Embed Size (px)
Citation preview

1
Second Advanced Training Course in
Thrombosis & Haemostasis
Course Manual
Second Advanced Training Course Manual.indd 1 2/27/14 10:23 AM

2 3
Table of Contents
Second Advanced Training Course in
Thrombosis & Haemostasis
4 Message from the Chairman
5 Meet the ISTH
6 Advanced Training Course Program
6 Thursday March 13
6 Friday March 14
7 Saturday March 15
8 Sunday March 16
9 Speakers
12 Program Abstracts
12 S1 & S4 Blood Coagulation and its Regulation by Anticoagulant Pathways
16 S2 Challenges in the Diagnosis and Management of the Hemophilias
17 S3 Diagnosis and Management of von Willebrand Disease
18 S5 Hemostasis in Patients with Impaired Liver Function
19 S6 How to Approach a Patient with Bleeding
21 S7 Venous Thrombosis: Manifestations, Diagnosis and Therapy
23 S8 Antiphospholipid Syndrome
24 S9 Women Issues and Thrombosis
26 S10 Novel Antithrombotic Drugs
28 S11 How to Approach a Patient with Confirmed Venous Thrombosis
30 S12 Perioperative Management in Patients with Risk for Thrombosis
32 S13 Platelet Function
33 S14 Heparin-Induced Thrombocytopenia (HIT)
35 S15 Immune Thrombocytopenias
39 S16 Diagnosis and Treatment of Inherited Platelet Disorders
40 S17 Diagnosis and Treatment of Acquired Platelet Disorders
44 S18 Disseminated Intravascular Coagulation (DIC)
46 Sponsors
47 General Information
49 ISTH Membership
Second Advanced Training Course Manual.indd 2-3 2/27/14 10:23 AM

4 5
On behalf of the International Society on Thrombosis and Haemostasis (ISTH), it is a pleasure to welcome you to the Second Advanced Training Course of the ISTH in Cascais, Portugal.
The course is designed to provide the latest training in biological and clinical aspects of hemostasis and thrombosis. Over the next three days you will take part in an intense examination on the subjects of blood coagulation and bleeding disorders, platelets and venous thrombosis.
We are privileged to have some of the leading scientists and clinicians in our field take part as speakers. All speakers will deliver focused lectures followed by ample time for discussion and close interaction with the participants. There will be plenty of time in the afternoon and evenings for interactive sessions relating to the analysis of the topics discussed during the day. You will all have a chance to meet and talk with the experts.
We invite you to take advantage of this unique opportunity to actively interact with your fellow course participants and the faculty.
We wish you a successful meeting and hope you enjoy the course!
Sincerely,
Andreas GreinacherChairman, ISTH Education Committee
Andreas Greinacher
Message from theChairman & the ISTH Meet the ISTH
Andreas Greinacher
Chairman, ISTH Education Committee
The mission the ISTH is to advance the understanding, prevention, diagnosis and treatment of thrombotic and bleeding disorders.
The ISTH first and foremost stands for leading edge science. As a global membership community of specialists in bleeding and clotting disorders, we are dedicated to transformative scientific discoveries to advance clinical practice and improve the lives of millions of people worldwide.
Programs & Activities
• Journal of Thrombosis and Haemostasis (JTH)
• ISTH Biennial World Congresses
• ISTH SSC Annual Meetings
• E-learning Opportunities Year Round
• Networking and Career Development
• Educational and Training Courses
• Fellowships and Travel Grants
• Scientific Standardization and Nomenclature
• Professional Capacity Building in Developing Countries
Become a Member of the ISTH ISTH membership is an important investment in your own future at every stage in your career. To learn more about the rewarding benefits of membership, visit us online at www.isth.org.
Join Today!
Second Advanced Training Course Manual.indd 4-5 2/27/14 10:23 AM

6 7
Advanced Training Course Program
Thursday, March 13
Blood Coagulation And Bleeding Disorders
13:30 – 14:00 Welcome and introduction to the course Andreas Greinacher Germany
14:00 – 16:00 S1 Current Concepts of the Coagulation System Questions and discussions
Björn Dahlbäck Sweden
S2 Challenges in the Diagnosis and Management of Hemophilias Questions and discussions
David Lillicrap Canada
16:00 – 16:30 Coffee break & networking
16:30 – 18:30 S3 Diagnosis and Management of von Willebrand Disease Questions and discussions
David Lillicrap Canada
S4 Control of Coagulation Questions and discussions
Björn Dahlbäck Sweden
18:30 – 20:00 Dinner & networking
20:00 – 21:00 Meet the Experts
Friday, March 14
Blood Coagulation And Bleeding Disorders
08:30 – 10:30 S5 Hemostasis in Patients with Impaired Liver Function Questions and Discussions
Flip de Groot Netherlands
S6 How to Approach a Patient with Bleeding Questions and Discussions
Bernd Pötzsch Germany
All sessions will be held in the Longshot/Bogey room located on the first floor of the hotel.
10:30 – 11:00 Coffee break & networking
Venous Thrombosis
11: 00 – 13:00 S7 Venous Thrombosis: Manifestations, Diagnosis and Therapy Questions and discussions
Sam SchulmanCanada
S8 Antiphospholipid Syndrome Questions and discussions
Flip de GrootNetherlands
13:00 – 14:00 Lunch & networking
14:00 – 16:00 S9 Women Issues and Thrombosis Questions and discussions
Sabine EichingerAustria
16:00 – 16:30 Coffee break & networking
16:30 – 18:30 Interactive session with speakers and/or case presentations
18:30 – 20-:00 Dinner & networking
20:00 – 21:00 Meet the Expert
Saturday, March 15
Venous Thrombosis
08:30 – 10:30 S10 Novel Antithrombotic Drugs Questions and discussions
Sam SchulmanCanada
S11 How to Approach a Patient with Venous Thrombosis Questions and discussions
Bernd PötzschGermany
S12 Perioperative Management in Patients with Risk for Thrombosis Questions and discussions
Sabine EichingerAustria
10:30 – 11:00 Coffee break & networking
Platelets, Platelet Disorders
11:00 – 13:00 S13 Platelet Function Questions and discussions
Christian GachetFrance
S14 Heparin-induced Thrombocytopenia Questions and discussions
Ted Warkentin Canada
Second Advanced Training Course Manual.indd 6-7 2/27/14 10:23 AM

8 9
13:00 – 14:00 Lunch & networking
14:00 – 16:00 S15 Diagnosis and Management of Immune Thrombocytopenias Questions and discussions
Andreas GreinacherGermany
S16 Diagnosis and Treatment of Inherited Platelet Disorders Questions and discussions
Christian GachetFrance
16:00 – 16:30 Coffee break & networking
16:30 – 18:30 Interactive session with speakers and/or case presentations
18:30 – 20:00 Dinner & networking
20:00 – 21:00 Meet the Expert
Sunday, March 16
Platelets, Platelet Disorders
08:00 – 10:00 S17 Diagnosis and Treatment of Acquired Platelet Disorders Questions and discussions
Andreas GreinacherGermany
S18 Disseminated Intravascular Coagulation Questions and discussions
Ted WarkentinCanada
10:00 – 10:30 Coffee break & networking
10:30 – 11:45 Interactive session with the speakers and/or case presentations
11:45 – 12:00 Course evaluation and farewell
Björn Dahlbäck, Ph.D.Björn Dahlbäck is professor of Blood Coagulation Research at Lund University, Department of Laboratory Medicine, Skåne University Hospital, Malmö, Sweden. His research on the molecular mechanisms of the protein C anticoagulant system has focused on the role of activated protein C and its cofactor protein S in the degradation of coagulation factors V and VIII. Observations made during these studies lead into studies of other biologically important host defense systems, in particular the complement system and the interactions between the coagulation and complement systems. He described that protein S in blood circulates both as free protein and bound to the complement regulatory protein C4b-binding protein (C4BP) and demonstrated that analysis of free protein S is the method of choice to detect inherited protein S deficiency. He has also been interested in the genetics of thrombophilia and described APC resistance, caused by FV Leiden as the major inherited risk factor of thrombosis. Recently, he was involved in the elucidation of a mysterious autosomal dominant bleeding disorder from East Texas. The disease is caused by a point mutation in factor V, which induces alternative splicing and the creation of a short form of FV that causes the bleeding tendency via tissue factor pathway inhibitor (TFPI). Björn Dahlbäck is member of the ISTH Council, the Swedish Royal Academy of Science, and honorary member of the American Society of Hematology. He has published 305 original papers and 99 reviews, has been cited approx. 22,000 times and has an H-index of 75. He has received many awards for his research.
Sabine Eichinger, M.D.Sabine Eichinger is Associate Professor of Medicine at the Medical University of Vienna, Austria, and Head of the Anticoagulation Clinic at the Department of Medicine I of the Medical University Hospital in Vienna. Dr. Eichinger received her medical and scientific training at the Department of Medicine I, Division of Haematology and Haemostasis, University of Vienna, and at the Beth Israel Hospital, Harvard Medical School, Boston, USA. Dr. Eichinger’s anticoagulation research has included investigating the mechanism of action of pro-coagulants and anticoagulants in in vitro and in vivo models, and the pathophysiology of the coagulation system in women. She has undertaken research to evaluate the risk factors for thrombotic disease and biomarkers of the coagulation system. In 1992, she initiated one of the world’s largest studies in patients with venous thrombosis, the Austrian Study on Recurrent Venous Thromboembolism. She is the local principal investigator of several interventional studies in patients with venous thromboembolism and atrial fibrillation, and an internationally recognised expert in designing and conducting clinical studies. She is Chair-elect of the Scientific and Standardization Subcommittee (SSC) of the ISTH, and is the recipient of several academic awards.
Christian Gachet, M.D., Ph.D.Dr. Christian Gachet graduated as a medical doctor in 1985 and gained his Ph.D. in Pharmacology in 1991, both at the Université Louis Pasteur, Strasbourg, France. He became a Research Director at INSERM (Institut National de la Santé et de la Recherche Médicale) in 1998. Dr. Gachet is currently the Director of the INSERM Research Unit 949 and the Scientific Director at Etablissement Français du Sang-Alsace. The focus of Dr. Gachet’s work has long been the molecular mechanisms of ADP-induced platelet activation and its inhibition by the thienopyridine compounds. This led to the characterization and the identification of some of the platelet P2 receptors. He carried out extensive work on the P2Y1 platelet receptor and the P2X1 receptor. Dr. Gachet’s current interest is in general platelet physiology (formation of blood platelets, megakaryopoiesis, platelet activation) and pharmacology (new drugs, new targets), animal models of platelet defects (BSS, MYH9, P2 receptors), mouse models of arterial thrombosis in a context of atherosclerosis and clinical studies on the variability of the response to clopidogrel. Dr. Gachet has published over 200 papers in peer-reviewed journals and over 300 communications in his area of expertise. He also serves on the editorial boards of the Journal of Thrombosis and Haemostasis(JTH). Dr. Gachet has received several distinctions for his work in the field of atherothrombosis.
SpeakersSecond Advanced Training Course in
Thrombosis & Haemostasis
Second Advanced Training Course Manual.indd 8-9 2/27/14 10:23 AM

10 11
Andreas Greinacher, M.D. Andreas Greinacher is an M.D. with a specialization in transfusion medicine and hemostasis. His scientific career is focused on platelet disorders, bridging immuno-hematology and hemostasis. He works at the University Hospital Greifswald, Germany, where he is head of the Institute of Immunology and Transfusion Medicine, the clinical thrombosis and hemostasis service, the hemostasis out-patient clinic, the transfusion and stem cell service and the immuno-hematology laboratory. Aside from heparin-induced thrombocytopenia and drug dependent thrombocytopenia, he has a major interest in hereditary and acquired platelet disorders. His recent work is focusing on adopting nanotechnology and biophysical methods to investigate platelets and protein changes. He is section editor of several journals in the field of thrombosis and hemostasis and the current chairman of the education committee of the ISTH.
Philip (Flip) G. de Groot, Ph.D.Phillip G. de Groot is a professor of Biochemistry appointed at the University Hospital in Utrecht, the Netherlands. Presently, he is deputy head of the department of Clinical Chemistry and Haematology. Besides his responsibility for laboratory diagnostics, particularly in the field of haematology, he has run a research group on thrombosis and haemostasis for over 30 years. He has published over 400 peer-reviewed articles and has a Hirsch index of 66. This year he was awarded with the biennial Distinguished Career Award of the ISTH. One of the major interests of his laboratory is to understand why the presence of antiphospholipid antibodies increases the risk on thrombosis. Their approach to this problem was twofold; they aim to develop assays available that identify the subpopulation of antiphospholipid antibodies responsible for the thrombotic risk and, secondly, to understand the physiological function of the major antigen in this syndrome, b2-glycoprotein I. If they understand the protein, they will understand why antibodies directed against this protein are one of the most common acquired risk factors for thrombosis and pregnancy morbidity.
David Lillicrap, M.D., FRCPCDavid Lillicrap is a Professor in the Department of Pathology and Molecular Medicine at Queen’s University, Kingston, Canada. Since 2000, he has been the recipient of a Canada Research Chair in Molecular Hemostasis and is a past Career Investigator of the Heart and Stroke Foundation of Ontario. He has served on the Gene Therapy Working Group of the US National Hemophilia Foundation (NHF) and has been a member of NHF’s Medical and Scientific Advisory Committee. He is the Chair of the Research Committee of the World Federation of Hemophilia (WFH) and is a member of the Medical Advisory Board of the WFH. He recently completed a three-year term as the chairperson of the ISTH’s SSC Subcommittee on von Willebrand Factor, and is the current Chair of the Society’s SSC. He is an Associate Editor of the journal Blood, and a member of the editorial board of the British Journal of Haematology. His research program relates to molecular aspects of the hemostatic system with particular emphasis on novel therapeutic approaches and immunological complications of hemophilia A, and the genetics, biology and pathobiology of von Willebrand Factor.
Speakers
Bernd Pötzsch, M.D.Bernd Pötzsch received his medical education at Justus-Liebig-University in Giessen, Germany, 1981 – 1988, and his postgraduate training at the Heart Centre Kerckhoff-Clinik in Bad Nauheim, Germany. Since 1999 he is a full-term professor and senior physician at the University of Bonn, Germany, specialized in hemostasis and thrombosis. His focus of research is pathogenesis, diagnosis and treatment of thrombophilia and use of aptamers in diagnosis and treatment of coagulation disorders.
Sam Schulman, M.D.Sam Schulman graduated from Karolinska Institute, Stockholm, Sweden in 1977 and became a specialist in Internal Medicine in 1984, with subspecialties in Haematology and in Coagulation in 1985. That year he also received his Dr Med Sc with the thesis: “Studies on the Medical Treatment of Deep Vein Thrombosis.” He has worked within the field of coagulation disorders continuously since 1984, worked as a consultant at the national Hemophilia Center at Tel Hashomer, Israel from 1992-1996, and was director of the Hemophilia Treatment Center in Stockholm from 1996-2004. His major research activities have been clinical studies in venous thromboembolism, including several randomized trials and in hemophilia and its complications. He is currently involved in trials with new antithrombotic agents, such as the oral thrombin inhibitors. He has been a member of the Executive Committee of the World Federation of Hemophilia (2000-2004) and was chairman of the Subcommittee on Control of Anticoagulation of the SSC Subcommittee of ISTH from 2005-2008. Dr. Schulman is associate professor in Internal Medicine at Karolinska Institute and since September 2004 also a professor in Medicine at McMaster University. He is Director of the Thrombosis Service at HHS-General Hospital in Hamilton and Director of the Clinical Thromboembolism Program of McMaster University.
Theodore (Ted) E. Warkentin M.D., BSc (Med), FRCP(C), FACPTheodore E. Warkentin, M.D., is a Professor in the Department of Medicine and the Department of Pathology and Molecular Medicine at the Michael G. DeGroote School of Medicine, McMaster University, in Hamilton, Ontario, Canada. He is also Regional Director, Transfusion Medicine, of the Hamilton Regional Laboratory Medicine Program and Hematologist, Service of Clinical Hematology, at Hamilton Health Sciences, also in Hamilton. Dr. Warkentin received both his Bachelor of Science (in Medicine) and his M.D. from the University of Manitoba in Winnipeg, Manitoba, Canada. He completed a hematology research fellowship at McMaster University and postgraduate work in medicine and hematology at the University of Toronto and McMaster University, respectively. He was awarded the XVth Jean Julliard Prize at the XXIVth Congress of the International Society of Blood Transfusion (Makuhari, Japan; in 1996) and was a Research Scholar of the Heart and Stroke Foundation of Canada from 1993 to 1998. Dr Warkentin is the former Chair (four-year term ending 2007) of the Platelet Immunology SSC Subcommittee, and was the Chair of the 2004 and 2008 consensus conference guidelines on heparin-induced thrombocytopenia, written under the aegis of the American College of Chest Physicians (ACCP).
Second Advanced Training Course Manual.indd 10-11 2/27/14 10:23 AM

