Embed Size (px)
Citation preview

THE JOURNAL OF B I O ~ I C A L CHEMISTRY 0 1994 by The American Society for Bicchemistry and Molecular Biology, Inc.
Vol. 269, No. 5, lsaue of February 4, pp, 3469-3474, 1994 Printed in USA.
Residue at Position 331 in the IgG1 and IgG4 CH2 Domains Contributes to Their Differential Ability to Bind and Activate Complement*
(Received for publication, August 5, 1993, and in revised form, September 7, 1993)
Yuanyuan Xu$, Ray OomenB, and Michel H. KleinSlnIl From the Departments of $Immunology and IBiochemistry, University of Toronto, Toronto, Ontario, M5S lA8 Canada and the §Connaught Center for Biotechnology Research, Willowdale, Ontario, M2R 3T4 Canada
Aconserved proline residue is found at position 331 in the CH2 domains of human IgG subclasses which fix complement. This residue is replaced by a serine in IgG4 which is inactive. To determine the role of residue 331 in the differential ability of human IgGs to activate the complement cascade, a pair of genetically engineered anti-dinitrophenol IgGl and IgG4 antibodies with recip- rocal mutations at position 331 were tested for their he- molytic activity as well as for their ability to bind Clq, activate C1 and cleave C4. The IgGl Sel.9s1 mutant was virtually unable to mediate the lysis of trinitrobenzene- sulfonic acid-derivatized sheep red blood cells as a re- sult of a marked defect in Clq binding activity. In con- trast, the substitution of Pro for S e P 1 in IgG4 bestowed partial hemolytic activity (40%) to the IgG4 ProSS1 vari- ant. Under low ionic strength conditions, this mutant was found to be approximately 50 and 76% as active as wild-type IgGl in the Clq binding and C4b deposition assays, respectively. These results indicate that residue ProSS1, which folds into close proximity to a previously identified Clq binding motif (Duncan, A. R., and Winter, G. (1988) Nature 332,738-740), contributes to the archi- tecture of the IgGl Clq binding site and that its replace- ment by a serine residue in IgG4 is largely responsible for the functional inactivity of this isotype.
In spite of their extensive amino acid sequence similarities, human IgG subclasses markedly differ in their abilities to ac- tivate the classical pathway of complement. IgGl and IgG3 are the most efficient at binding and activating C1, IgG2 is signifi- cantly less active, and IgG4 is inactive (1). The observation that the Fcy4 fragment binds C1(2), whereas hinge-deleted proteins are unable to fm complement (3,4), led to the hypothesis that, in molecules with restricted hinges, the Fab arms are brought in closer apposition to the Fc fragment and thus render the Clq binding site less accessible. It was initially believed that the segmental flexibility of IgG molecules depended on the length of their hinge region and correlated with complement activa- tion (5,6). However, later studies (7, 8) using genetically engi- neered IgG3 and IgG4 antibodies with either modified or switched hinge regions clearly demonstrated that the hinge was not responsible for the differences observed in the comple- ment fming ability of human IgG isotypes.
Medical Research Council of Canada. The costs of publication of this * This work was supported by Grant MT-4259 (to M. H. K.) from the
article were defrayed in part by the payment of page charges. This
with 18 U.S.C. Section 1734 solely to indicate this fact. article must therefore be hereby marked “aduertisernent” in accordance
11 To whom correspondence should be addressed: Dept. of Immunology, Medical Sciences Bldg., University of Toronto, Toronto, Ontario M5S 1A8, Canada M5S 1A8.
Early fragmentation studies revealed that the Clq binding site was located to the IgGl cH2 domain (9). This finding was unambiguously confirmed by experiments which showed that IgG1’ and IgG3 (11) variants harboring an IgG4 cH2 domain were no longer able to activate C1, whereas the grafting of either an IgGl or IgG3 C H ~ domain onto IgG4 conferred C1 binding activity to the resulting hybrid molecule. In addition, the cH2 carbohydrate moieties were found to be critical in maintaining the functional integrity of the C1 binding site, since aglycosylated IgGs do not fm complement (12, 13). A Clq binding motif ( E M ) , including 3 charged residues, Glu3I8, LYS~~O, and Lys322 (Eu numbering) of the cH2 domain, was identified by site-directed mutagenesis of a murine comple- ment-fixing antibody (14). However, this motif is conserved among noncomplement-fixing antibodies. More recently, Tao et al . (11) engineered a set of IgGlflgG4 hybrid antibodies in which the C-terminal halves of the IgGl and IgG4 C H ~ domains were reciprocally exchanged and provided evidence that the C-terminal region of the cH2 domain (residues 292-340) con- tains the residues responsible for the isotype-specific differ- ences in complement activation. Interestingly, residues Ser330 and Sel-731 within this region are IgG4-specific, whereas resi- dues Ala330 and Pro331 are conserved among the other human IgG subclasses. Moreover, the analysis of the IgGl Fc crystal structure (15) reveals that Pro331 folds into proximity of the EKK Clq binding motif, and it has been shown that the mu- tation of its structural equivalent, residue Pro436, in IgM (16) to Ser markedly decreased the binding affinity of IgM for Clq (17). To determine the putative role of residue 331 in the IgG
ability to bind and activate C1, we have engineered anti-DNP2 chimeric IgGl and IgG4 with reciprocal amino acid substitu- tions at this position. We have shown that a Pro331 + Ser331 substitution virtually abolished IgGl ability to bind C1, whereas the reciprocal mutation bestowed significant, al- though partial, complement-f~ng activity to IgG4.
