Embed Size (px)
Citation preview

J . Chem. SOC., Faraday Trans. I , 1988, 84(3), 751-764
Photoluminescence Properties of MgO Powders with Coordinatively Unsaturated Surface Ions
Masakazu Anpo,* Yoshiaki Yamada and Yutaka Kubokawa Department of Applied Chemistry, College of Engineering, University of Osaka
Prefecture, Sakai, Osaka 591 , Japan
Salvatore Coluccia and Adriano Zecchina Istituto di Chimica Fisica, Universita di Torino, Corso Massimo d’Azeglio 48,
10125 Torino, Italy
Michel Che Laboratoire de Rkactivitk de Surface et Structure, Universite‘ P. et M . Curie, U. A . l106-CNRSl 4 Place Jussieu, Tour 54, 7.5252 Paris Cedex 0.5, France
The photoluminescence spectrum of MgO degassed at high temperature has been reinvestigated using a standard JRC-MgO sample because the contribution of low-coordination surface sites (MgZ+,-Ot;) to the observed photoluminescence of the degassed MgO samples has been recently questioned. The JRC-MgO-I sample exhibits two different types of photoluminescence, i.e. one short-lived with a lifetime of ca. lop4 s, the other long-lived with a lifetime of 1-104 s. The effect of the degassing temperature of the sample and of added quencher molecules indicates that the short-lived photoluminescence observed under U.V. excitation is a radiative decay process from the charge-transfer-excited complex (Mgt,-O;,)* with a lower coordination number of four. However, the luminescence observed after U.V. excitation, i.e. a long-lived emission, is a radiative recombination process of photo-produced electrons and holes via defects such as F+centres. Thus, both charge transfer and defect mechanisms account for the photoluminescence of the MgO degassed at high temperatures, although the long-lived emission is not directly measured in the present work owing to its much smaller contribution.
Tench and Pottl have found that degassed high-surface-area powders such as MgO photoluminesce when excited by U.V. light with an energy (ca. 4.6 eV) much lower than the band-gap of the bulk solids (ca. 8.7 eV), although the pure solids in single-crystal form show no absorption in the near-ultraviolet, and that such photoluminescence spectra are completely quenched by the admission of air or oxygen. They have also shown that the values for the surface band-gap, Esg, for different surface planes of MgO calculated from the expression of Levine and Mark by considering the surface Madelung constant are in good agreement with the excitation energies of the photoluminescence spectra of the powders. On the other hand, Zecchina and Stone2 have found that in the U.V. reflectance spectra of well degassed powdered MgO, CaO, SrO and BaO the abnormal absorption appears at much lower frequencies than those of bulk crystals.
More extensive s t u d i e P have shown that the additional absorption and the observed short-lifetime (1 0-6-1 0-3 s) photoluminescence spectra are associated with the charge- transfer transition on the numerous incompletely coordinated oxide ions located on the surfaces of the well degassed powders
hv
(Mg2t,-O:L)+ (Mg+,,-O,,)*.
75 1
hv’
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online / Journal Homepage / Table of Contents for this issue

752 Photoluminescence of MgO Powders
Previously the observed luminescence had been explained in terms of the existence of intrinsic defects, such as small impurities, or extrinsic lattice defects such as the F+ centre, an electron trapped at a surface anion v a ~ a n c y . ~ ' ~
Recently, it has been found that the cis-trans isomerization of but-2-ene is photocatalysed on degassed MgO powders. The correlation between the intensity of photoluminescence and the rate of photocatalysed isomerization indicates that coordinatively unsaturated surface ions play a significant role in the photocatalytic activity of the degassed MgO powder catalyst^.^^^ However, almost at the same time Shvets et aL9 observed photoluminescence with thermoevacuated MgO powder and claimed that the observed photoluminescence spectra seemed to be in better agreement with the presence of surface F+ centres rather than coordinatively unsaturated surface ions.9 Although their data are very limited and not sufficient to upset the charge-transfer mechanism proposed by Tench and C01uccia,~*~ and Stone and Zecchina,, it is necessary to reconsider the mechanism for the photoluminescence of MgO powders degassed at higher temperatures.
Fortunately, we have found that the standard JRC-MgO-I catalyst exhibits two different types of photoluminescence, i.e. emissions with short and with long lifetimes, which are associated with the charge-transfer processes and with the presence of surface F+ centres, respectively. Therefore, in this paper we discuss the features and mechanisms of the observed photoluminescence of MgO powders outgassed in vacuum at various temperatures.
Experimental Samples of MgO microcrystals (JRC-MgO-I) were supplied from the Catalysis Society of Japan as a standard catalyst.'' MgO samples were commercially produced from sea water (MgO purity 99.02 YO ; major impurities Ca, Si, and Fe; B.E.T. surface area ca. 40 m2 g-l; bulk specific density 0.42 g ~ m - ~ ) and were degassed for 2 h at the desired temperature. The rate of increase of the degassing temperature was ca. 1 K min-l and ultimate pressures of ca. Pa) were attainable. The photoluminescence spectra were recorded at 293-298 K using a Shimadzu RF-50 1 spectrofluorophotometer (equipped with 500 W Xe lamp as excitation source) with a resolution of 0.3 nm, equipped with colour filters to eliminate scattered light." E.s.r. measurements were carried out at 77 K using a JES-ME-1 @-band) spectrometer. Details of the experimental procedures have been described previously. 7 9 l2
The gases, H, and CO (Takachiho Kogyo Co., 99.9%) were used without further purification and passed through a liquid-nitrogen trap before use. Commercial oxygen was purified by low- temperature distillation. Deionized double-distilled H,O was degassed by alternate freezing and thawing in vacuo.
Torr (
Results Effect of the Degassing Temperatures on the Photoluminescence Properties
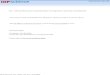
The degassed MgO sample exhibits a photoluminescence spectrum at ca. 340-450 nm when it is excited with U.V. light with 240-280 nm wavelength. Fig. 1 shows the photoluminescence spectrum of the MgO sample degassed at 873 K when it is excited at 280 and 240 nm, respectively. The photoluminescence spectrum of MgO is dependent on the excitation energy, i.e. photoluminescence recorded under excitation at 240 nm is much higher in intensity and much shorter in wavelength than that obtained at 280 nm excitation. As suggested by Coluccia13 and Anpo et aZ.,14 these results indicate that at least two different emitting sites might be involved. As will be described later, the emission observed when MgO is excited by 280 nm radiation seems to be associated with the surface OH groups;** l5 also, with this MgO sample, the photoluminescence yield
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online
M . Anpo et al. 753
wavelength/ nm
Fig. 1. Photoluminescence spectra at 298 K of the MgO sample degassed at 873 K for 2 h: (a) excitation 240& 10 nm, recording range 500 mV, (b) excitation 280+ 10 nm, recording range
50 mV.
