Embed Size (px)
Citation preview

Research ArticlePrediction of Protein-Protein Interaction Strength UsingDomain Features with Supervised Regression
Mayumi Kamada1 Yusuke Sakuma2 Morihiro Hayashida3 and Tatsuya Akutsu3
1 Department of Biosciences and Informatics Keio University 3-14-1 Hiyoshi Kohoku-ku Yokohama 223-8522 Japan2 Japan Ichiba Section Development Unit Rakuten Inc 4-12-3 Higashi-shinagawa Shinagawa-ku Tokyo 140-0002 Japan3 Bioinformatics Center Institute for Chemical Research Kyoto University Gokasho Uji Kyoto 611-0011 Japan
Correspondence should be addressed to Morihiro Hayashida morihirokuicrkyoto-uacjp
Received 3 April 2014 Accepted 30 May 2014 Published 24 June 2014
Academic Editor Loris Nanni
Copyright copy 2014 Mayumi Kamada et al This is an open access article distributed under the Creative Commons AttributionLicense which permits unrestricted use distribution and reproduction in any medium provided the original work is properlycited
Proteins in living organisms express various important functions by interacting with other proteins and molecules Thereforemany efforts have been made to investigate and predict protein-protein interactions (PPIs) Analysis of strengths of PPIs isalso important because such strengths are involved in functionality of proteins In this paper we propose several feature spacemappings from protein pairs using protein domain information to predict strengths of PPIs Moreover we perform computationalexperiments employing twomachine learningmethods support vector regression (SVR) and relevance vector machine (RVM) fordataset obtained from biological experiments The prediction results showed that both SVR and RVM with our proposed featuresoutperformed the best existing method
1 Introduction
In cellular systems proteins perform their functions byinteracting with other proteins and molecules and protein-protein interactions (PPIs) play various important rolesTherefore revealing PPIs is a key to understanding biologicalsystems and many investigations and analyses have beendone In addition a variety of computational methods topredict and analyze PPIs have been developed for examplemethods for predicting PPI pairs using only sequences infor-mation [1ndash5] for predicting amino acid residues contributingto PPIs [6ndash8] and for assessing PPI reliability in PPI networks[9 10] As well as studies of PPIs analyses of strengths ofPPIs are important because such strengths are involved infunctionality of proteins In terms of transcription factorcomplexes if a constituent protein has aweak binding affinitytarget genes may not be transcribed depending on intracellu-lar circumstance For example it is known that multi-subunitcomplex NuA3 in Saccharomyces Cerevisiae consists of fiveproteins Sas3 Nto1 Yng1 Eaf6 and Taf30 acetylates lysine14 of histone H3 and activates gene transcription Howeveronly Yng1 and Nto1 are found solely in the complex and
interaction strengths between each component protein arethought to be different and transient Hence Byrum et alproposed a biological methodology for identifying stable andtransient protein interactions recently [11]
Although many biological experiments have been con-ducted for investigating PPIs [12 13] strengths of PPIshave not been always provided Ito et al conducted large-scale yeast two-hybrid experiments for whole yeast proteinsIn their experiments yeast two-hybrid experiments wereconducted for each protein pair multiple times the numberof experiments that observe interactions or the number ofinteraction sequence tags (ISTs) was counted Consequentlythey decided that protein pairs having three or more ISTsshould interact and reported interacting protein pairs
The ratio of the number of ISTs to the total num-ber of experiments for a protein pair can be regarded asthe interaction strength between their proteins On thebasis of this consideration several prediction methods forstrengths of PPIs have been developed LPNM [14] is alinear programming-basedmethod ASNM [15] is a modifiedmethod from the association method [16] for predictingPPIs Chen et al proposed association probabilistic method
Hindawi Publishing Corporatione Scientific World JournalVolume 2014 Article ID 240673 7 pageshttpdxdoiorg1011552014240673
2 The Scientific World Journal
(APM) [17] which is the best existing method for predictingstrengths of PPIs as far as we know
These methods are based on a probabilistic model of PPIsand make use of protein domain information Domains areknown as structural and functional units in proteins andwell-conserved regions in protein sequences The information ofdomains is stored in several databases such as Pfam [18] andInterPro [19] The same domain can be identified in severaldifferent proteins In these prediction methods interactionstrengths between domains are estimated from known inter-action strengths between proteins and interaction strengthsfor target protein pairs are predicted from estimated strengthsof domain-domain interactions (DDIs)
On the other hand Xia et al proposed a feature-basedmethod using neural network with features based on con-stituent domains of proteins [20] and they compared theirmethod with the association method and the expectation-maximization method [21] For the feature-based predictionof PPI strengths we also utilize domain information andpropose several feature space mappings from protein pairsWe use supervised regression and perform threefold crossvalidation for dataset obtained from biological experimentsThis paper augments the preliminary work presented in con-ference proceedings [22] Specifically major augmentationsof this paper and differences from the preliminary conferenceversion are summarized as follows
(i) We employ two supervised regression methods sup-port vector regression (SVR) and relevance vectormachine (RVM)Note that we used only SVRwith thepolynomial kernel in the preliminary version [22]
(ii) The Laplacian kernel is used as the kernel function forSVR and RVM and kernel parameters are selected viafivefold cross validation
(iii) We prepare the dataset from WI-PHI dataset [23]with high reliability
The computational experiments showed that the averagerootmean square error (RMSE) by our proposedmethodwassmaller than that by the best existing method APM [17]
2 Materials and Methods
In this section we briefly review a probabilistic model andrelated methods and propose several feature space mappingsusing domain information
21 ProbabilisticModel of PPIs Based onDDIs There are somecomputational prediction methods for PPI strengths andthey are based on the probabilistic model of PPIs proposedby Deng et al [21]Thismodel utilizes DDIs and assumes thattwo proteins interact with each other if and only if at leastone pair of the domains contained in the respective proteinsinteracts Figure 1(a) illustrates an example of this interactionmodel In this example there are two proteins 119875
1and 119875
2
which consist of domains 1198631 1198632and domains 119863
2 1198633 1198634
respectively According to Dengrsquos model if 1198751and 1198752interact
at least one pair among (1198631 1198632) (1198631 1198633) (1198631 1198634) (1198632
1198632) (1198632 1198633) and (119863
2 1198634) interacts Conversely if a pair
for instance (1198632 1198634) interacts 119875
1and 119875
2interact From
the assumption of this model we can derive the followingsimple probability that two proteins 119875
119894and 119875
119895interact with
each other
Pr (119875119894119895= 1) = 1 minus prod
119863119898isin119875119894119863119899isin119875119895
(1 minus Pr (119863119898119899
= 1)) (1)
where 119875119894119895
= 1 indicates the event that proteins 119875119894and 119875
119895
interact (otherwise 119875119894119895= 0) 119863
119898119899= 1 indicates the event
that domains 119863119898and 119863
119899interact (otherwise 119863
119898119899= 0) and
119875119894and 119875
119895also represent the sets of domains contained in 119875
119894
and 119875119895 respectively Deng et al applied the EM (expectation
maximization) algorithm to the problem of maximizing log-likelihood functions the estimated probabilities that twodomains interact Pr(119863
119898119899= 1) and proposed a method for
predicting PPIs using the estimated probabilities ofDDIs [21]Actually they calculated Pr(119875
119894119895= 1) using (1) and determined
whether or not 119875119894and 119875
119895interact by introducing a threshold
120579 that is 119875119894and 119875
119895interact if Pr(119875
119894119895= 1) ge 120579 otherwise the
proteins do not interactAsDengrsquosmethod typical PPIs predictionmethods based
on domains have the following two steps First the interactionbetween domains contained in interacting proteins is inferredfrom existing protein interaction data And then an inter-action between new protein pairs is predicted on the basisof the inferred domain interactions using a certain modelFigure 1(b) illustrates the flow of this type of PPIs predictionSince interacting sites may not be always included in someknown domain region it can cause the decrease of predictionaccuracy in this framework
22 Association Method Inferring DDI from PPI Data Asdescribed previously probability of PPIs could be predictedbased on probabilities of DDIs In this subsection we willbriefly review related methods to estimate a probability ofinteraction for domain pair
23 Association Method Let P be a set of protein pairsthat have been observed to interact or not The associationmethod [16] gives the following simple score for two domains119863119898and119863
119899using proteins that include the following domains
ASSOC (119863119898 119863119899)
=
10038161003816100381610038161003816(119875119894 119875119895) isin P | 119863
119898isin 119875119894 119863119899isin 119875119895 119875119894119895= 1
1003816100381610038161003816100381610038161003816100381610038161003816(119875119894 119875119895) isin P | 119863
119898isin 119875119894 119863119899isin 11987511989510038161003816100381610038161003816
(2)
where |119878| indicates the number of elements contained inthe set 119878 This score represents the ratio of the number ofinteracting protein pairs including 119863
119898and 119863
119899to the total
number of protein pairs including 119863119898and 119863
119899 Hence it can
be considered as the probability that119863119898and119863
119899interact
24 Association Method for Numerical Interaction Data(ASNM) Originally the association method has been de-signed for inferring binary protein interactions To pre-dict numerical interactions such as interaction strengthsHayashida et al proposed the associationmethod for numer-ical interaction (ASNM) by the modification of the original
The Scientific World Journal 3
P1P2
D1
D2
D2
D3
D4
(a)
Interacting proteins with their domains
Predicting PPIs based on inferred DDIs
Inferring model for DDIs
middot middot middot
middot middot middot
Protein
Protein Protein
Protein
(b)
Figure 1 (a) Illustration of protein-protein interactions (PPIs) model based on domain-domain interactions (DDIs) (b) Schematic overviewof PPIs prediction based on DDIs
association method [15] This method takes strengths ofPPIs as input data Let 120588
119894119895represent the interaction strength
between 119875119894and 119875
119895 and we suppose that 120588
119894119895is defined for all
(119875119894 119875119895) isin P Then the ASNM score for domains 119863
119898and 119863
119899
is defined as the average strength over protein pairs including119863119898and119863
119899by
ASNM (119863119898 119863119899) =
sum(119875119894119875119895)isinP|119863
119898isin119875119894119863119899isin119875119895120588119894119895
10038161003816100381610038161003816(119875119894 119875119895) isin P | 119863
119898isin 119875119894 119863119899isin 11987511989510038161003816100381610038161003816
(3)If 120588119894119895always takes only 0 or 1 ASNM(119863
119898 119863119899) becomes
ASSOC(119863119898 119863119899)
25 Association Probabilistic Method (APM) AlthoughASNM is a simple average of strengths of PPIs Chen et alproposed the association probabilistic method (APM) byreplacing the strength with an improved strength [17] It isbased on the idea that the contribution of one domain pair tothe strength of PPI should vary depending on the number ofdomain pairs included in a protein pair They assumed thatthe interaction probability of each domain pair is equivalentin a protein pair and transformed (1) as follows
Pr (119863119898119899
= 1) = 1 minus (1 minus Pr (119875119894119895= 1))1|119875119894||119875119895|
(4)
Thus by substituting the numerator of ASNM APM isdefined by
APM (119863119898 119863119899)
=
sum(119875119894119875119895)isinP|119863
119898isin119875119894119863119899isin119875119895(1 minus (1 minus 120588
119894119895)1|119875119894||119875119895|
)
10038161003816100381610038161003816(119875119894 119875119895) isin P | 119863
119898isin 119875119894 119863119899isin 11987511989510038161003816100381610038161003816
(5)
They conducted some computational experiments andreported that APM outperforms existing prediction methodssuch as ASNM and LPNM
26 Proposed Feature Space Mappings from Protein PairsThe association methods including ASNM and APM arebased on the probabilistic model of PPIs defined by (1)and infer strengths of PPIs from estimated DDIs usinggiven frequency of interactions or interaction strengths ofprotein pairs On the other hand we can also infer PPIstrengths utilizing features obtained from given informationsuch as sequence and structure of proteins with machinelearning methods Xia et al proposed a method to inferstrengths of PPIs using artificial neural network with featuresfrom constituent domains of proteins [20] In this paperfor predicting strengths of PPIs we propose several featurespace mappings from protein pairs making use of domaininformation
27 Feature Based onNumber of Domains (DN) As describedabove constituent domains information is useful for infer-ring PPIs and also can be used as a representation of eachprotein Actually Xia et al represented each protein bybinary numbers indicating whether a protein has a domainor not based on the information of constituent domains andused them with the artificial neural network to predict PPIstrengths [20] Here it can be considered that the probabilitythat two proteins interact increases with a larger number ofdomains included in the proteins Therefore in this paperwe propose a feature space mapping based on the numberof constituent domains (called DN) from two proteins Thefeature vector of DN for two proteins 119875
119894and 119875
119895is defined by
119891(119898)
119894119895= 119872(119863
119898 119875119894) for 119863
119898isin 119875119894
119891(119879+119899)
119894119895= 119872(119863
119899 119875119895) for 119863
119899isin 119875119895
119891(119897)
119894119895= 0 for 119863
119897notin 119875119894cup 119875119895
(6)
4 The Scientific World Journal
D1 D2 D3
MKANGLDNDOARTRMERTDIDSEHPEAQPLLNNNHRTLGAGSANGPAVEGRDIE
LDNDOARTRMERIDSEHPEAQPLLNNNHRGSANGPAVE
Figure 2 Illustration of restricting an amino acid sequence towhichthe spectrum kernel is applied to the domain regions
where 119879 indicates the total number of domains over allproteins and 119872(119863
119898 119875119894) indicates the number of domains
identified as119863119898in protein 119875
119894
28 Feature by Restriction of Spectrum Kernel to DomainRegion (SPD) DN is based only on the number of constituentdomains of each protein while amino acid sequences ofdomains are also considered useful for inferring strengthof PPI Therefore we propose a feature space mapping byrestricting the application of the spectrum kernel [24] todomain regions (called SPD) LetA be the set of 21 alphabetsrepresenting 20 types of amino acids and others Althoughwe used the set of 20 alphabets to express 20 types of aminoacids in the preliminary conference version [22] we add onealphabet to take the ambiguous amino acids such as X intoconsideration Then A119896 (119896 ge 1) means the set of all stringswith length 119896 generated from A The 119896-spectrum kernel forsequences 119909 and 119910 is defined by
119870119896(119909 119910) = ⟨Φ
119896(119909) Φ
119896(119910)⟩ (7)
whereΦ119896(119909) = (120601
119904(119909))119904isinA119896 and 120601119904(119909) indicates the number of
times that 119904 occurs in 119909 To make use of domain informationwe restrict an amino acid sequence to which the 119896-spectrumkernel is applied to the domain regions Figure 2 illustrates therestriction In this example the protein consists of domains1198631 1198632 1198633 and each domain region is surrounded by a