12 13
Björn Dahlbäck Lund University, Department of Laboratory Medicine Malmö, Sweden
Primary haemostasis and blood coagulation have evolved as important defense mechanisms against bleeding. The initial occlusion of a vascular lesion by the platelet plug is temporally co-ordinated with the activation of coagulation. The coagulation pathway is carefully controlled by several anticoagulant mechanisms and under normal conditions they prevail over the procoagulant forces. Disturbances of the natural balance between the pro- and anticoagulant systems caused by genetic or acquired factors may result in bleeding or thrombotic diseases.
Primary haemostasis mediated by platelet-protein interactions Damage of the vascular wall exposes blood to subendothelial tissue, which triggers the primary haemostasis events. Multiple coordinated interactions between receptors on platelets, plasma proteins, and tissue components result in the initial sealing of the wounded area. The platelet plug formation is the result of a series of reactions including adhesion, aggregation, release of granule content, and morphological changes. Adhesion is dependent on the interaction between platelets and the von Willebrand factor (VWF), a high molecular weight plasma protein composed of multiple disulphide-linked subunits. Freshly synthesized VWF multimers, which can be >20 million Da in mass and 4 um in length, undergo proteolytic processing in plasma mediated by the metalloprotease ADAMTS 13. In the adhesion process, VWF serves as a bridge between collagen in the subendothelium and platelet membrane glycoprotein Ib-V-IX (GP1b-V-IX). The adhesion process functions better under high shear stress, i.e. it is more efficient in small arteriole than in veins because high shear unfolds the VWF thus exposing the binding sites for GPIb-V-IX. Platelets also contain receptors for collagen (Integrin α2β1 and GPVI and GPVI) that enforce the anchoring of the platelets to the damaged tissue. Platelets undergo major morphological changes with rearrangement of the membrane and exposure of negatively charged phospholipids and formation of extensive pseudopodia that help anchor the platelets. Release of thromboxane A2, ADP, calcium, and serotonin results in additional platelet activation and contraction of smooth muscle cells of the vessel wall. A conformational change of the platelet Integrin αIIβ3 exposes binding sites for the adhesive proteins fibrinogen, VWF, fibronectin and thrombospondin, the bridging between platelets resulting in platelet aggregation.
Activation and propagation of the blood coagulation system Generation of thrombin at sites of vascular injury is the result of a series of reactions referred to as blood coagulation. Thrombin is the key effector enzyme having important functions, including feedback amplification of coagulation by activating factor V (FV), factor VIII (FVIII), and factor XI (FXI). It also cleaves off fibrinopeptides A and B from fibrinogen, which results in the polymerization of fibrin monomers to a fibrin network, it activates the fibrin crosslinking factor XIII (FXIII), and it activates platelets by cleaving PAR-1 (protease activated receptor-1).
Exposure of tissue factor (TF) to blood triggers the initiation of the coagulation system by binding plasma factor VII (FVII). TF is normally not in contact with blood but abundantly
Program Abstracts (in program order)
Second Advanced Training Course in
Thrombosis & Haemostasis
present in cells surrounding the vasculature. A small amount of FVII in plasma is activated (FVIIa) and the FVIIa-TF-complex converts factor IX (FIX) and factor X (FX) into active enzymes (FIXa and FXa). FIXa and FXa may remain bound to the TF-bearing cell or bind to the negatively charged phospholipid membrane of activated platelets. FXa and its cofactor activated FV (FVa) assemble on the activated platelets to form the prothrombinase complex that activates prothrombin to thrombin. FV can be activated directly by FXa but the majority of FV is activated by thrombin. Thrombin also activates FVIII to FVIIIa, which serves as a cofactor to FIXa in the tenase complex that activated FX to FXa. FVIII circulates bound to the VWF and is freed after activation to join FIXa on the platelet membrane in the formation of the tenase complex. The assembly of the prothrombinase and tenase complexes on the phospholipid surface is a prerequisite for the propagation of the coagulation system as highly efficient in converting several thousand substrate molecules per minute, whereas the free enzymes FXa and FIXa are inefficient (Fig. 1 schematically represents coagulation process).
The negative charge of the phospholipid membrane is due to the presence of phosphatidyl serine, which under normal conditions is located in the inner-layer leaflet of the cell membrane but it is translocated to the outer layer during platelet activation. All the participating proteins of the tenase and prothrombinase complexes have affinity for the negatively charged phospholipid surface, the enzymes and the substrates via their amino-terminal domains, which contain g-carboxy glutamic acid (Gla) residues. The formation of Gla is the result of a vitamin K-dependent post-translational modification of glutamic acid residues. The Gla-residues bind calcium, which is important for the correct folding of the Gla domain. Vitamin K-antagonists that are commonly used to treat thrombosis, inhibit the post-translational modification, resulting in misfolded Gla domains that are unable to bind negatively charged phospholipid membranes.
Thrombin generation continues after the generation of the fibrin clot, which is important for activation of FXIII and the thrombin activatable fibrinolysis inhibitor (TAFI). Activated FXIII (FXIIIa) is a transglutaminase that catalyses covalent cross-linkage of fibrinogen. TAFI is a carboxypeptidase that removes the carboxy-terminal lysines from fibrin. As these lysines are important for the binding of fibrinolytic enzymes to the fibrin, TAFI inhibits fibrinolysis.
Blood Coagulation and Bleeding Disorders
S1 & S4 Blood Coagulation and its Regulation by Anticoagulant Pathways
Fig. 1 Activation of coagulation by TF on extravascular cells and propagation on platelets.
Continued next page
Second Advanced Training Course Manual.indd 12-13 2/27/14 10:23 AM

14 15
AT is intrinsically an inefficient serpin, but its activity is stimulated by heparin and by heparin-like molecules (heparan sulphates or chondroitine sulphates) that are present on the surface of endothelial cells. Homozygous antithrombin knockout mice have a lethal phenotype, demonstrating the importance of the protein for control of coagulation. The protein C anticoagulant system inhibits the cofactors FVIIIa and FVa (Fig. 2).
The key component is protein C, a vitamin K-dependent zymogen, is activated by thrombin bound to thrombomodulin (TM) on the surface of intact endothelial cells. The endothelial protein C receptor (EPCR) stimulates the activation of protein C. The thrombin-mediated activation of protein C demonstrates that thrombin can function both as a pro- and an anticoagulant enzyme. The procoagulant functions are expressed at sites of vascular disrupture, whereas the anticoagulant functions require intact vessel walls with endothelial cells containing TM. Activated protein C (APC) cleaves peptide bonds in each of the membrane-bound cofactors FVa and FVIIIa. The anticoagulant activity of APC is stimulated by the cofactor protein S, a vitamin K-dependent plasma protein. Protein S exists in two forms in human plasma, approximately 30% as free protein, the remainder being bound to the complement regulatory protein C4b-binding protein (C4BP). Protein S is instrumental for the localization of the C4BP to negatively charged phospholipid membranes, which is a unique way to provide local complement regulatory activity. The free form of protein S functions as cofactor in degradation of both FVIIIa and FVa. In regulation of the tenase complex, not only protein S serves as cofactor but also the intact form of FV, which works in synergy with protein S as APC cofactor. Thus, FV like thrombin has both pro- and anticoagulant functions.
The physiological importance of the protein C system is demonstrated by the severe thromboembolic disease that is associated with homozygous deficiency of protein C in both man and mice. Mice lacking the protein C or TM genes are affected by a lethal phenotype, the TM deficiency being particularly severe affecting the embryogenesis even before development of a functional cardiovascular system.
APC also has anti-inflammatory and anti-apoptotic properties. These affects are dependent on the binding of APC to the endothelial protein C receptor (EPCR), which changes the substrate specificity of APC. When bound to EPCR, APC can cleave PAR-1 but is less efficient in cleaving FVa and FVIIIa.
The activation of coagulation via TF, which is referred to as the extrinsic pathway, is initiated in vivo in response to trauma. An alternative activation pathway involves the contact phase proteins factor XII, high-molecular weight kininogen (HMWK), prekallikrein and FXI and results in the generation of FXIa, that in turn activates FIX. These reactions are collectively called the intrinsic pathway and their physiological importance is not fully understood. The intrinsic pathway is not important in trauma-initiated coagulation because inherited deficiency of FXII is not associated with bleeding problems. FXI deficiency on the other hand yields a moderately severe bleeding disorder. However, several discoveries point to an important physiological function of the contact phase system. Thus, it was found that mice lacking FXII are protected against arterial thrombosis diseases including stroke and myocardial infarction. In addition, polyphosphates released from platelet granules have been identified as a possible physiological activator of the contact phase in vivo.
The plasma concentrations of the coagulation factors are very different concentrations, which relate to their specific functions in the pathway. In general, the early components have lower concentrations than those that take part at later stages. This is consistent with the principal organisation of the system with multiple reactions and amplification potential. Thus, the fibrinogen concentration (10 uM) is ≈50.000-fold higher than that of FVIII (0.2 nM). The high level of fibrinogen is required for the formation of the fibrin clot, whereas the low concentration of FVIII is more than sufficient to support FIXa in the activation of FX. Among the vitamin K-dependent proteins, FVII (10 nM) is the least abundant, FIX and FX being at intermediate levels (100 nM) and prothrombin circulating at the highest concentration (2 uM).
Knockout mice technology have contributed to the elucidation of the relative importance of the various coagulation factors in vivo. The crucial importance of the TF pathway is demonstrated by the embryonic lethal phenotype associated with TF deficiency. In contrast, FVII-deficient mice develop normally in utero but succomb shortly after birth from severe bleeding. This difference in severity suggest a role for TF during embryogenesis beyond fibrin formation. Deficiencies of prothrombin and FV are associated with partial embryonic lethality and fatal haemorrhage. In contrast, FIX and FVIII deficient mice develop normally in utero but get haemophilia-like disease after birth. Mice deficient in fibrinogen suffer a moderate to severe bleeding phenotype similar to that of human fibrinogen deficiency. This suggests that thrombin generation is more important than fibrin deposition.
Anticoagulant pathways regulating blood coagulation Coagulation is regulated at multiple levels by different anticoagulant principles such as enzyme inhibition and proteolytic degradation of cofactors FVa and FVIIIa. The initial steps of the TF pathway are controlled by tissue factor pathway inhibitor (TFPI). In humans, there are no deficiency states of TFPI described. Mice lacking TFPI have a lethal phenotype with uncontrolled activation of coagulation and consumption of coagulation factors. TFPI circulates a low concentration bound to FV. Recently, the bleeding phenotype of the autosomal dominant East Texas Bleeding disorder was found to be caused by increased plasma concentrations of TFPI. The increase in TFPI was caused by an alternatively spliced FV isoform (caused by a point mutation in the FV gene) that bound TFPI with high affinity causing retention of TFPI in the circulation.
Several enzymes of the coagulation system are inhibited by the serine protease inhibitor (serpin) antithrombin (AT), which limits the coagulation process to sites of vascular injury.
Blood Coagulation and Bleeding Disorders
Fig. 2 Activation of protein C and inhibition of the coagulation system by APC.
Continued: S1 & S4 Blood Coagulation and its Regulation by Anticoagulant PathwaysRecommended reading:
1. Broze, GJ, Jr., and Girard, TJ. Tissue factor
pathway inhibitor: structure-function. Front
Biosci (Landmark Ed) 2012. 17:262-280.
2. Broze, GJ, Jr., and Girard, TJ. Factor V, tissue
factor pathway inhibitor, and east Texas bleeding
disorder. J Clin Invest 2013. 123:3710-3712.
3. Camire, RM, and Bos, MH. The molecular basis
of factor V and VIII procofactor activation. J
Thromb Haemost 2009. 7:1951-1961.
4. Dahlback, B. Advances in understanding
pathogenic mechanisms of thrombophilic
disorders. Blood 2008. 112:19-27.
5. Dahlback, B, and Villoutreix, BO. Regulation of
blood coagulation by the protein C anticoagulant
pathway: novel insights into structure-function
relationships and molecular recognition.
Arterioscler Thromb Vasc Biol 2005. 25:1311-
1320.
6. Griffin, JH, Fernandez, JA, Gale, AJ, and
Mosnier, LO. Activated protein C. J Thromb
Haemost 2007. 5 Suppl 1:73-80.
7. Lenting, PJ, Casari, C, Christophe, OD, and
Denis, CV. von Willebrand factor: the old, the
new and the unknown. J Thromb Haemost 2012.
10:2428-2437.
8. Morrissey, JH, Choi, SH, and Smith, SA.
Polyphosphate: an ancient molecule that links
platelets, coagulation, and inflammation. Blood
2012. 119:5972-5979.
9. Morrissey, JH, Tajkhorshid, E, Sligar, SG, and
Rienstra, CM. Tissue factor/factor VIIa complex:
role of the membrane surface. Thromb Res
2012. 129 Suppl 2:S8-10.
10. Mosesson, MW. Fibrinogen and fibrin structure
and functions. J Thromb Haemost 2005.
3:1894-1904.
11. Renne, T, Schmaier, AH, Nickel, KF, Blomback,
M, and Maas, C. In vivo roles of factor XII. Blood
2012. 120:4296-4303.
12. Sadler, JE. Von Willebrand factor, ADAMTS13,
and thrombotic thrombocytopenic purpura. Blood
2008. 112:11-18.
13. Segers, K, Dahlback, B, and Nicolaes, GA.
Coagulation factor V and thrombophilia:
background and mechanisms. Thromb Haemost
2007. 98:530-542.
14. Vincent, LM, Tran, S, Livaja, R, Bensend, TA,
Milewicz, DM, and Dahlback, B. Coagulation
factor V(A2440G) causes east Texas bleeding
disorder via TFPIalpha. J Clin Invest 2013.
123:3777-3787.
Second Advanced Training Course Manual.indd 14-15 2/27/14 10:23 AM

