Embed Size (px)
Citation preview

No cofactor e¡ect on equilibrium unfolding of Desulfovibrio desulfuricans£avodoxin
David Apiyo, Jesse Guidry, Pernilla Wittung-Stafshede *Chemistry Department, Tulane University, 6823 St. Charles Ave., New Orleans, LA 70118-5698, USA
Received 15 September 1999; received in revised form 21 December 1999; accepted 10 February 2000
Abstract
Flavodoxins are proteins with an K/L doubly wound topology that mediate electron transfer through a non-covalentlybound flavin mononucleotide (FMN). The FMN moiety binds strongly to folded flavodoxin (KD = 0.1 nM, oxidized FMN).To study the effect of this organic cofactor on the conformational stability, we have characterized apo and holo forms ofDesulfovibrio desulfuricans flavodoxin by GuHCl-induced denaturation. The unfolding reactions for both holo- and apo-flavodoxin are reversible. However, the unfolding curves monitored by far-UV circular dichroism and fluorescencespectroscopy do not coincide. For both apo- and holo-flavodoxin, a native-like intermediate (with altered tryptophanfluorescence but secondary structure as the folded form) is present at low GuHCl concentrations. There is no effect on theflavodoxin stability imposed by the presence of the FMN cofactor (vG = 20( þ 2) and 19( þ 1) kJ/mol for holo- and apo-flavodoxin, respectively). A thermodynamic cycle, connecting FMN binding to folded and unfolded flavodoxin with theunfolding free energies for apo- and holo-flavodoxin, suggests that the binding strength of FMN to unfolded flavodoxinmust be very high (KD = 0.2 nM). In agreement, we discovered that the FMN remains coordinated to the polypeptide uponunfolding. ß 2000 Elsevier Science B.V. All rights reserved.
Keywords: Flavodoxin; Protein folding; Cofactor; Flavin mononucleotide; Circular dichroism; Fluorescence
1. Introduction
A considerable body of work has demonstratedthat proteins fold to their native states with mecha-nisms of varying complexity and with kinetics rang-ing from microseconds to hours [1^3]. One key ¢nd-ing is that while many large proteins (s 100 amino
acid residues) populate folding intermediates [4,5],proteins smaller than this often fold directly to thenative state in apparent two-state reactions withoutpopulated intermediates [1,6]. The thermodynamicstabilization of folded proteins can be attributed tomany intra-molecular forces (such as van der Waalsinteractions, interactions between charged groups,between polar groups and between nonpolar groups)as well as to interactions with surrounding water. Inaddition, coordination of a cofactor may stabilize theprotein structure relative to the stability of the cor-responding apo-protein. In the case of redox-activecofactors, the extent of protein stabilization may fur-ther depend on the reduction state of the cofactor[7,8]. For example, heme proteins (cytochrome c, cy-
0167-4838 / 00 / $ ^ see front matter ß 2000 Elsevier Science B.V. All rights reserved.PII: S 0 1 6 7 - 4 8 3 8 ( 0 0 ) 0 0 0 3 2 - 7
Abbreviations: FMN, £avin mononucleotide; GuHCl, guani-dine hydrochloride; CD, circular dichroism; ANS, 1-anilino-naphthalene-8-sulfonate
* Corresponding author. Fax: +1-504-865-5596;E-mail : [email protected]
BBAPRO 36115 7-6-00
Biochimica et Biophysica Acta 1479 (2000) 214^224www.elsevier.com/locate/bba

tochrome b562 and myoglobin) are more stable to-wards unfolding when reduced than when oxidized[9^11], whereas copper proteins (azurin and the CuA
domain) are most stable in their oxidized forms[12,13]). Despite the abundance, and importance, ofproteins that coordinate prosthetic groups in biolog-ical systems [14], studies of the roles of cofactors inprotein-folding reactions have so far been sparse.
In addition to inherent structures preferred by var-ious peptide sequences [15], local non-random struc-tures in an unfolded protein may exist due to thepresence of a cofactor. Such structural restrictionsin the unfolded ensemble of protein molecules maydramatically decrease the entropy of the unfoldedstate as well as limit the conformational search forthe native state [15,16]. To date it is not clear to whatextent in vivo cofactors become coordinated to theircorresponding polypeptides before folding of theprotein [14]. Nevertheless, recent experimental ¢nd-ings have emphasized the signi¢cance of cofactorbinding to unfolded proteins in vitro [17,18], evenwhen no covalent bonds are present between the co-factor and the polypeptide. Coordination of thehemes in cytochrome b562 and myoglobin to the cor-responding unfolded polypeptides have been ob-served [10,11]. In the case of azurin and the CuA
domain [12,13,17], the reduced copper ions staybound to the unfolded polypeptides. Moreover, itwas reported that for the high-potential iron^sulfurprotein (HiPIP) from Chromatium vinosum, the poly-peptide unfolded before the Fe4S4 cluster was de-stroyed [19]. In the case of cytochrome c, it is be-lieved that in vivo the heme is inserted when thepolypeptide is still unfolded; only after heme inser-tion is folding of the protein possible [20]. Cofactorscould thus play important roles in protein-foldingreactions, serving as nucleation sites that direct thefolding of the polypeptides. In this study we havecharacterized the e¡ect on equilibrium unfoldingthat is imposed by the presence of an organic cofac-tor, the FMN group in £avodoxin.
Nine protein super-families, of which £avodoxinsconstitute one, share the £avodoxin-like structuralmotif [21]. All of these families exhibit little or nosequence similarity and comprise a broad range ofunrelated proteins with di¡erent functions (like cata-lases, chemotactic proteins, lipases, esterases and £a-vodoxins). Still, they are all characterized by a fold-
ing motif consisting of one ¢ve-stranded parallelL-sheet surrounded by two K-helices on each side(Fig. 1). The £avodoxins function as photosyntheticelectron-transport proteins and contain an organiccofactor, a £avin mononucleotide prosthetic group(FMN), non-covalently bound to the protein [22].Flavodoxins can transfer two electrons since theFMN has three redox states (oxidized, semiquinoneand hydroquinone); all forms have di¡erent spectralcharacteristics in the visible region [22]. In vitro, theFMN is oxidized in the resting state of the foldedprotein. We have investigated £avodoxin from thesulfate-reducing organism Desulfovibro desulfuricans[23]. This protein was recently crystallized [24]; it has146 residues including one tryptophan. The a¤nityof oxidized FMN to folded £avodoxin from D. de-sulfuricans is extremely strong; the dissociation con-stant is found to be as low as 0.1 nM [23]. Residuesin two peptide loops form the major portion of theFMN-binding site, which consist of a combination ofaromatic-stacking interactions, a nonpolar environ-
Fig. 1. Structure of D. vulgaris £avodoxin (PDB F2X), which isalmost identical to that of the D. desulfuricans protein (allbackbone atoms are superimposed with deviations of less than0.99 nm). The FMN and the Trp60 and Tyr98 are shown inblack.
