Embed Size (px)
Citation preview

REVIEW
Hyperphosphatemic familial tumoral calcinosis secondary to fibroblastgrowth factor 23 (FGF23) mutation: a report of two affected familiesand review of the literature
M. Chakhtoura1 & M.S. Ramnitz2 & N. Khoury3 & G. Nemer4 & N. Shabb5& A. Abchee6
& A. Berberi6 & M. Hourani3 &
M. Collins2 & S. Ichikawa7 & G. El Hajj Fuleihan1
Received: 24 January 2018 /Accepted: 14 May 2018 /Published online: 20 June 2018# International Osteoporosis Foundation and National Osteoporosis Foundation 2018
AbstractHyperphosphatemic familial tumoral calcinosis (HFTC), secondary to fibroblast growth factor 23 (FGF23) gene mutation,is a rare genetic disorder characterized by recurrent calcified masses. We describe young Lebanese cousins presentingwith HFTC, based on a retrospective chart review and a prospective case study. In addition, we present a comprehensivereview on the topic, based on a literature search conducted in PubMed and Google Scholar, in 2014 and updated inDecember 2017. While the patients had the same previously reported FGF23 gene mutation (homozygous c.G367Tvariant in exon 3 leading to a missense mutation), they presented with variable severity and age of disease onset (at4 years in patient 1 and at 23 years in patient 2). A review of the literature revealed several potential patho-physiologicpathways of HFTC clinical manifestations, some of which may be independent of hyperphosphatemia. Most availabletreatment options aim at reducing serum phosphate level, by stimulating renal excretion or by inhibiting intestinalabsorption. HFTC is a challenging disease. While the available medical treatment has a limited and inconsistent effecton disease symptomatology, surgical resection of calcified masses remains the last resort. Research is needed to determinethe safety and efficacy of FGF23 replacement or molecular therapy, targeting the specific genetic aberration.
* M. [email protected]
M.S. [email protected]
G. El Hajj [email protected]
1 Calcium Metabolism and Osteoporosis Program, WHOCollaborating Center for Metabolic Bone Disorders, AmericanUniversity of Beirut Medical Center, Beirut, Lebanon
2 Section on Skeletal Disorders and Mineral Homeostasis, NationalInstitute of Dental and Craniofacial Research, National Institutes ofHealth, Bethesda, MD, USA
3 Department of Radiology, American University of Beirut MedicalCenter, Beirut, Lebanon
4 Department of Biochemistry and Molecular Genetics, AmericanUniversity of Beirut Medical Center, Beirut, Lebanon
5 Department of Pathology and Laboratory Medicine, AmericanUniversity of Beirut Medical Center, Beirut, Lebanon
6 Department of Internal Medicine, American University of BeirutMedical Center, Beirut, Lebanon
7 Department of Medicine, Indiana University School of Medicine,Indianapolis, IN 46202, USA
Osteoporosis International (2018) 29:1987–2009https://doi.org/10.1007/s00198-018-4574-x

Hyperphosphatemic familial tumoral calcinosis is a rare genetic disorder characterized by recurrent calcified masses, inaddition to other visceral, skeletal, and vascular manifestations. It remains a very challenging disease.
Keywords Calcification . Familial tumoral calcinosis . Fibroblast growth factor 23 (FGF23) . Hyperphosphatemia
Introduction
Hyperphosphatemic familial tumoral calcinosis (HFTC)1 is arare group of genetic disorders, with shared clinical features. Itwas first described in 1898 by the French dermatologist Giard[1]. Initially reported in patients from African and MiddleEastern countries, cases of European and American descentwere described thereafter [2–4]. The disorder is characterizedby hyperphosphatemia and recurrent ectopic periarticular calci-fications, in addition to other visceral and vascular manifesta-tions, in the absence of any inflammatory or neoplastic disorder[5]. The disease is secondary to a mutation in the fibroblastgrowth factor 23 (FGF23), GALNT3, or KLOTHO genes,resulting in decreased FGF23 activity [3]. Since FGF23 playsa significant role in conditioning renal phosphate excretion, adecrease in its activity results in hyperphosphatemia. The latterdisturbance is, at least in part, responsible of HFTC clinicalmanifestations. These symptoms may start as early as in thefirst decade, even in the newborn period [6], or it can be delayeduntil the third decade.
Objectives
The objectives of this study are to describe the presentation oftwo first-degree cousins with HFTC, their prospective data inresponse to therapy, and results of their genetic mutation anal-yses and provide a comprehensive overview on the patho-physiology of the disease, its manifestations, and a summaryof the available treatment options.
Methods
HFTC patient presentation
The two patients were self-referred to our clinics.We collectedclinical information and laboratory data from the patients,
reviewed their previous records, and performed a physicalexamination at presentation. We obtained additional labo-ratory studies and scheduled follow-up visits as clinicallyindicated. Imaging studies were performed at theAmerican University of Beirut–Medical Center (AUB-MC)-Lebanon. Genetic studies were performed at our in-stitution and FGF23 levels were measured at the NationalInstitutes of Health, Bethesda, Maryland. Patients andfamily members provided written informed consent forevaluation and mutation analysis. The evaluation and con-sent form were approved by the Institutional ReviewBoard at AUB-MC.
Imaging
A bone radiologist (NK) reviewed the radiographs on patient1 and evaluated the characteristics of the periarticular lesions.Bone mineral density (BMD) by dual x-ray absorptiometry(DXA) in both patients was measured at the spine, hip, totalbody, and soft tissue buttock mass (the latter on patient 1only), on Hologic DXA machine Horizon A, V.13.5.3.1(Hologic, Bedford, MA, USA). Z-scores were derived bycomparison of the patient’s BMD to an age-matched popula-tion, using a Caucasian database. The mean coefficient ofvariation (CV) [standard deviation (SD)] of duplicate BMDmeasurements was 0.385 (0.252) at the lumbar spine, 0.483(0.232) at the total hip, 1.152 (0.822) at the femoral neck, and0.386 (0.183) at the radius. The BMD of the right buttock softtissue mass was compared to that of the rib (a well-definedtrabecular bone area), to the radius (a cortical bone area), andto the contralateral soft tissue region. We performed furtherevaluation of the cardiovascular system in both patients bymultiple detector cardiac computed tomography for coronaryartery calcium score (ICT 256 Philips Amsterdam,Netherlands), 2D-cardiac echocardiography (GE machineVivid E9), and carotid-femoral pulse wave velocity usingSphymocor CvMS V9.
Histopathology
Histopathology examination of resected calcified masses wasperformed on both cases, using H&E staining. While theslides from patient 1 were read at an outside institution(Saint George Hospital), the slides from patient 2 (left elbow)were reviewed at AUB-MC.
1 25(OH)D: 25-hydroxyvitamin D; 1;25(OH)2D: 1,25-dihydroxyviatmin D;AUB-MC:AmericanUniversity of Beirut–Medical Center; BMD: bonemineraldensity; CFGF23: C-terminal FGF23; CV: coefficient of variation; DXA: dualX-ray absorptiometry; FGF23: fibroblast growth factor 23; GFR: glomerularfiltration rate; HFTC: hyperphosphatemic familial tumoral calcinosis; IL-1: in-terleukin–1; iFGF23: intact FGF23; NaPi2: type II sodium-dependent phosphateco-transporters; MMP: metalloproteinases; ppGalNacT3: polypeptide N-acetyl-galactosaminyl-transferase 3; RIA: radio-immunoassay; SD: standard deviation;TRP: tubular reabsorption of phosphate; TmP: tubular maximum reabsorptionof phosphate; TRPV5: transient receptor potential vanilloid type 5 channel
1988 Osteoporos Int (2018) 29:1987–2009

FGF23 assay
The intact FGF23 (iFGF23) and C-terminal FGF23 (CFGF23)were measured at the Section on Skeletal Disorders andMineral Homeostasis, National Institute of Dental andCraniofacial Research, National Institutes of Health,Bethesda, MD, USA. This was done with second-generationELISA assays (Immutopics International, San Clemente, CA).The iFGF23 assay measures only the intact protein while theCFGF23 assay reflects both the intact and C-terminal frag-ments. Samples for the CFGF23 ELISA were diluted (1:4)using sample diluent, as recommended by the kit manufactur-er to assure that values were within the measurable range ofthe assay.
PTH and vitamin D metabolite levels
PTH levels were measured at an outside lab using ECLIA/Cobas 6000 Roche. Vitamin D metabolites were done atAUB-MC using radio-immunoassay (RIA).
FGF23 mutation analysis
DNA sequencing had already been performed at IndianaUniversity, for one of the HFTC patients (patient 2) andshowed a homozygous c.G367T variant in exon 3 ofFGF23. We targeted this mutation during DNA sequencingthat was performed at AUB-MC for both patients and theirdirect family members (parents and siblings). Specifically, thefollowing primers were used to amplify exon 3: 5 ′AGGAGGAGCTGGGGAGTG 3′ (F) and 5′ CCCTGTCACCTTTCCCATC 3′ (R). The amplicons were resolved on a1.2% agarose gel. PCR products were purified using theQiagen QIAEX II kit (Cat No./ID: 20021) as previously de-scribed [7] and then sequenced using the ABI3500 platform atthe American University of Beirut Molecular Core facilities.The sequences were compared to the online databases (thereference sequence ID for FGF23 is NM_020638).
Literature review
We initially conducted a literature search on PubMed andGoogle Scholar in 2014 and then updated the search inDecember 2017, without any time restriction. We used thefollowing terms: familial tumoral calcinosis, hyperostosishyperphosphatemic syndrome, calcinosis, FGF23,hyperphosphatemia, and calcification. We identified addition-al articles by reviewing the citations of published articles onthe topic and included additional ones as available in the au-thors’ library.
Results
Patient 1
A 26-year-old Lebanese woman presented for recurrentmasses at the elbow, shoulder, and hip joints. Her medicalhistory revealed that her symptoms started at the age of23 years, when she had a right elbow mass that was resected.At that time, she was discovered to have a high serum phos-phate level, 6.9 mg/dL (2.4–4.1 mg/dL), with a maximumlevel reported of 7.9 mg/dL. Four months later, she developedpainful debilitating left elbow and left hip masses that im-paired her normal range of motion and required surgical re-moval. She was intermittently treated thereafter with acetazol-amide and sevelamer (medication doses not reported by pa-tient). Despite medical treatment, her phosphate levelremained above 6mg/dL. Upon presentation to our institution,she was complaining of a large right buttock and right elbowmasses. Her height was 171 cm, and her weight was 62 kg.Physical examination revealed a 4 × 6 cm firm mass on theextensor surface of the right elbow and a large right buttockmass of 15 × 13 cm.
Other laboratory tests showed the following: creatinine0.6 mg/dL (reference range 0.4–1.2 mg/dL), calcium9.9 mg/dL (reference range 8.3–11 mg/dL), magnesium2.1 mg/L (reference range 1.5–2.7 mg/L), PTH 19.5 pg/mL(reference range 15–65 pg/mL), 25-hydroxyvitamin D[25(OH)D] 28 ng/mL (reference range 20–80 ng/mL), and1,25 dihydroxyvitamin D [1,25(OH)2D] 43 pg/mL (referencerange 20–46 pg/mL), inappropriately normal in the presenceof hyperphosphatemia. Twenty-four-hour urine studiesshowed the following: volume 1450 mL, phosphate 0.28 g/24 h (reference range 0.4–1.3 g/24 h), calcium 184 g/24 h(reference range 100–321 mg/24 h), and creatinine 681 mg/24 h (reference range 600–2000 mg/24 h). The creatinine-corrected phosphate was 0.41 g/g creatinine and thecreatinine-corrected calcium was 0.27 g/g creatinine.Calculated tubular maximum reabsorption of phosphate(TmP) to glomerular filtration rate (GFR) was 7.15 mg/dL(reference range 2.4–3.6 mg/dL) [8]. Tubular reabsorption ofphosphate (TRP) was estimated at 99%. A full workup forendocrine, auto-immune, and infectious diseases, includingthyroid and parathyroid disorders, rheumatoid arthritis, anky-losing spondylitis, sarcoidosis, brucellosis, hepatitis, and par-vovirus infection, was negative. Since her original visit atAUB-MC, the patient was prescribed a low-phosphate dietin consultation with the dietician and advised to takesevelamer (1600 mg two times a day, then increased to threetimes a day), probenecid (250 mg two times a day, thenincreased to 500 mg twice a day), and acetazolamide(250 mg twice a day), in an attempt to normalize the phos-phate level. Her initial phosphate level was 6.7 mg/dL.
Osteoporos Int (2018) 29:1987–2009 1989

