Embed Size (px)
Citation preview

Novel Vector Design and Hexosaminidase Variant EnablingSelf-Complementary Adeno-Associated Virus for the Treatmentof Tay-Sachs Disease
Subha Karumuthil-Melethil,1 Sahana Nagabhushan Kalburgi,1 Patrick Thompson,2
Michael Tropak,3 Michael D. Kaytor,4 John G. Keimel,4 Brian L. Mark,5 Don Mahuran,3,6
Jagdeep S. Walia,2 and Steven J. Gray1,7,*1Gene Therapy Center, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina; 2Medical Genetics/Departments of Pediatrics, Queen’s University,
Kingston, Ontario, Canada; 3Genetics and Genome Biology, SickKids, Toronto, Ontario, Canada; 4New Hope Research Foundation, North Oaks, Minnesota;5Department of Microbiology, University of Manitoba, Winnipeg, Manitoba, Canada; 6Department of Laboratory Medicine and Pathology, University of Toronto,
Toronto, Ontario, Canada; and 7Department of Ophthalmology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina.
GM2 gangliosidosis is a family of three genetic neurodegenerative disorders caused by the accumulation ofGM2 ganglioside (GM2) in neuronal tissue. Two of these are due to the deficiency of the heterodimeric (a–b),‘‘A’’ isoenzyme of lysosomal b-hexosaminidase (HexA). Mutations in the a-subunit (encoded by HEXA) leadto Tay-Sachs disease (TSD), whereas mutations in the b-subunit (encoded by HEXB) lead to Sandhoffdisease (SD). The third form results from a deficiency of the GM2 activator protein (GM2AP), a substrate-specific cofactor for HexA. In their infantile, acute forms, these diseases rapidly progress with mental andpsychomotor deterioration resulting in death by approximately 4 years of age. After gene transfer thatoverexpresses one of the deficient subunits, the amount of HexA heterodimer formed would empirically belimited by the availability of the other endogenous Hex subunit. The present study used a new variant ofthe human HexA a-subunit, l, incorporating critical sequences from the b-subunit that produce a stablehomodimer (HexM) and promote functional interactions with the GM2AP– GM2 complex. We report thedesign of a compact adeno-associated viral (AAV) genome using a synthetic promoter–intron combinationto allow self-complementary (sc) packaging of the HEXM gene. Also, a previously published capsid mu-tant, AAV9.47, was used to deliver the gene to brain and spinal cord while having restricted biodis-tribution to the liver. The novel capsid and cassette design combination was characterized in vivo in TSDmice for its ability to efficiently transduce cells in the central nervous system when delivered intrave-nously in both adult and neonatal mice. This study demonstrates that the modified HexM is capable ofdegrading long-standing GM2 storage in mice, and it further demonstrates the potential of this novelscAAV vector design to facilitate widespread distribution of the HEXM gene or potentially other similar-sized genes to the nervous system.
INTRODUCTION
TAY-SACHS DISEASE (TSD) is one of a group ofthree lysosomal storage diseases called GM2 gang-liosidosis, caused by defective catabolism of GM2
ganglioside (GM2). It is an autosomal recessiveneurodegenerative disease caused by mutationsin the a-subunit of the heterodimeric enzyme b-hexosaminidase A (HexA). Sandhoff disease (SD)is caused by mutations in the b-subunit of HexA.Both subunits can also form functional homo-dimers, HexS (a–a) and HexB (b–b), which in
humans cannot significantly turn over GM2. HexSis unstable and physiologically less significant,whereas HexB is highly stable and can degradesome neutral glycolipids, oligosaccharides, and ar-tificial substrates. The enzyme HexA (a–b) requiresa substrate-specific cofactor, called GM2 activatorprotein (GM2AP), to efficiently degrade GM2. Muta-tions to the gene encoding GM2AP lead to anotherrare variant of GM2 gangliosidosis, the AB-variantform. The characteristic histopathological feature ofthese diseases is the presence of swollen neurons
*Correspondence: Dr. Steven J. Gray, UNC Gene Therapy Center, 7119 Thurston Bowles Building, Campus Box 7352, Chapel Hill, NC 27599. E-mail: [email protected]
HUMAN GENE THERAPY, VOLUME 27 NUMBER 7 DOI: 10.1089/hum.2016.013 j 509ª 2016 by Mary Ann Liebert, Inc.

and neuronal distortion throughout the nervoussystem caused by accumulated GM2. TSD and SDare clinically variable, with severe forms showingprofound mental retardation and death within 2–3years of birth.1 Heterogeneity of the disease re-garding severity of clinical signs and age of onsetcan be attributed to the level of residual HexA ac-tivity allowed by different mutations. It has beenestimated that only between 10 and 15% of normalHexA activity is needed to prevent GM2 accumula-tion.2 At present, there are no effective treatmentoptions available for these diseases.
To study the disease mechanisms and to testvarious therapeutic approaches, mouse models havebeen developed for both these diseases. The mousemodel for TSD, developed by the targeted disruptionof the hexa gene encoding the a-subunit,3–5 has no ora mild behavioral phenotype but displays many ofthe neuropathological features characteristic ofTSD, in particular GM2 ganglioside accumulation.The absence of clinical signs in TSD mice is due tothe presence of a lysosomal mouse sialidase capableof hydrolyzing GM2 to its neutral asialo derivative,GA2, which then interacts with HexB to be furtherdegraded to glucosylceramide.5,6 This alternativemetabolic pathway helps TSD mice to keep GM2 ac-cumulation below an acutely toxic level and therebyescape clinical symptoms. The hexb–/– mouse modelfor Sandhoff disease exhibits a severe disease phe-notype as it is deficient in both HexA and HexB andhas extensive GM2 accumulation throughout thebrain and spinal cord with a humane end point ofapproximately 16 weeks of age.4,5
Several gene replacement therapies have beentested in the mouse models for these diseases. Theheterodimeric nature of HexA, which is the onlyisoenzyme capable of degrading GM2, makes thetreatment for TSD and SD more challenging. Ade-noviral gene therapy of TSD mice, using a vectorencoding HEXA alone, when injected intravenouslyproduced only a slight increase in HexA (or possiblyHexS mistaken for HexA) activity in the serum,whereas simultaneous injection of vectors encodingboth HEXA and HEXB increased the serum HexAactivity to 42% of wild-type levels in these mice.7 SDmice injected intracranially with a recombinantadeno-associated viral (rAAV) vector expressing theb-subunit, or a combination of vectors with a- andb-subunits, showed reduction in GM2 levels and in-creased survival.8 However, the effect of the single-subunit vector in this study could be attributed tohigher levels of HexB and the mouse sialidasedriving the alternative metabolic pathway forhydrolyzing GM2. A single striatal injection of amixture of vectors expressing the b-hexosaminidase
a- and b-subunits in SD mice prevented the neuronalloss in the brain.9 Cachon-Gonzalez and colleaguesreported that intracranial coinjection of rAAV vec-tors expressing the a- and b-subunits into 1-month-old SD mice prevented the disease pathologythroughout the brain and spinal cord, which helpedthe mice survive up to 2 years.10 In that study, aninfusion of an rAAV vector encoding the b-subunitalone resulted in detectable expression of only theHexB isozyme. AAVrh8 vectors encoding feline a-and b-subunits were injected intracranially into SDcats at a 1:1 ratio, and this resulted in a significantreduction in cerebrospinal fluid (CSF) biomarkers ofthe disease. It ultimately resulted in extension of thelife span for the treated cats.11 Intracranial stereo-taxic infusion of a mixture of rAAV2/1a and rAAV2/1b into 4-week-old SD mice extended the mediansurvival from 131 to 615 days.12
The gene replacement therapies so far discussedall used direct injections to the brain, using viralvectors. The possibility of translating this highly in-vasive delivery approach into humans is problematicsimply given the size differential between the brainsof animal models and humans, along with the limitedspread of AAV vectors from the injection site. Theemergence of AAV9 vectors for global gene transferinto the brain and spinal cord, due to its ability topenetrate the blood–brain barrier, has resulted intherapeutic approaches for multiple CNS diseasesvia intravascular gene delivery.13–17 Intravenousadministration of self-complementary AAV9 vectorshas been demonstrated as a viable and translatableapproach to target the CNS in mice, cats, and non-human primates,18–21 and this approach is beingapplied in a human trial for spinal muscular atrophy(clinicaltrials.gov identifier NCT02122952). Thesestudies demonstrating the ability of AAV9 to achievewidespread CNS transduction all relied on the moreefficient, self-complementary AAV (scAAV), vectordesign. scAAV vectors have been shown to be greaterthan 10-fold more efficient in transducing cells,compared with traditional single-stranded AAV(ssAAV) vectors when tested in vivo.18,22 The draw-back of using an scAAV vector is the restricted pack-aging capacity of approximately 2.2 kb of foreignDNA,making itdifficult topackage largergenesalongwith regulatory expression elements. In a review byPowell and colleagues,23 they listed the relativestrength and size of various promoters and poly-adenylation [poly(A)] signals. Among the widely usedubiquitous promoters, the cytomegalovirus (CMV)early enhancer/chicken b-actin (CBA or CAGGS)promoter (1600 bp), the related hybrid chickenb-actin(CBh) promoter (800 bp), and the CMV enhancer/promoter (800 bp) are all too large to accommodate a
