Embed Size (px)
Citation preview

GSK-3 Inhibition: A Novel Approach to Sensitization of Chemo-resistant Pancreatic Cancer Cells
By Shadi Mamaghani
A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy
Graduate Department of Laboratory Medicine and Pathobiology University of Toronto
© Copyright by Shadi Mamaghani 2011

ii
GSK-3 Inhibition: A Novel Approach to Sensitization of Chemo-resistant Pancreatic Cancer Cells
Shadi Mamaghani
Doctor of Philosophy
Department of Laboratory Medicine and Pathobiology University of Toronto
2011
Abstract
The aggressive nature of pancreatic cancer, characterized by invasiveness, resistance to
treatment, rapid progression, and its high prevalence in the population urges the need for
developing more effective treatments. Many studies have attributed resistance to
therapeutics of pancreatic cancer to activity of the transcription factor nuclear factor kappa B
(NF-κB). NF-κB is regulated by the serine/threonine kinase glycogen synthase kinase-3
(GSK-3). GSK-3 is a key mediator of pathways such as insulin, wnt, and PI3K/Akt and has
roles in proliferation, glucose metabolism, apoptosis, motility and neuroprotection.
Depending on the cellular context, GSK-3 activity can promote or inhibit cell survival.
GSK-3 inhibition was recently reported to have anti-cancer effects against pancreatic cancer
cells. This effect was in part attributed to suppression of NF-κB. In this thesis, I showed that
while blocking GSK-3 disrupts NF-κB, and has anti-survival effects on pancreatic cancer
cells, it does not sensitize to the chemotherapeutic drug gemcitabine. NF-κB inhibition by
curcumin also resulted in similar effects. These results questions previous reports that NF-κB
activation plays a major role in chemo-resistance of pancreatic cancer. The inhibition of NF-
κB by genetic disruption of GSK-3 was previously reported to sensitize mouse embryonic

iii
fibroblasts and hepatocytes to TNF-α cytotoxicity. I therefore tested whether GSK-3
inhibition could sensitize pancreatic cancer cells to apoptosis induced by the clinically
applicable member of the TNF-α family, TNF-α related apoptosis inducing ligand (TRAIL).
In contrast to the results obtained with gemcitabine, the combination of genetic or
pharmacological inhibition of GSK-3 and TRAIL was found to be highly synergistic in
apoptosis induction. Analysis of the apoptotic mechanisms, point towards effects of GSK-3
inhibition on caspase-8 activation, consistent with inhibition of the death receptor signalling
pathway. It was found that not only caspase-8 but also mitochondrial anti-apoptotic proteins
such as Bcl-XL and Mcl-1 were mediating the TRAIL sensitization. Furthermore, for the
first time the in vivo effects of GSK-3 inhibition in combination with TRAIL treatment was
investigated. The results indicate a significant enhancement of apoptosis in pancreatic cancer
xenografts with minimal toxic effects. Together, these studies provide a rationale for
developing combination treatments based on GSK3 inhibition and TRAIL death receptor
activation to treat pancreatic cancer.

iv
Acknowledgments
I am grateful, first and foremost, to David Hedley for providing an environment in which I
could have fun, interact scientifically and emerge as an independent investigator. Your
patience, tolerance and mentorship were impeccable! The conference trips we took to Banff
and Prague, smoked fish lunches in the lab, St. David’s day daffodils and leaks, Christmas
marzipan pigs and my birthday Shamrocks are the best memories of my life that I will carry
in my heart and soul forever. During roller coasters of my personal life, there was no place
better than your office to go and have someone to listen and share and get advice. You
presence in my life has made a huge impact on me and whoever that will benefit from my
education.
My committee members-Vuk Stambolic, Susan Done, Aaron Schimmer, and Jeremy Squire-
whom with their advice, enthusiasm, and scientific contribution, my project developed and
flourished. Our collaborator, Jim Woodgett’s flawless generous advice was the driving force
of the project.
Over the past six years, I have had the pleasure of working with many wonderful people
whom have made my experience in the lab pleasant. The past and present members of
Hedley lab, particularly Mary Cao, May Kwan, Sue Chow, and Trudey Nicklee whom I have
learned a great deal and were an enormous stand for me and for my project development.
I was fortunate to be in a department where I received emotional, educational, and financial
support. The faculty, staff, and the students of the department of Laboratory Medicine and
Pathobiology bear particular mention, specifically Harry Elsholtz, from whom I have learned
a lot and have always been a great source of inspiration. Financial support from University of
Toronto, the private donors, and PMH foundation had huge impact in my success and
provided a safe space during the roller coasters of my scientific adventure.
Over the years working in Ontario cancer Institute, I had the privilege of having a strong
network of people around me, without whom I could not complete my project. Of particular,
Satish Patel, Shekeb Khan, Aws Abdulwahid, who gave me their endless emotional and
scientific support and their presence, was an asset during all these years. When I felt I was in

v
a dark tunnel with no lights to be seen, they were the ones taking my hand and walking me
along the path. Also, special thanks go to two wonderful people who scientifically
contributed to my project-Bizhan Bandarchi, and Melania Pintilie.
For the past two years, the level of support I received from the coaches, participants, and the
trainings of The Landmark Education-specifically Introduction Leaders Program buddies-
were indispensable to my successful completion of the PhD project. Special
acknowledgements go to Lori Douglas, Lara Coombs, and Evan Kosiner who believed in me
and were a stand for finding a cure for cancer.
Big part of my accomplishments in life could not be achieved without the endless supports
from my family and friends. My mother has been a continuous source of inspiration for me
and without her encouragements; I would have not even dared to take the path I have chosen
in life. My brothers-Shahram and Shahriar- and their beautiful wives- Mona and Maryam-
have been a constant source of support, and I would have not been able to succeed without
them. My most profound thanks go to my friends Behrang, Mandana, Mitra, Ladan,
Mohammad, Reza, and Tanaz for their love, mentorship and support.
Lastly-and most importantly- I dedicate this thesis to three loved ones who are not amongst
us anymore -my father, and my cousin-Elena. Their memories are cherished forever and I am
sure they are all smiling at us from up in the heaven.

vi
Table of Contents
Abstract ii
Acknowledgments iv
Table of Contents vi
List of Figures x
List of Abbreviations xii
Chapter 1 Introduction 1
1.1 Clinical Implications of Pancreatic Cancer 2
1.1.1 Risk Factors: 2
1.2 Molecular Etiology of Pancreatic Ductal Adenocarcinoma 5
1.2.1 Histological Precursor Lesions 5
1.2.2 Chromosome Abnormalities 7
1.2.3 Genetic Modifications 7
1.2.4 Pancreatic Inflammation and its Link to Pancreatic Ductal Adenocarcinoma 8
1.3 Pathways of Apoptosis in Pancreatic Ductal Adenocarcinoma 8
1.3.1 The Intrinsic Apoptosis Pathway 10
1.3.2 The Extrinsic Apoptosis Pathway 13
1.4 Chemoresistance of Pancreatic Cancer 16
1.4.1 Role of Nuclear Factor Kappa B in Survival of Pancreatic Cancer and Chemo‐resistance 17
1.5 Role of Glycogen Synthase Kinase3 in Tumorigenesis of Pancreatic Adenocarcinoma 22
1.5.1 Regulation of GSK‐3 and Its Substrates 24
1.5.2 Homologs of GSK‐3 in Lower Eukaryotes 26

