Embed Size (px)
Citation preview

Self-assembly of metal nanostructureson binary alloy surfacesT. Dugueta,b, Yong Hanc, Chad Yuena,b, Dapeng Jinga, Barış Ünald,b, J. W. Evanse,f,b,1, and P. A. Thiela,d,b
aDepartments of Chemistry; dMaterials Science and Engineering; ePhysics and Astronomy; and fMathematics cInstitute of Physical Research andTechnology; and bAmes Laboratory–US Department of Energy (USDOE), Iowa State University, Ames, IA 50011
Edited by Charles T. Campbell, University of Washington, Seattle, WA, and accepted by the Editorial Board October 12, 2010 (received for review June 10, 2010)
Deposition of metals on binary alloy surfaces offers new possibili-ties for guiding the formation of functional metal nanostructures.This idea is explored with scanning tunneling microscopy studiesand atomistic-level analysis and modeling of nonequilibrium islandformation. For Au/NiAl(110), complex monolayer structures arefound and compared with the simple fcc(110) bilayer structurerecently observed for Ag/NiAl(110). We also consider a morecomplex codeposition system, ðNiþ AlÞ∕NiAlð110Þ, which offers theopportunity for fundamental studies of self-growth of alloysincluding deviations for equilibrium ordering. A general multisitelattice-gasmodel framework enables analysis of structure selectionand morphological evolution in these systems.
deposition ∣ epitaxial growth ∣ STM ∣ DFT ∣ KMC
Self-assembly involves the autonomous organization of compo-nents into structures (1). This process requires mobility of
aggregating components, and usually occurs on smooth surfacesor in fluids. Some degree of relaxation in the aggregated state istypically also operative. Self-assembly can be manifested in eithercomplex equilibrium structures, e.g., reflecting competing interac-tions, or in far-from-equilibrium growth structures (1). The lattercan be very different and more diverse than the equilibrium forms(2). Significantly, self-assembly provides a practical strategy forcreating ensembles of nanostructures with unique size and shapedependent properties, a central goal of nanotechnology.
A broad range of systems self-assemble, spanning hard and softmatter, with a wide variety of interactions and component sizes.We consider the formation of metal nanostructures motivatedby applications ranging from catalysis to plasmonics (3–5). Ourspecific focus is vapor deposition of metal atoms on single-crystalmetal surfaces under the well controlled conditions of ultrahighvacuum (UHV). This process leads to the self-assembly of metalnanostructures and growth of epitaxial metal films. Here, thenonaggregated components are rapidly diffusing adsorbed atoms(adatoms) which assemble into islands. Relaxation in the aggre-gated state can be achieved via diffusion of adatoms along islandedges or via detachment-reattachment. Understanding theseprocesses on the atomic scale facilitates guided formation offunctional nanostructures with tailored morphologies and (foralloys) compositions. Ideally, control over formation allows tun-ing of desired properties, e.g., for heterogeneous catalysis (6).
Despite the complexity of self-assembly processes, significantadvances are being made in the development of predictive mod-els even in soft material systems (7). Our focus on epitaxialgrowth on perfect single-crystal surfaces (hard materials) underUHV has a special advantage in facilitating extremely detailedand realistic atomistic-level modeling. Localization of adatoms toa periodic array of adsorption sites enables the use of lattice-gas(LG) models for which nonequilibrium evolution can be effi-ciently analyzed on the appropriate time- and length-scales viakinetic Monte Carlo (KMC) simulation (8, 9). Critical energeticinput parameters (adatom adsorption and interaction energies,and diffusion barriers) can be obtained or at least guided byab initio density functional theory (DFT) analysis.
Since the early 1990’s, there have been major advances inunderstanding the rich variety of far-from-equilibrium morphol-ogies observed in “simple” metal homoepitaxial growth (A on A)(8, 9). Of most relevance there was the observation for submono-layer growth of diverse two-dimensional island growth shapes,which included dendrites and fractals (reflecting inhibited edgediffusion) as well as compact structures. Island coalescence andmultilayer growth at higher coverages are also well understood.For metal heteroepitaxial growth (A on B) with lowmisfit, behaviorcan in fact be quite similar to homoepitaxy, particularly withregard to two-dimensional island growth shapes. However, therelative strengths of chemical interactions (A-A vs. A-B) as wellas strain due to lattice-mismatch (of A and B) generally controlthe multilayer growth mode, e.g., smooth vs. rough, which istraditionally assessed using equilibrium concepts (10). Recentstudies have revealed other growth modes, e.g., “electronicgrowth” of height-selected islands reflecting “quantum size ef-fects” (QSE) of electrons confined in the metal overlayer (11, 12).
For more complex metal heteroepitaxial systems with alloying,a diverse structural and compositional phase-space of island andadlayer configurations is possible. Heteroepitaxial growth of Aon B with intermixing produces ðA þ BÞ∕B binary alloy surfacestructures which can display stress domain patterns (13), unusualgrowth structures (14), and have applications for catalysis (15).Studies of heteroepitaxial code-position A þ B on C (16) haveexplored near-equilibrated patterns (e.g., stripes or dropletsanalogous to diblock copolymer systems) reflecting an interplaybetween chemical interactions and strain, as well as fundamentalissues such as the interplay between nonequilibrium AB-orderingand growth (17).