510 KARUMUTHIL-MELETHIL ET AL.

HEX gene in an scAAV vector. Small enhancerlesspromoters such as UbC, MeCP2, and GUSB couldaccommodate the size of the HEX gene, but wouldlikely provide inadequate levels of expression. Atpresent, no publication has described an scAAVvector design capable of packaging the approxima-tely 1.6-kb HEXA or HEXB gene to treat TSD or SD.
An ideal vector capable of treating TSD and SDwould express both HEXA and HEXB genes simul-taneously, but this would be beyond the packagingcapacity of scAAV, which is needed to achieve effi-cient global CNS gene transfer. Therefore, there is aconceptual advantage to creating an engineeredsubunit that could be delivered, using a single scAAVvector, and that could form a stable and functionalhomodimer when delivered in vivo and degradeGM2 in a GM2AP-dependent manner. The crystallo-graphic structures of human HexA and HexB havebeen elucidated in detail.24,25 Matsuoka and col-leagues attempted to design and develop a modifiedHexB reported to have GM2-degrading activity andGM2AP-binding ability, based on the amino acid ho-mology between the Hex a- and b-subunits.26 Theypurified the modified HexB recombinant enzymewith the altered substrate specificity, that is, theability to efficiently bind negatively charged sub-strates, and an a-loop sequence for enhancingits interaction with the GM2AP–GM2 complex.They administered the modified HexB by in-tracerebroventricular injection directly to theCNS of 10-week-old SD mice and observed a re-markable reduction in GM2 storage in the thalamus,cerebral cortex, and also the liver. Their resultsindicated that the modified HexB enzyme can func-tion in vivo if used in enzyme replacement therapy(ERT). However, Sinici and colleagues27 tested thisconstruct and another more extensive hybrid b-subunit construct in cellulo, using Tay-Sachs cellspreloaded with a fluorescent GM2 derivative, andfound that neither modified b-subunit wascapable ofsignificant human GM2AP-dependent hydrolysis ofGM2. Likely, the encouraging results reported byMatsuoka and colleagues are attributable to resto-ration of HexB-like activity that takes advantage ofthe alternative sialidase pathway in mice. As analternative approach to develop a hybrid subunit,Tropak and colleagues28 designed a modified a-subunit incorporating both the unique b-subunitsequences required to form a stable, HexB-like,homodimer and those sequences needed for thehomodimer to interact with the GM2AP–GM2 com-plex. The a-active site was retained in order tofacilitate the hydrolysis of negatively charged sub-strates, for example, GM2. This modified subunit (l)was confirmed to form a stable dimeric enzyme,
HexM, which could bind to the GM2AP–GM2 complex,and degrade GM2 in live human embryonic kidney(HEK) cells rendered HEXA–/– and HEXB–/– byCRISPR (clustered regularly interspaced short pal-indromic repeats) gene editing. In vivo experimentsin this study further demonstrated the ability ofHexM to reduce accumulated GM2 in Tay-Sachs andSandhoff mice. Therefore, this homodimer has HexA-like activity and can be expressed from a single genepackageable within an scAAV vector. This constructis discussed further in the present study.
It is important to note that systemic delivery ofAAV9 vectors results in substantially higher biodis-tribution to the liver compared with the CNS, whichcould lead to transgene-specific liver toxicity.15,29
Furthermore, some studies have reported incidenceof liver tumors with intravenous delivery of AAV9into neonates.14,30,31 A liver-detargeted version ofAAV9 with comparable levels of brain and spinal cordtransduction could be a safer reagent to treat neuro-logical disorders. Pulicherla and colleagues32 re-ported the generation of AAV9-derived vectorsdisplaying selective loss of liver tropism. One AAV9variant, denoted as AAV9.47 with four point muta-tions (S414N, G453D, K557E, and T582I), showed asignificant 110-fold decrease in the liver transductionin mice injected via the tail vein and analyzed 4 weekspostinjection by vector DNA biodistribution andtransgene (luciferase) expression. The transductionpattern in thebraindidnotshowanysignificantdrop,which suggests AAV9.47 as a good candidate for glo-bal gene delivery to the CNS via systemic injection.
In the present study, we discuss the functionalability of the new hexosaminidase variant HexMand the design of a novel expression cassette topackage it into an scAAV vector for in vivo applica-tions. We show that the novel cassette has the abilityto provide ubiquitous transduction throughout theCNS. We provide further characterization of theliver-detargeted AAV9.47 variant to demonstrateits ability to transduce neurons and glia throughoutthe CNS after intravenous injection into neonataland adult mice. We demonstrate that the HexMhomodimer is capable of degrading GM2 in a local-ized manner when injected intracranially intoadult TSD mice, and that the scAAV9.47/HEXMvector can mediate long-term GM2 reduction wheninjected intravenously into neonatal TSD mice.
MATERIALS AND METHODSPlasmid constructs
The plasmids carrying the transgene cassetterequired for packaging into scAAV vectors weredesigned for GFP, HEXA, and HEXM. This novel
GENE THERAPY VECTOR IMPROVEMENTS FOR TSD 511

design includes the combination of a short syntheticand ubiquitous promoter derived from the JeTpromoter33 and a synthetic intron (UsP, also re-ferred to as the JeTI promoter) along with the sim-ian virus 40 polyadenylation signal [SV40 poly(A)]to drive expression of the encoded gene (Fig. 1). AKozak consensus sequence was incorporated at thestart codon. Pilot in vitro studies showed that theUsP expression cassette had approximately 15% ofthe activity when using a CMV promoter (Supple-mentary Fig. S1; supplementary methods and dataareavailableonlineatwww.liebertpub.com/hum).Thenucleotide sequence of the synthetic JeT promoterand intron is provided in Supplementary Fig. S2.
The human HEXA and HEXM genes were codon-optimized by DNA2.0 (Menlo Park, CA). The con-struct expressing GFP from the CBh promoter22
with the bovine growth hormone (BGH) poly(A)was used as a reference standard for comparativegene expression for the newly designed constructs.
Vector preparationRecombinant AAV vectors were generated as
described,34 using proprietary methods developed atthe University of North Carolina Gene TherapyCenter Vector Core facility (Chapel Hill, NC). Thefollowing AAV9 or AAV9.47 vectors were producedand used for this study: AAV9/CBh-GFP, AAV9.47/CBh-GFP, AAV9/UsP-GFP, AAV9/UsP-HEXA, AAV9/UsP-HEXM, AAV9.47/UsP-GFP, and AAV9.47/UsP-HEXM. All of these were scAAV vectors. Peak frac-tions were dialyzed in phosphate-buffered saline(PBS) containing 5% D-sorbitol and 350 mM NaCl.Viral titers were determined by qPCR as described.35
Mouse studiesAll investigations were approved by the Uni-
versity of North Carolina–Chapel Hill Institutional
Animal Care and Use Committee. The Tay-Sachsdisease mouse model [strain B6;129S-hexa–/–]3 andwild-type C57BL/6 mice were purchased from theJackson Laboratory (Bar Harbor, ME) and bred atthe University of North Carolina.
