Embed Size (px)
Citation preview

ORIGINAL PAPER
T. Kimura á T. Komiyama á Y. Furuichi á Y. IimuraS. Karita á K. Sakka á K. Ohmiya
N -glycosylation is involved in the sensitivityof Saccharomyces cerevisiae to HM-1 killer toxin secretedfrom Hansenula mrakii IFO 0895
Received: 13 July 1998 /Received last revision: 18 September 1998 /Accepted: 2 October 1998
Abstract Saccharomyces cerevisiae rhk mutants werepreviously shown to have a phenotype that is resistant toHM-1 killer toxin secreted from Hansenula mrakii IFO0895. The RHK1/ALG3 gene encodes a mannosyl-transferase that is involved in the synthesis of anoligosaccharide in protein N-glycosylation. Previously,this gene was cloned and shown to complement the rhk1mutation. In this study, the RHK2 gene, which com-plements the rhk2 mutation, was cloned. The RHK2gene was found to be identical to the essential geneSTT3, which encodes a subunit of the oligosaccharyl-transferase complex. This complex transfers the coreoligosaccharide to proteins. The rhk2 mutants showedsupersensitivity to several drugs (Calco¯uor White,ca�eine and FK506), suggesting that these strains havecell-wall defects. Activity staining of invertase in anacrylamide gel indicated that it was underglycosylated.These results suggest that one or more mannoproteinsare involved in the cytocidal process of HM-1.
Introduction
The killer phenomenon is widespread among variousgenera and species of yeasts. Killer yeasts secrete intotheir culture medium polypeptides, known as killertoxins, that kill sensitive strains of yeast. Among thesetoxins, Saccharomyces cerevisiae K1 (Bussey 1991) andKluyveromyces lactis killer toxins (Stark et al. 1990) havebeen the most studied. They are encoded by RNA andDNA plasmids respectively. These cells have immunitysystems encoded on these plasmids that provide immu-nity to their own toxins. The action of the killer toxin ofK1 starts with the adsorption of the toxin to b-1,6-glu-can in the cell-wall of a susceptible yeast strain(Al-Aidroos and Bussey 1977), which then disruptscytoplasmic membrane function. The toxin produced byK. lactis binds the cell-wall chitin and then the toxin isinternalized into the cell. Finally, the c subunit of thetoxin causes arrest of the G1 phase, which leads to a lossof viability. Studies of the killing mechanisms of thesetoxins have aided the understanding of important yeastcell functions, especially the synthesis of cell-wall com-ponents that are the receptors of the individual killertoxins. Receptor analysis of the K1 killer toxin revealedthat the KRE genes are involved in the synthesis ofb-1,6-glucan and O-linked glycosylation of mannopro-teins in the yeast cell wall (Boone et al. 1990; Brownand Bussey 1993; Brown et al. 1993; Hill et al. 1992;Maeden et al. 1990; Rommer and Bussey 1991). Thegenes involved in the synthesis of chitin were isolatedby analyzing the K. lactis-killer-resistant mutants ofS. cerevisiae (Kawamoto et al. 1992; Takita and Cast-ilho-Valanicius 1993).
The HM-1 killer toxin of Hansenula mrakii IFO 0895has unique features. Biochemical and microbial studieshave demonstrated that it is a monomeric protein,containing 88 amino acid residues, that is rich in cys-teine, is stable against heat treatment (100 °C, 10 min)and a wide range of pH values (pH 2.0±11.0) (Yama-moto et al. 1986a), and exerts a strong cytocidal e�ect on
Appl Microbiol Biotechnol (1999) 51: 176±184 Ó Springer-Verlag 1999
T. Kimura (&) á K. Sakka á K. OhmiyaFaculty of Bioresources, Mie University,Tsu, Mie 514-8507, Japane-mail: [email protected].: +81-59-231-9606Fax: +81-59-231-9634
S. KaritaCenter for Molecular Biology and Genetics,Mie University, Tsu, Mie 514-8507, Japan
T. KomiyamaDepartment of Biochemistry, Niigata College of Pharmacy,Niigata 950-21, Japan
Y. FuruichiAGENE Research Institute, Kajiwara, Kamakura,Kanagawa 214, Japan
Y. IimuraFaculty of Engineering, Yamanashi University,Kofu, Yamanashi 400-0016, Japan

many yeast strains (Nomoto et al. 1984; Yamamoto et al.1988). HM-1 has been shown to inhibit the synthesisof b-1,3-glucans in vivo and in vitro (Yamamoto et al.1986b), which are the major components of the yeastcell-wall. These features contrast with those of otherkiller toxins reported so far. We reported the cloning ofthe gene encoding HM-1 (Kimura et al. 1993) and thatthe HM-1 killer toxin family is distributed in severalHansenula species (Kimura et al. 1995). Recently, HM-1was shown by nuclear magnetic resonance to consist oftwo twisted four-stranded antiparallel b-sheets (Antuchet al. 1996).
Two genes of S. cerevisiae that confer sensitivity toHM-1 (KNR4 and HKR1) have been reported (Honget al. 1994; Kasahara et al. 1994a; Yabe et al. 1996). Bothare involved in the synthesis of b-1,3-glucan. Disruptionof KNR4 resulted in HM-1 resistance and in a reducedlevel of b-1,3-glucan synthase. In contrast, over-expres-sion of HKR1 conferred HM-1 resistance and increasedthe b-1,3-glucan content in the cell-wall fraction. Usingscanning electron microscopy, we previously found thatcells treated with HM-1 burst out at the top of the budsand bud-neck, probably because of the osmotic pressure(Komiyama et al. 1996; Kimura et al. 1997). These®ndings also indicated that the ®nal target of HM-1 is astep in the synthesis of b-1,3-glucan. In fact, partiallypuri®ed b-1,3-glucan synthase was inhibited by HM-1(Takasuka et al. 1995). Despite these studies, little isknown about the killing process of HM-1. The cytocidale�ect of HM-1 on S. cerevisiae was suppressed by ad-dition of b-1,3-glucan, b-1,6-glucan and mannan, butwas not suppressed by addition of chitin (Kasahara et al.1994b). This suggests that the receptor for HM-1 is aglucan and/or mannan in the cell wall. Such receptorswork to concentrate the toxin around sensitive cells.Although the ®nal target of HM-1 was shown to beb-1,3-glucan synthase, no other cell-wall componentsinvolved in sensitivity to the toxin are known. Recently,we reported that the RHK1/ALG3 gene, which encodes amannosyltransferase that is responsible for theglycosylation of mannoprotein, is involved in HM-1sensitivity (Kimura et al. 1997). In this study, we reportfurther analyses of HM-1-resistant mutants of S. cere-visiae and that mannoproteins are involved in HM-1sensitivity.