200 250 300 300 350 400 450 500
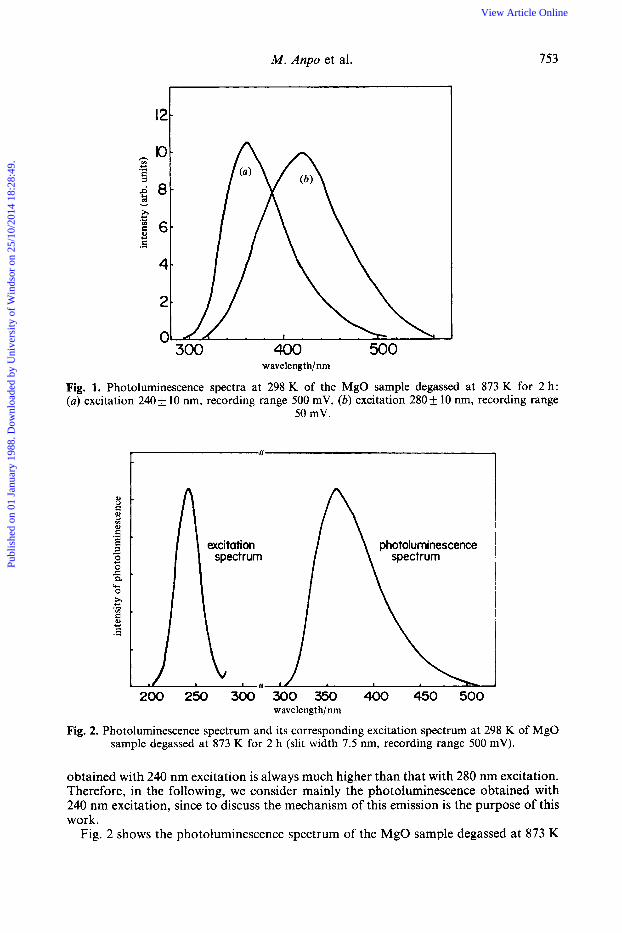
Fig. 2. Photoluminescence spectrum and its corresponding excitation spectrum at 298 K of MgO sample degassed at 873 K for 2 h (slit width 7.5 nm, recording range 500 mV).
wavelength/nm
obtained with 240 nm excitation is always much higher than that with 280 nm excitation. Therefore, in the following, we consider mainly the photoluminescence obtained with 240 nm excitation, since to discuss the mechanism of this emission is the purpose of this work.
Fig. 2 shows the photoluminescence spectrum of the MgO sample degassed at 873 K
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online
754 Photoluminescence of MgO Powders
200 250 300 350 400 450 500 wavelength/nm
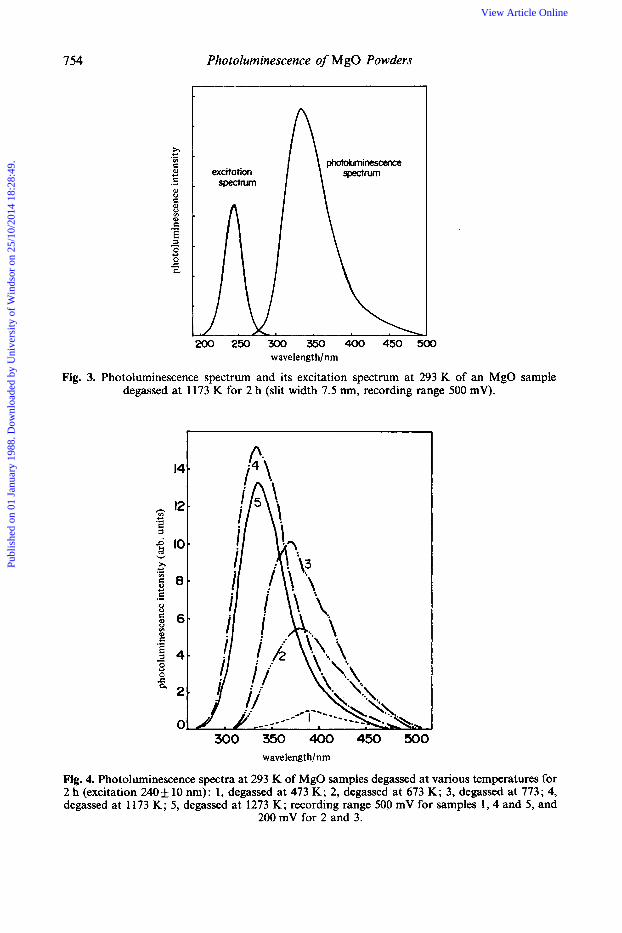
Fig. 3. Photoluminescence spectrum and its excitation spectrum at 293 K of an MgO sample degassed at 1173 K for 2 h (slit width 7.5 mn, recording range 500 mV).
300 350 400 450 500 wavelengt h/nm
Fig. 4. Photoluminescence spectra at 293 K of MgO samples degassed at various temperatures for 2 h (excitation 240& 10 nm): 1, degassed at 473 K; 2, degassed at 673 K; 3, degassed at 773; 4, degassed at 1 173 K; 5, degassed at 1273 K; recording range 500 mV for samples 1 , 4 and 5, and
200 mV for 2 and 3.
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online
M . Anpo et al. 755
273 473 673 873 1073 1273 degassing temperaturelK
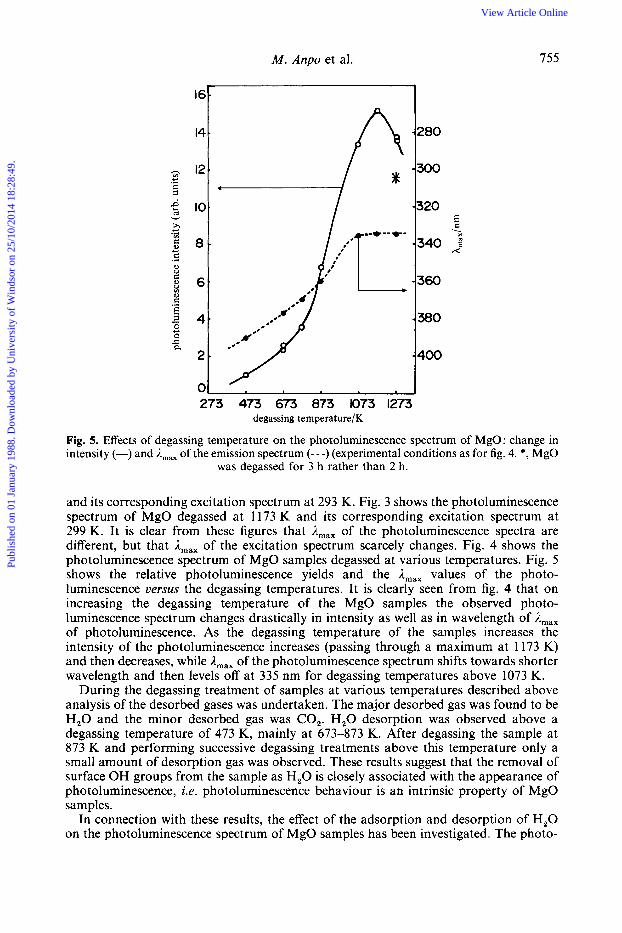
Fig. 5. Effects of degassing temperature on the photoluminescence spectrum of MgO: change in intensity (-) and A,,, of the emission spectrum (---) (experimental conditions as for fig. 4. *, MgO
was degassed for 3 h rather than 2 h.
