-
7/30/2019 Hematologic Pathology p1-23
1/23
Hematopathology Preview Fri. 10/15/2010
von Willebrand Disease platelet function problem (primary
hemostasis).
Bleeding time is elevated (the amount of time it takes for a
laceration toclot). PT is normal. vWF serves as a receptor for
platelets, and it is also thecarrier protein for Factor VIII if
abnormal vWF, may also haveabnormal/deficient Factor VIII would
give high PTT. This happensfrequently, but not always (so change
chart to high or normal).
Hemophilia A Factor VIII deficiency the PTT will be elevated.
DIC consumptive coagulopathy: bleeding clotting bleeding
clotting
etc = you use up everything. Bleeding time, PT, and PTT will be
high. If youmeasured fibrinogen, it would be high. If you measured
D dimer, it would be
high. War/HepIf you are monitoring coumadin or heparin therapy,
PT and PTT
will be abnormal. Clinically, following the PT forcoumadin
therapy worksbest, and following the PTT forheparin works best.
von Willebrand Disease, Bernard-Soulier Syndrome, and
GlanzmannThromasthenia:
- von Willebrand Disease deficiency of vW factor
Glycoprotein I = receptor for vW factorGlycoprotein II = the
receptor for fibrinogen
- Bernard-Soulier Syndrome deficiency of Glycoprotein I (vW
factor receptor)- Glanzmann Thrombasthenia deficiency of
Glycoprotein II (fibrinogen receptor)
Any of these can give rise to abnormal platelet structure. If a
person has a low platelet #,will have problems w/primary
hemostasis
Thrombosis: many factors that favor it andmany factors that
inhibit it.
Remember that in DIC, all the factors areconsumed. Patients are
clotting lysingclotting lysing. They use up all the
platelets, the factors, the fibrinogen everything gets consumed
and eventually theywill bleed out.
Factors that favor thrombosis:Tissue factor: produced by
endothelial cellsin response to cytokines (TNF, IL-1,
bacterialendotoxin); major activator of extrinsic
clottingcascadePAIs: produced by endothelial cells; inhibitorsof
plasminogen activator limit fibrinolysisand favor thrombosis
Factors that inhibit thrombosis:
Prostacyclin (PGI2) + NO: produced by endothelial cells to
impede platelet adhesionAdenosine diphosphatase: degrade
ADPAntithrombin III: with cofactor, inactivate
thrombinThrombomodulin: binds thrombin convert to anticoagulant via
ability to activate protein C
- Protein C inhibits clotting by inactivating Va + VIIIaTissue
Factor Pathway inhibitor(TFPI): cell surface protein that directly
inhibits TF VIIa and Xat-PA: protease cleaves plasminogen to form
plasmin plasmin cleaves fibrin to degrade thrombi
Remember that in DIC, all the factors are consumed. Patients are
clotting lysing clotting lysing. They use up all the platelets,the
factors, the fibrinogen everything gets consumed and eventually
they will bleed out.
Test q:A 66F comes to the ER 3 hours after the onset of chest
pain that radiates to her neck and left arm. She is diaphoretic and
hypotensive; theserum troponin I level is elevated. Thrombolytic
therapy is begun. Which of the following drugs is most likely to be
administered? Tissue plasminogenactivator. (Other choices: Aspirin,
Heparin, NO, Vit K)
or normal
-
7/30/2019 Hematologic Pathology p1-23
2/23
Fibrinolysis: Plasmin: breaks down fibrin and interferes with
its
polymerization FDPs: act as weak anti-coagulant factors Elevated
FDPs (fibrin derived D-dimers) can be used in
diagnosing abnormal thrombotic states, including DIC,
DVT,pulmonary embolism
Protein C + S: anti-coagulation factors; protein S is a
cofactorfor protein C; function to degrade activated factors VIII
and V
The Leiden mutation is a structural mutation in Factor V renders
Factor V resistant to the action of Protein S and activated Protein
C.Factor V still works, but it is no longer subject to the
limitations of activated Protein C. Leiden mutation can lead to
recurrent DVT.
Test q:A 25F has had multiple episodes of DVT during the past
10yr and one episode of pulmonary thromboembolism during the past
year. PT, PTT,platelet count, and platelet function studies are all
normal. Which of the following risk factors has most likely
contributed to the patients condition?Leiden mutation. (Other
choices: Antithrombin III deficiency, Mutation in protein C,
Hyperhomocysteinemia, Smoking cigarettes) REPEATED x3
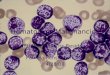
Above (both pictures): Thrombus causing microangiopathic
hemolytic anemia. Patients w/DIC have abnormal multimers of fibrin
inthe blood vessels shred RBCs as they pass through.
Microangiopathic, small blood vessels, hemolytic anemia. Destroy
RBCs, andthe fractured RBCs are called schistocytes. Schistocytes =
microangiopathic hemolytic anemia. May have abnormal heart valve
thatcould shred RBCs can also cause microangiopathic hemolytic
anemia. DIC and certain types of leukemia can also cause this.
Test q:A 45M has a 3-day history of f lank pain and fever. On
phys exam, his temp is 37.9C. There is right costovertebral angle
tenderness. Lab
studies include a urine culture that is positive forEscherichia
coli. The WBC count is 13,310/mm
3
. Two days later, he becomes hypotensive, and ablood culture is
positive forE. coli. He requires increasing pressor support to
maintain blood pressure. He develops a guaiac-positive stool
and
ecchymoses of the skin. CBC shows Hgb 9.2 g/dL, hematocrit
28.1%, and platelet count 70,000/mm3. Increased amounts of fibrin
split products are
identified in the blood (elevated D dimer). Which of the
following conditions is most likely responsible for the low
hematocrit? Microangiopathichemolytic anemia. (MAHA). Robbins
explanation: This patient has DIC, which can result from
gram-negative septicemia. This is a form of MAHA in
which there is deposition of fibrin strands in small vessels.