The focus of this contribution is the exploration ofmetal deposi-tion on a binary alloy surface. Such systems offer new opportunitiesand possibilities for guiding the formation ofmetal nanostructuresand thin films with desirable properties. Specifically, we analyzedeposition ofAu onNiAl(110), and compare its behavior with thatof Ag/NiAl(110). In addition, this paper explores an even morecomplex system, codeposition of Ni and Al on NiAl(110). Forthe latter, the equilibrium adlayer or thin film morphology is asimple perfect alloy overlayer for stoichiometric codeposition,but observed structures are quite different. There are actuallyfew previous studies of metal deposition on well-characterizedalloy surfaces. Some recent work manipulated adatoms with ascanning tunneling microscope (STM) tip to assemble Ag and Auinto atomic rows on NiAl(110) (18). In contrast, our focus is onspontaneous, diffusion-mediated self-assembly of island nano-structures during deposition. Working at or close to room tem-perature ensures that island structures are far-from-equilibriumand that the substrate is essentially frozen. In contrast, a previous
Author contributions: J.W.E. and P.A.T. designed research; T.D., Y.H., C.Y., D.J., andB.U. performed research; T.D., Y.H., C.Y., D.J., B.U., J.W.E., and P.A.T. analyzed data; andJ.W.E. and P.A.T. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. C.T.C. is a guest editor invited by theEditorial Board.1To whom correspondence should be addressed. E-mail: [email protected].
www.pnas.org/cgi/doi/10.1073/pnas.1008157107 PNAS ∣ January 18, 2011 ∣ vol. 108 ∣ no. 3 ∣ 989–994
CHEM
ISTR
YSP
ECIALFEAT
URE
Dow
nloa
ded
by g
uest
on
May
27,
202
0

study of Al deposition on NiAl(110) at 700–900 K revealed adynamic substrate with bulk defects impacting film growth (19).
Our strategy is to closely integrate experiment, theory, andmodeling in analyzing these complex alloy systems. We introducemultisite LG models which have the flexibility to treat multipleadsorption sites and diffusion pathways, as well as selection froma variety of competing structures. Because nanostructure forma-tion analyzed here is far-from-equilibrium, it is not sufficient forour modeling to just describe correctly the thermodynamics ofthe system (via adatom adsorption energies and interactions).The models must also provide a realistic and accurate treatmentof diffusion kinetics. Our approach has general applicability formulticomponent epitaxial surface nanostructures.
Thin Film Deposition: Key ConceptsFor epitaxial growth either on single-component or binary alloycrystalline substrates, the surfaces are usually prepared to havemorphologies with atomically flat broad terraces separated bymonoatomic steps. The key underlying atomistic processes are asfollows (8, 9): random deposition of single atoms; terrace diffu-sion; aggregation of diffusing atoms into new stable islands (islandnucleation); attachment of diffusing atoms to existing islands(island growth); edge diffusion; possible detachment from islands;and interlayer transport. It is the cooperative interplay betweenall these processes which determines thin film morphology.
On perfect terraces of single-component substrates, there isoften a single type of stable adsorption site for a metal adatom,e.g., the fourfold hollow site on body-centered-cubic (bcc) or face-centered-cubic (fcc) (100) surfaces. Metal atoms diffuse acrossterraces often via simple hopping between neighboring stable sites.For alloy surfaces, in general there are distinct types of stableadsorption sites with very different adsorption energies. This fea-ture can diversify the possibilities for terrace and edge diffusionpathways and energetics. This feature may also allow a variety ofrelatively stable configurations for small clusters or islands of ada-toms (such as ad-dimers), thus impacting the nucleation process.
The early stage of deposition (on broad terraces) is character-ized by a competition between nucleation of new islands andgrowth of existing islands. It is common to introduce a criticalsize, i, such that islands with >i atoms are stable (8, 9). Then i ¼1 (i > 1) corresponds to irreversible (reversible) island formation.For homogeneous nucleation on perfect terraces, traditionalmean-field rate equations are often used to describe the depen-dence of island density, N isl, on temperature T or deposition fluxF, but they fail to describe the island size distribution (ISD) (9).Alloy surfaces tend to be imperfect, e.g., due to surface segrega-tion which produces exchange defects which can constitute trapsfor heterogeneous nucleation of islands (20). Then, N isl can reflectthe trap density.
The later stages of deposition are characterized by island coales-cence, and subsequent formation of continuous multilayer films.Sometimes, island coalescence and smooth multilayer growth areinhibited by the presence of an Ehrlich-Schwoebel (ES) barrierrestricting downward transport from higher to lower layers (8, 9).
MethodsRegarding our experimental procedures, the UHV system, preparation of theNiAl(110) surface, the available surface analytic techniques, and depositionprocedures, are described in refs. 20–22. Sample preparation can producebroad terraces up to 1 μm wide. Film purity is checked with Auger electronspectroscopy and X-ray photoemission spectroscopy (XPS), and the absenceof intermixingwith the substrate is deduced from XPS. Filmmorphologies areprobed using an Omicron variable-T STM.
Wehave performed the extensive DFTanalysis using the plane-wave basedVASP package (23) using the projector augmented wave approach and thePerdew-Burke-Ernzerhof functional. DFT analysis is performed for: (i) theenergetics of various configurations of complete overlayers and multilayerthin films; (ii) single-atom adsorption energies at different adsorption siteson the substrate; (iii) adatom-adatom interaction energies; and (iv) terraceand edge diffusion barriers. These energies are incorporated into single- or
multisite LG models wherein atoms are deposited at and hop between peri-odic array(s) of adsorption sites. LG models are efficiently analyzed by KMCsimulation (8, 9). Accurate barriers, Eact, for Arrhenius hop rates must be pre-scribed not just for terrace diffusion, but also for edge diffusion, attachment-detachment, and interlayer transport for many step edge configurations.
