Embed Size (px)
Citation preview

Analysis of Membranes Photolabeled with Lipid Analogues REACTION OF PHOSPHOLIPIDS CONTAINING A DISULFIDE GROUP AND A NITRENE OR CARBENE PRECURSOR WITH LIPIDS AND WITH GRAMICIDIN A*
(Received for publication, September 20, 1979)
Josef BrunnerS and Frederic M. Richards From the Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut 06520
Analogues of fatty acids have been synthesized which contain an S-S bridge in the aliphatic chain and at the @-carbon the photoactivatable p-(3-trifluoromethyldi- azirino)phenyl or p-azidophenyl group as a carbene and nitrene precursor, respectively. These acids were used to acylate 1-palmitoyl phosphatidylcholine by the procedure described by Gupta et al. (Gupta, C. M., Radhakrishnan, R., and Khorana, H. G. (1977) Pmc. Natl. Acad Sci U. S. A. 74, 4315-4319). Multilamellar liposomes prepared from various radioactively labeled lipids and 2 to 10 mol% of one of the photosensitive phospholipid analogues were photolyzed and the extent of cross-linking was determined. The carbene-gener- ating probe labeled dipalmitoyl phosphatidylcholine in yields of 15 to 20%, while labeling with the nitrene probe was about 10 times less efficient. In contrast these probes were found to react with the unsaturated fatty acid of 1-palmitoyl-2-oleoyl phosphatidylcholine with almost equaI efficiency. The nitrene was 2 to 3 times more efficient than the carbene in cross-linking to gramicidin A. Amino acid analysis and Edman deg- radation of the labeled peptide indicated that both probes predominantly attacked tryptophan residues. No geometrical correlation was found between the ex- pected penetration of the reactive group of the probe within the bilayer and the labeled sites of the peptide. This result is discussed in terms of chemical selectivi- ties of the photoactivated intermediates.
After photocross-linking, the disulfide bridge can be cleaved to generate a free sulfhydryl group which sub- sequently can be utilized for preparative or analytical purposes. Photolabeled lipids and peptides can be sep- arated easily from the bulk of the unlabeled material.
These phospholipid analogues are substrates for the phospholipid exchange protein isolated from calf liver. Transfer from sonicated dispersions of the analogues into an acceptor membrane was demonstrated. Individ- ual monolayers can thus be labeled separately with these reagents.
Descriptions of the structure and interaction of proteins with membranes are in general restricted to terms such as “outside,” “inside,” or “intrinsic.” Most of this information
* This work was supported by the United States Public Health Service through Grant GM-21714 to the Membrane Research Center a t Yale University. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ Supported by fellowship from the Swiss National Science Foun- dation. Present address, Eidgenossische Technische Hochschule Zurich, Laboratorium fur Biochemie, Universitatstrasse 16, CH-8092 Zurich, Switzerland.
has been obtained from the chemical or biochemical modifi- cation of specific amino acid residues within definable com- partments of a membrane. The structural resolution which can be reached from these studies could be improved if labeling techniques were available which do not depend on the presence of specific functional groups. Nitrenes and car- benes photolytically derived from stable precursors are highly reactive species and allow modification of amino acid residues which are inert by normal peptide-chemical standards. There- fore, photochemical approaches have been of particular inter- est where general labeling (1) of proteins and not selective modification of specific functional groups is envisaged. In addition to other areas of application (2-8), photosensitive reagents have been used for the labeling and identification of those domains of membrane proteins which are embedded within the lipid core of the membrane (9-13).
Khorana and associates (14-18) and other investigators (19, 20) have outlined programs for the labeling of membranes within the defined geometry of the lipid core using probes of amphipathic molecular structure (for example, phospholipid analogues). In these reagents the reactive group is covalently bound to a fatty acyl chain a t a defined position. Upon photolysis, cross-linked products are formed with proteins and lipids which are in contact with the reaction intermediate.
Details concerning the folding of proteins within the bilayer may be derived from such studies provided a correlation exists between the position of the photosensitive group along the acyl chain and the depth of reaction within a membrane.
In any attempt to locate the sites of‘ reaction within an amino acid sequence, one has to consider that photolabeling will rarely lead to stoichiometric modification of an amino acid and that perhaps only a very small fraction of a mem- brane protein will indeed carry the photolabel at all. The scope of analysis would be increased if labeled proteins or peptides could be separated from unlabeled material and specifically manipulated.
These considerations lead us to the synthesis and investi- gation of lecithin analogues containing a photosensitive group which is bound via a disulfide bridge to a fatty acyl chain. Photolabeling followed by reduction thus generates free sulfhydryl groups in the target molecule at the sites where photocross-linking with the probe has occurred. These ”SH groups are then used for the separation of modified peptides or lipids and to assist in the subsequent sequence analysis of the peptides.
Lecithin analogues are more difficult to insert into native membranes than simple lipid-soluble probes. On occasion this may be accomplished biosynthetically where the fatty acid analogue is taken up and used without modification. Another possible route is use of phospholipid exchange proteins (21, 22) to transfer the reagent molecule from a carrier liposome to an acceptor membrane. The latter procedure has been
3319

3320 SS Containing Photolabile Phospholipid Analogues
examined with one of the reagents reported here. There are conflicting reports in the literature on the relative
ability of nitrenes and carbenes to insert into various bonds and to usefully label biologically interesting systems (see discussion in Refs. 13, 23, and 24). In this paper, the p - azidophenyl and p-(3-trifluoromethyldiazirino)phenyl groups are compared in their ability to label saturated and unsatu- rated lipids and gramicidin A upon photolysis in lipid vesicles. The nitrene and carbene generated from these two reagents are sterically very similar.
The structure of the gramicidin A pore has now been established with considerable confidence (25-27) and conse- quently represents a useful system for the study of many aspects of photolabeling of membrane-associated peptides. For the usual modification reagents, this peptide has no reactive groups except the COOH-terminal ethanolamide function which lies at the surface of the membrane. Thus, extent of labeling with a probe which generates the phenyl- nitrene group is of interest. The other reactive group, 3- trifluoromethyl-3-phenyldiazirine, has recently been devel- oped in this laboratory (28) and insertion of the carbene intermediate into the C-H bonds of cyclohexane in high yield has been documented. The reaction of the carbene and nitrene intermediates with this peptide is explored. The Edman deg- radation has been applied in an attempt to locate the site of reaction.
MATERIALS AND METHODS
All materials and solvents used in synthesis were from commercial sources and used without further purification. Thin layer chromatog- raphy was performed on precoated plates, Silica Gel 60 Fzs, (Merck). Compounds were visualized by fluorescence quenching, with iodine vapor, or, for reagents containing the azidophenyl group, by their color reaction after exposure to sunlight for 1 h. Ultraviolet, infrared, and ‘H NMR spectra were recorded with a Cary model 15, Perkin Elmer model 337, and Varian EM 360A spectrometer, respectively.
Free sulfhydryl or sulfenylthiocarbonates were determined with the Ellman reagent (29) in a buffer solution containing 40% acetone.
Free Sulfhydryl Groups Thiol (<0.2 pmol of “SH) was mixed with 5.0 ml of a reagent
solution prepared from 50 ml of 0.1 M Nap,, pH 8.0, 25 ml of H20, 50 ml of acetone, and 1 ml of a stock solution of 5,5-dithiobis(2-nitroben- zoic acid) (39.6 mg in 10 ml of Nap,, 0.1 M, pH 7.0). Absorbance at 412 nm was monitored ( E = 13,600 mo1”cm-’ for ”SH form of reagent).
Sulfenylthiocarbonates
Solutions of reduced Ellman reagent were prepared in Nap, ace- tone, pH 8.0, with an optical density 0.7 to 1.0 (412 nm). Sulfenylthio- carbonate (-0.1 pmol) was added to 5.0 ml of the reagent solution and kept a t room temperature for 1 h. The amount of sulfenylthio- carbonates was calculated from AA,u nm before and after addition of the sample based on the following reaction:
HOOC
6 0 I1
0, x ’ \ SH + R-S-S-COR - €412 = 13,600
1 : b S - S - R - + COS + R’OH
colorless
Both procedures gave a linear response with increasing concentrations of sulfhydryl or sulfenylthiocarbonate compounds.
General Method for the Synthesis of Phospholipid Analogues Containing a Photoactivatable Group and the Cleavable S-S Bond
The structure of three synthesized lecithin analogues is shown in Fig. 1. These were prepared following the general procedure described
y 2
0 I -o-p=o I 0 I
CH2- CH - CU2
0 0 I I R =
I lI m FIG. 1. Synthetic 1-palmitoyl-2-acyl phosphatidylcholines.
