Embed Size (px)
Citation preview

Postal address P.O. Box 26, SE-751 03 Uppsala, SWEDEN
Visiting address: Dag Hammarskjölds väg 42, Uppsala
Phone: +46 (0)18 17 46 00 Fax: +46 (0)18 54 85 66
Internet: www.lakemedelsverket.se E-mail: [email protected]
In-depth analysis of various alternative courses of action aimed at achieving the milestone target for environmental considerations in medicinal product legislation within the EU and internationally Report from the Medical Products Agency 15-12-2014 A translation of Fördjupad analys av olika handlingsalternativ för att nå etappmålet om miljöhänsyn i läkemedelslagstiftningen inom EU och internationellt, Rapport från Läkemedelsverket

Page 2 (96)

Page 3 (96)
List of contents 1. Summary of the report .............................................................................................................7
2. The assignment .......................................................................................................................15 2.1. Limitations of the assignment ..................................................................................................................... 16 2.2. Consultation ............................................................................................................................................... 16 2.3. Ongoing international activities ................................................................................................................. 17 2.4. The structure of the report .......................................................................................................................... 18
3. Assessment by the Swedish Environmental Protection Agency and the Swedish Chemicals Agency of damage to the environment and to human health that medicinal products give rise to and the associated costs ..................................................................................................................19
3.1. Emission of pharmaceutical ingredients into the environment ................................................................... 19 3.1.1. Treatment plants............................................................................................................................. 20 3.1.2. Individual sewage systems ............................................................................................................. 21 3.1.3. Emissions from production plants.................................................................................................. 22 3.1.4. Medicinal products for veterinary purposes ................................................................................... 22 3.1.5. Emissions into the environment via refuse .................................................................................... 23
3.2. Measuring emission concentrations in the environment ............................................................................ 23 3.2.1. Surface water ................................................................................................................................. 23 3.2.2. Drinking water ............................................................................................................................... 24 3.2.3. Concentrations in fish .................................................................................................................... 26 3.2.4. Concentrations in sediment ............................................................................................................ 26
3.3. Environmental impact (not resistance development) .................................................................................. 26 3.3.1. Effects on fish ................................................................................................................................ 27 3.3.2. Effects on other aquatic organisms ................................................................................................ 28 3.3.3. Effects on land animals .................................................................................................................. 29 3.3.4. Effects on humans .......................................................................................................................... 29 3.3.5. Cocktail effects in the environment ............................................................................................... 30
3.4. Development of resistance .......................................................................................................................... 30 3.5. Overall conclusions regarding damage to the environment and human health ......................................... 31 3.6. Broad estimate of costs ............................................................................................................................... 32
3.6.1. Costs of reducing emissions ........................................................................................................... 32 3.6.2. Macroeconomic costs of the effects of pharmaceutical substances (excluding resistance development). ............................................................................................................................................... 35 3.6.3. Macroeconomic costs of the spread of antibiotic resistance .......................................................... 36
4. Environmental considerations in current pharmaceutical legislation ..................................37 4.1. Marketing authorisation for medicinal products ........................................................................................ 37
4.1.1. Requirements for environmental data in conjunction with authorisation....................................... 38 4.1.2. Veterinary medicinal products ....................................................................................................... 38
4.2. Post authorisation control of medicinal products ...................................................................................... 39 4.3. Manufacturing authorisation for medicinal products................................................................................. 39 4.4. Manufacturing authorisations under EU environmental legislation .......................................................... 39
5. The four measures ..................................................................................................................41 6. Measure A: Testing requirements for medicinal products and the environmental risk assessments carried out in conjunction with an application for authorisation of medicinal products...........................................................................................................................................42

Page 4 (96)
6.1. Previous studies bearing on Measure A ..................................................................................................... 42 6.2. Limitations to Measure A ........................................................................................................................... 42 6.3. Background to Measure A .......................................................................................................................... 42 6.4. Courses of action for Measure A ................................................................................................................ 43
6.4.1. Course of Action A1: Review current limitations such that more pharmaceutical substances are covered by requirements for environmental risk assessment. ...................................................................... 43 6.4.2. Course of Action A2: Develop the testing requirements for environmental risk assessment such that the tests are better adapted to specific properties of the medicinal products. ........................................ 44
6.5. Effects of Measure A and respective courses of action............................................................................... 44 7. Measure B: Gathering environmental data on active pharmaceutical ingredients and making it accessible ........................................................................................................................46
7.1. Previous government assignments pertinent to Measure B ........................................................................ 46 7.2. Limitations to Measure B ........................................................................................................................... 46 7.3. Background to Measure B .......................................................................................................................... 46 7.4. Conditions for Measure B........................................................................................................................... 48
7.4.1. Relevant information in the database ............................................................................................. 48 7.4.2. Need for procedures for compiling, making accessible and updating information in the database. 49 7.4.3. Impact of Measure B ...................................................................................................................... 50
8. Measure C: Environmental aspects of the assessment of risks and benefits of medicinal products...........................................................................................................................................52
8.1. Previous studies pertinent to Measure C .................................................................................................... 52 8.2. Limitations to Measure C ........................................................................................................................... 52 8.3. Background to Measure C .......................................................................................................................... 52 8.4. Courses of action for Measure C ................................................................................................................ 53
8.4.1. Course of Action C1: Environmental data is seen as one factor in the risk-benefit balance .......... 53 8.4.2. Course of action C2: Environmental impact is compared with environmental impact of other medicinal products ....................................................................................................................................... 55 8.4.3. Need for revision of legislation and consequences of changes ...................................................... 57
8.5. Impact of Measure C .................................................................................................................................. 59 9. Measure D: A regulation of minimum requirements for conditions of manufacture for the sale of products in the EU market ..................................................................................................62
9.1. Previous government assignment for Measure D ....................................................................................... 62 9.2. Limitations to measure D ........................................................................................................................... 62 9.3. Background to Measure D .......................................................................................................................... 63
9.3.1. Authorisation requirements for manufacture in the EU ................................................................. 64 9.3.2. Good Manufacturing Practice (GMP) in the legislation ................................................................ 64 9.3.3. Good Manufacturing Practice (GMP) Global agreements ............................................................. 65
9.4. Suggested changes to the regulations ......................................................................................................... 66 9.5. Courses of action for Measure D................................................................................................................ 67
9.5.1. Course of Action D1: In the definition of GMP reference is made to a new EU regulation where emissions of active ingredients are regulated. .............................................................................................. 67 9.5.2. Course of action D2: In the definition of GMP, reference is made to the framework for the Industrial Emissions Directive (IED) where the emission limits of active pharmaceutical ingredients are regulated. ...................................................................................................................................................... 68

Page 5 (96)
9.6. Setting emission limits for the substances to be regulated ......................................................................... 69 9.6.1. Regulation of specific substances .................................................................................................. 70 9.6.2. Regulation with the help of the calculation model ......................................................................... 70 9.6.3. Regulation via maximum level ...................................................................................................... 71
9.7. Impact of Measure D .................................................................................................................................. 71 9.8. Other consequences of the proposal ........................................................................................................... 72
9.8.1. Supervisory control and inspection ................................................................................................ 72 9.8.2. Possibility of revoking authorisation to manufacture..................................................................... 73 9.8.3. Impact of application for marketing authorisation. ........................................................................ 73
10. The National Board of Trade's assessment of the impact of the measures on global trade in medicinal products......................................................................................................................74
10.1. General comments on the global trade in medicinal products ................................................................... 74 10.2. Statistics...................................................................................................................................................... 75
10.2.1. Comment on statistics ............................................................................................................... 77 10.3. WTO agreement on technical trade barriers to trade ................................................................................ 77 10.4. Existing bilateral trade agreements............................................................................................................ 79 10.5. Conclusions on global trade ....................................................................................................................... 79
11. Abbreviations .....................................................................................................................81
13. Definitions...........................................................................................................................82
14. References ...........................................................................................................................83 Appendix: 1 Testing requirements in accordance with guidelines for environmental risk assessment .......................................................................................................................................87 Appendix: 2 The Dental and Pharmaceutical Benefits Agency evaluation of pricing, benefits, cost-effectiveness, and the cost to society of medicinal products under the proposed Measures A-D ......................................................................................................................................................89
1. Aim and outline ......................................................................................................................89 1.1. Limitations .................................................................................................................................................. 89
2. Background - cost and price theory .......................................................................................89
3. Impact of Measure A ..............................................................................................................90
4. Impact of Measure B ..............................................................................................................91
5. Impact of Measure C ..............................................................................................................92 6. Impact of Measure D ..............................................................................................................93
7. Combined assessment of the effects of Measures A, B, C, and D ..........................................95

Page 6 (96)

Page 7 (96)
1. Summary of the report Following Parliament's adoption of milestone targets for the environment and medicinal products, the Government gave the Medical Products Agency the assignment to analyse four measures that might reduce the environmental impact of medicinal products during production and use, and thereby contribute to the attainment of these targets. The Medical Products Agency has identified possible courses of action based on the four measures, and what impact they would have on health and the environment, availability of medicinal products, development of new medicinal products and on the pharmaceutical companies. Under this assignment the Medicinal Products Agency does not recommend any courses of action but rather describes the advantages and disadvantages of each. In the report the Dental and Pharmaceutical Benefits Agency analyses possible impact on price, benefits, and cost effectiveness of medicinal products while the National Board of Trade analyses possible effects of cross-border trade, imports and international trade. The Swedish Environmental Protection Agency and the Swedish Chemicals Agency highlight damage to the environment and to human health caused by medicinal products as a result of their environmental impact and associated costs.
The environmental impact of medicinal products Medicinal products contain biologically active ingredients that sooner or later end up mainly in the water environment. Pharmaceutical substances are discharged into the environment at manufacturing sites and through excretion by patients via wastewater treatment plants. Individual sewage systems, improper disposal of medicinal products and leakage from sewage sludge can also contribute to contamination of the environment. The ability to remove pharmaceutical substances currently varies between wastewater treatment plants but a number of active ingredients are present in the outflow from wastewater treatment facilities. Many substances have been detected in the waterways and even in the open seas. Negative effects on biota caused by exposure to pharmaceutical substances at environmentally relevant concentrations, either as a single ingredient or mixtures of several substances, have been recorded in both field and laboratory studies. The identified effects include the poisoning of birds and reproductive and behavioural disorders in fish and other aquatic organisms. No adverse effects on public health due to the presence of medicinal products in drinking water or in contaminated fish or seafood have been shown. However, levels of pharmaceutical substances in water that are equal to therapeutic doses have been found at production facilities in third countries. The discharge of active pharmaceutical ingredients into the environment is one of the contributors to the spread of antibiotic resistance. In order to predict the long-term effects on human health and the environment, a greater understanding is needed of how pharmaceutical substances interact together and with other substances found in the environment. The Swedish Environmental Protection Agency and the Swedish Chemicals Agency have estimated the financial cost of upgrading sewage treatment plants. However, they found it impossible to calculate the total costs associated with the adverse effects on the environment and public health caused by the environmental impact of medicinal products. Although it is not possible to estimate the overall economic impact, it is known from experience that attempts to restore damaged ecosystems are both complex and costly. There is still a need to develop better tools to analyse how the impact of pharmaceuticals on ecosystems affects human health and the environment.

Page 8 (96)
Medicinal products from a global perspective Medicinal products in Europe are largely regulated by harmonised EU legislation. This includes the rules for the marketing authorisation, the follow up after authorisation, requirements for the manufacture of medicinal products and active ingredients as well as supervision and enforcement of regulatory compliance. Some of the courses of action the MPA has identified in the report would require changes to the EU regulatory framework. Pharmaceutical trade is global and benefits from harmonised international regulations. Therefore, it is recommended to initiate early discussions within relevant contexts regarding international regulations when considering making changes to the EU regulatory framework. Environmental considerations in the application for authorisation of medicinal products for human use Since 2006 there has been a requirement for applicants seeking marketing authorisation for medicinal products for human use to carry out an environmental risk assessment (ERA) for the active pharmaceutical ingredients. The applicant should examine the environmental impact of the medicinal product, submit the assessment of the potential risks and, if relevant, provide specific risk minimisation measures (RMM) aimed at reducing the estimated impact of the medicinal product on the environment. However, the identified environmental risks are not considered in the evaluation of the product’s risk-benefit balance and therefore have no decisive significance for the approval of new medicinal products. At present, environmental risk assessments are not collected in any standardised way in Sweden or within the EU. They are not generally accessible or searchable in a database. The same applies to Risk Minimisation Measures that have been identified. Also, the ERA data produced are not used in any analysis to get an overall picture of the environmental impact of active pharmaceutical ingredients. In certain cases, parts of the environmental information are published in the Public Assessment Report (PAR), whilst other patient information frequently only contains a standard text regarding the destruction of left-over medicinal products.
The four measures The aim of this governmental assignment is to identify and analyse different courses of action for the following measures in order to reach the milestone target for greater environmental considerations within pharmaceutical legislation0F
1:
Measure A: testing requirements for environmental risk assessments, Measure B: collection and accessibility of environmental data regarding active pharmaceutical ingredients, Measure C: environmental aspects of the assessment of the benefits and risks of medicinal products, and Measure D: emission limits for manufacturing of medicinal products.
The objective of the measures is to reduce the environmental impact caused by manufacture and use of medicinal products. Measures A and B aim to increase the knowledge about the environmental impact of pharmaceuticals. Measure C aims to limit the consequences following the use of environmentally dangerous medicinal products, and Measure D aims at limiting discharges and emissions into the environment of pharmaceutical substances from production sites both within and outside the EU.
1 The measures are part of a milestone target for medicinal products that the Riksdag adopted in 2014 as part of Sweden's policy on chemicals.

Page 9 (96)
Picture 1: Objectives of the respective measures and how they interact While these measures can be viewed individually it is important to recognise the strong links between them. Some of the measures are contingent for their implementation upon other measures being taken before or simultaneously. For example, information about a substance’s environmental impact must first be available in order to develop appropriate risk minimisation measures or calculate emission limits. Several international activities have been initiated which may concern the measures described in this report. For example, the EU Commission is working on a new strategic approach to medicinal products and the environment, and changes in the regulatory framework for veterinary medicinal products are being discussed. The identified courses of action can be implemented individually but they can also be combined, depending on the effect desired. Primarily, the impact on marketing authorisations of new medicinal products for human use has been analysed. The courses of action have been specified in general terms. In order to become more specific, the development of praxis, primarily by EU working groups, is required. Therefore, it is not yet possible to give a more defined description of the identified courses of action and their expected effects.
Measure A. Provide relevant environmental data Currently, the procedure for environmental risk assessments cannot be regarded as optimal since it is based on the procedure used for environmental risk assessments for industrial chemicals. Pharmaceutical substances, however, often possess different properties compared to such chemicals. At the EU-level, discussions are already ongoing regarding revising the guidelines for environmental risk assessment for medicinal products. Under current guidelines, the requirements for studies that form the basis for an environmental risk assessment are restricted if the expected concentration of a compound in water is below a certain concentration or if its ability to accumulate in fatty tissue is considered to be low. Two courses of action with the aim of providing better information about the environmental impact of pharmaceutical substances have been identified:
1. Review current limitations such that more pharmaceutical substances are covered by requirements for environmental risk assessment.
2. Develop the testing requirements for environmental risk assessments to better reflect the specific properties of medicinal products.

Page 10 (96)
These courses of action can also be combined. The result would be that more substances would be subject to an environmental risk assessment and that their environmental impact would be evaluated by more relevant studies. The proposal is a natural development of the ERA guidelines document. Consequently, it can be assumed that the proposed courses of action would have a limited effect on the development of new medicinal products or availability of medicines. More relevant testing requirements may make ERAs somewhat more expensive, even if the new studies would replace existing, less specific tests. There is also a risk that the new testing requirements might prolong application preparation time, but the risk might be reduced by running environmental studies in parallel with other studies. Because many older pharmaceutical substances lack environmental risk assessments, possible ways assessments might be made for these substances should be investigated. Relevant ERAs provide knowledge of the environmental impact of active pharmaceutical ingredients. This information is needed in order to guarantee proper use and development of appropriate risk minimisation measures. When implemented these measures will decrease negative environmental impact as well as contribute to better public health.
Measure B. Make the data on environmental impact of medicinal products accessible Environmental information produced within the process of applying for marketing authorisation is currently only accessible to a limited extent to the stakeholders who work with reduction of environmental impact of pharmaceuticals. As previously mentioned, there is no centralised collection of information that would make the data readily available and the information which does exist is not generally accessible. Consultation within the framework of this report has shown that there are many stakeholders who have a need for this information. Such stakeholders include water companies, wastewater treatment plants, environmental authorities responsible for the control and reduction of water pollution, authorities responsible for drinking water quality, health authorities and county councils. According to the governmental assignment, a database with information on the environmental impact of active pharmaceutical ingredients should be built and managed by the EMA, which is why the course of action for this measure is defined within the assignment. Several issues have been identified that need further investigation before a database can be put into operation. The following needs are highlighted in the report:
1. The database must contain relevant information. 2. Procedures are needed for compiling and updating information in the database, as well as for
making it accessible. Current pharmaceutical legislation needs to be revised to forge a common EU requirement regarding what environmental data should be collected and made accessible. A process for managing the collection and quality assurance of data would also need to be developed. The information should be linked to the specific substances and based on data from environmental risk assessments for medicinal products for both human and veterinary use. There also need to be requirements that the information is kept up-to-date and is updated as and when new information or scientific studies appear. In addition, there are information gaps on the environmental impact of existing active pharmaceutical ingredients, which makes the database incomplete. It is therefore important to start the work of acquiring this missing information.

Page 11 (96)
Collecting and making the environmental data accessible constitutes the basis for the development of appropriate risk minimisation measures, and facilitates overall analyses of the environmental impact of pharmaceutical substances.
Measure C. Consider environmental risk in the risk-benefit assessment of new medicinal products Under current pharmaceutical legislation, a medicinal product for human use can receive a marketing authorisation if it is considered to have a positive therapeutic effect in relation to the risks its use might have on a patient’s health or public health. The decision is based on a risk-benefit assessment of the product regarding its quality, safety and efficacy. The goal of Measure C is to include environmental considerations in the evaluation of the risk-benefit balance for medicinal products. This would require changes to legislation. Two different courses of action have been identified:
1. Environmental data is seen as a factor in the risk-benefit assessment. 2. The environmental impact of a new medicinal product is compared to the environmental
impact of existing authorised medicinal products. Course of Action 1 entails the environmental impact of a medicinal product being factored into the risk-benefit assessment along with other elements, namely quality, efficacy and safety. This approach is already applied in the legislation concerning veterinary medicinal products. If the use of a medicinal product leads to environmental risks then appropriate risk minimisation measures are identified and established. If the clinical risk-benefit balance were favourable for a new medicinal product, especially if it were to be used to treat a serious illness, or if there were only limited treatment alternatives, an application for authorisation of such a medicinal product would probably not be rejected on the grounds of environmental considerations. Risk minimisation measures would be implemented as far as possible instead, in order to minimise risk to the environment. If, on the other hand, the risk-benefit balance were to be challenged based on available data regarding quality, efficacy or safety, a serious environmental risk which has been identified, together with other risk factors might contribute to the application for authorisation being rejected. In rare cases, an application for marketing authorisation could be rejected solely based on environmental risk. Such a rejection would only be possible in cases when there are no appropriate measures available to minimise a serious environmental risk and when other medicinal products or medical treatments are in place to offer adequate treatment. Facilitating implementation of the course of action such that environmental risk is seen as a factor in the risk-benefit assessment requires not only changes to current legislation but also development of a common EU praxis regarding how environmental risk should be factored into the risk-benefit assessment. Should environmental considerations be taken into account each time a risk to the environment is identified, or only when the risk could lead to potentially serious effects for the environment or public health? Equally, there is a need to develop new appropriate risk minimisation measures, ranging from the restriction of indications and the restriction of use to selected patient groups and classifying the medicinal product as prescription only. The need for risk minimisation measures is also likely to be dependent on available wastewater treatment techniques. Course of Action 2 is a proposal that originally arose from previous investigations in this field. Under this course of action, the environmental risk of a new medicinal product should be compared to that of authorised medicinal products fulfilling the same medical need. If the use of the new medicinal

Page 12 (96)
product, after applying risk minimisation measures, is still determined to have a greater negative environmental impact than the comparable medicinal products, the application for marketing authorisation may be rejected. This Course of Action presumes that it is possible to select medicinal products that fulfil the same medical need, i.e. it is possible to compare different products’ therapeutic effects, and that environmental data for these medicinal products is available. Furthermore, Course of Action 2 presumes that there are procedures in place that allow for a comparison of the environmental impact of various medicinal products. At present, the comparisons of therapeutic effects of different medicinal products are made at the national level. One group doing this is the Drug and Therapeutics Committees working at the Swedish County Councils. However, their comparisons are made only for medicinal products which have been authorised and the procedure cannot exclude any medicinal product from the market. The total environmental risk of a medicinal product has different significance in different situations since this course of action is based on comparisons within similar therapeutic groups. Two substances with "the same" total environmental risk ought to be able to be dealt with in a different way, depending on if the comparison groups contain medicinal products which impose a heavy or light burden on the environment. Both courses of action would bring a new perspective to risk-benefit assessment since, in addition to the patient’s health, environmental concerns would also be factored in. This in turn would increase the complexity of the evaluations. Regardless of the course of action, the inclusion of environmental considerations in the risk-benefit assessment would probably entail the following major actions: revision of the guidelines for environmental risk assessments, better implementation of the evaluation of environmental risk assessments, making environmental information accessible and also including environmental considerations in post-marketing control. These courses of action may result in reduced availability of medicinal products even if risk minimisation measures are applied first. This proposal would most likely only result in rejection in exceptional cases where adequate treatments were guaranteed by existing medicinal products. The introduction of environmental considerations into evaluation of risk-benefit assessments would give environmental issues a higher priority in the application process. This in turn, would lead to better environmental risk assessments and well-formulated risk minimisation measures. In the long term this would lead to a situation whereby, when being granted a marketing authorisation, each new medicinal product would already have a plan for how its environmental impact could be minimised.
Measure D. Restrict emissions of active ingredients from pharmaceutical manufacturing sites Discharges and emissions of active ingredients from the manufacture of pharmaceuticals could cause considerable impact on the local environment. When considering antibiotics, discharges and emissions may contribute to growing antibiotic resistance, which is a major threat to global public health. The overall objective of Swedish environmental policy is to leave to the next generation a society in which the major environmental problems have been resolved without causing environmental and health problems beyond Sweden’s borders. This requires to be taken at the international level. Measure D is built on previous proposals to introduce environmental requirements within the framework of European pharmaceutical legislation. According to the proposal, within the framework of good manufacturing practices (GMP), there would be requirements imposed on those who manufacture medicinal products to comply with specific requirements limiting emissions of active ingredients. It is also proposed to amend the regulatory framework so that the definition of GMP is

Page 13 (96)
moved to the Human Medicinal Products Directive1F
2 and the Veterinary Medicinal Products Directive2 F
3. Two different courses of action have been identified:
1. In the definition of GMP reference is made to a new EU regulation where emissions of active ingredients are regulated.
2. In the definition of GMP reference is made to a regulation within the framework of the Industrial Emissions Directive3 F
4 (IED) where emissions of active ingredients are regulated. The report identifies three methods for setting emission limits:
• regulation of specific substances, • adoption of a computational model that could be applied to all substances, or • establishment of a common emission limit for all substances.
The difference between the courses of action is that in Course of Action 1, any of the methods specified above could be applied; one of them alone or in combination. According to the MPA, the most viable method for Course of Action 2 is to regulate specific substances. Both courses of action require a decision on how the control of emission levels should be formulated. Implementing Measure D would result in a genuine reduction of emissions of active pharmaceutical ingredients into the environment, thus having a positive impact on both the environment and human health, regardless of where in the world the manufacturing facility it is located.
Effects on price, benefits and cost-effectiveness of medicinal products According to the Dental and Pharmaceutical Benefits Agency’s assessment of the effects on medicinal products prices, benefits and cost-effectiveness, the initial outcome of implementing all the measures would be unlikely to have a direct influence on prices of medicinal products. However, fewer authorised medicinal products combined with risk minimisation measures would decrease competition and indirectly affect prices. The greatest impact of the measures would arguably be that benefits to patients would decrease because fewer medicinal products would be authorised and risk minimisation measures would further restrict their use. In the long term, patient benefits would also be affected by fewer incentives to develop new medicinal products and the chance that research resources would be reallocated to other pharmaceutical fields. However, it is difficult to quantify the effect on reduced patient benefits. This is because of the current uncertainty about factors such as the scale of costs for environmental testing and risk minimisation measures. This also means that it is difficult to gauge the scale of the various effects in relation to one another. It is reasonable to believe the effect on patient benefits from Measure A would be marginal. Measure D should not significantly affect patient benefits either. The greatest impact is likely to come from Measure C but as mentioned, the extent depends on how risk minimisation measures are formulated. The effects on other benefits to society when lowering environmental impact and the effects on the social costs arising from the environmental impact of medicinal products were not taken into account in the Dental and Pharmaceutical Benefits Agency analysis. The findings from the Dental and
2 Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. 3 Directive 2001/82/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to veterinary medicinal products. 4 Directive 2010/75/EU of the European Parliament and of the Council of 24 November 2010 on industrial emissions (integrated pollution prevention and control).

Page 14 (96)
Pharmaceutical Benefits Agency section of the report should be used together with estimates of the effects on other benefits and costs to society when drawing conclusions about whether the measures are justified from a social perspective or not.
Impact on global pharmaceuticals trade According to the National Board of Trade, there is a likelihood that Measure D (emission limits for manufacture of medicinal products) would have a greater impact on the international trading system than the other proposed measures. Measures stemming from GMP and GMP inspections may negatively impact existing international cooperation if commitments are not met due to the introduction of additional requirements. There might, therefore, be reason to argue for the implementation of the proposed measures being conducted through a comprehensive harmonisation of regulations, e.g. within the framework of existing international collaboration networks, the EU might undertake measures to influence developments in the direction of increased environmental considerations in GMP. It should also be noted that Measure C (environmental aspects included in the evaluation of the risk-benefit balance of medicinal products) could have an effect on trade with third countries as its outcome may increase the requirements. Measure A (environmental risk assessments) and B (collecting and making environmental data accessible) seem to entail the lowest impact under current trade regulations. In addition to the impact on trade in terms of commitments to the WTO and other countries that have signed trade agreements with the EU, it might be worthwhile discussing how the various measures might affect trading conditions in terms of actual trade. Considering the large number of imports into the EU, it is likely that many of the stakeholders in third countries who are trading with the EU would be affected by the measures. How and in which capacity is difficult to estimate in the context of this analysis. However, it may be worth taking into account how the perceived elevated safety levels in the EU may affect existing trade flows between the EU and third countries. A consequence could be that companies may choose to export their medicinal products to other markets with less restrictive market requirements. This is where the free trade negotiations with the United States ought to be considered given that the United States is the world's second largest pharmaceutical importer after the EU. In view of this argument as well as from a trade perspective, there are compelling reasons to examine the opportunities to establish equivalent environmental objectives or to extend the proposed measures through broad regulatory solutions that include more stakeholders than only the EU.