square Then the subsequence in each domain is extractedand all the subsequences in the protein are concatenated inthe same order as domains We apply the 119896-spectrum kernelto the concatenated sequence Let 120601(119903)
119904(119909) be the number of
times that string 119904 occurs in the sequence restricted to thedomain regions in protein 119909 in the abovemannerThe featurevector of SPD for proteins 119875
119894and 119875
119895is defined by
119891119897
119894119895= 120601(119903)
119904119897
(119875119894) for 119904
119897isin A119896
119891(21119896
+119897)
119894119895= 120601(119903)
119904119897
(119875119895) for 119904
119897isin A119896
(8)
It should be noted that 120601(119903)119904
for proteins having the samecomposition of domains can vary depending on the aminoacid sequences of their proteins That is even if 119875
119894and 119875
119895
have the same compositions as 119875119896and 119875
119897 respectively and
the feature vector of DN for 119875119894and 119875
119895is the same as that for
119875119896and 119875
119897 then the feature vector of SPD for 119875
119894and 119875
119895can be
different from that for 119875119896and 119875119897
29 Support Vector Regression (SVR) To predict strengths ofPPIs we employ support vector regression (SVR) [25] with
our proposed features In the case of linear functions SVRfinds parameters119908 and 119887 for 119891(119909) = ⟨119908 119909⟩ + 119887 by solving thefollowing optimization problem
minimize 1
21199082+ 119862sum
119894
(120585119894+ 1205851015840
119894)
subject to 119910119894minus ⟨119908 119909
119894⟩ minus 119887 le 120598 + 120585
119894
119910119894minus ⟨119908 119909
119894⟩ minus 119887 ge minus120598 minus 120585
1015840
119894
120585119894ge 0 120585
1015840
119894ge 0
(9)
where 119862 and 120598 are positive constants and (119909119894 119910119894) is a training
data Here the penalty is added only if the difference between119891(119909119894) and 119910
119894is larger than 120598 In our problem 119909
119894means a
protein pair and 119910119894means the corresponding interaction
strength
210 Relevance Vector Machine (RVM) In this paper wealso employ relevance vector machine (RVM) [26] to predictstrengths of PPIs RVM is a sparse Bayesian model utilizingthe same data-dependent kernel basis as the SVM Its frame-work is almost the same as typical Bayesian linear regressionGiven a training data 119909
119894 119910119894119873
119894=0 the conditional probability of
119910 given 119909 is modeled as
119901 (119910 | 119909 119908 120573) = N (119910 | 119908119879120601 (119909) 120573
minus1) (10)
where 120573 = 1205902 is noise parameter and 120601(sdot) is a typically
nonlinear projection of input features To obtain sparsesolutions in RVM framework a prior weight distribution ismodified so that a different variance parameter is assigned foreach weight as
119901 (119908 | 120572) = prod
119894=0
N (119908119894| 0 120572minus1
119894) (11)
where119872 = 119873+1 and 120572 = (1205721 120572
119872)119879 is a hyperparameter
RVM finds hyperparameter 120572 by maximizing the marginallikelihood 119901(119910 | 119909 120572) via ldquoevidence approximationrdquo Inthe process of maximizing evidence some 120572
119894approach
infinity and the corresponding 119908119894become zero Thus the
basis function corresponding with these parameters can beremoved and it leads sparse models In many cases RVMperforms better than SVM especially in regression problems
3 Results and Discussion
31 Computational Experiments To evaluate our proposedmethod we conducted computational experiments and com-pared with the existing method APM
32 Data and Implementation It is difficult to directlymeasure actual strengths of PPIs for many protein pairs bybiological and physical experiments Hence we used WI-PHI dataset with 50000 protein pairs [23] For each PPI WI-PHI contains a weight that is considered to represent somereliability of the PPI and is calculated from several different
The Scientific World Journal 5
Table 1 Results of average RMSE for training and test data
119862 = 1 119862 = 2 119862 = 5
Training Test Training Test Training TestSVR + DN 010472 012573 010656 012600 009982 012484RVM + DN 009210 012873 009178 012881 009474 012908SVR + SPD (119896 = 1) 008819 012699 008080 012954 007927 012903RVM + SPD (119896 = 1) 002848 012743 001504 012706 003276 012792SVR + SPD (119896 = 2) 008891 012654 008188 012782 008117 012909RVM + SPD (119896 = 2) 002529 012470 002301 012476 002243 012493SVR + APM 006846 013112 006795 013247 006791 013277RVM + APM 007052 013556 007037 013550 007032 013493APM Training = 006811 Test = 013517
kinds of PPI datasets in some statistical manner to rankphysical protein interactions As strengths of PPIs we usedthe value dividing the weight of PPI by the maximum weightforWI-PHIWe used dataset file ldquouniprot sprot fungidatgzrdquodownloaded from UniProt database [27] to get amino acidsequences information of domain compositions and domainregions in proteins In this experiment we used 1387 proteinpairs that could be extracted from WI-PHI dataset withcomplete domain sequence viaUniProt datasetThe extracteddataset contains 758 proteins and 327 domains Since thisdataset does not include protein pairs with interactionstrength 0 we randomly selected 100 protein pairs that do nothave any weights in the dataset and added them as proteinpairs with strength 0 Thus totally 1487 protein pairs wereused in this experiment We used ldquokernlabrdquo package [28]for executing support vector regression and relevance vectormachine and used the Laplacian kernel119870(119909 119910) = exp(minus120590119909minus119910) The dataset and the source code implemented by R areavailable upon request
To evaluate prediction accuracy we calculated the rootmean square error (RMSE) for each prediction RMSE isa measure of differences between predicted values 119910
119894and
actually observed values 119910119894and is defined by
RMSE = radic1
119873
119873
sum
119894=1
(119910119894minus 119910119894)2
(12)
where119873 is the number of test data
33 Results of Computational Experiments We preformedthreefold cross-validation calculated the average RMSE andcompared with APM [17] For APM method strengths ofPPIs are inferred based on APM scores for domain pairs thatconsist of target proteins However it is not always possible tocalculate APM scores for all domain pairs from training setTherefore as test set we used only protein pairs that consistof domain pairs with APM scores calculated via training set(In all cases about 40 of protein pairs in test set were used)For the Laplacian kernel employed in both SVR and RVMwe selected kernel parameter 120590 by fivefold cross-validationfrom candidate set 120590 isin 001 002 01 The parameter119862 for the regularization term in the Lagrange formulation isset to 119862 = 1 2 5 Additionally APM scores for each protein
pair also can be used as input featuresTherefore we also usedAPM scores as inputs for SVR and RVM and compared themodel using APM scores with the model using our proposedfeatures to confirm the usefulness of feature representationHere we used candidate set 120590 isin 30 31 32 90 forkernel parameter 120590 of RVM+APMmodel because themodelcould not be trained with 120590 values smaller than 3 On theother hand for 120590 of SVM + APM model we used the sameset as other models
Table 1 shows the results of the average RMSE by SVR andRVM with our proposed features (DN and SPD of 119896 = 1 2)and APM score and by APM for training and test datasetsFor training set the average RMSEs by RVM with SPD of119896 = 2were smaller than those by APM and others Moreoverfor test set all the average RMSEs by RVMwith SPD and DNwere smaller than those by APM The results suggested thatsupervised regression methods SVR and RVM with domainbased features are useful for prediction of PPI strengthsTaking all results together the model by RVM with SPD of119896 = 2was regarded as the best for prediction of PPI strengths
Since the average RMSEs of SVR with APM for bothtraining and test dataset were smaller than those of originalAPM SVR has potential to improve prediction accuraciesBy contrast the average RMSEs of RVM with APM becamelarger than those of original APM and all average RMSEsof the models with APM for test set were larger thanthose of the models with DN and SPD Accordingly theresults suggested that prediction accuracies were enhancedby feature representation and SPD is especially useful amongthese feature representations for predicting strengths of PPIsAlthough DN and SPD of 119896 = 1 have 654 and 42 dimensionsfor each protein pair respectively the average RMSEs withSPDof 119896 = 1 for training setwere smaller than thosewithDNIt implies that information of amino acid sequence in domainregions is more informative comparing with information ofdomain compositions to make a model fit in with dataset
In contrast the RMSEs by SVR with DN were smallerthan those by others in some cases of test set Table 2 showsthe numbers of relevance vectors and support vectors andthe 120590 values selected by fivefold cross-validation in all casesFor the models with DN and APM scores the numbers ofrelevance vectors were smaller than the numbers of supportvectors On the other hand the numbers of relevance vectorswere larger than the numbers of support vectors for the
6 The Scientific World Journal
Table 2The number of relevance vectors (RVs) and support vectors (SVs) for each model with DN SPD and APM and the selected 120590 valuesfor each fold
119862 = 1 119862 = 2 119862 = 5
SVR RVM SVR RVM SVR RVMSVs (120590 value) RVs (120590 value) SVs (120590 value) RVs (120590 value) SVs (120590 value) RVs (120590 value)
Fold 1
DN 271 (002) 113 (005) 271 (001) 123 (007) 308 (001) 74 (002)SPD (119896 = 1) 367 (001) 448 (002) 402 (002) 680 (005) 402 (002) 537 (003)SPD (119896 = 2) 392 (001) 502 (003) 409 (001) 628 (005) 421 (001) 628 (005)
APM 362 (008) 4 (500) 361 (010) 6 (480) 357 (004) 6 (580)
Fold 2
DN 280 (002) 94 (008) 281 (001) 92 (009) 314 (001) 82 (004)SPD (119896 = 1) 408 (001) 617 (004) 453 (004) 706 (006) 411 (001) 545 (003)SPD (119896 = 2) 430 (001) 558 (004) 435 (001) 618 (005) 495 (004) 654 (006)
APM 375 (010) 5 (650) 372 (010) 6 (690) 373 (004) 4 (550)
Fold 3
DN 321 (004) 107 (008) 289 (001) 107 (010) 330 (001) 107 (008)SPD (119896 = 1) 371 (001) 439 (002) 412 (003) 658 (005) 382 (001) 305 (001)SPD (119896 = 2) 387 (001) 625 (006) 418 (002) 529 (004) 398 (001) 529 (004)
APM 368 (008) 3 (710) 368 (004) 3 (670) 372 (001) 5 (420)
models with SPD feature in spite of the fact that usuallyRVM provides a sparse model compared with SVR In RVMframework sparsity of model is caused by distributions ofeach weight that is the number of relevance vectors isinfluenced by values and variances of each dimension offeatures rather than by the number of dimensions of featuresActually each dimension of SPD feature almost always haswidely varying values In contrast DN feature has manyzeros and APM score is inferred from training dataset andthereby has similar distribution Thus it is considered thatmany weights corresponding to features in RVM model didnot become zero and the RVM models with SPD featuretended to be complex and to overfit the training data
4 ConclusionsFor the prediction of strengths of PPIs we proposed featurespace mappings DN and SPD DN is based on the numberof domains in a protein SPD is based on the spectrumkernel and defined using the amino acid subsequences indomain regions In this work we employed support vec-tor regression (SVR) and relevance vector machine (RVM)with the Laplacian kernel and conducted threefold cross-validation using WI-PHI dataset For both training and testdataset the average RMSEs by RVM with SPD feature weresmaller than those by APMThe results showed that machinelearning methods with domain information outperformedexisting association method that is based on the probabilisticmodel of PPIs and implied that the information of aminoacid sequence is useful for prediction comparing with onlyinformation of domain compositions However the modelswith SPD feature tended to be complex and overfitted to thetraining data Therefore to further enhance the predictionaccuracy improving kernel functions combining physicalcharacteristics of domains and amino acids might be helpful
Conflict of InterestsThe authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgment
This work was partially supported by Grants-in-Aid nos22240009 and 24500361 fromMEXT Japan
References
[1] J R Bock and D A Gough ldquoPredicting protein-proteininteractions from primary structurerdquo Bioinformatics vol 17 no5 pp 455ndash460 2001
[2] Y Guo L Yu Z Wen and M Li ldquoUsing support vectormachine combined with auto covariance to predict protein-protein interactions from protein sequencesrdquo Nucleic AcidsResearch vol 36 no 9 pp 3025ndash3030 2008
[3] C Yu L Chou and D T Chang ldquoPredicting protein-proteininteractions in unbalanced data using the primary structure ofproteinsrdquo BMC Bioinformatics vol 11 article 167 2010
[4] J Xia X Zhao and D Huang ldquoPredicting protein-proteininteractions from protein sequences using meta predictorrdquoAmino Acids vol 39 no 5 pp 1595ndash1599 2010
[5] J Xia K Han and D Huang ldquoSequence-based prediction ofprotein-protein interactions by means of rotation forest andautocorrelation descriptorrdquo Protein and Peptide Letters vol 17no 1 pp 137ndash145 2010
[6] S J Darnell D Page and J CMitchell ldquoAn automated decision-tree approach to predicting protein interaction hot spotsrdquoProteins Structure Function and Bioinformatics vol 68 no 4pp 813ndash823 2007
[7] K Cho D Kim and D Lee ldquoA feature-based approach tomodeling protein-protein interaction hot spotsrdquo Nucleic AcidsResearch vol 37 no 8 pp 2672ndash2687 2009
[8] J Xia X Zhao J Song and D Huang ldquoAPIS accurate predic-tion of hot spots in protein interfaces by combining protrusionindexwith solvent accessibilityrdquoBMCBioinformatics vol 11 no1 article 174 2010
[9] O KuchaievM Rasajski D J Higham andN Przulj ldquoGeomet-ric de-noising of protein-protein interaction networksrdquo PLoSComputational Biology vol 5 no 8 Article ID e1000454 2009
[10] L Zhu Z You and D Huang ldquoIncreasing the reliability ofprotein-protein interaction networks via non-convex semanticembeddingrdquo Neurocomputing vol 121 pp 99ndash107 2013
The Scientific World Journal 7
[11] S Byrum S Smart S Larson and A Tackett ldquoAnalysis ofstable and transient protein-protein interactionsrdquo Methods inMolecular Biology vol 833 pp 143ndash152 2012
[12] P Uetz L Glot G Cagney et al ldquoA comprehensive analysisof protein-protein interactions in Saccharomyces cerevisiaerdquoNature vol 403 no 6770 pp 623ndash627 2000
[13] T Ito T Chiba R Ozawa M Yoshida M Hattori and YSakaki ldquoA comprehensive two-hybrid analysis to explore theyeast protein interactomerdquo Proceedings of the National Academyof Sciences of the United States of America vol 98 no 8 pp4569ndash4574 2001
[14] M Hayashida N Ueda and T Akutsu ldquoInferring strengthsof protein-protein interactions from experimental data usinglinear programmingrdquo Bioinformatics vol 19 no 2 pp ii58ndashii652003
[15] M Hayashida N Ueda and T Akutsu ldquoA simple methodfor inferring strengths of protein-protein interactionsrdquoGenomeInformatics vol 15 no 1 pp 56ndash68 2004
[16] E Sprinzak and H Margalit ldquoCorrelated sequence-signaturesas markers of protein-protein interactionrdquo Journal of MolecularBiology vol 311 no 4 pp 681ndash692 2001
[17] L Chen L Wu Y Wang and X Zhang ldquoInferring-proteininteractions from experimental data by association probabilisticmethodrdquo Proteins Structure Function and Genetics vol 62 no4 pp 833ndash837 2006
[18] R Finn J Mistry J Tate et al ldquoThe Pfam protein familiesdatabaserdquo Nucleic Acids Research vol 38 supplement 1 ppD211ndashD222 2009
[19] S Hunter P Jones A Mitchell et al ldquoInterPro in 2011 newdevelopments in the family and domain prediction databaserdquoNucleic Acids Research vol 40 no 1 pp D306ndashD312 2012
[20] J Xia B Wang and D Huang ldquoInferring strengths of protein-protein interaction using artificial neural networkrdquo in Proceed-ings of the International Joint Conference on Neural Networks(IJCNN rsquo07) pp 2471ndash2475 Orlando Fla USA August 2007
[21] M Deng S Mehta F Sun and T Chen ldquoInferring domain-domain interactions from protein-protein interactionsrdquoGenome Research vol 12 no 10 pp 1540ndash1548 2002
[22] Y Sakuma M Kamada M Hayashida and T Akutsu ldquoInfer-ring strengths of protein-protein interactions using supportvector regressionrdquo inProceedings of the International Conferenceon Parallel and Distributed Processing Techniques and Applica-tions 2013 httpworld-comporgp2013PDP2162pdf