The RBCs are damaged during passage between these strands.
Coagulation factors andplatelets are consumed, which does not occur
w/other forms of hemolytic anemia.
Tumor Markers:PSA Prostate cancer = prostate specific
antigen
CEA Colorectal/PancreaticCA 19-9 PancreaticAlpha-fetoprotein
Hepatic/Yolk sacBeta-HCG Choriocarcinoma/Molar pregnancy
CA 125 Ovarian cancerS-100 Melanoma/AstrocytomaThyroglobulin
Thyroid cancerCalcitonin Medullary carcinoma of
ThyroidCatecholamines Pheochromocytoma/NeuroblastomaTRAP stain
Hairy cell leukemia = tartrate resistant acid phosphatase
Alkaline phosphatase Mets to bones, Pagets disease of bone
Test q:A 76M has experienced lower back pain for the past year.
On phys exam, the physician palpates a firm nodule in the prostate.
Lab studiesshow an alkaline phosphatase level of 290 U/L and a
serum prostate specific antigen level of 17 ng/mL. A prostate
needle biopsy specimen shows a
moderately differentiated adenocarcinoma. Which of the following
mechanisms best accounts for these findings? Metastases to
vertebrae. (Otherchoices: Tumor extension to rectum; Paraneoplastic
syndrome; High tumor grade; and Tumor angiogenesis) REPEATED x3
Translocations:t (9;22) CML (bcr-abl)t (8;14) Burkitts lymphoma
(c-myc)t (14;18) Follicular lymphoma (bcl-2)t (15;17) AML M3
(promyelocytic
leukemia)
t (1;14) T-cell Lymphoblastic lymphomat (11; 22) Ewings sarcomat
(11; 14) Mantle cell lymphoma (bcl-1)t (11; 18) MALT lymphoma
(API-2)t (1; 14) MALT lymphoma (bcl-10)
-
7/30/2019 Hematologic Pathology p1-23
3/23
Test q:A 71M present to his primary care physician for a
check-up accompanied by his wife, who reports that she had to push
him out of the door togo to see a doctor as his last physical exam
was almost 8yr ago. He states that he has been feeling well other
than having some lower back pain. Phys
exam reveals a firm, nodular prostate. On further questioning,
the patient states that he has not noticed any change in urination.
Lab studies reveal amildly elevated serum prostate-specific antigen
level and a serum alkaline phosphatase level almost three times
normal. These findings are mostconsistent w/which of the following?
Metastatic prostate carcinoma.
Test q:A 77M has experienced increasing malaise and a 6kg weight
loss over the past year. He has noted more severe and constant back
pain for thepast 3 months. On phys exam, his temp is 38.7C. His
prostate is firm and irregular when palpated on digital rectal
exam. Lab studies include a urine
culture positive forEscherichia coli, serum glucose of 70 mg/dL,
creatinine 1.1 mg/dL, total bilirubin 1.0 mg/dL, alkaline
phosphatase 293 U/L, calcium10.3 mg/dL, phosphorus 2.6 mg/dL, and
PSA 25 ng/mL. CBC shows Hgb 9.1 g/dL, hematocrit 27.3%, MCV 94m
3, platelet count 55,600/mm
3, and
WBC count 3570/mm3
with 18% segmented neutrophils, 7% bands, 2% metamyelocytes, 1%
myelocytes, 61% lymphocytes, 11% monocytes, and 3
nucleated RBCs per 100 WBCs. Which of the following is the most
likely diagnosis? Metastatic carcinoma.
Test q: Thyroglobulin is used clinically as: a tumor marker.
Above (left): Schistocytes fragmented RBCs. Seen in DIC,
microangiopathic hemolytic anemia, mechanical heart valves.Above
(right) : Can do reticulocyte count lab will tell you how many
reticulocytes are present per 100. Useful when monitoringpeople
with iron deficiency on iron therapy patients should start
reticking. Reticking actively making red cells. People
w/Fedeficiency do not make enough RBCs.
Figure: Blood smear. Want to find Goldilocks zone not too thick,
not too thin, just right. Find area wherered cells are just barely
touching each other.
Above: -thalassemia. See RBC abnormalities. Howell-Jollybodies
nuclear remnants that remain in RBCs becausepatient does not have a
functional spleen. Seen in sickle cellanemia patients because they
infarct their spleen by age 6-7.These patients are also highly
susceptible to infection by S.pneumoniae, Salmonella, H. influenzae
(due to lack of spleen).
Above: Spherocytes RBCs are smaller and perfectlyround.
Hereditary spherocytosis abnormality inmembrane of RBC take very
small shape. These cellsdo not live 120 days (which classifies
hereditaryspherocytosis as a hemolytic anemia). Will see
nucleatedRBCs, spherocytes, and reticulocytes (aka polychromasia bi
blue cells ver oun er throc tes .
-
7/30/2019 Hematologic Pathology p1-23
4/23
Above (left): Iron deficiency anemia. See lymphocyte RBCs are
smaller than the lymphocyte (= microcytic). RBCs haveexaggerated
pale area in the middle (= hypochromic donut-looking, less color).
Microcytic, hypochromic anemia fits irondeficiency. Poikilocytosis
= abnormal shape. Anisocytosis = abnormal size. Anisopoikilocytosis
= abnormal size and shape (describesiron deficiency).Above (right):
megaloblastic anemia. Macrocytic anemia RBCs are larger than the
lymphocyte. They are often oval-shaped, not
round. Seen in folate/Vitamin B12 deficiency. In addition to
large RBCs, will also see multisegmented neutrophils.
Case 1. 70 y.o. female with fatigue. No hemoptosis or vaginal
bleeding. Normal
physical exam except for pallor.
When patients come in complaining of tiredness/have no energy,
may indicate anemia not enough RBCs, not distributing enough oxygen
to tissues. May also indicate apulmonary disease.