Periodic blood tests revealed a phosphate ranging between5.1 and 6.2 mg/dL, and calcium X phosphate product rang-ing between 46 and 62 mg2/dL2 (normal calcium X phos-phate product < 55 mg2/dL2). The patient had normal am-bulation and was maintaining normal activities of dailyliving.
Imaging findings
Bone mineral density findings Before resection of the rightbuttock mass in 2014, BMD results were as follows:right femoral neck 1.187 g/cm2 (Z-score + 3.0); righttotal hip 1.363 g/cm2 (Z-score + 3.5); left femoral neck1.037 g/cm2 (Z-score + 1.7); left total hip 1.236 g/cm2
(Z-score + 2.4); lumbar spine (L1–L4) BMD 1.166 g/cm2
(Z-score + 1.1); left forearm 0.847 (Z-score + 2.7); andtotal body 1.454 g/cm2 (Z-score + 3.8); interestingly,right buttock soft tissue BMD at the area of the enlarging
mass showed a BMD of 0.762 g/cm2, which was close tobone density at the right ribs 0.764 g/cm2 and right fore-arm 0.775 g/cm2. The left buttock soft tissue BMD was0 g/cm2, indicating the absence of calcifications. Theright hip calcification-estimated weight, based on bonemineral content (BMC) findings, was 0.45 kg.
Radiographs Several radiographs of the affected jointswere obtained at different time intervals and revealedlarge, periarticular, multilobulated, densely calcified softtissue masses of the left shoulder (Fig. 1a), bilateral hips(Fig. 1b–d), and left elbow (Fig. 1e). Brachial artery cal-cifications were present at the left elbow region (Fig. 1e).There were also interstitial calcifications at the level ofthe right ankle (Fig. 1f). Intervertebral disc calcificationswere detected on spine radiographs (Fig. 1g). In addition,extensive anterior rib cartilage calcifications and deformi-ties in the posterior arch of ribs at the costo-vertebral
a b
c d
Fig. 1 Radiographs of FTC lesions in patient 1. a–c Radiographs wereobtained at different time points. Typical features of tumoral calcinosisseen as periarticular, multilobulated calcified soft tissue lesions involvingthe left shoulder (a), left hip (b), and the right hip (c) which were less densethan the other lesions. d MRI of the right hip using short tau inversionrecovery sequence (STIR). The para-articular lesion shows dependentlow-signal intensity material in keeping with calcifications as well asfluid signal components. Findings are in keeping with calcium and fluidcontaining cystic spaces resulting in fluid-calcium levels (sedimentationsign) (arrow). e Lateral radiograph of the left elbow showing a largelobulated para-articular mass at the extensor surface of the elbow
(arrow). Also noted extensive brachial artery calcifications (arrowheads)extending to its bifurcation. f Lateral radiograph of the right ankle. Thereare thin subcutaneous interstitial calcifications (arrows) mainly at thedorsal (extensor) aspect of the distal leg and ankle. g Lateral radiographof the dorso-lumbar spine showing multilevel intervertebral disccalcifications (arrows). h Coronal reconstructed CT image of the chestwall, obtained during the calcium score study, showing extensiveanterior rib cartilage calcifications. i Axial CT cut of the chest, obtainedduring the calcium score study. There is deformity of the posterior arch of aright rib at the costo-vertebral junctions (arrow). Note the calcifications ofthe anterior rib cartilage (arrowhead)
1990 Osteoporos Int (2018) 29:1987–2009

junctions were detected on CT chest images obtained dur-ing CT calcium score study (Fig. 1h, i).
CT calcium score 256 multidetector computed tomography(MDCT) of the heart revealed a calcium score of zero. Therewas a tiny rim of calcification in the mitral annulus but nocalcification in the aortic valve.
2D-echocardiographyEchocardiography revealed normal sys-tolic and diastolic function and absence of valvular disease.
Pulse wave velocity study Pulse wave velocity (carotid-femoral) [mean (SD)] was at the upper limit of normal, mea-sured at 6.2 (0.6) m/s (normal range for age is 3–7.5 m/s).
Histopathologic findings
Right buttock mass revealed multiple large nodular foci ofamorphous calcified material bordered by an abundance ofmacrophages and multinucleated giant cells. Elbow mass re-vealed a fibrous cystic wall lined by a rim of macrophages andmultinucleated giant cells around the calcified material.
Patient 2
A14-year-old Lebanese girl with a history of a hard neckmassthat was surgically removed at 4 years of age presented to ourclinic for evaluation of recurrent calcified masses. Additionalhistory included a left elbow mass, which was resected twice
fe
g h
i
Fig. 1 (continued)
Osteoporos Int (2018) 29:1987–2009 1991

at the age of 7. It was not until the age of 11 years that she wasdiscovered to have a high blood phosphate level of 8.5–9.9 mg/dL (reference range 2.7–5.1 mg/dL). At the age of13, she underwent resection of left ankle calcification follow-ed by right wrist mass resection. Accordingly, over the courseof the 10 years, she underwent resection of five soft tissuemasses. Upon presentation to AUB-MC, she was symptomat-ic with a new left elbow lesion that was increasing in size. Herheight was 169 cm, and her weight was 56 kg. Physical examrevealed a 5 × 7 cm hard mass at the extensor surface of theleft elbow. Laboratory evaluation revealed the following: cre-atinine 0.4 mg/dL (reference range 0.4–1.2) mg/dL, calciumlevel 10 mg/dL (reference range 8.3–11 mg/dL), ionized cal-cium 1.3 mmol/L (reference range 1.18–1.3 mmol/L), magne-sium 2.1 mg/L (reference range 1.6–2.5 mg/L), PTH 21 pg/mL (reference range 15–65 pg/mL), 25(OH)D 16 ng/mL (ref-erence range 20–80 ng/mL), and 1,25(OH)2D 54.5 pg/mL(reference range 20–46 pg/mL). Urine studies were not ob-tained. She was treated with sevelamer and acetazolamide(doses not reported by the patient). Since her original eval-uation, she was placed on a low-phosphate diet, and anescalating regimen that included sevelamer (1600 mg twicedaily, then three times per day), probenecid (250 mg twiceper day, increased to 500 mg twice per day), and aluminumhydroxide (300 mg three times per day). She did not toler-ate acetazolamide, which caused severe lower extremitypain. She was taking aluminum hydroxide intermittently.While on treatment, her phosphate results have fluctuatedwith the lowest value achieved of 6.1 mg/dL. The calciumX phosphate product ranged between 57 and 85 mg2/dL2
(normal calcium X phosphate product < 55 mg2/dL2).The patient was non-adherent to medical therapy, in partdue to intolerance (as is the case with acetazolamide),despite multiple attempts at modifying her regimen.Compliance to a low-phosphate diet was not assessedformally, as the patient was not following up regularlywith a dietician. The patient had normal ambulation andwas able to maintain normal activities in most of the days,but occasionally, she suffered from severe acute exacerba-tions, for few days, during which she was not able to maintainher regular activities.
Imaging findings
Bone mineral density findings Left femoral neck BMD of0.659 g/cm2 (Z-score − 1.7), left total hip BMD of0.706 g/cm2 (Z-score − 1.9), and a total body BMD of1.029 (Z-score + 0.2).
CT calcium score 256 MDCT of the heart revealed a calciumscore of zero. CT images of the chest wall showed exten-sive anterior rib cartilage calcifications, unusual for the
patient’s age (Fig. 2a). The CT also showed symmetricaldeformity of the posterior arches of the ribs at the costo-vertebral junctions (Fig. 2b), as well as intervertebral disccalcifications (Fig. 2c).
2D-echocardiography The study revealed normal systolic anddiastolic function and absence of valvular disease.
Pulse wave velocity study Pulse wave velocity (carotid-femoral) [mean (SD)] was normal measured at 4.6 (0.4) m/s(normal range for her age is not available, normal range for theage of 20 years 3.5–7.5 m/s).
Histopathology of the resected left elbow lesion
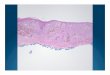
Granular calcium deposits associated with epithelioid histio-cytes and multinucleated giant cells were identified as shownin Fig. 3.
Mutation analyses on HFTC patients and their familymembers
The family pedigree is shown in Fig. 4. The patients werefirst-degree cousins and both their mothers and fathers wererespective siblings (two sisters marrying two brothers fromanother family, and the two families, to the best of our knowl-edge, are not consanguineous). DNA Sanger sequencing anal-ysis of the FGF23 coding exons revealed a homozygousc.G367T variant in exon 3 leading to a p.G123W amino acidmissense mutation in both affected probands (Fig. 5). Thisvariant was previously reported in a Caucasian female withtumoral calcinosis [9] . Both parents are heterozygous whilesiblings have either a normal genotype or a heterozygous ge-notype for the c.G367T variant. This variant is not found inthe whole exome and genome databases (GnomAD), and thep.G123Wmissense mutation is predicted to be probably dam-aging with a score of 1.00 according to the Polyphen 2 insilico prediction software [10].
Both parents and siblings in the two families, with theexception of the two patients, are reported to be asymptomat-ic, and no laboratory data, other than genetic studies, is avail-able on any of them at this point.
iFGF23 and CFGF23 measurement on HFTC patientsand their carrier family members
CFGF23 levels were elevated in both patients, reaching 5.5times the upper limit of normal in patient 1 and 9 times theupper limit of normal in patient 2 (Table 1). CFGF23 levelswere within the normal range in all other relatives, except forthe father of patient 2, in whom the level was 1.5 times theupper limit of normal. In the HFTC patients, iFGF23 levels
1992 Osteoporos Int (2018) 29:1987–2009