Materials and methods
Strains and media
The S. cerevisiae strains used in this study are described in Table 1.H. mrakii IFO 0895 was obtained from the Institute of Fermen-tation, Osaka, Japan. Yeast cultures were routinely grown in YPDmedium (1% Bacto yeast extract, 2% Bacto peptone, 2% glucose)or synthetic dextrose (SD) medium (0.67% Difco yeast nitrogenbase without amino acids, 2% glucose). HM-1 killer medium wasprepared as described previously (Kimura et al. 1997). Brie¯y,H. mrakii IFO 0895 was cultivated in SD medium for 24 h and theculture supernatant was concentrated to 1/200th of the originalvolume by lyophilization. This concentrated toxin was added tothe SD medium at a ratio of 1 ml/200 ml after the medium hadbeen autoclaved. Escherichia coli strain DH5a was used forrecombinant DNA procedures. Luria-Bertani medium was used forE. coli cultivation. S. cerevisiae strains for chromosome loss map-ping using 2l plasmid were obtained from the Yeast Genetic StockCenter (Berkeley, Calif., USA) and were used according to theirprotocol.
DNA recombinant method
Standard recombinant DNA procedures were carried out as de-scribed by Sambrook et al. (1989). DNA sequence analysis wasdone using the pBluescript II KS vector. DNA sequencing wasdone on a model 4000L automated DNA sequencer (LI-COR,Lincoln, Neb., USA) with appropriate dye primers. Yeast trans-formation was carried out by the method of Ito et al. (1983). Othergeneral yeast genetic methods were employed as described by Roseet al. (1990).
Analysis of killer toxin sensitivity to HM-1, K1and K. lactis killer toxins
To evaluate the extent of HM-1 resistance, YPD plates containingvarious amounts of HM-1 killer toxin were prepared. Each strainwas pre-grown in liquid YPD medium for 24 h and the cell densitywas adjusted to 103 cells/5 ll. A 5-ll sample of each diluted cellsuspension was placed on HM-1 killer plates. Growth was scoredafter 1 day of incubation at 30 °C.
For testing sensitivity to other killer toxins, a killer eclipseassay was employed as described in Kishida et al. (1996). Cellsuspensions of each HM-1-resistant strain, prepared as describedabove, were spotted on the killer assay plate (YPD and pH wasadjusted to 4.5 with sodium citrate bu�er). The killer yeastsH. mrakii IFO 0895 (K9 killer), K. lactis IFO 1267 and S. cerevisiaeATCC 66899 (K1 killer) were inoculated on the edge of the testcell spot. The plates were incubated for 48 h at 25 °C. The zoneof growth inhibition in the test cell spot formed by each killeryeast was examined.
Table 1 Yeast strains used inthis study Strains Description Source
BJ1824 MATa ura3 trp1 leu2 pep4 E. W. Jonesrhk1D isogenic to BJ1824, rhk1D::URA3 T. Kimura et al. (1997)KR1 HM-1 resistant mutant derived from BJ1824, rhk1-1 T. Kimura et al. (1997)KR7 HM-1 resistant mutant derived from BJ1824, rhk3-1 This studyKR9 HM-1 resistant mutant derived from BJ1824, rhk3-2 This studyKR11 HM-1 resistant mutant derived from BJ1824, rhk2-1 This studyKR13 HM-1 resistant mutant derived from BJ1824, rhk2-2 This studyKR17 HM-1 resistant mutant derived from BJ1824, rhk2-3 This studyKR18 HM-1 resistant mutant derived from BJ1824, rhk2-4 This studyKR19 HM-1 resistant mutant derived from BJ1824, rhk2-5 This study
177

Activity staining of invertase following gel electrophoresis
Invertase (encoded by SUC2) in the cell extract was separated bynondenaturing polyacrylamide gel electrophoresis. After electro-phoresis, the gel was incubated in 0.1 M sucrose solution (pH 5.2,sodium acetate bu�er) for 30 min and stained as described byBallou et al. (1986).
Assay for drug sensitivity
Ca�eine and Calco¯uor White (CFW) sensitivities of each HM-1-resistant strain were examined as described previously (Ram et al.1994). Brie¯y, the cell density of each strain pre-cultured in YPDfor 24 h was adjusted to an absorbance of 8.0 at 600 nm. Drops(5 ll) of each diluted cell suspension were spotted on YPD platescontaining each drug. Growth was scored after 2 days incubationat 28 °C.
Each HM-1-resistant mutant of KR, harboring the plasmidYCp50 or pKR02 (the YCp50-derived plasmid to which the RHK2gene was subcloned), was grown in SD medium supplemented withthe required amino acids. These cells were harvested at the mid-logphase, and suspended in SD medium at a density of 104 cells/ml.Aliquots (100 ll) of these cells were pipetted into microtiter wellsand 100 ll control solution, or the appropriate dilution of eachdrug, was added and mixed using a pipette tip. After the cells hadbeen cultured at 30 °C for 2 days, their growth was monitored bymeasuring the absorbance at 600 nm of the cell suspension, using aplate reader. The activity of a drug was expressed as the percentagereduction in growth of each strain with respect to the drug-freecontrol.