Structures of synthesized lecithin analogues.
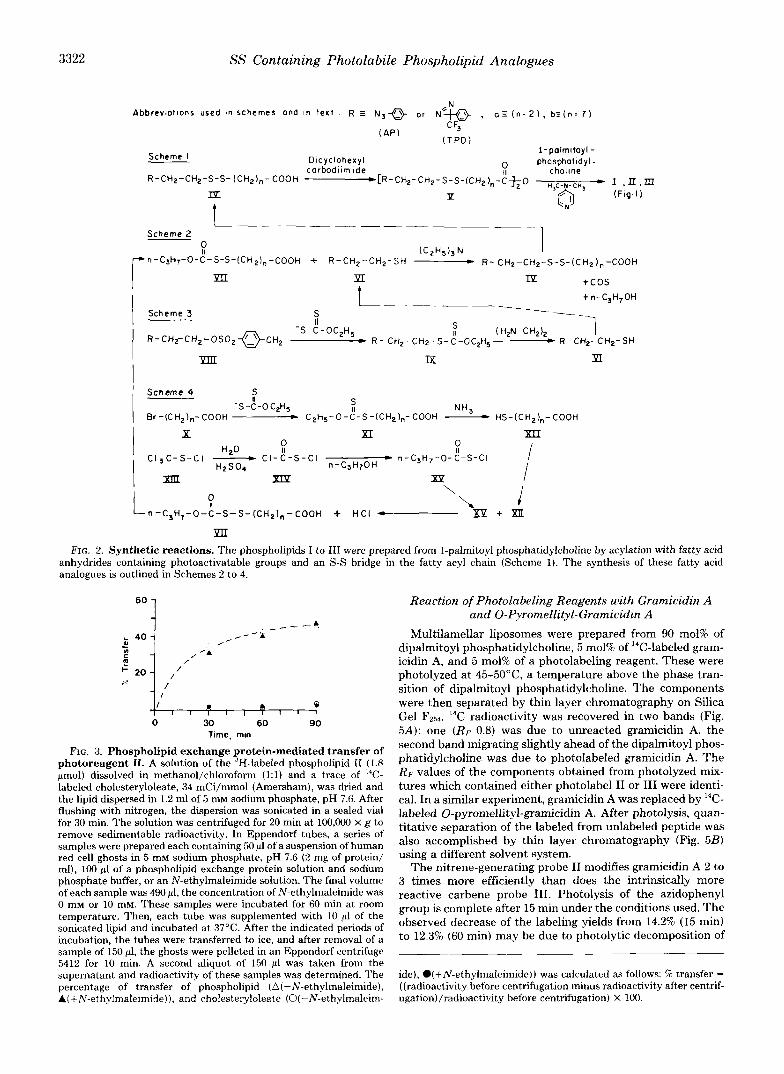
by Gupta et al. (16). The synthetic schemes are outlined in Fig. 2. Scheme 1-Fatty acid analogues (IV) containing a photosensitive
unit and an S-S bond substituting for two CH, groups of the acyl chain were converted to their anhydrides (V) using 0.55 eq of dicyclo- hexylcarbodiimide in carbon tetrachloride. 1-Palmitoyl phosphatidyl- choline was acylated in the presence of N,N-dimethyl-4-aminopyri- dine as an acylation catalyst.
Synthesis of the Fatty Acid Analogues (IV) Described in Schemes 2 to 4-Asymmetric disulfides IVa,b were prepared by the thiol- induced fragmentation of sulfenylthiocarbonates (VII) (30) according to Scheme 2. Thiols VIa,b were synthesized by a sequence of reactions shown in Scheme 3. This involved formation of 0-tosylates of 24p- azidopheny1)ethanol (VIII, R = AP’) and 2-[p-(3-trifluoromethyldi- azirino)phenyl]ethanol (VIII, R = TPD), their conversion to the corresponding xanthogenic acid esters (IX) and the ethylenediamine- mediated cleavage (31). Tosylates VIIIa,b were prepared according to procedures described earlier (28, 32). Sulfenylthiocarbonates VIIa,b (Scheme 2) were obtained by a reaction sequence depicted in Scheme 4.
Lecithin Analogues I to III Obtained from the Reaction Mixtures as Follows-Chloroform was evaporated on a rotavap at reduced pressure and the residues dissolved in 2 voIumes of methylene chlo- ride:methanol (1:l). These solutions were subjected to gel filtration on Sephadex LH-20 (2.5 X 50 cm) using methylene ch1oride:methanol (1:l) as the eluant. Lecithin and some unreacted 1-palmitoyl phos- phatidylcholine were eluted prior to free fatty acid, N,N-dimethyl-4- aminopyridine, and pyridine. Fractions containing lecithin and 1- palmitoyl phosphatidylcholine (detected by thin layer chromatogra- phy) were pooled and concentrated. Separation of the two compo- nents was achieved by column chromatography on silica gel (metha- nol:chloroform, 1:l) or by preparative thin layer chromatography (chloroform:methanol:water, 65:25:4).
The following lecithins and lecithin analogues were synthesized following this general procedure: 1) l-palmitoyl-2-[9,10-3H]palmitoyl phosphatidylcholine; 2 ) l-palmit0yl-2-[l-’~C]oleoyl phosphatidylcho- line; 3) l-palmitoyl-2-[7-(p-azidophenyl)4,5-dithiaheptanoyl] phos- phatidylcholine (I); 4) l-palmitoyl-2-[12-(p-azidophenyl)9,lO-dithia- dodecanoyl] phosphatidylcholine (11); 5) l-palmitoyl-2-[12-(p-azido- phenyl)[ll-”H]9,l0-diathiadodecanoyl]-phosphatidylcholine (W); 6) 1-palmitoyl-2-[12-(3-trifluoromethyldiazirinophenyl)-9,lO-dithiatri- decanoyl] phosphatidylcholine (111).
The purity of these lecithins was routinely checked by thin layer chromatography. All operations involving diazirines or azides were performed in the dark or under dim light. Treatment of the synthetic phospholipids with phospholipase AP from Crotalus adamanteus
’ The abbreviations used are: AP, aminopyridine; TPD, 3-trifluo- romethyl-3-phenyldiazirine; PTH, phenylthiohydantoin.

SS Containing Photolabile Phospholipid Analogues 3321
(Sigma) resulted in the formation of 1-palmitoyl phosphatidylcholine and of the fatty acid analogues as judged by their co-migration with authentic samples on thin layer chromatographic plates.
Multilamellar or small unilamellar liposomes were prepared in water according to standard procedures (33, 34) . Multilamellar lipo- somes containing a fraction of gramicidin A (or O-pyromellityl-gram- icidin A) were prepared as described by Chapman et al. (35).
A Hanovia 450-watt medium pressure mercury lamp was used as the light source for photolysis. Radiation was filtered through a water cooled Pyrex-glass jacket and 1.5-mm uranium glass. Photolyses were performed in Pyrex-glass test tubes under an air atmosphere. Further details are included under “Results.”
Phospholipid exchange protein was isolated from calf liver follow- ing the procedure by Kamp and Wirtz (36), except that the final step of purification (Sephadex G-50 gel filtration) was omitted. Phospho- lipid exchange protein activity was measured by the transfer of [“Hldipalmitoyl phosphatidylcholine from sonicated egg lecithin li- posomes containing traces of the tritium-labeled lipid and [‘4C]cho- lesteryloleate (Amersham) to red cell ghosts. Red cell membranes were prepared by the method of Steck and Kant (37). Sonicated lipid (-1 pg of egg lecithin containing 50 nCi of ‘H and 5 nCi of I4C) was incubated with red cell ghost (-100 pg of protein) and an appropriate aliquot of the exchange protein in 0.5 ml of 5 mM sodium phosphate buffer, pH 7.6. Incubation was carried out for 10 to 60 min at 37°C with constant agitation. Transfer of radioactivity from the liposomes to ghosts was measured after pelleting ghosts at 25,000 X g for 10 min. All measurements were based on relative activity. The absolute specific activity of this preparation was not established.
Synthetic Section
See supplementary material.’
RESULTS
Physicochemical and Biochemical Properties of Lecithin Analogues I to III
1. Dried fiims of these lecithin analogues spontaneously form dispersions in water or buffer solutions. Sonication a t room temperature produces clear or slightly opalescent solu- tions. High resolution ‘H NMR spectra of sonicated disper- sions obtained from these synthetic lipids indicate the pres- ence of liposomal structures of a size similar to those obtained from egg phosphatidylcholine.