were inappropriately low/normal. iFGF23 level was withinthe normal range in all other relatives with the exception ofthe mother of patient 1 in whom the level was undetectable.The ratios [iFGF23 + CFGF23]/iFGF23 were very high in pa-tients 1 and 2, of 125.6 and 87.1, respectively. In the otherfamily members, it ranged between 2.7 and 11.8 (Table 1),higher than the levels previously reported in healthy controls,0.9 (0.3) [11].
Discussion and review of literature
We presented two cases of HFTC who have the samegenetic mutation but a large discrepancy in the age of
onset of symptomatology, within the same family (4 yearsin patient 1 and 23 years in patient 2). HFTC is an auto-somal recessive disorder, requiring mutations in both al-leles; mutations can be the same (homozygous) or differ-ent (compound heterozygote). HFTC is characterized by adefect in the activity of FGF23 and can be caused by aloss-of-function mutation in the FGF23 gene (FGF23,12p 13.3) [12, 13] or a loss-of-function mutation in theGALNT3 gene (GALNT3, 2q24-q31), both leading to ex-cessive FGF23 proteolysis [14], or a loss of function mu-tation in the KLOTHO gene (KL,13q12), which encodes aco-receptor of FGF23 and results in FGF23 resistance[15]. Accordingly, mutations in FGF23 and GALNT3 re-sult in low iFGF23 levels but high CFGF23 levels, sec-ondary to excessive proteolysis of FGF23 [16], whereasmutations in KLOTHO result in elevation of both iFGF23and CFGF23 [17].
Hyperostosis-hyperphosphatemia syndrome (HHS) isanother disease caused by a loss-of-function mutation ineither FGF23 or GALNT3. Previous case reports of HHShave been described in Lebanon [18]. HHS subjects pre-sented with long bone pain and were found to havehyperphosphatemia and cortical hyperostosis demonstratedon limb x-rays [18, 19]. Subsequently, a number of sub-jects with clinical features of both HFTC and HHS weredescribed in the literature and found to have one of thethree known causative mutations of HFTC [20]. Thus,HFTC and HHS represent different manifestations of thesame disease. In HHS, long bone changes were reportedincluding diffuse medullary bone sclerosis showing diffuselow signal on T1-weighted MR sequence [21]. Mild dif-fuse cortical thickening and uni-lamellated periosteal reac-tion were also described, with surrounding soft tissue
a b
c
Fig. 2 Radiographs of FTC inpatient 2. a Coronal reconstructedCT image of the chest wall duringthe calcium score CT studyshowing extensive anterior ribcartilage calcifications, unusualfor the patient’s age. These areless than those of the other olderpatient. b Axial CT cut of thechest. There is symmetricaldeformity of the posterior archesof the ribs at the costo-vertebraljunctions (arrows). Note thecalcifications of the anterior ribcartilage (arrowheads). c Lateralreconstructed CT cut of the chestshowing intervertebral disccalcifications at the level of thedorsal spine (arrow)
Fig. 3 Histopathologic features of FTC lesion. Granular calcium deposits(long thin arrow) associated with epithelioid histiocytes (short thinarrows) and multinucleated giant cells (thick arrows). Hematoxylin andeosin, × 20
Osteoporos Int (2018) 29:1987–2009 1993

edema on MRI, in particular within the tibia, fibula, andulna [21, 22]. These changes show increased radionuclideuptake on bone scintigraphy [21, 22].
Insights into FGF23/GALNT3/Klotho physiologyand links to HFTC manifestations
The “Appendix” summarizes the phenotype-genotype of casereports of HFTC published to date, with mutations inGALNT3gene being the most common [3, 9, 11–13, 19, 20, 22–50].
After synthesis, FGF23 undergoes O-glycosylation and actsthrough fibroblast growth factor receptor 1 (FGFR1), andits signal transduction requires the presence of its co-receptor,α-Klotho [32, 51–56]. For details on FGF23/GALNT3/Klotho axis activation, please refer to Fig. 6 [14, 57–59].
The kidneys represent the target organ of FGF23 [51],where the main role of FGF23 is to inhibit phosphate reab-sorption through decreased expression of type II sodium-dependent phosphate co-transporters (NaPi2a and NaPi2c)[60]. In addition, FGF23 inhibits 1,25(OH)2D production(through decreased expression of CYP27B1) and increases1,25(OH)2D degradation (through increased expression ofCYP24A1) [51]. Decreases in 1,25(OH)2D result in decreasedphosphate absorption [61] through the vitamin D-regulatedphosphate co-transporter (NaPi2b) in the gastrointestinal tract[51] (Fig. 6).
Studies in animals suggested that FGF23 may targetother organs, including the parathyroid glands [62, 63],the heart [64], and the bone [65]. In the parathyroidglands, FGF23 results in the inhibition of PTH secretion[51, 61]. In the heart, FGF23 results in left ventricularhypertrophy [64]. In fact, in chronic kidney disease pa-tients, high FGF23 levels were found to be an indepen-dent predictor of left ventricular hypertrophy [66]. Theregulatory effect on bone can be derived from FGF23ablation or overexpression studies in animal models andis still not clearly defined [67, 68]. FGF23 deficiencyresulted in a decrease in mature osteoblasts and in bonedensity [67]. Skeletal mineralization showed a variablepattern, with areas of increased mineralization and areasof unmineralized osteoid accumulation, leading to fragilebone [67]. FGF23 overexpression also inhibits bone min-eralization [68] (Fig. 6).
Subcutaneous and other tissue calcifications
HFTC is characterized by recurrent cutaneous and subcutane-ous calcifications (calcium phosphate or hydroxyapatite
Fig. 4 Family pedigree. Circles represent female subjects; squaresrepresent male subjects; diamonds represent children from familieswithout HFTC. The number inside the diamond represents the numberof children per family. Black-filled symbols denote affected individuals
(patients), homozygous for FGF23 mutation. Symbols with hashed linesrepresent carriers (heterozygous for FGF23 mutation). Empty symbolsrepresent healthy subjects. Symbols with an oblique line represent adeceased family member
Fig. 5 DNA Sanger sequencing analysis of the FGF23 coding exons inthe HFTC patients and their family members
1994 Osteoporos Int (2018) 29:1987–2009

crystals), typically on the extensor surfaces of the periarticularskin of large joints, most commonly the hip, followed by theelbow, shoulder, foot, and wrist, and less commonly at thescalp, spine, and sacrum [13]. Their location in theperiarticular region, in specific, has been related to theperiarticular forces that result in vascular injury and sy-novial metaplasia, with mononuclear and osteoclast-likelining, as demonstrated on histopathologic studies [4, 69].These lesions are thought to occur in response to repeti-tive trauma that can be very minimal and often unnoticed[4, 69]. These calcified masses enlarge (may reach >1 kg) to become painful, compressing the surroundingtissue, and preventing normal joint range of motion.They may ulcerate and can be complicated by infections[12, 13, 17]. Multiple surgical resections are the last re-sort [12]. On histopathology, familial tumoral calcinosislesions are usually situated in the subcutaneous fat anddeeper structures. They are characterized by intracellularand extracellular calcium deposits which appear as vari-ably sized, amorphous, basophilic granular material [5].The calcium is associated with epithelioid histiocytes andmultinucleated giant cells [5, 44]. Chronic inflammationand fibrosis are also present [5, 70].
One possible mechanism behind cutaneous calcificationsin HFTC may be illustrated as follows (Fig. 6): increasedphosphate level may directly stimulate FGF7, resulting insubcutaneous calcifications [14]. Furthermore, in HFTCpatients with GALNT3 mutations, the inhibitory effect ofthe polypeptide N-acetyl-galactosaminyl-transferase 3(ppGalNacT3) on FGF7 is lacking, potentiating furtherFGF7 activation and enhancing ectopic calcifications[14]. Therefore, we can speculate that treatment of
hyperphosphatemia per se in patients with GALNT3 muta-tion may not be enough to completely resolve dermal le-sions. Whether the same mechanism applies to patientswith FGF23 and KLOTHO mutations remains unknown.The wide discrepancy in the aggressiveness of the recur-rence of additional calcified soft tissue masses betweenour patients remains unexplained and may in part be relat-ed to high calcium X phosphate product (> 55 mg2/dL2),known to be associated with high mortality in chronickidney disease patients [71] and difference in FGF7 levels,in addition to trauma, inflammation, or other potential un-identified modulators of this biochemical-physicalprocess.
In addition to subcutaneous calcifications, other areasof tissue calcifications were described in the literature(“Appendix”). In particular, subcutaneous interstitial sep-tal calcifications were noted in the calves [3]. Our firstpatient had this finding at her right ankle region (Fig.1f). Cartilage calcification may also occur. Calcificationof costal cartilage of lower ribs was previously describedbriefly by Dumitrescu et al. [3]. Our report is the secondin the literature describing severe costo-chondral calcifi-cations in a young adolescent with HFTC, most likelyrelated to the mineral metabolic disorder. In fact, costo-chondral calcifications are rarely seen in healthy adoles-cent girls, and when they occur, they are usually mild[72]. Severe costo-chondral calcifications are related toinfectious etiologies, metabolic disorders, specially hyper-thyroidism and renal failure, malignancy, and genetic fac-tors [73]. Other areas of calcifications have also beenreported, including thyroid and costal cartilage, as in ourpatients [3]; calcifications of the nucleus pulposus;
Table 1 Measurement of intactFGF and FGF23 fragments inindex cases and their familymembers identified as carriers ongenetic screening
C-terminal FGF23 (CFGF23)(3 months–17 years ≤ 230 RU/mL;≥ 18 years ≤ 180 RU/mL)
Intact FGF23(iFGF23)(22–63 pg/mL)
Ratio2
CFGF23þiFGF23½ �iFGF23
Index case 1 997 8 125.6
Case 1 father 79 19 5.2
Case 1 mother 100 11 10.1
Case 1 brother 103 29 4.5
Case 1 sister 166 59 3.8
Index case 21 2152 25 87.1
Case 2 mother 89 0 NA
Case 2 father 280 26 11.8
Case 2 brother 96 56 2.7
Case 2 brother1 129 125 2.0
NA not applicable1 Age < 18 years2 Ratio in affected subjects reported to range between 10 and 72, and in control subjects, reported values are mean(SD) 0.99 (0.3) [11]
Osteoporos Int (2018) 29:1987–2009 1995

annulus fibrosus [3]; and dural calcifications [29]. Onbone scintigraphy, such lesions show increased uptake[21]. Fluid-fluid level reflecting calcium layering influid-filled cysts within the lesions has been described asa diagnostic finding of HFTC [5, 21, 74] and has beenreferred to as the “sedimentation sign,” as in our patient(Fig. 1d). Although this can be seen on radiographs, it ismuch better characterized on CT or MR imaging. On MRimaging, HFTC lesions show homogeneous or heteroge-neous low signal on T1-weighted sequence. On T2-weighted sequence, they may appear of diffusely low sig-nal or heterogeneous with both high fluid signal intensityareas due to fluid and low-signal areas due to the presenceof calcium, with sedimentation sign [5, 21, 74]. Reactive
soft tissue edema may be encountered around the lesions[21] (Fig. 1d).
Vascular and visceral calcifications
Vascular involvement has been reported in HFTC, includingcalcifications in the aorta and aortic valve [12], carotid arteries[41], femoral vessels [24], and arteries in the extremities [12].The presentation may overlap with pseudo-xanthomaelasticum that also may present with angioid streaks of theretina and vascular and skin calcifications, similar to HFTC,but with more prominent cerebral aneurysmal vasculopathy[75]. Other manifestations including ocular involvement withcalcifications of eyelid, cornea, retina, and conjunctiva [20,
Co-receptor: Klotho
High Phosphate
FGF73
ppGalNaCT3 O-glycosylation
O-
Matrix Metalloproteinase
Subcutaneous calcifications
Kidneys
GI tract
Parathyroid glands
Bone1
↓ Phosphate reabsorption ↓ 1,25 (OH)D2 synthesis ↑ 1,25 (OH)D2degradation
↓ Phosphate absorption
↓ PTH secretion
Regulates bone formation
FGF23-independent effects: Kidney: ↑ Phosphate excretion ↑ Calcium reabsorption ↓ Calcium excretion
Vessels and Leftventricle2
Regulates vascular calcification and LV hypertrophy
Targets of available HFTC medical therapy
Inhibitory effect
Stimulatory effect
Biochemical
FGFR1
Active FGF23
Fig. 6 Molecular pathways illustrating key genes, molecules, andtarget organs involved in the physiology of phosphate and thepathophysiology of HFTC. After synthesis in the osteocytes, theintact FGF23 (iFGF23), the active form, undergoes O-glycosylationby a glycosyl-transferase (polypeptide N-acetyl-galactosaminyl-transferase 3: ppGalNacT3) that protects FGF23 from proteolysisby pro-protein convertases into inactive fragments (N-terminals andC-terminals). iFGF23 requires Klotho co-receptor, a facilitator ofintracellular signal transduction to be able to act on its target organsincluding the kidneys, the gastrointestinal tract, the parathyroidglands, and the bones. Conversely, iFGF23 possibly actsindependently of Klotho co-receptor, on the vessels and leftventricle, through biochemical pathways illustrated on the right sideof the figure. Furthermore, Klotho may have FGF23-independentactivity in the kidneys, resulting in increased renal calciumreabsorption and phosphate excretion, through the inhibition of thetransient receptor potential vanilloid type 5 channel (TRPV5), andthe NaPi2a, respectively. In addition, it modulates the action ofseveral hormones including insulin, insulin growth factor 1 (IGF-1), and tissue growth factor β (TGF-β), therefore described as an
“anti-aging gene.” A high phosphate level stimulates FGF23,ppGalNaCT3, and FGF7. In healthy individuals, FGF23 stimulationactivates the kidney, GI tract, and the parathyroid gland pathways, asa negative feedback to decrease phosphate level. Stimulation ofppGalNaCT3 results in FGF23 glycosylation and inhibition ofFGF7; the latter being a protein specifically produced by dermalfibroblasts, it stimulates matrix metalloproteinase (MMP8collagenase, MMP9 gelatinase, and MMP3) and results insubcutaneous calcification, as shown in one in vitro study.Therefore, stimulation of ppGalNaCT3 inhibits subcutaneouscalcifications, through inhibition of FGF7, while its downregulationincreases FGF7 excretion [14, 57–59]. Continuous arrows representconfirmed FGF23 effects; dashed arrows represent FGF23 effectsthat are still controversial. FGF7 fibroblast growth factor 7, FGFR1fibroblast growth factor receptor 1, GI gastrointestinal tract,ppGalNacT3 polypeptide N-acetyl-galactosaminyl-transferase.1
Independent of phosphate level.2 Independent of Klotho, possiblyalso independent of hyperphosphatemia.3 Based on one study only.4
Effect of FGF23 still controversial
1996 Osteoporos Int (2018) 29:1987–2009