HM-1 binding analysis
Intact cells of each strain in the mid-log phase (108 cells) weresuspended in 0.5 ml citrate bu�er (0.1 M, pH 4.5). This cell sus-pension was mixed with 0.5 ml HM-1 killer toxin solution andincubated at 4 °C for 60 min. After incubation, cells were removedby centrifugation (8000 g, 5 min, 4 °C) and ®ltration through a®lter membrane (0.2 lm pore size). The residual toxin activity inthe ®ltrate was determined by the agar-well di�usion method asfollows. The ®ltrate was loaded into a well (0.5 mm diameter) on aYPD plate, containing 106 cells of HM-1-sensitive H. anomala IFO0569, and incubated at 30 °C for 24 h. The growth-inhibitory zonearound the well was measured. Residual activity was calculatedrelative to a control (HM-1 killer toxin without cells).
Results
Characterization of HM-1-resistant mutants
In the previous report, we described the isolation ofHM-1-resistant mutants from S. cerevisiae BJ1824(Kimura et al. 1997). We designated these mutants asKR (killer-resistant) and the locus as rhk (resistant toHansenula killer). Genetic analyses of these mutantswere carried out. Each mutant was crossed with wild-type 5073 (MATa, arg4 leu1 trp1 thr4) to ®nd HM-1-resistant phenotypes that segregated 2:2 in meiosis. Alltetrads showed 2:2 segregation, indicating that eachmutant contains a mutation in a single gene. Comple-mentation analysis of these mutants revealed threecomplementation groups (rhk1, rhk2, rhk3), all of whichare recessive for HM-1 resistance. We carried out achromosome-loss mapping analysis to map the rhk1,rhk2 and rhk3 genes. The rhk1 gene was mapped on
chromosome II and the rhk2 gene was mapped onchromosome VII. However, the location of the rhk3gene could not be determined by this method.
We analyzed the killer sensitivity of eight mutants(KR1, KR7, KR9, KR11, KR13, KR17, KR18 andKR19) in more detail. Although strains KR11, KR13,KR17, KR18, KR19, which have di�erent alleles ofrhk2, showed slightly weak HM-1 resistance on a 1/10dilution of the concentrated HM-1 plate, they had moreresistance than did S. cerevisiae wild strain BJ1824(Fig. 1).
The KR mutants were examined for sensitivity toK. lactis killer toxin and S. cerevisiae K1 killer toxin, thekilling processes of which have been well characterized.A novel method to evaluate killer sensitivity, describedin Kishida et al. (1996), was employed. As shown inFig. 2, the KR mutants retained their sensitivity to killertoxins of K. lactis and S. cerevisiae, suggesting that KRmutants are distinct from other killer-resistant mutantsthat showed resistance to K. lactis killer toxin (Yajimaet al. 1997) and K1 toxin.
Fig. 1 HM-1 sensitivities of KR1 (rhk1-1), KR7 (rhk3-1), KR9 (rhk3-2), KR11 (rhk2-1), KR13 (rhk2-2), KR17 (rhk2-3), KR18 (rhk2-4),KR19 (rhk2-5), rhk1D, and wild strains of yeast. Cell densities of thestrains were adjusted and drops of each cell suspension were spottedon HM-1 killer plates as described in Materials and methods.Dilutions of HM-1 toxin are indicated above the panel. YPD yeast/peptone/glucose medium
178

Isolation of the RHK2 gene
We recently reported the cloning of the RHK1 gene,which complements the rhk1-1 mutation (Kimura et al.1997). Here we attempted to clone the wild RHK2 andRHK3 genes that complement the rhk2 and rhk3 muta-tions respectively. To isolate the wild-type RHK2 gene,the rhk2-1 mutant KR11 was transformed with a genelibrary prepared using the centromeric vector YCp50from wild-type S. cerevisiae genomic DNA. Approxi-mately 10 000 transformants were screened for the HM-1-sensitive phenotype by replicating onto killer/SDplates as described previously (Kimura et al. 1997). Fivetransformants were found to have restored HM-1 sen-sitivity. These plasmids harbored the same DNA frag-ments of about 8 kb. A 4.3-kb fragment from a partialBamHI digestion was able to complement the rhk2mutation. The restriction map for this DNA fragmentwas established and a XbaI-SalI fragment of about3.5 kb was found to complement the rhk2 mutation(Fig. 3), indicating that this area encodes the wild RHK2gene. In contrast to RHK2, RHK3 could not be clonedsince the transformation e�ciencies of KR7 and KR9were low.
The nucleotide sequence of the 3.5-kb fragment re-vealed an open reading frame encoding a polypeptidecomposed of 718 amino acids. A DNA homology searchusing the BLAST program on the NCBI mail servershowed a striking match to a yeast genome DNA se-quence deposited in Genbank (accession D28952,YGL022w), which maps to chromosome VII. The resultshowed good agreement with chromosome loss mappingof the rhk2 locus. This gene was reported to be theessential STT3 gene, which complements the staur-
osporine- and temperature-sensitive mutation stt3(Yoshida et al. 1995), and the product of this gene(Stt3p) was found to be a component of the oligosac-charyltransferase complex (Zu�erey et al. 1995; Spiriget al. 1997).
