Embed Size (px)
Citation preview

Nucleozin targets cytoplasmic trafficking of vRNP-Rab11 1
complexes in influenza A virus infection 2
Maria Joao Amorim1,2,# Richard Kao3 and Paul Digard1,4 3
4
1Division of Virology, Department of Pathology, University of Cambridge, Tennis 5
Court 6
Road, Cambridge, CB2 1QP, United Kingdom. 7
2 Cell biology of Viral Infection, Instituto Gulbenkian de Ciência, Rua da Quinta 8
Grande, 6, 2780-156 Oeiras, Portugal. 9
3Department of Microbiology and Research Center of Infection and Immunity, LKS 10
Faculty of Medicine, The University of Hong Kong, Hong Kong. 11
4 The Roslin Institute, University of Edinburgh, Easter Bush, Midlothian EH25 9RG, 12
United Kingdom. 13
# email: [email protected] 14 15 Classification: BIOLOGICAL SCIENCES; microbiology; antiviral therapy 16
Abstract word count: 250 17
Main text word count: circa 5541 not counting references or figure legends 18
Running title: nucleozin targets influenza A virus RNP trafficking 19
20
21
Copyright © 2013, American Society for Microbiology. All Rights Reserved.J. Virol. doi:10.1128/JVI.03123-12 JVI Accepts, published online ahead of print on 13 February 2013
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

2
Abstract: 22
Novel antivirals are needed to supplement existing control strategies for 23
influenza A virus (IAV). A promising new class of drug, exemplified by the 24
compound nucleozin, has recently been identified that targets the viral nucleoprotein 25
(NP). These inhibitors are thought to act as ‘molecular staples’ that stabilise 26
interactions between NP monomers, promoting the formation of non-functional 27
aggregates. Here we detail the inhibitory mechanism of nucleozin, finding that the 28
drug has both early- and late-acting effects on the IAV lifecycle. When present at the 29
start of infection, it inhibited viral RNA and protein synthesis. However, when added 30
at later time points, it still potently blocked the production of infectious progeny but 31
without affecting viral macromolecular synthesis. Instead, nucleozin blocked the 32
cytoplasmic trafficking of RNPs that had undergone nuclear export, promoting the 33
formation of large perinuclear aggregates of RNPs along with cellular Rab11. This 34
effect led to the production of much reduced amounts of often markedly smaller virus 35
particles. We conclude that the primary target of nucleozin is the viral RNP, not NP 36
and this work also provides proof of principle that IAV replication can be effectively 37
inhibited by blocking cytoplasmic trafficking of the viral genome. 38
39
40
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

3
Introduction: 41
The influenza A virus (IAV) genome consists of eight segments of negative sense, 42
single stranded RNA (vRNAs) that encode 10-14 identified proteins (29, 49, 50). 43
Each vRNA is separately encapsidated into ribonucleoprotein (RNP) particles by the 44
trimeric viral RNA dependent RNA polymerase (PB1, PB2 and PA) and single-strand 45
RNA-binding protein NP (39). At the start of infection, RNPs are released into the 46
cytoplasm and imported into the nucleus by the host importin alpha/beta pathway (5). 47
Once in the nucleus the vRNAs are transcribed by the viral polymerase to produce 48
capped and polyadenylated mRNAs, as well as replicated via an alternative plus sense 49
transcript (cRNA). Both the cRNA and progeny vRNA molecules are encapsidated 50
into RNPs, necessitating the nuclear import of newly translated NP, again via 51
interactions with importin alpha (5). Once levels of the viral M1 and NS2/NEP 52
polypeptides are sufficient, newly synthesised RNPs are exported to the cytoplasm 53
utilising the cellular CRM1/exportin 1 pathway (5). We and others have recently 54
reported that RNPs accumulate at the perinuclear recycling endosome (RE) after 55
leaving the nucleus. Here, they piggyback onto Rab11 positive vesicles to access the 56
microtubule network for transit through the cytoplasm (1, 3, 20, 33). The 57
assembly/budding phase then starts as RNPs join with the three viral membrane 58
proteins (HA, NA and M2) as well as with the M1 matrix protein and small amounts 59
of NS2 and the RNPs to form new enveloped virus particles at the apical PM (43). 60
Influenza transmission and disease are partially controlled by a global vaccination 61
program and antiviral therapy. The available antivirals target the ion channel protein 62
M2 (amantadine/rimantadine), or NA (oseltamivir/zanamivir), responsible for 63
entrance and exit into the host cell respectively. However, both therapies are rendered 64
less effective by resistance (15). New antivirals developed against other IAV targets 65
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

4
are therefore required. One attractive strategy is to target NP, since this protein plays 66
many essential roles in viral RNA synthesis, genome trafficking and virus assembly 67
(27, 39). Recently, several groups identified NP-interacting molecules able to inhibit 68
virus replication (12, 13, 23, 30, 46). These compounds, nucleozin or related 69
derivatives, bind to at least two sites on NP and appear to act as ‘molecular staples’ 70
that promote the formation of higher order oligomers or aggregates by stabilizing 71
monomer interactions. Evidence for this comes from various in vitro biophysical 72
analyses of purified NP, co-crystallization studies with the inhibitor, as well as 73
fluorescent microscopy of infected, drug-treated cells (12, 23, 30, 46). Further support 74
for this model comes from the identification of several resistance mutations in NP 75
that map to the separate drug-binding pockets (23, 30, 46). The drug-induced 76
aggregation evidently interferes with NP function, because the compounds potently 77
inhibit virus replication in tissue culture and infected animals (12, 23, 30, 46). 78
However, the precise mechanism(s) of action have not been investigated in detail. 79
While the compounds clearly inhibit viral transcription, either from RNPs 80
reconstituted in cells by plasmid-transfection or from RNPs purified from virus (23, 81
30, 46), discrepancies between the IC50 values for inhibition of overall virus 82
replication compared to transcription raise the possibility of more than one 83
mechanism of action (13). 84
Here we describe a distinct effect of nucleozin on the later stages of viral 85
infection, finding that it inhibits the cytoplasmic transport of Rab11-RNP complexes. 86
This is a novel mechanism of action for any antiviral, which provides proof of 87
principle for new strategies to interfere with viral replication. 88
89
90
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

5
Materials and Methods 91
Cells, viruses, antisera and drug 92
Human embryonic kidney 293T, Madin-Darby Canine Kidney (MDCK), and 93
human alveolar basal epithelial cells (A549) were cultured as previously described 94
(9). Reverse genetics-derived A/Puerto Rico/8/34 (PR8; H1N1) was used as a model 95
virus (16) and titrated according to (32). Mutations to segment 5 (NP Y289H) were 96
made and viruses rescued as described in (28). Infections were carried out at an MOI 97
of 3 in serum free medium (SFM). After 30 min, infected cells were overlaid with 98
SFM and 0.14% BSA. Antibodies used included mouse monoclonal antibodies 99
against GFP (JL8; Clontech, 632380), GM130 (BD Transduction laboratories 100
610822), calnexin, EEA1 and Clathrin (BD Transduction laboratories 610524, 610457 101
and 610499), LAMP1 (BD Pharmigen, 553792), influenza M2 (14C2; AbCam, 102
ab5416) and NA (7D8; a gift from Susanna Colaco and Dr. Phil Stevenson), rat 103
monoclonal anti-alpha tubulin (YL1/2 MCA77G; AbD-Serotec) and sheep polyclonal 104
antibody to TGN46 (AbD-Serotec; AHP500G). Rabbit polyclonal antisera were: to 105
whole PR8 virions, PR8 M1 (2), PB1 (V19), PA (V35) (18), PB2 (2N580) (38) and 106
NP (2915) (36), Anti-NS2 was a gift from Dr A. Portela. All secondary antibodies 107
used were from the AlexaFluor range (Invitrogen). Nucleozin (30) was dissolved in 108
DMSO and used at a concentration of 1μM unless otherwise stated. 109
110
Plasmids and transfections 111
Plasmids encoding green fluorescent protein (GFP)-tagged NP (31) or red 112
fluorescent protein (RFP)-tagged WT Rab11a (1) have been previously described. 113
pCDNA3 plasmids used to synthesise FISH probes to detect vRNA from segments 1, 114
4 and 7 were described in (2, 25, 34) and to detect mRNA for segment 5 in (40). To 115
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