MATERIALS AND METHODS Cell Lines-The murine myeloma A chain-producing variant MOPC
315.26 cell line was maintained as described previously (18). Plasmid Constructions-The 3.2-kilobase HindIII-BarnHI Cy1 and
Cy4 constant region genes were lundly provided by Dr. L. Hood (Cali- fornia Institute of Technology). The genes were cloned into the pEMBL19 vector. Al l molecular cloning techniques were performed ac- cording to Sambrook et al. (19). The reciprocal exchanges of residues
ton, and M. Klein, unpublished data. Y. Xu, M. Everett, S. Chappel, C. Home, D. Isenman, K. J. Domng-
* The abbreviations used are: DNP, dinitrophenol; GVB2+, isotonic Verona1 buffer containing 0.1% gelatin, 0.15 m~ CaC12, and 0.5 m~ MgC12; PBS, phosphate-buffered saline; SRBCs, sheep red blood cells; TNBS, trinitrobenzenesulfonic acid; C, constant region: H, heavy chain; L, light chain; V,, heavy chain variable region gene segment.
3469

3470 Role of C,2 Residue 331 in Ig( pr0331(CCC + TCC) in IgGl and Se+YTCC + CCCj) in IgG4 (Eu numbering) we; performed by site-directed mutagenesis of their re- spective codons using the method of Tayloret al. (20) (Amersham Corp.). The mutations were confirmed by DNA sequence analysis. The wild- type and mutated Cy genes were then subcloned into the mammalian expression vector pSV2neo-VH315, which contains the IgA MOPC 315 rearranged VH gene placed under the control of its own promoter (18). The resulting expression vectors encode an entire heavy chain contain- ing the murine MOPC 315 VH domain and a human IgG constant region. All constructions were then introduced into MOPC 315.26 cells by electroporation (18). The transfected cells were grown and selected in a minimum essential medium supplemented with 10% fetal calf serum, 2 m glutamine, 10 m HEPES, 100 pg of streptomycidml, and 100 units of penicillidml in the presence of 600 pg of G418/ml (Life Tech- nologies, Inc.). The production of the chimeric IgG transfectomas was detected by an IgG-specific capture enzyme-linked immunosorbent as- say, using purified human myeloma IgGl and IgG4 as standards.
Purification of Chimeric Anti-DNP ZgG Proteins-All recombinant IgG molecules were purified from culture supernatants by DNP-lysine affinity chromatography. After extensive dialysis against PBS, pH 7.2, the antibodies were further purified by gel chromatography on a Sepha- dex G-200 column. Purified antibodies (5 pg) were electrophoresed on a 10% SDS-polyacrylamide gel under both reducing and nonreducing con- ditions. The gel was stained with PAGE Blue 83 "Electran" (BDH, Toronto) and analyzed by scanning densitometry using an LKB Ultros- can XL densitometer. Antibody preparations were ultracentrifuged at 100.000 x g for 1 h to remove potential aggregates before all functional assays. Antibody concentrations were measured by the micro-BCA pro- tein assay (Pierce Chemical Co.), using purified human myeloma IgGl as a standard.
Preparation of TNBS-SRBCs-The preparation of TNBS-sensitized SRBCs was carried out as described by Rittenberg and Pratt (21). SR- BCs (Woodlyn Labs, Guelph, Ontario, Canada) were washed three times with ice-cold PBS. Then, 20 mg of TNBS (Sigma) in 5 ml of PBS were added dropwise to a 1-ml suspension of 5 x lo9 SRBCs. The mixture was incubated for 20 min a t room temperature with constant mixing. TNBS-coated cells were washed four times with ice-cold PBS and stored a t a density of 1 x lo9 celldml at 4 "C in the dark.
Complement-mediated Hemolysis-TNBS-SRBCs (2 x 10') were washed three times with isotonic GVB2* (Verona1 buffer containing 0.1% gelatin, 0.15 m CaCI2, and 0.5 II~M MgC12) (p = 0.15) and incu- bated with increasing amounts of purified IgGs (0.06-2 pg) for 30 min a t 25 "C. GVB2+ alone was used as negative control. Unbound antibod- ies were removed by centrifuging the cells a t 1,300 x g for 5 min a t 4 "C and washing the cell pellets twice with 1 ml of ice-cold GVBZ+. IgG- sensitized TNBS-SRBCs were then resuspended in 250 pl of GVB2+ and reacted with 100 pl of a 1/30 dilution of guinea pig complement (Dia- medix, Miami, FL) preadsorbed with TNBS-SRBCs. Cell lysis was al- lowed to occur a t 37 "C for l h. The reaction was terminated by adding 1 ml of ice-cold GVB2+ containing 10 m EDTA. Unlysed TNBS-SRBCs were pelleted by centrifugation, and the degree of cell lysis was deter- mined by measuring the optical density of the supernatant at 412 nm. All assays were performed in duplicate. 2 values (2 = average number of hemolytic sitedcell) are expressed as the means of three independent determinations.
