Embed Size (px)
Citation preview

Publications of the University of Eastern Finland
Dissertations in Health Sciences
isbn 978-952-61-0909-1
Publications of the University of Eastern FinlandDissertations in Health Sciencesd
issertation
s | 133 | Nico
lae-C
ostin
Dia
con
u | M
ast Cells in H
uman E
pithelial C
ancers
Nicolae-Costin DiaconuMast Cells in Human
Epithelial Cancers
Nicolae-Costin Diaconu
Mast Cells in Human Epithelial Cancers
The possible role of MC mediators
was investigated in SCC and BCC
carcinomas. Novel information on
the MC proteinases tryptase and
chymase in SCC and BCC carcinomas
is presented and their potential to
degrade the extracellular matrix is
discussed. Moreover, the presence
MCs cytokines and their counter-
part receptors is also described. The
results demonstrate that chymase
may release viable tumoral cells
that tolerate the cytolytic action of
TNF-α and histamine leading to the
spread of SCC. In BCC, MCs express
TNF-α and CD30 ligand but the lack
of CD40L may be responsible of the
inefficient antitumoral response.
brought to you by COREView metadata, citation and similar papers at core.ac.uk
provided by UEF Electronic Publications

NICOLAE-COSTIN DIACONU
Mast Cells in Human Epithelial Cancers
To be presented by permission of the Faculty of Health Sciences, University of Eastern
Finland for public examination in auditorium of Kuopio University Hospital, Kuopio,
on Friday, October 12th 2012, at 13 o`clock
Publications of the University of Eastern Finland Dissertations in Health Sciences
Number 133
Department of Dermatology, School of Medicine, Faculty of Health Sciences, University of Eastern Finland Kuopio
2012

Kopijyvä Oy Kuopio, 2012
Series Editors:
Professor Veli-Matti Kosma, M.D., Ph.D. Institute of Clinical Medicine, Pathology
Faculty of Health Sciences
Professor Hannele Turunen, Ph.D. Department of Nursing Science
Faculty of Health Sciences
Professor Olli Gröhn, Ph.D. A.I. Virtanen Institute for Molecular Sciences
Faculty of Health Sciences
Distributor: University of Eastern Finland
Kuopio Campus Library P.O.Box 1627
FI-70211 Kuopio, Finland http://www.uef.fi/kirjasto
ISBN (print): 978-952-61-0909-1 ISBN (pdf): 978-952-61-0910-7
ISSN (print): 1798-5706 ISSN (pdf): 1798-5714
ISSN-L: 1798-5706

III
Author’s address: School of Medicine
University of Eastern Finland KUOPIO FINLAND
Supervisors: Professor Ilkka T. Harvima, M.D., Ph.D.
Department of Dermatology/School of Medicine University of Eastern Finland KUOPIO FINLAND
Reviewers: Docent Pekka Autio, M.D., Ph. D., Assoc. prof. Department of Dermatology University of Oulu OULU FINLAND
Ulrich Blank, Ph.D. Inserm U699 Faculty of Medicine Denis Diderot University of Paris 7 PARIS FRANCE
Opponent: Professor Antti Lauerma, M.D., Ph.D.
Department of Dermatology University of Helsinki HELSINKI FINLAND

IV

V
Diaconu, Nicolae-Costin Mast Cell in Human Epithelial Cancers, 78 p. University of Eastern Finland, Faculty of Health Sciences, 2012 Publications of the University of Eastern Finland. Dissertations in Health Sciences, 133. 78 p. ISBN (print): 978-952-61-0909-1 ISBN (pdf): 978-952-61-0910-7 ISSN (print): 1798-5706 ISSN (pdf): 1798-5714 ISSN-L: 1798-5706 ABSTRACT Mast cells (MCs) release a variety of biologically potent mediators exerting their effects either locally or systemically. Many of these are preformed mediators stored in the cytoplasmic granules, while others are produced de novo upon MC activation. During the last 15 years, human MCs have also been recognized as a source of cytokines and they can express a wide range of cell surface molecules. All these molecules may participate in carcinoma development through matrix degradation, immunomodulation, angiogenesis or tumor cell proliferation. In this work, the possible role of MC mediators histamine, heparin, tryptase, chymase, TNF-α and other TNF superfamily ligands CD30L and CD40L was investigated in squamous (SCC) and/or basal cell (BCC) carcinomas. The expression of these factors in SCC and BCC and the possible involvement of mediators in the growth, migration and adherence of uterine cervical SCC cells (SiHa and ME-180 cell lines) were studied by using histochemical and cell culture techniques. The results show that in superficial spreading BCC, the number of tryptase- and chymase-positive MCs (MCTC cells) is increased, possibly as a consequence of Kit receptor activation by the stem cell factor. Even though tryptase was apparently fully active in the BCC lesion, chymase was partially inactivated possibly due to action of α1-proteinase inhibitor and α1-antichymotrypsin localized to mast cells. However, residual chymase activity may still cause matrix degradation and other effects in the lesion. In addition, MCs express CD30L and TNF-α, but not substantially CD40L, in the BCC lesion. The counterpart receptors of all these ligands are present in the lesional epidermis or dermis, but basal buds were negative for TNFRI and CD40. In uterine cervical SCC specimens, MCTC cells were detected in substantial numbers in the peritumoral stroma, and in vitro especially chymase was capable of detaching viable and growing cervical SCC cells from substratum. The amount of chymase inhibitor SCCA-2 in cervix cancer cells is low as only weak immunopositivity for this antigen was found. In contrast to normal keratinocytes where TNF-α and histamine together had a profound growth-inhibitory and cytolytic effect, these mediators did not affect markedly the cervical SCC cells. Based on these results, chymase may release viable SCC cells from a tumor leading to spreading of SCC. In addition, SCC cells may have lost their responsivity to the cytolytic action of TNF-α and histamine, a conclusion based partly on the result that SiHa and ME-180 cells and cervix SCC expressed only relatively low levels of TNFRI and II. In the BCC lesion, significant changes were found in MCTC cells pointing to an intimate involvement in pathogenesis. Decreased chymase activity may not be able to degrade TNF-α or cleave the malignant epithelium away, and the lack of CD40L in MCs may result in inefficient antitumoral response. National Library of Medicine Classification:

VI
Medical Subject Headings: Mast cell; Basal Cell Carcinoma; Squamous Cell Carcinoma; Chymase; Tryptase; TNF-α; Histamine; TNFRI; TNFRII; TNF ligand; CD30; CD30L; CD40; CD40L

VII

VIII
Acknowledgements
This study was conducted at the Department of Dermatology, School of Medicine, University of Eastern Finland, Kuopio, during the years 2004-2012. I would like to express my deepest gratitude to my supervisor, Professor Ilkka Harvima, MD, PhD, for giving me the opportunity to be part of the scientific community, for his support, continuous guidance, meticulous suggestions and astute criticism during the practical phase, for his kindness and inexhaustible patience during the correction phase of this dissertation. I am also deeply grateful to Docent Rauno Harvima, MD, PhD, for providing me with the opportunities for research work, for his unique skill to always create a positive atmosphere at work, being there when I needed help and support. I greatly appreciate the help provided by Docent Anita Naukkarinen PhD, and Jaana Rummukainen MD, PhD, for preparing and performing the evaluation of histological samples. All their help and advice is appreciated and valued. I am also deeply grateful to Mikko Mättö, Ph.D, for his guidance and help with flow cytometric analysis. I wish to express my gratitude to Professor Gunnar Nilsson PhD, for his invaluable help and guidance. I thank the official reviewers of this thesis, Docent Pekka Autio, MD, PhD and Ulrich Blank, PhD, for their constructive comments and valuable aid in the final preparation of the work. I wish to express my gratitude to all the co-authors and other collaborators of the published manuscripts: Renata Kaminska, MD, PhD, for his work and help on articles and Professor Jukka Pelkonen, MD, PhD for providing valuable comments on the manuscripts. I greatly appreciate the help provided by Ewen MacDonald, Pharm. D., for correcting the English language of this thesis. I am also deeply grateful to Anne Koivisto and Katja Dufva, for their excellent technical assistance and friendship, for their time spent and never-failing interest in this research during all these years. I also express my warmest thanks to Hannele Virnes and Marianne Tokola for their support during these years and friendship. My sincere gratitude goes to everyone in the MCCID-team for their positive encouragement and warm support. I owe my warmest thanks to the collegues and the personnel of the Department of Dermatology, Kuopio University Hospital, for supporting me and my work with great attitude.

IX
I also express my warmest thanks to all my relatives. I owe my sincere thanks to my parents, Dinica and Nicolae, for all the help, never-ending love, enormous support and encouragement during my whole life and just being there for me whenever I needed. Thank You Camelia, Ana-Maria and Sânziana for being the great joy of my life. I thank to my friends for their long-lasting loyalty and endless support as well as enjoyable and memorable moments, especially to Rio Dumitrascu, Corina and Jussi Pennanen who helped me in my hardest moments. This work was supported by Marie Curie Mobility Actions for Early Stage Researcher by the sixth framework of the EU commission (project no. 504926), the Finnish Cultural Foundation, Cancer Center of Eastern Finland and Kuopio University Hospital, which I gratefully acknowledge. Kuopio, October 2012
Nicolae-Costin Diaconu

X
List of the original publications ´This thesis is based on the following original publications which will be referred to by their Roman numerals:
I Diaconu N-C, Kaminska R, Naukkarinen A, Harvima RJ, Harvima IT. The increase in tryptase- and chymase-positive mast cells is associated with partial inactivation of chymase and increase in protease inhibitors in basal cell carcinoma. J Eur Acad Dermatol Venereol 2007; 21: 908–915.
II Diaconu N-C, Kaminska R, Naukkarinen A, Harvima RJ, Nilsson G, Harvima IT. Increase in CD30 ligand/CD153 and TNF-α expressing mast cells in basal cell carcinoma. Cancer Immunol Immunother 2007; 56: 1407-1415.
III Diaconu N-C, Rummukainen J, Mättö M, Naukkarinen A, Harvima RJ, Pelkonen
J, Harvima IT. Cervical squamous carcinoma cells are resistant to the combined action of tumor necrosis factor-α and histamine whereas normal keratinocytes undergo cytolysis. BMC Cancer 2008; 8: 46.
IV Diaconu NC, Rummukainen J, Naukkarinen A, Mättö M, Harvima RJ, Pelkonen J, Harvima IT. Mast cell chymase is present in uterine cervical carcinoma and it detaches viable and growing cervical squamous carcinoma cells from substratum in vitro. Arch Dermatol Res 2011; 303: 499-512.
The publications were adapted with the permission of the copyright owners.

XI

XII
Contents
1 INTRODUCTION ...................................................................................................................... 1
2 REVIEW OF THE LITERATURE ............................................................................................ 3 2.1 General characteristics of MCs ............................................................................................................... 3
2.1.1 Activation of MCs .............................................................................................................. 5 2.2 MC mediators ................................................................................................................................................. 6
2.2.1 Histamine ............................................................................................................................ 6 2.2.2 Heparin ................................................................................................................................ 8 2.2.3 TNF-α ................................................................................................................................... 9 2.2.4 Tryptase ............................................................................................................................. 10 2.2.5 Chymase ............................................................................................................................ 12 2.2.6 IL-8 ...................................................................................................................................... 14 2.2.7 Other MC mediators ........................................................................................................ 15
2.3 Non-melanoma skin cancers .................................................................................................................. 15
2.3.1 BCC ..................................................................................................................................... 16 2.3.2 SCC ..................................................................................................................................... 17
2.4 Cervical squamous cell carcinoma ...................................................................................................... 19
2.5 MCs and their mediators in cancer ..................................................................................................... 20
2.6 TNF ligand and receptor superfamily ............................................................................................... 23
2.6.1 CD30 ligand (CD30L) and CD30 receptor .................................................................... 24 2.6.2 CD40 ligand (CD40L) and CD40 receptor .................................................................... 25 2.6.3 TNF receptors I and II ..................................................................................................... 28
2.7 Serine (or cysteine) proteinase inhibitor (SCCA 1 and SCCA 2) ........................................... 29
3 AIMS OF THE STUDY ............................................................................................................ 31
4 MATERIALS AND METHODS ............................................................................................. 33 4.1 Tissue samples .............................................................................................................................................. 33
4.2 Immunohistochemistry ............................................................................................................................ 34
4.3 Cell cultures ................................................................................................................................................... 36
4.3.1 Apoptosis assays .............................................................................................................. 36 4.3.2 Cell growth determination by fluorometric DNA assay ............................................ 37 4.3.3 Determination of keratinocyte growth and migration ............................................... 37 4.3.4 Determination of SiHa cell migration and invasion using an in vitro transwell assay ............................................................................................................................................ 38 4.3.5 Adherence onto fibronectin-coated plastic surface assay .......................................... 38 4.3.6 Determination of cell viability and cytotoxicity .......................................................... 39 4.3.7 Determination of cell cycle by flow-cytometry ............................................................ 39
4.4 Cytokines and MC mediators ................................................................................................................ 40
5 RESULTS .................................................................................................................................... 41

XIII
5.1 Tryptase- and chymase-positive MCs are increased in number and are associated with protease inhibitors in BCC (I) ............................................................................................................ 41
5.2 BCC lesions display increased CD30L/CD153 and TNF-α expressing MCs (II) ............ 41
5.3 TNF-α and histamine do not influence the growth of squamous carcinoma cells from uterine cervix whereas normal keratinocytes undergo cytolysis (III) ....................................... 43
5.4 MC chymase is present in uterine cervical carcinoma and is capable of detaching viable and growing cervical squamous carcinoma cells from substratum (IV) .................... 45
6 DISCUSSION ............................................................................................................................ 48 6.1 Partial inactivation of chymase and increase in protease inhibitors in BCC ................... 48
6.2 TNF-α and CD30L but not CD40L immunopositive MCs in BCC lesion ......................... 51
6.3 TNF-α and histamine have a significant growth-inhibitory and cytolytic effect on normal keratinocytes but not on cervix carcinoma cell lines SiHa and ME-180 ................... 53
6.4 Is MC chymase able to release SCC cells from tumor leading to spreading of malignant cells? ................................................................................................................................................... 56
7 CONCLUSIONS ........................................................................................................................ 61
REFERENCES ................................................................................................................................ 63
APPENDIX I: ORIGINAL PUBLICATIONS I-IV
XIV
Abbreviations
α1-AC α1-Antichymotrypsin α1-PI α1-Proteinase inhibitor BCC Basal cell carcinoma bFGF Basic fibroblast growth factor BPE bovine pituitary extract BSA Bovine serum albumin CD30L CD30 ligand CD40L CD40 ligand CIN Cervical intraepithelial neoplasia CIS Carcinoma in situ CRD Cysteine rich domain D-PBS Dulbecco's Phosphate Buffered Saline DcR1 Decoy receptor 1 DFP Diisopropyl fluorophosphate DMEM Dulbecco's Modified Eagle Medium DMSO Dimethyl sulfoxide DNA Deoxyribonucleic acid Ecs Endothelial cells EDTA Ethylenediaminetetraacetic acid EGF Epidermal growth factor FCS Fetal calf serum FGF-2 Fibroblast growth factor-2 HPV Human papilloma virus ICAM-1 Intercellular adhesion molecule-1 IL Interleukin MC/MCs Mast cell/Mast cells MCC Mast cell chymase positive MCT Mast cell tryptase positive MCTC Mast cell chymase and tryptase positive MEM Minimum essential medium mRNA Messenger ribonucleic acid MTT (3-(4,5-Dimethylthiazol-2-yl)-
2,5- diphenyltetrazolium bromide NK cells Natural killer cells OCT Optimal cutting temperature medium OPG Osteoprotegerin PAR-2 Protease-activated receptor-2 PDGF Platelet-derived growth factor PLAD Pre-ligand binding assembly domain PUVA psoralen + UVA photochemotherapy Rh-chymase Recombinant human chymase Rh-TNF-α Recombinant human TNF-α SCC Squamous cell carcinoma SCCA Squamous cell carcinoma antigen SBTI Soybean trypsin inhibitor SCF Stem cell factor SDS-PAGE Sodium dodecyl sulfate polyacrylamide gel electrophoresis SERPINB3 Serpin peptidase inhibitor, clade B (ovalbumin), member 3 SLPI Secretory leukocyte protease inhibitor TLCK Tosyl-L-lysine chloromethyl ketone TLR Toll-like receptor TNF-α Tumor necrosis factor-α TNFRI Tumor necrosis factor receptor type I TNFRII Tumor necrosis factor receptor type II TGF Transforming growth factor TR6 TNF receptor-like-6 UV Ultraviolet

XV
VEGF Vascular endothelial growth factor

XVI

1 Introduction
The two most common skin cancer types are basal cell carcinoma (BCC) and squamous cell
carcinoma (SCC). The incidence of these epithelial and non-melanoma cancer types has
been increased in the Australian`s white population in an almost epidemic fashion during
the last 30 years [1]. There is clear epidemiological evidence for a role for UV radiation in
the development of SCC. The increased incidence of SCC in sunny climates is related to the
skin type [2]. Basal cell keratinocytes are more tolerant to UV radiation than squamous cells
because of their stem cell type properties [3]. Intermittent intense sun exposure has been
considered to be one significant risk factor for BCC [4,5], whereas in the case of SCC, the
cumulative sun exposure over the lifetime has been incriminated [6,5]. Moreover, a history
of sunburn in childhood has been described as a significant risk factor for the development
of BCC [7,8].
The most common form of cervical cancers of the uterus is SCC which usually arises from
the metaplastic squamous mucosa in the region of the transformation zone [9]. The risk of
spreading increases with the depth of the invasion and is less than 1% in the early stages.
Virtually all cervical SCCs are positive for human papilloma viruses (HPV) and HPV is
considered to be the main causal factor for the development of cervical cancer [10]. Only a
limited number of viral types of the family of the HPVs are associated with the etiology of
SCC. Co-factors that increase the risk among HPV DNA positive women include smoking,
high parity, the use of oral contraceptives for five or more years and previous exposure to
other sexually transmitted diseases such as Chlamydia trachomatis and Herpes simplex Virus
type 2. Human immunodeficiency virus (HIV) increases also the risk for HPV infection,
HPV DNA persistency and progression of HPV lesions to cervical cancer [11].
It is well-known that mast cells (MCs) can be found in high numbers in a variety of cancer
types, especially in the invasion zone [12,13]. Moreover, it is known that MCs are involved
in cancer invasion directly by their own proteases and indirectly via interactions with other

2
cells [14]. However, little is known of the significance of MCs in epithelial cancer survival,
growth and invasion. Therefore, the purpose of this study was to investigate MCs and their
major mediators in BCC and SCC. In the first studies, I and II, the main focus was the
expression of MC tryptase and chymase as well as TNF superfamily ligands in MCs in
BCC. In the works III and IV, the possible role and function of MC mediators, including
histamine, heparin, TNF-α, tryptase and chymase, were studied in uterine cervix SCC.

3
2 Review of the literature
2.1 GENERAL CHARACTERISTICS OF MCS
MCs were first reported by von Recklinghausen and coworkers in 1863 and were identified
in 1877 by Paul Ehrlich who observed cells with metachromatically staining granules in
connective tissue stained with an aniline dye and named them "mastzellen", meaning "well-
fed-cells". In particular, during the last decades, MCs have attracted the interest of many
investigators throughout the world. MCs are large (10 to 15 µm in diameter), ovoid,
fusiform or triangular mononuclear cells which in their cytoplasm display specific exocrine
granules enclosed by a single membrane that are particularly avid for metachromatic dyes,
such as toluidine blue, Giemsa stain, and methylene blue [15]. MCs are derived from
pluripotent haematopoetic stem cells in bone marrow [16,17], from where undifferentiated
MC progenitor cells subsequently migrate and circulate in the blood and lymphatic system
before migrating to their target tissues, especially to various mucosal surfaces and skin
where they proliferate, differentiate and mature into histologically identifiable MCs
[18,19,20]. MC migration and differentiation is influenced by several cytokines, such as
interleukins (IL), IL-3, IL-4, IL-6 and IL-9, and also by nerve growth factor [21,22] and SCF,
which are secreted locally by stromal cells, such as fibroblasts [23]. These are the only
cytokines that promote the development of MCs in adult human system [24,17]. MCs,
ubiquitous cells located in all mammalian connective tissues, are often found in the
proximity of blood and lymph vessels, in the skin, and in the mucosa of the upper and
lower respiratory tracts and the gastrointestinal tract [25,26,18], where they play their
primary defense role as the “sentinel cells” at the interface of the host and environment.
MC subpopulations can be defined on the basis of response specificity [27], tissue migration

4
patterns [25,28] or granule content [28]. Two MC phenotypes, based on their proteinase-
content, tryptase and chymase, have been described in humans: MCT cells contain only
tryptase as their neutral serine proteinase and MCTC cells have both tryptase and chymase
and they have primarily been found in the skin and gastrointestinal submucosa. [29,25,30].
Chymase-positive and tryptase-negative MCC MCs which are rich in chymase and
carboxypeptidase have been identified at several body sites, though not markedly in
normal human skin and, in fact many investigators have failed to detect them [30,31,32,33].
In addition, there is experimental evidence from skin organ cultures that the chymase-
positive and tryptase-negative MC may represent an apoptotic MC from which tryptase has
dissolved away whereas chymase has remained at the site of the dying cell [34,35].
However, it has been speculated that there is much more heterogeneity due to the large
family of proteases that are differentially expressed in MCs. Furthermore, the relative
amount of tryptase and chymase in MCs may be regulated by cytokines, such as IL-4 [36].
Bonini et al. has described MC heterogeneity with respect to Toll-like receptor (TLR)
expression [37].
MC stimulation can lead to different responses: exocytosis of secretory granules containing
proteoglycans and other preformed mediators of immediate hypersensitivity; synthesis and
secretion of newly-generated mediators, typically produced during IgE-mediated activation
such as arachidonic acid metabolites, principally leukotriene C4 (LTC4) and prostaglandin
D2 (PGD2); and synthesis and secretion of cytokines such as tumor necrosis factor-α (TNF-
α), IL-4, IL-5 and IL-6 [38].
MCs are involved in various physiological, immunological and pathological processes,
particularly in allergic reactions [39], but also in lipoprotein metabolism [40], in the defense
against parasites and bacteria [41], in gastric acid secretion [42], in autoimmune diseases
[43], MC disorders (mastocytosis), tissue remodelling and these cells are believed to be
involved also in tumorigenesis and skin cancer progression [14].