Page 15 (96)
2. The assignment On 16 April 2014 (S2014/3530/FS4F
5) the government assigned the Medical Products Agency (MPA) with the task of carrying out an in-depth analysis of various alternative courses of action aimed at achieving the milestone target for environmental considerations in medicinal product legislation within the EU and internationally. Government decision: The Government assigned the Medical Products Agency the task of producing, in collaboration with the Dental and Pharmaceutical Benefits Agency, an in-depth report to help formulate the basis of the Government's approach to how the environmental quality objective milestone of a “poison-free environment” (introducing expanded environmental considerations into medicinal product legislation) shall be driven forward within the EU and internationally. The assignment involves identifying different courses of action and analysing what impact these courses of action would have on health and the environment and on availability of medicinal products in addition to the costs to society of medicinal products. The assignment shall include the following elements in order to achieve the milestone target: a) testing requirements for medicinal products and the environmental risk assessments carried out in conjunction with an application for authorisation of a medicinal product, b) collection of environmental data regarding active pharmaceutical ingredients and making this accessible, c) environmental aspects of the assessment of the risks and benefits of medicinal products, and d) regulation of minimum requirements regarding conditions of production for the sale of products on the EU-market. The analysis should place a particular focus on the possible consequences of point c. The analysis of the consequences of alternative courses of action should be based on a broad analysis of the needs for medicinal products within health and medical care. However, particular stress should be placed on the following areas:
-the impact on the development of new medicinal products -the impact on availability of medicinal products, - the impact on the price and benefits of medicinal products, - the impact on the cost-effectiveness of medicinal products, and - the impact on the pharmaceutical companies.
The assignment includes highlighting the effects of different courses of action on human health and the environment. As a part of the analysis the authorities will provide an overview of what damage may be caused to the environment by medicinal products as well as potential negative impact on human health. The review should include harm to nature and its animal life, and to the health of humans. The scale of the cost to society of this damage, including disruption of the drinking water supply, should be estimated. The analysis of the consequences of various alternative courses of action should be based on Swedish conditions while being set in an EU and international perspective. The assignment should also highlight potential impacts on cross-border trade and imports and potential interaction with international commitments in the business area.
5 The Ministry of Health and Social Affairs. Assignment regarding in-depth analysis of various alternative courses of action aimed at achieving the milestone target for environmental considerations in medicinal product legislation within the EU and internationally. S2014/3530/FS Search: http://www.regeringen.se

Page 16 (96)
The Medical Products Agency will assume overall responsibility for the assignment. The Dental and Pharmaceutical Benefits Agency shall assume responsibility for the assignment in those parts which concern pricing and benefits of medicinal products, cost-effectiveness and cost to society of medicinal products. The Medical Products Agency shall hold dialogues with concerned parties at the national, EU and international levels during the course of the assignment. The assignment shall be carried out in collaboration with the Swedish Environmental Protection Agency and the Swedish Chemicals Agency in those parts which concern what harm to the environment and human health medicinal products give rise to and the costs of such. The assignment shall be carried out in conjunction with the Swedish National Board of Health and Welfare and the Swedish National Board of Trade. In addition, the Medical Products Agency shall collaborate with the Swedish Association of Local Authorities and Regions, representatives of patient organisations and trade unions, Svenskt Vatten, the Swedish Association of the Pharmaceutical Industry, the Association for Generic Pharmaceuticals in Sweden and the Swedish Association of Pharma Traders. The review should contain a summary in English. The outcome of this assignment is to be submitted to the Swedish Government Offices (Department of Health) by latest December 31, 2014 under the reference number of this decision.
2.1. Limitations of the assignment Harmonisation of the limits of the assignment has been carried with the Department of Health and the Department of Environment on two separate occasions. – The focus of the assignment is to analyse the emission of active pharmaceutical ingredients into
the environment as a result of consumption or production, but generally not as a result of destruction or disposal.
– The report will take into account medicinal products and active pharmaceutical ingredients as these are covered by medicinal product legislation. Other substances and solvents used in the manufacture of medicinal products are principally dealt with in chemicals legislation.
– The possibility of comparative assessment of environmental risks from medicinal products at times other than when authorisation is given are not analysed.
– Previously authorised medicinal products and veterinary medicinal products are generally not covered. Some are covered, however, in certain of the measures. See limitations under the respective measures.
– The Medical Products Agency or the collaborating partners shall not recommend any of the alternative courses of action.
– The English summary of the report shall be added after the report has been delivered to the Department, i.e. in January 2015.
– The report from the Dental and Pharmaceutical Benefits Agency will review possible tendencies for how the price, benefits and cost-effectiveness of medicinal products might be influenced by the proposed measures. The aim is not to present exact figures and costs
2.2. Consultation The Medical Products Agency (MPA) has overall responsibility for the assignment. The Dental and Pharmaceutical Benefits Agency has responsibility for the parts of the assignment connected to pricing and benefits of medicinal products, cost-effectiveness, and the cost to society of medicinal products. Those parts of the assignment related to what damage medicinal products might cause to the environment and to human health and the cost of this damage were carried out in collaboration with

Page 17 (96)
the Swedish Environmental Protection Agency, and the Swedish Chemicals Agency. In addition, the National Board of Trade has highlighted potential effects on cross-border trade and imports and possible interaction with international trade and commerce commitments. Regular harmonisation meetings have been held. In line with the assignment these authorities contributed the analyses requested from them, see chapters 3,0 and Appendix 2. There was also consultation with the National Board of Health and Welfare Meetings with LIF (the Swedish Association of the Pharmaceutical Industry) and LOK (the board of chairpersons of the Drug and Therapeutics Committees) were held, as were discussions with Svenskt Vatten and MistraPharma. Coordination meetings were held to which the above organisations and other authorities, (the National Food Agency, the Swedish Agency for Marine and Water Management, the Swedish Consumer Agency, the Public Health Agency, professional and trade organisations SLS (the Swedish teachers Association), Läkare för miljön (Doctors for the Environment) and the Swedish Pharmaceutical Society, the Swedish Association of Local Authorities and Regions plus all the patient organisations were invited. The Swedish Association of Pharma Traders, the Swedish Association of the Pharmaceutical Industry, and the Association for Generic Pharmaceuticals in Sweden were also invited. Of those invited representatives from the Dental and Pharmaceutical Benefits Agency, the Swedish Environmental Protection Agency, the Swedish Association of the Pharmaceutical Industry, LOK (see above), Svenskt Vatten, Mistra Pharma, the National Food Agency, the Public Health Agency, Läkare för miljön, the Swedish Pharmaceutical Society, the Swedish Association of Local Authorities and Regions, the Swedish Association of Pharma Traders, the Swedish Association of the Pharmaceutical Industry, the Association for Generic Pharmaceuticals in Sweden and one of the patient organisations (Willis-Ekbom Disease förbundet) attended. The assignment was also presented on county council and municipality environment days. DG Sanco and DG Environment as well as the Swedish permanent representation in Brussels were also informed about the assignment. The Medicinal Products Agency presented the fundamentals of the assignment at a workshop in Brussels arranged by EFPIA and Health Care Without Harm. Internally at MPA the progress of the project has been regularly run past a reference group with the aim of ensuring diversity in the assignment.
2.3. Ongoing international activities In the area of medicinal products and the environment a number of activities are being carried out which have a link to the measures in this report. Below are listed activities which should be monitored as their outcomes could have an impact on the ability to design and implement the measures. • EU water policy framework directive5 F
6. This work includes the creation of environmental quality norms for substances which are thought to contribute to water pollution. Three pharmaceutical substances have already been included in a monitor list and one further substance has been suggested for inclusion.
6 Directive 2000/60/EC of the European Parliament and of the Council of 23 October 2000 establishing a framework for Community action in the field of water policy, amended by Directive 2008/105/EC of the European Parliament and of the Council of 16 December 2008 on environmental quality standards in the field of water policy, amending and subsequently repealing Council Directives 82/176/EEC, 83/513/EEC, 84/156/EEC, 84/491/EEC, 86/280/EEC and amending Directive 2000/60/EC of the European Parliament and of the Council.

Page 18 (96)
• Strategy to counteract contamination of water by medicinal products. Directive 2013/39/EU as regards priority substances in the field of water policy6F
7 stipulates that the Commission shall by latest 2015 come back with a "strategic approach to the contamination of water by pharmaceuticals". A knowledge-gathering initiative was established at the behest of GD Health and Consumer Issues (DG Sanco) prior to the beginning of the strategy work (Bio Intelligence Service, 2013). The report sets out the areas of measures this initiative is aimed at. The formulation of the Commission’s strategy will be crucial in the ability to implement the Swedish measures.
• "EcoRiskPrediction" In the autumn of 2013 the Commission and the Innovative Medicine Initiative (IMI) put out a call for "EcoRiskPrediction", whose aim is to develop new methods of assessing the environmental impact of pharmaceuticals7 F
8. These new methods may influence the design of environmental risk assessments in the future (compare Measure A). The consortium who won the call will in all probability begin their project in January 2015. The project will last for four years.
• Proposal for EU legislation for veterinary medicinal products. Veterinary medicinal products are not part of the brief of this report, but the Commission's proposal for new EU legislation8F
9 is important, as one of its proposals is a database of veterinary medicinal products. This would have an influence on the formulation and implementation of requirements concerning the ways in which environmental data is collected and made accessible (compare Measure B). Within the framework for this continuing work on legislation, minimum requirements for production may also be raised (compare Measure D).
2.4. The structure of the report The report begins with the background, summarising studies about harm to the environment and human health that medicinal products give rise to and providing a broad estimate of the costs associated with this. Chapter 3 was written by the Swedish Environmental Protection Agency and the Swedish Chemicals Agency. After a short description of the regulatory framework for medicinal products from an environmental perspective there is an analysis by the Medical Products Agency of measures A-D (chapters 4-9). In addition to the courses of action, this section also discusses the general impact, the impact on pharmaceuticals companies and on the development and availability of medicinal products as well as the consequences for human health and environment that the courses of action would give rise to. The National Board of Trade highlights the impact on cross-border trade and commerce and imports and the interaction with international commitments in the trade and commerce field (chapter 10). Finally, based on the above analyses, the Dental and Pharmaceutical Benefits Agency describes the impact of the measures on the price of medicinal products, benefits to patients and cost-effectiveness (appendix 2).
7 Directive 2013/39/EU of the European Parliament and of the Council of 12 August 2013 amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the field of water policy Text with EEA relevance. 8 EcoRiskPrediction, IMI's 11th call webinars, http://www.imi.europa.eu 9 COM(2014) 558 final, 2014/0257 (COD).

Page 19 (96)
3. Assessment by the Swedish Environmental Protection Agency and the Swedish Chemicals Agency of damage to the environment and to human health that medicinal products give rise to and the associated costs
One requirement of the assignment is for the Swedish Environmental Protection Agency and the Swedish Chemicals Agency to highlight the impact of different courses of action on health and the environment. This should include an overview of what environmental damage pharmaceutical substances could give rise to, as well as their negative impact on human health. The extent of the costs to society of this damage, including disruption of the drinking water supply, should also be estimated, if possible.
3.1. Emission of pharmaceutical ingredients into the environment Active ingredients in medicinal products for human use reach the environment via emissions from production plants, after we have consumed them, when they, intact or as metabolites, are expelled by urine and faeces and end up in our sewage systems as well as when they end up in our refuse. Our towns and cities are quantitatively the main source of medicinal products in the environment. They most often reach the environment via treatment plants and individual sewage systems. In treatment plants downstream of towns and cities substances can meet a variety of fates. To a certain extent most substances are broken down or altered into different products which can sometimes give rise to similar effects on the environment as the original substance or have another effect. Some pass through the treatment plant intact, however. The remaining ingredients finish up in the purified outflow wastewater (surface water), or in the sludge. Some of the sludge it is spread on arable land.
Picture 2: The flow of medicinal products for human use into the environment (from Larsson and Lööf, 2014) Note: By "watercourse" in the figure is meant both surface water and groundwater.
Page 20 (96)
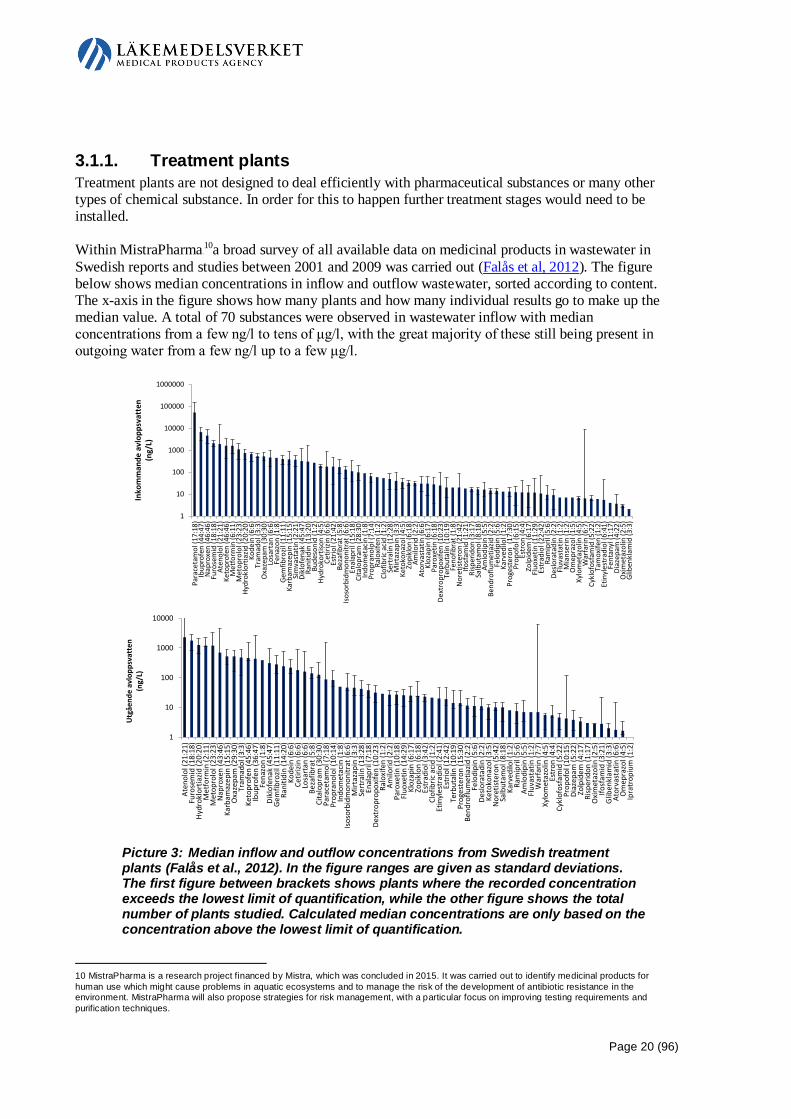
3.1.1. Treatment plants Treatment plants are not designed to deal efficiently with pharmaceutical substances or many other types of chemical substance. In order for this to happen further treatment stages would need to be installed. Within MistraPharma9 F
10a broad survey of all available data on medicinal products in wastewater in Swedish reports and studies between 2001 and 2009 was carried out (Falås et al, 2012). The figure below shows median concentrations in inflow and outflow wastewater, sorted according to content. The x-axis in the figure shows how many plants and how many individual results go to make up the median value. A total of 70 substances were observed in wastewater inflow with median concentrations from a few ng/l to tens of μg/l, with the great majority of these still being present in outgoing water from a few ng/l up to a few μg/l.
Picture 3: Median inflow and outflow concentrations from Swedish treatment plants (Falås et al., 2012). In the figure ranges are given as standard deviations. The first figure between brackets shows plants where the recorded concentration exceeds the lowest limit of quantification, while the other figure shows the total number of plants studied. Calculated median concentrations are only based on the concentration above the lowest limit of quantification.
10 MistraPharma is a research project financed by Mistra, which was concluded in 2015. It was carried out to identify medicinal products for human use which might cause problems in aquatic ecosystems and to manage the risk of the development of antibiotic resistance in the environment. MistraPharma will also propose strategies for risk management, with a particular focus on improving testing requirements and purification techniques.
1
10
100
1000
10000
100000
1000000
Para
ceta
mol
(17:
18)
Ibup
rofe
n (4
4:47
)N
apro
xen
(46:
46)
Furo
sem
id (1
8:18
)At
enol
ol (2
1:21
)Ke
topr
ofen
(46:
46)
Met
form
in (6
:11)
Met
opro
lol (
23:2
3)Hy
drok
lort
iazid
(20:
20)
Kode
in (6
:6)
Tram
adol
(3:3
)O
xaze
pam
(30:
30)
Losa
rtan
(6:6
)Fe
nazo
n (1
:8)
Gem
fibro
zil (1
1:11
)Ka
rbam
azep
in (1
5:15
)Si
mva
stat
in (2
:21)
Dikl
ofen
ak (4
5:47
)Ra
nitid
in (1
3:20
)Bu
deso
nid
(1:2
)Hy
drok
ortis
on (4
:5)
Cetir
izin
(6:6
)Es
trio
l (21
:42)
Beza
fibra
t (5:
8)Is
osor
bidm
onon
itrat
(6:6
)En
alap
ril (1
5:18
)Ci
talo
pram
(28:
30)
Indo
met
acin
(1:8
)Pr
opra
nolo
l (7:
14)
Ralo
xife
n (1
:2)
Clof
ibric
aci
d (1
:2)
Sert
ralin
(12:
28)
Mirt
azap
in (3
:3)
Keto
kona
zol (
4:5)
Zopi
klon
(6:1
8)Am
ilorid
(2:2
)At
orva
stat
in (6
:6)
Kloz
apin
(6:1
7)Pa
roxe
tin (6
:18)
Dext
ropr
opox
ifen
(10:
23)
Terb
utal
in (1
0:19
)Fe
nofib
rat (
1:8)
Nor
etist
eron
(21:
42)
Ifosf
amid
(1:2
1)Ri
sper
idon
(3:1
7)Sa
lbut
amol
(8:1
8)Am
lodi
pin
(5:5
)Be
ndro
flum
etia
zid (2
:2)
Felo
dipi
n (5
:6)
Karv
edilo
l (1:
2)Pr
oges
tero
n (1
7:30
)Pr
opof
ol (6
:15)
Estr
on (4
:4)
Zolp
idem
(6:1
7)Fl
uoxe
tin (1
1:29
)Es
trad
iol (
22:4
2)Ra
mip
ril (5
:6)
Deslo
rata
din
(2:2
)Fl
uvas
tatin
(1:2
)M
ians
erin
(1:2
)O
mep
razo
l (1:
5)Xy
lom
etaz
olin
(4:5
)W
arfa
rin (6
:7)
Cykl
ofos
fam
id (5
:22)
Tam
oxife
n (1
:2)
Etin
yles
trad
iol (
6:41
)Fe
ntan
yl (1
:17)
Diaz
epam
(4:2
2)O
xim
etaz
olin
(2:5
)G
liben
klam
id (3
:3)
Inko
mm
ande
avl
opps
vatt
en(n
g/L)
1
10
100
1000
10000
Aten
olol
(21:
21)
Furo
sem
id (1
8:18
)Hy
drok
lort
iazid
(20:
20)
Met
form
in (2
:11)
Met
opro
lol (
23:2
3)N
apro
xen
(43:
46)
Karb
amaz
epin
(15:
15)
Oxa
zepa
m (2
9:30
)Tr
amad
ol (3
:3)
Keto
prof
en (4
5:46
)Ib
upro
fen
(36:
47)
Fena
zon
(1:8
)Di
klof
enak
(45:
47)
Gem
fibro
zil (1
1:11
)Ra
nitid
in (1
4:20
)Ko
dein
(6:6
)Ce
tirizi
n (6
:6)
Losa
rtan
(6:6
)Be
zafib
rat (
5:8)
Cita
lopr
am (3
0:30
)Pa
race
tam
ol (7
:18)
Prop
rano
lol (
10:1
4)In
dom
etac
in (1
:8)
Isos
orbi
dmon
onitr
at (6
:6)
Mirt
azap
in (3
:3)
Sert
ralin
(13:
28)
Enal
april
(7:1
8)De
xtro
prop
oxife
n (1
0:23
)Ra
loxi
fen
(1:2
)Am
ilorid
(2:2
)Pa
roxe
tin (1
0:18
)Fl
uoxe
tin (1
4:29
)Kl
ozap
in (6
:17)
Zopi
klon
(6:1
8)Es
trad
iol (
3:42
)Cl
ofib
ric a
cid
(1:2
)Et
inyl
estr
adio
l (2:
41)
Estr
iol (
12:4
2)Te
rbut
alin
(10:
19)
Prog
este
ron
(15:
30)
Bend
roflu
met
iazid
(2:2
)Fe
lodi
pin
(5:6
)De
slora
tadi
n (2
:2)
Keto
kona
zol (
3:5)
Nor
etist
eron
(5:4
2)Sa
lbut
amol
(8:1
8)Ka
rved
ilol (
1:2)
Ram
ipril
(5:6
)Am
lodi
pin
(5:5
)Fl
uvas
tatin
(1:2
)W
arfa
rin (7
:7)
Xylo
met
azol
in (4
:5)
Estr
on (4
:4)
Cykl
ofos
fam
id (5
:22)
Prop
ofol
(10:
15)
Diaz
epam
(5:2
2)Zo
lpid
em (4
:17)
Risp
erid
on (1
:17)
Oxi
met
azol
in (2
:5)
Ifosf
amid
(3:2
1)G
liben
klam
id (3
:3)
Ator
vast
atin
(6:6
)O
mep
razo
l (4:
5)Ip
ratr
opiu
m (1
:2)
Utg
åend
e av
lopp
svat
ten
(ng/
L)

Page 21 (96)
There are a number of studies of the amounts of pharmaceutical substances released into the environment via treatment plants. Given here is one example of a study which also shows in a representative way the volumes being released. In a screening study carried out in 2010 a total of 101 pharmaceutical substances were measured. A total of 121 samples from different treatment plants were included in the study. Of the 101 substances included in the study 92 were detected in inflow wastewater in at least one waste treatment plant, with concentrations from low ng/l up to 540 ng/l. In outflow wastewater 85 of the substances were detected in at least one treatment plant in quantities from low ng/l up to 4 µg/l10 F
11. Median concentration was 35 ng/l. In the sludge 73 of the substances studied were detected. Content varied from a few ng/kg up to mg/kg levels, with a median content of 20 µg/kg. What proportion of the pharmaceutical substances is extracted in the treatment plant varies greatly, depending on what the substances is – from 0 to 100% disappears depending on the substance (Fick et al, 2011). This study demonstrated pharmaceutical substances at a total level of 58 µg/l in the outflow from the Henriksdal treatment plant in Stockholm, 74 µg/l in the outflow from the treatment plant in Umeå and 58 µg/l in the outflow from the treatment plant in Skövde. Equivalent total content levels for sludge were 5800 µg/kg in Stockholm, 5500 µg/kg in Umeå and 4600 µg/kg in Skövde. These totals refer to the total content of substance above the limit of detection. The following example calculation may give an idea of the scale of the amounts of pharmaceutical substances released into the environment in towns and cities via our sewage treatment plants. Taking the rough estimate that 1.2 billion m³ water is released from sewage treatment plants in Sweden, an average content of 58 µg/l in all outgoing water would mean emissions of 70 metric tons of pharmaceutical substances per year from outflow from Swedish sewage treatment plants. If we then take a rough estimate that 200,000 tons of sludge are produced per year, then, for example, a total content of pharmaceutical substances of 5800 µg/kg would mean that an annual total of 1200 kg of pharmaceutical substances are emitted via all the sludge which is produced11 F
12. About 25% of this is spread on our agricultural fields. (Statistiska meddelanden, 2012). The degree of magnitude is also in line with data from European studies, for example the EU coordinated FATE-study of measures of chemicals in outgoing water from treatment plants (JRC, 2012). A larger number of samples was analysed for, among other things, the presence of pharmaceutical substances. In many cases the content is also higher than that found in Sweden. Pharmaceutical substances were present in most of the samples. The highest median content levels found were for example carbamazepine (752 ng/l), tramadol ((218 ng/l), venlafaxine (97 ng/l), irbesartan (85 ng/l), ciprofloxacin (82 ng/l), oxazepam (64 ng/l), fexofenadine (59 ng/l), clindamycin (46 ng/l), caffeine (35 ng/l), citalopram (21 ng/l), and codeine (21 ng/l).
3.1.2. Individual sewage systems A number of projects have been carried out to see if and in what amounts pharmaceutical substances are emitted from individual sewage systems. In Sweden there are in the region 700,000 properties with individual sewage systems, of which approximately 75% consist of ground-based installations where
11 Higher levels of an active pharmaceutical ingredient may sometimes be measured in the outflow from treatment plants than in its incoming water. One explanation for this may be that metabolites, transformation products from medicinal products created and expelled from human bodies, are transformed back into the original form of the medicinal product during the processes in the treatment plant. 12 The 200,000 tons of sludge per year refers to DM (dry matter). The analyses in the screening study are expressed in X and refer to dewatered sludge. Depending on the technique used the dewatered sludge may vary from 3%-4% DM to around perhaps 30% DM (when there is a high DM content it is usually called dry sludge). This has not been taken into account in the calculation, which means that the amounts of pharmaceutical substances in the sludge are in all probability significantly higher. It depends on how "dewatered" the sludge was.

Page 22 (96)
the purified wastewater reaches surface water receptacles either directly or via the groundwater. What is released from individual waste systems depends very much on who is using the property. In another study it was estimated that approximately 10 times more carbamazepine (for epilepsy) and diclofenac (anti-inflammatory) were released from individual sewage systems throughout Sweden than from Henriksdal treatment plant. Henriksdal treatment plant is the largest in Stockholm, treating sewage from approximately 780,000 people. Paracetamol (contained in painkillers) is the substance responsible for the greatest amounts of emissions from individual sewage systems, estimated at approximately 12 metric tons in 2010. After paracetamol come carbamazepine and naproxen (anti-inflammatory) with emissions of 633kg and 377kg respectively (Ejhed et al, 2012). These substances are also among the highest ranking in the broad survey of all available data on pharmaceutical substances in wastewater reported in section 3.1.1 above (Falås et al, 2012) Research initiatives which have been put in place for such things as seeing how emissions from individual sewage systems can be reduced are OPTITREAT1 2F
13 and RED-MIC1 3F
14.
3.1.3. Emissions from production plants There is no legal requirement to measure emissions of pharmaceutical substances from production plants. This means that no statistics have been gathered for emissions, so all that can be done is to give examples of what may be released into the environment and any measurements taken were thus done so on the initiative of the enterprise. Emissions from a Swedish production plant Work has been carried out on "Environmental Reference Concentrations" or "ERC." In this, threshold values were created for pharmaceutical substance emissions in water outside Swedish manufacturing units in order to monitor emissions of pharmaceutical substances. The threshold values of the assessed pharmaceutical substances are thus set such that it appears that concentrations in water are unlikely to have a negative impact on the environment or humans (see, for example Murray-Smith et al, 2012). Adherence to the threshold values is regularly controlled. Emissions from production plants A large number of medicinal products in Europe are manufactured in third countries, often in countries with low wages, in order to make medicines cheaper. However, studies have shown that purification in these factories can be very poor and that enormously high concentrations of pharmaceutical residues are released into nature with severe consequences in the form of impact on the environment and possibly also on human health (Larsson and Lööf, 2014). In certain cases, higher concentrations of the substance have been shown in the water than the concentration in the blood of a patient treated with the relevant product. In the number of countries, emissions of antibiotics have also given rise to increased incidence of resistance factors in bacterial communities downstream from production plants. A compilation with examples of reports of emissions of active pharmaceutical ingredients from production plants can be found in Larsson (2012).
3.1.4. Medicinal products for veterinary purposes A large number of medicinal products are used for both animals and humans. The same pharmaceutical substances that end up in the environment through their use as medicinal products for humans can thus, in many cases, reach the environment through use as veterinary medicinal products. 13 Optitreat (http://www.bonusportal.org/bonus_projects/innovation_projects/optitreat) 14 RED-MIC (http://www.redmic.se/)