For intravenous delivery into wild-type adult fe-male C57BL/6 mice, 200 ll of scAAV9/GFP (2 · 1011
VG) or AAV9.47/GFP (2 · 1011 VG) was injected andexpression/biodistribution was analyzed 3–4 weekspostinjection. Adult TSD knockout mice were in-jected via the tail vein with scAAV9.47/GFP (5 · 1011
VG) in a volume of 200 ll when 4–5 months old andwere analyzed 6 months postinjection.
Neonatal facial vein injections into TSD micewere performed on postnatal days 0–2 (P0–P2), us-ing scAAV9.47/UsP-GFP (5 · 1010 VG) or scAAV9.47/UsP-HEXM (5 · 1010 VG) in a volume of 20ll. Theinjections were carried out before the genotypes ofthe mice were determined and as such included bothheterozygous and knockout mice. They were eutha-nized when 15 months old.
Intracranial injections were performed as de-scribed35 into 15-month-old TSD knockout mice,using the following stereotaxic coordinates, relativeto bregma: rostral/caudal, +0.5 mm (toward rostral);left/right: left, +3 mm; up/down, -4 mm. These micereceived a mixture of AAV9/GFP (1 · 109 VG) andeither AAV9/HEXM (1 · 109 VG) or AAV9/HEXA(1 · 109 VG) in a total volume of 1 ll delivered over10 min. Two mice were injected per vector mix-ture. They were analyzed 4 weeks postinjection.At sacrifice, the mice were transcardially perfusedwith PBS containing heparin at 1 U/ml (Abraxis,Schaumburg, IL) and samples were collected fordownstream analysis.
ImmunohistochemistryAfter 48 hr of fixation in freshly made PBS con-
taining 4% paraformaldehyde, the entire brain andportions of the cervical and lumbar spinal cords weresectioned at 40lm, using a Leica vibrating micro-tome. Every fifth section was processed for immu-nohistochemistry (IHC). Sections were treated withhydrogen peroxide to remove endogenous peroxi-dase activity before blocking. Samples were incu-bated for 1 hr at room temperature in blockingsolution (10% goat serum, 0.1% Triton X-100,1 · PBS) and then incubated for 48–72 hr at 4�C inprimary antibody solution (3% goat serum, 0.1%Triton X-100, 1 · PBS, and primary antibody). Pri-mary antibodies were as follows: rabbit anti-GFP(diluted 1:1000; #AB3080, Millipore, Bedford, MA)or the humananti-GM2 gangliosideantibody (diluted1:1000; KM966, gift from Kyowa Hakko Kirin, To-kyo, Japan). After incubating with the appropriate
Figure 1. Adeno-associated viral (AAV) construct diagrams. The expres-sion cassette design for packaging GFP and codon-optimized versions ofhuman HEXA and HEXM included a novel synthetic promoter–intron com-bination (UsP) along with the simian virus 40 polyadenylation signal (SV40polyA) to drive expression. These elements are flanked by the mutant in-verted terminal repeat (ITR) on the 5¢ end and the wild-type (WT) AAV2 ITRon the 3¢ end to package into a self-complementary AAV vector. Colorimages available online at www.liebertpub.com/hum
512 KARUMUTHIL-MELETHIL ET AL.

secondary antibody for 1 hr at room temperature,color development was performed with a VECTAS-TAIN Elite ABC kit (#PK-6100; Vector Laboratories,Burlingame, CA) with 3,3¢-diaminobenzidine tetra-chloride (DAB) (#04008; Polysciences, Warrington,PA) substrate and nickel–cobalt intensification ofthe reaction product.
DAB-processed brain sections were digitized witha ScanScope slide scanner (Aperio Technologies,Vista, CA). Virtual slides were viewed with theImageScope software package (version 10.0; AperioTechnologies).
For the data provided in SupplementaryFig. S3B, a region of interest was created in threerepresentative images of an identical cortex area ofthe brain per vector treatment, and the number ofastrocytes and neurons (identified by morphology)were manually counted. The results from two in-dependent counters were averaged. Data are ex-pressed as number of cells per millimeter squared.
Quantitative PCR analysis of vector genomesQuantitative PCR (qPCR) was used for vector bio-
distribution studies. Tissue DNA was purified andquantified as described.18 Data are reported as thenumber of double-stranded GFP or HEXM DNAmolecules per two double-stranded copies of the mu-rine LaminB2 locus, or in other words, the number ofvector DNA copies per diploid mouse genome.
Biochemical detection of GM2 gangliosidesGangliosides were extracted by a modification of
a method described earlier.36 Briefly, previouslyfrozen murine mid-brain sections were weighedand sonicated in 0.5 ml of methanol at amplitude15–20% for 3 · 10 sec pulses with cooling on icebetween pulses (Sonic Dismembrator model 500;Fisher Scientific, Waltham, MA). Samples werecentrifuged for 15 min at 10,000 rpm at 4�C, and0.4 ml of supernatant was removed for enzyme ac-tivity studies; the remainder was suspended (in-cluding the pellet) up to a final volume of 1.5 ml ofmethanol added to 2.5 ml of chloroform. Acidic gan-gliosides were isolated using C18-E columns (Phe-nomenex, Torrance, CA) and eluted sequentially in2 ml of methanol, and then in 2 ml of a 1:1 methanol–chloroform mixture.The eluate was then evaporatedunder a stream of nitrogen, and the residue was re-suspended in 1:1 methanol–chloroform and thenapplied to an HPTLC silica gel 60 plate (Merck,Darmstadt, Germany) by applying 10ll at a timeand drying between applications. Plates were de-veloped with running buffer (chloroform–methanol–CaCl2, 0.22% [55:45:10]). After staining with orcinol(Sigma-Aldrich, St. Louis, MO) and baking at 105�C
for 10 min, plates were immediately scanned forsubsequent spot densitometric analysis (ImageJsoftware), with a mix of monosialogangliosidestandards (Matreya, Pleasant Gap, PA) alongside.The ganglioside ratios are presented as GM2:totalgangliosides.
RESULTSLiver-detargeted AAV9.47 capsid transducesneural cells after intravenous administrationin adult mice
AAV9.47 capsid was engineered as described.32
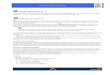
It was categorized as a liver-detargeted version ofthe AAV9 capsid, with a significant 110-fold de-crease in liver transduction relative to the natu-rally occurring AAV9 capsid. The level of overallbrain transduction was comparable to that of theoriginal AAV9 capsid, making it a good candidatefor targeted delivery to brain and other CNS organsvia the intravenous route while avoiding the highbiodistribution of AAV9 to the liver. The liver-detargeted characteristics of the AAV9.47 vectorreported previously were confirmed in our study bya biodistribution study (Supplementary Fig. S3A).However, previous studies did not identify whichcell types were transduced by AAV9.47 within theCNS. In our study, we observed that the capsidis able to transduce neural cells, predominantlyneurons, when delivered intravenously into adultwild-type mice (Fig. 2B and Supplementary Fig. S4).GFP expression was visible throughout various re-gions of the brain and spinal cord. When comparedwith the transduction profile of AAV9 capsid pack-aged with the same transgene cassette containingGFP under the control of the CBh promoter andinjected into wild-type mice (Fig. 2A), there was anapproximately 4-fold reduction in the number ofastrocytes transduced by the AAV9.47 capsid andan approximately 50% increase in the number oftransduced neurons in the cortex (SupplementaryFig. S3B). Overall, AAV9.47 transduced approxi-mately 20% less cells than AAV9 in the cortex.
Synthetic UsP promoter–intron drivesubiquitous expression in the brain
Previously, our laboratory described the use ofa modified b-actin promoter with a shortenedMVM (minute virus of mice) intron to drive sus-tained high levels of transgene expression inboth neurons and glia throughout the CNS.22
Combined with the BGH poly(A) signal, thisscAAV construct can package a transgene of upto approximately 1.2 kb in length. The HEXA andHEXM transgenes are approximately 1.6 kb inlength, requiring a novel expression cassette to
GENE THERAPY VECTOR IMPROVEMENTS FOR TSD 513
provide efficient and ubiquitous expression withinthe contexts of an scAAV genome. The newly de-signed expression cassette (Fig. 1), using the syn-thetic UsP promoter–intron combination, pairedwith the SV40 poly(A) signal, is capable of wide-spread expression in the CNS (Fig. 2C and Sup-plementary Fig. S4) after intravenous delivery inwild-type mice. The intensity of GFP expression inastrocytes is noticeably decreased for the UsP-GFPconstruct, compared with the CBh-GFP construct,when both were packaged in the AAV9 capsid.However, the novel design has the advantage of
being capable of packaging a transgene up to ap-proximately 1.7 kb within an scAAV vector, whichwill accommodate the HEXA or HEXM transgene.