16 17
David Lillicrap Queen’s University, Department of Pathology and Molecular Medicine Kingston, Ontario, Canada
Hemophilia A and B are the two most common forms of severe inherited bleeding disease with incidence rates of approximately 1 in 5,000 and 1 in 25,000 male births, respectively. Both conditions are transmitted as X-linked recessive traits, with the majority of affected subjects being males. However, due to variations in the pattern of X inactivation, approximately 20% of female carriers of hemophilia can also express a mild bleeding tendency, often manifest as menorrhagia.
The diagnosis of hemophilia is made through a combination of clinical and laboratory features. In approximately 60% of cases there will be a preceding family history of the condition. The clinical manifestations of hemophilia are very similar for deficiencies of factor VIII (FVIII) and factor IX (FIX), although there is some suggestion that severe FIX deficiency is less problematic than severe hemophilia A. Severe disease (factor levels <1%) results in frequent episodes of spontaneous musculoskeletal bleeding (~20-30/yr) with clinical events starting around 1-2 years of age. The long-term outcome of these bleeding episodes, if not adequately treated, is a chronic disabling arthropathy most often affecting the ankles, knees and elbows. Patients with moderately severe disease (factor levels 1-5%) usually do not manifest spontaneous bleeding. Finally, patients with mild hemophilia (factor levels 5-40%) only bleed on provocation and can sometimes present late in adult life at the time of a surgical or dental intervention.
The laboratory diagnosis of hemophilia requires an accurate assessment of FVIII or FIX levels with one-stage clotting assays. There is no advantage to using an alternate factor assay (ie. a chromogenic substrate assay) for diagnosis, unless there is concern about a discrepancy between the severity of the patient’s bleeding symptoms and the results of the one-stage assay. In many centers, the initial phenotypic diagnosis of hemophilia is complemented with molecular genetic testing to identify the causative FVIII or FIX mutation. Hemophilia mutation information can be used for family planning purposes by the patient (carrier detection and possible prenatal testing), and the type of mutation can be incorporated into the risk profile for developing an immune response to replacement therapy.
Current treatment of hemophilia involves various forms of protein replacement therapy. In the past, a wide variety of plasma-derived FVIII and FIX concentrates have been extensively used, and since the institution of donor screening programs and effective viral inactivation protocols the transmission of infectious agents by these concentrates has been eliminated. Since the early 1990s, there has been an increasing utilization of recombinant clotting factor concentrates and we are now in the midst of a period where new concentrates with prolonged circulating half-lives are being introduced. In addition, other novel approaches to hemophilia therapy are also being explored and include the generation of inhibitory protocols for the natural anticoagulant pathways and the development of a FVIII mimetic molecule. Finally, after many examples of successful pre-clinical studies of hemophilia gene therapy there is now evidence that AAV-mediated gene transfer is feasible and results in long-term expression of therapeutic FIX levels in hemophilia B.
S2 Challenges in the Diagnosis and Management of the Hemophilias
Recommended reading:
1. The hemophilias--from royal genes to gene
therapy. Mannucci PM, Tuddenham EG. N Engl J Med. 2001 Jun 7;344(23):1773-9
2. Coagulation factor concentrates: past,
present, and future. Key NS, Negrier C.
Lancet. 2007 Aug 4;370(9585):439-48.
3. How we choose factor VIII to treat
hemophilia. Mannucci PM, Mancuso
ME, Santagostino E. Blood. 2012 May
3;119(18):4108-1410.
4. How we treat a hemophilia A patient with a
factor VIII inhibitor.Kempton CL, White GC
2nd. Blood. 2009 Jan 1;113(1):11-7.
5. Adenovirus-associated virus vector-
mediated gene transfer in hemophilia B.
Nathwani AC et al, N Engl J Med. 2011
Dec 22;365(25):2357-65.
S3 Diagnosis and Management of von Willebrand Disease
David Lillicrap Queen’s University, Department of Pathology and Molecular Medicine Kingston, Ontario, Canada
The index case of von Willebrand disease was described on one of the Aland Islands in the Baltic Sea in 1926. This teenage girl bled to death with her 4th menstrual period. Subsequent investigation of this family and others in the Aland archipelago, some 80 years later, demonstrated that they were affected by type 3 von Willebrand disease (VWD).
von Willebrand disease is the most common inherited bleeding disorder in humans, with a prevalence of symptomatic subjects of approximately 1 in 1,000. In all documented VWD populations, females outnumber males by 2:1 due presumably to the enhanced likelihood of manifesting excessive mucocutaneous bleeding at the time of menses and childbirth. There are three subtypes of VWD: type 1 disease is a quantitative deficiency of functionally normal VWF. In most populations this accounts for ~65% of VWD cases. Type 2 VWD represents a group of qualitative VWF variants (types 2A, 2B, 2M and 2N) comprising approximately 30% of VWD and finally, type 3 VWD is the virtual absence of VWF (with accompanying very low levels of FVIII) occurring in approximately 1 in 1 million of the population.
The diagnosis of VWD requires consideration of three components: a personal history of excessive mucocutaneous bleeding, laboratory test results consistent with VWD, and finally, a family history of the condition. Recently, there has been a resurgence of interest in the utility of bleeding assessment tools (bleeding scores) to quantify bleeding symptoms. These tools have now been thoroughly validated in the diagnosis of VWD. In the laboratory, the key tests to make this diagnosis are the VWF:Ag and a VWF activity assay, which until very recently has usually involved performance of the VWF:RCo test. The temporal variability of VWF can sometimes make these assessments problematic, and repeat testing is often required to confirm or refute the diagnosis of type 1 VWD, especially. Recently, the VWF:RCo assay, that is infamously difficult to standardize, has started to be replaced with direct GPIb binding assays that appear to be more consistent and sensitive than the VWF:RCo test.
Over the past 15 years, substantial progress has been made in terms of the molecular genetic pathology responsible for VWD. The mutations responsible for the various type 2 subtypes of VWD have now been well documented, and the incorporation of molecular testing for confirmation of these variants is increasingly being employed. Similarly, the genetic background of type 3 VWD has also been extensively studied and can now be applied to questions concerning family planning and prenatal diagnosis. In contrast, the molecular genetics of type 1 disease remains challenging. Studies of >500 index cases of type 1 VWD indicate that only 65% of cases have candidate mutations in the coding regions and splice junctions of the VWF gene, and the pathogenic nature of some of these variants remains in question. Thus, the application of molecular analysis for the routine diagnosis of type 1 VWD is currently not recommended.
The prevention and treatment of bleeding in VWD has changed very little over the past two decades. Many cases of type 1 VWD and some type 2 cases can be treated with desmopressin, and the remaining cases will require infusion with plasma-derived VWF-FVIII concentrates.
Recommended reading:
1. Principles of care for the diagnosis and
treatment of von Willebrand disease.
Castaman G, Goodeve, Eikenboom J;
European Group on von Willebrand
Disease. Haematologica. 2013
May;98(5):667-74.
2. Making a diagnosis of VWD. Branchford
BR, Di Paola J. Hematology Am Soc Hematol Educ Program. 2012;2012:161-
7.
3. The molecular characterization of von
Willebrand disease: good in parts. James
PD, Lillicrap D. Br J Haematol. 2013
Apr;161(2):166-76.
4. Treatment of von Willebrand disease with
FVIII/VWF concentrates. Castaman G.
Blood Transfus. 2011 May; 9 Suppl 2:s9-
13.
Blood Coagulation and Bleeding Disorders
Second Advanced Training Course Manual.indd 16-17 2/27/14 10:23 AM

18 19
S5 Hemostasis in Patients with Impaired Liver Function
Philip G. de Groot Laboratory of Clinical Chemistry and Haematology, University Medical Center Utrecht, the Netherlands
Chronic liver disease is a major cause of mortality and morbidity in many countries. Chronic or acute liver failure results in substantial changes in the hemostatic system. The liver is involved in the synthesis of most of the clotting factor proteins and reduced amounts of these proteins are found in the circulation with the exception of factor VIII. Moreover, the liver has lost partly its capacity to clear activated clotting factors-inhibitor complexes. Liver failure also results in a reduced platelet count and platelet function. All these defects are counterbalanced by a concomitant defect in anticoagulant and pro-fibrinolytic factors. Moreover, a decreased platelet function is counterbalanced by elevated levels of von Willebrand factor. The classic assays to detect a bleeding disorder, the prothrombin time (PT) and the activated partial thromboplastin time (APTT) do not correlate with a bleeding tendency because these assays do not measure the reduced activity of the physiological inhibitors such as antithrombin. The thrombin generation assay, an assay that is sensitive for these inhibitors, is often within a normal range in patients with liver cirrhosis, indicating that in these patients the hemostasic system is rebalanced. This rebalance is represented by a limited bleeding during surgery, including liver transplantation and by the thrombotic complications regularly seen after surgery. In this lecture I will discuss this rebalance and explain that this balance is less stable. The balance easily tip towards a hyper-or hypocoagulable state. Coagulopathy in patients with critical liver dysfunction is complex and can quickly decompensate to bleeding as well as to thrombosis. Liver cirrhosis is a unique clinical setting in which bleeding and thrombosis coexist.
Recommended reading:
1. Lisman T, Leebeek FW & de Groot PG. (2002) Haemostatic abnormalities in patients with liver
disease. J Hepatol 37: 280-287.
2. Lisman T, Caldwell SH, Burroughs AK, Northup PG, Senzolo M, Stravitz RT, Tripodi A, Trotter
JF, Valla DC, Porte RJ; Coagulation in Liver Disease Study Group (2010) Hemostasis and
thrombosis in patients with liver disease: the ups and downs. J Hepatol. 53: 362-71.
3. Nowatari T, Murata S, Fukunaga K, Ohkohchi N. (2013) Role of platelets in chronic liver
disease and acute liver injury. Hepatol Res. Jul 11 [Epub ahead of print].
4. Arshad F, Lisman T, Porte RJ.(2013) Hypercoagulability as a contributor to thrombotic
complications in the liver transplant recipient. Liver Int. 33: 820-7.
5. Northup PG, Argo CK, Shah N, Caldwell SH (2012) Hypercoagulation and thrombophilia in
nonalcoholic fatty liver disease: mechanisms, human evidence, therapeutic implications, and
preventive implications. Semin Liver Dis. 32: 39-48.
6. Tripodi A, Primignani M, Chantarangkul V, et al. (2009) An imbalance of pro- vs
anticoagulation factors in plasma from patients with cirrhosis. Gastroenterology 137: 2105–
11.
Bernd Pötzsch Institute of Experimental Hematology and Transfusion Medicine University Hospital Bonn Bonn, Germany
Bleeding disorders can be inherited or acquired and include coagulation factor deficiencies, hyperfibrinolysis, platelet deficiencies and/or dysfunctions, and von Willebrand´s disease (vWD). The initial evaluation of a patient with a suspected bleeding disorder should include a comprehensive medical and bleeding history, a complete family history, a detailed physical examination and selected laboratory tests.
The bleeding history may provide important clues about the likelihood of a bleeding disorder and the type of the bleeding disorder. For example mucocutaneous bleeding such as petechiae, bruising, epistaxis, gastrointestinal bleeding and/or menorrhagia suggests disorders of platelets, von Willebrand factor (vWF) or a vascular bleeding disorder whereas bleeding into muscles and joints, soft tissues and delayed surgical bleeding suggests disorders of coagulation factors. The use of standardized scores to quantitate bleeding disorders is recommended. Standardized and validated bleeding questionnaires are available (Biss 2010).
Physical examination should evaluate the localization, size, and age of hematomas and the presence of any signs of bleeding such as hemarthroses or evidence of chronic joint abnormalities. Signs of coexisting illness may be indicative for acquired bleeding disorders. For example lymphadenopathy and/or organomegaly suggest an infiltrative process such as malignancy while signs of liver failure suggest acquired coagulation factor deficiencies.
Initial tests to screen for bleeding disorders should include a complete blood count (CBC), blood film, whole blood platelet function testing, prothrombin time (PT), activated partial thromboplastin time (APTT), and factor XIII testing. The CBC is performed to exclude thrombocytopenia and to detect additional pathologies of white blood cells and red cells. The blood film provides further information regarding platelet and leukocyte morphology. Several point-of-care tests measuring the platelet function in whole blood are commercially available. For example the platelet function analyzer (PFA) provides a simple and rapid assessment of high shear-dependent platelet function. This test is successfully used in the screening for vWD but lacks sensitivity for some congenital platelet disorders such as patients with P2Y12 deficiency and patients with granule deficiencies. The PT and APTT measure the activity of all coagulation factors that are involved in the generation of thrombin and thrombin-dependent fibrin formation. Clotting times of both assays within the age-specific reference ranges make the presence of a clinical relevant clotting factor deficiency unlikely although it should be noted that an aPTT within the reference range does not reliably exclude mild FVIII, FIX or FXI deficiency. Therefore, factor assays should be performed if the bleeding history or the family history suggests a mild bleeding disorder. Testing of FXIII activity is included into the laboratory screen because the PT and aPTT are not sensitive for FXIII.
S6 How to Approach a Patient with Bleeding
Blood Coagulation and Bleeding Disorders
Continued next page
Second Advanced Training Course Manual.indd 18-19 2/27/14 10:23 AM

20 21
Based on the initial test findings, plus the degree of clinical evidence, further evaluation may or may not be required. A negative bleeding history together with screening tests within the reference ranges make the presence of a bleeding disorder most unlikely and no further testing is recommended. Abnormal results of the initial screen require additional testing. For example, a prolonged closure time in the PFA together with a slightly prolonged APTT requires further testing for vWD including vWF-Ag ELISA and functional vWF assays. In patients with a positive bleeding history and no evidence of pathological laboratory tests it is difficult to establish a final diagnosis. In those cases a diagnosis of a bleeding disorder of unknown causes should be made.
Recommended reading:
1. Rodeghiero E, Kadir RA, Tosetto A, James PD. Relevance of quantitative assessment of
bleeding in haemorrhagic disorders. Haemophilia 2008; 14 (suppl 3): 68
2. Biss TT, Blanchette VS, Clark DS, Bowman M, Wakefield CD, Silva M, Lillicrap D, James PD,
Rand ML. Quantitation of bleeding symptoms in children with von Willebrand disease: use of a
standardized pediatric bleeding questionnaire. J Thromb Haemost 2010; 8: 950
3. Harrison P, Mackie I, Mumford A, Briggs C, Liesner R, Winter M, Machin S. Guidelines for the
laboratory investigation of heritable disorders of platelet function. Br J Haematol 2011; 155:
30
4. Hayward CP. Diagnosis and management of mild bleeding disorders. Hematology Am Soc Hematol Educ Program 2005; 2005: 423
Continued: S6 How to Approach a Patient with Bleeding
Venous Thrombosis
S7 Venous Thrombosis: Manifestations, Diagnosis and Therapy
Blood Coagulation and Bleeding Disorders
Sam Schulman McMaster University, Clinical Thromboembolism Program Hamilton, Ontario, Canada
The incidence of venous thromboembolism (VTE) has been reported from many population studies as approximately 100 per 100,000 but is influenced by selection of adults or certain age range and the use of autopsy. The incidence has often been quoted as lower in Asia, but there may be confounding by suspicion bias. The best estimate of difference between races is derived from a very large population cohort in California, demonstrating a decreasing rate from African Americans via whites and Hispanics to the lowest among Asian and Pacific islanders. Whereas there is a well-described exponential increase with age, the influence by sex is controversial. Most cohort data indicate a higher risk for young women and possibly for old men. A seasonal variation with peaks in December-January has been ascribed to variations in the fibrinogen level; in turn a possible result of respiratory tract infections.
More specifically, the incidence of deep vein thrombosis (DVT) is often stated to be 50-100% higher than that of pulmonary embolism (PE). This is logical since PE is considered as almost always originating from the leg veins. Patients with venographically confirmed DVT have asymptomatic pulmonary embolism on lung-scan in 30-50% whereas 70% of carefully investigated patients with PE turn out to have silent DVT. Typical, yet unspecific symptoms of DVT are pain deep in the calf or thigh, and unilateral swelling. Increased skin temperature, tenderness and discoloration are even less specific. In case of essentially total obstruction of venous return the leg becomes cyanotic and very painful – phlegmasia cerulea dolens – and with massive edema a compartment syndrome with closure also of the arterial circulation the leg becomes pale and very painful – phlegmasia alba dolens. Both conditions may lead to loss of the limb. For PE the most common symptoms in decreasing order are pleuritic pain (65%), dyspnea (20%), syncope (10%) and hemoptysis (few cases). The diagnosis of VTE using clinical symptoms and signs has low sensitivity and specificity. A clinical probability assessment is, however, valuable as part of a diagnostic algorithm. For low clinical probability a negative D-dimer test usually excludes the diagnosis. For high clinical probability, imaging diagnostic techniques are required, typically compression ultrasound for suspected DVT and computed tomography of pulmonary arteries for suspected PE, although in some cases ventilation-perfusion scan is advantageous. The standard initial treatment today is subcutaneous low-molecular-weight heparin (LMWH) without monitoring. Patients with DVT can generally be managed as outpatients. For those with PE a risk stratification tool should be used to select for outpatient treatment. A vitamin K antagonist should be started simultaneously, overlapping at least 5 days and then continue for 3-6 months. At that point an assessment of risk vs. benefit of long-term anticoagulation should be performed. For patients with massive PE and hemodynamic instability thrombolytic therapy is indicated. Patients with massive DVT should be assessed for catheter-directed thrombolysis +/- mechanical removal.
Over 10 years about 30% of patients with unprovoked VTE will have a recurrence and DVT will almost always recur as lower extremity thrombosis. Conversely, PE usually recurs as PE. (Continued next page)
Second Advanced Training Course Manual.indd 20-21 2/27/14 10:23 AM