6
GFP-tag influenza RNPs, 1x105 293T or A549 cells were transfected with GFP-NP 116
(200 ng) using Lipofectamine Plus and LTX (Invitrogen) according to the 117
manufacturer’s instructions, incubated overnight and superinfected with PR8. Cells 118
were imaged 8 h later (unless stated). 119
120
Microscopy 121
FISH methods to detect vRNA were described in (2) and to detect mRNA in 122
(40). Immunofluorescence (IF) was carried out as in (45). Samples were imaged using 123
a Leica SP5 confocal microscope and post-processed using Adobe Photoshop and 124
ImageJ (NIH). Single optical sections are shown. 125
For live imaging, cells were grown in chambered glass bottomed dishes (Lab-126
Tek) and maintained at 37°C in Leibovitz L-15 CO2-independent medium (Gibco) 127
during analysis. Cells were transfected with GFP-NP and 12 h later infected with 128
PR8. When treated with nucleozin, images were acquired for 5 min, then nucleozin 129
was added to a final concentration of 2 μM before imaging for around 20-30 min (as 130
stated). Images were acquired at 0.25 or 0.71 frames/sec and processed using ImageJ. 131
132
Pull downs, western blotting and primer extension analysis 133
Pulldowns of GFP-tagged proteins were performed using GFP-Trap beads 134
(Chromotek) as described in (1). Confluent 6-well dishes of 293T cells were 135
transfected with 500 ng of each plasmid (GFP or GFP-NP) and where applicable, 136
infected 12 h later with virus. All samples were collected at 8h p.i.. At the end of the 137
process bound proteins were eluted by boiling in SDS-PAGE sample buffer, while 138
bound RNA was extracted by adding 1 ml of Trizol (Invitrogen) and 200 µl 139
chloroform directly to the Trap beads and recovered by ethanol precipitation. Specific 140
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

7
RNA species were detected by reverse transcriptase-radiolabelled primer extension 141
followed by UREA-PAGE and autoradiography as described previously (41). Western 142
blotting was performed according to standard procedures and imaged using a LiCor 143
Biosciences Odyssey near-infrared platform (8). 144
145
Transmission Electron Microscopy (TEM) 146
A549 cells were seeded onto 24 well plates, transfected and infected as above. 147
Cells were washed in 0.1 M HEPES, pH 7.4 and incubated for 2h at 4°C in 0.1 M 148
HEPES, pH 7.4, 2% glutaraldehyde, 0.1% H2O2. The monolayer was washed with 149
HEPES buffer, and cells were scraped into buffer and harvested by centrifugation at 150
14,000g for 10 min. The supernatant was replaced with fresh HEPES buffer and 151
processed by the Cambridge University Multi-Imaging Centre. Images were acquired 152
using a FEI Philips CM100 TEM equipped with a Deben AMT digital camera and 153
EDAX Genesis XM4 EDX system. 154
155
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

8
Results: 156
Nucleozin has distinct effects on virus replication depending on time of addition 157
As a first test of how nucleozin inhibits IAV replication, we examined the 158
effects of adding drug at different times post-infection on the titre of replicated virus, 159
utilising wild type (WT) PR8 virus as well as a derivative engineered to contain the 160
nucleozin-resistance mutation NP Y289H. In the absence of drug, WT virus release 161
was detectable by 6h p.i. but was around 20-fold higher at 8h p.i.. However, addition 162
of the drug at 0.5, 2.5 or 4h p.i. decreased titres at 8h p.i. by around 1000-fold and on 163
average, by 10 fold if added at 6h p.i. (Fig. 1A). The NP Y289H mutant reached 164
similar titres in the absence or presence of the drug, but had an overall growth defect 165
of about 10 fold when compared to WT, as reported for the A/WSN/33 strain (30). 166
Thus nucleozin is an effective inhibitor of WT virus replication even when added at 167
relatively late times in the replication cycle. 168
We next tested the effect of nucleozin on WT viral macromolecular synthesis 169
over a time course of infection. Expression of viral proteins increased over time in 170
untreated cells, as expected (Fig 1B, lanes 1-4). When nucleozin was added at 0.5h 171
p.i., viral polypeptide synthesis was much reduced but not completely abolished, with 172
small amounts of NP, M1 and PB2 detected at 8h p.i. (Fig 1B lanes 5-8). Inhibition of 173
viral protein synthesis was much less noticeable when the drug was added at 2.5h p.i. 174
(lanes 9-11) and was not significant when added at 4h p.i. (lanes 12 and 13), where 175
protein expression reached the same levels of untreated cells. These alterations in 176
polypeptide accumulation were reflected in drug-induced changes to the abundance of 177
viral mRNA, when PA/segment 3 and NP/segment 5 transcripts were examined as 178
examples. In untreated cells, viral mRNA peaked at 4h p.i. and declined thereafter 179
(Fig 1D, lanes 1-4), as previously observed (34, 41). When drug was added at the 180
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

9
beginning of infection, transcript accumulation was much delayed and reduced (lanes 181
5-8), but this effect was not seen when the compound was added at 2.5h or 4h p.i. 182
(lanes 9-13). Notably, this failure to inhibit mRNA production when drug was added 183
later was seen even when compound was added before substantial amounts of 184
transcription had occured (compare lanes 1 and 2 with 9 and 10). Synthesis of vRNA 185
followed a similar kinetic pattern to that of the viral proteins, with detectable and 186
increasing synthesis from 4h p.i. in normal cells (Fig 1C, lanes 1-4) that was 187
markedly reduced when nucleozin was added at 0.5h p.i. (lanes 5-8). Limited (~ 25% 188
of normal) vRNA synthesis was seen when the drug was added at 2.5h p.i., that 189
peaked relatively early at 4h p.i. and showed little increase thereafter (lanes 9-11). 190
For later time points of drug addition, only minimal inhibition of vRNA synthesis was 191
seen; quantification of replicate experiments with WT virus showed less than a two-192
fold decrease when the drug was added at 4 or 6h p.i. (Figs 1C, D). The NP Y289H 193
mutant produced similar levels of vRNA at 8h p.i., independently of the time of 194
addition of drug (Fig 1D). 195
Therefore nucleozin showed two distinct effects on virus replication 196
depending on the time of addition. When added early in infection it inhibited 197
synthesis of all classes of viral RNA and (if added early enough to block 198
transcription), viral proteins. However, when added later in infection, at 4h p.i. or 199
later, viral macromolecular synthesis seemed near normal and yet the drug still 200
profoundly inhibited the release of infectious viral particles; a differential effect that 201
could be clearly seen when the relative effects of time of drug addition on vRNA 202
accumulation and titre of released virus was compared (Figs 1A, D). 203
204
205
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