and its corresponding excitation spectrum at 293 K. Fig. 3 shows the photoluminescence spectrum of MgO degassed at 1173 K and its corresponding excitation spectrum at 299 K. It is clear from these figures that A,,, of the photoluminescence spectra are different, but that Amax of the excitation spectrum scarcely changes. Fig. 4 shows the photoluminescence spectrum of MgO samples degassed at various temperatures. Fig. 5 shows the relative photoluminescence yields and the Amax values of the photo- luminescence versus the degassing temperatures. It is clearly seen from fig. 4 that on increasing the degassing temperature of the MgO samples the observed photo- luminescence spectrum changes drastically in intensity as well as in wavelength of A,,, of photoluminescence. As the degassing temperature of the samples increases the intensity of the photoluminescence increases (passing through a maximum at 1173 K) and then decreases, while A,,, of the photoluminescence spectrum shifts towards shorter wavelength and then levels off at 335 nm for degassing temperatures above 1073 K.
During the degassing treatment of samples at various temperatures described above analysis of the desorbed gases was undertaken. The major desorbed gas was found to be H,O and the minor desorbed gas was CO,. H,O desorption was observed above a degassing temperature of 473 K, mainly at 673-873 K. After degassing the sample at 873 K and performing successive degassing treatments above this temperature only a small amount of desorption gas was observed. These results suggest that the removal of surface OH groups from the sample as H,O is closely associated with the appearance of photoluminescence, i.e. photoluminescence behaviour is an intrinsic property of MgO samples.
In connection with these results, the effect of the adsorption and desorption of H,O on the photoluminescence spectrum of MgO samples has been investigated. The photo-
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online
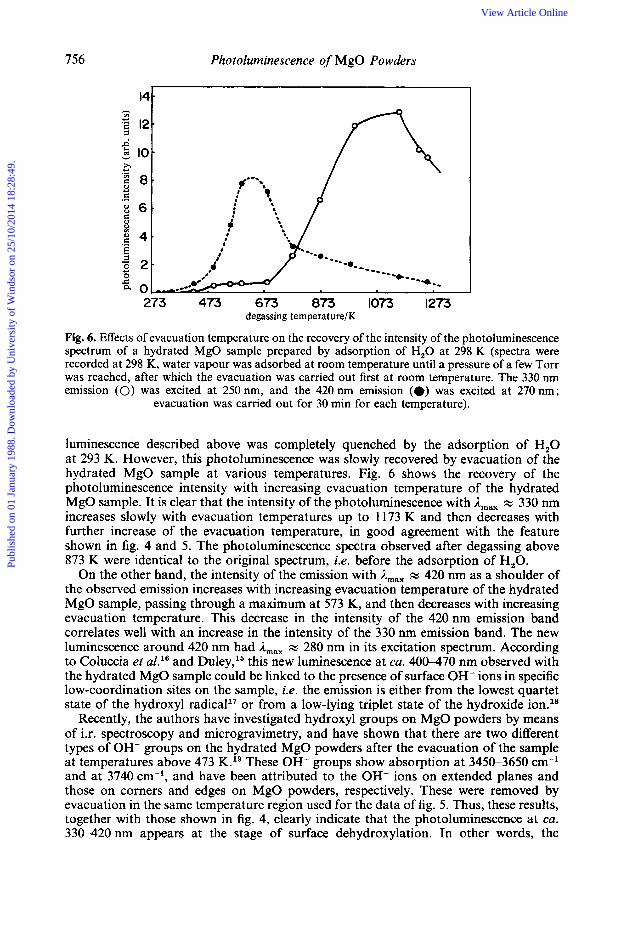
756 Photoluminescence of MgO Powders
273 473 673 073 1073 1273 degassing tempera t we/ K
Fig. 6. Effects of evacuation temperature on the recovery of the intensity of the photoluminescence spectrum of a hydrated MgO sample prepared by adsorption of H,O at 298 K (spectra were recorded at 298 K, water vapour was adsorbed at room temperature until a pressure of a few Torr was reached, after which the evacuation was carried out first at room temperature. The 330 nm emission (0) was excited at 250 nm, and the 420 nm emission (*) was excited at 270 nm;
evacuation was carried out for 30 min for each temperature).
luminescence described above was completely quenched by the adsorption of H,O at 293 K. However, this photoluminescence was slowly recovered by evacuation of the hydrated MgO sample at various temperatures. Fig. 6 shows the recovery of the photoluminescence intensity with increasing evacuation temperature of the hydrated MgO sample. It is clear that the intensity of the photoluminescence with A,,, x 330 nm increases slowly with evacuation temperatures up to 1173 K and then decreases with further increase of the evacuation temperature, in good agreement with the feature shown in fig. 4 and 5. The photoluminescence spectra observed after degassing above 873 K were identical to the original spectrum, i.e. before the adsorption of H,O.
