Embed Size (px)
Citation preview

1
APPLICATION FOR A RESEARCH & DEVELOPMENT PROJECT PROPOSAL UNDER CATEGORY II OF CEIB SCHEME
(Broad subject area: Human pathogen and Structural Biology)
SUBMITTED TO
Department of Biotechnology (DBT) Government of India
Molecular dissection of and inhibitor discovery against motors
associated with protein translation in malaria parasites
Amit Sharma
Head, Structural and Computational Biology Group
International Centre for Genetic Engineering and Biotechnology Aruna Asaf Ali Marg, New Delhi - 110067
July 2012

2
PROFORMA – I
PROFORMA FOR SUBMISSION OF PROJECT PROPOSALS ON RESEARCH AND
DEVELOPMENT, PROGRAMME SUPPORT
(To be filled by the applicant)
PART I: GENERAL INFORMATION
1. Name of the Institute/University/Organization submitting the Project Proposal:
International Centre for Genetic Engineering and Biotechnology (ICGEB) 2. State: New Delhi 3. Status of the Institute: International
4. Name and designation of the Executive Authority of the Institute/University forwarding the application: Dr. V. S. Chauhan, Director, ICGEB, New Delhi
5. Project Title:
Molecular dissection of and inhibitor discovery against motors
associated with protein translation in malaria parasites
6. Category of the Project (Please tick) : R&D/Programme Support (Category II: OSRP) 7. Specific Area (Please see Annexure - II): Basic Research
Subject Area/Key Words: Human pathogen and Structural Biology
8. Duration : 5 Years
9. Total Cost (Rs.) 573.04 lakhs
10. Is the project Single Institutional or Multiple-Institutional (S/M) ? : S
11. If the project is multi-institutional, please furnish the following :
Name of Project Coordinator : NA

3
12. Scope of application indicating anticipated product and processes
Structural studies on proteins from infectious organisms provide bases for rational drug
design. Malaria is a major infectious disease worldwide. Components of the molecular machinery
that are responsible for functioning of the parasite are therefore target of both vaccine and drug
discovery. The non-ribosomal components of the protein translation apparatus are largely
unexplored as drug targets against Plasmodium falciparum (Pf) malaria. Our group has established
a program on in-depth dissection of molecular motors crucial for protein translation in malaria
parasites. Over the past decade, several of our publications on this theme are beginning to highlight
pivotal avenues which need to be explored in greater detail over a sustained period of engagement
(5-10 years). We have already made considerable progress in generating a wealth of reagents,
including target-specific antibodies and parasite protein expression systems required for this thrust
which will entail multi-disciplinary techniques of molecular biology. Our medium to longer terms
goals are twinned: (1) to understand the unique biology of malaria parasite protein translation
factors, and (2) to exploit this knowledge for development of specific inhibitors that may lead to
novel anti-malarials. For the latter objective, we have established links with pharmaceutical
partners. We plan to develop and eventually patent a whole battery of inhibitors that will target
translation machinery motors. The investigative tools which will lead to discovery of novel
inhibitors therefore will include - extensive biochemical characterization, confocal-based protein
localization, definition of protein-protein interactions partners, three-dimensional structure
determination, and structure-based inhibitor screening using parasite viability assays.

4
13. Project Summary (Not to exceed one page. Please use separate sheet).
Over the 5 years, we have published extensively on the unique and exploitable attributes of
protein translation motors in malaria parasites. This body of work has emanated from a vibrant on-
going program on structure-function studies on Pf aminoacyl-tRNA synthetases (aaRSs). We
already possess protein expression clones of all target proteins in this proposal, and indeed we have
also solved crystal structures of several aaRSs already (we remain the only laboratory in the world
with this information). Therefore, a strong foundation for our ideas has been laid through rigorous
planning and execution. We need support now to continue our molecular biology studies, and to
experimentally pursue protein targets ripe for inhibitor discovery. In addition to our on-going
investigations on malaria parasite aaRSs, we have recently initiated studies on 4 crucial translation
factors which are at the centre of this research proposal: (1) CCA-adding enzyme: performs addition
of CCA to 3’ end of tRNAs before they can be used for aminoacylation, (2) Leucyl/phenylalanine
transferase: regulates degradation of intracellular proteins using charged tRNAs as co-factors, (3)
Pyrophosphatase: performs hydrolysis of pyrophosphates generated by biochemical processes and
(4) P43: is involved in complex formation with one or several aminoacyl tRNA synthetases. Our
studies will involve both extensive basic biological investigations on these target proteins and a
structure-based thrust towards inhibitor discovery. Through these analyses we hope, to highlight
pivotal biological attributes of target proteins, and alongside list high scoring small molecule
compounds with potential to block active sites of these 4 target proteins. These compounds will be
evaluated for their ability to inhibit parasite growth in vivo, and will initiate an iterative process
where constant cross-talk with medicinal chemists from pharmaceutical sector may allow
convergence to lead-like compounds against malaria.
Specific objectives (to be pursued concurrently):
A. Production of recombinant target proteins for biochemistry and structural biology.
B. Three dimensional structure determination of targets.
C. Protein localization, tracking of post-translational modifications and interactome studies.
D. Structure-based inhibitor discovery and parasite viability assays.

5
PART II: PARTICULARS OF INVESTIGATORS
(One or more co-investigators are preferred in every project. Inclusion of co-investigator(s) is
mandatory for investigators retiring before completion of the project) Principal Investigator:
14. Name: Dr. Amit Sharma
Date of Birth: 12th
April 1968 Sex (M/F): M
Designation: Scientist and Group Leader
Department: Structural and Computational Biology Group
Institute/University: International Centre for genetic Engineering and Biotechnology
Address: Address: Aruna Asaf Ali Marg, New Delhi
PIN: 110067
Telephone: 26741731 Fax: 26741731 E-mail: [email protected]
Number of research projects being handled at present: Three

6
PART III : TECHNICAL DETAILS OF PROJECT
(Under the following heads on separate sheets)
16. Introduction (not to exceed 2 pages or 1000 words)
Structural studies on proteins from infectious organisms provide bases for rational drug
design. Our group has established a program on in-depth dissection of protein motors crucial for
protein translation in malaria parasites. Several of our publications on this theme are beginning to
highlight pivotal avenues which need to be explored in greater detail over a sustained period of
engagement (5-10 years). We have already made considerable progress in generating a wealth of
reagents, including antibodies and expression vectors required for this multi-disciplinary thrust. Our
medium to longer terms goals are twinned: (1) to understand the unique biology of malaria parasite
protein translation factors, and (2) to exploit this knowledge for development of specific inhibitors
that may lead to novel anti-malarials. For the latter objective, we have established links with
pharmaceutical partners like Jubiliant Biosciences. We plan to patent and then develop a whole
battery of inhibitors that will target translation machinery motors. The investigative tools which will
lead to discovery of novel inhibitors therefore will include - extensive biochemical characterization,
confocal-based protein localization, definition of protein-protein interactions partners, three-
dimensional structure determination, and structure-based inhibitor screening using parasite viability
assays.
We have published extensively on the unique and exploitable attributes of protein translation
motors in malaria parasites. This has emanated from a vibrant on-going program on structure-
function studies of Pf aminoacyl-tRNA synthetases (aaRSs). We already molecular clones of all
relevant target proteins in this proposal, and in each case soluble, recombinant proteins have already
been obtained – based on preliminary pilot scale experiments. Further, we have also solved crystal
structures of several malarial aaRSs already, and we remain the only laboratory in the world with
this information. Therefore, a strong foundation for our ideas has been laid through rigorous
planning and execution. We need support now to continue our molecular biology studies, and to
pursue parasite protein targets that are ripe for inhibitor discovery. Our structure-based approach to
inhibitor discovery will involve, as the first step, an in silico approach. Through these analyses, we
will select high scoring compounds that will be evaluated for their ability to inhibit parasite growth
in vivo. This will initiate an iterative process where constant cross-talk with medicinal chemists
from pharmaceutical sector will allow convergence on to lead-like compounds against malaria.
Eventual integration with medicinal and synthetic chemistry expertise (using our links with
pharmaceutical partners) will propel us towards our translational objectives.

7
16.1 Origin of the proposal
Malaria remains a worldwide human health problem despite global efforts to fight it.
Detailed structure-function studies of crucial malaria parasite proteins are critical in understanding
basic biology of the parasite. Aminoacyl-tRNA synthetases (aaRSs) and associated proteins (which
form part of this proposal) are central motors for protein translation in any cell. The four target
translation-associated proteins: CCA-adding enzyme (CCAse), leucyl/phenylalanine tRNA
transferase (LFT), pyrophosphatase (PPase) and P43 play crucial roles in protein translation.
Further, the target proteins either show very poor or no sequence homology with host proteins –
thereby partly validating them for pathogen-specific inhibitor development. Specifically, CCAse
performs the task of adding CCA to 3' ends of tRNAs so that these are competent for
aminoacylation by aaRSs. Intriguingly, only 6 out of 72 tRNAs encoded in P. falciparum genome
contain CCAs – in other words more than 90% of the transcribed tRNA pool requires the CCAse
activity for tRNA maturation. Leucyl/phenylalanyl-tRNA-protein transferase (LFT) regulates half-
lives of proteins in vivo using the N-end rule pathway. LFT initiates attachment of either leucine or
a phenylalanine from a pool of charged corresponding tRNAs to protein substrates which contains
N-terminal arginine or lysine. This marks the product proteins for degradation. Once again, by
extrapolation from other systems, we expect LFT to be an essential enzyme for normal protein
metabolism processes in malaria parasites. Soluble version of pyrophosphatase (PPase) catalyzes
hydrolysis of pyrophosphate to phosphate, and is essential for cycling of ATP for energy
generation. The essentiality of PPase in other biological systems has been established, and we
expect the malarial PPase to be another valuable target for inhibitor development. P43 is an
auxiliary factor associated with multi-synthetase complex (MSC), which in humans is composed of
nine different aaRS (leucyl-, lysyl-, prolyl-, isoleucyl-, methionyl-, glutamyl-, glutaminyl-, arginyl-,
and aspartyl-tRNA synthetases) enzymes and three non-enzyme factors, including P18, P38 and
P43. The existence and relevance of MSC in malaria parasites has not been addressed as yet, and
we expect P43 to be a very important aaRS-associated protein factor worthy of being targeted by
small molecule inhibitors. The above 4 target proteins therefore form part of the present proposal
and remain the key targets we wish to focus on. Detailed descriptions of the target proteins are
given later in the proposal.
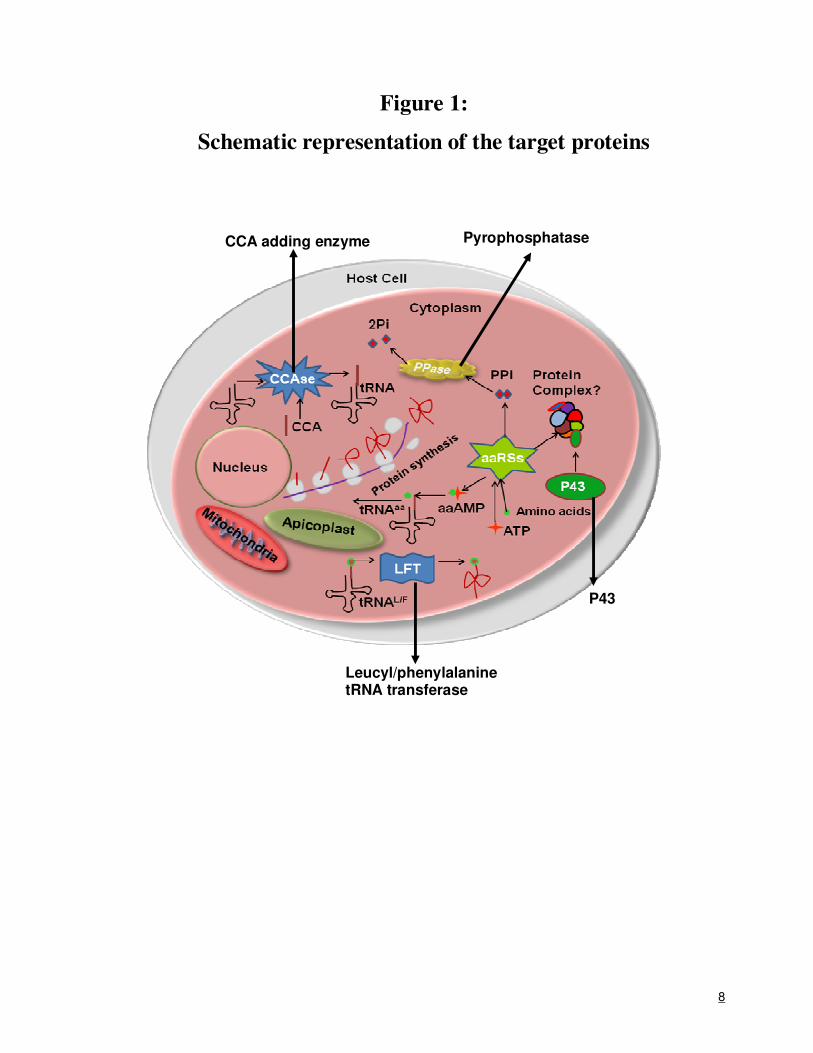
8
Figure 1:
Schematic representation of the target proteins
CCA adding enzyme Pyrophosphatase
Leucyl/phenylalanine tRNA transferase
P43
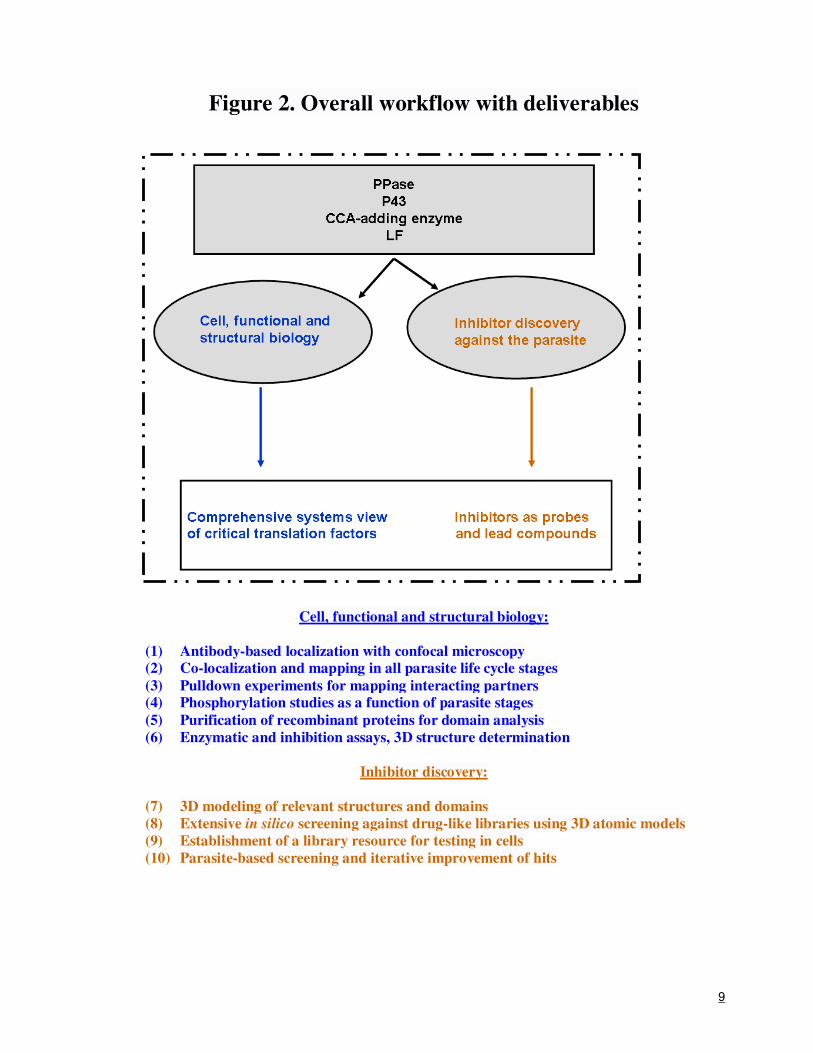
9
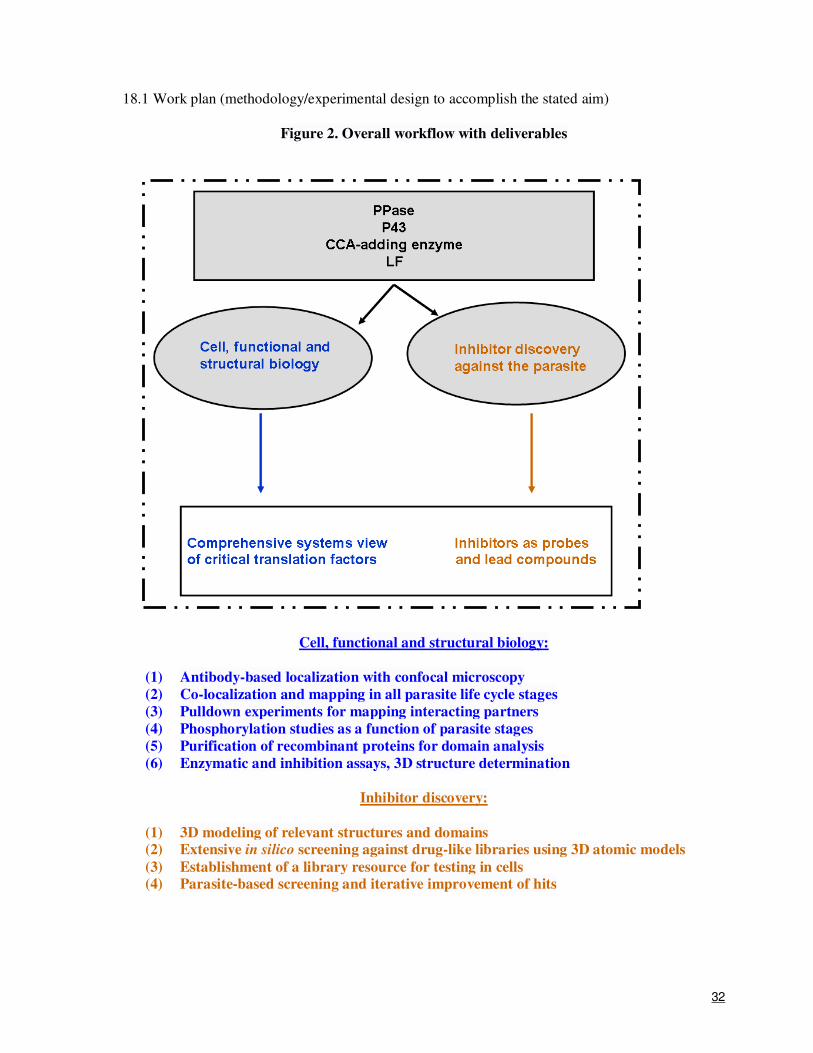
Figure 2. Overall workflow with deliverables
Cell, functional and structural biology:
(1) Antibody-based localization with confocal microscopy
(2) Co-localization and mapping in all parasite life cycle stages
(3) Pulldown experiments for mapping interacting partners
(4) Phosphorylation studies as a function of parasite stages
(5) Purification of recombinant proteins for domain analysis
(6) Enzymatic and inhibition assays, 3D structure determination
Inhibitor discovery:
(7) 3D modeling of relevant structures and domains
(8) Extensive in silico screening against drug-like libraries using 3D atomic models
(9) Establishment of a library resource for testing in cells
(10) Parasite-based screening and iterative improvement of hits