22 23
S7 Venous Thrombosis: Manifestations, Diagnosis and Therapy (Continued)
Case fatality is problematic to assess and influenced by the study methodology and autopsy rates. It is higher in PE, at least in the short-term perspective. At least 50, possibly 80% of patients with DVT develop venous insufficiency as part of the post-thrombotic syndrome and the severe form with venous ulcers has a linear increase, reaching 5% at 10 years.
The mortality in patients with VTE is higher than in controls matched for age and sex. It is also specifically higher than expected for cancer or myocardial infarction and ischemic stroke. Finally, the prevalence of VTE is projected to double until 2050.
Recommended reading:
1. Heit JA, Silverstein MD, Mohr DN, Petterson TM, Lohse CM, O’Fallon WM, Melton LJ, 3rd.
The epidemiology of venous thromboembolism in the community. Thromb Haemost. 2001; 86:
452-63.
2. Prandoni P, Lensing AW, Cogo A, Cuppini S, Villalta S, Carta M, Cattelan AM, Polistena P,
Bernardi E, Prins MH. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med. 1996; 125: 1-7.
3. Schulman S, Lindmarker P, Holmstrom M, Lärfars G, Carlsson A, Nicol P, Svensson E,
Ljungberg B, Viering S, Nordlander S, Leijd B, Jahed K, Hjorth M, Linder O, Beckman M.
Post-thrombotic syndrome, recurrence, and death 10 years after the first episode of venous
thromboembolism treated with warfarin for 6 weeks or 6 months. J Thromb Haemost. 2006; 4:
734-42.
4. Spencer FA, Emery C, Lessard D, Anderson F, Emani S, Aragam J, Becker RC, Goldberg RJ.
The Worcester Venous Thromboembolism study: a population-based study of the clinical
epidemiology of venous thromboembolism. J Gen Intern Med. 2006; 21: 722-7.
5. Kearon C, Akl EA, Comerota AJ, et al. Antithrombotic therapy for VTE disease: Antithrombotic
Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians
Evidence-Based Clinical Practice Guidelines. Chest 2012;141:e419S-94S.
6. Holbrook A, Schulman S, Witt DM, et al. Evidence-based management of anticoagulant
therapy. Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of
Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012;141:e89S-119S.
7. Garcia DA, Baglin TP, Weitz JI, et al. Parenteral anticoagulants. Antithrombotic therapy and
prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based
clinical practice guidelines. Chest. 2012;141:e24S-e43S.
8. Wells, PS, Anderson, DR, Rodger, M, et al. (2003). Evaluation of D-dimer in the diagnosis of
suspected deep-vein thrombosis. N Engl J Med 349, s. 1227-35.
9. Wells, PS, Anderson, DR, Rodger, M, et al. (2000). Derivation of a simple clinical model to
categorize patients probability of pulmonary embolism: increasing the models utility with the
SimpliRED D-dimer. Thromb Haemost 83, s. 416-20.
10. Bates SM, Jaeschke R, Stevens SM, Goodacre S, Wells PS, Stevenson MD, et al. Diagnosis of
DVT: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest
Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012;141:e351S-418S.
11. Prandoni P, Bilora F, Marchiori A, Bernardi E, Petrobelli F, Lensing AW, et al. An association
between atherosclerosis and venous thrombosis. N Engl J Med 2003;348:1435-1441.
Venous Thrombosis
S8 Antiphospholipid Syndrome
Philip G. de Groot Laboratory of Clinical Chemistry and Haematology, University Medical Center Utrecht, the Netherlands
The antiphospholipid syndrome is an auto-immune disease characterized by thrombotic complications in both arteries and veins as well as fetal losses in combination with the presence of so-called antiphospholipid antibodies in plasma of these patients. Antiphospholipid antibodies are a family of auto-antibodies that can be measured with different assays that determine the presence of closely related but not overlapping antibody populations. Although the presence of these autoantibodies is regarded as a common cause of thrombosis and pregnancy morbidity in individuals at an age under 50 years, the true frequency of clinical significant antiphospholipid antibodies in not known. The lack of large-scale prospective population studies, the multi-factorial nature of thrombosis and fetal loss and the lack of standardization of the assays to detect the presence of these antibodies are major hurdles to determine the magnitude of the anti-phospholipid antibody problem.
It is now generally accepted that the relevant auto-antibodies are not directed against negatively charged phospholipids but towards plasma proteins bound to these phospholipids. The most prominent antigen in APS is β2-Glycoprotein I (β2-GPI), a plasma protein with affinity towards anionic phospholipids. APS is an intriguing syndrome because we have difficulties to comprehend how the presence of auto-antibodies against β2-glycoprotein I increases the risk for thrombosis and fetal loss. β2-Glycoprotein I is a plasma protein without a clear function and individuals without this protein seem to be completely healthy. Moreover, the most relevant assay that we use to detect the presence of auto-antibodies against β2-glycoprotein I, a prolongation of a clotting assay named Lupus anticoagulant, express an opposite effect on coagulation as expected for a thrombotic risk. Prolongation of clotting assays points to a bleeding tendency, not a thrombotic tendency. Both the target of the auto-antibodies, β2-Glycoprotein I, and the detection method, lupus anticoagulant, do not give us a lead to the mechanism behind the increased thrombotic risk. Nevertheless, mouse models in which auto-antibodies against β2-glycoprotein I isolated from patients were used show abundantly clear that these auto-antibodies are the cause of the increased risk for thrombotic manifestations and pregnancy morbidity.
In the present lecture I will introduce the antiphospholipid syndrome and explain the difficulties in the diagnosis of the syndrome. I will discuss the relative importance of the different assay we have available to diagnose the syndrome. I will give some possible explanations why individuals with these auto-antibodies in their blood have such a high risk of severe thrombotic complications at a younger age. I will finish with the different treatment options.
Recommended reading:
1. Giannakopoulos B, Krilis SA. (2013) The
pathogenesis of the antiphospholipid
syndrome N. Engl. J. Med. 368: 1033-44
2. de Groot PG, Urbanus RT (2012) The
significance of auto-antibodies against β2-
Glycoprotein I. Blood 120: 266-74
3. de Groot PG, Meijers JC. (2011) β(2)
-Glycoprotein I: evolution, structure and
function. J. Thromb. Haemostas. 9: 1275-
1284.
4. Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza
I, Brey R, Crowther M, Derksen R, Erkan
D, Krilis S, Machin S, Pengo V, Pierangeli
S, Tektonidou M, Khamashta M. (2011)
Evidence-based recommendations for the
prevention and long-term management of
thrombosis in antiphospholipid antibody-
positive patients: report of a task force
at the 13th International Congress on
antiphospholipid antibodies. Lupus 20:
206-18
5. de Groot PG, Urbanus RT (2012) The
future of antiphospholipid antibody testing.
Sem. Thromb. Hemostas. 38: 412-20.
6. Barbhaiya M, Erkan D. (2013) Top
10 clinical research developments
in antiphospholipid syndrome. Curr Rheumatol Rep. 15: 367
7. Lockshin MD. (2013) Pregnancy and
antiphospholipid syndrome. Am J Reprod Immunol. 69: 585-7
8. Andreoli L, Chighizola CB, Banzato A,
Pons-Estel GJ, de Jesus GR, Erkan D;
on behalf of APS ACTION. (2013) The
estimated frequency of antiphospholipid
antibodies in patients with pregnancy
morbidity, stroke, myocardial infarction,
and deep vein thrombosis. Arthritis Care Res (Hoboken). Jul 16. [Epub ahead of
print]
Second Advanced Training Course Manual.indd 22-23 2/27/14 10:23 AM

24 25
Venous Thrombosis
S9 Women Issues and Thrombosis
Sabine Eichinger Medical University of Vienna, Department of Internal Med Vienna, Austria
Women are subject to specific hormonal changes which influence the coagulation and fibrinolytic systems and put them at increased thrombotic risk. During reproductive age use of hormone contraceptives, profertility ovarian stimulation or pregnancy alter the pro- and anticoagulant forces, while after menopause age-related aspects or hormone replacement therapy contribute to a hypercoagulable state.
Hormone contraceptive use increases the risk of venous thromboembolism (VTE) about 2- to 6-fold (1). The increased risk is related to the dose of estrogen but is also influenced by the type of progestogen (2). Non-oral hormonal contraceptives including the transdermal patch or the contraceptive vaginal ring are also associated with an increased risk of VTE (3). Evidence regarding the cardiovascular safety of progestogen-only methods of contraception is limited. A systematic review and meta-analysis of published data concluded that the use of progestogen-only contraceptives was not associated with an increased risk of VTE compared with non-users of hormonal contraception. However, the potential association between injectable progestogens and thrombosis requires further study (4).The relative risk of VTE compared with non-users among women using the levonorgestel-releasing intrauterine system (Mirena®) was low (RR 0.57 (95% CI; 0.41-0.81) (3).
Ovarian hyper-stimulation does increase the risk of thromboembolic disorders and peaks dramatically in pregnant women with the ovarian hyper-stimulation syndrome requiring hospital admission. Pregnancy is a major risk factor for thrombosis. The risk of thrombosis is increased throughout pregnancy and is particularly high after delivery. Anticoagulant thromboprophylaxis is prescribed with analogy to prophylaxis outside pregnancy and is not standardized. Low-molecular-weight heparin (LMWH) is the drug of choice for preventing pregnancy-related VTE, whereas in puerperium the oral anticoagulants can be alternatively used. Women who are candidates for antithrombotic prophylaxis are those with a previous VTE or with a known severe inherited or acquired thrombophilia. Which type of thrombophilia does increase significantly the risk of VTE during pregnancy, suggesting the appropriateness of LMWH prophylaxis, is matter of debate. For the treatment of acute VTE in pregnant women fixed-dose, weight-adjusted subcutaneous LMWH is the anticoagulant of choice and should be given at a therapeutic dose throughout pregnancy (5). LMWH should be discontinued 24 hours before induction of labour or caesarean section, re-started at a reduced dose when it is safe to do so and continued for an additional six to eight weeks.
Menopause is accompanied by processes of physiological aging which is associated with increased plasma levels of many proteins of blood coagulation, alterations of platelets and fibrinolysis impairment. Hormone replacement therapy poses a specific thrombotic risk to women. Hormone replacement therapy contains estrogen and is combined with a progestogen in women who still have their uterus. Hormone replacement therapy during menopause is associated with a two- to four-fold increased risk of deep vein thrombosis (6). There is evidence that the thrombotic risk depends on the route of estrogen administration. In a population based study using the data set of about one million women, a higher risk for venous thrombosis was seen in women using oral compared to transdermal hormone
replacement therapy and a risk being greatest in users of oral formulations containing medroxyprogesterone acetate (7). Hormone replacement therapy also confers an increased risk of recurrent venous thrombosis. Oral hormone replacement therapy increases not only the risk of venous thrombosis but also of stroke (8). The risk is higher with advancing age and if additional risk factors, such as obesity, previous thromboembolic disease, smoking, and immobility are present. In otherwise healthy women younger than 60 years the absolute risk of thromboembolic disease is low.
Recommended reading:
1. van Hylckama Vlieg A, Middeldorp S. Hormone therapies and venous thromboembolism: where
are we now? J Thromb Haemost 2011;9:257-66.
2. Lidegaard Ø, Nielsen LH, Skovlund CW, Skjeldestad FE, Løkkegaard E. Risk of venous
thromboembolism from use of oral contraceptives containing different progestogens and
oestrogen doses: Danish cohort study, 2001-9. BMJ 2011;343:d6423.
3. Lidegaard O, Nielsen LH, Skovlund CW, Løkkegaard E. Venous thrombosis in users of non-oral
hormonal contraception: follow-up study, Denmark 2001-10. BMJ 2012;344:e2990.
4. Mantha S, Karp R, Raghavan V, Terrin N, Bauer KA, Zwicker JI. Assessing the risk of venous
thromboembolic events in women taking progestin-only contraception: a meta-analysis. BMJ. 2012;345:e4944
5. Arya R. How I manage venous thromboembolism in pregnancy. Br J Haematol. 2011;153:698-
708
6. Hickey M, Elliott J, Davison SL. Hormone replacement therapy. BMJ. 2012;344:e763
7. Sweetland S, Beral V, Balkwill A, Liu B, Benson VS, Canonico M, Green J, Reeves GK; Million
Women Study Collaborators1. Venous thromboembolism risk in relation to use of different
types of postmenopausal hormone therapy in a large prospective study. J Thromb Haemost. 2012;10:2277-86
Second Advanced Training Course Manual.indd 24-25 2/27/14 10:23 AM

26 27
Sam Schulman McMaster University, Clinical Thromboembolism Program Hamilton, Ontario, Canada
During the past decade many studies on highly specific, orally available anticoagulants in the treatment of venous thromboembolism (VTE) and for stroke prophylaxis in atrial fibrillation (SPAF) have been published. This story started with the first oral thrombin inhibitor, ximelagatran, but the drug was withdrawn early from the market due to liver toxicity. In 2009 the phase III studies on the next oral thrombin inhibitor were published, showing in comparison with warfarin similar or improved efficacy for SPAF and similar efficacy in treatment of VTE. There was also a reduction of some bleeding outcomes. Subsequently, the pattern has been repeated with oral direct factor Xa inhibitors. The phase III program of the first three of those agents has been fully presented and the drugs has been approved for SPAF in many jurisdictions and one drug, rivaroxaban, also for treatment and for extended secondary prophylaxis of VTE.
These new agents appear to provide the same efficacy as the combination of low-molecular-weight heparin overlapping with a vitamin K antagonist for the typical patients with deep vein thrombosis and pulmonary embolism. It is therefore anticipated that the new agents will slowly take over the market for this indication. The safety is an important component in this development. All new anticoagulants showed a reduced risk for intracranial hemorrhage compared to vitamin K antagonists in the SPAF studies, and this is probably the same in the smaller venous thromboembolism trials. Intracranial hemorrhage is the most feared complication of anticoagulation and therefore the new agents should lead to a lower resistance against treating patients appropriately for longer periods. Some of the drugs were started in the trials with only one or two initial doses of parenteral therapy, whereas others had a full week of overlap. It is not unlikely that physicians will prefer the initial parenteral therapy for patients with extensive deep vein thrombosis with significant pain and swelling of the leg. Likewise, patients with large pulmonary emboli may also be treated with parenteral therapy in the hospital until they are completely stable.
In the extended treatment, typically beyond 6 months trials have assessed low-dose warfarin, new anticoagulants and aspirin. There seems to be a trade-off between efficacy and safety that can be used to tailor the treatment according to the preferences and concerns of each patient. It can thus be anticipated that a patient fearing mostly a recurrence of venous thromboembolism will receive the most effective anticoagulant. Conversely, a patient with primarily fear of bleeding will be treated long-term with an anticoagulant drug with slightly lower efficacy but no increase of bleeding versus placebo or with low-dose acetylsalicylic acid if there is an increased risk for arterial thromboembolism.
If the anticoagulant treatment will be extended indefinitely, the risk for recurrent venous thromboembolism will be reduced and thereby reasonably also the postthrombotic syndrome, with becomes very burdensome and costly for some patients. Progress in this field may reduce days off from work and treatment expenditures for management of venous ulcers.
Special patient groups that demonstrate extreme hypercoagulability were hardly included in these studies. Thus, patients with active cancer constituted only 4-6% of the study
Venous Thrombosis
populations. There has not been any signal that the new anticoagulants are less effective than vitamin K antagonists in this subset. The standard treatment for patients with active cancer and thrombosis is, however, low-molecular-weight heparin for 3-6 months and the new anticoagulants should be evaluated against this comparator to convince prescribing physicians. Furthermore, patients with antiphospholipid syndrome can also be very hypercoagulable and need to be studied with the new agents.
A concern with the new and very convenient new anticoagulants is that family practitioners may bypass the diagnostic imaging in case of high degree of suspicion and directly prescribe an oral thrombin- or factor Xa inhibitor. Another issue is that non-hematologist physicians may not take their time to explain to the patients the rationale for anticoagulant treatment, the potential consequences of poor compliance and actions to take in case of side effects. If the result is that the patients at an early stage drop the anticoagulant treatment, the result will be an increase of the recurrent thromboembolic events. Education and educational tools is therefore key for the success of these new agents.
Recommended reading:
1. Weitz JI, Eikelboom JW, Samama MM. New antithrombotic drugs: Antithrombotic Therapy
and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based
Clinical Practice Guidelines. Chest 2012;141:e120S-51S.
2. Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial
fibrillation. N Engl J Med. 2009;361:1139-1151.
3. Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute
venous thromboembolism. N Engl J Med. 2009;361:2342-2352.
4. Wolowacz SE, Roskell NS, Plumb JM, et al. Efficacy and safety of dabigatran etexilate for
the prevention of venous thromboembolism following total hip or knee arthroplasty: a meta-
analysis. Thromb Haemost. 2009;101:77-85.
5. Patel MR, Mahaffey KW, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365:883-91.
6. Bauersachs R, Berkowitz SD, Brenner B, et al. Oral rivaroxaban for symptomatic venous
thromboembolism. N Engl J Med. 2010;363:2499-510.
7. Buller HR, Prins MH, Lensing AW, et al. Oral rivaroxaban for the treatment of symptomatic
pulmonary embolism. N Engl J Med 2012;366:1287-97.
8. Granger CB, Alexander JH, McMurray JJ, et al. Apixaban versus warfarin in patients with atrial
fibrillation. N Engl J Med. 2011;365:981-92.
9. Connolly SJ, Eikelboom J, Joyner C, et al. Apixaban in patients with atrial fibrillation. N Engl J Med. 2011;364:806-17.
10. Agnelli G, Buller HR, Cohen A, et al. Oral apixaban for the treatment of acute venous
thromboembolism. N Engl J Med. 2013;369:799-808.
11. Giugliano RP, Ruff CT, Braunwald E, et al. Edoxaban versus warfarin in patients with atrial
fibrillation. N Engl J Med. 2013;369:2093-104.
12. The Hokusai-VTE Investigators. Edoxaban versus warfarin for the treatment of symptomatic
venous thromboembolism. N Engl J Med. 2013;369:1406-15.
S10 Novel Antithrombotic Drugs
Second Advanced Training Course Manual.indd 26-27 2/27/14 10:23 AM