On the other hand, the intensity of the emission with A,,, x 420 nm as a shoulder of the observed emission increases with increasing evacuation temperature of the hydrated MgO sample, passing through a maximum at 573 K, and then decreases with increasing evacuation temperature. This decrease in the intensity of the 420 nm emission band correlates well with an increase in the intensity of the 330 nm emission band. The new luminescence around 420 nm had A,,, x 280 nm in its excitation spectrum. According to Coluccia et aZ.16 and Duley,15 this new luminescence at ca. 400-470 nm observed with the hydrated MgO sample could be linked to the presence of surface OH- ions in specific low-coordination sites on the sample, i.e. the emission is either from the lowest quartet state of the hydroxyl radical1' or from a low-lying triplet state of the hydroxide ion.18
Recently, the authors have investigated hydroxyl groups on MgO powders by means of i.r. spectroscopy and microgravimetry, and have shown that there are two different types of OH- groups on the hydrated MgO powders after the evacuation of the sample at temperatures above 473 K.19 These OH- groups show absorption at 3450-3650 cm-l and at 3740 cm-l, and have been attributed to the OH- ions on extended planes and those on corners and edges on MgO powders, respectively. These were removed by evacuation in the same temperature region used for the data of fig. 5. Thus, these results, together with those shown in fig. 4, clearly indicate that the photoluminescence at ca. 330-420nm appears at the stage of surface dehydroxylation. In other words, the
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online

M . Anpo et al. 757
intrinsic surface sites associated with the photoluminescence are formed by the removal of the surface OH- groups as in the following reaction:
Garrone et aL20 have reported the results of U.V. reflectance spectroscopy which show that the bands related to the sites in a specific low-coordination state appear at the final stages of surface dehydroxylation. This is in good agreement with the results mentioned above.
Coluccia et aL21 have found that highly dispersed MgO, prepared by thermal decomposition of high-purity hydroxide or basic carbonate in vacuo, and outgassed at 1200 K for 1 h (100-200 m2 g-l), exhibits photoluminescence with A,,, = 390 nm wavelength when excited at ca. 230 nm. They have also found that the MgO sample exhibits another emission at ca. 470 nm when it is excited at 274 nm, which is easily quenched by added H,. They assigned the former excitation to the charge-transfer- excitation processes on lower-coordination surface sites, including Mgtg, 0;; and the latter on sites including Mgi;, Oi;, respectively. The JRC-MgO-I sample seems to be less stable on degassing treatment at higher temperature, 22 because after outgassing at higher temperatures the photoluminescence merely decreased without appearance of any new emission to indicate the formation of further low-coordination surface sites.
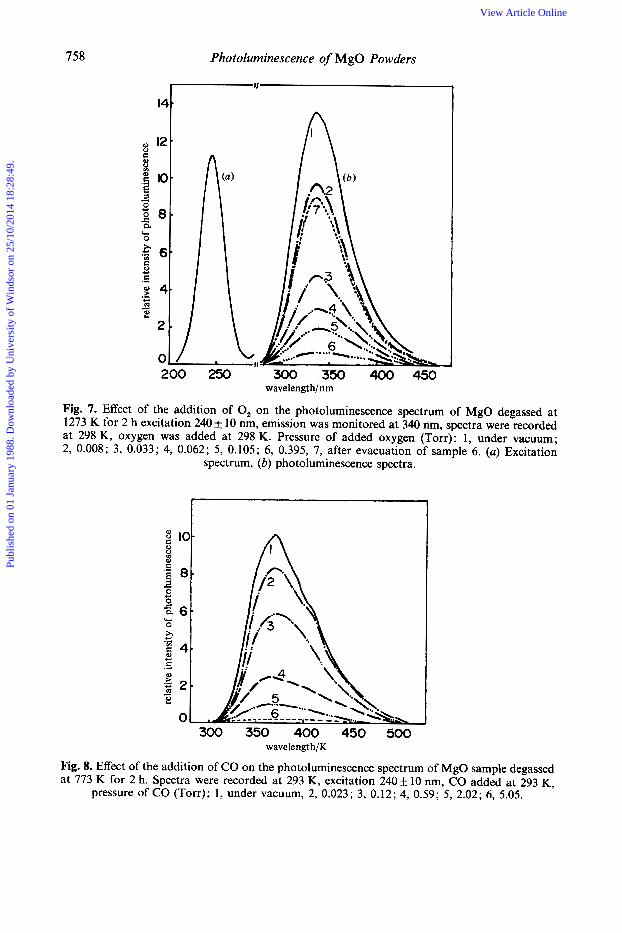
Quenching of the Photoluminescence with added O,, CO and H, As shown in fig. 7, the addition of 0, at 293-298 K onto the MgO sample which was degassed at 1273 K leads to efficient quenching of the photoluminescence intensity without any change of the shape. For example, the addition of oxygen at only 0.033 Torr quenches the photoluminescence by ca. 70%, the addition of ca. 1 Torr of oxygen leading a complete quenching of the emission. As shown in fig. 6, the evacuation of oxygen at 298 K for 20 min, after complete quenching of the emission, leads to a recovery of most of the emission, but not to complete recovery. It was found that the recovery of the emission seems to depend on the exposure time of the sample to the U.V. excitation beam in the presence of oxygen.
Fig. 8 shows the effect of the addition of CO molecules at 293 K on the photoluminescence of an MgO sample degassed at 773 K. With increasing pressure of CO the photoluminescence decreases in intensity without any change in the shape of the emission. Evacuation of the sample at 293 K for 30 min, after complete quenching of the photoluminescence with CO at ca. 5.1 Torr, leads to recovery of most of the pho t oluminescence.
Almost identical quenching of the photoluminescence with added 0, or CO molecules was observed with other MgO samples degassed at different temperatures.22 As described previous1y,2*11 two different mechanisms may be involved in the quenching: (1) collisional (or weak-interaction) quenching, whereby gaseous molecules interact with the emitting sites in their metastable excited state; (2) quenching due to the formation of an adsorbed complex between the adsorbed molecules and the excited active emitting sites with different pathways. These two mechanisms might operate in the quenching processes to give a non-radiative deactivation pathway, i.e. to enhance the intersystem crossing to the ground state. For MgO samples, the former mechanism seems to be predominant since the reversible nature of the quenching, i.e. recovery of the photoluminescence after the evacuation of the MgO sample at room temperature for 20-30 min, suggests that the added molecules interact weakly with the active surface sites on the MgO sample, except for some small part being irreversible.