37, 45, 76]; renal medullary calcinosis [12]; and testicularmicrolithiasis have been reported [34, 40]. Despite the occur-rence of vascular calcifications, premature coronary arterydisease has not been reported in the literature. Vascular diseaseseems to be more often described in HFTC patients withFGF23 or KLOTHO gene mutations, and only few patientswith GALNT3 mutations were reported to have vascular in-volvement (“Appendix”). Noteworthy, not all reported caseshave systematically assessed for the presence or absence ofvascular involvement (“Appendix”), therefore, vascular calci-fications may have been underreported.
It is well known that vascular calcifications represent acommon complication of elevated calcium and/or phos-phate levels and increased calcium X phosphate product[77]. However, several other factors regulate this processincluding Fetuin A, FGF23, Matrix GLA protein, andPTH [51, 77]. Accordingly, in addition to high phosphatelevels, HFTC patients have FGF23 deficiency, which may,by itself, potentiate vascular calcifications [51]. Anotherpotential mechanism has been recently elucidated in afamily with HFTC, presenting with severe vascular calci-f icat ions [29]. Mutations in other genes (Wnt5 ,TNFRSF11B, SFRP1), known to interact and to encodeproteins that inhibit vascular calcification, were identified,co-segregating with FGF23 mutation, and acting as genemodifiers [29]. Such findings might explain the differ-ences in phenotypes among family members with thesame mutation. Given these potential pathways, vascularcalcifications in HFTC patients may not be completelyprevented by lowering phosphate levels.
Dental and oral abnormalities
Dental abnormalities have been reported with bothFGF23 and GALNT3 mutations and include pulp stones,absence of pulp chambers, and short bulbous roots(“Appendix”). These are secondary to dentin abnormal-ities [78], as a result of hyperphosphatemia [79]. Inaddition, gingivitis and other intra-oral mucosa andextra-oral abnormalities have been described in aJewish family [80]. Our patients did not have any ofthese findings. Prominent dental abnormalities are com-monly seen on panoramic radiographs, includinghypercalcification of multiple tooth roots and thistleshape of the dental pulp [3, 5, 19].
Bone abnormalities
HHS and HFTC represent different manifestations of thesame underlying disorder [19]. Therefore, long bone hy-perostosis can be frequently present in HFTC patients andmay go undiagnosed as imaging is not often routinely
performed to search for such lesions [19]. Two previousreports assessed bone density in patients with GALNT3gene mutations and showed normal results [3, 36]. Inour cohort, Case 1 had a high BMD Z-score, ranging from1 to 2.8. Indeed, this could have been artifactually in-creased secondary to subcutaneous calcifications, withpossible residual microscopic material, even after surgicalresection. Patient 2 had a BMD that was within normal forage with a Z-score ranging from − 1.9 to 0.2; no specificpattern for cortical or trabecular bone effect could beidentified.
Overview of available therapeutic options
Medical treatment options are limited with inconsistentimprovement, and often failure to normalize phosphatelevel or decrease the size of the calcified soft tissuemasses [57]. Evidence behind the use of various drugshas been provided in case reports and case series.Decreasing gastrointestinal absorption of phosphate andincreasing phosphate renal excretion are the basis of thevarious drug therapies that are currently used (Fig. 6;Table 2). A combination of two or more drugs is com-monly prescribed.
A low-phosphate diet is an essential first step, al-though this recommendation may be very difficult toimplement as phosphate is ubiquitous in the diet [88].Phosphate binders decrease phosphate absorption in thegut. Sevelamer [9, 20, 36, 41, 81] and the older phos-phate binder aluminum hydroxide [2, 9, 19, 35] mayresult in a decrease in serum phosphate level, but theresponse in the described cases in the literature wasinconsistent [9, 20, 35, 36, 41, 81]. One report on theuse of lanthanum carbonate in combination withibandronate showed a decrease in subcutaneous calcifi-cations [49]. The use of calcium-based phosphatebinders has been described [82], but they are not rou-tinely used given the risk of increasing serum calciumand therefore the increased risk of calcification.Phosphaturic drugs are also described. Acetazolamideis a carbonic anhydrase inhibitor, frequently prescribedin HFTC. It increases phosphate loss in urine by renaltubular acidification and, therefore, may decrease phos-phate level and/or improve HFTC symptomatology [3,9, 19, 36, 81, 83, 84]. Probenecid competitively inhibitsphosphate reabsorption and may help decrease serumphosphate or reduce calcifications [9, 19]. Similar tophosphate binders, the response to phosphaturic agentsis also variable [3, 19, 36, 81, 83, 84]. Scarce data areavailable on the efficacy of bisphosphonates, calcitonin,and nicotinamide on decreasing phosphate levels or
Osteoporos Int (2018) 29:1987–2009 1997

reducing the growth of calcified masses [3, 49, 84–86,89]. The use of topical or systemic steroids [2] andtopical/intra-lesional sodium thiosulfate [31, 48, 90]has been described. One reported case presenting withblurred vision, secondary to retinal angioid streaks, wastreated with intravitreal ranibizumab (a vascular epider-mal growth factor receptor antagonist) [87]. In a recent-ly published report, treatment of HFTC patients showingevidence of systemic inflammation with interleukin–1(IL-1) antagonist (canakinumab) or recombinant IL-1receptor antagonist (anakinra) resulted in an improve-ment in their symptomatology [11].
Conclusion
HFTC is a rare group of disorders of calcium/phosphatehomeostasis. It is characterized by recurrent, painful cal-cified masses that often require surgical resection, whenthe response to medical treatment is unsatisfactory.Despite insight into the molecular basis for the patho-physiology of the disease, the wide variation in the clin-ical presentation, severity, and progress remains in large
part unexplained. Research is needed to determine thesafety and efficacy of FGF23 replacement and moleculartherapy directly targeting the specific genetic aberration,in FGF23 or other related genes. While awaiting theprogress in therapeutic options, mutation analysis re-mains an important step for counseling in families whereHFTC cases are identified.
Acknowledgments The authors would like to thank Nguyen Sydney M.for her help in performing genetic sequencing at Dr. Ichikawa’s Lab.
Funding information This work was supported by a grant from theMedical Resource Plan at the American University of Beirut and madepossible thanks to the Scholars in HeAlth Research Program (SHARP).Research reported in this publication was supported by the FogartyInternational Center and Office of Dietary Supplements of the NationalInstitutes of Health under Award Number D43TW009118.
Compliance with ethical standards
Patients and family members provided written informed consent for eval-uation and mutation analysis. The evaluation and consent form wereapproved by the Institutional Review Board at AUB-MC.
Conflicts of interest None.
Table 2 Drug therapy in FTC, as described in case reports and case series
Drug Starting dose Maximum dose
Phosphate binders in the gastrointestinal tract
Sevelamer [9, 20, 35, 36, 41, 81] 800 mg 3 times daily 1600 mg 3 times daily
Aluminum hydroxide [2, 9, 19] 300 mg 3 times daily 600 mg 3 times daily
Lanthanum carbonate [49] 750 mg daily 3000 mg daily
Glycine and calcium carbonate [82] Reported dose: glycine 180 mg and calcium carbonate 420 mg 2 tablets 3 times daily
Phosphaturic drugs
Probenecid1 [9, 19] 250–500 mg BID Increase 500 mg every 4 weeks to amaximum of2 g/day
Acetazolamide2 [3, 19, 36, 81, 83, 84]. 250 mg BID 500 mg BID
Nicotinamide [3] 500 mg BID
Calcitonin [84–86] 100 IU daily to 200 IU daily given as 100 IU BID s/c or as continuous s/c infusion (pump)
Parathyroid hormone [84] Intravenous injection over a 15-min period in two doses (30 U at 09:00 h and 300 U at 11:30 h)
Other treatment options reported in the literature
Bisphosphonate risedronate/ibandronate [2, 49] Risedronate 35 mg weeklyIbandronate 150 mg once monthly
Sodium thiosulfate [31, 48] 370–833 g daily application
Interleukin–1 (IL-1) antagonist (canakinumab)/recombinant IL-1 receptor antagonist (anakinra)[11]
Canakinumab 2 mg/kg every 8 weeksAnakinra 100 mg subcutaneously
daily
Canakinumab 3 mg/kg every 8 weeks (100 mg)Anakinra 100 mg subcutaneously twice daily
One study reported on the use of intravitreal ranibizumab, resulting in an improvement of ocular disease [87]
BID two times daily, IL-1 interleukin–1, s/c subcutaneous1Uric acid levels at baseline and follow-up can be used as a dose titration marker. Goal is to lower the serum uric acid to below the normal range2Monitor serum bicarbonate and titrate acetazolamide up to as high as 500mg twice daily with a goal of pushing the bicarbonate into the lower end of thenormal range or just below the normal range
1998 Osteoporos Int (2018) 29:1987–2009