[23] L Kiemer S Costa M Ueffing and G Cesareni ldquoWI-PHIa weighted yeast interactome enriched for direct physicalinteractionsrdquo Proteomics vol 7 no 6 pp 932ndash943 2007
[24] C Leslie E Eskin and W Noble ldquoThe spectrum Kernel astring Kernel for SVM protein classi fi cationrdquo in Proceedingsof Pacific Symposium on Biocomputing (PSB rsquo02) pp 564ndash575Lihue Hawaii USA January 2002
[25] VN VapnikTheNature of Statistical LearningTheory Springer1995
[26] M E Tipping ldquoSparse bayesian learning and the relevancevector machinerdquo Journal of Machine Learning Research vol 1no 3 pp 211ndash244 2001
[27] T U Consortium ldquoReorganizing the protein space at theuniversal protein resource (UniProt)rdquo Nucleic Acids Researchvol 40 no D1 pp D71ndashD75 2012
[28] A Karatzoglou K Hornik A Smola and A Zeileis ldquokernlabmdashan S4 package for kernel methods in Rrdquo Journal of StatisticalSoftware vol 11 no 9 pp 1ndash20 2004
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

2 The Scientific World Journal
(APM) [17] which is the best existing method for predictingstrengths of PPIs as far as we know
These methods are based on a probabilistic model of PPIsand make use of protein domain information Domains areknown as structural and functional units in proteins andwell-conserved regions in protein sequences The information ofdomains is stored in several databases such as Pfam [18] andInterPro [19] The same domain can be identified in severaldifferent proteins In these prediction methods interactionstrengths between domains are estimated from known inter-action strengths between proteins and interaction strengthsfor target protein pairs are predicted from estimated strengthsof domain-domain interactions (DDIs)
On the other hand Xia et al proposed a feature-basedmethod using neural network with features based on con-stituent domains of proteins [20] and they compared theirmethod with the association method and the expectation-maximization method [21] For the feature-based predictionof PPI strengths we also utilize domain information andpropose several feature space mappings from protein pairsWe use supervised regression and perform threefold crossvalidation for dataset obtained from biological experimentsThis paper augments the preliminary work presented in con-ference proceedings [22] Specifically major augmentationsof this paper and differences from the preliminary conferenceversion are summarized as follows
(i) We employ two supervised regression methods sup-port vector regression (SVR) and relevance vectormachine (RVM)Note that we used only SVRwith thepolynomial kernel in the preliminary version [22]
(ii) The Laplacian kernel is used as the kernel function forSVR and RVM and kernel parameters are selected viafivefold cross validation
(iii) We prepare the dataset from WI-PHI dataset [23]with high reliability
The computational experiments showed that the averagerootmean square error (RMSE) by our proposedmethodwassmaller than that by the best existing method APM [17]
2 Materials and Methods
In this section we briefly review a probabilistic model andrelated methods and propose several feature space mappingsusing domain information
21 ProbabilisticModel of PPIs Based onDDIs There are somecomputational prediction methods for PPI strengths andthey are based on the probabilistic model of PPIs proposedby Deng et al [21]Thismodel utilizes DDIs and assumes thattwo proteins interact with each other if and only if at leastone pair of the domains contained in the respective proteinsinteracts Figure 1(a) illustrates an example of this interactionmodel In this example there are two proteins 119875
1and 119875
2
which consist of domains 1198631 1198632and domains 119863
2 1198633 1198634
respectively According to Dengrsquos model if 1198751and 1198752interact
at least one pair among (1198631 1198632) (1198631 1198633) (1198631 1198634) (1198632
1198632) (1198632 1198633) and (119863
2 1198634) interacts Conversely if a pair
for instance (1198632 1198634) interacts 119875
1and 119875
2interact From
the assumption of this model we can derive the followingsimple probability that two proteins 119875
119894and 119875
119895interact with
each other
Pr (119875119894119895= 1) = 1 minus prod
119863119898isin119875119894119863119899isin119875119895
(1 minus Pr (119863119898119899
= 1)) (1)
where 119875119894119895
= 1 indicates the event that proteins 119875119894and 119875
119895
interact (otherwise 119875119894119895= 0) 119863
119898119899= 1 indicates the event
that domains 119863119898and 119863
119899interact (otherwise 119863
119898119899= 0) and
119875119894and 119875
119895also represent the sets of domains contained in 119875
119894
and 119875119895 respectively Deng et al applied the EM (expectation
maximization) algorithm to the problem of maximizing log-likelihood functions the estimated probabilities that twodomains interact Pr(119863
119898119899= 1) and proposed a method for
predicting PPIs using the estimated probabilities ofDDIs [21]Actually they calculated Pr(119875
119894119895= 1) using (1) and determined
whether or not 119875119894and 119875
119895interact by introducing a threshold
120579 that is 119875119894and 119875
119895interact if Pr(119875
119894119895= 1) ge 120579 otherwise the
proteins do not interactAsDengrsquosmethod typical PPIs predictionmethods based
on domains have the following two steps First the interactionbetween domains contained in interacting proteins is inferredfrom existing protein interaction data And then an inter-action between new protein pairs is predicted on the basisof the inferred domain interactions using a certain modelFigure 1(b) illustrates the flow of this type of PPIs predictionSince interacting sites may not be always included in someknown domain region it can cause the decrease of predictionaccuracy in this framework
22 Association Method Inferring DDI from PPI Data Asdescribed previously probability of PPIs could be predictedbased on probabilities of DDIs In this subsection we willbriefly review related methods to estimate a probability ofinteraction for domain pair
23 Association Method Let P be a set of protein pairsthat have been observed to interact or not The associationmethod [16] gives the following simple score for two domains119863119898and119863
119899using proteins that include the following domains
ASSOC (119863119898 119863119899)
=
10038161003816100381610038161003816(119875119894 119875119895) isin P | 119863
119898isin 119875119894 119863119899isin 119875119895 119875119894119895= 1
1003816100381610038161003816100381610038161003816100381610038161003816(119875119894 119875119895) isin P | 119863
119898isin 119875119894 119863119899isin 11987511989510038161003816100381610038161003816
(2)
where |119878| indicates the number of elements contained inthe set 119878 This score represents the ratio of the number ofinteracting protein pairs including 119863
119898and 119863
119899to the total
number of protein pairs including 119863119898and 119863
119899 Hence it can
be considered as the probability that119863119898and119863
119899interact
24 Association Method for Numerical Interaction Data(ASNM) Originally the association method has been de-signed for inferring binary protein interactions To pre-dict numerical interactions such as interaction strengthsHayashida et al proposed the associationmethod for numer-ical interaction (ASNM) by the modification of the original
The Scientific World Journal 3
P1P2
D1
D2
D2
D3
D4
(a)
Interacting proteins with their domains
Predicting PPIs based on inferred DDIs
Inferring model for DDIs
middot middot middot
middot middot middot
Protein
Protein Protein
Protein
(b)
Figure 1 (a) Illustration of protein-protein interactions (PPIs) model based on domain-domain interactions (DDIs) (b) Schematic overviewof PPIs prediction based on DDIs
association method [15] This method takes strengths ofPPIs as input data Let 120588
119894119895represent the interaction strength
between 119875119894and 119875
119895 and we suppose that 120588
119894119895is defined for all
(119875119894 119875119895) isin P Then the ASNM score for domains 119863
119898and 119863
119899
is defined as the average strength over protein pairs including119863119898and119863
119899by
ASNM (119863119898 119863119899) =
sum(119875119894119875119895)isinP|119863
119898isin119875119894119863119899isin119875119895120588119894119895
10038161003816100381610038161003816(119875119894 119875119895) isin P | 119863
119898isin 119875119894 119863119899isin 11987511989510038161003816100381610038161003816
(3)If 120588119894119895always takes only 0 or 1 ASNM(119863
119898 119863119899) becomes
ASSOC(119863119898 119863119899)
25 Association Probabilistic Method (APM) AlthoughASNM is a simple average of strengths of PPIs Chen et alproposed the association probabilistic method (APM) byreplacing the strength with an improved strength [17] It isbased on the idea that the contribution of one domain pair tothe strength of PPI should vary depending on the number ofdomain pairs included in a protein pair They assumed thatthe interaction probability of each domain pair is equivalentin a protein pair and transformed (1) as follows
Pr (119863119898119899
= 1) = 1 minus (1 minus Pr (119875119894119895= 1))1|119875119894||119875119895|
(4)
Thus by substituting the numerator of ASNM APM isdefined by
APM (119863119898 119863119899)
=
sum(119875119894119875119895)isinP|119863
119898isin119875119894119863119899isin119875119895(1 minus (1 minus 120588
119894119895)1|119875119894||119875119895|
)
10038161003816100381610038161003816(119875119894 119875119895) isin P | 119863
119898isin 119875119894 119863119899isin 11987511989510038161003816100381610038161003816
(5)
They conducted some computational experiments andreported that APM outperforms existing prediction methodssuch as ASNM and LPNM
26 Proposed Feature Space Mappings from Protein PairsThe association methods including ASNM and APM arebased on the probabilistic model of PPIs defined by (1)and infer strengths of PPIs from estimated DDIs usinggiven frequency of interactions or interaction strengths ofprotein pairs On the other hand we can also infer PPIstrengths utilizing features obtained from given informationsuch as sequence and structure of proteins with machinelearning methods Xia et al proposed a method to inferstrengths of PPIs using artificial neural network with featuresfrom constituent domains of proteins [20] In this paperfor predicting strengths of PPIs we propose several featurespace mappings from protein pairs making use of domaininformation
27 Feature Based onNumber of Domains (DN) As describedabove constituent domains information is useful for infer-ring PPIs and also can be used as a representation of eachprotein Actually Xia et al represented each protein bybinary numbers indicating whether a protein has a domainor not based on the information of constituent domains andused them with the artificial neural network to predict PPIstrengths [20] Here it can be considered that the probabilitythat two proteins interact increases with a larger number ofdomains included in the proteins Therefore in this paperwe propose a feature space mapping based on the numberof constituent domains (called DN) from two proteins Thefeature vector of DN for two proteins 119875
119894and 119875
119895is defined by
119891(119898)
119894119895= 119872(119863
119898 119875119894) for 119863
119898isin 119875119894
119891(119879+119899)
119894119895= 119872(119863
119899 119875119895) for 119863
119899isin 119875119895
119891(119897)
119894119895= 0 for 119863
119897notin 119875119894cup 119875119895
(6)
4 The Scientific World Journal
D1 D2 D3
MKANGLDNDOARTRMERTDIDSEHPEAQPLLNNNHRTLGAGSANGPAVEGRDIE
LDNDOARTRMERIDSEHPEAQPLLNNNHRGSANGPAVE
Figure 2 Illustration of restricting an amino acid sequence towhichthe spectrum kernel is applied to the domain regions
where 119879 indicates the total number of domains over allproteins and 119872(119863
119898 119875119894) indicates the number of domains
identified as119863119898in protein 119875
119894
28 Feature by Restriction of Spectrum Kernel to DomainRegion (SPD) DN is based only on the number of constituentdomains of each protein while amino acid sequences ofdomains are also considered useful for inferring strengthof PPI Therefore we propose a feature space mapping byrestricting the application of the spectrum kernel [24] todomain regions (called SPD) LetA be the set of 21 alphabetsrepresenting 20 types of amino acids and others Althoughwe used the set of 20 alphabets to express 20 types of aminoacids in the preliminary conference version [22] we add onealphabet to take the ambiguous amino acids such as X intoconsideration Then A119896 (119896 ge 1) means the set of all stringswith length 119896 generated from A The 119896-spectrum kernel forsequences 119909 and 119910 is defined by
119870119896(119909 119910) = ⟨Φ
119896(119909) Φ
119896(119910)⟩ (7)
whereΦ119896(119909) = (120601
119904(119909))119904isinA119896 and 120601119904(119909) indicates the number of
times that 119904 occurs in 119909 To make use of domain informationwe restrict an amino acid sequence to which the 119896-spectrumkernel is applied to the domain regions Figure 2 illustrates therestriction In this example the protein consists of domains1198631 1198632 1198633 and each domain region is surrounded by a
square Then the subsequence in each domain is extractedand all the subsequences in the protein are concatenated inthe same order as domains We apply the 119896-spectrum kernelto the concatenated sequence Let 120601(119903)
119904(119909) be the number of
times that string 119904 occurs in the sequence restricted to thedomain regions in protein 119909 in the abovemannerThe featurevector of SPD for proteins 119875
119894and 119875
119895is defined by
119891119897
119894119895= 120601(119903)
119904119897
(119875119894) for 119904
119897isin A119896
119891(21119896
+119897)
119894119895= 120601(119903)
119904119897
(119875119895) for 119904
119897isin A119896
(8)
It should be noted that 120601(119903)119904
for proteins having the samecomposition of domains can vary depending on the aminoacid sequences of their proteins That is even if 119875
119894and 119875
119895
have the same compositions as 119875119896and 119875
119897 respectively and
the feature vector of DN for 119875119894and 119875
119895is the same as that for
119875119896and 119875
119897 then the feature vector of SPD for 119875
119894and 119875
119895can be
different from that for 119875119896and 119875119897
29 Support Vector Regression (SVR) To predict strengths ofPPIs we employ support vector regression (SVR) [25] with
our proposed features In the case of linear functions SVRfinds parameters119908 and 119887 for 119891(119909) = ⟨119908 119909⟩ + 119887 by solving thefollowing optimization problem
minimize 1
21199082+ 119862sum
119894
(120585119894+ 1205851015840
119894)
subject to 119910119894minus ⟨119908 119909
119894⟩ minus 119887 le 120598 + 120585
119894
119910119894minus ⟨119908 119909
119894⟩ minus 119887 ge minus120598 minus 120585
1015840
119894
120585119894ge 0 120585
1015840
119894ge 0
(9)
where 119862 and 120598 are positive constants and (119909119894 119910119894) is a training
data Here the penalty is added only if the difference between119891(119909119894) and 119910
119894is larger than 120598 In our problem 119909
119894means a
protein pair and 119910119894means the corresponding interaction
strength
210 Relevance Vector Machine (RVM) In this paper wealso employ relevance vector machine (RVM) [26] to predictstrengths of PPIs RVM is a sparse Bayesian model utilizingthe same data-dependent kernel basis as the SVM Its frame-work is almost the same as typical Bayesian linear regressionGiven a training data 119909
119894 119910119894119873
119894=0 the conditional probability of
119910 given 119909 is modeled as
119901 (119910 | 119909 119908 120573) = N (119910 | 119908119879120601 (119909) 120573
minus1) (10)
where 120573 = 1205902 is noise parameter and 120601(sdot) is a typically
nonlinear projection of input features To obtain sparsesolutions in RVM framework a prior weight distribution ismodified so that a different variance parameter is assigned foreach weight as
119901 (119908 | 120572) = prod
119894=0
N (119908119894| 0 120572minus1
119894) (11)
where119872 = 119873+1 and 120572 = (1205721 120572
119872)119879 is a hyperparameter
RVM finds hyperparameter 120572 by maximizing the marginallikelihood 119901(119910 | 119909 120572) via ldquoevidence approximationrdquo Inthe process of maximizing evidence some 120572
119894approach