2. It was found that l-palmitoyl-2-[12-(p-azidophenyl)9,10- dithiadodecanoyl] phosphatidylcholine I1 is a substrate for the phospholipid-exchange protein. Sonicated dispersions of the tritiated lecithin analogue I1 containing trace amounts of [‘4C]cholesteryloleate as a nonexchangeable marker were added to resealed ghosts from human erythrocytes in a re- agent:protein ratio of 1:lOO (w/w). Addition of phospholipid- exchange protein resulted in an immediate transfer of ‘H (but not of I4C) from the sonicated lipid into the ghost membranes. This is shown in Fig. 3. The exchange rate was found to be similar to that measured for the transfer of “H-labeled dipal- mitoyl phosphatidylcholine from sonicated egg lecithin lipo- somes to ghost membranes. “Near equilibrium” values indi- cated that approximately 50% of the lipid initially present in the sonicated liposomes (corresponding to -75% present in the outer monolayer of small liposomes) are exchanged. The presence of 10 mM N-ethylmaleimide in the incubation me- dium had no effect on the activity of the exchange protein. The ability to carry out the exchange procedure in the absence of “SH groups markedly reduces the possibility of any un- wanted SS exchange reactions involving the photolabile POUP.
’ The “Experimental Procedures” are presented in miniprint at the end of this paper. Miniprint is easily read with the aid of a standard magnifying glass. Full-size photocopies are available from the Journal of Biological Chemistry, 9650 Rockville Pike, Bethesda, Maryland 20014. Request Document No. 79M-1922, cite author(s), and include a check or money order for $1.35 per set of photocopies.
Reaction of Photolabeling Reagents with Phospholipids Multilamellar liposomes were prepared from 95 mol% of 1-
pa~it0y1-2-[l-~~C]oleoyl phosphatidylcholine, 0.1 mol% of 1- palmitoyl-2-[9,l0-”H]palmitoyl phosphatidylcholine, and 5 mol% of photoreagent I1 or I11 (1ipid:reagent molar ratio = 2 0 1) and were photolyzed, Intermolecular cross-linking of the reagent with lipid results in the formation of radioactive compounds which contain a disulfide bond. Disulfides were then cleaved with tributylphosphine (see miniprint supple- ment) and sulfhydryls covalently bound to a derivatized po- rous glass containing an N-substituted maleimido function as shown in Fig. 4.
Reaction of the nitrene or carbene with saturated lipid can be estimated from the amount of the glass-immobilized ma- terial containing tritium. Since the principal lipid, l-palmitoyl- 2-[1-14C]-oleoyl phosphatidylcholine, can formally be regarded as a dipalmitoyl phosphatidylcholine containing in addition an olefinic double bond, cross-linking to I4C-lipid includes reaction of the reagent with the saturated moiety of the lipid and addition to the double bond.
The amount of “H and I4C radioactivity bound to glass can be used to estimate reaction of the nitrene or carbene with the saturated lipid and with the olefinic bond. These results are shown in Table I. Both nitrene (11) and carbene (111) react with saturated and unsaturated lipid. However, the carbene shows a greater tendency to react with the saturated chains than does the nitrene. In all cases the yields of intermolecular cross-linking are less than 10%.
Attachment to glass is not quantitative (see below and miniprint supplement) and the actual labeling yields are prob- ably at least twice as large as the numbers in Table I indicate. Quantitative data, however, were obtained after separation of cross-linked from unreacted lipid by a procedure described previously (17) using Sephadex LH-20 gel filtration. These data are shown in Table 11.
In a control experiment it was shown that photolysis of pure [3H]dipalmitoyl phosphatidylcholine liposomes for 60 min did not lead to any detectable (<0.02%) cross-linked products. The nitrene generating reagent (11) reacted with saturated lipid in yields of 2 to 3%. In contrast, the carbene type reagent (111) was found much more efficient and yields up to 20% were measured. As the molar ratio of reagent to lipid is decreased from 1:lO to 1:40 a slight increase in the percentage yields of radioactive cross-linked products was found. This concentration dependence suggests that a fraction of the carbene may be consumed in a bimolecular reaction with the photolabel itself.
The detailed chemical reaction which leads to cross-linking of the carbene with saturated lipids has not yet been investi- gated. However, we have previously demonstrated that 3- trifluoromethyl-2-phenyldiazirine (15 mM) when photolyzed in cyclohexane yields the C-H insertion product in 250% yield (28). It is therefore plausibie that insertion of the carbene into C-H bonds of the palmitoyl chains is the principal reaction type which leads to cross-linking:” In the case of the nitrene- generating reagent, C-H insertion is less certain and a radical- induced mechanism should also be considered.
:I I Y F NMR spectroscopy of sonicated egg lecithin liposomes con- taining photoreagent I11 (1ecithin:reagent molar ratio = 20.1) upon photolysis gave a sharp resonance at 0 ppm uersus CFaCOOH) and a broad signal centered at 14 ppm. The resonance at 14 ppm can in fact be assigned to products which resulted from C-H insertion of the carbene intermediate into aliphatic chains of the lecithin, since the insertion product of the carbene into C-H bonds of cyclohexane has a chemical shift of 14.2 ppm (28). The major signal at 0 ppm corre- sponds to the reaction product of the carbene with water.
3322 SS Containing Photolabile Phospholipid Analogues
Abbrevlotlons used in schemes ond I” text
CAP) CF3
(TPD)
Scheme I Dlcyclohexyl l-polm~toyl-
9- phosphotidyl-
challne CarOodllm ride R-Ct+-CH2-S-S- (CH2),- COOH ~CR-CH2-CH,-S-~-(~~Z),-~f;~ - I .n.m
I!z P (Flg.1)
t
a(n=2), bz(n=7)
L--- -__p--p _____ Scheme 2
P (CZH~)~N r n-CaH,-0-C-S-S-(CH&-COOH + R-CH,-CH,-SH p R- CH2-CH2-S-S-ICH,),-COOH
YII PT IY +cos
t t n-C3H,0H
____ Scheme 3
---
i ------
R-CH,-Cl-i,-OSO,+&, -S-C-OC,H, S
(H,N-CH,), -I * R-CH,-CHZ-S-~-OC~H~---- R-CHe-CH,-SH
YllI ix XI
’ Scheme 4 s
-S-C-O&H, : Br-(CH2Jn-~00~ p
NH3 C,H,-0-C-S-(CH,),-COOH - HS-(CH2),-COOH
X XI
“2“ P tl CI,C-S-Cl - Cl-C-S-Cl
H2S04 n-C3H70H - n-C3H,-O-C-S-Cl
L ? n-C3H,-0-C-S-S-KH,l,-COOH + HCI 4 23 +
FIG. 2. Synthetic reactions. The phospholipids I to III were prepared from I-palmitoyl phosphatidylcholine by acylation with fatty acid anhydrides containing photoactivatable groups and an S-S bridge in the fatty acyl chain (Scheme 1). The synthesis of these fatty acid analogues is outlined in Schemes 2 to 4.
60 1 4
I' I * 4 1 I t I I, 11 1 I
0 30 60 90 Time, mm
FIG. 3. Phospholipid exchange protein-mediated transfer of photoreagent II. A solution of the “H-labeled phospholipid II (1.8 pmol) dissolved in methanol/chloroform (1:l) and a trace of 14C- labeled cholesteryloleate, 34 mCi/mmol (Amersham), was dried and the lipid dispersed in 1.2 ml of 5 mM sodium phosphate, pH 7.6. After flushing with nitrogen, the dispersion was sonicated in a sealed vial for 30 min. The solution was centrifuged for 20 min at 100,000 x g to remove sedimentable radioactivity. In Eppendorf tubes, a series of samples were prepared each containing 50 ~1 of a suspension of human red cell ghosts in 5 mM sodium phosphate, pH 7.6 (2 mg of protein/ ml), 100 pl of a phospholipid exchange protein solution and sodium phosphate buffer, or an N-ethylmaleimide solution. The final volume of each sample was 490 ~1, the concentration of N-ethylmaleimide was 0 mM or 10 mM. These samples were incubated for 60 min at room temperature. Then, each tube was supplemented with 10 ~1 of the sonicated lipid and incubated at 37°C. After the indicated periods of incubation, the tubes were transferred to ice, and after removal of a sample of 150 ~1, the ghosts were pelleted in an Eppendorf centrifuge 5412 for 10 min. A second aliquot of 150 ~1 was taken from the supernatant and radioactivity of these samples was determined. The percentage of transfer of phospholipid (A(-N-ethylmaleimide), A(+N-ethylmaleimide)), and cholesteryloleate (O(-N-ethylmaleim-
Reaction of Photolabeling Reagents with Gramicidin A and 0-Pyromellityl-Gramicidin A
Multilamellar liposomes were prepared from 90 mol% of dipalmitoyl phosphatidylcholine, 5 mol% of ‘?-labeled gram- icidin A, and 5 mol% of a photolabeling reagent. These were photolyzed at 45-50°C a temperature above the phase tran- sition of dipalmitoyl phosphatidylcholine. The components were then separated by thin layer chromatography on Silica Gel Fgs?. 14C radioactivity was recovered in two bands (Fig. 5A): one (RF 0.8) was due to unreacted gramicidin A, the second band migrating slightly ahead of the dipalmitoyl phos- phatidylcholine was due to photolabeled gramicidin A. The RF values of the components obtained from photolyzed mix- tures which contained either photolabel II or III were identi- cal. In a similar experiment, gramicidin A was replaced by ‘C- labeled 0-pyromellityl-gramicidin A. After photolysis, quan- titative separation of the labeled from unlabeled peptide was also accomplished by thin layer chromatography (Fig. 5B) using a different solvent system.