10
Presently, no significant biological or any structural information is available on these 4
target proteins in context of malaria parasite biology. This therefore provides a platform for launch
of in-depth, comprehensive structure-function investigations on these targets over an extended
period of research time. The 4 targets in this proposal, along with Pf aaRSs (already on-going and
funded by DBT), cover important non-ribosomal translation proteins in malaria parasites. We
therefore expect our proposed efforts to have a deep impact in the field of protein translation in
pathogens. We have already initiated the proposed studies in the expectation of attracting research
funding. This jump start has provided us with a toolkit which contains molecular clones, expression
profiles, purified proteins and most other preliminary reagents (like enzymatic assay protocols) for
all 4 target proteins proposed in this work. We are therefore at a stage where funding support will
directly enhance the pace of research, and we expect to fulfill most of the proposed aims. Our
objectives are to enlighten the field of malaria parasite biology with a focused thrust at exploiting
the protein translation machinery motors for discovery of novel anti-malarials.
16.2 (a) Rationale of the study supported by cited literature
Malaria control requires constant discovery of new anti-malarials due to the proclivity of
malaria parasites to develop drug resistance. In this on-going battle, new targets for anti-malarial
development need to be continually identified and validated. Quinine is an age old recipe for
malaria. Chloroquine was introduced in the late 1940s and used on a massive scale for malaria
treatment and prevention (AlKadi, 2007). Mefloquine and drug combinations of sulfadoxine–
pyrimethamine (Fansidar) were introduced in 1970s after the emergence of resistance to
chloroquine (Trenholme et al., 1975). Primaquine is currently the only approved therapy for the
treatment of P. vivax hypnozoite liver stages (Wells et al., 2010). Since the emergence of resistance
to almost all quinolone and antifolate drugs in P. falciparum malaria, artemisinin and its derivatives
have been used (Eastman and Fidock, 2009). Components of protein synthesis machinery represent
very attractive targets for development of new anti-infectives (Jackson et al., 2011). The antibiotic
compound doxycycline is a tetracycline compound derived from oxytetracycline that inhibits the
process of protein synthesis by binding to the 30S ribosomal subunit thus preventing the 50s and
30s units from interacting. Doxycycline is used in combination with quinine where the chloroquine
resistance exists (Eastman and Fidock, 2009).

11
Several effective anti-malarial drugs are currently available but the problem as ever is of
emerging drug resistance in the parasites (Fidock et al., 2005, Eastman and Fidock, 2009).
Resistance has been documented for plethora of front-line malaria drugs including chloroquine and
artemisinin (Hyde, 2007; Chavchich et al., 2010). Indeed, drug development in malaria using
modern molecular biology tools is heavily reliant on best possible ways to avoid the rather
inescapable, inevitable mutations which occur in parasites. Therefore, a deep molecular
understanding of drug resistance at the level of interacting crucial proteins is clearly essential – both
from a basic mechanistic perspective and also from view of providing insights which could propel
newer generation of anti-malarials. In this context, it is noteworthy that the PIs laboratory is at the
forefront of investigating and exploring a family of malaria parasite proteins (aaRSs and the 4
proteins in this proposal) which in principle can serve as leading new candidates for drug
development. Therefore, focusing on development of inhibitors which target non-ribosomal protein
translation components in malaria parasites may be rewarding.
The malaria parasite (like other apicomplexans) has three foci for ribosome-driven
translation: cytoplasm, apicoplast and the mitochondria (see Figure 1). Therefore, protein-specific
inhibitors against the target proteins may abrogate normal protein metabolism in all three parasite
compartments – thereby severely affecting parasite growth. We plan a comprehensive assault on
delineating and exploring the potential of 4 target proteins for development of small molecule
inhibitors. CCA-adding enzymes (CCAses) are polymerases synthesize the CCA at tRNA 3′-ends
(Pan et al., 2010; Toh et al., 2008, 2009; Li et al., 2002; Augustin et al., 2003), and the malarial
version seems to be significantly different from its human counterpart in terms of domain structures
and sequence divergence. Leucyl/phenylalanyl-tRNA-protein transferase (LFT) regulates half-lives
of proteins in vivo using the N-end rule pathway (Varshavsky, 1996). LFT initiates attachment of
either leucine or a phenylalanine from a pool of charged corresponding tRNAs to protein substrates
which contains N-terminal arginine or lysine. This marks the product proteins for degradation.
Inorganic pyrophosphatases (PPase) exist in oligomeric states and require conformational flexibility
for their essential role in converting inorganic pyrophosphate into phosphate (Kankare et al., 1996;
Sivula et al., 1991; Oksanen et al., 2007). P43 is an auxiliary factor associated with multi-synthetase
complex (MSC), which in humans is composed of nine different aaRS (leucyl-, lysyl-, prolyl-,
isoleucyl-, methionyl-, glutamyl-, glutaminyl-, arginyl-, and aspartyl-tRNA synthetases) enzymes
and three non-enzyme factors, including P18, P38 and P43 (Quevillon et al., 1999; Robinson et al.,
2000). The essential nature of the 4 target proteins and a general paucity of biological information
on these in pathogen context together provide a very strong rationale for this research proposal.

12
(b) Hypothesis and (c) key questions
Based on published evidence and our initial experimental data, we hypothesize the following:
1) CCAse activity must be present in the parasite cytoplasm and its apicoplast – possibly by virtue of dual localization. The mechanistic basis of dual localization and its molecular mechanism are presently unaddressed.
2) Inhibition of CCAse should have a drastic effect on parasite growth as this single enzyme is
an absolute requirement for addition of CCA to more than 90% of all tRNAs in the parasite. The subdomain structure of malarial CCAse suggests crucial mechanistic differences with its human counterpart – again an unexplored area.
3) How does LFT function for the apicoplast proteins if it is only present in the parasite
cytoplasm (as suggested by lack of targeting signals)? Does LFT localize in multiple compartments without any obvious signal sequence motifs?
4) Inhibition of LFT must also be detrimental to the parasite as protein degradation processes
will be subverted. This remains untested.
5) What are the LFT-associated components of the protein degradation machinery in Pf? 6) Is the malarial LFT unique in its mechanism given its phylogenetic uniqueness? 7) Malarial PPase contains an N-terminal extension which seems to be required for its enzyme
activity (based on our unpublished data). Therefore, the mechanism of action of malarial PPase should be subtly or drastically different from that of humans PPase. This remains unexplored.
8) P43 may or may not form part of the multi-synthetase complex (MSC) in malaria parasites.
The central question here is whether apicomplexans like Plasmodium contain an MSC?

13
16.5 Current status of research and development in the subject (both international and national status)
Genome sequencing projects have provided the field of structural biology with new
opportunities and challenges to broaden our understanding of the molecular basis of life. Among
protozoan parasites, the phylum of Apicomplexa includes organisms that cause malaria,
toxoplasmosis and cryptosporidiosis. The field of structural parasitology faces different challenges
since parasite proteins of variable evolutionary backgrounds present unusual problems. Parasite
proteins, particularly those from Plasmodium genomes, have proven to be more difficult than most
to express recombinantly in heterologous expression systems. The complicating factors include
AT-richness of Pf genome and prevalence of low complexity insertions. Structure genomics
consortiums (SGCs), along with other laboratories (like ours) have overcome challenges for some
parasitic proteins and have solved a number of 3D structures to date. A total of 370 entries (135
from SGCs and 235 from various labs) are available for Plasmodium falciparum. However, the total
numbers remain low compared to other species (Pf proteome has ~5300 proteins). Structural
information on proteins from other human parasites is also scarce. Thus, structural parasitology
remains a largely unexplored field where we feel that India can make a valuable contribution.
In the modern day competitive research environment, research on malaria is well supported,
both in India and abroad. A plethora of high quality research laboratories exist worldwide, with
varying foci of research. However, there are still very few international (or national) laboratories
whose forte is structural parasitology, and indeed the PIs lab is one that is well established in this
field as evidenced by publications on the subject. A unique aspect of this proposal is the focus on
protein translation machinery in malaria parasites – in this subfield very few national and
international investigators have active research programs. We therefore feel that we are highly
competitive in the identified microcosm, and hope that continued support from DBT will enable
realization of our strategic objectives.
16.6 The relevance and expected outcome of the proposed study
Malaria remains a major disease in India, and although it is responsible for considerably
more deaths in Africa, it still extracts a heavy price in India due to malaria-associated morbidity.
Therefore, we and other malarialogists in India have a continuing commitment to understanding and
controlling this disease. The field of protein translation research is also strong in India, with several
major international investigators contributing high quality work from CDRI, CCMB, IISc and other