infinity and the corresponding 119908119894become zero Thus the
basis function corresponding with these parameters can beremoved and it leads sparse models In many cases RVMperforms better than SVM especially in regression problems
3 Results and Discussion
31 Computational Experiments To evaluate our proposedmethod we conducted computational experiments and com-pared with the existing method APM
32 Data and Implementation It is difficult to directlymeasure actual strengths of PPIs for many protein pairs bybiological and physical experiments Hence we used WI-PHI dataset with 50000 protein pairs [23] For each PPI WI-PHI contains a weight that is considered to represent somereliability of the PPI and is calculated from several different
The Scientific World Journal 5
Table 1 Results of average RMSE for training and test data
119862 = 1 119862 = 2 119862 = 5
Training Test Training Test Training TestSVR + DN 010472 012573 010656 012600 009982 012484RVM + DN 009210 012873 009178 012881 009474 012908SVR + SPD (119896 = 1) 008819 012699 008080 012954 007927 012903RVM + SPD (119896 = 1) 002848 012743 001504 012706 003276 012792SVR + SPD (119896 = 2) 008891 012654 008188 012782 008117 012909RVM + SPD (119896 = 2) 002529 012470 002301 012476 002243 012493SVR + APM 006846 013112 006795 013247 006791 013277RVM + APM 007052 013556 007037 013550 007032 013493APM Training = 006811 Test = 013517
kinds of PPI datasets in some statistical manner to rankphysical protein interactions As strengths of PPIs we usedthe value dividing the weight of PPI by the maximum weightforWI-PHIWe used dataset file ldquouniprot sprot fungidatgzrdquodownloaded from UniProt database [27] to get amino acidsequences information of domain compositions and domainregions in proteins In this experiment we used 1387 proteinpairs that could be extracted from WI-PHI dataset withcomplete domain sequence viaUniProt datasetThe extracteddataset contains 758 proteins and 327 domains Since thisdataset does not include protein pairs with interactionstrength 0 we randomly selected 100 protein pairs that do nothave any weights in the dataset and added them as proteinpairs with strength 0 Thus totally 1487 protein pairs wereused in this experiment We used ldquokernlabrdquo package [28]for executing support vector regression and relevance vectormachine and used the Laplacian kernel119870(119909 119910) = exp(minus120590119909minus119910) The dataset and the source code implemented by R areavailable upon request
To evaluate prediction accuracy we calculated the rootmean square error (RMSE) for each prediction RMSE isa measure of differences between predicted values 119910
119894and
actually observed values 119910119894and is defined by
RMSE = radic1
119873
119873
sum
119894=1
(119910119894minus 119910119894)2
(12)
where119873 is the number of test data
33 Results of Computational Experiments We preformedthreefold cross-validation calculated the average RMSE andcompared with APM [17] For APM method strengths ofPPIs are inferred based on APM scores for domain pairs thatconsist of target proteins However it is not always possible tocalculate APM scores for all domain pairs from training setTherefore as test set we used only protein pairs that consistof domain pairs with APM scores calculated via training set(In all cases about 40 of protein pairs in test set were used)For the Laplacian kernel employed in both SVR and RVMwe selected kernel parameter 120590 by fivefold cross-validationfrom candidate set 120590 isin 001 002 01 The parameter119862 for the regularization term in the Lagrange formulation isset to 119862 = 1 2 5 Additionally APM scores for each protein
pair also can be used as input featuresTherefore we also usedAPM scores as inputs for SVR and RVM and compared themodel using APM scores with the model using our proposedfeatures to confirm the usefulness of feature representationHere we used candidate set 120590 isin 30 31 32 90 forkernel parameter 120590 of RVM+APMmodel because themodelcould not be trained with 120590 values smaller than 3 On theother hand for 120590 of SVM + APM model we used the sameset as other models
Table 1 shows the results of the average RMSE by SVR andRVM with our proposed features (DN and SPD of 119896 = 1 2)and APM score and by APM for training and test datasetsFor training set the average RMSEs by RVM with SPD of119896 = 2were smaller than those by APM and others Moreoverfor test set all the average RMSEs by RVMwith SPD and DNwere smaller than those by APM The results suggested thatsupervised regression methods SVR and RVM with domainbased features are useful for prediction of PPI strengthsTaking all results together the model by RVM with SPD of119896 = 2was regarded as the best for prediction of PPI strengths
Since the average RMSEs of SVR with APM for bothtraining and test dataset were smaller than those of originalAPM SVR has potential to improve prediction accuraciesBy contrast the average RMSEs of RVM with APM becamelarger than those of original APM and all average RMSEsof the models with APM for test set were larger thanthose of the models with DN and SPD Accordingly theresults suggested that prediction accuracies were enhancedby feature representation and SPD is especially useful amongthese feature representations for predicting strengths of PPIsAlthough DN and SPD of 119896 = 1 have 654 and 42 dimensionsfor each protein pair respectively the average RMSEs withSPDof 119896 = 1 for training setwere smaller than thosewithDNIt implies that information of amino acid sequence in domainregions is more informative comparing with information ofdomain compositions to make a model fit in with dataset
In contrast the RMSEs by SVR with DN were smallerthan those by others in some cases of test set Table 2 showsthe numbers of relevance vectors and support vectors andthe 120590 values selected by fivefold cross-validation in all casesFor the models with DN and APM scores the numbers ofrelevance vectors were smaller than the numbers of supportvectors On the other hand the numbers of relevance vectorswere larger than the numbers of support vectors for the
6 The Scientific World Journal
Table 2The number of relevance vectors (RVs) and support vectors (SVs) for each model with DN SPD and APM and the selected 120590 valuesfor each fold
119862 = 1 119862 = 2 119862 = 5
SVR RVM SVR RVM SVR RVMSVs (120590 value) RVs (120590 value) SVs (120590 value) RVs (120590 value) SVs (120590 value) RVs (120590 value)
Fold 1
DN 271 (002) 113 (005) 271 (001) 123 (007) 308 (001) 74 (002)SPD (119896 = 1) 367 (001) 448 (002) 402 (002) 680 (005) 402 (002) 537 (003)SPD (119896 = 2) 392 (001) 502 (003) 409 (001) 628 (005) 421 (001) 628 (005)
APM 362 (008) 4 (500) 361 (010) 6 (480) 357 (004) 6 (580)
Fold 2
DN 280 (002) 94 (008) 281 (001) 92 (009) 314 (001) 82 (004)SPD (119896 = 1) 408 (001) 617 (004) 453 (004) 706 (006) 411 (001) 545 (003)SPD (119896 = 2) 430 (001) 558 (004) 435 (001) 618 (005) 495 (004) 654 (006)
APM 375 (010) 5 (650) 372 (010) 6 (690) 373 (004) 4 (550)
Fold 3
DN 321 (004) 107 (008) 289 (001) 107 (010) 330 (001) 107 (008)SPD (119896 = 1) 371 (001) 439 (002) 412 (003) 658 (005) 382 (001) 305 (001)SPD (119896 = 2) 387 (001) 625 (006) 418 (002) 529 (004) 398 (001) 529 (004)
APM 368 (008) 3 (710) 368 (004) 3 (670) 372 (001) 5 (420)
models with SPD feature in spite of the fact that usuallyRVM provides a sparse model compared with SVR In RVMframework sparsity of model is caused by distributions ofeach weight that is the number of relevance vectors isinfluenced by values and variances of each dimension offeatures rather than by the number of dimensions of featuresActually each dimension of SPD feature almost always haswidely varying values In contrast DN feature has manyzeros and APM score is inferred from training dataset andthereby has similar distribution Thus it is considered thatmany weights corresponding to features in RVM model didnot become zero and the RVM models with SPD featuretended to be complex and to overfit the training data
4 ConclusionsFor the prediction of strengths of PPIs we proposed featurespace mappings DN and SPD DN is based on the numberof domains in a protein SPD is based on the spectrumkernel and defined using the amino acid subsequences indomain regions In this work we employed support vec-tor regression (SVR) and relevance vector machine (RVM)with the Laplacian kernel and conducted threefold cross-validation using WI-PHI dataset For both training and testdataset the average RMSEs by RVM with SPD feature weresmaller than those by APMThe results showed that machinelearning methods with domain information outperformedexisting association method that is based on the probabilisticmodel of PPIs and implied that the information of aminoacid sequence is useful for prediction comparing with onlyinformation of domain compositions However the modelswith SPD feature tended to be complex and overfitted to thetraining data Therefore to further enhance the predictionaccuracy improving kernel functions combining physicalcharacteristics of domains and amino acids might be helpful
Conflict of InterestsThe authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgment
This work was partially supported by Grants-in-Aid nos22240009 and 24500361 fromMEXT Japan
References
[1] J R Bock and D A Gough ldquoPredicting protein-proteininteractions from primary structurerdquo Bioinformatics vol 17 no5 pp 455ndash460 2001
[2] Y Guo L Yu Z Wen and M Li ldquoUsing support vectormachine combined with auto covariance to predict protein-protein interactions from protein sequencesrdquo Nucleic AcidsResearch vol 36 no 9 pp 3025ndash3030 2008
[3] C Yu L Chou and D T Chang ldquoPredicting protein-proteininteractions in unbalanced data using the primary structure ofproteinsrdquo BMC Bioinformatics vol 11 article 167 2010
[4] J Xia X Zhao and D Huang ldquoPredicting protein-proteininteractions from protein sequences using meta predictorrdquoAmino Acids vol 39 no 5 pp 1595ndash1599 2010
[5] J Xia K Han and D Huang ldquoSequence-based prediction ofprotein-protein interactions by means of rotation forest andautocorrelation descriptorrdquo Protein and Peptide Letters vol 17no 1 pp 137ndash145 2010
[6] S J Darnell D Page and J CMitchell ldquoAn automated decision-tree approach to predicting protein interaction hot spotsrdquoProteins Structure Function and Bioinformatics vol 68 no 4pp 813ndash823 2007
[7] K Cho D Kim and D Lee ldquoA feature-based approach tomodeling protein-protein interaction hot spotsrdquo Nucleic AcidsResearch vol 37 no 8 pp 2672ndash2687 2009
[8] J Xia X Zhao J Song and D Huang ldquoAPIS accurate predic-tion of hot spots in protein interfaces by combining protrusionindexwith solvent accessibilityrdquoBMCBioinformatics vol 11 no1 article 174 2010
[9] O KuchaievM Rasajski D J Higham andN Przulj ldquoGeomet-ric de-noising of protein-protein interaction networksrdquo PLoSComputational Biology vol 5 no 8 Article ID e1000454 2009
[10] L Zhu Z You and D Huang ldquoIncreasing the reliability ofprotein-protein interaction networks via non-convex semanticembeddingrdquo Neurocomputing vol 121 pp 99ndash107 2013
The Scientific World Journal 7
[11] S Byrum S Smart S Larson and A Tackett ldquoAnalysis ofstable and transient protein-protein interactionsrdquo Methods inMolecular Biology vol 833 pp 143ndash152 2012
[12] P Uetz L Glot G Cagney et al ldquoA comprehensive analysisof protein-protein interactions in Saccharomyces cerevisiaerdquoNature vol 403 no 6770 pp 623ndash627 2000
[13] T Ito T Chiba R Ozawa M Yoshida M Hattori and YSakaki ldquoA comprehensive two-hybrid analysis to explore theyeast protein interactomerdquo Proceedings of the National Academyof Sciences of the United States of America vol 98 no 8 pp4569ndash4574 2001
[14] M Hayashida N Ueda and T Akutsu ldquoInferring strengthsof protein-protein interactions from experimental data usinglinear programmingrdquo Bioinformatics vol 19 no 2 pp ii58ndashii652003
[15] M Hayashida N Ueda and T Akutsu ldquoA simple methodfor inferring strengths of protein-protein interactionsrdquoGenomeInformatics vol 15 no 1 pp 56ndash68 2004
[16] E Sprinzak and H Margalit ldquoCorrelated sequence-signaturesas markers of protein-protein interactionrdquo Journal of MolecularBiology vol 311 no 4 pp 681ndash692 2001
[17] L Chen L Wu Y Wang and X Zhang ldquoInferring-proteininteractions from experimental data by association probabilisticmethodrdquo Proteins Structure Function and Genetics vol 62 no4 pp 833ndash837 2006
[18] R Finn J Mistry J Tate et al ldquoThe Pfam protein familiesdatabaserdquo Nucleic Acids Research vol 38 supplement 1 ppD211ndashD222 2009
[19] S Hunter P Jones A Mitchell et al ldquoInterPro in 2011 newdevelopments in the family and domain prediction databaserdquoNucleic Acids Research vol 40 no 1 pp D306ndashD312 2012
[20] J Xia B Wang and D Huang ldquoInferring strengths of protein-protein interaction using artificial neural networkrdquo in Proceed-ings of the International Joint Conference on Neural Networks(IJCNN rsquo07) pp 2471ndash2475 Orlando Fla USA August 2007
[21] M Deng S Mehta F Sun and T Chen ldquoInferring domain-domain interactions from protein-protein interactionsrdquoGenome Research vol 12 no 10 pp 1540ndash1548 2002
[22] Y Sakuma M Kamada M Hayashida and T Akutsu ldquoInfer-ring strengths of protein-protein interactions using supportvector regressionrdquo inProceedings of the International Conferenceon Parallel and Distributed Processing Techniques and Applica-tions 2013 httpworld-comporgp2013PDP2162pdf
[23] L Kiemer S Costa M Ueffing and G Cesareni ldquoWI-PHIa weighted yeast interactome enriched for direct physicalinteractionsrdquo Proteomics vol 7 no 6 pp 932ndash943 2007
[24] C Leslie E Eskin and W Noble ldquoThe spectrum Kernel astring Kernel for SVM protein classi fi cationrdquo in Proceedingsof Pacific Symposium on Biocomputing (PSB rsquo02) pp 564ndash575Lihue Hawaii USA January 2002
[25] VN VapnikTheNature of Statistical LearningTheory Springer1995
[26] M E Tipping ldquoSparse bayesian learning and the relevancevector machinerdquo Journal of Machine Learning Research vol 1no 3 pp 211ndash244 2001
[27] T U Consortium ldquoReorganizing the protein space at theuniversal protein resource (UniProt)rdquo Nucleic Acids Researchvol 40 no D1 pp D71ndashD75 2012
[28] A Karatzoglou K Hornik A Smola and A Zeileis ldquokernlabmdashan S4 package for kernel methods in Rrdquo Journal of StatisticalSoftware vol 11 no 9 pp 1ndash20 2004
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

The Scientific World Journal 3
P1P2
D1
D2
D2
D3
D4
(a)
Interacting proteins with their domains
Predicting PPIs based on inferred DDIs
Inferring model for DDIs
middot middot middot
middot middot middot
Protein
Protein Protein
Protein
(b)
Figure 1 (a) Illustration of protein-protein interactions (PPIs) model based on domain-domain interactions (DDIs) (b) Schematic overviewof PPIs prediction based on DDIs
association method [15] This method takes strengths ofPPIs as input data Let 120588
119894119895represent the interaction strength
between 119875119894and 119875
119895 and we suppose that 120588
119894119895is defined for all
(119875119894 119875119895) isin P Then the ASNM score for domains 119863
119898and 119863
119899
is defined as the average strength over protein pairs including119863119898and119863
119899by
ASNM (119863119898 119863119899) =
sum(119875119894119875119895)isinP|119863
119898isin119875119894119863119899isin119875119895120588119894119895
10038161003816100381610038161003816(119875119894 119875119895) isin P | 119863
119898isin 119875119894 119863119899isin 11987511989510038161003816100381610038161003816
(3)If 120588119894119895always takes only 0 or 1 ASNM(119863
119898 119863119899) becomes
ASSOC(119863119898 119863119899)
25 Association Probabilistic Method (APM) AlthoughASNM is a simple average of strengths of PPIs Chen et alproposed the association probabilistic method (APM) byreplacing the strength with an improved strength [17] It isbased on the idea that the contribution of one domain pair tothe strength of PPI should vary depending on the number ofdomain pairs included in a protein pair They assumed thatthe interaction probability of each domain pair is equivalentin a protein pair and transformed (1) as follows
Pr (119863119898119899
= 1) = 1 minus (1 minus Pr (119875119894119895= 1))1|119875119894||119875119895|
(4)
Thus by substituting the numerator of ASNM APM isdefined by
APM (119863119898 119863119899)
=
sum(119875119894119875119895)isinP|119863
119898isin119875119894119863119899isin119875119895(1 minus (1 minus 120588
119894119895)1|119875119894||119875119895|
)
10038161003816100381610038161003816(119875119894 119875119895) isin P | 119863