The rhk1 and rhk2 mutants have a defect in the N-glycosylation of extracellular invertase. Since RHK1and RHK2 were found to be ALG3 and STT3, whoseproducts Alg3p and Stt3p were reported to be involvedin protein N-glycosylation, we examined the size of theglycosylated protein in the cell wall of KR mutants.A defect in the N-glycosyl structure of mannoprotein
Fig. 2 Killer eclipse assay forKR1 (rhk1-1), KR7 (rhk3-1),KR9 (rhk3-2), KR11 (rhk2-1),KR13 (rhk2-2), KR17 (rhk2-3),KR18 (rhk2-4), KR19 (rhk2-5),rhk1D, and wild strain. Eachstrain was assayed for sensitivityto three killer yeasts. The namesof the tested strains and killerstrains are shown on the left andtop respectively
Fig. 3 Restriction map and subclones of the RHK2 gene. The openreading frame (ORF) of the RHK2, deduced from the nucleotidesequence, is shown in the shaded box and the direction of translation isshown by an arrow. The nucleotide sequence of the ORF is identicalto that of YGL022w, which is located on chromosome VII betweenthe nucleotide coordinates 452400 and 454556. The complementingactivity of various fragments is shown on the right: +, ), HM-1-sensitive and HM-1-resistant respectively. Restriction enzymes:BamHI (B), SalI (S), XbaI (X)
179
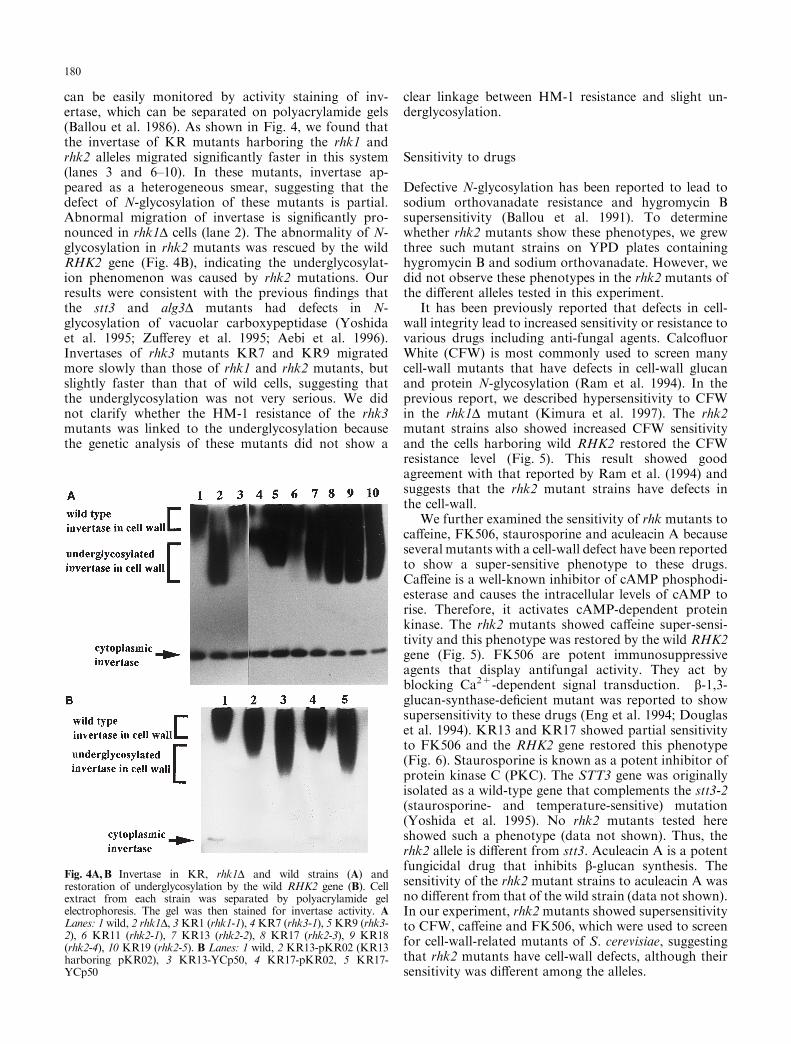
can be easily monitored by activity staining of inv-ertase, which can be separated on polyacrylamide gels(Ballou et al. 1986). As shown in Fig. 4, we found thatthe invertase of KR mutants harboring the rhk1 andrhk2 alleles migrated signi®cantly faster in this system(lanes 3 and 6±10). In these mutants, invertase ap-peared as a heterogeneous smear, suggesting that thedefect of N-glycosylation of these mutants is partial.Abnormal migration of invertase is signi®cantly pro-nounced in rhk1D cells (lane 2). The abnormality of N-glycosylation in rhk2 mutants was rescued by the wildRHK2 gene (Fig. 4B), indicating the underglycosylat-ion phenomenon was caused by rhk2 mutations. Ourresults were consistent with the previous ®ndings thatthe stt3 and alg3D mutants had defects in N-glycosylation of vacuolar carboxypeptidase (Yoshidaet al. 1995; Zu�erey et al. 1995; Aebi et al. 1996).Invertases of rhk3 mutants KR7 and KR9 migratedmore slowly than those of rhk1 and rhk2 mutants, butslightly faster than that of wild cells, suggesting thatthe underglycosylation was not very serious. We didnot clarify whether the HM-1 resistance of the rhk3mutants was linked to the underglycosylation becausethe genetic analysis of these mutants did not show a
clear linkage between HM-1 resistance and slight un-derglycosylation.
Sensitivity to drugs
Defective N-glycosylation has been reported to lead tosodium orthovanadate resistance and hygromycin Bsupersensitivity (Ballou et al. 1991). To determinewhether rhk2 mutants show these phenotypes, we grewthree such mutant strains on YPD plates containinghygromycin B and sodium orthovanadate. However, wedid not observe these phenotypes in the rhk2 mutants ofthe di�erent alleles tested in this experiment.