14
institutes. Indeed, we have already witnessed crystallization of parallel lines of investigation in the
field of Leishmania from Dr. Madhubala in JNU, New Delhi (in collaboration with us). We
therefore feel that our exploratory insights will additionally be valuable in understanding other
pathogenic systems along with providing a base for future Indian investigators.
The proposed structural, functional and inhibitor discovery studies on Pf protein translation
motors are expected to have multiple outcomes, which have been summarized in Figure 2. Briefly,
we expect to deliver for each target: antibody-based cellular co-localization and spatial mapping in
all parasite life cycle stages, protein-protein pulldown data for mapping of interacting partners
within the parasite, phosphorylation studies as a function of parasite life stages, recombinant target
proteins, enzymatic and inhibition assays data, three-dimensional structure information, and short
lists of target-specific inhibitors. We expect that parasite-based inhibitor screening and iterative
improvement of the identified anti-parasitic compounds will provide a launch pad for possible
design of a new generation of anti-malarials.
16.7 Preliminary work done so far Over the past decade, the PI has established a very strong and highly productive structural
biology group within ICGEB which has expertise and state-of-the-art facilities for protein X-ray
crystallography, NMR spectroscopy and electron microscopy. The group has tremendous synergy in
terms of tackling biological problems with a wide array of techniques. PI’s research program
involves biochemistry, structural biology, cell biology and structure-based inhibitor discovery. Over
the past few years, the PI's team has primarily focused on translational machinery motors from
malaria parasites. We have characterized numerous aminoacyl-tRNA synthetases (aaRSs) in P.
falciparum, solved several three-dimensional structures of Pf aaRSs and advanced this subfield
significantly. We have recently published on the peculiar distribution of Pf aaRSs and their editing
domains – an effort that validates targeting this family of enzymes for development of novel anti-
malarials (Khan et al., 2011). We discovered that 4 out of 36 aaRSs (AlaRS, GlyRS, CysRS and
ThrRS) are single gene copies that are dual localized proteins in parasitic compartments – raising
the possibility of exploiting them for development of translation inhibitors at multiple translation
foci. Three-dimensional crystallographic structures of TyrRS, TrpRS, LysRS and D-tyrosyl-
tRNATyr deacylase (an editing domain) have been determined in our laboratory (Bhatt et al., 2010,
2011; Yogavel et al., 2010), and we are actively pursuing structure-based inhibitor development

15
where appropriate. To date, more than 600 drug-like compounds have been screened for their ability
to inhibit parasite growth in our laboratory, and several compounds with low µM IC50 have been
discovered (part of this work has been published, Khan et al., 2011). Presently, hit optimization,
structure-activity studies, and identification of scaffold-based screening using natural and synthetic
compounds are being pursued.
The methods mentioned in this grant (like protein purification, crystallization, structure
determination, inhibitor docking studies, confocal microscopy, protein pulldown assays, parasite
viability assays and various enzymatic assays) are all already well established in the PIs laboratory
as evidenced from our publications (Chandra et al., 2005; Bhatt et al., 2010, 2011, Sharma et al.,
2011, Khan et al., 2011). Therefore, for sake of brevity, these methods have not been described in
detail, and may be taken as established laboratory methodologies. Below, we provide details of the
4 proposed proteins that will be studied under the aegis of this grant:
Target 1: The malarial CCA-adding enzyme (CCAse)
Background:
For successful translation of proteins in any organism, the transcribed tRNAs must terminate their
3’ ends with the invariant sequence of CCA. Latter is essential for subsequent aminoacylation steps
and for binding of tRNAs with ribosomes (Allen et al., 2005; Agirrezabala and Frank, 2009). All
cells therefore contain CCA-adding enzymes (CCAses) that add terminal RNA sequence of cytidine
(C) -cytidine-adenosine (A) to the 3' ends of tRNAs. Eubacterial and eukaryotic CCAses studied so
far have included enzymes from Archaeoglobus fulgidus, Thermotoga maritima, Geobacillus
stearothermophilus and human mitochondrial (Pan et al., 2010; Toh et al., 2008, 2009; Li et al.,
2002; Augustin et al., 2003). The addition of CCA to 3’ end of tRNA is required in cases where the
naturally transcribed tRNA lacks this processing. Strikingly, in Pf only 6 out of 72 tRNAs have an
intact terminal CCA and therefore the malarial CCAse is an absolutely critical component for
production of mature tRNAs which can be utilized by the parasite translation machinery. Clearly, in
the absence of efficient CCA addition, most malarial tRNAs will remain unprocessed and therefore
will be unable to serve as substrates for charging by aminoacyl-tRNA synthetases (aaRSs). This is
expected to halt protein translation immediately. In this context, targeting the P. falciparum CCAse
(Pf CCAse) should severely impair protein translation within the parasites. Interestingly, CCAses

16
recognizes ATP and CTP within the same active site pocket in a reaction which also requires two
divalent metal cations (Pan et al., 2010). CCAses are considered atypical polymerases as they add
CCA onto tRNAs in absence of a template (Weiner, 2004). The catalytic cores of CCAases come in
two distinct flavours: class I are represented by archaeal CCAses and class II by
eubacterial/eukaryotic enzymes (Yue et al., 1996). The two classes share their overall 3D structures
in terms of significant domains (called head, neck, body and tail) despite a general lack of sequence
homology (Li et al., 2002; Augustin et al., 2003; Okabe et al., 2003; Hoffmeier et al., 2010). For
class II CCA-adding enzymes, crystal structures from prokaryotic and human sources are available
(Li et al., 2002; Augustin et al., 2003), thereby setting up the stage for comparative structural
dissection between the malarial and homologous enzymes.
Unique features of Pf CCAase (see Figure 3):
(1) Pf CCAase is much larger than its human homolog
(2) Pf CCAase has signal sequence for possible multiple localization
(3) Pf CCAase is unique in the parasite proteome
(4) roles of N- and C-terminal extensions in Pf CCAase are unknown
(5) the interacting partners of Pf CCAase in the parasite are unknown
Objectives and progress:
Our laboratory has been successful in cloning and expression of the catalytic core of Pf CCAse
(Figure 3). The relevant domain (Figure 4) has been expressed in E. coli in soluble form in fusion
with maltose-binding-protein (MBP). The purified protein has already been used for generation of
polyclonal sera for organelle localization and protein pulldown studies. Proposed localization
experiments will be critical in understanding how CCA may be added to the apicoplast-based
tRNAs, which is translationally independent. We aim to generate both full length and relevant
fragments of Pf CCAse using similar cloning and protein expression strategies. These efforts will
help in understanding the significance/role of additional N- and C- terminal regions in Pf CCAse.
Catalytic assays on the conserved domain as well as on larger fragments (and indeed full length Pf
CCAse) will be performed to delineate any possible role of the extra sequences in the malarial
version of the enzyme. We have already generated a 3D model of the catalytic core and performed
an initial round of structure-based inhibitor screening (Figure 5). The short listed inhibitors will
soon be tested in parasite assays to test their effect on growth of the pathogen.

17
95 kDa
72 kDa
Figure 3: Domain analysis of PfCCA-adding enzyme (PF3D7_1120500, also known as tRNA nucleotidyltransferase) along with a comparison with Hs homolog. The Pf enzyme is clearly much larger than the human counterpart, and we have initiated our studies by first focusing on the conserved domain. The Pf enzyme shares ~ 30% sequence identity with H. sapiens homolog.
Figure 4: SDS-PAGE analysis of expression/purification of conserved domain of CCA-adding enzyme from Pf. The protein is expressed in fusion with MBP in a soluble form, and since been purified to homogeneity.
Figure 5: Homology model of CCA-adding enzyme and docking modes of potential inhibitors.
.

18
Target 2: Malarial Leucyl/Phenylalanine tRNA transferase (LFT)
Background:
Fidelity in protein translation is ensured at many steps including during the aminoacylation reaction,
editing and during regulated protein degradation (Varshavsky, 1996). Addition of an amino acid to
protein N-terminus marks it for degradation by the N-end rule pathway (Varshavsky, 1996). This
rule correlates protein half-life in vivo with the 1st N-terminal amino acid. An enzyme called
leucyl/phenylalanyl tRNA protein transferase initiates attachment of either leucine or a
phenylalanine amino acid from a pool of charged tRNAs to protein substrates which contains N-
terminal arginine or lysine (Figure 6; Varshavsky, 1996). The protein product of this reaction is
proteolysed by the ATP-dependent proteasome-like motor ClpAP. In eukaryotes, significance of
this degradation pathway is underscored by studies which indicate importance of N-rule pathway in
diverse sets of phenomena like heart development (Kwon et al., 2002), apoptosis (Varshavsky,
2003) and G-protein signaling (Hamilton et al., 2003). Indeed in eukaryotes the ubiquitin system
regulates import, chromosomal separation and nitric oxide responses (Hu et al., 2005, Turner et al.,
2000, Rao et al., 2001). The N-terminal recognition is more complicated in eukaryotes with Arg,
Lys, or His representing primary while Phe, Leu, Trp, Tyr or Ile are the secondary destabilizing
residues in the substrates. Either way, Ub ligases ubiquitylate products of LFT which are then
shunted to the 26S proteasome for hydrolysis (Meinnel et al., 2006). Other N-terminal residues like
Asp, Glu, Cys etc are targeted along similar lines for degradation in some cases by R-transferases
which add an arginine to the N-terminal (Kwon et al., 2002; Hu et al., 2005). Pf genome has a
single gene for eukaryotic LFT homolog that resembles a eukaryotic R-transferase (Figure 7;
Graciet et al., 2006). Several critical residues conserved in typical LFTs are divergent in Pf LFT,
while those that interact with tRNAs are retained.
The molecular motors and details of the N-end rule pathway remain unexplored in malarial
parasites (or indeed in any pathogen, it seems). We therefore plan a comprehensive assault on
delineating Pf LFT and exploring its potential as a target of small molecule inhibitors. Limited
structural information is available for LFTs. Intriguingly, it has been shown that LFT binds to the
aminoacyl-adenylate analog puromycin (aminonucleoside antibiotic produced by Streptomyces
alboniger, Suto et al., 2006). Puromycin is highly toxic to most cells; however it remains to be
addressed whether puromycin is capable of directly binding to Pf LFT, thereby confirming LFT as a
potential target for this antibiotic. We and others have shown that puromycin is a very potent
inhibitor of malaria parasites but the link between puromycin and Pf LFT remains unaddressed.

19
Unique features of Pf LFT:
(1) Pf LFT is much larger than homologs found in other organisms
(2) Pf LFT does not have a sequence homolog in humans
(3) Pf LFT is unique in parasite proteome and therefore likely to be pivotal in protein metabolism
(4) role of N- terminal extension in Pf LFT is unknown
(5) interacting protein partners for Pf LFT are unknown
Objectives and progress:
Our laboratory has been successful in cloning and over-expression of the conserved
domain of Pf LFT. The relevant domain has been expressed in E. coli in soluble form in fusion with
histidine tag (Figure 8). The purified protein is already in crystallization trials, and presently we are
producing polyclonal sera against Pf LFT for cellular localization studies and protein-pulldown
experiments. We aim to produce full length Pf LFT to study possible roles for its N-terminal
extension. Lack of a sequence homolog of Pf LFT in humans marks this protein as a special
potential target for development of selective inhibitors. Towards this, we have generated a 3D
model of the conserved LFT domain and after an initial round of structure-based inhibitor screening
we have procured the short listed compounds for further testing both in vitro and ex vivo (Figure 9).
Figure 6: Catalytic mechanism of Leucyl/Phenylalanine tRNA transferase (LFT).
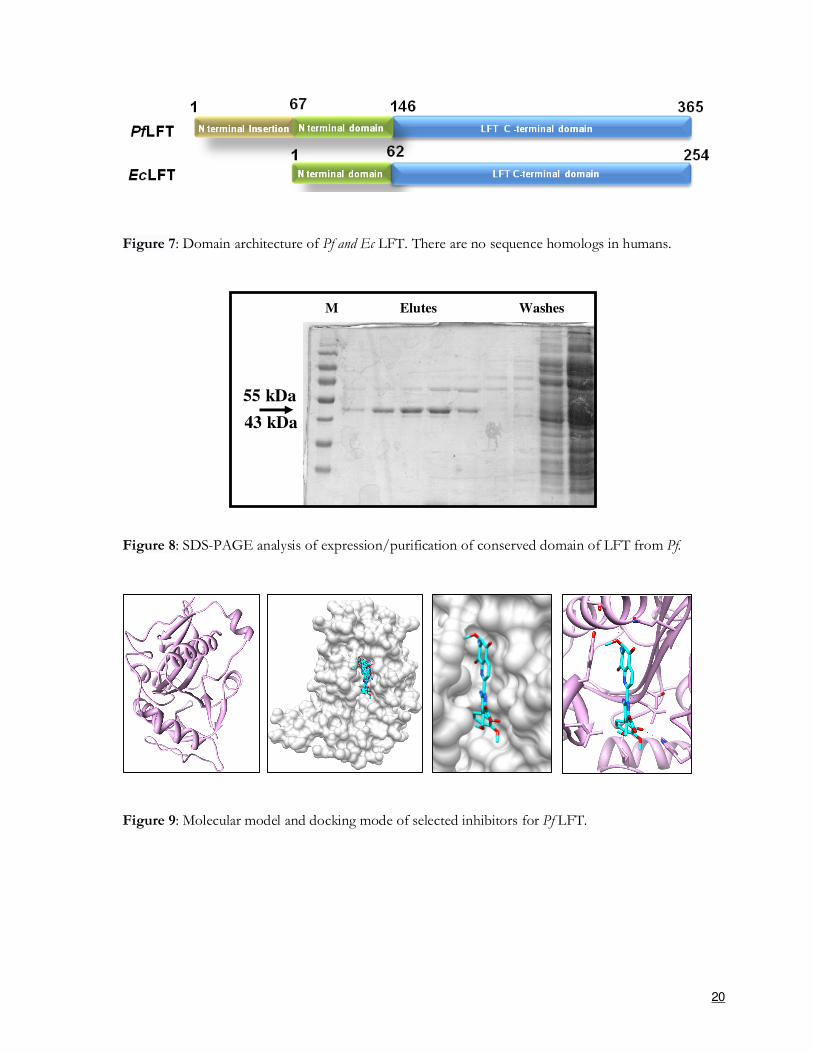
20
Figure 7: Domain architecture of Pf and Ec LFT. There are no sequence homologs in humans.
Figure 8: SDS-PAGE analysis of expression/purification of conserved domain of LFT from Pf.
Figure 9: Molecular model and docking mode of selected inhibitors for Pf LFT.
55 kDa
M Elutes Washes FT
43 kDa