Blood smear: see lymphocyte compare RBC size microcytic. Some
arehypochromic. Also see abnormal shapes. Anisopoikilocytosis. See
platelets so noproblem w/the platelets.
CBC: Normal:Hgb 5.9 ~15 (man), ~14 (woman) Low. Not enough
RBCs.MCV 56.2 80-100 MCV = volume of RBCs. Patients low value =
microcytic anemia.RDW 20.2 Teens (13-19) Measure of size variation
in RBCs. Patients is high.
WBC 5,900 4000-8000 Normal.Plt 383,000 200,000-400,000
Normal.
Normally, hematocrit = 3x hemoglobin, so normal hemotocrit for
man = 45; woman = 42.
1a. The RBCs show?A. MacrocytosisB. HyperchromasiaC.
SpherocytosisD. SchistocytosisE. Anisopoikilocytosis too small and
some are funny-shaped.
1c. Before the patient leaves your office you should?
A. Order TSH and T3B. Order TSH and T4C. Prescribe multivitamins
with ironD. Perform stool exam for occult blood ALWAYS check if
patient is an older person w/iron deficiency anemia.E. Perform
stool exam for hookworms and other parasites
Test q:A 70F complains of fatigue. She cites no history of
hemoptosis or vaginal bleeding. Phys exam is normal except for
pallor. CBC shows: Hgb
5.9 g/dL, MCV 56.2 m3, RDW 20.2, WBC 5900/mm
3, Plt 383,000/mm
3, anisopoikilocytosis. What lab result would you expect? Low
serum iron and
high TIBC. Before the patient leaves your office you should?
Perform stool exam for occult blood. BOTH PARTS REPEATED x2
Test q:A 73M has been healthy all his life. He takes no meds and
has had no major illnesses or surgeries. However, for the past
year, he has becomeincreasingly tired and listless and appears
pale. Phys exam shows no hepatosplenomegaly and no deformities. CBC
shows Hgb 9.7 g/dL, hematocrit29.9%, MCV 69.7mm
3(one year, MCV was 59.0), RBC count 4.28 million/mm
3, platelet count 331,000/mm
3, and WBC count 5500/mm
3. Which of the
following is the most likely underlying condition causing this
patients findings? GI bleed. REPEATED x2 (One year, answer was
occult malignancy)
1b. What lab result would you expect?A. Elevated serum ferritin
B. Elevated serum ironC. High serum iron and low TIBCD. Low serum
iron and high TIBCE. Low serum iron and low TIBC
D High Total Iron Binding Capacity becausepatient is trying to
compensate.
-
7/30/2019 Hematologic Pathology p1-23
5/23
Case 2. 34 y.o. female with 2-day history of fever, petichiae
and hematuria; alsoheadaches and mental confusion.
Symptoms are characteristic ofthrombotic thrombocytopenic pupura
(TTP). Itis a consumptive coagulopathy (like DIC) bleeding clotting
bleeding clotting. TTP has a pentad of symptoms (5 things): fever,
thrombocytopenia(obvious because of petechiae), renal disease
(hematuria), headache (CNSdisease), and microangiopathic hemolytic
anemia (MAHA).
Blood smear: Platelets missing. See schistocytes, spherocytes,
but platelets are
used up. Microangiopathic hemolytic anemia. Fits TTP.
CBC:Hgb 7.0 Low = anemic.MCV 77 Not too lowRDW 23 High
consistent w/the spherocytes, schistocytes, size variation.WBC
17,000 HighPlt 14,000 Very low
2a. Describe the RBCs:A. NormochromicB. MicrocyticC. Spherocytes
and schistocytesD. Drepanocytes and schistocytes
E. Howell-Jolly bodies
Schistocytes are classic in MAHA, but you also usuallysee some
spherocytes.
2b. Diagnosis?A. Sickle cell anemiaB. Hereditary spherocytosisC.
Thrombotic thrombocytopenic purpura (TTP)D. Anemia of chronic
disease secondary tohypothyroidismE. Folate deficiency
C. TTP unknown cause. Some kind of autoimmunedisease can detect
auto-antibodies, but the trigger isnot known. Much more common in
females. Treatmentis exchange phoresis (trade plasma). Used to
beuniversally fatal now can save 90% of patients. Tendsto
recur.
2c. What test result would you expect?A. D dimer elevatedB.
Haptoglobin elevatedC. Plasma free hemoglobin decreasedD. BUN
normal
E. Urine protein negative
A is correct. If you have a consumptive coagulopathy(clotting
lysing clotting lysing), expect to see Ddimer.B: Haptoglobin is a
protein in blood that combinesw/any free hemoglobin. If youre
destroying RBCs(and releasing hemoglobin) it will complex
w/haptoglobin haptoglobin level goes down in hemolytic anemia.C: If
you are rupturing RBCs and releasing hemoglobininto the blood,
plasma free Hgb will be increased.D: BUN is a measure of renal
function patient hasrenal disease, so BUN will be abnormal.
E: Patient will have proteinuria because of renal disease
Test q:A 30F mother of 4 exhibits a dramatic fall in hematocrit
after atrip to West Africa. Plasmodium falciparum is identified on
a Wright
stain of blood. In this patient: Haptoglobin is decreased;
plasmafree Hgb is increased.
Test q:A 54F sees her physician because of sudden onset of
headaches and photophobia. This condition has been worsening for
the past 2 days. Onphys exam, she has a temperature of 38C and is
disoriented. CBC shows Hgb 11.2 g/dL, hematocrit 33.7%, MCV 94m
3, platelet count 32,000/mm
3,
and WBC count 9900/mm3. The peripheral blood smear shows
schistocytes. The serum urea nitrogen level is 38 mg/dL, and the
creatinine level is 3.9
mg/dL. Which of the following is the most likely diagnosis?
Thrombotic thrombocytopenic purpura.