As described above, the photoluminescence of the MgO degassed at high temperature is easily quenched by added quencher molecules such as 0,. Thus the photophysical
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online
rela
tive i
nten
sity
of p
hoto
lum
ines
cenc
e
:
rela
tive
inte
nsity
of
phot
olum
ines
cenc
e
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online
M . Anpo et al. 759
10 i I
8
e 8 '06
4
2
1
0 0.02 0.04 0.06 0.08 0.1 0.1; pressure of O,/Torr
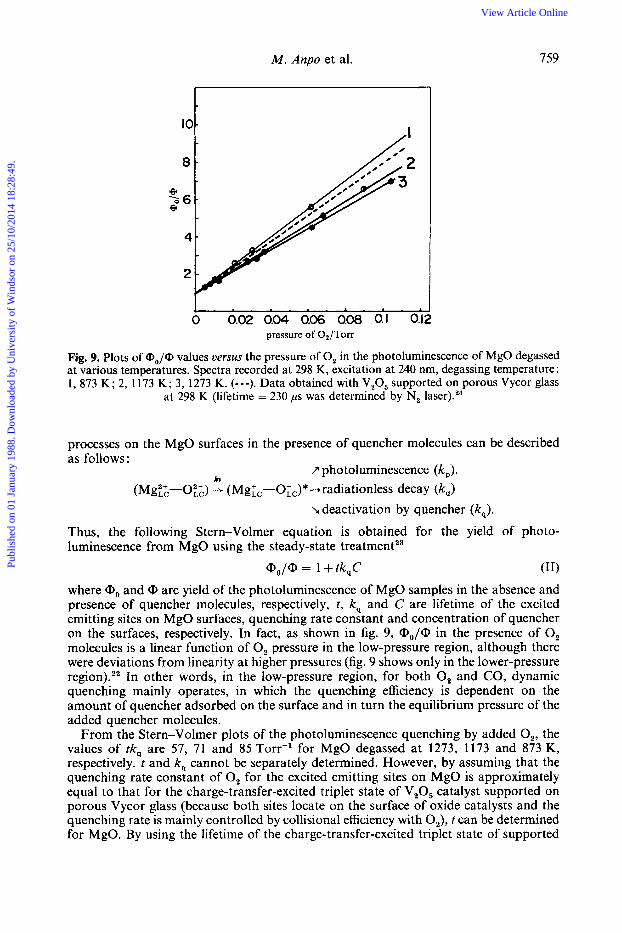
Fig. 9. Plots of (Do/@ values uerms the pressure of 0, in the photoluminescence of MgO degassed at various temperatures. Spectra recorded at 298 K, excitation at 240 nm, degassing temperature : 1, 873 K; 2, 1173 K; 3, 1273 K. (---). Data obtained with V,O, supported on porous Vycor glass
at 298 K (lifetime = 230 ps was determined by N, laser).24
processes on the MgO surfaces in the presence of quencher molecules can be described as follows:
hv 7 photoluminescence (kp).
(Mgz-0;;) -+ (MgL,-O;,)* -+ radiationless decay (k,) L deactivation by quencher (kJ .
Thus, the following Stern-Volmer equation is obtained for the yield of photo- luminescence from MgO using the steady-state treatment23
(11) Qo/@ = 1 + tk,C where <Do and <D are yield of the photoluminescence of MgO samples in the absence and presence of quencher molecules, respectively, t , k, and C are lifetime of the excited emitting sites on MgO surfaces, quenching rate constant and concentration of quencher on the surfaces, respectively. In fact, as shown in fig. 9, a,-,/@ in the presence of 0, molecules is a linear function of 0, pressure in the low-pressure region, although there were deviations from linearity at higher pressures (fig. 9 shows only in the lower-pressure region).,, In other words, in the low-pressure region, for both 0, and CO, dynamic quenching mainly operates, in which the quenching efficiency is dependent on the amount of quencher adsorbed on the surface and in turn the equilibrium pressure of the added quencher molecules.
From the Stern-Volmer plots of the photoluminescence quenching by added O,, the values of tk, are 57, 71 and 85 Torr-' for MgO degassed at 1273, 1173 and 873 K, respectively. t and k, cannot be separately determined. However, by assuming that the quenching rate constant of 0, for the excited emitting sites on MgO is approximately equal to that for the charge-transfer-excited triplet state of V,O, catalyst supported on porous Vycor glass (because both sites locate on the surface of oxide catalysts and the quenching rate is mainly controlled by collisional efficiency with 0,), t can be determined for MgO. By using the lifetime of the charge-transfer-excited triplet state of supported
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online
760 Photoluminescence of MgO Powders
U.V. light off
0 0.4 0.8 12 1.6 0 20 40 60 80 100 pressure of H2/Torr U.V. irradiation time/min
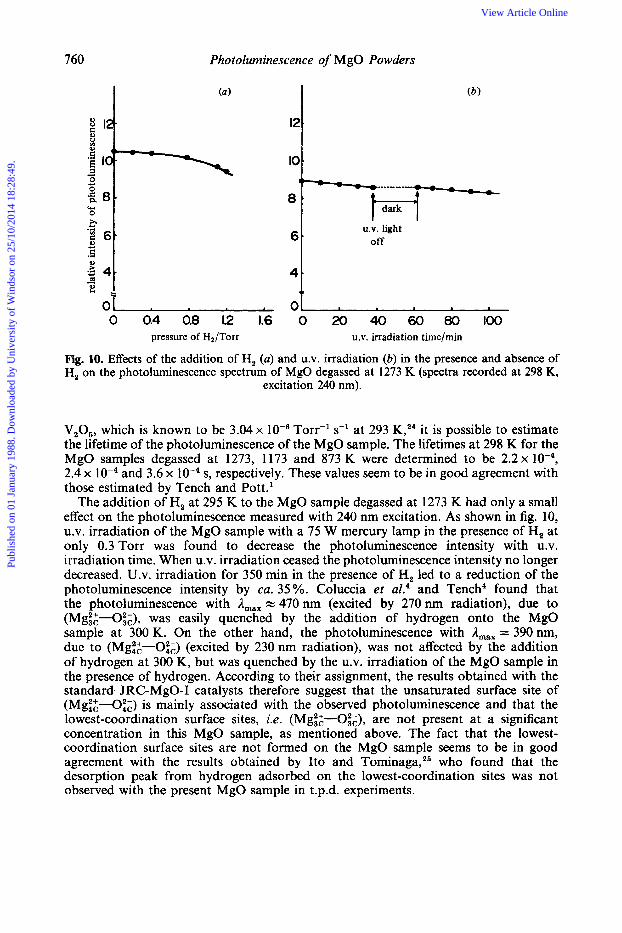
Fig. 10. Effects of the addition of H, (a) and U.V. irradiation (6) in the presence and absence of H, on the photoluminescence spectrum of MgO degassed at 1273 K (spectra recorded at 298 K,
excitation 240 nm).