126Z-Clq BindingAssay-Human Clq was prepared by the method of Tenner et al. (22). Purified Clq was iodinated with carrier-free N a T (specific activity, 459.8 MBq/pg; Amersham Corp.) using the lactoper- oxidase method as described by Wright et al. (17) to a specific activity of 710,000 cpdpg. To prepare IgG-sensitized TNBS-SRBCs, 2 x lo7 TNBS-SRBCs were mixed with 2.5 pg of anti-DNP IgG in 200 pl of GVB2+ and kept on ice for 1 h with occasional shaking. Excess IgG was removed by washing the cells with ice-cold GVB2+ twice. Sensitized TNBS-SRBCs were then resuspended in 100 p1 of low ionic strength SGVB2+ buffer containing 48.6% sucrose, 0.1% gelatin, 0.15 m CaCl,, and 0.5 m MgCI, (p = 0.06) and incubated with increasing amounts (0.01-1 pg) of 1261-Clq in 100 p1 of SGVB2+ a t 30 "C for 30 min. The cell suspension was then layered onto a 2 0 0 4 dibutyl phthalate oil (Fisher) cushion in 400-pl microcentrifuge tubes. The tubes were centrifuged at 10,000 x g for 30 s, and the tips were cut. Cell-bound '2sI-Clq was measured by counting the cell pellet radioactivity in a y counter. Non- specific binding was determined using TNBS-SRBCs incubated with equivalent amounts of human myeloma IgG1. Each assay was per- formed in duplicate.
12sZ-C4b Deposition Assay-Purified human C1 and C4 molecules were kindly donated by Dr. D. Isenman (University of Toronto). C4 was iodinated by the lactoperoxidase method to a specific activity of 840,000 cpdpg. The lZ5I-C4b deposition assay was performed according to the
Complement Binding Ability
+ 68 Kd
+ 30 Kd
hc. 1. SDSpolyacrylamide gel analysis of purified wild-type and mutated anti-DNP antibodies. Chimeric antibodies were puri- fied by DNP affinity and Sephadex G-200 gel filtration chromatography
(5 pg) were analyzed by 10% SDS-PAGE under nonreducing conditions. from the supernatant of MOPC 315.26 transfectants. Purified samples
The electrophoretic mobilities of IgG tetramers and intermediate spe- cies are indicated.
protocol used for the 1261-Clq binding assay, except that 100 pl of 3-fold dilutions (0.4-100 ng) of purified C1 in SGVB2+ were added to 100 pl of IgG-TNBS-SRBCs. After a 30-min incubation a t 30 "C, the target cells were washed in SGVB2+ at 25 "C and reacted with a fixed amount of 12s11-C4 (800,000 cpm) in 200 pl of SGVB2+ at 30 "C for 30 min. Free Iz5I-C4 was separated by centrifugation through an oil cushion, and the cell pellets were counted for radioactivity. Nonspecific binding was de- termined from the residual counts associated with TNBS-SRBCs incu- bated with equal amounts of human myeloma IgGl. Each assay was performed in duplicate. The amount of cell-bound lZsI-C4b was ex- pressed as the mean of two independent experiments.
Quantitation of Chimeric Antibodies Bound to TNBS-SRBCs- Purified protein A (20 pg) (Pharmacia LKB Biotechnology Inc.) was labeled with NaIz5I using the chloramine-T method to a specific activity of 1.5 x lo7 cpdpg. Free NalZ5I was removed by repeated centrifuga- tions through Sephadex G25 spin columns. To estimate the quantities of chimeric IgGs bound to TNBS-SRBCs, increasing amounts of chi- meric IgGs (0.025-2 pg) were incubated with 2 x lo7 TNBS-SRBCs in 200 pl of GVB2+ as described for the complement-mediated lysis assay. IgGTNBS-SRBCs were washed with 1 ml of ice-cold GVB2+, pelleted, and resuspended in 100 p1 of GVB2+ containing a saturating amount of 'TI-protein A (250,000 cpm). After incubation for 30 min at 25 "C, cells were centrifuged through an oil cushion and pellets counted for radio- activity. Nonspecific binding was determined using TNBS-SRBCs mixed with an equal volume of GVB2+. All determinations were per- formed in duplicate.
Molecular Modeling-One cH2 domain (Brookhaven Protein Data Bank entry lfcl) (15) was used for energy minimization analysis of the
+ SeF'l mutation, including residues 238-340 and the 9 crys- tallographically resolved carbohydrate residues of the N-linked sugar moieties. A - S e F 1 mutant was constructed by replacing the prolyl side chain with that of serine using the INSIGHT I1 software program (BIOSYM Technologies Inc., San Diego, CA). Both mutated and starting structures were minimized using conjugate gradients until the gradient of the system total energy was less than 0.01 Kcal.