5
2.1.1 Activation of MCs
An important function of MCs is their ability to release inflammatory mediators by
degranulation, which involves a complex interaction of signaling molecules. FcεRI
receptors that bind the Fc portion of immunoglobulin (IgE) antibody with high affinity are
expressed on the MC surface [44]. Over 70% of the dry weight and 50% of its volume [45] is
represented by granules, the most specific morphologic feature of MCs [46]. MCs can be
activated by several diverse stimuli: via IgE-FcεRI binding and antigen recognition, which
leads to receptor crosslinking and aggregation [47], by tryptic proteases interacting with the
PAR-2 receptor on skin MCs including the involvement of tryptase [48], by histamine
releasing factors secreted by neighboring T lymphocytes [49] or macrophages [50], by
components of the complement system (C3a, C5a) [42], cytokines (e.g., SCF, TNF-α, IFN-γ),
adenosine, TLR ligands, neuropeptides and hyperosmolality [51]. A novel mechanism for
degranulation-independent secretion of chemokines from MCs via reverse signaling upon
CD30L-CD30 interaction has recently been described [52]. However, the mechanisms for
MC activation in chronic inflammatory diseases have not been completely elucidated (Fig.
1) [51].
Figure 1. Mast cell activation after interaction with different ligands, cytokines and T cells (see
reference [53] for background)

6
MC activation is followed by the process of exocytosis called degranulation when the cytoplasmic
secretory granule content is rapidly expelled into the extracellular milieu. The exocytosis mechanism
involves the trafficking of secretory granules to release sites. The granule’s trafficking is controlled
by a cytoskeletal barrier composed of actin and myosin which undergoes extensive reorganization to
allow secretory granules to dock and fuse with the plasma membrane to release their soluble granule
contents [54,55,56]. Finally, the vesicles are recycled by clathrin-mediated endocytosis and the lost
preformed mediators through rapid onset of de novo synthesis allowing formation of new secretory
granules [57]. MCs release bioactive molecules including those contained in cytoplasmic granules
that elicit immediate reactions and those synthesized de novo upon stimulation that exert rather long-
lasting effects [58,59,60,61].
2.2 MC MEDIATORS
Numerous powerful MC mediators, such as serine proteinases tryptase and chymase,
histamine, heparin, leukotriene C4, prostaglandin D2, and a wide range of cytokines,
chemokines and growth factors, enable MC participation in both adaptive and innate
immune responses [59,60,61]. The release of cytokines in injured skin may have an
important role in the development of many inflammatory skin disorders [62]. For example,
in chronic cutaneous inflammatory diseases, MCs exhibit increased levels of IL-4, TNF-α,
and interferon-gamma immunoreactivity [62,63,64]. Furthermore, MCs are the
predominant TNF-α positive cell type in psoriasis and atopic dermatitis and the number of
TNF-α positive MCs was found to be increased in the upper dermis of these skin diseases
[62]. Human MCs evidently express also many other members of the TNF superfamily,
such as CD30L, CD40L, 4-1BBL and OX40L [65,66,67].
2.2.1 Histamine
Histamine [2-(4-imidazolyl)-ethylamine], a part of imidazoleamine group, is a biogenic
amine derived from the decarboxylation of the amino acid L-histidine, a reaction catalyzed
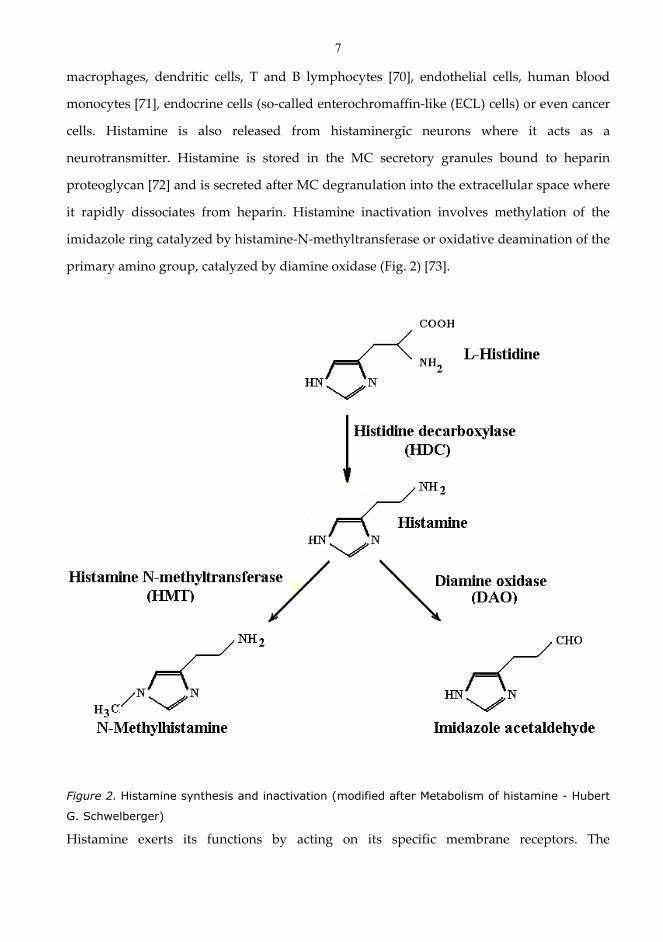
by the enzyme L-histidine decarboxylase (Fig. 2). The major sources of histamine in the
tissues are MCs [68,69] but there are also other histamine secretors like basophils,
7
macrophages, dendritic cells, T and B lymphocytes [70], endothelial cells, human blood
monocytes [71], endocrine cells (so-called enterochromaffin-like (ECL) cells) or even cancer
cells. Histamine is also released from histaminergic neurons where it acts as a
neurotransmitter. Histamine is stored in the MC secretory granules bound to heparin
proteoglycan [72] and is secreted after MC degranulation into the extracellular space where
it rapidly dissociates from heparin. Histamine inactivation involves methylation of the
imidazole ring catalyzed by histamine-N-methyltransferase or oxidative deamination of the
primary amino group, catalyzed by diamine oxidase (Fig. 2) [73].
Figure 2. Histamine synthesis and inactivation (modified after Metabolism of histamine - Hubert
G. Schwelberger)
Histamine exerts its functions by acting on its specific membrane receptors. The

8
inflammatory wheal and flare response is attributable to the H1 receptor [74]. The H2
receptor controls typically gastric acid secretion in the gut [75]. The H3 receptor is involved
in neurotransmitter release in the central nervous system [76]; The H4 receptor, primarily
expressed in immune cells, has been shown to be involved in chemotaxis and mediator
release in various types of immune cells, including MCs, eosinophils, dendritic cells and T
cells [77]. Histamine, a potent vasoactive agent, a constrictor of bronchial smooth muscle
and the major mediator of the acute inflammatory and immediate hypersensitivity
responses [78], can affect also chronic inflammation and regulate several essential events in
the immune response.
Histamine is the best known endogenous agent that evokes itch [79]. In acute urticaria
histamine is the major mediator released from MC [80]. Even though the essential role of
histamine in acute dermatological skin diseases is well-known, the role of histamine in the
epidermis is still largely unknown in chronic skin diseases.
2.2.2 Heparin
Heparin, a highly sulfated member of the glycosaminoglycan (GAG) family, is a well
known anticoagulan. It also possesses several non-anticoagulant properties including
modulation of various proteases [81,82], inhibition of cell growth [83,84] reduction of
inflammatory responses [85,86] and recently an anti-cancer effect in experimental animal
models [87], modulations of angiogenesis [88], tumor metastasis [89,90,91,92], and viral
invasion [93,94,95,96,97,98].
Heparin is stored, together with histamine, MC proteases and other mediators, in the
secretory granules of MCs and it is essential for the storage of specific granule proteases in
MCs [99,100,101]. Histamine is stored in MC granules by electrostatic interactions with the
highly negatively charged heparin, binding site-specifically to it [102,103], even though
intragranular histamine is mobile [104]. The details of the interaction between heparin and
positively charged MC proteases are not known, but it is thought that the interaction is
utilized by MCs to ensure that only properly folded proteases are targeted to the secretory

9
granule [99]. The release of heparin from these granules in response to injury and its
subsequent entry into the bloodstream leads to inhibition of blood clotting.
Heparin is expressed in connective-tissue type MCs, where it is biosynthesized as heparin
proteoglycan [105]. The binding of heparin to MC proteinases and other protein mediators
can have pronounced effects on the physicochemical and biological functions of the
mediators. Heparin is essential for keeping tryptase stabile as an enzymatically active
tetramer [106,107]. Heparin also binds to chymase modulating the detachment of cultured
keratinocytes [108]. Furthermore, heparin binds efficiently to TNF-α potentiating its
growth-inhibitory action on cultured keratinocytes [109].
Heparin has many properties impacting on carcinogenesis and metastasis including
mitogenic effect on endothelial cells [110,111], stimulation of the migration of cultured
capillary endothelial cells [112] and an anticoagulant effect preventing microthrombi
formation in the new vessels, which helps propagation of metastases.
2.2.3 TNF-α
TNF-α was isolated as a soluble factor released by lymphocytes and macrophages that
caused the lysis of a transplanted tumor (sarcoma Meth A) [113]. TNF-α is a
multifunctional cytokine originally described as a molecule with antitumor properties and
it is involved in numerous physiological and pathophysiological processes, being the
prototype of an inflammatory cytokine [114,115].
TNF-α is mainly produced by macrophages, but also by a diversity of other cells including
lymphoid cells, MCs, endothelial cells, fibroblasts and neuronal tissue. The MC is the only
cell type that can store preformed TNF-α in granules and release it rapidly upon activation
[116]. MCs can also produce large amounts of TNF-α in response to activation. MC TNF-α
increases vascular permeability [117], stimulates the expression of adhesion molecules
(ICAM-1 and VCAM-1) on endothelial cells facilitating leukocyte migration to the site of
inflammation [118,119], provides maturation and migration signals to dendritic cells,
enhancing the development of immune responses [120,121] and activates macrophages and

10
neutrophils for inflammatory mediator production [122,123]. In addition, TNF-α induces
apoptosis by a cytotoxic effect mediated via TRAIL (TNF related apoptosis-inducing
ligand) in a variety of tumor cells, but generally not in normal cells [124]. Moreover, MC-
derived TNF also stimulates T-lymphocyte secretion of the gelatinase MMP-9, which
cleaves type IV collagen and may facilitate the migration of leukocytes between tissue
compartments [125].
TNF-α has a large spectrum of bioactivities representing a major proinflammatory
mediator, with an optional capacity to induce apoptosis. Under certain conditions, TNF-α
has a distinctive functional duality, being involved in opposite processes such as tissue
regeneration and destruction [126]. Together with other cytokines, TNF-α is recognized as
to be a key player in the development of septic shock as high concentrations of TNF induce
shock-like symptoms. On the other hand, the prolonged exposure to low concentrations of
TNF can result in the wasting syndrome, cachexia, experienced by tumor patients [127].
TNF-α is identified as a key mediator in UV-induced local immunosuppression and its
levels are increased in UV-exposed skin where it may act by altering Langerhans cell
morphology and function [128]. Furthermore, large amounts of MC TNF-α are stored and
released upon activation in UV-induced systemic immunosuppression [129].
TNF-α has frequently been detected in human cancer biopsies, produced either by
epithelial or stromal tumour cells. The production of TNF-α has been associated with a
poor prognosis, loss of hormone responsiveness and asthenia [130,131]. Detected in serum
of some cancer patients, TNF-α concentrations are elevated in relation to the extent of
disease. Moreover, TNF-α concentrations are also correlated with serum concentrations of
IL-8 [132].
2.2.4 Tryptase
Tryptase was discovered in 1960 as a trypsin-like activity in MCs [133], and it is the major
type of proteinase being stored in all human MC granules in a fully active form [134,135].
Tryptase, which is enzymatically active in a tetrameric form [81], becomes inactive when

11
the tetramer dissociates into monomers in the extracellular fluid by cleavage or in the
absence of heparin, resulting in an enzymatically inactive monomeric form of tryptase
[136]. Since tryptase has four enzymatically active centers in the subunits it has been found
to be an allosteric enzyme following sigmoidal enzyme kinetics at high salt concentrations
[137]. The active centers are, however buried in the ring-like tetrameric molecule and they
are directed towards the central pore [138].
Four different types of human MC tryptase have been identified. β-Tryptase, the main form
of tryptase stored in MC granules, is not normally released into the circulation but
increased levels of β-tryptase can be found in serum during extensive reactions such as
systemic anaphylaxis. α-Tryptase exhibits to have low activity compared to β-tryptase and
low levels of α-protryptase have been detected in the circulation even in the absence of MC
degranulation suggesting that it is released constitutively. The last two types of human
tryptase, γ- and δ-tryptase, are both expressed in the MC-like cell line (HMC-1) but also in
airway MCs (γ-tryptase) or colon, lung and heart MCs (δ-tryptase) [135].
A distinctive attribute of tryptase is its resistance to inhibition by most natural protein
inhibitors of serine proteinases [139]. According to current concepts, there are no known
physiological inhibitors to this proteinase. However, serpin B6 has been found to form
complexes with monomeric beta-tryptase but it is not known whether the tryptase
monomer is inhibited by this inhibitor [140].
MC tryptase is believed to exert numerous effects. For example, tryptase up-regulates IL-1
and IL-8 secretion, mediates accumulation of neutrophils and eosinophils, induces MC
activation and histamine release, enhances the presence of intercellular adhesion
molecules/selectins on endothelial cells, produces vascular leakage by fibrinogen
inactivation, is a moderately potent mitogen for epithelial cells, fibroblasts, and smooth
muscle cells in vitro leading to increased synthesis and secretion of collagen and it may
increase the contractility of pulmonary smooth muscle to histamine [141,142,33]. Moreover,
tryptase is also involved in angiogenesis, degradation of extracellular matrix components

12
by activation of prostromelysin, cleavage of type IV collagen, fibronectin, elastase, and
proteoglycans, release of matrix-associated growth factors or by indirect activation of
matrix-degrading metalloproteinases and urinary type plasminogen activator
[143,144,145,146]. In the skin, tryptase has been found to induce focal dermis-epidermis
separation at the level of lamina lucida and to degrade fibronectin in the basement
membrane [147]. Furthermore, it has been suggested that tryptase may be involved in
matrix destruction in order to make space for migration of tumor cells, and also in
angiogenesis it is believed to be crucial for tumor survival and growth [143,145,146,148]. In
addition, tryptase has the capability of diffusing through the matrix and therefore it can
reach more distant sites after its release from MCs [34,35]. In lesional psoriatic skin,
tryptase-positive MCs are increased in number throughout the dermis but especially
beneath the epidermis [149] and the number of tryptase-positive MCs was increased in
lesional compared with nonlesional skin in AD [62,64] .
2.2.5 Chymase
Chymase, a neutral serine proteinase with a chymotrypsin-like activity exclusively located
in the MC granules similarly to tryptase, can be released together with other preformed
mediators [150]. Chymase is synthesized in an inactive form (pro-chymase) in the MC
secretory granules where it is stored as a macromolecular complex with heparin
proteoglycan. Chymase is activated by a thiol proteinase, dipeptidylpeptidase I (DPPI). MC
chymase is very stable in situ and can be activated immediately after degranulation.
Chymase hydrolyzes the peptide bonds of proteins, typically after the C-terminal side of an
amino acid residue with an aromatic structure, such as phenylalanine, tyrosine and
tryptophan [151].
Increased levels of chymase inhibitors, α1-AC and α1-PI, have been found in MCs in a
variety of skin diseases like psoriasis, herpes zoster and blistering skin diseases [149,
152,153]. Although these inhibitors have been postulated to be a part of the MC secretory
granules, it is not known whether MCs can synthesize these inhibitors or whether they are
derived from blood circulation. [154]. Furthermore, secretory leukocyte protease inhibitor

13
(SLPI), which can also inhibit chymase has been shown to be produced by human MC
[155]. In addition, the light chain of inter-α-trypsin inhibitor that is assumed to be taken up
by MCs from plasma, has been shown to stain positively in human MCs [156]. Squamous
cellular carcinoma antigen-2 (SCCA-2), a part of the superfamily of high molecular weight
serine proteinase inhibitors, inhibits chymotrypsin-like serine proteinases, cathepsin G, and
MC chymase. In contrast, SCCA-1 is a cross-class inhibitor of papain-like cysteine
proteinases, such as cathepsins L, S, and K [157]. Moreover, there is a series of chemical
products able to inhibit serine proteinases. SBTI, a potent inhibitor of trypsin, inhibits both
chymase and cathepsin G but aprotinin inhibits only cathepsin G and not chymase and, in
contrast, tryptase is not inhibited by either inhibitor [106,143,34,35]. LBTI (lima bean trypsin
inhibitor) is a potent inhibitor of chymase but a less potent inhibitor of tryptase [158].
Similarly to tryptase, chymase also induces degradation of the extracellular matrix and
basal membrane components, either indirectly by activating MMP-1 pro-collagenase (pro-
MMP-1) [144] or directly by its ability to degrade a variety of extracellular matrix substrates
[159,160]. Moreover, chymase induces the proliferation of fibroblasts, degrades
neuropeptides SP and VIP and also acts as an IL-1β convertase, by cleaving inactive
precursor IL-1β to yield the active molecule. In contrast to tryptase, chymase can stimulate
indirectly the proliferation of keratinocytes by inducing the formation of angiotensin II. On
the other hand, it acts similarly to tryptase and inhibits the EGF and α-thrombin induced
keratinocyte proliferation and subsequently is potentially capable of affecting epidermal
wound healing [136]. Moreover, chymase also may contribute to the interruption of axon-
reflex-mediated neurogenic inflammation via a negative feedback mechanism of MC-
induced neurogenic activation, since chymase can degrade substance P, CGRP, and other
neuropeptides [161,162]. Human chymase can induce dermis-epidermis separation in skin
biopsies at the level of lamina lucida without causing any apparent degradation of laminin
[163]. Furthermore, chymase can efficiently detach monolayer keratinocytes and
keratinocyte epithelium from a plastic surface [108].
In the upper dermis of lesional AD chymase apparently loses its activity and the enzyme

14
partially lacks the capability to suppress inflammation, such as degradation of
neuropeptides and proteins. In addition, the dysregulation of this proteinase can be
detected already in non-lesional skin of AD [164]. However, the apparent inactivation of
chymase is not a specific feature of any distinct skin disease, since it has been observed in
many pathological skin conditions such as psoriasis [149], blistering diseases [165], chronic
ulcers [166], allergic prick-test reaction [167], and even in skin organ cultures [34,35]. Only
in irradiation-induced cutaneous fibrosis, any increase in the number of MCs with chymase
activity has been detected [168].
2.2.6 IL-8
IL-8, first purified as a chemotactic factor for neutrophils, has also chemotactic activities for
T-lymphocytes, basophils and eosinophils [169]. MC tryptase stimulates de novo synthesis
of IL-8 and may have a role in initiating MC-induced inflammation, though also thrombin,
a similar protease, can induce the release of this cytokine. Furthermore, TNF-α, a more
potent stimulus for endothelial IL-8 release than tryptase, has been proposed to be a key
mediator in granulocyte recruitment at sites of MC activation [170]. MC produced IL-8, a
major source of this cytokine, promotes rapid accumulation of neutrophils to sites of
inflammation. Furthermore, it has been speculated that recruited neutrophils may produce
cytokines such as IL-10, indirectly regulating immune responses [171].
It has been speculated that IL-8 may be involved in angiogenesis in response to tissue
injury and may regulate neovascularization and metastasis in tumor. IL-8 has the capability
of stimulating human keratinocyte migration and proliferation in vitro as keratinocytes
express both of the IL-8 receptors. Moreover, dermal fibroblasts produce IL-8 even though
IL-8 mRNA expression by fibroblasts has not been detected at sites of wound healing [172].
Additionally, increased number of IL-8-positive MCs in psoriatic and atopic dermatitis
lesional skin demonstrates that MCs are a marked source of IL-8 in these skin diseases.
Together with MIP-1α and MIP-1β, IL-8 is capable of recruiting most of the cell types found
in lesional tissues of these diseases, e.g., accumulation of neutrophils and maturation and
activation of granulocytes [52,170].

15
2.2.7 Other MC mediators
Upon activation, the MC has the capability of releasing a wide array of mediators to fulfill
its biological functions, including neutral proteases, enzymes other than tryptase and
chymase (acidic hydrolases, cathepsin G, carboxypeptidase, and metalloproteinases),
leukotrienes, prostanoids and platelet activating factor (PAF), cytokines, chemokines and
growth factors. The pattern of produced cytokines depends on the MC type and stimulus
[173,136]. Thus, MCs synthesize and secrete a diversity of mediators, which are vasoactive
or regulate inflammation and cellular growth.
Eicosanoids, a group of newly generated mediators of MCs produced from arachidonic
acid, are rapidly oxidized along either of two pathways – via cyclooxygenase to form PGD2
and along the lipoxygenase pathway to produce LTC4. PAF, a product of phospholipid
metabolism in MCs, is also a newly generated mediator.
The MC cytokines and chemokines, preformed and newly synthesized mediators, include
IL-1β, IL-3, IL-4, IL-5, IL-6, IL-8, IL-9, IL-10, IL-13, IL-16, IL-18, IL-25, TNF-α, granulocyte-
macrophage colony-stimulating factor (GM-CSF), SCF, macrophage chemotactic peptide
(MCP)-1, 3, 4, regulated on activation of normal T cell-expressed and secreted protein
(RANTES) and eotaxin [174,150,173,136]. However, the list of cytokines and chemokines
produced by MCs is continuing to increase.
2.3 NON-MELANOMA SKIN CANCERS
Commonly referred to as non-melanoma skin cancers, BCC and SCC are the most common
form of cancers in Caucasian populations and the incidence continues to rise due to
increased exposure to UV-radiation as a result of decreasing ozone levels. About 75% of
nonmelanoma skin cancers are BCCs, and the majority of the remaining cases are SCCs
[175].

16
Many genetic and environmental factors contribute to the pathogenesis of these neoplasms.
Exposure to UV radiation is considered to be the major causal factor for developing BCC
and SCC [176,177], but the exposure time is also crucial. UV radiation induces skin cancer
by DNA mutations or lesions induced by the absorption of UV photons, which may
produce damage to various immune mechanisms. The damage of the cellular proteins and
cell membrane carbohydrates and fatty acids may also be a result of UV-induced free
radicals, which may influence the process of carcinogenesis through altered cellular
communication, injuries to cell receptor functioning, and alterations in DNA repair systems
and cell proliferation pathways [178].
2.3.1 BCC
BCC, first described by Jacob in 1827, is the most commonly observed neoplasm in the
Caucasian population. It is usually manifested on sun-exposed areas such as the face, the
head and neck, and its incidence will surely increase in the future [179]. Even though BCC
is evidently curable when the diagnosis is made in its early phase, it represents a vast
financial burden on the health care system [180] and anxiety on an individual level. BCC,
the most common form of skin cancer in white-skinned races [181,182], is very rare in
darkly pigmented people [183,184] and its frequency is slightly higher in males. Other risk
factors include geographic locations with high solar intensity, exposures to ionizing
radiation [185], ingestion of arsenic alone or in combination with other risk factors [186,187]
and immunosuppression [188]. Genetic studies show that the formation of sporadic basal
cell tumors may be induced by the loss-of-function mutations in the tumor-suppressor gene
patched, or gain-of-function mutations in the smoothened gene [189,190].
BCC arises from the epidermis and the appendages, which resemble the basal layer of the
epidermis and is associated with a characteristic stroma [191]. It tends to grow slowly and
over many years, it invades nearby tissue and spread along the plane of least resistance
such as periosteum, perichondrium, fascia and tarsal plate [14] which eventually leads to
ulceration. Even if the tumor has an infiltrating and destructive growth, it does not usually

17
metastasize [7]. Metastasis has been observed in men with large neglected ulcerated
primary BCC lesions. The aggressive histologic BCC forms like morpheic, basosquamous,
infiltrating or metatypical showed a higher incidence of metastasis [192].
Histopathologically, the tumor appears as proliferating basaloid cells forming cords and
islands invading into the dermis in most of the cases with a characteristic “palisading”
arrangement of the peripheral cells and connections between the tumor islands and the
epidermis. Another characteristic feature of BCC is the retraction of the tumor islands away
from the adjacent stroma - an artifact which occurs during tissue processing [193]. BCC is
usually asymptomatic unless ulceration occurs and the carcinoma is characteristically slow
growing after a period of months to years. It most frequently occurs on sun-exposed (face
and upper trunk) skin sites and is rare on the palms and soles [194].
There are five clinical types: nodular, ulcerating, sclerosing (cicatricial), pigmented, and
superficial.
Nodular BCC exhibits papule or nodule, translucent or "pearly". In the ulcerating BCC form
the ulcer is often covered with a crust having a rolled border (rodent ulcer). The sclerosing
BCC appears as a small patch of morphea or a superficial scar whitish but also with
peppery pigmentation. In this infiltrating type of BCC there is an excessive amount of
fibrous stroma. Pigmented BCC may be brown to blue or black and consequently may be
indistinguishable from superficial spreading or nodular melanoma. It has a smooth,
glistening surface. Superficial spreading BCC exhibits flat, well-demarcated plaques with a
rolled edge, which can bleed easily when scratched. It is slow growing, erythematous, with
minimal induration and located primarily on the trunk e.g. on regions which are most of
the time covered from the UV exposure. The lesions have a characteristic horizontal growth
pattern, and often present in multiples [195]. Histologically, superficial spreading BCC
exhibits characteristically tumour cells budding down from the lower layer of the
epidermis and palisading around the periphery of the tumour nest [196].
2.3.2 SCC
SCC, a malignant tumor arising from epidermal or appendageal keratinocytes or from the

18
squamous mucosal epithelium, carries an overall metastatic rate of 2–6%, with the
metastasis generally occuring to lymph nodes [197,198,14]. The total number of new cases
in Finland in 2008 was over 1200, and the annual death rate about 45 [199].
A history of damage by exogenous carcinogenic agents such as sunlight, ionizing radiation,
local irritants, or arsenic ingestion is often found in the etiology of SCC. A closer correlation
between SCCs and chronic cumulative sun exposure than between sun exposure and BCC
has been observed, in close connection with the latitude gradient [200]. An extremely rare
but a frecvently reported complication is the development of SCC in chronic burn wounds
and scars [201]. Mutations in the p53 tumor-suppressor gene have been found in a number
of SCCs, [202]. An increased risk to develop SCC has been found in patients who have
received UVB or psoralen plus UVA (PUVA) for the treatment of skin diseases, such as
psoriasis or mycosis fungoides [203,204,205]. Moreover, a much higher incidence of SCC
has been reported in immunosuppressed patients [206], e.g., in renal [207] or heart
transplant recipients [208]. Actinic keratosis is the most common precursor lesion of SCC of
the skin. The actual percentage of actinic keratosis that transform to invasive squamous cell
carcinoma varies from 0.1% to 5.0% [209]. Furthermore, SCC in situ also known as Bowen’s
disease, Erythroplasia of Queyrat or leukoplakia is considered to be another premalignant
lesion of SCC [195].
Histopathologically, in the intraepithelial (in situ) carcinoma stage, the limit between
epidermis and dermis remains sharp throughout the lesion. Subsequently, in the invasive
stage, the tumor cells are seen to proliferate downwards in cords and single cells into the
dermis [193]. The tumor may have different degrees of differentiation determined by extent
of the tumor, with the spindle-cell squamous cells representing the least differentiated form
[193].
SCC usually expands faster than BCC. The intraepidermal SCC, the initial lesion, typically
has a sharply demarcated but irregular outline and appears as a scaly, erythematous plaque
on sun-exposed areas. Invasive SCC in most of the cases arises from a pre-existing

19
premalignant lesion or from an in situ carcinoma [210]. Regional lymphadenopathy as a
response to infection of the ulcerated primary lesion or from metastases may be present in
the invasive form [193]. For didactic reasons, two types can be distinguished: highly
differentiated SCCs, which practically always show signs of hyperkeratosis of the tumor
and poorly differentiated SCCs, without any signs of keratinization [195].
2.4 CERVICAL SQUAMOUS CELL CARCINOMA
Cervical cancer of the uterus, the third most common cancer among women worldwide
after breast and colorectal cancer and the third most common neoplasm of the female
genital tract, has long been proposed to have a sexually transmitted etiology. The total
number of new cases in Finland in 2008 was over 150, and the annual death rate about 55
[199]. The causal relationship between human papillomavirus (HPV) and cervical neoplasia
supports an etiologic role for some types of HPV [211]. More than 35 out of over 78 types of
HPV that have been described are associated with anogenital disease, and 30 or more are
associated with cancer [212]. Intermediate and high-risk (16, 18, 31, 33, 35, and 45) HPV
types have been identified in about 77% of high-grade squamous intraepithelial lesions
(CIN 2 and 3) and in 84% of invasive lesions. HPV 16, the most prevalent HPV type in
women with cervical neoplasia, being present in up to 50% of high-grade squamous
intraepithelial lesions and invasive lesions, is the most common HPV type identified in
cytologically normal women [211,213,214]. Even though a significant role for HPV infection
in the etiology of cervical neoplasia has been clearly established, other cofactors (sexual and
reproductive history, smoking, hormonal and dietary factors) are necessary for the
development and progression of the disease [214].
Histopathologically, two types of cervix SCC are described: nonkeratinizing carcinoma,
which is characterized by squamous cells with somewhat hyperchromatic nuclei and a
moderate amount of cytoplasm growing in discrete nests separated by stroma and
keratinizing carcinoma characterized by cells with very hyperchromatic nuclei and densely

20
eosinophilic cytoplasm growing in irregular invasive nests which in many cases may have
central “pearls” that contain abundant keratin [215,216].
2.5 MCS AND THEIR MEDIATORS IN CANCER
As extensively reviewed by many authors [14, 217], the properties of MCs to be present in
the vicinity of malignant tumors has attracted as much interest and controversy as the
debate about the functional role of the MC and its wide-ranging and sometimes opposing
effects in cancer. However, the majority of studies support a secondary role for MCs in
cancer development and progression (Fig. 3).
Figure 3. The accessory roles of MCs in the skin cancer progression and spreading (Modified
from Ch'ng S et al., 2006 [14])
MCs accumulate within and around the tumor. Increased MCs density (MCD) has been
shown in many cancer types including BCC and melanoma [217]. Furthermore, some
researchers have demonstrated a consistent decrease in MC numbers during tumor