AAV9.47 capsid is capable of widespreadgene transfer to the brain after intravenousadministration in both adult and neonatalTay-Sachs mice
To assess the ability of the UsP construct tomediate long-term sustained expression in theTay-Sachs mouse model, we packaged the newlydesigned UsP-GFP construct within the AAV9.47
Figure 2. CNS transduction pattern of AAV9/CBh-GFP, AAV9.47/CBh-GFP, and AAV9/UsP-GFP after intravenous administration in wild-type mice. Adult wild-type mice were injected intravenously with 2 · 1011 vector genomes (VG) of scAAV9/CBh-GFP (A), scAAV9.47/CBh-GFP (B), or scAAV9/UsP-GFP (C) and thenwere euthanized 3 weeks postinjection to assess green fluorescent protein (GFP) transduction levels in various parts of the brain and spinal cord. Scale bar:500 lm. Insets: An additional · 5 magnification. All images are representative of at least three mice.
514 KARUMUTHIL-MELETHIL ET AL.
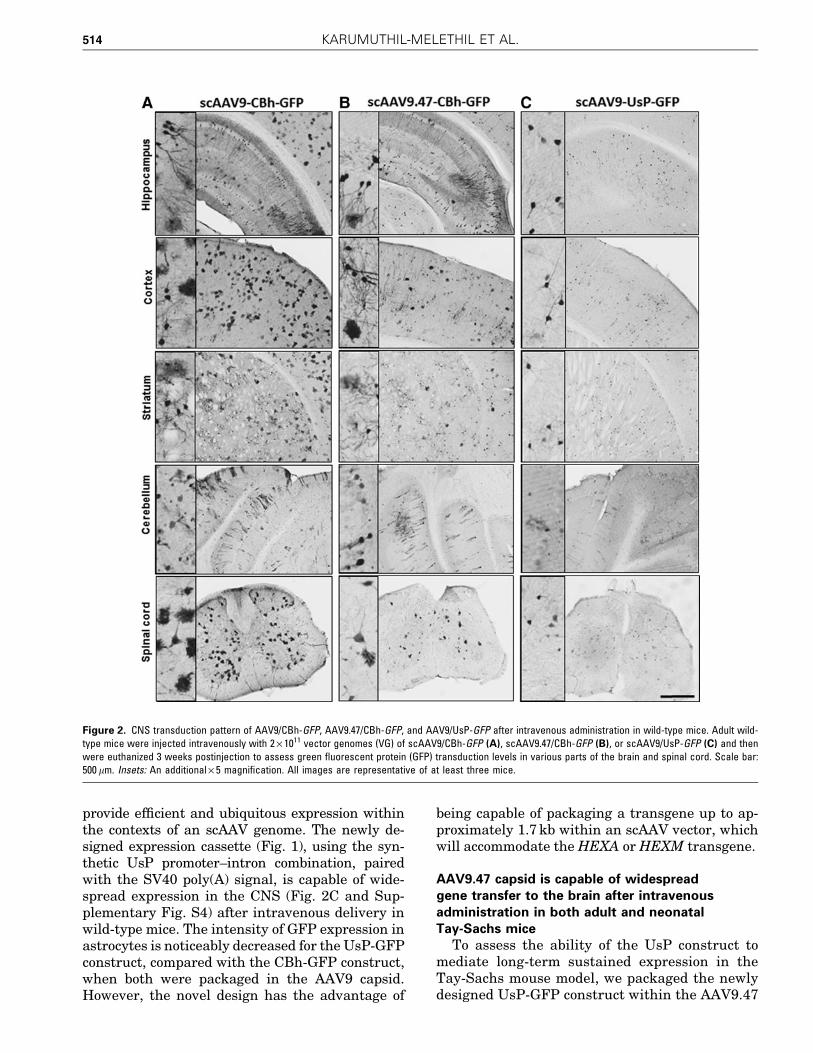
capsid and injected it intravenously into adultTSD mice at 4–5 months of age and into neonatalmice, consisting of both TSD and their heterozy-gous littermates, at age P0–P2. These mice wereeuthanized 6 and 15 months postinjection, re-spectively, and were analyzed for the biodis-tribution of the AAV vector by qPCR and for CNStransduction by IHC against GFP. As previouslyreported, the vector was significantly detargetedfrom the liver compared with the biodistributionpattern of AAV9,18,32 while retaining biodistribu-tion to the brain and spinal cord (Figs. 3A and 4A).IHC images of the brain clearly showed that theAAV9.47 capsid can deliver the gene to all regionsof brain and facilitate expression in neurons and
glia within the CNS of hexa–/– and hexa+/– mice,and that this expression persisted for 15 months(Figs. 3B and 4B).
The hexosaminidase variant, HexM, can clearGM2 storage similar to HexA after intracranialadministration into aged Tay-Sachs mice
To directly test the expression of the new hexos-aminidase gene variant, HEXM, to reduce long-standing accumulated GM2 in the TSD mouse brain,compared with that of the unmodified HEXA gene,these were packaged into scAAV9 vectors and in-jected stereotaxically into 15-month-old TSD micealong with an identical titer of scAAV9/UsP-GFPvector to track vector spread. Each experimental
Figure 3. Biodistribution and brain transduction after intravenous administration of AAV9.47/UsP-GFP in adult Tay-Sachs mice. Adult Tay-Sachs knockoutmice were injected intravenously when 4–5 months old with 5 · 1011 VG of scAAV9.47/UsP-GFP and analyzed 6 months postinjection. (A) The average numberof GFP vector genomes in major organs from six mice. Error bars indicate the SEM. (B) Immunohistochemical (IHC) images from various regions of the brainfrom TSD mice showing GFP expression. Scale bar: 500 lm. Insets: An additional · 5 magnification. The images are representative of four mice.
