Embed Size (px)
Citation preview
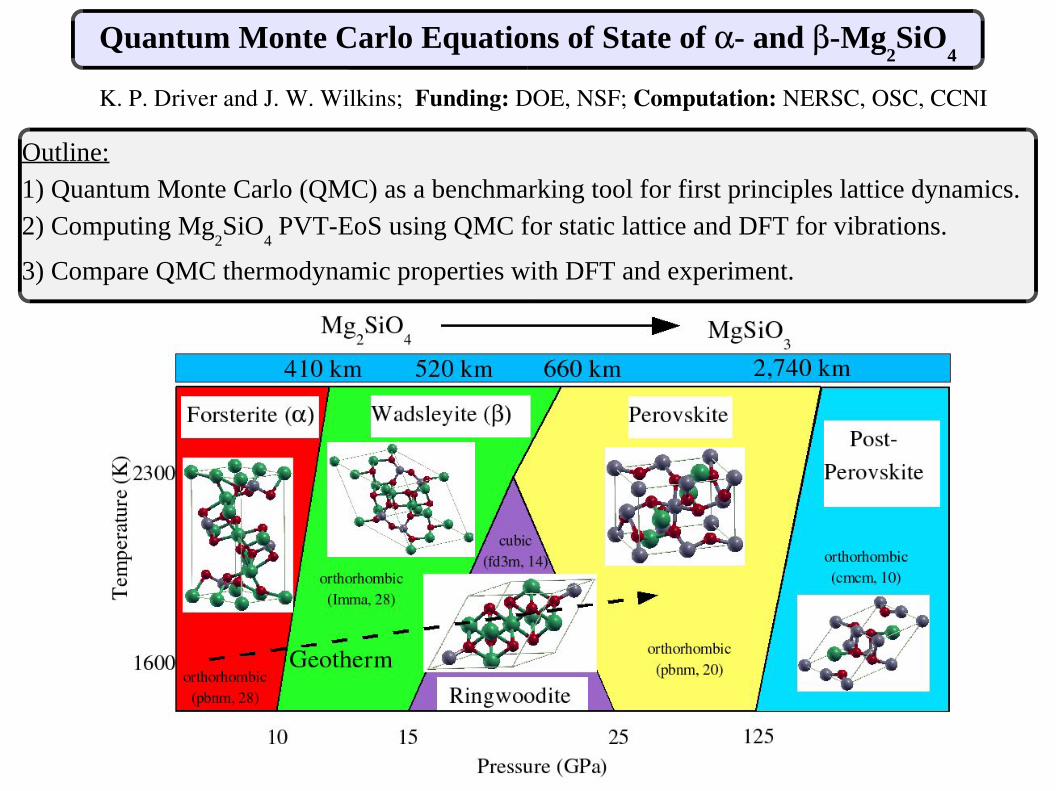
Quantum Monte Carlo Equations of State of - and-Mg2SiO
4
K. P. Driver and J. W. Wilkins; Funding: DOE, NSF; Computation: NERSC, OSC, CCNI
Outline:1) Quantum Monte Carlo (QMC) as a benchmarking tool for first principles lattice dynamics. 2) Computing Mg
2SiO
4 PVT-EoS using QMC for static lattice and DFT for vibrations.
3) Compare QMC thermodynamic properties with DFT and experiment.

DFT generally great, but can fail:●LDA underpredicts quartz-stishovite and forsterite-wadsleyite transition by 6 GPa.●GGA's may improve transition, but may worsen other properties.
●Extreme materials research demands pushing the frontier of first-principle simulations.●DFT is current workhorse, but exchange-correlation (XC) functionals leave room for doubt.●QMC avoids XC approximation, but is too expensive replace DFT anytime soon. ●Use QMC as a benchmark tool to spot-check important results.
Introduction and Motivation
Tem
pera
ture
(K)
0
2000
1000
0 25
Pressure (GPa)6
Figure from:Yu et al., Earth Planet. Sci. Lett. 273, 115 (2008)Recent DFT work:Li et al., J. Geophys. Res. 112, B05206 (2007)Wu et al., J. Geophys. Res. 112, B12202 (2007)
DFT(LDA)
DFT(GGA)
12
Goals of this work:●Explore feasibility of using QMC with DFPT for high pressure/temperature phase transitions.
●Check QMC/DFPT performance for first ternary solid oxide Mg
2SiO
4. (Number of QMC calculations on
solid oxides is tiny: (MgO, NiO, MnO, FeO, SiO2)
to Mg2SiO4

QMC1)Explicit manybody method.2)Use DFT's relaxed crystal structures.3)Optimize DFT wavefunction (fixed nodes).4)Compute energy stochastically.
DFT1)Singleparticle theory in effective potential.2)Choose XCfunctional and pseudopotential.3)Relax crystal structures.4)Compute energy and wavefunction.
Density Functional Theory and Quantum Monte Carlo
●WCGGA functional and potentials ●smallcore Mg potential (Opium code)●28/56 atom cells
●MPC, 112 atom simulation cells, ~1.5 mH FSE●Bspline basis, 0.005 time step●Jastrow with 2 & 3body, PW (109 paramaters)●~4 million CPU hours(NERSC, CCNI, OSC)
Experiment 123129 289292QMC 136(14) 309(1)WCGGA 112 297.8LDA 126.4 289.5
Experiment 160175 535539QMC T=0 K 156(12) 544(3.4)WCGGA T=0K 159.5 545.9LDA 166.7 541.35
Forsterite Bulk Modulus (GPa) Volume (Ang3)
Wadsleyite
DFT
Properties at T=300 K, P= 0 GPa