The nitrene-generating probe II modifies gramicidin A 2 to 3 times more efficiently than does the intrinsically more reactive carbene probe III. Photolysis of the azidophenyl group is complete after 15 min under the conditions used. The observed decrease of the labeling yields from 14.2% (15 min) to 12.3% (60 min) may be due to photolytic decomposition of
ide), l (+N-ethylmaleimide)) was calculated as follows: W transfer = ((radioactivity before centrifugation minus radioactivity after centrif- ugation)/radioactivity before centrifugation) x 100.

SS Containing Photolabile Phospholipid Analogues 3323
the labeled gramicidin A. Although the diazirine I11 is photo- lyzed faster than the nitrene (not shown), an increase in the labeling yields from 3.5% (15 min) to 5.4% (60 min) was observed. This must be assigned to the carbene derived from the slowly photolyzing diazoisomer which is formed as a product of photoisomerization of the diazirine (28). The num- bers in Table I11 do in fact agree with the previous observation that 35% of the diazirine does not photolyze to yield the carbene directly.
It has been shown that most of the gramicidin A molecules
1.
2.
R < / S
S \
R < / S
S \
ATTACHMEN: -Nbs-R 0
H
FIG. 4. Covalent attachment of a photolabeled target based on the sulfhydryl function generated by cleavage of the disul- fide bond of the probe. The photolabeling reaction is shown with the carbene generating probe as a C-H insertion (line I ) . Reduction (line 2) was performed in 95% n-propanol with tributylphosphine. Free sulfhydryls (R-SH) were immobilized by reaction with a deriv- atized porous glass (70 A) containing N-substituted maleimide arms (line 3).
TABLE I Cross-linking ofphotolabel 11 and 111 with phospholipids-yields
bused on coupling of the reaction products to glass Ten milligrams of ["C]palmitoyloleyl phosphatidylcholine, 0.1
mol% of [3H]dipalmitoyl phosphatidylcholine, and photosensitive probe I1 or 111 (molar ratio of phospholipidlabel = 201) were freeze- dried from 95% benzene, 5% methanol, dispersed in 2 ml water a t 45"C, and photolyzed for 45 min at wavelength >300 nm. After freeze- drying the lipids were dissolved in 800 pl of 95% n-propanol and extensively flushed with nitrogen. Disulfide was reduced with a 2- to 3-fold excess of tributylphosphine and sulfhydryls trapped with 100 mg of porous glass containing the N-alkyl-maleimido group (see Fig. 3). Adsorbed radioactivity was removed by repeated extraction of the beads with methanokformic acid (98:2). Bound radioactivity was determined after release from glass by acid hydrolysis.
Counts bound to glass (% of total) Labeling yields (%)"
Photolabel 3H
Reaction with Reaction with I4C " ~ ~ ~ ~ ~ d double bonds
I1 0.095 0.385 1.9 I11
5.8 0.320 0.535 6.4 4.3
a If every reagent molecule reacted with one lipid molecule and if the binding of the "SH groups in the reduced samples were quanti- tative, the expected numbers of 3H and 14C counts bound to the glass beads was defined as a labeling yield of 100% for each isotope.
TABLE I1 Cross-linking ofphotolabels with phospholipids-yields based on
separation ofphotolysis products by gel filtration on Sephudex
Multilamellar liposomes were prepared according to standard pro- cedures from radioactive phospholipids and photolabel I or I1 as specified below. Photolysis was performed at 45°C for 60 min. Samples were freeze-dried, dissolved in lml of chloroform/methanol (l:l), and subjected to gel filtration on Sephadex LH-20 (2 X 90 cm). Solvent ch1oroform:methanol (1:l); flow rate, 39 m1.h". Fractions (1.3 m l ) within an elution volume from 104 to 115 ml contained cross-linked lipids. Unreacted lipids were recovered within a second pool (120 to 140 ml elution volume).
Counts eluted with pool
Composition of lipo- (5% of total)
(molar ratio) somesa
LH-20
of cross-linked lipids Labeling yieldsh
Reaction Reaction 3H I4C with satu- with dou-
rated b i d ble bond
[3H]DPPC t0.02
[3H]DPPC:photola- (control)
bel I1 (201) 0.14 2.8
[3H]DPPC:photola- bel I11
(1O:l) 1.55 15.5 (20:l) 0.92 18.5 (40:l) 0.49
[3H]DPPC:['4C]- POPC photola- bel I11
19.5
(0.1:201) 1.03 1.67 20.6 12.6 a Abbreviations: DPPC = dipalmitoyl phosphatidylcholine; POPC
= 1-palmitoyl-2-oleoyl phosphatidylcholine. If every reagent molecule reacted with one radioactive lipid mol-
ecule, the expected numbers of 3H and 14C counts found in the pool of the cross-linked lipids was defined as a labeling yield of 100% for each isotope.
A B
4 0 a D 3
' O O Q W D - 8 6 96- -83
m '4
I P O Q I
1 I 2 3 1 4 5 6
FIG. 5. Product separation by thin layer chromatography. Solvent A ch1oroform:methanolwater = 65254 (v/v). Solvent B: butanolacetic acidwater = 63:27:10 (v/v). 1, [I4C]gramicidin + lipid photolyzed in the absence of photolabel II; 2, [14C]gramicidin A + photolabel I1 + lipid, photolyzed; 3, photolabel I1 + lipid, photolyzed; 4, ['4C]O-pyr~mellityl-gramicidin A + lipid, photolyzed in the absence of photolabel 11; 5, ['4C]O-pyromellityl-gramicidin A + photolabel I1 + lipid photolyzed; 6, photolabel I1 + lipid photolyzed.
when incorporated in lipid bilayer membranes form dimeric ion conducting pores (25-27). However, a dynamic equilibrium between the dimeric form of the pore and alternative struc- tures (orientations) of the peptide within the membrane may exist. If this is the case, particularly reactive residues may be transiently exposed within the bilayer while not being acces- sible to the reagent in the pore structure. The hydroxyl function of the COOH-terminal ethanolamide group, espe- cially, must be considered as such a potentially reactive site

3324 SS Containing Photolabile Phospholipid Analogues
TABLE 111 Cross-linking ofphotolabel ZZ and ZZZ with gramicidin A and 0- pyromellityl-gramicidin A in dipalmitoyl phosphatidylcholine
liposomes Phospholipid, reagent, and ["Clgramicidin A or [''C]pyromellityl-
gramicidin A were lyophilized from 95% benzene, 5% methanol. The residues (10 mg of lipid) were dispersed in 2 ml of water a t 45-50'C and photolyzed. After lyophilization the residue was dissolved in 0.5 ml of methanol and an aliquot of 100 p1 was subjected to thin layer chromatography as described in Fig. 5. Quantitation of cross-linking is based on radioactivity recovered in the eluted bands.
Observed labeling yields (%)h
yields (56)' as- Expected
Composition of li- suming equal posomes Photolysis Photolysis Photolysis insertion prob-
(molar ratio)" time 15 time 20 t i zn60 lipid and pep- abilities in all
tide C-H mm min
DPPC: photola- bel 111: gramicidin A
(20:l:l) 3.5 5.4 1.5 DPPC:photola-
bel 11: gram- icidin A
DPPC:photola- bel 11: O-py- romellityl- gramicidin A
(20:l:l) 14.2 14.3 12.3 0.22
(20:l:l) 13.8 0.22
Abbreviation: DPPC = dipalmitoyl phosphatidylcholine. * Since the molar ratio of photolabekpeptide was 1:1, the numbers
(%) represent both the fraction of reagent and peptide which formed cross-linked products.