We employ two strategies for prescription of Eact whichmust be consistentwith detailed balance. The first approximate strategy is geared to single-com-ponent systems with simpler island shapes (20, 21). DFT is used to determinebarriers for terrace and straight edge configurations. A general form forEact ¼ Eo − Ei is developed which recovers these barriers and satisfies detailedbalance. Ei is the total lateral interaction in the initial state (with attractionsbeing negative). For all intralayer attachment, detachment, and terracediffusion processes, Eo is the terrace diffusion barrier for an isolated adatom.For edge diffusion with the atom at the edge both before and after thehop, Eo is selected to recover DFT barriers for straight edges. For interlayerdiffusion, Eo must have the same value for both upward and downwardhops. Modification is required to account for an ES barrier and for differingadsorption energies in different layers.
A powerful second general strategy can handle much more complexsystems. We use DFT to guide selection of two distinct sets of adatom inter-actions, one for both adatoms at adsorption sites, and another for one ada-tom at relevant transition states (TS) for hopping. Then, given adsorptionenergies at both types of sites, one can calculate the total energy for theadatom at the initial site, Einit, and at the transition state, ETS. These energiesare simply the sum of the adsorption energy (taken as negative) and the totallateral interaction energy (with attractions being negative). Then, the barrierfor hopping satisfies Eact ¼ ETS − Einit. We assume pairwise additive interac-tions, and that the location of the TS for hopping does not depend stronglyon the local environment. Relevant schematics are shown below.
Previous formulations for Eact in surface processes (either epitaxial filmgrowth or surface reactions) consider adatom interactions only with adatomsat adsorption sites and just estimate behavior at the TS. In contrast, ourapproach directly determining interactions affecting ETS yields precise Eact
for vast numbers of edge configurations in complex multisite, multicompo-nent systems. Again, this precision is key for reliable description of nonequi-librium nanostructure formation.
Au on NiAl(110) and Comparison with Ag on NiAl(110)A natural motivation for this study comes from the relevance ofboth Au and Ag for catalysis, e.g., Ag for ethylene epoxidationand Au for oxidation reactions (6, 24). The two metals are similar.An appealing and unifying feature for our study is that Au and Aghave almost identical fcc lattice constants, and furthermore thereis an almost perfect match between rectangular surface unitcells for Au(110) and Ag(110) and that for NiAl(110). Thus,one might achieve film growth in the absence of lateral mismatchstrain, although growth of fcc metals on a substrate with CsCl bccstructure still must involve some strain. Also given the similarelectronic (as well as structural) properties of Au and Ag, and theanticipated similarity in adatom interactions, one might expectsimilar island formation and film growth modes. However, onesurprise of our study is the discovery of strong differences. Wecan explain these variations and elucidate the detailed form ofgrowth structures by exploiting high-level atomistic modeling.
There are subtle differences between Au and Ag that mightbe anticipated to influence their growth on NiAl(110), at leastfor thicker films. Bulk Au(110) is well known for its 2 × 1 (orsometimes 3 × 1) missing row reconstruction, a feature preservedin heteroepitaxy on well-matched fcc(110) substrates (25, 26).DFT predicts similar 2 × 1 and 3 × 1 energies, actually with3 × 1 slightly lower ignoring entropy contributions (27). In con-trast, Ag(110) does not display such reconstructions. However,our study reveals that differences in the interaction of Au andAg with the NiAl(110) substrate are more important.
Experimental Observations. STM studies of the deposition of Auat 200 K and 300 K are now described and contrasted with ourprevious observations for Ag deposition on NiAl(110) (20–22).First, consider island heights. The height of first level Au islandsfrom line profiles is 0.25 nm at 200 K and 0.23–0.25 nm at 300 K(depending on tip bias) implying a monolayer structure, thedetails of which will be described further below. (See Fig. 1 A
990 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1008157107 Duguet et al.
Dow
nloa
ded
by g
uest
on
May
27,
202
0
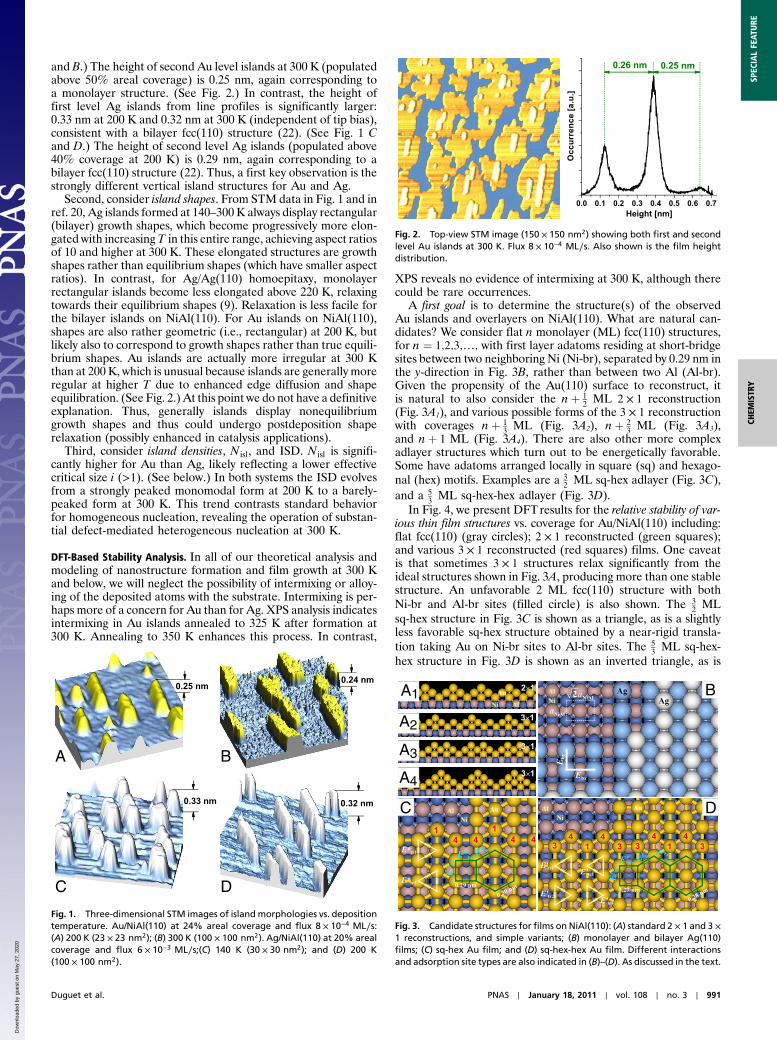
and B.) The height of second Au level islands at 300 K (populatedabove 50% areal coverage) is 0.25 nm, again corresponding toa monolayer structure. (See Fig. 2.) In contrast, the height offirst level Ag islands from line profiles is significantly larger:0.33 nm at 200 K and 0.32 nm at 300 K (independent of tip bias),consistent with a bilayer fcc(110) structure (22). (See Fig. 1 Cand D.) The height of second level Ag islands (populated above40% coverage at 200 K) is 0.29 nm, again corresponding to abilayer fcc(110) structure (22). Thus, a first key observation is thestrongly different vertical island structures for Au and Ag.