GENE THERAPY VECTOR IMPROVEMENTS FOR TSD 515
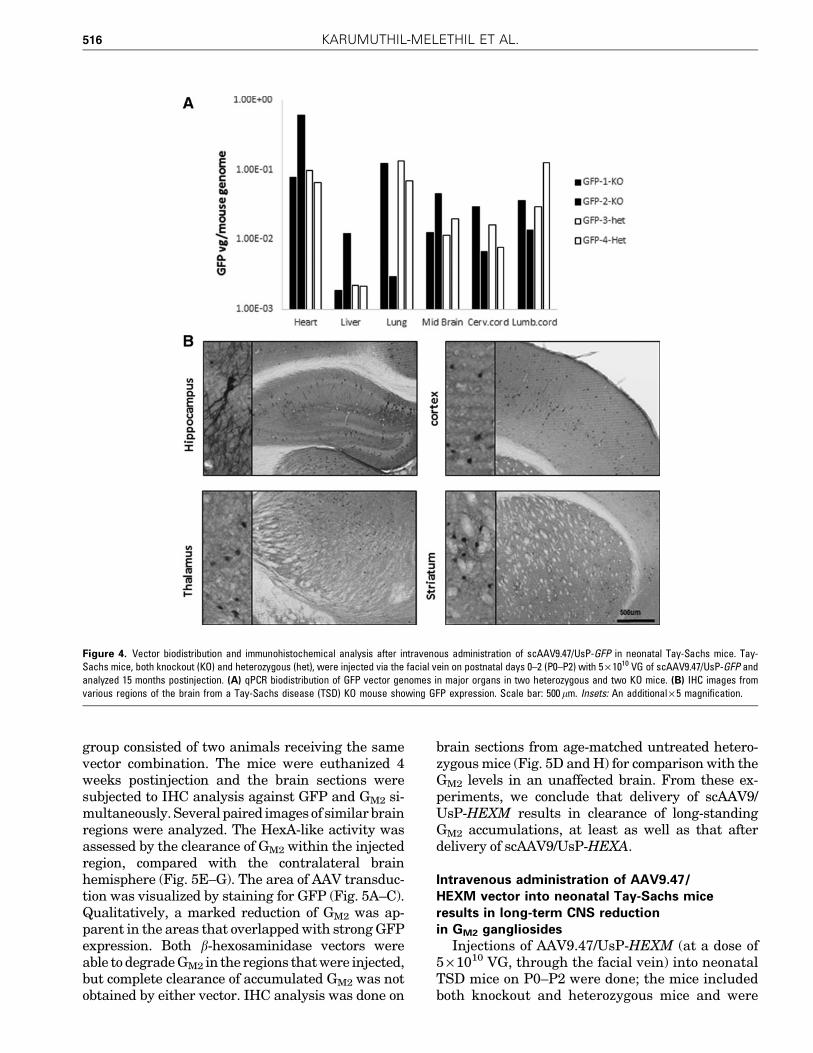
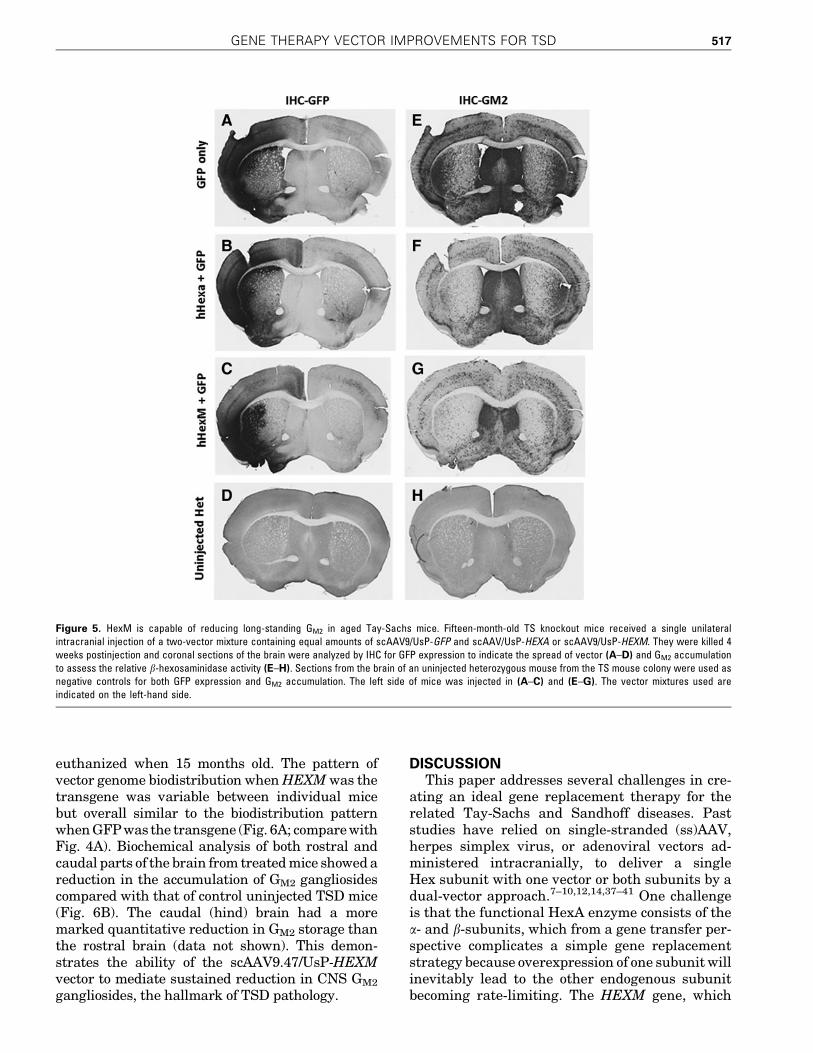
group consisted of two animals receiving the samevector combination. The mice were euthanized 4weeks postinjection and the brain sections weresubjected to IHC analysis against GFP and GM2 si-multaneously. Several paired images of similar brainregions were analyzed. The HexA-like activity wasassessed by the clearance of GM2 within the injectedregion, compared with the contralateral brainhemisphere (Fig. 5E–G). The area of AAV transduc-tion was visualized by staining for GFP (Fig. 5A–C).Qualitatively, a marked reduction of GM2 was ap-parent in the areas that overlapped with strong GFPexpression. Both b-hexosaminidase vectors wereable to degrade GM2 in the regions that were injected,but complete clearance of accumulated GM2 was notobtained by either vector. IHC analysis was done on
brain sections from age-matched untreated hetero-zygous mice (Fig. 5D and H) for comparison with theGM2 levels in an unaffected brain. From these ex-periments, we conclude that delivery of scAAV9/UsP-HEXM results in clearance of long-standingGM2 accumulations, at least as well as that afterdelivery of scAAV9/UsP-HEXA.
Intravenous administration of AAV9.47/HEXM vector into neonatal Tay-Sachs miceresults in long-term CNS reductionin GM2 gangliosides
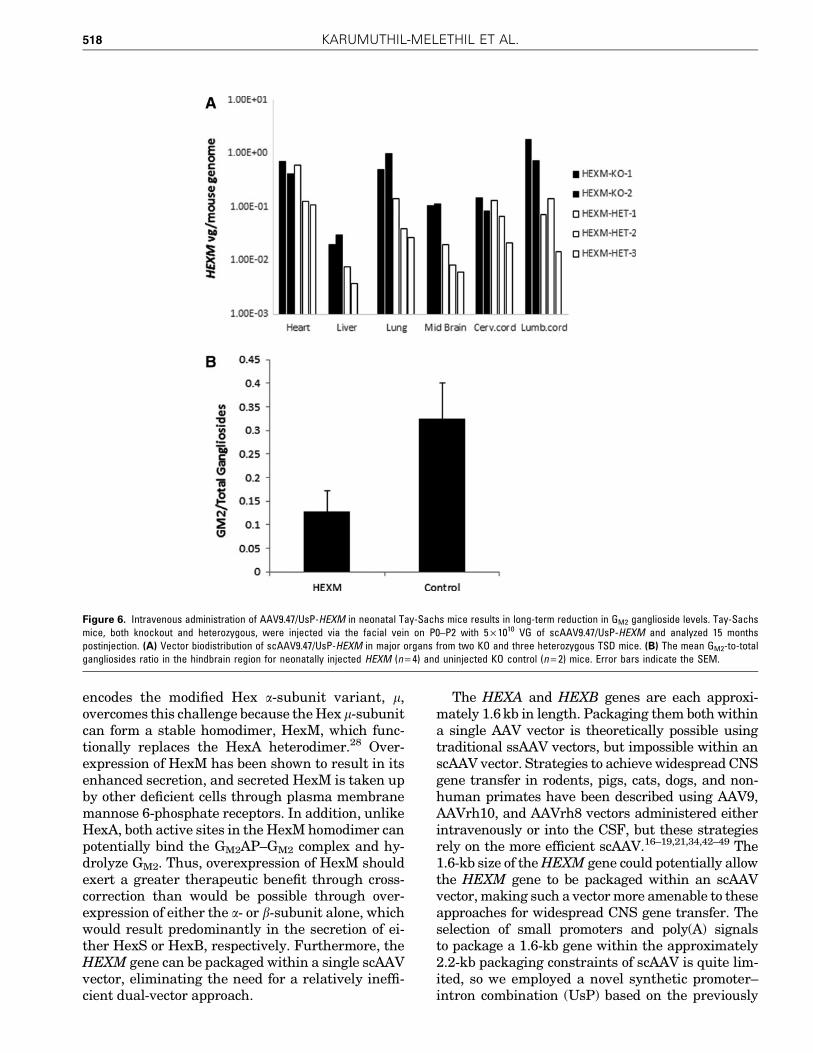
Injections of AAV9.47/UsP-HEXM (at a dose of5 · 1010 VG, through the facial vein) into neonatalTSD mice on P0–P2 were done; the mice includedboth knockout and heterozygous mice and were
Figure 4. Vector biodistribution and immunohistochemical analysis after intravenous administration of scAAV9.47/UsP-GFP in neonatal Tay-Sachs mice. Tay-Sachs mice, both knockout (KO) and heterozygous (het), were injected via the facial vein on postnatal days 0–2 (P0–P2) with 5 · 1010 VG of scAAV9.47/UsP-GFP andanalyzed 15 months postinjection. (A) qPCR biodistribution of GFP vector genomes in major organs in two heterozygous and two KO mice. (B) IHC images fromvarious regions of the brain from a Tay-Sachs disease (TSD) KO mouse showing GFP expression. Scale bar: 500 lm. Insets: An additional · 5 magnification.