21
progression [218], a decrease which may be due to degranulation of MCs and the
appearance of phantom cells. However, increased MCD has not always been associated
with solid tumors and poor prognosis in humans.
MCs accumulate around BCC lesions and may contribute to cancer growth by inducing
immunosuppression. The MCs present in BCC express VEGF, IL-8 and RANTES [219,14]. A
higher dermal MC prevalence in non-sunexposed buttock skin has been found in Danish
patients with a history of sporadic BCC [220]. In clear contrast, there was no significant
difference in dermal MC prevalence between SCC patients and control subjects. [221]. MCs
appear to be able to promote tumor development through many different mechanisms:
immunosuppression, induction of angiogenesis, degradation of the extracellular matrix
components and promotion of tumor cell mitosis [14]. TNF-α and histamine, which are
important for the local and systemic effects, respectively, are believed to be the critical MC
products involved in UV-induced immunosuppression [222].
Angiogenesis is a crucial mechanism implicated in growth, maintenance, and metastasis of
solid tumors [217]. Several authors have reported the concept of MC-induced angiogenesis.
Takeda et al. demonstrated that connective tissue MCs are associated with small vessels
[223]. Moreover, MCs have been found in many angiogenesis-dependent situations
including wound healing, hemangioma and neoplasia [217]. In the initiation of
angiogenesis, degradation of the extracellular matrix components is a critical step and
tryptase may play a key role in this process by activating metalloproteinases and
plasminogen activatior. Moreover, in combination with heparin, tryptase may stimulate the
migration and division of vascular endothelial cells [224]. Additionally, a positive
correlation between cervical cancer progression and the number of tryptase-positive MCs
has been demonstrated [217]. IL-8 increases the expression of ICAM-1, which is responsible
for cell adhesion during angiogenesis [218]. In addition, IL-8 has been found to be inhibited
by squamous cell carcinoma antigen-2 (SCCA-2), a tumor cell product, and this kind of IL-8
inhibition has been considered to be involved in the progression of advanced SCC [225].
Furthermore, histamine, chymase, bFGF and particularly VEGF have also angiogenic

22
properties and regulate endothelial cell proliferation and function. In addition, VEGF, bFGF
and platelet-derived growth factor (PDGF) induce MC migration to sites of
neovascularisation [218,217]. Controversially, some studies have shown that MC-induced
neoangiogenesis could be associated with greater survival in oral squamous carcinoma or a
better response to chemotherapy in ovarian cancer. Tryptase-induced fibrosis and
angiogenesis have been postulated as survival factors in advanced ovarian cancer [225].
The proteolytic activity promotes aggressive tumor infiltration to the surrounding tissues
[218]. Moreover, the higher number of MCs in primary tumors (metastases-free tumors) can
facilitate the infiltration of lymphatic vessels [218].
Histamine may promote or inhibit tumor cell proliferation by acting through its receptors
[219]. This has been documented in some in vitro studies suggesting that certain anti-
histaminic compounds promote proliferation of cultured cancer cells. Furthermore, it has
been shown that a high histamine concentration inhibits primary melanoma cell
proliferation most likely by acting through H1 receptors, whereas a low concentration had
proliferatory effect acting via H2 receptors. Thus, H2 receptor antagonist might be
beneficial by blocking histamine-induced immunosuppression [219]. Moreover, other MC
mediators including TNF-α, IL-1, IL-6 and interferon-γ have been reported to inhibit
melanoma cell growth [14]. Heparan sulfate proteoglycans prevented neovascularization
by blocking the binding of heparin binding growth factors to the cell surface and MC
chondroitin sulfate may inhibit tumor metastasis by acting as a decoy [219]. MC chymase
can act as an apoptotic factor on different target cells and chemoattractant for macrophages,
neutrophils and other inflammatory cells in vivo [225,226,227]. The wide variation of MC
effects in cancer is still difficult to completely understand their function, although the
majority of authors do recognise an accessory role for MCs during malignant tumor
development and progression.
Studies performed in human uterine cervix have revealed that MCT and MCTC are normal
constituents of the tissue, with the MCT being the predominant phenotype [228]. In both
benign and malignant lesions of the uterine cervix, these two types of MC have been found

23
and, as is also the case in other types of human tumors [164,229,230], it appears that the
MCT is the predominant phenotype observed. A constant total number of MC has been
found during the earlier stages of the carcinogenesis in human uterine cervix (CIN 1–3, CIS)
but there is an increased number of MCT in invasive carcinomas compared with normal
tissues [228].
2.6 TNF LIGAND AND RECEPTOR SUPERFAMILY
All cells in the human body have membrane receptors that induce certain actions in a cell.
Some stimulate the cell growth, some stimulate cell division, whereas others cause cell
death. Obviously, those receptors on cancer cells that cause cell death need more attention.
These receptors are called TNF family receptors.
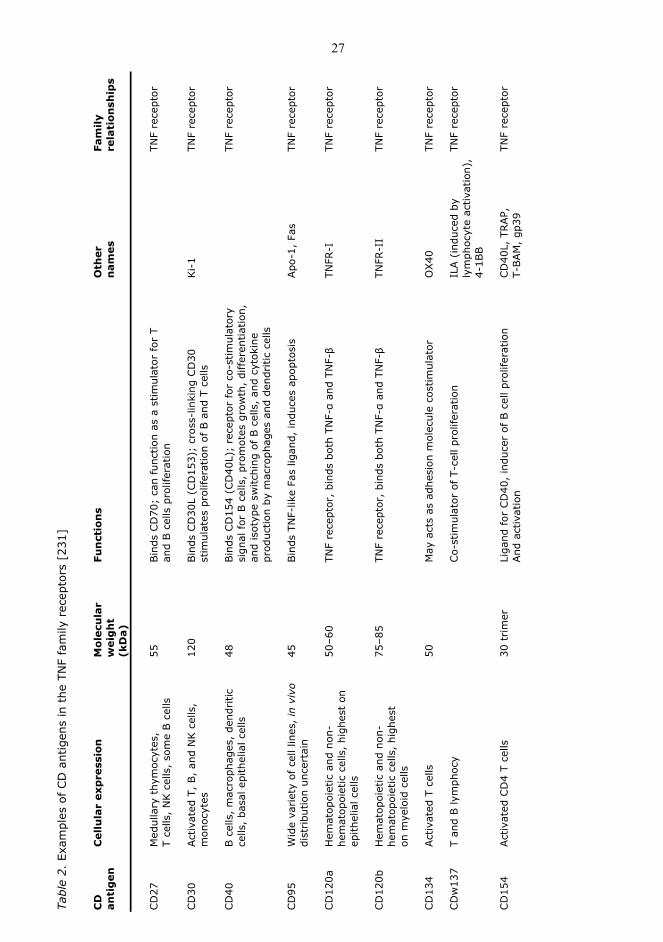
The TNF ligand and receptor superfamily (Table 1 and 2) has been found to be involved in
the regulation of innate and adaptive immunity, the induction of apoptosis, but also in the
reversible formation of secondary lymph organs, the development of sweat glands, teeth
and hair follicles and regulation of bone homeostasis [231,232]. During the past two
decades, 19 ligands and 29 receptors that belong to the TNF superfamily have been
identified. Members of the TNF superfamily mediate hematopoiesis and morphogenesis,
are believed to be involved in tumorigenesis and tumour metastasis, in transplant rejection,
septic shock, viral replication, bone resorption, osteoporosis, rheumatoid arthritis and
diabetes and they act as factors regulating inflammation and autoimmune responses. These
cytokines or receptor ligands either induce cellular proliferation, survival, differentiation or
apoptosis having a “double-edged swords” role [233].
TNF receptor members, with a few exceptions (OPG, TR6 and DcR1), have a single
transmembrane domain and a cytoplasmic portion without enzymatic activity [231]. All
members of the TNF receptor family contain at least one copy of the so-called cysteine rich
domain (CRD) in their extracellular part, a structure of about 40 amino acids stabilized by

24
three cysteine bridges [234]. Usually, one or two CRDs form the contact sites to the
respective ligands within a single receptor molecule. In some receptor members, e.g. Fas,
TNFR1, TNFR2, TRAIL-R1 and CD40, the membrane distal CRD serves as a pre-ligand
binding assembly domain (PLAD) [235,236]. Crosslinking studies with bivalent chemical
agents have indicated that PLAD-containing receptors occur as homotrimers on the cell
surface [235,236] and binding of the trimerized ligand might lead to structural changes in
the PLAD-trimerized receptors rather than to a de novo trimerization per se.
Most ligands of the TNF superfamily are expressed as soluble proteins (cytokines) or as
type II transmembrane proteins. One characteristic of the ligands is the formation of non-
covalently linked homotrimers with a unique structure, as indicated by crosslinking studies
with bivalent chemical agents, but also simply by the fact that these molecules possess
bioactivity [237,238,239]. Most of the TNF cytokine family members interact with more than
one receptor of the corresponding superfamily of cognate receptors and vice versa.
2.6.1 CD30 ligand (CD30L) and CD30 receptor
CD30L and CD30, members of the TNF ligand and receptor superfamily, respectively,
interact on the cell surfaces and have important immune regulatory functions [240,241].
CD30L has been reported to be expressed on a variety of human cell types, including MCs,
medullary thymic epithelial cells, eosinophils, neutrophils and peripheral blood B cells
[242]. However, only distinct subsets of B cells from human lymphoid tissue can express
CD30L constitutively or in response to ex vivo activation [243]. In contrast, in Hodgkin’s
lymphoma, at least 50% of the MCs display CD30L immunoreactivity, these being the
predominant CD30L-positive cells. Moreover, the CD30L-CD30 interaction between MCs
and Hodgkin’s lymphoma may promote the growth of tumor cells, and in this way
participate in the tumorigenesis process [244]. Increased numbers of CD30L expressing
MCs have also been found in cutaneous inflammatory diseases, atopic dermatitis and
psoriasis [52].
CD30, a marker of anaplastic large cell lymphoma, Hodgkin's disease and other lymphoid
malignancies [245,246], is also present at inflammatory sites in several human diseases,

25
including atopic dermatitis [247,248], rheumatoid arthritis [249], chronic graft versus host
disease [250], systemic sclerosis [250,251], seminoma and embryonal carcinoma [252]. It is
typically expressed on subpopulations of T (Th1 and Th2 cells) and B cells and is observed
in normal human tissues only on activated blasts in parafollicular areas of lymphoid tissues
and in the thymic medulla, mainly around the Hassall's corpuscles [246]. Soluble CD30 can
be detected in the serum of most normal individuals and can be used as a marker of
tumour extension in Hodgkin's disease and anaplastic large cell lymphoma [253,254,255].
Moreover, CD30 was found to be expressed in MC in aggressive Systemic mastocytosis and
mast cell leukemia [256].
The CD30L structure, with a C-terminal extracellular domain and a short intracellular N-
terminal domain makes it capable of acting as a receptor and transmitting a downstream
reverse signal that leads to diverse effects, such as cytokine production. This capacity may
explain the ability of MCs to produce a wide variety of inflammatory mediators via
activation of CD30L [52]. It has been reported that CD30L is capable of inducing
proliferation and IL-6 production by T-cells, IL-8 production and oxidative burst in
neutrophils [257], impaired immunoglobulin isotype switching in B cells [243], and
chemokine release in MCs, via CD30L reverse signaling [52].
2.6.2 CD40 ligand (CD40L) and CD40 receptor
CD40, identified as a surface marker on bladder carcinomas and on B cells in 1985 [258], has
a critical role in B cell activation in thymus-dependent humoral responses. Its natural
ligand, CD40L, a Type II 39-kDa membrane glycoprotein, was first isolated in activated T
cells in 1992 [259]. A vast array of normal hematopoietic and nonhematopoietic cell types
has been found to express CD40 and CD40L. Cells with a high proliferative potential, such
as epithelial and endothelial cells, hematopoietic progenitors, activated monocytes,
activated B lymphocytes and dendritic cells express CD40 [260]. Moreover, eosinophils,
CD8+ T cells, fibroblasts [261], cortical, medullary epithelia and interdigiting cells of the
thymus also express CD40 [262]. CD40 expression has been also found in B-cell
malignancies, melanomas, and a variety of carcinoma cell types [263,264].

26
Tabl
e 1.
Exa
mpl
es o
f th
e TN
F fa
mily
liga
nds
and
rec
epto
rs [
231]
Cyt
okin
e/
Pro
du
cer
cells
R
ecep
tors
S
ize
(no.
of
amin
o ac
ids)
A
ctio
ns
O
ther
nam
es
Lig
and
s
an
d f
orm
TN
F-α
M
acro
phag
es,
MCs
p55/
CD
120a
, p7
5/CD
120b
15
7, t
rim
ers
Loca
l inf
lam
mat
ion,
NK &
T c
ells
cel
ls
endo
thel
ial a
ctiv
atio
n TN
F-β
T ce
lls,
B c
ells
p5
5/CD
120a
, p7
5/CD
120b
17
1, t
rim
ers
Kill
ing,
end
othe
lial
lym
phot
oxin
, LT
, LT
-α
Act
ivat
ion
LT-β
T
cells
, B c
ells
LTβR
or
HVEM
Tr
ansm
embr
ane,
trim
eriz
es
Lym
ph n
ode
with
TN
F-β,
(LT
-α)
deve
lopm
ent
CD
40 li
gand
T
cells
, M
Cs
CD
40
Trim
ers
B-c
ell a
ctiv
atio
n, c
lass
CD
40L
Sw
itchi
ng
Fas
ligan
d T
cells
, st
rom
a?
CD
95 (
Fas)
Tr
imer
s Apo
ptos
is,
Ca2+
- Fa
sL
inde
pend
ent
cyto
toxi
city
CD
27 li
gand
T
cells
CD
27
Trim
ers
(?)
Stim
ulat
es T
-cel
l CD
27L
prol
ifera
tion
CD
30 li
gand
T
cells
, M
Cs
CD
30
Trim
ers
(?)
Stim
ulat
es T
-and
B-
CD
30L
cell
prol
ifera
tion
4-
1BBL
T ce
lls,
MCs
4-1B
B
Trim
ers
(?)
Co-
stim
ulat
es T
and
B
cells
Tr
ail
T ce
lls,
mon
ocyt
es
DR4,
DR5
DCR1,
DCR2
281,
aa
trim
ers
Apo
ptos
is o
f ac
tivat
ed
and
OPG
T ce
lls a
nd t
umor
cel
ls
OPG
-L
Ost
eobl
asts
, T
cells
RAN
K/O
PG
316
aa t
rim
ers
Stim
ulat
es o
steo
clas
ts
RAN
K-L
an
d b
one
reso
rptio
n
27
in aggressive Systemic mastocytosis and mast cell leukemia [265].
Tabl
e 2.
Exa
mpl
es o
f CD
ant
igen
s in
the
TN
F fa
mily
rec
epto
rs [
231]
C
D
C
ellu
lar
exp
ress
ion
M
olec
ula
r
Fun
ctio
ns
O
ther
Fa
mily
an
tig
en
wei
gh
t
n
ames
re
lati
onsh
ips
(kD
a)
CD
27
Med
ulla
ry t
hym
ocyt
es,
55
Bin
ds C
D70
; ca
n fu
nctio
n as
a s
timul
ator
for
T
TN
F re
cept
or
T ce
lls,
NK c
ells
, so
me
B c
ells
an
d B c
ells
pro
lifer
atio
n CD
30
Act
ivat
ed T
, B,
and
NK c
ells
,
120
Bin
ds C
D30
L (C
D15
3);
cros
s-lin
king
CD
30
Ki-
1
TNF
rece
ptor
mon
ocyt
es
st
imul
ates
pro
lifer
atio
n of
B a
nd T
cel
ls
CD
40
B c
ells
, m
acro
phag
es,
dend
ritic
48
Bin
ds C
D15
4 (C
D40
L);
rece
ptor
for
co-
stim
ulat
ory
TNF
rece
ptor
ce
lls,
basa
l epi
thel
ial c
ells
si
gnal
for
B c
ells
, pr
omot
es g
row
th,
differ
entia
tion,
and
isot
ype
switc
hing
of
B c
ells
, an
d cy
toki
ne
pr
oduc
tion
by m
acro
phag
es a
nd d
endr
itic
cells
CD
95
Wid
e va
riet
y of
cel
l lin
es,
in v
ivo
45
Bin
ds T
NF-
like
Fas
ligan
d, in
duce
s ap
opto
sis
Apo
-1,
Fas
TNF
rece
ptor
di
stribu
tion
unce
rtai
n CD
120a
H
emat
opoi
etic
and
non
-
50
–60
TN
F re
cept
or,
bind
s bo
th T
NF-α
and
TNF-β
TNFR
-I
TNF
rece
ptor
he
mat
opoi
etic
cel
ls,
high
est
on
epith
elia
l cel
ls
CD
120b
H
emat
opoi
etic
and
non
-
75
–85
TN
F re
cept
or,
bind
s bo
th T
NF-α
and
TNF-β
TNFR
-II
TNF
rece
ptor
he
mat
opoi
etic
cel
ls,
high
est
on
mye
loid
cel
ls
CD
134
Act
ivat
ed T
cel
ls
50
M
ay a
cts
as a
dhes
ion
mol
ecul
e co
stim
ulat
or
O
X40
TN
F re
cept
or
CD
w13
7 T
and
B ly
mph
ocy
Co-
stim
ulat
or o
f T-
cell
prol
ifera
tion
IL
A (
indu
ced
by
TN
F re
cept
or
lym
phoc
yte
activ
atio
n),
4-1B
B
CD
154
Act
ivat
ed C
D4
T ce
lls
30 t
rim
er
Li
gand
for
CD
40,
indu
cer
of B
cel
l pro
lifer
atio
n
CD
40L,
TRAP,
TNF
rece
ptor
And
act
ivat
ion
T-BAM
, gp
39

28
Despite its surface receptor, CD40L is expressed transiently on activated leukocytes, MCs,
eosinophils, and IL-2 activated NK cells [266]. Furthermore, an increased CD40L expression
in the T cells of patients with myasthenia gravis [267], multiple sclerosis [268] and systemic
lupus erythematosus [269] has been found and it may produce a proinflammatory signal
contributing to disease pathogenesis. In rheumatoid arthritis or atheroslerosis, CD40L
induces and activates metalloproteinases, which function in matrix degradation and tissue
remodeling [260].
In 1998, Young et al. postulated that there is an increased tumor susceptibility or
uncontrolled chronic infection and inflammation due to the loss of CD40L-dependent
epithelial cell growth regulation [270]. As the majority of oral basal epithelium cells co-
express CD40 and CD40L and in SCC, a loss of CD40L expression and maintained
expression of CD40 have been observed, it has been proposed that the CD40-CD40L
interaction could contribute to epithelial tumorigenesis [271].
2.6.3 TNF receptors I and II
TNF-α acts via two distinct receptors [272]. Even though the affinity for TNFRI (p55,
CD120a) is five times lower than that for TNFRII (p75, CD120b) [273], TNFRI is responsible
for the majority of the biological activities and is expressed on all cell types, while TNFRII
expression is mainly restricted to immune cells [233]. In contrast to TNFRII, TNFRI is an
important member of the death receptor family being capable of inducing apoptotic cell
death due to its death domain (DD) [274]. TNF-α activates via TNFRI both survival and
proliferation pathways along with apoptotic pathways. The separate pathways are well
defined, while the survival-apoptosis balance regulation is still poorly understood
[275,276]. Like other death receptors, TNFRI is able to signal cell death via its cytoplasmic
death domain in a variety of cell lines. However, in vivo TNF-induced apoptosis seems to
have only a minor role compared to its overwhelming function in the regulation of
inflammatory processes.

29
The interaction between TNF-α and its receptors plays a critical role in cellular
communication during host defense, inflammation and organogenesis but also in many
other pathological situations, including cancer, which is characterized by an imbalance
between survival and apoptosis signals. TNFRII is incapable of inducing apoptosis since it
does not contain a death domain. However, agonistic anti-TNFRII antibodies have clearly
shown that in some cells exclusive triggering of this receptor is sufficient to induce cell
death [277,278]. The stimulation of TNFRII induces apoptosis indirectly by induction of
endogenous membrane TNF, which stimulates the death receptor TNFRI [279].
Remarkably, the stimulation of TNFRII produces an enhanced action of the endogenously
produced membrane-bound TNF on TNFRI. Similarly, the other TNF receptor family
members lacking a death domain are also able to produce endogenous TNF and to
indirectly induce cell apoptosis [279].
2.7 SERINE (OR CYSTEINE) PROTEINASE INHIBITOR (SCCA 1 AND SCCA
2)
Kato and Torigoe (1977, when immunizing a rabbit with a crude extract prepared from
cervix SCC), described for the first the so called tumor antigen 4 (TA-4). Subsequently it
was called squamous cellular carcinoma antigen (SCCA). Further analyses revealed that
SCCA, also called SERPINB3, belongs to the superfamily of high molecular weight serine
proteinase inhibitors (serpins) [280]. Furthermore, Schneider et al. (1995) found two
tandemly arrayed genes, the more telomeric and the more centromeric gene, corresponding
to SCCA-1 and SCCA-2, respectively.
SCCA-1 and SCCA-2 are almost identical members of serpin superfamily and encode for
proteins with a 98% homology at the nucleotide level and 92% homology at the amino acid
sequences level. However, significant differences in their reactive site loops suggest that
they inhibit different classes of proteinases. SCCA-1 is an inhibitor of papain-like cysteine
proteinases such as cathepsin L, S and K, whereas SCCA-2 inhibits chymotrypsin-like

30
serine proteinases, cathepsin G and MC chymase [281,157]. These findings suggest a role
for these inhibitors in the control of proteinases, and they may be involved in tumor cell
motility, invasion, growth and death.
Although clinically used as a serological marker for the management of SCC [282], SCCA is
also expressed in normal squamous epithelium of the uterine cervix [283]. However,
significantly increased SCCA-2 mRNA levels, but not of SCCA-1 mRNA levels, have been
reported in cervical SCC tissue when compared with normal squamous epithelial tissue
[284]. Moreover, SCCA-1/SCCA-2 mRNA ratios significantly differ between SCC and
keratinocyte cell line [285]. On the contrary, previous reports revealed by
immunohistochemical studies using specific antibodies that SCCA-1 and SCCA-2 are
expressed equally and colocalize in normal and malignant squamous epithelial tissues
[286]. In order to resolve this discrepancey, further investigation is required on SCCA
isoform expression in normal and malignant squamous epithelial tissue.

31
3 Aims of the study
The role and significance of MCs in epithelial cancers, or cancers in general, are rather
obscure. MCs are typically increased in number in the peritumoral stroma but it is not
known whether MCs promote or inhibit cancer cell growth and invasion. MCs can produce
and release powerful preformed and newly-synthesized mediators, such as histamine,
heparin and TNF-α and they also contain serine proteinases tryptase and chymase that are
stored in the secretory granules of MCs. Upon MC activation by different stimuli, these
mediators are released and can affect simultaneously the surrounding cells and epithelium.
Therefore, this thesis work focused on elucidating the possible roles and function of MCs
and their mediators in human epithelial cancers. Specifically, the aims were:
1. To study whether there are characteristic changes in MC numbers, MC
types and tryptase and chymase activities in superficial spreading BCC, and
whether there are changes in different protease inhibitors (α1-PI, α1-AC,
SCCA-2) known to regulate chymase activity.
2. To examine whether MCs in superficial spreading BCC express different
TNF superfamily ligands (TNF-α, CD30L, CD40L) and whether the
corresponding TNF family receptors are also present in the epidermis
and/or dermis in this epithelial carcinoma type.
3. To investigate the influence of histamine and TNF-α on the growth,
migration and viability of uterine cervical SCC cells in vitro in comparison
to normal keratinocytes, and to clarify whether TNF-α and its receptors are
present in uterine cervical SCC.
4. To determine whether tryptase or chymase is capable of influencing the
growth, migration or viability of uterine cervical SCC cells in vitro in

32
comparison to normal keratinocytes, and to clarify whether these serine
proteinases are present in uterine cervical SCC.

