Embed Size (px)
Citation preview

1
Varicella-zoster virus glycoprotein I is essential for spread in dorsal root ganglia and 1
facilitates axonal localization of structural virion components in neuronal cultures 2
Jenna Christensen1, Megan Steain
1, Barry Slobedman
1,2* and Allison Abendroth
1,2*# 3
1. Discipline of Infectious Diseases and Immunology, The University of Sydney, Australia; 4
2. The Centre for Virus Research, Westmead Millennium Institute, Sydney, Australia 5
#Address for correspondence: 6
A/Prof Allison Abendroth 7
Discipline of Infectious Diseases and Immunology 8
The University of Sydney, New South Wales, Australia 2006. 9
* Equal contribution 10
11
Running Title: Role of VZV gI in neuronal cell infection 12
13
14
15
16
17
18
JVI Accepts, published online ahead of print on 9 October 2013J. Virol. doi:10.1128/JVI.02293-13Copyright © 2013, American Society for Microbiology. All Rights Reserved.
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

2
ABSTRACT 19
Neurons of the sensory ganglia are the major site of varicella-zoster virus (VZV) latency and 20
may undergo productive infection during reactivation. Although the VZV gE/gI complex is 21
known to be critical for neurovirulence, few studies have assessed the roles of these proteins 22
during DRG infection due to the high human specificity of the virus. Here, we show that 23
VZV glycoprotein I is an important neurotropic gene responsible for mediating the spread of 24
virus in neuronal cultures and explanted dorsal root ganglia (DRG). In comparison with the 25
parental strain (VZV rOka), inoculation of differentiated SH-SY5Y neuronal cell cultures 26
with a VZV gI deletion strain (VZV rOka∆gI) showed a large reduction in the percentage of 27
cells infected and significantly smaller plaque sizes. In contrast VZV rOka∆gI was not 28
significantly attenuated in fibroblast cultures, demonstrating a cell-type specific role of VZV 29
gI. Analysis of rOka∆gI protein localization by immunofluorescent staining revealed aberrant 30
localization of viral glycoprotein and capsid proteins, with little or no staining present in the 31
axons of differentiated SH-SY5Y cells infected with rOka∆gI, yet axonal vesicle trafficking 32
was not impaired. Further studies utilizing explanted human DRG indicated that VZV gI is 33
required for spread of virus within DRG. These data demonstrate a role for VZV gI in cell-to-34
cell spread of virus during productive replication in neuronal cells and a role in facilitating 35
access of virion components to axons. 36
37
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

3
INTRODUCTION 38
Varicella-zoster virus (VZV) is the etiological agent of the human disease varicella 39
(chickenpox) (1). Following primary infection, the virus establishes latency within neurons of 40
the sensory ganglia (2, 3) from where it may reactivate to result in the secondary clinical 41
disease herpes zoster (shingles) (4). Herpes zoster may be followed by a state of debilitating, 42
long-term pain known as post-herpetic neuralgia (PHN) which is commonly resistant to many 43
traditional pain therapies (3, 5, 6). 44
VZV glycoprotein I (gI) is a type I membrane protein encoded by open reading frame 67 45
(ORF67) and functions primarily as a heterodimer with the most abundant VZV protein gE 46
(7). Although gI is dispensable in cell culture, deletion results in an impairment of syncytia 47
formation, delayed replication and a decrease in infectious virus yields within melanoma cells 48
(8). In the context of neuronal infection, gI has been shown to be dispensable in a rat model 49
of persistent VZV infection, although this host is non-permissive (9). In a severe combined 50
immunodeficiency human (SCIDhu) mouse model of VZV infection utilizing human 51
xenografted dorsal root ganglia (DRG), infection with a VZV mutant lacking gI (rOka∆gI) 52
resulted in prolonged replication within the DRG and an inability to transition to a persistent 53
state normally characterized by limited viral transcription (10). In addition, mutation of the 54
cysteine rich region in VZV gE responsible for gE/gI heterodimer formation impaired cell-to-55
cell spread of virus in DRG (11). 56
Studies assessing the herpes simplex virus (HSV) and pseudorabiesvirus (PRV) homologs of 57
gI have reported important functions in mediating anterograde axonal egress during neuronal 58
infection. HSV infection of neurons using compartmentalized chamber systems and rat 59
models have demonstrated that gI is essential for anterograde spread of virus (12). Studies of 60
HSV have demonstrated that the gE/gI heterodimer is required for transport of the viral 61
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

4
proteins gB and gD as well as viral capsids (13). PRV gE/gI deletion viruses are also 62
impaired in anterograde spread in rat models of retinal infection, despite no impairment of 63
retrograde transport or replication within retinal ganglia neurons (14, 15). Similarly, deletion 64
of PRV gI results in impairment of anterograde spread using compartmentalized chamber 65
systems (16, 17). However, VZV interactions within neuronal axons have been poorly 66
defined to date due to the high human specificity of the virus, posing limitations on the 67
models available to assess VZV neuronal infection (1, 18). 68
A number of cell culture models have recently been developed for the assessment of VZV-69
neuronal interactions. VZV infection of differentiated human neural stem cells (19) and 70
terminally differentiated neurons derived from induced pluripotent stem cells (20, 21) have 71
been reported as persistent models of VZV infection. In contrast, human embryonic stem cell 72
(22) and differentiated human neuroblastoma (23) derived neuronal cells induced productive 73
VZV infection under the study conditions utilized. The differentiated SH-SY5Y 74
neuroblastoma cell culture model previously developed by our laboratory may be particularly 75
useful to assess the neurovirulence of VZV deletion or mutant strains during productive VZV 76
infection (23). Here, we utilize this model to detail the requirements of gI during VZV 77
neuronal infection using parental strain rOka and the gI deletion strain rOka∆gI (8). Flow 78
cytometry and infectious center assays were used to demonstrate a cell type specific role for 79
VZV gI in mediating spread of virus between neuronal cells. Furthermore, 80
immunofluorescent staining and confocal microscopy identified a role for VZV gI in 81
facilitating access of virion proteins to axons. Experiments utilizing primary human fetal 82
explanted DRG (24) to assess the role of VZV gI in an ex vivo setting where the natural 83
architecture of the ganglia is preserved support the hypothesis that gI is an important VZV 84
protein required for cell-to-cell spread of virus between ganglionic cells. 85
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