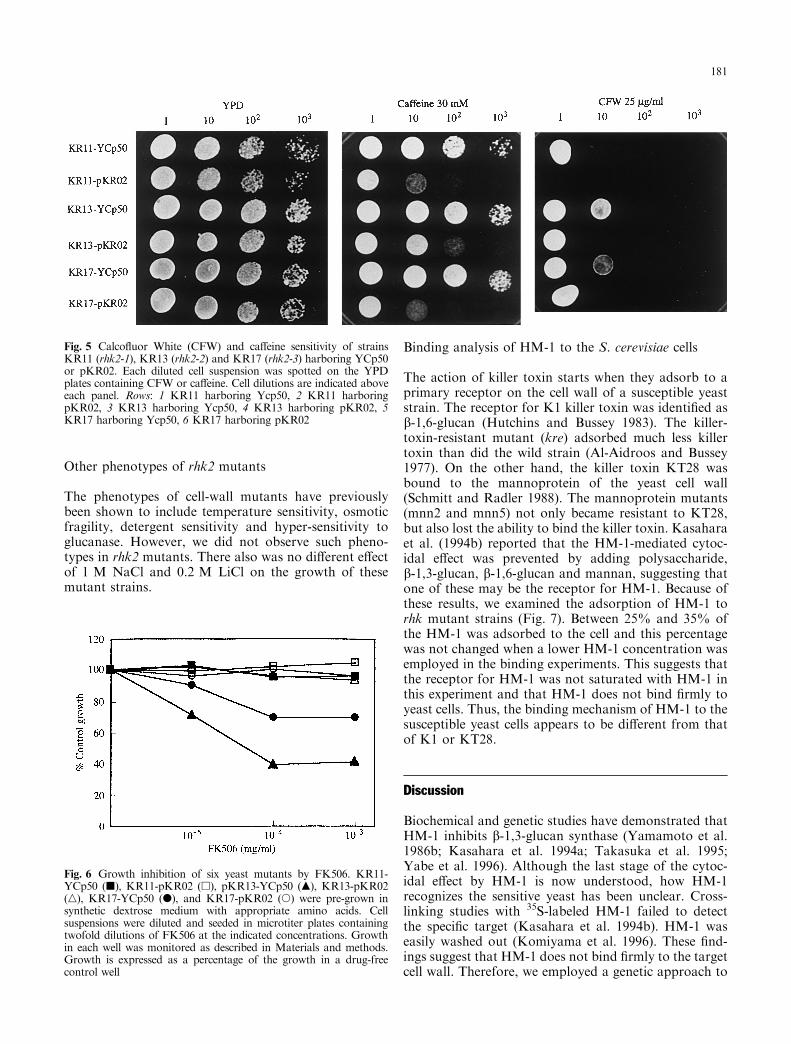
It has been previously reported that defects in cell-wall integrity lead to increased sensitivity or resistance tovarious drugs including anti-fungal agents. Calco¯uorWhite (CFW) is most commonly used to screen manycell-wall mutants that have defects in cell-wall glucanand protein N-glycosylation (Ram et al. 1994). In theprevious report, we described hypersensitivity to CFWin the rhk1D mutant (Kimura et al. 1997). The rhk2mutant strains also showed increased CFW sensitivityand the cells harboring wild RHK2 restored the CFWresistance level (Fig. 5). This result showed goodagreement with that reported by Ram et al. (1994) andsuggests that the rhk2 mutant strains have defects inthe cell-wall.
We further examined the sensitivity of rhk mutants toca�eine, FK506, staurosporine and aculeacin A becauseseveral mutants with a cell-wall defect have been reportedto show a super-sensitive phenotype to these drugs.Ca�eine is a well-known inhibitor of cAMP phosphodi-esterase and causes the intracellular levels of cAMP torise. Therefore, it activates cAMP-dependent proteinkinase. The rhk2 mutants showed ca�eine super-sensi-tivity and this phenotype was restored by the wild RHK2gene (Fig. 5). FK506 are potent immunosuppressiveagents that display antifungal activity. They act byblocking Ca2+-dependent signal transduction. b-1,3-glucan-synthase-de®cient mutant was reported to showsupersensitivity to these drugs (Eng et al. 1994; Douglaset al. 1994). KR13 and KR17 showed partial sensitivityto FK506 and the RHK2 gene restored this phenotype(Fig. 6). Staurosporine is known as a potent inhibitor ofprotein kinase C (PKC). The STT3 gene was originallyisolated as a wild-type gene that complements the stt3-2(staurosporine- and temperature-sensitive) mutation(Yoshida et al. 1995). No rhk2 mutants tested hereshowed such a phenotype (data not shown). Thus, therhk2 allele is di�erent from stt3. Aculeacin A is a potentfungicidal drug that inhibits b-glucan synthesis. Thesensitivity of the rhk2 mutant strains to aculeacin A wasno di�erent from that of the wild strain (data not shown).In our experiment, rhk2 mutants showed supersensitivityto CFW, ca�eine and FK506, which were used to screenfor cell-wall-related mutants of S. cerevisiae, suggestingthat rhk2 mutants have cell-wall defects, although theirsensitivity was di�erent among the alleles.
Fig. 4A,B Invertase in KR, rhk1D and wild strains (A) andrestoration of underglycosylation by the wild RHK2 gene (B). Cellextract from each strain was separated by polyacrylamide gelelectrophoresis. The gel was then stained for invertase activity. ALanes: 1 wild, 2 rhk1D, 3KR1 (rhk1-1), 4KR7 (rhk3-1), 5KR9 (rhk3-2), 6 KR11 (rhk2-1), 7 KR13 (rhk2-2), 8 KR17 (rhk2-3), 9 KR18(rhk2-4), 10 KR19 (rhk2-5). B Lanes: 1 wild, 2 KR13-pKR02 (KR13harboring pKR02), 3 KR13-YCp50, 4 KR17-pKR02, 5 KR17-YCp50
180
Other phenotypes of rhk2 mutants
The phenotypes of cell-wall mutants have previouslybeen shown to include temperature sensitivity, osmoticfragility, detergent sensitivity and hyper-sensitivity toglucanase. However, we did not observe such pheno-types in rhk2 mutants. There also was no di�erent e�ectof 1 M NaCl and 0.2 M LiCl on the growth of thesemutant strains.
Binding analysis of HM-1 to the S. cerevisiae cells
The action of killer toxin starts when they adsorb to aprimary receptor on the cell wall of a susceptible yeaststrain. The receptor for K1 killer toxin was identi®ed asb-1,6-glucan (Hutchins and Bussey 1983). The killer-toxin-resistant mutant (kre) adsorbed much less killertoxin than did the wild strain (Al-Aidroos and Bussey1977). On the other hand, the killer toxin KT28 wasbound to the mannoprotein of the yeast cell wall(Schmitt and Radler 1988). The mannoprotein mutants(mnn2 and mnn5) not only became resistant to KT28,but also lost the ability to bind the killer toxin. Kasaharaet al. (1994b) reported that the HM-1-mediated cytoc-idal e�ect was prevented by adding polysaccharide,b-1,3-glucan, b-1,6-glucan and mannan, suggesting thatone of these may be the receptor for HM-1. Because ofthese results, we examined the adsorption of HM-1 torhk mutant strains (Fig. 7). Between 25% and 35% ofthe HM-1 was adsorbed to the cell and this percentagewas not changed when a lower HM-1 concentration wasemployed in the binding experiments. This suggests thatthe receptor for HM-1 was not saturated with HM-1 inthis experiment and that HM-1 does not bind ®rmly toyeast cells. Thus, the binding mechanism of HM-1 to thesusceptible yeast cells appears to be di�erent from thatof K1 or KT28.