RESULTS
Expression and Purification of Native and Mutated Chimeric ZgGl and ZgG4 Antibodies-High-producer transfectants se- creting wild-type anti-DNP IgGl and IgG4 as well as their reciprocal IgGl Se?31 and IgG4 P r ~ ~ ~ l m u t a n t s a t levels be- tween l and 16 pg/ml were maintained in G418-containing medium. SDS-PAGE analysis (Fig. 1) of the DNP afinity-puri- fied antibodies revealed that all recombinant IgGs were >95% pure. IgGls were essentially secreted as covalent HzL2 tetra- mers, although the trace amounts of H2 and H2L intermediates were detected. Wild-type IgG4 and the IgG4 mutant
Role of CH2 Residue 331 in IgG Complement Binding Ability 3471
+ lgGl S331
0
+ lgG1 5331 u lgG4 wl + lgG4 P331
r
0
8 10’ I
0 1 .o 2.0 0 1 .o 2.0
IgG ADDED (ug) IgG ADDED (ug)
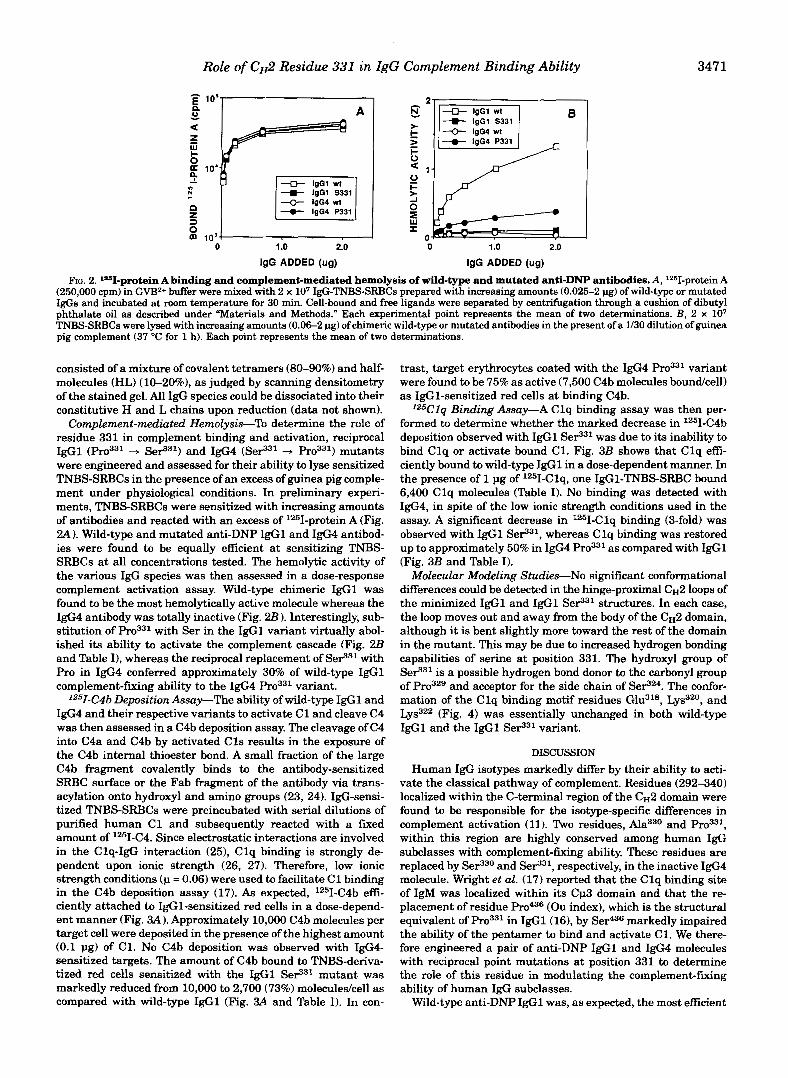
(250,000 cpm) in GVB2+ buffer were mixed with 2 x lo7 IgG-TNBS-SRBCs prepared with increasing amounts (0.025-2 pg) of wild-type or mutated FIG. 2. lsI-protein A binding and complement-mediated hemolysis of wild-type and mutated anti-DNP antibodies. A, lZ6I-protein A
IgGs and incubated at room temperature for 30 min. Cell-bound and free ligands were separated by centrifugation through a cushion of dibutyl
TNBS-SRBCs were lysed with increasing amounts (0.06-2 pg) of chimeric wild-type or mutated antibodies in the present of a 1/30 dilution of guinea phthalate oil as described under “Materials and Methods.” Each experimental point represents the mean of two determinations. B, 2 x lo7 pig complement (37 “C for 1 h). Each point represents the mean of two determinationa.
consisted of a mixture of covalent tetramers (80-90%) and half- molecules (HL) (10-20%), as judged by scanning densitometry of the stained gel. All IgG species could be dissociated into their constitutive H and L chains upon reduction (data not shown).