5
MATERIALS AND METHODS 86
Cell and virus culture 87
SH-SY5Y cells were differentiated as previously described and cultures established by 88
inoculating 1 x 105 cells/coverslip in 24-well culture plates (23). In parallel, cultures of 89
human foreskin fibroblasts (HFFs) were similarly established by inoculating 1 x 105
90
cells/coverslip in 24-well culture plates. HFFs were cultured in Dulbecco’s Modified Eagle’s 91
Medium (DMEM; Gibco, Australia) containing fetal calf serum (FCS, 10% v/v; In Vitro 92
Technologies, Australia) and penicillin-streptomycin (50 IU/mL; Gibco, Australia) and were 93
allowed to adhere overnight prior to infection. 94
VZV rOka and rOka∆gI (8) were propagated in HFFs in DMEM containing FCS (10% v/v) 95
and penicillin-streptomycin (50 IU/mL) and were harvested for infection experiments when 96
80-90% of the monolayers were exhibiting cytopathic effect (CPE). Infected or uninfected 97
(mock) cultures were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; 98
Sigma-Aldrich, Australia) and then inoculated onto either differentiated SH-SY5Y or HFF 99
cultures at a density of 1 x 105 cells/well, or a final 1:1 ratio of infected cells to uninfected 100
cells. Coverslips were harvested at various times p.i. for analysis. 101
Immunostaining and flow cytometry 102
Triplicate wells containing differentiated SH-SY5Y cells or HFFs inoculated with CFSE 103
labeled mock, rOka or rOka∆gI infected HFFs were pooled together at either 24, 48 or 72 104
hours p.i. and fixed in 1% paraformaldehyde (PFA; BD Biosciences, Australia) for 20 105
minutes. Prior to immunostaining, cells were permeabilized in saponin buffer (PBS 106
containing 1% FCS, 0.2% sodium azide (Sigma-Aldrich, Australia), 10 mM EDTA (Gibco, 107
Australia) and 0.3% saponin (Sigma-Aldrich, Australia)) for 10 minutes. To quantify the 108
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

6
proportion of VZV antigen positive cells, samples were separately stained in saponin buffer 109
with a mouse anti-VZV primary antibody specific to the immediate-early IE62 protein (clone 110
IE(62) 1:50; Meridian Life Science, USA) or a mouse anti-VZV primary antibody specific to 111
late gEgI proteins (clone SG1 1:200, Meridian Life Science, USA). The gEgI specific 112
antibody used in this study is documented to individually react with both the precursor and 113
mature forms of gE and gI. Cells were incubated at 4°C for an hour, washed in PBS and then 114
incubated with a goat anti-mouse Alexa Fluor 647 conjugated antibody (1:200; Life 115
Technologies, Australia) in saponin buffer at 4°C for an hour. Cells were washed in PBS, 116
filtered and processed using a FACS Canto cytometer and analyzed using FACS Diva 117
software. Specificity of staining was confirmed by assessing isotype antibody treated cultures 118
at corresponding concentrations. All statistical analysis was performed using a paired 2-tailed 119
student’s t-test. 120
Infectious Center Assay 121
Coverslips containing cultures of either differentiated SH-SY5Y cells or HFFs were 122
inoculated with serial 1 in 10 dilutions of mock, rOka or rOka∆gI infected HFFs. Cultures 123
were left to incubate for 96 hours and were then fixed in 4% PFA for 20 minutes. Single 124
immunofluorescent staining was performed using a primary antibody against VZV gEgI 125
(1:600; Meridian Life Sciences, USA) followed by a donkey anti-mouse Alexa Fluor 594 126
conjugated secondary antibody (1:200; Life Technologies, Australia). Specificity of staining 127
was confirmed by assessing isotype antibody treated cultures at corresponding concentrations. 128
Coverslips were mounted with Prolong Gold antifade reagent containing 4',6-Diamidino-2-129
Phenylindole, Dihydrochloride (DAPI; Life Technologies, Australia). Infectious centers were 130
imaged and counted using a Zeiss LSM 510 Meta confocal microscope. Approximately 10 131
infectious centers per sample were randomly selected and area quantified using Zeiss 132
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

7
Axiovision software. All statistical analysis was performed using a paired 2-tailed student’s t-133
test. 134
Immunofluorescence staining and confocal microscopy 135
Coverslips containing differentiated SH-SY5Y cells inoculated with CFSE labeled mock, 136
rOka or rOka∆gI infected HFFs were fixed in 4% PFA for 20 minutes. Cells were washed in 137
PBS, permeabilized and blocked in PBS containing 0.1% Triton X and 20% normal donkey 138
serum (NDS) for 30 minutes at room temperature. For single immunofluorescent staining, 139
cells were incubated with mouse anti-VZV primary antibodies against gH (clone SG3 1:500; 140
Meridian Life Science, USA), gB (clone SG2-E6, 1:50; Meridian Life Science, USA), gEgI 141
(1:600, Meridian Life Science, USA) or pORF40 (clone NCP-1 1:500; Meridian Life Science, 142
USA) at 37°C for an hour under humidified conditions. VZV antibodies were detected by 143
incubation with a donkey anti-mouse Alexa Fluor 546 conjugated secondary antibody (1:200; 144
Life Technologies, Australia) for a further 30 minutes at 37°C. Specificity of staining was 145
confirmed by assessing isotype control antibody treated cultures at corresponding 146
concentrations. Coverslips were mounted with Prolong Gold antifade reagent containing 147
DAPI. 148
Dual immunofluorescent staining was performed using the VZV antibodies utilized in single 149
immunofluorescent experiments in combination with rabbit antibodies specific to syntaxin-6 150
(clone C34B2 1:100; Cell signaling, USA), synaptophysin (clone Z66 1:10; Invitrogen, 151
Australia) or chromogranin A (N-terminus 1:200; Abcam, UK). Coverslips incubated with 152
syntaxin-6 antibody were incubated overnight at 4°C or coverslips incubated with 153
synaptophysin or chromogranin A were incubated for an hour at room temperature, under 154
humidified conditions. Primary antibodies were detected with donkey anti-rabbit Alexa Fluor 155
647 conjugated (1:200; Life Technologies, Australia) and donkey anti-mouse Alexa Fluor 156
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

8
546 conjugated secondary antibodies (1:200; Life Technologies, Australia) applied for 30 157
minutes at 37°C. Specificity of staining was confirmed by using isotype antibody treated 158
cultures at corresponding concentrations. All coverslips were mounted with Prolong Gold 159
antifade reagent containing DAPI. 160
Fluorescence images were acquired using a Zeiss LSM 510 Meta confocal microscope. Areas 161
containing abundant axonal processes were identified by transmitted light imaging with DIC 162
and were chosen for further analysis. Image analysis was performed using Zeiss Axiovision 163
software. 164
Explant DRG culture and immunofluorescence staining 165
Human fetal spinal tissue was obtained at 15-20 weeks gestational period after informed 166
consent and approval by the University of Sydney Human Ethics Committee. DRG were 167
dissected and explanted as previously described (24). 4 days post-plating, DRG exhibiting 168
extensive axonal outgrowth were infected with either mock, rOka or rOka∆gI infected HFFs 169
at a density of 1 x105 HFFs/well. DRG were formalin fixed at 96 hours p.i., paraffin 170
embedded and 5µm sections microtomed then collected onto microscope slides. 171
Immediately preceding immunofluorescent staining of DRG, sections were dewaxed in 172
histolene and rehydrated in a gradient of decreasing ethanol dilutions. Unmasking was 173
performed in citrate buffer (pH 6) at 95°C. Sections were blocked in 20% NDS in tris-174
buffered saline (TBS) for an hour at room temperature. VZV infection was detected using a 175
rabbit anti-VZV pORF62 specific antibody (1:200; kindly provided by P. Kinchington, 176
University of Pittsburgh) or a mouse anti-VZV gE specific antibody (1:10; Millipore, 177
Australia). In conjunction, a mouse anti-NCAM specific antibody (clone 123C3, 1:20; Cell 178
Signaling, Australia) or a rabbit anti-S100B specific polyclonal antibody (predilute; Dako, 179
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