Page 23 (96)
One type of plant where veterinary medicinal products are used is in farming of different types of aquatic creatures (fish, mussels, crayfish etc.). Here the medicinal product reaches the environment directly via the water. In Europe 14 pharmaceuticals are permitted for use in this type of husbandry. They include seven different antibiotics/antimicrobials, six biocides/antiparasitic substances and one anaesthetic (Executive Agency for Health and Consumers, 2013). Often, however, the dissemination route for veterinary medicinal substances is more complex, and some are spread mostly by land, for example they can end up on agricultural land (Larsson and Lööf, 2014). Studies have also been done regarding to what extent the medical veterinary use of antibiotics and antiparasitics in agricultural areas causes spread to the environment, and whether this spread may cause toxic effects in the environment (Sternbeck et al, 2007). The study was focused on farms with large numbers of fattening pigs and milk cows, and includes measurements in slurry, fertilised land, groundwater, surface water and surface sediment. Samples were taken in farms, in their immediate vicinity and in watercourses downstream of the farms. In total 63 samples were analysed for 50 different substances. None of the samples contained any substance in sufficiently high concentrations to be demonstrated.
3.1.5. Emissions into the environment via refuse Pharmaceutical residues in the form of unused medicine can also be a burden on the environment if they are thrown in the refuse or end up in an insecure waste management centre. For this reason, medicinal products which have not been used should be dealt with by pharmacies or in another environmentally safe manner in accordance with the product information leaflet. The total quantity of medicinal products discarded in 2011 in Sweden is estimated to total approximately 1500 metric tons (including packaging). Of this approximately 250 metric tons is estimated to be of the kind the public placed in the household refuse or discarded in another way (Medical Products Agency, 2012). Coordination is required to make it simpler for the public to dispense with pharmaceutical waste - leftover medicines, including waste with sharp edges or sharp points in the form of needles and syringes etc. - in a simple and safe way. Rules on the disposal of medicinal products are insufficient for the minimisation of environmental damage (Swedish Environmental Protection Agency 2013).
3.2. Measuring emission concentrations in the environment
3.2.1. Surface water Of course, the further from the point of emission the lower the concentration of pharmaceutical substances measured. Stockholm Vatten carried out a study to investigate concentrations of pharmaceutical substances in the Baltic Sea (Östersjön) which is a recipient for both Henriksdal and Bromma treatment plants, see the figure below (Hörsing et al, 2014). Centralbron bridge is a sampling point located upstream of the emissions from the treatment plants. Here overflows and individual sewage systems in Lake Mälaren can cause pharmaceutical substances to be detected. Samples were then taken along a gradient following the path waste takes from the emission points in the centre of the city, north through the Stockholm Archipelago. In the first sample (Blockhusudden) after the emission point, over 30 of the 75 analysed substances detected in the outgoing wastewater were found. Concentrations then gradually diminished, presumably due to dilution. NV Eknö serves as a reference point and should not be impacted by the wastewater stream.
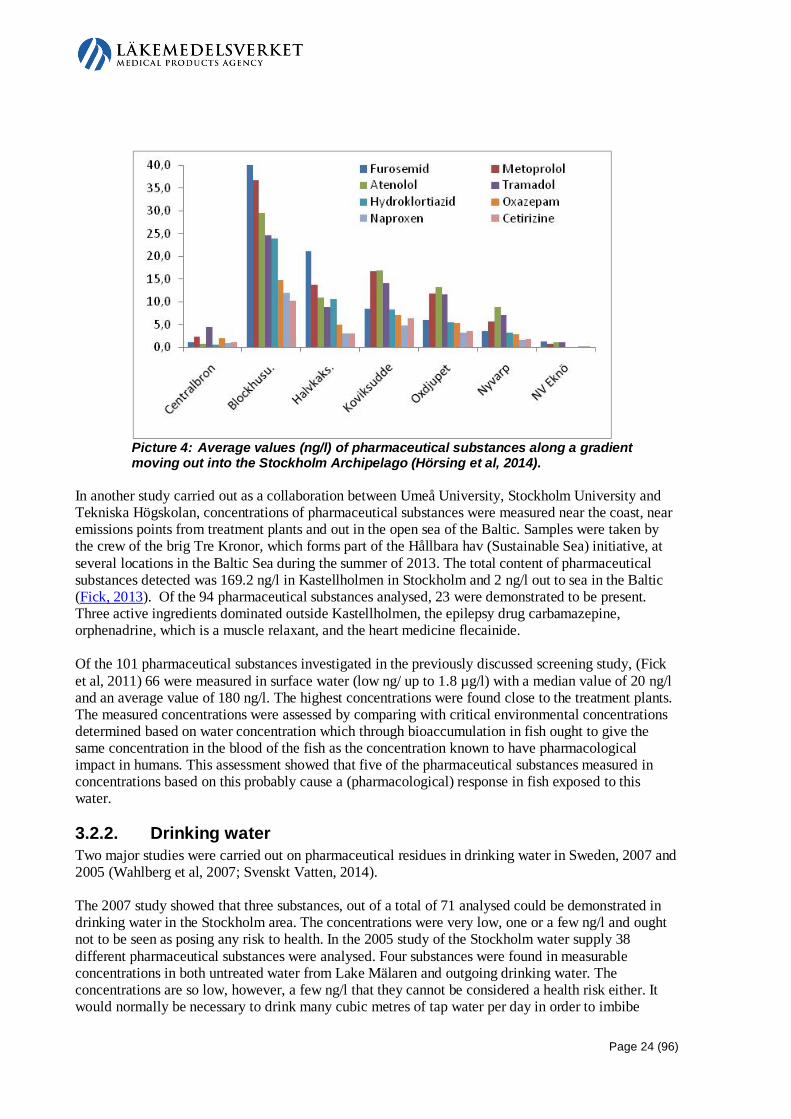
Page 24 (96)
Picture 4: Average values (ng/l) of pharmaceutical substances along a gradient moving out into the Stockholm Archipelago (Hörsing et al, 2014).
In another study carried out as a collaboration between Umeå University, Stockholm University and Tekniska Högskolan, concentrations of pharmaceutical substances were measured near the coast, near emissions points from treatment plants and out in the open sea of the Baltic. Samples were taken by the crew of the brig Tre Kronor, which forms part of the Hållbara hav (Sustainable Sea) initiative, at several locations in the Baltic Sea during the summer of 2013. The total content of pharmaceutical substances detected was 169.2 ng/l in Kastellholmen in Stockholm and 2 ng/l out to sea in the Baltic (Fick, 2013). Of the 94 pharmaceutical substances analysed, 23 were demonstrated to be present. Three active ingredients dominated outside Kastellholmen, the epilepsy drug carbamazepine, orphenadrine, which is a muscle relaxant, and the heart medicine flecainide. Of the 101 pharmaceutical substances investigated in the previously discussed screening study, (Fick et al, 2011) 66 were measured in surface water (low ng/ up to 1.8 µg/l) with a median value of 20 ng/l and an average value of 180 ng/l. The highest concentrations were found close to the treatment plants. The measured concentrations were assessed by comparing with critical environmental concentrations determined based on water concentration which through bioaccumulation in fish ought to give the same concentration in the blood of the fish as the concentration known to have pharmacological impact in humans. This assessment showed that five of the pharmaceutical substances measured in concentrations based on this probably cause a (pharmacological) response in fish exposed to this water.
3.2.2. Drinking water Two major studies were carried out on pharmaceutical residues in drinking water in Sweden, 2007 and 2005 (Wahlberg et al, 2007; Svenskt Vatten, 2014). The 2007 study showed that three substances, out of a total of 71 analysed could be demonstrated in drinking water in the Stockholm area. The concentrations were very low, one or a few ng/l and ought not to be seen as posing any risk to health. In the 2005 study of the Stockholm water supply 38 different pharmaceutical substances were analysed. Four substances were found in measurable concentrations in both untreated water from Lake Mälaren and outgoing drinking water. The concentrations are so low, however, a few ng/l that they cannot be considered a health risk either. It would normally be necessary to drink many cubic metres of tap water per day in order to imbibe
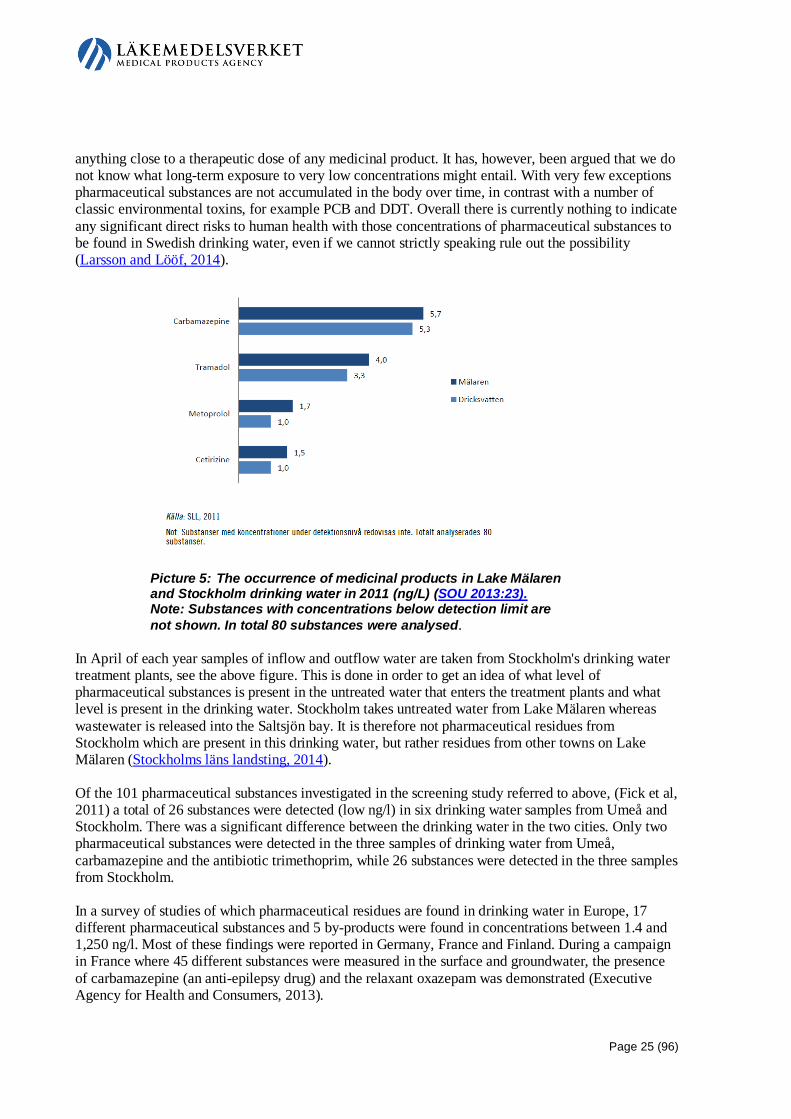
Page 25 (96)
anything close to a therapeutic dose of any medicinal product. It has, however, been argued that we do not know what long-term exposure to very low concentrations might entail. With very few exceptions pharmaceutical substances are not accumulated in the body over time, in contrast with a number of classic environmental toxins, for example PCB and DDT. Overall there is currently nothing to indicate any significant direct risks to human health with those concentrations of pharmaceutical substances to be found in Swedish drinking water, even if we cannot strictly speaking rule out the possibility (Larsson and Lööf, 2014).
Picture 5: The occurrence of medicinal products in Lake Mälaren and Stockholm drinking water in 2011 (ng/L) (SOU 2013:23). Note: Substances with concentrations below detection limit are not shown. In total 80 substances were analysed.
In April of each year samples of inflow and outflow water are taken from Stockholm's drinking water treatment plants, see the above figure. This is done in order to get an idea of what level of pharmaceutical substances is present in the untreated water that enters the treatment plants and what level is present in the drinking water. Stockholm takes untreated water from Lake Mälaren whereas wastewater is released into the Saltsjön bay. It is therefore not pharmaceutical residues from Stockholm which are present in this drinking water, but rather residues from other towns on Lake Mälaren (Stockholms läns landsting, 2014). Of the 101 pharmaceutical substances investigated in the screening study referred to above, (Fick et al, 2011) a total of 26 substances were detected (low ng/l) in six drinking water samples from Umeå and Stockholm. There was a significant difference between the drinking water in the two cities. Only two pharmaceutical substances were detected in the three samples of drinking water from Umeå, carbamazepine and the antibiotic trimethoprim, while 26 substances were detected in the three samples from Stockholm. In a survey of studies of which pharmaceutical residues are found in drinking water in Europe, 17 different pharmaceutical substances and 5 by-products were found in concentrations between 1.4 and 1,250 ng/l. Most of these findings were reported in Germany, France and Finland. During a campaign in France where 45 different substances were measured in the surface and groundwater, the presence of carbamazepine (an anti-epilepsy drug) and the relaxant oxazepam was demonstrated (Executive Agency for Health and Consumers, 2013).

Page 26 (96)
WHO states in its latest Guidelines for drinking-water quality that they do not see any acute toxic danger for human health linked to exposure of pharmaceutical residues in drinking water (WHO, 2011; WHO, 2012). In the long term, on the other hand, there may be reason to take account of low concentrations of pharmaceutical residues currently present in the environment along with currently unknown combination effects of various types of pharmaceutical residues.
3.2.3. Concentrations in fish Fish and other aquatic animals breathe using gills through which large amounts of water pass. They may therefore be exposed to pharmaceutical residues in the water to a greater extent than air breathing animals who only drink water. In certain cases, fish may bioconcentrate pharmaceutical substances in concentrations which are significantly higher than in the surrounding water. The studies which have been carried out so far on levels of pharmaceutical substances in fish flesh do not, however, show any particular risk for significant human exposure as a result of consuming fish (Larsson and Lööf, 2014). The extent to which pharmaceutical substances accumulate in fish has also been studied in combination with other research studies (e.g. Fick et al, 2010). Trout were exposed to outgoing wastewater for 14 days and then concentrations of 25 different pharmaceutical substances in the fish were analysed. 16 substances were found. Levonorgestrel (synthetic hormone) was demonstrated to be present in fish plasma in concentrations of 8 – 12 ng/l, which is higher than the concentration which affects humans. Of the 101 pharmaceutical substances investigated in the previously mentioned screening study (Fick et al., 2011) a total of 23 substances were detected in the biota samples which were included in the study. The concentrations were low (low µg/kg levels) and the highest detected levels were found in fish captured in proximity to the Kungsängsverket treatment plant emission point in Fyrisån in Uppsala. Concentrations measured tallied with previously published data on pharmaceutical residues in biota (living organisms).
3.2.4. Concentrations in sediment Many chemical substances which reach the environment sooner or later end up in the sediment of lakes, oceans, and watercourses. Measurements of pharmaceutical substances in sediment have been taken as part of a number of different investigations of their levels in the environment (e.g. Andersson et al, 2006). Certain pharmaceutical residues were found, but the concentrations of most were below the level of detection.
3.3. Environmental impact (not resistance development) Even if (so far) concentrations of pharmaceutical substances in fish have not been found such that they cause concern over the impact on human health when the fish are eaten, there are a large number of studies describing the impact on organisms in the environment. It should also be noted that there are a rising number of reports saying that endocrine disruptive chemicals in general have an effect on hormone regulated systems and reproduction in humans and animals. We are receiving more and more information saying that diseases can be related to hormone effects. Around 800 chemicals have been identified as being capable of affecting the hormone systems of animals and humans. Only a very small number of these have been studied in any significant manner (UNEP, 2012). One group of compounds which belongs here is pharmaceutical substances. Reports are beginning to arrive about different impacts which have been observed after animals were

Page 27 (96)
exposed to pharmaceutical substances in concentrations found in the environment. However, only a small number of substances have been studied. Medicinal products have an effect on various processes in our body, usually by means of the active ingredients more or less specifically binding to different proteins, for example receptors or enzymes. Many of these proteins are present in other species too, in particular other vertebrates that are relatively closely related to us, for example fish. With sufficiently high exposure to pharmaceutical substances there is, therefore, a risk that these species will also be affected. In addition to the effects of individual substances there is also the risk of cocktail effects, that is, where the substances which an organism is exposed to can combine and have an effect. A survey with examples of reports about environmental effects linked to exposure to wastewater from pharmaceutical production may be found in Larsson (2012).
3.3.1. Effects on fish Ethinylestradiol, which commonly occurs in oral contraceptives, is the pharmaceutical substance whose potential effects on the environment has been the subject of the best studies. Ethinylestradiol is very potent for aquatic vertebrates (fish and frogs) at levels of around 1 ng/l and has, according to several studies, effects on gender differentiation (development of testicles and ovaries) as well as fertility. In purified wastewater in Sweden around 1 ng/l ethinylestradiol is often found. In those places where the initial dilution is great, downstream of a treatment plant it appears as if the levels of ethinylestradiol in the surface water are very often well under 1 ng/l, in other words levels at which ethinylestradiol alone probably does not have any significant effects. Nevertheless, there are a number of different substances in the wastewater which bind to the same receptors as ethinylestradiol, and which therefore combine. These include natural oestrogens (oestradiol and oestrone) which only have barely one 10th the hormone effect of ethinylestradiol but which nevertheless are usually present in high concentrations in wastewater. Certain oestrogens, which are used for treatment of menopause symptoms, have also been measured in waste water at levels which can have an impact. In addition to this there is a range of weak oestrogen industrial chemicals, including nonylphenol and bisphenol A, which can contribute to the total oestrogen effect. In Great Britain, where the most studies have been done, it was found that downstream of many treatment plants fish with both testicles and ovary tissue in the same individual occur commonly, the fertility of these fish being impaired (Parrot and Blunt, 2005; Tyler et al, 2009: Larsson and Lööf, 2014) In a comprehensive, 7 year-long study in Canada ethinylestradiol was added to an experimental lake in order to investigate such things as what population effects a low chronic exposure might give rise to. The concentration of ethinylestradiol in the lake was 5-6 ng/l, which caused feminised vitellogenin-producing1 4F
15 males of the fish species “fathead minnow” (Pimephales promelas). The females demonstrated prolonged vitellogenin production after the spawning season. Interference with the development of the sexual organs occurred in both genders. As was the case with the British treatment plants, see above, the same individuals had both testicles and ovary tissue. Eventually the population collapsed and the species was almost completely eradicated from the lake (Kidd et al, 2007). Levonorgestrel, which is also common in oral contraceptives, also presents an environmental risk. Reproduction has been affected in fish in laboratory tests at concentrations as low as 0.8 ng/l (Zeilinger et al, 2009). Those are concentrations which are found in municipal wastewater and surface 15 Vitellogenin is an egg yolk protein which is normally only produced by sexually mature female fish.

Page 28 (96)
water. Fish that were exposed during the experiment to purified Swedish municipal wastewater accumulated levonorgestrel in their blood in concentrations that in certain cases exceeded levels in the blood of women who have taken oral contraceptives(Fick et.al, 2010). Levonorgostrel also had an effect on the normal reproduction cycle of sticklebacks in concentrations of a few or several ng/l, which might well be found in water which is affected by emissions from treatment plants (Svensson et al, 2014). Perch which in laboratory experiments were given the anxiety reducing substance oxazepam in concentrations of a few µg/l ate quicker, took more risks, and became less sociable than those which had not been exposed. This is behaviour which in various ways could lead to the composition of species in the water changing, and disturbances to the ecological balance. Relatively high levels of oxazepam have been found in wild perch, which means that this is probably something that takes place in the environment (Brodin et al, 2013). The anti-inflammatory drug diclofenac often passes through treatment plants without being broken down to any great extent and the concentrations in Swedish purified wastewater are often around 1 µg /l. There are reports that this concentration, in aquarium experiments, leads to histological changes in a number of fish organs (Triebskorn et al, 2004). The environmentally hazardous properties of diclofenac have been particularly noted in the EU where Directive 2013/39/EU has meant it being placed on the Water Framework Directive watch list.15 F
16 There are many different anti-inflammatory drugs and there is a risk that they act together out in the environment. Other substances are more water-soluble than diclofenac and therefore accumulate in low quantities in fish (Brown et al, 2007). A study which was published in 2014 also suggests that the presence of psychotropic drugs in the water lengthens the lives of perch (Klaminder et al, 2014). This effect of changing behaviour is not necessarily only positive. In the long term it can impact on the whole ecosystem with results that we cannot predict today.
3.3.2. Effects on other aquatic organisms Antihistamines are a group of pharmaceutical substances which are used to reduce allergic problems. They have been found in the environment in concentrations from 1-10 ng/l in surface water (Stackelberg et al, 2007; Kosonen and Kronberg, 2009). Aquatic insects use histamine as neurotransmitters. This means that it is conceivable that there could be effects of the antihistamine drugs in wastewater. After exposure to two common drugs (Hydroxyzine and Fexofenadine, 360± 42 and 2,200 ± 43 ng/l respectively) affected the behaviour of insects and they accumulated pharmaceutical substances in the body (Jonsson et al, 2014). Levonorgestrel, a synthetic hormone, affects frogs also. It can be found in wastewater in concentrations of up to 30 ng/l. When frogs were exposed to wastewater containing levonorgestrel for 7 or 28 days, egg formation was affected. 28 days of exposure caused a significant reduction in the numbers of egg cells (Säfholm et al, 2012).
Blue mussels were exposed to diclofenac, ibuprofen and propranolol to investigate how they were affected by pharmaceutical substances in water. The normal physiology of the mussels was affected by substances in concentrations of around 100 to 1000 times higher than normally found in outgoing
16 Directive 2013/39/EU of the European Parliament and of the Council of 12 August 2013 amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the field of water policy Text with EEA relevance.

Page 29 (96)
water from treatment plants. The study drew the conclusion that impact locally, where the concentrations may be high, cannot be ruled out. (Ericson et al, 2010).
Where algae were exposed to the fungicide clotrimazole in concentrations of a few hundred pmol/litre effects were measured. These concentrations could conceivably occur in the environment (Porsbring et al, 2009; OSPAR, 2013).
A number of different antibiotic drugs have also been shown to be toxic against bacteria and algae in the environment (Halling-Sørensen et al, 2002; Halling-Sørensen, 2000).
3.3.3. Effects on land animals Vulture populations on the Indian subcontinent have been made virtually extinct as a consequence of exposure to the anti-inflammatory pharmaceutical diclofenac. This is an excellent example of a widespread, devastating and yet hard to predict effect on the environment that the dissemination of a pharmaceutical substance can give rise to, in this case in the form of a veterinary medicinal product. The vultures imbibe small amounts of diclofenac by eating the corpses of creatures which have previously been treated with it. Certain species of vulture find it particularly difficult to break down diclofenac, which means that their kidney function is destroyed, resulting in death. Currently diclofenac is banned as a veterinary medicinal product in this region, but is still used illegally (Oaks et. al, 2004).
3.3.4. Effects on humans The levels of pharmaceutical substances measured in Sweden are low, both in absolute terms and in comparison with concentrations measured in Central Europe. As an example, it may be estimated that, based on concentrations of carbamazepine measured in drinking water, it would be necessary to drink 19 million litres of water to reach one standard dose (100 mg). It should also be pointed out that there is no knowledge on the long-term effects on humans of exposure to low concentrations over a long period, in particular during childhood growth (SOU 2013:23). Neither is it possible to rule out effects of long-term exposure to various pharmaceutical substances in very low concentrations. We do not know whether or how the development of the foetus is affected. Neither do we know how growing children are affected. There is, however, nothing which currently indicates that there is any direct risk to human health from the concentrations of pharmaceutical substances to be found in Swedish drinking water. The studies which have been carried out hitherto on concentrations of pharmaceutical substances in fish flesh do not show any risk of significant human exposure following consumption of fish, though the scientific basis for this is still limited, see for example: (Ramirez et.al. 2009). The same applies to potential exposure via other aquatic animals, and to exposure to pharmaceutical substances when we eat meat, for example. The information we have for assessing such risks is poor. Another possible means of exposure might be via the earth. This would be, for example, when children put mud in their mouth. The greatest concentrations of pharmaceutical substances are found in areas where sludge is spread as filling material or is mixed with the earth which is spread out in, for example, parks or gardens, which is not at all uncommon. The calculations made do, however, show that it would be necessary to consume large amounts of earth for it to pose any risk – from 200g to 1kg would be necessary to be the equivalent of a daily dose of a medicinal product (Executive Agency for Health and Consumers, 2013).

Page 30 (96)
3.3.5. Cocktail effects in the environment Humans and the environment are continually exposed to mixtures of different chemical substances and a variety of pharmaceutical residues can be present in the environment in relatively low concentrations. There is currently a good deal of research information showing that the combined effects of a chemical mixture, known as the "cocktail effect" often have a higher toxic impact than the original substances alone, even in the case of the most poisonous substances. There are also studies that show that even if individual components in a mixture are present in concentrations which would not be expected to produce harmful effects in themselves, the mixture might have harmful effects (Backhaus et al, 2010; Kortenkamp et al, 2009; Executive Agency for Health and Consumers, 2013). In the case of pharmaceutical substances with the same mode of action the risk of cocktail effects is manifest. This is the case for example, as previously mentioned, with oestrogen substances which bind to the same receptor. As it is practically impossible in the laboratory to test every different combination which could conceivably be found in the environment, researchers have instead developed various calculation models of the toxicity of combinations of chemicals (SWECO, 2008). Current knowledge unanimously demonstrates that the toxicity of combinations of active ingredients in medicinal products which have similar modes of action within a target organism can be calculated using concentration addition, CA (Kortenkamp et al, 2009). This is where the substances making up a mixture contribute toxicity in proportion to their concentration. What's more there are a number of studies which indicate that concentration addition may be used for substances with different modes of action, for example (Backhaus et al, 2011; Fen al, 2006; Cedergreen et al, 2009). It has been demonstrated that even for substances with different modes of action concentration addition is generally a good means of evaluating the toxicity of mixtures and even in those cases where the toxicity is overestimated this is only by a relatively small amount. Concentration addition has been held to be a suitable first choice and a good tool for hazard and risk assessment of mixtures of pharmaceutical substances (Kortenkamp et al, 2009; Backhaus, 2012). In this context account must also be taken of the fact that emissions in the environment resulting from the use of a medicinal product become a mixture of the active ingredient, metabolites from usage and degradation products. Further, a number of other chemical substances from various human activities also form part of the mixtures present in the environment. How to deal with the problem of cocktail effects is seen as a highly topical and very serious issue which has attracted attention within the chemicals field, including in the development of regulations, both within the EU and internationally16 F
17.
3.4. Development of resistance Resistance means power to withstand and there are doubtless an enormous number of resistance properties among all the bacteria on the planet. Resistance properties can provide important advantages in the competition between different microorganisms. Resistance often arises via mutations of the bacteria's DNA or by means of exchange of genetic material between a resistant and a non-resistant bacterium. Mutations may arise by chance but it has been shown that the more a particular antibiotic is used the greater the risk that strains of bacteria develop resistance against that antibiotic. Nowadays many types of antibiotic contain substances which are not broken down by the body but which are rather disseminated into the environment. There they become active and bacteria are able to develop resistance. These genes then remain due to the fact that the resistant bacteria have an advantage over non-resistant ones for survival.
17 Combination effects of chemicals European Commission, http://ec.europa.eu

Page 31 (96)
The development of resistance to antibiotics among bacteria is one of the great public health problems of our time. The lack of effective antibiotics makes treatment of commonly occurring infections more difficult and slower. Increased mortality, longer care times and increased welfare costs are placing a great burden on healthcare services (WHO, 2014). Development of bacterial resistance is not a new phenomenon. Resistant bacteria occur naturally since other microorganisms are always producing antibacterial substances. The resistance genes in bacteria in the environment are therefore thought to be a reservoir for the transfer to pathogenic bacteria. Water is the most important medium in the environment where antibiotic resistance can arise and spread. The probability increases that resistance is transmitted between bacteria and spread when different bacteria meet and are continuously exposed to antibiotics. In the case of emissions of antibiotics there is partly a direct impact on local microbial communities, and partly an indirect risk to humans. The risk is that emissions drive the development and spread of resistant bacteria. This is a significant problem both in Sweden and internationally since most treatment plants today are not adapted to deal with antibiotic residues (or pharmaceuticals in general). Resistant bacteria tend to spread across land – where the resistance develops is therefore less significant in the long run. Also, accumulations of antibacterial substances in aquatic environments probably favour the development of resistant bacteria. Research is being carried out to find out more information about to what extent emissions of antibacterial substances and metals produce increases in the spread of antibiotic resistance in the environment. If a bacteria community is exposed to an antibacterial substance the bacteria which are resistant to this precise substance will survive. If the plasmid, a bacterial DNA molecule with genetic information, also carries genes which are resistant to antibiotics, the spread of antibiotic resistance can be favoured in different environments where the concentrations of biocides are higher, such as in and around treatment plants. It is clear that the rapid development of resistance globally is a result of the high and sometimes incorrect use of antibiotics. In recent years antibiotic residues in the environment have been increasingly regarded as a possible risk factor for the development of resistance alongside the use on animals and humans
3.5. Overall conclusions regarding damage to the environment and human health
Very large quantities of pharmaceutical substances are released into the environment. Concentrations in the water in Sweden are still so low that they hardly have any direct influence on humans when considering the effects of one substance at a time. At production plants in other countries, on the other hand, concentrations in the water can sometimes be so high that in these places therapeutic doses of pharmaceutical substances are present in the water. One of the stipulations of the general “generational goal” in the Swedish environmental objective system is also that the consumption patterns, for example of medicinal products, should cause as little health and environmental problems for other countries as possible. There are many other uncertainties and reasons for misgivings. Pharmaceuticals are designed to have a biological effect and the impact when substances are disseminated in the environment are difficult to predict. Our understanding of how pharmaceutical substances in the cocktail of compounds which are present in the environment interact with each other and with other substances is poor, but it is becoming increasingly clear that attention must be paid to cocktail effects and that the risk they pose to human health and the environment needs to be assessed.

Page 32 (96)
There is also an increasing number of reports that show or suggest that pharmaceutical substances may have an effect on the ecosystem in those concentrations which already exist in the environment, including in Sweden. Behaviour, reproduction and survival of fish, insects and other animals are being affected. The environment is also an important source of the spread of antibiotic resistance. These are effects which are not tested for when a medicinal product is approved. At this moment in time we know very little about the large-scale effects of this on the environment and in the long run on human society. WHO states in its latest Guidelines for drinking-water quality that they do not see any acute toxic danger for human health linked to exposure of pharmaceutical residues in drinking water (WHO, 2011; WHO, 2012). In the long term, on the other hand, there may be reason to pay attention to low concentrations of pharmaceutical residues currently present in the environment along with currently unknown combination effects of various types of pharmaceutical residues. As has previously been reported, the damage to the environment and human health which we may fear will result from pharmaceutical residues in the environment is linked to long-term effects on the receptor or the hormone system that the medicine in question is designed to affect (Svenskt Vatten, 2005). There is a risk that other organisms will develop changes to their behaviour or physical development. Fish and other animals have receptors which have similarities to human receptors and are therefore particularly sensitive to residues from our medicinal products. The most sensitive time is when the embryo is developing, when even moderate exposure may have serious consequences. When hormone disruptive substances damage the reproductive abilities of wild animals the situation becomes particularly serious. When such damage occurs, the population may diminish at a rapid pace leading to possible extinction. There is also, therefore, a great probability that other parts of the ecosystem which are dependent on those populations will be affected, leading to major chain reactions. There is no way of judging the extent of the macroeconomic consequences of these combined effects but models of ecosystem changes where various top predators become extinct usually show changes to our societies. Common to these changes is that it is difficult if not impossible, and thus very expensive, for societies to find resources to retrospectively redress damage to the ecosystem.
3.6. Broad estimate of costs One part of the assignment relates to estimating the costs of the effects on the environment and human health brought about by emissions of pharmaceutical residues into the environment, which is touched upon in sections 3.6.2 - 3.6.3 below. Supplementary to this, it may also be appropriate to estimate the costs involved in ensuring that additional products are not released into the environment where they can cause damage. In chapters 6-10 and in appendix 2 there is a description of the consequences and costs of the different courses of action for increased environmental considerations in medicinal product legislation which the assignment aims at developing and describing. What's more such preventative measures require measures to improve efficiency at treatment plants and individual sewage systems if emissions of pharmaceutical residues into the environment are to be remedied within a reasonable time period. The costs of taking measures in the treatment plants are described in section 3.6.1 below.
3.6.1. Costs of reducing emissions The most efficient way of reducing the environmental effects of pharmaceutical products is probably to reduce emissions into the environment by implementing measures on individual sewage systems and by upgrading treatment plants.