Complement-mediated Hemolysis-% determine the role of residue 331 in complement binding and activation, reciprocal IgGl + SeP31) and IgG4 (SeS3l + Pro331) mutants were engineered and assessed for their ability to lyse sensitized TNBS-SRBCs in the presence of an excess of guinea pig comple- ment under physiological conditions. In preliminary experi- ments, TNBS-SRBCs were sensitized with increasing amounts of antibodies and reacted with an excess of 1251-protein A (Fig. 2A). Wild-type and mutated anti-DNP IgGl and I@ antibod- ies were found to be equally efficient at sensitizing TNBS- SRBCs at all concentrations tested. The hemolytic activity of the various IgG species was then assessed in a dose-response complement activation assay. Wild-type chimeric IgGl was found to be the most hemolytically active molecule whereas the IgG4 antibody was totally inactive (Fig. 2 B ) . Interestingly, sub- stitution of with Ser in the IgGl variant virtually abol- ished its ability to activate the complement cascade (Fig. 2B and Table I), whereas the reciprocal replacement of SeP31 with Pro in IgG4 conferred approximately 30% of wild-type IgGl complement-fixing ability to the IgG4 variant.
Iz51-C4b Deposition Assay-The ability of wild-type IgGl and IgG4 and their respective variants to activate C1 and cleave C4 was then assessed in a C4b deposition assay. The cleavage of C4 into C4a and C4b by activated C l s results in the exposure of the C4b internal thioester bond. A small fraction of the large C4b fragment covalently binds to the antibody-sensitized SRBC surface or the Fab fragment of the antibody via trans- acylation onto hydroxyl and amino groups (23, 24). IgG-sensi- tized TNBS-SRBCs were preincubated with serial dilutions of purified human C1 and subsequently reacted with a fixed amount of 1251-C4. Since electrostatic interactions are involved in the Clq-IgG interaction (25), Clq binding is strongly de- pendent upon ionic strength (26, 27). Therefore, low ionic strength conditions (p = 0.06) were used to facilitate C1 binding in the C4b deposition assay (17). As expected, IZ5I-C4b effi- ciently attached to IgG1-sensitized red cells in a dose-depend- ent manner (Fig. 3A) . Approximately 10,000 C4b molecules per target cell were deposited in the presence of the highest amount (0.1 pg) of C1. No C4b deposition was observed with IgG4- sensitized targets. The amount of C4b bound to TNBS-deriva- tized red cells sensitized with the IgGl SeFj31 mutant was markedly reduced from 10,000 to 2,700 (73%) moleculedcell as compared with wild-type IgGl (Fig. 3A and Table I). In con-
trast, target erythrocytes coated with the IgG4 variant were found to be 75% as active (7,500 C4b molecules boundcell) as IgG1-sensitized red cells at binding C4b.
IZ5Clq Binding Assay-A Clq binding assay was then per- formed to determine whether the marked decrease in lZ51-C4b deposition observed with IgGl SeP31 was due to its inability to bind Clq or activate bound C1. Fig. 3B shows that Clq efi- ciently bound to wild-type IgGl in a dose-dependent manner. In the presence of 1 pg of lZ5I-Clq, one IgG1-TNBS-SRBC bound 6,400 Clq molecules (Table I). No binding was detected with IgG4, in spite of the low ionic strength conditions used in the assay. A significant decrease in ‘251-Clq binding (3-fold) was observed with IgGl SeP31, whereas Clq binding was restored up to approximately 50% in IgG4 Pro331 as compared with IgGl (Fig. 3B and Table I).
Molecular Modeling Studies-No significant conformational differences could be detected in the hinge-proximal C H ~ loops of the minimized IgGl and IgGl SeP31 structures. In each case, the loop moves out and away from the body of the CH2 domain, although it is bent slightly more toward the rest of the domain in the mutant. This may be due to increased hydrogen bonding capabilities of serine at position 331. The hydroxyl group of SeP31 is a possible hydrogen bond donor to the carbonyl group of Pro3” and acceptor for the side chain of SePz4. The confor- mation of the Clq binding motif residues Glu318, LYS~~O, and Lys322 (Fig. 4) was essentially unchanged in both wild-type IgGl and the IgGl SeP31 variant.
DISCUSSION Human IgG isotypes markedly differ by their ability to acti-
vate the classical pathway of complement. Residues (292-340) localized within the C-terminal region of the CH2 domain were found to be responsible for the isotype-specific differences in complement activation (11). Two residues, Ala330 and within this region are highly conserved among human IgG subclasses with complement-fixing ability. These residues are replaced by Ser330 and Ser331, respectively, in the inactive IgG4 molecule. Wright et al. (17) reported that the C lq binding site of IgM was localized within its Cp3 domain and that the re- placement of residue Pro436 (Ou index), which is the structural equivalent of Pro331 in IgGl(16), by Ser436 markedly impaired the ability of the pentamer to bind and activate C1. We there- fore engineered a pair of anti-DNP IgGl and IgG4 molecules with reciprocal point mutations at position 331 to determine the role of this residue in modulating the complement-f~ng ability of human IgG subclasses.