V,O,, which is known to be 3.04 x T0rr-l s-l at 293 K,24 it is possible to estimate the lifetime of the photoluminescence of the MgO sample. The lifetimes at 298 K for the MgO samples degassed at 1273, 1173 and 873 K were determined to be 2.2 x lo-', 2.4 x and 3.6 x low4 s, respectively. These values seem to be in good agreement with those estimated by Tench and P0tt.l
The addition of H, at 295 K to the MgO sample degassed at 1273 K had only a small effect on the photoluminescence measured with 240 nm excitation. As shown in fig. 10, U.V. irradiation of the MgO sample with a 75 W mercury lamp in the presence of H, at only 0.3 Torr was found to decrease the photoluminescence intensity with U.V. irradiation time. When U.V. irradiation ceased the photoluminescence intensity no longer decreased. U.V. irradiation for 350 min in the presence of H, led to a reduction of the photoluminescence intensity by ca. 35 %. Coluccia et aL4 and Tench4 found that the photoluminescence with A,,, = 470 nm (excited by 270 nm radiation), due to (Mg::-Oi;), was easily quenched by the addition of hydrogen onto the MgO sample at 300 K. On the other hand, the photoluminescence with A,,, = 390 nm, due to (Mgt:-Oi;) (excited by 230 nm radiation), was not affected by the addition of hydrogen at 300 K, but was quenched by the U.V. irradiation of the MgO sample in the presence of hydrogen. According to their assignment, the results obtained with the standard JRC-MgO-I catalysts therefore suggest that the unsaturated surface site of (Mgi:-Oi;) is mainly associated with the observed photoluminescence and that the lowest-coordination surface sites, i.e. (Mg::-O&), are not present at a significant concentration in this MgO sample, as mentioned above. The fact that the lowest- coordination surface sites are not formed on the MgO sample seems to be in good agreement with the results obtained by Ito and To~n inaga ,~~ who found that the desorption peak from hydrogen adsorbed on the lowest-coordination sites was not observed with the present MgO sample in t.p.d. experiments.
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online
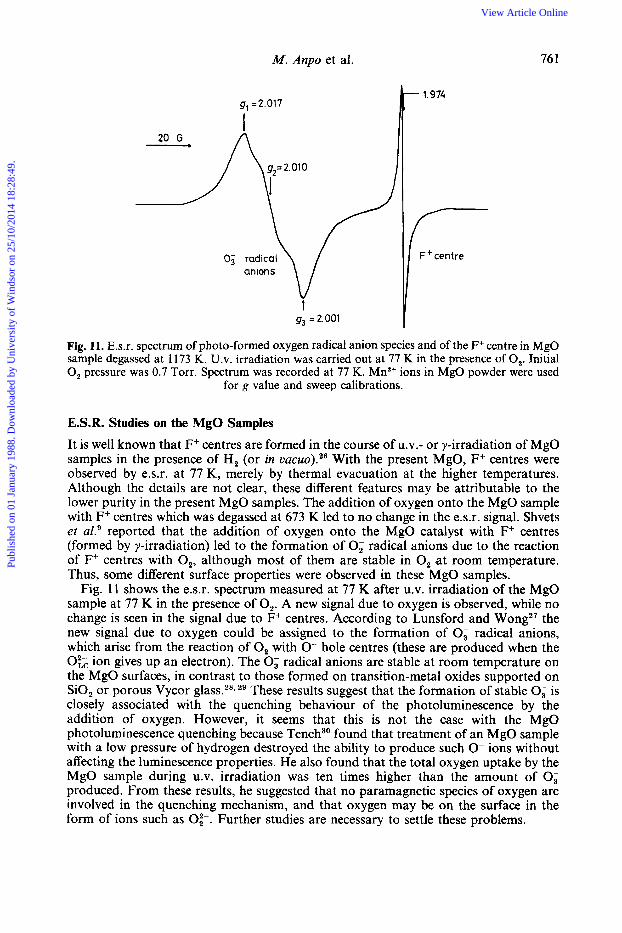
M. Anpo et al. 76 I
9, =2.017
1 1.974 t
Fig. 11. E.s.r. spectrum of photo-formed oxygen radical anion species and of the F+ centre in MgO sample degassed at 1173 K. U.V. irradiation was carried out at 77 K in the presence of 0,. Initial 0, pressure was 0.7 Torr. Spectrum was recorded at 77 K. Mn2+ ions in MgO powder were used
for g value and sweep calibrations.
E.S.R. Studies on the MgO Samples It is well known that F+ centres are formed in the course of u.v.- or y-irradiation of MgO samples in the presence of H, (or in vacuo).26 With the present MgO, F+ centres were observed by e.s.r. at 77 K, merely by thermal evacuation at the higher temperatures. Although the details are not clear, these different features may be attributable to the lower purity in the present MgO samples. The addition of oxygen onto the MgO sample with F+ centres which was degassed at 673 K led to no change in the e.s.r. signal. Shvets et a1.' reported that the addition of oxygen onto the MgO catalyst with F+ centres (formed by y-irradiation) led to the formation of 0; radical anions due to the reaction of F+ centres with 0,, although most of them are stable in 0, a t room temperature. Thus, some different surface properties were observed in these MgO samples.
Fig. 11 shows the e.s.r. spectrum measured at 77 K after U.V. irradiation of the MgO sample at 77 K in the presence of 0,. A new signal due to oxygen is observed, while no change is seen in the signal due to F+ centres. According to Lunsford and Wong2' the new signal due to oxygen could be assigned to the formation of 0; radical anions, which arise from the reaction of 0, with 0- hole centres (these are produced when the 0;; ion gives up an electron). The 0; radical anions are stable at room temperature on the MgO surfaces, in contrast to those formed on transition-metal oxides supported on SiO, or porous Vycor glass.28129 These results suggest that the formation of stable 0; is closely associated with the quenching behaviour of the photoluminescence by the addition of oxygen. However, it seems that this is not the case with the MgO photoluminescence quenching because Tench3' found that treatment of an MgO sample with a low pressure of hydrogen destroyed the ability to produce such 0- ions without affecting the luminescence properties. He also found that the total oxygen uptake by the MgO sample during U.V. irradiation was ten times higher than the amount of 0; produced. From these results, he suggested that no paramagnetic species of oxygen are involved in the quenching mechanism, and that oxygen may be on the surface in the form of ions such as 0;-. Further studies are necessary to settle these problems.
