUMBER 9 JOURNAL OF BIOLOGICAL CHEMISTRY 5797
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

35. Imaeda, A., Kaneko, T., Aoki, T., Kondo, Y., Nakamura, N., Nagase, H.,and Yoshikawa, T. (2002) Antioxidative effects of fluvastatin and its me-tabolites against DNA damage in streptozotocin-treated mice. FoodChem. Toxicol. 40, 1415–1422
36. Blasiak, J., Sikora, A., Wozniak, K., and Drzewoski, J. (2004) Genotoxicityof streptozotocin in normal and cancer cells and its modulation by freeradical scavengers. Cell Biol. Toxicol. 20, 83–96
37. Andican, G., and Burçak, G. (2005) Oxidative Damage to Nuclear DNA inStreptozotocin-Diabetic Rat Liver. Clin. Exp. Pharmacol. Physiol. 32,663– 666
38. Suzuki, S., Hinokio, Y., Komatu, K., Ohtomo, M., Onoda, M., Hirai, S.,Hirai, M, Hirai, A., Chiba, M., Kasuga, S., Akai, H., and Toyota, T. (1999)Oxidative damage to mitochondrial DNA and its relationship to diabeticcomplications. Diabetes Res. Clin. Pract. 45, 161–168
39. Tyrberg, B., Anachkov, K., Dib, S., Wang-Rodriguez, J., Yoon, K., Levine, F.(2002) Islet expression of the DNA repair enzyme 8-oxoguanosine DNAglycosylase (OGG1) in human type 2 diabetes. BMC Endocrine Disorders2, 2
40. Zhang, C., McFarlane, C., Lokireddy, S., Bonala, S., Ge, X., Masuda, S.,Gluckman, P., Sharma, M., and Kambadur, R. (2011) Myostatin-deficientmice exhibit reduced insulin resistance through activating the AMP-acti-vated protein kinase signalling pathway. Diabetologia 54, 1491–1501
41. Sofer, A., Lei, K., Johannessen, C. M., and Ellisen, L. W. (2005) Regulationof mTOR and Cell Growth in Response to Energy Stress by REDD1. Mol.
Cell. Biol. 25, 5834 –584542. Frost, R. A., Huber, D., Pruznak, A., and Lang, C. H. (2009) Regulation of
REDD1 by insulin-like growth factor-I in skeletal muscle and myotubes.J. Cell. Biochem. 108, 1192–1202
43. Lin, Y.-L., Sengupta, S., Gurdziel, K., Bell, G. W., Jacks, T., and Flores, E. R.(2009). p63 and p73 Transcriptionally Regulate Genes Involved in DNARepair. PLoS Genet 5, e1000680
44. Narciso, L., Fortini, P., Pajalunga, D., Franchitto, A., Liu, P., Degan, P.,Frechet, M., Demple, B., Crescenzi, M., and Dogliotti, E. (2007) Terminallydifferentiated muscle cells are defective in base excision DNA repair andhypersensitive to oxygen injury. Proc. Natl. Acad. Sci. U.S.A. 104,17010 –17015
45. Turinetto, V., Orlando, L., Sanchez-Ripoll, Y., Kumpfmueller, B., Storm,M. P., Porcedda, P., Minieri, V., Saviozzi, S., Accomasso, L., Cibrario Roc-chietti, E., Moorwood, K., Circosta, P., Cignetti, A., Welham, M. J., andGiachino, C. (2012) High Basal �H2AX Levels Sustain Self-Renewal ofMouse Embryonic and Induced Pluripotent Stem Cells. Stem Cells 30,1414 –1423
46. Wang, Q., Guo, T., McPherron, A. (2011) Inhibition of myostatin signal-ing increases glucose in insulin-deficient diabetic mice. BMC Proc. 6, P52
47. Zhang, C., Tan, C. K., McFarlane, C., Sharma, M., Tan, N. S., and Kamba-dur, R. (2012) Myostatin-null mice exhibit delayed skin wound healingthrough the blockade of transforming growth factor-� signaling bydecorin. Am. J. Physiol. 302, C1213-C1225
Myostatin-induced DNA Damage in Skeletal Muscle
5798 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 289 • NUMBER 9 • FEBRUARY 28, 2014
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Mridula SharmaMonica Senna Salerno, Ravi Kambadur andPrasanna Kumar Juvvuna, Craig McFarlane, Sandhya Sriram, Subha Subramanian, Type 1 Diabetic MiceSkeletal Muscle of Streptozotocin-induced Myostatin Induces DNA Damage inCell Biology:
doi: 10.1074/jbc.M113.483115 originally published online January 14, 20142014, 289:5784-5798.J. Biol. Chem.
10.1074/jbc.M113.483115Access the most updated version of this article at doi:
.JBC Affinity SitesFind articles, minireviews, Reflections and Classics on similar topics on the
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/289/9/5784.full.html#ref-list-1
This article cites 47 references, 11 of which can be accessed free at
at Cadm
us Professional Com
munications on O
ctober 5, 2015http://w
ww
.jbc.org/D
ownloaded from
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Monica Senna Salerno, Ravi Kambadur and Mridula SharmaSandhya Sriram, Subha Subramanian, Prasanna Kumar Juvvuna, Craig McFarlane,
Type 1 Diabetic MiceMyostatin Induces DNA Damage in Skeletal Muscle of Streptozotocin-induced
doi: 10.1074/jbc.M113.483115 originally published online January 14, 20142014, 289:5784-5798.J. Biol. Chem.
10.1074/jbc.M113.483115Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/289/9/5784.full.html#ref-list-1
This article cites 47 references, 10 of which can be accessed free at
by guest on July 13, 2018http://w
ww
.jbc.org/D
ownloaded from





