33
4 Materials and methods
This chapter covers the materials and methods used in this thesis. A detailed description of
materials and methods is given in the original publications I-IV. All methods used in this
study were approved by the Ethics Committee of Kuopio University Hospital, Kuopio,
Finland.
4.1 TISSUE SAMPLES
Biopsy specimens of the BCC lesion and from the healthy-looking skin at least 2 cm distant
of the lesion were obtained from patients recruited from the out-patient units of the
Department of Dermatology, Kuopio University Hospital, Kuopio, Finland or Department
of Dermatology, Central-Bothnia Central Hospital, Kokkola, Finland (I,II). Two 4-mm
punch biopsies were taken, one from the peripheral and palpable area of the BCC lesion
and the other one from the healthy-looking skin at least 2 cm away from the lesion. Two
biopsy pairs were taken from one patient with several BCCs. The biopsies were taken
under local anesthesia with lidocaine-adrenaline. After removal, the skin biopsies were
immediately embedded in optimal cutting temperature medium (OCT) compound (Miles
Scientific, Naperville, IL) followed by freezing in isopentane cooled with a mixture of
absolute ethanol and dry ice.
For publications III and IV, formalin-fixed and paraffin-embedded tissue specimens from
the uterine cervix were obtained and used without identification data from the archives of
diagnostic specimens of Department of Clinical Pathology, Kuopio University Hospital.
Biopsy specimens from the uterine cervical SCC and control cervical samples (11 samples)
with non-specific inflammation had been taken either during the routine Papanicolaou test
or during surgery and were known to have non-specific abundant inflammatory

34
lymphocyte infiltrates based on the histopathologic examination, without alterations
attributable to HPV infection. The diagnostic carcinoma biopsies used in the study were
from 9 patients with uterine cervical SCC verified in the histopathologic examination.
4.2 IMMUNOHISTOCHEMISTRY
Sequential double-staining method was used on cryosections for the localization of CD30L,
CD40L, TNF-α, IL-8, α1-PI and α1-AC in tryptase-positive MCs (I,II). Briefly, tryptase-
positive MCs were first identified enzyme-histochemically by using Z-Gly-Pro-Arg-MNA
as the substrate and Fast Garnet GBC as the chromogen. Thereafter, at least 6 adjacent
photographs per sample lining the epidermis and extending approximately 0.4 mm down
to the dermis were taken and, thereafter, the red azo dye was dissolved away with 15%
Tween 20 overnight. In the next step, the same sections were fixed again in cold acetone
and stained immunohistochemically with the primary antibody, using the Vectastain Elite
ABC kit for visualization. After re-photographing at exactly the same sites as the previous
pictures, the positively stained cells were counted by comparing the photographs side by
side. The results are expressed as the mean percentage of tryptase-positive MCs exhibiting
immunoreactivity for the primary antibody. Psoriasis sections were used as positive control
samples since psoriatic lesions have been shown to display the immunoreactivity for these
antigens (CD30L, CD40L, IL-8, TNF-α) [62].
Immunostaining of the sections from formalin-fixed and paraffin-embedded specimens was
performed by the avidin-biotin-peroxidase complex technique using Vectastain ABC Kit
(Vector Laboratories, Inc., Burlingame, CA) or DAKO Kit (DAKO, A/S, Glostrup, Denmark)
(III, IV). Samples were deparaffinized, dehydrated and endogenous peroxidase was
blocked with hydrogen peroxidase. Normal serum was used to block non-specific staining.
Sections were pre-treated with trypsin (10 mg/ml) or by micro-waving in citrate buffer (pH
6.0). Primary antibodies (Table 3) were incubated in a humidified atmosphere for 1-3 h at 37
°C or overnight at 4oC. Diaminobenzidine was used as chromogen and hematoxylin as the
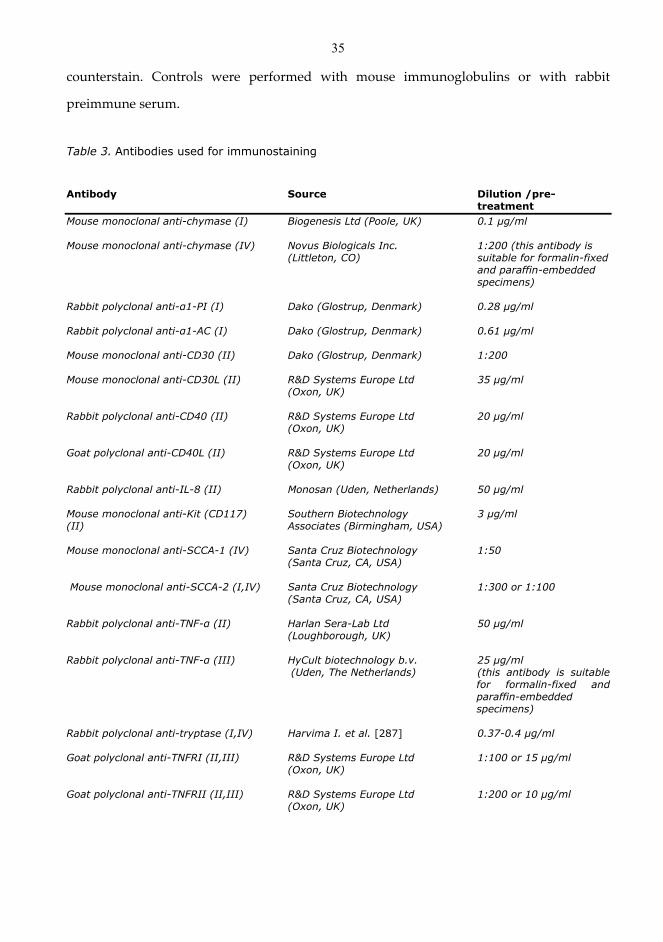
35
counterstain. Controls were performed with mouse immunoglobulins or with rabbit
preimmune serum.
Table 3. Antibodies used for immunostaining
Antibody Source Dilution /pre-
treatment Mouse monoclonal anti-chymase (I) Biogenesis Ltd (Poole, UK) 0.1 µg/ml Mouse monoclonal anti-chymase (IV) Novus Biologicals Inc. 1:200 (this antibody is (Littleton, CO) suitable for formalin-fixed
and paraffin-embedded specimens)
Rabbit polyclonal anti-α1-PI (I) Dako (Glostrup, Denmark) 0.28 µg/ml Rabbit polyclonal anti-α1-AC (I) Dako (Glostrup, Denmark) 0.61 µg/ml Mouse monoclonal anti-CD30 (II) Dako (Glostrup, Denmark) 1:200 Mouse monoclonal anti-CD30L (II) R&D Systems Europe Ltd 35 µg/ml (Oxon, UK) Rabbit polyclonal anti-CD40 (II) R&D Systems Europe Ltd 20 μg/ml (Oxon, UK) Goat polyclonal anti-CD40L (II) R&D Systems Europe Ltd 20 μg/ml (Oxon, UK) Rabbit polyclonal anti-IL-8 (II) Monosan (Uden, Netherlands) 50 μg/ml Mouse monoclonal anti-Kit (CD117) Southern Biotechnology 3 µg/ml (II) Associates (Birmingham, USA) Mouse monoclonal anti-SCCA-1 (IV) Santa Cruz Biotechnology 1:50 (Santa Cruz, CA, USA) Mouse monoclonal anti-SCCA-2 (I,IV) Santa Cruz Biotechnology 1:300 or 1:100 (Santa Cruz, CA, USA) Rabbit polyclonal anti-TNF-α (II) Harlan Sera-Lab Ltd 50 μg/ml (Loughborough, UK) Rabbit polyclonal anti-TNF-α (III) HyCult biotechnology b.v. 25 μg/ml
(Uden, The Netherlands) (this antibody is suitable for formalin-fixed and paraffin-embedded specimens)
Rabbit polyclonal anti-tryptase (I,IV) Harvima I. et al. [287] 0.37-0.4 µg/ml Goat polyclonal anti-TNFRI (II,III) R&D Systems Europe Ltd 1:100 or 15 μg/ml (Oxon, UK) Goat polyclonal anti-TNFRII (II,III) R&D Systems Europe Ltd 1:200 or 10 μg/ml (Oxon, UK)

36
4.3 CELL CULTURES
Primary human keratinocytes were isolated from human foreskin specimens (III,IV).
Subcutaneous fat and deep dermis were removed, and the remaining tissue was incubated
overnight in 0.25% trypsin in solution A (Gibco BRL, Life Technologies). Following the
incubation, keratinocytes were scraped off from the epidermis with a scalpel and
suspended in Keratinocyte-SFM medium (GIBCO™-Life Technologies Ltd, Paisley, UK),
supplemented with 5 ng/ml epidermal growth factor (EGF) and 50 µg/ml bovine pituitary
extract (BPE) (supplied by the vendor). Keratinocytes were maintained in Keratinocyte-
SFM medium supplemented with EGF and BPE, and passages 1 to 5 were used in the
experiments. For immunostaining, primary keratinocytes were also cultured on Nunc Lab-
Tek™ chamber slides (plastic, fibronectin or collagen coated) and immunostained with
antibodies as described for tissue samples.
SiHa and ME180 cells, two cervical SCC cell lines were obtained from the American Type
Culture Collection (Rockville, MD., USA) (III,IV). SiHa cells were cultured in minimum
essential medium (MEM) containing 10% FCS, 0.1 mM non-essential amino acids, 1.0 mM
sodium pyruvate, 1.5 g/L sodium bicarbonate, 100 U/ml penicillin and 100 µg/ml
streptomycin at 5% CO2 and 37°C. ME-180 cells were cultured in McCoy's 5a medium
supplemented with 10% FCS. The SCC cells were examined in subcultures 5 to 10 and they
appeared homogenous in visual inspection.
4.3.1 Apoptosis assays
The TACS TdT in situ apoptosis detection kit (R&D Systems) was used to determine any
possible apoptotic changes in SiHa cells and keratinocytes [35] (IV). The cells were cultured
in 6-well plates until the optimal density was achieved. Subsequently, the medium was
changed to a basal one and 5 µg/ml rh-chymase was added (Table 4). The detached cells
were collected and chymase was inactivated with SBTI. In the next step, the viability was
analyzed with the trypan-blue exclusion method and cytospin preparations were made. In

37
control wells, the cells were briefly incubated with trypsin-EDTA. Afterwards, trypsin was
inactivated before processing the cells for cytospin preparations. After a short formalin
fixation, the cytospin preparations were stained using the apoptosis detection kit. The cells
were counted and the apoptotic index (the ratio of apoptotic cell number to total cell
number) was calculated.
4.3.2 Cell growth determination by fluorometric DNA assay
The cell growth was determined using a fluorometric DNA assay and Hoechst 33258 as
described previously [288,109,289] (III,IV). The cells were seeded in wells of a 24-well plate
(Nunc, Roskilde, Denmark) and incubated until the optimal cell density was achieved.
Next, the medium was replaced and various concentrations of modulating agents or
diluent control were added for 2–3 days. Subsequently, a solution consisting of 8 M urea,
0.04% sodium dodecyl sulphate, 0.154 M NaCl, and 0.015 M sodium citrate, pH 7.0 was
added into the wells at 37°C for 1 h to dissolve the DNA of cells. Thereafter, 1.0 µg/ml of
Hoechst 33258 was added and the fluorescence of the solution was measured using Spectra
Fluor reader (Q-Lab Pty Ltd, Eagle Farm, Australia).
4.3.3 Determination of keratinocyte growth and migration
To determine the growth and migration of normal human keratinocytes under high-
calcium conditions, metallic cylinders were placed (6 mm inner diameter) on the bottom of
each well of an uncoated 6-well plate (Falcon, Becton Dickinson, Plymouth, UK) [109] (III).
The wells and cylinders were equilibrated in 5 ml of complete Keratinocyte-SFM® medium
at 37°C and 5% CO2. Then, about 30,000 keratinocytes, suspended in the same medium,
were added cautiously into each cylinder. The cells were allowed to adhere onto the plastic
surface overnight and practically complete confluence of keratinocytes was achieved. Next,
the metallic cylinders were removed and Keratinocyte-SFM® was changed to 5 ml of
DMEM, 10% FCS, 100 U/ml penicillin and 100 µg/ml streptomycin, a medium which creates
high-calcium conditions. Under these conditions, the keratinocytes monolayer begins to
differentiate and form an epithelium multilayer. After equilibration for 1–2 h, MC
mediators were added in varying combinations and concentrations. The medium and

38
agents were changed every 2–3 days until the epithelium border almost reached the wall of
the well. Afterward, overnight fixation of the epithelium with 4% formaldehyde followed
by staining with Mayer's hematoxylin was made.
4.3.4 Determination of SiHa cell migration and invasion using an in vitro transwell assay
BD Biocoat™ Matrigel™ Invasion chambers and control chambers (BD Biosciences Europe,
Erembodegem, Belgium) for 24-well plates were used to study the effect of MC mediators
on the migration and invasion of SiHa cells (III,IV). In this method, cells were seeded into
the transwells using incomplete DMEM. The lower well contained complete DMEM. After
adherence of the cells at 37°C and 5% CO2 for 1 hour, the agents were added in the upper
transwell chamber. After overnight incubation, the non-migrating and non-invading cells
were removed from the upper surface of the membrane by "scrubbing". The cells on the
lower surface of the membrane were stained with hematoxylin and counted under the
microscope at 200× magnification using an ocular grid [290].
4.3.5 Adherence onto fibronectin-coated plastic surface assay
The method has been described previously [288,109,108] (IV). First, the wells of 24-well
plates were either uncoated or coated by adding 250 µl of 50 µg/ml plasma-fibronectin in
D-PBS to the wells overnight. Thereafter, excess fibronectin was washed away and the MC
mediators were added in varying combinations to the wells overnight. On the following
day, the solution was washed away with PBS and 1% heat-inactivated BSA was added to
the wells for 2 h to block non-specific binding. The wells were washed again with D-PBS
and 1 ml of serum-free DMEM was added to the wells. After 30 min of incubation, about 2
x 105 SiHa cells in serum-free DMEM were added and the cells were allowed to adhere onto
the plastic surface for 60 min followed by washing non-adherent cells away with D-PBS
twice. The SiHa cell adherence was quantitated by measuring the amount of solubilized
DNA of adherent cells using a fluorometric DNA assay [289]. The analyses were performed
at least three times using triplicate wells.

39
4.3.6 Determination of cell viability and cytotoxicity
Cell viability and the cytotoxic effects of different modulating agents was determined with
the MTT assay as described [109,288] (III). The cells were seeded in wells of a 96-microwell
plate at a density of about 4,000 cells/well using complete medium. On the next day, the
medium was replaced with fresh medium. The adhering cells were treated with various
concentrations of modulating agents for 6–8 h. Then 0.33 mg/ml MTT in incomplete
Keratinocyte-SFM was added into the wells at 37°C and 5% CO2 for 2 h. Subsequently, the
MTT solution was removed and the intracellular dye was solubilized by incubating with
DMSO for 15–20 min. The absorbance of the solution was measured at 550 nm using a
micro-ELISA reader (SLT-Labinstruments GmbH, Salzburg, Austria). The cultures and
analyses were made with eight parallel wells and each experiment was performed at least
four times.
4.3.7 Determination of cell cycle by flow-cytometry
SiHa cells were seeded in 6-well plates using complete MEM medium (III). The next day,
the medium was replaced by either complete or incomplete MEM and different modulating
agents were added for 24 h. Subsequently, the conditioned medium containing possible
detached cells was collected into a test tube. The attached cells were released by incubating
in trypsin-EDTA for 10 min. After trypsin inactivation with 10% FCS, the cell suspension
was combined with the spontaneously detached cells. The final cell suspension was
centrifuged and washed with D-PBS. For cell fixation, cells were suspended in 0.5 ml of
cold D-PBS and then were added cautiously, drop by drop, to 5 ml of ice-cold 70% ethanol
under continuous mixing of the solution as described earlier [109]. After a minimum
fixation time of 1 day, cells were centrifuged and suspended in D-PBS, treated with 0.15
mg/ml RNAse at 50°C for 1 h, and incubated in 16 µg/ml propidium iodide at 37°C for 2 h.
In the final step, the cells were analyzed using a FACScan (BD Biosciences, USA) flow
cytometer. The experiment was performed three times.

40
4.4 CYTOKINES AND MC MEDIATORS
In order to study the growth modulation and cytotoxicity of different cytokines,
keratinocytes were cultured on 24-well tissue culture plates. 70-80% confluent cells were
repeatedly washed with PBS and incubated overnight in Keratinocyte-SFM medium with
supplements. Subsequently, keratinocytes were treated with different cytokines or growth
factors for 24 h (Table 4). All treatments were conducted in Keratinocyte-SFM medium with
or without supplements. After 24 h, total DNA was extracted from the cells with untreated
cells being used as a control.
Table 4. MC mediators used in the study
Cytokine or MC mediator Source Heparin sodium salt Fluka BioChemica (Buchs, Germany) Histamine diphosphate monohydrate Fluka BioChemica (Buchs, Germany) Rh-TNF-α R&D Systems Europe Ltd (Oxon, UK) Rh-chymase R&D Systems Europe Ltd (Oxon, UK) Tryptase-chymase preparation Prepared in our laboratory [108]

41
5 Results
5.1 TRYPTASE- AND CHYMASE-POSITIVE MCS ARE INCREASED IN
NUMBER AND ARE ASSOCIATED WITH PROTEASE INHIBITORS IN BCC (I)
BCC lesions showed more MCs than healthy-looking skin (I, Fig. 1). In the BCC lesions, the
numbers of MCs with tryptase activity and those with chymase immunoreactivity were
significantly increased by 2.2-2.3-fold when compared to the situation in the healthy-
looking skin samples. However, the ratio of cells with chymase activity to those with
chymase immunoreactivity was significantly decreased in BCC lesion (I, Table 1), possibly
due to chymase inhibitory effects of α1-PI, α1-AC or SCCA-2.
To clarify further whether tryptase and chymase are inactivated in the BCC lesion, the
sequential double-staining method for tryptase and immunoreactivity for chymase
inhibitors was utilized. The results showed that only occasional tryptase-immunopositive
cells (about 1%) did not display tryptase enzyme activity, and the expression of chymase
inhibitors α1-AC and α1-PI was significantly increased (I, Table 1). SCCA-2
immunoreactivity was strongly increased in the malignant BCC epithelium, mostly in the
suprabasal layers. Interestingly, the entire BCC epithelium was stained except for the basal
part, which appeared to be either unstained or weakly stained (I, Fig. 3). Large basal buds
attached to the BCC epithelium were negative for SCCA-2 (I, Fig. 3).
5.2 BCC LESIONS DISPLAY INCREASED CD30L/CD153 AND TNF-α
EXPRESSING MCS (II)
The upper dermis of BCC lesions display over twofold higher amounts of tryptase-positive
MCs, many of them in close apposition to the BCC epithelium. Moreover, a significantly

42
increased number of cells positive for Kit was found, the latter being an important growth
and survival receptor on MCs (II, Table 1).
A statistically significant increase in the percentage of MCs displaying CD30L in the BCC
lesions is evidence that CD30L is up-regulated in these MCs. In addition, MCs in BCC
lesions were one of the predominant cell types exhibiting CD30L immunoreactivity (II, Fig.
1). Furthermore, the counterpart, CD30 immunopositive cells, was found to be increased in
number in the upper dermis of BCC lesion (II, Table 1; Fig. 2). In contrast, the epidermis of
the healthy-looking skin and the BCC epithelial compartment showed only occasional
scattered CD30-positive cells (II, Fig. 2).
MCs were practically devoid of CD40L immunopositivity and only rare occasional MCs
displayed slight CD40L staining in the BCC lesion (II, Fig. 5). The counterpart, CD40
positive cells, showed significantly higher scores in the upper dermis of the BCC lesion
compared to the situation in healthy-looking skin (3 ± 0 versus 1.8 ± 0.5, p = 0.00012,
respectively). The basal layer of the epidermis of the healthy-looking skin and BCC
epithelium displayed CD40 immunoreactivity but not the upper layers (II, Fig. 6). Many
larger basal buds were mostly negative for CD40 in the BCC lesion even in the basal layers
(II, Fig. 6).
An increase, although statistically non-significant, in the percentage of TNF-α-positive MCs
in the BCC lesions was observed when compared with the healthy-looking skin (36.7%
versus 27.0%) (II, Table 2). However, the increased number TNF-α-positive MCs in the BCC
lesion appeared to be a result of the increased number of tryptase-positive MCs in the
upper dermis. In addition, MCs appeared to be the predominant cell type displaying TNF-
α immunopositivity in the BCC lesion (II, Fig. 3). The counterpart TNFRI- and TNFRII-
positive cells were found to be significantly increased in number in the BCC lesional dermis
when they were semiquantitatively analyzed (II, Table 2). Both TNFRs were detected
similarly in the viable healthy epidermis and BCC epithelium (II, Fig. 4) though larger basal
buds in 5 cases showed no TNFRI.

43
The proinflammatory cytokine IL-8 has recently been described to be produced by MCs in
psoriasis and atopic dermatitis [52]. Using a double-staining method for IL-8 we found a
significantly higher percentage of IL-8-positive MCs in the upper dermis of the BCC lesions
compared to healthy-looking skin (5.5 ± 6.5 versus 1.1 ± 1.2%, P = 0.037, respectively),
although the percentage values were generally low.
5.3 TNF-α AND HISTAMINE DO NOT INFLUENCE THE GROWTH OF
SQUAMOUS CARCINOMA CELLS FROM UTERINE CERVIX WHEREAS
NORMAL KERATINOCYTES UNDERGO CYTOLYSIS (III)
Histamine alone did not evoke any stimulatory effect on SiHa or ME-180 cell growth after
cultivation in complete or incomplete medium (III, Fig. 1). In incomplete medium, TNF-α
alone significantly inhibited the growth of SiHa or ME-180 cells. The combination of
histamine (0.1 and 1 mM) and TNF-α (10 and 50 ng/ml) did not produce any additional
inhibitory effect over that of TNF-α alone on SiHa or ME-180 cell growth. In contrast, the
growth of normal keratinocytes was significantly inhibited by histamine (1 mM) or TNF-α
(10 or 50 ng/ml) and, furthermore, the inhibitory effect was found to be cumulative in both
complete and incomplete Keratinocyte-SFM medium (III, Fig. 3).
The combination of 0.1 or 1 mM histamine and 10 or 50 ng/ml TNF-α produced significant
cytotoxic effects on normal keratinocytes (III, Fig. 5a). In contrast, neither histamine nor
TNF-α, nor both in combination, could induce any marked cytotoxic effect on SiHa or ME-
180 cells (III, Fig. 5b, c). After treatment with emetine, a protein synthesis inhibitor, which
has the capacity to render SiHa cells susceptible to the cytotoxic effects of TNF-α [291], the
combination of histamine and TNF-α did not cause any increase in cytotoxicity on SiHa
cells when compared to TNF-α alone (III, Fig. 6). In addition, neither TNF-α at 10 and 50
ng/ml, histamine at 0.1 and 1 mM nor their combination of both could induce apoptotic
events above the control level (3.9% in incomplete and 0.9% in complete medium), although

44
TNF-α alone slightly induced cell cycle arrest at G0/G1 phase especially in incomplete
medium.
Histamine alone (0.1 or 1 mM) was unable to modulate SiHa cell migration. In contrast,
TNF-α produced a significant increase in cell migration. Combination of histamine (0.1 or 1
mM) and TNF-α (10 or 50 ng/ml) could not induce any additional effect over the one
achieved by TNF-α alone (III, Fig. 8). Furthermore, neither histamine nor TNF-α alone or in
combination, using the same mediator concentrations as in the migration assay, induced
any significant changes on SiHa cell invasion by using the Matrigel transwell assay.
To explain the TNF-α effects on keratinocytes and cancer cells, the presence of the
counterpart receptors, TNFRI and TNFRII, was confirmed in these cells using
immunostaining techniques. Overall, 52% and 36% of the keratinocytes were positive for
TNFRI and TNFRII (III, Fig. 9), respectively. In clear contrast, SiHa and also ME-180 cells
displayed only weak immunopositivity, having far less intensity for both TNFRs than
keratinocytes.
Immunostaining was used to evaluate the presence of TNF-α and its receptors in cervix
carcinoma samples. Clear immunopositivity for TNF-α in cervix carcinoma samples was
found in seven out of eight cases in the tumor cells and peritumoral stroma (III, Fig. 11b).
Control samples showed clear immunopositivity for TNF-α inside the epithelium (III, Fig.
11a). In addition, control samples displayed immunopositivity with TNFRI localized in the
basal layers of epidermis, whereas one sample showed positivity in the upper keratinocytes
(III, Table 2). Half of the carcinoma samples had lost their positivity for TNFRI. In contrast,
TNFRII showed positivity in the upper layers of epidermis in all control samples, whereas
tumor cells were considered to be negative in two cases. Interestingly, two samples were
negative for both TNFRs (III, Fig. 12).

45
5.4 MC CHYMASE IS PRESENT IN UTERINE CERVICAL CARCINOMA AND IS
CAPABLE OF DETACHING VIABLE AND GROWING CERVICAL SQUAMOUS
CARCINOMA CELLS FROM SUBSTRATUM (IV)
The numbers of chymase-immunopositive cells were found to be similar in the upper
subepithelial stroma of control tissue and in the peritumoral stroma of SCC (IV, Fig.1a)
(69±21 versus 62±58 cells/mm2, respectively). Furthermore, similar numbers of tryptase-
positive cells were found in the peritumoral stroma of SCC (81±60 cells/mm2) and in
controls (99±54 cells/mm2) (IV, Fig. 1b). Interestingly, the ratio of the number of chymase-
positive cells to that of tryptase-positive cells was similar in all areas – 75±29% in the
peritumoral stroma, 79±18% in the normal-looking tissue of SCC samples, and 74±22% in
control samples. Tumor cells and the epithelium of control samples did not show
immunopositivity for tryptase and chymase.
Immunohistochemical staining for SCC antigens revealed only weak immunopositivity for
SCCA-2 in 4 cervical SCC specimens and clear immunopositivity in only one specimen (IV,
Fig. 2a, Table 1). However, no SCCA-1 staining was detected in any of the SCC specimens.
Instead, immunopositivity for SCCA-1 in the squamous layer of the epithelium was
detected in all control specimens (IV, Fig. 2b, Table 1), though these specimens did not
express SCCA-2.
The tryptase-chymase preparation, in the absence of heparin, caused effective detachment
of SiHa cells from the plastic surface (IV, Fig. 3a) when the cells were cultivated in
incomplete medium. Furthermore, SBTI, an inhibitor of both chymase and cathepsin G,
completely prevented the changes induced by the tryptase-chymase preparation whereas
aprotinin, which inhibits only cathepsin G [108] had no effect. In addition, neither
histamine nor heparin was able to affect the cell detachment in the presence of tryptase-
chymase preparation. Interestingly, over 86% of the SiHa cells detached by the tryptase-
chymase preparation were found to be viable by the trypan-blue exclusion method. This

46
finding was confirmed when detached cells were transferred to another 6-well plate and
found to continue their growth in an apparently normal fashion.
In clear contrast to SCC cells, prolonged treatment with rh-chymase reduced the cell
viability and induced apoptosis in detached normal keratinocytes. The viability of the cells
was estimated using two different methods: trypan-blue exclusion and cytospin
preparations stained with Apoptosis detection kit (IV, Fig. 8).
The detachment and/or reduced adherence of SiHa cells onto substratum by tryptase
and/or chymase was a result of matrix protein cleavage, since SiHa cell adherence onto the
pretreated fibronectin-coated plastic surface was inhibited by overnight treatment with the
tryptase-chymase preparation. Moreover, DFP, a serine proteinase inhibitor, when
combined with the enzyme preparation prevented the cell detachment. In contrast, TLCK,
an effective inhibitor of tryptase [108] had no marked effect on cell adherence. In addition,
aprotinin did not prevent the effect of the tryptase-chymase preparation, which effectively
decreased the cell adherence. In contrast, SBTI markedly inhibited the effects of the
tryptase-chymase preparation, indicating that it was chymase, which was responsible for
the decreased adherence of SiHa cells to fibronectin (IV, Fig. 5). Furthermore, rh-chymase
degraded fibronectin in a dose-dependent manner as confirmed using the SDS-PAGE assay
(IV, Fig 6).
In addition to the tryptase-chymase preparation, rh-chymase was capable of inhibiting the
cell growth probably due to its ability to detach viable cells from the plastic surface. The rh-
chymase effect was a result of its catalytic properties since SBTI completely prevented the
changes induced by rh-chymase, though SBTI on its own had no effect on cell detachment
(IV, Fig. 7). Also, it was found that rh-chymase could inhibit SiHa cell migration in a dose-
dependent manner, probably owing to it´s capability of detaching cells, and SBTI
completely prevented this effect (IV, Fig. 9).