Second, consider island shapes. From STM data in Fig. 1 and inref. 20, Ag islands formed at 140–300 K always display rectangular(bilayer) growth shapes, which become progressively more elon-gated with increasingT in this entire range, achieving aspect ratiosof 10 and higher at 300 K. These elongated structures are growthshapes rather than equilibrium shapes (which have smaller aspectratios). In contrast, for Ag/Ag(110) homoepitaxy, monolayerrectangular islands become less elongated above 220 K, relaxingtowards their equilibrium shapes (9). Relaxation is less facile forthe bilayer islands on NiAl(110). For Au islands on NiAl(110),shapes are also rather geometric (i.e., rectangular) at 200 K, butlikely also to correspond to growth shapes rather than true equili-brium shapes. Au islands are actually more irregular at 300 Kthan at 200 K, which is unusual because islands are generally moreregular at higher T due to enhanced edge diffusion and shapeequilibration. (See Fig. 2.) At this point we do not have a definitiveexplanation. Thus, generally islands display nonequilibriumgrowth shapes and thus could undergo postdeposition shaperelaxation (possibly enhanced in catalysis applications).
Third, consider island densities, N isl, and ISD. N isl is signifi-cantly higher for Au than Ag, likely reflecting a lower effectivecritical size i (>1). (See below.) In both systems the ISD evolvesfrom a strongly peaked monomodal form at 200 K to a barely-peaked form at 300 K. This trend contrasts standard behaviorfor homogeneous nucleation, revealing the operation of substan-tial defect-mediated heterogeneous nucleation at 300 K.
DFT-Based Stability Analysis. In all of our theoretical analysis andmodeling of nanostructure formation and film growth at 300 Kand below, we will neglect the possibility of intermixing or alloy-ing of the deposited atoms with the substrate. Intermixing is per-haps more of a concern for Au than for Ag. XPS analysis indicatesintermixing in Au islands annealed to 325 K after formation at300 K. Annealing to 350 K enhances this process. In contrast,
XPS reveals no evidence of intermixing at 300 K, although therecould be rare occurrences.
A first goal is to determine the structure(s) of the observedAu islands and overlayers on NiAl(110). What are natural can-didates? We consider flat n monolayer (ML) fcc(110) structures,for n ¼ 1;2;3;…; with first layer adatoms residing at short-bridgesites between two neighboring Ni (Ni-br), separated by 0.29 nm inthe y-direction in Fig. 3B, rather than between two Al (Al-br).Given the propensity of the Au(110) surface to reconstruct, itis natural to also consider the nþ 1
2ML 2 × 1 reconstruction
(Fig. 3A1), and various possible forms of the 3 × 1 reconstructionwith coverages nþ 1
3ML (Fig. 3A2), nþ 2
3ML (Fig. 3A3),
and nþ 1 ML (Fig. 3A4). There are also other more complexadlayer structures which turn out to be energetically favorable.Some have adatoms arranged locally in square (sq) and hexago-nal (hex) motifs. Examples are a 3
2ML sq-hex adlayer (Fig. 3C),
and a 53ML sq-hex-hex adlayer (Fig. 3D).
In Fig. 4, we present DFTresults for the relative stability of var-ious thin film structures vs. coverage for Au/NiAl(110) including:flat fcc(110) (gray circles); 2 × 1 reconstructed (green squares);and various 3 × 1 reconstructed (red squares) films. One caveatis that sometimes 3 × 1 structures relax significantly from theideal structures shown in Fig. 3A, producing more than one stablestructure. An unfavorable 2 ML fcc(110) structure with bothNi-br and Al-br sites (filled circle) is also shown. The 3
2ML
sq-hex structure in Fig. 3C is shown as a triangle, as is a slightlyless favorable sq-hex structure obtained by a near-rigid transla-tion taking Au on Ni-br sites to Al-br sites. The 5
3ML sq-hex-
hex structure in Fig. 3D is shown as an inverted triangle, as is
A B
C D
Fig. 1. Three-dimensional STM images of island morphologies vs. depositiontemperature. Au/NiAl(110) at 24% areal coverage and flux 8 × 10−4 ML∕s:(A) 200 K (23 × 23 nm2); (B) 300 K (100 × 100 nm2). Ag/NiAl(110) at 20% arealcoverage and flux 6 × 10−3 ML∕s;(C) 140 K (30 × 30 nm2); and (D) 200 K(100 × 100 nm2).
Fig. 2. Top-view STM image (150 × 150 nm2) showing both first and secondlevel Au islands at 300 K. Flux 8 × 10−4 ML∕s. Also shown is the film heightdistribution.