vii
1.5.3 Mammalian GSK‐3 Homologs 27
1.5.4 Tumor Suppressor Role of GSK‐3 in Substrate Regulation 29
1.5.5 Tumor Promoting Role of GSK‐3 in Substrate Regulation 33
1.6 Inhibition of GSK3: A Double Edged Sword in Chemotherapy 35
1.7 Clinical Management of Pancreatic Cancer 38
1.7.1 Current State of Clinical Trials 38
1.7.2 Ongoing Clinical Trials and the Future of Molecular Targeted Therapeutics 39
1.8 Thesis Overview 40
Chapter 2 Glycogen Synthase Kinase3 Inhibition Disrupts Nuclear FactorkappaB Activity in Pancreatic Cancer, but Fails to Sensitize to Gemcitabine Chemotherapy 42
2.1 Background 44
2.2 Materials and Methods 45
2.2.1 Reagents and Antibodies 45
2.2.2 Cell Lines and Media 46
2.2.3 Cell Treatments, Lysate Preparation, and Immunoblotting 46
2.2.4 Proliferation Assay 47
2.2.5 Transient Transfection and Luciferase Assay 47
2.2.6 Genetic Knockdown of GSK‐3 48
2.2.7 Clonogenic Assay 49
2.2.8 Statistical Analysis 49
2.3 Results 49
2.3.1 Proliferation and Colony‐Forming Capacity of Pancreatic Cancer Cells is Decreased after Pharmacological Inhibition of GSK‐3 49
2.3.2 GSK‐3 Mediates NF‐κB Activation in Pancreatic Cancer Cells 51
2.3.3 Genetic Knockdown of GSK‐3 Abolishes NF‐κB Activity in Pancreatic Cancer Cells 53
2.3.4 GSK‐3 Inhibition does not Enhance the Anti‐Tumor Effects of Gemcitabine in Pancreatic Cancer In vitro 55
2.4 Discussion 60

viii
2.5 Conclusions 63
Chapter 3 GSK3 Inhibition Sensitizes Pancreatic Cancer Cells to TRAILinduced Apoptosis 64
3.1 Abstract 65
3.2 Background 66
3.3 Materials and Methods 68
3.3.1 Cell Lines and Reagents 68
3.3.2 Stable Transfections 69
3.3.3 Survival Assay 69
3.3.4 Flow Cytometry Analysis of Apoptosis 70
3.3.5 Immunoblotting 71
3.3.6 Genetic Knockdown of GSK‐3 71
3.3.7 Statistical Analysis 71
3.4 Results 71
3.4.1 Effects of GSK‐3 Inhibition and TRAIL in Human Prostate Cancer Cell Lines In vitro 71
3.4.2 GSK‐3 Inhibition Sensitizes TRAIL‐ Resistant Pancreatic Cancer Cells to Apoptosis 72
3.4.3 GSK‐3 Inhibition Enhances TRAIL‐Induced Cell Death in a Time‐Dependent Manner 74
3.4.4 Apoptotic Nature of TRAIL Sensitization after GSK‐3 Inhibition by AR‐18 74
3.4.5 Caspase‐dependency of TRAIL Sensitivity by GSK‐3 Inhibition 78
3.4.6 Effects of Genetic Knockdown of GSK‐3 on TRAIL Sensitization of Pancreatic Cancer Cells 78
3.4.7 TRAIL Sensitization upon GSK‐3 Inhibition is Mediated through Mitochondria 81
3.4.8 Molecular Mechanism of TRAIL Sensitization by GSK‐3 Inhibition 83
3.5 Discussion 85
3.6 Conclusion 90
Chapter 4 GSK3 Inhibition in Combination with TRAIL Promotes Apoptosis in PANC1 Xenografts in Mice 91

ix
4.1 Abstract 92
4.2 Background 92
4.3 Materials and Methods 93
4.3.1 Cells and Reagents 93
4.3.2 Pancreatic Cancer Xenograft Model 93
4.3.3 Treatment Procedure and Drug Schedule 94
4.3.4 Immunoblotting 94
4.3.5 Immunohistochemistry 96
4.3.6 Image Capture 96
4.3.7 Quantification of Cleaved Caspase‐3 96
4.3.8 Statistical Analysis 96
4.4 Results 97
4.4.1 Preliminary Studies Established A Non‐toxic Dose Schedule for Acute Treatment of AR‐18 97
4.4.2 In vivo Inhibition of GSK‐3 Results in Increased β‐catenin Levels 97
4.4.3 Combination Therapy Increases Cleaved Caspase‐3 In vivo 100
4.4.4 Tumor Sensitization to TRAIL by AR‐18 did not Significantly Increase Host Toxicity 100
4.4.5 Discussion 102
4.5 Conclusion 103
Chapter 5 Discussion and Future Directions 104
5.1 Summary and Key Findings 105
5.2 Future Work 105
5.3 Concluding Remarks 108
Appendix 109
References 111