9
Denmark) were utilized. Primary antibodies were applied for 2 hours at room temperature 180
under humidified conditions. Donkey anti-mouse Alexa Fluor 488 conjugated and donkey 181
anti-rabbit Alexa Fluor 594 conjugated secondary antibodies (1:200; Life Technologies, 182
Australia) were used for detection by incubation at room temperature for 30 minutes. 183
Specificity of staining was confirmed by using isotype antibody treated sections at 184
corresponding concentrations. Sections were mounted with Prolong Gold antifade reagent 185
containing DAPI and sealed with coverslips. Images were acquired using a Zeiss LSM 510 186
Meta confocal microscope and were analyzed using Zeiss Axiovision software. 187
RESULTS 188
VZV infection of neuronal cells is significantly impaired in the absence of gI. 189
To assess the kinetics of VZV infection in neuronal cultures, HFFs infected with the VZV gI 190
deletion virus rOkaѐgI (8), parental virus rOka or mock HFFs were CFSE labeled and 191
inoculated onto differentiated SH-SY5Y neuronal cultures as previously described (23). The 192
percentage of infected cells in inoculating cultures ranged from 77-91%, with viral strains 193
within 5% infection for each replicate experiment. In parallel, cultures of HFFs were 194
inoculated at the same ratio of infection. Immunostaining was performed for cultures at 24, 195
48 and 72 hours p.i. using antibodies for Immediate Early (IE62) and Late (gEgI) class VZV 196
proteins and the proportion of VZV antigen positive (CFSE negative) cells were quantified 197
by flow cytometry. Whilst the gEgI antibody reacts with both gE and gI, we did not observe 198
any difference in staining efficiency of cells infected with parental or gI deletion viruses. 199
Results are the collation of data from three independent experiments, with the same 200
inoculating cultures used to infect both HFF and SH-SY5Y cells in parallel experiments. 201
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

10
In HFF cultures, very little difference in the percentage of cells infected with rOka and 202
rOkaѐgI was observed at 24 hours p.i. However, a slight reduction in VZV infection was 203
observed in cultures inoculated with rOka∆gI when compared with rOka for both antibodies 204
tested at 48 and 72 hours p.i. Representative flow cytometry scatter plots are shown for VZV 205
IE62 staining of HFFs at 72 hours p.i. inoculated with mock, rOka and rOka∆gI infected 206
HFFs (Fig. 1A). By 72 hours p.i., the average percentage of VZV IE62 positive cells in rOka 207
inoculated cultures was 35% (Fig. 1B), with the gEgI specific antibody detecting 48% HFFs 208
VZV positive (Fig. 1C). In rOka∆gI inoculated cultures, the average proportion of VZV IE62 209
positive cells at 72 hours p.i. was 23% (Fig. 1B), with 34% cells VZV gEgI positive (Fig. 1C). 210
No VZV specific staining was observed in mock infected cultures (Fig. 1A). 211
In neuronal cell cultures, a statistically significant difference in the percentage of cells 212
infected was observed when infection was compared between the two VZV strains rOka and 213
rOka∆gI. Representative flow cytometry scatter plots are shown for VZV IE62 staining of 214
SH-SY5Ys at 72 hours p.i. inoculated with CFSE labeled mock, rOka and rOka∆gI infected 215
HFFs (Fig. 1D). By 72 hours p.i., an average of 48% SH-SY5Y cells inoculated with rOka 216
were VZV IE62 positive (Fig. 1E), with 69% cells VZV gEgI positive (Fig. 1F). Conversely, 217
only 12% of SH-SY5Y cells inoculated with rOka∆gI were VZV IE62 positive (Fig. 1E) and 218
only 20% of SH-SY5Y cells were VZV gEgI positive (Fig. 1F). No VZV specific staining 219
was observed in mock infected cultures (Fig. 1D). Overall, the reduction in the percentage of 220
VZV IE62 antigen positive cells infected with rOka∆gI compared with rOka at 48 and 72 221
hours p.i. correlated to a significant 3-fold and 4-fold decrease, respectively. Similarly, the 222
percentage of VZV gEgI positive cells in rOka∆gI inoculated cultures was found to amount 223
to a 3.5-fold significant difference at 72 hours p.i. when compared with rOka infected cells. 224
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