A1
A2
A3
A4
C D
B
Fig. 3. Candidate structures for films on NiAl(110): (A) standard 2 × 1 and 3 ×1 reconstructions, and simple variants; (B) monolayer and bilayer Ag(110)films; (C) sq-hex Au film; and (D) sq-hex-hex Au film. Different interactionsand adsorption site types are also indicated in (B)–(D). As discussed in the text.
Duguet et al. PNAS ∣ January 18, 2011 ∣ vol. 108 ∣ no. 3 ∣ 991
CHEM
ISTR
YSP
ECIALFEAT
URE
Dow
nloa
ded
by g
uest
on
May
27,
202
0
a slightly less favorable translated sq-hex-hex structure. Theremay also be other complex low energy structures.
The relative surface energy in Fig. 4 is defined as the totalenergy of N atoms in the film per NiAl(110) unit cell less N timesthe bulk cohesive energy. Thus, the plot provides a simple way tocompare the relative stability of different structures. To assesswhether a structure is stable against “bifurcation” into a pair ofstructures with higher and lower coverage, one draws a tie-linebetween the pair and checks whether the energy of the structureof interest is below (stable) or above (unstable) this line. (How-ever, not all energetically preferred bifurcations occur duringgrowth due to kinetic limitations, particularly for pairs with far-separated coverages.) Our conclusion based on available results isthat the sq-hex and sq-hex-hex structure are the most favorablefor local coverages between 1 ML and 2 ML, and thus maycorrespond to those observed in experiment.
Why does Ag on NiAl(110) prefer a fcc(110) bilayer structurefor low coverages in contrast to Au? Actually, both Ag and Aufcc(110) films on NiAl(110) display strong bilayer oscillations inthe relative surface energy, with the even layer films being moststable. (See Fig. 4 and refs. 20–22.) These bilayer oscillationsreflectQSE, the half Fermiwavelength of confined electrons beingroughly commensurate with a bilayer film thickness. One differ-ence is that a monolayer Ag(110) film is unstable against bifurca-tion into a bilayer film and a bare substrate, in contrast to Au.In addition, nonfcc(110) structures with lower energy exist forAu as shown in Fig. 4, although analogous structures could becompetitive for Ag. Thus, calculation of these total energies fordifferent film structures does not provide complete understandinginto differences betweenAuandAg film growth onNiAl(110), andprovides little insight into the kinetics of film growth. This uncer-tainty prompts the additional atomistic-level analysis below.
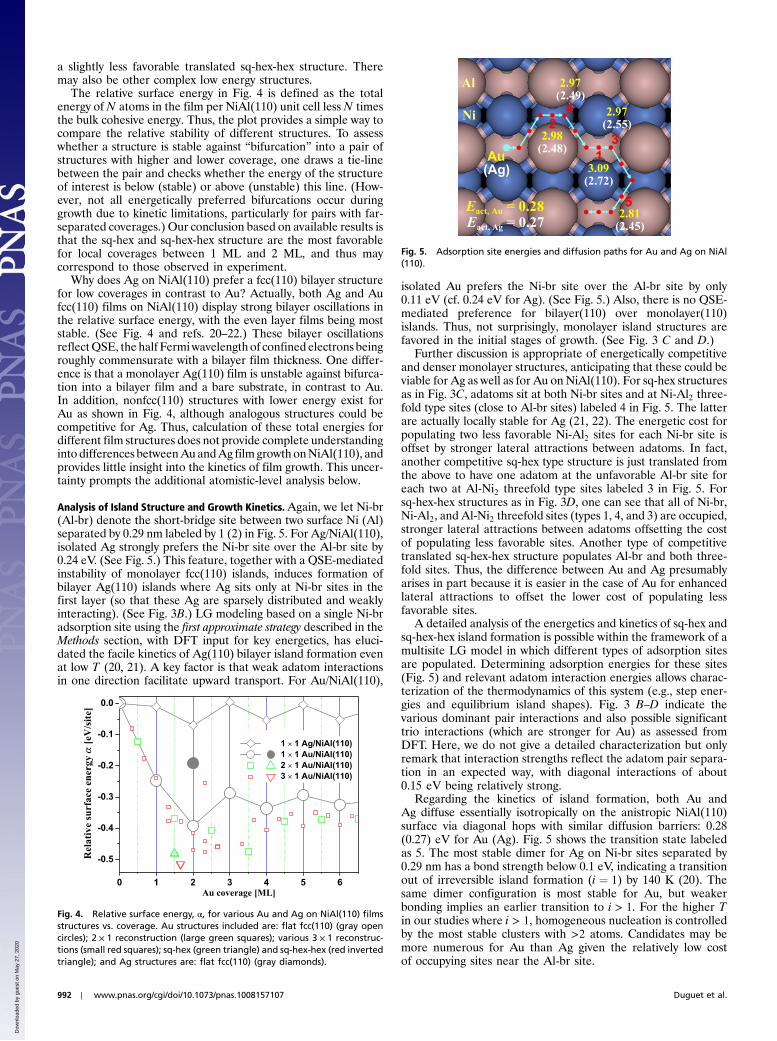
Analysis of Island Structure and Growth Kinetics.Again, we let Ni-br(Al-br) denote the short-bridge site between two surface Ni (Al)separated by 0.29 nm labeled by 1 (2) in Fig. 5. For Ag/NiAl(110),isolated Ag strongly prefers the Ni-br site over the Al-br site by0.24 eV. (See Fig. 5.) This feature, together with a QSE-mediatedinstability of monolayer fcc(110) islands, induces formation ofbilayer Ag(110) islands where Ag sits only at Ni-br sites in thefirst layer (so that these Ag are sparsely distributed and weaklyinteracting). (See Fig. 3B.) LG modeling based on a single Ni-bradsorption site using the first approximate strategy described in theMethods section, with DFT input for key energetics, has eluci-dated the facile kinetics of Ag(110) bilayer island formation evenat low T (20, 21). A key factor is that weak adatom interactionsin one direction facilitate upward transport. For Au/NiAl(110),
isolated Au prefers the Ni-br site over the Al-br site by only0.11 eV (cf. 0.24 eV for Ag). (See Fig. 5.) Also, there is no QSE-mediated preference for bilayer(110) over monolayer(110)islands. Thus, not surprisingly, monolayer island structures arefavored in the initial stages of growth. (See Fig. 3 C and D.)
