Embed Size (px)
Citation preview

POSTGRAD. MED. J. (1965), 41, 313
SOME BIOCHEMICAL ASPECTS OFTHE MYOPATHIES
B. P. HUGHES, B.Sc., Ph.D.Department of Chemical Pathology, The National Hospital, Queen Square, W.C.I.
SEVERAL general accounts of the biochemistryof the myopathies have recently appeared(e.g. Dreyfus and Schapira, 1962; Schapira andDreyfus, 1963; Pennington, 1964). Consequentlythis article will not attempt a comprehensivereview of the subject but will rather treatcertain topics in greater detail, especially thosewhich may have significant practical impli-cations, for instance the value of serum enzymeassays. A number of recent developments inthe field will also be mentioned.
Despite the considerable volume of publishedwork, elucidation of the fundamental bio-chemical abnormalities which underlie thedegenerative changes seen in the primarymyopathies remains an unsolved problem. Amajor difficulty has been to pick out anabnormality which is a specific accompanimentof a single disease process, even when it is aquestion of distinguishing between a primarymyopathy and a neurogenic muscle disorder.In this last respect, however, some progresshas resulted from a realisation that the findingof elevated serum activities of various tissueenzymes in some types of primary muscledisease serves to distinguish them fairly sharplyfrom the secondary myopathies. The distinctionis not always clear-cut, but is sufficiently markedto be of practical value, in particular for suchpurposes as the early diagnosis of Duchennemuscular dystrophy and for detecting carriersof the form which is inherited as a sex-linkedrecessive characteristic.Examples of muscle disorders in which there
appear to be specific biochemical changes areMcArdle's syndrome and certain other diseasesof carbohydrate metabolism.Most of this article will be devoted to a
discussion of these topics, but to provide thenecessary biochemical background it is neces-sary to deal briefly with some of the less specificbiochemical changes that occur.
Changes in Creatine and CreatinineMetabolism
It has been realised for more than 50 yearsthat some muscle disorders are accompaniedby an increased urinary output of creatine
(Levene and Kriszteller, 1909) and a decreasedoutput of its anhydride creatinine.
It has been shown that creatine is storedin the muscle as creatine phosphate but issynthesised elsewhere. Production of creatinephosphate is mediated by the enzyme creatine-adenosine triphosphate phosphotransferase, anenzyme, subsequently referred to by its trivialname, phosphocreatine kinase, which catalysesthe following reaction:Creatine + ATP , Creatine phosphate + ADPthe so-called Lohman reaction (Lohman, 1934).Creatine is formed by the breakdown, probablyspontaneous, of creatine phosphate. Whenmuscle degeneration occurs, capacity to storecreatine as the phosphate is reduced, whereasnormally the rate of synthesis is maintained.The resultant creatinaemia provokes a creatin-uria when the renal threshold of this substanceis exceeded. On the other hand, since creatinineis formed from creatine phosphate the urinaryoutput of creatinine falls. Because the creatin-uria is chiefly a result of reduced muscle mass,it is a pretty non-specific accompaniment ofmuscle wasting and of little help in diagnosis;thus Van Pilsum and Wolin (1958) found amarked creatinuria both in pseudohypertrophicmuscular dystrophy and in poliomyelitis.Before leaving the topic of creatine metabolismit may be noted that reversibility of thephosphocreatine kinase reaction provides ameans by which ATP can be resynthesisedfrom creatine phosphate. As it now seems fairlycertain that ATP breakdown is the source ofchemical energy needed for the functioningof the contractile system in muscle (e.g. Gergely,1964) creatine phosphate and phosphocreatinekinase play an extremely important part in thesupply of energy.Muscle Enzymes
Because an adequate supply of chemicalenergy is essential to the function of muscle,many investigators have compared the tissueconcentrations of enzymes involved in energyproduction in normal and diseased muscle.
Figure 1 indicates schematically the way inwhich chemical energy in the form of ATP
copyright. on A
pril 3, 2022 by guest. Protected by
http://pmj.bm
j.com/
Postgrad M
ed J: first published as 10.1136/pgmj.41.476.313 on 1 June 1965. D
ownloaded from

POSTGRADUATE MEDICAL JOURNAL
GLUCOSEHexokinasel
G - - P
FRUCTOSE Hexokinose F-PF .6-P
ATP
Phosphoglucomutase G -I-P
Acids]9 Stoges
PhosphoryloseDebronching Enzyme
. GLYCOGEN.
* H,0, CO,
Pyruvote
Lactate
FIG. 1.-Sources of energy in muscle. A schematicrepresentation of some major pathways for theproduction of ATP.
(adenosine triphosphate) is produced, startingwith glycogen.Glycqgen is a highly branched molecule
formed of glucose residues. The branchesconsist, of glucose units joined together in the1-4 positions (essentially a head-to-tail arrange-ment of the cyclic form of the glucose molecule).The branching points are produced byadditional links in the 1-6 positions. Theinitial degradation of the structure to glucose1-phosphate requires two enzymes, phosphory-lase to attack 1-4 links as far as a branch point,and the debranching enzyme (amylo-1,6-glucosidase) to attack the 1-6 links in orderthat further phosphorylase action can occur.As Fig. 1 indicates, glucose itself and fructosecan also enter the pathway of anaerobicglycolysis (the Emden-Myerhof pathway).Under conditions of maximal activity
anaerobic breakdown of carbohydrate, i.e.muscle glycogen, to lactate supplies most ofthe energy, but in resting muscle, contrary towhat was once thought to be the case, it seemslikely that aerobic oxidation of fatty acids orlipids meets a good deal of the tissue energyrequirements (e.g. Andres, Cader and Zierler,1956; Fritz, Davis, Holtrop and Dundee, 1958;Bass and Hudlicka, 1964).
If carbohydrate is fully oxidised much moreATP is produced per glucose residue than ifmetabolism is terminated at the pyruvate stage;
thus on complete oxidation of a glucose residue38 molecules of ATP are produced, whereasif the process stops at pyruvate only 2 moleculesare formed (Slater, 1960). The aerobic processis thus energetically much more efficient, butmay be limited during intense activity by therate at which oxygen can be transferred to thesite of ATP synthesis. Aerobic metabolismof carbohydrate and fatty acid requires theparticipation of the mitochondrion, in particularthe mitochondrial electron transport system,and terminates in reaction with oxygen trans-ported within the cell by myoglobin. Becauseof its greater efficiency, in muscles whosefunction is sustained moderate activity ratherthan short intense activity, the aerobic processpredominates, whereas in short activity musclethe anaerobic phase may be more important.This is reflected in the increased numbers ofmitochondria and increased concentration ofoxidative enzymes in the former. In animalssuch as the rabbit, the different types of muscle,red and white, are clearly differentiated,although the difference is really a statisticalone depending on the relative amounts ofmitochondria-rich red fibres, and white fibres,poor in mitochondria but rich in enzymes suchas phosphorylase (Stein and Padykula, 1962).In man the difference between muscles is lessobvious, but the existence of the two fibretypes has been demonstrated histochemically
314 June, 1965copyright.
on April 3, 2022 by guest. P
rotected byhttp://pm
j.bmj.com
/P
ostgrad Med J: first published as 10.1136/pgm
j.41.476.313 on 1 June 1965. Dow
nloaded from

HUGHES: Biochemical Aspects of the Myopathies(Dubowitz and Pearce, 1960) and it is possiblethat their reactions to disease may be somewhatdifferent, since in muscular dystrophy the fibresrich in oxidative enzymes tend to atrophy andthe phosphorylase-rich fibres tend to be hyper-trophied (Dubowitz and Pearce, 1961).The capability of muscle to supply its energy
requirements from anaerobic carbohydratebreakdown is one of its special biochemicalfeatures, so that the enzymes of the anaerobicphase of carbohydrate metabolism in normaland diseased muscle have received considerableattention, notably from Dreyfus, Schapira andtheir collaborators (e.g. Dreyfus, Schapira,Schapira and Demos, 1956). Other studieswhich may be cited are those of Vignos andLefkowitz (1959), Ronzoni, Berg and Landau(1960) and the more recent extensive inves-tigation of Heyck, Laudahn and Liiders (1963).