119898isin 119875119894 119863119899isin 11987511989510038161003816100381610038161003816
(5)
They conducted some computational experiments andreported that APM outperforms existing prediction methodssuch as ASNM and LPNM
26 Proposed Feature Space Mappings from Protein PairsThe association methods including ASNM and APM arebased on the probabilistic model of PPIs defined by (1)and infer strengths of PPIs from estimated DDIs usinggiven frequency of interactions or interaction strengths ofprotein pairs On the other hand we can also infer PPIstrengths utilizing features obtained from given informationsuch as sequence and structure of proteins with machinelearning methods Xia et al proposed a method to inferstrengths of PPIs using artificial neural network with featuresfrom constituent domains of proteins [20] In this paperfor predicting strengths of PPIs we propose several featurespace mappings from protein pairs making use of domaininformation
27 Feature Based onNumber of Domains (DN) As describedabove constituent domains information is useful for infer-ring PPIs and also can be used as a representation of eachprotein Actually Xia et al represented each protein bybinary numbers indicating whether a protein has a domainor not based on the information of constituent domains andused them with the artificial neural network to predict PPIstrengths [20] Here it can be considered that the probabilitythat two proteins interact increases with a larger number ofdomains included in the proteins Therefore in this paperwe propose a feature space mapping based on the numberof constituent domains (called DN) from two proteins Thefeature vector of DN for two proteins 119875
119894and 119875
119895is defined by
119891(119898)
119894119895= 119872(119863
119898 119875119894) for 119863
119898isin 119875119894
119891(119879+119899)
119894119895= 119872(119863
119899 119875119895) for 119863
119899isin 119875119895
119891(119897)
119894119895= 0 for 119863
119897notin 119875119894cup 119875119895
(6)
4 The Scientific World Journal
D1 D2 D3
MKANGLDNDOARTRMERTDIDSEHPEAQPLLNNNHRTLGAGSANGPAVEGRDIE
LDNDOARTRMERIDSEHPEAQPLLNNNHRGSANGPAVE
Figure 2 Illustration of restricting an amino acid sequence towhichthe spectrum kernel is applied to the domain regions
where 119879 indicates the total number of domains over allproteins and 119872(119863
119898 119875119894) indicates the number of domains
identified as119863119898in protein 119875
119894
28 Feature by Restriction of Spectrum Kernel to DomainRegion (SPD) DN is based only on the number of constituentdomains of each protein while amino acid sequences ofdomains are also considered useful for inferring strengthof PPI Therefore we propose a feature space mapping byrestricting the application of the spectrum kernel [24] todomain regions (called SPD) LetA be the set of 21 alphabetsrepresenting 20 types of amino acids and others Althoughwe used the set of 20 alphabets to express 20 types of aminoacids in the preliminary conference version [22] we add onealphabet to take the ambiguous amino acids such as X intoconsideration Then A119896 (119896 ge 1) means the set of all stringswith length 119896 generated from A The 119896-spectrum kernel forsequences 119909 and 119910 is defined by
119870119896(119909 119910) = ⟨Φ
119896(119909) Φ
119896(119910)⟩ (7)
whereΦ119896(119909) = (120601
119904(119909))119904isinA119896 and 120601119904(119909) indicates the number of
times that 119904 occurs in 119909 To make use of domain informationwe restrict an amino acid sequence to which the 119896-spectrumkernel is applied to the domain regions Figure 2 illustrates therestriction In this example the protein consists of domains1198631 1198632 1198633 and each domain region is surrounded by a
square Then the subsequence in each domain is extractedand all the subsequences in the protein are concatenated inthe same order as domains We apply the 119896-spectrum kernelto the concatenated sequence Let 120601(119903)
119904(119909) be the number of
times that string 119904 occurs in the sequence restricted to thedomain regions in protein 119909 in the abovemannerThe featurevector of SPD for proteins 119875
119894and 119875
119895is defined by
119891119897
119894119895= 120601(119903)
119904119897
(119875119894) for 119904
119897isin A119896
119891(21119896
+119897)
119894119895= 120601(119903)
119904119897
(119875119895) for 119904
119897isin A119896
(8)
It should be noted that 120601(119903)119904
for proteins having the samecomposition of domains can vary depending on the aminoacid sequences of their proteins That is even if 119875
119894and 119875
119895
have the same compositions as 119875119896and 119875
119897 respectively and
the feature vector of DN for 119875119894and 119875
119895is the same as that for
119875119896and 119875
119897 then the feature vector of SPD for 119875
119894and 119875
119895can be
different from that for 119875119896and 119875119897
29 Support Vector Regression (SVR) To predict strengths ofPPIs we employ support vector regression (SVR) [25] with
our proposed features In the case of linear functions SVRfinds parameters119908 and 119887 for 119891(119909) = ⟨119908 119909⟩ + 119887 by solving thefollowing optimization problem
minimize 1
21199082+ 119862sum
119894
(120585119894+ 1205851015840
119894)
subject to 119910119894minus ⟨119908 119909
119894⟩ minus 119887 le 120598 + 120585
119894
119910119894minus ⟨119908 119909
119894⟩ minus 119887 ge minus120598 minus 120585
1015840
119894
120585119894ge 0 120585
1015840
119894ge 0
(9)
where 119862 and 120598 are positive constants and (119909119894 119910119894) is a training
data Here the penalty is added only if the difference between119891(119909119894) and 119910
119894is larger than 120598 In our problem 119909
119894means a
protein pair and 119910119894means the corresponding interaction
strength
210 Relevance Vector Machine (RVM) In this paper wealso employ relevance vector machine (RVM) [26] to predictstrengths of PPIs RVM is a sparse Bayesian model utilizingthe same data-dependent kernel basis as the SVM Its frame-work is almost the same as typical Bayesian linear regressionGiven a training data 119909
119894 119910119894119873
119894=0 the conditional probability of
119910 given 119909 is modeled as
119901 (119910 | 119909 119908 120573) = N (119910 | 119908119879120601 (119909) 120573
minus1) (10)
where 120573 = 1205902 is noise parameter and 120601(sdot) is a typically
nonlinear projection of input features To obtain sparsesolutions in RVM framework a prior weight distribution ismodified so that a different variance parameter is assigned foreach weight as
119901 (119908 | 120572) = prod
119894=0
N (119908119894| 0 120572minus1
119894) (11)
where119872 = 119873+1 and 120572 = (1205721 120572
119872)119879 is a hyperparameter
RVM finds hyperparameter 120572 by maximizing the marginallikelihood 119901(119910 | 119909 120572) via ldquoevidence approximationrdquo Inthe process of maximizing evidence some 120572
119894approach
infinity and the corresponding 119908119894become zero Thus the
basis function corresponding with these parameters can beremoved and it leads sparse models In many cases RVMperforms better than SVM especially in regression problems
3 Results and Discussion
31 Computational Experiments To evaluate our proposedmethod we conducted computational experiments and com-pared with the existing method APM
32 Data and Implementation It is difficult to directlymeasure actual strengths of PPIs for many protein pairs bybiological and physical experiments Hence we used WI-PHI dataset with 50000 protein pairs [23] For each PPI WI-PHI contains a weight that is considered to represent somereliability of the PPI and is calculated from several different
The Scientific World Journal 5
Table 1 Results of average RMSE for training and test data
119862 = 1 119862 = 2 119862 = 5
Training Test Training Test Training TestSVR + DN 010472 012573 010656 012600 009982 012484RVM + DN 009210 012873 009178 012881 009474 012908SVR + SPD (119896 = 1) 008819 012699 008080 012954 007927 012903RVM + SPD (119896 = 1) 002848 012743 001504 012706 003276 012792SVR + SPD (119896 = 2) 008891 012654 008188 012782 008117 012909RVM + SPD (119896 = 2) 002529 012470 002301 012476 002243 012493SVR + APM 006846 013112 006795 013247 006791 013277RVM + APM 007052 013556 007037 013550 007032 013493APM Training = 006811 Test = 013517
kinds of PPI datasets in some statistical manner to rankphysical protein interactions As strengths of PPIs we usedthe value dividing the weight of PPI by the maximum weightforWI-PHIWe used dataset file ldquouniprot sprot fungidatgzrdquodownloaded from UniProt database [27] to get amino acidsequences information of domain compositions and domainregions in proteins In this experiment we used 1387 proteinpairs that could be extracted from WI-PHI dataset withcomplete domain sequence viaUniProt datasetThe extracteddataset contains 758 proteins and 327 domains Since thisdataset does not include protein pairs with interactionstrength 0 we randomly selected 100 protein pairs that do nothave any weights in the dataset and added them as proteinpairs with strength 0 Thus totally 1487 protein pairs wereused in this experiment We used ldquokernlabrdquo package [28]for executing support vector regression and relevance vectormachine and used the Laplacian kernel119870(119909 119910) = exp(minus120590119909minus119910) The dataset and the source code implemented by R areavailable upon request
To evaluate prediction accuracy we calculated the rootmean square error (RMSE) for each prediction RMSE isa measure of differences between predicted values 119910
119894and
actually observed values 119910119894and is defined by
RMSE = radic1
119873
119873
sum
119894=1
(119910119894minus 119910119894)2
(12)
where119873 is the number of test data
33 Results of Computational Experiments We preformedthreefold cross-validation calculated the average RMSE andcompared with APM [17] For APM method strengths ofPPIs are inferred based on APM scores for domain pairs thatconsist of target proteins However it is not always possible tocalculate APM scores for all domain pairs from training setTherefore as test set we used only protein pairs that consistof domain pairs with APM scores calculated via training set(In all cases about 40 of protein pairs in test set were used)For the Laplacian kernel employed in both SVR and RVMwe selected kernel parameter 120590 by fivefold cross-validationfrom candidate set 120590 isin 001 002 01 The parameter119862 for the regularization term in the Lagrange formulation isset to 119862 = 1 2 5 Additionally APM scores for each protein
pair also can be used as input featuresTherefore we also usedAPM scores as inputs for SVR and RVM and compared themodel using APM scores with the model using our proposedfeatures to confirm the usefulness of feature representationHere we used candidate set 120590 isin 30 31 32 90 forkernel parameter 120590 of RVM+APMmodel because themodelcould not be trained with 120590 values smaller than 3 On theother hand for 120590 of SVM + APM model we used the sameset as other models
Table 1 shows the results of the average RMSE by SVR andRVM with our proposed features (DN and SPD of 119896 = 1 2)and APM score and by APM for training and test datasetsFor training set the average RMSEs by RVM with SPD of119896 = 2were smaller than those by APM and others Moreoverfor test set all the average RMSEs by RVMwith SPD and DNwere smaller than those by APM The results suggested thatsupervised regression methods SVR and RVM with domainbased features are useful for prediction of PPI strengthsTaking all results together the model by RVM with SPD of119896 = 2was regarded as the best for prediction of PPI strengths
Since the average RMSEs of SVR with APM for bothtraining and test dataset were smaller than those of originalAPM SVR has potential to improve prediction accuraciesBy contrast the average RMSEs of RVM with APM becamelarger than those of original APM and all average RMSEsof the models with APM for test set were larger thanthose of the models with DN and SPD Accordingly theresults suggested that prediction accuracies were enhancedby feature representation and SPD is especially useful amongthese feature representations for predicting strengths of PPIsAlthough DN and SPD of 119896 = 1 have 654 and 42 dimensionsfor each protein pair respectively the average RMSEs withSPDof 119896 = 1 for training setwere smaller than thosewithDNIt implies that information of amino acid sequence in domainregions is more informative comparing with information ofdomain compositions to make a model fit in with dataset
In contrast the RMSEs by SVR with DN were smallerthan those by others in some cases of test set Table 2 showsthe numbers of relevance vectors and support vectors andthe 120590 values selected by fivefold cross-validation in all casesFor the models with DN and APM scores the numbers ofrelevance vectors were smaller than the numbers of supportvectors On the other hand the numbers of relevance vectorswere larger than the numbers of support vectors for the
6 The Scientific World Journal
Table 2The number of relevance vectors (RVs) and support vectors (SVs) for each model with DN SPD and APM and the selected 120590 valuesfor each fold
119862 = 1 119862 = 2 119862 = 5
SVR RVM SVR RVM SVR RVMSVs (120590 value) RVs (120590 value) SVs (120590 value) RVs (120590 value) SVs (120590 value) RVs (120590 value)
Fold 1
DN 271 (002) 113 (005) 271 (001) 123 (007) 308 (001) 74 (002)SPD (119896 = 1) 367 (001) 448 (002) 402 (002) 680 (005) 402 (002) 537 (003)SPD (119896 = 2) 392 (001) 502 (003) 409 (001) 628 (005) 421 (001) 628 (005)
APM 362 (008) 4 (500) 361 (010) 6 (480) 357 (004) 6 (580)
Fold 2
DN 280 (002) 94 (008) 281 (001) 92 (009) 314 (001) 82 (004)SPD (119896 = 1) 408 (001) 617 (004) 453 (004) 706 (006) 411 (001) 545 (003)SPD (119896 = 2) 430 (001) 558 (004) 435 (001) 618 (005) 495 (004) 654 (006)
APM 375 (010) 5 (650) 372 (010) 6 (690) 373 (004) 4 (550)
Fold 3
DN 321 (004) 107 (008) 289 (001) 107 (010) 330 (001) 107 (008)SPD (119896 = 1) 371 (001) 439 (002) 412 (003) 658 (005) 382 (001) 305 (001)SPD (119896 = 2) 387 (001) 625 (006) 418 (002) 529 (004) 398 (001) 529 (004)
APM 368 (008) 3 (710) 368 (004) 3 (670) 372 (001) 5 (420)
models with SPD feature in spite of the fact that usuallyRVM provides a sparse model compared with SVR In RVMframework sparsity of model is caused by distributions ofeach weight that is the number of relevance vectors isinfluenced by values and variances of each dimension offeatures rather than by the number of dimensions of featuresActually each dimension of SPD feature almost always haswidely varying values In contrast DN feature has manyzeros and APM score is inferred from training dataset andthereby has similar distribution Thus it is considered thatmany weights corresponding to features in RVM model didnot become zero and the RVM models with SPD featuretended to be complex and to overfit the training data
4 ConclusionsFor the prediction of strengths of PPIs we proposed featurespace mappings DN and SPD DN is based on the numberof domains in a protein SPD is based on the spectrumkernel and defined using the amino acid subsequences indomain regions In this work we employed support vec-tor regression (SVR) and relevance vector machine (RVM)with the Laplacian kernel and conducted threefold cross-validation using WI-PHI dataset For both training and testdataset the average RMSEs by RVM with SPD feature weresmaller than those by APMThe results showed that machinelearning methods with domain information outperformedexisting association method that is based on the probabilisticmodel of PPIs and implied that the information of aminoacid sequence is useful for prediction comparing with onlyinformation of domain compositions However the modelswith SPD feature tended to be complex and overfitted to thetraining data Therefore to further enhance the predictionaccuracy improving kernel functions combining physicalcharacteristics of domains and amino acids might be helpful
Conflict of InterestsThe authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgment
This work was partially supported by Grants-in-Aid nos22240009 and 24500361 fromMEXT Japan
References
[1] J R Bock and D A Gough ldquoPredicting protein-proteininteractions from primary structurerdquo Bioinformatics vol 17 no5 pp 455ndash460 2001
[2] Y Guo L Yu Z Wen and M Li ldquoUsing support vectormachine combined with auto covariance to predict protein-protein interactions from protein sequencesrdquo Nucleic AcidsResearch vol 36 no 9 pp 3025ndash3030 2008