28 29
Bernd Pötzsch Institute of Experimental Hematology and Transfusion Medicine, University Hospital Bonn Bonn, Germany
Once the diagnosis of venous thrombosis has been established and initial anticoagulant treatment has been started using low molecular weight heparin or rivaroxaban/epixaban the physician is faced with the question how long the anticoagulant therapy should be continued and which type of oral anticoagulant should be used.
In patients developing a deep venous thrombosis (DVT) during a typical risk situation such as surgical intervention the majority of guidelines such as the 2012 American College of Physicians Evidence-based Clinical Practice Guidelines recommend anticoagulant treatment for 3 months. There is further consensus that patients developing DVT outside a typical risk situation will benefit from extended anticoagulant treatment. However, it is still a matter of debate if those patients should be tested for endogenous thrombophilic risk factors including APC resistance/FV-Leiden mutation, protein C/S, antithrombin, prothrombin-G20210A-mutation, lupus anticoagulant/antiphospholipid antibodies and PNH. Although it has been shown that the development of unprovoked DVT by itself indicates a high risk of recurrence and therefore justifies extended anticoagulant treatment, the results of the thrombophilia screen give further information on the overall risk situation and might be helpful in tailoring the duration of the anticoagulant treatment. For example, patients tested positive for antiphospholipid antibodies should receive anticoagulant treatment until stable remission of the antiphospholipid antibodies occurs but do not require life-long anticoagulant treatment. Furthermore, relatives carrying the same mutation as the index patient might benefit from thromboprophylaxis when undergoing typical risk situations.
Another important question is that of testing for occult cancer. Although unprovoked thrombosis is a classical symptom of a paraneoplastic syndrome there is no evidence that patients benefit from an extensive cancer search. Therefore it is recommended to restrict the cancer search to patients with other cancer symptoms such as unexplained weight loss, fatigue, fever, etc..
As randomized clinical trials focused on the anticoagulant treatment of thrombosis at uncommon sites are lacking recommendations of the anticoagulant treatment of thrombosis of the cerebral veins and sinus, gastrointestinal tract, portal veins and renal veins are based on personal experience and retrospective studies. Pros and cons of long-term anticoagulant treatment in these patients will be discussed.
Two cohorts of patients represent major treatment challenges. These are thrombosis patients with underlying diseases that predispose them to a high risk of bleeding and patients who develop thrombosis while under oral anticoagulant treatment. Typical examples of patients who are at high risk of bleeding are patients with ulcerative colitis. In those patients the use of oral anticoagulants is associated with a high bleeding risk and parenteral anticoagulants such as low molecular weight heparins or fondaparinux are preferred. Patients developing thrombosis while under treatment with anticoagulants should undergo an extended screening for an underlying malignant disease. A d-dimer based algorithm for increasing the anticoagulant intensity in those patients will be discussed.
Finally the management of thrombosis in childhood will be discussed, although compared to adults, venous thrombosis in children are relatively uncommon. In most children the venous thrombosis result from secondary complications of primary underlying diseases such as infection, cancer, congenital heart disease, inflammatory conditions or are related to therapeutic interventions such as central venous catheters.
Recommended reading:
1. Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G. Oral anticoagulant
therapy – antithrombotic therapy and prevention of thrombosis, 9th ed: American College of
Chest Physicians evidence-based clinical practice guidelines. Chest 2012; 141: e44S – e88S
2. Kearon C, Akl EA, Comerota AJ, Prandoni P, Bounameaux H, Goldhaber SZ, Nelson ME, Wells
PS, Gould MK, Dentali F, Crowther M, Kahn SR. Antithrombotic therapy for VTE disease –
Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest
Physicians evidence-based clinical practice guidelines. Chest 2012; 141: e419S-e494S
3. Kleinjan A, Aggarwal A, van de Geer A, Faselis C, Büller HR, Di Nisio M, Rickles FR,
Kamphuisen PW. A worldwide survey to assess the current approach to the treatment of
patients with cancer and venous thromboembolism. Thromb Haemost 2013; 110: 959-965
4. Lee AY, Peterson EA. Treatment of cancer-associated thrombosis. Blood 2013; 122: 2310-
2317
5. Goodin S. Selecting an anticoagulant for recurrent venous thromboembolism in cancer. Am J Health Syst Pharm 2005; 62: S10-S13
6. Liew A, Eikelboom JW, O´Donnell M, Hart RG. Assessment of anticoagulation intensity and
management of bleeding with old and new oral anticoagulants. Can J Cardiol 2013; 29:
S34-S44
7. Cardiovascular disease educational and research trust, European venous forum, north American
thrombosis forum, international union of angiology, union international du phlebologie.
Prevention and treatment of venous thromboembolism: international consensus statement
(guidelines according to scientific evidence). Clin Appl Thromb Hemost 2013; 19: 116-118
8. Liew A, Douketis J. Initial and long-term treatment of deep venous thrombosis: recent clinical
trials and their impact on patient management. Expert Opin Pharmacother 2013; 14: 385-396
9. Schulman S. Optimal duration of anticoagulant therapy. Semin Thromb Hemost 2013; 39:
141-146
Venous Thrombosis
S11 How to Approach a Patient with Confirmed Venous Thrombosis
Second Advanced Training Course Manual.indd 28-29 2/27/14 10:23 AM

30 31
Sabine Eichinger Medical University of Vienna, Department of Internal Med Vienna, Austria
Patients at risk of thrombosis who need to undergo surgery or other invasive procedures are not only at an increased risk of thrombosis but also of bleeding. The thrombotic risk is composed by the patient’s intrinsic risk and the risk that is specific for the respective procedure. In analogy, the bleeding risk is influenced by patient characteristics and the type of surgery. Thus, for optimal perioperative management the thrombotic risk needs to be weighed against the bleeding risk.
Invasive procedures with low bleeding risk (including tooth extractions and other dental procedures, minor dermatologic surgical interventions, endoscopy with low bleeding risk, cataract surgery) should be performed without discontinuing of anticoagulant treatment (1). Of note, in case of treatment with a vitamin K antagonist the international normalized ratio (INR) should be around 2.5 (not higher than 2.8) on the day of the procedure. Patients who are treated with a direct oral anticoagulant should take the medication after the procedure rather than before.
Patients who undergo surgical interventions with a higher bleeding risk (including, organ biopsies, thoracal, urogenital or abdominal surgery) need to stop their anticoagulant treatment. Because of their long half-life vitamin K antagonists must be discontinued already several days before surgery which then can be safely performed once the INR is below 1.5 (2). Administration of vitamin K will enhance the normalization of the INR. Requirement of perioperative bridging with low molecular weight heparin depends on the patient’s thrombotic risk. In patients with low thrombotic risk (low risk atrial fibrillation, mechanic heart valve in aortic position without risk factors, VTE more than 12 months ago) preoperative heparin bridging should not be performed. All other patients should receive low molecular weight heparin at therapeutic (or half therapeutic doses in case of increased bleeding risk) already before surgery. Heparin at therapeutic doses needs to be stopped 24 hours before intervention and should not be given before 48 hours after surgery because of an otherwise increased bleeding risk. In a metaanalysis the risk of major bleeding in patients receiving heparin bridging was almost 4 times higher (Odds Ratio 3.60; 95% CI 1.52– 8.50) compared to the non-bridged cohort (3). As there was no difference in thromboembolic complications between the two groups, heparin bridging should be performed with caution and only in moderate and high risk patients.
Because of their short half-life, perioperative management of direct oral anticoagulant is less complex compared to vitamin K antagonists (4). In case of direct factor Xa inhibitors (rivaroxaban, apixaban), treatment should be interrupted 24 hours before surgery in patients without impaired renal function. The direct thrombin inhibitor dabigatran is primarily cleared by the kidneys. The time of discontinuation of the drug before an invasive procedures depends on the renal function of the patient and the bleeding risk and ranges between 24 hours and up to 5 days (in case of a creatinine clearance of less than 50 ml/min and a surgery with high bleeding risk) (5). Preoperative bridging with low molecular heparin is required for none of the direct oral anticoagulants.
Venous Thrombosis
S12 Perioperative Management in Patients with Risk for Thrombosis
Postoperatively, low molecular weight heparin at thromboprophylactic dose should be given except in patients undergoing total hip- or knee replacement in which case direct oral anticoagulants are licensed also for postoperative thromboprophylaxis. Oral anticoagulants can be started once hemostasis has been achieved and the surgeon considers it as safe. Notably, the maximum anticoagulant effect of direct oral anticoagulants is reached already two hours after administration.
Recommended reading:
1. Douketis JD, Spyropoulos AC, Spencer FA, Mayr M, Jaffer AK, Eckman MH, Dunn AS, Kunz R;
American College of Chest Physicians. Perioperative management of antithrombotic therapy:
Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest
Physicians Evidence-Based Clinical Practice Guidelines.Chest. 2012;141(2 Suppl):e326S-
50S
2. Spyropoulos AC, Douketis JD. How I treat anticoagulated patients undergoing an elective
procedure or surgery. Blood. 2012;120:2954-62.
3. Siegal D, Yudin J, Kaatz S, Douketis JD, Lim W, Spyropoulos AC. Periprocedural heparin
bridging in patients receiving vitamin K antagonists: systematic review and meta-analysis of
bleeding and thromboembolic rates. Circulation. 2012 ;126:1630-9.
4. Wysokinski WE, McBane RD 2nd. Periprocedural bridging management of anticoagulation.
Circulation. 2012;126:486-90.
5. Weltermann A, Brodmann M, Domanovits H, Eber B, Gottsauner-Wolf M, Halbmayer WM,
Hiesmayr JM, Kyrle PA, Längle F, Roithinger FX, Watzke H, Windhager R, Wolf C, Zweiker R.
Dabigatran in patients with atrial fibrillation: perioperative and periinterventional management.
Wien Klin Wochenschr. 2012;124:340-7
Second Advanced Training Course Manual.indd 30-31 2/27/14 10:23 AM

32 33
Christian Gachet Inserm, Université de Strasbourg Alsace, France
The essential role of blood platelets is to maintain vascular integrity and to ensure primary haemostasis, which means cessation of bleeding, upon vascular injury. The main platelet functions required to fulfil this role are 1) adhesion at sites of vascular damage through interaction of major membrane glycoproteins (the GPIb-V-IX complex), the Willebrand Factor circulating adhesive protein, and subendothelial matrix proteins (collagens, laminins, and fibronectin, among others); 2) activation triggered by multiple receptors and signalling pathways including integrins (α2β1, α6β1, αIIbβ3, αvβ3), immunoglobulin-like receptors (GPVI, CLEC-2) and G protein-coupled receptors for ADP (P2Y1, P2Y12), thrombin (PAR1, PAR4), thromboxane A2, serotonin, and adrenaline; 3) secretion of various alpha and dense granule contents such as adhesive proteins, coagulation and growth factors, nucleotides and serotonin among others; 4) aggregation via ligation of soluble fibrinogen to the αIIbβ3 integrin; 5) procoagulant activity by exposure of phosphatidylserine where coagulation factors bind and thrombin is generated. Activated platelets also expose proteins which allow interaction with leukocytes and endothelial cells.
Most of these steps occur almost simultaneously although they can be distinguished and described in a sequential way. The same mechanisms are triggered (involved, put into effect) when a thrombus forms in arteries while on the other end each of these functions can be altered to give rise to haemorrhagic diathesis.
Beyond haemostasis and its pathological counterpart thrombosis, platelets are also involved in many other physiological and pathophysiological processes, including tissue repair, angiogenesis, innate and acquired immunity, embryonic development, vascular inflammation, atherosclerosis and metastatic dissemination. The mechanisms involved in these roles are less well known and some of them may be specific such as the role of CLEC-2 in the organisation of the lymphatic vasculature during embryogenesis, TLR receptors during sepsis or particular integrins in cancer dissemination.
Platelets, Platelet Disorders
S13 Platelet Function
Ted Warkentin Hamilton Regional Laboratory Medical Program, Hematology Department Hamilton, Ontario, Canada
HIT is a prothrombotic (RR ~12.0) drug reaction caused by platelet-activating IgG that recognize multimolecular platelet factor 4 (PF4)/heparin complexes. Platelet activation is triggered when the PF4-heparin-IgG complexes cross-link FcβIIa receptors. Thrombotic effects arise from: (a) platelet activation, (b) procoagulant platelet-derived microparticles, (c) monocyte tissue factor expression, (d) endothelial activation (not proven), and (e) neutralization of heparin by PF4 released from activated platelets.
The typical clinical picture is an otherwise unexplained drop in the platelet count that begins 5-10 days into an immunizing heparin exposure. About 50-75% develop clinically evident thrombosis, most often deep-vein thrombosis (DVT) ± pulmonary embolism (PE). Less common venous thromboses include: adrenal vein thrombosis (= adrenal hemorrhage, either uni- or bilateral [risk of fatal adrenal failure]), mesenteric vein thrombosis, and cerebral venous dural sinus thrombosis. Upper-limb DVT occurs in association with central venous catheters. Arterial thrombosis most often causes acute limb ischemia (thrombosis of large limb arteries) > stroke > myocardial infarction. Rarely, skin necrosis occurs at sites of subcutaneous (sc) heparin injection. Patients can develop anaphylactoid reactions (fever/chills, hypertension, dyspnea, cardiac arrest, transient global amnesia) with 30min of an IV heparin bolus or within 2h of sc LMWH.
About 10-20% of patients develop overt DIC (elevated INR, greatly increased D-dimer, and [relative] hypofibrinogenemia: fibrinogen <3.0 g/L can indicate DIC, as post-operative patients usually have fibrinogen >4.5 g/L). Although severe HIT-associated DIC can cause acral limb necrosis in a limb with DVT, most HIT-associated venous limb ischemia (gangrene with pulses) occurs due to warfarin. Vitamin K antagonism is the most common cause of limb loss in HIT, due to profoundly disturbed procoagulant-anticoagulant balance: greatly increased thrombin generation (HIT) plus severe protein C depletion (warfarin); patients exhibit a supratherapeutic INR (>3.5; surrogate marker for severe protein C depletion via parallel severe depletion of factor VII).
HIT’s immune nature, featuring unusual antibody (Ab) transience, leads to its striking temporal features. HIT begins 5-10 days after into an immunizing course of heparin (first dose = day 0), irrespective of whether this is the first exposure, or whether many previous exposures have occurred. However, if a patient underwent a recent immunizing heparin exposure (within the past 100 days), then restarting heparin can cause abrupt platelet count fall (“rapid-onset HIT”), a profile seen in ~30% of cases. “Delayed-onset” (“auto-immune-like”) HIT features onset (or progression) of thrombocytopenia after heparin has been stopped; patients often have DIC and can fail therapy due to “PTT confounding.”
HIT frequency is variable, depending on (a) type of heparin (UFH > LMWH), (b) patient type (surgical > medical > obstetric/pediatric), (c) heparin duration (10 or more days > 5-10 days > less than 5 days), (d) sex (females > males). The role of dosing is complex: although prophylactic-dose (vs therapeutic-dose) heparin may be more immunogenic (due to PF4-heparin stoichiometry), the relationship is confounded by surgical (vs medical) S14
S14 Heparin-Induced Thrombocytopenia (HIT)
Continued next page
Second Advanced Training Course Manual.indd 32-33 2/27/14 10:23 AM