11
The detection of both the immediate early VZV protein IE62 and late proteins gEgI by flow 225
cytometry in neuronal cell cultures infected with rOka∆gI was indicative of a complete, 226
productive VZV infection in these cells. Thus it was hypothesized that the reduction of 227
infection in neuronal cell cultures inoculated with VZV rOka∆gI observed by flow cytometry 228
was not due to an impediment of virus entry and/or replication. 229
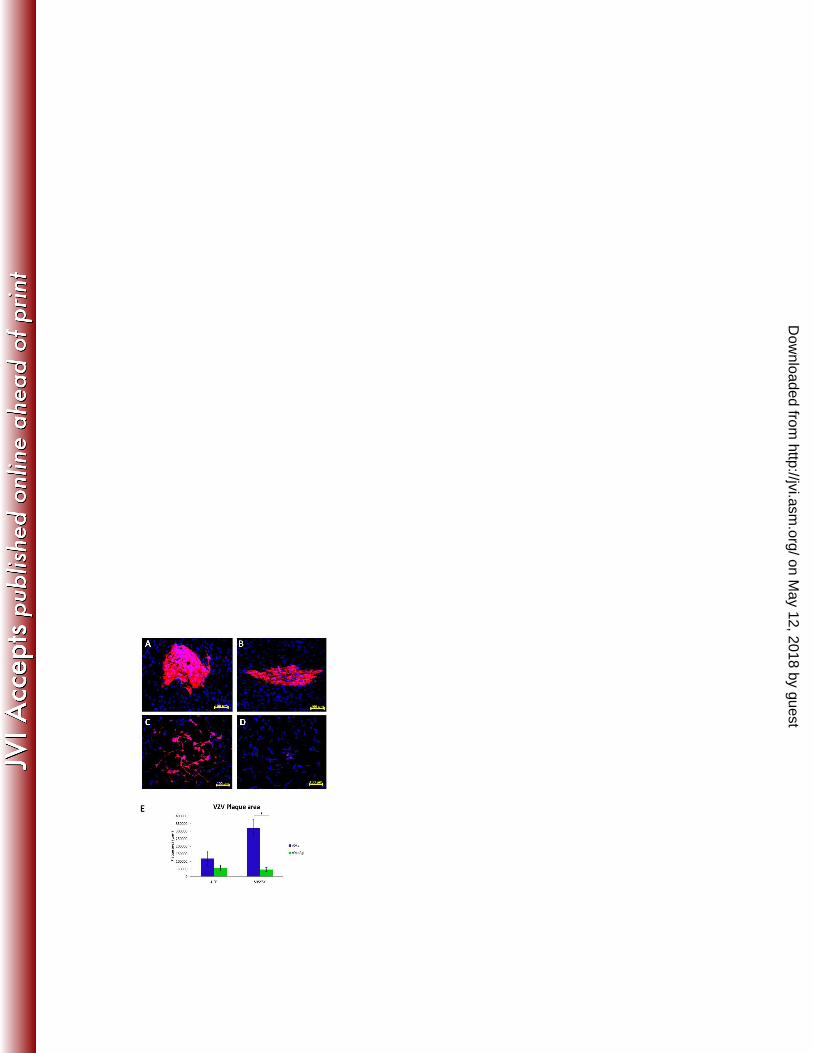
VZV spread in neuronal cultures is significantly impaired in the absence of gI 230
To assess the ability of VZV rOka∆gI to spread in neuronal cultures, we performed a series 231
of infectious center assays by inoculating HFFs infected with either rOka or rOka∆gI onto 232
monolayers of both HFFs and SH-SY5Y cells. Cultures were assessed at 96 hours p.i. by 233
immunofluorescent staining using a VZV gEgI specific primary antibody. Approximately 10 234
infectious centers per culture were randomly selected and plaque areas quantified using Zeiss 235
Axiovision software. Results were averaged for three independent experiments. HFFs 236
inoculated with rOka∆gI (Fig. 2B) exhibited a 1.9-fold average reduction in the size of 237
infectious centers compared with rOka inoculated cultures, although this decrease was not 238
statistically significant (Fig. 2A and E). However, SH-SY5Y cells were found to support 239
spread of rOka (Fig. 2C) much more efficiently than rOka∆gI (Fig. 2D) with a 6.8-fold 240
average decrease in infectious center area observed in rOka∆gI inoculated cultures when 241
compared with rOka (Fig. 2E). Thus, it was concluded from these experiments that gI may 242
play an important role in facilitating virus spread between neuronal cells. 243
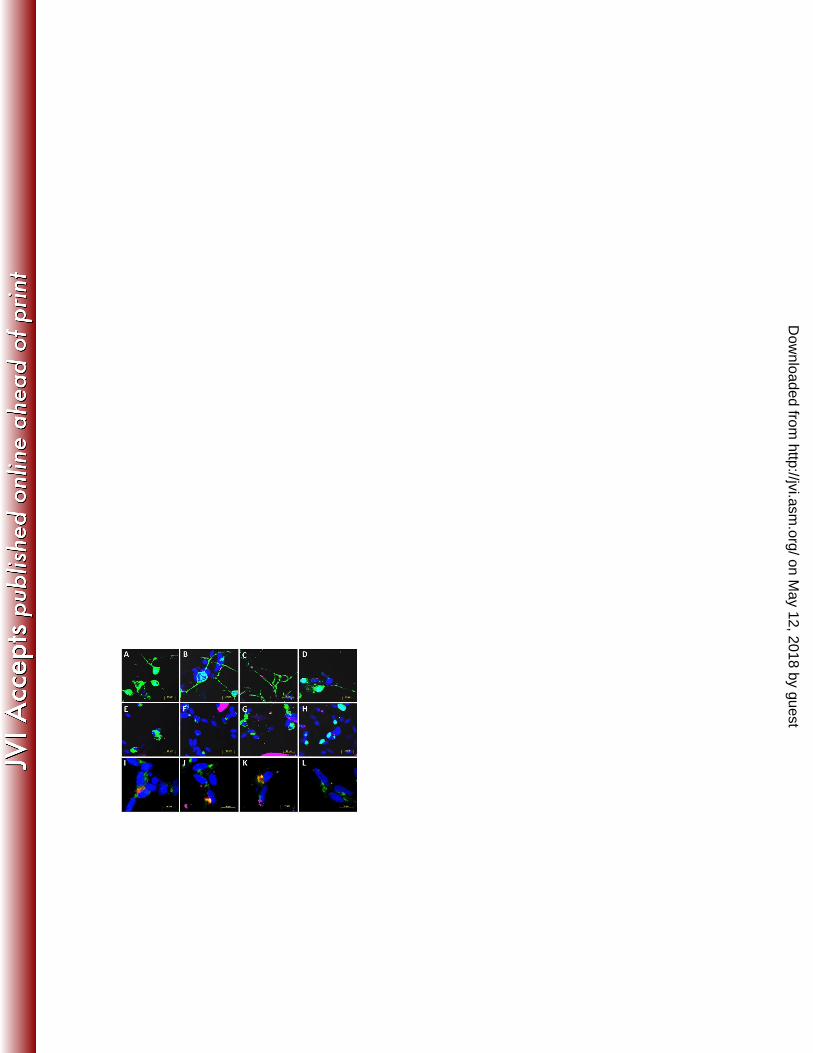
VZV gI is required for viral glycoprotein and capsid protein localization to neuronal 244
axons 245
As neuronal cells form extensive networks of axonal projections to facilitate cellular contact, 246
we hypothesized that axons in these cultures might be important for efficient neuron-to-247
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

12
neuron spread of infection. We therefore sought to assess the axonal localization of structural 248
virion proteins by performing immunofluorescent staining of differentiated neuronal cultures 249
inoculated with either mock, rOka or rOka∆gI infected HFFs. At 48 hours p.i. coverslips 250
containing cells were incubated with antibodies specific to VZV gB, gH, gEgI and the major 251
capsid protein pORF40 and were analyzed by confocal microscopy. In order to confirm that 252
the regions assessed within these cultures contained abundant axons, transmitted light/DIC 253
imaging was utilized to identify regions abundant in axonal processes and cells containing 254
visible axons were randomly selected for analysis. 255
In rOka infected neuronal cells, staining for VZV gB (Fig. 3A), gH (Fig. 3B) and gEgI (Fig. 256
3C) was readily observed with antigen localized predominantly to the cell surface and 257
cytoplasm of the cell body. Clear punctate staining for these VZV proteins were also visible 258
within the neuronal axons. pORF40 staining was predominately nuclear, with some 259
cytoplasmic staining and axonal puncta also clearly visible (Fig. 3D). In rOka∆gI infected 260
neuronal cells, staining for VZV gB (Fig. 3E), gH (Fig. 3F) and gEgI (Fig. 3G) showed an 261
aberrant localization of viral antigen within the cell cytoplasm. Specifically, VZV 262
glycoproteins were found to accumulate in a nuclear adjacent site, with limited expression on 263
the neuronal surface. Although VZV glycoproteins were detected as a puncta within a small 264
number of neuronal axons, many axons projecting from infected cells contained little or no 265
VZV glycoprotein specific staining. Similarly, neuronal cells infected with rOka∆gI were 266
found to exhibit pORF40 antigen within only the nucleus of infected cells, with little or no 267
antigen detected within either the cytoplasm or axons of infected neurons (Fig. 3H). 268
To further examine the aberrant sub-cellular localization of VZV glycoprotein retention 269
within the cytoplasm of neuronal cells infected with rOka∆gI, dual immunofluorescent 270
staining was performed using antibodies specific for VZV gB, gH and gEgI in conjunction 271
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