[3] C Yu L Chou and D T Chang ldquoPredicting protein-proteininteractions in unbalanced data using the primary structure ofproteinsrdquo BMC Bioinformatics vol 11 article 167 2010
[4] J Xia X Zhao and D Huang ldquoPredicting protein-proteininteractions from protein sequences using meta predictorrdquoAmino Acids vol 39 no 5 pp 1595ndash1599 2010
[5] J Xia K Han and D Huang ldquoSequence-based prediction ofprotein-protein interactions by means of rotation forest andautocorrelation descriptorrdquo Protein and Peptide Letters vol 17no 1 pp 137ndash145 2010
[6] S J Darnell D Page and J CMitchell ldquoAn automated decision-tree approach to predicting protein interaction hot spotsrdquoProteins Structure Function and Bioinformatics vol 68 no 4pp 813ndash823 2007
[7] K Cho D Kim and D Lee ldquoA feature-based approach tomodeling protein-protein interaction hot spotsrdquo Nucleic AcidsResearch vol 37 no 8 pp 2672ndash2687 2009
[8] J Xia X Zhao J Song and D Huang ldquoAPIS accurate predic-tion of hot spots in protein interfaces by combining protrusionindexwith solvent accessibilityrdquoBMCBioinformatics vol 11 no1 article 174 2010
[9] O KuchaievM Rasajski D J Higham andN Przulj ldquoGeomet-ric de-noising of protein-protein interaction networksrdquo PLoSComputational Biology vol 5 no 8 Article ID e1000454 2009
[10] L Zhu Z You and D Huang ldquoIncreasing the reliability ofprotein-protein interaction networks via non-convex semanticembeddingrdquo Neurocomputing vol 121 pp 99ndash107 2013
The Scientific World Journal 7
[11] S Byrum S Smart S Larson and A Tackett ldquoAnalysis ofstable and transient protein-protein interactionsrdquo Methods inMolecular Biology vol 833 pp 143ndash152 2012
[12] P Uetz L Glot G Cagney et al ldquoA comprehensive analysisof protein-protein interactions in Saccharomyces cerevisiaerdquoNature vol 403 no 6770 pp 623ndash627 2000
[13] T Ito T Chiba R Ozawa M Yoshida M Hattori and YSakaki ldquoA comprehensive two-hybrid analysis to explore theyeast protein interactomerdquo Proceedings of the National Academyof Sciences of the United States of America vol 98 no 8 pp4569ndash4574 2001
[14] M Hayashida N Ueda and T Akutsu ldquoInferring strengthsof protein-protein interactions from experimental data usinglinear programmingrdquo Bioinformatics vol 19 no 2 pp ii58ndashii652003
[15] M Hayashida N Ueda and T Akutsu ldquoA simple methodfor inferring strengths of protein-protein interactionsrdquoGenomeInformatics vol 15 no 1 pp 56ndash68 2004
[16] E Sprinzak and H Margalit ldquoCorrelated sequence-signaturesas markers of protein-protein interactionrdquo Journal of MolecularBiology vol 311 no 4 pp 681ndash692 2001
[17] L Chen L Wu Y Wang and X Zhang ldquoInferring-proteininteractions from experimental data by association probabilisticmethodrdquo Proteins Structure Function and Genetics vol 62 no4 pp 833ndash837 2006
[18] R Finn J Mistry J Tate et al ldquoThe Pfam protein familiesdatabaserdquo Nucleic Acids Research vol 38 supplement 1 ppD211ndashD222 2009
[19] S Hunter P Jones A Mitchell et al ldquoInterPro in 2011 newdevelopments in the family and domain prediction databaserdquoNucleic Acids Research vol 40 no 1 pp D306ndashD312 2012
[20] J Xia B Wang and D Huang ldquoInferring strengths of protein-protein interaction using artificial neural networkrdquo in Proceed-ings of the International Joint Conference on Neural Networks(IJCNN rsquo07) pp 2471ndash2475 Orlando Fla USA August 2007
[21] M Deng S Mehta F Sun and T Chen ldquoInferring domain-domain interactions from protein-protein interactionsrdquoGenome Research vol 12 no 10 pp 1540ndash1548 2002
[22] Y Sakuma M Kamada M Hayashida and T Akutsu ldquoInfer-ring strengths of protein-protein interactions using supportvector regressionrdquo inProceedings of the International Conferenceon Parallel and Distributed Processing Techniques and Applica-tions 2013 httpworld-comporgp2013PDP2162pdf
[23] L Kiemer S Costa M Ueffing and G Cesareni ldquoWI-PHIa weighted yeast interactome enriched for direct physicalinteractionsrdquo Proteomics vol 7 no 6 pp 932ndash943 2007
[24] C Leslie E Eskin and W Noble ldquoThe spectrum Kernel astring Kernel for SVM protein classi fi cationrdquo in Proceedingsof Pacific Symposium on Biocomputing (PSB rsquo02) pp 564ndash575Lihue Hawaii USA January 2002
[25] VN VapnikTheNature of Statistical LearningTheory Springer1995
[26] M E Tipping ldquoSparse bayesian learning and the relevancevector machinerdquo Journal of Machine Learning Research vol 1no 3 pp 211ndash244 2001
[27] T U Consortium ldquoReorganizing the protein space at theuniversal protein resource (UniProt)rdquo Nucleic Acids Researchvol 40 no D1 pp D71ndashD75 2012
[28] A Karatzoglou K Hornik A Smola and A Zeileis ldquokernlabmdashan S4 package for kernel methods in Rrdquo Journal of StatisticalSoftware vol 11 no 9 pp 1ndash20 2004
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

4 The Scientific World Journal
D1 D2 D3
MKANGLDNDOARTRMERTDIDSEHPEAQPLLNNNHRTLGAGSANGPAVEGRDIE
LDNDOARTRMERIDSEHPEAQPLLNNNHRGSANGPAVE
Figure 2 Illustration of restricting an amino acid sequence towhichthe spectrum kernel is applied to the domain regions
where 119879 indicates the total number of domains over allproteins and 119872(119863
119898 119875119894) indicates the number of domains
identified as119863119898in protein 119875
119894
28 Feature by Restriction of Spectrum Kernel to DomainRegion (SPD) DN is based only on the number of constituentdomains of each protein while amino acid sequences ofdomains are also considered useful for inferring strengthof PPI Therefore we propose a feature space mapping byrestricting the application of the spectrum kernel [24] todomain regions (called SPD) LetA be the set of 21 alphabetsrepresenting 20 types of amino acids and others Althoughwe used the set of 20 alphabets to express 20 types of aminoacids in the preliminary conference version [22] we add onealphabet to take the ambiguous amino acids such as X intoconsideration Then A119896 (119896 ge 1) means the set of all stringswith length 119896 generated from A The 119896-spectrum kernel forsequences 119909 and 119910 is defined by
119870119896(119909 119910) = ⟨Φ
119896(119909) Φ
119896(119910)⟩ (7)
whereΦ119896(119909) = (120601
119904(119909))119904isinA119896 and 120601119904(119909) indicates the number of
times that 119904 occurs in 119909 To make use of domain informationwe restrict an amino acid sequence to which the 119896-spectrumkernel is applied to the domain regions Figure 2 illustrates therestriction In this example the protein consists of domains1198631 1198632 1198633 and each domain region is surrounded by a
square Then the subsequence in each domain is extractedand all the subsequences in the protein are concatenated inthe same order as domains We apply the 119896-spectrum kernelto the concatenated sequence Let 120601(119903)
119904(119909) be the number of
times that string 119904 occurs in the sequence restricted to thedomain regions in protein 119909 in the abovemannerThe featurevector of SPD for proteins 119875
119894and 119875
119895is defined by
119891119897
119894119895= 120601(119903)
119904119897
(119875119894) for 119904
119897isin A119896
119891(21119896
+119897)
119894119895= 120601(119903)
119904119897
(119875119895) for 119904
119897isin A119896
(8)
It should be noted that 120601(119903)119904
for proteins having the samecomposition of domains can vary depending on the aminoacid sequences of their proteins That is even if 119875
119894and 119875
119895
have the same compositions as 119875119896and 119875
119897 respectively and
the feature vector of DN for 119875119894and 119875
119895is the same as that for
119875119896and 119875
119897 then the feature vector of SPD for 119875
119894and 119875
119895can be
different from that for 119875119896and 119875119897
29 Support Vector Regression (SVR) To predict strengths ofPPIs we employ support vector regression (SVR) [25] with
our proposed features In the case of linear functions SVRfinds parameters119908 and 119887 for 119891(119909) = ⟨119908 119909⟩ + 119887 by solving thefollowing optimization problem
minimize 1
21199082+ 119862sum
119894
(120585119894+ 1205851015840
119894)
subject to 119910119894minus ⟨119908 119909
119894⟩ minus 119887 le 120598 + 120585
119894
119910119894minus ⟨119908 119909
119894⟩ minus 119887 ge minus120598 minus 120585
1015840
119894
120585119894ge 0 120585
1015840
119894ge 0
(9)
where 119862 and 120598 are positive constants and (119909119894 119910119894) is a training
data Here the penalty is added only if the difference between119891(119909119894) and 119910
119894is larger than 120598 In our problem 119909
119894means a
protein pair and 119910119894means the corresponding interaction
strength
210 Relevance Vector Machine (RVM) In this paper wealso employ relevance vector machine (RVM) [26] to predictstrengths of PPIs RVM is a sparse Bayesian model utilizingthe same data-dependent kernel basis as the SVM Its frame-work is almost the same as typical Bayesian linear regressionGiven a training data 119909
119894 119910119894119873
119894=0 the conditional probability of
119910 given 119909 is modeled as
119901 (119910 | 119909 119908 120573) = N (119910 | 119908119879120601 (119909) 120573
minus1) (10)
where 120573 = 1205902 is noise parameter and 120601(sdot) is a typically
nonlinear projection of input features To obtain sparsesolutions in RVM framework a prior weight distribution ismodified so that a different variance parameter is assigned foreach weight as
119901 (119908 | 120572) = prod
119894=0
N (119908119894| 0 120572minus1
119894) (11)
where119872 = 119873+1 and 120572 = (1205721 120572
119872)119879 is a hyperparameter
RVM finds hyperparameter 120572 by maximizing the marginallikelihood 119901(119910 | 119909 120572) via ldquoevidence approximationrdquo Inthe process of maximizing evidence some 120572
119894approach
infinity and the corresponding 119908119894become zero Thus the
basis function corresponding with these parameters can beremoved and it leads sparse models In many cases RVMperforms better than SVM especially in regression problems
3 Results and Discussion
31 Computational Experiments To evaluate our proposedmethod we conducted computational experiments and com-pared with the existing method APM
32 Data and Implementation It is difficult to directlymeasure actual strengths of PPIs for many protein pairs bybiological and physical experiments Hence we used WI-PHI dataset with 50000 protein pairs [23] For each PPI WI-PHI contains a weight that is considered to represent somereliability of the PPI and is calculated from several different
The Scientific World Journal 5
Table 1 Results of average RMSE for training and test data
119862 = 1 119862 = 2 119862 = 5
Training Test Training Test Training TestSVR + DN 010472 012573 010656 012600 009982 012484RVM + DN 009210 012873 009178 012881 009474 012908SVR + SPD (119896 = 1) 008819 012699 008080 012954 007927 012903RVM + SPD (119896 = 1) 002848 012743 001504 012706 003276 012792SVR + SPD (119896 = 2) 008891 012654 008188 012782 008117 012909RVM + SPD (119896 = 2) 002529 012470 002301 012476 002243 012493SVR + APM 006846 013112 006795 013247 006791 013277RVM + APM 007052 013556 007037 013550 007032 013493APM Training = 006811 Test = 013517
kinds of PPI datasets in some statistical manner to rankphysical protein interactions As strengths of PPIs we usedthe value dividing the weight of PPI by the maximum weightforWI-PHIWe used dataset file ldquouniprot sprot fungidatgzrdquodownloaded from UniProt database [27] to get amino acidsequences information of domain compositions and domainregions in proteins In this experiment we used 1387 proteinpairs that could be extracted from WI-PHI dataset withcomplete domain sequence viaUniProt datasetThe extracteddataset contains 758 proteins and 327 domains Since thisdataset does not include protein pairs with interactionstrength 0 we randomly selected 100 protein pairs that do nothave any weights in the dataset and added them as proteinpairs with strength 0 Thus totally 1487 protein pairs wereused in this experiment We used ldquokernlabrdquo package [28]for executing support vector regression and relevance vectormachine and used the Laplacian kernel119870(119909 119910) = exp(minus120590119909minus119910) The dataset and the source code implemented by R areavailable upon request
To evaluate prediction accuracy we calculated the rootmean square error (RMSE) for each prediction RMSE isa measure of differences between predicted values 119910
119894and
actually observed values 119910119894and is defined by
RMSE = radic1
119873
119873
sum
119894=1
(119910119894minus 119910119894)2
(12)
where119873 is the number of test data
33 Results of Computational Experiments We preformedthreefold cross-validation calculated the average RMSE andcompared with APM [17] For APM method strengths ofPPIs are inferred based on APM scores for domain pairs thatconsist of target proteins However it is not always possible tocalculate APM scores for all domain pairs from training setTherefore as test set we used only protein pairs that consistof domain pairs with APM scores calculated via training set(In all cases about 40 of protein pairs in test set were used)For the Laplacian kernel employed in both SVR and RVMwe selected kernel parameter 120590 by fivefold cross-validationfrom candidate set 120590 isin 001 002 01 The parameter119862 for the regularization term in the Lagrange formulation isset to 119862 = 1 2 5 Additionally APM scores for each protein
pair also can be used as input featuresTherefore we also usedAPM scores as inputs for SVR and RVM and compared themodel using APM scores with the model using our proposedfeatures to confirm the usefulness of feature representationHere we used candidate set 120590 isin 30 31 32 90 forkernel parameter 120590 of RVM+APMmodel because themodelcould not be trained with 120590 values smaller than 3 On theother hand for 120590 of SVM + APM model we used the sameset as other models
Table 1 shows the results of the average RMSE by SVR andRVM with our proposed features (DN and SPD of 119896 = 1 2)and APM score and by APM for training and test datasetsFor training set the average RMSEs by RVM with SPD of119896 = 2were smaller than those by APM and others Moreoverfor test set all the average RMSEs by RVMwith SPD and DNwere smaller than those by APM The results suggested thatsupervised regression methods SVR and RVM with domainbased features are useful for prediction of PPI strengthsTaking all results together the model by RVM with SPD of119896 = 2was regarded as the best for prediction of PPI strengths
Since the average RMSEs of SVR with APM for bothtraining and test dataset were smaller than those of originalAPM SVR has potential to improve prediction accuraciesBy contrast the average RMSEs of RVM with APM becamelarger than those of original APM and all average RMSEsof the models with APM for test set were larger thanthose of the models with DN and SPD Accordingly theresults suggested that prediction accuracies were enhancedby feature representation and SPD is especially useful amongthese feature representations for predicting strengths of PPIsAlthough DN and SPD of 119896 = 1 have 654 and 42 dimensionsfor each protein pair respectively the average RMSEs withSPDof 119896 = 1 for training setwere smaller than thosewithDNIt implies that information of amino acid sequence in domainregions is more informative comparing with information ofdomain compositions to make a model fit in with dataset
In contrast the RMSEs by SVR with DN were smallerthan those by others in some cases of test set Table 2 showsthe numbers of relevance vectors and support vectors andthe 120590 values selected by fivefold cross-validation in all casesFor the models with DN and APM scores the numbers ofrelevance vectors were smaller than the numbers of supportvectors On the other hand the numbers of relevance vectorswere larger than the numbers of support vectors for the
6 The Scientific World Journal
Table 2The number of relevance vectors (RVs) and support vectors (SVs) for each model with DN SPD and APM and the selected 120590 valuesfor each fold
119862 = 1 119862 = 2 119862 = 5
SVR RVM SVR RVM SVR RVMSVs (120590 value) RVs (120590 value) SVs (120590 value) RVs (120590 value) SVs (120590 value) RVs (120590 value)
Fold 1
DN 271 (002) 113 (005) 271 (001) 123 (007) 308 (001) 74 (002)SPD (119896 = 1) 367 (001) 448 (002) 402 (002) 680 (005) 402 (002) 537 (003)SPD (119896 = 2) 392 (001) 502 (003) 409 (001) 628 (005) 421 (001) 628 (005)
APM 362 (008) 4 (500) 361 (010) 6 (480) 357 (004) 6 (580)
Fold 2