Case 3. 34 y.o. male in for routine physical has pallor and
icterus;splenomegaly on PE
Pale = not enough RBCs, not enough oxygenation. Icteric breaking
downRBCs and releasing Hb.
Blood smear:See lymphocyte, platelets. Lots of spherocytes
(small, roundRBCs), polychromasia (reticulocytes). Everything fits
hemolytic anemia.
CBC:Hgb 11MCV 84WBC 6,000Plt 350,000 Hgb low. Rest normal.
-
7/30/2019 Hematologic Pathology p1-23
6/23
3a. Describe the RBCs.A. NormochromicB. SchistocytosisC.
MicrocyticD. Spherocytosis perfectly round and dark redE.
Hypochromic
3b. What test should you order to confirm your diagnosis?A.
Serum ironB. Osmotic fragilityC. Reticulocyte countD. Malaria
screenE. D dimer
Osmotic fragility is a test for hereditary spherocytosis
(spherocytes die quickly in salt water).
In this case, the spleen is large because the spleen destroys
spherocytes. Spleen recognizes that they are not normalshape or
size and destroys them they do not live 120 days (definition of a
hemolytic anemia). Hemolytic anemia canoccur in the blood vessels
or in the spleen (intravascular vs. extravascular).
Case 4. 37 y.o. female treated lifelong for a seizure disorder
complains of fatigueand lightheadedness.
A lot of seizures medications predispose to vitamin B12
deficiency. These patientsfrequently develop macrocytic anemia.
Blood smear: See hypersegmented neutrophil. No lymphocytes
present for sizecomparison (RBCs), but these are large, oval RBCs =
macrocytosis.
CBC: fits w/macrocytic anemia.Hgb 6 LowMCV 130 HighWBC 6,000
NormalPlt 220,000 Normal
4a. All of the following correctly describe the RBCsEXCEPT:A.
NormochromicB. MacrocyticC. AnisocytosisD. OvalocytosisE.
Spherocytosis not this. Would look small.
A bone marrow aspirate was performed On bone marrow aspirates,
tend to see giant band neutrophils. The final stage inneutrophil
development is the segmented neutrophil, and the step
immediatelybefore that is the band neutrophil. If you have >5
segments in a neutrophil, toomany = macrocytic anemia. The labeled
band at the top is huge should be thesame size as a neutrophil, but
this is 2-3x bigger. Giant bands, hypersegmentedneutrophils in bone
marrow = macrocytic anemia.
4b. What findings on the bone marrow aspirate suggest folate
deficiency?A. Hyposegmented neutrophilsB. HypocellularityC. Giant
bands and hypersegmented neutrophilsD. Miniature bands and
hypersegmented neutrophils
E. Hypersegmented blasts and promyelocytesTest q:A 37F has been
treated lifelong for a seizure disorder and now complains of
fatigue and lightheadedness. CBC shows: Hgb 6g/dL, MCV 130
mm3, WBC 6000/mm
3, Plt 220,000/mm
3, ovalocytosis, hypersegmented neutrophils. You suspect:
Dilantin treatment and folate deficiency.
LEUKEMIA:AcuteALL and AMLHi/Low WBC (unpredictable)Rapidly fatal
if untreated (matter of weeks); anemic andthrombocytopenic (almost
always. Patient is very sick.)Curable
ChronicCLL and CMLAlways high WBCSlowly progressive- patient
lives many yearsDifficult to cure
M = myeloid (granulocytic); L = lymphoid (as in ALL, AML, CLL,
CML).
-
7/30/2019 Hematologic Pathology p1-23
7/23
Acute Leukemias:
5a. In Acute Leukemias you will almost always find:A. Normal
hct, normal plateletsB. Normal hct, decreased plateletsC. Decreased
hct, increased paltelets
D. Anemia, thrombocytopeniaE. WBC count >100,000
D: Patient is always really sick.
Myeloid PrecursorsSegmented PMN normal, mature cell. Band isstep
immediately before. In a normal patient,should not see any blasts.
Blast is very earlystage if you see lots of blasts, it is an
acuteleukemia.
There are two stem cells types in the bone marrow
lymphocytic/plasma cell lineage
andRBC/megakaryocyte/granulocyte/monocyte (everythingelse)
lineage.
ALL has only one type (lymphocyte). AML has manyvarieties.
Promyelocyte M3 version. Produces MAHAand DIC. Unique presentation,
unique treatment.
5b. In Chronic Leukemias you will almost alwaysfind:A. Anemia,
thrombocytopeniaB. WBC count >50,000
C. Normal hct, increased plateletsD. Anemia, thrombocytosisE.
Circulating blasts
Above: lymphoblasts. Not normal lymphocytes very large(compare
to RBCs). Have nucleoli all over the place, veryactive-looking
chromatin (so they are blasts). Cannot tell bylooking at this
whether they are lymphoblasts or myeloblasts can definitely tell
they are blasts, however. = Acute leukemia.
Must do more testing to figure out which one.
Above: ALL (PAS stain). ALLs (lymphoid in origin)are PAS + and
myeloperoxidase (MPO) negative.Makes sense since MPO is made by
granules ingranulocytes so AMLs are MPO positive.
-
7/30/2019 Hematologic Pathology p1-23
8/23
Test q:A 37F visits her physician because of a cough and fever
of one weeks duration. On phys exam, her temp is 38.3 degrees C.
She has diffuse
crackles in all lung fields. A chest radiograph shows bilateral
extensive infiltrates. CBC shows Hgb 13.9 g/dL, hematocrit 42%, MCV
89m
3
, plateletcount 210,000/mm
3, and WBC count 56,000/mm
3with 63% segmented neutrophils, 15% bands, 6% metamyelocytes,
3% myelocytes, 1% blasts, 8%
lymphocytes, 2% monocytes, and 2% eosinophils. The peripheral
blood leukocyte alkaline phosphatase score is increased. Which of
the following isthe most likely diagnosis? Leukemoid reaction.