Further discussion is appropriate of energetically competitiveand denser monolayer structures, anticipating that these could beviable for Ag as well as for Au on NiAl(110). For sq-hex structuresas in Fig. 3C, adatoms sit at both Ni-br sites and at Ni-Al2 three-fold type sites (close to Al-br sites) labeled 4 in Fig. 5. The latterare actually locally stable for Ag (21, 22). The energetic cost forpopulating two less favorable Ni-Al2 sites for each Ni-br site isoffset by stronger lateral attractions between adatoms. In fact,another competitive sq-hex type structure is just translated fromthe above to have one adatom at the unfavorable Al-br site foreach two at Al-Ni2 threefold type sites labeled 3 in Fig. 5. Forsq-hex-hex structures as in Fig. 3D, one can see that all of Ni-br,Ni-Al2, and Al-Ni2 threefold sites (types 1, 4, and 3) are occupied,stronger lateral attractions between adatoms offsetting the costof populating less favorable sites. Another type of competitivetranslated sq-hex-hex structure populates Al-br and both three-fold sites. Thus, the difference between Au and Ag presumablyarises in part because it is easier in the case of Au for enhancedlateral attractions to offset the lower cost of populating lessfavorable sites.
A detailed analysis of the energetics and kinetics of sq-hex andsq-hex-hex island formation is possible within the framework of amultisite LG model in which different types of adsorption sitesare populated. Determining adsorption energies for these sites(Fig. 5) and relevant adatom interaction energies allows charac-terization of the thermodynamics of this system (e.g., step ener-gies and equilibrium island shapes). Fig. 3 B–D indicate thevarious dominant pair interactions and also possible significanttrio interactions (which are stronger for Au) as assessed fromDFT. Here, we do not give a detailed characterization but onlyremark that interaction strengths reflect the adatom pair separa-tion in an expected way, with diagonal interactions of about0.15 eV being relatively strong.
Regarding the kinetics of island formation, both Au andAg diffuse essentially isotropically on the anistropic NiAl(110)surface via diagonal hops with similar diffusion barriers: 0.28(0.27) eV for Au (Ag). Fig. 5 shows the transition state labeledas 5. The most stable dimer for Ag on Ni-br sites separated by0.29 nm has a bond strength below 0.1 eV, indicating a transitionout of irreversible island formation (i ¼ 1) by 140 K (20). Thesame dimer configuration is most stable for Au, but weakerbonding implies an earlier transition to i > 1. For the higher Tin our studies where i > 1, homogeneous nucleation is controlledby the most stable clusters with >2 atoms. Candidates may bemore numerous for Au than Ag given the relatively low costof occupying sites near the Al-br site.
Fig. 4. Relative surface energy, α, for various Au and Ag on NiAl(110) filmsstructures vs. coverage. Au structures included are: flat fcc(110) (gray opencircles); 2 × 1 reconstruction (large green squares); various 3 × 1 reconstruc-tions (small red squares); sq-hex (green triangle) and sq-hex-hex (red invertedtriangle); and Ag structures are: flat fcc(110) (gray diamonds).
Fig. 5. Adsorption site energies and diffusion paths for Au and Ag on NiAl(110).
992 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1008157107 Duguet et al.
Dow
nloa
ded
by g
uest
on
May
27,
202
0

In summary, we have found that for both Au and Ag on NiAl(110), a rich variety of energetically competitive structures areavailable. Thus, slight differences in interaction of Au and Agwith the substrate can guide different selections. Characterizationof these structures in terms of multisite LG models, and analysisof relevant adatom adsorption energies and interactions, canenable detailed atomistic-level modeling and KMC simulation.
Niþ Al on NiAl(110): Nonequilibrium Alloy NanostructuresNext, we turn more briefly to a system that presents even greaterchemical and structural complexity because it consists of twoelemental metals deposited on the NiAl(110) binary alloy surface.We choose Ni and Al as the adsorbates because this lends insightinto yet another basic issue: the pathways and kinetics of alloyself growth. As noted in the Introduction, one appealing featureof this system is that the equilibriummorphology corresponds to asimple perfect alloy overlayer for stoichiometric codeposition.
Our DFT analysis shows that isolated Al and Ni both preferthe Ni-br site over the Al-br site by 0.50 eV for Al and0.15 eV for Ni. Interestingly, Ni favors the Ni-br site despite itbeing the “wrong” site for Ni in an ordered alloy overlayer. How-ever, for deposition of either Al alone or Ni alone, dense islandsform with both Ni-br and Al-br sites populated (contrasting Agand Au). The local coverage is twice as high as for the first layerof Ag(110) films on NiAl(110) (defined as 1 ML), and also higherthan that for the sq-hex (3
2ML) and sq-hex-hex (5
3ML) structures
for Au/NiAl(110). The energetic penalty of populating the unfa-vorable sites is offset by stronger lateral attractions. DetailedSTM studies have been performed that characterize the growthshapes of these Al islands and Ni islands. The observed behaviorhas been recovered via multisite atomistic LG modeling usingthe second general strategy described above. Analysis of the ISDfor Ni and Al on NiAl(110) indicates that heterogeneous nuclea-tion becomes prominent at 300 K, just as for Au and Ag.