DN 280 (002) 94 (008) 281 (001) 92 (009) 314 (001) 82 (004)SPD (119896 = 1) 408 (001) 617 (004) 453 (004) 706 (006) 411 (001) 545 (003)SPD (119896 = 2) 430 (001) 558 (004) 435 (001) 618 (005) 495 (004) 654 (006)
APM 375 (010) 5 (650) 372 (010) 6 (690) 373 (004) 4 (550)
Fold 3
DN 321 (004) 107 (008) 289 (001) 107 (010) 330 (001) 107 (008)SPD (119896 = 1) 371 (001) 439 (002) 412 (003) 658 (005) 382 (001) 305 (001)SPD (119896 = 2) 387 (001) 625 (006) 418 (002) 529 (004) 398 (001) 529 (004)
APM 368 (008) 3 (710) 368 (004) 3 (670) 372 (001) 5 (420)
models with SPD feature in spite of the fact that usuallyRVM provides a sparse model compared with SVR In RVMframework sparsity of model is caused by distributions ofeach weight that is the number of relevance vectors isinfluenced by values and variances of each dimension offeatures rather than by the number of dimensions of featuresActually each dimension of SPD feature almost always haswidely varying values In contrast DN feature has manyzeros and APM score is inferred from training dataset andthereby has similar distribution Thus it is considered thatmany weights corresponding to features in RVM model didnot become zero and the RVM models with SPD featuretended to be complex and to overfit the training data
4 ConclusionsFor the prediction of strengths of PPIs we proposed featurespace mappings DN and SPD DN is based on the numberof domains in a protein SPD is based on the spectrumkernel and defined using the amino acid subsequences indomain regions In this work we employed support vec-tor regression (SVR) and relevance vector machine (RVM)with the Laplacian kernel and conducted threefold cross-validation using WI-PHI dataset For both training and testdataset the average RMSEs by RVM with SPD feature weresmaller than those by APMThe results showed that machinelearning methods with domain information outperformedexisting association method that is based on the probabilisticmodel of PPIs and implied that the information of aminoacid sequence is useful for prediction comparing with onlyinformation of domain compositions However the modelswith SPD feature tended to be complex and overfitted to thetraining data Therefore to further enhance the predictionaccuracy improving kernel functions combining physicalcharacteristics of domains and amino acids might be helpful
Conflict of InterestsThe authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgment
This work was partially supported by Grants-in-Aid nos22240009 and 24500361 fromMEXT Japan
References
[1] J R Bock and D A Gough ldquoPredicting protein-proteininteractions from primary structurerdquo Bioinformatics vol 17 no5 pp 455ndash460 2001
[2] Y Guo L Yu Z Wen and M Li ldquoUsing support vectormachine combined with auto covariance to predict protein-protein interactions from protein sequencesrdquo Nucleic AcidsResearch vol 36 no 9 pp 3025ndash3030 2008
[3] C Yu L Chou and D T Chang ldquoPredicting protein-proteininteractions in unbalanced data using the primary structure ofproteinsrdquo BMC Bioinformatics vol 11 article 167 2010
[4] J Xia X Zhao and D Huang ldquoPredicting protein-proteininteractions from protein sequences using meta predictorrdquoAmino Acids vol 39 no 5 pp 1595ndash1599 2010
[5] J Xia K Han and D Huang ldquoSequence-based prediction ofprotein-protein interactions by means of rotation forest andautocorrelation descriptorrdquo Protein and Peptide Letters vol 17no 1 pp 137ndash145 2010
[6] S J Darnell D Page and J CMitchell ldquoAn automated decision-tree approach to predicting protein interaction hot spotsrdquoProteins Structure Function and Bioinformatics vol 68 no 4pp 813ndash823 2007
[7] K Cho D Kim and D Lee ldquoA feature-based approach tomodeling protein-protein interaction hot spotsrdquo Nucleic AcidsResearch vol 37 no 8 pp 2672ndash2687 2009
[8] J Xia X Zhao J Song and D Huang ldquoAPIS accurate predic-tion of hot spots in protein interfaces by combining protrusionindexwith solvent accessibilityrdquoBMCBioinformatics vol 11 no1 article 174 2010
[9] O KuchaievM Rasajski D J Higham andN Przulj ldquoGeomet-ric de-noising of protein-protein interaction networksrdquo PLoSComputational Biology vol 5 no 8 Article ID e1000454 2009
[10] L Zhu Z You and D Huang ldquoIncreasing the reliability ofprotein-protein interaction networks via non-convex semanticembeddingrdquo Neurocomputing vol 121 pp 99ndash107 2013
The Scientific World Journal 7
[11] S Byrum S Smart S Larson and A Tackett ldquoAnalysis ofstable and transient protein-protein interactionsrdquo Methods inMolecular Biology vol 833 pp 143ndash152 2012
[12] P Uetz L Glot G Cagney et al ldquoA comprehensive analysisof protein-protein interactions in Saccharomyces cerevisiaerdquoNature vol 403 no 6770 pp 623ndash627 2000
[13] T Ito T Chiba R Ozawa M Yoshida M Hattori and YSakaki ldquoA comprehensive two-hybrid analysis to explore theyeast protein interactomerdquo Proceedings of the National Academyof Sciences of the United States of America vol 98 no 8 pp4569ndash4574 2001
[14] M Hayashida N Ueda and T Akutsu ldquoInferring strengthsof protein-protein interactions from experimental data usinglinear programmingrdquo Bioinformatics vol 19 no 2 pp ii58ndashii652003
[15] M Hayashida N Ueda and T Akutsu ldquoA simple methodfor inferring strengths of protein-protein interactionsrdquoGenomeInformatics vol 15 no 1 pp 56ndash68 2004
[16] E Sprinzak and H Margalit ldquoCorrelated sequence-signaturesas markers of protein-protein interactionrdquo Journal of MolecularBiology vol 311 no 4 pp 681ndash692 2001
[17] L Chen L Wu Y Wang and X Zhang ldquoInferring-proteininteractions from experimental data by association probabilisticmethodrdquo Proteins Structure Function and Genetics vol 62 no4 pp 833ndash837 2006
[18] R Finn J Mistry J Tate et al ldquoThe Pfam protein familiesdatabaserdquo Nucleic Acids Research vol 38 supplement 1 ppD211ndashD222 2009
[19] S Hunter P Jones A Mitchell et al ldquoInterPro in 2011 newdevelopments in the family and domain prediction databaserdquoNucleic Acids Research vol 40 no 1 pp D306ndashD312 2012
[20] J Xia B Wang and D Huang ldquoInferring strengths of protein-protein interaction using artificial neural networkrdquo in Proceed-ings of the International Joint Conference on Neural Networks(IJCNN rsquo07) pp 2471ndash2475 Orlando Fla USA August 2007
[21] M Deng S Mehta F Sun and T Chen ldquoInferring domain-domain interactions from protein-protein interactionsrdquoGenome Research vol 12 no 10 pp 1540ndash1548 2002
[22] Y Sakuma M Kamada M Hayashida and T Akutsu ldquoInfer-ring strengths of protein-protein interactions using supportvector regressionrdquo inProceedings of the International Conferenceon Parallel and Distributed Processing Techniques and Applica-tions 2013 httpworld-comporgp2013PDP2162pdf
[23] L Kiemer S Costa M Ueffing and G Cesareni ldquoWI-PHIa weighted yeast interactome enriched for direct physicalinteractionsrdquo Proteomics vol 7 no 6 pp 932ndash943 2007
[24] C Leslie E Eskin and W Noble ldquoThe spectrum Kernel astring Kernel for SVM protein classi fi cationrdquo in Proceedingsof Pacific Symposium on Biocomputing (PSB rsquo02) pp 564ndash575Lihue Hawaii USA January 2002
[25] VN VapnikTheNature of Statistical LearningTheory Springer1995
[26] M E Tipping ldquoSparse bayesian learning and the relevancevector machinerdquo Journal of Machine Learning Research vol 1no 3 pp 211ndash244 2001
[27] T U Consortium ldquoReorganizing the protein space at theuniversal protein resource (UniProt)rdquo Nucleic Acids Researchvol 40 no D1 pp D71ndashD75 2012
[28] A Karatzoglou K Hornik A Smola and A Zeileis ldquokernlabmdashan S4 package for kernel methods in Rrdquo Journal of StatisticalSoftware vol 11 no 9 pp 1ndash20 2004
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

The Scientific World Journal 5
Table 1 Results of average RMSE for training and test data
119862 = 1 119862 = 2 119862 = 5
Training Test Training Test Training TestSVR + DN 010472 012573 010656 012600 009982 012484RVM + DN 009210 012873 009178 012881 009474 012908SVR + SPD (119896 = 1) 008819 012699 008080 012954 007927 012903RVM + SPD (119896 = 1) 002848 012743 001504 012706 003276 012792SVR + SPD (119896 = 2) 008891 012654 008188 012782 008117 012909RVM + SPD (119896 = 2) 002529 012470 002301 012476 002243 012493SVR + APM 006846 013112 006795 013247 006791 013277RVM + APM 007052 013556 007037 013550 007032 013493APM Training = 006811 Test = 013517
kinds of PPI datasets in some statistical manner to rankphysical protein interactions As strengths of PPIs we usedthe value dividing the weight of PPI by the maximum weightforWI-PHIWe used dataset file ldquouniprot sprot fungidatgzrdquodownloaded from UniProt database [27] to get amino acidsequences information of domain compositions and domainregions in proteins In this experiment we used 1387 proteinpairs that could be extracted from WI-PHI dataset withcomplete domain sequence viaUniProt datasetThe extracteddataset contains 758 proteins and 327 domains Since thisdataset does not include protein pairs with interactionstrength 0 we randomly selected 100 protein pairs that do nothave any weights in the dataset and added them as proteinpairs with strength 0 Thus totally 1487 protein pairs wereused in this experiment We used ldquokernlabrdquo package [28]for executing support vector regression and relevance vectormachine and used the Laplacian kernel119870(119909 119910) = exp(minus120590119909minus119910) The dataset and the source code implemented by R areavailable upon request
To evaluate prediction accuracy we calculated the rootmean square error (RMSE) for each prediction RMSE isa measure of differences between predicted values 119910
119894and
actually observed values 119910119894and is defined by
RMSE = radic1
119873
119873
sum
119894=1
(119910119894minus 119910119894)2
(12)
where119873 is the number of test data
33 Results of Computational Experiments We preformedthreefold cross-validation calculated the average RMSE andcompared with APM [17] For APM method strengths ofPPIs are inferred based on APM scores for domain pairs thatconsist of target proteins However it is not always possible tocalculate APM scores for all domain pairs from training setTherefore as test set we used only protein pairs that consistof domain pairs with APM scores calculated via training set(In all cases about 40 of protein pairs in test set were used)For the Laplacian kernel employed in both SVR and RVMwe selected kernel parameter 120590 by fivefold cross-validationfrom candidate set 120590 isin 001 002 01 The parameter119862 for the regularization term in the Lagrange formulation isset to 119862 = 1 2 5 Additionally APM scores for each protein
pair also can be used as input featuresTherefore we also usedAPM scores as inputs for SVR and RVM and compared themodel using APM scores with the model using our proposedfeatures to confirm the usefulness of feature representationHere we used candidate set 120590 isin 30 31 32 90 forkernel parameter 120590 of RVM+APMmodel because themodelcould not be trained with 120590 values smaller than 3 On theother hand for 120590 of SVM + APM model we used the sameset as other models
Table 1 shows the results of the average RMSE by SVR andRVM with our proposed features (DN and SPD of 119896 = 1 2)and APM score and by APM for training and test datasetsFor training set the average RMSEs by RVM with SPD of119896 = 2were smaller than those by APM and others Moreoverfor test set all the average RMSEs by RVMwith SPD and DNwere smaller than those by APM The results suggested thatsupervised regression methods SVR and RVM with domainbased features are useful for prediction of PPI strengthsTaking all results together the model by RVM with SPD of119896 = 2was regarded as the best for prediction of PPI strengths
Since the average RMSEs of SVR with APM for bothtraining and test dataset were smaller than those of originalAPM SVR has potential to improve prediction accuraciesBy contrast the average RMSEs of RVM with APM becamelarger than those of original APM and all average RMSEsof the models with APM for test set were larger thanthose of the models with DN and SPD Accordingly theresults suggested that prediction accuracies were enhancedby feature representation and SPD is especially useful amongthese feature representations for predicting strengths of PPIsAlthough DN and SPD of 119896 = 1 have 654 and 42 dimensionsfor each protein pair respectively the average RMSEs withSPDof 119896 = 1 for training setwere smaller than thosewithDNIt implies that information of amino acid sequence in domainregions is more informative comparing with information ofdomain compositions to make a model fit in with dataset
In contrast the RMSEs by SVR with DN were smallerthan those by others in some cases of test set Table 2 showsthe numbers of relevance vectors and support vectors andthe 120590 values selected by fivefold cross-validation in all casesFor the models with DN and APM scores the numbers ofrelevance vectors were smaller than the numbers of supportvectors On the other hand the numbers of relevance vectorswere larger than the numbers of support vectors for the
6 The Scientific World Journal
Table 2The number of relevance vectors (RVs) and support vectors (SVs) for each model with DN SPD and APM and the selected 120590 valuesfor each fold
119862 = 1 119862 = 2 119862 = 5
SVR RVM SVR RVM SVR RVMSVs (120590 value) RVs (120590 value) SVs (120590 value) RVs (120590 value) SVs (120590 value) RVs (120590 value)
Fold 1
DN 271 (002) 113 (005) 271 (001) 123 (007) 308 (001) 74 (002)SPD (119896 = 1) 367 (001) 448 (002) 402 (002) 680 (005) 402 (002) 537 (003)SPD (119896 = 2) 392 (001) 502 (003) 409 (001) 628 (005) 421 (001) 628 (005)
APM 362 (008) 4 (500) 361 (010) 6 (480) 357 (004) 6 (580)
Fold 2
DN 280 (002) 94 (008) 281 (001) 92 (009) 314 (001) 82 (004)SPD (119896 = 1) 408 (001) 617 (004) 453 (004) 706 (006) 411 (001) 545 (003)SPD (119896 = 2) 430 (001) 558 (004) 435 (001) 618 (005) 495 (004) 654 (006)
APM 375 (010) 5 (650) 372 (010) 6 (690) 373 (004) 4 (550)
Fold 3
DN 321 (004) 107 (008) 289 (001) 107 (010) 330 (001) 107 (008)SPD (119896 = 1) 371 (001) 439 (002) 412 (003) 658 (005) 382 (001) 305 (001)SPD (119896 = 2) 387 (001) 625 (006) 418 (002) 529 (004) 398 (001) 529 (004)
APM 368 (008) 3 (710) 368 (004) 3 (670) 372 (001) 5 (420)
models with SPD feature in spite of the fact that usuallyRVM provides a sparse model compared with SVR In RVMframework sparsity of model is caused by distributions ofeach weight that is the number of relevance vectors isinfluenced by values and variances of each dimension offeatures rather than by the number of dimensions of featuresActually each dimension of SPD feature almost always haswidely varying values In contrast DN feature has manyzeros and APM score is inferred from training dataset andthereby has similar distribution Thus it is considered thatmany weights corresponding to features in RVM model didnot become zero and the RVM models with SPD featuretended to be complex and to overfit the training data
4 ConclusionsFor the prediction of strengths of PPIs we proposed featurespace mappings DN and SPD DN is based on the numberof domains in a protein SPD is based on the spectrumkernel and defined using the amino acid subsequences indomain regions In this work we employed support vec-tor regression (SVR) and relevance vector machine (RVM)with the Laplacian kernel and conducted threefold cross-validation using WI-PHI dataset For both training and testdataset the average RMSEs by RVM with SPD feature weresmaller than those by APMThe results showed that machinelearning methods with domain information outperformedexisting association method that is based on the probabilisticmodel of PPIs and implied that the information of aminoacid sequence is useful for prediction comparing with onlyinformation of domain compositions However the modelswith SPD feature tended to be complex and overfitted to thetraining data Therefore to further enhance the predictionaccuracy improving kernel functions combining physicalcharacteristics of domains and amino acids might be helpful
Conflict of InterestsThe authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgment
This work was partially supported by Grants-in-Aid nos22240009 and 24500361 fromMEXT Japan
References
[1] J R Bock and D A Gough ldquoPredicting protein-proteininteractions from primary structurerdquo Bioinformatics vol 17 no5 pp 455ndash460 2001
[2] Y Guo L Yu Z Wen and M Li ldquoUsing support vectormachine combined with auto covariance to predict protein-protein interactions from protein sequencesrdquo Nucleic AcidsResearch vol 36 no 9 pp 3025ndash3030 2008