' The two fatty acyl chains of 1 DPPC molecule contain 62 C-H bonds, and 20 such molecules 1240 bonds. One gramicidin A molecule has 100 aromatic and aliphatic C-H bonds. From Table I1 the expected extent of insertion of nitrene probe I1 into saturated bonds is 2.8% X 100 + 1240 = 0.22%, for carbene probe 111 the expected insertion is 18.5% X 100 f 1240 = 1.5%.
within the gramicidin A molecule. In order to exclude the possibility of modification of the hydroxy group by the reagent and to further support the assertion that gramicidin A reacted in its pore form, 0-pyromellityl-gramicidin A was used for similar labeling studies. In this derivative the 0 - H group is acylated and the location of the negatively charged pyromel- lityl group is restricted to an aqueous compartment (38, 39). Labeling results using photolabel I1 and O-pyromellityl-gram- icidin A in dipalmitoyl phosphatidylcholine liposomes are given in Table 111. Essentially identical yields were found for gramicidin A and its 0-acylated derivative. Thus, whatever the sites are of modification they must be accessible to the probe equally well in both peptides. Based on this result, it appears unlikely that in the course of labeling gramicidin A, this peptide existed within the bilayer in a transient structure which positioned the ethanolamide group entirely within lipid core of the membrane. The starting sample of pyromellityl- gramicidin A showed a single spot in the solvent B thin layer chromatograpic system. As shown in Fig. 5B, approximately 4% of 0-pyromellityl-gramicidin A is degraded under the conditions of photolysis.
Immobilization on glass of labeled [14C]gramicidin A by the general procedure outlined in Fig. 3 was also examined. After photolysis the liposomes were lyophilized and the residue dissolved in 95% n-propanol. Reduction of the disulfide and coupling of the sulfhydryl group to derivatized glass was performed as described above. Repeated extraction of the glass with methano1:formic acid (98:2) showed an exponen- tially decreasing release of radioactivity probably due to ad- sorbed material. After five extraction steps, radioactivity re-
leased from the glass was only slightly above background levels and remained constant during additional extraction consistent with similar observations by others (40). Photolysis of gramicidin A in the absence of the photosensitive probe did not lead to detectable changes in the Uv spectrum of the peptide ruling out the possibility that attachment to glass could be the result of direct reaction of photolytically de- graded gramicidin A with the glass. Table IV demonstrates that photolysis is a prerequisite in order to bind gramicidin A to the porous glass. Evidence that a sulfhydryl specific reac- tion is involved was provided by an experiment in which the reduced photolysis products were supplemented with a 20- fold excess p-mercaptoethanol before the derivatized glass was added. Under these conditions, P-mercaptoethanol com- petes with the thiol groups derived from the reagent to react with the maleimide thereby decreasing the yields of covalently bound label from about 50% to 12.4% and that of bound gramicidin A from approximately 5% to 0.74%. That the reduction in binding was not exactly a factor of 20 may be due to differences in reactivity of the " S H groups of the photo- label and mercaptoethanol, or due to an excess of maleimido groups over the sulfhydryls derived from the photolabel.
The Sites of Cross-linking within Gramicidin A Amino Acid Composition-We have already indicated (Ta-
ble 111) that reaction of the nitrene and carbene with aliphatic amino acid residues (C-H insertion) cannot account for the observed labeling yields. Further, the fact that gramicidin A and 0-pyromellityl-gramicidin A were labeled to the same extent does also exclude the possibility that insertion of the reagent into the 0-H bond of the COOH-terminal ethanolam- ide group had occurred. Insertion of the carbene or nitrene into aliphatic C-H bonds would result in the formation of a C- N or a C-C bond predicted to be stable under the conditions of acid hydrolysis of peptides. Since in the glass-bound gam-
TABLE IV Binding to porous glass of reduced products obtained from the
photolysis of liposomes containing photolabel I Z and gramicidin A Multilamellar liposomes were prepared from egg lecithin, photo-
label 11, and gramicidin A in a molar ratio of 20:l:l in 1 mM EDTA, pH 7, and photolyzed for 10 min. The lyophilized material was dissolved in 95% n-propanol and reduced with tributylphosphine. Binding to glass was studied under various experimental conditions.
Adsorbed and bound" Covalently boundh (R of total) (I of total)
Experimental condi- tions 'H ' T 'H "C
bel)' A) bel)' A) (photola- (gramicidin (photola- (gramicidin
___ 1. Without photoly- 81 8.4 55 (0.05
2. With photolysis 86 14 45 5.4 84 11 49 4.5
3. Binding to glass in 18 4 12 0.7
sis
the presence of mercaptoetha- nol"
" The extent of adsorbed and covalently bound photolysis products to glass was determined by comparison of the radioactivity in aliquots of n-propanol solution before and after addition of the porous glass. ' Solely adsorbed products were released from glass by extensive
washing with methanol-formic acid (98:2). Covalently bound radio- activity was determined after methanolysis in 1.5 IVI HCl/methanol overnight at 60°C.
"The term "photolabel" stands for all products containing 'H radioactivity.
The ratio of reduced photolysis products to porous glass remained the same as in 1 and 2. However, about 20 times more sulfhydryl groups were available for reaction with the derivatized glass due in addition of P-mercaptoethanol (25 mo1/100 mg of glass).

SS Containing Photolabile Phospholipid Analogues 3325
TABLE V Amino acid composition of gramicidin A andphotolabeled
gramicidin A Samples of gramicidin A or photolabeled gramicidin A bound to
porous glass (10 to 20 nmol) were hydrolyzed in 6 M HC1 at llO°C for 48 h and subjected to amino acid analysis (Durrum D-500).
Amino acid“ Hydrolysates of gramicidin A labeled with
(Unlabeled) Probe I Probe I1 Probe 111
Glycineh 1.07 1.20 1.15 1.16 Alanineh 1.98 2.16 2.12 2.18 Valine 3.99 4.09 3.98 4.21 Leucine 4.00 4.00 4 .OO 4.00 Isoleucine 0.15 0.16 0.16 0.15
* Tryptophan was not determined in these analytical runs. Note that destruction of tryptophan in these analyses, where
hydrolysis is carried out in 6 M HCI at llO”C, does not produce glycine and alanine or breakdown products. These are produced in the back hydrolysis of the PTH derivative in 55% HI at 150°C.
icidin A 1 amino acid residue (out of 15) is chemically modi- fied, this should be reflected in the amino acid compositions of hydrolyzed samples when compared with .the amino acid composition of pure gramicidin A. However, as is shown in Table V, essentially identical amino acid ratios were found in all of these hydrolysates. Tryptophan contents could not be included in this analysis since ninhydrin-positive components were released from the derivatized glass which interfered with the quantitation of this amino acid and in addition partial degradation of tryptophan was expected under these condi- tions of hydrolysis. These results further support that C-H insertion into aliphatic residues was not the principal reaction and in particular that valine at position 1 of the peptide was not the main site of attack by the carbene and nitrene. (The tertiary C-H bond of valine had initially been considered as a likely site for reaction since it is more reactive than primary and secondary C-H bonds and also presumed readily accessi- ble for the probes I1 and 111.)
Edman Degradation of Photolabeled Gramicidin A Edman degradation of gramicidin A using conventional
techniques (spinning cup) is not possible since the peptide is completely extracted after 2 to 3 degradation cycles even if a carrier material such as “polybrene” is p r e ~ e n t . ~ Solid phase techniques require appropriate functional groups in a peptide, preferentially the COOH-terminal carboxyl group, which al- low covalent attachment of the peptide to a solid support. Various chemical modifications of the COOH-terminal ethan- olamide group of gramicidin A have been described; however, none of them appeared appropriate for the attachment of gramicidin A to a solid support. (An attempt to substitute the hydroxy group by an amino acid group was unsuccessful.) As a result of this, gramicidin A covalently bound to glass beads via the sulfhydryl arm of the photolabel was used for Edman degradation studies. It was assumed that (a) for statistical reasons each gramicidin A molecule was modified at a single site and therefore attachment to glass occurred via a single amino acid residue, and (b ) due to the absence of any charged residues the gramicidin A or its fragments would be reasonably soluble in many organic solvents.
If the modified amino acid residue through which gramici- din A is bound to the solid support is at position M within the polypeptide chain, Edman degradation should proceed nor- mally through step M - 1 thereby exposing the NH2 terminus of the modified amino acid M’. The following cycle cleaves between the labeled amino acid M’ and M + 1. However, since the remaining peptide (M + 1 to the COOH terminus) is not
P. Fletcher, personal communication.
attached to glass, this peptide will be released and extracted while the photolabeled amino acid M’ will remain bound to the glass. Thus, quantitation and identification of material released from each cycle should provide information concern- ing the sites of modification.