21
Target 3: The malarial pyrophosphase (PPase)
Background:
Inorganic pyrophosphatase (PPase) catalyzes hydrolysis of energy rich pyrophosphate (PPi) to
release phosphate and energy. It is a ubiquitous enzyme and crucial for normal functioning of many
biosynthetic reactions in all cells including protein metabolism.
In general, PPases can be soluble/cytosolic ones or membrane-bound H+ translocating enzymes.
These two have no homology with each other and the soluble version is responsible for activity in
the cytosol. Absence of PPase will generally lead to build up of PPi to toxic levels (Pace et al.,
2011), and consistent with this its essentiality has been proven in bacteria and yeast (Chen et al.,
1990; Lundin et al., 1991). In pathogenic T. gondii it has been shown that PPase gene knock out is
not viable (Pace et al., 2011). Previous studies have confirmed the existence of two functional V-
H+ PPase in P. falciparum, but no study till date has addressed the soluble enzyme. Our preliminary
bioinformatics analysis has identified a putative 380aa PPase protein in Pf (Figure 10A). Sequence
alignment of Pf PPase with type I soluble PPases shows conservation in active site residues but
intriguingly also insertions of sequences in key structural elements of Pf PPase (Figure 10B). The
role of these additional insertions in Pf PPase remains unknown and understanding their
significance is a key objective. Our data suggests that the extra N-term region in Pf PPase is
important for function as the purified recombinant Pf PPase devoid of N-terminal is non-functional.
Using colorimetric assays, we have been able to detect PPase activity in Pf lysates. This activity is
almost completely abolished in presence of 1 mM EDTA, indicating necessity of divalent cations
for activity. Further, the addition of sodium fluoride, a general phosphatase inhibitor also
diminished PPase activity. Therefore, our preliminary biochemical studies provide evidence for
existence of a functional Pf PPase in malaria parasites. Interestingly, unlike in humans, there is only
a single predicted Pf PPase gene, and there seems to be no additional copy of Pf PPase dedicated to
the parasite apicoplast or its mitochondria. The parasite apicoplast is a center for significant
biochemical activity including DNA/protein synthesis and fatty acid metabolism – processes which
liberate or consume PPi. This raises curiosity on how PPi is utilized or quelled from accumulating
to toxic levels in the parasite apicoplast. It is possible that the parasite uses dual targeting strategy
for its unique Pf PPase protein - a phenomenon well appreciated in apicomplexans where a protein
PPase PPi + H2O 2Pi + Energy

22
translated from a single gene can be targeted to distinct cellular compartments by utilizing alternate
translation starts or alternative splicing mechanisms. This central issue remains unexplored for Pf
PPase. On this note, it is also worth contemplating and investigating the possibility of alternative
factors in the apicoplast that can result resolve the problem of PPi usage and accumulation. One
such factor could be a novel enzyme that can metabolize PPi or a transporter that will release excess
PPi into the cytosol.
Interestingly, blood–stages of P. falciparum are unusual in that oxidative phosphorylation
seems not a significant source of energy/ATP (Sherman, 1998; Nina et al., 2011). And this opens a
very intriguing question on whether “PPi is the major donor of energy and phosphate donor for
various functional processes during blood stages”. If the premise is true, it means PPi can be a
potential signaling molecule or a phosphate donor to protein kinases of signaling pathways. This
also renders the possibility of PPase as a crucial regulatory component of several multi-protein
complexes in P. falciparum. Therefore, identification of such complexes is very important as it can
unravel previously unknown regulatory mechanisms of metabolism. Finally, PPase is an essential
and well conserved enzyme, which makes it a druggable target. An in-depth analysis of its three-
dimensional X-ray structure combined with biochemical investigations will assist in validating Pf
PPase as a potential target for inhibitor discovery.
Unique features of Pf PPase:
(1) Pf PPase is larger than homologs found in humans
(2) Pf PPase is unique in the parasite proteome and therefore may be multi-tasking
(3) role of N- terminal extension in Pf PPase is unknown
(4) interacting protein partners of Pf PPase in the parasite are unknown
(5) mechanism of PPi hydrolysis in apicoplast remains unknown
(6) effect of inhibiting the single copy Pf PPase should be wide ranging and needs to be evaluated

23
A
B
Figure 10: (A) PPase P. falciparum and humans showing the conserved core region and a peculiar
extension (green) at N-terminus of P. falciparum PPase homolog. (B) Multiple sequence alignment of
PPases from pathogens and humans. The essential active site residues conserved in all PPases are
highlighted in turquoise blue. The peculiar N-terminal extension in P. falciparum enzyme is shaded green
while notable insertions in the malaria enzyme are in red.
PPase
1 82 380
1 289
P. falciparum
H. Sapiens PPase
24
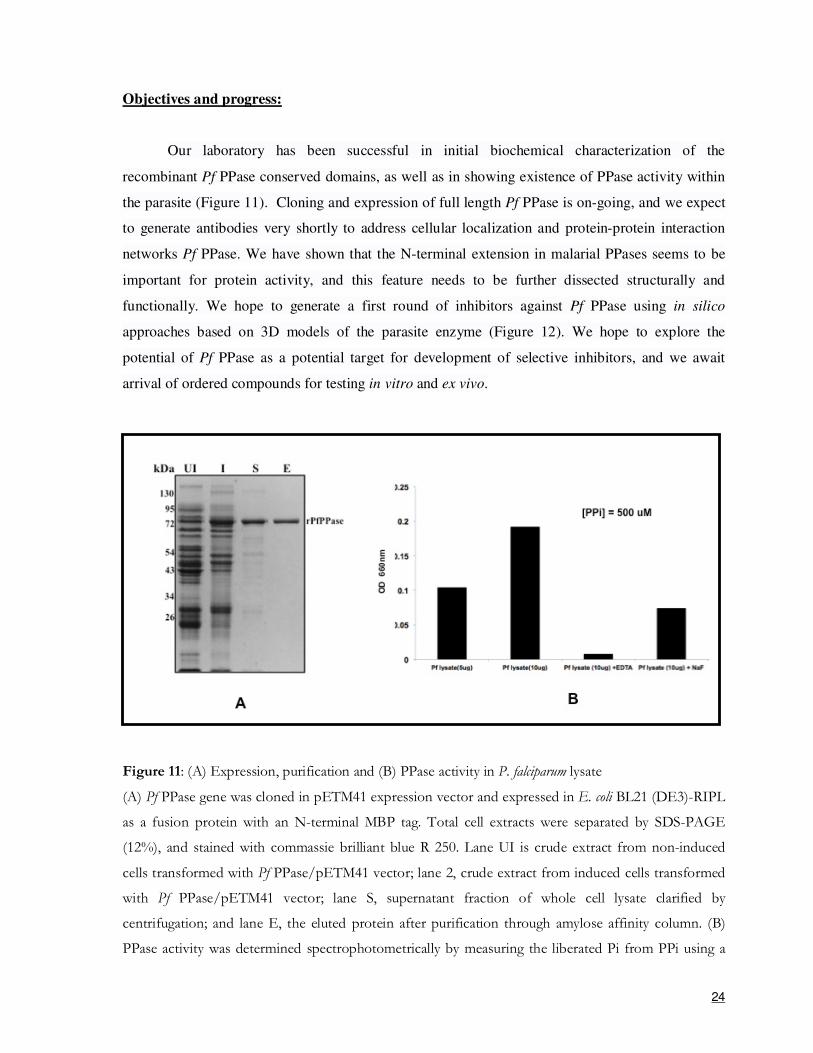
Objectives and progress:
Our laboratory has been successful in initial biochemical characterization of the
recombinant Pf PPase conserved domains, as well as in showing existence of PPase activity within
the parasite (Figure 11). Cloning and expression of full length Pf PPase is on-going, and we expect
to generate antibodies very shortly to address cellular localization and protein-protein interaction
networks Pf PPase. We have shown that the N-terminal extension in malarial PPases seems to be
important for protein activity, and this feature needs to be further dissected structurally and
functionally. We hope to generate a first round of inhibitors against Pf PPase using in silico
approaches based on 3D models of the parasite enzyme (Figure 12). We hope to explore the
potential of Pf PPase as a potential target for development of selective inhibitors, and we await
arrival of ordered compounds for testing in vitro and ex vivo.
Figure 11: (A) Expression, purification and (B) PPase activity in P. falciparum lysate
(A) Pf PPase gene was cloned in pETM41 expression vector and expressed in E. coli BL21 (DE3)-RIPL
as a fusion protein with an N-terminal MBP tag. Total cell extracts were separated by SDS-PAGE
(12%), and stained with commassie brilliant blue R 250. Lane UI is crude extract from non-induced
cells transformed with Pf PPase/pETM41 vector; lane 2, crude extract from induced cells transformed
with Pf PPase/pETM41 vector; lane S, supernatant fraction of whole cell lysate clarified by
centrifugation; and lane E, the eluted protein after purification through amylose affinity column. (B)
PPase activity was determined spectrophotometrically by measuring the liberated Pi from PPi using a

25
molybdate blue-based colorimetric assay. The cell homogenate was tested under standard conditions.
The assay was started by adding 5 µg and 10 µg of cleared cell homogenate to the standard mixture at
37° C. In addition to standard conditions, a reaction containing EDTA (1 mM) and NaF (5 mM) was
done to test inhibition of PPase activity. Hydrolysis was measured using a spectrophotometer at an
optical density of 660 nm.
Figure 12: Molecular model and docking mode of selected inhibitors for Pf PPase.

26
Target 4: The malarial multi-synthetase complex protein (P43)
Background:
In several studied eukaryotes, a multi-synthetase complex (MSC) exists which is a macromolecular
assembly of several amino-acyl tRNA synthetases (aaRSs) along with factors including P43. The
role of MSC is not clearly delineated and many reports have suggested diverse roles for this
complex (Quevillon et al., 1999; Robinson et al., 2000, Lee et al., 2004; Park et al., 2005; Guzzo and
Yang, 2008; Raina et al., 2012). Mammalian cells contain a >1 MDa MSC which is composed of the
following nine aaRS : leucyl-, lysyl-, prolyl-, isoleucyl-, methionyl-, glutamyl-, glutaminyl-,
arginyl-, and aspartyl-tRNA synthetases) (Quevillon et al., 1999; Robinson et al., 2000). In the
mammalian MSC, along with these aaRSs are found 3 factors called p38, p43 and p18 (Quevillon
and Mirande, 1996; Quevillon et al., 1997; Kaminska et al., 2009). The 3D structure of MSC remains
elusive, partly because the complex can be transient and variable. Electron microscopy has revealed
that mammalian MSC particles are V-shaped structures (Norcum, 1989, 1999; Norcum &
Warrington, 1998). MSC associated protein p38 may help in promoting protein–protein interactions
(Robinson et al., 2000; Kim et al., 2002; Ahn et al., 2003), while the C-terminus of p43 contains an
endothelial monocyte-activating polypeptide II (EMAPII)-like domain which confers a general
tRNA-binding property (Deinert et al., 2001). Intriguingly, the components of MSC vary from
species to species and simpler organisms and archae seem to require smaller MSCs (by reducing the
number of aaRSs that associate with accessory proteins, see Figure 13). This variable architecture of
MSCs complicates extending functional attributes across phylogenetically diverse organisms. In
particular, so far there is no evidence of MSC in any of the apicomplexan parasites including T.
gondii and of course malaria parasites. There are no homologs of Pf P43 in humans as the latter
contains 3 different subunits (p43, p38 and p18) which associate with MSC (Figure 14). In malaria
parasites, no homologs for p38 and p18 are present, suggesting that either a simpler or no MSC
exists in malaria parasites. However, there is no experimental information on this hypothesis and we
are poised to address this question directly via the proposed studies on Pf P43.
Unique features of Pf P43:
1) a homolog of Pf P43 is absent from humans
2) no structures for domains of Pf P43 are available
3) existence of MSC in malaria parasites or related apicomplexans remains unknown
4) no data on any interacting protein partners for Pf P43

27
Objectives and progress:
Our laboratory has been successful in cloning and protein over-expression of the intact Pf
P43. It has been expressed in E. coli in soluble form in fusion with histidine tag, and Pf P43 has
been further purified using cation exchange chromatography (Figure 15). Pf P43 is already in
crystallization trials and we have obtained some initial proteins crystals which diffract beyond 3 Å
resolution. We have generated polyclonals in rabbits for both cellular localization studies and
pulldown assays. Our initial pulldown experiments from parasite lysates suggest association of Pf
P43 with malarial lysine tRNA synthetase (unpublished data). Lack of a sequence homolog of Pf
P43 in humans marks this protein as a potential target for development of selective inhibitors. To
test this idea, we have already generated a 3D model of the tRNA binding domain of P43, and after
an initial round of structure-based inhibitor screening we have procured relevant compounds for
further testing both in vitro and ex vivo (Figure 16).
Figure 13: Schematic representation of multi-synthetases complexes (MSC) in various organisms.

28
Figure 14: Domain architecture of Pf P43 along with human counterparts.
Figure 15: SDS-PAGE gel showing the expression/purification of Pf P43.
Figure 16: Model and docking mode of selected inhibitors for C-terminal domain of P43.
MW Cation exchange elutions
55 kDa
43 kDa

29
Conclusion:
The strategic ideas outlined in this proposal are embedded within a decade-old program in
the PIs laboratory which has a structural parasitology thrust in malaria research. The present
proposal specifically entails expansion into non-ribosomal protein targets within the parasite protein
translation machinery for in-depth biological investigations and exploration of their therapeutic
utility. Several key, hitherto unexplored, protein translation motors that are very likely to serve as
sources of deep insights into parasite biology form an integral part of the proposed research. These
include CCA-adding enzyme, L/F transferase, Pyrophosphatase and P43 – these 4 (along with
aaRSs) cover many pivotal non-ribosomal protein translation apparatus targets in malaria parasites.
In order to facilitate our investigations, we are requesting augmentation and enhancement
of our research facilities along with an extended and generous period of financial support. We seek
encouragement and funding for our aims of furthering protein translation research with a concurrent
focus on development of new sets of parasite inhibitors which could be future anti-malarials.

30
17. Specific objectives (should be written in bulleted form, a short paragraph indicating the methods to be followed for achieving the objective and verifiable indicators of progress should follow each specific objective)
1. Production of recombinant target proteins for biochemistry and structural biology.
Genes for all 4 target proteins have been cloned in expression vectors, and in all cases recombinant
proteins are available in soluble forms. This is already a considerable achievement given the
difficulty of obtaining malaria parasite proteins recombinantly. Purification protocols have been
tested and finalised on a pilot scale for all targets.
Verifiable indicators: Availability of recombinant, soluble domains of target proteins in milligram
amounts along with polyclonal sera against them.
2. Three dimensional structure determination of targets.
Efforts to produce protein crystals of full length or relevant subdomains of the 4 target proteins will
be made. Structures will be solved using either molecular replacement or SAD phasing techniques
as has been done in the PIs laboratory over the past decade.
Verifiable indicators: 3D structural structures using modeling and experimental data.
3. Cell biology experiments including protein localization, tracking of post-translational
modifications, determination of the protein interactome.
Confocal microscopy will be used for cellular localization of target proteins. For identification of
interacting partners, protein pulldown experiments along with mass spectroscopy-based protein
identification will be used. Phosphorylation status of target proteins will be addressed using
phospho-specific antibodies. Where required, phosphorylation sites may be verified using
mutagenesis, and relevant kinases may be identified using kinase-specific inhibitors.
Verifiable indicators: Biochemical data on protein interaction partners for target proteins.
4. Structure-based inhibitor discovery and parasite viability assays in a cyclical fashion.
In silico screening using docking and virtual screening packages will be performed. Libraries
enriched in drug-like molecules (which are additionally commercially available) will be used for
initial repetitive rounds of in vitro screening until valuable scaffolds have been obtained. Latter will
then be developed/optimized using links with partners like Jubiliant Biosciences.
Verifiable indicators: List of best anti-parasitic, target-specific compound(s).