x
List of Figures
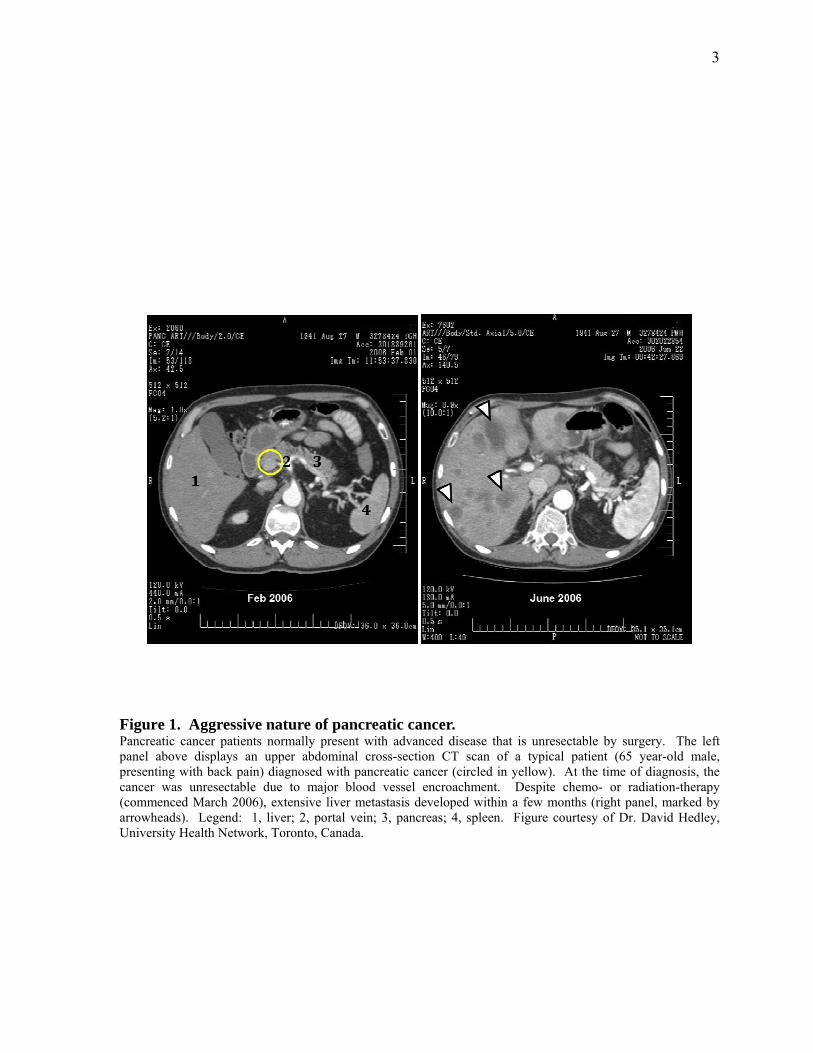
Figure 1. Aggressive nature of pancreatic cancer. 3
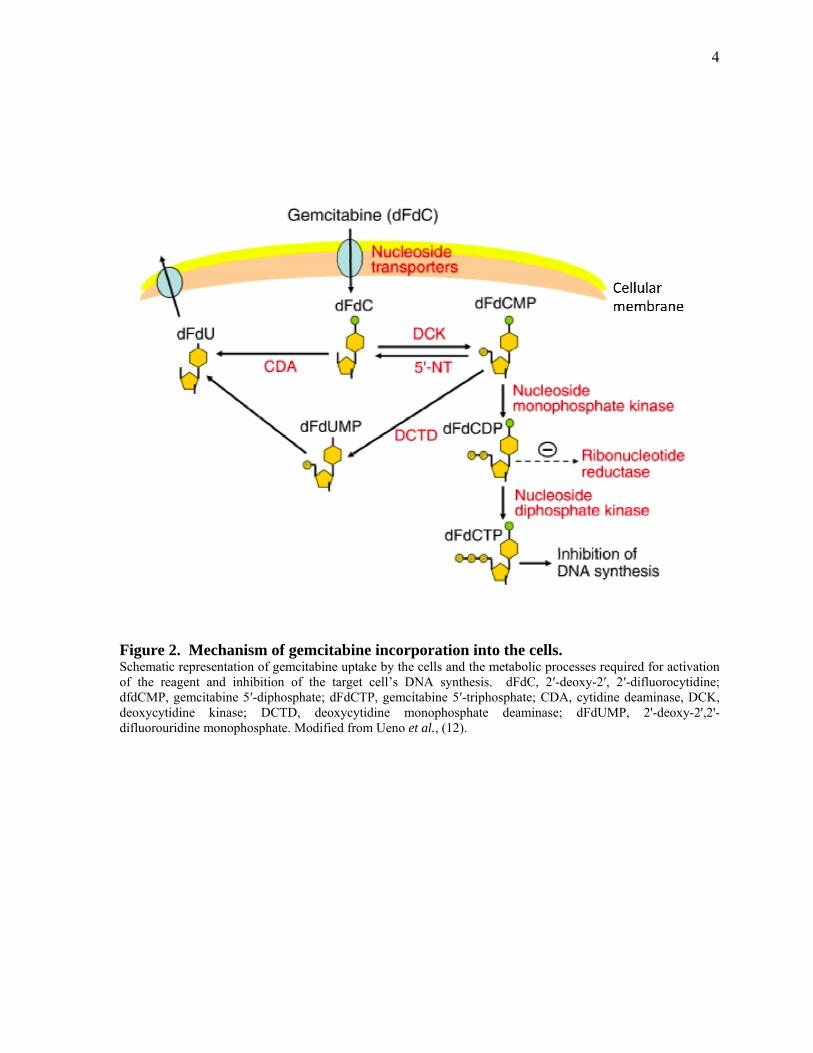
Figure 2. Mechanism of gemcitabine incorporation into the cells. 4
Figure 3. Schematic representation of development of pancreatic cancer. 6
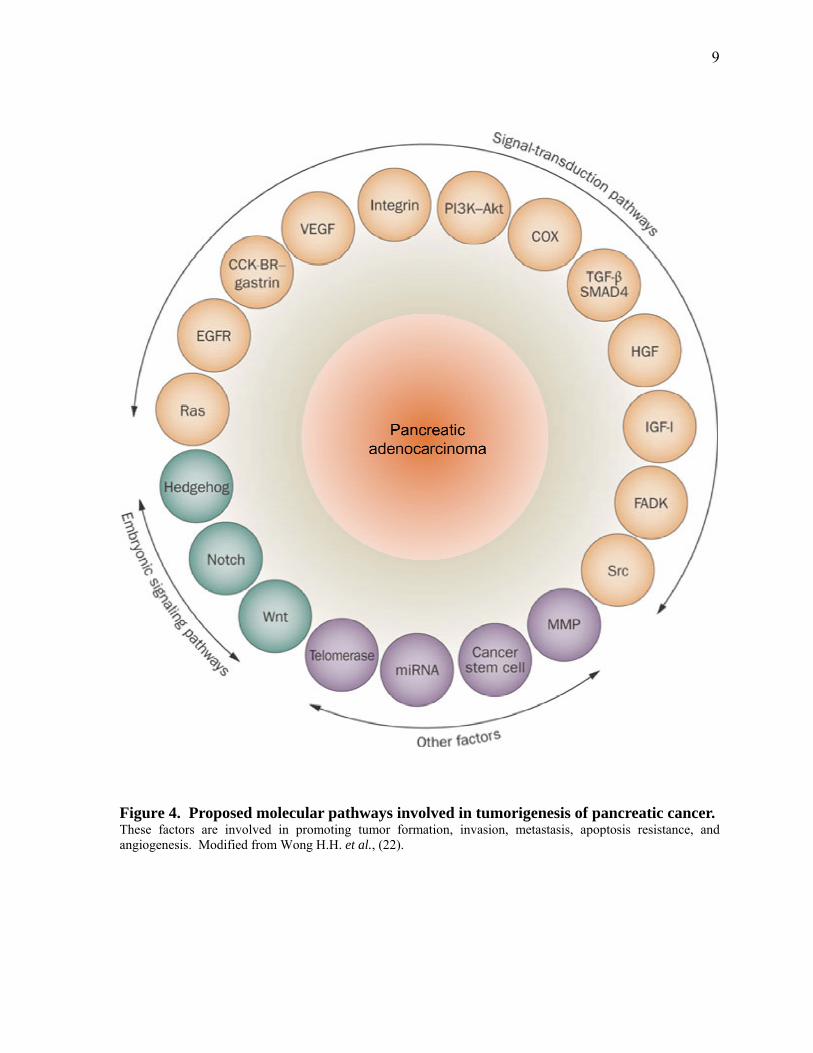
Figure 4. Proposed molecular pathways involved in tumorigenesis of pancreatic cancer. 9
Figure 5. Pathways of apoptosis. 11
Figure 6. Role of GSK‐3 in multiple cellular pathways. 23
Figure 1. Pro‐ and Anti‐apoptotic roles of GSK‐3. 31
Figure 8. Inhibition of GSK‐3 decreases proliferation and clonogenic survival of pancreatic cancer cells in a dose‐ and time‐dependent manner. 50
Figure 9. Inhibition of GSK‐3 disrupts NF‐κB activity in pancreatic cancer cells in a dose‐dependent manner. 52
Figure 10. Genetic knockdown of GSK‐3 by siRNA results in disruption of NF‐kB activity. 54
Figure 11. Effects of AR‐18 on gemcitabine sensitivity. 56
Figure 12. Effects of gemcitabine combined with GSK‐3 inhibition on NF‐κB. 58
Figure 13. NF‐κB inhibition by curcumin does not increase sensitivity to gemcitabine in pancreatic cancer cells. 59
Figure 14. GSK‐3 inhibition sensitizes TRAIL‐resistant prostate cancer cells to apoptosis. 73
Figure 15. GSK‐3 inhibition sensitizes TRAIL‐resistant pancreatic cancer cells to apoptosis. 75
Figure 16. GSK‐3 inhibition enhances TRAIL‐induced cell death in a time‐dependent manner. 76
Figure 17. GSK‐3 inhibition enhances TRAIL sensitization through PARP and caspase‐3 cleavage. 77
Figure 18. TRAIL sensitization through inhibition of GSK‐3 is caspase‐dependent. 79

xi
Figure 19. Genetic knockdown of GSK‐3 renders cells sensitive to TRAIL‐induced apoptosis. 80
Figure 20. TRAIL sensitization involves mitochondrial loop. 82
Figure 21. BCL‐2, MCL‐1 and CrmA are the anti‐apoptotic proteins involved in TRAIL sensitization of pancreatic cancer cells. 84
Figure 22. BCL‐2, Bcl‐XL, MCL‐1 and CrmA are the anti‐apoptotic proteins involved in TRAIL sensitization of prostate cancer cells. 86
Figure 23. Antitumor effect of GSK‐3 inhibition in vivo. 95
Figure 24. Increased β‐catenin levels in tumors treated with AR‐18. 98
Figure 25. Synergistic interaction of GSK‐3 and TRAIL in apoptosis induction in vivo. 99
Figure 26. Measurement of body weight. 101