Table3
Summaryof
genotype/phenotype
inreported
caseswith
familialtumoralcalcinosisandhyperostosishyperphosphatemicsyndrome
Authoryear
(reference)
Genetic
mutation
Phenotype
Hom
ozygousvs
compound
heterozygous
Consanguinity
Age
ofonseto
fsymptom
sSu
bcutaneous
and
musculo-skeletallesions
Dentallesions
Ocular,periorbital,
andneurologiclesions
Visceral
calcification
FGF23
gene
mutation
Benet-Pages
2005
[13]
FGF23
HFT
CHom
ozygous
No
12years
Swellin
gatelbowand
tibia
Pulp
stones
NR
NR
Araya
2005
[23]
FGF23
HFT
CHom
ozygous
Yes
Childhood
Subcutaneous
nodules2
NR
NR
NR
Chefetz2005
[12]
FGF23
HHS/HFT
CHom
ozygous
No
5years
Calcified
fociin
oral
mucosa.Massesin
wrists,ankle,knees.
Osteosclerosisand
absent
medullary
cavity
Delayed
eruptio
nof
perm
anentteeth;
diminishedroot
length
Calcified
fociin
lower
eyelids
Calcinosisof
renalm
edulla
Larsson
2005
(C1,
C2)
[24]
FGF23
HFT
CHom
ozygous
Yes
44and51
years
Massesin
upperand
lower
limbs.
Calcificatio
nof
vertebrald
iscs
NR
NR
NR
Garringer2008
(C1)
[36]
FGF23
HFT
CHom
ozygous
No
20years
Massesatelbow,hip,
knee
NR
NR
NR
Garringer2008
(C2)
[36]
FGF23
HFT
CHom
ozygous
Yes
11years
Massatbigtoe(following
trauma)
andelbow
NR
NR
NR
Garringer2008
(C3)
[36]
FGF23
HFT
CHom
ozygous
Yes
5years
Massatbigtoe(w
ithout
trauma)
NR
NR
NR
Lam
moglia
2009
[9]
FGF23
HHS/HFT
CHom
ozygous
No
3years
Massatelbow;
fluid-filledcystsin
the
arm
(shownby
CT)
NR
NR
NR
Bonescan:increased
activ
ityatelbowand
long
bones
Bergw
itz2009
[26]
FGF23
HHS/HFT
CHom
ozygous
NR
10months
Periarticular
eruptio
nsdisplaying
chalky
drainage
(44%
calcium
phosphate,12%
calcium
carbonate,
44%
protein)
athand,
knee,fingerjoint;
calcificmyelitis
NR
NR
NR
Masi2
009(C1)
[27]
FGF23
HFT
CHom
ozygous
Yes
25years
Massesatshoulder,
glutealarea,pelvis
NR
NR
NR
Appen
dix
Osteoporos Int (2018) 29:1987–2009 1999

Tab
le3
(contin
ued)
Authoryear
(reference)
Genetic
mutation
Phenotype
Hom
ozygousvs
compound
heterozygous
Consanguinity
Age
ofonseto
fsymptom
sSu
bcutaneous
and
musculo-skeletallesions
Dentallesions
Ocular,periorbital,
andneurologiclesions
Visceral
calcification
Masi2009(C2)
[27]
FGF23
HFT
CHom
ozygous
No
25years
Massinbutto
ckandother
ectopiccalcifications
NR
NR
NR
Abbasi2
014[28]
FGF23
HHS
Hom
ozygous
Yes
4years
Diaphysealh
yperostosis
NR
Decreased
visual
acuity
NR
Shah
2014
(C1,C2,
C3)
[29]
FGF23
HFT
CHom
ozygous
NR
34yearsforC1
NRforC2
andC3
Tendon
ossificatio
n.Ear
andfinger
joints
calcifications
NR
NR
NR
Ghafouri-Fard
2015
(C1)
[30]
FGF23
HHS/HFT
CNR
NR
NR
Diaphyseal
hyperostosison
x-ray;
largejoint
calcifications
NR
Facialpalsyand
decreasedvisual
acuity
NR
Ghafouri-Fard
2015
(C2)
FGF23
HHS/HFT
CNR
NR
NR
Diaphysealh
yperostosis
onx-ray
NR
Facialpalsyand
decreasedvisual
acuity
NR
Jost2016
(C1)
[31]
FGF23
HFT
CHom
ozygous
NR
42years
Lefttibiaandrightbuttock
NR
NR
NR
GALN
T3gene
mutation
Topaz2004
(F1)
[32]
GALN
T3HHS/HFT
CHom
ozygous
Yes
NR
Subcutaneousmasses
around
hipandknee
NR
NR
NR
Topaz2004
(F2)
[32]
GALN
T3HHS/HFT
CCom
pound
heterozygous
No
4years
Recurrent
massesathip,
knee,shoulder,elbows,
hands,thighs,feet
Gingivitis
NR
long-lastin
grash
reminiscent
ofvasculitis
Ichikawa2005
(3cases)[33]
GALN
T3HFTC
Com
pound
heterozygous
NA
NR
Tum
orouscalcific
masses2
Shortb
ulbous
roots,
pulp
stones,
radiculardentin
depositedin
swirls
NR
NR
Cam
pagnoli2
006
(C1)
[34]
GALN
T3HFTC
Hom
ozygous
Yes
6years
Massesatelbow,hip3
NR
NR
Testicular
microlithiasis
Cam
pagnoli2
006
(C2)
[34]
GALN
T3HFT
CHom
ozygous
Yes
6years
Elbow
,ulna,tib
ia,
metacarpalb
ones,
radius,and
hip
NR
NR
NR
Frishberg2005
(C1)
[35]
GALN
T3HHS
Hom
ozygous
Yes
7years
Bilateraltibialp
ainand
recurrentp
ainful
episodes
atvarious
locatio
ns
NR
NR
NR
Frishberg2005
(C2)
[35]
GALN
T3HHS/HFT
CHom
ozygous
Yes
9years
Bilateraltibialp
ainand
recurrentp
ainful
episodes
atvarious
locatio
ns,
subcutaneous
nodules
NR
NR
NR
Garringer
2006
[5]
GALN
T3HFT
CHom
ozygous
NR
26years
Recurrent
inflam
matory
polyarthritis
atshoulders,hands,and
feet
NR
NR
NR
2000 Osteoporos Int (2018) 29:1987–2009

Tab
le3
(contin
ued)
Authoryear
(reference)
Genetic
mutation
Phenotype
Hom
ozygousvs
compound
heterozygous
Consanguinity
Age
ofonseto
fsymptom
sSu
bcutaneous
and
musculo-skeletallesions
Dentallesions
Ocular,periorbital,
andneurologiclesions
Visceral
calcification
Ichikawa2006
[37]
GALN
T3HFT
CCom
pound
heterozygous
NR
22years
NR
NR
Whitedotson
her
eyelids,
calcifications
with
inthesuperficial
derm
is
NR
Specktor
2006
[38]
GALN
T3HFT
CHom
ozygous
No
Childhood
Massesat
acromio-clavicularand
elbowjoints
Thindentalenam
el,
shortb
lunt
roots,
tauro-dontismof
the
pre-molar/m
olar
teethand
obliterationof
the
pulp
cavitiesin
mostteeth
Conjunctiv
aldeposits
NR
Barbieri2
007[39]
GALN
T3HFT
CCom
pound
heterozygous
No
Firstd
ecade
Massive
semi-calcific
lesions2
NR
NR
NR
Garringer2007
(C1)
[40]
GALN
T3HFT
CHom
ozygous
No
17years
Calcificmassatdistal
femur
NR
Noeyeinvolvem
ent
Notesticular
microlithiasis
Garringer2007
(C2)
[40]
GALN
T3HFT
CHom
ozygous
No
45years
Massatglutealm
uscle,
irregularitiesat
acetabulum
,erosionsat
femoralneck
NR
Noeyeinvolvem
ent
Testis
microlithiasis
Ichikawa2007
[41]
GALN
T3HHS
Com
pound
heterozygous
NR
5years
Swellin
gof
tibiaand
forearm
NR
NR
NR
Laleye2008
(C1
andC2)
[42]
GALN
T3HFT
CHom
ozygous
Yes
17and19
years
Massesathipand
shoulders
Gingivitis
andteeth
decay
NR
NR
Olauson
2008
[43]
GALN
T3HHS
Hom
ozygous
NR
19years
Low
erextrem
itypain
NR
NR
NR
Dum
itrescu
2009
[3]
GALN
T3HHS/HFT
CCom
pound
heterozygous
NR
12years
Kneesw
ellin
gand
corticalhyperostosis.
Calcificatio
nof
thyroid
andanterior
costal
cartilagesof
thelower
ribs,vertebraldisc,
nucleuspulposus,
annulusfibrosis,and
interstitialseptain
the
calves
Calcificatio
nof
roots,
abnorm
alities
ofdentin
NR
NR
Gok
2009
[44]
GALN
T3HHS
Com
pound
heterozygous
Yes
8years
Leg
pain,w
ithdiaphyseal
periostealreactio
nNR
NR
NR
Ichikawa2010
(C1)
[19]
GALN
T3HFT
CHom
ozygous
Yes
13years
Glutealmass,
asym
ptom
aticcalcified
massesathip,knee,
andfoot
NR
NR
NR
Osteoporos Int (2018) 29:1987–2009 2001

Tab
le3
(contin
ued)
Authoryear
(reference)
Genetic
mutation
Phenotype
Hom
ozygousvs
compound
heterozygous
Consanguinity
Age
ofonseto
fsymptom
sSu
bcutaneous
and
musculo-skeletallesions
Dentallesions
Ocular,periorbital,
andneurologiclesions
Visceral
calcification
Ichikawa2010
(C2)
[19]
GALN
T3HFT
CHom
ozygous
Yes
4.5years
Low
erlegandelbow
mass;tib
ial
hyperostosis
NR
NR
NR
Ichikawa2010
(C3)
[19]
GALN
T3HFT
CHom
ozygous
No
8years
Tibialp
ainand
diaphyseal
hyperostosis
NR
NR
NR
Ichikawa2010
(C4)
GALN
T3HFT
CHom
ozygous
No
15years
Massattib
ia,
subperiosteal
ossificatio
n
Absence
ofpulp
cham
bersandroot
canals,and
short
bulbousrootsin
almostallteeth
Calcificatio
nsof
basal
ganglia
NR
Joseph
2010
(C1)
[22]
GALN
T3HHS/HFT
CCom
pound
heterozygous
No
2years
Calcified
nodulesat
butto
ckandother
sites.1Tibialp
ainwith
thicksclerosed
medullary
cavity
NR
NR
NR
Joseph
2010
(C2)
[27]
GALN
T3HHS
Com
pound
heterozygous
No
13years
Diaphysealm
edullary
sclerosis,avariable
periostealreactio
n,at
ulna
andtib
ia
NR
NR
NR
Yancovitch2011
(C1)
[45]
GALN
T3HFT
CHom
ozygous
Yes
13years
Massesatelbow,gluteal
region,and
popliteal
joint
Absence
ofpulp
cham
bers,short
bulbousroots,root
resorptio
n,andteeth
loss
Retinalangioidstreaks
NR
Yancovitch2011
(C2)
[45]
GALN
T3HFT
CHom
ozygous
Yes
9years
Massesatfoot
andhand
Abnormalgum
root
NR
NR
ElD
emellawy2014
[46]
GALN
T3HFT
CHom
ozygous
Yes
8years
Bilateralelbow
masses,
chronicrecurrent
multifocal
osteom
yelitis
NR
NR
NR
Krstevska
2012
[47]
GALNT3
HFTC
NR
NR
14years
Calcificatio
nof
lefthip
Gingivalo
vergrowth,
enlargem
ento
fher
maxillaryand
mandibularprocess
andthebody
ofthe
lower
jaw.B
ony
sclerosisof
mandible,maxilla
andskull,increased
trabeculae
and
decreasedmarrow
spaces,hyperostosis
NR
NR
2002 Osteoporos Int (2018) 29:1987–2009