13
with the trans-Golgi network (TGN) marker syntaxin-6. Confocal microscopy confirmed that 272
there was abundant syntaxin-6 expression in mock infected differentiated neuronal cells 273
adjacent to the nucleus, indicative of a TGN specific localization (Fig. 3L). An identical 274
localization of staining was observed for syntaxin-6 in cells infected with rOka∆gI (Fig. 3I to 275
K). Furthermore, co-localization of syntaxin-6 was observed with each of the VZV 276
glycoprotein specific antibodies gB (Fig. 3I), gH (Fig. 3J) and gEgI (Fig. 3K) within infected 277
cells. No VZV glycoprotein staining was observed in mock infected cultures (Fig. 3L). 278
The combined data from three independent experiments demonstrated that gI is required for 279
axonal localization of VZV glycoprotein and capsid proteins within infected neuronal cells. 280
Furthermore, VZV glycoproteins localized aberrantly to the TGN of cells infected with VZV 281
rOka∆gI. 282
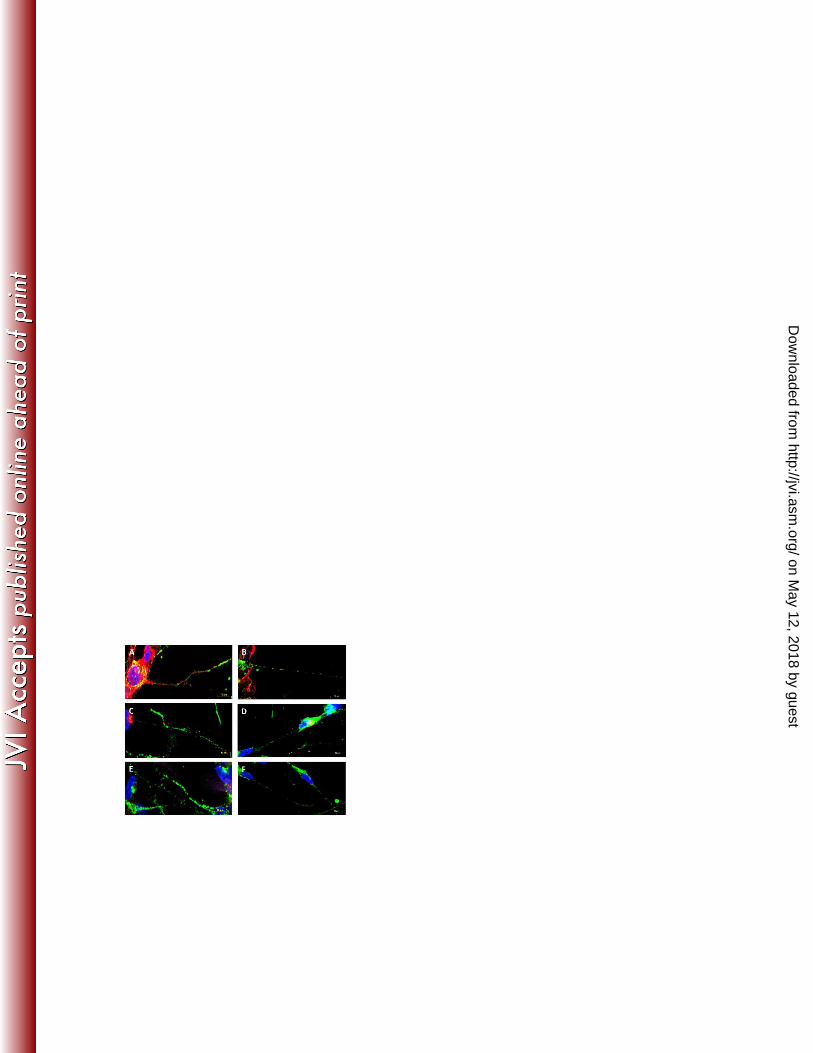
Axonal vesicle trafficking is not impaired in VZV rOka∆gI infected neuronal cells 283
To establish whether VZV glycoprotein retention within the TGN of rOka∆gI infected 284
neuronal cells was imparting a global effect on cellular TGN vesicle-derived trafficking 285
pathways, we sought to identify whether cellular vesicle proteins could be detected within the 286
axons of neuronal cells. Mock, rOka or rOka∆gI infected SH-SY5Y cultures were assessed at 287
48 hours p.i. by dual immunofluorescent staining using antibodies against VZV gH in 288
conjunction with the large dense-core vesicle marker chromogranin A (25) or the synaptic 289
vesicle marker synaptophysin (26). 290
Mock infected neuronal cells exhibited punctate staining within the axons for both 291
chromogranin A (Fig. 4E) and synaptophysin (Fig. 4F). Similar localization of staining was 292
observed in the axons of neuronal cells infected with VZV rOka (Fig. 4A and B). As 293
expected, VZV rOka infected cells additionally contained punctate gH staining along the 294
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

14
length of the axons. As determined previously neuronal cells infected with VZV rOka∆gI 295
contained little or no gH specific staining within the axons of infected cells (Figure 3F). 296
However, chromogranin A (Fig. 4C) and synaptophysin (Fig. 4D) were detectable as puncta 297
within the axons of infected cells, with a distribution and intensity of staining 298
indistinguishable from VZV rOka infected cells (Fig. 4A and B). The results from three 299
independent experiments indicated that VZV glycoprotein retention within the TGN of 300
neuronal cells infected with rOka∆gI does not appear to impair normal cellular vesicle 301
trafficking pathways. 302
VZV cell-to-cell spread within human DRG is impaired in the absence of gI 303
To examine the role of gI in facilitating infection in the context of intact human DRG, we 304
utilized a model of human fetal DRG explanted in vitro. Briefly, fetal DRG were harvested, 305
explanted and infected with either VZV rOka or rOka∆gI as previously described (24). DRG 306
were formalin fixed and paraffin embedded at 96 hours p.i. for assessment by 307
immunofluorescent staining and confocal microscopy. 308
To assess the localization of VZV antigen within the DRG at 96 hours p.i., sections were 309
immunostained with a VZV pORF62 specific antibody in conjunction with a neural cell 310
adhesion molecule (NCAM) antibody. DRG infected with rOka exhibited areas of extensive 311
VZV antigen positivity, suggesting that VZV was readily undergoing cell-to-cell spread (Fig. 312
5A). In contrast, DRG infected with rOka∆gI revealed limited pORF62 staining, which was 313
consistently restricted to isolated cells within the DRG (Fig. 5B). Staining and imaging of 314
multiple consecutive sections verified that these isolated pORF62 antigen positive cells were 315
not in contact with any clusters of VZV positive cells which may have undergone infection 316
via cell-to-cell contact with inoculating fibroblasts at the DRG periphery. No VZV specific 317
staining was observed within mock infected DRG (Fig. 5C). To determine which cell types 318
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