Experiments on Sequential Codeposition. In the current work, wefocus on nanostructure formation by sequential codeposition ofNi and Al on NiAl(110) at 300 K. Discrimination in mixed-com-position islands between Ni and Al regions broader than a few nmacross can be achieved because Ni islands always image with aheight of 0.2 nm, but the height of Al islands varies from0.2 nm for −2 V tip bias to 0.3 nm for þ2 V bias. Deposition ofAl first, then Ni, produces islands with a simple ring structurewhich is clearly visible with þ2 V bias. (See Fig. 6 A and B.) Niaggregates around the Al core with little disruption of the core.The shape of the outer boundary of the Ni ring is similar to thatof pure Ni islands. In contrast, deposition of Ni first, then Al,produces an irregular bumpy ring around the Ni core for þ2 Vbias. Also, second layer Al islands appear towards the center ofthe island. (See Fig. 6 D and E.) Quantitative analysis revealsthat the amount of second layer Al corresponds closely to thetotal amount of Al deposited directly on top of the growingisland. Thus, there is negligible downward transport, a featurethat can be traced to strong adsorption of Al on top of the Nicore (as confirmed by DFT).
For comparison, in Fig. 6, we also show the simulation resultscorresponding to the experimental deposition protocol. Fordeposition of Al first, then Ni, it is clear that the aggregating Nihas barely changed the Al core from the original Al island (alsoshown as an inset). In contrast, for deposition of Ni first, then Al,the Al aggregating around a Ni core extracts some edge Ni atoms,creating an irregular defective ring which likely relaxes beforeimaging and which images bumpily. (The simulation in Fig. 6Fdoes not show second layer Al.) Thus, our simulations are suc-cessful in recovering the key features of experiment. However,an even greater advantage of the modeling is the detailed atomis-tic-level insight that it provides into observed behavior, asdiscussed below.
Multisite LG Modeling of Nanostructure Formation. Codepositionof Ni and Al on NiAl(110) can be analyzed using our multisiteLG model, necessarily adopting the second general strategy de-scribed above, given the complexity of this system. An extensiveset of input energies is required. Fig. 7A summarizes the variousadsorption site energies and distinct diffusion paths for Ni and Alon NiAl(110). Fig. 7 B–D illustrate some of the adatom interac-tions which impact edge diffusion and detachment, solid linescorresponding to both atoms at adsorption sites, and dashed linesto one atom at a transition state. Fig. 7B shows diffusion of Nialong the diagonal edge of an alloy island. The calculated barriersreveal such diffusion is facile. More relevant for interpretationof our experiments, Fig. 7C reveals that it is difficult to extract Alfrom a typical Al island edge even in the presence of one or morenearby Ni (smaller green circles). In contrast, Fig. 7D reveals thatextraction of Ni from a typical diagonal Ni island edge is facilegiven the presence of just a single neighboring Al (smaller graycircle). (A separate analysis with Al below the Al-br site Ni yieldsan even lower extraction barrier of 0.46 eV.) What underlies thedifferent behavior? For Ni extraction, the Ni-Al attraction ismuch stronger in the TS relative to the initial state. Thus, ouratomistic modeling has also succeeded in explaining the subtlebehavior in this comparative sequential codeposition study.
Our model can also be used to explore simultaneous stoichio-metric codeposition. We have not yet performed such experi-ments, but simulation might guide them. For deposition athigh enough T, one should form islands with near-perfect alloyordering. However, at 300K there is significant disorder, although
A D
B E
C FFig. 6. STM images (25 × 25 nm2) of Al first then Ni deposition (left), and Nifirst then Al deposition (right) at 300 K: Upper is top-view, and center is three-dimensional. KMC simulation results (lower). Insets show island structureafter deposition of first element. Fluxes are around 3 × 10−3 ML∕s.
Duguet et al. PNAS ∣ January 18, 2011 ∣ vol. 108 ∣ no. 3 ∣ 993
CHEM
ISTR
YSP
ECIALFEAT
URE
Dow
nloa
ded
by g
uest
on
May
27,
202
0

quantitative analysis reveals more adatoms on the correct sitesthan the incorrect ones. (See Fig. 8.)
In summary, our experimental and simulated codepositionstudies at 300 K reveal a variety of distinct nonequilibriumstructures depending on the deposition protocol. None are closeto displaying perfect equilibrium alloy ordering which is onlyachieved by deposition at higher T.
Summary and OutlookWe have provided detailed atomistic-level insight into the forma-tion of single- and multicomponent metal nanostructures duringepitaxial thin film growth on a binary alloy surface. Our successrelies on an integrated combination of STM experiments anddevelopment of realistic atomistic-level multisite LG models(with DFT input). A number of concepts and findings have gen-eral applicability.
Alloy surfaces present complications for characterizing epitax-ial growth due to multiple adsorption sites and adatom diffusionpathways. The former can lead to a variety of possible (thermo-
dynamically competitive) atomic arrangements within islands andnanostructures. However, multisite LG models have the flexibil-ity to treat these complications. Another general feature is thatdeposition at lower T drives systems far-from-equilibrium therebyaccessing a vast phase-space of nanostructure morphologies,and of compositions for multicomponent systems. Islands canhave extreme geometric shapes, e.g., highly elongated rectangles,or subtle irregularities, both cases reflecting the details of edgediffusion and detachment-reattachment kinetics. Similarly, thedegree of alloy ordering for codeposition is controlled by subtlekinetic details. However, it is the diversity of this phase-space ofstructures which offers such great potential for tuning desiredproperties of these nanostructures.