31
18. Work Plan: should not exceed 3-4 pages (the section can be divided according to the specific aims and under each specific aim, the following should be stated clearly as sub headings)
The experimental and intellectual expertise required for this proposal has been carefully
nurtured and developed in the PIs laboratory over the past decade. We prefer to take multi-
disciplinary approaches towards obtaining a more complete biological picture, and this necessarily
includes techniques outside of X-ray crystallography like protein biochemistry, in silico docking,
confocal microscope-based subcellular localization, protein-protein pulldown assays, parasite
viability assays and inhibitor screening. These additional facets are now well entrenched in our
laboratory, as evidenced from PIs publications in reputed international journals (Chandra et al.,
2005, 2007, Singh et al., 2006; Navagi et al., 2006; Sharma et al., 2011a, 2011b 2011c; Yogavel et
al., 2007, 2008, 2009, 2010a, 2010b; Bhatt et al., 2009, 2010, 2011; Khan et al., 2011).
32
18.1 Work plan (methodology/experimental design to accomplish the stated aim)
Figure 2. Overall workflow with deliverables
Cell, functional and structural biology:
(1) Antibody-based localization with confocal microscopy
(2) Co-localization and mapping in all parasite life cycle stages
(3) Pulldown experiments for mapping interacting partners
(4) Phosphorylation studies as a function of parasite stages
(5) Purification of recombinant proteins for domain analysis
(6) Enzymatic and inhibition assays, 3D structure determination
Inhibitor discovery:
(1) 3D modeling of relevant structures and domains
(2) Extensive in silico screening against drug-like libraries using 3D atomic models
(3) Establishment of a library resource for testing in cells
(4) Parasite-based screening and iterative improvement of hits

33
Experimental design and methods:
1) Cloning, expression and purification
We have already generated molecular clones of the relevant protein domains from the four targets.
These clones have been tested, and encouragingly we have produced soluble, recombinant proteins
for each of the targets. Therefore, this stage of the project is already completed and we are well on
track to pursue subsequent proposed studies. Full length protein constructs and smaller subdomains
will be made using the recombinant technologies. For all clones, we will screen the entire protein
expression space (using different vector systems, temperatures, inducer concentrations and different
host strains) to identify conditions where protein expression is optimal. In all cases, established
purification protocols will be used to ensure homogeneous protein quality.
2) Biochemical experiments: Localization and protein-pulldown experiments
To address cellular localization of target proteins, polyclonal antibodies will be generated in mice
and rabbits. These antibodies will be labeled with suitable fluorescent tags and used in conjunction
with markers of parasite nucleus, apicoplast, cytoplasm and mitochondria. Reagents for the latter
are already available in the PIs laboratory. Enzymatic assays will be conducted in line with
published protocols, and in presence of identified inhibitors. This will address issues of inhibitor
selectivity. Confocal microscopy, protein pulldown experiments along with mass spectroscopy-
based protein identification protocols will be used. Phosphorylation status of target proteins will be
addressed using phospho-specific antibodies. Where required, phosphorylation sites may be verified
using mutagenesis, and relevant kinases may be identified using kinase-specific inhibitors.
3) Crystallization, data processing and structure determination
Highly purified and concentrated proteins will be used for extensive screening of crystallization
conditions by hanging drop vapor diffusion method. X-ray analysis of crystals will be carried out at
ICGEB, New Delhi using existing X-ray diffraction facility. Depending on the quality and extent of
diffraction, data will either be collected at the home source or at a synchrotron facility. All data
processing, analysis, and structure determination will be carried out in ICGEB New Delhi. Standard
protocols in structure determination and model refinement will be followed. Crystallographic
phases will be obtained by employing either molecular replacement or or SAD (single wavelength
anomalous dispersion) techniques, both of which have been successfully employed previously at
ICGEB in my group. Coordinates and structure factors will be deposited in public domain structure
databases.

34
4) In-silico inhibitor screening using commercial drug libraries
Molecular docking and in silico virtual screening will be performed using 3D structures in docking
programs like AutoDock vina (Trott and Olson, 2010) and Schrödinger suite. Commercial small
molecule drug libraries will be used as input. The libraries will be prepared using Ligprep module
in Schrodinger software suite to ensure proper partial charges and protonation states.
Physiochemical properties will be predicted using Qikprop module in Schrödinger suite. For in
silico screening, we will apply lead-like filters derived from Lipinski’s rule of five. The top docking
poses will be isolated from each output file for further analysis. Top ligands will be selected based
on docking scores and hydrogen bond interactions with residues in the target protein.
5) In vitro parasite growth inhibition assays
Top inhibitors selected via the above procedure will be tested on malaria parasite P. falciparum 3D7
strain. Parasites will be synchronized using sorbitol and starting with ring stages these parasites will
be cultured in 96 well plates. Initial screening of all top compounds will be performed by growing
parasites at 100 µM inhibitor concentration. After 48 hours of growth, parasitemia will be measured
by fluorescence assays as described earlier (Smilkstein et al., 2004). To further validate the anti-
malarial activity of top compounds from this first screen, experiments will be repeated in a broader
range of inhibitor concentrations ranging from 1 nM to 1 mM. These data will be used to calculate
IC50 values for each inhibitor. We have established these as standard screening protocols in our
laboratory, and published our results on related systems using the same.
18.2 Connectivity of the participating institutions and investigators (in case of multi-institutional projects only) NA
18.3 Alternate strategies (if the proposed experimental design or method does not work what is the alternate strategy)
The experimental and intellectual expertise required for this proposal has been carefully
nurtured and developed in the PIs laboratory over the past decade. Multi-disciplinary approaches
towards obtaining a more complete biological picture preferred by us necessarily includes
techniques outside of X-ray crystallography like protein biochemistry, in silico docking, confocal
microscope-based subcellular localization, protein-protein pulldown assays, parasite viability assays
and inhibitor screening. These additional facets are now well entrenched in the laboratory, as
evidenced from our series of research papers in reputed international journals (Chandra et al., 2005,

35
2007, Singh et al., 2006; Navagi et al., 2006; Sharma et al., 2011a, 2011b 2011c; Yogavel et al.,
2007, 2008, 2009, 2010a, 2010b; Bhatt et al., 2009, 2010, 2011; Khan et al., 2011). Therefore, for
sake of brevity, these methods have not been described in detail, and may be taken as established
methodologies in the PIs laboratory.
For protein over-expression, we already possess clones that are able to produce recombinant
target proteins. Libraries used for inhibitor screening can be expanded easily and therefore we aim
to continue testing small molecule compounds in 100s or 1000s until we arrive at sub-micromolar
IC50 value inhibitors against the target proteins. We have extensive experience in use of confocal
and other advanced microscopic systems and therefore we expect to accomplish protein localization
of the target proteins. Protein-interaction networks will have to be detailed using pulldown
experiments, which indeed can be tricky given the paucity of starting parasite material. However,
once again, our laboratory has recently been successful in highlighting novel interactions between
P43 and aaRSs using pulldown experiments. We therefore feel that we can replicate these protocols
for other target proteins. Finally, in cases, where we are unable to obtain crystals using hanging-
drop vapour-diffusion method, we will use sitting drop and micro batch methods with various
screening kits for growing native crystals and/or crystals of complexes.
References
Agirrezabala, X., Frank, J. 2009. Elongation in translation as a dynamic interaction among the ribosome, tRNA, and elongation factors EF-G and EF-Tu. Q. Rev. Biophys. 42, 159-200. Ahn, H. C., Kim, S., Lee, B. J 2003. Solution structure and p43 binding of the p38 leucine zipper motif: coiled-coil interactions mediate the association between p38 and p43. FEBS Lett. 542, 119-124.
Allen, G. S., Zavialov, A., Gursky, R., Ehrenberg, M., Frank, J. 2005. The cryo-EM structure of a
translation initiation complex from Escherichia coli. Cell, 121, 703-712.
AlKadi, H.O. (2007) Antimalarial drug toxicity: a review. Chemotherapy, 53, 385–391. Augustin, M. A., Reichert, A. S., Betat, H., Huber, R., Mörl, M., Steegborn, C. 2003. Crystal structure of the human CCA-adding enzyme: insights into template-independent polymerization. J. Mol. Biol. 328, 985-994. Balabaskaran Nina P., Morrisey, J. M., Ganesan, S. M., Ke, H., Pershing, A. M., Mather, M. W., Vaidya, A. B (2011). ATP synthase complex of Plasmodium falciparum: dimeric assembly in mitochondrial membranes and resistance to genetic disruption. J. Biol. Chem. 286, 41312-41322. Bhatt, T. K., Kapil, C., Khan, S., Jairajpuri, M. A., Sharma, V., Santoni, D., Silvestrini, F., Pizzi, E., Sharma, A. 2009. A genomic glimpse of aminoacyl-tRNA synthetases in malaria parasite Plasmodium falciparum. BMC Genomics, 10:644.

36
Bhatt, T. K., Khan, S., Dwivedi, V. P., Banday, M. M., Sharma, A., Chandele, A., Camacho, N., de Pouplana, L. R., Wu, Y., Craig, A. G., Mikkonen, A. T., Maier, A. G., Yogavel, M., Sharma, A. 2011. Malaria parasite tyrosyl-tRNA synthetase secretion triggers pro-inflammatory responses. Nature Commun.. 2:530. Bhatt, T. K., Yogavel, M., Wydau, S., Berwal, R., Sharma, A. 2010. Ligand-bound structures provide atomic snapshots for the catalytic mechanism of D-amino acid deacylase. J. Biol. Chem. 285, 5917-5930. Chandra, B. R., Olivieri, A., Silvestrini, F., Alano, P., Sharma, A. 2005. Biochemical characterization of the two nucleosome assembly proteins from Plasmodium falciparum. Mol. Biochem. Parasitol. 142, 237-247. Chandra, B. R., Yogavel, M., Sharma A. 2007. Structural analysis of ABC-family periplasmic zinc binding protein provides new insights into mechanism of ligand uptake and release. J. Mol. Biol. 367, 970-982. Chavchich, M., Gerena, L., Peters, J., Chen, N., Cheng, Q., Kyle, D. E. 2010. Role of pfmdr1 amplification and expression in induction of resistance to artemisinin derivatives in Plasmodium falciparum. Antimicrob. Agents Chemother. 54, 2455-2464. Chen, J., Brevet, A., Fromant, M., Lévêque, F., Schmitter, J. M., Blanquet, S., Plateau, P. 1990.
Pyrophosphatase is essential for growth of Escherichia coli. J. Bacteriol. 172, 5686–5689.
Deinert, K., Fasiolo, F., Hurt, E. C., Simos, G. 2001. Arc1p organizes the yeast aminoacyl-tRNA
synthetase complex and stabilizes its interaction with the cognate tRNAs. J. Biol. Chem. 276, 6000-6008.
Eastman, R.T. and Fidock, D.A. 2009. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nat. Rev. Microbiol. 7, 864–874.
Fidock, D. A., Rosenthal, P. J., Croft, S. L., Brun, R., Nwaka, S. 2004. Antimalarial drug discovery:
efficacy models for compound screening. Nat. Rev. Drug Discov. 3, 509-520.
Graciet, E., Hu, R. G., Piatkov, K., Rhee, J. H., Schwarz, E. M., Varshavsky, A. 2006. Aminoacyl-
transferases and the N-end rule pathway of prokaryotic/eukaryotic specificity in a human pathogen.
Proc. Natl. Acad. Sci. USA, 103, 3078-3083.
Guzzo, C. M., Yang, D. C. 2008. Lysyl-tRNA synthetase interacts with EF1α, aspartyl-tRNA synthetase and p38 in vitro. Biochem. Biophys. Res. Commun. 365, 718-23.
Hamilton, M. H., Cook, L. A., McRackan, T. R., Schey, K. L., Hildebrandt, J. D. 2003. γ2 subunit of G protein heterotrimer is an N-end rule ubiquitylation substrate. Proc. Natl. Acad. Sci. USA, 100, 5081-5086. Hoffmeier, A., Betat, H., Bluschke, A., Günther, R., Junghanns, S., Hofmann, H. J., Mörl, M. 2010. Unusual evolution of a catalytic core element in CCA-adding enzymes. Nucleic Acids Res. 38, 4436-4447. Hu, R. G., Sheng, J., Qi, X., Xu, Z., Takahashi, T. T., Varshavsky, A. 2005. The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature, 437, 981-986.