REPEATED x4 (Once, was a 50M instead)
Above: Immature granulocytes (leftshift). See lots of
granulocytes ininfections body produces lots ofgranulocytes in
response to bacteria(WBC count very high). Will see
normalgranulocytes, bands, metamyelocytes, butNO BLASTS. If you
have an infection anda high neutrophil count, will get a leftshift
lots of immature forms. Benign
condition.
Above: CML. Also see spectrum of cellstages, but especially see
lots ofBLASTSw/nucleoli. When a patient comes in with ahigh
granulocyte count, your differential mustinclude both benign
condition and leukemia.If WBC count >50,000, leukemia. If
lessthan 50, usually leukemoid reaction (left
shift).
Above: LAP stains. LAP = leukocyte
alkaline phosphatase. Made bynormal granulocytes, not CML cells.
Ifleft shift (leukemoid reaction - benignconditions), LAP score
will be >20. IfCML, they dont make it at all, so LAPscore will
approach 0. Left: alkalinephosphatase stain. Benign cells.Right:
LAP, CML.
Above: The real diagnosis of CML is made by
cytogeneticsdemonstrating that a small piece of chromosome 22
breaks off
and attaches to chromosome 9 9 22 translocation .
Above: FISH can also diagnose if ABL (green) and BCR(red) genes
are on the same chromosome, will see a yellow
color ri ht .
Above: CML and AML. See many blasts. CML amixture of forms
(segmented neutrophils, bands,metamyelocytes, blasts but relatively
few blasts).AML pretty much all blasts (no spectrum of forms).
Above: Left = AML. Can tell they are blasts because they are
4xthe size of RBCs and have prominent nucleoli. Know it is
acuteleukemia, but cannot tell which one. Right: Presence of Auer
rod(little red rod) = AML. Auer rods are crystallized
myeloperoxidase same thing as on MPO stain. Auer rods are only seen
in AML,but you dont have to have Auer rods to call it AML. Most
AMLs
do not have Auer rods.
-
7/30/2019 Hematologic Pathology p1-23
9/23
M0-M7 stages classified based on how the cells look:
M1 Myeloblastic. M2 Myeloid. M3 look like promyelocytes
(intermediate stage in granulocyte production), sonamed
promyelocytic leukemia. Lots of granules = DIC in patient (cause
MAHA). Auer rods common. Very fastprogression dead in a few days
w/o treatment. For promyelocytic leukemia treatment, use retinoic
acid.M4 myelomonocytic (in between granulocyte and monocyte).
Test q: The myeloperoxidase stain is generally positive in: M2,
M3, M4.
Above: M6 erythroid (RBC). M7 megakaryocytic version.
Below: Myeloperoxidase (MPO)AMLs are MPO positive.
Above: Sudan Black stain. AMLsare MPO positive. If moving
towardmonocyte side of spectrum, may
also be Sudan black + (fat stain).May be fat stain + or
non-specific
esterase +.
Above: Nonspecific esterase. IfSudan black + and
nonspecificesterase +, is either amyelomonocytic cell or
monocyticcell.
Above: M5 monocytic leukemia. Patientspresent with ULCERS
(deposits of these cellsin the mouth). If they have mouth ulcers
orhyperplasia of the gums = monocyticleukemia. If patient presents
w/MAHA, it ispromyelocytic leukemia.
-
7/30/2019 Hematologic Pathology p1-23
10/23
Case 6. 25 y.o. male with fever, recurrent URIs and
submandibular swelling;also poor wound healing.
Submandibular swelling indicates problems in the mouth.
Blood smear: See fold in cell (top right) monocyte-like. Also
see Auer rod incell (bottom right). This is an AML but going toward
the monocytic series.
CBC:Hgb 9.9MCV 106.3WBC 7.9 (70% abnormal cells)Plt 50,000
Platelets are low, patient is anemic. WBC w/acute leukemias
remember:may or may not be high.
6a. Diagnosis?A. ALLB. AML because Auer rod is present.C. CLLD.
CMLE. Hereditary spherocytosis
Test q:A 25M has rever, recurrent URIs and submandibular
swelling; also poor wound healing. The peripheral blood smear shows
WBCs w/high
nucleus-to-cytoplasm ratio, prominent nucleoli, and red,
needle-like cytoplasmic inclusions. Diagnosis? AML (M2). Which test
on a bone marrowaspirate confirms your diagnosis? MPO(+).
Case 7. 6 y.o. male with URIs, headache, bone pain and easy
bruising;axillary and cervical lymphadenopathy;
hepatosplenomegaly.
Easy bruising = low platelets (thrombocytopenia). If a young
child, usuallyhave ALL.
Blood smear: Top middle cell prominent fold. Right top boxing
glove.
Cells look monocytic. Cleaves in cells butt cells or plumber
cells. Bigcells, nucleoli = acute leukemia. Cannot tell which one
just by looking.Because the patient is 6 years old = ALL.
CBC:Hgb 9MCV 82WBC 92,000 (86% abnormal)Plt 18,000
7a. Diagnosis?A. ALLB. AML
C. CLLD. CML
7b. What test results are expected on a bone marrow aspirate?A.
MPO (+), PAS (+)B. MPO (-), PAS (-)
C. TdT (+), Precursor B cell (+)D. TdT (-), Precursor T cell
(+)E. Sudan black B (+); CD 10 (-)
C. TdT positive and precursor B cell positive. Would be Sudan
black + if we sawmonocytic cells. CD10 (cluster designation) ALL
marker (aka CALLA: common ALLantigen). If patient has ALL, will be
CD10+.
6b. Which test on a bone marrow aspirateconfirms your
diagnosis?A. MPO (-)B. MPO (+)C. PAS (+)D. Iron stain (Prussian
blue)
E. Leukocyte alkaline phosphatase = 20
-
7/30/2019 Hematologic Pathology p1-23
11/23
Case 8. 15 y.o. male with flulike symptoms; axillary and
cervicallymphadenopathy; gums are thick and bleeding.