[3] C Yu L Chou and D T Chang ldquoPredicting protein-proteininteractions in unbalanced data using the primary structure ofproteinsrdquo BMC Bioinformatics vol 11 article 167 2010
[4] J Xia X Zhao and D Huang ldquoPredicting protein-proteininteractions from protein sequences using meta predictorrdquoAmino Acids vol 39 no 5 pp 1595ndash1599 2010
[5] J Xia K Han and D Huang ldquoSequence-based prediction ofprotein-protein interactions by means of rotation forest andautocorrelation descriptorrdquo Protein and Peptide Letters vol 17no 1 pp 137ndash145 2010
[6] S J Darnell D Page and J CMitchell ldquoAn automated decision-tree approach to predicting protein interaction hot spotsrdquoProteins Structure Function and Bioinformatics vol 68 no 4pp 813ndash823 2007
[7] K Cho D Kim and D Lee ldquoA feature-based approach tomodeling protein-protein interaction hot spotsrdquo Nucleic AcidsResearch vol 37 no 8 pp 2672ndash2687 2009
[8] J Xia X Zhao J Song and D Huang ldquoAPIS accurate predic-tion of hot spots in protein interfaces by combining protrusionindexwith solvent accessibilityrdquoBMCBioinformatics vol 11 no1 article 174 2010
[9] O KuchaievM Rasajski D J Higham andN Przulj ldquoGeomet-ric de-noising of protein-protein interaction networksrdquo PLoSComputational Biology vol 5 no 8 Article ID e1000454 2009
[10] L Zhu Z You and D Huang ldquoIncreasing the reliability ofprotein-protein interaction networks via non-convex semanticembeddingrdquo Neurocomputing vol 121 pp 99ndash107 2013
The Scientific World Journal 7
[11] S Byrum S Smart S Larson and A Tackett ldquoAnalysis ofstable and transient protein-protein interactionsrdquo Methods inMolecular Biology vol 833 pp 143ndash152 2012
[12] P Uetz L Glot G Cagney et al ldquoA comprehensive analysisof protein-protein interactions in Saccharomyces cerevisiaerdquoNature vol 403 no 6770 pp 623ndash627 2000
[13] T Ito T Chiba R Ozawa M Yoshida M Hattori and YSakaki ldquoA comprehensive two-hybrid analysis to explore theyeast protein interactomerdquo Proceedings of the National Academyof Sciences of the United States of America vol 98 no 8 pp4569ndash4574 2001
[14] M Hayashida N Ueda and T Akutsu ldquoInferring strengthsof protein-protein interactions from experimental data usinglinear programmingrdquo Bioinformatics vol 19 no 2 pp ii58ndashii652003
[15] M Hayashida N Ueda and T Akutsu ldquoA simple methodfor inferring strengths of protein-protein interactionsrdquoGenomeInformatics vol 15 no 1 pp 56ndash68 2004
[16] E Sprinzak and H Margalit ldquoCorrelated sequence-signaturesas markers of protein-protein interactionrdquo Journal of MolecularBiology vol 311 no 4 pp 681ndash692 2001
[17] L Chen L Wu Y Wang and X Zhang ldquoInferring-proteininteractions from experimental data by association probabilisticmethodrdquo Proteins Structure Function and Genetics vol 62 no4 pp 833ndash837 2006
[18] R Finn J Mistry J Tate et al ldquoThe Pfam protein familiesdatabaserdquo Nucleic Acids Research vol 38 supplement 1 ppD211ndashD222 2009
[19] S Hunter P Jones A Mitchell et al ldquoInterPro in 2011 newdevelopments in the family and domain prediction databaserdquoNucleic Acids Research vol 40 no 1 pp D306ndashD312 2012
[20] J Xia B Wang and D Huang ldquoInferring strengths of protein-protein interaction using artificial neural networkrdquo in Proceed-ings of the International Joint Conference on Neural Networks(IJCNN rsquo07) pp 2471ndash2475 Orlando Fla USA August 2007
[21] M Deng S Mehta F Sun and T Chen ldquoInferring domain-domain interactions from protein-protein interactionsrdquoGenome Research vol 12 no 10 pp 1540ndash1548 2002
[22] Y Sakuma M Kamada M Hayashida and T Akutsu ldquoInfer-ring strengths of protein-protein interactions using supportvector regressionrdquo inProceedings of the International Conferenceon Parallel and Distributed Processing Techniques and Applica-tions 2013 httpworld-comporgp2013PDP2162pdf
[23] L Kiemer S Costa M Ueffing and G Cesareni ldquoWI-PHIa weighted yeast interactome enriched for direct physicalinteractionsrdquo Proteomics vol 7 no 6 pp 932ndash943 2007
[24] C Leslie E Eskin and W Noble ldquoThe spectrum Kernel astring Kernel for SVM protein classi fi cationrdquo in Proceedingsof Pacific Symposium on Biocomputing (PSB rsquo02) pp 564ndash575Lihue Hawaii USA January 2002
[25] VN VapnikTheNature of Statistical LearningTheory Springer1995
[26] M E Tipping ldquoSparse bayesian learning and the relevancevector machinerdquo Journal of Machine Learning Research vol 1no 3 pp 211ndash244 2001
[27] T U Consortium ldquoReorganizing the protein space at theuniversal protein resource (UniProt)rdquo Nucleic Acids Researchvol 40 no D1 pp D71ndashD75 2012
[28] A Karatzoglou K Hornik A Smola and A Zeileis ldquokernlabmdashan S4 package for kernel methods in Rrdquo Journal of StatisticalSoftware vol 11 no 9 pp 1ndash20 2004
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology
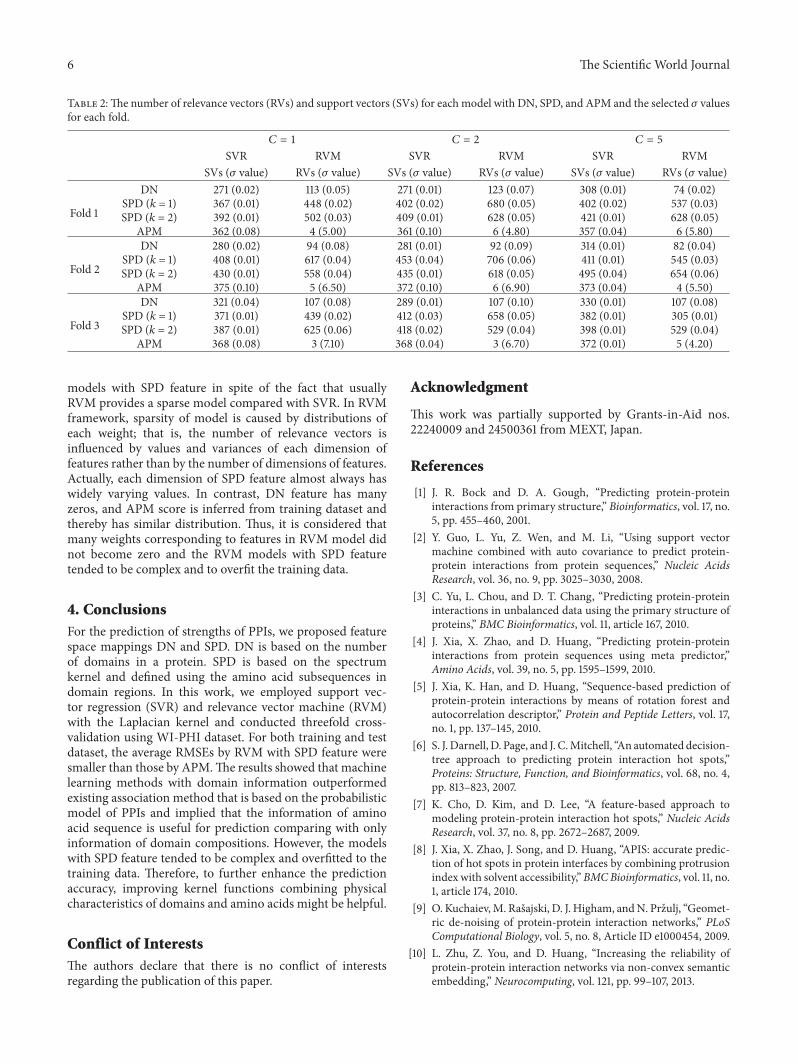
6 The Scientific World Journal
Table 2The number of relevance vectors (RVs) and support vectors (SVs) for each model with DN SPD and APM and the selected 120590 valuesfor each fold
119862 = 1 119862 = 2 119862 = 5
SVR RVM SVR RVM SVR RVMSVs (120590 value) RVs (120590 value) SVs (120590 value) RVs (120590 value) SVs (120590 value) RVs (120590 value)
Fold 1
DN 271 (002) 113 (005) 271 (001) 123 (007) 308 (001) 74 (002)SPD (119896 = 1) 367 (001) 448 (002) 402 (002) 680 (005) 402 (002) 537 (003)SPD (119896 = 2) 392 (001) 502 (003) 409 (001) 628 (005) 421 (001) 628 (005)
APM 362 (008) 4 (500) 361 (010) 6 (480) 357 (004) 6 (580)
Fold 2
DN 280 (002) 94 (008) 281 (001) 92 (009) 314 (001) 82 (004)SPD (119896 = 1) 408 (001) 617 (004) 453 (004) 706 (006) 411 (001) 545 (003)SPD (119896 = 2) 430 (001) 558 (004) 435 (001) 618 (005) 495 (004) 654 (006)
APM 375 (010) 5 (650) 372 (010) 6 (690) 373 (004) 4 (550)
Fold 3
DN 321 (004) 107 (008) 289 (001) 107 (010) 330 (001) 107 (008)SPD (119896 = 1) 371 (001) 439 (002) 412 (003) 658 (005) 382 (001) 305 (001)SPD (119896 = 2) 387 (001) 625 (006) 418 (002) 529 (004) 398 (001) 529 (004)
APM 368 (008) 3 (710) 368 (004) 3 (670) 372 (001) 5 (420)
models with SPD feature in spite of the fact that usuallyRVM provides a sparse model compared with SVR In RVMframework sparsity of model is caused by distributions ofeach weight that is the number of relevance vectors isinfluenced by values and variances of each dimension offeatures rather than by the number of dimensions of featuresActually each dimension of SPD feature almost always haswidely varying values In contrast DN feature has manyzeros and APM score is inferred from training dataset andthereby has similar distribution Thus it is considered thatmany weights corresponding to features in RVM model didnot become zero and the RVM models with SPD featuretended to be complex and to overfit the training data
4 ConclusionsFor the prediction of strengths of PPIs we proposed featurespace mappings DN and SPD DN is based on the numberof domains in a protein SPD is based on the spectrumkernel and defined using the amino acid subsequences indomain regions In this work we employed support vec-tor regression (SVR) and relevance vector machine (RVM)with the Laplacian kernel and conducted threefold cross-validation using WI-PHI dataset For both training and testdataset the average RMSEs by RVM with SPD feature weresmaller than those by APMThe results showed that machinelearning methods with domain information outperformedexisting association method that is based on the probabilisticmodel of PPIs and implied that the information of aminoacid sequence is useful for prediction comparing with onlyinformation of domain compositions However the modelswith SPD feature tended to be complex and overfitted to thetraining data Therefore to further enhance the predictionaccuracy improving kernel functions combining physicalcharacteristics of domains and amino acids might be helpful
Conflict of InterestsThe authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgment
This work was partially supported by Grants-in-Aid nos22240009 and 24500361 fromMEXT Japan
References
[1] J R Bock and D A Gough ldquoPredicting protein-proteininteractions from primary structurerdquo Bioinformatics vol 17 no5 pp 455ndash460 2001
[2] Y Guo L Yu Z Wen and M Li ldquoUsing support vectormachine combined with auto covariance to predict protein-protein interactions from protein sequencesrdquo Nucleic AcidsResearch vol 36 no 9 pp 3025ndash3030 2008
[3] C Yu L Chou and D T Chang ldquoPredicting protein-proteininteractions in unbalanced data using the primary structure ofproteinsrdquo BMC Bioinformatics vol 11 article 167 2010
[4] J Xia X Zhao and D Huang ldquoPredicting protein-proteininteractions from protein sequences using meta predictorrdquoAmino Acids vol 39 no 5 pp 1595ndash1599 2010
[5] J Xia K Han and D Huang ldquoSequence-based prediction ofprotein-protein interactions by means of rotation forest andautocorrelation descriptorrdquo Protein and Peptide Letters vol 17no 1 pp 137ndash145 2010
[6] S J Darnell D Page and J CMitchell ldquoAn automated decision-tree approach to predicting protein interaction hot spotsrdquoProteins Structure Function and Bioinformatics vol 68 no 4pp 813ndash823 2007
[7] K Cho D Kim and D Lee ldquoA feature-based approach tomodeling protein-protein interaction hot spotsrdquo Nucleic AcidsResearch vol 37 no 8 pp 2672ndash2687 2009
[8] J Xia X Zhao J Song and D Huang ldquoAPIS accurate predic-tion of hot spots in protein interfaces by combining protrusionindexwith solvent accessibilityrdquoBMCBioinformatics vol 11 no1 article 174 2010
[9] O KuchaievM Rasajski D J Higham andN Przulj ldquoGeomet-ric de-noising of protein-protein interaction networksrdquo PLoSComputational Biology vol 5 no 8 Article ID e1000454 2009
[10] L Zhu Z You and D Huang ldquoIncreasing the reliability ofprotein-protein interaction networks via non-convex semanticembeddingrdquo Neurocomputing vol 121 pp 99ndash107 2013
The Scientific World Journal 7
[11] S Byrum S Smart S Larson and A Tackett ldquoAnalysis ofstable and transient protein-protein interactionsrdquo Methods inMolecular Biology vol 833 pp 143ndash152 2012
[12] P Uetz L Glot G Cagney et al ldquoA comprehensive analysisof protein-protein interactions in Saccharomyces cerevisiaerdquoNature vol 403 no 6770 pp 623ndash627 2000
[13] T Ito T Chiba R Ozawa M Yoshida M Hattori and YSakaki ldquoA comprehensive two-hybrid analysis to explore theyeast protein interactomerdquo Proceedings of the National Academyof Sciences of the United States of America vol 98 no 8 pp4569ndash4574 2001
[14] M Hayashida N Ueda and T Akutsu ldquoInferring strengthsof protein-protein interactions from experimental data usinglinear programmingrdquo Bioinformatics vol 19 no 2 pp ii58ndashii652003
[15] M Hayashida N Ueda and T Akutsu ldquoA simple methodfor inferring strengths of protein-protein interactionsrdquoGenomeInformatics vol 15 no 1 pp 56ndash68 2004
[16] E Sprinzak and H Margalit ldquoCorrelated sequence-signaturesas markers of protein-protein interactionrdquo Journal of MolecularBiology vol 311 no 4 pp 681ndash692 2001
[17] L Chen L Wu Y Wang and X Zhang ldquoInferring-proteininteractions from experimental data by association probabilisticmethodrdquo Proteins Structure Function and Genetics vol 62 no4 pp 833ndash837 2006
[18] R Finn J Mistry J Tate et al ldquoThe Pfam protein familiesdatabaserdquo Nucleic Acids Research vol 38 supplement 1 ppD211ndashD222 2009
[19] S Hunter P Jones A Mitchell et al ldquoInterPro in 2011 newdevelopments in the family and domain prediction databaserdquoNucleic Acids Research vol 40 no 1 pp D306ndashD312 2012
[20] J Xia B Wang and D Huang ldquoInferring strengths of protein-protein interaction using artificial neural networkrdquo in Proceed-ings of the International Joint Conference on Neural Networks(IJCNN rsquo07) pp 2471ndash2475 Orlando Fla USA August 2007
[21] M Deng S Mehta F Sun and T Chen ldquoInferring domain-domain interactions from protein-protein interactionsrdquoGenome Research vol 12 no 10 pp 1540ndash1548 2002
[22] Y Sakuma M Kamada M Hayashida and T Akutsu ldquoInfer-ring strengths of protein-protein interactions using supportvector regressionrdquo inProceedings of the International Conferenceon Parallel and Distributed Processing Techniques and Applica-tions 2013 httpworld-comporgp2013PDP2162pdf
[23] L Kiemer S Costa M Ueffing and G Cesareni ldquoWI-PHIa weighted yeast interactome enriched for direct physicalinteractionsrdquo Proteomics vol 7 no 6 pp 932ndash943 2007
[24] C Leslie E Eskin and W Noble ldquoThe spectrum Kernel astring Kernel for SVM protein classi fi cationrdquo in Proceedingsof Pacific Symposium on Biocomputing (PSB rsquo02) pp 564ndash575Lihue Hawaii USA January 2002
[25] VN VapnikTheNature of Statistical LearningTheory Springer1995
[26] M E Tipping ldquoSparse bayesian learning and the relevancevector machinerdquo Journal of Machine Learning Research vol 1no 3 pp 211ndash244 2001
[27] T U Consortium ldquoReorganizing the protein space at theuniversal protein resource (UniProt)rdquo Nucleic Acids Researchvol 40 no D1 pp D71ndashD75 2012
[28] A Karatzoglou K Hornik A Smola and A Zeileis ldquokernlabmdashan S4 package for kernel methods in Rrdquo Journal of StatisticalSoftware vol 11 no 9 pp 1ndash20 2004
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

The Scientific World Journal 7
[11] S Byrum S Smart S Larson and A Tackett ldquoAnalysis ofstable and transient protein-protein interactionsrdquo Methods inMolecular Biology vol 833 pp 143ndash152 2012
[12] P Uetz L Glot G Cagney et al ldquoA comprehensive analysisof protein-protein interactions in Saccharomyces cerevisiaerdquoNature vol 403 no 6770 pp 623ndash627 2000
[13] T Ito T Chiba R Ozawa M Yoshida M Hattori and YSakaki ldquoA comprehensive two-hybrid analysis to explore theyeast protein interactomerdquo Proceedings of the National Academyof Sciences of the United States of America vol 98 no 8 pp4569ndash4574 2001
[14] M Hayashida N Ueda and T Akutsu ldquoInferring strengthsof protein-protein interactions from experimental data usinglinear programmingrdquo Bioinformatics vol 19 no 2 pp ii58ndashii652003
[15] M Hayashida N Ueda and T Akutsu ldquoA simple methodfor inferring strengths of protein-protein interactionsrdquoGenomeInformatics vol 15 no 1 pp 56ndash68 2004
[16] E Sprinzak and H Margalit ldquoCorrelated sequence-signaturesas markers of protein-protein interactionrdquo Journal of MolecularBiology vol 311 no 4 pp 681ndash692 2001
[17] L Chen L Wu Y Wang and X Zhang ldquoInferring-proteininteractions from experimental data by association probabilisticmethodrdquo Proteins Structure Function and Genetics vol 62 no4 pp 833ndash837 2006
[18] R Finn J Mistry J Tate et al ldquoThe Pfam protein familiesdatabaserdquo Nucleic Acids Research vol 38 supplement 1 ppD211ndashD222 2009
[19] S Hunter P Jones A Mitchell et al ldquoInterPro in 2011 newdevelopments in the family and domain prediction databaserdquoNucleic Acids Research vol 40 no 1 pp D306ndashD312 2012
[20] J Xia B Wang and D Huang ldquoInferring strengths of protein-protein interaction using artificial neural networkrdquo in Proceed-ings of the International Joint Conference on Neural Networks(IJCNN rsquo07) pp 2471ndash2475 Orlando Fla USA August 2007
[21] M Deng S Mehta F Sun and T Chen ldquoInferring domain-domain interactions from protein-protein interactionsrdquoGenome Research vol 12 no 10 pp 1540ndash1548 2002
[22] Y Sakuma M Kamada M Hayashida and T Akutsu ldquoInfer-ring strengths of protein-protein interactions using supportvector regressionrdquo inProceedings of the International Conferenceon Parallel and Distributed Processing Techniques and Applica-tions 2013 httpworld-comporgp2013PDP2162pdf
[23] L Kiemer S Costa M Ueffing and G Cesareni ldquoWI-PHIa weighted yeast interactome enriched for direct physicalinteractionsrdquo Proteomics vol 7 no 6 pp 932ndash943 2007
[24] C Leslie E Eskin and W Noble ldquoThe spectrum Kernel astring Kernel for SVM protein classi fi cationrdquo in Proceedingsof Pacific Symposium on Biocomputing (PSB rsquo02) pp 564ndash575Lihue Hawaii USA January 2002
[25] VN VapnikTheNature of Statistical LearningTheory Springer1995
[26] M E Tipping ldquoSparse bayesian learning and the relevancevector machinerdquo Journal of Machine Learning Research vol 1no 3 pp 211ndash244 2001
[27] T U Consortium ldquoReorganizing the protein space at theuniversal protein resource (UniProt)rdquo Nucleic Acids Researchvol 40 no D1 pp D71ndashD75 2012
[28] A Karatzoglou K Hornik A Smola and A Zeileis ldquokernlabmdashan S4 package for kernel methods in Rrdquo Journal of StatisticalSoftware vol 11 no 9 pp 1ndash20 2004
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology





