Discussion
Biochemical and genetic studies have demonstrated thatHM-1 inhibits b-1,3-glucan synthase (Yamamoto et al.1986b; Kasahara et al. 1994a; Takasuka et al. 1995;Yabe et al. 1996). Although the last stage of the cytoc-idal e�ect by HM-1 is now understood, how HM-1recognizes the sensitive yeast has been unclear. Cross-linking studies with 35S-labeled HM-1 failed to detectthe speci®c target (Kasahara et al. 1994b). HM-1 waseasily washed out (Komiyama et al. 1996). These ®nd-ings suggest that HM-1 does not bind ®rmly to the targetcell wall. Therefore, we employed a genetic approach to
Fig. 5 Calco¯uor White (CFW) and ca�eine sensitivity of strainsKR11 (rhk2-1), KR13 (rhk2-2) and KR17 (rhk2-3) harboring YCp50or pKR02. Each diluted cell suspension was spotted on the YPDplates containing CFW or ca�eine. Cell dilutions are indicated aboveeach panel. Rows: 1 KR11 harboring Ycp50, 2 KR11 harboringpKR02, 3 KR13 harboring Ycp50, 4 KR13 harboring pKR02, 5KR17 harboring Ycp50, 6 KR17 harboring pKR02
Fig. 6 Growth inhibition of six yeast mutants by FK506. KR11-YCp50 (j), KR11-pKR02 (h), pKR13-YCp50 (m), KR13-pKR02(n), KR17-YCp50 (d), and KR17-pKR02 (s) were pre-grown insynthetic dextrose medium with appropriate amino acids. Cellsuspensions were diluted and seeded in microtiter plates containingtwofold dilutions of FK506 at the indicated concentrations. Growthin each well was monitored as described in Materials and methods.Growth is expressed as a percentage of the growth in a drug-freecontrol well
181

determine the receptor structure for HM-1. The resultsindicated that two genes (RHK1/ALG3 encoding amannosyltransferase and RHK2/STT3 encoding asubunit of the oligosaccharyltransferase complex) areinvolved in HM-1 sensitivity, suggesting that proteinN-glycosylation is important for the cytocidal process ofHM-1.
In the protein N-glycosylation process, the coreoligosaccharide Glc3Man9GlcNAc2 is known to be as-sembled on the lipid carrier dolichol pyrophosphate(dol-PP), and then transferred to an asparagine residueof secretory proteins by the oligosaccharyltransferasecomplex (Fig. 8) (Orlean 1997). When the core
oligosaccharide is synthesized, two N-acetylglucosamineresidues and ®ve mannose residues are assembled in astepwise fashion on the cytoplasmic side of the endo-plasmic reticulum membrane. The resulting dolichol-linked-Man5GlcNAc2 (Man5GlcNAc2-PP-Dol) is thentranslocated to the lumenal side of the endoplasmicreticulum membrane, where four mannose residues andthree glucose residues are added. It has been sug-gested that Alg3p functions as a mannosyltransferasecatalyzing the addition of the sixth mannose residue toMan5GlcNAc2-PP-Dol. Deletion of ALG3 resulted inaccumulation of Man5GlcNAc2-PP-Dol and under-glycosylation of N-glycoproteins such as vacuolar car-boxypeptidase Y (Aebi et al. 1996). Stt3p is thought toregulate the oligosaccharyltransferase activity or modifythe substrate speci®city of the enzyme, and the stt3mutant also showed underglycosylation of carboxypep-tidase Y (Yoshida et al. 1995; Zu�erey et al. 1995; Spiriget al. 1997). We also indicated that rhk2 mutants hadunderglycosylated external invertase (Fig. 4). Zu�ereyet al. (1995) reported that the double mutant alg3 stt3shows pronounced underglycosylation of secretoryproteins and temperature-sensitive growth. They usedthis phenotype to clone ALG3 and STT3. It is interestingthat the same genes were cloned in our screen and theseresults suggest a strong functional correlation betweenAlg3p and Stt3p in N-glycosylation.
The results presented here suggest the involvement ofone or more mannoproteins of the cell wall in the pro-cess of HM-1 action. We consider the following twomechanisms that might explain the HM-1 resistance ofrhk mutants. (1) The reduced N-glycosylation inmannoproteins could alter the cell-wall structure, in-tegrity, hydrophobicity or charge and cause a pertur-bation of HM-1 adsorption or permeability. (2) Areceptor for HM-1 may be a mannoprotein or its car-bohydrate moiety, and alteration of these structurescould cause the cells to become insensitive to HM-1. The®rst model is supported by the result that rhk2 mutantsshowed super-sensitivity to several drugs (Figs. 5, 6),phenotypes that often indicate cell-wall defects (Ramet al. 1994). Mannoproteins, which consist of as much as50% carbohydrate, form a complex network betweenglucan and chitin. It is therefore possible that HM-1resistance may be due to secondary e�ects of cell-walldefects. The second model raises the possibility that aspeci®c receptor for HM-1, possibly a glycosyl chain ofwall mannoprotein or a glycoprotein in the plasmamembrane, exists in the cell-wall. This model is sup-ported by the fact that spheroplasts of rhk mutants re-tained HM-1 resistance (Kimura et al. 1997). It ispossible that the b-1,3-glucan synthase itself may be areceptor. Although glycosylation of this enzyme has notyet been examined, there are six possible N-glycosyl siteson its deduced amino acid sequence (Mazur et al. 1995).However, our preliminary result appeared to eliminatethis possibility since glucan synthase in crude mem-branes extracted from rhk1, rhk2 and wild cells had thesame speci®c activity and were inhibited by HM-1 to the
Fig. 7 Binding of HM-1 to KR, rhk1D and wild strains. Residualkiller activity was measured after HM-1 had been mixed with eachtype of cells as described in Materials and methods. Each residualkiller activity was the average of the results of three independentexperiments. Bar standard deviation of each residual killler activity
Fig. 8 Schematic representation of an oligosaccharide. Shadedmannose residues indicate those present in an alg3D strain. The``black'' mannose residue and the three glucose residues are notpresent on the oligosaccharide of N-glycoprotein since these residuesare removed before transfer to Asn on the protein (Orlean 1997). Theouter mannose chain is added after transfer to a protein by theMNN9product
182

same extent. Furthermore, it has been reported that notonly mannan but also b-1,3-glucan and b-1,6-glucanwere able to mask the action of HM-1 (Kasahara et al.1994b), suggesting that the mannoprotein is not the onlyreceptor for HM-1. Given the current observations, wefavor the ®rst model, which proposes that the change ofcell-wall structure causes the HM-1 resistance to thesensitive cells. However, we can not exclude the secondmodel in which HM-1 recognizes the precise structure ofan oligosaccharide, because the mnn9 mutant was sen-sitive to HM-1 in our preliminary experiment. HM-1binding experiments suggested that the adsorptionmechanism of HM-1 to the cell wall is di�erent fromthat of K1 and K. lactis killer toxin, and other methodsfor evaluating the binding of HM-1 to the target cell wallshould be developed to determine the mechanism ofHM-1 killing.