Blood smear: Boxing-glove nuclei. Look like monocytes.
CBC:Hgb 9MCV 90WBC 42,000 (88% abnormal)Plt 22,000 Patient is
anemic; platelets are low.
BM Bx results:MPO (-)PAS (-)Sudan Black B (-)Non-specific
esterase (++) marker formonocytic series.Serum lysozyme 162 (4-15
normal)
8. Diagnosis:A. AML (FAB M1)B. AML (FAB M3)C. AML (FAB M5)D. ALL
(L1)E. Infectious mononucleosis
Test q:A 15M present w/flu-like symptoms; axillary and cervical
lymphadenopathy; gums are thick and bleeding. CBC shows: Hgb 9g/dL,
MCV 90mm3,
WBC 42,000/mm3
(88% abnormal), Plt 22,000/mm3, blasts present. Bone marrow
biopsy results: MPO (-), PAS (-), Sudan Black B (-),
non-specific
esterase (++), serum lysozyme 162 (4-15 normal). Diagnosis? AML
(FAB M5).
Test q:A 25F visits her physician because she has had bleeding
gums for the past 3 weeks. Phys exam shows that her gingivae are
thickened and
friable. She has hepatosplenomegaly and generalized nontender
lymphadenopathy. CBC shows Hgb 11.2 g/dL, hematocrit 33.9%,
platelet count95,000/mm
3, and WBC count 4500/mm
3with 25% segmented neutrophils, 10% bands, 2% metamyelocytes,
55% lymphocytes, 8% monocytes, and 1
nucleated RBC per 100 WBCs. A bone marrow biopsy specimen shows
100% cellularity, with many large blasts that are myeloperoxidase
negative andnonspecific esterase positive. Which of the following
is the most likely diagnosis? Acute monocytic leukemia.
Test q:A 22y/o university student reports easy fatigability of
two months duration. On phys exam, she has no hepatosplenomegaly
orlymphadenopathy. Mucosal gingival hemorrhages are noted. CBC
shows Hgb 9.5 g/dL, hematocrit 28.2%, MCV 94m
3, platelet count 20,000/mm
3, and
WBC count 107,000/mm3. A bone marrow biopsy specimen shows that
the marrow is 100% cellular with few residual normal hematopoietic
cells. Most
of the cells in the marrow are large, w/nuclei having delicate
chromatin and several nucleoli. The cytoplasm of these cells has
azurophilic, peroxidase-positive granules. Which of the following
is the most likely diagnosis? Acute myelogenous leukemia.
THIS IS WHERE WE STOPPED IN CLASS. BELOW ARE THE REMAINING 26
SLIDES:
Case 9. 39-year-old female has routine workup for fibrocystic
disease of
breast; splenomegaly present.
CBC:Hgb 13MCV 96WBC 133,000 (seg 56, band 15, meta 13, myelo 4,
pro 3); baso 1, eo 1Plt 130,000
9. What test should you order on a bone marrow aspirate?A. MPOB.
PASC. Sudan black BD. LAPE. TRAP
10. 30-y/o. female; anemia, thrombocytopenia; CBC- blasts w.
Auer rods, schistocytes. Diagnosis?A. ALLB. AML, M1C. AML, M3D.
AML, M5E. TTP
-
7/30/2019 Hematologic Pathology p1-23
12/23
Quick Look at Lymphomas:
Hodgkin Non-HodgkinRS Cells Low grade- slowly progressive, rare
cureOften curable High grade- rapidly progressive; a minority are
curableStage important Grade importantFew malignant cells Mostly
lymphoma< cell-mediated < humoralLocalized radiation Systemic
chemotherapy
Follicular hyperplasia, lymph node. See benign Follicular
hyperplasia, lymph node. Seefollicles, mantle zone. benign
follicles, mantle zone.
Follicular hyperplasia, lymph node. See mantle Follicular
hyperplasia, lymph node. Seezone, dark zone, and light zone. See
tingible tingible body macrophages, mantle zone, andbody
macrophages. lymphoycytes.
Follicular lymphoma, lymph node. See follicles. Follicular
lymphoma, lymph node. See follicles.
-
7/30/2019 Hematologic Pathology p1-23
13/23
Follicular lymphoma, lymph node. See lymphocytes. SLL/SLL, lymph
node. See lymphocyte infiltration.
SLL/CLL, spleen see lymphocyte infiltration. SLL/CLL, lymph
node. See lymphocytes.
Burkitt Lymphoma, soft tissue. See tingible body Burkitt
Lymphoma, soft tissue. See stars and sky.macrophages (stars) and
lymphocytes (sky).
-
7/30/2019 Hematologic Pathology p1-23
14/23
Nodular Sclerosis Hodgkin Lymphoma, Lymph Node. Hodgkin
Lymphoma. See eosinophils, lymphocytes,See fibrous bands, nodules.
and Hodgkin cells.
Hodgkin Lymphoma, Spleen. See lymphocytes and Radiograph of
Plasma Cell (Multiple) Myeloma.Reed-Sternberg cells. See punched
out lesions.
Plasma cells and bone spicule. Plasma Cell (Multiple) Myeloma,
Bone. See plasma cells.
-
7/30/2019 Hematologic Pathology p1-23
15/23
Laboratory Session: Abnormalities of Hemostasis Wed. 10/13 or
Thurs. 10/14/10
This is the lab talk from the first lab week of this block I
usually dont post lab material, but there are some lecture testqs
from this material.
Primary Hemostasis:
1. Platelets bind via glycoprotein Ib (GpIb) receptors to von
Willebrandfactor (vWF) on exposed ECM and are activated, undergoing
shapechange and granular release
2. Released ADP and TxA2 induce additional platelet
aggregationthrough platelet GpIIb-IIIa receptor binding to
fibrinogen primaryhemostatic plug
vWF helps platelets stick to the endothelium. Plavix blocks the
ADPreceptor blocks adherence/aggregation of platelets.