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online
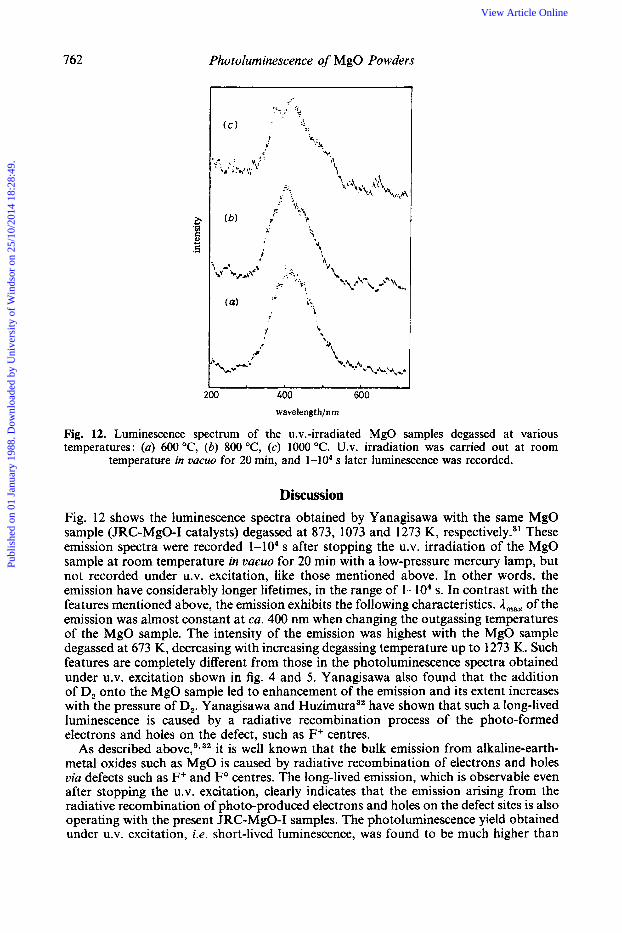
762 Photoluminescence of MgO Powders
.-...
i I ,\
0 400 600 wavelengt h/nm
Fig. 12. Luminescence spectrum of the u.v.-irradiated MgO samples degassed at various temperatures: (a) 600 "C, (b) 800 "C, (c) 1000 "C. U.V. irradiation was carried out at room
temperature in UQCUO for 20 min, and 1-104 s later luminescence was recorded.
Discussion Fig. 12 shows the luminescence spectra obtained by Yanagisawa with the same MgO sample (JRC-MgO-I catalysts) degassed at 873, 1073 and 1273 K, re~pectively.~~ These emission spectra were recorded 1-104 s after stopping the U.V. irradiation of the MgO sample at room temperature in vacuo for 20 min with a low-pressure mercury lamp, but not recorded under U.V. excitation, like those mentioned above. In other words, the emission have considerably longer lifetimes, in the range of 1-104 s. In contrast with the features mentioned above, the emission exhibits the following characteristics. A,,, of the emission was almost constant at ca. 400 nm when changing the outgassing temperatures of the MgO sample. The intensity of the emission was highest with the MgO sample degassed at 673 K, decreasing with increasing degassing temperature up to 1273 K. Such features are completely different from those in the photoluminescence spectra obtained under U.V. excitation shown in fig. 4 and 5. Yanagisawa also found that the addition of D, onto the MgO sample led to enhancement of the emission and its extent increases with the pressure of D,. Yanagisawa and H ~ z i m u r a ~ ~ have shown that such a long-lived luminescence is caused by a radiative recombination process of the photo-formed electrons and holes on the defect, such as F+ centres.
it is well known that the bulk emission from alkaline-earth- metal oxides such as MgO is caused by radiative recombination of electrons and holes via defects such as F+ and Fo centres. The long-lived emission, which is observable even after stopping the U.V. excitation, clearly indicates that the emission arising from the radiative recombination of photo-produced electrons and holes on the defect sites is also operating with the present JRC-MgO-I samples. The photoluminescence yield obtained under U.V. excitation, i.e. short-lived luminescence, was found to be much higher than
As described
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online

M . Anpo et al. 763
that of the emission observed after stopping the U.V. irradiation, i.e. long-lived emission, by ca. two orders of magnitude.33 This result suggests that the former process is much more important than the latter for the deactivation of the excited states of MgO powders.
Thus, all these results clearly show that there are two different radiative processes for the degassed JRC-MgO sample. The first radiative process is very sensitive to the added 0, and CO molecules but not to added H, molecules, and is only observable under U.V. excitation, i.e. its lifetime is very short and in the range of lop4 s. The second radiative process is rather insensitive to the added 0, and CO molecules (but added hydrogen enhances the intensity) and is observable even after stopping U.V. irradiation, i.e. its lifetime is long and in the range 1-104 s.
Shvets et al.’ observed the photoluminescence with the thermoevacuated powder MgO and claimed that the data are shown to be in better agreement with the attribution of the luminescence at 415 nm to surface F+ centres, rather than to coordinatively unsaturated surface ions, although their data are not better than those reported by Tench and coworker^.^'^ They reported that the adsorption of H, or 0, at room temperature and subsequent outgassing for 15-20 min did not change the luminescence spectrum. Their results are completely different from those obtained by Tench and c o ~ o r k e r s ~ * ~ and those obtained in the present work.? They explained that the disagreement with the results obtained by Tench and Coluccia could be caused by impurities in the H, used by them. However, such an explanation is not only unsound but is also untrue, as indicated in the present work. Although it is not possible to discuss the results reported by Shvets et al.’ because of a lack of experimental detail, it seems that they have observed only the long-lived luminescence, but not the short-lived photoluminescence. As a result, the data obtained by Shvets et al.’ might show better agreement with the defect luminescence mechanism. It seems likely that the yields of the short- and long-lived photoluminescence would change from sample to sample and could depend on the pretreatment of the samples.
It is well known that the lower the coordination around a surface oxide ion, the lower will be the frequency of the absorption of the charge-transfer transition associated with it. This is because the Madelung potential progressively decreases as the extent of unsaturation increases. According to Tench and coworker^^.^ the photoluminescences observed at ca. 390 and 470 nm are due to the radiative decay processes of the absorbed photon energy on the coordinatively unsaturated surface site of (Mg,2;-0:;) and that of (Mgi:--Ot;), respectively. With the present work, the photoluminescence at around 390-330 nm (excited at around 240 nm), which could be attributable to a radiative decay process associating with the surface unsaturated sites of (Mg:g-O:;), is observed, and the other photoluminescence, which is attributed to the presence of surface OH groups, is also observed at around 430nm (excited at around 280nm). However, photo- luminescence which is attributable to the radiative decay process associating with the much lower coordination site, (Mgi:-Oi,) was not observed, even with the MgO sample degassed at 1273 K. As described above, the MgO sample used in the present study is known not to be stable for degassing at higher temperatures. Therefore, the reason why the photoluminescence associating with the ( M g ~ ~ - O ~ J is not observed with this MgO might be connected with this instability of the sample. Further studies are in progress using other MgO samples involving a standard JRC-MgO-11.
M.A. thanks Prof. H. Hattori of Hokkaido University and Prof. T. Hattori of Nagoya University for supplying standard JRC-MgO samples, and Prof. Y.
The spectra obtained by Shvets et d’seem to be in better agreement with those obtained by Tench and coworker^^-^ than with the present work. This may be because spectra from the sample used in the present work are more sensitive to excitation energy and pretreatment of the sample.