xii
List of Abbreviations
AIF Apoptosis-inducing factor
AKT/PKB Protein kinase B
APAF-1 Apoptosis inducing factor-1
AR18 AR-A014418, a small molecule inhibitor of GSK-3
ATP Adenosine triphosphate
BCL-2 B-cell lymphoma-2
Bcl-XL B-cell lymphoma extra-large
BH Bcl-2 homology
BRCA2 Breast cancer-2 susceptibility protein
Ca+2 Calcium
CBP CREB-binding protein
CC3 Cleaved caspase 3
CDA Cytidine deaminase
CDK Cyclin dependant kinase
cFLIP Cellular FLICE (caspase-8)-inhibitory protein
CK-I Casein kinase-I
COX-2 Cyclooxygenase-2
CREB Cyclic AMP response element binding protein
CrmA Cytokine response modifier A
Cyt c Cytochrome c
DC Decoy receptor
DCK Deoxycytidine kinase
DCTD Deoxycytidine monophosphate deaminase;
DD Death domain
dFdC 2′-deoxy-2′, 2′-difluorocytidine (gemcitabine)
dfdCMP Gemcitabine 5′-diphosphate
dFdCTP Gemcitabine 5′-triphosphate
dFdUMP 2'-deoxy-2',2'-difluorouridine monophosphate.
DIABLO Direct inhibitor of apoptosis protein-binding protein with low PI
DISC Death-inducing signaling complex

xiii
DMSO Dimethyl sulfoxide
DNA Deoxynucleic acid
DR Death receptor
Dsh Disheveled
EGF Epidermal growth factor
EGFR Epidermal growth factor receptor
ELAM Endothelial-leukocyte adhesion molecule
EMT Epithelial mesenchymal transition
ER Endoplasmic reticulum
ERK Extracellular signal-regulated kinases
FACS Fluorescence-activated cell sorter
FADK Focal adhesion kinase
FGF Fibroblast growth factor
GBP GSK-3 binding protein
GMCSF Granulocyte macrophage colony stimulating factor
GS Glycogen synthase
GSK-3 Glycogen synthase kinase-3
HGF Hepatocyte growth factor
IAP Inhibitor of apoptosis
ICAM Inter-Cellular Adhesion Molecule-1
IGF-1 Insulin growth factors-1
IKK IκB-kinase
IL Interleukin
KO Knockout
LEF Lymphoid enhancer factor
LiCl Lithium chloride
MAPK Mitogen activated protein kinase
Mcl-1 Myeloid cell leukemia-1
MDM2 Murine double minute-2
MHC Major histocompatibility complex
miRNA Micro ribonucleic acid
MKK4 Mitogen-activated protein kinase kinase
MMP Mitochondrial membrane potential

xiv
MMP Matrix metalloproteinase
MSK-1 Mitogen- and stress-activated protein kinase-1
NF-κB Nuclear factor kappa-B
NIK NF-κB inducing kinase
NLS Nuclear localizing sequence
PanINs Pancreatic intraepithelial neoplasms
PARP Poly (ADP-ribose) polymerase
PDAC Pancreatic ductal adenocarcinoma
PI3K Phosphoinositide 3-kinase
PKA Protein kinase A
PKC Protein kinase C
PTPs Permeability transition pores
PYK-2 Proline-rich tyrosine kinase-2
RB1 Retinoblastoma-1
ROI Reactive oxygen intermediates
ROS Reactive oxygen species
SCID Severe combined immunodeficiency
Ser Serine
Shh Sonic hedgehog
siRNA Short interfering ribonucleic acid
Smac Second mitochondrial-derived activator of apoptosis
TCF T-cell factor
TGF-a Tumor growth factor-alpha
TGFb Transforming growth factor-beta
Thr Theronine
TNF-a Tumor necrosis factor-alpha
TP53 Tumor protein 53
TRAIL TNF-alpha related apoptosis inducing ligand
TWEAK TNF weak inducer of apoptosis
Tyr Tyrosine
VCAM Vascular cell adhesion molecule-1
VDAC Voltage dependent anion channels
VEGF Vascular-endothelial growth factor

xv
Wnt Wingless pathway
XIAP X-linked inhibitor of apoptosis

Chapter 1 Introduction

2
1.1 Clinical Implications of Pancreatic Cancer
Pancreatic cancer is one of the most fatal cancers, with a five year survival rate of less than
5% (1). Annually, there are about 4000 new cases in Canada, with similar number of deaths,
making it the fourth leading cause of cancer death in both men (4%) and women (5%) (2).
Surgery is the only curative treatment, but the majority of patients have metastatic disease or
an unresectable tumor at diagnosis and only 20% of pancreatic cancers cases are amenable to
surgical resection at presentation (Figure 1) (3, 4). The disease has a poor prognosis due to
delayed diagnosis, aggressive local invasion, early metastasis, and poor response to chemo-
and radiation-therapy (5, 6).
Gemcitabine (difluorodeoxycytidine; dFdC) is the standard chemotherapeutic drug for the
treatment of advanced pancreatic cancer (7). It is an analog of deoxycytidine, which is
transported into the cell as a pro-drug and requires to be phosphorylated initially by
deoxycytidine kinase to the active triphosphate (dFdCTP), which is incorporated into DNA
during S phase (Figure 2). This results in inhibition of DNA synthesis, arrest of the cell cycle
progression through the G1/S phase boundary, and induction of apoptosis (8). However, due
to pre-existing or acquired chemo-resistance, gemcitabine treatment has a marginal survival
benefit and yields an objective tumor response rate of less than 10% (9, 10).
1.1.1 Risk Factors:
A number of risk factors are associated with pancreatic cancer. Age plays an important role,
since it is mainly a disease of elderly age with the median age at diagnosis of 73 years (1).
Family history of pancreatic cancer is another risk factor, where individuals with a family
history of this disease have a much higher risk of developing the malignancy (1, 11).
Amongst other factors, cigarette smoking is known to be a cause of pancreatic cancer and
other factors such as chromosomal and genetic alterations, diets high in meats and fats, low
serum folate level, obesity, and diabetes mellitus play less important roles (1). Some studies
have also proposed a possible link between inflammatory conditions such as chronic
pancreatitis and development of pancreatic cancer, as the incidence of pancreatic cancer is
higher in these patients. However, the exact mechanism involved is unknown (5).
3
Figure 1. Aggressive nature of pancreatic cancer. Pancreatic cancer patients normally present with advanced disease that is unresectable by surgery. The left panel above displays an upper abdominal cross-section CT scan of a typical patient (65 year-old male, presenting with back pain) diagnosed with pancreatic cancer (circled in yellow). At the time of diagnosis, the cancer was unresectable due to major blood vessel encroachment. Despite chemo- or radiation-therapy (commenced March 2006), extensive liver metastasis developed within a few months (right panel, marked by arrowheads). Legend: 1, liver; 2, portal vein; 3, pancreas; 4, spleen. Figure courtesy of Dr. David Hedley, University Health Network, Toronto, Canada.
4
Figure 2. Mechanism of gemcitabine incorporation into the cells. Schematic representation of gemcitabine uptake by the cells and the metabolic processes required for activation of the reagent and inhibition of the target cell’s DNA synthesis. dFdC, 2′-deoxy-2′, 2′-difluorocytidine; dfdCMP, gemcitabine 5′-diphosphate; dFdCTP, gemcitabine 5′-triphosphate; CDA, cytidine deaminase, DCK, deoxycytidine kinase; DCTD, deoxycytidine monophosphate deaminase; dFdUMP, 2'-deoxy-2',2'-difluorouridine monophosphate. Modified from Ueno et al., (12).