47
To find out whether SiHa cells are capable of inhibiting chymase, the chymotryptic activity
of SiHa cells was first studied by sonicating a cell suspension in an ice bath. The result
showed that SiHa cell sonicate itself contained only weak, if any, chymotryptic activity.
Next it was found that the SiHa cell sonicate did not markedly inhibit rh-chymase activity
(IV, Fig. 10). Since SCCA-2 is a well known chymase inhibitor [280], it was studied whether
the SiHa cells contained any immunoreactivity for SCCA. Hence, SiHa cells cultured in
chamber slides were immunocytochemically stained for SCCA-1 and -2 using monoclonal
antibodies (IV). Only weak, if any, expression of SCC antigens in SiHa cells was found.

48
6 Discussion
MSc are usually increased in number in different cutaneous malignancies, and MCs are
thought to contribute to skin carcinogenesis by immunomodulation, induction of
angiogenesis, degradation of the extracellular matrix components, and promotion of
mitogenesis. The development of skin carcinomas requires malignant transformation and
compromised immune system [53]. UV radiation is the major causative factor for skin
carcinogenesis and MCs evidently have an essential role in UV-induced
immunosuppression using different mechanisms [5,129,181,202,53]. The recruitment of
immunomodulatory or immunosuppressive MCs to the skin tumor may be due to
carcinoma cell-derived SCF and Kit receptor on MCs [292] There is recent experimental
evidence to support this mechanism [293].
In addition to the regulatory role, MCs are often considered to be proinflammatory cells in
chronic tissue inflammation. This function of MCs can be in line with the concept that
cancer development is often accompanied by an inflammatory response and peritumoral
inflammatory cell infiltration. However, sufficient immunosuppression may be required to
prevent the harmful damages to the tumor by inhibiting the excessive inflammation
[293,294].
This background should be kept in mind when interpreting the results of the study. It is not
known in which situation MCs act as proinflammatory cells and in which conditions they
behave like immunosuppressive cells [53].
6.1 PARTIAL INACTIVATION OF CHYMASE AND INCREASE IN PROTEASE
INHIBITORS IN BCC

49
Previously, tryptase-positive MCs have been found in the adjacent stroma of the BCC lesion
[295] and other types of human cancers, such as breast cancer, oral SCC and malignant
melanoma [13,296,297]. This suggests that tryptase may be involved in matrix degradation,
an important factor for tumor survival and growth [143,145,146,148]. In our study, the
number of MCs with tryptase activity was increased by 2.2-fold in the BCC lesion. One
explanation for the accumulation of mast cells in the upper dermis close to the malignant
epithelium in BCC is the expression of α3-integrin on mast cells and thereby these cells can
bind to laminins LM-332 and LM-511 in the basement membrane [298].
MCs in the BCC lesion are probably in an activated form leading to increased degranulation
and exposure of serine proteinases tryptase and chymase to protease inhibitors at
physiological pH [299]. However, only about 1% of the tryptase-immunopositive cells in
the BCC lesions did not display any tryptase activity. These rare MCs with inactive tryptase
may be a result induced by the action of serpin B6 inhibitor or a result of a technical artifact
due to section processing [140]. Similar situation has been found in herpes zoster lesions
where only occasional tryptase-immunopositive cells were devoid of enzyme activity [153].
Consequently, tryptase is virtually in a fully active form, able to affect surrounding cells
and matrix components in the BCC lesion, a conclusion supported by the fact that there are
no known endogenous inhibitors for tetrameric tryptase. Independent of these
considerations, agents which dissociate tetrameric tryptase from heparin proteoglycan can
inactivate this enzyme since the tetrameric form dissociates into inactive monomers [300].
In contrast to tryptase-heparin proteoglycan complexes, the chymase-heparin proteoglycan
complexes are larger in size and tend to remain at the site of MC degranulation [34,35]. The
soluble form of chymase is effectively inactivated by α1-AC even though in the acidic MC
granules tightly packed chymase appears to be relatively insensitive to this protease
inhibitor [34]. Furthermore, chymase-immunopositive cell number was significantly
increased by 2.3-fold in the BCC lesion, suggesting a high number of the MCTC type of MCs.
In contrast, a 1.6-fold increase in number of MCs displaying chymase enzyme activity and a

50
significantly decreased ratio of cells with chymase activity to those with chymase-
immunopositivity are an evidence that chymase is partially inactivated in the BCC lesion.
Several different protease inhibitors are either synthesized in human MCs and/or taken up
from the extracellular environment [152,155,156]. In our study, the significant 1.7-fold
increase in the percentage of MCs showing α1-PI or α1-AC immunoreactivity in the BCC
lesion could well explain the apparent inactivation of chymase, even though SLPI [155]
could be another candidate for chymase inhibition. A similar inactivation of chymase to
that detected in the BCC lesion has been observed in other pathological skin conditions,
such as blistering diseases [165], psoriasis [149], chronic ulcers [166], allergic prick-test
reaction [167], and even in skin organ cultures [34,35]. Therefore. the findings of this study
are not specific to BCC, and partial inactivation of chymase may just reflect chronic
inflammation in this carcinoma type.
Chymase has potentially destructive effects to the epidermis and epithelium producing
epithelium detachment [163,108]. Therefore, the enzyme has to be in tight control in normal
skin to prevent epidermis-dermis separation every time mast cells activate and degranulate,
e.g., in urticarial wheal. Chymase is capable of degrading and inactivating it´s inhibitors α1-
PI and α1-AC [301]. In the chronic inflammation of BCC lesion, it is possible that an
equilibrium between chymase inactivation and degradation of its inhibitors may have been
reached [301]. However, chymase was not completely inactivated and the residual chymase
activity may still be sufficiently high to have a marked influence on the stromal cells,
basement membrane structures and BCC cells. It may even be possible that a fully active
chymase is needed for removing the malignant epithelium away, but chymase fails in this
task.
SCCA-2, a non-specific disease marker for squamous cell cancer, has also the capacity to act
as a chymase inhibitor. In this work, SCCA-2 was increasingly expressed in the areas of the
malignant BCC epithelium but larger basal buds, often surrounded by MCs, were negative

51
for this antigen. Thus, even though a strong expression of SCCA-2 was found in the BCC
lesion, it appears not to be as crucial chymase inhibitor as α1-PI or α1-AC.
6.2 TNF-α AND CD30L BUT NOT CD40L IMMUNOPOSITIVE MCS IN BCC
LESION
Previously, an increased number of MCs in upper dermis of psoriasis and BCC lesion has
been reported. Moreover, in psoriasis, MCs displayed immunoreactivity for TNF-α and
CD30L [62,63,52,295,302,303]. However, only limited data is available on the CD40L
expression in cutaneous MCs. In this study, the increased number of MCs in BCC lesion
was confirmed and MCs appeared to be one predominant TNF-α-positive cell type in BCC
lesion. However, the increased number of TNF-α-positive MCs in the upper dermis and the
percentage of TNF-α-positive MCs in the healthy-looking skin of BCC patients were similar
to those found previously in atopic dermatitis and psoriasis [62]. Since chymase can
degrade TNF-α to some extent [304] and the enzyme is partially inactivated in the BCC
lesion (I), it is possible that inactivation of chymase allows more effective actions of MC
TNF-α.
Markedly increased scores of TNFRI- and TNFRII-positive cells were found in the upper
dermis of the BCC lesion. These receptors may have the capability of regulating the tumor
cell death and inflammation just beneath the BCC epithelium when receptor-positive cells
are stimulated by soluble TNF-α released from MCs [305]. The staining pattern for TNFRI
and TNFRII in BCC resembled the staining in psoriatic epidermis showing perinuclear
accentuation but, in contrast to the positively stained parakeratotic area of the psoriatic
epidermis, the uppermost stratified area in the BCC epithelium was negative. Thus, based
on these histochemical results, MC TNF-α may interact directly with TNFR-positive cells in
the dermis and BCC epithelium. However, TNF-α may be captured before it reaches its
target cells by the serum levels of TNFRI, but not that of TNFRII, and as a result the
antitumoral response is impaired [306].

52
The specific biological effects of the CD30L-CD30 interaction are difficult to discern. One
important role of the CD30L-CD30 system is inhibition of function and apoptosis induction
in certain situations [307,241]. CD30 has been found on activated T, B and NK cells, as well
as in tumors of the immune system and in some nonhematolymphoid neoplasms [252].
Expression of the receptor is transient and certain effects appear to be opposed. CD30
engagement is costimulatory for the growth and differentiation of specific T cells [308,309].
CD30L has been found on macrophages [241], B and T cells [310,242], eosinophils [311],
erythrocyte precursors [312], leukemias cells [313] and mast cells [314]. CD30L stimulate the
expression of both CD30 and IL-4 by T cells [315]. In this study, MCs were the predominant
cell type expressing CD30L and the percentage of CD30L-positive MCs was significantly
increased in the upper dermis of the BCC lesion, in agreement with a previous finding in
psoriasis and atopic dermatitis [299]. In addition, even though the patient groups are not
directly comparable in these studies, the percentage of CD30L-positive MCs in the healthy-
looking skin of BCC patients was similar to the situation in the healthy-looking skin of
atopic dermatitis patients, but higher than in the healthy-looking psoriatic skin [52].
Furthermore, the colocation of CD30-positive cells with MCs in the upper dermis provides
morphological evidence for the interaction between these cells. In addition, the interaction
of soluble CD30 with MCs can activate them by reverse signaling and stimulate for
increased secretion of proinflammatory IL-8 and TNF-α [257,252,52]. Furthermore, this
interaction may explain the increased IL-8 expression in MCs in the BCC lesion. In clear
contrast to the situation in the upper dermis, but in agreement with the previous reports
[316], only few occasional cells of the BCC specimens showed CD30-immunopositivity in
the epithelium but these cells represent other cells than BCC cells. Thus, the findings on
CD30L and CD30 in BCC are not disease-specific and may be related to chronic
inflammation, like in the case of psoriasis and atopic dermatitis.
The CD40L-CD40 interaction regulates apoptosis positively or negatively, and may have
some remarkable antitumor effects [317,318,319,320]. Data on CD40L expression on skin
MCs is very limited. Previously it has been found that the BCC lesion does not show CD40L
expression [321]. In agreement with these studies we found that skin MCs were devoid of

53
CD40L in both healthy-looking skin and BCC lesion. Thus, it is possible that skin MCs have
lost their ability to express CD40L. However, the specificity of this finding needs to be
clarified in other chronic inflammatory conditions of the skin, e.g. in psoriasis, atopic
dermatitis and SCC.
Similarly to previous reports [322,321], the CD40 receptor was localized to basal layer of the
epidermis in healthy-looking skin. The situation was similar in superficial spreading BCC,
where the basal layer showed positivity for CD40 receptor. However, it was confirmed like
in the previous studies [322,321] that BCC cells, when protruding into the dermis forming
basal buds and nests, loose their capability of expressing marked CD40. A similar situation
was observed also for TNFRI, which showed faint immunopositivity in the larger basal
buds. As a result, without the CD40L-CD40 system and TNFRI, BCC cells may be free to
grow and invade into the dermis. Furthermore, it was also confirmed like in previous
reports that the CD40-positive cells in the BCC dermis can be dendritic cells, endothelial
cells and lymphocytes [322,321]. As the counterpart ligand CD40L is missing, the role of
these cells in dermis is still obscure.
MCs are the only cells expressing Kit receptor in the healthy and inflamed dermal skin
[106,323]. As an almost equal number of cells positive to Kit and tryptase in the dermis of
the healthy-looking skin and BCC lesion was found it can be concluded that all MCs
express Kit receptor. Thus, the SCF-Kit system can act in the BCC lesion as BCC cells and
peritumoral cells have previously been reported to express SCF [292].
6.3 TNF-α AND HISTAMINE HAVE A SIGNIFICANT GROWTH-
INHIBITORY AND CYTOLYTIC EFFECT ON NORMAL KERATINOCYTES BUT
NOT ON CERVIX CARCINOMA CELL LINES SIHA AND ME-180
Histamine together with TNF-α can induce a range of different effects on cells. Previously,
histamine has been shown to inhibit the mitosis of keratinocytes [324]. When acting in
conjuction on normal keratinocytes, histamine and TNF-α have shown synergism in the

54
induction of ICAM-1, probably through histamine H2-receptors [325]. In addition, ICAM-1
expression in keratinocytes is known to lead to T cell activation and to increased lysis of
keratinocytes by cytotoxic T cells [326]. In contrast, histamine may transiently also inhibit
the actions of TNF-α on cells [327]. In peripheral blood mononuclear cells it can suppress
gene expression and synthesis of TNF-α, again an effect mediated by H2-receptors [328]. In
the present study, histamine or TNF-α alone induced growth inhibition of keratinocytes in
a non-cytolytic manner. Furthermore, when histamine and TNF-α acted together, an
increased growth inhibitory and cytolytic effect on normal keratinocytes was observed.
Histamine and TNF-α in combination may activate and thereby induce ICAM-1 in
keratinocytes and presumably have also other effects. Thus, preactivation of keratinocytes
by histamine renders the cells more susceptible to the subsequent cytotoxic effects by TNF-
α or vice versa. Nevertheless, it is possible that there are still some unknown ways for the
tissue to control the growth of the epidermis or epithelium. Since heparin binds efficiently
TNF-α and augments the growth inhibition of keratinocytes induced by TNF-α [109], the
combination of histamine, heparin and TNF-α may be even more potent for causing
keratinocyte cytolysis.
TNF-α can cause cytolysis to some cancer cell lines or, oppositely, it may stimulate cancer
cell growth [329,330]. In cervical carcinomas caused by HPV viruses, TNF-α treatment
seemed rather to maintain the growth, or even stimulate cell growth in one study, rather
than cause growth inhibition and cytolysis of cervical carcinoma cells [331,332,333,291].
Furthermore, additional stressors such as protein synthesis inhibition or radiation are
needed to induce cytolysis in these carcinoma cells by TNF-α [291,334]. Moreover, EGF has
been found to be protective in ME180S cells, a cervical carcinoma cell line sensitive to TNF-
α, in TNF-α induced apoptosis [335]. In this study, TNF-α alone inhibited only slightly the
growth of SiHa cells in a non-cytolytic manner, an inhibition, which was probably
attributed to the slight growth arrest at G0/G1 phase of the cell cycle found in the serum-
free medium.

55
Previously, significantly decreased chemotactic migration of SiHa and CaSki cervical
carcinoma cell lines to laminin-1 by TNF-α has been shown, while migration towards type I
collagen was increased [336]. Moreover, TNF-α has a stimulatory effect on the migration of
SW756 cervical carcinoma cells [337]. Thus, the present finding that TNF-α increased SiHa
cell migration towards serum is in line with these previous findings. The weak growth
inhibitory effect and the slight cell cycle arrest at the G0/G1 of SiHa cells evoked by TNF-α
were associated with increased TNF-α induced SiHa cell migration. Thus, even though
carcinoma cell proliferation is slightly inhibited, the better motility of these cells induced by
TNF-α may enhance the spread of the tumor.
Histamine has been proposed to be involved in the regulation of cancer growth as MCs can
be found in increased numbers in the vicinity of tumors. In SCC cell lines HeLa and A431
histamine has been found to stimulate the growth and chemotactic migration through the
H1 receptor [338]. Moreover, histamine inhibited the production of interferon-induced
protein of 10 kDa in the SCC15 SCC cell line via the H2 receptor [339]. Even though we
confirmed the previous findings that histamine could inhibit the growth of monolayer
proliferating keratinocytes as well as the growth of keratinocyte epithelium [340], histamine
was not capable of influencing the growth, viability, cell cycle, migration and invasion of
SiHa SCC cells.
Emetine-sensitized SiHa cells have been shown to undergo cytolysis after treatment with
TNF-α [291,341,342]. Nevertheless, the combination of histamine and TNF-α did not evoke
any increased cytolysis effect in the SiHa cells pretreated with emetine. Similarly to the
situation in SiHa cells, ME-180 cells were not markedly affected by the combined action of
TNF-α and histamine suggesting that this may be a characteristic feature of SCC cells from
different origins.
TNFRI and TNFRII mediate the effect of TNF-α on the cell. In this study, these receptors
were markedly expressed in normal keratinocytes but, in contrast, SiHa and ME-180 cells
displayed weak immunoreactivity and only a few occasional cells appeared to clearly

56
display these receptors. This clear difference may provide one explanation why normal
keratinocytes react to TNF-α and histamine in a cytolytic fashion but SiHa and ME-180 cells
do not respond. Previously, immunoreactivity of TNF-α and TNFRI and II has been
detected in tumor cells of SCC from head and neck as well as from the oral cavity [343,344].
Moreover, SiHa cells have been shown to express TNF mRNA [345]. Since SiHa cells
displayed only weak TNFRI and II immunoreactivity in this study, TNF-α and TNFRs were
stained also in cervical carcinoma specimens. Only one cervix carcinoma specimen out of 8
revealed marked TNFRI or TNFRII immunopositivity and the same sample showed also
high positivity for TNF-α. Large areas among tumor cell sites did not show staining for
these receptors. Control samples showed higher scores of TNF-α and TNFRI or TNFRII.
Thus, cervix carcinoma cells appear to contain lower levels of TNFRI and TNFRII when
compared to the epithelium in samples with non-specific inflammation. Even though TNF-
α is expressed in cervix carcinoma, the tumor cells may not respond adequately due to lack
of receptors.
6.4 IS MC CHYMASE ABLE TO RELEASE SCC CELLS FROM TUMOR
LEADING TO SPREADING OF MALIGNANT CELLS?
Tryptase and chymase have been associated with a variety of human cancers [228;13].
Previously, it has been reported that there are increased numbers of MCs in the uterine
cervical carcinoma and they were found to be predominantly of the MCT type [228].
However, the enzyme-histochemical technique on formalin-fixed and paraffin-embedded
sections may not detect all chymase due to chymase inactivation during rough processing
conditions. In the present study, the ratio of the number of chymase-positive cells to that of
tryptase-positive cells was about 75-79 % in SCC specimens and about 74 % in control
specimens suggesting that about 21-25 % of the MCs were of the MCT type in the SCC
lesions. This percentage is much lower than the 80-83 % percentage of MCT cells described
previously [228]. Thus, the present results clearly show the presence of chymase in cervix
carcinoma, when chymase is stained with a mouse monoclonal antibody suitable for
formalin-fixed and paraffin-embedded specimens. However, the use of formalin-fixed and

57
paraffin-embedded specimens in this work does not allow studying the activity level of
chymase in a similar fashion as it was analyzed on cryosections from BCC samples (I).
SCCA-2, α1-PI, α1-AC and SLPI are effective inhibitors of chymase [281], whereas no
endogenous inhibitors are known for tetrameric tryptase and almost all tryptase-
immunopositive cells exhibit tryptase enzyme activity in cancer tissues [13]. Even though
there is high structural homology between SCCA-2 and SCCA-1, there are differences in
their capacity to inhibit proteinases: SCCA-1 inhibits cysteine proteinases and SCCA-2
inhibits chymotryptic serine proteinases [281]. SCCA-2, which is secreted by SCC cells, may
control the activity of chymase in the microenvironment between SCC cells and
peritumoral MCTC MCs. Interestingly, clear differences were found in SCCA-1 and SCCA-2
expression between SCC specimens and control specimens. The epithelium of control
specimens with non-specific inflammation was immunonegative for SCCA-2, but positive
for SCCA-1. In contrast, all SCC specimens were apparently immunonegative for SCCA-1.
One SCC specimen out of 9 showed significant SCCA-2 immunoreactivity, 4 specimens
revealed a few foci with some positive cells, and 4 specimens were negative. Thus, the
expression of SCCA-2 immunoreactivity was higher in SCC specimens than in control
specimens. However, the present finding of SCCA-2 and SCCA-1 expression in cervical
SCC and control specimens is in agreement with studies showing increased ratio of SCCA-2
mRNA to SCCA-1 mRNA in SCC in different tissues, including the uterine cervix [346;284].
Since MCTC cells were found to be predominant in SCC specimens and SCCA-2 expression
was unexpectedly low rather than high, it was decided to examine whether the partially
purified tryptase-chymase preparation from human skin [108] could influence the growth
and adherence of cervical carcinoma cells in vitro using a well characterized SiHa cervical
carcinoma cell line. Furthermore, in order to clarify which enzyme is responsible for the
effects, specific inhibitors were used to block either chymase and/or tryptase. As a result,
prolonged treatment with the tryptase-chymase preparation was found to induce the
detachment of viable SiHa cells from the plastic surface and these cells continued their
growth in an apparently normal fashion. Furthermore, the pretreatment of the fibronectin

58
coat with the tryptase-chymase preparation also resulted in reduced SiHa cell adherence
onto the fibronectin coat. The experiments with inhibitors strongly suggest that it is
chymase that induced SiHa cell detachment from substratum and one target of chymase is
fibronectin that was degraded by rh-chymase. Similar results of the crucial role of chymase
have previously been obtained with normal keratinocytes [108]. In fact, MC chymase has
also been found to induce epidermis-dermis separation at the level of lamina lucida in skin
biopsies in ex vivo experiments [163] and to cleave fibronectin, resulting in apoptosis in
conjunctival epithelial cells [347] and smooth muscle cells [159]. Tryptase can also degrade
fibronectin and induce focal epidermis-dermis separation in skin biopsies ex vivo [147].
However, the four active centers of a ring-like tetrameric tryptase are buried inside the
protein facing a central pore [138] and as a result they may not reach pericellular matrix
structures as efficiently as chymase [108]. Both enzymes are able also to activate latent
metalloproteinases which may further increase cell detachment. Tryptase may also have a
role in tumor growth by activating the protease-activated receptor-2 on tumor cells
[348;349].
In this study, the prolonged treatment with rh-chymase detached viable and growing SiHa
and ME-180 cells from the substratum but in contrast, in these conditions normal
keratinocytes undergo apoptosis, similar to the effects found previously in conjunctival
epithelial cells [347] and smooth muscle cells [159], probably due to a specific form of
apoptosis, anoikis. Previously, it has been found that chymase can inhibit in a dose-
dependent manner the growth and migration of keratinocyte epithelium and monolayer
keratinocytes [108] and it also inhibits the migration of monolayer conjunctival epithelial
cells [350]. In this study, rh-chymase inhibited SiHa cell migration in a dose-dependent
manner under the experimental conditions used. Since chymase pretreatment reduced the
adherence of SiHa cells onto the fibronectin-coated plastic surface, it is possible that
chymase interfered with cellular attachment process during migration or, alternatively,
chymase at high concentration simply prevented SiHa cell adherence onto the porous
transwell membrane. Previously, chymase has been found to release the hyaluronan
receptor CD44 from cells, an effect which may modulate cell adherence [351].

59
Chymase-heparin proteoglycan complexes within MC granules are larger in size than
tryptase-heparin proteoglycan complexes [352]. Thus, chymase tends to remain at the site
of MC when compared to solubilization of tryptase [34,35]. As a result, chymase-heparin
proteoglycan complexes may not diffuse efficiently to surround the cancer cell as is the case
in the cell culture well. In addition, heparin may cause steric hindrance and thereby
regulate the effects of chymase on carcinoma cells. However, chymase may be able to
detach viable single carcinoma cells or cell groups from the tumor, and they can continue
their growth as they migrate to more distant sites. On the other hand, in normal tissue, the
detachment of normal keratinocytes by chymase led to decreased viability and increased
apoptosis and thus it is possible that chymase attempts to control an excessive growth of
epidermis and epithelium. In the same fashion, MCTC MCs and chymase may also strive to
prevent the overgrowth of malignant epithelium by cleaving it off but ultimately they fail
in this task.
In previous studies, SiHa cells have been found to express SCCA-2 and its reduced
expression is associated with increased migration and invasion of these cells and decreased
E-cadherin expression [353,354]. Since chymase detached SiHa cells and inhibited their
migration, it was studied whether these cells were able to modulate the activity of chymase.
However, the sonicates of SiHa cells had no marked effect on chymase activity and only a
tiny proportion of these cells exhibited marked immunoreactivity towards SCCA-1 and
SCCA-2. Thus, even though SCCAs are expressed to some extent in SiHa cells, the amount
of active SCCA-2 protein is not enough to control chymase activity in culture. Nevertheless,
there is some discrepancy with the expression of SCCA-2, since its reduced expression in
cervical SCC cells in vitro has been correlated with increased migration and invasion
[353,354], but in contrast, an increased ratio of SCCA-2 mRNA to SCCA-1 mRNA in vivo is
related with poorer prognosis of cervical SCC [346].
Taken together, the findings raise the possibility that mast cells could participate in cancer
progression by affecting the growth, function, spreading and death of tumoral cells.

60
However, the role of other inflammatory cells and mediators in the process of cancer
evolution should not be underscored. Future experiments will be necessary to determine
the importance of the individual types of inflammatory cells in the onset, progression and
execution of the pathological processes ultimately culminating in the cancer progression.