Test q: The 3 factors involved in primary hemostasis are:
Platelets, endothelium, and von Willebrand factor.
Secondary Hemostasis:Local activation of the coagulation cascade
(involving tissue factor andplatelet phospholipids) results in
fibrin polymerization, cementing theplatelets into a definitive
secondary hemostatic plug.
Tissue factor (Factor III, thromboplastin):
membrane-boundprocoagulant glycoprotein synthesized by endothelial
cells. Acts inconjunction with factor VII as major in vivo
initiator of coagulationcascade eventually thrombin generation
Coagulation Cascade: Thrombin is the most important coagulation
factor At end of proteolytic cascade, thrombin converts fibrinogen
into fibrin monomers that polymerize into an insoluble gel.
Fibrin gel encases platelets Fibrin polymers are cross-linked
and stabilized by factor XIIIa
Binding of coagulation factors II, XII, IX, X to calcium depends
on addition of y-carboxyl groups to certain glutamic acidresidues.
Reaction uses Vitamin K as a cofactor and is antagonized by drugs
such as coumadin.
There is a balance of factors thatfavor thrombosis and factors
thatinhibit thrombosis. If the balance isthrown off, may have
bleeding ormay have thrombosis. (See page 1of this study guide for
complete infoon the pro- and anti-thromboticfactors)
-
7/30/2019 Hematologic Pathology p1-23
16/23
Test of Primary Hemostasis:
PFA-100: Objective test (vs Bleeding Time) Collagen + Epi
prolonged = ASPIRIN Collagen + Epi and collagen + ADP prolonged =
VON WILLEBRAND
Test q:A 51y/o obese woman w/a history diabetes mellitus had a
myocardial infarction three months ago. She is now taking a low
dose of aspirin toreduce the risk of arterial thrombosis. On which
of the following steps in hemostasis does aspirin have its greatest
effect? Aggregation of platelets.(Other choices: Adhesion of
platelets to collagen, Production of tissue factor, Synthesis of
vWF, and Synthesis of antithrombin III) REPEATED x3
review question from Exam 1 but showed up on 3 different years
of Exam 2, too.
Intrinsic pathway: Expose Hageman factor (XII) tothrombogenic
surfaces.
Extrinsic pathway: Most physiologically relevantpathway for
coagulation to occur following vasculardamage. Activated by tissue
factor, expressed at sites ofinjury. Ischemia and tissue damage
activate Factor VII complexes w/tissue factor.
Vitamin K Dependent Factors: II VII IX X Protein C Protein S
Fetal Bone Protein
Vitamin K deficiencies- liver disease, malabsorptivedisorder
Test q: Vitamin K dependent factors and proteins include all of
thefollowing EXCEPT: VIII. (Other choices: II, VII, X, Protein
S)
aspirin affectsplatelet aggregation
-
7/30/2019 Hematologic Pathology p1-23
17/23
Tests for Secondary Hemostasis:
Fibrinolysis: Plasmin: breaks down fibrin and interferes with
its
polymerization FDPs: act as weak anti-coagulant factors Elevated
FDPs (fibrin derived D-dimers) can be
used in diagnosing abnormal thrombotic states,including DIC,
DVT, pulmonary embolism
Protein C + S: anti-coagulation factors; protein Sis a cofactor
for protein C; function to degradeactivated factors VIII and V
INR (wantbetween 2-3)
used to measureCoumadin therapy
used to measureHeparin therapy
Abnormal PTT in hemophilia and VonWillebrands Disease.
Lupusanticoagulant
Leiden mutation resistantto contol by Prot C &
S(anticoagulant factors)
Lupus- anti-coagulant factor (in vitro) b/cincrease PTT
Leiden mutation: single-nucleotidemutation in factor V Recurrent
DVT Factor V resistant to cleavage by
Protein C (anti-coagulant factor)
-
7/30/2019 Hematologic Pathology p1-23
18/23
Case 1:
Bleeding into joint after minor fall + no family history of
bleeding disorderHemophilia A: most commonly familial but also
sporadic
The high APTT is consistent w/hemophilia. Normal PT.
Factor VIII mutation is most common.Factor IX is #2.
Below: Hemophilia shows bleeding into joints and/or large
muscles.
Test q:A 6yo child has a long history of a hereditary bleeding
disorder characterized by spontaneous nontraumatic hemorrhages into
joint spaces,skeletal muscle, and mucous membranes. Lab studies
reveal a normal PT, elevated PTT, very low factor VIII, normal
factor X, normal factor CI, andnormal platelet aggregation studies
w/ristocetin. Which of the following is the most likely diagnosis?
Hemophilia A.
Test q:A common complication in patients w/hemophilia is:
Hemarthrosis.
Test q:A 5y/o boy has had a history of easy bruising and blood
in his urine since infancy. Phys exam shows no organomegaly. He has
severalecchymoses of the skin on the lower extremities. Lab studies
show Hgb 13.1 g/dL, hematocrit 39.3%, platelet count 287,600/mm
3, WBC count
6830/mm3, PT 13s, PTT 54s, and less than 1% factor VIII activity
measured in plasma. If he does not receive transfusions of
recombinant factor VIII
concentrate, which of the following manifestations of this
illness is most likely to ensue? Hemarthroses. REPEATED x2
Above: symptoms of hemophilia. Includes easy
bruising, hepatitis, muscular hematomas, CNShemorrhages,
Pneumocystis pneumonia (in AIDS),operative/post-operative bleeding,
and spontaneoushemarthoses (bleeding into joints).
-
7/30/2019 Hematologic Pathology p1-23
19/23
Hemophilia: Usually X-linked but not always clear.
Case 2:
However, note that there is no bleeding into joints, muscles,
CNS.