Tab
le3
(contin
ued)
Authoryear
(reference)
Genetic
mutation
Phenotype
Hom
ozygousvs
compound
heterozygous
Consanguinity
Age
ofonseto
fsymptom
sSu
bcutaneous
and
musculo-skeletallesions
Dentallesions
Ocular,periorbital,
andneurologiclesions
Visceral
calcification
frontalis
interna,
sclerosisof
dental
pulp
cham
bers
Ratsimbazafy
2012
[48]
GALN
T3HFT
CNR
NR
12years
Massatelbow
NR
NR
NR
Favia2014
[49]
GALN
T3HFT
CHom
ozygous
NA
9years
Calcified
massesathip
andelbow
Enamelhypoplasia,
maxillaryand
mandibular
hypoplasi,and
crossbite
NR
NR
Finer2014
[81]
GALN
T3HFT
CCom
pound
heterozygous
No
7years
Buttock,hip,hem
i-pelvis,
mandible,forearm
(boneandbone
marrow)
NR
NR
NR
Rafaelsen
2014
(C1)
[20]
GALN
T3HFT
CHom
ozygous
Yes
14years
Elbow
mass,ischial
tuberosity,gluteal
area
Dentald
isease:
abscessesandteeth
loss
Conjunctiv
alirritatio
nNR
Rafaelsen
2014
(C1)
[20]
GALN
T3HHS/HFT
CHom
ozygous
Yes
3weeks
Scalp,gluteal,thigh,
masses.Tibial
sclerosisand
periostealreactio
n
NR
NR
NR
Masi2
015[27]
GALN
T3HHS/HFT
CHom
ozygous
No
3years
Glutealmasses,
diaphysitis
and
corticalhyperostosis;
intracranial
calcifications
NR
NR
NR
Jost2016
(C2)
[31]
GALN
T3HHS/HFT
CCom
pound
heterozygous
No
12years
Massattheleftelbow
NR
NR
NR
Ram
nitz2016
(C1-C8)
4[11]
GANT3
HHS/HFT
CHom
ozygousand
compound
heterozygous
No
20months
−45years
Periarticular
calcinosis
anddiaphysitis;
grow
thplateinvasion
anddestructionin
elbow.C
hestwalllarge
cysticmasseswith
fluidlevelscontaining
“milk
ofcalcium”
Short,bulbousroots
with
obliterationof
dentalpulp.
Inset-thistle-shaped
pulp
cham
bersand
apulp
stone
NR
Subm
ucosalgut
calcification
Sun2016
[50]
GALN
T3HFT
CHom
ozygous
No
2years
Periarticular
masses
(hip
andfeet)
NR
NR
NR
KLO
THOgene
mutation
Ichikawa2007
[15]
KLO
THO
HFT
CHom
ozygous
No
13years
Swellin
gatmalleolus
andthenar
eminence;
punctatecalcifications
ontheAchilles
tendon
NR
NR
NR
Osteoporos Int (2018) 29:1987–2009 2003

Tab
le3
(contin
ued)
Authoryear
(reference)
Vascularcalcification
Ph
TRP
1,25
(OH) 2D
FGF-23
levels1
Notes
C-
term
inal
Intact
Authoryear
(reference)
Vascularcalcification
Ph
TRP
1,25
(OH) 2D
FGF-23
levels1
Notes
C-
term
inal
Intact
FGF23
gene
mutation
Benet-Pages
2005
[13]
NR
↑↑
↑↑
NR
Araya
2005
[23]
NR
↑↑
↑↑
↓
Chefetz2005
[12]
Aortic
valve,aorticarch,and
femoralartery
NR
NR
NR
NR
NR
Reduced
trabecular
density
onCTof
theradius
Larsson
2005
(C1,
C2)
[24]
Placental,femoral,foot,ankle,popliteal
vessels
↑↑
NR
NR
NR
Mild
hypercalcemia
Garringer2008
(C1)
[36]
NR
↑NR
NR
NR
NR
Garringer2008
(C2)
[36]
NR
↑NR
NR
NR
NR
C2andC3arecousins
Garringer2008
(C3)
[36]
NR
↑NR
NR
NR
NR
C2andC3arecousins
Lam
moglia
2009
[9]
Nocerebralartery
occlusivediseaseon
angiography
↑↑
NR
NR
NR
Sisterof
indexcase
(7months)ishomozygousforthemutationandhashigh
phosphatebut
nosymptom
s(sym
ptom
sin
theindexcase
startedat2yearsof
age)
Bergw
itz2009
[26]
NR
↑↑
NL
↑↓
Described
initially
byYam
aghuchietal.(1995)
Masi2
009(C1)
[27]
NR
↑↑
NR
↑↓
Masi2009(C2)
[27]
NR
↑↑
NR
↑↓
Abbasi2
014[28]
Large
vesselcalcification
↑NR
NR
NR
NR
Shah
2014
(C1,C2,
C3)
[29]
Aorta,m
esenteri,renal,carotid,lim
b,heart,
andbrainvasculaturealongwith
ectopic
calcification
↑↑
NL
↑↓
C1hadaboveandbelowknee
amputatio
n.C2andC3hadsymptom
ssimilarto
C1,but
details
werenotreported
Ghafouri-Fard
2015
(C1)
[30]
Large
vesselcalcifications
↑NR
NR
NR
NR
Her
sister
hadthesamesymptom
atologyanddied
attheageof
7years
Ghafouri-Fard
2015
(C2)
Large
vesselcalcifications
↑NR
NR
NR
NR
Jost2016
(C1)
[31]
NR
↑NR
NR
NR
NR
GALN
T3gene
mutation
Topaz2004
(F1)
[32]
NR
↑NR
↑NR
NR
Described
initially
bySteinherzetal.(1985),Slavinetal.(1993),andMetzker
(1988)
Topaz2004
(F2)
[32]
NR
↑↑
↑NR
NR
Ichikawa2005
(3cases)[33]
Systemicandcerebralvascular
calcification
inonecase
↑NR
↑NR
NR
Described
initiallyby
McPhaulandLy
les(1961),and
Lylesetal.,(1985).Pseudo-autosomal
dominantp
attern:H
eterozygousmotherhadhigh
phosphate4.9mg/dl
(normal2.5–4.5)
andhigh
1,25(O
H) 2Dbuttypicalsymptom
sof
FTCwerenotreported
Cam
pagnoli2
006
(C1)
[34]
NR
NR
NR
↑NR
NR
Cam
pagnoli2
006
(C2)
[34]
NR
↑NR
↑NR
NR
Patient
developedalso
immunethrombocytopenicpurpura
NR
↑NR
NL
NR
NR
Tab
le3(contin
ued)
2004 Osteoporos Int (2018) 29:1987–2009

Tab
le3
(contin
ued)
Authoryear
(reference)
Vascularcalcification
Ph
TRP
1,25
(OH) 2D
FGF-23
levels1
Notes
C-
term
inal
Intact
Frishberg2005
(C1)
[35]
Frishberg2005
(C2)
[35]
NR
↑NR
NL
NR
NR
Garringer
2006
[5]
Novascular
calcifications
↑NR
↑↑
↓Bonemineraldensity
was
norm
al
Ichikawa2006
[37]
NR
↑↑
↑NR
NR
Specktor
2006
[38]
NR
↑↑
↑↑
NR
Barbieri2
007[39]
NR
↑↑
↑↑
NR
Garringer2007
(C1)
[40]
NR
↑NR
NR
↑↓
Garringer2007
(C2)
[40]
NR
↑NR
NR
↑↓
Ichikawa2007
[41]
NR
↑↑
↑↑
↓
Laleye2008
(C1
andC2)
[42]
NR
↑NR
NR
NR
NR
Olauson
2008
[43]
NR
↑↑
NL
↑↓
Dum
itrescu
2009
[3]
Nocoronary
calcification
↑↑
NL
↑↓
BMDZ-score:totalhip1.9,APspine0.3,leftforearm
1.0
Gok
2009
[44]
NR
↑NR
NR
↑NR
Ichikawa2010
(C1)
[19]
NR
↑NR
NL
NR
NR
Ichikawa2010
(C2)
[19]
NR
↑NR
NL
↑↓
Ichikawa2010
(C3)
[19]
NR
↑↑
NL
↑↓
Ichikawa2010
(C4)
NR
↑NR
NR
NR
↓
Joseph
2010
(C1)
[22]
NR
↑NR
NR
NR
NR
Pseudo-autosom
aldominantpattern:F
atherh
asslightlyhigh
Phof
5.4(upperlim
it5mg/dl)
Joseph
2010
(C2)
[27]
NR
↑NR
NR
NR
NR
Yancovitch2011
(C1)
[45]
NR
↑NR
NR
NR
NR
Yancovitch2011
(C2)
[45]
NR
↑NR
NR
NR
NR
ElD
emellawy2014
[46]
NR
↑NR
NL
NR
NR
x-ray:
sclerotic
leftfibulaandleftfemoralosteochondrald
efect
SPECT:focalincreaseduptake
inthelateralaspectoftheleftmaxilla,symphysismenti,left
ascendingramus
ofthemandible,leftmedialfem
oralcondyle,leftgreatertrochanter,left
medialm
alleolus,and
leftelbow
Krstevska
2012
[47]
NR
NR
NR
NR
NR
NR
Osteoporos Int (2018) 29:1987–2009 2005

Tab
le3
(contin
ued)
Authoryear
(reference)
Vascularcalcification
Ph
TRP
1,25
(OH) 2D
FGF-23
levels1
Notes
C-
term
inal
Intact
Ratsimbazafy
2012
[48]
NR
NR
NR
NR
NR
NR
Favia2014
[49]
NR
↑NR
NR
NR
NR
Finer2014
[81]
NR
↑↑
NR
NR
NR
Rafaelsen
2014
(C1)
[20]
NR
↑↑
NL
↑↓
BMDZ-scores:FN
1.9,TH2.2,LS0.5
Rafaelsen
2014
(C1)
[20]
Placentalcalcification
↑↑
NL
↑↓
Masi2
015[27]
Low
erlim
bvascular
calcifications
↑↑
NL
↑↓
Patient
developedhypogonadotropichypogonadism
andhypothyroidism
LS-BMD
[0.456
g/cm
2(LSZ-score
−5.1SD)]
Jost2016
(C2)
[31]
NR
↑NR
NR
NR
NR
Ram
nitz2016
(C1-C8)
4[11]
Calcificatio
nof
leftanterior
descending
and
aorta.Calcificatio
nof
papillary
muscle
↑↑
NLor
↑↑
↓BMDwas
with
in2SDof
themeanforZ-score
(adults
andchild
ren)
andT-score(adults
only)forallsubjectsbuto
neattheAPspine,totalh
ip,and
1/3radius
FTC4,who
presentedwith
severe,longstandinginflam
mation,hadan
APspineBMDof
0.667g/cm
2,T-score−3.9;totalhipBMDof
0.551g/cm
2,T-score−3.2;and1/3radius
BMDof
0.612g/cm
2,T-score−1.4
Sun2016
[50]
NR
↑NR
NR
NR
NR
KLO
THOgene
mutation
Ichikawa2007
[15]
Calcificatio
nof
theduraandcarotid
arteries,
with
outp
arenchym
alcalcifications
↑↑
↑↑
↑Onx-ray:
diffuseosteopeniapatchy
sclerosisin
hands,feet,longbones,andcalvaria.
Presenceof
metacarpalp
eriostealreaction
Reportedcasesandon
whom
genetic
studieswerenotp
erform
edarenotincludedin
thistable
1,25(O
H)2D1,25
dihydroxyvitaminDlevel,C1case
1,C2case
2,C3case
3(sym
ptom
sof
individualcasespresentedseparatelywhencasesarenotrelated),F1family
1,F2family
2,NLnorm
al,N
Rnot
reported,P
hphosphate,TR
Ptubularphosphatereabsorptio
n1↓FG
F23levelswhenlowor
inappropriatelynorm
al2Locationof
massesnotspecified
3C1andC2aresiblings
4Cohorto
fsixfamilies.N
ogenetic
mutationwas
identifiedin
case
7
2006 Osteoporos Int (2018) 29:1987–2009