37
Hyde, J. E. 2010. Drug-resistant malaria - an insight. FEBS J. 274, 4688-4698. Jackson, K. E., Habib, S., Frugier, M., Hoen, R., Khan, S., Pham, J. S., Ribas de Pouplana, L., Royo, M.,
Santos, M. A., Sharma, A., Ralph, S.A. 2011. Protein translation in Plasmodium parasites. Trends
Parasitol. 27, 467-476
Kaminska, M., Havrylenko, S., Decottignies, P., Le Marechal, P., Negrutskii, B., Mirande, M. 2009. Dynamic organization of aminoacyl-tRNA synthetase complexes in the cytoplasm of human cells. J. Biol. Chem. 284, 13746–13754 Kankare, J., Salminen, T., Lahti, R., Cooperman, B. S., Baykov, A. A, et al. 1996 Structure of Escherichia
coli inorganic pyrophosphatase at 2.2 A ° resolution. Acta Crystallogr D Biol Crystallogr 52, 551-563.
Kappe, S. H., Vaughan, A. M., Boddey, J. A., Cowman, A. F. 2010. That was then but this is now: malaria research in the time of an eradication agenda. Science, 328, 862-866. Khan, S., Sharma, A., Jamwal, A., Sharma, V., Pole, A. K., Thakur, K. K., Sharma, A. 2011. Uneven spread of cis- and trans-editing aminoacyl-tRNA synthetase domains within translational compartments of P. falciparum. Scientific Reports. 1:188.
Kim, J. Y., Kang, Y. S., Lee, J. W., Kim, H. J., Ahn, Y. H., Park, H., Ko, Y.G., Kim, S. 2002. p38 is
essential for the assembly and stability of macromolecular tRNA synthetase complex: implications for
its physiological significance. Proc. Natl. Acad. Sci. USA. 99, 7912-7926.
Kwon, Y, T., Kashina, A. S., Davydov, I. V., Hu, R. G., An, J. Y., Seo, J. W., Du, F., Varshavsky, A. 2002. An essential role of N-terminal arginylation in cardiovascular development. Science, 297, 96-99. Lee, S. W., Cho, B. H., Park, S. G., Kim, S. 2004. Aminoacyl-tRNA synthetase complexes: beyond
translation. J. Cell. Sci. 117, 3725-3734.
Li, F., Xiong, Y., Wang, J., Cho, H. D., Tomita, K., Weiner, A. M., Steitz, T. A. 2002. Crystal structures of the Bacillus stearothermophilus CCA-adding enzyme and its complexes with ATP or CTP. Cell, 111, 815-824. Lundin, M., Baltscheffsky, H., Ronne, H. 1991. Yeast PPA2 gene encodes a mitochondrial inorganic pyrophosphatase that is essential for mitochondrial function. J. Biol. Chem. 266, 12168-12172.
Meinnel, T., Serero, A., Giglione, C. 2006. Impact of the N-terminal amino acid on targeted protein degradation. Biol. Chem. 387, 839-351. Navadgi, V. M., Chandra, B. R., Mishra, P. C., Sharma, A. 2006. The two Plasmodium falciparum
nucleosome assembly proteins play distinct roles in histone transport and chromatin assembly. J. Biol.
Chem. 281, 16978-16984.
Norcum, M. T. 1989. Isolation and electron microscopic characterization of the high molecular mass
aminoacyl-tRNA synthetase complex from murine erythroleukemia cells. J. Biol. Chem. 264, 15043-
14051.

38
Norcum, M. T. 1999. Ultrastructure of the eukaryotic aminoacyl-tRNA synthetase complex derived
from two dimensional averaging and classification of negatively stained electron microscopic images.
FEBS Lett. 447, 217-222.
Norcum, M. T., Warrington, J. A. 1998. Structural analysis of the multienzyme aminoacyl-tRNA
synthetase complex: a three-domain model based on reversible chemical crosslinking. Protein Sci. 7, 79-
87.
Okabe, M., Tomita, K., Ishitani, R., Ishii, R., Takeuchi, N., Arisaka, F., Nureki, O., Yokoyama, S. 2003. Divergent evolutions of trinucleotide polymerization revealed by an archaeal CCA-adding enzyme structure. EMBO J. 22, 5918-5927. Oksanen, E., Ahonen, A. K., Tuominen, H., Tuominen, V., Lahti, R., et al. 2007. A complete structural description of the catalytic cycle ofyeast pyrophosphatase. Biochemistry, 46, 1228-1239.
Pace, D. A., Fang, J., Cintron, R., Docampo , M. D., Moreno, S. N. 2011. Overexpression of a cytosolic
pyrophosphatase (TgPPase) reveals a regulatory role of PP(i) in glycolysis for Toxoplasma gondii.
Biochem J. 440, 229-240.
Pan, B., Xiong, Y., Steitz, T. A. 2010. How the CCA-adding enzyme selects adenine over cytosine at position 76 of tRNA. Science, 330, 937-940.
Park, S. G., Ewalt, K. L., Kim S. 2005. Functional expansion of aminoacyl-tRNA synthetases and their
interacting factors: new perspectives on housekeepers. Trends Biochem Sci. 30, 569-574.
Quevillon, S., Mirande, M. 1996. The p18 component of the multisynthetase complex shares a protein
motif with the beta and gamma subunits of eukaryotic elongation factor 1. FEBS Lett. 395, 63-67
Quevillon, S., Agou, F., Robinson, J. C., Mirande, M. 1997. The p43 component of the mammalian
multi-synthetase complex is likely to be the precursor of the endothelial monocyte-activating
polypeptide II cytokine J. Biol. Chem. 272, 32573–32579.
Quevillon, S., Robinson, J. C., Berthonneau, E., Siateckam M., Mirande, M. 1999. Macromolecular
assemblage of aminoacyl-tRNA synthetases: identification of protein-protein interactions and
characterization of a core protein. J. Mol. Biol. 285, 183-195.
Raina, M., Elgamal, S., Santangelo, T. J., Ibba, M. (2012). Association of a multi-synthetase complex
with translating ribosomes in the archaeon Thermococcus kodakarensis. FEBS Lett. [Epub ahead of print]
Robinson, J. C., Kerjan, P., Mirande, M. 2000. Macromolecular assemblage of aminoacyl-tRNA
synthetases: quantitative analysis of protein-protein interactions and mechanism of complex assembly.
J. Mol. Biol. 304, 983-994.
Sharma, A., Sharma A. 2011a. Fatty acid induced remodeling within the human liver fatty acid-binding protein. Journal of Biological Chemistry. 286, 31924-31928.

39
Sharma, A., Sharma, A., Dixit, S., Sharma, A. 2011b. Structural insights into thioredoxin-2: a component of malaria parasite protein secretion machinery. Scientific Reports 1:179.
Sharma, A.; Yogavel, M.; Sharma, A. 2011c. Utility of anion and cation combinations for phasing of protein structures. J. struct. funct. genomics, In Press.
Sherman, I. W. 1998 in Malaria: Parasite Biology, Pathogenesis, and Protection (Sherman, I. W., ed) pp. 135–144, American Society for Microbiology, Washington, D. C. Singh, S. K., Hora, R., Belrhali, H., Chitnis, C. E., Sharma, A. 2006. Structural basis for Duffy recognition by the malaria parasite Duffy-binding-like domain. Nature, 439, 741-744.
Sivula, T., Salminen, A., Parfenyev, A. N., Pohjanjoki, P., Goldman, A., et al. 1999 Evolutionary aspects of inorganic pyrophosphatase. FEBS Lett. 454, 75-80. Smilkstein, M., Sriwilaijaroen, N., Kelly, J. X., Wilairat, P., Riscoe, M. 2004. Simple and inexpensive
fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents
Chemother. 48, 1803-1806.
Suto, K., Shimizu, Y., Watanabe, K., Ueda, T., Fukai, S., Nureki, O., Tomita, K. 2006. Crystal structures
of leucyl/phenylalanyl-tRNA-protein transferase and its complex with an aminoacyl-tRNA analog.
EMBO J. 25, 5942-5950.
Tan, K. R., Magill, A. J., Parise, M. E., Arguin, P. M. (2011). Doxycycline for malaria chemoprophylaxis
and treatment: report from the CDC expert meeting on malaria chemoprophylaxis. Am. J. Trop. Med.
Hyg. 84, 517-531.
Toh, Y., Takeshita, D., Numata, T., Fukai , S., Nureki, O., Tomita, K. 2009. Mechanism for the definition of elongation and termination by the class II CCA-adding enzyme. EMBO J. 28, 3353-3365. Toh, Y., Numata, T., Watanabe, K., Takeshita, D., Nureki, O., Tomita, K. 2008. Molecular basis for maintenance of fidelity during the CCA-adding reaction by a CCA-adding enzyme. EMBO J. 27, 1944-1952. Trenholme, C. M., Williams, R. L., Desjardins, R. E., Frischer, H., Carson, P. E., Rieckmann, K.H. and Canfield, C. J. 1975. Mefloquine (WR 142,490) in thetreatment of human malaria. Science (New York, NY) 190, 792–794. Trott, O., Olson, A. J. 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455-461. Uhlemann, A.C. and Krishna, S. 2005. Antimalarial multi-drug resistance in Asia: mechanisms and assessment. Curr. Top. Microbiol. Immunol. 295, 39-53. Varshavsky, A. 1996. The N-end rule: functions, mysteries, uses. Proc. Natl. Acad. Sci. USA.93, 12142-12149. Varshavsky, A. 2003. The N-end rule and regulation of apoptosis. Nat. Cell Biol. 5, 373-376. Weiner, A. M. 2004. tRNA maturation: RNA polymerization without a nucleic acid template. Curr. Biol. 14, R883-R885.