Wild-type anti-DNP IgGl was, as expected, the most efficient

3472 Role of CH2 Residue 331 in IgG Complement Binding Ability TABLE I
Relative hemolytic, Clq, and C4b binding activities of wild-type and mutated chimeric IgG antibodies
IgG species activity (2)- Hemolytic Bound Clqb
ne Moleculeelcell
C4b deposition'
ne Moleculeelcell
IgGl wt 1.000 98 2 4.2 67 2 3.3 10,000 2 493 (100%)
1 g ~ 4 ~ 0 3 3 1
IgGl Se1331 IgG4 wt
6,400 2 274 (100%) 1,900 2 85 (29%) 0.009
0.006 18 2 0.9 2,700 135 (27%)
0.281 48 f 5.3 3,100 -c 342 (48%) 50 -c 2.2 7,500 -c 330 (75%)
29 2 1.3 UDd UD UD UD
a Z values are calculated for 1 pg of IgG and expressed relative to that of wild-type IgGl taken as 1.000. * Amounts of cell-bound Clq per 2 x 10' TNBS-SRBCs sensitized with 2.5 pg of IgG antibodies and reacted with 1 pg of purified Clq. Results
e Amounts of cell-bound C4b per 2 x lo7 TNBS-SRBCs sensitized with 2.5 pg of IgG antibodies and reacted with 0.1 pg of purified C1. Assays are expressed as the means of two determinations -c S.E.
were performed in duplicate and results are expressed as the means of two independent determinations 2 S.E. UD, undetected.
A
- 40'
.1 1 10 100 loo0 .01 .1 1 10
C1 ADDED (ne) t 25 lGlq ADDED (ug)
FIG. 3. Deposition of '"I-C4b on and binding of '"I-Clq to IgG-TNBS-SRBCs. TNBS-SRBCs (2 x 10') in GVB2+ (p = 0.15) were preincubated with 2.5 pg of either wild-type or mutated chimeric IgG antibodies. A, TNBS-SRBCs were then mixed with serial dilutions (0.4-100.0 ng) of purified human Cl(30 "C, 30 min) and subsequently reacted with a fixed amount of Iz6I-C4 in SGVB2+ (p = 0.06) at 30 "C for 30 min. The amount of target-bound Iz6C4b was calculated as described under "Materials and Methods." Each assay was performed in duplicate, and each point represents the mean of two independent determinations. B , sensitized erythrocytes were reacted with increasing amounts of 1261-Clq (0.01-1 pg) in SGVBz+ at 30 "C for 30 min. The amount of bound Clq was determined as described under "Materials and Methods." Each point represents the mean of two determinations.
FIG. 4. A, ribbon diagram of the Fc do- main from Brookhaven Protein Data Bank entry lfcl with N-linked oligosac-
The lower hinge region (residues 229- charide separating the two cH2 domains.
237) is a model (R. Oomen, unpublished results) to indicate the approximate loca- tion of Leu234 relative to residue His266, G ~ u ~ ~ ~ , L ~ S ~ ~ " , Lys322, Ala330, and Van der Waals surfaces are shown for wzg6 on the opposite domain. B , all-atom (except hydrogens) detail of the boxed CH2 domain from A, in the same orientation. The carbohydrate moieties and labeled side chains are drawn with heavy lines. A
for the labeled residues using a 1.4 A solvent-accessible surface was calculated
probe according to Connolly (10). The sol- vent-accessible surface of residues Ala33o,
and those forming the EKK Clq binding motif are clearly contiguous.
A B +
antibody at lysing TNBS-SRBCs, whereas chimeric wild-type C4b deposition assays were performed in parallel to determine IgG4 was totally inactive. Since the degree of C1 activation and the mechanisms responsible for the changes in hemolytic ac- the extent of C4 cleavage depend upon the antibody isotype and tivities observed with the IgGl Ser331 and IgG4 mu- do not necessarily correlate (28, 29), both the Clq binding and tants. The Pro33 + SeS31 substitution in IgGl completely abol-

Role of CH2 Residue 331 in IgG Complement Binding Ability 3473 ished its hemolytic activity. Under low ionic strength conditions, the defective IgGl Se1331 mutant exhibited a marked decrease (73%) in its ability to cleave C4 as a result of a proportional reduction (71%) in Clq binding activity. In con- trast, the substitution of Se931 with Pro conferred 50% Clq binding activity to IgG4 relative to wild-type IgGl and an even more significant ability (75%) to fur C4b. This is remi- niscent of the findings by Bindon et al. (28,29) that, at compa- rable levels of C1 binding, some IgG isotypes either activate C4 faster or are better substrates for the covalent attachment of C4b. Taking into account the fact that reduced IgGs do not bind C1 (2) and that IgG4 Pro331 contained approximately 15% of half-molecules, its actual hemolytic activity was calculated to be 40% that of native IgG1.