Lab screen tests: Bleeding time is elevated. Could indicate the
absence ofvon Willebrand factor(defect in primary hemostasis).
Test q: Which of the following is responsible for a defect in
primary hemostasis? vonWillebrand factor. (Other choices: Factor V;
Factors II, VII, IX, and X; Factors VIII and IX;
Fibrinogen)
Symptoms include mucous membrane bleeding, ecchymoses, and
bleeding from venupuncture sites.
There is no bleeding into joints or muscles.
Platelet activation by ADP triggers conformational change in
theplatelet GpIIb-IIIa receptors that induces binding to
fibrinogen, a largeprotein that forms bridging interactions between
platelets promote
platelet aggregation.
Platelet adhesion to ECM is mediated largely via interactions
withvWF, which acts as a bridge between platelet surface
receptors(glycoprotein Ib) and exposed collagen.
vWF associations are needed to overcome the high shear forces
offlowing blood.
Bernard-Soulier syndrome: deficiency in vWF receptor (or
GpIb)von Willebrand disease: deficiency in vWFGlanzmann
thrombasthenia: absence of GPIIb-IIIa complex
-
7/30/2019 Hematologic Pathology p1-23
20/23
Case 3:
Chronic diarrhea = Malabsorbing fat = malabsorbing Vitamin
KSteatorrhea = fat in stool (abnormal). Sudan black (fat) stain
will be positive.
Intrinsic (II, IX, X) and extrinsic (VII) coagulation
cascadeabnormalities are present. Vitamin K deficiency.
Vitamin K Dependent Factors: II VII
IX X Protein C Protein S Fetal Bone Protein
Test q:A Caucasian infant presents w/failure to thrive and
chronic diarrhea. There isoozing from a recent skin laceration.
Stoll sample testing reveals steatorrhea.
Diagnosis: Vitamin K deficiency.
Test q:An infant born at term has Apgar scores of 8 and 9 at one
and five minutes. The infant appears healthy, but three days after
birth, there is
bleeding from the umbilical cord stump, and ecchymoses are
observed over the buttocks. Seizures soon develop. A deficiency of
which of the followingnutrients best accounts for these findings?
Vitamin K (Other choices: Iron, Vit E, Folic acid, Iodine)
Case 4:
Preeclampsia: hi BP, proteinuria, edema, predisposed to develop
DIC (constantly clotting and lysing using upcoagulation factors,
fibrinogen). Cause: tissue anoxia or ischemia trigger event.
Preeclampsia can also lead toeclampsia (life-threatening for mother
and baby).
Normal: PT, PTT because not full blown eclampsia
D-dimers increased: b/c constant clot and lyse; allfactors used
up eventually in DIC
-
7/30/2019 Hematologic Pathology p1-23
21/23
Schistocytes = chopped up RBCs.
Above (middle): Schistocyte in microangiopathic hemolytic
anemia, most commonly seen with DIC. Microvascularlesion that
results in luminal narrowing, often due to deposition of fibrin and
platelets. Vascular changes produce shearstresses that mechanically
injure passing red cells. Regardless of cause, traumatic damage
leads to appearance of redcell fragments (schistocytes) in blood
smear.Above (right): Child w/DIC secondary to H. influenzae sepsis.
DIC also in infectious disease. DIC may becomplication of anything
that goes wrong with placenta.
Test q:A 60M has developed widespread ecchymoses over the skin
in the past month. His medical history includes a diagnosis or
mucinous
adenocarcinoma of the rectum. On phys exam, he appears cachectic
and pale. An abdominal CT scan shows multiple hepatic masses. Lab
studiesshow PT 30s, PTT 55s, platelet count of 15,200/mm
3, fibrinogen level of 75 mg/dL, and fibrin split product levels
(D dimer) that are very elevated.
Which of the following morphologic findings is most likely to be
present on exam of his peripheral blood smear? Schistocytes.
REPEATED x2
Robbins: The patient has DICschistocytes.
Test q:A 23F in her 25th
week of pregnancy has felt no fetal movement for the past 3
days. Three weeks later, she still has not given birth and
suddenl
develops dyspnea w/cyanosis. On phys exam, her temp is 36.9C,
pulse 102/min, respirations 21/min, and blood pressure is 80/40 mm
Hg. She haslarge ecchymoses over the skin of her entire body. A
stool sample is positive for occult blood. Lab studies show an
elevated PT and PTT. The plateletcount is decreased, plasma
fibrinogen is markedly decreased, and fibrin split products are
detected. A blood culture is negative. Which of the following
is the most likely cause of the bleeding diathesis? Increased
consumption of clotting factors and platelets. (Other choices:
Increased vascularfragility, Toxic injury to the endothelium,
Reduced production of platelets, Defects in platelet adhesion and
aggregation.) Robbins: The presence ofthrombocytopenia, increased
PT/PTT/fibrin split products, and low fibrinogen concentration all
suggest DIC, which was most likely caused by a retained
dead fetus, an obstetric complication that can lead to DIC
through release of thromboplastins from the fetus. This release
causes widespreadmicrovascular thrombosis and consumes clotting
factors and platelets.
Case 5:
PFA-100 is somewhat decreased. Likely to thrombose.
-
7/30/2019 Hematologic Pathology p1-23
22/23
Counter-regulatory mechanisms, mediated by tissue
plasminogenactivator (t-PA, fibrinolytic product) and
thrombomodulin, confinehemostatic process to site of injury.
Thrombomodulin: bind to thrombin and converts it
fromprocoagulant into anticoagulant via its ability to activate
Protein C,which inhibits clotting by inactivating factors Va and
VIIIa.
DVT can break off to cause pulmonary embolus.
Above: Can do perfusion scan to check for pulmonary embolus.
-
7/30/2019 Hematologic Pathology p1-23
23/23
Lab preview for next week:
Goldilocks spot; not too thick and not too thin:
Iron deficiency anemia.
Sickle cell anemia is an example of a hemolytic anemia.