15
within the DRG center were undergoing productive VZV infection, DRG were 319
immunostained at 96 hours p.i. for VZV gE in conjunction with a S100B satellite cell marker. 320
DRG infected with rOka showed localization of gE to both satellite cells and adjacent 321
neurons (Fig. 5D). In contrast, in DRG infected with rOka∆gI gE staining was observed 322
almost exclusively in neurons, with satellite cells being almost universally gE negative (Fig. 323
5E). No VZV specific staining was observed within mock infected DRG (Fig. 5F). These 324
results indicate rOka∆gI is impaired in cell-to-cell spread of VZV within intact DRG. 325
NCAM expression is disrupted during VZV infection and is dependent on gI 326
As disruption of the gE/gI complex has previously been shown to inhibit VZV induced 327
membrane fusion of neuron-satellite cell complexes using the SCIDhu model of infection 328
(11), we sought to determine whether a similar phenotype could be observed in a DRG 329
explant model of VZV infection. Dual immunofluorescent staining of DRG inoculated with 330
mock, rOka or rOka∆gI infected HFFs was performed at 96 hours p.i. using antibodies 331
specific to VZV gE in conjunction with NCAM. In mock infected cells, continuous NCAM 332
staining was readily observed along the membrane of neurons, demarcating the neuron-333
satellite cell boundary (Fig. 6C). Quantification of mock infected neurons revealed that intact 334
NCAM staining was evident surrounding approximately 96% of neurons. In contrast, 53% of 335
rOka infected neurons exhibited discontinuous or dim NCAM staining, although adjacent 336
VZV gE negative neurons exhibited staining with comparable intensity and localization to 337
mock (Fig. 6A). Discontinuous NCAM staining was predominately observed when VZV gE 338
positive neurons were adjacent to infected satellite cells, as has been previously reported (11, 339
27). Neurons infected with rOka∆gI however, showed a reduced alteration in NCAM 340
localization and intensity when compared with rOka, with only 20% of cells exhibiting 341
aberrant NCAM staining (Fig. 6B). No specific staining was observed in isotype control 342
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

16
antibody treated cultures (Fig. 6D). These data illustrate that gI is required for VZV induced 343
neuron-to-satellite cell fusion during VZV infection of human explant DRG. 344
DISCUSSION 345
We assessed the role of VZV gI in mediating spread of infection in cultures of synaptically 346
linked neuronal cells and human fetal DRG explanted in vitro. A significant impairment of 347
rOkaѐgI spread was observed in both of these models, despite only a conservative reduction 348
in cell-to-cell spread within cultures of permissive HFFs. This report provides evidence that 349
gI is required for axonal localization of VZV virion proteins and suggests a role for gI in 350
facilitating axonal transmission of virus. 351
We observed that VZV glycoproteins localized aberrantly to the TGN in the absence of gI, in 352
agreement with a number of previous studies. Electron microscopy studies of infected human 353
embryonic lung fibroblasts showed that gI is required for VZV envelopment in the TGN, 354
with abnormal membranous TGN stacks formed during rOkaѐgI infection (28). Experiments 355
using SCIDhu xenografted DRG similarly confirmed this phenotype within rOkaѐgI infected 356
neurons, with distorted Golgi-like cisternae and disruption of gE trafficking observed (10). 357
We expanded on these findings to also show that the virion components gE, gH, gB and 358
pORF40 are retained within the TGN and trafficking of these protein to axons is disrupted 359
during rOkaѐgI infection of cultured neuronal cells. 360
Although a chamber system was not employed in this study to separate inoculating cells from 361
newly infected cells, these results indicate that there may be some impairment of VZV axonal 362
transport in the absence of gI. As neurons grown in culture have limited cell-to-cell body 363
contact, it is likely that viral infection is mediated at least in part by axonal transmission of 364
virus. Impairment of virion envelopment resulting from gI deletion suggests that the major 365
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

17
impediment in rOka∆gI transport may occur during anterograde viral spread, consistent with 366
the identified roles of gI during HSV and PRV infection (12, 13, 15-17, 29, 30). Additionally, 367
infection of explanted DRG with rOka∆gI showed that the ability to infect discrete individual 368
neurons within the ganglia center was retained. Although the possibility that infection within 369
these cells is initiated by infiltrating inoculating cells cannot be completely excluded, it is 370
likely that the large size of fibroblasts would prevent their entry into the DRG. It is therefore 371
probable that these cells become infected by axonal transport of virus to the DRG center. 372
The obstruction of virion protein trafficking to the axons of infected neuronal cells observed 373
is likely to be a consequence of the morphological changes to the TGN resulting from 374
rOkaѐgI infection (10, 28), although it remains possible that VZV gI is directly required to 375
facilitate virion axonal egress via interactions with vesicles or their molecular motor 376
machinery as is hypothesized for HSV (13, 31). Synaptophysin and chromogranin A puncta 377
were still readily observed along the length of the axons within our study, illustrating that the 378
functionality of TGN secretory pathways was at least partly retained. Although a model of 379
VZV retrograde transport has recently been reported (22, 32), a model assessing anterograde 380
spread of virus still remains in developmental stages. Thus, development of more suitable 381
models will be required before these fundamental questions can be directly addressed. 382
Efficient neuron-to-satellite cell spread of virus within the DRG was dependent on gI. 383
Inspection of rOka infected DRG at 96 hours p.i. revealed that extensive areas of pORF62 384
staining were visible. Extensive cell-to-cell spread was observed throughout the DRG, with 385
both neurons and S100B antigen positive satellite cells infected with VZV. In contrast, 386
pORF62 staining within rOkaѐgI infected DRG was mainly restricted to isolated neurons 387
within the DRG center, with limited evidence of any viral spread at 96 hours p.i. The 388
impairment of VZV spread in the absence of gI supports the findings of a previous study 389
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

18
using SCIDhu xenografted DRG, whereby single rOkaѐgI infected neurons were observed at 390
42 days p.i., similarly implicating impairment of virus spread to neighboring satellite cells 391
(10). Infection of these DRG with both rOkaѐgI and a virus lacking the cysteine rich gE/gI 392
binding domain resulted in impaired replication of VZV and an inability to establish 393
persistence for extended timepoints p.i. (10, 11). Thus the neurotropic role of gI characterized 394
in our study is likely to be integrally linked to its ability to heterodimerize with gE. 395
The SCIDhu model has been used to demonstrate that polykaryon formation between neurons 396
and satellite cell membranes is disrupted during VZV infection, with reduced NCAM 397
expression on the membranes of infected cells observed (11, 27). It was also shown that 398
deletion of gI or the cysteine rich region facilitating gE/gI heterodimer formation was able to 399
restore NCAM expression (11). In our study, we also found that rOka infected neurons 400
exhibited a downregulation of NCAM expression by IFA. In contrast, mock or rOka∆gI 401
infected DRG retained strong NCAM staining localized to the membrane of neuron-satellite 402
cell junctions, similarly indicating that rOka∆gI may be impaired for polykaryon formation. 403
The restriction of NCAM to neural tissue may assist in explaining the cell-type specific 404
impairment of infection resulting from rOka∆gI infection. Although a small reduction in 405
VZV rOka∆gI infection was observed in cultures of HFFs over a 72 hour timecourse when 406
compared with the parental virus, it has been shown that free nucleocapsids within the 407
cytosol are able to fuse with adjacent cells to potentiate infection (28). It is possible that the 408
gI mediated inhibition of NCAM downregulation inhibits cell fusion and therefore prevents 409
the transmission of these free cytosolic nucleocapsids to neighboring cells. 410
In summary, this report details a neurotropic role of VZV glycoprotein I in facilitating axonal 411
localization of virion proteins and virus spread between cultured neuronal cells. Additional 412
experiments performed using an explant DRG model of productive VZV infection confirmed 413
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