To assure optimal extraction of released peptides during the degradation process, the normal sequenator procedure was modified to include an extraction step with methanol. (Methanol is an excellent solvent for short hydrophobic pep- tides as well as for gramicidin A.)
Photolabeled [ 14C]gramicidin A bound to glass was initially treated at room temperature with 1.5 M HC1 in methanol for 4 to 6 h to release the NHz-terminal formyl group. Subsequent amino acid analysis of a glass sample demonstrated that this treatment did not release the bound peptide. Edman degra- dation was performed with three samples of gramicidin A labeled with the photoreagent I, 11, and 111. In each run, 100 mg of the porous glass were introduced containing 25 to 50 nmol of peptide. The methanol phase of each cycle containing the 2-anilino-5-thiazolinone derivative of the released amino acid (and released peptide) was analyzed according to stan- dard techniques. 1) Anilinothiazolinone amino acids were converted to their phenylthiohydantoin derivatives and these were analyzed by gas chromatography. 2) Samples were also hydrolyzed (55%, HI, 150”C, 4 h) and subjected to amino acid analysis. This latter step also allowed any peptide released and extracted into methanol to be recovered and analyzed as follows. During the conversion of the anilinothiazolinone amino acid to the phenylthiohydantoin derivative, the peptide was assumed not to be degraded and the subsequent ethyl acetate extraction was found to extract both the phenylthio- hydantoin amino acid and any peptide present. Therefore, under the conditions of “back hydrolysis” peptides would be hydrolyzed and could be analyzed together with the amino acid derived from its phenylthiohydantoin derivative.
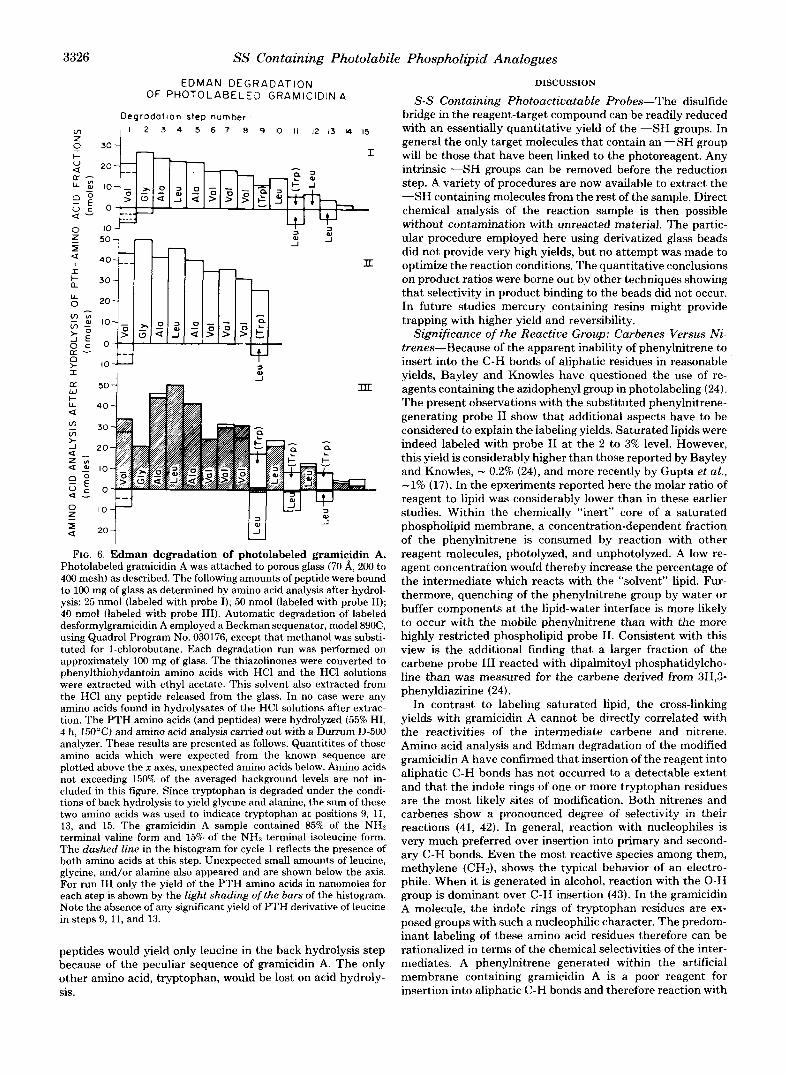
Gramicidin A labeled with probe I1 was degraded through 9 cycles, that labeled with probe I or I11 for 15 cycles. Fig. 6 shows amino acid analysis data for the hydrolysates of the ethylacetate extracts obtained from each degradation step. As explained above, each extract will contain a phenylthiohydan- toin amino acid and any COOH-terminal peptide (M + 1 to COOH terminus) released from the glass.
Through cycles 1 to 8 the Edman degradation proceeded normally and in particular there was no indication that any detectable amount of peptide was released. During cycle 9 (gramicidin A labeled with probe 111) and cycles 11 and 13 (gramicidin A labeled with probes I to 111) significant quanti- ties of leucine were found in the back hydrolysates. This extra amino acid could have originated from 1) PTH derivatives of leucine as a result of an “out-of-phase” degradation, or 2) from COOH-terminal peptides of residues 10 to 15 (Leu-Trp-Leu- Trp-Leu-Trp), 12 to 15 (Leu-Trp-Leu-Trp), and 14 and 15 (Leu-Trp). The former possibility, however, can be excluded since no PTH-leucine could be detected by gas chromatogra- phy and in addition the appearance of leucine at step 9 would not be easy to explain. These results can be rationalized, however, by assuming that gramicidin A was primarily bound to glass through tryptophan residues and that the origin of the leucine was in fact the peptides 10 to 15, 12 to 15, and 14 and 15. We conclude from this result that labeling of grami- cidin A has occurred primarily at the tryptophan residues. This conclusion is supported by the previous findings that (1) C-H insertion into aliphatic C-H bonds cannot account for the observed labeling yields and (2) that the COOH-terminal hydroxy group is unlikely to be the site of modification since both gramicidin A and 0-pyromellityl-gramicidin A gave iden- tical labeling yields. Any of the expected COOH-terminal
3326 SS Containing Photolabile Phospholipid Analogues
EDMAN DEGRADATION OF PHOTOLABELED GRAMICIDIN A
D e g r o d o t t o n step number , I 2 3 4 5 6 7 8 9 IO I1 1 2 1 3 14 15
, O i - T
u
I > lo-
FIG. 6. Edman degradation of photolabeled gramicidin A. Photolabeled gramicidin A was attached to porous glass (70 A, 200 to 400 mesh) as described. The following amounts of peptide were bound to 100 mg of glass as determined by amino acid analysis after hydrol- ysis: 25 nmol (labeled with probe I); 50 nmol (labeled with probe 11); 40 nmol (labeled with probe 111). Automatic degradation of labeled desformylgramicidin A employed a Beckman sequenator, model 890C, using Quadrol Program No. 030176, except that methanol was substi- tuted for 1-chlorobutane. Each degradation run was performed on approximately 100 mg of glass. The thiazolinones were converted to phenylthiohydantoin amino acids with HCl and the HCl solutions were extracted with ethyl acetate. This solvent also extracted from the HCl any peptide released from the glass. In no case were any amino acids found in hydrolysates of the HC1 solutions after extrac- tion. The PTH amino acids (and peptides) were hydrolyzed (55% HI, 4 h, 150’C) and amino acid analysis carried out with a Dunum D-500 analyzer. These results are presented as follows. Quantitites of those amino acids which were expected from the known sequence are plotted above the x axes, unexpected amino acids below. Amino acids not exceeding 150% of the averaged background levels are not in- cluded in this figure. Since tryptophan is degraded under the condi- tions of back hydrolysis to yield glycine and alanine, the sum of these two amino acids was used to indicate tryptophan at positions 9, 11, 13, and 15. The gramicidin A sample contained 85% of the NHz terminal valine form and 15% of the NH:! terminal isoleucine form. The dashed line in the histogram for cycle 1 reflects the presence of both amino acids at this step. Unexpected small amounts of leucine, glycine, and/or alanine also appeared and are shown below the axis. For run Il l only the yield of the PTH amino acids in nanomoles for each step is shown by t.he light shading of the burs of the histogram. Note the absence of any significant yield of PTH derivative of leucine in steps 9, 11, and 13.
peptides would yield only leucine in the back hydrolysis step because of the peculiar sequence of gramicidin A. The only other amino acid, tryptophan, would be lost on acid hydroly- sis.