516 KARUMUTHIL-MELETHIL ET AL.
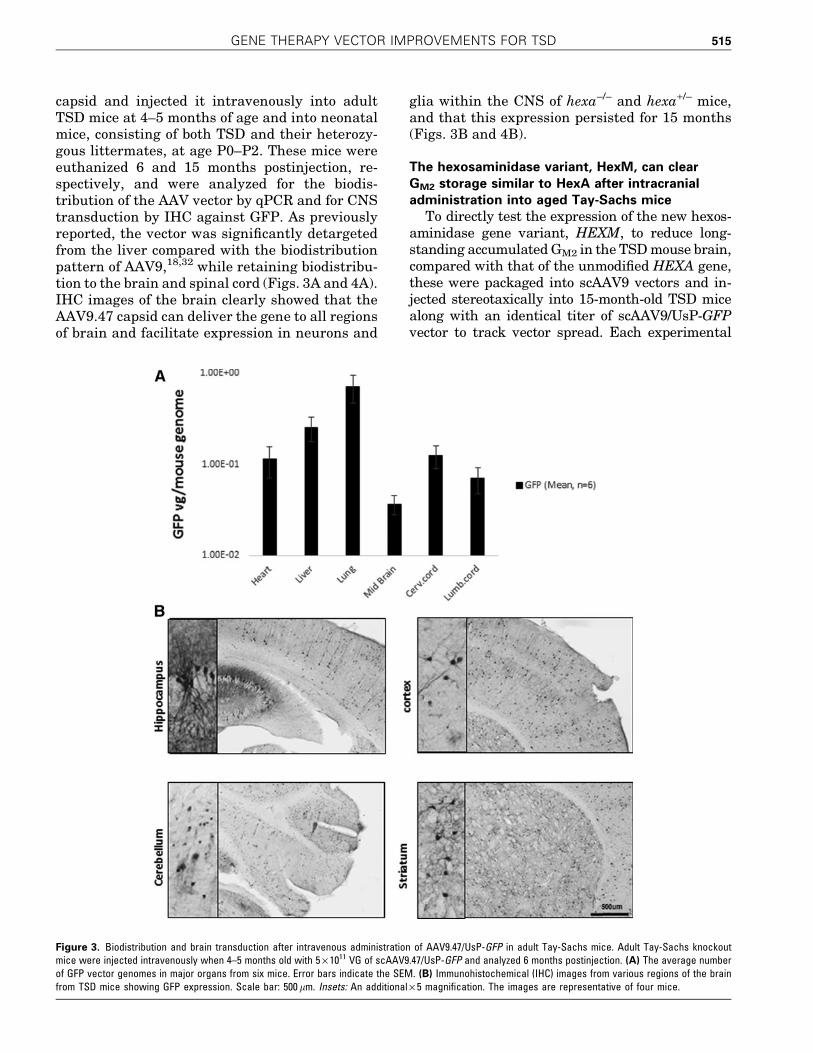
euthanized when 15 months old. The pattern ofvector genome biodistribution when HEXM was thetransgene was variable between individual micebut overall similar to the biodistribution patternwhen GFP was the transgene (Fig. 6A; compare withFig. 4A). Biochemical analysis of both rostral andcaudal parts of the brain from treated mice showed areduction in the accumulation of GM2 gangliosidescompared with that of control uninjected TSD mice(Fig. 6B). The caudal (hind) brain had a moremarked quantitative reduction in GM2 storage thanthe rostral brain (data not shown). This demon-strates the ability of the scAAV9.47/UsP-HEXMvector to mediate sustained reduction in CNS GM2
gangliosides, the hallmark of TSD pathology.
DISCUSSIONThis paper addresses several challenges in cre-
ating an ideal gene replacement therapy for therelated Tay-Sachs and Sandhoff diseases. Paststudies have relied on single-stranded (ss)AAV,herpes simplex virus, or adenoviral vectors ad-ministered intracranially, to deliver a singleHex subunit with one vector or both subunits by adual-vector approach.7–10,12,14,37–41 One challengeis that the functional HexA enzyme consists of thea- and b-subunits, which from a gene transfer per-spective complicates a simple gene replacementstrategy because overexpression of one subunit willinevitably lead to the other endogenous subunitbecoming rate-limiting. The HEXM gene, which
Figure 5. HexM is capable of reducing long-standing GM2 in aged Tay-Sachs mice. Fifteen-month-old TS knockout mice received a single unilateralintracranial injection of a two-vector mixture containing equal amounts of scAAV9/UsP-GFP and scAAV/UsP-HEXA or scAAV9/UsP-HEXM. They were killed 4weeks postinjection and coronal sections of the brain were analyzed by IHC for GFP expression to indicate the spread of vector (A–D) and GM2 accumulationto assess the relative b-hexosaminidase activity (E–H). Sections from the brain of an uninjected heterozygous mouse from the TS mouse colony were used asnegative controls for both GFP expression and GM2 accumulation. The left side of mice was injected in (A–C) and (E–G). The vector mixtures used areindicated on the left-hand side.
GENE THERAPY VECTOR IMPROVEMENTS FOR TSD 517
encodes the modified Hex a-subunit variant, l,overcomes this challenge because the Hex l-subunitcan form a stable homodimer, HexM, which func-tionally replaces the HexA heterodimer.28 Over-expression of HexM has been shown to result in itsenhanced secretion, and secreted HexM is taken upby other deficient cells through plasma membranemannose 6-phosphate receptors. In addition, unlikeHexA, both active sites in the HexM homodimer canpotentially bind the GM2AP–GM2 complex and hy-drolyze GM2. Thus, overexpression of HexM shouldexert a greater therapeutic benefit through cross-correction than would be possible through over-expression of either the a- or b-subunit alone, whichwould result predominantly in the secretion of ei-ther HexS or HexB, respectively. Furthermore, theHEXM gene can be packaged within a single scAAVvector, eliminating the need for a relatively ineffi-cient dual-vector approach.
The HEXA and HEXB genes are each approxi-mately 1.6 kb in length. Packaging them both withina single AAV vector is theoretically possible usingtraditional ssAAV vectors, but impossible within anscAAV vector. Strategies to achieve widespread CNSgene transfer in rodents, pigs, cats, dogs, and non-human primates have been described using AAV9,AAVrh10, and AAVrh8 vectors administered eitherintravenously or into the CSF, but these strategiesrely on the more efficient scAAV.16–19,21,34,42–49 The1.6-kb size of the HEXM gene could potentially allowthe HEXM gene to be packaged within an scAAVvector, making such a vector more amenable to theseapproaches for widespread CNS gene transfer. Theselection of small promoters and poly(A) signalsto package a 1.6-kb gene within the approximately2.2-kb packaging constraints of scAAV is quite lim-ited, so we employed a novel synthetic promoter–intron combination (UsP) based on the previously
Figure 6. Intravenous administration of AAV9.47/UsP-HEXM in neonatal Tay-Sachs mice results in long-term reduction in GM2 ganglioside levels. Tay-Sachsmice, both knockout and heterozygous, were injected via the facial vein on P0–P2 with 5 · 1010 VG of scAAV9.47/UsP-HEXM and analyzed 15 monthspostinjection. (A) Vector biodistribution of scAAV9.47/UsP-HEXM in major organs from two KO and three heterozygous TSD mice. (B) The mean GM2-to-totalgangliosides ratio in the hindbrain region for neonatally injected HEXM (n = 4) and uninjected KO control (n = 2) mice. Error bars indicate the SEM.
518 KARUMUTHIL-MELETHIL ET AL.

described synthetic JeT promoter,33 to provide amoderate level of expression when combined withthe SV40 poly(A) signal. When driving GFP, the UsPconstruct displays similar ubiquitous expressionthroughout the CNS compared with the previouslydescribed CBh promoter,22 although expression inastrocytes appears diminished somewhat. We con-clude that the UsP promoter–intron, combined withthe SV40 poly(A) signal, provides an ideal expres-sion construct for HEXM within an scAAV vector.