In this study, we have proposed two models gener-ating the HM-1-resistant phenotype. Determiningwhether one of these models is correct requires furtherexperiments involving biochemical analysis of HM-1-resistant mutants. Knowledge of the killing process ofHM-1 would enable us to understand the complexstructure of the fungal cell wall and to develop novelanti-mycotic agents.
Acknowledgement This investigation was ®nancially supported byBio-Oriented Technology Research Advancement Institution(BRAIN), Japan.
References
Aebi M, Gassenhuber J, Domdey H, Heesen S te (1996) Cloningand characterization of the ALG3 gene of Saccharomycescerevisiae. Glycobiology 6: 439±444
Al-Aidroos K, Bussey H (1977) Chromosomal mutants of Sac-charomyces cerevisiae a�ecting the cell wall binding site forkiller factor. Can J Microbiol 24: 228±237
Antuch W, Guntert P, Wuthrich K (1996) Ancestral bc-crystallinprecursor structure in a yeast killer toxin. Nat Struct Biol 3:662±665
Ballou L, Gopal P, Krummel B. Tammi M, Ballou CE (1986) Amutation that prevents glucosylation of the lipid-linkedoligosaccharide precursor leads to underglycosylation of se-creted yeast invertase. Proc Natl Acad Sci USA 83: 3081±3085
Ballou L, Hitzeman RA, Lewis MS, Ballou CE (1991) Vanadate-resistant yeast mutants are defective in protein glycosylation.Proc Natl Acad Sci USA 88: 3209±3212
Boone C, Sommer SS, Hensel A, Bussey H (1990) Yeast KRE genesprovide evidence for a pathway of cell wall b-glucan assembly.J Cell Biol 110: 1833±1843
Brown JL, Bussey H (1993) The yeast KRE9 gene encodes an O-glycoprotein involved in cell surface b-glucan assembly. MolCell Biol 13: 6346±6356
Brown JL, Kossaczka Z, Jiag B, Bussey H (1993) A mutationalanalysis of killer toxin resistance in Saccharomyces cerevisiaeidenti®es new genes involved in cell wall (1,6)-b-glucan syn-thesis. Genetics 133: 837±849
Bussey H (1991) K1 killer toxin, a pore-forming protein from yeast.Mol Microbiol 5: 2339±2343
Douglas CM, Marrinan JA, Kurtz MB (1994) A Saccharomycescerevisiae mutant with echinocandin-resistant 1,3-b-D-glucansynthase. J Bacteriol 176: 5686±5696
Eng W, Faucette L, McLaughlin MM, Ca�erkey R, Koltin Y,Morris RA, Young PR, Johnson RK, Livi JP (1994) The yeastFKS1 gene encodes a novel membrane protein, mutations inwhich confer FK506 and cyclosporin A hypersensitivity andcalcineurin-dependent growth. Gene 151: 61±71
Hill K, Boone C, Goebl M, Puccia R, Sdicu AM, Bussey H (1992)Yeast KRE2 de®nes a new gene family encoding probable se-cretory proteins, and is required for the correct N-glycosylationof proteins. Genetics 130: 273±283
Hong Z, Nathaniel PM, Brown H, Tran LE, Shaw KJ, Hare RS,DiDomenico B (1994) Cloning and characterization of KNR4, ayeast gene involved in (1,3)-b-glucan synthesis. Mol Cell Biol14: 1017±1025
Hutchins K, Bussey H (1983) Receptor for yeast killer toxin: In-volvement of 1,6-b-D-glucan. J Bacteriol 154: 161±169
Ito H, Fukuda Y, Murata K, Kimura A (1983) Transformation ofintact yeast cells treated with alkali cations. J Bacteriol 153:163±168
Kasahara S, Yamada H, Mio T, Shirotori Y, Miyamoto C, YabeT, Nakajima T, Ichishima E, Furuichi Y (1994a) Cloning of theSaccharomyces cerevisiae gene whose overexpression overcomesthe e�ects of HM-1 killer toxin, which inhibits b-glucan syn-thesis. J Bacteriol. 176: 1488±1499
Kasahara S, Inoue BS, Mio T, Yamada T, Nakajima T, IchishimaE, Furuichi Y, Yamada H (1994b) Involvement of cell wall b-glucan in the action of HM-1 toxin. FEBS Lett 348: 27±32
Kawamoto S, Nomura M, Ohno T (1992) Cloning and charac-terization of SKT5, a Saccharomyces cerevisiae gene that a�ectsprotoplast regeneration and resistance to killer toxin of Kluy-veromyces lactis. J Ferment Bioeg 74: 199±208
Kimura T, Kitamoto N, Matsuoka K, Nakamura K, Iimura Y,Kito Y (1993) Isolation and nucleotide sequences of the genesencoding killer toxins from Hansenula mrakii and H. saturnus.Gene 137: 265±270
Kimura T, Kitamoto N, Ohta Y, Kito Y, Iimura Y (1995) Struc-tural relationships among killer toxins secreted from the killerstrains of the genus Williopsis. J Ferment Bioeng 80: 85±87
Kimura T, Kitamoto N, Kito Y, Iimura Y, Shirai T, Komiyama T,Furuichi Y, Sakka K, Ohmiya K (1997) A novel yeast gene,RHK1, is involved in the synthesis of the cell wall receptor forthe HM-1 killer toxin that inhibits b-1,3-glucan synthesis. MolGen Genet 254: 139±147