Before discussing the results of theseinvestigations it must be pointed out that certaindifficulties of interpretation beset measurementsof tissue enzyme concentration if we want toknow anything about the composition of theactual muscle. In muscle disease an increase,relative or absolute, in the concentration ofconnective tissue and fat frequently occurs andthis would change the overall tissue com-position even if the remaining muscle werenormal, so that a reference base unaffected bysuch an increase is needed. Lilienthal, Zierler,Folk, Buka and Riley (1950) suggested non-collagen nitrogen as being suitable, and thissuggestion has been widely adopted.To summarise the results obtained by the
investigators cited above we can say thatseveral of the glycogenolytic enzymes referredto non-collagen nitrogen, and also overallglycolytic activity, are frequently reduced inprimary myopathies perhaps to a third of theirnormal value (Dreyfus and Schapira, 1962),although Ronzoni and others (1960) found thathexokinase was little affected and that lactatedehydrogenase was actually increased. Thislatter finding is not however supported by thework of Heyck and others (1963). As towhether the various myopathies differed in thepattern of enzyme change accompanying them,results seem conflicting (c.f. Vignos andLefkowitz, 1959 and Heyck and others, 1963).A possible source of confusion may have beenthe use of different clinical classifications.Besides being unable to differentiate clearlybetween the various primary myopathies theresults of these investigations do not provideany clear distinction even between primaryand secondary myopathies.
Enzymes of oxidative metabolismInvestigation of the enzymes of the oxidative
phase of metabolism e.g. succinate dehydro-genase, cytochrome oxidase, have revealed littleor no change, when analyses have been referredto non-collagen nitrogen (Dreyfus and Schapira,1962). This is rather surprising in view of thefact that electron microscope studies (e.g. VanBreemen, 1960; Pearce, 1963), have shownconsiderable mitochondrial abnormality anddestruction, and many of the oxidative enzymesare considered to be located within themitochondria.
Isoenzymes in muscle diseaseIsoenzymes are different proteins which have
similar, but not necessarily identical, enzymicactivity. In muscle disease most attention hasso far centred around the lactate dehydrogenaseisoenzymes which may be readily separated byelectrophoresis, on agar for example (Wieme,1959), and which may also be distinguished bydifferences in their enzymic behaviour(Plummer, Elliott, Cooke and Wilkinson, 1963;Dawson, Goodfriend and Kaplan, 1964). Innormal human skeletal muscle 5 isoenzymes canbe distinguished which are referred to asLDH1-5 in order of decreasing anodicmobility. In some mature muscles at least theslow moving isoenzymes usually predominate,whereas in human foetal muscle and in animalred muscle chiefly the fast moving enzymesare found. Various authors (e.g. Dreyfus, De-mos, Schapira and Schapira, 1962; Lauryssens,Lauryssens and Zondag, 1964) have found thatthe electrophoretic pattern of isoenzymes frompatients with Duchenne muscular dystrophyresembles that of the foetus, and recentlyEmery (1964) has reported loss of slow-movingisoenzymes in female carriers of the disease.Dreyfus and others (1962) suggested that thisindicates a failure of maturation. Howeverother workers (Lauryssens and others, 1963;Brody, 1964) have found similar abnormalitiesin human neurogenic atrophy as well as inprimary muscle disease, and Brody (1964) hasdemonstrated a progressive change followingexperimental denervation in the guinea pig.Dawson and others (1964) by a differenttechnique have also shown similar changesfollowing denervation in the rabbit. At themoment, therefore, it seems likely that thechange is a largely non-specific reaction of thetissue to metabolic or functional disturbance,and according to some authors (Lauryssens andothers, 1963; Dawson and others, 1964) may
June, 1965 315copyright.
on April 3, 2022 by guest. P
rotected byhttp://pm
j.bmj.com
/P
ostgrad Med J: first published as 10.1136/pgm
j.41.476.313 on 1 June 1965. Dow
nloaded from

POSTGRADUATE MEDICAL JOURNAL
reflect a decline in anaerobic glycolysis. How-ever this field of investigation is now veryactive and the question cannot be regardedas settled.
It is worth noting that comparison of normaland diseased human muscle must be interpretedwith caution since Dawson and others (1964)have shown considerable differences betweennormal muscles in their content of the varioustypes of lactate dehydrogenase.Other Abnormalities
In addition to the topics mentioned abovenumerous other biochemical abnormalities havebeen described in diseased muscle, some per-haps of very considerable importance forunderstanding the degenerative process, anexample 'being the marked increase of varioushydrolytic enzymes. Nevertheless since manyof these are discussed in the reviews citedalready, they will not be considered further.However one recent observation of considerableinterest has been described by Watts andHooton (1965) who found that a purifiedphosphocreatine kinase derived from dystrophicmice, despite having the same electrophoreticproperties as the enzyme from normal animals,was only half as active. Phosphocreatine kinasepossesses two sulphydryl groups (-SH) in themolecule which are concerned with activityand, as a possible explanation for their results,these authors suggested that one of the -SHgroups might be inactivated in vivo. Since anumber of enzymes, including myosin ATPase,require intact sulphydryl groups for activity,any increased tendency towards SH inactivationmight have profound metabolic consequences,and it will be of great interest to see if futurework confirms such a possibility in the mousedisease and in human myopathies.Familial Periodic ParalysisRecent accounts of the biochemical changes
found in both hypo-and hyperkalemic familialperiodic paralysis have been given by McArdle(1964) and by Pearson (1964), so that thesebiochemically interesting conditions will notbe discussed here.
Defects of Muscle Glycogen MetabolismMyophosphorylase deficiency(McArdle's syndrome)McArdle (1951) reported the case of a man,
aged 30, who since childhood had sufferedmuscle pain, weakness and stiffness broughton by slight exercise. On investigation no risein blood lactate or pyruvate following exercise
could be detected, particularly after ischaemicexercise of the forearm muscles, when a 3 to4-fold rise is normal. Several other cases withsimilar but not identical, clinical and bio-chemical signs have been described (Schmidand Mahler, 1959; Mommaerts, Illingworth,Pearson, Guillory and Seraydarian, 1959;Schmid and Hamaker, 1961; Mellick, Mahlerand Hughes, 1962; Thomson, McLaurin andPrinias, 1963; Rowland, Fahn and Schottland,1963; Hockaday, Downey and Mottram, 1964).Additional features in some cases weremyoglobinuria after excercise (Schmid andMahler, 1959; Schmid and Hamaker, 1961;Rowland and others, 1963), and the latedevelopment of a limb girdle myopathy (Schmidand Mahler, 1959; Schmid and Hamaker, 1961).In all individuals who were tested for thisenzyme, there was a severe myophosphorylasedeficiency, possibly a complete absence in themajority, as shown by various techniques e.g.biochemical, histochemical and immunological.In addition to phosphorylase deficiency mostpatients had excessive amounts of glycogen intheir muscles.The phosphorylase deficiency appeared to be
confined to muscle so that for example, epine-phrine injection provoked the expected risein blood sugar indicating the presence of hepaticphosphorylase and normal mobilization of liverglycogen. Reference to Fig. 1 shows thatphosphorylase together with the debranchingenzyme brings about the initial stage ofglycogenolysis, so that its absence blocks theprocess and interferes with energy productionfrom anaerobic glycolysis. Utilization of glucose(or fructose) and fatty acids is unaffected, sothat slight or moderate muscular activity canbe sustained by glucose or fatty acids extractedfrom the blood; consequently many of thesepatients were able to walk slowly on levelground without restriction. Fast walking orrunning however was soon halted by severediscomfort and weakness. Excercise capabilitiesvaried somewhat from patient to patient (c.f.Mommaerts and others, 1969; Schmid andMahler, 1959) and for the same patient fromday to day (McArdle, 1951; Mellick and others,1962). This variability may possibly reflectdifferences in the efficiency with which bloodsugar and fatty acids are utilized. In thisconnection it has been found that a short-termimprovement in excercise tolerance can beachieved by inducing hyperglycaemia by, forexample, oral or intravenous glucose andintravenous glucagon (Schmid and Mahler,1959; Schmid and Hamaker, 1961; Mellick