BBAPRO 36115 7-6-00
D. Apiyo et al. / Biochimica et Biophysica Acta 1479 (2000) 214^224 215

ment and electrostatic interactions. The iso-alloxa-zine ring of the FMN moiety is sandwiched betweentwo aromatic residues (the tryptophan, Trp60, and atyrosine, Tyr98) allowing for considerable Z-orbitaloverlap (FMN and these two residues are highlightedin Fig. 1).
To date, folding studies of £avodoxins have nottargeted holo-proteins; only a few apo-£avodoxincharacterizations have been performed. To directlyaddress the role of the FMN cofactor in proteinstability and folding, we here study equilibrium un-folding of holo (oxidized FMN) and apo D. desul-furicans £avodoxin. We ¢nd that unfolding of bothapo- and holo-£avodoxins are reversible. Unfoldingcurves, monitored far-UV circular dichroism (CD)and tryptophan £uorescence, di¡er from each otherin case of both apo- and holo-proteins. Thus, anintermediate state, with a structural change nearthe tryptophan but native-like secondary structure,is present during equilibrium unfolding of bothapo- and holo-£avodoxin. In contrast to many otherredox-active cofactors, the bound FMN confers nomeasurable e¡ect on the £avodoxin thermodynamicstability. The a¤nity of FMN to the unfolded poly-peptide must therefore be as high as to the folded£avodoxin, in accordance with our ¢nding of noFMN dissociation upon polypeptide unfolding.
2. Materials and methods
2.1. Chemicals
GuHCl (highest purity) and FMN were obtainedfrom Sigma.
2.2. Protein preparation
Flavodoxin from D. desulfuricans (ATCC 27774)was a gift from Dr. J. LeGall, University of Georgia,Athens, GA, USA. It was puri¢ed as described pre-viously [23,24]. Protein concentration was deter-mined from A452 = 10 300 M31 cm31 for the oxidizedholo-protein [23] and A280 = 11 460 M31 cm31 for theapo-protein (one tryptophan and four tyrosines).Apo-protein was prepared by lowering the pH to2.0 where FMN release is induced [25]. The holo-protein was ¢rst dialyzed against a 1000-fold larger
volume of 50 mM KCl, pH 2.0 for 36 h. (After 24 h,the yellow color disappeared, indicating that theFMN has been removed from the protein.) The col-orless sample was subsequently dialyzed against a1000-fold excess of 5 mM phosphate bu¡er, pH 7.0for 12 h. The apo-protein was characterized by ab-sorption, £uorescence and CD methods. Holo-£avo-doxin (in 3 M GuHCl) was dialyzed against a 1000-fold larger volume of a 3 M GuHCl solution for 24 h(10 kDa cut o¡), to check for possible cofactor dis-sociation in the unfolded state.
2.3. Spectroscopy
All reactions were carried out in the dark untilspectroscopic measurements were performed. Ab-sorption spectra were measured on a Cary 50 spec-trophotometer (1-cm cell). For far-UV CD measure-ments an OLIS spectropolarimeter (1-mm cell) wasused. In CD measurements the sample compartmentwas purged with nitrogen gas to avoid absorption ofO2. Fluorescence spectra (1-cm cell, excitation at285 nm (tryptophan); 1.5-nm excitation and 5-nmemission slits, respectively) were collected on a Shi-madzu £uorometer (Loyola College, New Orleans,USA). GuHCl titrations of apo- and holo-£avodoxinwere performed in 5 mM phosphate bu¡er, pH 7.0,at 20³C; in both cases monitored by CD and £uo-rescence. In the case of holo-£avodoxin, GuHCl ti-trations were also monitored by visible absorption(300^500 nm). In all experiments the protein hadattained equilibrium within less than 1 min and un-folding was shown to be reversible. Moreover, theunfolding reactions were not protein concentration(in the range of 3 to 20 WM) dependent. Fluorescenceexperiments using 1-anilinonaphthalene-8-sulfonate(1,8-ANS) as a probe for hydrophobic-surface-areaexposure were performed with apo-£avodoxin in var-ious GuHCl concentrations (ex: 360 nm, em: 450^550 nm; 6 WM apo-£avodoxin and 600 WM ANS).
2.4. Unfolding parameters
Each unfolding transition was analyzed using atwo-state model, fF+fU = 1, where fF and fU repre-sents the fraction of total protein in the folded andunfolded conformations, respectively. The equilibri-um constants, KU, and the free-energy changes, vGU,
BBAPRO 36115 7-6-00
D. Apiyo et al. / Biochimica et Biophysica Acta 1479 (2000) 214^224216

at each point in the unfolding reaction can be calcu-lated using:
KU � f U=�13f U� � f U=f F
vGU � 3RT lnKU
Moreover, the free-energy change can be expressedas:
vGU � vGU�H2O�3m�GuHCl�In this equation, m describes the `cooperativity', orthe steepness, of the unfolding transition [26,27] andvGU(H2O) represents the free energy of unfolding inaqueous solution. Direct ¢ts (in KaleidaGraph) tothe experimental unfolding curves (data presentedin Figs. 4 and 5) were performed using the followingequation (derived from the above equations):
Y obs �
�Y U3Y F�exp��vGU�H2O�3m�GuHCl��=RT���=
�1� �exp��vGU�H2O�3m�GuHCl��=RT���
Yobs, YU and YF are the observed signal (CD or£uorescence), unfolded-protein baseline and folded-protein baseline, respectively. From the ¢ts, valuesfor vGU(H2O) and m were directly determined foreach transition (listed in Table 1). The midpoints ofthe unfolding transitions were calculated asvGU(H2O)/m. Reported uncertainties for vGU andm values were obtained from the goodness of the ¢ts.