Kishida M, Tokunaga, M, Katayose Y, Yajima H, Kawamura-Watanabe A, Hishinuma F (1996) Isolation and genetic char-acterization of pGKL killer-sensitive mutants (iki) from Sac-charomyces cerevisiae. Biosci Biotechnol Biochem 60: 798±801
Komiyama T, Ohta T, Urakami H, Shirotori Y, Takasuka T,Satoh M, Watanabe T, Furuichi Y (1996) Pore formation onproliferating yeast Saccharomyces cerevisiae cell buds by HM-1killer toxin. J Biochem (Tokyo) 119: 731±736
Maeden P, Hill K, Wagner J, Slipetz D, Sommer SS, Bussey H(1990) The yeast KRE5 gene encodes a probable endoplasmicreticulum protein required for (1,6)-b-glucan synthesis andnormal cell growth. Mol Cell Biol 10: 3013±3019
Mazur P, Morin N, Baginsky W, EL-Sherbeini M, Clemas JA,Neilsen JB, Foor F (1995) Di�erential expression and functionof two homologous subunits of yeast 1,3-b-D-glucan synthase.Mol Cell Biol 15: 5671±5681
Nomoto H, Kitano K, Shimazaki K, Kodama K, Hara S (1984)Distribution of killer yeasts in the genus Hansenula. Agric BiolChem 48: 807±809
Orlean P (1997) Biogenesis of yeast wall and surface components.In: Pringle JR, Broach JR, Jones EW (eds) Molecular andcellular biology of the yeast Saccharomyces. Vol III. Cell cycleand cell biology. Cold Spring Harbor Laboratory Cold SpringHarbor, NY, pp 229±362
Ram AFJ, Wolters A, Hoopen RT, Klis RM (1994) A new ap-proach for isolating cell wall mutants in Saccharomyces cere-visiae by screening for hypersensitivity to Calco¯uor White.Yeast 10: 1019±1030
Rommer T, Bussey H (1991) Yeast b-glucan synthesis: KRE6 en-codes a predicted type II membrane protein required for glucan
183

synthesis in vivo and for glucan synthase activity in vitro. Proc.Natl Acad Sci USA 88: 11295±11299
Rose M, Winston F, Hieter P (1990) Methods in yeast genetics: alaboratory source manual Cold Spring Harbor Laboratory,Cold Spring Harbor, NY
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: alaboratory source manual Cold Spring Harbor Laboratory,Cold Spring Harbor, NY
Schmitt M, Radler F (1988) Molecular structure for killer toxinKT28 in Saccharomyces cerevisiae. J Bacteriol 170: 2192±2196
Spirig U, Glavas M, Bodmer D, Reiss G, Burda P, Lippuner V, deHeesen S, Aebi M (1997) The STT3 protein is a component ofthe yeast oligosaccharyltransferase complex. Mol Gen Genet256: 628±637
Stark MJ, Boyd A, Mileham J (1990) The plasmid-encoded killersystem of Kluyveromyces lactis: a review. Yeast 6: 1±29
Takasuka T, Komiyama T, Furuichi Y, Watanabe T (1995) Cellwall synthesis speci®c cytocidal e�ect of Hansenula mrakiitoxin-1 on Saccharomyces cerevisiae. Cell Mol Biol Res 41: 575±581
Takita MA, Castilho-Valanicius B (1993) Absence of cell wallchitin in Saccharomyces cerevisiae leads to resistance to Kluy-veromyces lactis killer toxin. Yeast 9: 589±598
Yabe T, Yamada-Okabe T, Kasahara S, Furuichi Y, Nakajima T,Ichishima E, Arisawa M, Yamada-Okabe H (1996) HKR1 en-codes a cell surface protein that regulates both cell wall b-glu-
can synthesis and budding pattern in the yeast Saccharomycescerevisiae. J Bacteriol 178: 477±483
Yajima H, Tokunaga M, Nakayama-Murayama A, Hishinuma F(1997) Characterization of IKI1 and IKI3 genes conferringpGLKL killer sensitivity on Saccharomyces cerevisiae. BiosciBiotechnol Biochem 61: 704±709
Yamamoto T, Imai M, Tachibana K, Mayumi M (1986a) Appli-cation of monoclonal antibodies to the isolation and charac-terization of a killer toxin secreted by Hansenula mrakii. FEBSLett 195: 253±257
Yamamoto T, Hiratani T, Hirata H, Imai M, Yamaguchi H(1986b) Killer toxin from Hansenula mrakii selectively in-hibits cell wall synthesis in a sensitive yeast. FEBS Lett 197:50±54
Yamamoto T, Uchida K, Hiratani T, Miyazaki T, Yagiu J,Yamaguchi H (1988) In vitro activity of the killer toxin fromyeast Hansenula mrakii against yeasts and molds. J Antibiot(Tokyo) 41: 398±403
Yoshida S, Ohya Y, Nakano A, Anraku Y (1995) STT3, a novelessential gene related to the PKC1/STT1 protein kinase path-way, is involved in protein glycosylation in yeast. Gene 164:167±172
Zu�erey R, Knauer R, Burda P, Stagljar I, Heesen S te, Lehle L,Aebi M (1995) STT3, a highly conserved protein required foryeast oligosaccharyl transferase activity in vivo. EMBO J 14:4949±4960
184





