316 June, 1965copyright.
on April 3, 2022 by guest. P
rotected byhttp://pm
j.bmj.com
/P
ostgrad Med J: first published as 10.1136/pgm
j.41.476.313 on 1 June 1965. Dow
nloaded from

HUGHES: Biochemical Aspects of the Myopathiesand others, 1962). Oral fructose may also havesimilar affect (Mellick and others, 1962;Thomson and others, 1963) but although it candirectly enter the glycolytic pathway (Fig. 1) itsprecise mode of action is a matter of discussion(Opie, Evans and Renold, 1962; Wolf, 1962).Despite the theoretical possibility of resultantincreased glycogen deposition in the muscle ithas been proposed that patients should takeoral fructose prior to unavoidable exertion(Mellick and others, 1962; McArdle, 1964),but this does not always seem to be effective(Hockaday and others, 1964).The demonstration of the presence of
excessive glycogen in conjunction with low orabsent phosphorylase has important biochemicalimplications. Originally the phosphorylasereaction was considered to be reversible, andcould therefore participate both in glycogensynthesis and breakdown. Although under theconditions used for assaying the enzyme in vitrothis is true (e.g. Sutherland and Wosilait, 1956),under physiological circumstances, glycogensynthesis is thought to take place by an irrever-sible process whereby glucose-l-phosphate anduridine triphosphate are converted to uridinediphosphate glucose and thence to glycogen,and which was first demonstrated by Leloirand Cardini (1957). It was later shown to occurin mammalian muscle by Robbins, Traut andLipmann (1959). As various authors havepointed out (e.g. Lamer and Villar-Palasi, 1959;Mommaerts and others, 1959) the excessglycogen usually present in McArdle's syndromeprovides the strongest evidence for the inde-pendence of muscle glycogen synthesis andbreakdown in man.Although in some cases muscle phosphory-lase seems to be entirely absent this may not
always be so, thus in the case reported byHockaday and others (1964) some inorganicphosphate was liberated when muscle homo-genates were incubated with glucose-l-phos-phate although much less than normal. Engel,Eyerman and Williams (1963) report two cases,brother and sister, with late development ofsymptoms and normal muscle glycogen in whomphosphorylase deficiency was complete in oneindividual but only partial in the other.
Other diseases of glycogen metabolismBesides phosphorylase deficiency, other rare
defects in glycogen metabolism have been re-ported, but only in some is muscle affected.These reports have been reviewed by Stettenand Stetten (1960) and also, for example, byRowland and others (1963).
Before leaving the subject of the glycogendiseases it is worth noting that the simplemeasurement of venous lactate rise afterischaemic exercise of the forearm will usuallyyield valuable information for diagnostic pur-poses, and the finding of a normal result oftenhelps to exclude the existence of a defect inanaerobic glycolysis which may sometimesbe suggested by the clinical picture. Additionalinformation as to whether any abnormality re-vealed in this way, is confined to muscle or ismore general, can be obtained by testing theblood sugar response to epinephrine orglucagon.
Other rare specific enzyme abnormalities inmuscle will doubtless come to light, but whetherdefects of this nature, not of course necessarilyin the sphere of carbohydrate metabolism, willeventually be found in the more common myo-pathies cannot be foreseen. However, the exist-ence of disorders such as the one just describedlends some encouragement to the search.
Serum EnzymesSibley and Lehninger (1949) whilst carrying
out a survey of serum aldolase levels in animaltissues noted that high serum levels were ob-served in two patients with progressive musculardystrophy. This observation was later followedup by various authors (e.g. Dreyfus, Schapiraand Schapira, 1958) who showed that besidesaldolase, enzymes such as lactate dehydro-genase, LDH, aspartate aminotransferase, AspT(formerly GOT), alanine aminotransferase,AIT (formerly GPT) and a number of otherswere also often raised in muscular dystrophyand in the myosites, particularly acute derma-tomyositis. By contrast in conditions involvingneurogenic atrophy, even when rapid as inpoliomyelitis, serum enzyme levels were normalor only transiently raised (Evans and Baker,1957; White, 1959). Comparison of resultsfor different enzymes indicated that serum aldo-lase levels were most consistently raised inprimary muscle disease. The proportion ofcases in whom abnormal levels were foundvaried according to their clinical classification,so that, for example, Thomson, Leyburn andWalton (1960) showed that all patients withmuscular dystrophy of the Duchenne type witha sex-linked recessive mode of inheritance, ex-cept very advanced cases had grossly abnormalserum aldolase levels, whereas the proportionwas smaller in limb-girdle cases and least ofall in the facio-scapulo-humeral form; in myo-tonic dystrophy, serum levels were not always
June, 1965 317copyright.
on April 3, 2022 by guest. P
rotected byhttp://pm
j.bmj.com
/P
ostgrad Med J: first published as 10.1136/pgm
j.41.476.313 on 1 June 1965. Dow
nloaded from

POSTGRADUATE MEDICAL JOURNAL
raised. Chung, Morton and Peters (1960) re-ported similar findings.Of considerable interest was the observation
of Thompson and others (1960) that inDuchenne muscular dystrophy the degree ofaldolasemia diminished with age or durationof symptoms, and in advanced cases reachednormal or near normal values. Although theevidence was less convincing, the same appearedto be true for the facio-scapulo-humeral formand for dystrophia myotonica, but for the limb-girdle form no such relationship between dura-tion of symptoms and degree of aldolasemiaexisted.An important advance in this field was made
possible by the observation of Ebashi, Toyo-kura, Momoi and Sugita (1959) that serumphosphocreatine kinase levels were also greatlyraised in muscular dystrophy patients. Resultsobtained by later workers (e.g. Dreyfus,Schapira and Demos, 1960; Hughes, 1962) fullyconfirmed this finding, and it has become ap-parent that there are certain advantages in usingphosphocreatine kinase for the study of muscledisease. Firstly it is found to be a moresensitive index of muscle disease even thanaldolase and it is also more specific. Thus,because it does not occur to any appreciableextent in liver, serum levels are not raised inliver disease, whereas aldolase may be grosslyabnormal. A more important practical ad-vantage follows from the fact that in man butnot in some other species, the erythrocytescontain extremely little phosphocreatine kinase(Solvonuk, McRae and Collier, 1956) so thatassays of activity in haemolysed specimens arepossible in circumstances when aldolase valueswould be valueless. Data illustrating thesepoints are given in Table 1.
Because of these advantages it is proposed todiscuss serum enzyme findings in muscle diseasechiefly in terms of phosphocreatine kinase, al-though whether phosphocreatine kinase is al-ways the enzyme of choice may still be opento doubt. First however we must considernormal levels.