5
1.2 Molecular Etiology of Pancreatic Ductal Adenocarcinoma
Pancreatic cancer arises through the accumulation of heterogenous genetic alterations ranging
from chromosomal abnormalities to point mutations and is classified into different
histological subtypes depending on the cell of origin in the pancreas. Pancreatic ductal
adenocarcinoma (PDAC) is the most predominant (80-90% of the cases) and aggressive type
of pancreatic cancer, with point mutations of the KRAS2 oncogene being the most prevalent
genetic alteration in this subtype (11). Due to the poor response to chemo- and radiation
therapies, the disease is highly lethal and is considered to be the fifth most frequent cause of
cancer related deaths in developed countries (13). PDAC is more prevalent in the ages of 60-
80 years old (11). The symptoms occur at a late stage typically with radiating abdominal and
back pain together with weight loss (11). The aggressive nature of PDAC is in accordance
with the infiltrating nature of the invasive cells into vascular, lymphatic, and perineural
tissues, and metastases to regional lymph nodes, the liver, and distant sites is very common
(1, 11). Lack of improvement in the survival rate reflects an urgent need for the development
of early diagnostic methods and sophisticated treatments (13).
1.2.1 Histological Precursor Lesions
Appearance of PDAC is believed to follow a “progressive model”, where pancreatic
intraepithelial neoplasms (PanINs) are considered as precursor lesions of fully developed
PDAC (Figure 3). These PanINs arise from non-invasive intraductal progressions and are
classified from low to high grade (PanIN 1-3) based on the their histological as well as
genetic alterations (11). Morphological changes in PanIN-1 and -2 include moderate
papillary atypia, nuclear hyperchromatism, pleomorphism, and nuclear stratification. These
histological changes in PanIN-1A and B are accompanied by activating mutations in KRAS2
and HER-2/NEU, and in PanIN-2 lesions with inactivation in CDKN2A/p16 tumor suppressor
genes (11). PanIN-3 is recognized by atypia in architecture and cytology accompanied by
inactivation mutations of MADH4/SMAD4/DPC4, BRCA2, and p53 genes (11). All these
stages are accompanied by shortening of the telomere lengths, rendering chromosomes
susceptible to abnormal chromosomal fusions and subsequently to chromosomal
rearrangements (11).

6
Figure 3. Schematic representation of development of pancreatic cancer. Progressive steps of pancreatic cancer formation involve formation of “pancreatic intraepithelial neoplasias” (PanINs), harbouring different stages of genetic and histological modifications required for resulting in full onset of disease. Modified from Hruban et al., (14).

7
1.2.2 Chromosome Abnormalities
These alterations can occur both in metastatic and primary lesions and involve numerical and
structural changes in chromosomes including translocations, deletions, gains, and
amplifications (11). While chromosome losses are more prevalent than gains and involve
chromosomes 4, 6, 9, 12, 13, 17, 18, 21, and Y; chromosome gains occur less frequently and
involve chromosomes 2, 7, 11, and 20. Chromosome losses occur mostly in regions with
tumor suppressor genes such as MADH4/SMAD4/DPC4 (18q21), BRCA2 (13q12),
CDKN2A/TP16/MTS1 (9p21), RB1 (13q14.2), TP53 (17p13), and the Peutz-Jeghers gene
(19p13.3) (11). With the help of studies such as cytogenetic, fluorescence in situ
hybridization technique (FISH), and comparative genomic hybridization (CGH) arrays,
chromosomal gains have been widely attributed to regions that harbour oncogenes, including
KRAS (12p12), MYB (6p24), AKT2 (19q13.1-q13.2), MDM2 (12q14.3-q15), AIB1 (20q12)
(11).
1.2.3 Genetic Modifications
This process can affect the activities of both tumor-suppressor genes as well as oncogenes.
The most frequently affected tumor-suppressor genes in PDAC are CDKN2A/p16/MTS1
(95%), TP53 (50-75%), and MADH4/SMAD4/DPC4 (55%). Other genes such as BRCA2,
MKK4, EP300, STK11, ALK4/ACVR1B, ACVR2, TGFBR1, and TGFBR2 are less affected.
Loss of these genes mostly occurs through homozygous deletion, and single allelic loss
together with a mutation in the second allele. Hyper-methylation in the promoter region of
the gene is also a less frequent cause of loss in the gene activity (11).
Activating mutations of oncogenes are a common phenomenon in PDAC. For instance,
KRAS2 mutations are reported in over 90% of PDAC (11). HER2/NEU over-expression is
another well identified factor correlating with the severity of dysplasia in PanIN lesions (11).
Additionally, up-regulation of certain tyrosine-kinase growth factor receptors (i.e. the
epidermal growth factor receptor; EGFR) and their protein ligands such as epidermal growth
factor (EGF), transforming growth factor-alpha (TGF-α), and amphiregulin are also
correlated with increased tumorigenicity and reduced survival in PDAC patients (11). Up-
regulation of other factors such as fibroblast growth factor (FGF) and its receptor, or insulin-
like growth factor I (IGF-I) and its receptor, as well as vascular endothelial growth factor

8
(VEGF) were also connected to a more invasive state of PDAC (11). A detailed diagram of
factors involved in promoting pancreatic cancer is illustrated in Figure 4.
1.2.4 Pancreatic Inflammation and its Link to Pancreatic Ductal Adenocarcinoma
There are controversial reports with regards to the role of chronic pancreatitis and the
development of PDAC. However, many studies emphasize the fact that there is an enhanced
risk for patients with pancreatitis to develop PDAC (5). For instance, in patients with
familial chronic pancreatitis, the duration of the disease correlates with the risk of cancer
development. In fact, 40% of patients with familial chronic pancreatitis develop PDAC by
the age of 70 years (15). Also, some risk factors such as smoking is shared between PDAC
and chronic pancreatic inflammation (5). Additionally, features of molecular changes in both
the diseases are similar. For example, mutations in a universal molecular marker of
pancreatic cancer such as K-ras are also found in 42% of chronic pancreatitis, or
inflammatory mediators such as: COX-2, lipoxygenase, inducible nitric oxide (iNOS), pro-
inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin (IL) 1-, -6, and
-8, reactive oxygen species (ROS), and nuclear factor kappa B (NF-κB) are overexpressed in
chronic pancreatitis as well as PDAC (5, 16). It is possible that the molecular changes
associated with inflammation lead to uncontrolled cell cycle progression that can eventually
lead to cancer initiation and progression (5). The mechanisms that drive pancreatic cancer
development can also promote cell survival, which is the main focus of this thesis.
1.3 Pathways of Apoptosis in Pancreatic Ductal Adenocarcinoma
Programmed cell death or apoptosis is a critical regulator of normal tissue homeostasis and
remodeling together with processes such as differentiation and proliferation (17). In contrast
to necrosis that results in inflammation, apoptosis is essential to maintaining a
physiologically balanced environment by removing aberrant, damaged, or infected cells, with
no harm to the neighboring cells (17-19). Apoptotic cells undergo DNA fragmentation,
cytoplasmic disruption, cell shrinkage, chromatin condensation, and blebbing (20). It is a
genetically encoded, tightly regulated process that occurs through an amplification process by
activating a protease cascade by a group of cysteine proteases; the caspases (21). In a
9
Figure 4. Proposed molecular pathways involved in tumorigenesis of pancreatic cancer. These factors are involved in promoting tumor formation, invasion, metastasis, apoptosis resistance, and angiogenesis. Modified from Wong H.H. et al., (22).

10
resting cell, caspases exist as pro-forms. Upon receiving an extrinsic (death receptor
mediated), or intrinsic (mitochondria mediated) death signal, the pro-forms are cleaved next
to aspartate residues, leading to activated caspases that can subsequently cleave numerous
cellular substrates such as nuclear lamins, DNase inhibitors, or cytoskeletal proteins and
finally lead to apoptosis as shown in Figure 5 (21). Caspases-8, 9, and -10 are named
initiator/executioner caspases, whereas caspases-3, -6, and -7 are known as effector caspases
(17, 21).
1.3.1 The Intrinsic Apoptosis Pathway
The key step in this pathway is mitochondrial outer membrane permeabilization which leads
to the release of cytochrome c into the cytosol and results in cytotoxicity (Figure 5) (23). It is
induced by cellular stress, growth factor deprivation, cytotoxic agents, and activation of
oncogenes (24). These stimuli can activate damage response proteins such as p53, which in
turn leads to the activation of BH3-only proteins (Puma and Noxa), followed by inhibition of
anti-apoptotic members of the Bcl-2 family of proteins (Bcl-2, Bcl-XL, Mcl-1) and subsequent
activation of pro-apoptotic Bcl-2 proteins (Bax, Bak, Bim) (23, 25, 26).
Activation of Bax and Bak is then followed by the perturbation of the integrity of the
mitochondrial outer membrane, release of cytochrome c and other apoptotic regulators such
as apoptosis-inducing factor (AIF), second mitochondrial-derived activator of apoptosis
(Smac), direct inhibitor of apoptosis protein (IAP)-binding protein with low PI (DIABLO),
endonuclease G, or the serine protease high temperature requirement protein Omi/HtrA2
from the inter-membraneous space of mitochondria (27). Cytochrome c, APAF-1, ATP, and
the initiator pro-caspase-9 form the apoptosome complex and result in cleavage and
activation of caspase-9 and subsequent cleavage and activation of executioner caspase-3 (27).
Meanwhile, Smac/DIABOLO, or Omi/HtrA2 inactivate the inhibitor of apoptosis (IAP)
proteins such as X-linked inhibitor of apoptosis (XIAP) (27). XIAP is recognized to block
caspase-3 auto-activation, hence preventing apoptosis (17).
Calcium (Ca2+) also plays a role in mediating mitochondria-dependent apoptotsis. Cellular
Ca2+ which is stored in the endoplasmic reticulum (ER), is released through the inositol 1,4,5-
trisphosphate (InsP3) receptor (InsP3R). These receptors are the principal ER Ca2+ release
channels in the majority of cells and their inhibition results in inhibition of Ca2+