References
1. Giard A (1898) Sur la calcification hibernale. CR Soc Biol 10:1013–1015
2. Metzker A, Eisenstein B, Oren J, Samuel R (1998) Tumoral calci-nosis revisited—common and uncommon features. Eur J Pediatr147(2):128–132
3. Dumitrescu CE, Kelly MH, Khosravi A, Hart TC, Brahim J, WhiteKE, Farrow EG, Nathan MH, Murphey MD, Collins MT (2009) Acas e o f f ami l i a l t umora l c a l c i nos i s / hype ro s to s i s–hyperphosphatemia syndrome due to a compound heterozygousmutation in GALNT3 demonstrating new phenotypic features.Osteoporos Int 20(7):1273–1278
4. Slavin RE, Wen J, Barmada A (2012) Tumoral calcinosis—a path-ogenetic overview a histological and ultrastructural study with areport of two new cases, one in infancy. Int J Surg Pathol 20(5):462–473
5. Olsen KM, Chew FS (2006) Tumoral calcinosis: pearls, polemics,and alternative possibilities 1. Radiographics 26(3):871–885
6. Polykandriotis EP, Beutel FK, Horch RE, Grünert J (2004) A caseof familial tumoral calcinosis in a neonate and review of the litera-ture. Arch Orthop Trauma Surg 124(8):563–567
7. El-Rassy I, Bou-Abdallah J, Al-Ghadban S, Bitar F, Nemer G(2008) Absence of NOTCH2 and Hey2 mutations in a familialAlagille syndrome case with a novel frameshift mutation inJAG1. Am J Med Genet A 146((7):937
8. TmP/GFR normal ranges. Available from (accessed in December2017): http://baspath.co.uk/test_directory/tindex/TmP%20Refrange.htm
9. Lammoglia JJ, Mericq V (2009) Familial tumoral calcinosis causedby a novel FGF23 mutation: response to induction of tubular renalacidosis with acetazolamide and the non-calcium phosphate bindersevelamer. Hormone Research in Paediatrics 71(3):178–184
10. Prediction of functional effects of human nsSNPs. Available from(accessed in December 2017): http://genetics.bwh.harvard.edu/pph2/
11. Ramnitz MS, Gourh P, Goldbach-Mansky R, Wodajo F, IchikawaS, Econs MJ, White KE, Molinolo A, Chen MY, Heller T, delRivero J, Seo-Mayer P, Arabshahi B, Jackson MB, Hatab S,McCarthy E, Guthrie LC, Brillante BA, Gafni RI, Collins MT(2016) Phenotypic and genotypic characterization and treatmentof a cohort with familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome. J Bone Miner Res 31(10):1845–1854
12. Chefetz I, Heller R, Galli-Tsinopoulou A, Richard G, Wollnik B,Indelman M, Koerber F, Topaz O, Bergman R, Sprecher E,Schoenau E (2005) A novel homozygous missense mutation inFGF23 causes Familial Tumoral Calcinosis associated with dissem-inated visceral calcification. Hum Genet 118(2):261–266
13. Benet-Pagès A, Orlik P, Strom TM, Lorenz-Depiereux B (2005) AnFGF23 missense mutation causes familial tumoral calcinosis withhyperphosphatemia. Hum Mol Genet 14(3):385–390
14. Chefetz I, KohnoK, Izumi H, Uitto J, Richard G, Sprecher E (2009)GALNT3, a gene associated with hyperphosphatemic familial tu-moral calcinosis, is transcriptionally regulated by extracellularphosphate and modulates matrix metalloproteinase activity.Biochim Biophys Acta (BBA)-Mol Basis Dis 1792(1):61–67
15. Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, SorensonAH, Goetz R, Mohammadi M, White KE, Econs MJ (2007) Ahomozygous missense mutation in human KLOTHO causes severetumoral calcinosis. J Clin Investig 117(9):2684–2691
16. Shawar SM, RamadanAR, Ali BR, AlghamdiMA, JohnA,HudaibFM (2016) FGF23–S129F mutant bypasses ER/Golgi to the circu-lation of hyperphosphatemic familial tumoral calcinosis patients.Bone 93:187–195
17. Farrow EG, Imel EA, White KE (2011) Hyperphosphatemic famil-ial tumoral calcinosis (FGF23, GALNT3 and αKlotho). Best PractRes Clin Rheumatol 25(5):735–747
18. Mikati MA, Melhem RE, Najjar SS (1981) The syndrome of hy-perostosis and hyperphosphatemia. J Pediatr 99(6):900–904
19. Ichikawa S, Baujat G, Seyahi A, Garoufali AG, Imel EA, PadgettLR et al (2010) Clinical variability of familial tumoral calcinosiscaused by novel GALNT3 mutations. Am J Med Genet A 152(4):896–903
20. Rafaelsen S, Johansson S, Ræder H, Bjerknes R (2014) Long-termclinical outcome and phenotypic variability in hyperphosphatemicfamilial tumoral calcinosis and hyperphosphatemic hyperostosissyndrome caused by a novel GALNT3 mutation; case report andreview of the literature. BMC Genet 15(1):98
21. Martinez S, Vogler J 3rd, Harrelson J, Lyles K (1990) Imaging oftumoral calcinosis: new observations. Radiology 174(1):215–222
22. Joseph L, Hing SN, Presneau N, O’Donnell P, Diss T, Idowu BD,Joseph S, Flanagan AM, Delaney D (2010) Familial tumoral calci-nosis and hyperostosis–hyperphosphataemia syndrome are differ-ent manifestations of the same disease: novel missense mutations inGALNT3. Skelet Radiol 39(1):63–68
23. Araya K, Fukumoto S, Backenroth R, Takeuchi Y, NakayamaK, ItoN, Yoshii N, Yamazaki Y, Yamashita T, Silver J, Igarashi T, Fujita T(2005) A novel mutation in fibroblast growth factor 23 gene as acause of tumoral calcinosis. J Clin Endocrinol Metab 90(10):5523–5527
24. Larsson T, Yu X, Davis SI, Draman MS, Mooney SD, Cullen MJ,White KE (2005) A novel recessive mutation in fibroblast growthfactor-23 causes familial tumoral calcinosis. J Clin EndocrinolMetab 90(4):2424–2427
25. Garringer HJ, Malekpour M, Esteghamat F, Mortazavi SM, DavisSI, Farrow EG et al (2008) Molecular genetic and biochemicalanalyses of FGF23 mutations in familial tumoral calcinosis. Am JPhysiol Endocrinol Metab 295(4):E929–EE37
26. Bergwitz C, Jüppner H (2010) Regulation of phosphate homeosta-sis by PTH, vitamin D, and FGF23. Annu Rev Med 61:91–104
27. Masi L, Gozzini A, Franchi A, Campanacci D, Amedei A, FalchettiA, Franceschelli F, Marcucci G, Tanini A, Capanna R, Brandi ML(2009) A novel recessive mutation of fibroblast growth factor-23 intumoral calcinosis. J Bone Joint Surg 91(5):1190–1198
28. Abbasi F, Ghafouri-Fard S, Javaheri M, Dideban A, Ebrahimi A,Ebrahim-Habibi A (2014) A newmissensemutation in FGF23 genein a male with hyperostosis–hyperphosphatemia syndrome (HHS).Gene 542(2):269–271
29. Shah A, Miller CJ, Nast CC, Adams MD, Truitt B, Tayek JA, TongL, Mehtani P, Monteon F, Sedor JR, Clinkenbeard EL, White K,Mehrotra R, LaPage J, Dickson P, Adler SG, Iyengar SK (2014)Severe vascular calcification and tumoral calcinosis in a family withhyperphosphatemia: a fibroblast growth factor 23 mutation identi-fied by exome sequencing. Nephrol Dial Transplant 29(12):2235–2243
30. Ghafouri-Fard S, Abbasi F, Azizi F, Javaheri M, Mehdizadeh M,Setoodeh A (2015) Hyperostosis-hyperphosphatemia syndrome(HHS): report of two cases with a recurrent mutation and reviewof the literature. J Pediatr Endocrinol Metab 28(1–2):231–235
31. Jost J, Bahans C, Courbebaisse M, Tran T-A, Linglart A, BenistanK et al (2016) Topical sodium thiosulfate: a treatment for calcifica-tions in hyperphosphatemic familial tumoral calcinosis? J ClinEndocrinol Metab. https://doi.org/10.1210/jc.2016-1087
32. Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P,Mizrachi M, Khamaysi Z, Behar D, Petronius D, Friedman V,Zelikovic I, Raimer S, Metzker A, Richard G, Sprecher E (2004)Mutations in GALNT3, encoding a protein involved in O-linkedglycosylation, cause familial tumoral calcinosis. Nat Genet 36(6):579–581
Osteoporos Int (2018) 29:1987–2009 2007