Normal individualsHughes (1962) reported that there was a
statistically significant difference between themean serum activities for normal men andwomen. This difference has not however beensubstantiated by other authors. Thus althoughGriffiths (1964) reports a rather higher meanactivity for male children and adults, as com-pared with females, the differences were smallerand not statistically significant. Pearce, Pen-
TABLE 1*ALDOLASE AND PHOSPHOCREATINE KINASE (PCK)IN HAEMOLYSED SERUM FROM NORMAL INDIVIDUALSAND IN SERUM FROM PATIENTS WITH NON-MUSCULAR
DISORDERS.Sex PCK Aldolase
(I.U./litre) (DHA units)Infective hepatitis M 15.0 22.0Obstructive jaundice M 26.7 48.0Aspirin poisoning F 40.0 58.8Normal (haemolysed serum) F 20.1 46.0Normal (haemolysed serum) F 18.4 52.5Units and normal ranges (x ± 2a)Phosphocreatine kinase (PC0K):
Units: International Units/litre of serum at 37°1 International Unit (I.U.) = 1 / mole creatineliberated/min.1 A mole/ml./hr. = 16.7 I.U./litreRanges: Males 10-67; Females 10-44
Aldolase:Units: Dihydroxyacetone (DHA) units perml. of serum (Friedman and Lapan, 1958)
Range: Both sexes 4.0-21.0* Data recalculated from Hughes (1963b)
nington and Walton (1964a) in a smaller seriesagain found no significant sex-difference. Themean values for women found by all these threeauthors, 1.5-1.7 pM/ml./hr. (25-28 I.U./litre),were however in good agreement and the some-what higher value for males found by Hughes(1962), even if confirmed, would seldom beof much practical significance.The authors quoted above were all using
very similar technical methods for assaying theenzyme; other workers (e.g. Colombo, Rich-terich and Rossi, 1962; Fowler and Pearson,1964) have used different techniques so thatcomparison of absolute values is not possible.However relative values found in normal sub-jects and in patients suffering from variousforms of muscle disease can be comparedunless the comparisons are vitiated by the lowsensitivity of the method employed. Fowlerand Pearson (1964) admit that this may havebeen the case in connection with some of theirearlier observations.
Various authors have reported increasedserum levels of a number of enzymes followingexercise (e.g. Schlang, 1961; Halonen and Kont-tinen, 1962) the rise being greater in untrainedas compared with trained subjects (Fowler,Chowdbury, Pearson, Gardner and Bratton,1962). If a substantial rise occurs after moder-ate exercise then obviously care needs to betaken when interpreting results obtained onuncontrolled subjects. Richterich, Rosin, Aebiand Rossi (1963) have reported that a markedrise of phosphocreatine kinase may also occurafter exercise, but Hughes (1963a) and Pearceand others (1964a) were unable to demonstrate
318 June, 1965copyright.
on April 3, 2022 by guest. P
rotected byhttp://pm
j.bmj.com
/P
ostgrad Med J: first published as 10.1136/pgm
j.41.476.313 on 1 June 1965. Dow
nloaded from
HUGHES: Biochemical Aspects of the Myopathies
any increase after a relatively short period ofvigorous exercise. However Hughes (1963a)suggested that prolonged periods of vigorousexercise might produce a moderate rise andalso that apparently normal subjects may some-times exhibit serum activities above the limitof normal. Except when the assay is beingused for the detection of carriers of the sex-linked recessive form of muscular dystrophyit seems unlikely that these considerations willbe of much practical importance.Duchenne muscular dystrophyThe term Duchenne muscular dystrophy re-
fers both to the type with the sex-linked re-cessive mode of inheritance and the usuallyrather more benign form, thought to be in-herited as an autosomal recessive trait(Dubowitz, 1960; Walton, 1962).
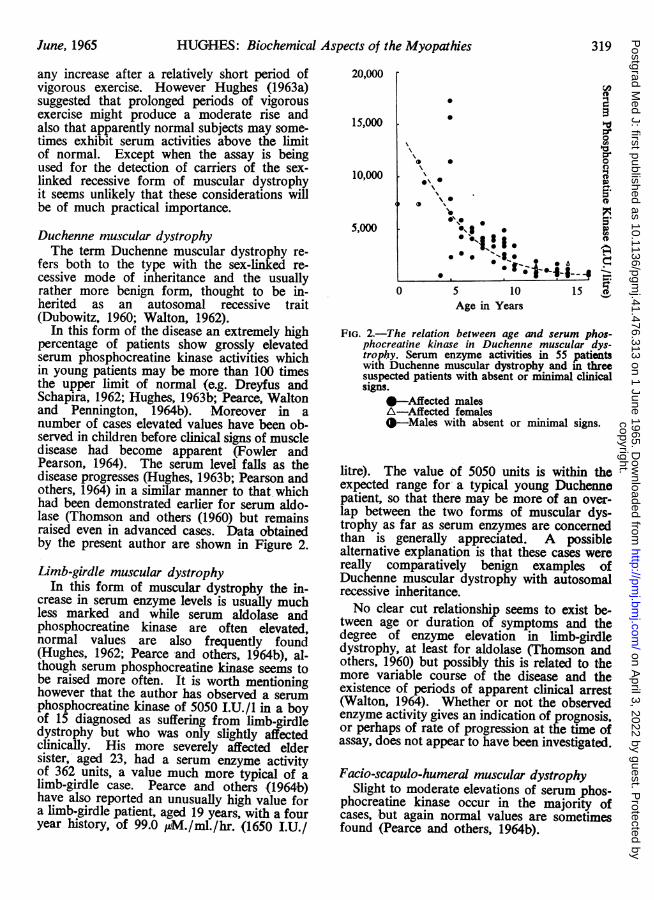
In this form of the disease an extremely highpercentage of patients show grossly elevatedserum phosphocreatine kinase activities whichin young patients may be more than 100 timesthe upper limit of normal (e.g. Dreyfus andSchapira, 1962; Hughes, 1963b; Pearce, Waltonand Pennington, 1964b). Moreover in anumber of cases elevated values have been ob-served in children before clinical signs of muscledisease had become apparent (Fowler andPearson, 1964). The serum level falls as thedisease progresses (Hughes, 1963b; Pearson andothers, 1964) in a similar manner to that whichhad been demonstrated earlier for serum aldo-lase (Thomson and others (1960) but remainsraised even in advanced cases. Data obtainedby the present author are shown in Figure 2.
Limb-girdle muscular dystrophyIn this form of muscular dystrophy the in-
crease in serum enzyme levels is usually muchless marked and while serum aldolase andphosphocreatine kinase are often elevated,normal values are also frequently found(Hughes, 1962; Pearce and others, 1964b), al-though serum phosphocreatine kinase seems tobe raised more often. It is worth mentioninghowever that the author has observed a serumphosphocreatine kinase of 5050 I.U./l in a boyof 15 diagnosed as suffering from limb-girdledystrophy but who was only slightly affectedclinically. His more severely affected eldersister, aged 23, had a serum enzyme activityof 362 units, a value much more typical of alimb-girdle case. Pearce and others (1964b)have also reported an unusually high value fora limb-girdle patient, aged 19 years, with a fouryear history, of 99.0 ptM./ml./hr. (1650 I.U./
20,000
15,000
10,000
5,000
* aO
* *0
\\ ·
*e· · '
O 5 10 15 0Age in Years
FIG. 2.-The relation between age and serum phos-phocreatine kinase in Duchenne muscular dys-trophy. Serum enzyme activities in 55 patientswith Duchenne muscular dystrophy and in threesuspected patients with absent or minimal clinicalsigns.
--Affected malesA-Affected females--Males with absent or minimal signs.
litre). The value of 5050 units is within theexpected range for a typical young Duchennepatient, so that there may be more of an over-lap between the two forms of muscular dys-trophy as far as serum enzymes are concernedthan is generally appreciated. A possiblealternative explanation is that these cases werereally comparatively benign examples ofDuchenne muscular dystrophy with autosomalrecessive inheritance.No clear cut relationship seems to exist be-
tween age or duration of symptoms and thedegree of enzyme elevation in limb-girdledystrophy, at least for aldolase (Thomson andothers, 1960) but possibly this is related to themore variable course of the disease and theexistence of periods of apparent clinical arrest(Walton, 1964). Whether or not the observedenzyme activity gives an indication of prognosis,or perhaps of rate of progression at the time ofassay, does not appear to have been investigated.