DISCUSSION
S-S Containing Photoactivatable Probes-The disulfide bridge in the reagent-target compound can be readily reduced with an essentially quantitative yield of the “SH groups. In general the only target molecules that contain an ”SH group will be those that have been linked to the photoreagent. Any intrinsic “ S H groups can be removed before the reduction step. A variety of procedures are now available to extract the ”SH containing molecules from the rest of the sample. Direct chemical analysis of the reaction sample is then possible without contamination with unreacted material. The partic- ular procedure employed here using derivatized glass beads did not provide very high yields, but no attempt was made to optimize the reaction conditions. The quantitative conclusions on product ratios were borne out by other techniques showing that selectivity in product binding to the beads did not occur. In future studies mercury containing resins might provide trapping with higher yield and reversibility.
Significance of the Reactive Group: Carbenes Versus Ni- trenes-Because of the apparent inability of phenylnitrene to insert into the C-H bonds of aliphatic residues in reasonable yields, Bayley and Knowles have questioned the use of re- agents containing the azidophenyl group in photolabeling (24). The present observations with the substituted phenylnitrene- generating probe I1 show that additional aspects have to be considered to explain the labeling yields. Saturated lipids were indeed labeled with probe I1 at the 2 to 3% level. However, this yield is considerably higher than those reported by Bayiey and Knowles, - 0.2% (24), and more recently by Gupta et al., -1% (17). In the epxeriments reported here the molar ratio of reagent to lipid was considerably lower than in these earlier studies. Within the chemically “inert” core of a saturated phospholipid membrane, a concentration-dependent fraction of the phenylnitrene is consumed by reaction with other reagent molecules, photolyzed, and unphotolyzed. A low re- agent concentration would thereby increase the percentage of the intermediate which reacts with the “solvent” lipid. Fur- thermore, quenching of the phenylnitrene group by water or buffer components at the lipid-water interface is more likely to occur with the mobile phenylnitrene than with the more highly restricted phospholipid probe 11. Consistent with this view is the additional finding that a larger fraction of the carbene probe I11 reacted with dipalmitoyl phosphatidylcho- line than was measured for the carbene derived from 3H,3- phenyldiazirine (24).
In contrast to labeling saturated lipid, the cross-linking yields with gramicidin A cannot be directly correlated with the reactivities of the intermediat.e carbene and nitrene. Amino acid analysis and Edman degradation of the modified gramicidin A have confirmed that insertion of the reagent into aliphatic C-H bonds has not occurred to a detectable extent and that the indole rings of one or more tryptophan residues are the most likely sites of modification. Both nitrenes and carbenes show a pronounced degree of selectivity in their reactions (41, 42). In general, reaction with nucleophiles is very much preferred over insertion into primary and second- ary C-H bonds. Even the most reactive species among them, methylene (CH,), shows the typical behavior of an electro- phile. When it is generated in alcohol, reaction with the 0 - H group is dominant over C-H insertion (43). In the gramicidin A molecule, the indole rings of tryptophan residues are ex- posed groups with such a nucleophilic character. The predom- inant labeling of these amino acid residues therefore can be rationalized in terms of the chemical selectivities of the inter- mediates. A phenylnitrene generated within the artificial membrane containing gramicidin A is a poor reagent for insertion into aliphatic C-H bonds and therefore reaction with

SS Containing Photolabile Phospholipid Analogues 3327
the tryptophans becomes virtually the exclusive reaction with the peptide. The more reactive carbene also favors the elec- tron-rich tryptophan residues, however, since also C-H inser- tion into the solvent lipid can occur, this latter reaction will consume a significant fraction of the carbene and lead to lower labeling yields of the peptide.
Reactivities and chemical selectivities are correlated with the lifetimes of the intermediates. The presumed longer life- time of the phenylnitrene compared with the carbene will allow more translational diffusion and consequently increase the probability of contact with reactive residues. The question of reactivities and chemical selectivities becomes crucial when the sites of labeling are to be correlated with the position of the reactive species along the length of a fatty acyl chain of an amphipathic reagent. Due to the fluidity of the lipid core of a membrane, the reactive group of the probe will not be localized exclusively at a specific depth within the membrane but will sample a rather broad distribution of positions. This is evident from the recent analysis of the sites of cross-linking within fatty acyl chains by Gupta et al. (18). The extent of labeling of a specific amino acid side chain localized at a particular depth in a membrane will therefore be a function of (i) the concentration of the reagent at the corresponding depth and (ii) the relative reactivities of this amino acid residue compared with other nearby groups. The finding that in gramicidin A tryptophan residues were preferentially attacked does not therefore imply that the carbene concentration was larger within the peripheral layer of the membrane, where the tryptophan residues lie, than in the central plane of the bilayer. However, it does imply that tryptophan residues are orders of magnitude more reactive than any other amino acid residue exposed in the gramicidin A pore. In these particular experiments noncovalent association of the aromatic rings of an indole group and of the reagent may additionally contribute to the observed reaction with tryptophan.
Whether or not the approach of labeling proteins in a membrane at a position defined by the length of the probe will be successful may depend on precursors which generate carbenes of such reactivity that within their lifetime diffusion can be neglected. Such a reagent would not distinguish at all between lipids and proteins and thus the labeling yield of any specific component might be extremely low. At the present time, little detailed information is available concerning life- times and chemical selectivities of most of the carbenes and nitrenes used in photolabeling.
Possible Improvement of the Structural Resolution from Photolabeling Studies-To restrict the location of photola- beling within a bilayer, one possible approach would be to attach the photosensitive group closer to the head group of a phospholipid molecule. In such a case, it could be assumed that only the outer, spatially better defined, layer of the hydrophobic core of a membrane would be accessible for reaction.
A second approach might be based on the present finding that l-palmitoyl-2-(12-p-azidophenyl-9,lO-dithiadodecanoyl) phosphatidylcholine was transferred by the phospholipid ex- change protein from sonicated dispersion of the reagent into the membrane of red blood cells. Membranes reconstituted from mixtures of phospholipids are in general closed vesicular systems of which the lipid components are uniformly or nearly uniformly distributed between the two monolayer halves. The protein-catalyzed exchange technique permits the incorpora- tion of this reagent (and presumably of other structurally related photosensitive lipids) specifically into the outer mono- layer of both synthetic and biological membranes. I t is rea- sonable to assume that a highly asymmetric distribution of the probe can be established and maintained both during the
exchange procedure and during photolysis. Labeling of mem- branes from within one monolayer a t a time should lead to an improvement of the structural resolution which can be derived from photolabeling studies.
Acknowledgments-Sincere thanks are addressed to Dr. Paul Fletcher and Gary Davis for peptide sequence analysis; Dr. Ruth Kramer for advice in the preparation and assays of the phospholipid exchange protein. We also acknowledge the help of Dr. J. Rosa, J . Mouning, and M. Lane in the preparation of the manuscript.
REFERENCES
1. Richards, F. M., and Brunner, J. (1979) Ann. N. Y. Acad. Sci. in
2. Kiehm, D. J., and Ji, T. H. (1977) J . Bwl. Chem. 252,8524-8531 3. Huang, C.-K., and Richards, F. M. (1977) J. Biol. Chem. 252,
5514-5521 4. Bayley, H., and Knowles, J. R. (1977) Methods Enzymol. 46,69-
114 5. Matheson, R. R., Van Wart, H. E., Burgess, A. W., Weinstein, L.
I., and Scheraga, H. A. (1977) Biochemistry 16, 396-403 6. Jelenc, P. C., Cantor, C. R., and Simon, S. R. (1978) Proc. Natl.