19
that neuron-to-satellite cell spread of virus and NCAM expression was impaired in the 414
absence of gI. These results will aid in our understanding of VZV neuropathogenesis and the 415
requirements for productive neuronal infection within the DRG characteristic of reactivation. 416
They also validate the combined use of SH-SY5Y and primary DRG models of infection to 417
more rapidly facilitate definition of the roles of VZV gene products in neuronal infection. 418
ACKNOWLEDGEMENTS 419
We would like to thank Dr. Louise Cole (University of Sydney) for assistance with 420
microscopy; Professor Ann Arvin (Stanford University) for providing the virus strains used in 421
this study; Professor Paul Kinchington (University of Pittsburgh) for the pORF62 antibody 422
and; Shanna Trollip and Sanaz Maleki (University of Sydney) for assistance with 423
histopathology. J.C. is a recipient of an Australian Postgraduate Award and University of 424
Sydney Merit Award. This work was supported by an Australian National Health and 425
Medical Research Council Project Grant funding awarded to A.A. and B.S. 426
427
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

20
REFERENCES 428
1. Arvin A. 2001. Varicella-zoster virus, p. 2731-2767. In Knipe DM, Howley P (ed.), 429
Fields Virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA. 430
2. Mueller NH, Gilden DH, Cohrs RJ, Mahalingam R, Nagel MA. 2008. Varicella 431
zoster virus infection: clinical features, molecular pathogenesis of disease, and latency. 432
Neurol Clin 26:675-697, viii. 433
3. Steiner I, Kennedy PG, Pachner AR. 2007. The neurotropic herpes viruses: herpes 434
simplex and varicella-zoster. Lancet Neurol 6:1015-1028. 435
4. Arvin AM. 1992. Cell-mediated immunity to varicella-zoster virus. J Infect Dis 166 436
Suppl 1:S35-41. 437
5. Hope-Simpson RE. 1975. Postherpetic neuralgia. J R Coll Gen Pract 25:571-575. 438
6. Watson PN, Evans RJ. 1986. Postherpetic neuralgia. A review. Arch Neurol 43:836-439
840. 440
7. Yao Z, Jackson W, Forghani B, Grose C. 1993. Varicella-zoster virus glycoprotein 441
gpI/gpIV receptor: expression, complex formation, and antigenicity within the vaccinia virus-442
T7 RNA polymerase transfection system. J Virol 67:305-314. 443
8. Mallory S, Sommer M, Arvin AM. 1997. Mutational analysis of the role of 444
glycoprotein I in varicella-zoster virus replication and its effects on glycoprotein E 445
conformation and trafficking. J Virol 71:8279-8288. 446
9. Grinfeld E, Sadzot-Delvaux C, Kennedy PG. 2004. Varicella-Zoster virus proteins 447
encoded by open reading frames 14 and 67 are both dispensable for the establishment of 448
latency in a rat model. Virology 323:85-90. 449
10. Zerboni L, Reichelt M, Jones CD, Zehnder JL, Ito H, Arvin AM. 2007. Aberrant 450
infection and persistence of varicella-zoster virus in human dorsal root ganglia in vivo in the 451
absence of glycoprotein I. Proc Natl Acad Sci U S A 104:14086-14091. 452
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

21
11. Zerboni L, Berarducci B, Rajamani J, Jones CD, Zehnder JL, Arvin A. 2011. 453
Varicella-zoster virus glycoprotein E is a critical determinant of virulence in the SCID 454
mouse-human model of neuropathogenesis. J Virol 85:98-111. 455
12. McGraw HM, Awasthi S, Wojcechowskyj JA, Friedman HM. 2009. Anterograde 456
spread of herpes simplex virus type 1 requires glycoprotein E and glycoprotein I but not Us9. 457
J Virol 83:8315-8326. 458
13. Snyder A, Polcicova K, Johnson DC. 2008. Herpes simplex virus gE/gI and US9 459
proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J Virol 460
82:10613-10624. 461
14. Enquist LW, Dubin J, Whealy ME, Card JP. 1994. Complementation analysis of 462
pseudorabies virus gE and gI mutants in retinal ganglion cell neurotropism. J Virol 68:5275-463
5279. 464
15. Husak PJ, Kuo T, Enquist LW. 2000. Pseudorabies virus membrane proteins gI and 465
gE facilitate anterograde spread of infection in projection-specific neurons in the rat. J Virol 466
74:10975-10983. 467
16. Ch'ng TH, Enquist LW. 2005. Neuron-to-cell spread of pseudorabies virus in a 468
compartmented neuronal culture system. J Virol 79:10875-10889. 469
17. Kratchmarov R, Kramer T, Greco TM, Taylor MP, Ch'ng TH, Cristea IM, 470
Enquist LW. 2013. Glycoproteins gE and gI Are Required for Efficient KIF1A-Dependent 471
Anterograde Axonal Transport of Alphaherpesvirus Particles in Neurons. J Virol 87:9431-472
9440. 473
18. Myers MG, Connelly BL. 1992. Animal models of varicella. J Infect Dis 166 Suppl 474
1:S48-50. 475
19. Pugazhenthi S, Nair S, Velmurugan K, Liang Q, Mahalingam R, Cohrs RJ, 476
Nagel MA, Gilden D. 2011. Varicella Zoster Virus Infection of Differentiated Human 477
Neural Stem Cells. J Virol 85:6678-6686. 478
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

22
20. Lee KS, Zhou W, Scott-McKean JJ, Emmerling KL, Cai GY, Krah DL, Costa 479
AC, Freed CR, Levin MJ. 2012. Human sensory neurons derived from induced pluripotent 480
stem cells support varicella-zoster virus infection. PLoS One 7:e53010. 481
21. Yu X, Seitz S, Pointon T, Bowlin JL, Cohrs RJ, Jonjic S, Haas J, Wellish M, 482
Gilden D. 2012. Varicella zoster virus infection of highly pure terminally differentiated 483
human neurons. J Neurovirol 19:75-81. 484
22. Markus A, Grigoryan S, Sloutskin A, Yee MB, Zhu H, Yang IH, Thakor NV, 485
Sarid R, Kinchington PR, Goldstein RS. 2011. Varicella zoster virus infection of neurons 486
derived from human embryonic stem cells: direct demonstration of axonal infection, transport 487
of VZV and productive neuronal infection. J Virol 85:6220-6233. 488
23. Christensen J, Steain M, Slobedman B, Abendroth A. 2011. Differentiated 489
neuroblastoma cells provide a highly efficient model for studies of productive varicella-zoster 490
virus infection of neuronal cells. J Virol 85:8436-8442. 491
24. Gowrishankar K, Slobedman B, Cunningham AL, Miranda-Saksena M, Boadle 492
RA, Abendroth A. 2007. Productive varicella-zoster virus infection of cultured intact human 493
ganglia. J Virol 81:6752-6756. 494
25. Taupenot L, Harper KL, O'Connor DT. 2003. The chromogranin-secretogranin 495
family. N Engl J Med 348:1134-1149. 496
26. Valtorta F, Pennuto M, Bonanomi D, Benfenati F. 2004. Synaptophysin: leading 497
actor or walk-on role in synaptic vesicle exocytosis? Bioessays 26:445-453. 498
27. Reichelt M, Zerboni L, Arvin AM. 2008. Mechanisms of varicella-zoster virus 499
neuropathogenesis in human dorsal root ganglia. J Virol 82:3971-3983. 500
28. Wang ZH, Gershon MD, Lungu O, Zhu Z, Mallory S, Arvin AM, Gershon AA. 501
2001. Essential role played by the C-terminal domain of glycoprotein I in envelopment of 502
varicella-zoster virus in the trans-Golgi network: interactions of glycoproteins with tegument. 503
J Virol 75:323-340. 504
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