Page 33 (96)
In 2011 Stockholm Vatten published the report “Läkemedelsrester i Stockholms vattenmiljö” (Pharmaceutical residues in Stockholm's water environment) and the main features of the cost calculations from the report are summarised in this section (Wahlberg et al, 2011). The report goes through various improved purification techniques. UV and hydrogen peroxide, activated carbon and membrane filtration which was tested at Henriksdal sewage treatment plant are covered in the report. Biological treatment methods such as activated sludge and membrane bioreactors are discussed in the study. However, the conclusion was that the latter treatment methods do not result in a sufficiently high reduction of pharmaceutical residues. The descriptions of functions and costs given in the Stockholm Vatten report and which are described below relate to a treatment plant providing waste purification for a population of over 100,000 people. The costs are given in SEK per cubic metre of purified wastewater. Oxidation treatment methods When ozone disintegrates in water hydroxy radicals (OH) are created which have good oxidisation properties and good effect for breaking down organic compounds, including, it has been shown, pharmaceutical substances. Ozonation also results in water being disinfected. Trials are being carried out with pharmaceutical substances in outgoing wastewater, where it was adjudged that ozonation was most effective at doses of 10 g/m³ with 92% reduction. With doses of 10g/m3 it was estimated that ozonation costs 0.1 SEK/m3 , with a total cost of 0.6 SEK/m³. Energy consumption for the plant is included in this calculation. UV-lamps result in the death of the bacteria and the creation of OH radicals. The combination of hydrogen peroxide and UV-light has shown itself to be able to give rise to more OH radicals, and with hydrogen peroxide concentrations of 40 g/m³ it is possible to remove approximately 84% of pharmaceutical substances from wastewater. However, it is judged that doses over 20 g/m³ should not be released into communal waste since this creates hydrogen peroxide surpluses which are not used. The energy cost for a UV dose of 400 Wh/m3 is calculated to be 0.4 SEK /m3. Hydrogen peroxide is reckoned to cost 0.24 SEK/m3. Storage and dosing equipment need to be added to this. The total cost for a treatment plant fitted with UV and hydrogen peroxide equipment is estimated to be 1.3 SEK per cubic metre of purified wastewater. Separation treatment methods Activated carbon (AC) has a high capacity for absorption of organic material. The used carbon which contains compounds which have been removed from the wastewater can either be incinerated or regenerated. With regeneration the pharmaceutical residues and 10% of the carbon are incinerated. The costs for a treatment plant that uses activated carbon treatment are estimated to be 1.0 and 3.1 SEK/m3respectively, depending on which of the two concentrations are tested (25 g/m³ and 140 g/m³ respectively). The purchase of carbon is included in these calculations, as is the cost for carbon filter tanks and the disposal of carbon after use. Nano filtration (NF) and reverse osmosis are different types of membrane filtration, the difference being that different types of membrane are used. The process results in a concentrate separated from the treated water during purification. Major cost items for these two filtration processes are electricity, staff, costs of membrane change and washing chemicals. But there are also costs for disposal of the concentrate which is created during treatment. The total cost for nano and osmosis filtration in treatment plants is estimated at 3.2 and 4.8 SEK/m3respectively.

Page 34 (96)
Picture 6: Illustrated cost for different purification measures and their effectiveness (Stockholm Vatten, 2011). Measurements from Henriksdal treatment plant; blue columns "Hdal in" show concentrations of 46 analysed pharmaceutical substances in the water flowing into the treatment plant and "Hdal ut" show the remaining concentration of the substances after the water has undergone standard purification measures. Other blue columns represent the remaining concentrations of pharma-ceutical substances after increased purification measures described in this section. The costs of "Hdal ut" represent the cost for standard treatment measures. The other costs represent the cost of treatment measures and ordinary purification at "Hdal ut".
A good breakdown of what the costs of upgrading the treatment plants and adding in extra purification steps would be was carried out by Wahlberg et al (2007). Considering the relatively rapid technological developments which are currently taking place it is probable that the costs would be higher today. A very rough estimate of operating, investment and capital costs for supplementary purification plants in Swedish conditions is based on the different parts of the operating, investment and capital costs. The costs for wastewater purification are also dependent on the size of the treatment plant. A small treatment plant costs relatively speaking more per capita both to build and to operate. This calculation is also carried out taking into account the actual distribution of the size of treatment plants in Sweden. The rough estimate of operating, investment and capital costs for Swedish conditions in their breakdown showed that the extra average cost for reducing pharmaceutical residues from wastewater stood at between 0.6 and 19 SEK/m3 depending on the choice of technology and the size of the treatment plant. When the costs were extrapolated to all the treatment plants in Sweden the resultant total cost amounted to between 1.2 and 5.7 billion SEK/year, which is the equivalent of 150-750 SEK per person per year. As things appear at the present time, with current technology, a large part of the increased costs would be made up of increased energy use. For example, the running of Sweden's water and sewage operations cost 14.3 billion SEK in 2003 (Svenskt Vatten, 2006). The reduction of pharmaceutical residues would at best involve a 10% increase in current costs, while the worst-case scenario would be an increase of 40%. As a comparison, the sales of medicinal products in Sweden in 2008 stood at 34 billion SEK.

Page 35 (96)
3.6.2. Macroeconomic costs of the effects of pharmaceutical substances (excluding resistance development).
As can be seen from the above there is good reason for concern regarding the effects in the environment that emissions of pharmaceutical residues might cause. There is evidence of local impact on organisms in the environment and reason to fear that long-term exposure in the environment to pharmaceutical substances, potentially in combination, may impact on the behaviour, reproduction and survival of fish, insects and other animals. This might lead to population changes which through sometimes complex chain reactions would impact on the ecosystem. There is no way of judging the extent of the macroeconomic consequences of these combined effects but models of ecosystem changes where various top predators become extinct usually show changes to our societies. Common to these changes is that it is difficult if not impossible, and thus very expensive, for societies to find resources to retrospectively redress damage to the ecosystem. It is therefore not currently possible to calculate the costs of the effects of pharmaceutical substances on the environment. The uncertainty stems more than anything from the fact that it is a growing phenomenon which is manifesting itself where society does not have the benefit of hindsight. With hindsight it would have been possible to create a macroeconomic cost of risk calculation of these effects, that is to say the benefits of dealing with pharmaceutical residues so that they do not cause side effects for the environment and human health, and to have weighed this calculation against the macroeconomic costs of measures to reduce these effects in order to create signposts for how society might be able to act in a cost efficient way. In such a context preventative measures in the here and now need to be weighed against long-term damage, (compare this with the reasoning within the field of climate change).
The cost of risk calculation consists of the following components: Exposure * probability of damage * cost of damage
Each of these components can be broken up into further sub factors at which point it becomes clear that research into the current situation is beginning to piece together the jigsaw at the level of these sub factors, but also that there is still much to do before a complete cost of risk can be calculated. Exposure: As previously discussed, knowledge does exist regarding to what extent humans are exposed to pharmaceutical residues via drinking water and food and indirectly via ecosystem services and how high concentrations are in most of these media (above all via drinking water). Probability of damage: Knowledge also exists of how great the therapeutic dose of the original pharmaceutical needs to be to provide the desired effect and we are beginning to understand how low the dose can be in order to exert certain acute effects on human health and how great the probability is of a human receiving such doses from the environment (very low probability). On the other hand, the probability of humans being harmed by low doses of pharmaceutical residues and cocktail effects of various pharmaceutical residue products is unknown. Regarding the concentrations of pharmaceuticals in drinking water, these concentrations are currently not sufficient to produce pharmacological impact even with very large intake. It is, however, unclear what lifelong exposure to these low concentrations might entail, particularly for individuals who are developing and growing. For obvious reasons experimental trials cannot be carried out. Cost of damage: The damage which occurs to humans may be thought to follow the same patterns observed in fish, birds and other animals. Negative impact on health and behaviour may be seen as bringing increased costs for society in offsetting this negative impact, particularly for healthcare

Page 36 (96)
services, but possibly also for schools and care services in general. In addition, it can be assumed that damage to the ecosystem may produce negative effects on, for example, the foodstuffs sector which may need to adapt its production, on open-air recreation if the local environment changes and also potentially on the property markets if the local environment becomes less attractive for buyers (for example if lakes become overgrown or coastal areas see increased algae). In a situation where the effects are irreversible, for example damage to human reproduction, which might lead in the near future to infinitely high costs – even if the probability of damage is seen to be relatively low, it becomes manifest that there is reason, whatever the circumstances, for preventative measures. Introducing an infinite number into a calculation is undesirable, but the argument highlights the fact that macroeconomic costs for damage to human reproductive abilities would be insurmountably expensive, even if the probability in the current situation is thought to be small but real. Following the precautionary principle supplementary measures should be put in place both upstream and downstream in order to limit the presence of chemicals which are hazardous to the environment and human health, including pharmaceuticals in drinking and wastewater.
3.6.3. Macroeconomic costs of the spread of antibiotic resistance Antibiotic resistance, for example ESBL producing E. coli bacteria which have already been shown to be present in untreated water in Sweden, is not part of standard water authority analyses of bacteria in untreated or drinking water. This makes it difficult to say anything about exposure to antibiotics in the ecosystem. The probability of damage, that is to say that antibiotic resistant bacteria develop as a consequence of this phenomenon, seems to be so great that the international community has raised the issue high up on the political agenda (WHO, 2012). In the same way as for other macroeconomic effects of pharmaceutical residues it is difficult to calculate the macroeconomic costs of damage, that is to say in the most acute cases antibiotic resistance has developed against diseases which affect humans where there is no alternative treatment. There is no way to survey the costs resulting from the acceleration of the development and spread of antibiotic resistance in such a way that even "milder" medical conditions cannot be treated with antibiotics with the risk that many diseases may become fatal.

Page 37 (96)
4. Environmental considerations in current pharmaceutical legislation
Below is a brief description of the pharmaceuticals regulations relevant to this assignment. A more detailed description may be found in the MPA report of 16-12-2009 "Report on government commission regarding the possibility of tightening environmental requirements for the manufacture of medicinal products and active ingredients". Medicinal products on the European market are regulated by a broad framework with the aim of ensuring quality, safety and medical efficacy. The regulations are to a great extent harmonised within the EU/EEA, partly through Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use (referred to herein as the Human Medicinal Products Directive), Directive 2001/82/EC of the European Parliament and of the Council on the Community code relating to veterinary medicinal products (referred to herein as the Veterinary Medicinal Products Directive) and Regulation (EC) No 726/2004 of the European Parliament and of the Council laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency. These regulations control both the EU body the European Medicines Agency, (EMA), and the work of the national authorities in such activities as authorisation and control of medicinal products. Within this regulatory framework medicinal products may be authorised in one member state, in all member states via a centralised authorisation process or alternatively one of the two procedures leading to marketing authorisation in several member states may be used (mutual recognition or decentralised procedure). The legislation requires, for example, that medicinal products be authorised within the EU in order to be marketed, that the company that holds the marketing authorisation takes responsibility for control of their medicinal products and that the production plant has authorisation for its activity. The medicinal products legislation is also applicable to the manufacture of active ingredients in medicinal products. Active ingredients in a medicinal product can in simple terms be described as the constituent part(s) of a medicinal product which give it its therapeutic function. In Sweden the Swedish Medical Products Agency (Läkemedelsverket) is the competent authority for the authorisation and control of medicinal products. The Medical Products Agency also grants authorisation for the manufacture of medicinal products in Sweden. Manufacturing authorisation under medicinal products legislation is totally independent from authorisation under environmental legislation.
4.1. Marketing authorisation for medicinal products In conjunction with an application for marketing authorisation for a medicinal product the company must provide the relevant European medicinal products authority with information about the medicinal product. The information and documents that should be submitted is laid out in both the medicinal products directives1 7F
18. Information is evaluated by the authorities and medicinal products which are adjudged to have a favourable risk-benefit balance are authorised. A favourable risk-benefit balance for medicinal products for human use signifies that the therapeutic effects of the medicinal product outweigh all the risks related to the product’s quality, safety or efficacy regarding the user's health or public health. 18 See article 8 in the Human Medicinal Products Directive and article 12 in the Veterinary Medicinal Products Directive.

Page 38 (96)
The authority evaluates the positive effects of the medicinal product in relation to the risks, but the authority may not, under current EU regulations, factor any potential environmental risks into the risk-benefit evaluation for medicinal products for human use. This follows from the fact that the concept of "risk-benefit balance" expressly excludes the expression "every risk of undesirable environmental impact".18 F
19
Under current legislation the application for marketing authorisation of a medicinal products for human use may, therefore, not be rejected due to the environmental effects which may result from use of the medicine, even though these effects must be reported in the application documents.
4.1.1. Requirements for environmental data in conjunction with authorisation
Since 2006 there has been a requirement that the applicant for marketing authorisation of a medicinal product for human use shall carry out an Environmental Risk Assessment, (ERA). The applicant should study and submit information on the potential environmental risks posed by the medicinal product and propose risk minimisation measures. By risk minimisation measures, RMM, is meant special measures to reduce environmental risks. The application shall also provide reasons for any potential precautionary and safety measures that should be taken during storage of the medicinal product, when administering it to patients and during disposal of waste products as well as information on any potential environmental risks the medicinal product might entail.19 F
20 Usually no environmental data is directly displayed in the summary of product characteristics or the package leaflet for authorised medicinal products for human use but there is a standard text for the destruction of unused medicines. Certain environmental information may appear in the published Public Assessment Reports, (PAR). Product information pertaining to the environment and medicinal products appears primarily in two contexts: i) information on risk minimisation measures for handling the product, e.g. destruction and disposal ii) information on environmental risk assessments. The amount of information varies, however, between different medicinal products. Generally, there is little information provided with products granted authorisation before the ERA requirement was introduced in 2006.
4.1.2. Veterinary medicinal products With veterinary medicinal products there is a larger requirement for environmental considerations compared with medicinal products for human use, as the application must state potential risks that the veterinary medicinal product might entail for the environment and for the health of humans, animals and plants As with medicinal products for human use the application must also report on the results of investigations to assess the potential environmental risks of a medicinal product; this environmental impact must be studied and special provisions to reduce it must be formulated in each individual case2 0F
21. If measures are proposed in the application process, with the purpose of reducing impact on the environment, this would appear in the product information included in the authorisation, presented in the form of a summary of product characteristics and a package leaflet, for example measures regulating the handling of manure and waste. In the risk-benefit assessment of medicinal products for veterinary use, unlike with the assessment of medicinal products for human use, each risk of undesirable environmental effects from the use of the medicinal product must be taken into account. These risks do not include environmental effects attributable to manufacture. In practice the authorities have not yet rejected any product for 19 See articles 1.28 and 1.28 a in the Human Medicinal Products Directive. 20 See articles 8.3 ca and 8.3 g in the Human Medicinal Products Directive. 21 See articles 12.3 g and 12.3 j, fourth indent of the Veterinary Medicinal Products Directive.

Page 39 (96)
environmental reasons, whereas certain species of animal or weight class have been excluded from treatment with certain products due to the risk of environmental impact. Work has begun to develop EU legislation for veterinary medicinal products. In current medicinal products regulations there are no requirements to report on environmental factors during the manufacture of medicinal products within the framework of the authorisation process, for medicinal products for veterinary or human use.
4.2. Post authorisation control of medicinal products Notwithstanding the large amount of data submitted with an application, the actual use of a medicinal product brings new information to light. In order to take account of this new information a comprehensive system of post authorisation control of medicinal products has been developed. Under current legislation on medicinal products for human use the risk to the environment is not seen as a factor in the risk-benefit balance so no requirements are placed on the company to follow up new findings related to the environmental risks of medicinal products.
4.3. Manufacturing authorisation for medicinal products The member states shall ensure that manufacture of medicinal products in their territories and the import of medicinal products from third countries only takes place with specific authorisation.2 1F
22 In Sweden such authorisation is issued by the Medical Products Agency. A more detailed account of current provisions regarding manufacture of medicinal products may be found in section 9.3. Neither in Swedish pharmaceutical legislation nor in EU legislation and guidelines regarding GMP for medicinal products is there any requirement concerning manufacture or import with the purpose of protecting the environment. It follows that issues related to environmental considerations in the manufacture of medicinal products have no part to play in the granting of manufacturing authorisation, and neither therefore may they constitute grounds for the withdrawal of previously granted authorisations.
4.4. Manufacturing authorisations under EU environmental legislation
Industrial manufacture within sectors which could have significant impact on the environment requires authorisation under environmental legislation which is harmonised at the European level. When issuing an authorisation, a range of factors should be considered, e.g. whether the current location is thought to be appropriate for the purpose, and various measures must be taken to limit any impact on the environment. Two important principles which must be applied here are the "polluter pays principle", whereby responsibility for limiting contamination falls on the party whose activity caused the contamination, and the "precautionary principle" whereby the part carrying out an operation which might cause harm to human health or the environment must choose the safe alternative over the non-safe alternative. Lack of knowledge may not be given as reason for not employing cost-effective protection measures. A certain evaluation of fairness arises here.
22 See article 44 in the Veterinary Medicinal Products Directive and article 40 in the Human Medicinal Products Directive.

Page 40 (96)
Directive 2010/75/EU of the European Parliament and of the Council on industrial emissions,2 2 F
23 a.k.a. the Industrial Emissions Directive or IED is a minimum directive, which means that the member states have the right to implement or uphold requirements which are more but not less stringent than those of the directive. The aim of the directive is for values of emission limits to be based upon Best Available Techniques (BAT). This principle entails using the established technique which is the most effective in achieving a high level of protection for the environment as a whole and which may be adapted within the relevant branch in an economical and technically comparable way, taking into account costs and benefits. BAT for an industrial sector is described in a BREF (BAT reference document) which is updated in the EU according to an established timetable. Those activities which are affected are industries within the energy sector, the metals and mineral industry, the chemicals industry, waste management and other operations such as the pulp and paper industry, the foodstuffs industry, large animal husbandry facilities and so on. Point 4.5 in the appendix to the directive stipulates that manufacture of medicinal products and also intermediate products is covered by the directive. This is subject to the condition that manufacture involves chemical or biological reactions and takes place on an industrial scale. This means that production plants which produce medicinal products and intermediate products for medicinal products which fall under these criteria must be evaluated for authorisation in accordance with IED. This testing must look at environmental impact from the plant, e.g. emissions into water, but currently the authorisation requirements are often in the form of aggregate parameters rather than in the form of emissions of individual substances. Only a small percentage of European medicinal product manufacturing sites with manufacturing authorisation are covered by IED. Both in Sweden and within the EU environmental legislation makes it possible when issuing authorisations to the pharmaceuticals industry to place requirements on what is emitted from production plants.
23 Directive 2010/75/EU of the European Parliament and of the Council of 24 November 2010 on industrial emissions (integrated pollution prevention and control).

Page 41 (96)
5. The four measures These measures were developed within the framework of work on the environmental quality objective “Poison-free environment". Together with 15 other objectives they comprise the basis of Swedish environmental policy. The environmental objectives are built upon the overall generational goal of passing to the next generation a society where the major environmental problems are resolved without causing environmental or health problems abroad. This goal is particularly important as the majority of production of medicinal products takes place outside Sweden. There are two types of measure which can result in reduced environmental impact from medicinal products: (i) "downstream measures", which are aimed at purification of wastewater and (ii) upstream measures, whose purpose is to prevent emissions of pharmaceutical substances into the waste. The two complement each other and both are necessary. The proposed measures can be seen as upstream measures, whose purpose is to reduce emissions of pollutants at source (Measure D) and at point of use (Measures A and C). With the aid of emission limits (Measure D), restricting emissions of active pharmaceutical ingredients into water, both in the EU and in third countries, is proposed. In conjunction with the assessment of marketing authorisation for medicinal products a weight of evidence approach is used, taking relevant information about the medicinal product in question and weighing its benefits against its risks. Measure C proposes that the potential environmental impact of medicinal products should be assessed as a risk and that relevant risk minimisation measures should be developed. This means that when they enter the market, authorised medicinal products already have a plan of measures to deal with their potential environmental effects.
Picture 7: Diagram of connections between the measures
There are connections between the four measures, see figure above. Some of the measures are contingent for their implementation upon other measures being taken before or simultaneously. The prerequisite for all the measures is the existence of adequate information about the environmental impact of pharmaceutical substances. Suitable environmental risk assessments (Measure A) are fundamental for the development of appropriate measures to prevent negative effects on the environment. These measures include both emission requirements during production of medicinal products (measure D) and creation of specific measures to minimise environmental exposure during use (Measure C). It is proposed (Measure B) that all the information generated in connection with environmental risk assessments be periodically collected together and made accessible to stakeholders (e.g. water and sewage treatment works, environmental authorities, health authorities, researchers etc.).

Page 42 (96)
6. Measure A: Testing requirements for medicinal products and the environmental risk assessments carried out in conjunction with an application for authorisation of medicinal products
6.1. Previous studies bearing on Measure A The testing requirements for environmental risk assessments have been discussed within the framework of work preparing for environmental objectives (see SOU 2012:38). The research project MistraPharma also investigated issues related to the suitability and effectiveness of current environmental risk assessments while also preparing proposals for changes.
6.2. Limitations to Measure A Regarding Measure A the report only deals with new medicinal products for human use.
6.3. Background to Measure A Directive 2001/83/EC2 3 F
24 requires that an environmental risk assessment (ERA) be carried out when there is an application for marketing authorisation of a medicinal product. Environmental risk assessments are carried out according to a guideline2 4F
25 and a "Question and Answer" document2 5F
26. These documents recommend studies which could form the basis of assessments of environmental risk from medicinal products during use (not during production or disposal). Conventional environmental risk assessment procedures primarily follow those carried out for chemicals in general under Reach legislation26 F
27. The guidelines were developed by the European Medicines Agency (EMA) in 2006 and have not been revised since, although a discussion on updating them is ongoing. For a detailed description of the testing requirements for environmental risk assessments as well as risk assessment procedures see Appendix 1. Current requirements for environmental risk assessments make exemptions for a number of types of substance such as proteins, vaccines, lipids and vitamins as these are not thought to have an impact on the environment. Moreover, substances which do not reach a given expected concentration in water ("action limits") or whose ability to accumulate in adipose tissue is deemed to be low do not require further testing regarding environmental risk. A survey initiated by the EMA in 2014 shows that in the year 2011-2012 fifty-nine pharmaceutical substances were assessed for authorisation under the centralised procedure (Caneva et al. 2014). Of these 17 (29%) were exempted from the requirement for environmental risk assessments and for 22 (37%) environmental risk assessment was terminated in the first phase without any environmental studies being carried out. 20 pharmaceutical substances (36%) were moved on to phase 2 and tested for their environmental impact. This shows that more substances would have been assessed if there had not been limitations to the recommendations for testing.
24 See article 8.3 ca) and the appendix to Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. 25 EMA/CHMP/SWP/4447/00 corr1 Environmental risk assessment of medicinal products for human use (June 2006) http://www.ema.europa.eu/ema/ 26 Q&A EMA/CHMP/SWP/44609/2010 Questions and answers on the guideline on the environmental risk assessment environmental risk assessment (April 2011) http://www.ema.europa.eu/ema/ 27 Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (Reach), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC.

Page 43 (96)
Regular revision of the guidelines is a part of the EMA programme. The decision on revision of guidelines may be made by the EMA and its Committee for Human Medicinal Products (CHMP). This follows existing legislation2 7F
28 stipulating that the guidelines shall be followed in the compilation of applications for marketing authorisation and thus the legislation would not need to be changed for Measure A. The courses of action for A may, therefore, be achieved by firstly revising the guidelines for environmental risk assessment. If Measure C (environmental considerations in the risk-benefit assessment) is implemented, it too will probably also entail the need for revision of the guidelines for environmental risk assessment.
6.4. Courses of action for Measure A In the table below courses of action identified for measure A are shown.
Table 1: Overview of Measure A with courses of action, measures and main effect.
Course of action Measure Effect
A1. Review current limitations such that more pharmaceutical substances are covered by requirements for environmental risk assessment.
Develop relevant scientific arguments for the updating of current limitations.
More pharmaceutical substances are included by the requirement to carry out environmental risk assessments.
A2. Develop the testing requirements for environmental risk assessment such that the tests are better adapted to specific properties of medical products.
Further development of the methodology for environmental risk assessments such that it reflects the specific properties of the medicinal product.
Better environmental information on those pharmaceutical substances which are to be tested.
6.4.1. Course of Action A1: Review current limitations such that more pharmaceutical substances are covered by requirements for environmental risk assessment.
In order for new pharmaceutical substances to undergo comprehensive environmental risk assessment, current limitations regarding testing requirements need to be revised. Above all the "action limits" (calculated concentrations of pharmaceutical substances in the water), which under the guidelines determine whether pharmaceutical substances need to undergo testing, should be removed or changed. The limitation based on the ability of a substance to accumulate in fat (which should reflect bioaccumulation ability) should be revised, since other mechanisms underlying bioaccumulation of pharmaceutical substances, such as protein binding, may be relevant.
28 See introduction to Appendix 1, point 4, directive 2001/83/EC.

Page 44 (96)
6.4.2. Course of Action A2: Develop the testing requirements for environmental risk assessment such that the tests are better adapted to specific properties of the medicinal products.
Under this course of action environmental risk assessment methodologies would be adapted to the specific type of substance that the active pharmaceutical ingredient is. The advantage of Course of Action A2 is that more relevant formation is produced about how medicinal products impact on the environment (and also whether in the long term they may impact on human health). This course of action consists of developing the procedure for environmental risk assessment such that all available information is taken into consideration, e.g. physical and chemical properties, mechanisms of action, various bioaccumulation mechanisms, metabolites and development of resistance. The changes which need to be made must be based on an analysis of desirable improvements and the availability of scientific solutions. At the current time such an analysis is not available. Neither are there currently any complete scientific solutions to many of the above mention issues. This means that developing the procedure for environmental risk assessment needs to happen step-by-step, as and when new knowledge appears. Changes do not necessarily entail further tests, but rather more specific studies replacing general ones. If production emission limits (Measure D) were to be implemented, the environmental information about the inherent properties of pharmaceutical substances developed in the ERA may comprise a part of the basis for the establishment of production emission limits.
6.5. Effects of Measure A and respective courses of action One or both of the courses of action may be implemented. Most information about the environmental impact of medicinal products would be obtained from both courses of action being applied together. In this way there would be basic environmental information on more pharmaceutical substances which would be more relevant in the assessment of their environmental impact than that provided by current testing requirements. The table below highlights which effects are deemed to be common between the measures and which effects may be expected from the individual courses of action.

Page 45 (96)
Table 2: Effects of Measure A and Courses of Action A1 and A2.
Course of Action A1: Review current limits such that more pharmaceutical substances are covered by requirement for environmental risk assessment.
Course of Action A2: Develop the testing requirements for environmental risk assessment such that the tests are better adapted to the specific properties of the medicinal products.
General effects
• Revised testing requirements lead to better information about the environmental impact of medicinal products and can therefore form a more appropriate basis for the development and use of risk minimisation measures. In the long term this would result in a better environment and consequently better public health.
• Environmental information about more pharmaceutical substances should be created.
• More relevant environmental information should be created regarding how pharmaceutical substances impact on the environment (and also if they might impact on human health in the long term).
• Conflicts with the EU policy on reduced testing on vertebrates Increasing testing requirements goes against the EU policy of reducing testing on vertebrate animals. In order to obtain further information about the impact of medicinal products on the environment and also to reduce unnecessary animal testing (above all studies on fish) solutions which favour the use of already available data should be developed.
Impact on the development of medicinal products
• There is a risk that development times for the medicinal products may be extended. This risk may be reduced if environmental studies are carried out in parallel with other studies.
Impact on availability of medicinal products
• It is not thought that increasing testing requirements would have an impact on availability, the potential impact would occur when these test results are evaluated, see Measure C.
Impact on pharmaceutical companies
• Increased costs if more environmental studies are required. The greatest impact (cost) would be on companies which have substances which place a burden on the environment, such as PBT (persistence, bioaccumulation and toxicity) or endocrine disruptors since it is probable that these substances need more comprehensive testing.
• More specific environmental risk assessments
More complex environmental risk assessments may require more or more specific studies. The preparation of environmental risk assessments may entail the need for new competences. Both situations may contribute to increased costs for the companies if they are to submit complete applications for authorisation of a medicinal product.