To prepare Fig. 6, assumptions were made that thechanges in far-UV CD (which are identical tochanges in visible absorption) re£ect the appearanceof the unfolded state, while the £uorescence datamonitors the disappearance of the native state. This
means that the folded and intermediate states haveidentical CD signals, whereas the intermediate andunfolded states have identical tryptophan £uores-cence spectra. Since the £uorescence transition is inprinciple complete at the GuHCl concentrationswhere the CD signal starts to change (this is thecase for both apo- and holo-£avodoxin, see Fig. 6),the assumptions are reasonable.
3. Results
3.1. FMN remains bound to unfolded holo-£avodoxin
In Fig. 2a, we present the emission spectra offolded and unfolded (in 2 M GuHCl) holo D. desul-furicans £avodoxin upon excitation (at 285 nm) ofthe tryptophan residue (Trp60). The tryptophanemission (around 350 nm) is highly quenched in thefolded state due to the presence of the FMN that iscoordinated within van der Waals distance [24]. Inaddition, the FMN absorbs light of 285 nm therebycreating an observed emission peak at 530 nm that isrelated both to direct excitation of FMN as well asto energy-transfer from the tryptophan residue (sinceFMN's major absorption peak is centered at 370 nm).The emission at 530 nm from holo-£avodoxin (ex285 nm) is much smaller than that detected fromfree FMN, in the protein it is quenched to about4% of that of free FMN (inset Fig. 2a). Upon £avo-doxin unfolding, the emission increases by almost25% at 347 nm (tryptophan), whereas the emissionat 530 nm (FMN) remains essentially the same. Theemission at 530 nm from free FMN under identicalGuHCl conditions and excitation at 285 nm is muchlarger, although it is lower than the emission from
Table 1Thermodynamic parameters for equilibrium unfolding obtained from direct ¢ts assuming apparent two-state transitions (see Section 2for details)
Flavodoxin [GuHCl]1=2 (M) m (kJ/mol, M) vG(H2O) (kJ/mol)
Holo £uorescence 0.95 9.8 ( þ 1.0) 9.3( þ 1.1)CD 1.77 11.3( þ 1.3) 20.0( þ 2.3)absorption 1.75 ^a ^a
Apo £uorescence 0.58 12.1( þ 0.9) 7.0( þ 0.6)CD 1.66 11.6( þ 0.6) 19.3( þ 0.9)
aThe visible-absorption change for FMN upon £avodoxin unfolding was small and accurate estimates for m and vG were not possi-ble.
BBAPRO 36115 7-6-00
D. Apiyo et al. / Biochimica et Biophysica Acta 1479 (2000) 214^224 217
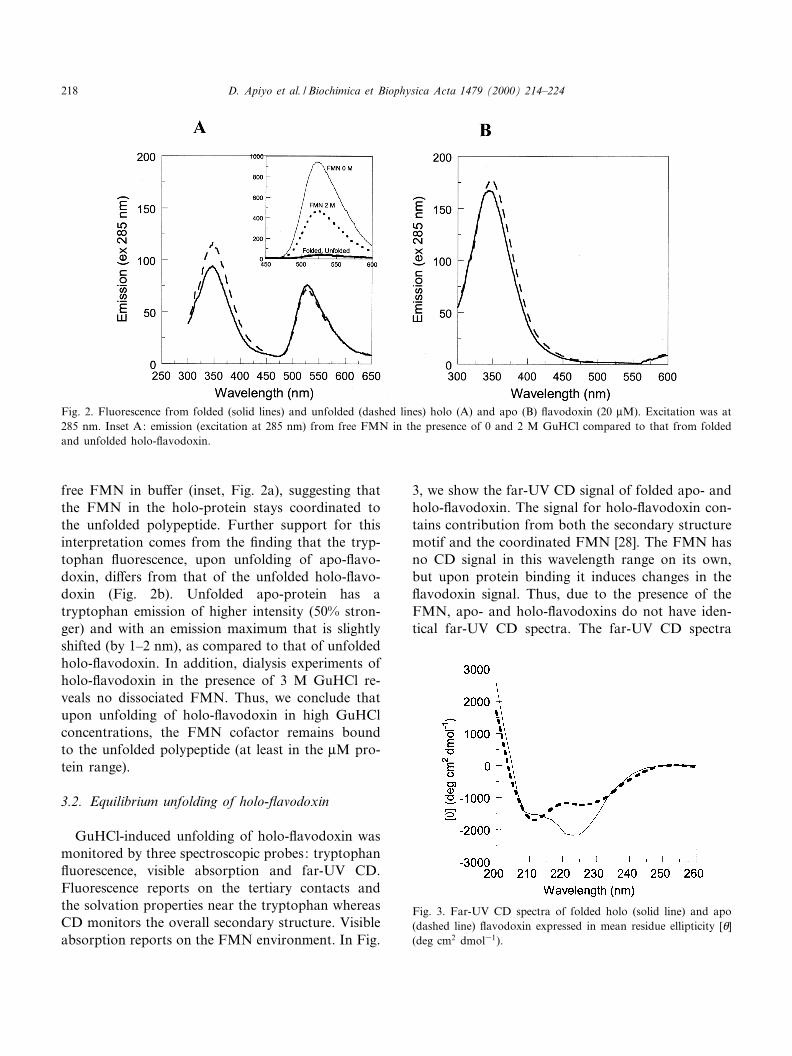
free FMN in bu¡er (inset, Fig. 2a), suggesting thatthe FMN in the holo-protein stays coordinated tothe unfolded polypeptide. Further support for thisinterpretation comes from the ¢nding that the tryp-tophan £uorescence, upon unfolding of apo-£avo-doxin, di¡ers from that of the unfolded holo-£avo-doxin (Fig. 2b). Unfolded apo-protein has atryptophan emission of higher intensity (50% stron-ger) and with an emission maximum that is slightlyshifted (by 1^2 nm), as compared to that of unfoldedholo-£avodoxin. In addition, dialysis experiments ofholo-£avodoxin in the presence of 3 M GuHCl re-veals no dissociated FMN. Thus, we conclude thatupon unfolding of holo-£avodoxin in high GuHClconcentrations, the FMN cofactor remains boundto the unfolded polypeptide (at least in the WM pro-tein range).