11
Figure 5. Pathways of apoptosis. Apoptosis is induced by either an internal signal sent to the mitochondria (A) or an external signal that has stimulated the death receptors (B). Both signals initiate a cascade of events that lead to activation of caspases and induction of apoptosis. Modfied from Beure E. et al., (28).

12
release (23). The released Ca2+ is then transferred into the mitochondria via a potential-
driven uniporter, where the excessive increase in Ca2+ levels can lead to the formation of the
voltage dependent anion channels (VDAC) called permeability transition pores (PTPs),
through which, cytochrome c is released into the cytoplasm (23, 29). The anti-apoptotic
action of Bcl-2 is by reducing Ca2+ stores in ER, while on the contrary Bcl-XL inhibits the
expression of InsP3R at the DNA level (23). In contrast, the pro-apoptotic protein Bax
increases Ca2+ loading in the ER leading to increased transmission of Ca2+ in the
mitochondria, hence supporting PTP formation (23).
The mechanism through which Mcl-1 (myeloid cell leukemia-1) promotes its anti-apoptotic
effect is poorly understood. However, studies suggest that Mcl-1 acts by inhibiting Ca2+
signaling directly within mitochondria and also through preventing Bak from forming its
active oligomer form. Activated Bak can enhance Ca2+ depletion from both ER and
mitochondria (23). Other studies suggest that it also binds and inhibits the pro-apoptotic
protein Bim on mitochondria and blocks its induction of apoptosis through Bax activation
(30).
Mcl-1 is a tightly regulated, high turn-over protein that has a differential expression when
compared to Bcl-2 or Bcl-XL proteins. Upstream regulation of Mcl-1 is not very well known.
However, studies suggest that glycogen synthase kinase (GSK)-3 has an important role in
regulating Mcl-1 (31, 32). It is known that Mcl-1 is acutely activated upon receiving signals
from survival factors such as granulocyte macrophage colony stimulating factor (GMCSF),
IL-3, epidermal growth factor (EGF), vascular-endothelial growth factor (VEGF). These
signals act through PI3-K/Akt, JAK/STAT, or MEK/MAPK signaling pathways to block
GSK-3 by phosphorylating it at Ser9 and Ser21, resulting in stabilization of Mcl-1 and
survival effects. On that note, apoptotic stimuli including growth factor withdrawal,
staurosporine, ionizing radiation, and DNA damage immediately phosphorylate and degrade
Mcl-1 in a GSK-3 dependent manner (32, 33). At the time of exposure to death inducing
signals such as chemotherapeutic agents, pancreatic cancer cells depend in large part on the
mitochondrial pathway to induce caspase-3 activation and apoptosis (34).

13
1.3.2 The Extrinsic Apoptosis Pathway
It is initiated by the cell surface death receptors of the tumor necrosis factor superfamily
including FAS/APO-1/CD95, TNF receptor-1, and TNF related apoptosis inducing ligand
(TRAIL) receptor-1(DR4) and -2 (DR5, also called APO-2), with all containing a ligand-
binding cystein-rich repeat in their N-terminal domain (Figure 5) (17). Death receptors are
type-1 transmembrane proteins, whereas their binding ligands are type-2 transmembrane
proteins that are either soluble (cleaved by metalloproteases), or membrane-embedded (17).
In order to induce a death signal, both ligands as well as their respective receptors need to
form an active dimer/trimer. Upon binding of ligand trimers to receptor complexes, these
receptors undergo a conformational change that aligns the intracellular death domains (DD)
in order to bind adaptor proteins, which finally leads to the recruitment of initiator caspases (-
8, or -10), formation of death-inducing signaling complex (DISC), and induction of apoptosis
cascade through activation of caspase-3 (17, 32, 35). DISC formation can be controlled at
multiple levels to avoid unwanted induction of apoptotic signals. A major regulatory process
involves an inhibitory protein called cellular FLICE (caspase-8)-inhibitory protein (cFLIP).
This protein binds adaptor proteins such as Fas-associated protein with DD (FADD) and pro-
caspase-8 via its DD, blocking recruitment and activation of caspase-8 in DISC complexes
(17).
According to downstream events of DISC formation, apoptotic cells are divided into type-I
and type-II cells (Figure 5). In type-I cells, DISC formation directly leads to caspase-8 and
subsequently caspase-3 activation. However, in type-II cells, the death receptor pathway may
also promote mitochondrial dysfunction via caspase-8-mediated, cleavage of the pro-
apoptotic molecule Bid into tBid, which then translocates into mitochondria, triggering outer
membrane permeabilization through facilitating Bax and Bak interaction (17). Previous
reports indicate that pancreatic cancer cells are type II cells, indicating that even for extrinsic
signals to be effective; they require amplification of apoptosis through the mitochondrial loop
(34, 36).
TNF signaling was first identified through its cytotoxic effects on tumor cells and induction
of necrosis (17). TNF ligand binds to two distinct receptors TNF-R1 and 2, each capable of
inducing separate pro- and anti-apoptotic outcomes. Apoptosis inducing effects are mainly

14
through TNF-R2, involving a multi-step process in which RIP and TRAF-2 molecules are
recruited to form subsequent complexes-I and –II, eventually leading to caspase-8 activation
and apoptosis (17, 37). In contrast, TRAF-2 and RIP molecules when recruited by TNF-R1
activate pro-survival effects via multiple pathways including: cIAP-1 and cIAP-2 mediated
inhibition of caspase-8; TRAF-2 induction of MAPK cascades followed by activation of JNK
and transcription of TNF-responsive genes; and also TRAF-2 mediated NF-κB activation and
transcription of survival proteins (17, 38).
CD95/FAS is a ubiquitously expressed system that when activated by its ligand, CD95L,
provides an efficient robust apoptotic system. Both ligand and the receptor exist in soluble as
well as membrane bound forms, and are expressed by activated T-cells. Ligand-receptor
binding results in DISC formation and enrollment of both caspases-8 and -10 to induce
apoptosis (17, 38). Although both TNF-R and CD95 represent a highly robust anti-tumor
mechanism, when used systemically in patients, they have severe toxic effects (17). In
contrast, TRAIL (Apo2L) has shown promising results as it specifically targets cancer cells
for apoptosis induction and yet it spares the normal cells through mechanisms that might
depend on oncogenes such as RAS or MYC (39). Full mechanism of this differential
regulation remains to be elucidated.
RhApo2L/TRAIL is a soluble homotrimeric protein known to bind five different receptors,
among which two are apoptosis activating receptors, mainly expressed on the surface of
tumor cells: TRAIL-R1 (DR4), TRAIL-R2 (DR5/APO-2/KILLER/TRICK2), and three are
decoy receptors: TRAIL-R3 (DcR-1), TRAIL-R4 (Dc-R2), and osteoprotegerin.
Osteoprotegerin is a soluble decoy receptor that is suggested to have a role in bone
morphogenesis by binding to the osteoclast differentiation factor (17). The apoptosis
inducing receptors are expressed on the surface of a variety of cancers including colorectal
cancer, lymphoma, and non-small-cell lung cancer, hence rendering these cells susceptible to
the apoptotic effects of TRAIL (39). These receptors have an intracellular DD domain that
transfers the death signals to the downstream signaling pathways, whereas decoy receptors
lack DD domain. TRAIL has a high sequence homology to CD95, and induces apoptosis in a
similar manner by recruiting FADD, caspases-8, -10, and DISC formation. However,
contrary to CD95, apoptosis induction by use of TRAIL is physiologically safe and does not