61
7 Conclusions
1. Significant changes in MCs take place in the superficial spreading BCC: MCTC cells
increase in number, but at the same time MCs contain increased levels of powerful chymase
inhibitors α1-PI and α1-AC. Tryptase remains active but chymase is partially inactivated and
therefore it may not be able to cleave the malignant epithelium away. However, residual
chymase activity may still have the power to affect the basement membrane zone and
SCCA-2 negative basal part and basal buds of the malignant epithelium. These changes in
mast cells, however, appear not to be specific to this BCC type, and they rather reflect
chronic inflammation.
2. The number of MCs is increased in the BCC lesion, possibly as a result of SCF-Kit
activation. In addition, MCs are increasingly immunopositive for CD30L and possibly for
TNF-α, but are almost negative for CD40L, in the BCC lesion. This suggests selective
expression and up-regulation of certain TNF superfamily ligands in MCs. An increase in
the number of positive cells for the receptors of CD30L and TNF-α in the upper dermis of
the BCC lesion suggests that MC CD30L and TNF-α may affect and modulate the dermal
immune system. In addition, the decrease of chymase activity may allow enhanced MC
TNF-α effects on dermal immune cells. The lack of CD40L in MCs and the lack of CD40 and
TNFRI in basal buds can be of importance for the inefficient antitumoral response in BCC.
However, the changes in MC CD30L and TNF-α are not disease-specific to BCC.
3. The combination of TNF-α and histamine has a profound growth-inhibitory and cytolytic
effect on normal keratinocytes but, in contrast, this combination showed no significant
effect on the growth, viability, cell cycle, migration and invasion of SiHa cells suggesting
that these cells have lost their capability of reacting to this endogenous cytolytic
mechanism, possibly due to their lower TNFRI and TNFRII expression. Similarly, another

62
SCC cell line, ME-180, was also resistant to the combined action of TNF-α and histamine.
TNF-α alone induced only slight growth inhibition and cell cycle arrest at the G0/G1 phase
in serum-free medium but was capable of increasing the migration of SiHa cells. Since TNF-
α was found in the cervix carcinoma tissue, it may promote the spread of carcinoma cells.
Furthermore, the expression level of TNFRI and TNFRII in the tumor cells of cervix
carcinoma may not be sufficient to evoke the cytolytic changes normally induced by TNF-α.
4. Significant numbers of MCTC cells are present in the peritumoral stroma of uterine
cervical SCC. A higher SCCA-2 expression was found in cervical SCC specimens than in
control specimens, though only one out of nine SCC specimens showed marked SCCA-2
immunoreactivity. When compared to the effect of tryptase in the tryptase-chymase
preparation, it appears that it is chymase that is capable of detaching viable and growing
SiHa and ME-180 SCC cells and therefore chymase may be able to release single SCC cells
or cell groups from a tumor leading to the spread of malignant cells.

63
References
1 Diepgen TL, Mahler V (2002) The epidemiology of skin cancer. Br J Dermatol. Suppl 61: 1-6.
2 Kricker A, Armstrong BK, English DR, Heenan PJ (1991) Pigmentary and cutaneous risk factors for non-melanocytic skin cancer-a case-control study. Int J Cancer. 48: 650-662.
3 Miller SJ (1991) Biology of basal cell carcinoma (Part II) J Am Acad Dermatol. 24: 161-175.
4 Kricker A, Armstrong BK, English DR, Heenan PJ (1995) A dose-response curve for sun exposure and basal cell carcinoma. Int J Cancer. 60: 482-488.
5 Armstrong BK, Kricker A (2001) The epidemiology of UV induced skin cancer. J Photochem Photobiol B. 63: 8-18.
6 Strom SS, Yamamura Y (1997) Epidemiology of nonmelanoma skin cancer. Clin Plast Surg. 24: 627-636.
7 Erb P, Ji J, Wernli M, Kump E, Glaser A, Buchner SA (2005) Role of apoptosis in basal cell and squamous cell carcinoma formation. Immunol Lett. 100: 68-72.
8 Grimbaldeston MA, Skov L, Finlay-Jones JJ, Hart PH (2002) Increased dermal mast cell prevalence and susceptibility to development of basal cell carcinoma in humans. Methods. 28: 90-96.
9 Tiltman AJ (2005) The pathology of cervical tumours. Best Pract Res Clin Obstet Gynaecol. 19: 485-500.
10 Tjalma WAA, Van Waes TR, Van den Eeden LEM, Bogers JJPM (2005) Role of human papillomavirus in the carcinogenesis of squamous cell carcinoma and adenocarcinoma of the cervix. Best Pract Res Clin Obstet Cynaecol 19: 469-483.
11 Bosch FX, de Sanjosé S (2007) The epidemiology of human papillomavirus infection and cervical cancer. Dis Markers. 23: 213-227.
12 Dabbous MK, Woolley DE, Haney L, Carter LM, Nicolson GL (1986) Host-mediated effectors of tumor invasion: role of mast cells in matrix degradation. Clin Exp Metastasis. 4: 141-152.
13 Kankkunen JP, Harvima IT, Naukkarinen A (1997) Quantitative analysis of tryptase and chymase containing mast cells in benign and malignant breast lesions. Int J Cancer 72: 385–388.
14 Ch'ng S, Wallis RA, Yuan L, Davis PF, Tan ST (2006) Mast cells and cutaneous malignancies. Mod Pathol. 19: 149-159.
15 Benditt EP, Lagunoff D (1964) The mast cell: its structure and function. Prog Allergy. 8: 195-223.
16 Rodewald HR, Dessing M, Dvorak AM, Galli SJ (1996) Identification of a committed precursor for the mast cell lineage. Science. 271: 818-822.
17 Li L, Krilis SA (1996) Mast-cell growth and differentiation. Allergy. 54: 306-312.
18 Galli SJ (1990) New insights into "the riddle of the mast cells": microenvironmental regulation of mast cell development and phenotypic heterogeneity. 62: 5-33.
19 Kitamura Y, Fujita J (1989) Regulation of mast cell differentiation. Bioessays. 10: 193-196.
20 Wasserman SI (1990) Mast cell biology. J Allergy Clin Immunol. 86: 590-593.
21 Madden KB, Urban JF Jr, Ziltener HJ, Schrader JW, Finkelman FD, Katona IM (1991) Antibodies to IL-3 and IL-4 suppress helminth-induced intestinal mastocytosis. J Immunol. 147: 1387-1391.
22 Mekori YA, Metcalfe DD (2000) Mast cells in innate immunity. Immunol Rev. 173: 131-140.
23 Galli SJ, Iemura A, Garlick DS, Gamba-Vitalo C, Zsebo KM, Andrews RG (1993) Reversible expansion of primate mast cell populations in vivo by stem cell factor. J Clin Invest. 91: 148-152.
24 Valent P, Sillaber C, Bettelheim P (1991) The growth and differentiation of mast cells. Prog Growth Factor Res. 3: 27-41.
25 Irani AA, Schechter NM, Craig SS, DeBlois G, Schwartz LB (1986) Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci U S A. 83: 4464-4468.

64
26 Kitamura Y (1989) Heterogeneity of mast cells and phenotypic change between subpopulations. Annu Rev Immunol. 7: 59-76.
27 Lawrence ID, Warner JA, Cohan VL, Hubbard WC, Kagey-Sobotka A, Lichtenstein LM (1987) Purification and characterization of human skin mast cells. Evidence for human mast cell heterogeneity. J Immunol. 139: 3062-3069.
28 Schwartz LB, Irani AA, Roller K, Castells MC, Schechter NM (1987) Quantitation of histamine, tryptase and chymase in dispersed human T and TC mast cells. J Immunol. 138: 2611.
29 Schwartz LB (1991) Mast cells and their role in urticaria. J Am Acad Dermatol. 25: 190-203.
30 Irani AM, Bradford TR, Kepley CL, Schechter NM, Schwartz LB (1989) Detection of MCT and MCTC types of human mast cells by immunohistochemistry using new monoclonal anti/tryptase and anti/chymase antibodies. J Histochem Cytochem. 37: 1509-1515.
31 Weidner N, Austen KF (1993) Heterogeneity of mast cells at multiple body sites. Fluorescent determination of avidin binding and immunofluorescent determination of chymase, tryptase, and carboxypeptidase content. Pathol Res Pract. Mar;189: 156-162.
32 Schechter NM, Irani AM, Sprows JL, Abernethy J, Wintroub B, Schwartz LB (1990) Identification of a cathepsin G-like proteinase in the MCTC type of human mast cell. J Immunol. 145: 2652-2661.
33 Harvima IT, Horsmanheimo L, Naukkarinen A, Horsmanheimo M (1994) Mast cell proteinases and cytokines in skin inflammation. Arch Dermatol Res. 287: 61-67.
34 Kivinen PK, Kaminska R, Naukkarinen A, Harvima RJ, Horsmanheimo M, Harvima IT (2001) Release of soluble tryptase but only minor amounts of chymase activity from cutaneous mast cells. Exp Dermatol. 10: 246-255.
35 Kivinen PK, Nilsson G, Naukkarinen A, Harvima IT (2003) Mast cell survival and apoptosis in organ-cultured human skin. Exp Dermatol. 12: 53-60.
36 Toru H, Eguchi M, Matsumato R, Yanagida M, Yata J, Nakahata T (1998) Interleukin-4 promotes the development of tryptase and chymase double-positive human mast cells accompanies by cell maturation. Blood. 91: 187-195.
37 Bonini S, Micera A, Iovieno A, Lambiase A, Bonini S (2005) Expression of Toll-like receptors in healthy and allergic conjunctiva. Ophthalmology. 112: 1528-159.
38 Metcalfe DD, Kaliner MA (1993) The Mast Cell in Health and Disease. New York, Dekker. 1- 89.
39 Williams CM, Galli SJ (2000) The diverse potential effector and immunoregulatory roles of mast cells in allergic disease. J Allergy Clin Immunol. 105: 847-859.
40 Kovanen PT (1995) Role of mast cells in atherosclerosis. Chem Immunol. 62: 132-170.
41 Pejler G, Abrink M, Ringvall M, Wernersson S (2007) Mast cell proteases. Adv Immunol. 95: 167-255.
42 Metcalfe DD, Kaliner M, Donlon MA (1981) The mast cell. Crit Rev Immunol. 3: 23-74.
43 Benoist C, Mathis D (2002) Mast cells in autoimmune disease. Nature. 420: 875-878.
44 Kinet JP (1989) The high affinity receptor for IgE. Curr. Opin. Immunol. 2: 499–505.
45 Lloyd AG, Bloom GD, Balazs EA, Haegermark O (1967) Combined biochemical and morphological ultrastructure studies on mast-cell granules. Biochem J. 103: 75-76.
46 Michels NA (1963) The mast cells. In: Handbook of Hematology, Vol. 1, H. Downey, Ed. (Hoeber, New York 1938) Reproduced in Ann. N.Y. Acad. Sci. 103: 231-372.
47 Ishizaka T, Ishizaka K (1984) Activation of mast cells for mediator release through IgE receptors. Prog Allergy. 34: 188-235.
48 Moormann C, Artuc M, Pohl E, Varga G, Buddenkotte J, Vergnolle N, Brehler R, Henz BM, Schneider SW, Luger TA, Steinhoff M (2006) Functional characterization and expression analysis of the proteinase-activated receptor-2 in human cutaneous mast cells. J Invest Dermatol. 126: 746-755.
49 Sedgwick JD, Holt PG, Turner KJ (1981) Production of a histamine-releasing lymphokine by antigen- or mitogen-stimulated human peripheral T cells. Clin Exp Immunol. 45: 409-418.
50 Liu MC, Proud D, Lichtenstein LM, MacGlashan DW Jr, Schleimer RP, Adkinson NF Jr, Kagey-Sobotka A, Schulman ES, Plaut M (1986) Human lung macrophage-derived histamine-releasing activity is due to IgE-dependent factors. J Immunol. 136: 2588-2595.

65
51 Bradding P, Walls AF, Holgate ST (2006) The role of the mast cell in the pathophysiology of asthma. J Allergy Clin Immunol. 117: 1277-1284.
52 Fischer M, Harvima IT, Carvalho RF, Möller C, Naukkarinen A, Enblad G, Nilsson G (2006) Mast cell CD30 ligand is upregulated in cutaneous inflammation and mediates degranulation-independent chemokine secretion. J Clin Invest. 116: 2748-2756.
53 Harvima IT, Nilsson G (2011) Mast cells as regulators of skin inflammation and immunity. Acta Derm Venereol. 91: 644-50.
54 Ludowyke RI, Peleg I, Beaven MA, Adelstein RS (1989) Antigen induced secretion of histamine and the phosphorylation of myosin by protein kinase C in rat basophilic leukemia cells. J Biol Chem 264: 12492–12501.
55 Spudich A (1994) Myosin reorganization in activated RBL cells correlates temporally with stimulated secretion. Cell Motil Cytoskeleton. 29: 345–353.
56 Lang T, Wacker I, Wunderlich I, Rohrbach A, Giese G, Soldati T (2000) Role of actin cortex in the subplasmalemmal transport of secretory granules in PC-12 cells. Biophys J. 78: 2863–2877.
57 Dvorak AM, Schleimer RP, Schulman ES, Lichtenstein LM (1986) Human mast cells use conservation and condensation mechanisms during recovery from degranulation. In vitro studies with mast cells purified from human lungs. Lab Invest. 54: 663-678.
58 Metcalfe DD, Baram D, Mekori YA (1997) Mast cells. Physiol Rev 77: 1033-1079.
59 Galli SJ, Nakae S, Tsai M (2005) Mast cells in the development of adaptive immune responses. Nat Immunol. 6: 135-142.
60 Henz BM, Maurer M, Lippert U, Worm M, Babina M (2001) Mast cells as initiators of immunity and host defense. Exp Dermatol. 10: 1-10.
61 Marshall JS, Jawdat DM (2004) Mast cells in innate immunity. J Allergy Clin Immunol. 114: 21-27.
62 Ackermann L, Harvima IT (1998) Mast cells of psoriatic and atopic dermatitis skin are positive for TNF-α and their degranulation is associated with expression of ICAM-1 in the epidermis. Arch Dermatol Res. 290: 353-359.
63 Ackermann L, Harvima IT, Pelkonen J, Ritamäki-Salo V, Naukkarinen A, Harvima RJ, Horsmanheimo M (1999) Mast cells in psoriatic skin are strongly positive for interferon-gamma. Br J Dermatol 140: 624-633.
64 Horsmanheimo L, Harvima IT, Järvikallio A, Harvima RJ, Naukkarinen A, Horsmanheimo M (1994) Mast cells are one major source of interleukin-4 in atopic dermatitis. Br J Dermatol. 131: 348-353.
65 Kashiwakura J, Yokoi H, Saito H, Okayama Y (2004) T cell proliferation by direct cross-talk between OX40 ligand on human mast cells and OX40 on human T cells: comparison of gene expression profiles between human tonsillar and lung-cultured mast cells. J Immunol. 173: 5247-5257.
66 Ottaiano A, Pisano, C, De Chiara A, Ascierto PA, Botti G, Barletta E, Apice G, Gridelli, C, Iaffaioli VR (2002) CD40 activation as potential tool in malignant neoplasms. Tumori. 88: 361-366.
67 Pawankar R, Okuda M, Yssel H, Okumura K, Ra C (1997) Nasal mast cells in perennial allergic rhinitis exhibit increased expression of the FcεRI, CD40L, IL-4, and IL-13, and can induce IgE synthesis in B cells. J Clin Invest. 99: 1465-1466.
68 Riley JF, West GB (1953) The presence of histamine in tissue mast cells. J Physiol. 120: 528–537.
69 Code CF, Mitchell RG (1957) Histamine in eosinophils and basopohils in the blood. J Physiol. 136: 449–468.
70 Aoi R, Nakashima I, Kitamura Y, Asai H, Nakano K (1989) Histamine synthesis by mouse T lymphocytes through induced histidine decarboxylase. Immunology. 66: 219–223.
71 Zwadlo-Klarwasser G, Braam U, Muhl-Zurbes P, Schmutzler W (1994) Macrophages and lymphocytes: alternative sources of histamine.Agents Actions. 41: 99-100.
72 Röhlich P, Anderson P, Uvnäs B (1971) Mast cell degranulation as a process of sequential exocytoses. Acta Biol Acad Sci Hung. 22: 197-213.
73 Maslinski C, Fogel WA (1996) Catabolism of Histamine. In: Uvnäs B (ed) Handbook of experimental pharmacology. Vol. 97. Histamine and histamine antagonists. Springer, Berlin-Heidelberg, 1991: 165-189.

66
74 Owen DA, Poy E, Woodward DF, Daniel D (1980) Evaluation of the role of Histamine H1- and H2-receptors in cutaneous inflammation in the guinea-pig produced by histamine and mast cell degranulation. Br J Pharmacol. 69: 615-623.
75 Pounder RE, Russell RG, Williams JG, Misiewicz JJ, Milton-Thompson GJ (1975) Proceedings: New histamine H2-receptor antagonist inhibits food-stimulated gastric acid secretion. Gut. 16: 397-398.
76 Garbarg M, Schwartz JC (1985) Histamine receptors in the brain. N Engl Reg Allergy Proc. 6: 195-200.
77 Zhang M, Venable JD, Thurmond RL (2006) The histamine H4 receptor in autoimmune disease. Expert Opin Investig Drugs. 15: 1443-1452.
78 Repka-Ramirez MS, Baraniuk JN (2002) Histamine in health and disease. Clin Allergy Immunol. 17: 1-25.
79 Shim WS, Oh U (2008) Histamine-induced itch and its relationship with pain. Mol Pain. 31: 4-29.
80 Nuutinen P, Harvima IT, Ackermann L (2007) Histamine, but not leukotriene C4, is an essential mediator in cold urticaria wheals. Acta Derm Venereol. 87: 9-13.
81 Schwartz LB, Bradford TR (1986) Regulation of tryptase from human lung mast cells by heparin. J Biol Chem. 261: 7372-7379.
82 Sayama S, Iozzo RV, Lazarus GS, Schecter NM (1987) Human skin chymotrypsin like proteinase chymase: subcellular localization to mast cell granules and interaction with heparin and other glycosaminoglycans. J Biol Chem. 262: 6808-6815.
83 Castellot JJ, Cochran DL, Karnovsky MM (1985) Effect of heparin on vascular smooth muscle cell, I: cell metabolism. J Cell Physiol. 124: 21-28.
84 Wright TC, Cochran DL, Karnovsky MJ (1985) Inhibition of rat cervical epithelial cell growth by heparin and its reversal by EGF. J Cell Physiol. 125: 499-506.
85 Roemisch J, Gray E, Hoffmann JN, Wiedermann CJ (2002) Antithrombin: a new look at the actions of a serine protease inhibitor. Blood Coagul Fibrinolysis. 13: 657-670.
86 Okajima K (2001) Regulation of inflammatory responses by natural anticoagulants. Immunol Rev. 184: 258-274.
87 Niers TM, Klerk CP, DiNisio M, Van Noorden CJ, Buller HR, Reitsma PH, Richel DJ (2007) Mechanisms of heparin induced anti-cancer activity in experimental cancer models. Crit Rev Oncol Hematol. 61: 195-207.
88 Folkman J, Shing Y (1992) Control of angiogenesis by heparin and other sulfated polysaccharides. Adv Exp Med Biol. 313: 355-364.
89 Vlodavsky I, Mohsen M, Lider O, Svahn CM, Ekre HP, Vigoda M, Ishai-Michaeli R, Peretz T (1994) Inhibition of tumor metastasis by heparanase inhibiting species of heparin. Invasion Metastasis. 14: 290-302.
90 Parish CR, Freeman C, Brown KJ, Francis DJ, Cowden WB (1999) Identification of sulfated oligosaccharide-based inhibitors of tumor growth and metastasis using novel in vitro assays for angiogenesis and heparanase activity. Cancer Res. 59: 3433-3441.
91 Parish CR, Freeman C, Hulett MD (2001) Heparanase: a key enzyme involved in cell invasion. Biochim Biophys Acta. 1471: 99-108.
92 Borsig L, Wong R, Feramisco J, Nadeau DR, Varki NM, Varki A (2001) Heparin and cancer revisited: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc Natl Acad Sci U S A. 98: 3352-3357.
93 Herold BC, Gerber SI, Polonsky T, Belval BJ, Shaklee PN, Holme K (1995) Identification of structural features of heparin required for inhibition of herpes simplex virus type 1 binding. Virology. 206: 1108-1116.
94 Baba M, Pauwels R, Balzarini J, Arnout J, Desmyter J, De Clercq E (1988) Mechanism of inhibitory effect of dextran sulfate and heparin on replication of human immunodeficiency virus in vitro. Proc Natl Acad Sci U S A. 85: 6132-6136.
95 Rider CC, Coombe DR, Harrop HA, Hounsell EF, Bauer C, Feeney J, Mulloy B, Mahmood N, Hay A, Parish CR (1994) Anti-HIV-1 activity of chemically modified heparins: correlation between binding to the V3 loop of gp120 and inhibition of cellular HIV-1 infection in vitro. Biochemistry. 33: 6974-6980.
96 Howell AL, Taylor TH, Miller JD, Groveman DS, Eccles EH, Zacharski LR (1996) Inhibition of HIV-1 infectivity by low molecular weight heparin. Results of in vitro studies and a pilot clinical trial in patients with advanced AIDS. Int J Clin Lab Res. 26: 124-131.

67
97 Clayette P, Moczar E, Mabondzo A, Martin M, Toutain B, Marce D, Dormont D (1996) Inhibition of human immunodeficiency virus infection by heparin derivatives. Aids Res. Human Retrov. 12: 63–69.
98 Harrop HA, Rider CC (1998) Heparin and its derivatives bind to HIV-1 recombinant envelope glycoproteins, rather than to recombinant HIV-1 receptor, CD4. Glycobiology. 8: 131-137.
99 Humphries DE, Wong GW, Friend DS, Gurish MF, Qiu WT, Huang C, Sharpe AH, Stevens RL (1999) Heparin is essential for the storage of specific granule proteases in mast cells. Nature. 400: 769-772.
100 Forsberg E, Pejler G, Ringvall M, Lunderius C, Tomasini-Johansson B, Kusche-Gullberg M, Eriksson I, Ledin J, Hellman L, Kjellén L (1999) Abnormal mast cells in mice deficient in a heparin-synthesizing enzyme. Nature. 400: 773-776.
101 Zehnder JL, Galli SJ (1999) Mast-cell heparin demystified. Nature. 400: 714-715.
102 Chuang WL, Christ MD, Peng J, Rabenstein DL (2000) An NMR and molecular modeling study of the site-specific binding of histamine by heparin, chemically modified heparin, and heparin-derived oligosaccharides. Biochemistry. 39: 3542-3555.
103 Rabenstein DL, Bratt P, Schierling TD, Robert JM, Guo W (1992) The interaction of biological molecules with heparin and related glycosaminoglycans. 1. Identification of a specific heparin binding site for histamine. J. Am. Chem. Soc.114: 3278-3285.
104 Rabenstein DL, Ludowyke R, Lagunoff D (1987) Proton nuclear magnetic resonance studies of mast cell histamine. Biochemistry. 26: 6923-6926.
105 Rabenstein DL (2002) Heparin and heparan sulfate: structure and function. Nat Prod Rep. 19: 312-331.
106 Harvima IT, Schechter NM, Harvima RJ, Fräki JE (1988) Human skin tryptase: purification, partial characterization and comparison with human lung tryptase. Biochim Biophys Acta. 957: 71-80.
107 Harvima RJ, Harvima IT, Dull D, Dunder UK, Schwartz LB (1999) Identification and characterization of multiple forms of tryptase from human mast cells. Arch Dermatol Res. 291: 73-80.
108 Huttunen M, Harvima IT (2005) Mast cell tryptase and chymase in chronic leg ulcers: chymase is potentially destructive to epithelium and is controlled by proteinase inhibitors. Br J Dermatol. 152:1149-1160.
109 Harvima IT, Lappalainen K, Hirvonen MR, Mättö M, Kivinen PK, Hyttinen M, Pelkonen J, Naukkarinen A (2004) Heparin modulates the growth and adherence and augments the growth-inhibitory action of TNF-α on cultured human keratinocytes. J Cell Biochem. 92: 372-386.
110 Roche W (1985) Mast cells and tumor angiogenesis: the tumor-mediated release of an endothelial growth factor mast cells. Int J Cancer. 36: 721–728.
111 Duncan J, Brown F, McKinnon A, Long WF, Williamson FB, Thompson WD (1992) Patterns of angiogenic response to mast cell granule constituents. Int J Microcirc Clin Exp. 11: 21–33.
112 Poole T, Zetter B (1983) Stimulation of rat peritoneal mast cell migration by tumor-derived peptides. Cancer Res 43: 5857–5861.
113 Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B (1975) An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 72: 3666-3670.
114 Pfeffer K (2003) Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine Growth Factor Rev. 14: 185-191.
115 Ghezzi P, Cerami A (2005) Tumor necrosis factor as a pharmacological target. Mol Biotechnol. 31: 239-44.
116 Gordon JR, Burd PR, Galli SJ (1990) Mast cells as a source of multifunctional cytokines. Immunol Today. 11: 458-464.
117 Horvath CJ, Ferro TJ, Jesmok G, Malik AB (1988) Recombinant tumor necrosis factor increases pulmonary vascular permeability independent of neutrophils. Proc Natl Acad Sci U S A. 85: 9219-9223.
118 Klein LM, Lavker RM, Matis WL, Murphy GF (1989) Degranulation of human mast cells induces an endothelial antigen central to leukocyte adhesion. Proc. Natl Acad. Sci. USA. 86: 8972-8976.
119 Carlos TM, Schwartz BR, Kovach NL, Yee E, Rosa M, Osborn L, Chi-Rosso G, Newman B, Lobb R (1990) Vascular cell adhesion molecule-1 mediates lymphocyte adherence to cytokine-activated cultured human endothelial cells. Blood. 76: 965-970.