Page 46 (96)
7. Measure B: Gathering environmental data on active pharmaceutical ingredients and making it accessible
7.1. Previous government assignments pertinent to Measure B Making environmental information accessible in the form of a database is one of the proposals made by the All-Party Committee on Environmental Objectives (Miljömålsberedningen). The interim report from the All-Party Committee on Environmental Objectives SOU 2012:38 points out the need for gathering environmental data on active pharmaceutical ingredients and making it accessible.
7.2. Limitations to Measure B As the government assignment is focused on dealing with medicinal products for human use, the measures are based on this too. Nevertheless, the consequences of excluding medicinal products for veterinary use are touched upon. The usefulness of the measure is also negatively impacted if only newer pharmaceutical substances which have data from environmental risk assessments (ERA) are to be included. Fundamental to the report is that existing medicinal products which currently lack ERA data should be included. Relevant data may be obtained subsequently.
7.3. Background to Measure B Environmental risk assessments (ERA) are carried out in conjunction with the application for authorisation of the medicinal product, but these ERA results are not collected in any common EU database. Neither is the information made available by any national pharmaceutical authority or the EMA. The same applies to risk minimisation measures (RMM) that have been identified. This means that an overall analysis of data providing a general picture of the environmental impact of pharmaceutical substances cannot be carried out based on this environmental information. In certain cases, parts of the environmental information are published in the Public Assessment Report (PAR), whilst other patient information frequently only contains a standard text regarding the destruction of unused medicinal products. Access to environmental information which has been collected and quality assured is, therefore, of great value for ongoing environmental work with medicinal products. Qualitative data is of benefit, not only for pharmaceuticals authorities, but also for many other entities. This became very apparent at the coordination meetings carried out under the framework of this government assignment. The collaborating partners stressed that environmental information is needed by, for example, county councils, pharmaceuticals boards, water treatment companies, health and environment authorities as well as for research. Even if certain data for certain substances is already available from various sources, this information needs to be gathered together and made more complete and made official, searchable and easily comparable.

Page 47 (96)
Picture 8: Examples of stakeholders who have a need of environmental information
A current EU report states that qualitative data about environmental risks from medicinal products is either scarce or non-existent and that data compiled in the form of environmental risk assessments in conjunction with applications for marketing authorisation is often not available to others, being regarded as confidential (Bio Intelligence Service, 2013). The EU report also proposes that environmental information on pharmaceutical substances should be compiled and made accessible. Access to a database containing environmental information on active ingredients in medicinal products is also of great help in including environmental factors in the assessment of the benefits and risks of medicinal products (Measure C) and in the implementation of minimum requirements on conditions of production for the sale of products in the EU market (Measure D) and also when taking advantage of increased testing requirements for medicinal products (Measure A) in identifying environmental risks.
Existing databases Neither the EMA nor the national pharmaceuticals authorities has a general database where the companies are obliged to make environmental information available. A voluntary medicinal products database in Sweden is available via FASS.se2 8F
29. As substances used in medicinal products may also have other areas of use, they may also be present in other databases such as the Reach database2 9F
30 (industrial chemicals) and the EU pesticides database3 0F
31 (for plant protection products). The Reach database was produced under the framework for Regulation (EC) No 1907/2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH)31 F
32 The Reach regulation means that companies who manufacture or import chemicals within the EU are obliged to register substances they are using. However, certain substances, including medicinal products, are exempted from certain parts of Reach. If a pharmaceutical substance is also used as an industrial chemical then it is present in the Reach database; examples of such substances are acetylsalicylic acid, and paracetamol.
29 Miljöinformation i Fass.se, LIF, http://www.fass.se/ 30 Reach database, European chemicals agency, http://echa.europa.eu/information-on-chemicals/ 31 EU pesticides database, Directorate General for Health & Consumers, http://ec.europa.eu/sanco_pesticides 32 Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC.
Accessible environmental
information
Researchers
Pharmaceutical companies
Pharmaceutical authorities
Health and medical care
Treatment plants
Environmental authorities
Patients and consumers

Page 48 (96)
The database for pesticides was produced within the framework of Regulation (EC) No 1107/2009 of the European Parliament and of the Council concerning the placing of plant protection products on the market3 2F
33 and is managed by the European Food Safety Authority, EFSA. Information on all plant protection products which have been assessed within the EU is collated in this database which includes certain veterinary medicinal products which are also used as plant protection products. There should be an investigation into how much cooperation might take place with ECHA/EFSA regarding the establishment and maintenance of a database of pharmaceutical substances with the aim of reducing duplicate administration and increasing the amount of information accumulated. Environmental information on active ingredients in Swedish medicinal products has also been accessible via FASS.se since 2005. 29 The pharmaceutical companies voluntarily make available environmental information which has been compiled in conjunction with applications for authorisation. FASS.se does not contain information on any possible environmental considerations during the manufacture of medicinal products. The system was developed by the Swedish Association of the Pharmaceutical Industry (LIF) in collaboration with a number of other entities and is based on the pharmaceutical companies making available environmental information which has been audited by the external auditors IVL (Svenska Miljöinstitutet AB). For more information on what data is published, assessments and models see Fakta för förskrivare3 3F
34. This document is updated every third year and new information is published on FASS.se as and when new substances appear. WikiPharma, a literature database which compiles research results, is produced by the MistraPharma research project34 F
35.
Home for a database The fact that the European Medicines Agency is working toward harmonised information on pharmaceuticals makes it a natural home for environmental data, which is also proposed in SOU 2012:38. In proposals from the Commission rregarding new EU legislation for veterinary medicinal products3 5 F
36 several databases were proposed, to be administered by the EMA. Further developments in this area should therefore be borne in mind. Even if the EMA is the owner and administrator of the database there are various ways of publishing information and keeping the database up-to-date. Both in the database for industrial chemicals and Fass.se companies are responsible for this and they have some form of auditing process to ensure the quality of the contents. In the NPL (Swedish national register of medicinal products) the data has different owners depending on who it was supplied by.
7.4. Conditions for Measure B A decision is needed regarding what information the database should contain for it to be fit for purpose and regarding how quality assurance should be carried out. Information owners also need to be assigned.
7.4.1. Relevant information in the database In order to obtain the greatest possible benefit from the database as many substances/medicinal products as possible should be included but various boundaries can be adhered to. The fundamental
33 Regulation (EC) No 1107/2009 of the European Parliament and of the Council of 21 October 2009 concerning the placing of plant protection products on the market and repealing Council Directives 79/117/EEC and 91/414/EEC. 34 Fakta för förskrivare 2014-01-07, LIF, http://www.fass.se/LIF/healthcarefacts?headlineId=2432#document-top 35 http://www.wikipharma.org/welcome.asp 36 COM(2014) 558 final, 2014/0257 (COD)

Page 49 (96)
point is that new pharmaceutical substances for human and veterinary use which have data stemming from environmental risk assessments (ERA) should be included and the gathering of data should take advantage of this ERA data. If only newly authorised medicinal products were included in the environmental information it would take a very long time before sufficient quantities of data became accessible. As previously discussed under Measure A (section 6.3), in year 2011-12 fifty-nine substances were assessed for environmental impact under the central procedure. The total number of active ingredients in Europe is estimated to be in the region of 3,000. In order to have enough data on which to base emission limits, risk minimisation measures and so on, the data on previously authorised pharmaceutical substances needs to be included as and when such data is obtained. Studies of how to proceed with the compilation of environmental information for these medicinal products need to be started. It is also necessary to analyse which environmental requirements might be placed on renewal of, major changes to, and broadening of authorisation etc. with the aim of, over time, gathering environmental information on all authorised pharmaceutical substances. Initiatives towards this have been taken by the Commission’s EcoRiskPrediction (ERP) project. This work should be monitored. Information may be compiled by substance or by medicinal product. In the Reach database environmental data on each substance is published. The pharmaceuticals authorities in Germany and France are currently working actively on developing "eco-monographs" for veterinary medical pharmaceutical substances. It is preferable to have information based on the substance in a database too. This is mainly to facilitate use of the same data for medicinal products containing the same active ingredients. This is also preferable for researchers and other entities who wish to monitor the impact of those substances in the environment. Certain information specific to a medicinal product probably needs to be linked to this substance data so that information is usable by entities who focus on the medicinal product itself, for example for procurement and pricing.
7.4.2. Need for procedures for compiling, making accessible and updating information in the database.
The objective is for environmental information on every active ingredient, which is an ingredient of an approved medicinal product and which has an ERA, to be gathered in one place, made accessible and searchable for different entities so that they can use the information for further work. The information has to be quality-assured and up-to-date. Certain companies currently publish this information while others view it as a trade secret. This data is dealt with in different ways in secrecy legislation in the different member states. Current pharmaceutical legislation needs to be revised to forge a common EU requirement regarding what environmental data should be collected and made available. Within the EU opinions are often divided regarding what should be transparent. In order that pharmaceuticals authorities make the same assessments, the legislation needs to set out what information should be made available in the database. In order to ensure that every company provides the information, legislation is needed such that accessibility of the data is not wholly dependent on the goodwill of the companies/authorities. Such legislation also needs to make it clear that it is the duty of the company and/or authority to make this data available in a given database and that this data must be updated as and when new knowledge arises. The creation of environmental data may require that companies carry out studies which may be demanding on financial and other resources. It is of great importance that the companies' ownership of

Page 50 (96)
data that warrants protection is respected in the EU legislation, if that information is to be made accessible in a database. Special services for access control and other protective measures should also be considered. Initially the database would probably mainly consist of information created in conjunction with applications/authorisation of medicinal products. If environmental considerations are inserted into the risk-benefit assessment (Measure C) the environmental aspects should be included in the post authorisation control system. Requirements would thereby be created for companies to keep themselves additionally informed from an environmental perspective about developments of their substances and thus to update the database as and when new information arises. A process for the collection and quality assurance of data needs to be developed. The methodology must ensure that both new and previously authorised pharmaceutical substances can be processed and that the database is able to be updated as and when new knowledge arises. Within the pharmaceuticals field the EMA and EDQM have experience of processes for creating monographs and within chemicals legislation the Reach database has such experience. Common to these processes is that they are often seen as consuming both time and effort but on the plus side they create the opportunity to have quality assured, harmonised data. Work must be done to produce environmental information on medicinal products authorised before the requirement for ERA was introduced. Methodology is required for environmental risk assessments of substances where a number of companies, both research and generic drug companies, are in possession of authorisation for medicinal products containing the same substance. The Reach legislation solved the equivalent problem by obliging the companies to collaborate in producing necessary data. When producing monographs for plant substances and suchlike at the EMA the pharmaceuticals authorities compile existing information and request the companies to provide supplementary information before a working group harmonises the data.
A simpler type of database or purchase of information structure from existing databases, for example the Reach database, the database of plant protection products or biocides, should be considered in order to obtain a suitable database with justifiable development costs. The opportunity for companies to benefit from each other's information should be encouraged, for example through trading of completed studies. These could then be reused for fresh applications. This is contingent upon data protection being established in the legislation. There is an equivalent trade in clinical trials, for example.
Companies have expressed how it is desirable to be able to market medicinal products as "green pharmaceuticals". Currently possibilities for such marketing are limited, because there is no entity which makes such assessments and because the legislation does not provide space for the marketing to provide information which does not have an equivalent in SmPC. A database with accessible, validated, harmonised data would contribute to a solution to this problem.
7.4.3. Impact of Measure B Regular collection of environmental data which was created during the authorisation process (as well as relevant risk minimisation measures), which is then made accessible, is a fundamental condition of the use and development of relevant risk minimisation measures. In the long term this would result in reduced environmental impact, more efficient use of resources and better public health due to reduced environmental problems.

Page 51 (96)
Impact on the development of new medicinal products and access to medicinal products The fact of companies providing data which they are already required to create prior to authorisation application does not appear to have any notable effect on the development of new medicinal products. When data on substances is available this facilitates work on the development of medicinal products which contain those substances.
Impact on the pharmaceutical companies The measure entails increased administrative costs. Depending on how the requirement is formulated regarding the database, companies may have to submit data to the database. If the environmental information to be included is to be processed by the authorities, however, the latter would be responsible for relevant administration. More studies may be reused if a company is able to make use of information from the database or purchase information from the company that carried out the relevant study.

Page 52 (96)
8. Measure C: Environmental aspects of the assessment of risks and benefits of medicinal products
8.1. Previous studies pertinent to Measure C The inclusion of environmental considerations in risk-benefit assessments regarding whether a medicinal product shall be authorised for sale was proposed by the All-Party Committee on Environmental Objectives (see SOU 2012:38) and comprises a part of the Parliamentary milestone target for medicinal products and the environment.
8.2. Limitations to Measure C In accordance with the assignment this report only deals with the processing of new medicinal products for human use.
8.3. Background to Measure C Extract from government assignment: "according to the decision on the milestone target one important basis for the chemicals policy is to tackle the problems of environmentally hazardous chemicals primarily at source. This should also, as far as possible, be a fundamental point regarding processing environmentally hazard chemicals in medicinal products for human use. However, it should be possible to make exceptions in such cases where the measures are deemed to have an undesirable impact on access to treatment with a medicinal product or on the cost to society of medicinal products. In certain cases, it should also be possible for society to, under certain circumstances and in accordance with clear criteria, refuse authorisation for medicinal products for human use on the grounds of serious environmental impact or if there is a threat to human life." If environmental risks attributable to the use of a medicinal product are to be taken into consideration in the assessment of the risk-benefit balance during the assessment of an application for marketing authorisation, those environmental risks may contribute to the rejection of the application.
Risk-benefit balance under current legislation In article 1.28 of Directive 2001/83/EC36 F
37 (the Human Medicinal Products Directive), risk related to the use of a medicinal product is defined as:
— any risk relating to the quality, safety or efficacy of the medicinal product as regards patients' health or public health.
— any risk of undesirable effects on the environment. According to article 1.28 the risk-benefit balance is defined as an assessment of the positive therapeutic effects of a medicinal product in relation to the risk according to the definition in point 28, first indent. As the second indent is not included in this article the risks of undesired environmental effects cannot be factors in the risk-benefit balance for medicinal products for human use. Pursuant to article 26.1 of the same directive, marketing authorisation shall not be granted if, after investigation of the information and documents submitted in support of the application, the risk-benefit balance is not deemed to be favourable. Despite this, under article 8.3 ca) of the Human Medicinal Products Directive an assessment of potential risks of the medicinal product should be included in an application for marketing authorisation for that product. Moreover, environmental impact should be studied and special
37 Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use.

Page 53 (96)
measures to reduce it should be developed in each case. The results of these studies may not, however, pursuant to what is stated above, be included in the risk-benefit balance for medicinal products for human use. Praxis for risk-benefit assessment The risk-benefit assessment is not described in any specific guidelines but is rather carried out based on experience and praxis developed by the EMA Committee for Medicinal Products for Human Use (CHMP) and the national authorities. Assessments are always made during evaluation of an application for marketing authorisation for a medicinal product. The assessment does not include any relative risk-benefit assessment where the therapeutic effects of different medicinal products are compared with each other, but account is taken of whether alternative treatments are available.
8.4. Courses of action for Measure C The table below shows the courses of action identified for Measure C.
Table 3: Overview of Measure C with courses of action, assessment and main effects.
Course of action Assessment Effect
C1 environmental information is seen as a factor in the risk-benefit assessment
The ERA and RMM are studied and weighed against other factors.
If the clinical risk-benefit balance is favourable for a new medicinal product, especially if it provides treatment for a serious disease or if there are only limited alternative treatments, this medicinal product would probably not be rejected on grounds of environmental risk. Risk minimisation measures would be implemented as far as possible instead, in order to minimise risk to the environment. If, on the other hand, the clinical risk-benefit balance is in question then serious environmental risk, which would not be able to be adequately contained with use of risk minimisation measures, would be able to contribute to the decision to reject an application for authorisation.
C2 Environmental impact of new medicinal products compared with medicinal products which have already been authorised
The ERA and the RMM are investigated. The environmental impact of a medicinal product is compared with the environmental impact of existing medicinal products which meet the same medicinal needs.
If there are other medicinal products which meet the same medicinal needs, a new medicinal product may be rejected in cases whereby serious environmental hazards remain after RMM have been taken into account and environmental risk is greater than for the medicinal product which has already been authorised.
8.4.1. Course of Action C1: Environmental data is seen as one factor in the risk-benefit balance
The purpose of this course of action is to take environmental risk and suitable risk minimisation measures into account in the risk-benefit assessment for each individual medicinal product. This assessment mostly takes into account environmental risks arising from the use of medicinal products. Under this course of action, the data on the environmental impact of a pharmaceutical substance becomes a factor to be taken into account in the risk-benefit assessment together with other factors which are currently part of the evaluation; quality, efficacy and safety. If the use of a medicinal product leads to environmental risks then appropriate risk minimisation measures are established. This

Page 54 (96)
ERA and RMM are investigated and considered
approach is reminiscent of the approach adapted for veterinary medicinal products, where risk minimisation measures are decided upon during the authorisation process.
Picture 9: Suggestion environmental data would be a factor in the risk-benefit analysis Changes in EU legislation would be required in order to implement this course of action since one condition of the proposal is that the above described articles in the Human Medicinal Products Directive should be supplemented, such that environmental risks will be taken into account in the risk-benefit assessment, see section 8.4.3. Praxis needs to be developed regarding when and how environmental risks will be included in the risk-benefit assessment. There are a number of different alternatives regarding when environmental data might be taken into our account in the risk-benefit assessment. The environmental data might be included each time a risk to the environment has been identified, or only when the risk would lead to potentially serious environmental or health effects (as a consequence of exposure to pharmaceutical substances via the environment, such as drinking water or contaminated food). As environmental considerations comprise a new factor in the assessment, praxis is needed for how it should be evaluated in relation to the other risks, especially when environmental risk assessment shows that the use of a medicinal product might cause serious environmental damage even after proposed risk minimisation measures have been factored in. If the clinical risk-benefit balance is favourable for a new medicinal product, especially if it provides treatment for a serious disease, or if there are only limited treatment alternatives, an application for authorisation of such a medicinal product would probably not be rejected on the grounds of environmental risk, since the benefit of the medicinal product would be valued more highly. Risk minimisation measures would be implemented as far as possible instead, in order to minimise risk to the environment. If, on the other hand, the clinical risk-benefit balance is questionable based on available data regarding quality, efficacy or safety, a serious environmental risk which has been identified (which remains after risk minimisation measures have been factored in) together with other risk factors might contribute to the application for authorisation being rejected. In rare cases the new medicinal product might be rejected exclusively on the grounds of environmental risk, but only where there is no possibility of minimising the serious environmental risk of the new
Medicinal products of human use 2001/83/EEC
Positive therapeutic effects in relation to the risks as regards patients' health or public healthand any risk of undesirable effects on the environment.
QualitySafety
EfficacyEnvironmental risk

Page 55 (96)
medicinal product and where there exist other medicines/treatments which ensure availability of adequate treatment. The Medical Products Agency deems that this course of action would probably seldom lead to a medicinal product being rejected solely on account of its risk to the environment. On the contrary, this course of action would lead to an increased application of risk minimisation measures. Appropriate risk minimisation measures need to be developed, principally because they need to be proportional to the environmental risks that they should minimise. Current pharmaceuticals legislation also states that appropriate risk minimisation measures should be developed, see article 8.3 ca) in the Human Medicinal Products Directive. This would, however, need to be further expanded and supplemented with new legislation such as restrictions on prescriptions, use of certain medicinal products exclusively for inpatient care, use of environmentally harmful medicinal products for restricted groups of patients (e.g. those who have not responded to other treatments a.k.a. second-line patients) or selection of other forms of the same medicine. In addition to this, risk minimisation measures developed by wider society, such as water treatment techniques should be taken into account. The development of risk minimisation measures ought to be driven on the initiative of the companies but should also be able to be harmonised as with veterinary medicinal products.
8.4.2. Course of action C2: Environmental impact is compared with environmental impact of other medicinal products
Environmental risk caused by the use of a specific medicinal product is compared with the equivalent environmental risk from other medicinal products which meet the same medical needs and which are already available on the market. This course of action has been put forward several times before including by the All-Party Committee on Environmental Objectives (SOU 2012:38).
Picture 10: Environmental impact of a new medicinal product compared with environmental impact of existing medicinal products
The environmental impact of a medicinal product subject to a marketing authorisation application is compared with the environmental impact medicinal products which have already been authorised. It follows that this type of environmental consideration would be enacted for medicinal products that have at least one equivalent on the market. For many new medicinal products, the effect and use of which is unique, this type of environmental consideration would not apply.
New medicinal products
Positive therapeutic effects in relation to the risks as regards patients' health or
public health
Environmental risk
Existing medicinal products, with the same
medical need
Positive therapeutic effects in relation to the risks as regards patients' health or
public health
Environmental risk

Page 56 (96)
Changes in EU legislation would also be required in order to implement this course of action. The Human Medicinal Products Directive must be changed in order to allow environmental risks to be included in the risk-benefit assessment, see section 8.4.3. Besides changes to pharmaceuticals legislation this course of action would require
• it to be possible to select medicinal products which are seen as meeting the same medicinal needs,
• environmental data for these medicinal products to be available, • there to be guidelines for the comparison of environmental impact between medicinal
products, and • risk minimisation measures to have been developed.
There is currently no method for selecting which medicinal products might be seen as "meeting the medicinal need in question". Under current legislation a risk-benefit assessment is carried out for the specific medicinal product for which marketing authorisation is sought. This assessment does not include a relative risk-benefit assessment which compares medicinal products with one another (on an EU basis). Establishing which medicinal products "meet the medicinal need in question" and then comparing these with one another entails completely new aspects to the risk-benefit assessment and would entail development of new praxis. In addition, the diversity of medicinal products sold in different EU countries means that it would be difficult to decide which medicinal products should be included in the comparisons for "meeting the same medicinal need". Comparison of the outcomes of the risk-benefit assessment for different medicinal products is not carried out within the framework of existing EU procedures for authorisation. On the other hand, comparisons are made by different national bodies in Sweden for various purposes after authorisation has been granted for the medicinal product. Comparisons between different medicinal products (without environmental considerations) are made during the development of treatment recommendations. The Medical Products Agency carries out an assessment on possible interchangeability of medicinal products. One example of where authorised medicinal products are compared with each other is the creation of the pharmaceuticals committees' recommendations lists, as well as those of the Drug and Therapeutics Committees working at the Swedish County Councils (Stockholms läns läkemedelskommitté, SLK). SLK draws up a list of recommendations, “Kloka Listan”, which contains medicinal products which are recommended for the treatment of common diseases. These recommendations are based on scientific documentation regarding the efficacy and safety, pharmaceutical suitability, cost efficiency and environmental aspects. In the assessments from SLK account is taken of all available information about the medicinal product in question, for example comparative studies, clinical experience, treatment recommendations, new areas of use and reported side effects. This knowledge is wider than the knowledge used in the risk-benefit assessment of the medicinal product which takes place in conjunction with the authorisation process and is thus mainly based on results from clinical studies. The Dental and Pharmaceutical Benefits Agency also compares various medicinal products whereby the cost-effectiveness (measured as cost per quality-adjusted life year) of a new medicinal product is assessed in relation to existing medicinal products. The assessment does not include any consideration of any potential environmental impact of the medicinal product. The total environmental risk of a medicinal product has different weight in different situations since this course of action is based on comparisons within similar therapeutic groups. Two substances with "the same" total environmental risk ought to be able to be dealt with in a different way, depending on if the comparison groups contain medicinal products which impose a heavy or light environmental

Page 57 (96)
burden. If a new substance imposes a medium level environmental burden, risk minimisation measures would be required or it might be banned when it is compared with substances imposing a low burden. On the other hand, it would be seen as better from an environmental standpoint when compared with pharmaceutical substances which impose a heavy environmental burden. This relative comparison basis might be perceived as an unjust treatment of similar substances. It might also cause companies to experience barriers to competitivity. Guidelines for comparison of the environmental impact of different medicinal products with one another would need to be drawn up. Experience from other chemicals legislation might be used here. Risk minimisation measures need to be developed for this course of action too. Under the proposal from the All-Party Committee on Environmental Objectives medicinal products with a negative impact on the environment could be denied authorisation if there are already other medicinal products on the market which meet the medicinal need in question. In view of the fact that denying authorisation is a drastic measure, the Medical Products Agency stipulates that risk minimisation measures should also be the measure of first resort in assessments under this course of action. Also see section 8.4.1. Under this course of action an application for authorisation of a new medicinal product could be rejected if there is already another medicine/treatment on the market which can be given to the patient and if there are no risk minimisation measures which could minimise the serious environmental risk that the use of the new pharmaceutical product would entail. The difference from course of action C1 is that in C2 the environmental risk of a medicinal product is evaluated based on a comparison with existing medicinal products and not on the need for measures due to actual environmental impact of the medicinal product.
8.4.3. Need for revision of legislation and consequences of changes In order for environmental considerations to be taken into account in the risk-benefit assessment, changes must be made to the legislation. References to the below articles are to the Human Medicinal Products Directive37 F
38 unless otherwise stated. In order to achieve successful implementation of Measure C a change would need to be made to article 1.28a so that the subsection on undesirable environmental effects in article 1.28 would be included in the risk-benefit balance. If environmental risks are included in the risk-benefit balance then it follows that the environmental effects must be taken into consideration and monitored during the whole of the life-cycle of the medicinal product. If undesirable environmental effects are included in the risk-benefit assessment, these effects may contribute to the risks being seen as outweighing the benefits for any specific medicinal product and thus contribute to the marketing authorisation application being rejected, see article 26.1 a. Under article 23.2 the holder of a marketing authorisation must at the earliest opportunity submit to the national competent authority any new information which might entail changes to the information and documentation referred to in article 8.3 and appendix 1 of the directive and elsewhere. The environmental risk assessment is covered by these references. In addition, the holder of the authorisation must inform the relevant national authority of any new information which might influence the assessment of the risks and benefits of the medicinal product in question. It is expressly stated that the information shall contain both positive and negative outcomes of clinical trials or other
38 Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use.

Page 58 (96)
studies in all indications and populations, regardless of whether this is included in the marketing authorisation, as well as data on the use of the medicinal product where this use is not covered by the conditions stated in the marketing authorisation. A national competent authority may under article 23.4, in order to be able to continuously assess the risk-benefit balance, require at any time the holder of the marketing authorisation to submit information showing that the risk-benefit balance is still favourable. It is also stipulated that the holder of the marketing authorisation must fully and without delay respond to such a request. If the environmental risk is included in the risk-benefit balance, these articles will have a direct significance regarding changed circumstances referring to knowledge about the environmental effects of the medicinal product. Something that should be considered is making it possible for environmental risks that a concerned member state sees in Community procedures to occasion an action in accordance with article 29.1. In other words, that an environmental risk that the member state believes has not been taken sufficiently into consideration during the investigation or during previous authorisation (mutual recognition or decentralised procedure) should be able to lead to, as a last resort, referral for a final decision on what applies to the medicinal product in question. Under article 116 the national competent authorities should temporarily revoke, suspend or change the marketing authorisation, if, for example they believe that said medicinal product is harmful, that it lacks therapeutic effect, or that the risk-benefit balance is not favourable. Further, the marketing authorisation may also be temporarily revoked, suspended or changed if the data submitted in support of the application under article 8 (and elsewhere) is erroneous or has not been changed in accordance with article 23. It follows from article 117 that the member states shall take suitable measures to ensure that the supply of a given medicinal product is banned and that said product is withdrawn from the market if it is deemed that the risk-benefit balance is not favourable. The table below shows schematically how the decisions concerning risk-benefit assessments for medicinal products for human use may be changed in order to introduce environmental considerations under Measure C. The proposal would mean that the risk-benefit assessment would be similar to the assessment carried out for veterinary medicinal products. Each course of action has, however, specific ways of taking into account environmental risks in the risk-benefit assessment.
Table 4: Risk-benefit ratio under each piece of legislation. Proposals which apply to Measure C are written in bold
Medicinal products for human use 2001/83/EEC
Veterinary medicinal products 2001/82/EEC
Positive therapeutic effects in relation to risk to the health of the user or public health and undesirable environmental impact Factors:
• Quality • Safety • Efficacy • Environmental risk*
Positive therapeutic effects in relation to risk to the health of humans and animals and undesirable environmental impact Factors:
• Quality • Safety • Efficacy • Environmental risk
*Environmental and/or health risk risks due to human exposure to medicinal products via the environment

Page 59 (96)
According to the All-Party Committee on Environmental Objectives it would be desirable to have the risk-benefit assessment include the impact on human health due to exposure to medicinal products via the environment (e.g. through the consumption of contaminated drinking water or fish). This in turn would require the pharmaceutical companies to carry out a health risk assessment, which is not a requirement under current legislation. As this aspect is not included in this assignment no further analysis has been made but it might need to be studied more closely. If Course of Action C2 is chosen, under which the investigation of an application for marketing authorisation would compare the harmful environmental effects of a medicinal product with those of other medicinal products which meet the same medicinal need, the method of selection of such medicinal products would need to be laid down in the legislation. Under the Medical Products Agency proposal environmental risk which is identified and included in the risk-benefit assessment should be that risk which remains after implementation of risk minimisation measures. Use of such measures needs to be developed, both from the applicants' side and regarding measures which can be taken in conjunction with authorisation. Measures which can be taken in conjunction with authorisation of medicinal products should be created such that they apply in common across the EU so that their application is harmonised. Changes to legislation may also be required. This would apply to, for example, the proposal that one risk minimisation measure would be to make a medicinal product prescription only. Article 71 stipulates when a given medicinal product shall be prescription only. The factor that the medicinal product might pose a risk to the environment is not present there. The corresponding article in Directive 2001/82/EC on the Community code relating to veterinary medicinal products does contain such a factor (see article 67 in the Directive). If the Medical Products Agency proposal is to be fully implemented, article 71 ought then to be amended such that environmental risk may also constitute grounds for designating a medicinal product for human use prescription only.
8.5. Impact of Measure C Regardless of the course of action, the inclusion of environmental considerations in the risk-benefit assessment entails the following major actions: revision of the guideline document for environmental risk assessments, better implementation of the evaluation of environmental risk assessments, making environmental information accessible and also including environmental considerations in post authorisation control work. The inclusion of environmental considerations in the risk-benefit assessment ought also to lead to a certain change in perspective, since not only the interests of patients but also those of the environment will be taken into account. In addition, the complexity of the risk-benefit assessment would increase, which might cause the authorisation process to consume greater resources. Below is a description of the above-mentioned effects in more detail as well as effects specific to the different courses of action.