10
Nucleozin aggregates outgoing vRNPs and Rab11 in the cytoplasm 206
Previous work has suggested two mechanisms of action for nucleozin: direct 207
inhibition of RNP transcription, or a block to NP trafficking, postulated to be prior to 208
nuclear import and RNP assembly (12, 23, 30, 46). However, neither mechanism 209
seemed a plausible explanation of the late-acting drug effect, since viral RNA 210
synthesis and therefore by extrapolation, RNP formation, was near normal. 211
Furthermore, after ~5h p.i., the major flow of NP is from the nucleus to the cytoplasm 212
(6, 21). Accordingly, to better understand the effect of nucleozin on NP/RNP 213
trafficking, we infected A549 cells with WT and NP Y289H mutant viruses and 214
followed the localization of NP (used as a proxy for vRNPs) by indirect 215
immunofluorescence during the later stages of infection. We also double-stained cells 216
for Rab11, since vRNPs use Rab11-positive vesicles to reach the PM (1, 3, 20, 33). In 217
untreated cells infected with WT virus, NP localized in the nucleus early in infection, 218
and then accumulated in the cytoplasm where it co-localised with Rab11 both at 6 and 219
8h p.i. (Fig 2A; high magnification images shown in Fig S1), as previously observed 220
(1, 3, 20, 33). When WT virus was treated with nucleozin at 4h p.i. (and fixed at 8h), 221
NP accumulation in the cytoplasm was retarded and largely confined to the 222
perinuclear region, where it co-localized with a small proportion of Rab11. The 223
majority of Rab11-staining was however, distributed throughout the cytoplasm. 224
Similar nuclear retention of NP was seen when nucleozin was added at 6h p.i. and 225
incubated for a further 2h prior to fixing the cells, but with the striking addition of 226
large juxtanuclear accumulations of cytoplasmic staining that co-localised strongly 227
with Rab11 at low magnification (Fig 2A). At higher magnification, NP staining was 228
more pronounced towards the exterior of these structures and Rab11 staining in their 229
interior (Fig S1). However, the distribution of Rab11 was not altered by the presence 230
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

11
of the drug in uninfected cells, even when added for 4h (Figs 2A, S1). Nor were any 231
differences in the localization of either NP and Rab11 observed for the NP Y289H 232
mutant in the presence and absence of nucleozin (Fig S1). 233
Nucleozin-induced cytoplasmic aggregates of NP have been noted before but 234
interpreted to be newly synthesised NP on the way into the nucleus (30, 46)(12). 235
However, the timing of the effect we observed, as well as the inclusion of Rab11 in 236
the aggregates (Rab11 interacts with RNPs but not non-RNP NP; (1, 33)) suggested 237
that the drug was altering the trafficking of RNPs. To test this, we used fluorescence 238
in situ hybridization (FISH) to examine the localization of vRNA. As expected, in 239
untreated cells, the 3 genomic segments examined (1, 4 and 7), progressed from early 240
accumulation in the nucleus at 4h p.i. to a later punctate distribution in the cytoplasm 241
with a perinuclear focus (Fig 2B). However, as observed for NP, when nucleozin was 242
added at either 4 or 6h p.i. (and cells fixed at 8h p.i.), prominent cytoplasmic 243
aggregates of vRNA were seen surrounding the nucleus (Fig 2B). Furthermore, this 244
effect was specific for vRNA, because when similarly treated cells were double-245
stained by FISH for segment 3 vRNA and segment 7 mRNA, no drug-induced 246
alterations to mRNA localisation were apparent and nor was there any significant 247
cytoplasmic colocalisation of the two senses of viral RNA under any condition (Fig 248
S2). Thus nucleozin has a late-acting effect of causing the cytoplasmic aggregation of 249
the viral genomic RNPs along with cellular Rab11. 250
The prominent cytoplasmic aggregates of NP and vRNA clearly reflected the 251
disruption of normal RNP trafficking, so we next tested if this resulted from the drug 252
‘dismantling’ assembled RNPs. For this, we used a system in which cells are 253
transfected with GFP-NP and superinfected with virus to produce GFP-tagged RNPs 254
that can be affinity purified or followed by live-cell microscopy (1). Cells were 255
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

12
infected with PR8 virus 12h after GFP or GFP-NP transfection, treated (or not) with 256
nucleozin at 6 hpi and lysed at 8 hpi. Aliquots of the cell lysates were then examined 257
by western blotting for proteins of interest or by radioactive reverse transcriptase 258
primer extension reactions for RNA before or after affinity fractionation over GFP-259
Trap beads. Examination of unfractionated cell lysate showed that GFP and GFP-NP 260
were expressed as expected and that all viral proteins tested were present in the 261
supernatant of infected cells in equal quantities, as was the cellular protein α-tubulin 262
(Fig 3, lanes 1-6). Primer extension reactions confirmed that segment 3 vRNA and 263
cellular 5S rRNA were also present in approximately equal quantities. When GFP-NP 264
was affinity purified (but not GFP), the NP and polymerase components of RNPs 265
were co-precipitated equally in the absence and presence of nucleozin (lanes 7-9, 11). 266
Moreover, primer extension analysis detected vRNA but not 5S rRNA when GFP-NP 267
was purified. Thus the system successfully detected RNP formation and this was not 268
substantially changed by nucleozin treatment. We also tested for co-precipitation of 269
other viral structural proteins; M1 and NS2, both known to interact with RNPs during 270
nuclear export and virion assembly (5) and M2. A small proportion of M1 was 271
selectively purified by GFP-NP (Fig 7, lanes 7 and 8), but no detectable amounts of 272
NS2 or M2 were precipitated with or without drug treatment. Nucleozin therefore 273
induces the cytoplasmic aggregation of RNPs but not other viral proteins. 274
275
Nucleozin impairs the trafficking of circulating RNPs in the cytoplasm 276
Nucleozin induces aggregation of vRNPs in the perinuclear RE area, a region 277
of the cell hypothesised to represent a ‘way station’ for transport of the virus genome 278
from nucleus to the apical plasma membrane (1, 20, 33). To test if nucleozin blocked 279
onward transport from the RE, we acquired images of living virus-infected cells 280
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