68
120 Cumberbatch M, Fielfing I, Kimber I (1994) Modulation of epidermal Langerhans’ cell frequency by tumor necrosis factor-α. Immunology. 81: 395-401.
121 Strieter RM, Remick DG, Lynch JP 3rd, Spengler RN, Kunkel S (1989) Interleukin-2-induced tumor necrosis factor-α (TNF-α) gene expression in human alveolar macrophages and blood monocytes. Am Rev Respir Dis. 139: 335-342.
122 Sirois J. Menard G, Moses AS, Bissonnette EY (2000) Importance of histamine in the cytokine network in the lung through H2 and H3 receptors: Stimulation IL-10 production. J. Immunol. 164: 2964-2970.
123 Butler DM, Maini RN, Feldmann M, Brennan FM (1995) Modulation of proinflammatory cytokine release in rheumatoid synovial membrane cell cultures. Comparison of monoclonal anti TNF-α antibody with the interleukin-1 receptor antagonist. Eur Cytokine Netw. 6: 225-230.
124 Wang S, El-Deiry WS (2003) TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 22: 8628-8633.
125 Walsh LJ (2003) Mast cells and oral inflammation. Crit Rev Oral Biol Med. 14: 188-198.
126 Wajant H, Pfizenmaier K, Scheurich P (2003) Tumor necrosis factor signaling. Cell Death Differ. 10:45-65.
127 Mannel DN, Echtenacher B (2000) TNF in the inflammatory response. Chem. Immunol. 74: 141-161
128 Walsh L (1995) Ultraviolet B irradiation of skin induces mast cell degranulation and release of tumor necrosis factor-α. Immunol Cell Biol. 73: 226–233.
129 Hart P, Townley S, Grimbaldeston M, Khalil Z, Finlay-Jones JJ (2002) Mast cells, neuropeptides, histamine, and prostaglandins in UV-induced systemic immunosuppression. Methods 28: 79–89.
130 Balkwill F. (2002) Tumor necrosis factor or tumor promoting factor? Cytokine & Growth Factor Reviews. 13: 135–141.
131 Szlosarek P, Balkwill F (2003) Tumour necrosis factor-α: A potential target in the therapy of solid tumors. Lancet Oncology. 4: 565–573.
132 Pfitzenmaier J, Vessella R, Higano CS, Noteboom J L, Wallace D Jr, Corey E (2003) Elevation of cytokine levels in cachectic patients with prostate carcinoma. Cancer. 97: 1211–1216.
133 Glenner GG, Cohen LA (1960) Nature. 185: 846–847.
134 Schwartz LB, Lewis RA, Seldin D, Austen KF (1981) Acid hydrolases and tryptase from secretory granules of dispersed human lung mast cells. J Immunol. 126: 1290–1294.
135 Hallgren J, Pejler G (2006) Biology of mast cell tryptase. An inflammatory mediator. FEBS J. 273: 1871-1895.
136 Artuc M, Hermes B, Steckelings UM, Grutzkau A, Henz BM (1999) Mast cells and their mediators in cutaneous wound healing--active participants or innocent bystanders? Exp Dermatol. 8: 1-16.
137 Harvima IT, Harvima RJ, Eloranta TO, Fräki JE (1988) The allosteric effect of salt on human mast cell tryptase. Biochim Biophys Acta. 956: 133-139.
138 Pereira PJ, Bergner A, Macedo-Ribeiro S, Huber R, Matschiner G, Fritz H, Sommerhoff CP, Bode W (1998) Human beta-tryptase is a ring-like tetramer with active sites facing a central pore. Nature. 392: 306-311.
139 Smith TJ, Hougland MW, Johnson DA (1984) Human lung tryptase. Purification and characterization. J Biol Chem. 259: 11046-11051.
140 Strik MC, Wolbink A, Wouters D, Bladergroen BA, Verlaan AR, van Houdt IS, Hijlkema S, Hack CE, Kummer JA (2004) Intracellular serpin SERPINB6 (PI6) is abundantly expressed by human mast cells and forms complexes with beta-tryptase monomers. Blood. 103: 2710-2717.
141 Steinhoff M, Buddenkotte J, Shpacovitch V, Rattenholl A, Moormann C, Vergnolle N, Luger TA, Hollenberg MD (2005) Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev.26: 1-43.
142 Schwartz LB (1990) Tryptase, a mediator of human mast cells. J Allergy Clin Immunol. 86: 594-598.
143 Lohi J, Harvima I, Keski-Oja J (1992) Pericellular substrates of human mast cell tryptase: 72,000 dalton gelatinase and fibronectin. J Cell Biochem. 50: 337-349.
144 Saarinen J, Kalkkinen N, Welgus HG, Kovanen PT (1994) Activation of human interstitial procollagenase through direct cleavage of the Leu83-Thr84 bond by mast cell chymase. J Biol Chem. 269: 18134-18140.

69
145 Gruber BL, Marchese MJ, Suzuki K, Schwartz LB, Okada Y, Nagase H, Ramamurthy NS (1989) Synovial procollagenase activation by human mast cell tryptase: dependence upon matrix metalloproteinase 3 activation. J Clin Invest. 84: 1657-1662.
146 Stack MS, Johnson DA (1994) Human mast cell tryptase activates single-chain urinary-type plasminogen activator (pro-urokinase) J Biol Chem. 269: 9416-9419.
147 Kaminska R, Helisalmi P, Harvima RJ, Naukkarinen A, Horsmanheimo M, Harvima IT (1999) Focal dermal-epidermal separation and fibronectin cleavage in basement membrane by human mast cell tryptase. J Invest Dermatol.113: 567-573.
148 Blair RJ, Meng H, Marchese MJ, Ren S, Schwartz LB, Tonnesen MG, Gruber BL (1997) Human mast cells stimulate vascular tube formation: tryptase is a novel, potent angiogenic factor. J Clin Invest. 99: 2691-2700.
149 Harvima IT, Naukkarinen A, Paukkonen K, Harvima RJ, Aalto ML, Schwartz LB, Horsmanheimo M (1993) Mast cell tryptase and chymase in developing and mature psoriatic lesions. Arch Dermatol Res. 285: 184-192.
150 He SH (2004) Key role of mast cells and their major secretory products in inflammatory bowel disease. World J Gastroenterol. 10: 309-318.
151 Miyazaki M, Takai S, Jin D, Muramatsu M (2006) Pathological roles of angiotensin II produced by mast cell chymase and the effects of chymase inhibition in animal models. Pharmacol Ther. 112: 668-676.
152 Harvima IT, Haapanen L, Ackermann L, Naukkarinen A, Harvima RJ, Horsmanheimo M (1999) Decreased chymase activity is associated with increased levels of protease inhibitors in mast cells of psoriatic lesions. Acta Derm Venereol. 79: 98-104.
153 Kaminska R, Harvima IT, Naukkarinen A, Nilsson G, Horsmanheimo M (1996) Alterations in mast cell proteinases and protease inhibitors in the progress of cutaneous herpes zoster infection. J Pathol. 180: 434-440.
154 Akiyama M, Watanabe Y, Nishikawa T (1991) Immunohistochemical characterization of human cutaneous mast cells in urticaria pigmentosa (cutaneous mastocytosis) Acta Pathol Jpn. 41: 344-349.
155 Westin U, Polling A, Ljungkrantz I, Ohlsson K (1999) Identification of SLPI (secretory leukocyte protease inhibitor) in human mast cells using immunohistochemistry and in situ hybridisation. Biol Chem. 380: 489-493.
156 Ide H, Itoh H, Yoshida E, Kobayashi T, Tomita M, Maruyama H, Osada Y, Nakahata T, Nawa Y. (1999) Immunohistochemical demonstration of inter-α-trypsin inhibitor light chain (bikunin) in human mast cells. Cell Tissue Res. 297:149-154.
157 Schick C, Pemberton PA, Shi GP, Kamachi Y, Cataltepe S, Bartuski AJ, Gornstein ER, Brömme D, Chapman HA, Silverman GA (1998) Cross-class inhibition of the cysteine proteinases cathepsins K, L, and S by the serpin squamous cell carcinoma antigen 1: a kinetic analysis. Biochemistry. 37: 5258-5266.
158 Muramatsu M, Katada J, Hattori M, Hayashi I, Majima M (2000) Chymase mediates mast cell-induced angiogenesis in hamster sponge granulomas. Eur J Pharmacol. 402: 181-191.
159 Leskinen MJ, Lindstedt KA, Wang Y, Kovanen PT (2003) Mast cell chymase induces smooth muscle cell apoptosis by a mechanism involving fibronectin degradation and disruption of focal adhesions. Arterioscler Thromb Vasc Biol. 23: 238-243.
160 Vartio T, Seppä H, Vaheri A (1981) Susceptibility of soluble and matrix fibronectins to degradation by tissue proteinases, mast cell chymase and cathepsin G. J Biol Chem. 256: 471-477.
161 Welle M (1997) Development, significance and heterogeneity of mast cells with particular regard to the mast cell-specific proteases chymase and tryptase. J Leukocyte Biol. 61: 233–245.
162 Caughey GH, Leidig F, Viro NF, Nadel JA (1988) Substance P and vasoactive intestinal polypeptide degradation by mast cell tryptase and chymase. J Pharmacol Exp Ther. 244: 133–137.
163 Briggaman RA, Schechter NM, Fraki J, Lazarus GS (1984) Degradation of the epidermal-dermal junction by proteolytic enzymes from human skin and human polymorphonuclear leukocytes. J Exp Med. 160: 1027-1042.
164 Jarvikallio A, Naukkarinen A, Harvima IT, Aalto ML, Horsmanheimo M (1997) Quantitative analysis of tryptase- and chymase-containing mast cells in atopic dermatitis and nummular eczema. Br J Dermatol. 136: 871-877.
165 Kaminska R, Naukkarinen A, Glinski W, Horsmanheimo M, Harvima IT (1999) Mast cells in developing subepidermal bullous diseases: emphasis on tryptase, chymase and protease inhibitors. Acta Derm Venereol. 79: 351-355.

70
166 Huttunen M, Aalto M-L, Harvima RJ, Horsmanheimo M, Harvima IT (2000) Alterations in mast cells showing tryptase and chymase activity in epithelializating and chronic wounds. Exp Dermatol. 9: 258-265.
167 Saarinen JV, Harvima RJ, Naukkarinen A, Horsmanheimo M, Harvima IT (2001) The release of histamine is associated with the inactivation of mast cell chymase during immediate allergic wheal reaction in the skin. Clin Exp Allergy 31: 593-601.
168 Riekki R, Harvima IT, Jukkola A, Risteli J, Oikarinen A (2004) The production of collagen and the activity of mast-cell chymase increase in human skin after irradiation therapy. Exp Dermatol. 13: 364-371.
169 Mukaida N, Harada A, Matsushima K (1998) Interleukin-8 (IL-8) and monocyte chemotactic and activating factor (MCAF/MCP-1), chemokines essentially involved in inflammatory and immune reactions. Cytokine Growth Factor Rev. 9: 9-23.
170 Compton SJ, Cairns JA, Holgate ST, Walls AF (1998) The role of mast cell tryptase in regulating endothelial cell proliferation, cytokine release, and adhesion molecule expression: tryptase induces expression of mRNA for IL-1 beta and IL-8 and stimulates the selective release of IL-8 from human umbilical vein endothelial cells. J Immunol. 161: 1939-1946.
171 Endoh I, Di Girolamo N, Hampartzoumian T, Cameron B, Geczy CL, Tedla N (2007) Ultraviolet B irradiation selectively increases the production of interleukin-8 in human cord blood-derived mast cells. Clin Exp Immunol. 148: 161-167.
172 Gillitzer R, Goebeler M (2001) Chemokines in cutaneous wound healing. J Leukoc Biol. 69: 513-521.
173 Hart PH (2001) Regulation of the inflammatory response in asthma by mast cell products. Immunol Cell Biol. 79: 149-153.
174 Holgate ST (2000) The role of mast cells and basophils in inflammation. Clin Exp Allergy. 30: 28-32
175 Samarasinghe V, Madan V (2012) Nonmelanoma skin cancer. J Cutan Aesthet Surg. 5: 3-10.
176 Rosso S, Zanetti R, Martinez C, Tormo MJ, Schraub S, Sancho-Garnier H, Franceschi S, Gafà L, Perea E, Navarro C, Laurent R, Schrameck C, Talamini R, Tumino R, Wechsler J (1996) The multicentre south European study 'Helios'. II: Different sun exposure patterns in the aetiology of basal cell and squamous cell carcinomas of the skin. Br J Cancer. 73: 1447-1454.
177 English DR, Armstrong BK, Kricker A, Winter MG, Heenan PJ, Randell PL (1998) Case-control study of sun exposure and squamous cell carcinoma of the skin. 77: 347-353.
178 McNaughton SA, Marks GC, Green AC (2005) Role of dietary factors in the development of basal cell cancer and squamous cell cancer of the skin. Cancer Epidemiol Biomarkers Prev. 14: 1596-1607.
179 Pinkus H (1965) Epithelial and fibroepithelial tumors. Arch Dermatol 91: 24-37.
180 Crowson AN (2006) Basal cell carcinoma: biology, morphology and clinical implications. Mod Pathol.19: 127-147.
181 Cole CA, Forbes D, Davoes RE (1986) An action spectrum for UV photocarcinogenesis. Photochem Photobiol 43: 275-284.
182 Scotto J, Fears TR, Fraumeni JF (1983) Incidence of nonmelanoma skin cancer in the United States. Washington, DC: US Dept Health and Human Services. Publication No (NIH) 82–2433.
183 Abreo F, Sanusi ID (1991) Basal cell carcinoma in North American blacks: clinical and histopathologic study of 26 patients. J Am Acad Dermatol. 25: 1005-1011.
184 Halder RM, Bang KM (1988) Skin cancer in blacks in the United States. Dermatol Clin. 6: 397-405.
185 Pousti A (1979) Malignant tumors of the scalp resulting from x-ray treatment of tinea capitis. Br J Plast Surg. 32: 52-54.
186 Fabbrocini G, Triassi M, Mauriello MC, Torre G, Annunziata MC, Vita VD, Pastore F, D’Arco V, Monfrecola G (2010) Epidemiology of Skin Cancer: Role of Some Environmental Factors. Cancers 2: 1980-1989.
187 Johnson TM, Rowe DE, Nelson BR, Swanson NA (1992) Squamous cell carcinoma of the skin (excluding lip and oral mucosa) J Am Acad Dermatol. 26: 467-484.
188 Abel EA (1989) Cutaneous manifestations of immunosuppression in organ transplant recipients. J Am Acad Dermatol. 21: 167-179.

71
189 Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH Jr, Scott MP (1996) Human homolog of patched, a candidate gene for the basal cell carcinoma syndrome. Science. 272: 1668-1671.
190 Lam CW, Xie J, To KF, Ng HK, Lee KC, Yuen NW, Lim PL, Chan LY, Tong SF, McCormick F (1999) A frequent activated smoothened mutation in sporadic basal cell carcinomas. Oncogene. 18: 833-836.
191 Hashimoto K, Lever WF (1968) Appendage tumors of the skin. Springfield: C. C. Thomas.
192 Ting PT, Kasper R, Arlette JP (2005) Metastatic basal cell carcinoma: report of two cases and literature review. J Cutan Med Surg. 9: 10-5.
193 Kufe D W, Holland JF, Frei E (2003) Cancer medicine 6 (6th ed.) Hamilton, Ont.: BC Decker.
194 Urbach E, Davies RE, Forbes PD (1966) Ultraviolet radiation and skin cancer in man. ln: Montagna, W & Dobson, RL., eds, Advances in Biology ofSkin, VoL. VII, Carcinogenesis, Oxord, Pergamon Press, 195-214.
195 Wolff K, Johnson RA, Suurmond D, Fitzpatrick TB (2005) Precancerous Lesions and Cutaneous Carcinomas. Fitzpatrick's color atlas and synopsis of clinical dermatology (5th ed., pp. 150-169) New York: McGraw-Hill Medical Pub. Division.
196 Rhodes LE (2004) The hazards of sunbeds. Dermatol Pract 111: 6–8.
197 Katz AD, Urbach F, Lilienfeld AM (1957) The frequency and risk of metastases in squamous cell carcinoma of the skin. Cancer 10: 1162-1166.
198 Rowe DE, Carroll RJ, Day CL (1992) Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip: implications for treatment modality selection. J Am Acad Dermatol 26: 976-990.
199 NORDCAN, Cancer stat fact sheets, D:\Web\DEP\CancerMondial\NORDCAN\English\logo_000.prn. (n.d.) CANCER Mondial. Retrieved December 31, 2011, from http://www-dep.iarc.fr/NORDCAN/English/ StatsFact.asp?cancer=300&country=246.
200 Fears TR, Scotto J, Schneiderman MA (1977) Mathematical models of age and ultraviolet effects on the incidence of skin cancer among whites in the United States. Am J Epidemiol. 105: 420-427.
201 Lindelöf B, Krynitz B, Granath F, Ekbom A (2008) Burn injuries and skin cancer: a population-based cohort study. Acta Derm Venereol. 88: 20-2.
202 Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, Halperin AJ, Pontén J (1991) A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA. 88: 10124-10128.
203 Roenigk HH, Caro WA (1981) Skin cancer in the PUVA-48 cooperative study. J Am Acad Dermatol. 4: 319-324.
204 Chuang T-Y, Heinrich LA, Schultz MD, Reizner GT, Kumm RC, Cripps DJ (1992) PUVA and skin cancer: a historical cohort study on 492 patients. J Am Acad Dermatol 26: 173-177.
205 Stern RS, Lange R (1988) Nonmelanoma skin cancer occurring in patients treated with PUVA five to ten years after first treatment. J Invest Dermatol. 91: 120-124.
206 Hoxtell EO, Mandel JS, Murray SS, Schuman LM, Goltz RW (1977) Incidence of skin carcinoma after renal transplantation. Arch Dermatol. 113: 436-438.
207 Gupta AK, Cardella CJ, Haberman HF (1986) Cutaneous malignant neoplasms in patients with renal transplants. Arch Dermatol. 122: 1288-1293.
208 España A, Redondo P, Fernández AL, Zabala M, Herreros J, Llorens R, Quintanilla E (1995) Skin cancer in heart transplant recipients. J Am Acad Dermatol. 32: 458-465.
209 Quatresooz P, Piérard-Franchimont C, Paquet P, Hubert P, Delvenne P, Piérard GE (2008) Crossroads between actinic keratosis and squamous cell carcinoma, and novel pharmacological issues. Eur J Dermatol. 18: 6-10.
210 Bendl BJ, Graham JH (1968) New concepts on the origin of squamous cell carcinomas of the skin: solar (senile) keratosis with squamous cell carcinoma—a clinicopathologic and histochemical study. In JB Lippincott (Ed.) Proceedings of the Sixth National Cancer Conference, Philadelphia: Lippincott.
211 IARC Monographs- Monographs available in PDF format. (n.d.) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Retrieved January 2, 2012, from http://monographs.iarc.fr/ENG/Monographs.

72
212 HPV Sequence Database WWW Home Page. (n.d.) HPV Sequence Database WWW Home Page. Retrieved January 2, 2012, from http://www.stdgen.lanl.gov/.
213 Bosch FX, Manos MM, Munoz N, Sherman M, Jansen AM, Peto J, Schiffman MH, Moreno V, Kurman R, Shah KV (1995) Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. International Biological Study on Cervical Cancer (IBSCC) Study Group [comments]. J Natl Cancer Inst 87: 796-802.
214 Munoz N, Bosch F, Shah K, Schneider A, Koutsky L (1992) Natural history and epidemiological features of genital HPV infection. The epidemiology of cervical cancer and human papillomavirus [Workshop, November 1991, Brussels] (pp. 25–52) Lyon: IARC.
215 Scully, R. E., & Poulsen, H. E. (1994) Histological typing of female genital tract tumours (2nd ed.) Berlin: Springer-Verlag.
216 Wright TC, Kurman RJ, Ferenczy A (1994) Precancerous lessions of the cervix. Pathology of the female Genital Tract. (4th ed., pp. 233-241) Kurman RJ. Springer-Verlag, New York.
217 Hiromatsu Y, Toda S (2003) Mast cells and angiogenesis. Microsc Res Tech. 60: 64-69.
218 Nienartowicz A, Sobaniec-Lotowska ME, Jarocka-Cyrta E, Lemancewicz D (2006) Mast cells in neoangiogenesis. Med Sci Monit. 12: 53-56.
219 Theoharides TC, Conti P (2004) Mast cells: the Jekyll and Hyde of tumor growth. Trends Immunol. 25: 235-241.
220 Grimbaldeston MA, Skov L, Baadsgaard O, Skov BG, Marshman G, Finlay-Jones JJ, Hart PH (2000) Communications: high dermal mast cell prevalence is a predisposing factor for basal cell carcinoma in humans. J Invest Dermatol. 115: 317-320.
221 Grimbaldeston MA, Skov L, Finlay-Jones JJ, Hart PH (2002) Squamous cell carcinoma is not associated with high dermal mast cell prevalence in humans. J Invest Dermatol. 119: 1204-1206.
222 Hart P, Grimbaldeston M, Finlay-Jones J (2001) Sunlight, immunosuppression and skin cancer: role of histamine and mast cells. Clin Exp Pharmacol Physiol. 28: 1–8.
223 Takeda K, Hatamochi A, Ueki H (1989) Increaseed collagen synthesis in linear localized scleroderma. Arch Dermatol Res. 281: 288-290.
224 Ribatti D, Crivellato E, Roccaro AM, Ria R, Vacca A (2004) Mast cell contribution to angiogenesis related to tumour progression. Clin Exp Allergy. 34: 1660-1664.
225 Ozdemir O (2006) Mast cells and the tumor-associated neoangiogenesis. Comment on: Med Sci Monit. 12: 53-56.
226 Tani K, Ogushi F, Kido H, Kawano T, Kunori Y, Kamimura T, Cui P, Sone S (2000) Chymase is a potent chemoattractant for human monocytes and neutrophils. J Leukoc Biol. 67: 585-589.
227 Leskinen MJ, Heikkilä HM, Speer MY, Hakala JK, Laine M, Kovanen PT, Lindstedt KA (2006) Mast cell chymase induces smooth muscle cell apoptosis by disrupting NF-kappaB-mediated survival signaling. Exp Cell Res. 312: 1289-1298.
228 Cabanillas-Saez A, Schalper JA, Nicovani SM, Rudolph MI (2002) Characterization of mast cells according to their content of tryptase and chymase in normal and neoplastic human uterine cervix. Int J Gynecol Cancer. 12: 92-98.
229 Beil WJ, Fureder W, Wiener H, Grossschmidt K, Maier U, Schedle A, Bankl HC, Lechner K, Valent P (1998) Phenotypic and functional characterization of mast cells derived from renal tumor tissues. Exp Hematol. 26: 158–169.
230 Ribatti D, Vacca A, Marzullo A, Nico B, Ria R, Roncali L, Dammacco F (2000) Angiogenesis and mast cell density with tryptase activity increase simultaneously with pathological progression in B-cell non-Hodgkin’s lymphomas. Int J Cancer.85: 171–175.
231 Janeway, C., Janeway, P., Walport, M., & Shlomchik, M. (2001) Immunobiology (5th Ed.) New York: Garland.
232 Locksley RM, Killeen N, Lenardo MJ (2001) The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 104: 487-501.
233 Aggarwal BB (2003) Signaling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 3: 745-756.
234 Naismith JH, Sprang SR (1998) Modularity in the TNF-receptor family. Trends Biochem Sci. 23: 74-79.

73
235 Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, Lynch D, Tsien RY, Lenardo MJ (2000) Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 288: 2354-2357.
236 Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ (2000) A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 288: 2351–2354.
237 Eck MJ, Sprang SR (1989) The structure of tumor necrosis factor-α at 2.6 Å resolution. Implications for receptor binding. J. Biol. Chem. 264: 17595–17605.
238 Cha SS, Kim MS, Choi YH, Sung BJ, Shin NK, Shin HC, Sung YC, Oh BH (1999) 2.8 Å resolution crystal structure of human TRAIL, a cytokine with selective antitumor activity. Immunity. 11: 253–261.
239 Karpusas M, Hsu YM, Wang JH, Thompson J, Lederman S, Chess L, Thomas D (1995) 2 Å crystal structure of an extracellular fragment of human CD40 ligand. Structure. 3: 1031–1039.
240 Dürkop H, Latza U, Hummel M, Eitelbach F, Seed B, Stein H (1992) Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin's disease. Cell. 68: 421–427.
241 Smith CA, Gruss HJ, Davis T, Anderson D, Farrah T, Baker E, Sutherland GR, Brannan CI, Copeland NG, Jenkins NA (1993) CD30 antigen, a marker for Hodgkin's lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell. 73: 1349–1360.
242 Younes A, Consoli U, Zhao S, Snell V, Thomas E, Gruss HJ, Cabanillas F, Andreeff M (1996) CD30 ligand is expressed on resting normal and malignant human B lymphocytes. Br J Haematol. 93: 569–571.
243 Cerutti A, Schaffer A, Goodwin RG, Shah S, Zan H, Ely S, Casali P (2000) Engagement of CD153 (CD30 ligand) by CD30+ T cells inhibits class switch DNA recombination and antibody production in human IgD+ IgM+ B cells. J Immunol. 165: 786–794.
244 Molin D, Fischer M, Xiang Z, Larsson U, Harvima I, Venge P, Nilsson K, Sundström C, Enblad G, Nilsson G (2001) Mast cells express functional CD30 ligand and are the predominant CD30L-positive cells in Hodgkin’s disease. Br J Haematol 114: 616–623.
245 Stein H, Foss HD, Dürkop H, Marafioti T, Delsol G, Pulford K, Pileri S, Falini B (2000) CD30+ anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 96:3681–3695.
246 Falini B, Pileri S, Pizzolo G, Dürkop H, Flenghi L, Stirpe F, Martelli MF, Stein H (1995) CD30 (Ki-1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood. 85: 1–14.
247 Caproni M, Bianchi B, D'Elios MM, De Carli M, Amedei A, Fabbri P (1997) In vivo relevance of CD30 in atopic dermatitis. Allergy. 52: 1063–1070.
248 Dummer W, Rose C, Bröcker EB (1998) Expression of CD30 on T helper cells in the inflammatory infiltrate of acute atopic dermatitis but not of allergic contact dermatitis. Arch Dermatol Res. 290: 598–602.
249 Gerli R, Pitzalis C, Bistoni O, Falini B, Costantini V, Russano A, Lunardi C (2000) CD30+ T cells in rheumatoid synovitis: mechanisms of recruitment and functional role. J Immunol. 164: 4399–4407.
250 D'Elios MM, Romagnani P, Scaletti C, Annunziato F, Manghetti M, Mavilia C, Parronchi P, Pupilli C, Pizzolo G, Maggi E, Del Prete GF, Romagnani S (1997) In vivo CD30 expression in human diseases with predominant activation of Th2-like T cells. J Leukoc Biol. 61: 539–544.
251 Mavalia C, Scaletti C, Romagnani P, Carossino AM, Pignone A, Emmi L, Pupilli C, Pizzolo G, Maggi E, Romagnani S (1997) Type 2 helper T-cell predominance and high CD30 expression in systemic sclerosis. Am J Pathol. 151: 1751–1758.
252 Bengtsson Å (2001) The role of CD30 in atopic disease. Allergy 56: 593–603.
253 Pizzolo G, Vinante F, Chilosi M, Dallenbach F, Josimovic-Alasevic O, Diamantstein T, Stein H (1990) Serum levels of soluble CD30 molecule (Ki-1 antigen) in Hodgkin's disease: relationship with disease activity and clinical stage. Br J Haematol. 75: 282–284.
254 Nadali G, Vinante F, Stein H, Todeschini G, Tecchio C, Morosato L, Chilosi M, Menestrina F, Kinney MC, Greer JP (1995) Serum levels of the soluble form of CD30 molecule as a tumor marker in CD30+ anaplastic large-cell lymphoma. J Clin Oncol. 13: 1355–1360.