Page 60 (96)
Table 5: Breakdown of effects of Measure C and effects of the respective courses of action Course of Action C1: Environmental risk is seen as one factor in the risk-benefit balance
Course of Action C2: The environmental risk of a new medicinal product is compared with environmental risks of existing products
General effects
• New perspective in risk-benefit assessments The inclusion of environmental considerations might lead to a change in perspective, since not only the interests of patients but also those of the environment will be taken into account.
• Increased complexity of the risk-benefit assessment The inclusion of environmental considerations should mean that a new factor is introduced into the risk-benefit assessment. This new factor which contains both environmental risk assessment and appropriate risk minimisation measures would probably lead to greater complexity in the risk-benefit assessment. Before a common EU approach has been established this process may be both time-consuming and difficult to harmonise. This in turn might result in the authorisation process consuming greater resources.
• Revision of guideline document for environmental risk assessments If environmental risk assessments are to be factored in, they must be fit for purpose, containing relevant information. Current methodology has been criticised for not reflecting the specific environmental impact of pharmaceutical substances. It would therefore require revision before environmental considerations could be factored into the risk-benefit assessment.
• Better implementation and evaluation of environmental risk assessments If environmental data is to be a factor into the risk-benefit assessment, it may be assumed that environmental risk assessments would be better carried out by companies and evaluated by authorities.
• Stronger requirement for environmental data to be made available The significance of environmental information in the risk-benefit assessment ought to be transparent and may well entail environmental information on medicinal products being made available.
• Development of risk minimisation measures The introduction of environmental considerations into the risk-benefit assessment might lead to the development of new and improved risk minimisation measures.
• Environmental considerations in post authorisation control work The quality, efficacy and safety of a medicinal product is monitored in a post control system. Among other things this means that if it becomes apparent that the use of the medicinal product leads to new knowledge, e.g. side effects which affect the risk-benefit balance, the assessment might have to be re-evaluated. The introduction of environmental considerations into the risk-benefit assessment would result in environmental considerations also being included in this post authorisation control system.
• Competence requirements for assessing environmental considerations in the risk-benefit assessment The introduction of environmental considerations into the risk-benefit assessment would create the need for new competences. Environmental competences would probably be needed in the companies and the authorities.
• If only new medicinal products are covered by the proposal there would be a great danger of existing, more environent-ally hazardous medicinal products remaining on the market.
Impact on the development of medicinal products
• The introduction of environmental considerations into the risk-benefit assessment sends a signal to the companies. It is difficult to predict how this signal might influence the pharmaceutical companies and their approach to the development of medicinal products. It might appear that certain risk minimisation methods could limit sales of a medicinal product, but on the other hand the risk minimisation measures might make it possible for medicinal products with serious impact on the environment to gain authorisation.
• As additional medicinal products which place a heavy burden on the environment would be the
• As the bases for comparison are dependent on the environmental impact of existing medicinal products,

Page 61 (96)
Course of Action C1: Environmental risk is seen as one factor in the risk-benefit balance
Course of Action C2: The environmental risk of a new medicinal product is compared with environmental risks of existing products
target for more or more stringent RMM (e.g. restricted prescription) this might lead to more restricted profits for the companies. This might result in a given medicinal product with certain properties, e.g. PBT or endocrine disruptive substances becoming less interesting prospects for prescription and for development.
new medicinal products which impose the same environmental burden might be evaluated differently, see the above discussion. This may result in the company expressing uncertainty in the development of medicinal products.
• If only new medicinal products are covered, then there would be no change to the current situation regarding previously authorised medicinal prod-ucts, whose use entails environmental risks, and which have not undergone the same environ-mental assessment or been covered by the same requirements for risk minimisation measures in conjunction with the authorisation process.
• If only new medicinal products are covered by this requirement, it may lead to the development of new medicinal products being impeded. A new mechanism would therefore be needed here, which would phase the old medicinal products into the system.
• It is possible that the new requirements would result in a developmental niche for the companies, where they might invest in developing new medicinal products with less environmental impact than at present.
Impact on availability of medicinal products
• If a new medicinal product does not receive authorisation, no information will be obtained on its possible future positive impact on new indications, except for those the company initially sought authorisation for. For a number of medicinal products new positive effects and possible uses in other therapies first become apparent during use.
• New innovative medicinal products with high medicinal demand would not be affected by this course of action.
• New innovative medicinal products for which there are no comparable medicinal products would not be affected by this course of action.
• In rare cases (presenting serious environmental hazards) this course of action might result in authorisation of a medicinal product being declined.
Impact on the pharmaceutical companies
• The introduction of environmental considerations into the risk-benefit assessment might entail higher costs for the companies partly due to revised testing requirements (Measure A) and partly due to the increased complexity of the application procedure for marketing authorisation.
• The impact on the medicinal product could vary depending on what praxis is developed for environmental considerations in the risk-benefit assessment. Including a new factor in the risk-benefit assessment might create uncertainty for the companies.
• The impact on the medicinal product might vary depending on other medicinal products on the market and comparison criteria. This might create uncertainty for the companies.
• This might impact on market share for medicinal products which place a high burden on the environment as companies could compete by marketing their product as "environmentally friendly".
Impact on human health and the environment
• Introduction of environmental considerations into the risk-benefit assessment might result in a number of effects that together contribute to increased priority for environmental issues. This may lead to a medicinal product having a list of relevant measures aimed at reducing its environmental impact the moment it is placed on the market. This is generally a positive thing for the environment and for public health. In individual cases, when a medicinal product has not been authorised for marketing or when its use is restricted to certain groups of patients, this may result in diminished patient health.

Page 62 (96)
9. Measure D: A regulation of minimum requirements for conditions of manufacture for the sale of products in the EU market
9.1. Previous government assignment for Measure D The Medical Products Agency previously issued the assignment in the 2009 appropriation directions, to lay out the possibilities of tightening up environmental requirements on the manufacture of medicinal products and active ingredients, with various proposals for measures to the government (Medical Products Agency, 2009). The main proposal submitted was the requirement for emissions of active pharmaceutical ingredients to be inserted into legislation on GMP (Good Manufacturing Practice). In the appropriation directions of 2011 the MPA was tasked with, based on the authority’s 2009 report on the possibilities of tightening up environmental requirements for manufacturing, laying the groundwork to facilitate the initiation of a revision of EU legislation on Good Manufacturing Practice. The assignment was divided into two stages: 1. “Analysis of how work on a revision of GMP should be structured in order to have the greatest chance of success” (Medical Products Agency 2011a) and 2. “Groundwork to facilitate the initiation of a revision of EU legislation on Good Manufacturing Practice, with the aim that legislation will also cover environmental considerations” (Medical Products Agency 2011b). In April 2014 the Swedish Environmental Protection Agency was tasked by the government with assignment (M 2014/991/Ke) in conjunction with the Medical Products Agency to document and analyse how regulations and guidance documents that will succeed directive 2010/75/EU of the European Parliament and Council on industrial emissions of 24 November 2010 can be adapted to regulate emissions of pharmaceutical substances and potential opportunities and obstacles the directive might bring in relation to the MPA’s previous proposals on the regulation of emissions through EU directives which regulate good manufacturing practice of pharmaceuticals for human and veterinary use (Swedish Environmental Protection Agency 2014). These four reports with government assignments issued have constituted the basis for the Medical Products Agency’s ongoing work with current assignments.
9.2. Limitations to measure D The wording in the government assignment “regulation of minimum requirements on production conditions for the sale of products on the EU market” renders only legislation that regulates production of medicinal products within the framework of medical products regulation and current GMP since it is here that the connection to the sale of medicinal products exists. For this reason, other possible voluntary measures for the reduction of environmental impact from industry are not described and an account of the possibilities of placing environmental requirements on public procurement in county councils and municipalities and the like are not set out. Something that could be investigated, but which is not part of the assignment, is whether it is possible to include a general text in the European Pharmacopoeia3 8F
39 to the effect that medicinal products and 39 The European Pharmacopoeia (Ph. Eur.) is the official collection of regulations and methods that concern the design, manufacture and quality of medicinal products for human and veterinary use, which is published by the Council of Europe. Ph.Eur. is administered by the European Directorate for the Quality of Medicines and Healthcare (EDQM).

Page 63 (96)
pharmaceutical substances must be manufactured in accordance with current environmental requirements. Such a text could be published in the European Pharmacopoeia as the official standards published there provide a legal and scientific basis for quality control during the development, manufacturing and marketing processes. All manufacturers of medicinal products and/or substances for pharmaceutical use must apply these quality standards in order to be able to market their products in the states that have signed the Council of Europe Convention on the Elaboration of a European Pharmacopoeia.
9.3. Background to Measure D During the last decade the manufacture of medicinal products for the European market has increasingly been transferred outside Sweden and Europe. This means that the manufacture of medicinal products and active ingredients and the resultant emissions takes place to a great extent in countries where the majority of these medicinal products are not consumed. Manufacture often takes place at the behest of companies in countries which are not their corporate domicile. One very relevant question, therefore, is what the legal situation is regarding influencing manufacture in a third country. This is important not least if we wish not to export environmental problems which stem from consumption within the EU. The overarching objective of Swedish environmental policy is defined by the “Generational Goal”. The goal is to pass to the next generation a society where the major environmental problems are resolved without causing environmental and health problems abroad. If emissions from Swedish production are simply moved outside Sweden and the EU then this goes against this “Generational Goal”. An important principal which exists within environmental legislation is to strive towards the Polluter Pays Principle. It is appropriate for a similar principal to apply to the minimisation of emissions of active ingredients from the manufacture of medicinal products harmful to health or the environment. The production chain for medicinal products is often long and covers a series of steps which often involve a number of companies and countries. The chain involves everything from the development of raw materials such as oil products, minerals and other natural products, further manufacture of intermediates, synthesis of pharmaceutical substances through to formulation and manufacture of pharmaceutical preparations, packaging and distribution. From manufacture of substances and later in the chain there is a risk that active pharmaceutical ingredients will reach the environment. As the compilation in chapter 3 shows, there are studies that indicate that significant emissions occur with certain medical products industries in EU as well as in third countries. In brief, this shows that medicinal products are disseminated not only by use but also via production in a way that demonstrably harms the local environment and contributes to the global antibiotic resistance problem. The common EU medicinal product legislation currently lacks manufacturing rules linked to environmental requirements such as emissions of pharmaceutical substances and their impact on the environment. Medical product manufacture is, however, defined as a chemical industry and is to a certain extent covered by EU chemical product legislation such as Reach3 9F
40 and the CLP regulation40 F
41.
40 Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC. 41 Regulation (EC) No 1272/2008 on classification, labelling and packaging of substances and mixtures, amending and repealing Directives 67/548/EEC and 1999/45/EC, and amending Regulation (EC) No 1907/2006.

Page 64 (96)
In the manufacture of medicinal products within the EU, the Industrial Emissions Directive (IED)4 1 F
42 and the Water Framework Directive42 F
43 can be applicable to regulate emissions of pharmaceutical substances harmful to the environment. One basis for the course of action described below is the previous government assignment proposals (16-06-2011) that a review of the directive regulating Good Manufacturing Practice, GMP, should be implemented such that they include requirements regarding environmental considerations in the manufacture of medicinal products and active ingredients. Therefore, there is a brief description below of the provisions which are significant for the previous proposals.
9.3.1. Authorisation requirements for manufacture in the EU As previously described in the report there are two central directives for medicinal products, Directive 2001/82/EC of the European Parliament and of the Council on the Community code relating to veterinary medicinal products and Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use (referred to below as the medicinal products directives). Many of the provisions described below occur in both directives. When an article is referred to without a particular reference then the first article cited refers to Directive 2001/82/EC and the second article cited refers to Directive 2001/83/EC. The Medicinal Products Directives stipulate that an authorisation is required from a competent medicinal products authority in the EU in order to manufacture (articles 44 and 40) and sell (articles 5 and 6) medicinal products within the EU. The requirements in place for a prospective medicinal products manufacturer to gain authorisation are stipulated along with what requirements the manufacturer must adhere to. Such authorisation is required regardless of whether said manufacture involves whole or partial production of the medicinal product, certain stages in the manufacturing process or various procedures for division, packaging, or wrapping/presentation. Authorisation for manufacture can apply to particular medicinal products or to all. Special authorisation is also required for the import of medicinal products from third countries and the directive’s rules of manufacture also apply to imported medicinal products. Thus, the same manufacturing requirements apply no matter where manufacture takes place.
9.3.2. Good Manufacturing Practice (GMP) in the legislation Those granted authorisation for the manufacture or import of medicinal products must conform to a number of requirements listed in the directives. One of these is to conform to the principals and guidelines for good manufacturing practice, GMP, for medicinal products and thereby only use as starting material active ingredients manufactured in accordance with good manufacturing practice for active ingredients (articles 50f and 46f). In articles 51 and 47 the Commission is assigned the task of adopting, in the form of a directive, the principals and guidelines of good manufacturing practice set out in articles 50 and 46. As a consequence of this the Commission has adopted Directive 91/412/EEC laying down the principles and guidelines of good manufacturing practice for veterinary medicinal products and Directive 2003/94/EC laying down the principles and guidelines of good manufacturing practice in respect of medicinal products for human use and investigational medicinal products for human use. In article 51 of the Veterinary Medicinal Products Directive the Commission is delegated the task of adopting detailed guidelines for good manufacturing practice for active ingredients used as starting materials.
42 Directive 2010/75/EU of the European Parliament and of the Council of 24 November 2010 on industrial emissions (integrated pollution prevention and control) 43 Directive 2000/60/EC of the European Parliament and of the Council of 23 October 2000 establishing a framework for Community action in the field of water policy.

Page 65 (96)
Such guidelines have also been adopted43 F
44. Article 47 of the Human Medicinal Products Directive stipulates that the Commission shall adopt by means of delegated acts the principles and guidelines of good manufacturing practice for active ingredients. With the support of this article the Commission has adopted a delegated regulation on this, (EU) No 1252/201444 F
45. The Commission will also adopt guidelines for good distribution practice for active ingredients. Article 2 of both the Commission directives on GMP contains a definition of good manufacturing practice. Good manufacturing practice is that part of quality assurance that is intended to ensure that the products are always made and subjected to controls in such a way that they meet the quality requirements appropriate for their intended use. According to the medicinal products directives the manufacturer’s conformity with the rules of manufacture and GMP should be inspected by the competent authorities, articles 80 and 111. After inspection has been carried out the authority shall issue a GMP certificate. If there are deficiencies in adherence to good manufacturing practice the authorities may issue a “non-compliance GMP statement”. This certificate is published in a common EU database, EudraGMDP, so that the information is disseminated to all member states. Inspections carried out are continuously entered in to this database by the competent authorities in the EU member states. This applies to inspections carried out both within and outside the territory of the member state carrying out the inspection. See articles 80.6 and 80.7 plus articles 111.6 and 111.7. According to articles 85.2 and 118.2 authority to manufacture may be limited or revoked, if, for example, GMP is not complied with.
Picture 11: Fundamental requirements for manufacturers and importers of active ingredients and medicinal products.
9.3.3. Good Manufacturing Practice (GMP) Global agreements Medicinal products legislation for GMP is not only harmonised within the EU but there is also close cooperation regarding work in this area between the EU and PIC/S (the Pharmaceutical Inspection Co-operation Scheme). PIC/S is an association of currently 45 medicinal products authorities from all over the world who have adopted a joint or equivalent GMP guide. At the current time the content of these GMP documents is closely harmonised. This cooperation aims, as far as is possible, to strengthen the extent to which changes in good manufacturing practice are harmonised on the global level. This integrated work further strengthens the impact in adjusted requirements and recommendations. A harmonised GMP regulatory framework benefits both the medicinal products industry and the medicinal products authorities.
44 The Rules Governing Medicinal Products in the European Union, Volume 4, Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use, Part II: Basic Requirements for Active Substances used as Starting Materials See http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm 45 Commission Delegated Regulation (EU) No 1252/2014 of 28 May 2014 supplementing Directive 2001/83/EC of the European Parliament and of the Council with regard to principles and guidelines of good manufacturing practice for active substances intended for medicinal products for human use.
Manufacturers/importers of active ingredients• Must give notification of activities to competent authorities in the member state where said activities are established.
•Must comply with GMP for starting materials set out in the guidelines and delegated acts issued by the Commission.
Manufacturers/importers of finished medicinal products (or parts of finished medicinal products)•Must be in possession of authorisation to manufacture in accordance with the medicinal products directives.
•Must comply with GMP set out in directives issued by the Commission.

Page 66 (96)
There is also an exchange of information with WHO which is the third major player with its own GMP regulatory framework. There are very good reasons for initiating discussions with international cooperation constellations, in parallel with further investigation of environmental considerations in the definition of GMP at the EU level, in order to avoid unilateral hikes in GMP requirements. The objective should be to identify possible common ground on the global level.
9.4. Suggested changes to the regulations Rendered in brief below are the Medical Products Agency’s previous suggestions (report 16-06-2011) on how the regulations might be changed in order to achieve the objective that environmental requirements will be included in the definition of GMP. GMP legislation is currently constructed such that the medicinal products directives stipulate that manufacture of medicinal products and active ingredients should comply with GMP. The definition of the concept of Good Manufacturing Practice along with principles and guidelines for GMP are set out in the Commission directives. There are special guidelines for GMP in the manufacture of active ingredients. In a previous report the Medical Products Agency proposed that the definition of GMP be moved from the Commission directives and placed in article 1 of each of the medicinal products directives. Article 1 of both medicinal products directives consists of several definitions. The Medical Products Agency has further proposed the definition should also state that a new legislative act regulating emissions levels for certain specific environmentally hazardous substances shall be followed when such substances are manufactured. Moving the definition of GMP to the medicinal products directives and including the stipulation than the new legislative act shall be followed achieves the goal of introducing environmental control into GMP. A legislative act shall as a rule be complied with by those to whom regulation applies. It might therefore appear strange for the definition to stipulate that another legislative act should be adhered to. Such a solution is necessary, however, in order to achieve the aim of the proposal, namely that environmental requirements should also have an impact on the manufacture of medicinal products and active ingredients in a third country intended for the European market. Since manufacture of active ingredients and medicinal products in third countries must at least fulfil the requirements of the GMP in effect in the EU, the proposal has the desired effect. Exactly how that new legislative act should be written in the definition needs to be specially studied. The Medical Products Agency proposal of 16-06-2011 proposed the following. Proposed new wording (new text in bold):
good manufacturing practice is that part of quality assurance that is intended to ensure that the products are always made and subjected to controls in such a way that they meet the quality requirements appropriate for their intended use and the requirements set out in “the legislative act”.
The exact form of the definition will also be dependent on what course of action is chosen, see section 9.5. The courses of action contain proposals for different legislative acts in which emission levels can be regulated and also different ways to regulate emission levels.

Page 67 (96)
Picture 12: Proposal for amended legislation to insert environmental requirements in the definition of GMP.
9.5. Courses of action for Measure D In order to be able to implement the above proposals the decision must be taken regarding in which legislative act emissions should be regulated. The following two courses of action both result in the requirement that environmental considerations are inserted into the definition of GMP.
Table 6: Overview of alternative courses of action for Measure D
Course of Action Measure Effect
D1: In the definition of GMP reference is made to a new EU regulation where emissions of active ingredients are regulated.
An EU regulation is drawn up where emission limits are set out.
Emissions of active pharmaceutical ingredients in the regulation are covered. Could conceivably cover all substances.
D2: In the definition of GMP, reference is made to the framework for the Industrial Emissions Directive (IED) where the emission limits of active pharmaceutical ingredients are regulated.
The relevant best available techniques (BAT) in the industry are developed. Emission limits for active ingredients are established.
Emissions of active pharmaceutical ingredients regulated in the IED are covered. Could probably cover all substances.
9.5.1. Course of Action D1: In the definition of GMP reference is made to a new EU regulation where emissions of active ingredients are regulated.
In this course of action, it is proposed that a new legislative act is adopted in the form of an EU regulation. In this regulation emission limits for the manufacture of medicinal products and active ingredients are set out. All manufacturers of such products in the EU are directly covered by the regulation. If they fail to comply with the requirements of the new regulation then according to the proposal they will additionally be regarded as not having fulfilled good manufacturing practice. By
Move the current definition of good
manufacturing practice from the Commission
directives on GMP to the medicinal products
directives
Add a supplement to the definition to the effect that requirements on emissions set out in
another legislative act shall be complied with so that good manufacturing
practice is followed.
Adopt limits for emissions for the manufacture of medicinal products and active ingredients in a
legislative act referred to in the GMP definition.

Page 68 (96)
having the emissions requirements covered by the GMP definition, manufacture in third countries will also be covered by the requirement to adhere to the emission limits. The emission limits may, for example, be formulated as a fixed general limit, be individually assigned for specific active pharmaceutical ingredients or be decided by a calculation model. Depending on the desired effect, the different means may be combined, see section 9.6. All emissions of active pharmaceutical ingredients may, with this course of action, be regulated with minimum requirements
9.5.2. Course of action D2: In the definition of GMP, reference is made to the framework for the Industrial Emissions Directive (IED) where the emission limits of active pharmaceutical ingredients are regulated.
As with course of action D1 the definition of GMP is changed in the medicinal products directives but in course of action D2 the definition refers to emission limits established within the framework of the IED4 5 F
46 rather than in a new legislative act. As for course of action D1 the emission limits may be formulated in different ways (individually assigned for specific active pharmaceutical ingredients or by a calculation model) in order to achieve the desired effect. The idea is not for the whole IED directive to be included in GMP but only the emission limits for pharmaceutical substances which are established within the IED regulatory framework. In order to be able to implement this course of action the regulations within the IED framework need to be changed or supplemented such that the emission limits for active pharmaceutical ingredients are given. These changes mean that every manufacturer of medical products in the EU who is covered by the IED has to adhere to the emissions requirements. By having the emissions requirements also covered by the GMP definition, as per the MPA proposal, other manufacturers of medicinal products and active ingredients in the EU and third countries (whose manufactured products are intended for the common market) will also be covered by the requirement to comply with the emission limits. The IED covers activities with high potential for pollution. Included here are industries within the energy sector, the metal and mineral industries, the chemicals industry, waste management and others such as the pulp and paper industry, the foodstuffs industry and large facilities managing livestock. Point 4.5 in the appendix means that manufacture of medicinal products as well as intermediate products is4 6 F
47 covered by the directive. This is subject to the condition that manufacture includes chemical or biological reactions and takes place on an industrial scale. This means that production plants that produce medicinal products and intermediate products that meet these criteria shall undergo an authorisation procedure in accordance with the IED. This authorisation procedure shall oversee environmental impact from the production plant, e.g. emissions into water. The objective of IED is an integrated approach to environmental management at the European level and is a minimum directive which means that the member states have the right to implement or maintain more but not less stringent requirements than those that come from the directive. For interpretation of the best available techniques (BAT), the EU has produced special interpretation documents, best available techniques reference documents (BREFs). These are the documents drawn up for specified activities and which in particular describe applied techniques, current emissions and consumption levels, techniques taken into account for the establishment of best available techniques and BAT conclusions. For activities included in BREFs there is information on and examples of BAT 46 Directive 2010/75/EU of the European Parliament and of the Council on industrial emissions (integrated pollution prevention and control). 47 The IED does not define the term intermediate but according to REACH an intermediate (product) is defined as a substance that is manufactured for and consumed in or used for chemical processing in order to be transformed into another substance or product.

Page 69 (96)
which, if used, leads to the achievement of a certain lowest possible emission level. In order to create a basis for information on available techniques and emission levels work was initiated by an EU task group surveying the best available techniques within the relevant branches. The conclusions which include emission levels (BAT Associated Emission Levels, BAT-AEL) have been given a special status in the directive such that they must be adhered to by latest four years after the publication of the conclusions regarding the plant’s main IED activities, unless special dispensation has been granted. The fact that emission requirements established for active pharmaceutical ingredients within the framework of the IED are also covered by the GMP definition (by reference to the emission limits in the IED) means that compliance with these emission requirements becomes a condition for fulfilment of good manufacturing practice. More manufacturers are covered by the requirements for GMP than fall under the scope of the IED as the definition of manufacture of medicinal products is broader in the medicinal products directives. By means of this proposal the emissions requirements established within the framework of the IED (which are relevant in the manufacture of medicinal products) will therefore be applicable for more producers than are covered by IED. As for Course of Action D1 the emissions requirements will also be applicable to the manufacture of medicinal products and active ingredients in third countries when the products are intended for the EU market, since such products must at least meet the requirements in force in the EU in order to be imported. In its review of government assignment (M 2014/991/Ke), in conjunction with the Medical Products Agency, the Swedish Environmental Protection Agency analysed how the successor legislation and guide documents to the IED can be adapted to regulate emissions of pharmaceutical substances. The conclusion is that there are a number of possible courses of action for the regulation of emissions of active ingredients in the manufacture of pharmaceuticals, including implementing requirements through GMP as well as via IED and BREFs for the medicinal products industry. The Swedish Environmental Protection Agency sees no significant disadvantages or obstacles in the implementation of regulations and requirements through both the IED and GMP, where the regulations ought to be able to complement each other. It is important for parties involved in the various processes to consult with one another and that there is coordination in the work in order to prevent conflicting requirements. In order to pass a regulation on emissions of pharmaceutical substances in the forthcoming BREF it is necessary for active preparation of the issues to begin now.
9.6. Setting emission limits for the substances to be regulated An important part of both courses of action is to fix a procedure for how emission limits shall be established for the substances to be regulated. The formulation of the regulations should anticipate and prevent the circumvention of the emission limits by, for example, moving uncleaned water from the production plant to an external purification plant. It is also important that emission limits be given per unit produced, so that dilution is not an alternative way for producers to meet environmental requirements. In general, requirements for emission limits within the framework of medicinal product legislation are formulated as minimum requirements, so that they do not take precedence over more stringent emissions requirements that may fall upon the manufacturing plant within the framework of environmental legislation. As such requirements are adapted to the environment of the production plant the limits must have precedence over the (probably) more general emission limits within the framework of medicinal product

Page 70 (96)
legislation. If, on the other hand, the limits set under the medicinal products legislation are more stringent in any individual circumstance, these should apply. There are various ways to regulate emissions content. Below are given three ways which may be combined to achieve the desired effect. Some deeper analyses of their formulation are needed but this lies outside the scope of the current report.
9.6.1. Regulation of specific substances The environmental hazards presented by certain active pharmaceutical ingredients are already well known. This is the case, for example, with antibiotics with their risk of the development of resistance and with certain hormones which may affect some species of animals which are exposed to these substances in the wild. One possible way to regulate emissions from production is therefore to stipulate emissions content for particular environmentally hazardous active pharmaceutical ingredients. Such a solution brings the task of determining which substances are to be regulated and which emission levels should be set. With this option new active ingredients/new chemical entities (NCE) cannot be covered by any emission limits until they have been included in the next revision of the legislative act. The option also has the condition that there should be regular monitoring to assess whether more substances should be specifically regulated or if emission limits need to be adjusted. Some kind of organ with environmental competence will probably be needed within the EU or the EMA to manage documentation and questions around investigation of which substances should be covered by emission limits and to set appropriate emission levels for these substances. The procedure with a candidate list such as Reach has can be a starting point for considerations of how new substances which need to be controlled shall be selected and considered for emissions regulation. Environmental risk information for the selection of substances will probably need to be obtained from a number of different sources especially for older substances which do not have ERAs. Sources might be information from the authorities’ environmental monitoring and research results. This option can appropriately be combined with a maximum value for emissions of substances which do not have a specifically assigned emissions limit. This is described more closely in section 9.6.3 below. If regulation of specific substances takes place within the framework for the IED, then the established procedure in place for the establishment of best available techniques (BAT) and the emissions levels reached thereby are used, see section 9.5.2. This way of regulating emission limits can be used regardless of which of the courses of action is chosen. This means that emission limits are transparent and are stated clearly in the legislation.
9.6.2. Regulation with the help of the calculation model Another option for the setting of acceptable emissions levels is to create a calculation model where different parameters for current pharmaceutical substances are used to calculate the highest acceptable emission levels. Starting from, for example, the environmental information which is created in the drafting of ERAs, critical parameters for active ingredients are established (for example whether the substance is persistent, bioaccumulative and/or toxic). The model takes into account levels which are critical for the environment and by using these values in a calculation formula maximum emissions levels for problematic active ingredients can be established.