40
Wells, T. N., Burrows, J. N. and Baird, J. K. 2010. Targeting the hypnozoite reservoir of Plasmodium
vivax: the hidden obstacle to malaria elimination. Trends Parasitol. 26, 145-151. Yogavel, M., Gill, J., Mishra, P. C., Sharma, A. 2007. SAD phasing of a structure based on cocrystallized iodides using an in-house Cu Kalpha X-ray source: effects of data redundancy and completeness on structure solution. Acta Crystallographica Section D: Biological Crystallography, 63, 931-934. Yogavel, M., Gill, J., Sharma, A. 2009. Iodide-SAD, SIR and SIRAS phasing for structure solution of a nucleosome assembly protein. Acta Crystallographica Section D: Biological Crystallography, 65, 618-622. Yogavel, M., Khan, S., Bhatt, T. K., Sharma, A. 2010a. Structure of D-tyrosyl-tRNATyr deacylase using home-source Cu Kalpha and moderate-quality iodide-SAD data: structural polymorphism and HEPES-bound enzyme states. Acta Crystallogr D Biol Crystallogr. 66, 584-592.
Yogavel, M., Nithya, N., Suzuki, A., Sugiyama, Y., Yamane, T., Velmurugan, D., Sharma A. 2010b. Structural analysis of actinidin and a comparison of cadmium and sulfur anomalous signals from actinidin crystals measured using in-house copper- and chromium-anode X-ray sources. Acta Crystallographica Section D: Biological Crystallography, 66,. 1323-1333.
Yue, D., Maizels, N., Weiner, A. M. 1996. CCA-adding enzymes and poly(A) polymerases are all members of the same nucleotidyltransferase superfamily: characterization of the CCA-adding enzyme from the archaeal hyperthermophile Sulfolobus shibatae. RNA, 2, 895-908.
41
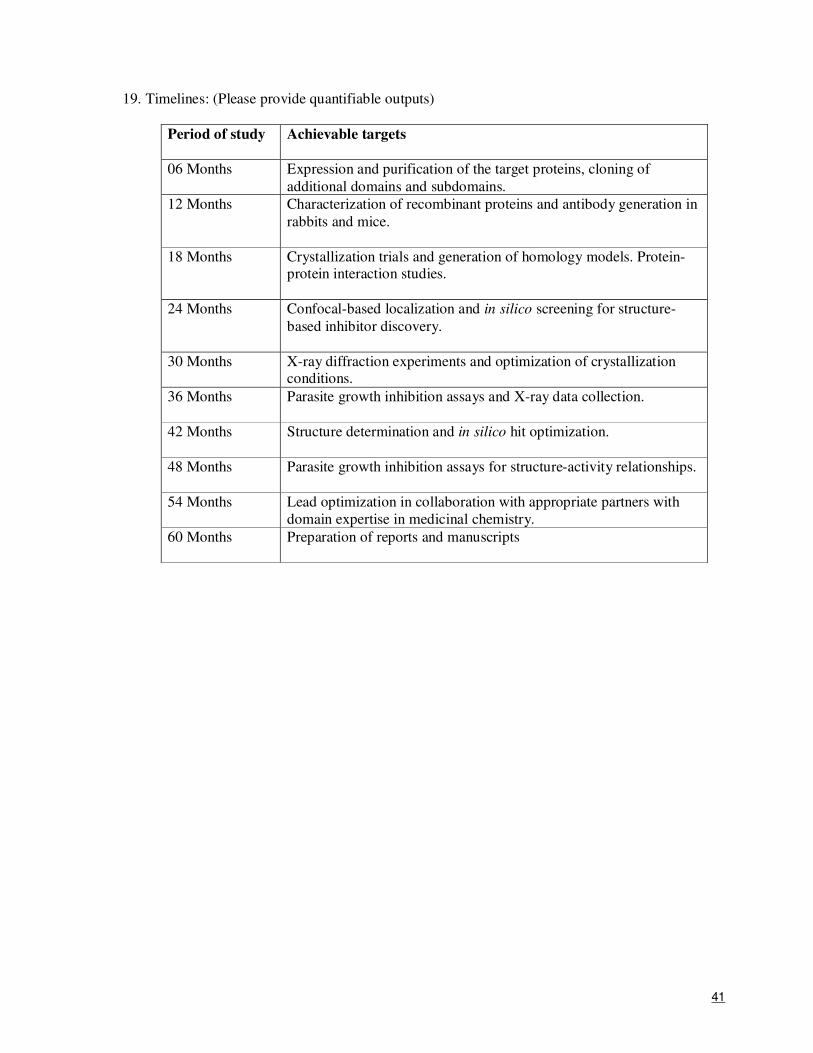
19. Timelines: (Please provide quantifiable outputs)
Period of study Achievable targets
06 Months
Expression and purification of the target proteins, cloning of additional domains and subdomains.
12 Months Characterization of recombinant proteins and antibody generation in rabbits and mice.
18 Months Crystallization trials and generation of homology models. Protein-protein interaction studies.
24 Months Confocal-based localization and in silico screening for structure-based inhibitor discovery.
30 Months
X-ray diffraction experiments and optimization of crystallization conditions.
36 Months
Parasite growth inhibition assays and X-ray data collection.
42 Months
Structure determination and in silico hit optimization.
48 Months
Parasite growth inhibition assays for structure-activity relationships.
54 Months
Lead optimization in collaboration with appropriate partners with domain expertise in medicinal chemistry.
60 Months
Preparation of reports and manuscripts
42
20. Name and address of 5 experts in the field
Sr.No. Name Designation Address 1
Dr. R. Sankaranarayanan Scientist Centre for Cellular and Molecular Biology (CCMB) Uppal Road, Hyderabad - 500 007 40-2719 (or) 2832-2835 E-mail: [email protected], [email protected]
2
Dr. Saman Habib
Scientist Division of Molecular and Structural Biology, Central Drug Research Institute, Lucknow E-Mail: [email protected]
3
Pushkar Sharma
Scientist Cell Signaling Group Eukaryotic Gene Expression Laboratory, NII E-Mail: [email protected]
4
Dr. G. Padmanaban
Hon. Professor
Indian Institute of Science Bangalore, India-560012 Phone : 91-80-22932473 Fax : 91-80-23600814 Email : [email protected]
5
Dr. Utpal S. Tatu Professor Indian Institute of Science Bangalore, India-560012 Phone : 91-80-22932473 Fax : 91-80-23600814 Email : [email protected]
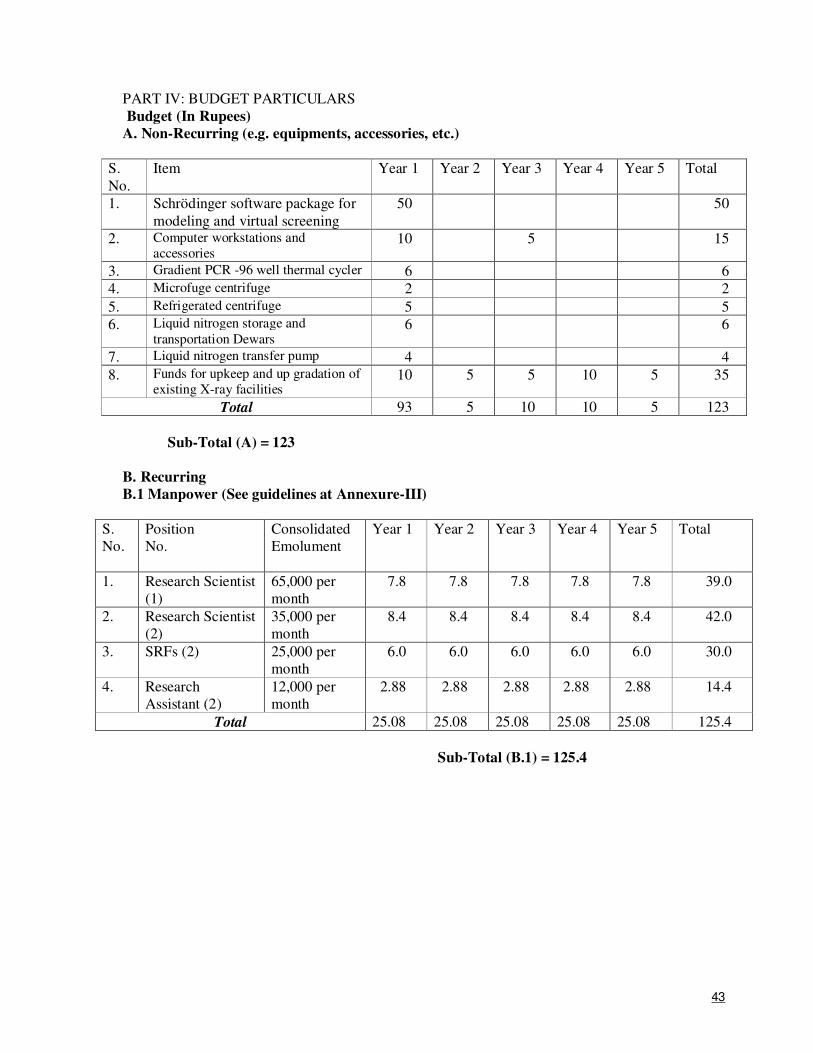
43
PART IV: BUDGET PARTICULARS Budget (In Rupees)
A. Non-Recurring (e.g. equipments, accessories, etc.)
S. No.
Item Year 1 Year 2 Year 3 Year 4 Year 5 Total
1. Schrödinger software package for modeling and virtual screening
50 50
2. Computer workstations and accessories
10 5 15
3. Gradient PCR -96 well thermal cycler 6 6 4. Microfuge centrifuge 2 2 5. Refrigerated centrifuge 5 5 6. Liquid nitrogen storage and
transportation Dewars 6 6
7. Liquid nitrogen transfer pump 4 4 8. Funds for upkeep and up gradation of
existing X-ray facilities 10 5 5 10 5 35
Total 93 5 10 10 5 123
Sub-Total (A) = 123
B. Recurring
B.1 Manpower (See guidelines at Annexure-III)
S. No.
Position No.
Consolidated Emolument
Year 1 Year 2 Year 3 Year 4 Year 5 Total
1. Research Scientist (1)
65,000 per month
7.8 7.8 7.8 7.8 7.8 39.0
2. Research Scientist (2)
35,000 per month
8.4 8.4 8.4 8.4 8.4 42.0
3. SRFs (2) 25,000 per month
6.0 6.0 6.0 6.0 6.0 30.0
4. Research Assistant (2)
12,000 per month
2.88 2.88 2.88 2.88 2.88 14.4
Total 25.08 25.08 25.08 25.08 25.08 125.4
Sub-Total (B.1) = 125.4
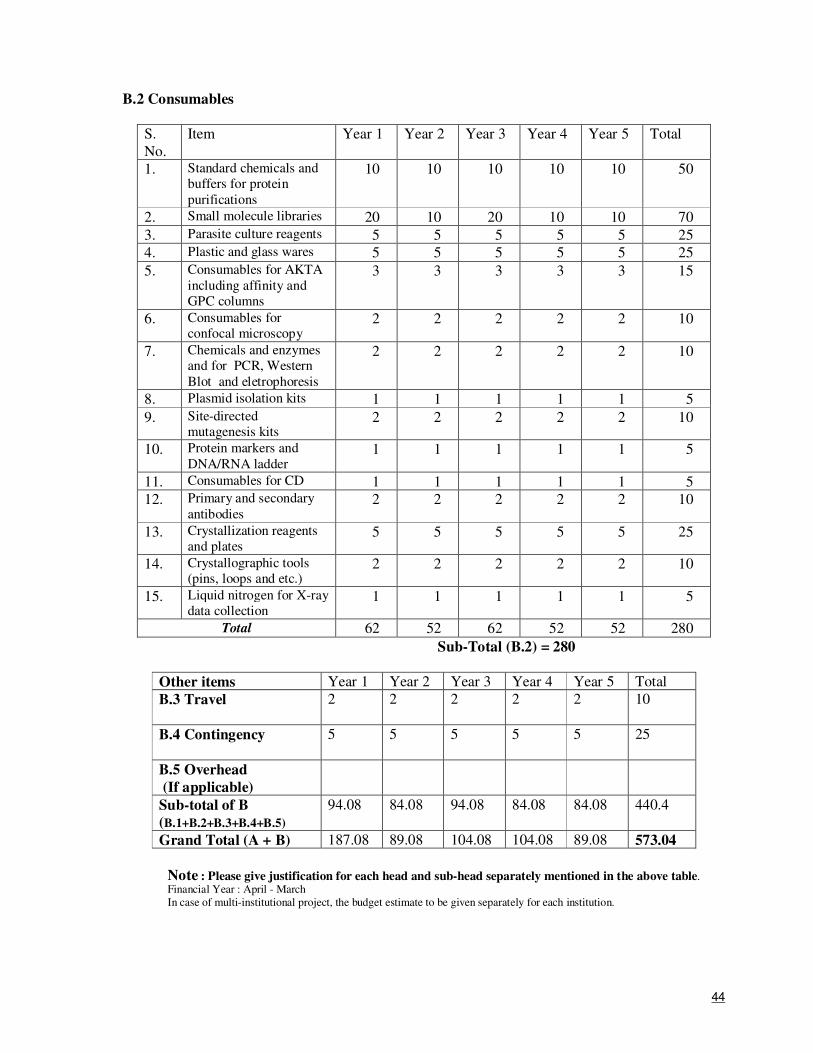
44
B.2 Consumables
S. No.
Item
Year 1 Year 2 Year 3 Year 4 Year 5 Total
1. Standard chemicals and buffers for protein purifications
10 10 10 10 10 50
2. Small molecule libraries 20 10 20 10 10 70 3. Parasite culture reagents 5 5 5 5 5 25 4. Plastic and glass wares 5 5 5 5 5 25 5. Consumables for AKTA
including affinity and GPC columns
3 3 3 3 3 15
6. Consumables for confocal microscopy
2 2 2 2 2 10
7. Chemicals and enzymes and for PCR, Western Blot and eletrophoresis
2 2 2 2 2 10
8. Plasmid isolation kits 1 1 1 1 1 5 9. Site-directed
mutagenesis kits 2 2 2 2 2 10
10. Protein markers and DNA/RNA ladder
1 1 1 1 1 5
11. Consumables for CD 1 1 1 1 1 5 12. Primary and secondary
antibodies 2 2 2 2 2 10
13. Crystallization reagents and plates
5 5 5 5 5 25
14. Crystallographic tools (pins, loops and etc.)
2 2 2 2 2 10
15. Liquid nitrogen for X-ray data collection
1 1 1 1 1 5
Total 62 52 62 52 52 280 Sub-Total (B.2) = 280
Other items Year 1 Year 2 Year 3 Year 4 Year 5 Total B.3 Travel
2 2 2 2 2 10
B.4 Contingency
5 5 5 5 5 25
B.5 Overhead
(If applicable)
Sub-total of B
(B.1+B.2+B.3+B.4+B.5)
94.08 84.08 94.08 84.08 84.08 440.4
Grand Total (A + B) 187.08 89.08 104.08 104.08 89.08 573.04
Note : Please give justification for each head and sub-head separately mentioned in the above table. Financial Year : April - March In case of multi-institutional project, the budget estimate to be given separately for each institution.

45
A. Justification equipments
The resources generated by this grant will be managed by the PI in an egalitarian fashion such that
they are accessible to other scientists at ICGEB. The PI will be entirely responsible for
procurement, installation, maintenance and upkeep of all scientific instruments bought on this grant.
The PI will regulate and ensure equitable distribution and utilization of equipments. All equipments
requested are essential for the proposed work, and no duplication or redundancy is evident in any
case.
Schrödinger software package
For molecular modeling and structure-based inhibitor design we require this state-of-the-art
software packages.
Computer workstations and accessories
Work stations are required for in silico virtual screening, molecular dynamics, X-ray data
collection, data processing, structure determination, refinement and analysis. Accessories like
backup devices are required for data storage.
Gradient PCR -96 well thermal cycler
Molecular cloning of AT-rich Pf genes can be difficult and therefore we are requesting a 96-well
format Gradient PCR machine.
Microfuge centrifuge and refrigerated Centrifuge
Centrifuge will be needed for plasmid preparation, protein concentration and routine biochemical
work.
Liquid nitrogen storage, transportation dewars and liquid nitrogen transfer pump
Dewars are required for crystal storage and transportation to synchrotron facility for data collection.
The LN2 transfer pump is required for transfer of liquid nitrogen from transportation dewars to
experimental dewars. Associated equipment and dewars are required to ensure proper functioning
of the existing X-ray facility.
Funds for upkeep and up gradation of existing X-ray facilities
For continuous operation of the X-ray diffraction facility we require funds for spare parts and
maintenance.

46
B.1. Manpower
The present project is interdisciplinary in its nature and therefore we will require highly talented
staff to cover the proposed aspects of research. Expertise required will span from basic cell biology
skills to inhibitor development. We therefore request series of well paid scientist positions in this
grant. We very much hope that trimming of this very important component of the proposal will not
be considered as it is clearly not possible to conduct high quality research with short-termed and
poorly paid personnel.
B.2. Consumables
The present proposal involves recombinant protein production, biochemical characterization, three-
dimensional structure determination and structure-based inhibitory discovery of 4 targets from the
protein translational machinery. Molecular cloning and protein expression experiments require
restriction enzymes, primers, fine chemicals, primers etc. Protein purification requires
NiNTA/amylose beads, cation and anion affinity columns, S75 and S200 gel filtration columns etc.
Cell biology and protein pulldown reagents are costly but essential for the stated objectives of the
grant. Procurement of compounds from libraries for testing in parasite viability assays will be
necessary, and can be an expensive enterprise. Generation of series of structural analogs (natural
and/or synthetic) of active compounds will require SAR studies. Finally, parasite culture work
requires media and plastic wares which are ever so expensive.
B.3. Travel
For attending national workshops and conferences.
B.4. Contingency
Contingency fund would be utilized for secretarial assistance and to fund transport of materials via FedEx and DHL couriers etc.
B.5. Overhead Charges As we understand, this is not applicable to ICGEB anymore.
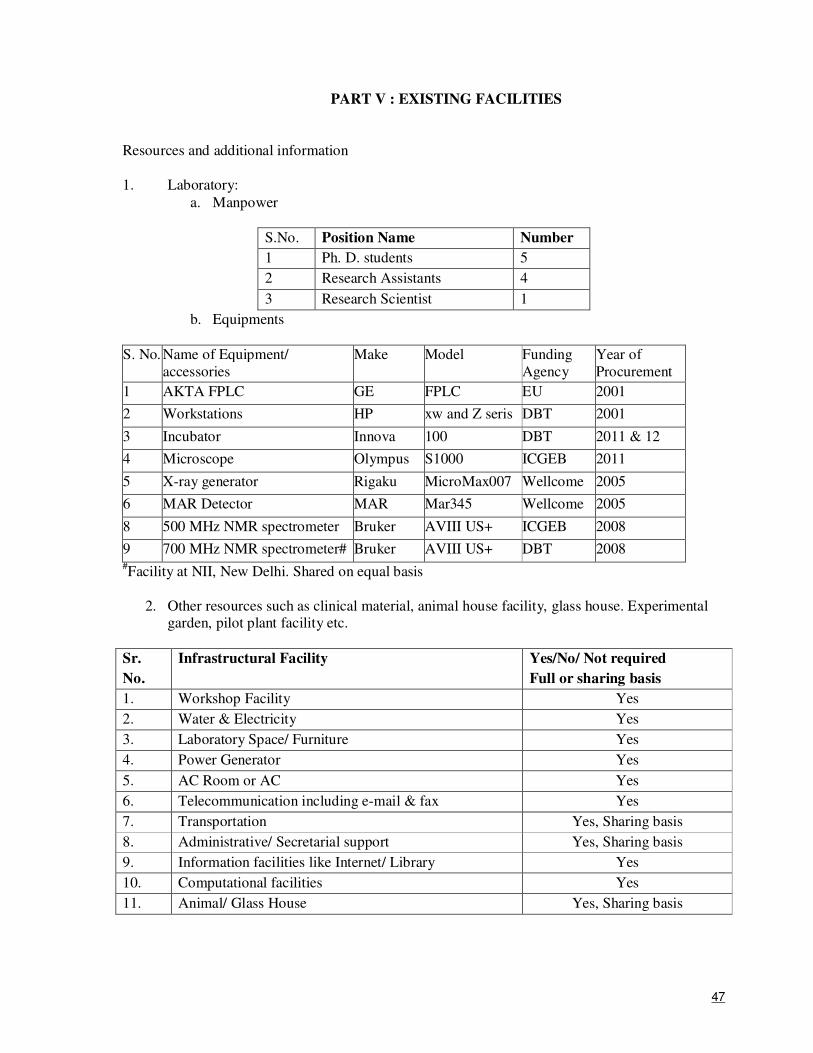
47
PART V : EXISTING FACILITIES
Resources and additional information 1. Laboratory:
a. Manpower
S.No. Position Name Number
1 Ph. D. students 5 2 Research Assistants 4 3 Research Scientist 1
b. Equipments S. No. Name of Equipment/
accessories Make Model Funding
Agency Year of Procurement
1 AKTA FPLC GE FPLC EU 2001
2 Workstations HP xw and Z seris DBT 2001
3 Incubator Innova 100 DBT 2011 & 12
4 Microscope Olympus S1000 ICGEB 2011
5 X-ray generator Rigaku MicroMax007 Wellcome 2005
6 MAR Detector MAR Mar345 Wellcome 2005
8 500 MHz NMR spectrometer Bruker AVIII US+ ICGEB 2008
9 700 MHz NMR spectrometer# Bruker AVIII US+ DBT 2008 #Facility at NII, New Delhi. Shared on equal basis
2. Other resources such as clinical material, animal house facility, glass house. Experimental garden, pilot plant facility etc.
Sr.
No.
Infrastructural Facility Yes/No/ Not required
Full or sharing basis
1. Workshop Facility Yes 2. Water & Electricity Yes 3. Laboratory Space/ Furniture Yes 4. Power Generator Yes 5. AC Room or AC Yes 6. Telecommunication including e-mail & fax Yes 7. Transportation Yes, Sharing basis 8. Administrative/ Secretarial support Yes, Sharing basis 9. Information facilities like Internet/ Library Yes 10. Computational facilities Yes 11. Animal/ Glass House Yes, Sharing basis