33. Ichikawa S, Lyles KW, Econs MJ (2005) A novel GALNT3 muta-tion in a pseudoautosomal dominant form of tumoral calcinosis:evidence that the disorder is autosomal recessive. J ClinEndocrinol Metab 90(4):2420–2423
34. Campagnoli M, Pucci A, Garelli E, Carando A, Defilippi C, Lala R,Ingrosso G, Dianzani I, Forni M, Ramenghi U (2006) Familialtumoral calcinosis and testicular microlithiasis associated with anew mutation of GALNT3 in a white family. J Clin Pathol 59(4):440–442
35. Frishberg Y, Topaz O, Bergman R, Behar D, Fisher D, Gordon D,Richard G, Sprecher E (2005) Identification of a recurrent mutationin GALNT3 demonstrates that hyperostosis-hyperphosphatemiasyndrome and familial tumoral calcinosis are allelic disorders. JMol Med 83(1):33–38
36. Garringer HJ, Fisher C, Larsson TE, Davis SI, Koller DL, CullenMJ, Draman MS, Conlon N, Jain A, Fedarko NS, Dasgupta B,White KE (2006) The role of mutant UDP-N-acetyl-α-d-galactos-amine-polypeptide N-acetylgalactosaminyltransferase 3 in regulat-ing serum intact fibroblast growth factor 23 and matrix extracellularphosphoglycoprotein in heritable tumoral calcinosis. J ClinEndocrinol Metab 91(10):4037–4042
37. Ichikawa S, Imel EA, Sorenson AH, Severe R, Knudson P, HarrisGJ, Shaker JL, Econs MJ (2006) Tumoral calcinosis presentingwith eyelid calcifications due to novel missense mutations in theglycosyl transferase domain of the GALNT3 gene. J ClinEndocrinol Metab 91(11):4472–4475
38. Specktor P, Cooper JG, Indelman M, Sprecher E (2006)Hyperphosphatemic familial tumoral calcinosis caused by a muta-tion in GALNT3 in a European kindred. J Hum Genet 51(5):487–490
39. Barbieri AM, Filopanti M, Bua G, Beck-Peccoz P (2007) Twonovel nonsense mutations in GALNT3 gene are responsible forfamilial tumoral calcinosis. J Hum Genet 52(5):464–468
40. Garringer HJ, Mortazavi SMJ, Esteghamat F, Malekpour M,Boztepe H, Tanakol R et al (2007) Two novel GALNT3 mutationsin familial tumoral calcinosis. Am J Med Genet A 143((20):2390–2396
41. Ichikawa S, Guigonis V, Imel EA, Courouble M, Heissat S, HenleyJD, Sorenson AH, Petit B, Lienhardt A, Econs MJ (2007) NovelGALNT3 mutations causing hyperostosis-hyperphosphatemia syn-drome result in low intact fibroblast growth factor 23 concentra-tions. J Clin Endocrinol Metab 92(5):1943–1947
42. Laleye A, AlaoM, Gbessi G, AdjagbaM,MarcheM, Coupry I et al(2007) Tumoral calcinosis due to GALNT3C. 516-2A> Tmutationin a black African family. Genet Couns (Geneva, Switzerland)19(2):183–192
43. Olauson H, Krajisnik T, Larsson C, Lindberg B, Larsson TE (2008)A novel missense mutation in GALNT3 causing hyperostosis–hyperphosphataemia syndrome. Eur J Endocrinol 158(6):929–934
44. Gok F, Chefetz I, Indelman M, Kocaoglu M, Sprecher E (2009)Newly discovered mutations in the GALNT3 gene causing autoso-mal recessive hyperostosis-hyperphosphatemia syndrome. ActaOrthop 80(1):131–134
45. Yancovitch A, Hershkovitz D, IndelmanM, Galloway P, WhitefordM, Sprecher E, Kılıç E (2011) Novel mutations in GALNT3 caus-ing hyperphosphatemic familial tumoral calcinosis. J Bone MinerMetab 29(5):621–625
46. El Demellawy D, Chang N, de Nanassy J, Nasr A (2015) GALNT3genemutation-associated chronic recurrent multifocal osteomyelitisand familial hyperphosphatemic familial tumoral calcinosis. ScandJ Rheumatol 44(2):170–172
47. Krstevska A, Gale S, Blair F (2011) Tumoral calcinosis: a dentalliterature review and case report. Dental Update 39(6):416–418 21
48. Ratsimbazafy V, Bahans C, Guigonis V (2012) Dramatic diminu-tion of a large calcification treated with topical sodium thiosulfate.Arthritis Rheum 64(11):3826
49. Favia G, Lacaita MG, Limongelli L, Tempesta A, Laforgia N,Cazzolla AP et al (2014) Hyperphosphatemic familial tumoral cal-cinosis: odontostomatologic management and pathological fea-tures. Am J Case Rep 15:569
50. Sun L, Zhao L, Du L, Zhang P, Zhang M, Li M et al (2016)Identification of two novel mutations in the GALNT3 gene in aChinese family with hyperphosphatemic familial tumoral calcino-sis. Bone Res 4:16038
51. Olauson H, Vervloet MG, Cozzolino M, Massy ZA, Torres PU,Larsson TE (2014) New insights into the FGF23-Klotho axis.Semin Nephrol 34(6):586–597
52. Razzaque MS (2009) FGF23-mediated regulation of systemicphosphate homeostasis: is Klotho an essential player? Am JPhysiol Ren Physiol 296(3):F470–F4F6
53. Bhattacharyya N, Chong WH, Gafni RI, Collins MT (2012)Fibroblast growth factor 23: state of the field and future directions.Trends Endocrinol Metab 23(12):610–618
54. HuMC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, RazzaqueMS, Rosenblatt KP, Baum MG, Kuro-o M, Moe OW (2010)Klotho: a novel phosphaturic substance acting as an autocrine en-zyme in the renal proximal tubule. FASEB J 24(9):3438–3450
55. Chang Q, Hoefs S, Van Der Kemp A, Topala C, Bindels R,Hoenderop J (2005) The ß-glucuronidase klotho hydrolyzes andactivates the TRPV5 channel. Science 310(5747):490–493
56. Wang Y, Sun Z (2009) Current understanding of klotho. AgeingRes Rev 8(1):43–51
57. Folsom LJ, Imel EA (2015) Hyperphosphatemic familial tumoralcalcinosis: genetic models of deficient FGF23 action. CurrOsteoporos Rep 13(2):78–87
58. auf dem Keller U, Krampert M, Kümin A, Braun S, Werner S(2004) Keratinocyte growth factor: effects on keratinocytes andmechanisms of action. Eur J Cell Biol 83(11):607–612
59. Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J,Shimkets R (2005) Fibroblast growth factor 7: an inhibitor of phos-phate transport derived from oncogenic osteomalacia-causing tu-mors. J Clin Endocrinol Metab 90(2):1012–1020
60. Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML,Goetz R, Mohammadi M, Baum M (2009) FGF23 decreases renalNaPi-2a and NaPi-2c expression and induces hypophosphatemia invivo predominantly via FGF receptor 1. Am J Physiol Ren Physiol297(2):F282–FF91
61. Razzaque MS (2009) The FGF23–Klotho axis: endocrine regula-tion of phosphate homeostasis. Nat Rev Endocrinol 5(11):611–619
62. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M,Mohammadi M, Sirkis R, Naveh-Many T, Silver J (2007) Theparathyroid is a target organ for FGF23 in rats. J Clin Invest117(12):4003–4008
63. Krajisnik T, Björklund P, Marsell R, Ljunggren Ö, Åkerström G,Jonsson KB et al (2007) Fibroblast growth factor-23 regulates para-thyroid hormone and 1α-hydroxylase expression in cultured bovineparathyroid cells. J Endocrinol 195(1):125–131
64. Faul C, Amaral AP, Oskouei B, Hu M-C, Sloan A, Isakova T,Gutiérrez OM, Aguillon-Prada R, Lincoln J, Hare JM, Mundel P,Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS,Rosas SE, Nessel L, Townsend RR, Feldman HI, St. John SuttonM, Ojo A, Gadegbeku C, di Marco GS, Reuter S, Kentrup D,Tiemann K, Brand M, Hill JA, Moe OW, Kuro-o M, Kusek JW,Keane MG, Wolf M (2011) FGF23 induces left ventricular hyper-trophy. J Clin Invest 121(11):4393–4408
65. Sitara D, Kim S, Razzaque MS, Bergwitz C, Taguchi T, Schüler C,Erben RG, Lanske B (2008) Genetic evidence of serum phosphate-independent functions of FGF-23 on bone. PLoS Genet 4(8):e1000154
66. Gutiérrez OM, Januzzi JL, Isakova T, Laliberte K, Smith K,Collerone G et al (2009) Fibroblast growth factor 23 and left
2008 Osteoporos Int (2018) 29:1987–2009

ventricular hypertrophy in chronic kidney disease. Circulation119(19):2545–2552
67. Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, ErbenRG, J+APw-ppner H, Lanske B (2004) Homozygous ablation offibroblast growth factor-23 results in hyperphosphatemia and im-paired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol 23(7):421–432
68. Wang H, Yoshiko Y, Yamamoto R, Minamizaki T, Kozai K, TanneK, Aubin JE, Maeda N (2008) Overexpression of fibroblast growthfactor 23 suppresses osteoblast differentiation and matrix minerali-zation in vitro. J Bone Miner Res 23(6):939–948
69. Slavin RE, Wen J, Kumar D, Evans EB (1993) Familial tumoralcalcinosis: a clinical, histopathologic, and ultrastructural study withan analysis of its calcifying process and pathogenesis. Am J SurgPathol 17(8):788–802
70. Hatori M, Oomamiuda K, Kokubun S (1996) Hydroxyapatite crys-tals in tumoral calcinosis: a case report. Tohoku J Exp Med 180(4):359–364
71. Block GA, Hulbert-Shearon TE, Levin NW, Port FK (1998)Association of serum phosphorus and calcium x phosphate productwith mortality risk in chronic hemodialysis patients: a nationalstudy. Am J Kidney Dis 31(4):607–617
72. Senac Jr M, Lee F, Gilsanz V (1985) Early costochondralcalcification in adolescent hyperthyroidism. Radiology156(2):375–377
73. Sariyildiz MA, Batmaz I, Aydin F, Azboy I (2012) Early onsetidiopathic costochondral calcification can simulate costal fracture.Int J Basic Clin Stud 1(II):69–74
74. Tsang WK, Tam KFG (2014) A mildly painful wrist mass. SkeletRadiol 43(3):413–415
75. Adams W, Laitt R, Davies M, O’Donovan D (1999) Familial tu-moral calcinosis: association with cerebral and peripheral aneurysmformation. Neuroradiology 41(5):351–355
76. Ghanchi F, Ramsay A, Coupland S, Barr D, LeeWR (1996) Oculartumoral calcinosis: a clinicopathologic study. Arch Ophthalmol114(3):341–345
77. Shanahan CM (2005) Vascular calcification. Curr Opin NephrolHypertens 14(4):361–367
78. Bailleul-Forestier I, Berdal A, Vinckier F, de Ravel T, Fryns JP,Verloes A (2008) The genetic basis of inherited anomalies of theteeth. Part 2: syndromes with significant dental involvement. Eur JMed Genet 51(5):383–408
79. Rangiani A, Cao Z-G, Liu Y, Rodgers AV, Jiang Y, Qin C-L et al(2013) Dentin matrix protein 1 and phosphate homeostasis are crit-ical for postnatal pulp, dentin and enamel formation. Int J Oral Sci4(4):189–195
80. Gal G, Metzker A, Garlick J, Gold Y, Calderon S (1994) Head andneck manifestations of tumoral calcinosis. Oral Surgery, OralMedicine, Oral Pathology 77(2):158–166
81. Finer G, Price HE, Shore RM, White KE, Langman CB (2014)Hyperphosphatemic familial tumoral calcinosis: response to acet-azolamide and postulated mechanisms. Am J Med Genet A164A(6):1545–1549
82. Alkhooly AZA (2009) Medical treatment for tumoral calcinosiswith eight years of follow-up: a report of four cases. J OrthopSurg 17(3):379–382
83. Yamaguchi T, Sugimoto T, Imai Y, Fukase M, Fujita T, Chihara K(1995) Successful treatment of hyperphosphatemic tumoral calci-nosis with long-term acetazolamide. Bone 16(4):S247–SS50
84. Lufkin EG, Wilson DM, Smith LH, Bill NJ, Deluca HF, Dousa TPet al (1980) Phosphorus excretion in tumoral calcinosis: response toparathyroid hormone and acetazolamide. J Clin Endocrinol Metab50(4):648–653
85. Kallmeyer J, Seimon L, MacSearraigh E (1978) The effect of thy-rocalcitonin therapy and phosphate deprivation on tumoral calcino-sis. S Afr Med J 54:963–966
86. Candrina R, Cerudelli B, Braga V, Salvi A (1989) Effects of theacute subcutaneous administration of synthetic salmon calcitonin intumoral calcinosis. J Endocrinol Investig 12(1):55–57
87. McGrath E, Harney F, Kinsella F (2010) Rare disease: an ocularpresentation of familial tumoral calcinosis. BMJ Case Rep. 2010
88. Phosphorus and Your CKD Diet. Available from (accessedDecember 2017): https://www.kidney.org/atoz/content/phosphorus
89. Ichikawa S, Sorenson AH, Austin AM, Mackenzie DS, Fritz TA,Moh A, Hui SL, Econs MJ (2009) Ablation of the Galnt3 geneleads to low-circulating intact fibroblast growth factor 23 (FGF23)concentrations and hyperphosphatemia despite increased FGF23expression. Endocrinology 150(6):2543–2550
90. Goossens J, Courbebaisse M, Caudron E, Bahans C, Vacquerie V,Melchior J, Salle PV, Moesch C, Daudon M, Frocht V, Richette P,Ea HK, Guigonis V (2017) Efficacy of intralesional sodium thio-sulfate injections for disabling tumoral calcinosis: two cases. SeminArthritis Rheum 47:451–455
Osteoporos Int (2018) 29:1987–2009 2009