Facio-scapulo-humeral muscular dystrophySlight to moderate elevations of serum phos-phocreatine kinase occur in the majority of
cases, but again normal values are sometimesfound (Pearce and others, 1964b).
June, 1965 319copyright.
on April 3, 2022 by guest. P
rotected byhttp://pm
j.bmj.com
/P
ostgrad Med J: first published as 10.1136/pgm
j.41.476.313 on 1 June 1965. Dow
nloaded from

POSTGRADUATE MEDICAL JOURNAL
Dystrophia myotonicaMarkedly raised levels of serum phospho-
creatine kinase are seldom observed but a slightto moderate degree of elevation is frequentlyseen. Thus Pearce and others (1964b) reportabnormal values in 4 out of 7 cases and Ihave found the same in 4 out of 8 patients.The proportion of patients showing abnormal
values of serum phosphocreatine kinase maybe greater than is found for other enzymes.Thus Thomson and others (1960) found only2 out of 12 cases who showed a serum aldolaseabove the upper limit of normal.
Ocular myopathyThere appears to be little information about
serum enzyme levels in this disorder. However,in serum examined in our laboratory frompatients diagnosed as suffering from this con-dition, slight to moderate elevation was foundin 4 out of 7 specimens.Polymyositis and dermatomyositisThere are conflicting reports on the frequency
with which raised serum values of phospho-creatine kinase are found in patients withvarious forms of polymyositis and dermatomy-ositis. Thus Pearce and others (1964b) findthat the level is raised so infrequently in adultsas to be of no diagnostic value. HoweverHughes (1962) has reported very high levelsfor phosphocreatine kinase and aldolase intwo cases and Richterich and others (1963)claim that activities of as much as 500 timesthe upper limit of normal are encountered.Recently the author has observed a value of17000 I.U./1. in a case of dermatomyositis whichfollowing steroid therapy fell to 28.5 units,i.e., well within normal limits (Table 1); and ina chronic case with a 4-year history who im-proved on treatment the enzyme level fell fromjust above normal limits in the pre-treatmentperiod to a level again well within normal limitsafterwards. Fowler and Pearson (1964) havepresented data on successive serum phospho-creatine kinase estimations during the courseof treatment and showed a progressive fall tonormal limits. Pearson (1964) comments thatboth aldolase and creatine kinase are sensitiveindicators of the disease process in acute poly-myositis. Thus although there is no doubt thatserum phosphocreatine kinase may be greatlyelevated in cases of dermatomyositis and poly-myositis, and in the author's limited experiencesome elevation is frequently found, its relativediagnostic efficiency as compared with, forexample, aldolase, requires further study.
Fortunately as a means of assessing theeffects of treatment it appears likely that anyenzyme whose serum level is initially foundto be raised may be employed. Thus besidesphosphocreatine kinase, serum AspT (deMoragas, Parry and Fleicher, 1957) andaldolase (Thompson and Vignos, 1959; Gautierand Richterich, 1963) have been shown todecline towards normal values when therapy isattended by clinical improvement, althoughimprovement in muscle strength may follownormalization of the serum enzyme picture onlyafter an interval of 3 to 4 weeks and likewisea subsequent rise may anticipate a clinicalrelapse by some weeks (Pearson, 1964).Thyroid diseaseVarious workers (e.g. Griffiths, 1963; Saito,
Hibi, Kawazura and Fukuyama, 1963) havereported that serum phosphocreatine kinase isfrequently elevated in hypothyroidism andreturns to normal following replacement therapy(Saito and others, 1963). This is of some interestas myopathic symptoms may be associatedwith myxoedema (McArdle, 1964). Whetheror not serum enzyme abnormalities are com-monly found in hyperthyroid conditions doesnot appear to have been systematically studied.Unpublished observations of the author'sindicate however that normal values may beobserved in acute thyrotoxicosis with markedweakness, but are too few to permit anygeneralization.Neurogenic muscular atrophy
Raised levels of serum enzymes, such asaldolase and the transaminases, are seldom afeature of disorders in which muscle weaknessor wasting is secondary to a neurogenic lesion(see for example Evans and Baker, 1957;Dreyfus, Schapira and Schapira, 1958). Hughes(1963b) and Pearson and others (1964b) haveshown that the same is true of phosphocreatinekinase. However it should be noted that thedistinction between primary and secondarymyopathies in this respect is not absolute.Hughes (1963b) presented data for threepatients in whom serum phosphocreatine kinasewas raised, in one by a substantial amount,but in whom histological and other evidencestrongly suggested denervation.
Further experience has confirmed theimpression that there exists a minority ofpatients with muscle disorders in whom strongevidence of a neurogenic lesion exists, whonevertheless show a serum enzyme elevation.Although no obvious common denominator is
320 June, 1965copyright.
on April 3, 2022 by guest. P
rotected byhttp://pm
j.bmj.com
/P
ostgrad Med J: first published as 10.1136/pgm
j.41.476.313 on 1 June 1965. Dow
nloaded from

HUGHES: Biochemical Aspects of the Myopathies
yet evident most of these patients exhibitedunusual clinical features, whereas enzymeabnormalities in classical cases of neurogenicatrophy are rare. Of interest in this connectionis a report by Chung and others (1960) of apatient with a myopathic electromyogram,neuropathic biopsy and fibrillation who showeda definite hyperaldolasemia and was consideredto represent a mixed myopathy and Charcot-Marie-Tooth disease.The practical value of serum enzyme
estimations in muscle disease can be summarisedas follows:
(a) They provide a powerful means of earlydiagnosis of Duchenne muscular dystrophyeven before clinical signs are apparent and,because elevations in neurogenic atrophy arerelatively uncommon, allow a fairly certaindifferential diagnosis in young children betweenthis condition and, for example, Werdnig-Hoffmann's disease. The ability to recogniseearly muscular dystrophy by allowing inves-tigation of the first changes to occur in themuscle, before they are obscured by theproliferation of secondary changes, providesconsiderable hope that the essential geneticdefect may eventually be uncovered. For thisapplication, apart from phosphocreatine kinase,various enzymes are suitable and the choicecan depend largely on convenience, providedtheir lower specificity is remembered.
(b) In other primary muscle diseasesalthough serum enzyme determinations do nothave such a decisive value, nevertheless inconjunction with the clinical evidence they canbe of considerable help in arriving at adiagnosis. Whereas phosphocreatine kinase isprobably the most sensitive index of muscledisease, this may not necessarily be so in thecase of acquired myopathies i.e. the myosites.
(c) Successive enzyme estimations are usefulin assessing the effect of therapy in poly-myositis and dermatomyositis. Here again thechoice of enzyme may often be one of con-venience e.g. availability of a laboratory ableto carry out the assay.Detection of Carriers of the Sex-linkedDuchenne Muscular Dystrophy
In 1960 Chung and others reported that asmall proportion (3 out of 21) of certaincarriers of the sex-linked Duchenne form ofmuscular dystrophy showed a raised serumaldolase activity; carriers of the limb-girdleform were quite normal in this respect.Schapira, Dreyfus, Schapira and Demos (1960)found that both aldolase and phosphocreatine
kinase was raised in a proportion of the mothersof children with the sex-linked form and thatraised values were found more often in caseswhere the family history was positive. Leyburn,Thomson and Walton (1961), however, wereunable to detect any serum aldolase abnor-malities in known and possible carriers.