We have also provided two powerful strategies enabling pre-dictive modeling of far-from-equilibrium nanostructure forma-tion. These incorporate accurate adatom hopping barriers intoour LG models, a feature critical to their success. One strategyis just suited for single-component islands with simple shapes,e.g., Ag/NiAl(110). However, the other is completely generaland can describe extremely complex multicomponent epitaxialsystems. This general approach probes via DFTadatom interac-tions at transition states as well as at adsorption sites enablingprecise description of hopping barriers for any complex step edgemorphology and any local composition.
ACKNOWLEDGMENTS. This work was supported by National ScienceFoundation Grant CHE-0809472. Teragrid provided computational support.The work was performed at Ames Laboratory operated for the USDOE byIowa State University under Contract No. DE-AC02-07CH11358.
1. Whitesides GM, Grzybowski B (2002) Self-assembly at all scales. Science295:2418–2421.
2. Viswanath B, Kundu P, Halder A, Ravishankar N (2009) Mechanistic aspects ofshape selection and symmetry breaking during nanostructure growth by wet chemicalmethods. J Phys Chem C 113:16866–16883.
3. Narayanan R, El-Sayed MA (2002) Catalysis with transition metal nanoparticles incolloidal solution. J Phys Chem B 109:12663–12676.
4. Kamat PV (2002) Photophysical, photochemical, and photocatalytic aspects of metalnanoparticles. J Phys Chem B 106:7729–7744.
5. Feldman DL, Foss CA, eds. (2002)Metal nanoparticles: synthesis, characterization, andapplications (CRC Press, New York).
6. ChenM, GoodmanDW (2006) Catalytically active gold: from nanoparticles to ultrathinfilms. Acc Chem Res 39:739–746.
7. McCullagh M, Prytkova T, Tonzani S, Winter ND, Schatz GC (2008) Modeling of self-assembly processes driven by nonbonded interactions in soft materials. J Phys Chem B112:10388–10398.
8. Michely T, Krug J (2004) Islands, mounds, and atoms (Springer, Berlin).9. Evans JW, Thiel PA, Bartelt MC (2006) Morphological evolution during epitaxial
thin film growth: formation of 2D islands and 3D mounds. Surf Sci Rep 61:1–128.10. Bauer E (1958) Phaenomenologische theorie der kristallabscheidung an oberflaechen.
Z Kristallogr 110:372–394.11. Zhang Z, Niu Q, Shih CK (1998) Electronic growth of metallic overlayers on semicon-
ductor substrates. Phys Rev Lett 80:5381–5384.12. Miller T, Chou MY, Chiang TC (2009) Phase relations with 1D shell effects in thin
metal films. Phys Rev Lett 102:236803.13. van Gastel R, Bartelt NC, Kellogg GL (2006) Reversible shape transitions of Pb islands
on Cu(111). Phys Rev Lett 96:036106.14. Rougemaille N, et al. (2007) Labyrinthine island growth during Pd/Ru(0001) hetetoe-
pitaxy. Phys Rev Lett 99:106101.
15. Behm RJ (2009) Interaction of hydrogen with bimetallic surfaces. Z Phys Chem223:9–36.
16. Tober ED, et al. (1998) Self-assembled lateral multilayers from thin film alloys ofimmiscible metals. Phys Rev Lett 81:1897–1900.
17. Volkmann T, Much F, Biehl M, Kotrla M (2005) Interplay of strain relaxation andchemically induced diffusion barriers. Surf Sci 586:157–173.
18. Nilius N, Wallis TM, Ho W (2005) Tailoring the electronic properties of atomic chainsassembled by STM. Appl Phys A-Mater 80:951–956.
19. Pierce JP, Bartelt NC, McCarty KF (2007) Evolution of a reactive surface via subsurfacedefect dynamics. Phys Rev Lett 99:026101.
20. Han Y, et al. (2010) Formation and coarsening of Ag(110) bilayer islands on NiAl(110):STM analysis and atomistic lattice-gas modeling. Phys Rev B 81:115462.
21. Han Y, et al. (2008) Kinetics of facile bilayer island formation at low temperature:Ag/NiAl(110). Phys Rev Lett 100:116105.
22. Unal B, et al. (2007) Scanning tunneling microscopy and density functional theorystudy of initial bilayer growth of Ag films on NiAl(110). Phys Rev B 76:195410.
23. Kresse G, Hafner J (1993) Ab initio molecular dynamics for liquid metals. Phys Rev B47:558–561.
24. Liu XY, Madix RJ, Friend CM (2008) Unraveling molecular transformations onsurfaces: a critical comparison of oxidation reactions on coinage metals. Chem SocRev 37:2243–2261.
25. Rousset S, Chiang S, Fowler DE, Chambliss DD (1992) Intermixing and 3D islands inepitaxial growth of Au on Ag(110). Phys Rev Lett 69:3200–3203.
26. Schmitz PJ, Leung W-Y, Kang HC, Thiel PA (1991) Reconstructions of Au films onPd(110). Phys Rev B 43:8834–8840.
27. LandmannM, Rauls E, Schmidt WG (2009) First-principles calculations of clean Au(110)surfaces and chemisorption of atomic oxygen. Phys Rev B 79:045412.
A B
C D
Fig. 7. (A) Adsorption energies and diffusion paths for Ni and Al; (B) Diffu-sion of Ni along a diagonal edge of a perfect alloy island; (C) Difficult Ni-as-sisted extraction of Al from the vertical edge of an Al island; and (D) FacileAl-assisted extraction of Ni from the diagonal edge of a Ni island.
A B CFig. 8. KMC results for island growth during simultaneous stoichiometriccodeposition at 300 K with equal Ni and Al fluxes of 10−2 ML∕s.
994 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1008157107 Duguet et al.
Dow
nloa
ded
by g
uest
on
May
27,
202
0





