Page 71 (96)
Having the calculation model in the legislation means new substances can be included as and when environmental information about these substances becomes available. This is a dynamic regulation where only inspection of the calculation model itself needs to be carried out as and when new knowledge becomes available. The calculation model should not cover substances which are excepted from requirements on ERA. The option with a calculation model should be useable in both courses of action.
9.6.3. Regulation via maximum level A further option for the establishment of emissions levels for active pharmaceutical ingredients is the determination of a general threshold value. A general maximum value for emissions can be needed where access to environmental data is deficient, such that a substance-specific threshold cannot be set with sufficient reliability or where the calculation model cannot be applied. Accessible environmental information on medicinal products may be limited as medicinal products approved before 2006 often lack an ERA. The need for an investigation on how environmental information should be created for these medicinal products was also described in Measure A. Regulation via maximum level should not cover substances which are excepted from ERA requirements. It is probably not possible to use a general maximum level within the framework of Course of Action D2, as production of medicinal products is included in the BREF which also applies to a range of other industrial production activities within the chemicals field. It should be possible to use the option in Course of Action D1.
9.7. Impact of Measure D The aim of this measure is to limit negative environmental impact which stems from production of medicinal products for the European market. Control of emissions of pharmaceutical substances will result in a cleaner water environment. In the long run this will bring a reduction in undesirable impact on the environment in the EU and in third countries. As emission limits also include emissions of, for example, antibiotics, the limits are a victory for public health, since the risk of development of antibiotic resistance is reduced. Against the background of the far-reaching harmonisation of GMP, not only in the EU but also internationally, it is important to implement discussions within international cooperation constellations before planned changes to the regulatory framework, in order to maintain the harmonisation.
Impact on development of and access to medicinal products The Medical Products Agency judgement stipulated that this measure will not affect the development of medicinal products to any significant extent. Neither have there been any such misgivings from representatives of the pharmaceuticals industry during this collaborative work. Neither is the proposal deemed to have had any significant impact on access to medicinal products under the condition that a relevant transition time for implementation of the requirement is allocated.
Impact on pharmaceutical companies The proposals for both courses of action create a clear system by having emission limits stipulated in legislation. This also means that companies can anticipate what requirements will apply. The limits should apply to both existing production and the manufacture of new pharmaceutical substances and medicinal products. With the implementation of the proposals medicinal products directives should state, by way of proposed transition regulations, at what point in time it is appropriate for the new emission thresholds to begin to apply.

Page 72 (96)
In order to conform to the emission limits requirements, the companies can optimise the whole chain of the manufacturing processes, including water conservation measures, which are often the most cost-effective way of avoiding emissions. The companies may also need to consider installing or upgrading purification equipment. Such equipment can be very energy intensive to operate. If the requirements on production cover all manufacture of medicinal products for the European market, then this results in competitive neutrality. The consultation has produced viewpoints to the effect that it is positive from an environmental and public health perspective if emissions of active ingredients are limited. Another opinion that has been put forward is that it is important not to move the focus away from quality within GMP. The Association for Generic Pharmaceuticals in Sweden state on their website that they support the proposal to insert environmental requirements in the definition of GMP 47 F
48.
9.8. Other consequences of the proposal If the proposal to insert environmental requirements into the definition of GMP is implemented then this would have further consequences and the advantages of the proposal would be reinforced by means of existing provisions about GMP in the medicinal products directives. These provisions may need to be supplemented or changed somewhat in order to make it clear that environmental requirements are also to be covered by the provisions. Below is a brief breakdown taken from the Medical Products Agency report of 16-06-2011.
9.8.1. Supervisory control and inspection When new requirements for control are brought in it is also important that the requirements are adhered to. Inspection of this needs to be done such that infringements are prevented and countermeasures taken. One positive aspect of adding environmental requirements to the definition of GMP is that there is an established and well-functioning inspection system for the control of manufacture and GMP. The competent authorities have both the right and the duty to carry out inspection in order to control, among other things, how GMP is adhered to by the manufacturers (see articles 80.6 and 111). The inspections are carried out both within the EU and in third countries. Adding environmental requirements to the definition of GMP means that the inspection regulations will also apply to checks that environmental requirements are complied with. No new such system need therefore be constructed. If the proposal is implemented it will be controlled within the framework of the inspections which competent authorities can and must carry out under the existing regulations on inspection of manufacturers. There needs to be control of compliance with the environmental requirements. The details of how this control should be carried out should be studied and developed. In order to maintain the manner in which inspections are carried out under the current system, a medicinal products inspector who controls compliance with GMP, for example, may examine some kind of certificate issued by, for example, an accredited environmental company who has carried out inspections of emissions at production plants. The way in which inspection is carried out will differ depending on which of the courses of action is chosen. For both courses of action D1 and D2 the existing system of inspections of GMP can be
48 http://www.generikaforeningen.se/wp-content/uploads/2014/02/Miljokonsekvenser-vid-lakemedelstillverkning.pdf

Page 73 (96)
applied if it is supplemented with special checks that emission limits are respected, both in the EU and in third countries. The difference is that under Course of Action D2 the inspection of emissions of active ingredients is carried out within the EU by the environmental authorities of the respective countries for production plants covered by IED. The implementation of the proposal therefore requires cooperation between a number of authorities.
9.8.2. Possibility of revoking authorisation to manufacture. In order to be able to run effective inspections a competent authority should be able to act if it comes to light that a manufacturer is no longer fulfilling environmental requirements. As the medicinal products directives are currently formulated, authorisation for manufacture may be temporarily suspended or revoked if something happens or something comes to light after the authorisation was granted, which means that GMP is no longer complied with, articles 85.2 and 118.2. If there are deficiencies in adherence to good manufacturing practice the authorities may also issue a “non-compliance GMP statement”. This regulation should also include that situation whereby the environmental requirements are not met under the terms of the described proposal, since they are part of the GMP definition. It should be considered whether, for the sake of clarity, it should be added to the articles that these measures may also be taken if requirements for emissions levels are not complied with.
9.8.3. Impact of application for marketing authorisation. Common to the proposed course of action is the further strengthening of the impact achieved since companies must show that their products are manufactured in line with GMP before their products are given authorisation for sale in Europe. In order for a medicinal product to be permitted to be sold in a member state the medicinal product must, as well as being manufactured in line with the details given above, be approved in the member state. Such marketing authorisation may either be communicated in line with one of the medicinal products directives or in accordance with regulation (EC) no 726/2004, see medicinal products directives article 5.1 and article 6.1. Authorisation is thus a precondition of the medicinal product being marketed. There are certain exceptions to this principal rule but they are of no importance in this context.
Requirements for documentation regarding manufacture in applications for marketing authorisation The proposed changes regarding GMP will entail stricter requirements on manufacturers of medicinal products. In order to have a comprehensive system for how controls on medicinal products are formulated, certain new requirements should also be placed on those seeking marketing authorisation. Thus, a supplement should be inserted in articles 12.3 and 8.3 regulating which information and which documentation should be included in an application for authorisation of a medicinal product. This should be done in order to impose on those who wish to have a medicinal product approved for sale the obligation to provide documents that show that the manufacturer(s) has/have complied with environmental requirements. Such requirements for documents will also apply to those who apply for marketing authorisation of a medicinal product that the applicant is manufacturing themselves. Such a regulation also makes it possible to reject an application for marketing authorisation of a medicinal product if environmental requirements for manufacture are not complied with (article 30 first para. and article 26.2).

Page 74 (96)
10. The National Board of Trade's assessment of the impact of the measures on global trade in medicinal products
Within the framework of this government assignment the National Board of Trade was assigned the role of collaborative partner with the Medical Products Agency and the Dental and Pharmaceutical Benefits Agency. In-line with the assignment the National Board of Trade played its part in those aspects which are attributable to "potential effects that the measures might have on cross-border trade and imports and any possible interface with international commercial or trade commitments".4 8 F
49 In line with this and based on its assignment the National Board of Trade has pledged to take responsibility for issues concerning foreign trade, the EU internal market and EU trade policy. The framework of this assignment includes working towards free trade. This means that the National Board of Trade is working for free movement on the internal markets and full liberalisation of trade between the EU and its neighbours, as well as globally.
10.1. General comments on the global trade in medicinal products Medicinal products are traded on what is an increasingly global market. In recent years there has been a tendency for the manufacture of medicinal products to be moved to third countries, not infrequently developing countries. It is also clear that the major players in the medicinal products sector in the EU, USA and Switzerland continue to hold a central position in the global pharmaceuticals trade. The global trade in medicinal products is facilitated by the fact that more countries follow harmonised international frameworks. International collaboration in production, authorisation and inspection of medicinal products means that regulatory authorities in different countries now work under regulatory frameworks which to some extent resemble one another. Noteworthy here are: The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) and The Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme (jointly known as PIC/S). International regulatory cooperation creates the conditions for consensus on how national regulations should be formulated. Similar regulatory conditions between countries facilitates global trade and contributes to the removal of trade barriers and prevents new barriers arising. In addition to regulatory cooperation in the field of pharmaceuticals there is also generally regulatory collaboration within the framework of the World Trade Organisation (WTO) and the Agreement on Technical Barriers to Trade, a.k.a. the TBT agreement. Several of the measures proposed in this report will be affected by the TBT agreement and covered by the requirements under the TBT agreement to notify the WTO of new technical rules. Some statistics on pharmaceutical trade are presented below. Besides the fact that statistics can be of general interest in gaining an overview of the international pharmaceuticals trade, it may also be important to have a clear knowledge of which countries export and import the most medicinal products. Some of the proposed measures may have an impact on manufacturing methods and production in other countries. Countries with a high level of exports may therefore be particularly affected by the placing of emission limits on production (Measure D).
49 Assignment regarding in-depth analysis of various alternative courses of action aimed at achieving the milestone target for environmental considerations in medicinal product legislation within the EU and internationally, p. 2.

Page 75 (96)
10.2. Statistics
Source: UN Comtrade
Diagram 1. Swedish trade in pharmaceuticals (SITC 54) Diagram one shows how Swedish exports of medicinal products have fallen in recent years. This was accompanied by slight increase in the import of medicinal products. Net trade fell by 20% over a five-year period.
Source: UN Comtrade
Diagram 2. EU trade in pharmaceuticals (SITC 54)
Diagram 2 shows that EU exports of pharmaceuticals increased rapidly over the last five years. Imports also increased but not at the same rate as exports. Net trade increased by almost 35% over a five-year period.
0123456789
10
2009 2010 2011 2012 2013
USD
(bill
ions
)
Export
Import
HandelsnettoNet trade
0
20
40
60
80
100
120
140
160
2009 2010 2011 2012 2013
USD
(bill
ions
)
Import
Export
HandelsnettoNet trade

Page 76 (96)
Source: UN Comtrade
Diagram 3. The eight largest exporters of medicinal products (SITC 54)
Diagram 3 shows the development of exports for the eight largest exporters of medicinal products over five years. Countries are arranged according to 2013 values, the largest exporter on the left and the smallest exporter on the right. It is noteworthy that India's pharmaceutical exports have increased by over 120% over five years, representing the most rapid increase in percentage terms. It is also noteworthy that India moved from 6th place in 2009 to 4th place in 2013. Behind them, China has seen an increase of 43%, Switzerland 40% in the EU of almost 30%.
Source: UN Comtrade
Diagram 4: The eight largest importers of medicinal products (SITC 54)
Diagram 4 shows the development of exports for the eight largest importers of medicinal products over five years. Countries are arranged according to 2013 values, the largest importer on the left and
0
20
40
60
80
100
120
140
160
EU Schweiz USA Indien Kina Singapore Israel Kanada
USD
(bill
ions
) 2009
2010
2011
2012
2013
0
10
20
30
40
50
60
70
80
90
EU USA Schweiz Japan Kina Ryssland Kanada Australien
USD
(bill
ions
) 2009
2010
2011
2012
2013

Page 77 (96)
the smallest importer on the right. China's pharmaceutical exports increased by almost 140% over five years, representing the most rapid increase in percentage terms. After that come Russia 72%, Japan with 54% and Switzerland with 38%.
10.2.1. Comment on statistics These trade statistics clearly show that the EU, USA and Switzerland are the biggest traders in medicinal products. The developing countries India and China have seen strong increases in exports. China has also experienced strong increases in imports. These increases started from low levels, however. The proposed measures, especially Measure D (production emission limits), indicate that it might be of interest to take into account the export market share of each country as well as how well those countries might be expected to meet increased minimum production requirements. Exports can give an indication of the size of a country's production, as they indicate that country's own market is saturated and thus has lower demand relative to what the country itself produces. It is, however, important to note in this context that exports and production are not the same thing. A country which exports large amounts of medicinal products does not necessarily produce a large amount itself. This is partly because the country may function as a transit country for pharmaceuticals, and therefore having large exports and imports, and partly because one country may primarily carry out the final stages of a long production chain, such as packaging and sales and therefore purchases constituent goods produced in other countries. Switzerland's large imports in relation to its relatively small population might point to such a situation. Conversely, a country might have high exports with low production and a country with low export potential might have a high production, e.g. in the sale and consumption of generic medicinal products in its internal market. Finally, it should be noted that diagrams 2, 3 and 4 contain information from both EU 27 and EU 28 due to Croatia joining. Croatia contributes so little, however, that no changes occurs in the diagram.
10.3. WTO agreement on technical trade barriers to trade The TBT agreement regulates the creation and application of technical regulations including technical provisions, standards and procedures for the assessment of conformance. In brief, the agreement covers the rules which in various ways regulate the nature and appearance of products, e.g. quality, characteristics, size as well as how they will be marked, produced and certified. The agreement covers most types of goods including medicinal products.
The main aim of the TBT agreement is to remove and counteract the creation of rules whose effect is to raise unnecessary barriers to global trade. By unnecessary trade barriers is meant trade barriers which cannot be justified under the protection interests detailed in the agreement, e.g. environmental and health protection. In order to investigate which barriers may be acceptable based on legitimate protection interests the agreement provides a notification procedure for drafts of new technical regulations. Under this notification procedure proposals for new technical regulations are submitted to the WTO and then subjected to a three-month investigation period. During those three months other WTO members provide comments on the proposal and raise objections if they feel that the proposal might create an unnecessary trade barrier. Comments received will be taken to into account as far as possible by the notifying member.
The TBT agreement means that the WTO will be notified of proposals for technical regulations where either the regulations are not covered by/do not conform with an international standard, and/or would have a major impact on international trade. The TBT agreement means that it is probable that

Page 78 (96)
notification of all of the proposed measures here would have to be submitted to the WTO. In particular Measure C (environmental considerations in the risk-benefit balance) and D (production emission limits) since these would clearly entail new requirements being placed on products. Measure A (amendments to environmental risk assessments) and B (accessibility of environmental information) might be covered by the TBT agreement, depending on how they are formulated in practice. Notification of technical regulations may be done nationally (in Sweden by the National Board of Trade) or by the EU Commission. The decision regarding whether notification of the proposal is made by the national authority or by the EU Commission has to do with whether the proposal is a target for harmonisation within the EU or whether the proposed regulations were created in the non-harmonised area, i.e. where the member state has national regulation competence. In the first case notification is carried out by the EU Commission and in the second case by the member state. As all proposed measures here suggest regulations at the EU level it would fall upon the EU commission to notify the WTO of future proposed regulations. A search of the WTO register of notifications (IMS) would reveal 122 notifications under the search word "pharmaceuticals". According to the EU TBT database one EU notification has received comments from one other WTO member. This case concerned a Japanese reaction regarding Directive 2001/83/EU on the Community code relating to medicinal products for human use. The reaction was aimed at specific parts linked to EU measures for preventing sale of false medicinal products and was therefore not really the type of proposed regulation in question here. The EU Commission responded to the Japanese reaction by explaining that the proposal would not impact on agreements which had been reached within the framework of ICH and related equivalence assessments regarding GMP. Japan accepted the EU Commission's explanation and the proposal was subsequently adopted. As well as the notification procedures set out above, which for transparency reasons necessitate notification of proposed regulations regardless of whether they entail trade barriers or not, the TBT agreement also contains a procedure for dealing with potential trade barriers in the TBT Committee. This is where WTO members can put forward what are known as specific trade concerns (STCs) related to proposals for regulations or existing old regulatory frameworks which they experience as being barriers to trade and in conflict with the TBT agreement. A search of the WTO register of STCs would reveal a trade barrier relating to pharmaceutical trade between Turkey and USA (IMS ID 264). The trade barrier had to do with a Turkish requirement that the Turkish Health Ministry should inspect factories producing pharmaceuticals before medicinal products manufactured outside Turkey could be marketed in Turkey. Approved inspection results would lead to a GMP certificate. Countries with mutual recognition agreements were not affected by this requirement. Turkey attracted comments from USA, EU and Switzerland. The USA was particularly critical of how in practice the regulations would mean that American GMP controls were not accepted. Another criticism has been targeted at how these procedures would lead to time-consuming administration which would diminish patient benefit in the long term. Similar reasoning may also be noted in a Brazilian notification (G/TBT/BRA/328) relating to regulations on health products and GMP certification. This regulation stipulates that the Brazilian health authorities must inspect production plants before a GMP certificate can be issued. Pretty much since 2010 the EU and the USA have been expressing the viewpoint that Brazil ought to accelerate its inspection times and that Brazil ought to accept existing GMP certification or contract accredited bodies in the EU and USA to carry out their inspections. The above STCs give a good indication of how these proposed measures might be received within the WTO. The criticisms expressed are related more to production emission limits (Measure D) than the

Page 79 (96)
other measures as this measure could mean that GMP certificates from other countries would not be accepted on the grounds of insufficient consideration of the environment. Adherence to the convention for pharmaceutical inspections might be made more difficult and in the long run this might lead to trust issues between pharmaceuticals authorities.
10.4. Existing bilateral trade agreements In addition to the multilateral collaboration in the WTO the EU has a number of free trade agreements with different countries which prescribe more long-term bilateral trade collaboration. A number of Mutual Recognition Agreements (MRA) contain provisions regarding GMP for medicinal products. For example, the EU has entered into agreements with Australia, Israel, Japan, Canada, New Zealand and Switzerland for mutual recognition of one another's GMP inspections. If the EU implements more long-term rules, meaning that mutual recognition of other countries' inspections is no longer possible, then MRAs such as the ones above might be undermined. For this reason, it is important that, as far as possible, the proposed measures take into account agreements which have already been entered into and that attention is paid to the fact that the EU has committed itself to recognise other countries' GMP inspections. Hence those measures aimed at developing more environmental information (Measure A), collecting and making environmental data accessible (Measure B) and environmental considerations in risk-benefit assessments (Measure C), have the advantage that they would not necessarily affect existing agreements on recognition of other countries' GMP in the same way as the reduction of production emission limits (Measure D) would. It should also be borne in mind that the EU is negotiating a comprehensive free trade and investment agreement with the USA (TTIP) and that discussions on mutual recognition of GMP inspections for pharmaceuticals are currently being held. If and when any of the proposed measures comes into force it should be updated in line with the outcome of these negotiations so that the measures are synchronised with a transatlantic market as it is expected to be.
10.5. Conclusions on global trade From the above it appears that the introduction of emission limits in production (Measure D) might have a greater impact on the above-mentioned trade policy frameworks than the other proposals. Measures attributable to GMP and GMP inspections may negatively impact existing collaboration if they entail previously agreed obligations not being met due to the implementation of increased requirements. There might, therefore, be reason to argue for the proposed measures to be implemented through a comprehensive harmonisation of regulations e.g. within the framework of existing international collaboration networks, the EU might undertake measures to influence developments in the direction of increased environmental considerations in GMP. It should also be emphasised that introduction of environmental considerations into risk-benefit assessments (Measure C) might have an impact on trade conditions with third countries, since it may have the effect of increasing requirements. Changes to environmental risk assessments (Measure A) and availability of environmental information (Measure B) appear to have the least impact on current trade regulatory frameworks. In addition to the trade impact, in terms of commitments to the WTO and other countries that have signed trade agreements with the EU, it might be worthwhile discussing how the various measures might affect trading conditions in terms of actual trade. To this end, these statistics aim to give a general picture of current trade in pharmaceuticals.

Page 80 (96)
Considering the large number of imports into the EU, it is likely that the majority of the stakeholders in third countries who are trading with the EU would be affected by the measures. How and in which capacity is difficult to estimate in the context of this analysis. However, it may be worth taking into account how the perceived elevated safety levels in the EU may affect existing trade flows between the EU and third countries. A consequence could be that companies may choose to export their medicinal products to other markets with less restrictive market requirements. This is where the free trade negotiations with the United States ought to be considered given that the United States is the world's second largest pharmaceutical importer after the EU. Might, for example, increased protection levels in the EU mean that more companies choose to export to the USA rather than the EU, and how might changes in the trade flow like this impact on aspects such as price and range of medicinal products? How, for example, can onward exports from third countries via the USA to the EU be integrated with the Transatlantic Trade and Investment Partnership (TTIP)? In view of this argument as well as of a trade perspective, there are compelling reasons to examine the opportunities to establish equivalent environmental objectives or to extend the proposed measures through broad regulatory solutions that include more stakeholders than only the EU.

Page 81 (96)
11. Abbreviations API Active Pharmaceutical Ingredient BAT Best Available Techniques (bästa tillgängliga teknik) BAT-AEL BAT Associated Emission Levels CHMP Committee for Human Medicinal Products DG SANCO Directorate-General for Health and Consumers at European Commission EC European Commission ECHA European Chemicals Agency (Europeiska kemikaliemyndigheten) EDQM European Directorate for the Quality of Medicines and Healthcare EFSA European Food Safety Agency (Europeiska myndigheten för livsmedelssäkerhet) EEA European Economic Area EMA European Medicines Agency (Europeiska läkemedelsmyndigheten) ERA Environmental Risk Assessment (miljöriskbedömning) ERC Environmental Reference Concentrations. ERP EcoRiskPrediction (projekt som driva av IMI och Europeiska kommissionen) EUDRA European Database GMPD FDA Food and Drug Administration Gl. Guideline (riktlinje) GMP Good Manufacturing Practice (god tillverkningssed) IED Industrial Emissions Directive (industriutsläppsdirektivet) IMI Innovative Medicine Initiative ISPE International Society for Pharmaceutical Engineering KemI Swedish Chemicals Agency (Kemikalieinspektionen) KK National Board of Trade (Kommerskollegium) LVFS Medical Products Agency Code of Statutes (Läkemedelsverkets författningssamling) MAH Marketing Authorisation Holder (innehavare av godkännande för försäljning
av läkemedel) MPA Medical Products Agency NCE New Chemical Entity (ny kemisk substans) NV Swedish Environmental Protection Agency (Naturvårdsverket) PAR Public Assessment Report. (Utredningsrapport) PBT Persistent, bioaccumulative and toxic substances PIC/S Pharmaceutical Inspection Co-operation Scheme PRAC Pharmacovigilance Risk Assessment Committee Reach Registration, Evaluation, Authorisation and restriction of Chemicals RMM Risk Minimisation Measures (riskminskningsåtgärder) RMP Risk Management Plans (riskhanteringsplaner) SKL Swedish Association of Local Authorities and Regions (Sveriges Kommuner och
Landsting) SoS The National Board of Health and Welfare (Socialstyrelsen) TBT Technical Barriers to Trade (tekniska handelshinder) TLV Dental and Pharmaceutical Benefits Agency (Tandvårds- och
läkemedelsförmånsverket) TTIP Transatlantic Trade and Investment Partnership WTO World Trade Organization (Världshandelsorganisationen)

Page 82 (96)
13. Definitions Action limit: Calculated concentration of a pharmaceutical substance in water. Stated in guidelines
to environmental risk assessments25 Active ingredient /pharmaceutical ingredient
From Directive 2001/83/EC, Article 1.3a Any substance or mixture of substances intended to be used in the manufacture of a medicinal product and that, when used in its production, becomes an active ingredient of that product intended to exert a pharmacological, immunological or metabolic action with a view to restoring, correcting or modifying physiological functions or to make a medical diagnosis.
Excipient: From Directive 2001/83/EG, Article 1.3b Any constituent of a medicinal product other than the active ingredient and the packaging material.
Medicinal product: From Directive 2001/83/EC, Article 1.2 a) any substance or combination of substances presented as having properties for treating or preventing disease in human beings, or b) any substance or combination of substances which may be used in or administered to human beings either with a view to restoring, correcting or modifying physiological functions by exerting a pharmacological, immunological or metabolic action, or to making a medical diagnosis.
Benefit-risk balance Risk-benefit balance
From Directive 2001/83/EC, Articles 1.28 and 1.28a An evaluation of the positive therapeutic effects of the medicinal product in relation to any risk relating to the quality, safety or efficacy of the medicinal product as regards patients' health or public health. In the directives this is referred to (in English) as the risk-benefit balance but in other sources it is often referred to as the benefit-risk balance.
Manufacture: From Directive 2001/83/EC, Article 40.2 Manufacture may refer to both total and partial manufacture, and for the various processes of dividing up, packaging or presentation.

Page 83 (96)
14. References Andersson J., Woldegiorgis A., Remberger M., Kaj L., Ekheden Y., Dusan B., Svensson A. och Brorström-Lundén E. (2006) Results from the Swedish National Screening Programme 2005, IVL rapport B1689, Subreport 1: Antibiotics, antiinflammatory substances and hormones.
Backhaus, T., Blanck och H., Faust, M. (2010) Hazard and Risk Assessment of Chemical Mixtures under REACH - State of the Art, Gaps and Options for Improvement. Swedish Chemicals Agency, PM 3/2010.
Backhaus, T. och Faust, M. (2012) Predictive Environmental Risk Assessment of Chemical Mixtures: A Conceptual Framework. Environ. Sci. Technol., 2012, 46 (5), 2564–2573.
Backhaus T., Porsbring T., Arrhenius Å., Brosche S., Johansson P. och Blanck H. (2011) Single-substance and mixture toxicity of five pharmaceuticals and personal care products to marine periphyton communities. Environmental Toxicology and Chemistry , 30(9), 2030–2040.
Bio Intelligence Science (2013) Study on the environmental risks of medicinal products, Executive Agency for Health and Consumers.
Brodin T., Fick J., Jonsson M. och Klaminder J. (2013) Dilute Concentrations of a Psychiatric Drug Alter Behavior of Fish from Natural Populations, Science, 339(6121), 814-815.
Brown, J.N., Paxéus, N., Förlin, L. och Larsson, J., (2007) Variations in bioconcentration of human pharmaceuticals from sewage effluents into fish blood plasma. Environmental Toxicology and Pharmacology, 24(3), 267–274.
Caneva L., Bonelli M., Papaluca-Amati M., Vidal J.-M. (2014) Critical Review on the Environmental Risk Assessment of medical products for human use in the centralised procedure. Regulatory Toxicology and Pharmacology, 68(3), 312-6.
Cedergreen N., Christensen A.M., Kamper, A., Kudsk P., Mathiassen S.K., Streibig, J.C. och Sørensen H., (2008) A review of independent action compared to concentration addition as reference models for mixtures of compounds with different molecular target sites. Environmental Toxicology and Chemistry Volume 27(7), 1621–1632.
Ejhed H., Magner J., Olshammar M., Remberger M., Norström K., Lilja K., Momina B., (2012) Enskilda avlopp som källa till läkemedelsrester och andra kemikalier. IVL rapport B2070
Ericson H., Thorsén G. och Kumblad L., (2010) Physiological effects of diclofenac, ibuprofen and propranolol on Baltic Sea blue mussels. Aquatic Toxicology, 99(2), 223–231.
Executive Agency for Health and Consumers (2013) Study on the environmental risks of medicinal products, Final report 12 December 2013.
Falås P., Baillon-Dhumez A., Andersen H.R., Ledin A., and la Cour Jansen J., (2012) Suspended biofilm carrier and activated sludge removal of acidic pharmaceuticals. Water Res. 46(4), 1167-75.
Fent K., Escher C. och Caminada D. (2006) Estrogenic activity of pharmaceuticals and pharmaceutical mixtures in a yeast reporter gene system. Reprod Toxicol. 22(2), 175-85.
Fick J. (2013) Presentation på Hållbara Hav: http://www.youtube.com/watch?v=cRt_IeyXClM
Fick J., Lindberg R.H., Parkkonen J., Arvidsson B., Tysklind M. och Larsson, J., (2010) Therapeutic Levels of Levonorgestrel Detected in Blood Plasma of Fish: Results from Screening Rainbow Trout Exposed to Treated Sewage Effluents. Environ. Sci. Technol., 44 (7), 2661–2666.