3.2. Equilibrium unfolding of holo-£avodoxin
GuHCl-induced unfolding of holo-£avodoxin wasmonitored by three spectroscopic probes: tryptophan£uorescence, visible absorption and far-UV CD.Fluorescence reports on the tertiary contacts andthe solvation properties near the tryptophan whereasCD monitors the overall secondary structure. Visibleabsorption reports on the FMN environment. In Fig.
3, we show the far-UV CD signal of folded apo- andholo-£avodoxin. The signal for holo-£avodoxin con-tains contribution from both the secondary structuremotif and the coordinated FMN [28]. The FMN hasno CD signal in this wavelength range on its own,but upon protein binding it induces changes in the£avodoxin signal. Thus, due to the presence of theFMN, apo- and holo-£avodoxins do not have iden-tical far-UV CD spectra. The far-UV CD spectra
Fig. 2. Fluorescence from folded (solid lines) and unfolded (dashed lines) holo (A) and apo (B) £avodoxin (20 WM). Excitation was at285 nm. Inset A: emission (excitation at 285 nm) from free FMN in the presence of 0 and 2 M GuHCl compared to that from foldedand unfolded holo-£avodoxin.
Fig. 3. Far-UV CD spectra of folded holo (solid line) and apo(dashed line) £avodoxin expressed in mean residue ellipticity [a](deg cm2 dmol31).
BBAPRO 36115 7-6-00
D. Apiyo et al. / Biochimica et Biophysica Acta 1479 (2000) 214^224218
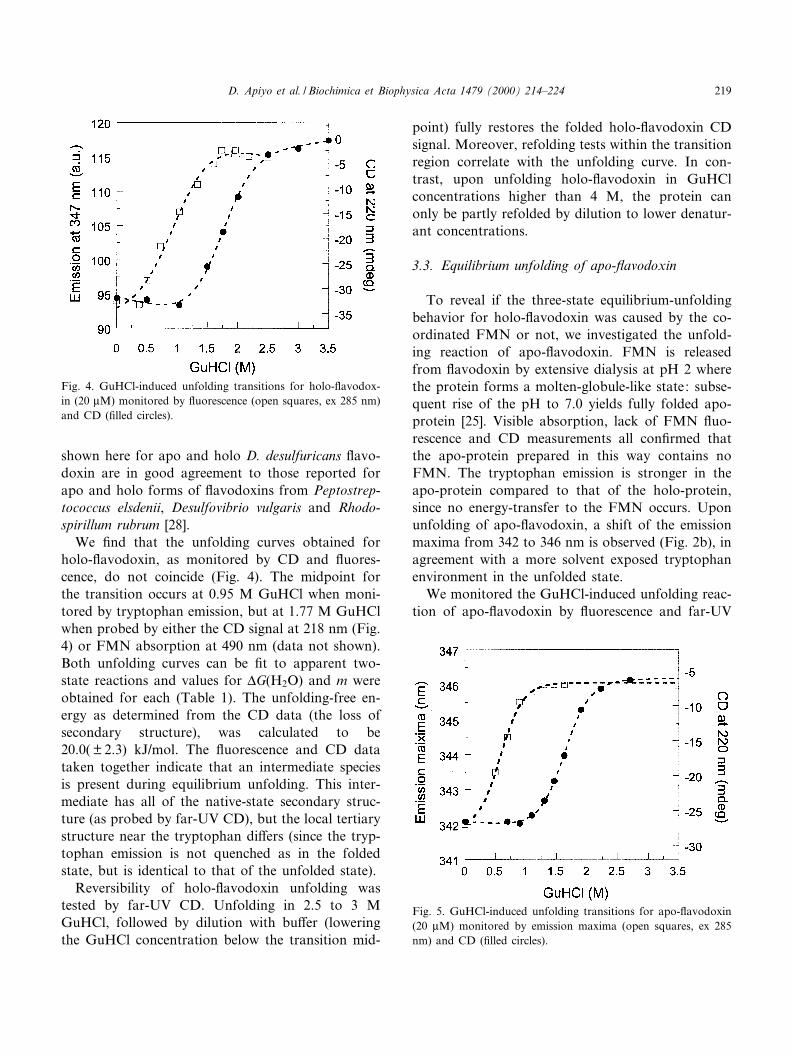
shown here for apo and holo D. desulfuricans £avo-doxin are in good agreement to those reported forapo and holo forms of £avodoxins from Peptostrep-tococcus elsdenii, Desulfovibrio vulgaris and Rhodo-spirillum rubrum [28].