13
containing GFP-NP tagged RNPs as well as RFP-Rab11. A549 cells were transfected 281
with GFP-NP and RFP-Rab11, 12 hours later infected (or mock infected) with PR8 282
virus and imaged at 8h p.i.. Inspection of still images from uninfected cells showed 283
that GFP-NP localised mostly to the nucleus, while RFP-Rab11 was dispersed 284
throughout the cytoplasm but with a prominent perinuclear concentration, probably 285
representing the RE (Fig 4A). Examination of time-lapse movies showed that both the 286
nucleoplasmic GFP-NP and the cytoplasmic RFP-Rab11 were highly mobile but did 287
not coincide substantially (Supplementary Movie 1; first 4.5 minutes). When cells 288
were infected, a distinct population of GFP-NP was seen as punctate objects in the 289
cytoplasm that co-localized with Rab11 (Fig 4B). Time lapse movies showed that 290
these too were highly mobile bodies, often forming filamentous or tubular structures. 291
This pattern of movement was stable under imaging conditions for at least 25 minutes 292
(Fig 4B, Supplementary Movie S2). Based on prior work from our laboratory and 293
others (1, 20, 33), this pattern was likely detecting GFP-NP that had been 294
incorporated into RNPs which, following nuclear export, were undergoing in part 295
microtubule-mediated transport courtesy of interactions with Rab11-positive vesicles. 296
In confirmation of this, when cells were fixed after imaging and stained by FISH for 297
segment 4 and 7 vRNAs, the signal co-localised with GFP-NP (data not shown). We 298
then tested the effect of adding nucleozin. In mock infected cells, drug addition had 299
little apparent effect, both in terms of the overall localisation patterns of GFP-NP and 300
RFP-Rab11 (Fig 4A) or their movement in time-lapse movies (Supplementary Movie 301
S1, minutes 4.5-29). However, when drug was added to infected cells, the result was 302
a dramatic loss of the small, highly mobile GFP-NP and RFP-Rab11 positive bodies, 303
to be replaced by increasingly large cytoplasmic aggregates that still contained both 304
proteins but that in many cases collapsed back onto the perinuclear region (Fig 4C, 305
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

14
supplementary movie S3). High magnification movies of vRNP-decorated vesicles 306
moving in the cytoplasm of infected cells, showed that they were highly mobile and 307
that while there were apparently continual interactions amongst them, these were 308
transient (Fig S3A, see white arrowheads and supplementary movie S4). However, in 309
the presence of nucleozin, interacting vesicular structures tended to remain attached 310
over time, thus forming large agglomerations. (Fig S3B, see white arrowheads and 311
supplementary movie S5). Thus nucleozin exerts a general block to cytoplasmic 312
transport of RNP-Rab11 complexes by sticking vRNPs and their associated transport 313
machinery together, most likely via NP-NP interactions. 314
315
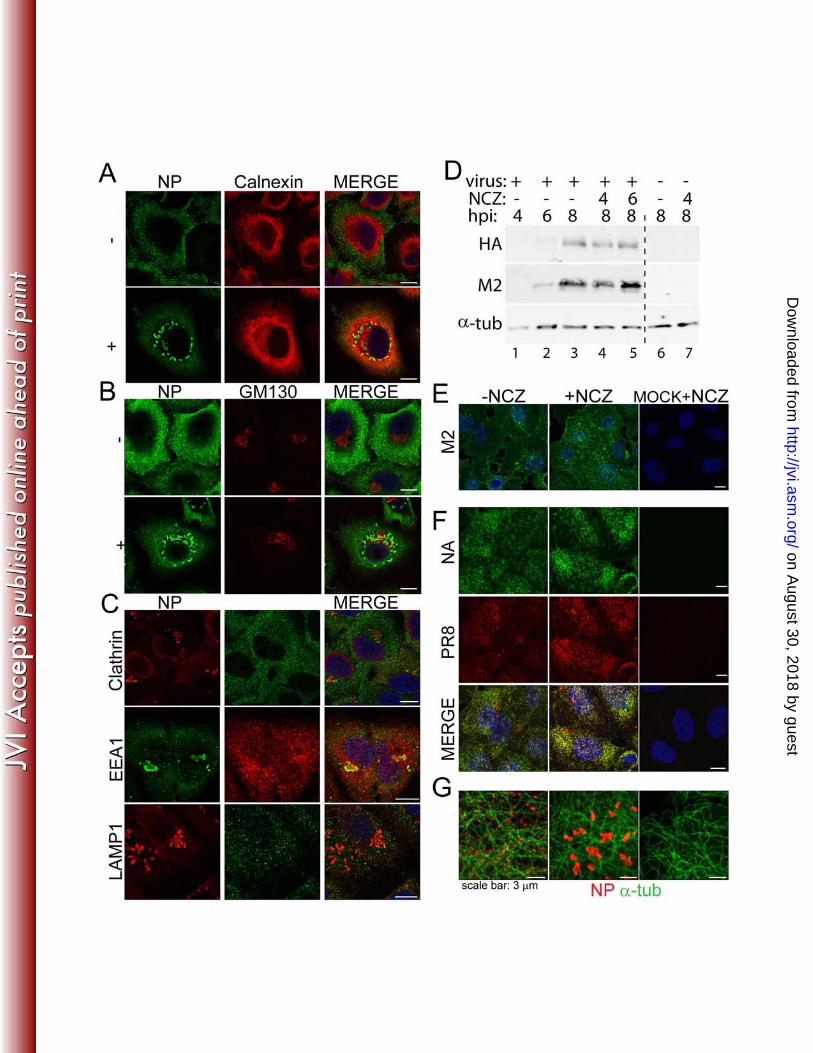
Nucleozin does not widely disrupt the exocytic pathway in infected cells 316
The components of the secretory pathway are interconnected in an intricate 317
network whereby disruption of one pathway can have secondary effects on other 318
vesicular pathways (4, 24, 26). Since nucleozin disrupted Rab11 localisation in 319
infected cells, we tested whether it had a generalized effect on the exocytic pathway; 320
and as a consequence, a drug-induced disruption of trafficking of viral membrane 321
proteins. First, we tested whether the localization of standard cellular marker proteins 322
for the secretory pathway was altered by the addition of nucleozin at late times post 323
infection. Neither endosomes (highlighted with EEA1, clathrin and LAMP1), the 324
endoplasmic reticulum (as assessed by calnexin), the Golgi (GM130 marker), or the 325
trans Golgi network (marked with TGN 46, data not shown), aggregated in treated 326
cells (Figs 5A-C) or indeed, noticeably changed localisation at all after drug addition 327
(Figs 5A, B and data not shown for C). In addition, double staining for NP confirmed 328
that the drug affected RNP localisation as expected, but in contrast to the result seen 329
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

15
with Rab11 (Fig 2A), little to no colocalisation was seen between the viral protein and 330
the cellular markers (Figs 5A, B, C). 331
Since M1 and NS2/NEP are involved in RNP nuclear export (5) and therefore 332
might accompany the genome while crossing the cytoplasm, we also examined 333
whether nucleozin affected their localisation. NS2 localization did not dramatically 334
change in the presence of the drug (data not shown), and nor did that of M1 (data not 335
shown). 336
Next, we tested the effect of nucleozin on the synthesis and accumulation at 337
the PM of the viral transmembrane proteins M2, NA and HA. By western blotting, 338
expression of M2 and HA proteins increased up to 8h p.i. in a typical untreated time 339
course (Fig 5D, lanes 1-3). The addition of nucleozin at either 4 or 6h p.i. had little 340
effect on the accumulation of either protein at 8h p.i. (lanes 4 and 5), consistent with 341
previous data showing the general insensitivity of viral protein expression to drug 342
addition after early times post infection (Fig 1C). NA accumulation could not be 343
similarly assessed because of the lack of an appropriate antibody. We then tested HA, 344
M2 and NA localisation at the PM by immunofluorescent confocal microscopy. 345
Staining of non-permeabilized cells for the three transmembrane proteins showed 346
readily detectable levels of cell surface protein at 8h p.i. (Figs 5E, F). However, no 347
diminution of staining intensity or obvious differences in localisation were seen after 348
treatment with nucleozin from 6h p.i. onwards. No effect on cell surface HA or M2 349
amounts were seen when cells were examined by fluorescent activated cell sorting 350
(data not shown). Thus the effects of nucleozin on viral and cellular trafficking 351
pathways are apparently specific to the conjunction of RNPs and Rab11 in infected 352
cells. 353
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