These results clearly indicate that plays an important role in conferring complement-furring ability to IgG isotypes. This residue of the hinge-proximal C H ~ loop folds adjacent to residue Lys322 which is part of the EKK Clq binding motif (15) (Fig. 4B). Molecular modeling supports the premise that the
+ SeS3' substitution did not dramatically alter the secondary structure of this loop, but suggests that serine may introduce local interactions such as hydrogen bonding, which may preclude contact with a complementary surface on Clq. Although residues Glu3lS, LYS~~O, and Lys322 are likely involved in the formation of electrostatic bonds between Clq and IgGl (25,301, their functional role in Clq binding must be modulated by other residues, since they are conserved among both comple- ment-furing and complement nodixing antibodies. The func- tional role of could then be to facilitate Clq docking. However, substitution of Ser331 with Pro could not confer a full IgG1-like hemolytic activity to IgG4 Hinge exchange experiments revealed that the grafting of the short IgG4 hinge region onto IgG3 restricted its segmental flexibility (7) but did not alter the ability of either hinge-modified IgG3 (7, 8,31) or IgG1' antibodies to activate complement. Therefore, other IgG1-specific residues within the CH2 domain must contribute to the architecture of the Clq binding site. We have shown that replacement of Leu234 in the IgGl "hinge link" with the IgG4- specific residue Phe234 dramatically reduced IgGl complement- fixing activity; however, the reciprocal substitution did not con- fer C1 binding to the IgG4 mutant.'
Besides Pro331, other residues on the surface of the C H ~ domain are thought to contribute to Clq binding. One such residue is the neighboring IgGl amino acid Ala330, which is replaced with a serine in IgG4. Ala330, which is solvent-ex- posed, could either interact directly with Clq or play a role at presenting the side chain of residue 331 in the proper orienta- tion. The greater (4, W) space of alanine, as compared with residues with longer side chains, allows the main chain to adopt conformations that can accommodate subsequent steri- cally restricting proline residues. Residues 330 and 331 extend the contiguous patch?f the solvent-exposed surface of the EKK motif to about 280 A (Fig. 4). Flexibility in the lower hinge could also bring the Leu234 side chain into proximity with the Pr~~~l-containing loop, thereby extending the surface even more. However, this hypothesis cannot account for the fact that IgG2, which contains both the Ala330-Pro33' dipeptide and the EKK motif, has an extremely low affinity for Clq (28, 32).
The tyrosine found a t position 296 in IgGl and IgG3 sub- classes is replaced by phenylalanine in the complement non- furing IgG2 and IgG4 isotypes. It has thus been proposed (11) that wS, which is located on the X face of the CH2 domain opposite to the Clq binding site (Fig. a), might contribute to its architecture, since both CH2 domains were believed to be required for C1 activation (33). However, more recent studies with hybrid IgGs suggest that only one H chain is necessary for C1 binding (34).
Thus, residues 234,331, and perhaps 330 contribute to the Clq binding site and are, a t least in part, responsible for the isotype-specific differences in complement activation. Together with the EKK binding motif they form a solvent-exposed patch likely recognized by the globular Clq heads. This contiguous patch may be partially hindered in hinge-deleted IgGl and IgG3 molecules which lack inter-heavy chain disulfide bridges (3, 4) by the closer apposition of the Fab arms relative to Fc region. However, the recent finding that a hinge-deleted IgG3 with engineered disulfide bonds bridging its heavy chains can fur complement (35) confirms the early hypothesis (2) that a covalent linkage between immunoglobulin heavy chains is re- quired for complement-fixing activity. Previous studies have clearly demonstrated that complement furation was abolished or 50% reduced in aglycosylated immunoglobulins (12, 13) and IgGs lacking cH3 (33, 36), respectively. In this regard, struc- tural perturbations in the vicinity of the Hisz6' (Fig&?) re- porter group were detected by 'H NMR in aglycosylated IgG3 molecules (37, 38), indicating that the removal of the N-linked oligosaccharides induces a certain degree of distortion in the cH2 hinge-proximal loop. Taken together, these results suggest that both the clipping of the cH2 domains by the hinge disul- fides and the paired CH3 domains and the presence of carbo- hydrates are necessary to stabilize their native orientation and to preserve the functional integrity of the Clq binding site.
Acknowledgments-We thank Dr. D. Isenman for having generously provided purified human C1 and C4 and for helpful suggestions, Dr. Leroy Hood for having kindly donated the original yl and y4 constant region genes, Dr. Mine Rinfret for the construction of the pSV2neo- V,315 expression vector, Dr. Alex Marks for donating the MOPC 315.26 variant cell line, and William Bradley for synthesizing the mutagenic oligonucleotides.
2. 1.
3.
4.
5.
6.
7.
8.
9.
10.