74
255 Nadali G, Tavecchia L, Zanolin E, Bonfante V, Viviani S, Camerini E, Musto P, Di Renzo N, Carotenuto M, Chilosi M, Krampera M, Pizzolo G (1998) Serum level of the soluble form of the CD30 molecule identifies patients with Hodgkin's disease at high risk of unfavorable outcome. Blood. 91: 3011–3016.
256 Chiu A, Orazi A (2012) Mastocytosis and related disorders. Semin Diagn Pathol. 29:19-30.
257 Wiley SR, Goodwin RG, Smith CA (1996) Reverse signaling via CD30 ligand. J. Immunol. 157: 3635–3639.
258 Paulie S, Rosén A, Ehlin-Henrikisson B Braesch-Andersen S, Jakobson E, Koho H, Perlmann P (1989) The human B lymphocyte and carcinoma antigen, CDw40, is a phosphoprotein involved in growth signal transduction. J Immunol. 142: 590-595.
259 Armitage RJ, Sato TA, Macduff BM, Clifford KN, Alpert AR, Smith CA, Fanslow WC (1992) Identification of a source of biologically active CD40 ligand. Eur J Immunol 22: 2071-2076.
260 Van Kooten C, Banchereau J (2000) CD40-CD40 ligand. J Leukoc Biol 2000 67: 2-17.
261 Biancone L, Cantaluppi V, Camussi G (1999) CD40-CD154 interactions in experimental and human disease. Int J Mol Med 3: 343-353.
262 Ruggiero G, Caceres EM, Voordouw A, Noteboom E, Graf D, Kroczek RA, Spits H (1996) CD40 expressed on thymic epithelial cells provides costimulation for proliferation but not for apoptosis of human thymocytes. J Immunol. 156: 3737-3746.
263 Hess S, Engelmann H (1996) A novel function of CD40: induction of cell death in transformed cells. J Exp Med. 183: 159-167.
264 Thomas WD, Smith MJ, Si Z, Hersey P (1996) Expression of the co-stimulatory molecule CD40 on melanoma cells. Int J Cancer. 68: 795-801.
265 Chiu A, Orazi A (2012) Mastocytosis and related disorders. Semin Diagn Pathol. 29:19-30.
266 Gauchat JF, Aubry JP, Mazzei G, Life P, Jomotte T, Elson G, Bonnefoy JY (1993) Human CD40 ligand: molecular cloning, cellular distribution and regulation of expression by factors controlling IgE production. FEBS Lett . 315: 259-266.
267 Im SH, Barchan D, Maiti PK, Fuchs S, Souroujon MC (2001) Blockade of CD40 ligand suppresses chronic experimental myasthenia gravis by down-regulation of Th1 differentiation and up-regulation of CTLA-4. J Immunol. 166: 6893-6898.
268 Jensen J, Krakauer M, Sellebjerg F (2001) Increased T cell expression of CD154 (CD40-ligand) in multiple sclerosis. Eur J Neurol. 8: 321-328.
269 Brams P, Black A, Padlan EA, Hariharan K, Leonard J, Chambers-Slater K, Noelle RJ, Newman R (2001) A humanized anti-human CD154 monoclonal antibody blocks CD154-CD40 mediated human B cell activation. Int Immunopharmacol. 1: 277-294.
270 Young LS, Eliopoulos AG, Gallagher N, Dawson CW (1998) CD40 and epithelial cells: across the great divide. Immunol Today. 19: 502-506.
271 Loro LL, Ohlsson M, Vintermyr OK, Liavaag PG, Jonsson R, Johannessen AC (2001) Maintained CD40 and loss of polarized CD40 ligand expression in oral squamous cell carcinoma. Anticancer Res. 21: 113-117.
272 Vandenabeele P, Declercq W, Beyaert R, Fiers W (1995) Two tumour necrosis factor receptors: structure and function. Trends Cell Biol 5: 392–399.
273 Tartaglia LA, Goeddel DV (1992) Two TNF receptors. Immunol Today 13: 151–153.
274 Ashkenazi A, Dixit VM (1998) Death receptors: signaling and modulation. Science. 281: 1305–1308.
275 Baker SJ, Reddy EP (1998) Modulation of life and death by the TNF receptor superfamily. Oncogene. 17: 3261-3270.
276 Muppidi JR, Tschopp J, Siegel RM (2004) Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity. 21: 461–465.
277 Bigda J, Beletsky I, Brakebusch C, Varfolomeev Y, Engelmann H, Bigda J, Holtmann H, Wallach D (1994) Dual role of the p75 tumor necrosis factor (TNF) receptor in TNF cytotoxicity. J. Exp. Med. 180: 445–460.
278 Medvedev AE, Sundan A, Espevik T (1994) Involvement of the tumor necrosis factor receptor p75 in mediating cytotoxicity and gene regulating activities. Eur. J. Immunol. 24: 2842–2849.

75
279 Grell M, Zimmermann G, Gottfried E (1999) Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J. 18: 3034–3043.
280 Suminami Y, Kishi F, Sekiguchi K, Kato H (1991) Squamous cell carcinoma antigen is a new member of the serine protease inhibitors. Biochem Biophys Res Commun. 181: 51-58.
281 Schick C, Kamachi Y, Bartuski AJ, Cataltepe S, Schechter NM, Pemberton PA, Silverman GA (1997) Squamous cell carcinoma antigen 2 is a novel serpin that inhibits the chymotrypsin-like proteinases cathepsin G and mast cell chymase. J Biol Chem. 272: 1849-1855.
282 Sell, S., & Kato, H. (1992) Squamous cell carcinoma antigen. Serological cancer markers (pp. 437-451) Totowa, N.J.: Humana Press.
283 Crombach G, Scharl A, Vierbuchen M, Würz H, Bolte A (1989) Detection of squamous cell carcinoma antigen in normal squamous epithelia and in squamous cell carcinomas of the uterine cervix. Cancer. 63: 1337-1342.
284 Murakami A, Suminami Y, Sakaguchi Y, Nawata S, Numa F, Kishi F, Kato H (2000) Specific detection and quantification of SCC antigen 1 and SCC antigen 2 mRNAs by Fluorescence-based asymmetric semi-nested reverse transcription PCR. Tumour Biol. 21: 224-234.
285 Hamada K, Hanakawa Y, Hashimoto K, Iwamoto M, Kihana T, Hirose S, Nakamura M, Ito M (2001) Gene expression of human squamous cell carcinoma antigens 1 and 2 in human cell lines. Oncol Rep. 8: 347-354.
286 Cataltepe S, Gornstein ER, Schick C, Kamachi Y, Chatson K, Fries J, Silverman GA, Upton MP (2000) Co-expression of the squamous cell carcinoma antigens 1 and 2 in normal adult human tissues and squamous cell carcinomas. J Histochem Cytochem. 48: 113-122.
287 Harvima IT, Naukkarinen A, Harvima RJ, Fräki JE (1988) Immunoperoxidase and enzyme-histochemical demonstration of human skin tryptase in cutaneous mast cells in normal and mastocytoma skin. Arch Dermatol Res. 280: 363-370.
288 Harvima IT, Heikura H, Hyttinen M, Naukkarinen A (2006) Hyaluronic acid inhibits the adherence and growth of monolayer keratinocytes but does not affect the growth of keratinocyte epithelium. Arch Dermatol Res. 298: 207-219.
289 Otto FJ (1994) High-resolution analysis of nuclear DNA employing the fluorochrome DAPI. Methods Cell Biol 41: 211-217.
290 De Angelis T, Noè A, Chatterjee M, Mulholland J (2002) Stromelysin-1 activation correlates with invasiveness in squamous cell carcinoma. J Invest Dermatol. 118: 759-766.
291 Mutch DG, Powell CB, Kao MS, Collins JL (1992) Resistance to cytolysis by tumor necrosis factor α in malignant gynecological cell lines is associated with the expression of protein(s) that prevent the activation of phospholipase A2 by tumor necrosis factor α. Cancer Res. 52: 866-872.
292 Yamamoto T, Katayama I, Nishioka K (1997) Expression of stem cell factor in basal cell carcinoma. Br J Dermatol 137:709–713.
293 Huang B, Lei Z, Zhang G-M, Li D, Song C, Li B, Liu Y, Yuan Y, Unkeless J, Xiong H, Feng Z-H (2008) SCF-mediated mast cell infiltration and activation exacerbate the inflammation and immunosuppression in tumor microenvironment. Blood 112: 1269-1279.
294 Blatner NR, Bonertz A, Beckhove P, Cheon EC, Krantz SB, Strouch M, Weitz J, Koch M, Halverson AL, Bentrem DJ, Khazaie K (2010) In colorectal cancer mast cells contribute to systemic regulatory T-cell dysfunction. Proc Natl Acad Sci (USA) 107: 6430-6435.
295 Aoki M, Pawankar R, Niimi Y, Kawana S (2003) Mast cells in basal cell carcinoma express VEGF, IL-8 and RANTES. Int Arch Allergy Immunol 130: 216-223.
296 Iamaroon A, Pongsiriwet S, Jittidecharaks S, Pattanaporn K, Prapayasatok S, Wanachantararak S (2003) Increase of mast cells and tumor angiogenesis in oral squamous cell carcinoma. J Oral Pathol Med. 32: 195–199.
297 Ribatti D, Vacca A, Ria R, Marzullo A, Nico B, Filotico R, Roncali L, Dammacco F (2003) Neovascularisation, expression of fibroblast growth factor-2, and mast cells with tryptase activity increase simultaneously with pathological progression in human malignant melanoma. Eur J Cancer. 39: 666–674.
298 Sime W, Lunderius-Andersson C, Enoksson M, Rousselle P, Tryggvason K, Nilsson G, Harvima I, Patarroyo M (2009) Human mast cells adhere to and migrate on epithelial and vascular basement membrane laminins LM-332 and LM-511 via α3β1 integrin. J Immunol. 183: 4657-4665.

76
299 Harvima IT, Nilsson G, Suttle MM, Naukkarinen A (2008) Is there a role for mast cells in psoriasis? Arch Dermatol Res. 300:461-478.
300 Rice KD, Tanaka RD, Katz BA, Numerof RP, Moore WR (1998) Inhibitors of tryptase for the treatment of mast cell-mediated diseases. Current Pharm Design. 4: 381-396.
301 Schechter NM, Sprows JL, Schoenberger OL, Lazarus GS, Cooperman BS, Rubin H (1989) Reaction of human skin chymotrypsin-like proteinase chymase with plasma proteinase inhibitors. J Biol Chem. 264: 21308-21315.
302 Humphreys TR, Monteiro MR, Murphy GF (2000) Mast cells and dendritic cells in basal carcinoma stroma. Dermatol Surg. 26: 200-203.
303 Janowski P, Strzelecki M, Brzezinska-Blaszczyk E, Zalewska A (2001) Computer analysis of normal and basal cell carcinoma mast cells. Med Sci Monit. 7: 260-265.
304 Zhao W, Oskeritzian CA, Pozez AL, Schwartz LB (2005) Cytokine production by skin-derived mast cells: endogenous proteases are responsible for degradation of cytokines. J Immunol 175: 2635-2642.
305 Gibbs BF, Wierecky J, Welker P, Henz BM, WolV HH, Grabbe J (2001) Human skin mast cells rapidly release preformed and newly generated TNF-α and IL-8 following stimulation with anti-IgE and other secretagogues. Exp Dermatol. 10: 312–320.
306 Malejczyk M, Jozwiak J, Jablonska S, PWster H, Majewski S, Malejczyk J (2005) Circulating soluble tumour necrosis factor receptors in patients with epidermodysplasia verruciformis as compared to patients with cutaneous tumours in the general population. Oncol Rep. 13: 151–155.
307 Muta H, Boise LH, Fang L, Podack ER (2000) CD30 signals integrate expression of cytotoxic effector molecules, lymphocyte trafficking signals, and signals for proliferation and apoptosis. J Immunol. 165: 5105-5111.
308 Dang NH, Hagemeister FB, Pro B, McLaughlin P, Romaguera JE, Jones D, Samuels B, Samaniego F, Younes A, Wang M, Goy A, Rodriguez MA, Walker PL, Arredondo Y, Tong AT, Fayad L (2004) Phase II study of denileukin diftitox for relapsed/refractory B-Cell non-Hodgkin's lymphoma. J Clin Oncol. 22: 4095-4102.
309 Gruss HJ, Boiani N, Williams DE, Armitage RJ, Smith CA, Goodwin RG (1994) Pleiotropic effects of the CD30 ligand on CD30-expressing cells and lymphoma cell lines. Blood. 83: 2045-2056.
310 Younes A, Zhao S, Zhang X, Snell V, Clodi K, Kliche KO, Thomas E, Cabanillas F, Andreeff M (1997) CD30-ligand and CD40-ligand expression in lymph nodes involved with Hodgkin's disease. Ann Oncol. 8: 97-100.
311 Pinto A, Aldinucci D, Gloghini A, Zagonel V, Degan M, Improta S, Juzbasic S, Todesco M, Perin V, Gattei V, Herrmann F, Gruss HJ, Carbone A (1996) Human eosinophils express functional CD30 ligand and stimulate proliferation of a Hodgkin's disease cell line. Blood. 88: 3299-32305.
312 Gattei V, Degan M, Rossi FM, de Iuliis A, Mazzocco FT, Serraino D, Zagonel V, Aldinucci D, Pinto A (1999) CD30 ligand (CD30L)-expressing acute myeloid leukemias: a new model of paracrine interactions for the regulation of blast cells proliferation. Leuk Lymphoma. 35: 21-35.
313 Gattei V, Degan M, Gloghini A, De Iuliis A, Improta S, Rossi FM, Aldinucci D, Perin V, Serraino D, Babare R, Zagonel V, Gruss HJ, Carbone A, Pinto A (1997) CD30 ligand is frequently expressed in human hematopoietic malignancies of myeloid and lymphoid origin. Blood. 89: 2048-2059.
314 Fisher CA, Colman Jr. JH, Doroshow, Rayner AA, Hawkins MJ, Mier JW, Wiernik P, McMannis JD, Weiss GR, Margolin KA (1988) Metastatic renal cancer treated with interleukin-2 and lymphokine activated killer cells. A phase II clinical trial. Ann Intern Med. 108: 518-523.
315 Rossi FM, Degan M, Mazzocut-Zecchin L, Di Francia R, Aldinucci D, Pinto A, Gattei V (2001) CD30L up-regulates CD30 and IL-4 expression by T cells. FEBS Lett. 508: 418–422.
316 Poniecka AW, Alexis JB (1999) An immunohistochemical study of basal cell carcinoma and trichoepithelioma. Am J Dermatopathol. 21: 332–336.
317 Funakoshi S, Longo DL, Beckwith M, Conley DK, Tsarfaty G, Tsarfaty I, Armitage RJ, Fanslow WC, Spriggs MK, Murphy WJ (1994) Inhibition of human B-cell lymphoma growth by CD40 stimulation. Blood. 83: 2787-2794.
318 Hill SC, Youde SJ, Man S, Teale GR, Baxendale AJ, Hislop A, Davies CC, Luesley DM, Blom AM, Rickinson AB, Young LS, Eliopoulos AG (2005) Activation of CD40 in cervical carcinoma cells facilitates CTL responses and augments chemotherapy-induced apoptosis. J Immunol. 174: 41-50.

77
319 Hirano A, Longo DL, Taub DD, Ferris DK, Young LS, Eliopoulos AG, Agathanggelou A, Cullen N, Macartney J, Fanslow WC, Murphy WJ (1999) Inhibition of human breast carcinoma growth by a soluble recombinant human CD40 ligand. Blood. 93: 2999-3007.
320 von Leoprechting A, van der Bruggen P, Pahl HL, Aruffo A, Simon JC (1999) Stimulation of CD40 on immunogenic human malignant melanomas augments their cytotoxic T lymphocyte-mediated lysis and induces apoptosis. Cancer Res. 59: 1287-1294.
321 Viac J, Schmitt D, Claudy A (1997) CD40 expression in epidermal tumors. Anticancer Res. 17: 569–572.
322 Kooy AJW, Prens EP, van Heukelum A, Vuzevski VD, van Joost T, Tank B (1999) Interferon-γ-induced ICAM-1 and CD40 expression, complete lack of HLA-DR and CD80 (B7.1), and inconsistent HLA-ABC expression in basal cell carcinoma: a possible role for interleukin-10? J Pathol. 187: 351–357.
323 Huttunen M, Naukkarinen A, Horsmanheimo M, Harvima IT (2002) Transient production of stem cell factor in dermal cells but increasing expression of Kit receptor in mast cells during normal wound healing. Arch Dermatol Res. 294: 324–330.
324 Flaxman BA, Harper RA (1975) In vitro analysis of the control of keratinocyte proliferation in human epidermis by physiologic and pharmacologic agents. J Invest Dermatol. 65: 59-68.
325 Mitra RS, Shimizu Y, Nickoloff BJ (1993) Histamine and cis-urocanic acid augment tumor necrosis factor-α mediated induction of keratinocyte intercellular adhesion molecule-1 expression. J Cell Physiol. 156: 348-357.
326 Symington FW, Santos EB (1991) Lysis of human keratinocytes by allogeneic HLA class I-specific cytotoxic T cells. Keratinocyte ICAM-1 (CD54) and T cell LFA-1 (CD11a/CD18) mediate enhanced lysis of IFNgamma-treated keratinocytes. 146: 2169-2175.
327 Wang J, Al-Lamki RS, Zhang H, Kirkiles-Smith N, Gaeta ML, Thiru S, Pober JS, Bradley JR (2003) Histamine antagonizes tumor necrosis factor (TNF) signaling by stimulating TNF receptor shedding from the cell surface and Golgi storage pool. J Biol Chem. 278: 21751-21760.
328 Vannier E, Miller LC, Dinarello CA (1991) Histamine suppresses gene expression and synthesis of tumor necrosis factor α via histamine H2 receptors. J Exp Med. 174: 281-294.
329 Szlosarek P, Charles KA, Balkwill FR (2006) Tumour necrosis factor-α as a tumour promoter. Eur J Cancer. 42: 745-750.
330 Mocellin S, Rossi CR, Pilati P, Nitti D (2005) Tumor necrosis factor, cancer and anticancer therapy. Cytokine Growth Factor Rev.16: 35-53.
331 Woodworth CD, McMullin E, Iglesias M, Plowman GD (1995) Interleukin 1 α and tumor necrosis factor α stimulate autocrine amphiregulin expression and proliferation of human papillomavirus-immortalized and carcinoma-derived cervical epithelial cells. Proc Natl Acad Sci USA. 92: 2840-2844.
332 Gaiotti D, Chung J, Iglesias M, Nees M, Baker PD, Evans CH, Woodworth CD (2000) Tumor necrosis factor-α promotes human papillomavirus (HPV) E6/E7 RNA expression and cyclin-dependent kinase activity in HPV-immortalized keratinocytes by a ras-dependent pathway. Mol Carcinog. 27: 97-109.
333 Rosen EM, Goldberg ID, Liu D, Setter E, Donovan MA, Bhargava M, Reiss M, Kacinski BM (1991) Tumor necrosis factor stimulates epithelial tumor cell motility. Cancer Res. 51: 5315-5321.
334 Herzog TJ, Nelson PK, Mutch DG, Wright WD, Kao MS, Collins JL (1992) Effects of radiation on TNF-α-mediated cytolysis of cell lines derived from cervical carcinomas. Gynecol Oncol. 47: 196-202.
335 Akca H, Akan SY, Yanikoglu A, Ozes ON (2003) Suppression of TNF-α mediated apoptosis by EGF in TNF-α sensitive human cervical carcinoma cell line. Growth Factors. 21: 31-39.
336 Van den Brule FA, Clausse N, Delvenne P, Franzen-Detrooz E, Castronovo V (2000) Combined interferon-α and tumor necrosis factor-α treatment differentially affects adhesion and migration of keratinocyte-derived cells to laminin-1. Cell Adhes Commun. 7: 321-329.
337 Hidalgo K, Rojas IG, Penissi AB, Rudolph MI (2005) TNFα increases in vitro migration of human HPV18- positive SW756 cervical carcinoma cells. Biocell. 29: 303-311.
338 Tilly BC, Tertoolen LGJ, Ladoux A, Verlaan I, de Laat SW, Moolenaar WH (1990) Histamine as a growth factor and chemoattractant for human carcinoma and melanoma cells: action through Ca2+ -mobilizing H1 receptors. J Cell Biol. 110: 1211-1215.

78
339 Kanda N, Watanabe S (2002) Histamine inhibits the production of interferon-induced protein of 10 kDa in human squamous cell carcinoma and melanoma. J Invest Dermatol. 119: 1411-1419.
340 Huttunen M, Hyttinen M, Nilsson G, Butterfield JH, Horsmanheimo M, Harvima IT (2001) Inhibition of keratinocyte growth in cell culture and whole skin culture by mast cell mediators. Exp Dermatol. 10:184-192.
341 Powell CB, Scott JH, Collins JL (1998) Comparison of TNF-α and TNF-α cytolytic mechanisms in human ovarian and cervical carcinoma cell lines. Gynecol Oncol. 71: 258-265.
342 Massad LS, Mutch DG, Kao M-S, Powell CB, Collins JL (1991) Inhibition of protein synthesis enhances the lytic effects of tumor necrosis factor α and interferon α in cell lines derived from gynecological malignancies. Cancer Immunol Immunother. 33: 183-188.
343 Younes F, Quartey EL, Kiguwa S, Partridge M (1996) Expression of TNF and the 55-kDa TNF receptor in epidermis, oral mucosa, lichen planus and squamous cell carcinoma. Oral Dis. 2: 25-31.
344 von Biberstein SE, Spiro JD, Lindquist R, Kreutzer DL (1995) Enhanced tumor cell expression of tumor necrosis factor receptors in head and neck squamous cell carcinoma. Am J Surg. 170: 416-422.
345 Ding SY, Wu R (2002) Detection of tumor necrosis factor mRNA expression in human cervix and endometrium by in situ hybridization. Di Yi Jun Yi Da Xue Xue Bao. 22: 244-246.
346 Hsu KF, Huang SC, Shiau AL, Cheng YM, Shen MR, Chen YF, Lin CY, Lee BH, Chou CY (2007) Increased expression level of squamous cell carcinoma antigen 2 and 1 ratio is associated with poor prognosis in early-stage uterine cervical cancer. Int J Gynecol Cancer. 17: 174-181.
347 Ebihara N, Takai S, Miyazaki M, Murakami A (2005) Mast cell chymase induces conjunctival epithelial cell apoptosis by a mechanism involving degradation of fibronectin. Curr Eye Res. 30: 429-435.
348 Nyberg P, Ylipalosaari M, Sorsa T, Salo T (2006) Trypsins and their role in carcinoma growth. Exp Cell Res. 312: 1219-1228.
349 Yoshii M, Jikuhara A, Mori S, Iwagaki H, Takahashi HK, Nishibori M, Tanaka N (2005) Mast cell tryptase stimulates DLD-1 carcinoma through prostaglandin- and MAP kinase-dependent manners. J Pharmacol Sci. 98: 450-458.
350 Ebihara N, Funaki T, Murakami A, Takai S, Miyazaki M (2005) Mast cell chymase decreases the barrier function and inhibits the migration of corneal epithelial cells. Curr Eye Res. 30: 1061-1069.
351 Lazaar AL, Plotnick MI, Kucich U, Crichton I, Lotfi S, Das SK, Kane S, Rosenbloom J, Panettieri RA Jr, Schechter NM, Puré E (2002) Mast cell chymase modifies cell-matrix interactions and inhibits mitogen-induced proliferation of human airway smooth muscle cells. J Immunol. 169: 1014-1020.
352 Goldstein SM, Leong J, Schwartz LB, Cooke D (1992) Protease composition of exocytosed human skin mast cell protease-proteoglycan complexes. Tryptase resides in a complex distinct from chymase and carboxypeptidase. J Immunol. 148: 2475-2482.
353 Murakami A, Nakagawa T, Kaneko M, Nawata S, Takeda O, Kato H, Sugino N (2006) Suppression of SCC antigen promotes cancer cell invasion and migration through the decrease in E-cadherin expression. Int J Oncol. 29: 1231-1235.
354 Iwasaki M, Nishikawa A, Akutagawa N, Fujimoto T, Teramoto M, Sakaguchi Y, Kato H, Ito M, Yoshida K, Kudo R (2004) E1AF/PEA3 reduces the invasiveness of SiHa cervical cancer cells by activating serine proteinase inhibitor squamous cell carcinoma antigen. Exp Cell Res. 299: 525-532.

Publications of the University of Eastern Finland
Dissertations in Health Sciences
isbn 978-952-61-0909-1
Publications of the University of Eastern FinlandDissertations in Health Sciences
dissertatio
ns | 133 | N
icola
e-Co
stin D
iaco
nu
| Mast C
ells in Hum
an Epith
elial Can
cers
Nicolae-Costin DiaconuMast Cells in Human
Epithelial Cancers
Nicolae-Costin Diaconu
Mast Cells in Human Epithelial Cancers
The possible role of MC mediators
was investigated in SCC and BCC
carcinomas. Novel information on
the MC proteinases tryptase and
chymase in SCC and BCC carcinomas
is presented and their potential to
degrade the extracellular matrix is
discussed. Moreover, the presence
MCs cytokines and their counter-
part receptors is also described. The
results demonstrate that chymase
may release viable tumoral cells
that tolerate the cytolytic action of
TNF-α and histamine leading to the
spread of SCC. In BCC, MCs express
TNF-α and CD30 ligand but the lack
of CD40L may be responsible of the
inefficient antitumoral response.





