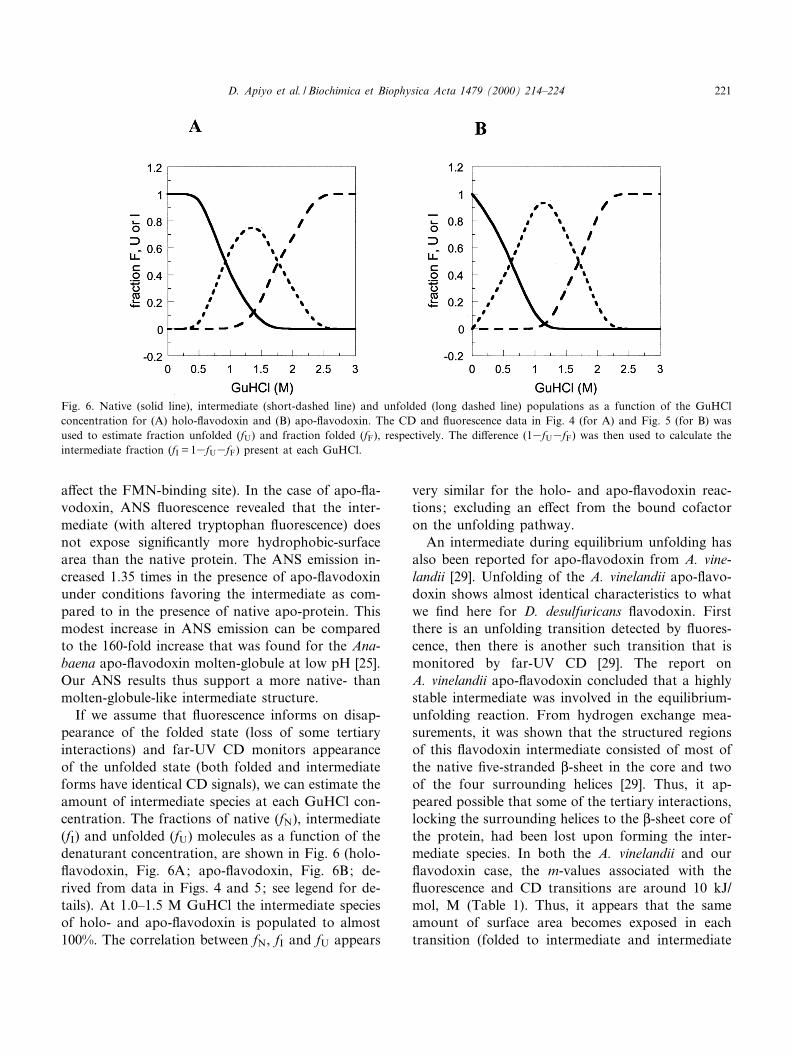
We ¢nd that the unfolding curves obtained forholo-£avodoxin, as monitored by CD and £uores-cence, do not coincide (Fig. 4). The midpoint forthe transition occurs at 0.95 M GuHCl when moni-tored by tryptophan emission, but at 1.77 M GuHClwhen probed by either the CD signal at 218 nm (Fig.4) or FMN absorption at 490 nm (data not shown).Both unfolding curves can be ¢t to apparent two-state reactions and values for vG(H2O) and m wereobtained for each (Table 1). The unfolding-free en-ergy as determined from the CD data (the loss ofsecondary structure), was calculated to be20.0( þ 2.3) kJ/mol. The £uorescence and CD datataken together indicate that an intermediate speciesis present during equilibrium unfolding. This inter-mediate has all of the native-state secondary struc-ture (as probed by far-UV CD), but the local tertiarystructure near the tryptophan di¡ers (since the tryp-tophan emission is not quenched as in the foldedstate, but is identical to that of the unfolded state).
Reversibility of holo-£avodoxin unfolding wastested by far-UV CD. Unfolding in 2.5 to 3 MGuHCl, followed by dilution with bu¡er (loweringthe GuHCl concentration below the transition mid-
point) fully restores the folded holo-£avodoxin CDsignal. Moreover, refolding tests within the transitionregion correlate with the unfolding curve. In con-trast, upon unfolding holo-£avodoxin in GuHClconcentrations higher than 4 M, the protein canonly be partly refolded by dilution to lower denatur-ant concentrations.
3.3. Equilibrium unfolding of apo-£avodoxin
To reveal if the three-state equilibrium-unfoldingbehavior for holo-£avodoxin was caused by the co-ordinated FMN or not, we investigated the unfold-ing reaction of apo-£avodoxin. FMN is releasedfrom £avodoxin by extensive dialysis at pH 2 wherethe protein forms a molten-globule-like state: subse-quent rise of the pH to 7.0 yields fully folded apo-protein [25]. Visible absorption, lack of FMN £uo-rescence and CD measurements all con¢rmed thatthe apo-protein prepared in this way contains noFMN. The tryptophan emission is stronger in theapo-protein compared to that of the holo-protein,since no energy-transfer to the FMN occurs. Uponunfolding of apo-£avodoxin, a shift of the emissionmaxima from 342 to 346 nm is observed (Fig. 2b), inagreement with a more solvent exposed tryptophanenvironment in the unfolded state.
We monitored the GuHCl-induced unfolding reac-tion of apo-£avodoxin by £uorescence and far-UV
Fig. 4. GuHCl-induced unfolding transitions for holo-£avodox-in (20 WM) monitored by £uorescence (open squares, ex 285 nm)and CD (¢lled circles).
Fig. 5. GuHCl-induced unfolding transitions for apo-£avodoxin(20 WM) monitored by emission maxima (open squares, ex 285nm) and CD (¢lled circles).
BBAPRO 36115 7-6-00
D. Apiyo et al. / Biochimica et Biophysica Acta 1479 (2000) 214^224 219

CD (Fig. 5). The parameters obtained from direct ¢tsof the unfolding curves are reported in Table 1. Firsta transition is detected by £uorescence (midpoint at0.58 M GuHCl), then there is a second transitionthat is detected by CD (midpoint at 1.66 M GuHCl).There is no protein-concentration dependence forthese transitions and refolding tests (monitored byfar-UV CD) show that the unfolding of apo-£avo-doxin is fully reversible. The intermediate apo-£avo-doxin state was probed for exposure of hydrophobicsurfaces by ANS £uorescence. ANS is known to in-teract favorably with hydrophobic surfaces and whenso it may increase its quantum yield 1000-fold ascompared to free in aqueous solution. The £uores-cence of ANS in the presence of apo-£avodoxin in1.0^1.5 M GuHCl showed only a small increase (by afactor of 1.35) as compared to in the presence ofnative apo-£avodoxin. Moreover, ANS emissionwas similar in the presence of apo-protein in 1.0 Mand unfolded apo-protein in 3 M GuHCl (data notshown).
The GuHCl-induced unfolding results for apo-£a-vodoxin are very similar to those for holo-£avodox-in, although the cofactor is removed in one case.There is a ¢rst transition, detected by £uorescenceat low GuHCl concentrations, which is followed bya second transition at higher GuHCl concentrations,with the disappearance of the secondary structure(Figs. 4 and 5). Both £uorescence and CD transitionsoccur at slightly lower GuHCl concentrations (0.1^0.3 M) for apo than for holo-£avodoxin. Because ofthe similar m-values for the transitions, the corre-sponding free energies also appear somewhat smallerfor the apo than for the holo-protein (Table 1). Sincethe di¡erences in free energies are too small to besigni¢cant with regard to the magnitude of possibleerror, we conclude that the thermodynamic stabilitiesof apo- and holo-£avodoxin are identical: 20( þ 2)kJ/mol. Thus, coordination of FMN to £avodoxindoes not stabilize the protein structure towardschemically induced unfolding.