REFERENCES
Burton, D. R., and Woof, J. M. (1992)Adu. Immunol. 51, 1-84 Isenman, D. E., Dorrington, K. J., and Painter, R. H. (1975) J. Immunol. 114,
1726-1729 Klein, M. H., HaeRner-Cavaillon, N.. Isenman, D. E., Rivat, C., Navia, M.,
Davies, D. R., and Domngton, K. J. (1981) Proc. Natl. Acad. Sci. U. S. A. 78,
Michaelsen, T. E., Aase, A., Westby, Y. C., and Sandlie, I. (1990) Scad. J. 524-528
Oi, V. T., Vuong. T. M., Hardy, R., Reidler, J., Dangl, J.. Herzenberg, L. A., and Immunol. 32,517528
Dangl, J. L., Wensel, T. G., Morrison, S. L., Stryer, L., Herzenberg, L. A., and Stryer, L. (1984) Nature 306, 136-140
Tan, L. K., Shopes, R. J., Oi, V. T., and Morrison, S . L. (1990) Proc. Nail. Acad. Oi, V. T. (1988) EMBO J. 7, 1989-1994
Norderhaug, L., Brekke, 0. H., Bremmes, B., Sandin, R., Aase, A., Michaelsen, Sci. U. S. A. 87, 162-166
Yasmeen, D., Ellerson, J. R., Dorrinnton, K J., and Painter. R. H. (1976) J. T. E., and Sandlie, I. (19911 Euc J. Immunol. 21, 2379-2384
Immunol. 116,518-526 ~ ~ Connolly, M. L. (1983) Science 221,709-713 11. Tao, M., Canfield, S . M., and Momson, S . L. (1991) J. Exp. Med. 175, 1025-
12. Nose, M., and Wizgell, H. (1983) P r o c . Natl. Acad. Sci. U. S. A 80,66324636 13. Tao, M., and Morrison, S . L. (1989) J. Immunol. 143, 2595-2601 14. Duncan, A. R., and Winter, G . (1988) Nature 332, 738-740 15. Deisenhofer, J. (1981) Biochemistry 20, 2361-2370 16. Perkins. S . J., Nealis, A. S., Sutton, B. J., and Feinstein,A. (1991) J. Mol. Biol.
1028
221. 1345-1366 17. Wright, J. F., Shulman, M. J., Isenman, D., and Painter, R. H. (1988) J. Biol.
I ~ ~ ~~~~
Chem. 263. 11221-11226 18. Rinfret, A., Horne, C.. Boux, H., Marks, A., Dorrington, K. J.. and Klein, M.
(1990) J. Zmmunol. 146,925-931 19. Sambrook, R., Fritsch, E. F., and Maniatis, J. (1989) Molecular Cloning; A
laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring
20. Taylor, J. W., Ott, J., and Eckstein, F. (1985) Nucleic Acids Res. 15,8764-8785 Harbor, NY
21. Rittenberg, M., and Pratt, K (1969) Proc. Soc. Exp. Biol. Med. 132.575-581 22. Tenner, A. J.. Lesavre, P. H., and Cooper, N. R. (1981) J. Zmmunol. 127,
23. Campbell, R. D., Dodds, A. W., and Porter, R. R. (1980) Biochem. J. 189,6740 24. Law, S . K. A,, and Reid, K B. M. (1988) Complement, IRL Press Ltd.. Oxford 25. Burton, D. R., Boyd, J., Brampton, A., Easterbrook-Smith, S., Emmanuel, E.
J., Novotny, J., Rademacher, T. W., Schravendijk, M. R., Sternberg, M. J. E.. and Dwek, R. A. (1980) Nature 288,338344
26. HughesJones, N. C. (1977) Immunology 32.191-198 27. Poon, P. H.. Phillips, M. L., and Schumaker, V. N. (1985) J. Biol. Chem. 260,
648-653
9357-9365

3474 Role of C,2 Residue 331 in IgG Complement Binding Ability 28. Bindon, C. I., Hale, C., Bruggemann, M., and Waldmann, H. (1988) J. Exp.
Med. 166. 127-142 29. Bindon, C. I.; Hale, C., and Waldmann, H. (1990)Eur: J. Zmmunol. 20.277-281 30. Schumaker, V. N., Calcott, M. A., Spiegelberg, H. L., and Muller-Eberhard, H.
31. Sandlie, I., Aase, A,, Westby, C., and Michaelsen, T. E. (1989)Eur: J. Zmmunol. J. (1976) Biochemistry 15,51755181
32. Briiggemann, M., Williams, G. T., Bindon, C. I., Clark, M. R., Walker, M. R., 19, 1599-1603
Jefferis, R.. Waldmann, H., and Neuberger, M. S . (1987) J. Exp. Med. 166, 1351-1361
33. Utsumi, S. M., Okada, K, Udaka, K, and Amano, T. (1985) Mol. Zmmunol. 22,
34. Clark, M., Bindon, C., Dyer, M., Friend, P., Hale, G., Cobbold, S., Caine, R., and
35. Brekke, 0. H., Michaelsen, T. E., Sandin, R., and Sandlie, I. (1993) Nature 563,
36. Colomb, M., and Porter, R. R. (1975) Biochem. J. 146,177-183 37. Lund, J., Tanaka, T., Takahashi, N., Sarmay. G., Arata. Y., and Jefferis, R.
(1990) Mol. Zmmunol. 27,1145-1153 38. Matsuda, H., Nakamura, S., Ichikawa, Y., Kozai, K, Takano, R.,
Nose, M., Endo, S., Nishimura, Y., and Arata, A (1990) Mol. Zmmunol. 27, 571-579
811-817
Waldmann, H. (1989) Eur: J. Zmmunol. 19,381388
628-630