15
lead to toxicity; indicating that the basal mechanism through which CD95 and TRAIL induce
cell death might be quite different (17).
Studies involving TRAIL ligand and its two receptors, DR4 and DR5, have shown little or no
toxic effects in vivo (40). This unique feature of TRAIL is beneficial for anti-cancer therapy
where selective targeting of tumor cells is desired with low toxicity for normal cells.
However, recent studies suggest that while tumor cell lines might be sensitive to TRAIL,
primary tumor cells could still be resistant. Combining TRAIL with chemotherapeutic agents
or irradiation is proposed to induce synergistic sensitization of the resistant cells to apoptosis
with little or no side effects (17, 41). This could specifically be beneficial to cases where
overexpression of apoptosis resistance factors such as Nuclear factor kappa-B (NF-κB), JNK,
and MAPK renders cells resistant to TRAIL induced apoptosis (17, 42, 43). In a study
where a patient xenograft model of pancreatic cancer in SCID mouse was used, a
recombinant form of TRAIL could induce apoptosis only in a subset of patient tumors and
combining TRAIL with gemcitabine could partially overcome the resistant tumors (44). The
sensitive tumors showed loss of pro-caspase-3, -8 and Bid along with lower levels of Bcl-XL
when compared to the resistant cells, indicating that the mechanism of TRAIL resistance
might be Bcl-XL dependent (44). Another report by Hinz and co-workers also suggests that
Bcl-XL might be responsible for TRAIL resistance in pancreatic cancer cells PANC-1 and
PancTu-1 (45).
In other studies, overexpression of XIAP in pancreatic cancer cells appears to be the key
cause of TRAIL resistance in pancreatic cancer cells and its inhibition by siRNA or small
molecule inhibitors has been shown to synergistically downregulate survival and clonogenic
properties of the cells both in vitro and in vivo in a Bcl-2-independent manner (34, 46). In
some other reports, the TRAIL resistance of pancreatic cancer cell lines PANC-1 and
HS766T is attributed to lower expression of DR4 and DR5 TRAIL receptors and inhibition of
NF-κB with bortezomib, PS-1145, or curcumin has been shown to reverse the resistance by
downregulating XIAP and Bcl-XL (43). All this supporting evidence indicates that TRAIL
treatment in combination with chemotherapy agents could be a promising strategy to
overcome apoptosis resistance of pancreatic cancer which is the main focus of Chapters 3 and
4 of this thesis.

16
It is of importance to note that there are also conflicting reports indicating that care should be
taken while studying the effects of TRAIL in pancreatic cancer, as the treatment of pancreatic
tumor cells with TRAIL has been shown to lead to enhanced tumorigenesis. In a study by
Trauzold et al., in vitro and in vivo treatment of pancreatic cancer cells induced secretion of
pro-inflammatory factors such as IL-8, and monocyte chemoattractant protein-1, and led to
enhancement of survival, invasion, and liver metastasis in orthotopic xenograft model,
suggesting that unpredicted side-effects of TRAIL treatment should be taken into
consideration (47). Accordingly, other studies involving TRAIL treatment of pancreatic
cancer cell lines COLO357, and Panc89, induced inflammation, proliferation, and invasion
markers such as uPA, IL-8, MMP-7, and -9 in a NF-κB dependent manner (48). Although
variation in pancreatic cancer cell lines, and also the experimental conditions, might be a
contributing factor to the above controversies, yet they all consistently point to a key role for
the activation of NF-κB or its downstream targets as a contributing factor to the existing or
acquired TRAIL resistance. As a result, downregulation of NF-κB before TRAIL treatment,
could be beneficial in both downregulating TRAIL resistant markers such as XIAP or Bcl-2
proteins, and also prevent potential growth promoting effects of TRAIL. In our study, we
have sensitized pancreatic cancer cells by inhibiting NF-κB through a combinational strategy
that involves co-treatment of a GSK-3 inhibitor and TRAIL. The results are reported and
discussed in Chapters 3 and 4 of this thesis respectively.
1.4 Chemo-resistance of Pancreatic Cancer
Evasion of cell death is the characteristic of nearly all malignant cancers and is responsible
for tumor formation and resistance to therapy (24). Owing to the pre-existing or acquired
chemo-resistance of pancreatic cancer cells, gemcitabine, the only clinically active drug in
the therapy of pancreatic adenocarcinoma, has been shown to exhibit a small advantage in
terms of increasing the survival of the patients (49). Multiple mechanisms are recruited to
evade apoptosis in pancreatic adenocarcinoma including: altered interactions of
chemotherapeutic agents with intracellular targets; enhanced expression of detoxification
mechanisms such as MDR-genes and antioxidants; or defective activation or degradation of
the agents resulting in insufficient intracellular concentration of the cytotoxic agents;
mutations and modifications of proto-oncogenes, anti-apoptotic genes or tumor supressors
(49). These anti-apoptotic mechanisms that have been linked to chemo-resistance of

17
pancreatic cancer, act through a multi-level system that includes death receptors, the
mitochondria, IAP family of apoptosis inhibitors, or survival pathways such as PI3K/protein
kinase B (PKB/AKT), and nuclear factor kappa-B (NF-κB) (24, 49).
1.4.1 Role of Nuclear Factor Kappa B in Survival of Pancreatic Cancer and Chemo-resistance
Numerous lines of evidence propose that the pro-inflammatory transcription factor NF-κB,
can provoke inflammation, tumorigenesis, metastasis, angiogenesis, survival, and chemo-
resistance in many inflammation associated cancers including pancreatic cancer (50-55). In a
range of tumors, overexpression of NF-κB or its constitutive activation is correlated with
tumor formation or chemo-resistance, and hence its inhibition is proposed as a means for the
prevention and treatment of cancer (51, 53, 56).
NF-κB was first discovered by Baltimore et al., in 1986 as a transcription factor that binds to
the promoter of the kappa light chain of immunoglobulin in the nucleus of immune B-cells
(57). It was later shown to be ubiquitously present in the cytoplasm of all cells and is
expressed in all eukaryotes ranging from Drosophila to human (53). NF-κB is comprised of a
family of protein homo-or hetero-dimers, the so-called Rel protein family, that exhibit a N-
terminal conserved region comprised of about 300 residues called Rel homology domain
(RHD) (57, 58). RHD is a region through which Rel proteins dimerize, involve in DNA
binding, and interact with IκB molecules (58, 59). In mammals, there are five members of
Rel family of proteins: Rel A (p65), Rel B, NF-κB1 (p50 and its precursor p105), Rel (c-Rel),
and NF-κB2 (p52 and its precursor p100). The most prevalent form of NF-κB dimer is
comprised of the heterodimer of p65/p50 subunits (58-60).
In resting cells, the inactive form of NF-κB is sequestered in the cytoplasm in association
with members of the inhibitory IκB family such as IκB-α, IκB-β, and IκB-ε (60). Upon
activation by a wide variety of agents including cytokines (for instance, Tumor necrosis
factor (TNF)-α, interleukin (IL)-1), viruses, bacteria, free radicals, inflammatory stimuli,
stress, cigarette smoke, growth factors, carcinogens, tumor promoters, and endotoxins, IκB
proteins are subjected to phosphorylation at N-terminal serine residues by a cellular kinase