A potential challenge related to intravenous ad-ministration of AAV vectors (AAV9, AAVrh10, orAAVrh8) targeting the CNS is that the high vectordoses required also result in extraordinarily highloads of gene transfer to the liver. This createsthe potential for deleterious liver-specific T cell re-sponses,50,51 possible toxicity related to transgeneoverexpression, and hepatocellular carcinoma whenadministered to neonates.30,31 By using the AAV9.47capsid, biodistribution to the liver was highly re-duced relative to AAV9, while preserving the abilityof AAV9 to cross the blood–brain barrier. An ap-parent reduction in astrocyte transduction withAAV9.47 was noted relative to AAV9, which maynecessitate a slightly higher overall dose. However,in diseases that benefit from cross-correction, pri-mary expression in neurons should be sufficient.Regarding the potential for hepatocellular carci-noma, our use of an enhancer-less expression con-struct would be expected to reduce the potential foroncogenesis. Whether as a result of the reduced liverbiodistribution or the lack of an enhancer, our co-horts injected intravenously as neonates did notdevelop tumors up to their planned end point of 15months old. Although this observation should beexamined more carefully, it is encouraging. The timecourse of infantile TSD and SD in humans is rapid,and the earliest possible age of intervention wouldbeideal. Although it is unclear whether the risk of he-patocellular carcinoma with AAV vector observed inmice could translate to humans, our vector designpotentially reduces that hypothetical risk.
The efficacy of gene therapy in mouse and catmodels of GM2 gangliosidosis is susceptible to misin-terpretation because of the sialidase metabolic by-pass pathway, which could significantly reduce GM2
storage levels with any increase in hexosaminidaseactivity (i.e., HexA, HexB, HexS, or HexM). This by-pass pathway does not exist in humans. The studiesreported here were conducted in the TSD model,which is not ideal to evaluate treatment efficacy be-cause of the mild phenotype of the mice. This paperprovides information on the development of the re-agents, along with evidence supporting the hypothe-sis that this has a likelihoodofproviding a therapeutic
benefit. A companion article by Osmon and col-leagues,52 in this issue of Human Gene Therapy (seep. 497), describes the application of this approach andvector design with a mouse model of SD, which doesdisplay a severe disease phenotype, and is more ap-propriate for evaluating treatment efficacy.
Several technological advances were combined togenerate a reagent to treat Tay-Sachs and Sandhoffdiseases. This single scAAV vector design enablesdelivery approaches for widespread CNS genetransfer. Key to making use of scAAV are the novelhexosaminidase variant, HEXM, and the shortsynthetic promoter–intron UsP. Finally, by using anenhancerless expression construct and a liver-detargeted capsid, we suggest that the possible riskof hepatocellular carcinoma with this vector designwould be greatly diminished. In conclusion, the col-lective body of work presented herein describes anovel vector design that greatly enhances the po-tential success of a gene transfer strategy to treatboth Tay-Sachs and Sandhoff diseases. This vectordesign may also be useful to deliver other genes ofsimilar size to treat CNS disorders.
ACKNOWLEDGMENTS
This research was funded by the New Hope Re-search Foundation (http://newhoperesearch.org;S.G., J.W., D.M., S.K.M, S.N.K., and P.T.) and do-nations from the Uger Estate (M.T. and D.M.). In-direct administrative support for S.G. was providedby Research to Prevent Blindness to the UNC De-partment of Ophthalmology. B.M. is supported by aManitoba Research Chair (Research Manitoba). Theauthors thank the Animal Study Core at UNC Cha-pel Hill for help in delivering the intracranial injec-tions into TSD mice. The authors acknowledge CliffHeindel and Violeta Zaric for technical assistance inimmunohistochemistry and biodistribution studies.The authors thank Bentley Midkiff in the UNCTranslational Pathology Laboratory (TPL) for experttechnical assistance. The UNC Translational Pa-thology Laboratory is supported, in part, by grantsfrom the National Cancer Institute (3P30CA016086)and the UNC University Cancer Research Fund(UCRF). The authors thank Kyowa Hakko KirinCompany for providing the KM966 antibody.
AUTHOR DISCLOSURE
S.G. has received patent royalties from AsklepiosBiopharma that are not directly related to thesestudies. D.M. and B.M. are inventors on a pendingpatent for the sequence and potential uses of HexM.Credit is given to S.G., J.K., and M.K. for the designand initial characterization of the UsP construct. J.K.and M.K. are inventors on a pending patent for UsP.
GENE THERAPY VECTOR IMPROVEMENTS FOR TSD 519

REFERENCES
1. Sandhoff K, and Christomanou H. Biochemistryand genetics of gangliosidoses. Hum Genet 1979;50:107–143.
2. Leinekugel P, Michel S, Conzelmann E, et al.Quantitative correlation between the residualactivity of b-hexosaminidase A and arylsulfataseA and the severity of the resulting lysosomalstorage disease. Hum Genet 1992;88:513–523.
3. Yamanaka S, Johnson MD, Grinberg A, et al.Targeted disruption of the Hexa gene results inmice with biochemical and pathologic features ofTay-Sachs disease. Proc Natl Acad Sci U S A1994;91:9975–9979.
4. Sango K, Yamanaka S, Hoffmann A, et al. Mousemodels of Tay-Sachs and Sandhoff diseases differin neurologic phenotype and ganglioside metab-olism. Nat Genet 1996;11:170–176.
5. Phaneuf D, Wakamatsu N, Huang JQ, et al.Dramatically different phenotypes in mousemodels of human Tay-Sachs and Sandhoff dis-eases. Hum Mol Genet 1996;5:1–14.
6. Yuziuk JA, Bertoni C, Beccari T, et al. Specificityof mouse GM2 activator protein and b-N-acetylhexosaminidases A and B: similarities anddifferences with their human counterparts in thecatabolism of GM2. J Biol Chem 1998;273:66–72.
7. Guidotti JE, Mignon A, Hasse G, et al. Adenoviralgene therapy of the Tay-Sachs disease in hex-osaminidase A-deficient knock-out mice. Hum MolGenet 1999;8:831–838.
8. Cachon-Gonzalez MB, Wang SZ, Lynch A, et al.Effective gene therapy in an authentic model ofTay-Sachs-related diseases. Proc Natl Acad SciU S A 2006;103:10373–10378.
9. Sargeant TJ, Wang S, Bradley J, et al. Adeno-associated virus-mediated expression of b-hexosaminidase prevents neuronal loss in theSandhoff mouse brain. Hum Mol Genet 2011;20:4371–4380.
10. Cachon-Gonzalez MB, Wang SZ, McNair R, et al.Gene transfer corrects acute GM2 gangliosidosis—potential therapeutic contribution of perivascularenzyme flow. Mol Ther 2012;20:1489–1500.
11. Bradbury AM, Gray-Edwards HL, Shirley JL, et al.Biomarkers for disease progression and AAVtherapeutic efficacy in feline Sandhoff disease.Exp Neurol 2015;263:102–112.
12. Cachon-Gonzalez MB, Wang SZ, Ziegler R, et al.Reversibility of neuropathology in Tay-Sachs-relateddiseases. Hum Mol Genet 2014;23:730–748.
13. Weismann CM, Ferreira J, Keeler AM, et al.Systemic AAV9 gene transfer in adult GM1
gangliosidosis mice reduces lysosomal storage inCNS and extends lifespan. Hum Mol Genet 2015;24:4353–4364.
14. Walia JS, Altaleb N, Bello A, et al. Long-termcorrection of Sandhoff disease following intrave-nous delivery of rAAV9 to mouse neonates. MolTher 2015;23:414–422.
15. Gadalla KK, Bailey ME, Spike RC, et al. Improvedsurvival and reduced phenotypic severity follow-ing AAV9/MECP2 gene transfer to neonatal andjuvenile male Mecp2 knockout mice. Mol Ther2013;21:18–30.
16. Fu H, Dirosario J, Killedar S, et al. Correction ofneurological disease of mucopolysaccharidosisIIIB in adult mice by rAAV9 trans-blood–brainbarrier gene delivery. Mol Ther 2011;19:1025–1033.