16
The normal trafficking of cellular and additional viral proteins suggests that 354
nucleozin did not affect the function of the microtubule network. Consistent with this, 355
α-tubulin staining in treated and untreated cells was identical (Fig 5G). 356
357
Virions budding from PM in the presence of nucleozin exhibit marked defects 358
Nucleozin impedes the viral genome from reaching the PM in the later stages 359
of viral infection. To understand the implications of this for virion formation, WT and 360
NP Y289H mutant viruses were analyzed by TEM, with and without drug treatment. 361
Budding virions were not detected at 4h p.i. with either strain (Fig 6A). By 6h p.i., 362
cells infected with both viruses displayed the characteristic features of budding virus; 363
ellipsoidal structures surrounded by a glycoprotein fringe, generally attached to the 364
plasma membrane by the tip of their shorter axis. When the number of budding events 365
were counted in WT virus-infected cells at 8h p.i., the numbers varied around a mean 366
of 4-5 particles per µm of plasma membrane (Fig 6B), while measurement of the 367
lengths of budding virus showed a mean long axis dimension of 132 ±2.9 nm (Fig 6C; 368
gallery of representative images shown in Fig 6D). When nucleozin was added at 369
either 4 or 6h p.i. to cells infected with the nucleozin resistant virus NP Y289H, little 370
effect was apparent on the budding (Fig 6A, right hand panels) or size of the virus 371
particles formed at 8h p.i. (Figs 6C, D), with sizes of 113.9±2.1; 124.9±2.5 and 372
122.1±2.8 nm for no drug, and nucleozin added at 4 or 6h.p.i.. The WT virus, 373
however showed marked and distinct defects. When drug was added at 4h p.i., very 374
few virions were detected and in the rare cases where they were observed, they had an 375
altered morphology, appearing much rounder, with an average height of around 376
71.5±3.8 nm (Figs 6C, D In addition, they were often still attached to the PM by wide 377
necks in a manner similar to that seen when virus budding was inhibited through 378
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

17
siRNA-mediated depletion of Rab11 (7). When the drug was added at 6h p.i. and the 379
cells fixed at 8h p.i. , the number of budding events was still very low compared to 380
untreated cells (Fig 6B) and although some normal particles were seen, the 381
morphology of many others was abnormal; many virions were shorter than in 382
untreated cells (105.4±5.9 nm), while others lacked an electron-dense content (Figs 383
6A, D). Thus the late-acting effects of nucleozin on viral RNP trafficking and titre 384
output can be correlated with matching effects on virus particle formation, suggesting 385
a direct link between RNP localization and virus budding. 386
387
Discussion 388
Independent work from several laboratories has identified nucleozin and 389
related compounds as drugs that promote the formation of cytoplasmic NP 390
aggregates, inhibit viral RNA synthesis and effectively interfere with virus replication 391
(12, 23, 30, 46). Structural analyses suggest that the drugs act as ‘molecular staples’ 392
that stabilise NP-NP interactions; presumably either by inducing functionally 393
inappropriate binding modes that do not normally occur, or by locking ordinarily 394
transient interactions in place so that they become inhibitory. The EC50 of nucleozin 395
for overall virus replication is around 15-fold lower than the corresponding 396
concentration required to inhibit transcription by isolated RNPs (23), suggesting that 397
direct inhibition of viral RNA synthesis is not the primary block to virus replication. 398
Instead, initial studies hypothesised that the drug caused the cytoplasmic aggregation 399
of newly translated NP, thereby blocking its nuclear import and ‘starving’ viral 400
genome replication of the building blocks required for encapsidation (12, 23, 30, 46). 401
However, based on our time of addition experiments and detailed analyses of 402
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

18
individual steps within viral replication, we propose that RNPs, not NP, are the 403
primary target of the drug and that the sensitive step is their cytoplasmic transport. 404
We show that nucleozin shows distinct effects on virus replication, depending 405
on the time of addition. When added early in infection it inhibited synthesis of viral 406
proteins and all classes of RNA (Fig 1). While failure to produce vRNA could result 407
from a depleted pool of intranuclear NP, viral transcription and translation are 408
independent of this (39), and a distinct inhibitory mechanism must operate here. 409
Furthermore, the inhibitory effect of nucleozin on overall virus replication could be 410
decoupled from any reduction in the synthesis of viral mRNA, proteins or vRNA by 411
adding the drug later in infection. Tellingly, adding drug at 4h p.i. had no effect on 412
viral protein synthesis, only caused a two-fold drop in vRNA accumulation and yet 413
reduced output titre by around 100-fold compared to untreated cells (Fig 1). Instead, 414
the drug caused a striking and rapid aggregation of cytoplasmic RNPs (Figs 2, 4, 5, 415
S1, S2). This blockade to RNP transport also correlated with a marked reduction in 416
the amount of virus budding as well as the formation of abnormally small virus 417
particles (Fig 6). Notably however, we saw little effect of drug treatment on 418
intranuclear NP localisation, either on GFP-NP with or without RNP formation or on 419
authentic NP (Fig 2A). We therefore conclude that the late-acting effect of nucleozin 420
on virus replication is directly attributable to effects on RNP localisation. 421
Regarding the undoubted ability of nucleozin to inhibit earlier steps in the 422
virus lifecycle, given that the drug did not inhibit mRNA synthesis when added at 423
2.5h p.i. (before large amounts of mRNA have accumulated) and that we were 424
working at drug concentrations substantially below those required to inhibit in vitro 425
transcription by isolated RNPs, we think it plausible that the transcriptional inhibition 426
observed when the drug was added 30 minutes p.i. can also be explained by inhibition 427
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

19
of cytoplasmic RNP transport, but on the way into the nucleus rather than away from 428
it. Thus although NP is primarily regarded as part of the viral RNA synthesis 429
machinery, this study provides proof of principle that targeting its role in intracellular 430
trafficking of the virus genome can lead to effective inhibition of virus replication. 431
This mechanism has parallels with the block to RNP nuclear export caused by MEK 432
inhibitors (19, 37) and it would be interesting to test these compounds for synergy 433
with nucleozin. 434
A further striking outcome of the late-acting effect of nucleozin was the 435
inclusion of Rab11 in the cytoplasmic aggregates of RNPs (Figs 2, 4 S1). Since 436
Rab11 trafficking was not affected by nucleozin in the absence of viral RNPs, it 437
seems likely that the cellular protein is secondarily recruited to the aggregates via its 438
interaction with RNPs. This argues against the simple hypothesis that nucleozin 439
disrupts RNP trafficking by breaking interactions with Rab11. Taken all into account, 440
suggests a model in which, Rab11-vRNP decorated vesicles get “stapled” together via 441
persistent RNP-RNP interactions that override ordinarily transient interactions 442
between vesicular cargo (Fig S3 and supplementary movies S4-S5). Transfection of 443
dominant negative GFP-Rab11 or depletion of endogenous Rab11 siRNA did not 444
prevent these accumulations in cells infected with WT viruses (data not shown), 445
suggesting that Rab11 may not be essential for aggregate formation. While this is 446
consistent with data suggesting Rab11 interacts with the viral polymerase rather than 447
NP (1, 33), it leaves the question open as to why the aggregates cluster near the 448
nucleus. The speed with which this occurs suggests that the RNP aggregates remain 449
attached to microtubules (also see Fig 5G), but that the net flow becomes biased 450
towards the inwards (minus end) direction. Why the increase in size of the RNP-451
Rab11 cargo complexes should favour this direction of travel is not obvious but may 452
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