Page 84 (96)
Fick J., Lindberg J.H., Kaj L. och Brorström-Lundén E., (2011) Results from the national screening programme 2010. IVL rapport B2014, Subreport Pharmaceuticals.
Flaherty C.M. och Dodson S.I., (2005) Effects of pharmaceuticals on Daphnia survival, growth, and reproduction. Chemosphere., 61(2), 200-7.
Halling-Sørensen B., (2000) Algal toxicity of antibacterial agents used in intensive farming. Chemosphere., 40(7), 731-9.
Halling-Sørensen B., Sengeløv G. och Tjørnelund J., (2002) Toxicity of tetracyclines and tetracycline degradation products to environmentally relevant bacteria, including selected tetracycline-resistant bacteria. Arch Environ Contam Toxicol. , 42(3), 263-71.
Hörsing M., Wahlberg C., Falås P., Hey G., Ledin A. och la Cour Jansen J., (2014) Reduktion av läkemedel i svenska avloppsreningsverk – kunskapssammanställning, Svenskt vatten utveckling rapport 2014-16.
Jonsson M., Fick J., Klaminder J. and Brodin T. (2014) Antihistamines and aquatic insects: Bioconcentration and impacts on behavior in damselfly larvae (Zygoptera), Science of The Total Environment, 472, 108–111.
JRC (2012) EU Wide Monitoring Survey on Waste Water Treatment Plant Effluents. (http://publications.jrc.ec.europa.eu/repository/handle/111111111/26927
Kidd K.A, Blanchfield P. J., Mills K. H., Palace V. P., Evans R.E., Lazorchak J. M. och Flick, R.W. 2007. Collapse of a fish population after exposure to a synthetic estrogen. Proceedings of the National Academy of Sciences of the United States of America, 104(21), 8897–901.
Klaminder, J., Jonsson, M., Fick, J., Sundelin A. and Brodin T. (2014) The conceptual imperfection of aquatic risk assessment tests: highlighting the need for tests designed to detect therapeutic effects of pharmaceutical contaminants Environmental Research Letters, 9(8).
Kortenkamp A., Backhaus T. and Faust., (2009) State of the Art Report on Mixture Toxicity. Directorate General for the Environment, EU Commission (Report).
Kosonen J. and Kronberg L. (2009) The occurrence of antihistamines in sewage waters and in recipient rivers. Environ Sci Pollut Res Int., 16(5), 555-64. doi: 10.1007/s11356-009-0144-2. Epub 2009 Apr 1.
Larsson J., (2012) Utsläpp från Läkemedelsindustri påverkar miljön. Läkartidningen, 14.
Larsson J. and Lööf L., (2014) Läkemedel i miljön. From Läkemedelsboken 2014. www.läkemedelsboken.se
Medical Products Agency (2009) Report on government commission regarding the possibility of tightening environmental requirements for the manufacture of medicinal products and active ingredients.
Medical Products Agency (2011a) Analysis of how work on a revision of GMP should be structured in order to have the greatest chance of success.
Medical products agency (2011b) Groundwork to facilitate the initiation of a revision of EU legislation on Good Manufacturing Practice, with the aim that legislation will also cover environmental considerations. Search: http://www.lakemedelsverket.se/overgripande/Publikationer/Rapporter/Avslutade-regeringsuppdrag/

Page 85 (96)
Läkemedelsverket (2012) Ytterligare åtgärder som kan vidtas på nationell nivå för att minska kassationen av läkemedel och begränsa miljöpåverkan av läkemedelsanvändningen. Search: http://www.lakemedelsverket.se/overgripande/Publikationer/Rapporter/Avslutade-regeringsuppdrag/
Murray-Smith R.J., Coombe V.T., Haag Grönlund M., Waern F. and Baird J.A. (2012) Managing Emissions of Active Pharmaceutical Ingredients from Manufacturing Facilities: An Environmental Quality Standard Approach., Integrated Environmental Assessment and Management, 8(2), 320–330.
Swedish Environmental Protection Agency (2013) Producentansvar för medicinal products. Redovisning 2013-10-17 NV-00335-13. Search: http://www.naturvardsverket.se/Miljoarbete-i-samhallet/Miljoarbete-i-Sverige/Regeringsuppdrag/Redovisade-2013/Producentansvar-for-lakemedel/
Swedish Environmental Protection Agency (2014) Miljökrav vid produktion av läkemedel inom EU. Search: http://www.naturvardsverket.se/Miljoarbete-i-samhallet/Miljoarbete-i-Sverige/Regeringsuppdrag/Redovisade-2014/Miljokrav-vid-produktion-av-lakemedel-inom-EU/
Oaks J.L., Gilbert M., Virani M.Z., Watson R.T., Meteyer C.U, Rideout B.A., Shivaprasad H.L., Ahmed S., Chaudhry M.J., Arshad M., Mahmood S., Ali A. and Khan A.A., (2004) Diclofenac residues as the cause of vulture population decline in Pakistan. Nature. 427(6975):630-3.
OSPAR (2013) Background Document on Clotrimazole (2013 update)
Parrott J.L. and Blunt B.R., (2005) Life-cycle exposure of fathead minnows (Pimephales promelas) to an ethinylestradiol concentration below 1 ng/L reduces egg fertilization success and demasculinizes males. Reprod Toxicol. 20(2), 131-41.
Porsbring T., Blanck H., Tjellström H. och Backhaus T., (2009) Toxicity of the pharmaceutical clotrimazole to marine microalgal communities, Aquat Toxicol, 91(3), 203-11.
Ramirez A.J., Brain R.A., Usenko S., Mottaleb M.A., O'Donnell J.G., Stahl L.L., Wathen J.B., Snyder B.D., Pitt J.L., Perez-Hurtado P., Dobbins L.L., Brooks B.W. och Chambliss C.K., (2009)
Occurrence of pharmaceuticals and personal care products in fish: results of a national pilot study in the United States. Environ Toxicol Chem. 28(12), 2587-97.
SOU 2012:38 (2013) Minska riskerna med farliga ämnen! Strategi för Sveriges arbete för en giftfri miljö. Delbetänkande av All Party Committee on Environmental Objectives http://www.regeringen.se/sb/d/108/a/195014
SOU 2013:23 (2013) Ersättning vid läkemedelsskador och miljöhänsyn i läkemedelsförmånerna, del 1, del 2, http://www.regeringen.se/sb/d/108/a/213991
Stackelberg P.E., Gibs J., Furlong E.T., Meyer M.T., Zaugg S.D. and Lippincotte R.L., (2007) Efficiency of conventional drinking-water-treatment processes in removal of pharmaceuticals and other organic compounds. Science of The Total Environment. 377( 2–3), 255–272.
Sternbeck J., Österås A.-H., Josefsson K., Andreasson F. and Kreuger J., (2007) Screening of veterinary medicines in agricultural areas. WSP. Rapport till den nationella miljöövervakningen.
Stockholms läns landsting (2014) Provtagning av läkemedelsrester i vatten. http://www.tmr.sll.se/Miljoutmaning-2016/Halsoframjande-miljoarbete/Lakemedel/Provtagning-av-lakemedelsrester-i-vatten/
Svenskt Vatten (2014) Läkemedelsrester i dricksvatten http://www.svensktvatten.se/Vattentjanster/Dricksvatten/Takt-till-kran/Kemiska-amnen/Lakemedel/ Svensson J., Fick J., Brandt I. och Brunström B. (2014) Environmental concentrations of an

Page 86 (96)
androgenic progestin disrupts the seasonal breeding cycle in male three-spined stickleback (Gasterosteus aculeatus) Aquatic Toxicology, 147, 84–91.
Sveriges Officiella Statistiska meddelanden (2012) Utsläpp till vatten och slamproduktion 2012. MI22 SM 1401.
Säfholm M., Norder A., Fick J., and Berg C., (2012) Disrupted oogenesis in the frog Xenopus tropicalis after exposure to environmental progestin concentrations. Biol Reprod. 27;86(4), 126.
Triebskorn R., Casper H., Heyd A., Eikemper R., Köhler H.R. and Schwaiger J., (2004) Toxic effects of the non-steroidal anti-inflammatory drug diclofenac. Part II: cytological effects in liver, kidney, gills and intestine of rainbow trout (Oncorhynchus mykiss). Aquat Toxicol., 68(2), 151-66.
Tyler C.R., Filby A.L., Bickley L.K., Cumming R.I., Gibson R., Labadie P., Katsu Y., Liney K.E., Shears J.A., Silva-Castro V., Urushitani H., Lange A., Winter M.J., Iguchi T.H. and Hill E.M., (2009) Environmental health impacts of equine estrogens derived from hormone replacement therapy. Environ Sci Technol., 43(10), 3897-904.
UNEP (2012) State of the Science of endocrine disrupting chemicals – 2012. Ed: Bergman A., Heindel J.J., Jobling S., Kidd K.A., and Zoeller T.
Wahlberg C., Björlenius B., and Paxeus N., (2007) Läkemedelsrester i Stockholms vattenmiljö. Förekomst, förebyggande åtgärder och rening av avloppsvatten. Stockholm Vatten ISBN 978-91-633-6642-0.
WHO (2011) Guidelines for drinking-water quality, fourth edition
http://www.who.int/water_sanitation_health/publications/2011/dwq_guidelines/en/
WHO (2012) Pharmaceuticals in drinking-water . http://www.who.int/water_sanitation_health/publications/2012/pharmaceuticals/en/
WHO (2014) Antimicrobial resistance: global report on surveillance 2014. http://www.who.int/drugresistance/documents/surveillancereport/en/
Zeilinger J., Steger-Hartmann T., Maser E., Goller S., Vonk R., and Länge R., (2009) Effects of synthetic gestagens on fish reproduction. Environ Toxicol Chem. 10.1897/08-485.1. Epub 2009 May 26.

Page 87 (96)
Appendix: 1 Testing requirements in accordance with guidelines for environmental risk assessment
Stage in regulatory evaluation
Stage in risk assessment
Objective Method Test /Data requirements
Phase I Pre-screening Estimation of exposure
Action limit: if PECSURFACEWATER ≥0.01 µg/L or of concern, e.g. suspected endocrine disrupter proceed to Phase II A
Or if log Kow>4.5 perform PBT assessment
Consumption data
log KOW
Phase II Tier A
Pre-screening
Initial prediction of risk
Risk assessment: if risk proceed to Phase II B
Base set on fate and effects: Ready biodegradability (OECD 301) test or Aerobic and Anaerobic Transformation in Aquatic Sediment System (OECD 308) Adsorption-Desorption (OECD 106/121/OPPTS) Fish Bioconcentration study (OECD 305) Activated Sludge, Respiration Inhibition Test (OECD 209) Algae, Growth Inhibition Test (OECD 201) Daphnia sp. Reproduction Test (OECD 211) Fish, Early Life Stage Toxicity Test (OECD 209) Tailored hazard assessment for Endocrine Disrupters
Phase II Tier B
Extended Substance and compartment-specific refinement and risk assessment
Risk assessment
Extended data set on emission, fate and effects Refinement of PEC surface water Aerobic and anaerobic transformation in soil OECD 307 Soil Microorganisms OECD 216 Terrestrial Plants, Growth Test OECD 208 Earthworm, Acute Toxicity Test OECD 207 Collembola, Reproduction Test ISO 11267 Sediment dwelling organism OECD 308

Page 88 (96)
Within ERAs studies are carried out in part to assess environmental risk and in part to assess whether the substance is a PBT substance. PBT assessment is a hazard assessment which investigates whether a substance has the properties of a typical environmental poison, i.e. if it is broken down in the environment (Persistent), accumulates in living organisms (Bioaccumulative) and is toxic (Toxic) 49 F
50. ERA is carried out in two steps (see table above) In the first stage of ERA (known as the pre-screening phase) the expected concentration of the active ingredient in the environment is calculated and its lipophilicity is estimated. If the anticipated concentration falls below the limit of 0.01 µg/l the ERA is terminated at this point. In the same way the PBT assessment is terminated if the lipophilicity of the substance is below Log Kow <4.5. This means that unless systematic calculation of the anticipated concentration in the water environment and an assessment of the lipophilicity are carried out, no further information on this medicinal product will be obtained. There will be no information on the specific environmental properties and environmental impact of the medicinal product. The environmental assessments of a (large?) proportion of medicinal products are terminated after this phase. Those medicinal products which exceed the action limit of 0.01 µg/l continue onto the next step (phase II A) where the fundamental data on their environmental impact is compiled and a first rough environmental risk assessment is carried out. Similarly, if the lipophilicity of the substance exceeds Log Kow>4.5 then the studies required to decide whether the substance is a PBT substance go forward. These studies correspond to studies in phase II.
50 The PBT criteria are specified in appendix XIII of REACH. Assessment is carried out in accordance with the European Chemicals Agency guideline document.

Page 89 (96)
Appendix: 2 The Dental and Pharmaceutical Benefits Agency evaluation of pricing, benefits, cost-effectiveness, and the cost to society of medicinal products under the proposed Measures A-D Under the governmental assignment the Dental and Pharmaceutical Benefits Agency has been tasked with looking at those aspects which concern the pricing and benefits of medicinal products, their cost-effectiveness and their cost to society.
1. Aim and outline The aim of the Dental and Pharmaceutical Benefits Agency report is thus to provide an account of how the proposed measures (A-D) with related courses of action might impact on the price, benefits and cost-effectiveness of medicinal products. The report begins by defining the limitations of the analysis. There follows a background section which contains a brief description of macroeconomic theory on pricing of goods and services, which serves as the foundation for the analysis of the effects. In section 3-6 below, the effects of the respective measures are set out. Section 7 provides a joint assessment of all the measures.
1.1. Limitations The Dental and Pharmaceutical Benefits Agency assessment only takes into account the potential effects of the price, benefits and cost effectiveness of medicinal products in the wake of the introduction of the various measures. The results were presented to show in what direction prices, benefits and cost effectiveness might head, subject to limitations stipulated in the description of the Dental and Pharmaceutical Benefits Agency's assignment. If the proposed measures were to have any impact on prices, cost effectiveness and incentive for research, this would in all probability be a negative impact. Effects of benefits to society in the form of reduced environmental impact and effects on cost to the economy as a result of environmental impacts of medicinal products are taken up in this section. Pronouncing on the size of this impact in relation to environmental externalities and potential costs for damage to the environment are, due to the limitations mentioned above, difficult to make, and need more extensive evaluation. The analysis does not compare the total value to society of implementing the measures against not implementing them. This part of the report can, therefore, not be used on its own to draw conclusions on whether the measures would be justified from a societal perspective or not.
2. Background - cost and price theory The relationship between costs and pricing of medicinal products is analysed based on the fundamental assumptions of economic theory. One of these assumptions is that participants in the markets, i.e. individuals and companies, are rational. The assumption is that rational companies strive to maximise profits. The profits of a company are defined as the difference between the revenue and the costs of that company. If the company's costs increase, then profits will fall.
Profit = total revenue - total costs

Page 90 (96)
The total costs are composed of variable costs and fixed costs. Variable costs may be characterised as costs which vary according to the number of units produced. Fixed costs may be characterised as costs which do not vary according to the number of units produced. Economic theory states that rational companies only pay attention to variable costs when setting prices. The company will produce goods/services for as long as marginal revenue5 0F
51 exceeds marginal cost5 1 F
52. Thus, the company will set a price whereby the marginal cost is the same as the marginal revenue, since at this breakeven point the company will maximise its profits. Companies maximise profits when the marginal cost is the same as the marginal revenue regardless of whether the market is characterised by perfect competition, oligopoly or monopoly. The pharmaceuticals market for patent-protected products is characterised by a monopoly situation. In a monopoly situation the companies can set prices higher than when there is perfect competition. When the patent on the medicinal product expires generic competition arises which often leads to a fall in the price of the medicinal product. In both situations the company will attempt to maximise profits. Studies have been carried out which investigate how pharmaceutical companies set their prices on new patent-protected medicinal products. These studies indicate that pricing is governed by demand factors, in particular which therapeutic benefits the new medicinal product can provide compared with its alternatives, see e.g. Lu and Comanor (1998)5 2F
53.
3. Impact of Measure A Measure A proposes a change in testing requirements for environmental risk assessment. The medical products agency has developed two potential courses of action to implement this:
• A1: Review current limitations such that more pharmaceutical substances are covered by the requirement for environmental risk assessment.
• A2: Develop the testing requirements for environmental risk assessments to better reflect the specific properties of medicinal products.
Environmental risk assessments are already carried out and it is important to stress that proposal A2 entails developing the requirements for testing. It is difficult to determine how broad this will be and it may, in some cases, be marginal, simply a part of a normal development. The Dental and Pharmaceutical Benefits Agency postulates that the direct effect of Measure A is that the revised environmental testing would lead to increased fixed costs for the companies. The effects of the increased costs are examined via two scenarios. In the basic scenario, which is thought to be the most probable, the price of medicinal products would not be affected by increased costs due to environmental tests. The rationale for this comes from section 2. The alternative scenario looks at the consequences of the prices of medicinal products rising as a result of Courses of Action A1 and A2.
Basic Scenario The Dental and Pharmaceutical Benefits Agency deems that changing the testing requirements for ERAs (Courses of Action A1 and A2) would entail increased fixed costs for companies which probably would not impact on the price of medicinal products (see section 2). On the other hand, those increased fixed costs would signify a reduction in profits for the companies. The size of this reduction is difficult to quantify, but in the long term it might lead to lowered incentives to develop medicinal products. That would in turn lead to long-term diminishing of patient health due to a reduced range of medicinal products. 51 The revenue which the production of one more unit brings. 52 The cost of producing one more unit. 53 Lu Z.J. and Comanor W.S., (1998) Strategic Pricing of new Pharmaceuticals, Review of Economics and Statistics, 80(1), 108-118.

Page 91 (96)
Given that Measure A would reduce incentives to develop medicinal products, one consequence might be that the companies would redistribute their pharmaceutical development budget and invest more in the development of substances which require less extensive environmental testing. Such a redistribution of development investment, focusing less on patient benefits and more on environmental considerations, might then lead to the medicinal products that are developed being of less benefit to patients overall5 3F
54. The combined effect is that Measure A would probably not have an effect on the price of medicinal products but patient benefit might be diminished in the long term. This would mean a long-term reduction in the cost-effectiveness of medicinal products. The Dental and Pharmaceutical Benefits Agency considers that these effects would probably happen regardless of choice of course of action. There is some uncertainty around the extent of the potential effect, partly due to the size of the costs in relation to the total budget of the pharmaceutical companies. The cost of environmental testing is probably low relative to the total cost of developing new medicinal products, and the implementation of Measure A would therefore have a minor impact. It might even be the case that companies which produce the same active ingredients could share the cost of testing, given that the same substance would only need to be tested once. That would mean a lower increase in costs for the companies.
Alternative scenario An alternative scenario assumes that the companies would set the prices of medicinal products higher as a consequence of the increased costs associated with environmental testing. If the companies raise the price of medicinal products it might in turn might lead to reduced patient benefit, if the increased prices were to lead to medicinal products which had previously been subsidised now being denied their subsidy. Increasing prices for medicinal products may in the long term lead to medicinal products being squeezed out of the benefit system due to budgetary concerns. This alternative scenario would entail reduced cost-effectiveness due to increased prices for medicinal products and also in the long term reduced patient benefit. If the price of medicinal products were to rise due to increased fixed costs then the incentive to develop new products would not be reduced. Both in the basic scenario and the alternative scenario one other effect on patient benefit is conceivable. If an increase in testing requirements were to lead to slower authorisation of medicinal products this would have a negative effect on patient benefit. This effect would be possible seeing that the implementation of testing necessarily consumes resources. Nevertheless, the effect on patient benefit would probably be small.
4. Impact of Measure B Measure B: collection and accessibility of environmental data regarding active pharmaceutical ingredients. In the judgement of the Dental and Pharmaceutical Benefits Agency this measure would not have any impact on the price or benefits of medicinal products. This is assuming that environmental information would be processed and administered by the authorities. The companies may encounter an administrative burden when information about environmental data has to be sent to the authorities, but the judgement is that this would not have any impact on the price of medicinal products, their benefits or their cost effectiveness since the company already submits information to the authorities in the course of applying for authorisation for medicinal products. If the introduction of a database with accessible environmental data were to permit the results of environmental testing to be reused, this would influence the impact of Measure A. In that case Measure B would have a neutralising effect on Measure A. This is because the increase in cost of 54 The concept of patient benefits refers to benefits that a medicinal product provides for patients in the form of improved quality of life and extended lifespan.

Page 92 (96)
expanded testing requirements would be less if it was possible to reuse the tests. Companies who produced the same active ingredients would share the cost of testing since the same substance only needs to be tested once, or trading of test results might develop.
5. Impact of Measure C Measure C proposes that environmental considerations be taken into account in the risk-benefit assessment of medicinal products. It is the impact on the environment during use that would be tested. The Medical Products Agency has developed two potential courses of action to implement this:
• C1: Environmental data is seen as a factor in the risk-benefit assessment. • C2: Environmental impact of a new medicinal product is compared with environmental impact
of existing medicinal products
Under Measure C1 environmental risk and suitable risk minimisation measures will be taken into account in the risk-benefit assessment for each individual medicinal product. In the case of medicinal products where alternative treatments are limited or in the case of serious diseases medicinal products would probably not be rejected on the grounds of environmental risk. Risk minimisation measures would instead be prescribed. Under Measure C2 environmental risk caused by the use of a specific medicinal product is compared with the equivalent environmental risk from other medicinal products which fulfil the same medical needs and which are already available on the market. An application might then be rejected if there was already another medicine/treatment on the market which could be given to the patient and if there were no risk minimisation measures which could minimise the serious environmental risk that the use of the new pharmaceutical product would entail. Two principal effects were identified in both courses of action in Measure C.
i) Reduced quantities of medicinal products available on the market ii) Increased costs to the pharmaceutical companies
These two effects and their consequences are described below.
i) Reduced quantities of medicinal products available on the market The reduction in quantities of medicinal products available on the market could have two causes. The first cause is that risk minimisation measures might consist of measures which in different ways limit the sales of a medicinal product. There might be a limit on the use of second-line treatment, or a form of preparation may be excluded, or marketing might be restricted to certain groups of patients. This would result in a reduced range of medicinal products, which might lead to a smaller number of patients being able to benefit from the medicinal product in question, or in other words diminished patient benefit. The second cause of reduced quantities of medicinal products is when a medicinal product is refused authorisation, or when authorisation is withdrawn because risk minimisation measures have not been successfully applied. A reduction in the quantity of medicinal products available would have two effects. The first would be a direct effect in the form of diminished patient benefit. The second would be an indirect effect in the form of a worsening of the competitive situation. The fewer the medicinal products with the same effect, the less the incentive for companies to compete on price. This could lead to increased prices for medicinal products. If there are fewer alternatives for the same benefits the company might be granted subsidies to provide a higher price for future medicinal products.
ii) Increased costs to the pharmaceutical companies The effect of increased costs to the pharmaceutical companies is examined in two scenarios. These are the same two scenarios described for Measure A. In the basic scenario, which is adjudged to be the most probable, the price of medicinal products would not be influenced by the increased costs due to

Page 93 (96)
environmental testing. In the alternative scenario the consequences of a company raising the price of medicinal products as a result of increased costs are examined.
Basic Scenario Risk minimisation measures would entail increased costs, either for the pharmaceutical companies or in the form of increased public expenditure. If the costs were to fall to the public purse, the cost effectiveness of medicinal products would be influenced negatively. If the costs were to fall to the pharmaceuticals companies they would probably be in the form of fixed costs, i.e. costs which are not dependent on how many units are produced. Since pricing only pays heed to variable costs, the price of the medicinal product would not be affected 2. If the prices of medicinal products were constant while a company's fixed costs increased there would be a negative impact on the pharmaceutical company's profits. One possible effect of this would be a lower incentive to develop new medicinal products, which would reduce patient benefits in the long term.
Alternative scenario To begin with, therefore, Measure C would have no impact on the price of medicinal products. The alternative scenario assumes that the increased fixed costs would lead to increased medicinal product prices. If the companies raised the price of medicinal products this might in turn might lead to reduced patient benefit, if the increased prices were to lead to medicinal products which had previously been subsidised now being denied their subsidy. Increased medicinal product prices could also have a long-term effect on budgetary considerations – meaning that other medicinal products would be squeezed out of the benefit system, reducing patient benefits. This alternative scenario would also bring increased medicinal product prices and in the long term diminished patient benefit. Given that increased fixed costs influence medicinal product prices, Measure C would be unlikely to have the effect of reducing incentives to develop medicinal products Both effects i) and ii) might lead to certain pharmaceutical substances becoming less attractive as subjects of research. If the danger of risk minimisation measures and/or the danger of being denied authorisation was to be greater in certain areas than others, companies might re-prioritise their research investment. The same would apply if certain areas entailed greater costs to the company for risk minimisation measures than others. A redistribution of development investment such that less attention was paid to cost effectiveness and more to environmental aspects might lead to those medicinal products which are developed offering diminished overall patient benefit. Altogether it might be stated that Measure C would lead to reduced cost-effectiveness due to reduced patient benefit and, also, in the long term increased medicinal product prices. The extent of the effects depends primarily on the number of medicinal products for which authorisation would be denied/withdrawn as a result of Courses of Action C1 and C2 and the extent of the impact of potential risk minimisation measures on the range of additional products available.
6. Impact of Measure D Measure D would entail regulation of emissions levels during the production of active pharmaceutical ingredients. The Medical Products Agency has proposed two possible courses of action to implement this:
• D1: 1. In the definition of GMP reference is made to a new EU regulation where emissions of active ingredients are regulated.
• D2: In the definition of GMP, reference is made to the framework for the Industrial Emissions Directive (IED) where the emission limits of active pharmaceutical ingredients are regulated.

Page 94 (96)
This is the only one of the measures that the Dental and Pharmaceutical Benefits Agency considers might also impact on non-patent protected additional products. Effects on pricing might be quite different depending on whether a substance is patent-protected or not. For substances that are patent-protected the companies would probably not set the price based on production costs, particularly not on fixed costs. Demand would be the decisive factor here. On the generic market, however, if there is strong competition pricing would be governed in a much clearer way by production costs. For this reason we discuss the two types of market separately.
Patent-protected medicinal products The Dental and Pharmaceutical Benefits Agency judgement is that Measure D (both Courses of Action D1 and D2) would lead to increased fixed costs, in particular for companies with high emission levels during production. This assessment is based on the assumption that companies would need to develop their medicinal product manufacturing processes in order to reduce emission levels. The difference between the two courses of action is that D1 would probably cover all pharmaceutical substances, which is not the case with D2. This would mean that D1 would lead to increased costs for more companies than D2 would. As with Measure A it is assumed that medicinal product prices would not be influenced by increased fixed costs but would rather be completely based on demand. As with Measure A the increased fixed costs would also lead to a long-term reduction in profits for the companies, which might lead to reduced incentives to develop new medicinal products. That would in turn lead to diminished patient health due to a reduced range of medicinal products. Given that Measure A would reduce incentives to develop medicinal products, one consequence might be that the companies would redistribute their pharmaceutical development budgets. Course of Action D2 might lead to companies investing more in the development of substances where the requirement for lower emissions is less stringent. A redistribution of development investment, focusing less on patient benefits and more on environmental considerations, might lead to the medicinal products that are developed being of less benefit to patients overall. The assessment of the Dental and Pharmaceutical Benefits Agency is that both courses of action could lead to reduced cost-effectiveness in the long term, due to reduced patient benefits (though there is some uncertainty as to how significant this might be), but that neither of the courses of action would impact on medicinal product prices. In the alternative scenario, whereby medicinal product prices are impacted by increased fixed costs, this would lead to reduced cost-effectiveness due to higher medicinal product prices, and also due to weaker competition.
Non-patent-protected medicinal products In the long term increased fixed costs might also lead to reduced profits for companies who manufacture non-patent-protected medicinal products. However, the alternative scenario whereby medicinal product prices are affected by increased fixed costs is more likely for the generic companies, who to a great extent deal in non-patent-protected products, since they have lower profit margins and are subject to stronger competition. This scenario might also be manifested if additional costs for increased minimum requirements on production are not completely made up of fixed costs, but rather to some extent by variable costs. Since production costs constitute a major part of the costs for generic companies compared with original product/research companies the effect of cost increases due to Measure D would be greater than that due to the other measures (A, B, C) which would not affect generic companies. This would then lead to reduced cost-effectiveness due to higher medicinal product prices.

Page 95 (96)
In summary, there is some uncertainty around the extent of the potential effect, partly due to the size of the costs of regulating emissions in relation to the total development and production costs of the pharmaceutical companies. If the cost of regulating emissions was low in relation to the pharmaceutical companies' total costs then the implementation of minimum requirements in production would probably have only a minor overall impact on patient benefit.
7. Combined assessment of the effects of Measures A, B, C, and D
The Dental and Pharmaceutical Benefits Agency’s assessment of the effects on medicinal product prices, benefits and cost-effectiveness, is that the overall effect of implementing all the measures would be unlikely to have a direct influence on prices of medicinal products. However, fewer authorised medicinal products combined with risk minimisation measures would decrease competition and indirectly affect prices. The greatest impact of the measures would arguably be that benefits to patients would decrease because fewer medicinal products would be authorised and risk minimisation measures would further restrict their use. In the long-term, patient benefits would also be affected by fewer incentives to develop new medicinal products and the chance that research resources would be reallocated to other pharmaceutical fields. However, it is difficult to quantify the effect on reduced patient benefits. This is because of the current uncertainty about factors such as the scale of costs for environmental testing and risk minimisation measures. This also means that it is difficult to gauge the scale of the various effects in relation to one another. It is reasonable to believe the effect from Measure A on patient benefit would be marginal. Measure D should not significantly affect patient benefits either. The greatest impact is likely to come from Measure C but as mentioned, the extent depends on how risk minimisation measures are formulated. Other societal benefits in the form of reduced environmental impact and the effects on the social costs arising from the environmental impact of medicinal products have not been analysed in this part. The findings from the Dental and Pharmaceutical Benefits Agency section of the report should be used together with estimates of the effects on other benefits and costs to society when drawing conclusions about whether the measures are justified from a social perspective or not.

Page 96 (96)





