4. Discussion
Proteins containing prosthetic groups perform cru-cial cellular functions such as electron transfer, oxy-gen delivery and catalysis. Coordination of the co-
factor often confers structural and functionalintegrity to the holo-protein. Revealing the originsof structure and stability of such proteins, as wellas the determinants for speci¢city of the cofactorbinding, are important goals. Here we report the ¢rststep towards understanding what role the organicFMN cofactor has in folding of D. desulfuricans £a-vodoxin, a protein with an K/L topology [22^24].There have been no other folding studies of a £avo-doxin protein where the FMN cofactor has beenpresent, although a few studies of apo-£avodoxins(from Anabaena and Azotobacter vinelandii) havebeen reported [25,29]. In agreement with our presentresults, it has been shown that apo-£avodoxin foldsalso in the absence of FMN; in fact, NMR experi-ments have shown that apo- and holo-£avodoxinstructures are identical [30].
4.1. Presence of a native-like folding intermediate
As seen in Figs. 4 and 5 (and Table 1), the unfold-ing pro¢les obtained for the GuHCl-induced unfold-ing (of both apo- and holo-£avodoxin) as monitoredby CD (and visible absorption in case of holo) and£uorescence spectroscopy do not coincide. Therefore,in both cases, unfolding cannot be described by sim-ple two-state mechanisms. Since the £uorescence-de-tected transition is complete at the GuHCl concen-trations where the CD-monitored transition occurs,we have analyzed each transition separately using atwo-state approximation. Importantly, the deviationfrom classical two-state behavior is evident for bothapo- and holo-£avodoxin unfolding reactions; there-fore, the presence of the FMN does not cause thise¡ect. Although we cannot rule out other possibil-ities, the reaction behavior can be described underthe assumption that the unfolding occurs throughan intermediate that has an altered structure nearthe tryptophan residue (deduced from £uorescence),but with all secondary structure still present (con-cluded from far-UV CD). It is likely that the £uores-cence change (leading to the intermediate) corre-sponds to a local change near the tryptophan butthe tertiary structure still remains in its native top-ology. In accordance, when using FMN absorptionas a probe of holo-£avodoxin unfolding, the sametransition as that monitored by far-UV CD wasfound (thus formation of the intermediate does not
BBAPRO 36115 7-6-00
D. Apiyo et al. / Biochimica et Biophysica Acta 1479 (2000) 214^224220
a¡ect the FMN-binding site). In the case of apo-£a-vodoxin, ANS £uorescence revealed that the inter-mediate (with altered tryptophan £uorescence) doesnot expose signi¢cantly more hydrophobic-surfacearea than the native protein. The ANS emission in-creased 1.35 times in the presence of apo-£avodoxinunder conditions favoring the intermediate as com-pared to in the presence of native apo-protein. Thismodest increase in ANS emission can be comparedto the 160-fold increase that was found for the Ana-baena apo-£avodoxin molten-globule at low pH [25].Our ANS results thus support a more native- thanmolten-globule-like intermediate structure.
If we assume that £uorescence informs on disap-pearance of the folded state (loss of some tertiaryinteractions) and far-UV CD monitors appearanceof the unfolded state (both folded and intermediateforms have identical CD signals), we can estimate theamount of intermediate species at each GuHCl con-centration. The fractions of native (fN), intermediate(fI) and unfolded (fU) molecules as a function of thedenaturant concentration, are shown in Fig. 6 (holo-£avodoxin, Fig. 6A; apo-£avodoxin, Fig. 6B; de-rived from data in Figs. 4 and 5; see legend for de-tails). At 1.0^1.5 M GuHCl the intermediate speciesof holo- and apo-£avodoxin is populated to almost100%. The correlation between fN, fI and fU appears
very similar for the holo- and apo-£avodoxin reac-tions; excluding an e¡ect from the bound cofactoron the unfolding pathway.
An intermediate during equilibrium unfolding hasalso been reported for apo-£avodoxin from A. vine-landii [29]. Unfolding of the A. vinelandii apo-£avo-doxin shows almost identical characteristics to whatwe ¢nd here for D. desulfuricans £avodoxin. Firstthere is an unfolding transition detected by £uores-cence, then there is another such transition that ismonitored by far-UV CD [29]. The report onA. vinelandii apo-£avodoxin concluded that a highlystable intermediate was involved in the equilibrium-unfolding reaction. From hydrogen exchange mea-surements, it was shown that the structured regionsof this £avodoxin intermediate consisted of most ofthe native ¢ve-stranded L-sheet in the core and twoof the four surrounding helices [29]. Thus, it ap-peared possible that some of the tertiary interactions,locking the surrounding helices to the L-sheet core ofthe protein, had been lost upon forming the inter-mediate species. In both the A. vinelandii and our£avodoxin case, the m-values associated with the£uorescence and CD transitions are around 10 kJ/mol, M (Table 1). Thus, it appears that the sameamount of surface area becomes exposed in eachtransition (folded to intermediate and intermediate
Fig. 6. Native (solid line), intermediate (short-dashed line) and unfolded (long dashed line) populations as a function of the GuHClconcentration for (A) holo-£avodoxin and (B) apo-£avodoxin. The CD and £uorescence data in Fig. 4 (for A) and Fig. 5 (for B) wasused to estimate fraction unfolded (fU) and fraction folded (fF), respectively. The di¡erence (13fU3fF) was then used to calculate theintermediate fraction (fI = 13fU3fF) present at each GuHCl.
BBAPRO 36115 7-6-00
D. Apiyo et al. / Biochimica et Biophysica Acta 1479 (2000) 214^224 221

to unfolded) and therefore the intermediate would behalf unfolded. It is however possible that the ¢rsttransition, leading to the intermediate, exposes lesssurface area than the second transition (in accord-ance with our other results suggesting a native-likestructure) but that instead more aromatic residues(contributing favorably to the size of m) are involved[31]. Our ¢ndings (on both apo- and holo-proteins)and the A. vinelandii apo-study [29] strongly suggestthat the observed three-state unfolding behavior maybe an inherent property of the £avodoxin doublywound K/L topology (and independent of presence/absence of cofactor). This conclusion is however incontrast with the observation that urea unfolding ofAnabaena apo-£avodoxin follows a simple two-statemechanism, although this protein was shown toadopt an intermediate (molten-globule like) structureat low pH [25].