20
become clearer when the motor proteins involved in RNP transport are identified. 453
Dynein has however been implicated in the transport of “aggresomes”, created by 454
misfolded proteins, so it is possible that the same mechanism operated here (48). 455
Nucleozin treatment also inhibited virus budding, leading to the production of 456
markedly fewer and smaller virus particles (with the latter potentially because of 457
reduced RNP content) (Fig 6). For normal virus budding, all virion components need 458
to meet at the PM from where a membrane curvature protrudes, followed by 459
formation of a neck and finally membrane scission. All three viral integral membrane 460
proteins have been implicated in this process (11, 42, 51). Despite the major effect 461
nucleozin had on Rab11 localization (and thus presumably traffic throughout the 462
recycling endosomal pathway) we did not detect any drug-induced alterations to the 463
standard exocytic pathway or to localisation of HA, NA and M2 (Fig 5). We therefore 464
favour the hypothesis that correct trafficking of the viral genome is specifically 465
required to allow efficient virus assembly and/or budding. This is consistent with our 466
previous findings of defective budding and RNP trafficking in the absence of normal 467
levels of Rab11 (1, 7). Although the effect of nucleozin does not help distinguish 468
between a budding defect resulting primarily from disruption of Rab11 function and a 469
secondary effect on RNP localisation, observations that viruses lacking an individual 470
segment or containing disrupted segment-specific packaging signals produce reduced 471
numbers of particles (17, 22, 28, 35), favours a role for RNP organisation in normal 472
virion formation. 473
The Rab11 pathway has been implicated in the transport of progeny RNA from 474
other negative strand viruses, notably Sendai virus and human respiratory syncytial 475
virus, members of the Paramyxoviridae family (10, 47); hantavirus (44) as well as 476
hepatitis C virus, a positive sense RNA virus from the Flaviviridae family (14). The 477
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

21
common exploitation of a pathway by several circulating virus families able to infect 478
mammals and on occasion cause severe disease in humans, suggests that the 479
inhibitory strategy herein described could be applied to other viruses. 480
481
Acknowledgments 482
We thank Susanna Colaco and Dr. Phil Stevenson for the gift of reagents. This 483
work was supported by a grant from the MRC (no G0700815) to PD. MJA was 484
supported by a Newton Trust grant from Trinity College, University of Cambridge, 485
Cambridge, UK. RK was supported by GRF 768010M from RGC Hong Kong. We 486
also thank the multiimaging centre from the University of Cambridge for processing 487
images for TEM. 488
489
Figure legends 490
Fig 1. Time of addition experiments examining the effect of nucleozin on IAV 491
replication and macromolecular synthesis. A549 cells were infected with either WT or 492
NP Y289H (NP 289) mutant viruses and 1 μM nucleozin added at the indicated time 493
points (A). Supernatants were collected and plaque titrated in MDCK cells. Virus 494
titres (white bars) are expressed as the mean ± SD % (n = 3). (B) Cell lysates were 495
analysed by western blotting for the indicated proteins. (C) Total cellular RNA was 496
extracted and analysed by radioactive primer extension, urea-PAGE and 497
autoradiography for mRNA and vRNA from segments 3 and 5 as well as 5S 498
ribosomal RNA. (D) vRNA accumulation was quantified by densitometry of primer 499
extension autoradiographs using Image J and plotted as the mean±SD (n = 3) 500
percentages of the amount from untreated cells at 8h p.i.. 501
502
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

22
Fig 2. Nucleozin induces cytoplasmic aggregates containing RNP and Rab11. A549 503
cells were infected (or mock infected) with WT virus and either left untreated or 504
treated with 1 μM nucleozin as labelled. Samples were fixed at at the times shown 505
and (A) stained for Rab11 and NP or (B) processed for FISH for the indicated vRNAs 506
before imaging by confocal microscopy. Merged images include a DAPI channel 507
shown in blue. Scale bar represents 10 μm. Images are representative of three 508
independent experiments. 509
510
Fig 3. Nucleozin treatment does not disrupt RNPs. 293T cells were transfected with 511
plasmids expressing GFP alone or GFP-NP, incubated for 24 h and infected (or mock 512
infected) with WT virus. At 6h p.i. one set of cells were treated with 1μM nucleozin. 513
Cell lysates prepared at 8h p.i. were analysed by western blotting for the indicated 514
polypeptides after GFP-TRAP affinity selection into supernatant and bound fractions. 515
Sample loading is such that the supernatants are equivalent to 1/10th of the bound 516
fractions. RNA was analysed by primer extension for segment 3 vRNA and 5S 517
ribosomal RNA before or after GFP-TRAP selection. The experimental procedure 518
was performed twice. 519
520
Fig 4. Live cell trafficking of GFP-NP and RFP-Rab11. A549 cells were transfected 521
GFP-NP and RFP-Rab11 and 12 h later, infected (or mock infected) with WT PR8 522
before imaging under time-lapse conditions (approximately every 4 seconds) at 8h 523
p.i.. Selected still images are shown while arrows indicate the time of drug addition (1 524
µM). (A) Mock infected cell. (B) Infected cell without drug treatment. (C) Infected 525
cell with nucleozin treatment. Scale bars represent 10 μm. Images were acquired 526
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

23
using a SPE confocal microscope and images processed using LAS AF Lite. See 527
Supplementary Movies S1-3. 528
529
Fig 5. Nucleozin does not disrupt the main exocytic pathway. (A-C, E, F) A549 cells 530
were infected with WT virus and at 6h p.i., 1μM nucleozin was added to one set. 531
Samples were fixed at 8h p.i. and stained for (A) NP and calnexin; (B) NP and 532
GM130, (C) NP and clathrin or EEA1 or LAMP1 in the presence of nucleozin and as 533
labeled on the sides, (E) M2, (F) HA and NA (G) NP and alpha-tubulin [α-tub]. 534
Merged images include a DAPI channel shown in blue. Scale bar represents 10 μm, 535
unless mentioned. In (D), lysates from cells treated and harvested as indicated were 536
analysed by western blotting for HA, M2 and (as a loading control, alpha-tubulin [α-537
tub]). Images are representative of three independent experiments. 538
539
Fig 6. TEM visualization of budding virions in nucleozin-treated cells. A549 cells 540
were infected with WT or NP Y289H mutant virus, treated with drug and fixed at 541
time points as labelled before imaging by TEM. White arrows indicate defective 542
budding events (A) Representative images. Scale bar, 100 nm. (B) Number of WT 543
budding virions were calculated over the whole surface of one side of the cell. (C) 544
Sizes of budding virions from cells infected with WT PR8 were determined (as 545
shown) for at least 30 particles from 3 independent experiments and plotted as 546
average ± SEM. Nonparametric statistical analysis values using ANOVA Krustal-547
Wallis with significance of 95% were calculated using GraphPad Prism. (D) Gallery 548
of representative budding events with or without nucleozin treatment (1 µM, added at 549
the indicted times p.i.). Black arrows indicate defective particles. Images are compiled 550
from 3 independent experiments. 551
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