Acad. Sci. U. S. A. 75,3564-3568 7. Benisek, W. F., and Smith, S. B. (1979) Ann. N. Y. Acad. Sci. in
press 8. Sperling, J. (1979) Ann. N. Y. Acad. Sci. in press 9. Klip, A,, and Gitler, C. (1974) Biochem. Biophys. Res. Commun.
press
60, 1155-1162 10. Bercovici, T., and Gitler, C. (1978) Biochemistry 17, 1484-1489 11. Kahane, I., and Gitler, C. (1978) Science 201, 351-352 12. Goldman, D. W., Pober, J. S., White, J., and Bayley, H. (1979)
13. Gitler, C., and Bercovici, T. (1979) Ann. N. Y. Acad. Scz. in press 14. Chakrabarti, P., and Khorana, H. G. (1975) Biochemistry 14,
15. Greenburg, G. R., Chakrabarti, P., and Khorana, H. G. (1976) Proc. Natl. Acad. Sci. U. S. A. 73, 86-90
16. Gupta, C. M., Radhakrishnan, R., and Khorana, H. G. (1977) Proc. Natt. Acad. Sci. U. S. A. 74,4315-4319
17. Gupta, C. M., Radhakrishnan, R., Gerber, G. E., Olsen, W. L., Quay, S. C., and Khorana, H. G. (1979) Proc. Natl. Acad. Sci. U. S. A. 76,2595-2599
18. Gupta, C. M., Costello, C. E., and Khorana, H. G. (1979) Proc. Natl. Acad. Sci. U. S. A. 76,3139-3143
19. Stoffel, W., Schreiber, C., and Scheefers, H. (1978) Hoppe-Seyler’s Z. Physiol. Chem. 359,923-931
20. Bramhall, J., Ishida, B., and Wisnieski, B. (1978) J . Supramol. Struct. 9,399-406
21. Wirtz, K. W. A., Van Golde, L. M. G., and Van Deenen, L. L. M. (1970) Biochim. Biophys. Acta 218, 176-179
22. Wirtz, K. W. A., Kamp, H. H., and Van Deenen, L. L. M. (1972) Biochim. Biophys. Acta 274, 606-617
23. Bayley, H., and Knowles, J. R. (1978) Biochemistry 17,2414-2419 24. Bayley, H., and Knowles, J. R. (1978) Biochemistry 17,2420-2423 25. Veatch, W. R., Mathies, R., Eisenberg, M., and Stryer, L. (1975)
26. Veatch, W. R., and Stryer, L. (1977) J. Mol. Biol. 113, 89-102 27. Weinstein, S., Wallace, B. A,, Blout, E. R., Morrow, J . S., and
Veatch, W. (1979) Proc. Natl. Acad. Sci. U. S. A. 76,4230-4234 28. Brunner, J., Senn, H., and Richards, F. M. (1980) J. Biol. Chem.
255,3313-3318 29. Ellman, G. L. (1959) Arch. Biochem. Biophys. 82, 70-77 30. Brois, S. J., Pilot, J . F., and Barnum, H. W. (1970) J. Am. Chem.
31. Mori, K., and Nakamura, Y. (1969) J . Org. Chem. 34,4170-4172 32. DeGraw, J. I., and Engstrom, J. S. (1975) J. Labelled Compounds
33. Bangham, A. D. (1968) Progr. Biophys. Mol. Biol. 18,29-95 34. Huang, C. (1969) Biochemistry 8, 344-352 35. Chapman, D., Cornell, B. A., Eliasz, A. W., and Perry, A. (1977)
36. Kamp, H. H., and Wirtz, K. W. A. (1974) Methods Enzymol. 32,
37. Steck, T. L., and Kant, J . A. (1974) Methods Enzymol. 31, 172-
38. Apell, H.-J., Bamberg, E., Alpes, H., and Lauger, P. (1977) Membr.
Nature 280, 841-843
5021-5033
J. Mol. Biol. 99, 75-92
SOC. 92, 7629-7631
XI, 233-239
J. Mol. Biol. 113, 517-538
140-146
180
Biol. 31, 171-188

3328 SS Containing Photolabile Phospholipid Analogues
39. Bamberg, E., Apell, H. J., and Alpes, H. (1977) Proc. Natl. Acad. Sci. U. S. A. 74, 2402-2406
40. Powers, D. A. (1975) in Solid Phase Methods in Protein Sequence Analysis (Laursen, R., ed) pp. 299-320, Pierce Chemical Co., Pub., Rockford, Ill.
41. Lwowski, W. (1970) Nitrenes, Interscience Publishers, New York
SUPPLEMENTARY MATERIAL
TO
ANALYSIS OF MEMBRANES PHOTOLAMELED WITH L I P I D ANALOGUES. REACTION OF PHOSPHOLIPIDS CONTAINING A DISULFIDE GROUP AND A NITRENE OR CARMENE PRECURSOR WITH LIPIDS AND U I T H
G R A M I C l U l N A
MY
Jo ief M r u n n e ~ dnd F reder l c M. Richard5
EXPERIMENTAL PROCEDURES
3 - ~ E t h a x u t h l o ~ a r b a n y l ) t h l o l p r o p a n o l c dc ld , XI1 A IO lU t lOn O f 5 3 . 5 y ( 0 . 3 5 moler) o f 3-
bromo-pTOpanOlC a c l d ( ICN) , 56.1 y (0.35 naler) O f p o t a ~ ~ l ~ r n e thy l xdn thogenate (F lukd)
d l l s o l v e d ~n 350 m1 o f I M NdOH was stirred a t room temperature for 24 h o u r i . The yellow
solution was c h l l l e d to 0" (Cryitalllne p r e c l p l t d t e j and a c l d l f i e d w1th COnc. HCI to pH 2
m.p. 66-68' (Me i ls te ln , 111, p 300: 71'1, IH-NMR ( C D C 1 3 ) 1 .42 ( t . J=7 HI, 3 H , ) , 7.83
The p r e c l p l t a t e i s rerOOwd by f l l t r d t l o n and washed Severdl t l rner m t h h a t e r . 62.4 g (=92.),
( t , J=7 HZ. 2 H ) . I 3.37 ( t , J.7 HI. 2 H ) . \ 4 .65 ( g , J=7 HZ, 2H) .
8 - [ ( E t h ~ r y t h l o c a r b o n y l l t h l ~ t a n o i c acid. X l b A I O l U t l O n o f 8 0 9 I 3 6 m l O l e i ) o f 8-bromo-
o c t m o l c dcld ( I C N ) , 14.3 rnl o f 2 . 5 M NdOH and 10 rnl O f w t e r was coo led ~n a n 1ce bath.
Then, 5.75 g p o t d l i l m e thy l xan thogend te d> iso l ved ~n 10 8nI O f water was added dnd the mix-
t d t e WhlCn war e x t r a c t e d i n t o 100 rnl O f ether The o rgan ic layer "35 washed twice Wlth Water
tule k e p t a t 4 ~ ' f o r 2 d a y s . A d d l t l o l i o f c o n c e n t r a t e d HCI ( f l n a l pH 1 - 2 1 g d v e dn D l l y preclpi-
and d r led over MySDq Evaporation o f the SUlVerlt gave 8 P 9 ( 9 5 " ) A f t e l I r y l t d l l l i d t l D n
f r o r h o t Il-heXd!$e, 7.65 g (81.5:) o f t he tltir compOu!#d wd5 ob ta ! red nl p 29-31- IH-NMR
ZH). ( C D C 1 3 ) 1 . 2 - 1 9 (rn,lOH), 2 . 3 ( t , J.7 H i , ? H I , ' 3.05 (t. J=i H i , 2H1, i 4.60 ( q , J - 7 H i ,
42. Kirmse, W. (1964) Carbene Chemistry, Academic Press, New
43. Kerr, J. A., O'Grady, B. V., and Trotman-Dickenson, A. F. (1967)
Additional references are found on p. 3329.
York
J . Chem. SOC. (A) 897-898

SS Containing Photolabile Phospholipid Analogues
I . 2. 3. 4.
5. 6 . 7 . 8. 9.
10
Zumach, G. and KUhle. E . ( 1 9 7 0 ) Anqew. Chern. i n t e r n a t . E d i t . 9, 54-63.
DeGraw. J.I. and Engr tmm, J.S. 11975) J . Labe l l ed Compounds, V o l . g , NO. 2 , 233-239. Brunner. J . , Senn, H. and Richards. F.M. ( 1 9 7 9 ) Gupta, C . H . , Radhakrirhnan. R. and Khorand, H.G. ( 1 9 7 7 ) Proc. N a t l . Rcad. Sci. U S R 74. 4315-4319.
Chen. P . S . Tar iba ra , T.Y. and Warner, H. (1956) 4na l . Chem. 28. 1756-1758. Sarger. R and Witkop, B. (1965) J. &. p e ~ . %. 8 7 , 2011-2020.
Apel l . H. -J . , Barnberg, E., Alpes. H . and Llluger, P . (19771 h b r a n e B i o 1 . 31, 171-188. Keller. 0. and Rudinger, J . (1975) H I I Y . Chim. Acta E. 531-54.
Robinson. P.J . . Dunnh i l l . P. and L l l l y , M.D. ( 1 9 7 1 ) Blochim. Biophys. Acta 212, 659-661. RUegg, U.T. and Rudinger. J . ( 1 9 7 7 ) i n : H e t h o d r i n E n z m l a IHirs, C.H.U. and T i m r h e f f , S . N . . eds.1. Val. XLVII, pp. 111-126. Academic Prerr, New Y%,





