23
29. Dingwell KS, Doering LC, Johnson DC. 1995. Glycoproteins E and I facilitate 505
neuron-to-neuron spread of herpes simplex virus. J Virol 69:7087-7098. 506
30. Howard PW, Howard TL, Johnson DC. 2013. Herpes simplex virus membrane 507
proteins gE/gI and US9 act cooperatively to promote transport of capsids and glycoproteins 508
from neuron cell bodies into initial axon segments. J Virol 87:403-414. 509
31. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus 510
egress. Nat Rev Microbiol 9:382-394. 511
32. Grigoryan S, Kinchington PR, Yang IH, Selariu A, Zhu H, Yee M, Goldstein RS. 512
2012. Retrograde axonal transport of VZV: kinetic studies in hESC-derived neurons. J 513
Neurovirol 18:462-470. 514
515
516
517
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

24
FIGURE LEGENDS 518
Figure 1. Kinetic analysis of rOka and rOka∆gI infection in HFFs and neuronal cells by 519
flow cytometry. Either mock, rOka or rOka∆gI infected HFFs were labeled with CFSE and 520
inoculated onto neuronal cells. Cultures were harvested for immunostaining and flow 521
cytometry at 24, 48 and 72 hours p.i. In parallel, cultures of HFFs were infected and analyzed 522
using the same method. (A) Flow cytometry scatter plots of HFFs inoculated with mock, 523
rOka or rOkaѐgI infected HFFs and stained with a VZV IE62 specific antibody at 72 hours 524
p.i. Red boxes show the total non-inoculum (CFSE negative) population, with the percentage 525
of VZV positive cells within this population given. (B) The average percentage of VZV IE62 526
positive HFFs from three independent experiments are shown over a timecourse of infection 527
comparing rOka (blue) and rOkaѐgI (green). (C) The average percentage of VZV gEgI 528
positive HFFs from three independent experiments are shown over a timecourse of infection 529
comparing rOka (blue) and rOkaѐgI (green). (D) Flow cytometry scatter plots of neuronal 530
cells inoculated with mock, rOka or rOkaѐgI infected HFFs and stained with a VZV IE62 531
specific antibody at 72 hours p.i. Red boxes show the total non-inoculum (CFSE negative) 532
population, with the percentage of VZV positive cells within this population given. (E) The 533
average percentage of VZV IE62 positive neuronal cells from three independent experiments 534
are shown over a timecourse of infection comparing rOka (blue) and rOkaѐgI (green). (F) 535
The average percentage of VZV gEgI positive neuronal cells from three independent 536
experiments are shown over a timecourse of infection comparing rOka (blue) and rOkaѐgI 537
(green). Values are means, with error bars showing the standard error of the mean from three 538
independent experiments. Significant difference was determined using a paired two-tailed 539
student’s t-test, (*P, < 0.05, **P, < 0.01). 540
541
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

25
Figure 2. Assessment of rOka and rOka∆gI spread in HFFs and neuronal cells by 542
infectious center assay. Serial dilutions of either rOka or rOka∆gI infected HFFs were 543
inoculated onto monolayers of either HFFs or differentiated neuronal cells. At 96 hours p.i. 544
coverslips were harvested and immunofluorescent staining was performed using an antibody 545
specific to VZV gEgI (red) and a DAPI counterstain (blue). Representative infectious centers 546
are shown following infection of HFFs with either (A) rOka or (B) rOka∆gI, or following 547
infection of neuronal cells with either (C) rOka or (D) rOka∆gI. (E) Infectious center areas 548
were quantified using Zeiss Axiovision software and the mean values from three independent 549
experiments determined. Error bars show the standard error of the mean and significant 550
difference was determined using a paired two-tailed student’s t-test, (*P, < 0.05). 551
Figure 3. Assessment of viral capsid and glycoprotein localization in neuronal cells 552
infected with rOka and rOka∆gI. Either (A-D) rOka, (E-K) rOka∆gI or (L) mock infected 553
HFFs were labeled with CFSE (pseudocoloured purple) and inoculated onto cultures of 554
neuronal cells. (A-H) Cultures were harvested at 48 hours p.i. and immunostained using an 555
antibody against (A,E) VZV gB, (B,F) gH, (C,G) gEgI, or (D,H) pORF40 (pseudocoloured 556
green) and were counterstained with DAPI (blue). Areas containing abundant axons were 557
selected for imaging using transmitted light/DIC overlay (data not shown). (I-L) Cultures 558
were immunostained for syntaxin-6 (pseudocoloured green), with dual immunofluorescent 559
staining performed using antibodies specific for (I) VZV gB, (J) gH or (K) gEgI (red). Cell 560
nuclei were visualized using a DAPI counterstain (blue). (L) No VZV specific staining was 561
observed in mock infected cultures. 562
Figure 4. Analysis of axonal secretory vesicle proteins in rOka and rOka∆gI infected 563
neuronal cells. (A-B) rOka, (C-D) rOka∆gI or (E-F) mock infected HFFs were CFSE labeled 564
(pseudocoloured purple) and inoculated onto cultures of neuronal cells. Cultures were 565
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

26
harvested at 48 hours p.i. and immunostained for either (A,C,E) chromogranin A or (B,D,F) 566
synaptophysin (pseudocoloured green). VZV antigen was detected with a gH specific 567
antibody (red). Cells were counterstained with DAPI (blue). 568
Figure 5. Assessment of viral spread within explant dorsal root ganglia. (A,D) rOka, 569
(B,E) rOkaѐgI or (C,F) mock infected DRG were harvested at 96 hours p.i. and assessed by 570
dual immunofluorescent staining. (A-C) DRG were stained for NCAM (green) in conjunction 571
with a VZV pORF62 specific antibody (red). (D-F) DRG were stained for S100B (green) in 572
conjunction with a VZV gE specific antibody (red). Cells were counterstained with DAPI 573
(blue). 574
Figure 6. NCAM and VZV antigen detection within explant dorsal root ganglia. (A,D) 575
rOka, (B) rOkaѐgI or (C) mock infected DRG were harvested at 96 hours p.i. and assessed by 576
dual immunofluorescent staining. (A-C) DRG were stained for NCAM (green) in conjunction 577
with a VZV pORF62 specific antibody (red) or with (D) isotype control antibodies. Cells 578
were counterstained with DAPI (blue). Arrows depict discontinuous NCAM staining on the 579
membranes of VZV infected neurons adjacent to infected satellite cells. 580
on May 12, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from













![7KHPROHFXODUFKDUDFWHUL]DWLRQV …jvi.asm.org/content/early/2016/03/31/JVI.00180-16.full.pdf · tut Receptor binding analyses of the H5Nx HAs tuu 7KH+$UHFHSWRUELQGLQJVLWH 5%6 FRQWULEXWHVWRKRVWUDQJH](https://img.pdfslide.us/doc/110x75/5a89c0367f8b9a78648b7c4c/7khprohfxodufkdudfwhuldwlrqv-jviasmorgcontentearly20160331jvi00180-16fullpdftut.jpg)