24
References 552
1. Amorim, M. J., E. A. Bruce, E. K. Read, A. Foeglein, R. Mahen, A. D. 553 Stuart, and P. Digard. 2011. A Rab11- and microtubule-dependent 554 mechanism for cytoplasmic transport of influenza A virus viral RNA. J Virol 555 85:4143-56. 556
2. Amorim, M. J., E. K. Read, R. M. Dalton, L. Medcalf, and P. Digard. 557 2007. Nuclear export of influenza A virus mRNAs requires ongoing RNA 558 polymerase II activity. Traffic 8:1-11. 559
3. Avilov, S. V., D. Moisy, S. Munier, O. Schraidt, N. Naffakh, and S. 560 Cusack. 2012. Replication-competent influenza A virus that encodes a split-561 green fluorescent protein-tagged PB2 polymerase subunit allows live-cell 562 imaging of the virus life cycle. J Virol 86:1433-48. 563
4. Bard, F., and V. Malhotra. 2006. The formation of TGN-to-plasma-564 membrane transport carriers. Annu Rev Cell Dev Biol 22:439-55. 565
5. Boulo, S., H. Akarsu, R. W. Ruigrok, and F. Baudin. 2007. Nuclear traffic 566 of influenza virus proteins and ribonucleoprotein complexes. Virus Res 567 124:12-21. 568
6. Breitenfeld, P. M., and W. Schafer. 1957. The formation of fowl plague 569 virus antigens in infected cells, as studied with fluorescent antibodies. 570 Virology 4:328-45. 571
7. Bruce, E. A., P. Digard, and A. D. Stuart. 2010. The Rab11 pathway is 572 required for influenza A virus budding and filament formation. J Virol 573 84:5848-59. 574
8. Bruce, E. A., L. Medcalf, C. M. Crump, S. L. Noton, A. D. Stuart, H. M. 575 Wise, D. Elton, K. Bowers, and P. Digard. 2009. Budding of filamentous 576 and non-filamentous influenza A virus occurs via a VPS4 and VPS28-577 independent pathway. Virology 390:268-78. 578
9. Carrasco, M., M. J. Amorim, and P. Digard. 2004. Lipid raft-dependent 579 targeting of the influenza A virus nucleoprotein to the apical plasma 580 membrane. Traffic 5:979-92. 581
10. Chambers, R., and T. Takimoto. 2010. Trafficking of Sendai virus 582 nucleocapsids is mediated by intracellular vesicles. PLoS One 5:e10994. 583
11. Chen, B. J., G. P. Leser, E. Morita, and R. A. Lamb. 2007. Influenza virus 584 hemagglutinin and neuraminidase, but not the matrix protein, are required for 585 assembly and budding of plasmid-derived virus-like particles. J Virol 586 81:7111-23. 587
12. Cheng, H., J. Wan, M. I. Lin, Y. Liu, X. Lu, J. Liu, Y. Xu, J. Chen, Z. Tu, 588 Y. S. Cheng, and K. Ding. 2012. Design, Synthesis, and in Vitro Biological 589 Evaluation of 1H-1,2,3-Triazole-4-carboxamide Derivatives as New Anti-590 influenza A Agents Targeting Virus Nucleoprotein. J Med Chem 55: 591 2144−2153. 592
13. Cianci, C., S. W. Gerritz, C. Deminie, and M. Krystal. 2012. Influenza 593 Nucleoprotein: Promising Target for Antiviral Chemotherapy. Antivir Chem 594 Chemother Jul 26. doi: 10.3851/IMP2235. 595
14. Coller, K. E., N. S. Heaton, K. L. Berger, J. D. Cooper, J. L. Saunders, 596 and G. Randall. 2012. Molecular determinants and dynamics of hepatitis C 597 virus secretion. PLoS Pathog 8:e1002466. 598
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

25
15. Das, K., J. M. Aramini, L. C. Ma, R. M. Krug, and E. Arnold. 2010. 599 Structures of influenza A proteins and insights into antiviral drug targets. Nat 600 Struct Mol Biol 17:530-8. 601
16. de Wit, E., M. I. Spronken, T. M. Bestebroer, G. F. Rimmelzwaan, A. D. 602 Osterhaus, and R. A. Fouchier. 2004. Efficient generation and growth of 603 influenza virus A/PR/8/34 from eight cDNA fragments. Virus Res 103:155-604 61. 605
17. de Wit, E., M. I. Spronken, G. F. Rimmelzwaan, A. D. Osterhaus, and R. 606 A. Fouchier. 2006. Evidence for specific packaging of the influenza A virus 607 genome from conditionally defective virus particles lacking a polymerase 608 gene. Vaccine 24:6647-50. 609
18. Digard, P., V. C. Blok, and S. C. Inglis. 1989. Complex formation between 610 influenza virus polymerase proteins expressed in Xenopus oocytes. Virology 611 171:162-9. 612
19. Droebner, K., S. Pleschka, S. Ludwig, and O. Planz. 2011. Antiviral 613 activity of the MEK-inhibitor U0126 against pandemic H1N1v and highly 614 pathogenic avian influenza virus in vitro and in vivo. Antiviral Res 92:195-615 203. 616
20. Eisfeld, A. J., E. Kawakami, T. Watanabe, G. Neumann, and Y. 617 Kawaoka. 2011. RAB11A is essential for transport of the influenza virus 618 genome to the plasma membrane. J Virol 85:6117-26. 619
21. Elton, D., M. J. Amorim, L. Medcalf, and P. Digard. 2005. 'Genome 620 gating'; polarized intranuclear trafficking of influenza virus RNPs. Biol Lett 621 1:113-7. 622
22. Fujii, Y., H. Goto, T. Watanabe, T. Yoshida, and Y. Kawaoka. 2003. 623 Selective incorporation of influenza virus RNA segments into virions. Proc 624 Natl Acad Sci U S A 100:2002-7. 625
23. Gerritz, S. W., C. Cianci, S. Kim, B. C. Pearce, C. Deminie, L. Discotto, B. 626 McAuliffe, B. F. Minassian, S. Shi, S. Zhu, W. Zhai, A. Pendri, G. Li, M. 627 A. Poss, S. Edavettal, P. A. McDonnell, H. A. Lewis, K. Maskos, M. Mortl, 628 R. Kiefersauer, S. Steinbacher, E. T. Baldwin, W. Metzler, J. Bryson, M. 629 D. Healy, T. Philip, M. Zoeckler, R. Schartman, M. Sinz, V. H. Leyva-630 Grado, H. H. Hoffmann, D. R. Langley, N. A. Meanwell, and M. Krystal. 631 2011. Inhibition of influenza virus replication via small molecules that induce 632 the formation of higher-order nucleoprotein oligomers. Proc Natl Acad Sci U 633 S A 108:15366-71. 634
24. Gonnord, P., C. M. Blouin, and C. Lamaze. 2011. Membrane trafficking 635 and signaling: Two sides of the same coin. Semin Cell Dev Biol. 636
25. Harman, A., H. Browne, and T. Minson. 2002. The transmembrane domain 637 and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role 638 in membrane fusion. J Virol 76:10708-16. 639
26. Hsu, V. W., and R. Prekeris. 2010. Transport at the recycling endosome. 640 Curr Opin Cell Biol 22:528-34. 641
27. Huang, T. S., P. Palese, and M. Krystal. 1990. Determination of influenza 642 virus proteins required for genome replication. J Virol 64:5669-73. 643
28. Hutchinson, E. C., M. D. Curran, E. K. Read, J. R. Gog, and P. Digard. 644 2008. Mutational analysis of cis-acting RNA signals in segment 7 of influenza 645 A virus. J Virol 82:11869-79. 646
29. Jagger, B. W., H. M. Wise, J. C. Kash, K. A. Walters, N. M. Wills, Y. L. 647 Xiao, R. L. Dunfee, L. M. Schwartzman, A. Ozinsky, G. L. Bell, R. M. 648
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