Noteworthy, the £avodoxin from Anabaena wasshown to form a stable, partly folded structureupon excision of the C-terminal helix [32]. The prop-erties of this £avodoxin fragment are similar to thosefound for the D. desulfuricans and A. vinelandii un-folding intermediates. Speci¢cally, the truncated apo-£avodoxin has native-like far-UV CD but more un-folded-like tryptophan emission and its unfoldingtransition is cooperative despite a low thermodynam-ic stability [32]. Thus, the Anabaena £avodoxin frag-ment shares common features with the D. desulfuri-cans and A. vinelandii £avodoxin intermediates. TheAnabaena fragment study supports the hypothesisthat only minor changes in the tertiary structure(such as helix dissociation from the L-sheet core)occur upon forming the intermediates of the full-length proteins.
4.2. No e¡ect of FMN cofactor on £avodoxin stability
We ¢nd no e¡ect on the protein stability arisingfrom the bound FMN cofactor since the unfolding-free energies from the CD-monitored transitions(which report on global unfolding, Table 1) indicatea di¡erence of only 1( þ 2) kJ/mol between apo andholo forms. Thus there is no stabilizing e¡ect to-wards GuHCl-induced unfolding on the £avodoxinstructure that is imposed by the coordinated FMNgroup. This ¢nding is somewhat surprising since ithas been shown for proteins coordinating inorganic
cofactors, that the folded proteins are signi¢cantlystabilized by the cofactor [18,33^35]. For example,cytochrome b562 is at least 14 kJ/mol more stabletowards unfolding when the oxidized heme ispresent, as compared to when in its apo form [18].When the heme is reduced, the stability of holo cy-tochrome b562 increases further [10]. In the case ofthe copper-binding protein azurin, the holo form isstabilized by almost 20 kJ/mol, as compared to thestability of the apo-protein, against GuHCl-inducedunfolding [33].
The holo form of a protein will always be morestable than the apo form if the folded form has ahigher a¤nity for the cofactor than the unfoldedprotein. A thermodynamic cycle, illustrating this re-lationship between cofactor binding and unfolding-free energy of apo and holo forms, is shown in Fig. 7with values for D. desulfuricans £avodoxin. Since theunfolding-free energies do not di¡er for apo- andholo-£avodoxin reactions, it follows from the schemethat the binding constants for the FMN to the foldedand the unfolded polypeptides must also be essen-tially the same. Using the free energies for the lossof secondary structure (the far-UV CD transitions)and the dissociation constant for FMN to foldedD. desulfuricans £avodoxin (KD = 0.1 nM; [23]), weestimated the KD for the FMN in the unfolded stateto be 0.2 nM. Thus FMN is predicted to have a veryhigh a¤nity for unfolded £avodoxin; in good agree-ment with our observation that FMN remains coor-dinated to the polypeptide upon unfolding (at WM
Fig. 7. Thermodynamic cycle connecting folding-free energiesfor apo- and holo-£avodoxin with binding strength of FMN tofolded and unfolded states. Using the unfolding-free energiesdetermined from the CD transitions, and the previously re-ported KD [23] for FMN binding to folded £avodoxin, we esti-mated the dissociation constant for FMN binding to unfolded£avodoxin.
BBAPRO 36115 7-6-00
D. Apiyo et al. / Biochimica et Biophysica Acta 1479 (2000) 214^224222

protein concentrations). Most of the interactions be-tween the FMN and the protein are through twopeptide loops, both of which are not involved ineither the helical or the L-sheet parts characteristicof the folded £avodoxin structure, although thephosphate tail of FMN interacts with the end ofone helix [23,24]. We speculate that most of theseinteractions between the FMN and the native proteincan remain intact when the secondary structure un-ravels upon £avodoxin unfolding and, therefore, thestrength of the FMN binding stays high also in theunfolded state. In contrast, the binding constant ofthe heme group in unfolded cytochrome b562 is in themicromolar range; this is 1000-fold weaker than thebinding to folded cytochrome b562 [18]. In the cyto-chrome b562 case, upon unfolding the heme loses 50%(one of two) of its ligands (the methionine), o¡eringone explanation to why the heme-binding strengthdecreases upon protein unfolding [10].
Since the FMN appears to have such a high a¤n-ity to unfolded £avodoxin, it is possible that afterribosomal translation in vivo, the cofactor binds tothe £avodoxin polypeptide before folding takesplace. Although not stabilizing the native £avodoxinstructure, coordination of the FMN in the unfoldedstate may a¡ect the folding kinetics. This e¡ect couldappear as a decrease in entropy of unfolded holo-£avodoxin, limiting the conformational search forthe native state so that folding progresses faster.
4.3. Conclusions
The in£uence of the organic cofactor FMN on£avodoxin folding has been investigated. Equilibriumstudies of apo and holo D. desulfuricans £avodoxinshow that both unfolding reactions are reversible andboth proceed through native-like intermediates. Thethermodynamic stabilities of £avodoxin with andwithout FMN are identical, indicating that theFMN must bind as strongly to the unfolded stateas it does to native £avodoxin. In agreement, uponunfolding of holo-£avodoxin, we ¢nd the FMN toremain bound to the unfolded polypeptide.
Acknowledgements
We thank Dr. J. LeGall (University of Georgia,
Athens, GA, USA) for providing £avodoxin andDr. W. Walkenhorst (Loyola University, New Or-leans, LA, USA) for the use of his £uorometer. ADreyfus New Faculty Award, a Louisiana Board ofRegents Research Grant and the Keck Foundationare acknowledged for ¢nancial support.