48
49
PART VII: PROFORMA FOR BIOGRAPHICAL SKETCH OF INVESTIGATORS
Provide the following information for the key personnel in the order listed on PART II. Follow this format for each person. DO NOT EXCEED THREE PAGES
Name: Amit Sharma
Designation: Group Leader
Department/Institute/University: Structural and Computational Biology, ICGEB
Date of Birth: 12.04.1968 Sex (M/F): M SC/ST: NA
Education (Post-Graduation onwards & Professional Career) Sl No. Institution
Place Degree Awarded
Year Field of Study
1. Northwestern University, Evanston, USA
PhD 1995 Protein Crystallography
2. Oxford University, UK Postdoctoral Fellow
2000 Structural Biology
A. Position and Honors
Position and Employment (Starting with the most recent employment)
Sl No. Institution Place
Position From (Date) To (date)
1. Structural and Computational Biology Group, ICGEB, New Delhi, India
Group Leader 2004 Continuing
2. India Research Group of the ICGEB in New Delhi, India
Wellcome Trust International Senior Research Fellow
2001 2006
3. Malaria Research Group, ICGEB, New Delhi, India
Research Scientist/Principal Investigator
2001 2004
4. Institute of Molecular Medicine, Laboratory of Molecular Biophysics and the Wellcome Trust Centre for Genomic Medicine, Oxford University, Oxford, UK
Lecturer and Junior Research Fellow
1995 2000
5. Northwestern University, Evanston, USA
National Institute of Health, National Research Scholar
1993 1995
6. Purdue University, USA Howard Hughes Medical Institute Undergraduate Summer Research Awardee
1989 1990
50
Honors/Awards
2011 Shanti Swarup Bhatnagar Prize for Biological Sciences 2010 ISCA Platinum Jubilee Lecture Award 2009 Goyal Young Scientist Prize 2008 National Bioscience Award, Dept. of Biotechnology 2008 S. Jaswant Singh Rai Memorial Award in Life Sciences 2007 Prof. Umakant Sinha Memorial Award of Indian Science Congress 2007 Awarded the LNJ Bhilwara Bhilwara Research Fellowship 2006 Bachawat Memorial Award in Biomedical Sciences 2006 B. M. Birla Prize in Biological Sciences 2006 Elected Fellow of the National Academy of Sciences, Allahabad, India 2005 MOT Iyengar National Science Award for Biomedical Research 2001-2006 Awarded Wellcome Trust International Senior Research Fellowship, UK 1996 -2000 Elected as Junior Research Fellow (Lecturer) in Biochemistry, St. John’s College,
Oxford University, UK
1993-1995 Awarded National Institute of Health National Research Scholarship, Northwestern University, USA
1989-1990 Awarded Howard Hughes Medical Institute Undergraduate Summer Research Fellowship, Purdue Univ., USA
Professional Experience and Training relevant to the Project
Dr. Sharma and his group uses multi-disciplinary techniques within modern biology -
including biochemistry, bioinformatics, cell biology, molecular biology, parasitology and protein
crystallography to unravel the mechanism of action of important parasite proteins. They aim to
highlight the principles that govern biological function of some key parasite proteins which play
crucial biological roles. The current focus of research includes critical parasite processes like
invasion of liver and red blood cells, nucleosome assembly, gametocytogenesis and cytoadherence.
They have elucidated crystal structures of key proteins from the sporozoite, asexual and sexual
parasite stages. At present, they have a ‘state-of-the-art’ protein crystallography unit at ICGEB,
New Delhi, where diffraction data are collected and processed for these analyses. They have also
initiated in silico inhibitor discovery program for the parasite proteins of interest. The objective is to
utilize crystal structure information for discovery of novel inhibitors using structure-based
computational approaches. They aim to close the experimental loop by testing the 'lead' compounds
directly against the malaria parasite. In summary, the research program spans experimental work
starting from parasite genomics to target and inhibitor discovery, with the aim of providing
potentials leads as anti-malarial.

51
B. Publications (Numbers only) 48
Selected peer-reviewed publications (Ten best publications in chronological order from India)
1. Bhatt, T. K., Khan, S., Dwivedi, V. P, Banday, M. M., Sharma, A., Chandele, A., Camacho, N., de Pouplana, L. R., Wu, Y., Craig, A. G., Mikkonen, A. T., Maier, A, G., Yogavel, M., Sharma, A. (2011) Malaria parasite tyrosyl-tRNA synthetase secretion triggers pro-inflammatory responses. Nature Communications, 2,530
2. Sharma, A., Sharma, A., Dixit, S., Sharma, A. (2011) Structural insights into thioredoxin-2: a
component of malaria parasite protein secretion machinery. Scientific Reports, 1:179
3. Bhatt, T. K., Yogavel, M., Wydau, S., Berwal, R., Sharma, A. (2010) Ligand-bound structures provide atomic snapshots for the catalytic mechanism of D-amino acid deacylase. Journal of
Biological Chemistry, 285, 5917-5930
4. Gill J., Yogavel M., Kumar A., Belrhali H., Jain S.K., Rug M., Brown M, Maier A.G., Sharma
A. (2009) Crystal structure of malaria parasite nucleosome assembly protein, distinct modes of protein localization and histone recognition. Journal of Biological Chemistry 284, 10076-10087
5. Hora, R., Bridges, D. J., Craig, A., Sharma, A. (2009) Erythrocytic casein kinase II regulates cytoadherence of Plasmodium falciparum-infected red blood cells. Journal of Biological
Chemistry, 284, 6260-6269 6. Sharma, A., Yogavel, M., Akhouri, R.R., Gill, J., Sharma, A., (2008) Crystal structure of
soluble domain of malaria sporozoite protein UIS3 in complex with lipid. Journal of Biological
Chemistry 283:24077-24088 7. Chandra, B.R., Yogavel, M., Sharma, A., (2007) Structural analysis of ABC-family
periplasmic Zinc binding protein provides new insights into mechanism of ligand uptake and release. Journal of Molecular Biology 367:970-982
8. Navagi, V.M., Mishra, P.C., Chandra, B.R., Sharma, A. (2006) The two Plasmodium
falciparum nucleosome assembly proteins play distinct roles in histone transport and chromatin assembly. Journal of Biological Chemistry 281:16978-16984
9. Singh, S., Hora, R., Belrhali, H., Chitnis, C.E., Sharma, A. (2006) Structural basis for
recognition of Duffy receptor by malaria parasite Duffy-binding-like domain. Nature 439:741-744
10. Sharma, A., Sharma, I., Kogkasuriyachai, D., Kumar, N. (2003) Structure of a gametocyte protein essential for sexual development in Plasmodium falciparum. Nature Structural Biology, 10, 197-203
.

52
List maximum of five of your recent publications relevant to the proposed area of work.
1. Khan, S., Sharma, A., Jamwal, A., Sharma, V., Pole, A. K., Thakur, K. K., Sharma, A. (2011)
Uneven spread of cis- and trans-editing aminoacyl-tRNA synthetase domains within translational compartments of P. falciparum. Scientific Reports, 1,188
2. Bhatt, T. K., Yogavel, M., Wydau, S., Berwal, R., Sharma, A. (2010) Ligand-bound structures provide atomic snapshots for the catalytic mechanism of D-amino acid deacylase. Journal of Biological Chemistry, 285, 5917-5930
3. Bhatt, T. K., Khan, S., Dwivedi, V. P, Banday, M. M., Sharma, A., Chandele, A., Camacho, N., de Pouplana, L. R., Wu, Y., Craig, A. G., Mikkonen, A. T., Maier, A, G., Yogavel, M., Sharma, A. (2011) Malaria parasite tyrosyl-tRNA synthetase secretion triggers pro-inflammatory responses. Nature Communications, 2,530
4. Gill J., Yogavel M., Kumar A., Belrhali H., Jain S.K., Rug M., Brown M, Maier A.G., Sharma A. (2009) Crystal structure of malaria parasite nucleosome assembly protein, distinct modes of protein localization and histone recognition. Journal of Biological Chemistry 284, 10076-10087.
5. Singh, S., Hora, R., Belrhali, H., Chitnis, C.E., Sharma, A., (2006) Structural basis for recognition of Duffy receptor by malaria parasite Duffy-binding-like domain. Nature 439:741-744
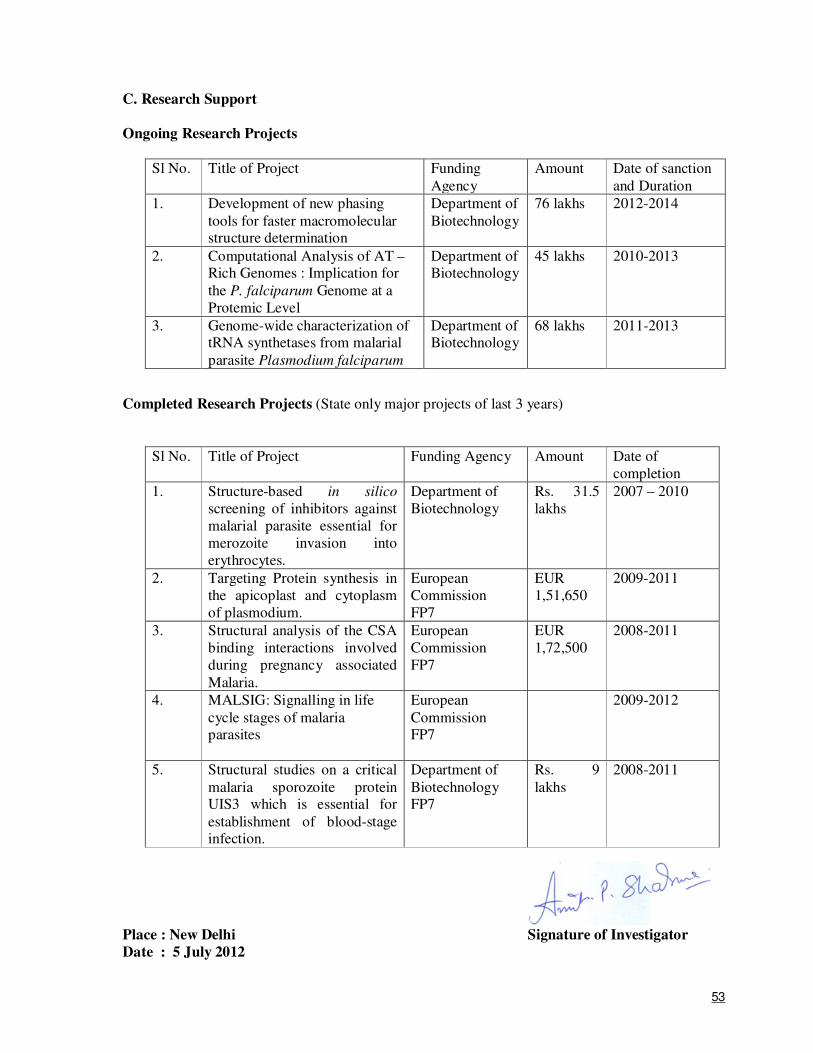
53
C. Research Support
Ongoing Research Projects
Sl No. Title of Project Funding
Agency Amount Date of sanction
and Duration 1. Development of new phasing
tools for faster macromolecular structure determination
Department of Biotechnology
76 lakhs 2012-2014
2. Computational Analysis of AT – Rich Genomes : Implication for the P. falciparum Genome at a Protemic Level
Department of Biotechnology
45 lakhs 2010-2013
3. Genome-wide characterization of tRNA synthetases from malarial parasite Plasmodium falciparum
Department of Biotechnology
68 lakhs 2011-2013
Completed Research Projects (State only major projects of last 3 years)
Sl No. Title of Project Funding Agency Amount Date of completion
1. Structure-based in silico screening of inhibitors against malarial parasite essential for merozoite invasion into erythrocytes.
Department of Biotechnology
Rs. 31.5 lakhs
2007 – 2010
2. Targeting Protein synthesis in the apicoplast and cytoplasm of plasmodium.
European Commission FP7
EUR 1,51,650
2009-2011
3. Structural analysis of the CSA binding interactions involved during pregnancy associated Malaria.
European Commission FP7
EUR 1,72,500
2008-2011
4. MALSIG: Signalling in life cycle stages of malaria parasites
European Commission FP7
2009-2012
5. Structural studies on a critical malaria sporozoite protein UIS3 which is essential for establishment of blood-stage infection.
Department of Biotechnology FP7
Rs. 9 lakhs
2008-2011
Place : New Delhi Signature of Investigator
Date : 5 July 2012


![Dissection-BKW · 2018. 6. 1. · Dissection. Wereplaceournaive c -sumalgorithmbymoreadvancedtime-memorytechniqueslike Schroeppel-Shamir[34]anditsgeneralization,Dissection[11],toreducetheclassicrunningtime.Wecall](https://img.pdfslide.us/doc/110x75/5ffc5cc4c887922f656f708b/dissection-bkw-2018-6-1-dissection-wereplaceournaive-c-sumalgorithmbymoreadvancedtime-memorytechniqueslike.jpg)


