34 35
S14 Heparin-Induced Thrombocytopenia (HIT) Continued
patients being more likely to receive prophylactic-dose heparin. Nevertheless, a post-orthopedic surgery female patient receiving 10 days of UFH has HIT risk of ~5%, whereas a patient with a brief, highly-immunizing UFH exposure (cardiac surgery) but without ongoing postoperative heparin exposure has risk 0.02-0.05%, although the course likely will be severe (“delayed-onset HIT”).
“Spontaneous” HIT features a clinical and serological picture identical to HIT, except that no proximate heparin exposure is identified. Affected patients are either post-knee replacement (perhaps cartilage glycosaminoglycans substitute for heparin?) or have preceding infection (bacterial cell walls bind PF4 and can trigger anti-PF4/heparin Abs).
The modern concept of HIT is that patients MUST have detectable anti-PF4/heparin antibodies with platelet-activating properties. Although non-PF4-dependent antigens (e.g., IL-8) are proposed, these are not established. PF4-dependent EIAs have 99-100% sensitivity, though specificity is poor (20-50%); strength of optical density (OD) predicts for platelet-activating Abs (OD > 2.00 units ~90% frequency of a positive washed platelet serotonin-release assay [SRA]). Washed platelet activation assays (SRA, HIPA) performed by experienced labs is the gold standard. Many other PF4-dependent assays are becoming available, e.g., particle gel immunoassay, instrumentation-based assays.
Treatment principles of strongly-suspected HIT: 2 Do’s (stop heparin, begin alternative non-heparin anticoagulation), 2 Don’ts (don’t give warfarin [give vit K if VKA already given], avoid platelet transfusions, 2 Diagnostics (test for HIT Abs, image for lower-limb DVT). Two general approaches: (a) direct thrombin inhibitors (DTIs; argatroban, hirudin [NB: lepirudin discontinued], bivalirudin, [?dabigatran]), (b) Xa inhibitors (danaparoid, fondaparinux, [?rivaroxaban]). The Author favors Xa inhibitors (danap, fonda), as they (a) have prophylactic/therapeutic dosing regimens, (b) are effective in Ab+ HIT (whereas efficacy data for argatroban in Ab+ HIT is lacking), and (c) avoid PTT confounding (failure of PTT-adjusted anticoagulation due to misleading PTTs in setting of HIT-DIC).
Heparin re-exposure despite previous HIT is reasonable, e.g., need for cardiac surgery: (a) show that platelet-activating Abs are no longer present, (b) use UFH only intra-operatively, (c) use a non-heparin agent post-operatively, if needed (e.g., fonda). Recurrent HIT beginning 5-10d post-op is possible if the patient makes Abs that activate platelets without need for heparin (delayed-onset HIT), a scenario that has been reported.
Platelets, Platelet Disorders
Recommended reading:
1. Krauel K, ... Greinacher A. Platelet factor
4 binds to bacteria – inducing antibodies
cross-reacting with the major antigen in
heparin-induced thrombocytopenia. Blood 117:1370–8, 2011.
2. Warkentin TE. Fondaparinux: does it cause
HIT? can it treat HIT? Exp Rev Hematol
3:567, 2010.
3. Warkentin TE. HIT paradigms and
paradoxes. J Thromb Haemost 9 (Suppl
1):105-117, 2011.
4. Warkentin TE, Kelton JG. Temporal aspects
of HIT. N Engl J Med 344:1286-92,
2001.
5. Warkentin TE, Sheppard JI. Serological
investigation of patients with a
previous history of heparin-induced
thrombocytopenia who are re-exposed to
heparin. Blood 2014 in press.
6. Warkentin TE, et al. Fondaparinux
treatment of acute HIT confirmed by the
serotonin-release assay: a 30-month,
16-patient case series. J Thromb Haemost 9:2389-96, 2011.
7. Warkentin TE, Greinacher A, et al.
Laboratory testing for HIT: a conceptual
framework and implications for diagnosis.
J Thromb Haemost 9:2498-2500, 2011
Andreas Greinacher Institut für Immunologie und Transfusionsmedizin Universitätsmedizin Greifswald, Germany
Immune mediated thrombocytopenias are a heterogeneous group of platelet disorders caused by antibody- or T-lymphocyte-mediated platelet destruction. The underlying cause can be autoimmune disease (autoimmune thrombocytopenia [ITP]), alloantibody mediated (neonatal alloimmune thrombocytopenia [NAIT] and transfusion induced alloimmune thrombocytopenia), a mixed form with both characteristics (post transfusion purpura [PTP]), or drug dependent. Drug induced thrombocytopenias can be distinguished into several distinct groups with different pathogenesis: drug-dependent immune thrombocytopenia (dITP; e.g. by quinine), GPIIbIIa inhibitor induced thrombocytopenias (fibans and abciximab), hapten induced (e.g. penicillin) and heparin-induced thrombocytopenia (HIT). Rarely drugs (e.g. gold) can induce platelet autoantibodies and thereby trigger ITP.
Diagnostic approachesTests for platelet antibodies measure free antibodies in patient serum/plasma or platelet-bound antibodies. With the exception of HIT (and post transfusion purpura), laboratory diagnosis of immune mediated thrombocytopenias is limited due to the relatively low sensitivity of the available tests.
ITP: Platelet autoantibody testing has not been endorsed in recent evidence-based guidelines of the management of ITP patients due to low sensitivity of the assays (~60% for chronic ITP; for T-cell mediated platelet destruction no practicable assay is currently available). However, an evolving theme is that the ‘ITP syndrome’ is a disorder which may comprise groups of patients with distinct clinical and serological profiles. To better understand the diagnostic, prognostic and pathogenic role of platelet autoantibodies in ITP, further systematic evaluation is required. Direct tests for platelet autoantibodies, which measure antibodies bound to patient’s platelets, have higher sensitivity than indirect tests, which measure free antibodies in plasma or serum. Assays assessing glycoprotein-specific antibodies (e.g. against against GPIb/IX/V and GPIIb/IIIa), like MAIPA or monoclonal antigen capture EIA (MACE) are much more specific than assays measuring platelet-associated IgG e.g. by flow cytometry. Assays using platelet eluates are commercially available. They may improve the sensitivity of the laboratory diagnosis of ITP, but require further validation.
Alloimmune thrombocytopenias: the assays of choice measure platelet alloantibodies by indirect glycoprotein specific assays in the patient’s plasma/serum, or, in case of NAIT, antibodies in the maternal blood. Assays measuring antibody binding to whole platelets have a lower specificity (false positive signals caused by HLA antibodies) and sensitivity (antibodies against antigens with low copy numbers). Some alloantibodies are of low affinity, although they can cause significant platelet destruction in vivo. These low affinity antibodies are washed away during the washing steps in the current EIAs but can be detected by techniques not requiring washing such as surface plasmon resonance.
dITP and GPIIb/IIIa inhibitor induced antibodies: drug dependent antibodies are measured in the patient’s plasma/serum by indirect assays using whole platelets in the presence or absence of the drug. The read out is antibody binding measured by EIA or flow cytometry.
S15 Immune Thrombocytopenias
Continued next page
Second Advanced Training Course Manual.indd 34-35 2/27/14 10:23 AM

36 37
S15 Immune Thrombocytopenias Continued
These assays require appropriate controls, as the drug itself can cause false positive and false negative results by interfering with the assay reagents. It is important to add the drug to all assay steps, including to the washing solution, as the labile complex between drug, platelet glycoprotein and antibody will dissociate if the concentration of the drug becomes too low. In several cases not the pharmaceutical form of the drug but a metabolite causes the immune reaction. This is a major limitation for laboratories, as the metabolites are usually not available in the laboratory.
PathogenesisITP is not a single disease. Rather it describes several different causes which result in autoimmune mediated decrease of the platelet count. Increased platelet destruction resulting in shortened platelet survival is a well-accepted cause of the low platelet count in ITP. Beside classic antibody mediated platelet phagocytosis in the reticuloendothelial system, complement activation may lead to intravascular lysis of platelets (or CD35-mediated phagocytosis), and impaired platelet production may also contribute to the low platelet count. Megakaryocytes cultured in plasma of ITP patients show abnormal growth and increased apoptosis, and primed T-cells may directly attack the megakaryocytes. Another relatively newly evolving observation is the increased prevalence of arterial and especially venous thrombosis in ITP patients compared to the normal population. The reasons are not well understood. Intravascular destruction of platelets and resulting platelet microparticles may be one reason. In secondary ITP (see below) also indirect effects caused by the underlying disease may increase the risk for thrombosis.
ITP can occur in an acute and transient form or in a chronic form in which thrombocytopenia persists for >12 months. Acute ITP is often triggered by viral infections which, especially in children, typically precede the onset of bleeding symptoms by 1-2 weeks. Although platelet counts often decrease to very low values (<10,000/µL), associated with bleeding symptoms, acute ITP is usually self-limiting, has an excellent prognosis and often does not require specific treatment. Platelet antibody tests are negative in acute, postinfectious ITP of childhood, indicating a different pathogenesis than chronic ITP. Chronic ITP occurs at all age groups but typically affects more adults and shows a female preponderance. Platelet counts can vary widely and spontaneous bleeding symptoms often manifest at very low platelet counts only. ITP can be primary or secondary. In primary ITP no obvious other comorbidity is present, while secondary ITP (10-20% of ITPs) includes all other forms of ITP. Secondary ITP can be associated with other auto-immune disorders, most frequently systemic lupus erythematodes, antiphospholipid syndrome, and autoimmune thyreoditis. Thrombocytopenia may precede the other clinical manifestations by months or even years. Chronic lymphatic leukemia, or chronic infections (HCV, HIV, H. pylory) are also frequently associated with ITP. In chronic infections, antibodies against the pathogen may cross-react with structures on platelet glykoproteins; e.g. in HIV platelet autoantibodies cross react with the HIV receptor precursor GP160 and bind to AA 49-66 of GPIIIa. Similarly in HCV and H. pylory infections cross-reacting antibodies, which react with platelets have been described. An evolving and very interesting concept is that some autoantibodies crosslink GPIb, thereby inducing an outside-in signal and release of neuraminidase from platelet granules. This enzyme deglykosylates GPIb and exposes the binding side for the Ashwell receptor in the liver to which platelets bind and are phagocytosed independently of Fc-receptors. Taken together, the pathogenesis of different forms of ITP differs considerably. Prospective studies on ITP should include systematic assessment of platelet antibodies to better understand a potential correlation between certain antibodies, their correlation with the clinical course, and potentially even the efficacy of specific treatments.
Platelets, Platelet Disorders
In alloimmune thrombocytopenia, antibodies of an individual who has been immunized against a human platelet alloantigen (HPA) are transferred to another individual. The most frequent situation is pregnancy in which maternal antibodies against HPA-1a lead to thrombocytopenia in the fetus. Very rarely platelet alloantibodies are transfused with therapeutic plasma into a patient causing passive acute thrombocytopenia. A special form of alloantibody induced thrombocytopenia is post transfusion purpura (PTP). Patients who had been preimmunized against HPA-1a (typically women during previous pregnancies) are boosted by transfusion of blood from a HPA-1a positive donor, often many years after the immunizing event. The boosted antibodies show very high titers and, potentially by epitope spreading of B-cells, bind not only to the transfused HPA-1a positive platelets but also to HPA-1a negative platelets, showing autoantibody like activity.
dITP and GPIIbIIIa inhibitor induced thrombocytopenia: in dITP a complex is formed between an epitope on a platelet glycoprotein, the drug (or one of its metabolites) and antibodies. For some dITP antibodies it has been shown that the drug binds to the antibody, thereby changing its specificity in a way that the antibody binds with high affinity to an epitope expressed on platelets. Importantly, 50% of GPIIbIIIa inhibitor induced thrombocytopenias are laboratory artefacts resulting from pseudothrombocytopenia which occurs in EDTA AND in citrated blood.
Treatment of immune thrombocytopenias:ITP: the most important principle is not to treat platelet count numbers but to treat clinical bleeding symptoms. Many ITP patients are over treated. Often a wait and watch strategy is appropriate, especially if an effective treatment to which the individual patient responds is known. In the acute bleeding patient a combination of steroids and ivIgG is most effective. In life threatening bleeding, platelet transfusions can be given, but then repeated transfusions might be required. While steroids are often effective in increasing the platelet count, treatment duration with steroids should be limited as the adverse effects of long term steroids often cause a higher morbidity than ITP by itself. Splenectomy results in complete remission in about 60% of chronic ITP patients but has adverse effects (perioperative risk, increased risk of severe infection, probably increased risk for malignancies). Rituximab can be effective but the increase in platelet count is delayed. Thrombopoietin receptor agonists are very effective to increase the platelet count. Long term treatment seems to be associated with little toxicity. Most important adverse effects are the increased risk for venous thrombosis, which is dose related and increased bone marrow fibrosis. Whether these drugs should be given before splenectomy, or restricted to otherwise treatment refractory patients is currently a matter of debate. One of the reasons are the high costs of these drugs. Treatment is usually long term as the thrombopoietin receptor agonists are only symptomatic treatment.
Secondary ITP requires treatment of the underlying disease.
Alloimmune thrombocytopenias: in NAIT treatment of the mother with ivIgG + steroids from on the 20th week of gestation is currently the standard. In affected newborns platelet transfusions should be given at platelet counts <30,000/µL to prevent intracranial hemorrhage. If no antigen compatible platelet concentrates are available, treatment must not be delayed and random platelets should be given as soon as possible (most postpartum major bleedings occur within the first 36hs). For PTP ivIgG is most appropriate.
dITP: cessation of the drug and, in case of bleeding symptoms, ivIgG are the most effective treatments. GPIIbIIIa inhibitor induced thrombocytopenia usually requires no treatment and any prothrombotic intervention must be balanced against the risk for acute coronary occlusions.
Continued next page
Second Advanced Training Course Manual.indd 36-37 2/27/14 10:23 AM
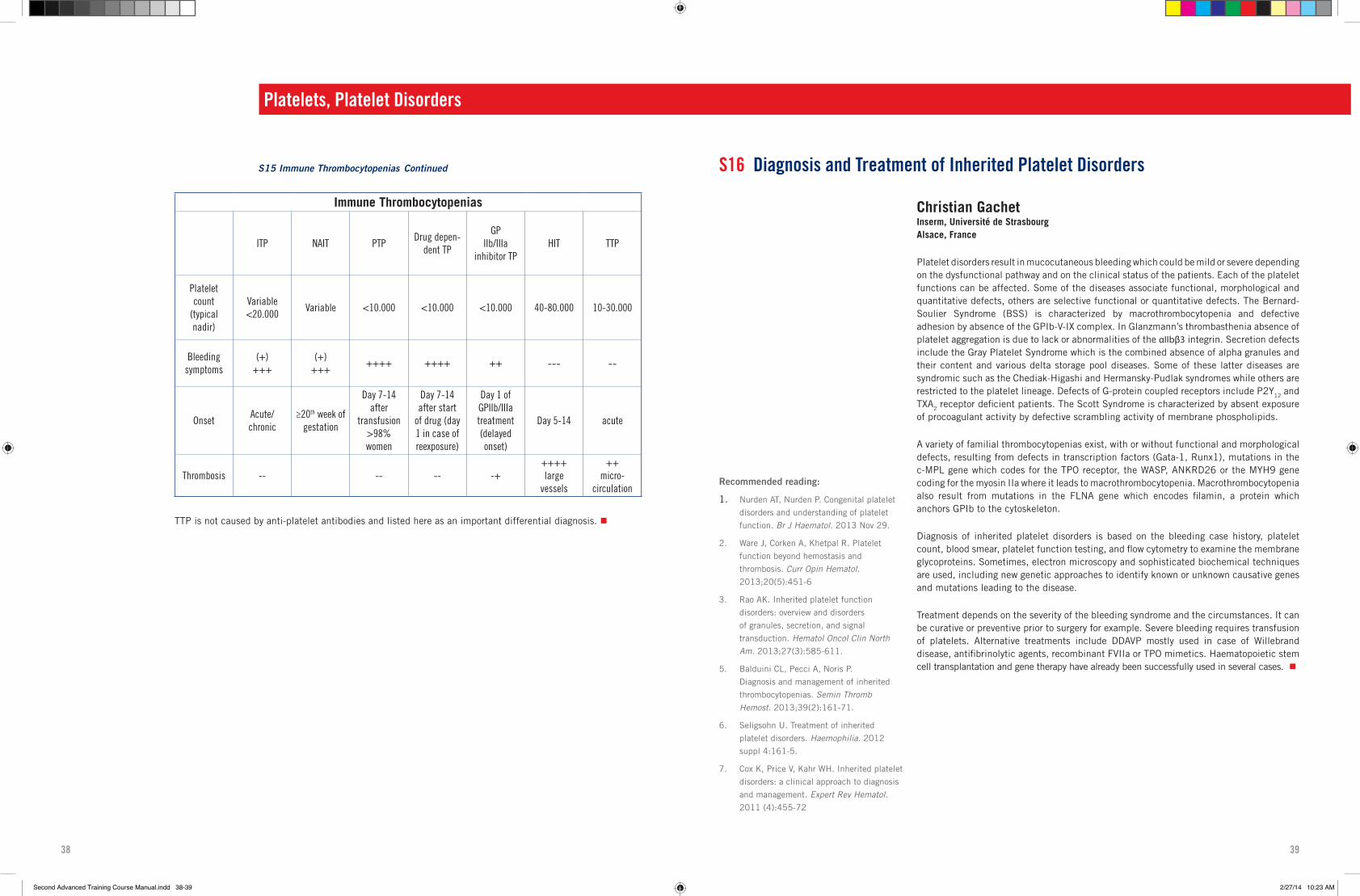
38 39
Immune Thrombocytopenias
ITP NAIT PTPDrug depen-
dent TP
GP IIb/IIIa
inhibitor TPHIT TTP
Platelet count
(typical nadir)
Variable<20.000
Variable <10.000 <10.000 <10.000 40-80.000 10-30.000
Bleeding symptoms
(+)+++
(+)+++
++++ ++++ ++ --- --
OnsetAcute/chronic
≥20th week of gestation
Day 7-14 after
transfusion >98% women
Day 7-14 after start
of drug (day 1 in case of reexposure)
Day 1 of GPIIb/IIIa treatment (delayed onset)
Day 5-14 acute
Thrombosis -- -- -- -+++++large
vessels
++micro-
circulation
TTP is not caused by anti-platelet antibodies and listed here as an important differential diagnosis.
Platelets, Platelet Disorders
Christian Gachet Inserm, Université de Strasbourg Alsace, France
Platelet disorders result in mucocutaneous bleeding which could be mild or severe depending on the dysfunctional pathway and on the clinical status of the patients. Each of the platelet functions can be affected. Some of the diseases associate functional, morphological and quantitative defects, others are selective functional or quantitative defects. The Bernard-Soulier Syndrome (BSS) is characterized by macrothrombocytopenia and defective adhesion by absence of the GPIb-V-IX complex. In Glanzmann’s thrombasthenia absence of platelet aggregation is due to lack or abnormalities of the αIIbβ3 integrin. Secretion defects include the Gray Platelet Syndrome which is the combined absence of alpha granules and their content and various delta storage pool diseases. Some of these latter diseases are syndromic such as the Chediak-Higashi and Hermansky-Pudlak syndromes while others are restricted to the platelet lineage. Defects of G-protein coupled receptors include P2Y12 and TXA2 receptor deficient patients. The Scott Syndrome is characterized by absent exposure of procoagulant activity by defective scrambling activity of membrane phospholipids.
A variety of familial thrombocytopenias exist, with or without functional and morphological defects, resulting from defects in transcription factors (Gata-1, Runx1), mutations in the c-MPL gene which codes for the TPO receptor, the WASP, ANKRD26 or the MYH9 gene coding for the myosin IIa where it leads to macrothrombocytopenia. Macrothrombocytopenia also result from mutations in the FLNA gene which encodes filamin, a protein which anchors GPIb to the cytoskeleton.
Diagnosis of inherited platelet disorders is based on the bleeding case history, platelet count, blood smear, platelet function testing, and flow cytometry to examine the membrane glycoproteins. Sometimes, electron microscopy and sophisticated biochemical techniques are used, including new genetic approaches to identify known or unknown causative genes and mutations leading to the disease.
Treatment depends on the severity of the bleeding syndrome and the circumstances. It can be curative or preventive prior to surgery for example. Severe bleeding requires transfusion of platelets. Alternative treatments include DDAVP mostly used in case of Willebrand disease, antifibrinolytic agents, recombinant FVIIa or TPO mimetics. Haematopoietic stem cell transplantation and gene therapy have already been successfully used in several cases.
S16 Diagnosis and Treatment of Inherited Platelet Disorders
Recommended reading:
1. Nurden AT, Nurden P. Congenital platelet
disorders and understanding of platelet
function. Br J Haematol. 2013 Nov 29.
2. Ware J, Corken A, Khetpal R. Platelet
function beyond hemostasis and
thrombosis. Curr Opin Hematol. 2013;20(5):451-6
3. Rao AK. Inherited platelet function
disorders: overview and disorders
of granules, secretion, and signal
transduction. Hematol Oncol Clin North Am. 2013;27(3):585-611.
5. Balduini CL, Pecci A, Noris P.
Diagnosis and management of inherited
thrombocytopenias. Semin Thromb Hemost. 2013;39(2):161-71.
6. Seligsohn U. Treatment of inherited
platelet disorders. Haemophilia. 2012
suppl 4:161-5.
7. Cox K, Price V, Kahr WH. Inherited platelet
disorders: a clinical approach to diagnosis
and management. Expert Rev Hematol. 2011 (4):455-72
S15 Immune Thrombocytopenias Continued
Second Advanced Training Course Manual.indd 38-39 2/27/14 10:23 AM