26
Dalton, A. Lo, S. Efstathiou, J. F. Atkins, A. E. Firth, J. K. Taubenberger, 649 and P. Digard. 2012. An overlapping protein-coding region in influenza A 650 virus segment 3 modulates the host response. Science 337:199-204. 651
30. Kao, R. Y., D. Yang, L. S. Lau, W. H. Tsui, L. Hu, J. Dai, M. P. Chan, C. 652 M. Chan, P. Wang, B. J. Zheng, J. Sun, J. D. Huang, J. Madar, G. Chen, 653 H. Chen, Y. Guan, and K. Y. Yuen. 2010. Identification of influenza A 654 nucleoprotein as an antiviral target. Nat Biotechnol 28:600-5. 655
31. Loucaides, E. M., J. C. von Kirchbach, A. Foeglein, J. Sharps, E. Fodor, 656 and P. Digard. 2009. Nuclear dynamics of influenza A virus 657 ribonucleoproteins revealed by live-cell imaging studies. Virology 394:154-658 63. 659
32. Matrosovich, M., T. Matrosovich, W. Garten, and H. D. Klenk. 2006. New 660 low-viscosity overlay medium for viral plaque assays. Virol J 3:63. 661
33. Momose, F., T. Sekimoto, T. Ohkura, S. Jo, A. Kawaguchi, K. Nagata, 662 and Y. Morikawa. 2011. Apical Transport of Influenza A Virus 663 Ribonucleoprotein Requires Rab11-positive Recycling Endosome. PLoS One 664 6:e21123. 665
34. Mullin, A. E., R. M. Dalton, M. J. Amorim, D. Elton, and P. Digard. 2004. 666 Increased amounts of the influenza virus nucleoprotein do not promote higher 667 levels of viral genome replication. J Gen Virol 85:3689-98. 668
35. Muramoto, Y., A. Takada, K. Fujii, T. Noda, K. Iwatsuki-Horimoto, S. 669 Watanabe, T. Horimoto, H. Kida, and Y. Kawaoka. 2006. Hierarchy 670 among viral RNA (vRNA) segments in their role in vRNA incorporation into 671 influenza A virions. J Virol 80:2318-25. 672
36. Noton, S. L., E. Medcalf, D. Fisher, A. E. Mullin, D. Elton, and P. Digard. 673 2007. Identification of the domains of the influenza A virus M1 matrix protein 674 required for NP binding, oligomerization and incorporation into virions. J Gen 675 Virol 88:2280-90. 676
37. Pleschka, S., T. Wolff, C. Ehrhardt, G. Hobom, O. Planz, U. R. Rapp, and 677 S. Ludwig. 2001. Influenza virus propagation is impaired by inhibition of the 678 Raf/MEK/ERK signalling cascade. Nat Cell Biol 3:301-5. 679
38. Poole, E., D. Elton, L. Medcalf, and P. Digard. 2004. Functional domains of 680 the influenza A virus PB2 protein: identification of NP- and PB1-binding sites. 681 Virology 321:120-33. 682
39. Portela, A., and P. Digard. 2002. The influenza virus nucleoprotein: a 683 multifunctional RNA-binding protein pivotal to virus replication. J Gen Virol 684 83:723-34. 685
40. Read, E. K., and P. Digard. 2010. Individual influenza A virus mRNAs show 686 differential dependence on cellular NXF1/TAP for their nuclear export. J Gen 687 Virol 91:1290-301. 688
41. Robb, N. C., M. Smith, F. T. Vreede, and E. Fodor. 2009. NS2/NEP protein 689 regulates transcription and replication of the influenza virus RNA genome. J 690 Gen Virol 90:1398-407. 691
42. Rossman, J. S., X. Jing, G. P. Leser, and R. A. Lamb. 2010. Influenza virus 692 M2 protein mediates ESCRT-independent membrane scission. Cell 142:902-693 13. 694
43. Rossman, J. S., and R. A. Lamb. 2011. Influenza virus assembly and 695 budding. Virology 411:229-36. 696
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

27
44. Rowe, R. K., J. W. Suszko, and A. Pekosz. 2008. Roles for the recycling 697 endosome, Rab8, and Rab11 in hantavirus release from epithelial cells. 698 Virology 382:239-49. 699
45. Simpson-Holley, M., D. Ellis, D. Fisher, D. Elton, J. McCauley, and P. 700 Digard. 2002. A functional link between the actin cytoskeleton and lipid rafts 701 during budding of filamentous influenza virions. Virology 301:212-25. 702
46. Su, C. Y., T. J. Cheng, M. I. Lin, S. Y. Wang, W. I. Huang, S. Y. Lin-Chu, 703 Y. H. Chen, C. Y. Wu, M. M. Lai, W. C. Cheng, Y. T. Wu, M. D. Tsai, Y. 704 S. Cheng, and C. H. Wong. 2010. High-throughput identification of 705 compounds targeting influenza RNA-dependent RNA polymerase activity. 706 Proc Natl Acad Sci U S A 107:19151-6. 707
47. Utley, T. J., N. A. Ducharme, V. Varthakavi, B. E. Shepherd, P. J. 708 Santangelo, M. E. Lindquist, J. R. Goldenring, and J. E. Crowe, Jr. 2008. 709 Respiratory syncytial virus uses a Vps4-independent budding mechanism 710 controlled by Rab11-FIP2. Proc Natl Acad Sci U S A 105:10209-14. 711
48. Wan, Y., Z. Yang, J. Guo, Q. Zhang, L. Zeng, W. Song, Y. Xiao, and X. 712 Zhu. 2012. Misfolded Gbeta is recruited to cytoplasmic dynein by Nudel for 713 efficient clearance. Cell Res 22:1140-54. 714
49. Wise, H. M., A. Foeglein, J. Sun, R. M. Dalton, S. Patel, W. Howard, E. C. 715 Anderson, W. S. Barclay, and P. Digard. 2009. A complicated message: 716 Identification of a novel PB1-related protein translated from influenza A virus 717 segment 2 mRNA. J Virol 83:8021-31. 718
50. Wise, H. M., E. C. Hutchinson, B. W. Jagger, A. D. Stuart, Z. H. Kang, N. 719 Robb, L. M. Schwartzman, J. C. Kash, E. Fodor, A. E. Firth, J. R. Gog, J. 720 K. Taubenberger, and P. Digard. 2012. Identification of a novel splice 721 variant form of the influenza a virus m2 ion channel with an antigenically 722 distinct ectodomain. PLoS Pathog 8:e1002998. 723
51. Yondola, M. A., F. Fernandes, A. Belicha-Villanueva, M. Uccelini, Q. 724 Gao, C. Carter, and P. Palese. 2011. Budding capability of the influenza 725 virus neuraminidase can be modulated by tetherin. J Virol 85:2480-91. 726
727 728 729 730
on August 30, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from