References
[1] S.E. Jackson, Fold. Des. 3 (1998) R81^R91.[2] K.W. Plaxco, C.M. Dobson, Curr. Opin. Struct. Biol. 6
(1996) 630^636.[3] C.M. Dobson, A. Sali, M. Karplus, Angew. Chem. Int. 37
(1998) 868^893.[4] H. Roder, W. Colon, Curr. Opin. Struct. Biol. 7 (1997) 15^
28.[5] R.L. Baldwin, Fold. Des. 1 (1996) R1^R8.[6] K.W. Plaxco, K.T. Simons, D. Baker, J. Mol. Biol. 277
(1998) 985^994.[7] J. Bixler, G. Bakker, G. McLendon, J. Am. Chem. Soc. 114
(1992) 6938^6939.[8] J.R. Winkler, P. Wittung-Stafshede, J. Leckner, B.G. Malm-
strom, H.B. Gray, Proc. Natl. Acad. Sci. USA 94 (1997)4246^4249.
[9] T. Pascher, J.P. Chesick, J.R. Winkler, H.B. Gray, Science271 (1996) 1558^1560.
[10] P. Wittung-Stafshede, H.B. Gray, J.R. Winkler, J. Am.Chem. Soc. 119 (1997) 9562^9563.
[11] P. Wittung-Stafshede, B.G. Malmstrom, J.R. Winkler, H.B.Gray, J. Phys. Chem. 102 (1998) 5599^5601.
[12] J. Leckner, P. Wittung, N. Bonander, B.G. Karlsson, B.G.Malmstrom, J. Biol. Inorg. Chem. 2 (1997) 368^371.
[13] P. Wittung-Stafshede, B. Malmstrom, D. Sanders, J. Fee, J.Winkler, H.B. Gray, Biochemistry 37 (1998) 3172^3177.
[14] R. Holm, E. Solomon, Chem. Rev. 96 (1996) 2237^3030.[15] D.L. Luisi, W.-J. Wu, D.P. Raleigh, J. Mol. Biol. 286 (1999)
395^407.[16] D.R. Shortle, Curr. Opin. Struct. Biol. 6 (1996) 24^30.[17] P. Wittung-Stafshede, M.G. Hill, E. Gomez, A. Di Bilio, G.
Karlsson, J. Leckner, J.R. Winkler, H.B. Gray, B.G. Malm-strom, J. Biol. Inorg. Chem. 3 (1998) 367^370.
[18] C.R. Robinson, Y. Liu, J.A. Thomson, J.M. Sturtevant,S.G. Sligar, Biochemistry 36 (1997) 16141^16146.
[19] I. Bertini, J.A. Cowan, C. Luchinat, K. Natarajan, M. Pic-cioli, Biochemistry 36 (1997) 9332^9339.
[20] P. Wittung-Stafshede, Biochim. Biophys. Acta 1382 (1998)324^332.
[21] S.E. Brenner, Curr. Opin. Struct. Biol. 7 (1997) 369^376.[22] S.G. Mayhew, G. Tollin, in: F. Muller (Ed.), Chemistry and
Biochemistry of Flavoenzymes, Vol. 3, CRC Press, BocaRaton, FL, 1992, pp. 389^426.
[23] J. Calderia, N. Palma, M. Regalla, J. Lamperia, J. Calvete,W. Schafer, J. LeGall, I. Moura, J. Moua, Eur. J. Biochem.220 (1994) 287^295.
BBAPRO 36115 7-6-00
D. Apiyo et al. / Biochimica et Biophysica Acta 1479 (2000) 214^224 223

[24] A. Romero, J. Caldeira, J. LeGall, I. Moura, J. Moura, M.Romao, Eur. J. Biochem. 239 (1996) 190^196.
[25] C.G. Genzor, A. Beldarrain, C. Gomez-Moreno, J.L. Lopez-Lacomba, M. Cortiljo, J. Sancho, Prot. Sci. 5 (1996) 1376^1388.
[26] M.M. Santoro, D.W. Bolen, Biochemistry 27 (1988) 8063^8068.
[27] N.C. Pace, B.A. Shirley, J.A. Thompson, in: T.F. Chreight-on (Ed.), Protein Structure: a Practical Approach, IRLPress, Oxford, 1990, pp. 311^330.
[28] J.A. D'Anna, G. Tollin, Biochemistry 11 (1972) 1073^1080.[29] C. van Mierlo, W. van Dongen, F. Vergeldt, W. van Berkel,
E. Steensma, Protein Sci. 7 (1998) 2331^2344.
[30] E. Steensma, C. van Mierlo, J. Mol. Biol. 282 (1998) 653^666.
[31] J.K. Myers, C.N. Pace, J.M. Scholtz, Protein Sci. 4 (1995)2138^2148.
[32] S. Maldonado, M.A. Jimenez, G.M. Langdon, J. Sancho,Biochemistry 37 (1998) 10589^10596.
[33] J. Leckner, N. Bonander, P. Wittung-Stafshede, B.G. Malm-strom, B.G. Karlsson, Biochim. Biophys. Acta 1342 (1997)19^27.
[34] C.J. Falzone, M.R. Mayer, E.L. Whiteman, C.D. Moore,J.T.G. Lecomte, Biochemistry 35 (1996) 6519^6526.
[35] M.S. Hargrove, J.S. Olson, Biochemistry 35 (1996) 11310^11318.
BBAPRO 36115 7-6-00
D. Apiyo et al. / Biochimica et Biophysica Acta 1479 (2000) 214^224224

![Mutualistic Growth of the SulfateReducer Desulfovibrio ...digital.csic.es/bitstream/10261/79980/1/Mutualistic growth of the... · Meyerhof–Parnas pathway [9, 10], the degradation](https://img.pdfslide.us/doc/110x75/5e68148dbef0cd325b1073c5/mutualistic-growth-of-the-sulfatereducer-desulfovibrio-growth-of-the-meyerhofaparnas.jpg)

















![Mutualistic Growth of the SulfateReducer Desulfovibrio ... · Meyerhof–Parnas pathway [9, 10], the degradation pathway of PTStransported sugars, indicates that mannose class sugars](https://img.pdfslide.us/doc/110x75/5e68148dbef0cd325b1073c4/mutualistic-growth-of-the-sulfatereducer-desulfovibrio-meyerhofaparnas-pathway.jpg)