40 41
Andreas Greinacher Institut für Immunologie und Transfusionsmedizin, Universitätsmedizin Greifswald, Germany
Acquired platelet disorders can affect platelet number (thrombocytopenia) or platelet function (thrombocytopathy); very often, both are affected.
Thrombocytopenia: Thrombocytopenia often complicates critical illness and is associated with increased morbidity and mortality. The most frequent cause of severe thrombocytopenia in hospitalized patients is chemotherapy-induced hypoproliferative thrombocytopenia. Several recent prospective trials in these patients show: a prophylactic transfusion regimen using a transfusion trigger of 10,000 platelets/µL primarily reduces WHO grade 2 bleeding (which may be important for quality of life), but not WHO grade 3 or 4 bleeding; patients with acute leukemias benefit from a prophylactic transfusion regimen, while autologous stem cell transplantation may justify a therapeutic transfusion regimen.
In the critically ill, non-chemotherapy patient thrombocytopenia is also frequent but more challenging from a diagnostic point of view because of the multifactorial pathogenesis of this disorder. Five major mechanisms can result in thrombocytopenia, which are listed in Table 1. Thrombocytopenia is both a pathologic entity and a sign of severe illness. Severely sick patients often lack the capacity of normal platelet production due to toxic effects on the bone marrow and in addition may present with several causes for increased platelet consumption such as bleeding or infection. Treatment should target the underlying disease. Platelet transfusions are indicated in bleeding patients, while there is no strong evidence supporting the usefulness of prophylactic transfusions in critically ill thrombocytopenic patients. In some patients thrombocytopenia may even indicate an increased risk for macrovascular (e.g. in heparin-induced thrombocytopenia [HIT]) or microvascular (e.g. in thrombotic thrombocytopenic purpura [TTP]) thrombosis. These prothrombotic causes of thrombocytopenia require different management strategies than other causes of thrombocytopenia. Therefore it is most important for treatment decisions to differentiate between a prohemorrhagic and a prothrombotic disorder.
Beside the absolute platelet count, interpretation of the platelet count course is helpful to narrow down the cause and to distinguish between different etiologies of thrombocytopenia. Whereas a moderate decrease in platelet counts within the first three days after major surgery or severe infection is rather typical, an absent or blunted platelet count increase after five days indicates continuing critical illness and a worse outcome.
Post-surgical patients: The platelet count typically declines after major surgery reaching a nadir between day 1 and day 4. The magnitude of the platelet count decrease during the first 3-4 days after surgery reflects the extent of the tissue trauma or blood loss and is primarily caused by platelet consumption. Thereafter, platelet counts increase constantly reaching the pre-surgery level between day 5 and day 7 and peak at around day 14 after surgery. A platelet count decrease in the second week of ICU-treatment should prompt further diagnostic workup, even if the absolute platelet count is still within a range not associated with a substantial increase in the bleeding risk, as this may be an indicator for immune mediated complications e.g. heparin-induced thrombocytopenia.
Platelets, Platelet Disorders
S17 Diagnosis and Treatment of Acquired Platelet Disorders Medical patients: The platelet course in medical ICU-patients depends on the underlying disease and is usually not influenced by an accidental or iatrogenic tissue trauma. Conditions predisposing for thrombocytopenia are sepsis, renal replacement therapy, use of extracorporal circuits, intravascular devices, disseminated intravascular coagulation, multi organ failure, and recent cardiopulmonary resuscitation. Medical ICU-patients in whom the underlying disease is successfully treated, typically show a moderate platelet count decrease after admission with a recovery after about five days. This may indicate.
Thrombocytopathies: By far the most common cause of an impaired platelet function is the intake of anti-platelet drugs. Beside thienopyridins (clopidogrel, prasugrel, ticagrelor) and non-steroidal antiphlogistic drugs (e.g. ASA, diclophenac), other drugs can also inhibit platelet function e.g. the serotonin reuptake inhibitors (antidepressants), or antiepileptic drugs. Especially acute coronary syndrome (ACS) or percutaneous coronary interventions with stent implantation require dual antiplatelet therapy for 3-12 months (after ACS or drug eluting stent [DES] insertion). If urgent surgery is required, the risk of perioperative bleeding is increased by up to 50% in patients receiving dual antiplatelet therapy. Transient ‘reversal’ of antiplatelet therapy by platelet transfusion is possible, based on the pharmacokinetic profile of the drug. As ASA and clopidogrel both have short half-lives, platelet transfusions 12-24 hours after last intake of these drugs provide hemostasis. ASA inhibits thromboxane formation, but not the thromboxane receptor. Thus, otherwise permanently ASA-inactivated platelets can be recruited by thromboxane which is generated by transfused platelets. After surgery ASA can be restarted once bleeding is controlled (often within 6 hours). Such a bridging strategy requires absence of active antiplatelet metabolites from the circulation prior to platelet transfusion, otherwise, the transfused platelets would also be rapidly inhibited. The half-life of the active metabolites of prasugrel is 8h and the drug needs to be stopped longer before platelet transfusions are fully effective. More importantly, ticagrelor is a reversible inhibitor reaching high plasma concentrations and needs to be stopped 96 h before surgery because the active metabolite is present in the patient’s blood for up to 72-96 hours after administration. While this strategy is promising and addresses an up to date unresolved and increasingly pertinent clinical problem, prospective clinical trials should be initiated to further assess this approach.
There are many other causes leading to acquired platelet disorders:• infectious diseases (malaria, Dengue hemorrhagic fever) cause thrombocytopenia;
• uremia, is associated with multiple platelet function defects, including reduced platelet adhesion to endothelial cells, alpha storage pool deficiency and dense granule deficiency, dysfunctional TXA2 production;
• in chronic liver disease platelets are altered by increased thrombin generation, which activates platelets, and by increased levels of fibrin split products which bind to platelets;
• myeloproliferative disorders (essential thrombocytemia, polycythemia vera) are associated with very heterogeneous platelet function defects and in paraproteinemia the pathologic proteins may block platelet receptors or impair the interaction of platelets and von Willebrand factor;
• chronic platelet activation, e.g. due to extracorporeal circuits, or cardiac assist devices leads to acquired storage pool deficiency, as well as shedding/degradation of platelet membrane proteins and receptors;
• immune thrombocytopenias and rarely anti-platelet antibodies which impair platelet function but do not cause thrombocytopenia.
Recommended reading:
1. Stanworth SJ et al. A no-prophylaxis
platelet-transfusion strategy
for hematologic cancers. NEJM 2013;368:1771-80.
2. Wandt H. et al. Therapeutic platelet
transfusion versus routine prophylactic
transfusion in patients with haematological
malignancies: an open-label,
multicentre, randomised study. Lancet. 2012;380:1309-16.
3. Lieberman L et al. Evidence-based focused
review of platelet transfusions for critically
ill patients with thrombocytopenia. Blood. 2013 Dec 12. [Epub].
4. Hui P, Cook DJ, Lim W, Fraser GA,
Arnold DM. The frequency and clinical
significance of thrombocytopenia
complicating critical illness: a systematic
review. Chest 2011;139(2):271-278.
5. Selleng S, Malowsky B, Strobel U,
et al. Early-onset and persisting
thrombocytopenia in post-cardiac surgery
patients is rarely due to heparin-induced
thrombocytopenia, even when antibody
tests are positive. J Thromb Haemost 2010;8(1):30-36.
6. Stansbury LG, Hess AS, Thompson K, et
al. The clinical significance of platelet
counts in the first 24 hours after severe
injury. Transfusion 2012.
7. Greinacher A, Warkentin TE. “Acquired
non-immune thrombocytopenia” In: Marder
VJ, Aird WC, Bennett JS eds, Hemostasis and thrombosis: Basic principles and clinical practice. 6 ed. Philadelphia:
Lippincott Williams & Wilkins; 2013.
8. Thiele T, Selleng K; Selleng S, Greinacher
A, Bakchoul T. Thrombocytopenia in the
Intensive Care Unit – Diagnostic Approach
and Management. Semin Thromb Hemost 2013 in press.
9. Koneti Rao, “Acquired disorders of
platelet function” in Platelets 3rd ed. A.
Michaelson editor; 2013.
10. Lieberman L. et al. Evidence-based
focused review of platelet transfusions
for critically ill patients with
thrombocytopenia. Blood. 2013 Dec 12.
[Epub ahead of print]Continued next page
Second Advanced Training Course Manual.indd 40-41 2/27/14 10:23 AM

42 43
Platelets, Platelet Disorders
Table 1: Different Mechanisms of Thrombocytopenia
Onset of Thrombocytopenia
At admissionAbsent platelet recovery orslow decrease after initial recovery
Rapid decrease after initial recovery
infusion of plasma expanders and fluids
Hemodilution
infusion of plasma expanders and fluids
transfusion of red blood cells and plasma
Increased platelet consumption
major blood loss persistent bleeding SIRS / sepsis
massive tissue trauma repeated major surgery DIC
DIC sepsis/nosocomial infection pulmonary embolism
SIRS/ sepsis multi-organ failure bleeding
severe pulmonary embolism dialysis
diabetic ketoacidosis extracorporeal circulation
HELLP-syndrome DIC
promyelocytic leukemia
malaria
microangiopathic desease
Increased sequestration
hepatosplenomegaly circulatory shock
hypothermia resuscitation
Decreased production
intoxication (drugs, toxins, herbals) Intoxication
acute leukemia viral infections
myelodysplasia metastatic bone marrow infiltration
metastatic bone marrow infiltration viral infections
chronic liver disease acute and chronic liver failure
viral infections (EBV,CMV,HCV,HIV) iron depletion
irradiation folate-deficiency
graft failure after HSCT vitamin-B12 deficiency
hereditary macrothrombocytopenia chemotherapy
hemophagocytosis
Platelet destruction
immune thrombocytopenia thrombotic thrombocytopenic purpura early onset HIT antiphospholipid syndrome
post-surgery TTP HIT
viral infections (HIV,HCV)drug induced immune thrombocy-topenia
drug induced immune thrombocyto-penia (out patients)
GPIIb/IIIa -inhibitor induced throm-bocytopeni
post-transfusion purpura!!! Post-transfusion purpura
autoimmune disease (lupus)passive transfusion of platelet alloantibodies
(S17 Diagnosis and Treatment of Acquired Platelet Disorders continued)
Second Advanced Training Course Manual.indd 42-43 2/27/14 10:23 AM