Later work (e.g. Hughes, 1962, 1963a;Barwick, 1963; Richterich and others, 1963)confirmed the observation of Schapira andothers (1960) that enzyme abnormalities werefound sufficiently frequently to allow thedetection of many Duchenne carriers by thismeans and indicated that phosphocreatinekinase was the most useful enzyme for thepurpose. Although results obtained in variouslaboratories differ somewhat, it appears that60-80% of certain or probable carriers of thesex-linked Duchenne muscular dystrophyexhibit a serum phosphocreatine kinase abnor-mality, but a minority are quite normal andcannot be detected in this way. Recentlyhowever Wilson, Evans and Carter (1965) usinga more refined statistical treatment claim tobe able to detect 90% of carriers on the basisof three separate assays on each individual.Although as a diagnostic test for the carrierstate the enzyme assay is not completelysuccessful, taken together with the known modeof inheritance, a reasonable assessment can bemade of the risk that a female relative, a sisterfor example, is a carrier. It must be emphasisedthat because healthy individuals may occasion-ally show a transient rise of serum enzymelevels above the upper limit of normal, it isadvisable to carry out the test on more thanone occasion when the results are to be usedto assist in genetic counselling (see also Wilsonand others, 1965). Other abnormalities inaddition to those of serum enzymes have beenreported to be present in carriers, thus van denBosch (1963) claimed that myopathic electro-myographic changes could be demonstrated, andit has recently become clear that histologicalchanges are sometimes found (Emery, 1963;Pearson, Fowler and Wright, 1963) and thatoccasionally clinical signs may be present(Emery, 1963). An explanation of these findings(Emery, 1964a; Pearson and others, 1963) hasbeen suggested on the basis of the so-calledLyon hypothesis (Lyon, 1961) of the biologicallyinactive X-chromosome.By combining serum enzyme, histological,
electrophysiological and other methods ofinvestigation the proportion of carriers who canbe reliably detected may prove to be con-siderably higher than is at present possible.
June, 1965 321copyright.
on April 3, 2022 by guest. P
rotected byhttp://pm
j.bmj.com
/P
ostgrad Med J: first published as 10.1136/pgm
j.41.476.313 on 1 June 1965. Dow
nloaded from

322 POSTGRADUATE MEDICAL JOURNAL June, 1965
Investigations on carriers of other types ofmuscular dystrophy have so far proved negative.General Considerations Arising FromSerum Enzyme AbnormalitiesWe have seen earlier that many tissue enzyme
levels are low or normal in both primary andsecondary muscle disease. This is certainlytrue of those enzymes most consistently foundto have increased activity in the serum ofmyopathic patients. Assuming that the rateof clearance of a particular enzyme from theserum is not diminished then an increasedoutflow from the cell is indicated, and thus anincreased permeability of the cell membrane,which seems to be particularly marked inmuscular dystrophy of the Duchenne type andin acute dermatomyositis. By contrast, inneurogenic atrophy the permeability increaseis at most slight.The fact that the serum concentration of
ussue enzymes falls as the disease progressescorrelates quite closely with the amount ofmuscle which remains (Thompson and Vignos,1959), although it is possible that outflow isalso reduced when physical activity becomesrestricted (Thomson, 1962) and that this is anadditional factor.Two general questions may be asked, firstly
is the loss of tissue enzymes a major causeof degeneration, and, secondly, does the in-creased permeability of the cell membrane de-note that the membrane is the site of the prim-ary, genetically determined defect? Pennington(1964) has pointed that although not all thenecessary information is available, the amountof aldolase lost from the affected muscle perday is probably only a very small fraction ofthe amount present. Consequently, although itmay be a factor it is unlikely that tissue enzymeloss is, in itself, a major cause of degeneration.Whether or not a primary membrane defect islikely to be the ultimate cause of muscledegeneration in any of the mvoDathies is opento argument since, as Zierler (1958) has shown,there is good evidence that disturbance causedby various factors such as anoxia, glucose lackand metabolic poisons can cause an increasedoutflow of intracellular enzymes. However,although it may be naive to sutpose that theserum enzyme findings denote the presence ofa primary membrane defect, the importanceof the various cellular membrane structures,e.g. the sarcolemma, the sarcoplasmic reticulumand the mitochondrial membranes, in the lightof recent work on muscle biochemistry, andon the biochemistry of other cells, is such that
a careful study of their chemical compositionin health and disease and its relation to functionmight well throw valuable light on the ultimatenature of the disease process.The author is a research fellow of the Muscular
Dystrophy Group of Great Britain to whom he isindebted for financial support.
REFERENCESANDRES, R., CADER, G., and ZIERLER, K. L. (1956):
Quantitatively Minor Role of Carbohydrate inOxidative Metabolism by Skeletal Muscle in IntactMan in the Basal State: Measurement of 02 andGlucose Uptake and 002 and Lactate Productionin the Forearm, J. clin. Invest., 35, 671.
BARWICK, D. D. (1963): Investigations of the CarrierState in the Duchenne Type of Dystrophy. Researchin Muscular Dystrophy. The Proceedings of theSecond Symposium January 1963, p. 10. London:Pitman Medical.
BASS, A., and HUDLICKA, 0. (1964): Interrelationsbetween Metabolism and Blood Flow in Normaland Denervated Dog Gastrocnemius Muscle at Restand During Stimulation, Physiol. bohemoslov., 13,48.
BRODY, I. A. (1964): The Significance of LactateDehydrogenase Isoenzymes in Abnormal HumanSkeletal Muscle, Neurology (Minneap.), 14, 1091.
CHUNG, C. S., MORTON, N. E., and PETERS, H. A.(1960): Serum Enzymes and Genetic Carriers inMuscular Dystrophy, Amer. J. hum. Genet., 12, 52.
COLOMBO, J. P., RICHTERICH, R., and Rossi, E.(1962): Serum Kreatin Phosphokinase: Bestim-mung und Diagnostische Bedeutung, Klin. Wschr.,40, 37.
DAWSON, D. M., GOODFRIEND, T. L., and KAPLAN,N. 0. (1964): Lactic Dehydrogenases: Functionsof the Two Types, Science, 143, 929.
DE MORAGAS, J. M., PERRY, H. 0., and FLEICHER,G. A. (1957): Serum Glutamic Oxalacetic Tran-saminase in Dermatomyositis, J. Amer. med. Ass.,165, 1936.
DREYFUS, J. C., DEMOS, J., SCHAPIRA, F., andSCHAPIRA, G. (1962): La Lacticod6shdyrogenaseMusculaire chez le Myopathe: PersistanceApparente du Type Foetal, C.R. Acad. Sci. (Paris),254, 4385.
DREYFUS, J. C., and SCHAPIRA, G. (1962): Bio-chemistry of Hereditary Myopathies, Springfield,Illinois: Charles C. Thomas.
DREYFUS, J. C., SCHAPIRA, G., and DEMOS, J. (1960):Etude de la Cr6atine Kinase Serique chez lesMyopathies et Leurs Families, Rev. franc. Etud.clin. biol., 5, 384.
DREYFUS, J. C., SCHAPIRA, G., and SCHAPIRA, F.(1958): Serum Enzymes in the Physiopathology ofMuscle, Ann. N.Y. Acad. Sci., 75, 235.
DREYFUS, J. C., SCHAPIRA, G., SCHAPIRA, F., andDEMOS, J. (1956): Activit6s Enzymatiques duMuscle Humain. Recherches sur la BiochimieCompar6e de l'Homme Normal et Myopathiqueet du Rat, Clin. chim. Acta, 1, 434.
DUBOWITZ, V. (1960): Progressive MuscularDystrophy of the Duchenne Type in Females andits Mode of Inheritance, Brain, 83, 432.
DUBOWITZ, V., and PEARSE, A. G. E. (1960): AComparative Histochemical Study of OxidativeEnzymes and Phosphorylase Activity in SkeletalMuscle, Histochemie, 2, 105.
copyright. on A
pril 3, 2022 by guest. Protected by
http://pmj.bm
j.com/
Postgrad M
ed J: first published as 10.1136/pgmj.41.476.313 on 1 June 1965. D
ownloaded from

June, 1965 HUGHES: Biochemical Aspects of the Myopathies 323
DUBOWITZ, V., and PEARSE, A. G. E. (1961): EnzymicActivity of Normal and Diseased Human Muscle:a Histochemical Study, J. Path. Bact., 81, 365.