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online

764 Photoluminescence of MgO Powders
Yanagisawa for his permission to use his data before publication. Thanks are due to The Ministry of Education of Japan (Grant-in-aid for Scientific Research, grant nos. 59470007, 59550558, and 61223022). M.A. thanks UniversitC P. et M. Curie for a position as an invited Professor at Paris, and Universita di Torino for an invitation to visit Torino.
References I A. J. Tench and G. T. Pott, Chem. Phys. Lett., 1974, 26, 590. 2 A. Zecchina, M. G. Lofthouse and F. S. Stone, J. Chem. SOC., Faraday Trans. I , 1975,71, 1476; F. S.
Stone and A. Zecchina, Proc. 6th Int. Congr. Catal. (The Chemical Society, London, 1976), A8; A. Zecchina and F. S . Stone, J. Chem. SOC., Faraday Trans. I , 1976, 72, 2364; 1978, 74, 2278.
3 S. Coluccia, J. F. Hemidy and A. J. Tench, J. Chem. SOC., Faraday Trans. I , 1978, 74, 2763. 4 S. Coluccia, M. Deane and A. J. Tench, J. Chem. SOC., Faraday Trans. I , 1978, 74, 2913; S. Coluccia
and A. J. Tench, Proc. 7th Int. Congr. Catal. (Kodansha, Tokyo, 1981), B, 1154. 5 J. Cunningham, in Comprehensive Chemical Kinetics, ed. C. H. Bamford and R. G. Compton (Elsevier,
Amsterdam, 1984), chap. 3; J. Nuran, J. Cunningham, A. M. Deane, E. A. Colbourn and W. C. Mackrodt, in Adsorption and Catalysis on Oxide Surfaces, ed. M. Che and G. C. Bond (Elsevier, Amsterdam, 1985), p. 83; J. Cunningham and C. P. Healy, J. Chem. SOC., Faraday Trans. 1, 1987,83, 2973.
6 M. Che, in Adrorption and Catalysis on Oxide Surfaces, ed. M. Che and G. C. Bond (Elsevier, Amsterdam, 1985), p. 11; M. Che and A. J. Tench, A h . Catal., 1982, 31, 77.
7 M. Anpo, Y. Yamada and Y. Kubokawa, J. Chem. SOC., Chem. Commun., 1986, 714. 8 M. Anpo and Y. Yamada, in Advances in Basic Solid Materials, ed. K. Tanabe (Elsevier, Sequoia,
1987), and unpublished data. 9 V. A. Shvets, A. V. Kuznetsov, V. A. Fenin and V. B. Kazansky, J. Chem. SOC., Faraday Trans. I ,
1985, 81, 2913. 10 A standard JRC-MgO-I sample was supplied from Catalysis Society of Japan. Various data about this
MgO sample are available from a Data Book of 9th Reference Meeting of Catalysis Society of Japan, (Toyama, Tokyo, 1985).
11 M. Anpo, C. Yun and Y. Kubokawa, J. Chem. SOC., Faraday Trans. I , 1980, 76, 1014. 12 M. Anpo, Y. Yamada and Y. Kubokawa, in a Data Book of 9th Reference Meeting of Catalysis Society
13 S . Coluccia, in Adsorption and Catalysis on Oxide Surfaces, ed. M. Che and G. C. Bond (Elsevier,
14 M. Anpo, M. Kondo, Y. Kubokawa, C. Louis, M. Che and S. Coluccia, Chem. Expr., 1987,2,61, the
15 W. W. Duley, J. Chem. SOC., Faraday Trans. 1, 1984, 80, 1173. 16 S. Coluccia, M. Deane and A. J. Tench, Proc. 6th Int. Congr. Catal. (The Chemical Society, London,
17 H. J. Maria and S. P. McGlynn, J. Chem. Phys., 1970, 52, 3402. 18 P. B. Merkel and W. H. Hamill, J. Chem. Phys., 1971, 55, 2174. 19 S. Coluccia, L. Marchese, S. Lavagnino and M. Anpo, Spectrochim. Acta, 1987, in press. 20 E. Garrone, A. Zecchina and F. S. Stone, Philos. Mag., Sect. B, 1980, 42, 683. 21 S. Coluccia, A. J. Tench and R. L. Segall, J. Chem. SOC., Faraday Trans. I , 1979, 75, 1769. 22 M. Anpo and Y. Yamada, unpublished data. 23 N. J. Turro, in Modern Molecular Photochemistry (Benjamin/Cummings, Menlo Park, 1978). 24 M. Anpo, I. Tanahashi and Y. Kubokawa, J. Phys. Chem., 1982, 86, 1; M. Anpo, T. Suzuki,
25 T. Ito and N. Tominaga, in a Data Book of 9th Reference Meeting of Catalysis Society of Japan
26 R. L. Nelson, A. J. Tench and B. J. Harmsworth, Trans. Faraday SOC., 1967, 63, 1427; R. L. Nelson
27 J. H. Lunsford, Catal. Rev., 1973, 8, 135; N. B. Wong and J. H. Lunsford, J. Phys. Chem., 1966, 44,
28 M. Anpo, N. Aikawa, Y. Kubokawa, M. Che, C. Louis and E. Giamello, J. Phys. Chem., 1985, 89,
29 M. Anpo, T. Fujii, S. Suzuki and Y. Kubokawa, J. Phys. Chem., 1984, 88, 2572. 30 A. J. Tench, Proc. 6th Int. Congr. Catal. (The Chemical Society, London, 1976), p. 182. 31 Y. Yanagisawa, in a Data Book of 9th Reference Meeting of Catalysis Society of Japan (Toyama,
32 Y. Yanagisawa and R. Huzimura, J. Phys. SOC. Jpn, 1984, 53, 66. 33 M. Anpo and Y. Yanagisawa, unpublished data.
of Japan, (Toyama, Tokyo, 1985), p. 25.
Amsterdam, 1985), p. 59.
revised manuscript was submitted to J, Phys. Chem.
1976), A9.
I. Tanahashi, M. Kondo and Y. Kubokawa, Shokubai (Catalysis), 1986, 28, 68.
(Toyama, Tokyo, 1985), p. 21.
and J. W. Hale, Discuss. Faraday SOC., 1971, 52, 77.
1487.
5689; M. Anpo, unpublished data.
Tokyo, 1985), p. 29.
Paper 71293; 17th February, 1987
Publ
ishe
d on
01
Janu
ary
1988
. Dow
nloa
ded
by U
nive
rsity
of
Win
dsor
on
25/1
0/20
14 1
8:28
:49.
View Article Online