44 45
Ted Warkentin Hamilton Regional Laboratory Medical Program, Hematology Department Hamilton, Ontario, Canada
DIC is a severe form of consumptive coagulopathy in which systemic activation of hemostasis leads to widespread intravascular fibrin deposition with associated depletion of prohemostatic and anticoagulant factors, with parallel secondary fibrinolysis, potentially leading to a wide range of adverse outcomes, including thrombotic occlusion of small/mid-sized vessels, organ dysfunction/failure, and/or bleeding. One devastating sequela is “symmetrical peripheral gangrene,” characterized by acral (distal extremity) necrosis of two (or four) limbs, despite palpable/doppler-identifiable arterial pulses.
DIC can be viewed as a clinical-pathological entity, in which the patient must have one (or more) disorders (listed in the next paragraph) associated with pathological activation of hemostasis, plus lab tests showing increased markers of fibrin formation and (usually) depletion of hemostatic factors, including platelets. A challenge is to discern when the clinical-laboratory picture truly indicates DIC versus “usual” hemostatic changes associated with critical illness, post-major surgery, thrombosis occurrence, etc.
Among the clinical disorders that trigger DIC: septicemia, severe inflammation reactions (adult respiratory distress syndrome [ARDS], systemic inflammatory response syndrome [SIRS]), cardiogenic shock, trauma (especially brain injury and/or hypovolemic shock), severe acidemia (e.g., lactic acidosis), cancer (e.g., metastatic adenocarcinoma), obstetric complications (placental abruption, puerperal sepsis, retained placental products, severe pre-/eclampsia, etc.), severe heparin-induced thrombocytopenia (particularly the variant known as “delayed-onset” or “autoimmune-like” HIT), envenomations, organ failure (necrotizing pancreatitis, acute liver failure), severe hemolysis (e.g., ABO incompatible transfusion, severe thrombotic thrombocytopenic purpura), pulmonary embolism/intra-cardiac thrombus, severe allergic reactions (e.g. severe transplant rejection).
A DIC-mimicking picture can result from localized intravascular coagulation due to aortopathy (e.g., abdominal/thoracic aneurysm with ulcerated endothelium and adherent thrombus, hemangioma/thrombocytopenia [Kasabach Merritt syndrome]). Another DIC-mimicking disorder is primary fibrinolysis, e.g., secondary to metastatic prostate cancer.
The ISTH scoring system for DIC evaluates: (a) platelet count, (b) prothrombin time (PT), (c) fibrin-specific marker(s) (e.g., fibrin monomer, fibrin D-dimer, fibrin split products), (d) fibrinogen. The last is the least useful: ~75% of DIC patients have normal (or even elevated) fibrinogen levels (fibrinogen is an acute phase reactant).
Besides elevated PT and fibrin-specific markers, thrombocytopenia, and (relative reduction in) fibrinogen, other lab abnormalities can include: (a) elevated partial thromboplastin time (PTT), (b) circulating schistocytes (red cell fragments) with increased lactate dehydrogenase (LD), (c) circulating nucleated red cells (normoblasts), and (d) reduced procoagulant factors (e.g., extrinsic/common pathway [VII, X, V, II], intrinsic/contact pathway [XII, PK, HMWK, XI, VIII, IX], and reduced natural anticoagulants (antithrombin, protein C). An elevated PTT is an important therapeutic problem if PTT-adjusted anticoagulant therapy (e.g., unfractionated heparin [UFH]) is given, as incorrect UFH dosing due to misleading
Platelets, Platelet Disorders
S18 Disseminated Intravascular Coagulation (DIC)
PTTs results (“PTT confounding”). Natural anticoagulant depletion is a risk factor for microvascular thrombosis. It is important to monitor results of coagulation assays serially, as DIC is a dynamic process that can worsen appreciably within a short period of time.
Various subtypes of DIC exist, including: marked hypofibrinogenemia of head trauma and obstetric triggers of DIC (potential need for fibrinogen concentrates/cryoprecipitate transfusion), shock with concomitant DIC and acute liver dysfunction (shock liver) with symmetric peripheral gangrene (acute DIC/liver necrosis-limb necrosis syndrome), and DIC with associated large-vessel thrombosis (adenocarcinoma- and HIT-associated DIC). Warfarin therapy is a risk factor for microthrombosis (acral limb gangrene) in DIC patients with HIT and adenocarcinoma; a supratherapeutic INR (>3.5) is characteristic.
Various diagnostic and treatment dilemmas exist: (a) should blood products be given? (prevent/treat bleeding vs risk of potentiating thrombosis); (b) should the patient be anticoagulated? (treat/prevent thrombosis, but risks bleeding; UFH vs low-molecular-weight heparin (LMWH) vs non-heparin anticoagulation?); (c) is it HIT vs (non-HIT) DIC? (HIT: avoid heparin, begin non-heparin anticoagulation; DIC: heparin is the anticoagulant of choice!); finally (d) in patients with severely deranged hemostasis (e.g., acute DIC/liver necrosis-limb necrosis syndrome), is effective treatment even possible?
Current therapeutic trends for DIC include (strength of evidence): (a) emphasizing treatment of the underlying condition that is the “trigger” of DIC (moderate); (b) avoiding prophylactic platelet or frozen plasma (FP) or cryoprecipitate administration unless hemostatic abnormalities are severe (e.g., platelet count <20) and patients judged to be at high risk of bleeding, and/or invasive procedures are required (low); (c) for bleeding patients, therapeutic platelet (plt <50), FP (1.5X increased PT and/or PTT), and/or fibrinogen concentrate/cryoprecipitate (<1.5 g/L) can be given (low); (d) therapeutic-dose heparin should be considered where thrombosis predominates (low), with LMWH preferred (when appropriate) over UFH (low); (e) prophylactic-dose heparin is recommended in critically-ill, non-bleeding patients with DIC (high); (f) adjunctive therapies (e.g., antithrombin concentrates) may be appropriate in special circumstances, although antifibrinolytic agents (e.g., tranexamic acid) should be restricted to hyper-fibrinolytic situations (e.g., prostate cancer/hyperfibrinolysis, acute head trauma).
Recommended reading:
1. Levi M, ten Cate H. Disseminated
intravascular coagulation. N Engl J Med 341: 586-92, 1999.
2. Levi M, Meijers JC. DIC: which laboratory
tests are most useful? Blood Rev 25:33-7,
2011.
3. Molos MA, Hall JC. Symmetrical peripheral
gangrene and disseminated intravascular
coagulation. Arch Dermatol 121:1057-61,
1985.
4. Siegal DM, Cook RJ, Warkentin TE. Acute
hepatic necrosis and ischemic limb
necrosis. N Engl J Med 367:879-81,
2012.
5. Takemitsu T, et al. Prospective evaluation
of three different diagnostic criteria for
disseminated intravascular coagulation.
Thromb Haemost 105:40-44, 2011.
6. Wada H, et al. Guidance for diagnosis and
treatment of disseminated intravascular
coagulation from harmonization of the
recommendations from three guidelines. J Thromb Haemost 11:761-7, 2013.
7. Warkentin TE. Anticoagulant failure in
coagulopathic patients: PTT confounding
and other pitfalls. Exp Opin Drug Saf 13:25-43, 2014.
Second Advanced Training Course Manual.indd 44-45 2/27/14 10:23 AM

46 47
Sponsors General InformationSecond Advanced Training Course in
Thrombosis & Haemostasis
The ISTH Second Advanced Training Course is supported by
unrestricted educational grants from:
Course Sponsor
Instrumentation Laboratory180 Hartwell Road
01730 Bedford USA
www.ilww.com
Instrumentation Laboratory (IL) is passionate about delivering the most innovative solutions to address a range of hemostasis testing needs. The advanced ACL™ family of systems brings complete automation to the hemostasis lab—including the ACL TOP® Family of Hemostasis Testing Systems, featuring the new ACL TOP 300 CTS; and, the ACL AcuStar®, the first fully automated, chemiluminescent analyzer for hemostasis specialty testing. Combined with the HemosIL line of reagents, a comprehensive panel of fully automated assays, IL offers complete disease state management for the hemostasis lab.
Course Benefactor
Stago9 rue des Frères Chausson
92600 Asnières sur Seine
France
www.stago.com
Stago, created in 1945, is an IVD Company which develops and markets reagents and automated systems for the investigation of blood coagulation disorders. Stago is a leading player in Haemostasis. Headquarters, as well as R&D, manufacturing and logistics activities are located mainly in the Paris area (France). In 2012, Stago significantly improved its direct presence by opening 8 new subsidiaries in Europe. Its products are also available in more than 110 countries throughout the world through a network of selected partners. In 2013, Stago had more than 2,000 employees worldwide.
AccommodationsAccommodations are not included with the meeting registration. Attendees are requested to settle all hotel charges upon departure directly with the hotel.
Business CenterThe business center is located on the second floor (above the reception area). It has two computers avaliable for use by hotel guests and basic printing capabilities.
Certification of AttendanceA certificate of attendance will be available at the registration desk as of Sunday, March 16 at 7:30.
CMEThe Second Advanced Training Course has been accredited by the European Hematology Association (EHA) with 24 credit points.
To obtain CME credits:
• Visit EHA’s website: eha-cme.org
• Create a professional account for yourself: simple and free of charge
• Please inform your details to the EHA-CME Administration ([email protected]), so they can add your credit points.
For further assistance please contact:EHA-CME UnitTel: +31 70 3020 099Email: [email protected] www.eha-cme.org
For U.S. Physicians Trying to Obtain CME Credits through the EHA are not reciprocal with the U.S. ACCME (AMA), and this course has not been accredited by European Accreditation Council for Continuing Medical Education (EACCME).
Coffee Breaks Complimentary refreshments will be served throughout the meeting. The times of the refreshment breaks are listed in the overall program and will take place outside the meeting room.
Course EvaluationThe post course evaluation can be found online at URL: http://isth.Portugalcourse.sgizmo.com/s3/. We kindly ask for you to complete the evaluation following the course.
InsuranceThe ISTH will not be held liable for illness, accidents or thefts suffered by participants or accompanying persons during the course or a person’s stay in Portugal before or after the course. Participants are advised to seek personal insurance coverage.
Notes
Second Advanced Training Course Manual.indd 46-47 2/27/14 10:23 AM

48 49
InternetInternet is available at a cost of €8 per user per day. It will work in all public areas of the hotel, sleeping rooms and meeting rooms.
LanguageAll presentations will be presented in English. There will be no simultaneous translation.
Lost and FoundFor any lost items, please see the front desk of the hotel.
Lunches and DinnersBuffet lunches and dinners are included with the meeting registration and will be served in the hotel’s Five Pines Restaurant. Please refer to the program schedule for meal times.
Prayer Room Please contact the ISTH staff if accommodations are needed for prayer.
Presentation SlidesSelected slides will be available after the conclusion of the course, attendees will receive instructions on downloading slides after the meeting.
Registration HoursThe ISTH registration desk is located outside the Longshot/Bogey Room located on the first floor of the hotel. The hours are as follows:
Thursday March 13 11:00-18:30
Friday March 14 7:00-18:30
Saturday March 15 7:30-18:30
Sunday March 16 7:30-13:00
TransportationTransportation to the Lisbon airport may be booked directly using a car service. Payment is made directly to the driver and reservations can be made by contacting Mr. Manuel Madeira at [email protected].
Other forms of transportation may be arranged at the hotel concierge.
VenueHotel Quinta Da Marinha Quinta da Marinha, 2750-005 Cascais, Portugal T: (+351) 214 860 100 F: (+351) 214 869 482
*Please note that all hotel charges must be settled by each attendee prior to leaving the meeting.
General Information
2014 ISTH Membership Renewal Form
International Society on Thrombosis and HaemostasisTelephone +1 919 929-3807 Fax +1 919 [email protected] | www.isth.org ISTH Tax ID#: 23-7428305
Membership Form Please fill out front & back of this form and fax or mail with payment to the address above
Full Name Email
Organization
Address
City/Town Country State/Location
Postal Code Work Phone Fax
Dues are for the calendar year. See member benefits and countries online at www.isth.org
2014 Regular Member US $160 $
Renew Membership for 2 years [2014 – 2015] US $320 $
Renew Membership for 3 years [2014 – 2016] US $480 $
Check this box if you do not require a printed copy of the journal and are prepared to donate your copy of the journal to a colleague in the developing world
2014 Associate Member [Student, Fellow, Trainee] US $35 $
Please attach a letter from your institution verifying your status for 2014
2014 Nurse US $35 $
2014 Allied Health Professional US $35 $
2014 Reach-the-World US $35 $
2014 Emeritus Member (non-dues paying) N/A
2014 Emeritus Member US $35 $ Includes online-only subscription to Journal of Thrombosis and Haemostasis
Voluntary Contributions $
Total Amount Paid $
Credit Card InformationVisa MasterCard (EuroCard) Amex
Name on Card (Please print)
Card Number CCV Number Expiration Date
Cardholder’s Full Address
Authorized Signature Data Privacy: All information requested is for use in compliance with applicable international privacy protection legislation. By completing this form, I request that ISTH use this data to keep me informed of ISTH activities, enhance my participation in ISTH, and make my full contact details available to other ISTH members, understanding that my contact and demographic details may be made available to third parties deemed appropriate by the ISTH and, since ISTH is an international organization, may be processed in any nation of the world. I also understand and confirm that my data can be exchanged between the ISTH and partners in order to provide me with access to ISTH membership benefits, including, but not limited to the Society Journal. Any exceptions to this request are noted below and are provided with the understanding that limiting use of my data may limit my access to member benefits.
Check here if you do not wish to be included in the ISTH membership directory
Check here if you do not wish to receive third party mailings
Check here if you do not wish to receive ISTH electronic news and updates (except renewal communications)
Please complete demographic data on reverse of this application
Notes
Second Advanced Training Course Manual.indd 48-49 2/27/14 10:23 AM

50 51
Notes
International Society on Thrombosis and HaemostasisTelephone +1 919 929-3807 Fax +1 919 [email protected] | www.isth.org ISTH Tax ID#: 23-7428305
Demographic Data Please complete the data below
Date of Birth [mm/dd/yyyy] Gender Male Female
Place of Work Government Institute Group or Private Practice Hospital Industry [Biotech, Pharma,
Device Manufacturer Research Institute University or Medical School Other:
Academic Faculty Not Applicable Professor Associate Professor Assistant Professor Instructor Post-Doctoral Fellow Research Associate Retired Student: MD Student: PhD Other:
Administration(Select all that apply) Not Applicable Business & Management Administration Scientific Administration Education Other:
Industry(Select all that apply) Not Applicable Business Administration Management Marketing/Sales Science/Research Other:
PrescriptionsAre you licensed and write prescriptions for medications? Yes No
Specialization(Select all that apply) Animal Models Atherosclerosis Bleeding Disorders Coagulation Proteins &
Inhibitors Endothelial Cell Biology Fibrinolysis Inflammation Platelets Thrombotic Disorders Other:
Primary Basic Research(Select all that apply) Not Applicable Biochemistry Biophysics Cell Biology Experimental Pathology Genetics/Genomics Immunology Molecular Biology Pharmacology Physical Biochemistry Physiology Structural Biology Other:
Patient Volume 0 1-5 patients per week 6-20 patients per week +20 patients per week
Primary Clinical Research Area Not Applicable Clinical Trials Epidemiology Outcomes Research Toxicology Studies Translational Research Other:
Primary Medical Specialty Area Not Applicable Cardiology Clinical Chemistry/Laboratory Medicine Epidemiology Internal Medicine Hematology Neurology/Stroke Pathology Oncology Orthopaedics Surgery Transfusion Medicine Vascular Medicine Other:
Do you have membership in other societies? If so, please list:
Journal subscription will be interrupted in the event unpaid annual dues at the end of the calendar year.
MAIL TO: International Society on Thrombosis and Haemostasis | 610 Jones Ferry Road, Suite 205 Carrboro, NC 27510 USA or FAX to +1 919 929 3935 Receipts available on request Email: [email protected]
2014 ISTH Membership Renewal Form
Second Advanced Training Course Manual.indd 50-51 2/27/14 10:23 AM

52
Program Now Online!
Second Advanced Training Course Manual.indd 52 2/27/14 10:23 AM





