EBASHI, S., TOYOKURA, Y., MAMOI, H., and SUGITA,H. (1959): High Creatine Phosphokinase Activityof Sera of Progressive Muscular Dystrophy Patients,J. Biochem. (Tokyo), 46, 103.
EMERY, A. E. H. (1963): Clinical Manifestations inTwo Carriers of Duchenne Muscular Dystrophy,Lancet, i, 1126.
EMERY, A. E. H. (1964a): Lyonisation of the X-chromosome, Lancet, i, 884.
EMERY, A. E. H. (1964b): Electrophoretic Patternof Lactic Dehydrogenase in Carriers and Patientswith Duchenne Muscular Dystrophy, Nature(Lond.), 201, 1044.
ENGEL, W. K., EYERMAN, E. L., and WILLIAMS, H. Z.(1963): Late-onset Type of Skeletal MusclePhosphorylase Deficiency: a New Familial Varietywith Completely and Partially Affected Subjects,New Engl. J. Med., 268, 135.
EVANS, J. H., and BAKER, R. W. R. (1957): SerumAldolase and the Diagnosis of Myopathy, Brain,80, 557.
FOWLER, W. M., CHOWDHURY, S. R., PEARSON, C. M.,GARDNER, G., and BRATTON, R. (1962): Changes inSerum Enzyme Levels after Exercise in Trainedand Untrained Subjects, J. appl. Physiol., 17, 943.
FOWLER, W. M., and PEARSON, C. M. (1964):Diagnostic and Prognostic Significance of SerumEnzymes II, Neurological Diseases other thanMuscular Dystrophy, Arch. phys. Med., 45, 125.
FRIEDMAN, M. M., and LAPAN, B. (1958): SerumAldolase in the Neonatal Period: including aColorimetric Determination of Aldolase byStandardization with Dihydroxyacetone, J. lab. clin.Med., 51, 745.
FRITZ, I. B., DAVIS, D. G., HOLTROP, R. H., andDUNDEE, H. (1958): Fatty Acid Oxidation bySkeletal Muscle during Rest and Activity, Amer.J. Physiol., 194, 379.
GAUTIER. E., and RICHTERICH, R. (1963): ValeurDiagnostique d'Activit6s Enzymatique du Serumen Paediatre II. Constance et Specificit6 desAnomalies Enzymatique, Helv. paediat. Acta, 18,32.
GERGELY, J. (1964): Biochemical Aspects of MuscleStructure and Function: in Disorders of VoluntaryMuscle, p. 86, ed. J. N. Walton. London: J. & A.Churchill.
GRIFFITHS, P. D. (1963): Creatine PhosphokinaseLevels in Hypothyroidism, Lancet, i, 894.
GRIFFITHS, P. D. (1964): Serum Levels of CreatinePhosphokinase, J. din. Path., 17, 56.
HALONEN, P. I., and KONTTINEN, A. (1962): Effectof Physical Excercise on Some Enzymes in theSerum, Nature (Lond.), 193, 942.
HEYCK, H., LAUDAHN, G., and LUDERS, C. J. (1963):Fermentaktivitatsbestimmungen in der GesundenMenschlichen Muskulatur und bei Myopathien II.Enzymaktivitatsveranderungen in Muskel beiDystrophia Musculorum Progressiva, Klin. Wschr.,41, 500.
HOCKADAY, T. D. R., DOWNEY, J. A., and MOTTRAM,R. F. (1964): A Case of McArdle's Syndrome witha Positive Family History, J. Neurol. Neurosurg.Psychiat., 27, 186.
HUGHES, B. P. (1962): A Method for the Estimationof Serum Creatine Kinase and its Use in ComparingCreatine Kinase and Aldolase Activity in Normaland Pathological Sera, Clin. chim. Acta, 7, 597.
HUGHES, B. P. (1963a): Serum Enzyme Studies withSpecial Reference to the Duchenne Type Dystrophy.In Research in Muscular Dystrophy: The Pro-ceedings of the Second Symposium January 1963,p. 167. London: Pitman Medical.
HUGHES, B. P. (1963b): Serum Enzyme Changes inMuscle Disease and their Relation to TissueChange, Proc. roy. Soc. Med., 56, 179.
LARNER, J., and VILLAR-PALASI, C. (1959): Enzymesin Glycogen Storage Myopathy, Proc. nat. Acad.Sci. (Wash.), 45, 1234.
LAURYSSENS, M. G., LAURYSSENS, M. J., and ZONDAG,H. A. (1964): Electrophoretic Distribution Patternof Lactate Dehydrogenase in Mouse and HumanMuscular Dystrophy, Clin. chim. Acta, 9, 276.
LELOIR, L. F., and CARDINI, C. E. (1957): Biosyn-thesis of Glycogen from Uridine DiphosphateGlucose, J. Amer. chem. Soc., 79, 6340.
LEVENE, P. A., and KRISZTELLER, L. (1909): FactorsRegulating the Creatine Output in Man, Amer.J. Physiol., 24, 45.
LEYBURN, P. THOMSON, W. H. S., and WALTON, J. N.(1961): An Investigation of the Carrier State inthe Duchenne Type Muscular Dystrophy, Ann.hum. Genet., 25, 41.
LILIENTHAL, J. L. jun., ZIERLER, K. L., FOLK, B. P.,BUKA, R., and RILEY, M. J. (1950): A ReferenceBase and System for Analysis of Muscle Con-stituents, J. biol. Chem., 182, 501.
LOHMAN, K. (1934): Uber die EnzymatischeAufspaltung der Kreatin-phosphorsaure: Zugleichein Beitrag zum Chemismus der Muskelkontraktion,Biochem. Z., 271, 264.
LYON, M. F. (1961): Gene Action in the X-chromosome of the Mouse (Mus musculus L.),Nature (Lond.), 190, 372.
MCARDLE, B. (1951): Myopathy Due to a Defect inMuscle Glycogen Breakdown, Clin. Sci., 10, 13.
MCARDLE, B. (1964): Metabolic and EndocrineMyopathies: In Disorders of Voluntary Muscle,p. 389, ed. J. N. Walton. London: J. & A. Churchill.
MELLICK, R. S., MAHLER, R. F., and HUGHES, B. P.(1962): McArdle's Syndrome: Phosphorylase-deficient Myopathy, Lancet, ii, 1045.
MOMMAERTS, W. F. H. M., ILLINGWORTH, B., PEARSON,C. M., GUILLORY, R. J., and SERAYDARIAN, K.(1959): A Functional Disorder of Muscle Associatedwith the Absence of Phosphorylase, Proc. nat.Acad. Sci. (Wash.), 45, 791.
OPIE, L. H., EVANS, J. R., and RENOLD, A. E. (1962):Fructose in McArdle's Syndrome, Lancet, ii, 358.
PEARCE, G. W. (1964): Tissue Culture and ElectronMicroscopy in Muscle Disease: in Disorders ofVoluntary Muscle, ed. J. N. Walton, p. 237.London: J. & A. Churchill.
PEARCE, J. M. S., PENNINGTON, R. J., and WALTON,J. N. (1964a): Serum Enzyme Studies in MuscleDisease I. Variations in Serum Creatine KinaseActivity in Normal Individuals, J. Neurol.Neurosurg. Psychiat., 27, 1.
PEARCE, J. M. S., PENNINGTON, R. J., and WALTON,J. N. (1964b): Serum Enzyme Studies in MuscleDisease II. Serum Creatine Kinase in MuscularDystrophy and in other Myopathic and Neuro-pathic Disorders, J. Neurol. Neurosurg. Psychiat.,27, 26.
PEARSON, C. M. (1964): The Periodic Paralyses:Differential Features and Pathological Observationsin Permanent Myopathic Weakness, Brain, 87, 341.