Computing PVT Equations of State
FQHA=E staticV F vibrationV ,T
Compute static lattice energy with QMC●Dominant energy contribution●Most accurate method available for solids●CASINO code
Compute vibrational free energy with DFT●Currently too costly for QMC●Vibrational energy is small●Typically well described in DFT●ABINIT, Linear Response, Quasiharmonic
●Compute quasiharmonic Helmholtz free energy
Dispersion data from Lam et al., Am. Mineral 75, 109 (1990)
T=0 K Transition Pressure (GPa)Experiment 8, 12QMC 10.5 (2.3)WCGGA 8.6PBE 12.6LDA 6.5
QMC Static Forsterite and Wadsleyite EoS
Freq
uenc
y (c
m1)
Forsterite Phonon Dispersion (P=0 GPa)

Forsterite and Wadsleyite EoS
●QMC equations of state still need work.●QMC volumes and curvature are not yet correct – off by several per cent.●Must reconsider finite size errors, pseudopotential, and DFT structures.●WCGGA and LDA calculations agree well with experiment.
P=− ∂FQHA
∂V T
Compute pressure from free energy
Forsterite Wadsleyite (T=0 only)
LDA Li et al., J. Geophys. Res. 112, B05206 (2007) and Wu et al., J. Geophys. Res. 112, B12202 (2007)

Bulk Modulus Temperature and Pressure Dependence
P=0 GPa
K=−V ∂P∂V
T
●QMC modulus variation with temperature agrees with experiment.●Small QMC variation with pressure reflects nearly constant P(V) slope.●DFT(WC) modulus falls too rapidly with temperature.●DFT(WC) modulus increases with pressure as expected.
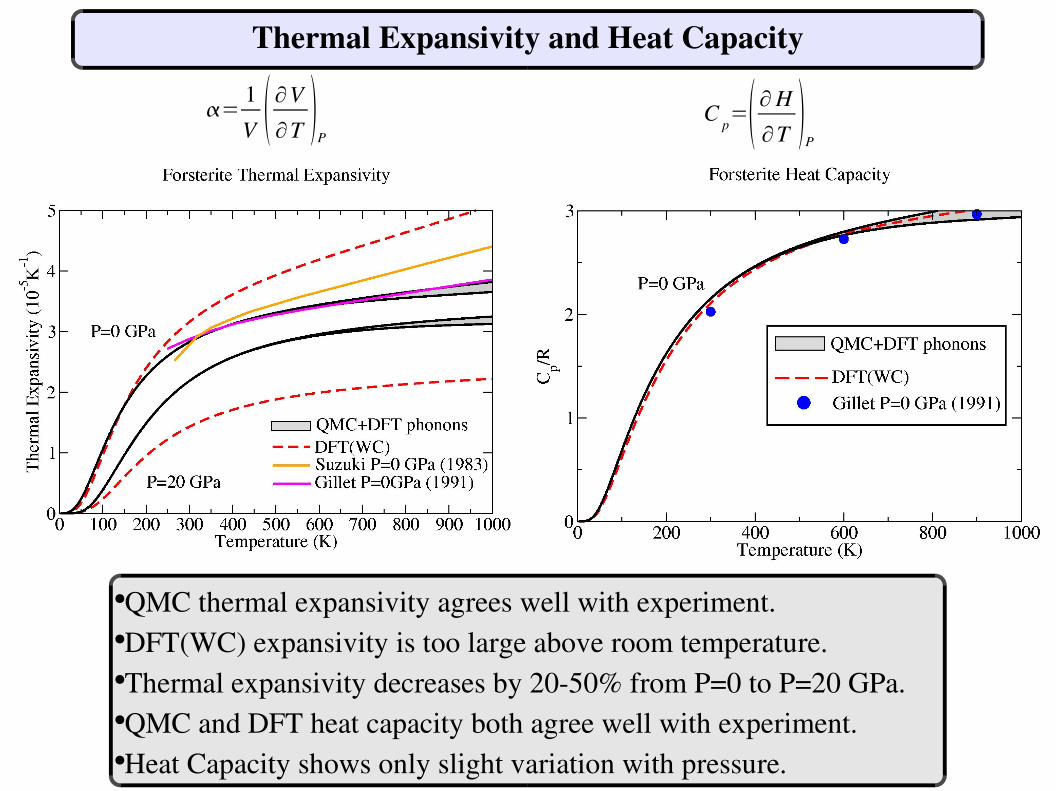
Thermal Expansivity and Heat Capacity
●QMC thermal expansivity agrees well with experiment.●DFT(WC) expansivity is too large above room temperature.●Thermal expansivity decreases by 2050% from P=0 to P=20 GPa.●QMC and DFT heat capacity both agree well with experiment.●Heat Capacity shows only slight variation with pressure.
=1V ∂V
∂T P
C p= ∂H∂T
P

Conclusions
●QMC with DFT phonons is a route to benchmark solid ab initio high P/T calculations.
●QMC solid calculations may be cumbersome and expensive with current resources.(How many DFT calculations did it take to make DFT what it is today?)
●Current QMC Mg2SiO
4 equation of state requires more work/thought to improve.
●QMC thermodynamic property variation with temperature is good despite the EoS.
●QMC thermodynamic property variation with pressure is poorly described due to poor quality of current EoS.