17. Ruzo A, Marco S, Garcia M, et al. Correction ofpathological accumulation of glycosaminoglycansin central nervous system and peripheral tissuesof MPSIIIA mice through systemic AAV9 genetransfer. Hum Gene Ther 2012;23:1237–1246.
18. Gray SJ, Matagne V, Bachaboina L, et al. Pre-clinical differences of intravascular AAV9 deliveryto neurons and glia: a comparative study of adultmice and nonhuman primates. Mol Ther 2011;19:1058–1069.
19. Duque S, Joussemet B, Riviere C, et al. In-travenous administration of self-complementaryAAV9 enables transgene delivery to adult motorneurons. Mol Ther 2009;17:1187–1196.
20. Mattar CN, Waddington SN, Biswas A, et al.Systemic delivery of scAAV9 in fetal macaquesfacilitates neuronal transduction of the centraland peripheral nervous systems. Gene Ther2013;20:69–83.
21. Foust KD, Nurre E, Montgomery CL, et al. In-travascular AAV9 preferentially targets neonatalneurons and adult astrocytes. Nat Biotechnol2009;27:59–65.
22. Gray SJ, Foti SB, Schwartz JW, et al. Optimizingpromoters for recombinant adeno-associatedvirus-mediated gene expression in the peripheraland central nervous system using self-complementaryvectors. Hum Gene Ther 2011;22:1143–1153.
23. Powell SK, Rivera-Soto R, and Gray SJ. Viral ex-pression cassette elements to enhance transgenetarget specificity and expression in gene therapy.Discov Med 2015;19:49–57.
24. Mark BL, Mahuran DJ, Cherney MM, et al. Crystalstructure of human b-hexosaminidase B: under-standing the molecular basis of Sandhoff and Tay-Sachs disease. J Mol Biol 2003;327:1093–1109.
25. Lemieux MJ, Mark BL, Cherney MM, et al. Crys-tallographic structure of human b-hexosaminidaseA: interpretation of Tay-Sachs mutations and lossof GM2 ganglioside hydrolysis. J Mol Biol 2006;359:913–929.
26. Matsuoka K, Tamura T, Tsuji D, et al. Therapeuticpotential of intracerebroventricular replacementof modified human b-hexosaminidase B for GM2
gangliosidosis. Mol Ther 2011;19:1017–1024.
27. Sinici I, Yonekawa S, Tkachyova I, et al. In celluloexamination of a b–a hybrid construct of b-hexosaminidase A subunits, reported to interactwith the GM2 activator protein and hydrolyze GM2
ganglioside. PLoS One 2013;8:e57908.
28. Tropak MB, Yonekawa S, Karumuthil-Melethil S,et al. Construction of a hybrid b-hexosaminidasesubunit capable of forming stable homodimersthat hydrolyze GM2 ganglioside in vivo. Mol TherMethods Clin Dev 2016;3:15057.
29. Zincarelli C, Soltys S, Rengo G, et al. Analysis ofAAV serotypes 1–9 mediated gene expressionand tropism in mice after systemic injection. MolTher 2008;16:1073–1080.
30. Donsante A, Miller DG, Li Y, et al. AAV vectorintegration sites in mouse hepatocellular carci-noma. Science 2007;317:477.
31. Chandler RJ, LaFave MC, Varshney GK, et al.Vector design influences hepatic genotoxicity af-ter adeno-associated virus gene therapy. J ClinInvest 2015;125:870–880.
32. Pulicherla N, Shen S, Yadav S, et al. Engineeringliver-detargeted AAV9 vectors for cardiac andmusculoskeletal gene transfer. Mol Ther 2011;19:1070–1078.
33. Tornøe J, Kusk P, Johansen TE, et al. Generationof a synthetic mammalian promoter library bymodification of sequences spacing transcriptionfactor binding sites. Gene 2002;297:21–32.
34. Gray SJ, Nagabhushan Kalburgi S, McCown TJ,et al. Global CNS gene delivery and evasion ofanti-AAV-neutralizing antibodies by intrathecal AAVadministration in non-human primates. Gene Ther2013;20:450–459.
35. Gray SJ, Choi VW, Asokan A, et al. Production ofrecombinant adeno-associated viral vectors anduse in in vitro and in vivo administration. CurrProtoc Neurosci 2011;Chapter 4:Unit 4.17.
36. Seyrantepe V, Lema P, Caqueret A, et al. Micedoubly-deficient in lysosomal hexosaminidase Aand neuraminidase 4 show epileptic crises andrapid neuronal loss. PLoS Genet 2010;6:e1001118.
37. Martino S, Marconi P, Tancini B, et al. A directgene transfer strategy via brain internal capsulereverses the biochemical defect in Tay-Sachsdisease. Hum Mol Genet 2005;14:2113–2123.
38. Gray-Edwards HL, Brunson BL, Holland M, et al.Mucopolysaccharidosis-like phenotype in felineSandhoff disease and partial correction after AAVgene therapy. Mol Genet Metab 2015;116:80–87.
39. Rockwell HE, McCurdy VJ, Eaton SC, et al. AAV-mediated gene delivery in a feline model ofSandhoff disease corrects lysosomal storagein the central nervous system. ASN Neuro2015;7(2).
40. McCurdy VJ, Rockwell HE, Arthur JR, et al.Widespread correction of central nervous systemdisease after intracranial gene therapy in a felinemodel of Sandhoff disease. Gene Ther 2015;22:181–189.
41. Bourgoin C, Emiliani C, Kremer EJ, et al. Wide-spread distribution of b-hexosaminidase activityin the brain of a Sandhoff mouse model aftercoinjection of adenoviral vector and mannitol.Gene Ther 2003;10:1841–1849.
520 KARUMUTHIL-MELETHIL ET AL.

42. Duque SI, Arnold WD, Odermatt P, et al. A largeanimal model of spinal muscular atrophy andcorrection of phenotype. Ann Neurol 2015;77:399–414.
43. Federici T, Taub JS, Baum GR, et al. Robust spinalmotor neuron transduction following intrathecaldelivery of AAV9 in pigs. Gene Ther 2011;19:852–859.
44. Meyer K, Ferrsiuolo L, Schmelzer L, et al. Im-proving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose–re-sponse study in mice and nonhuman primates.Mol Ther 2015;23:477–487.
45. Passini MA, Bu J, Richards AM, et al. Translationalfidelity of intrathecal delivery of self-complementaryAAV9-survival motor neuron 1 for spinal muscularatrophy. Hum Gene Ther 2014;25:619–630.
46. Samaranch L, Salegio EA, San Sebastian W, et al.Strong cortical and spinal cord transduction afterAAV7 and AAV9 delivery into the cerebrospinalfluid of nonhuman primates. Hum Gene Ther2013;24:526–532.
47. Samaranch L, Salegio EA, San Sebastian W, et al.Adeno-associated virus serotype 9 transduction inthe central nervous system of nonhuman prima-tes. Hum Gene Ther 2012;23:382–389.
48. Haurigot V, Marco S, Ribera A, et al. Whole bodycorrection of mucopolysaccharidosis IIIA by in-tracerebrospinal fluid gene therapy. J Clin Invest2013;123:3254–3271.
49. Bucher T, Colle MA, Wakeling E, et al. scAAV9intracisternal delivery results in efficient genetransfer to the central nervous system of a felinemodel of motor neuron disease. Hum Gene Ther2013;24:670–682.
50. Nathwani AC, Tuddenham EG, Rangarajan S,et al. Adenovirus-associated virus vector-mediatedgene transfer in hemophilia B. N Engl J Med 2011;365:2357–2365.
51. Mingozzi F, and High KA. Immune responses toAAV in clinical trials. Curr Gene Ther 2007;7:316–324.
52. Osmon KJ, Woodley E, Thompson P, et al. Sys-temic gene transfer of a hexosaminidase variantusing a scAAV9.47 vector corrects GM2 gang-liosidosis in Sandhoff mice. Hum Gene Ther 2016;27:497–508.
Received for publication January 27, 2016;accepted after revision May 14, 2016.
Published online: May 19, 2016.
GENE THERAPY VECTOR IMPROVEMENTS FOR TSD 521