PEARSON, C. M. (1964b): Polymyositis and RelatedDisorders: in Disorders of Voluntary Muscle,ed. J. N. Walton, p. 315. London: J. & A. Churchill.
copyright. on A
pril 3, 2022 by guest. Protected by
http://pmj.bm
j.com/
Postgrad M
ed J: first published as 10.1136/pgmj.41.476.313 on 1 June 1965. D
ownloaded from

324 POSTGRADUATE MEDICAL JOURNAL June, 1965
PEARSON, C. M., FOWLER, W. M., and WRIGHT, S. W.(1963): X-chromosome Mosaicism in Females withMuscular Dystrophy, Proc. nat. Acad. Sci. (Wash.),50, 24.
PENNINGTON, R. J. (1964): Biochemical Aspects ofMuscle Disease: in Disorders of Voluntary Muscle,ed. J. N. Walton, p. 255. London: J. & A. Churchill.
PLUMMER, D. T., ELLIOTr, B. A., COOKE, K. B., andWILKINSON, J. H. (1963): Organ Specificity andLactate Dehydrogenase Activity I. The RelativeActivities with Pyruvate and 2-oxobutyrate ofElectrophoretically Separated Fractions, Biochem.J., 87, 416.
RICHTERICH, R., ROSIN, S., AEBI, U., and Rossi, E.(1963): Progressive Muscular Dystrophy V. TheIdentification of the Carrier State in the DuchenneType by Serum Creatine Kinase Determination,Amer. J. hum. Genet., 15, 133.
ROBBINS, P. W., TRAUT, R. R., and LIPMANN, F.(1959): Glycogen Synthesis from Glucose, Glucose-6-phosphate and Uridine Diphosphate Glucose inMuscle Preparations, Proc. nat. Acad. Sci. (Wash.),45, 6.
RONZONI, E., BERG, L., and LANDAU, W. (1961):Enzyme Studies in Progressive Muscular Dystrophy,Res. Publ. Ass. nerv. ment. Dis., 38, 721.
ROWLAND, L. P., FAH, S., and SCHOTLAND, D. L.(1963): McArdle's Disease: Hereditary Myopathydue to Absence of Muscle Phosphorylase, Arch.Neurol. (Chic.), 9, 325.
SAITO, M., HIBI, I., KAWAZURA, M., and FUKUYAMA,Y. (1963): Creatine Phosphokinase Levels inHypothyroidism, Lancet, ii, 252.
SCHAPIRA, G., and DREYFUS, J. C. (1963): Bio-chemistry of Progressive Muscular Dystrophy: inMuscular Dystrophy in Man and Animals, ed.G. H. Bourne and N. Gollarz, p. 48. Basel: Karger.
SCHAPIRA, F., DREYFUS, J. C., SCHAPIRA, G., andDEMOS, J. (1960): Etude de l'aldolase et de laCreatine Kinase du Serum chez les Meres deMyopathes, Rev. franc. Etud. clin. biol., 5, 990.
SCHLANG, H. A. (1961): Effect of Physical Exerciseon Serum Transaminase, Amer. J. med. Sci., 242,338.
SCHMID, R., and HAMMAKER, L. (1961): HereditaryAbsence of Muscle Phosphorylase (McArdle'sSyndrome), New Engl. J. Med., 264, 223.
SCHMID, R., and MAHLER, R. (1959): ChronicProgressive Myopathy with Myoglobinuria:Demonstration of a Glycogenolytic Defect in theMuscle, J. clin. Invest., 38, 2044.
SIBLEY, J. A., and LEHNINGER, A. L. (1949): Aldolasein Serum and Tissues of Tumour-bearing Animals,J. nat. Cancer Inst., 9, 303.
SLATER, E. C. (1960): Biochemistry of Sarcosomes:in The Structure and Function of Muscle, ed. G. H.Bourne, p. 114. London: Academic Press.
SOLVONUK, P. F., MCRAE, S. C., and COLLIER, H.B.(1956): Creatine Phosphokinase Activity ofMammalian Erythrocytes, Canad. J. Biochem., 34,481.
STEIN, J. M., and PADYKULA, H. A. (1962): Histo-chemical Classification of Individual Skeletal
Muscle Fibres of the Rat, Amer. J. Anat., 110, 163.STEITEN, D., JUN., and STETEN, M. R. (1960): Gly-cogen Metabolism, Physiol. Rev., 40, 505.
SUTHERLAND, E. W., and WOSILAIT, W. D. (1956):The Relation of Epinephrine and Glucagon to liverPhosphorylase. I. Liver Phosphorylase; Preparationand Properties, J. biol. Chem., 218, 459.
THOMPSON, R. A., and VIGNOS, P. J. (1959): SerumAldolase in Muscle Disease, Arch. intern. Med.,103, 551.
THOMSON, W. H. S. (1962): Sources of Error in theBiochemical Diagnosis of Muscular Dystrophy,J. Neurol. Neurosurg. Psychiat., 25, 191.
THOMSON, W. H. S., LEYBURN, P., and WALTON, J. N.(1960): Serum Enzyme Activity in MuscularDystrophy, Brit. med. J., ii, 1276.
THOMSON, W. H. S., MACLAURIN, J. C., and PRINIAS,J. W. (1963): Skeletal Muscle Glycogenosis: anInvestigation of Two Dissimilar Cases, J. Neurol.Neurosurg. Psychiat., 26, 60.
VAN BREEMEN, V. L. (1960): Ultrastructure of HumanMuscle II. Observations on Dystrophic StriatedMuscle Fibres, Amer. J. Path., 37, 333.
VAN DEN BOSCH, J. (1963): Investigations of theCarrier State in the Duchenne Type Dystrophy.Research in Muscular Dystrophy. The Proceedingsof the Second Symposium January 1963, p. 23,London: Pitman Medical.
VAN PILSUM, J. F., and WOLIN, E. A. (1958):Guanidinium Compounds in Blood and Urine ofPatients Suffering from Muscle Disorders, J. Lab.clin. Med., 51, 219.
VIGNOS, P. J., JUN., and LEFKOWITZ, M. (1959): ABiochemical Study of Certain Skeletal MuscleConstituents in Human Progressive MuscularDystrophy, J. clin. Invest., 38, 873.
WALTON, J. N. (1963): Clinical Aspects of HumanMuscular Dystrophy: in Muscular Dystrophy inMan and Animals, ed. G. H. Bourne and N. Golarz,p. 263, Basel: Karger.
WALTON, J. N. (1964): Progressive Muscular Dys-trophy: in Disorders of Voluntary Muscle, ed.J. N. Walton, p. 286, London: J. & A. Churchill.
WATrS, D. C., and HOOTON, B. J. (1965): SomeProperties of Purified Creatine Kinase from theSkeletal Muscles of normal and Dystrophic BarHarbor Mice. Research in Muscular Dystrophy:The Proceedings of the Third Symposium January1965. London: Pitman Medical, in the press.
WHITE, L. P. (1959): Serum Enzymes. Variations ofActivity in Disease of Muscle, Calif. Med., 80, 1.
WIEME, R. J. (1959): Thesis: Studies on Agar GelElectrophoresis. Brussels.
WILSON, K. M., EVANS, K. A., and CARTER, C. O.(1965): Creatine Kinase Levels in Women whoCarry Genes for Three Types of Muscular Dys-trophy, Brit. med. J., i, 750.
WOLF, H. P. (1962): Fructose in McArdle's Syn-drome, Lancet ii, 937.ZIERLER, K. L. (1958): Increased Muscle Perme-
ability to Aldolase Produced by Depolarization andby Metabolic Inhibitors, Amer. J. Physiol., 193,534.
copyright. on A
pril 3, 2022 by guest. Protected by
http://pmj.bm
j.com/
Postgrad M
ed J: first published as 10.1136/pgmj.41.476.313 on 1 June 1965. D
ownloaded from





