18
complex called IκB-kinase (IKK), which leads to the subsequent ubiquitination and
degradation of IκB proteins and release of NF-κB (20).
Free NF-κB then translocates to the nucleus, binds to its coactivators, mainly CREB-binding
protein (CBP), and regulates expression of over 400 genes involved in the immune system,
growth, inflammation, cell survival, apoptosis inhibition, cell adhesion, and tumorigenesis.
These genes include but are not limited to: chemokines, pro-inflammatory cytokines such as
TNF-α, IL-1β, IL-2, IL-12, IL-6, IL-8, IL-2 receptor, interferon (IFN)-γ, inducible enzymes
such as cyclooxygenase-2, nitric oxide synthase, which is a regulator of the innate immune
response, major histocompatibility complex (MHC), growth factors including epidermal
growth factor (EGF), growth factor receptors such as HER2, cell adhesion molecules such as
VCAM-1, ICAM-1, ELAM-1, and E-selectin, transcription factor c-Myc, tissue invasion and
metastasis factors such as matrix metalloproteinases (MMPs), IL-8, urokinase type of
plasminogen activator (uPA), the chemokine receptors such as CXCR4, angiogenesis
promoting factors such as vascular endothelial growth factor (VEGF), anti-apoptotic proteins
such as members of the Bcl-2 family (Bcl-2, Bcl-XL) and IAP proteins (XIAP, cIAP-1, and
2), anti-apoptotic factors such as TNF receptor associated factor (TRAF)-1, and -2, c-FLIP,
GADD45β, and Ferritin Heavy Chain, angiogenic and pro-inflammatory molecule cox-2,
cell cycle regulator cyclin D1, and stem cell factors (20, 58, 59, 61).
Regulation of NF-κB can also occur through phosphorylation of its subunits by different
protein kinases in response to cytokines such as TNF-α, IL-1, or bacterial LPS, leading to
induction or inhibition of NF-κB activity (62). Phosphorylation of NF-κB mostly occurs at
multiple serine residues of the p65 subunit and has a key role in nuclear translocation, CBP
recruitment, DNA-binding activity, and regulation of its transcriptional activity (63). For
instance, both in vitro and in vivo studies by Wang et al., showed that TNF-α induces casein
kinase II (CK II) phosphorylation of p65 at serine 529 residue and this results in increased
transactivation of NF-κB (64). Further studies indicated that in response to TNF-α,
phosphorylation and activation of NF-κB could occur through other kinases such as protein
kinase A (PKA) at serine 276, IKK (at serine 536), protein kinase C (PKC)-ζ at serine 311,
mitogen-and stress-activated protein kinase-1 (MSK-1) at serine 276 (65-68). In a recent
study by Reber et al., it was shown that MAPK downstream nuclear kinase; MSK1, directly

19
phosphorylates the p65 subunit of NF-κB at Ser276, allowing its binding to the stem cell
factor (SCF) intronic enhancer, and driving pathophysiological SCF expression in
inflammation (61).
Although the molecular mechanisms and the biological consequences of p65 phosphorylation
are currently a point of interest for many researchers, nevertheless, the overall number of p65
phosphorylation sites and also the number of all potential p65 protein kinases is not yet
known (63, 69). For example, it was recently reported that besides IKK-α, at least three
other kinases including IKK-β, IKK-ε, TRAF family member-associated (TANK) binding
kinase-1 (TBK-1), converge on phosphorylating serine 536 of p65 (70, 71).
Phosphorylation of NF-κB may not always lead to its activation, and depending on the
cellular context it may even lead to its decreased activity. For instance, in a study by
Schwabe et al., in hepatocytes, it was shown that in response to TNF-α, glycogen synthase
kinase (GSK)-3β could phosphorylate p65 at the COOH-terminal transactivation domain
somewhere between residues 354-551, and enhance activity of NF-κB (72). Conversely, in
HeLa cells, the studies by Buss et al. , showed that phosphorylation of NF-κB at Serine 468
by GSK-3β resulted in decreased activity and negative regulation of NF-κB (71). The
regulatory phosphorylations of NF-κB is so important that cells lacking the protein kinases
GSK-3β (73), TRAF2-associated kinase (T2K, also called TBK1/NAK) (74, 75), IKKε (76)
NF-κB inducing kinase (NIK) (77) and PKCζ (78), showed an intact IκB phosphorylation but
a dysfunctional NF-κB, that led to impaired expression of target genes (71). Agents that can
suppress NF-κB activity include: Th2 cytokines (IL-4, IL-13, and IL-10), interferons,
endocrine hormones (LH, HCG, MSH, and GH), phytochemicals, corticosteroids, and
immunosuppressive agents.
The activation of NF-κB may have paradoxical effects. While a complete functionality is
essential to an appropriate immune response, improper NF-κB activity can lead to
inflammatory-mediated disease such as cancer. In fact, NF-κB is considered as the missing
link between inflammation and cancer (20, 50, 54). Constitutively active NF-κB has been
reported in the majority of tumors of different origin such as breast, prostate, pancreatic,
colon, ovarian, melanomas, and thyroid carcinomas, and its suppression has resulted in cell

20
cycle arrest, apoptosis, and tumor regression (20, 53, 79). Given that cancer is a multi-step
process during which normal cells undergo transformation by acquiring characteristics such
as: self sufficiency in growth, insensitivity to growth-inhibitory signals, apoptosis evasion,
unlimited proliferation, continuous angiogenesis, tissue invasion and metastasis, and the fact
that NF-κB can be a key player in transcribing genes responsible for all these processes,
strongly emphasizes an important role for NF-κB in tumorigenesis (20).
The exact mechanism behind up-regulation of the tightly regulated NF-κB in cancer cells is
not well determined and the reports seem inconsistent. The paradox stems from the fact that
while pro-inflammatory agents or the majority of carcinogens and oncogenic substances
activate NF-κB, in contrast, many chemotherapeutic agents or radiation-therapies also could
induce NF-κB activity, reflecting the role of NF-κB as part of the natural cellular self-
defense mechanism (53). Nonetheless, the constitutive activation of NF-κB in tumors can
also be linked to interruption of these stringent regulatory mechanisms including persistent
IKK activity, faulty IκBα activity, increased proteasomal activity, or consistent susceptibility
of cancer cells to autocrine/paracrine NF-κB stimulators (20).
NF-κB activation does not always lead to cell survival. Indeed, under specific circumstances,
it can also promote cell death and apoptosis (80). However, the signaling events leading to
this type of response are not well known, and it seems that it is highly depended on the nature
of the cell and the stimulus, as well as the type of NF-κB family member involved in the
process (81). For instance, c-Rel is considered pro-apoptotic, whereas RelA is shown to have
anti-apoptotic effects (82, 83). In studies by Farhana et al., and Jin et al., in prostate cancer
cells, it was reported that in the presence of retinoid related molecule, 3-CI-AHPC, or the
synthetic retinoid CD 437, activation of NF-κB leads to down-regulation of anti-apoptotic
proteins XIAP, cIAP1, and Bcl-XL and sensitizes the cells to apoptosis through up-regulation
of death receptors such as DR4, DR5, Fas, and Rip1 (82, 84). Other groups reported that
chemotherapeutic agents such as doxorubicin and danorubicin can activate NF-κB in a
fashion that the RelA molecule is deficient in phosphorylation and acetylation, leading to
repression of anti-apoptotic molecules and eventually promote apoptosis (85, 86).

21
Despite all the above mentioned contradictions, a number of reports provide supporting
evidence that NF-κB plays a key role in the development, survival, metastasis, and chemo-
resistance of pancreatic cancer (49, 87-89). Constitutively active NF-κB has been observed
in about 70% of pancreatic cancer patients as well as pancreatic cancer cell lines, but not in
normal pancreatic tissues or in immortalized pancreatic epithelial cells, indicating a sensitive
role that NF-κB plays in the tumorigenesis of pancreatic cancer (87, 88, 90). It has been
proposed that constitutive activation of NF-κB is linked to persistent induction through either
MAP kinases or K-ras; oncogenes that have been shown to hold an activating point mutation
in 80-95% of pancreatic adenocarcinomas (90). In addition, studies suggest that enhanced
proliferation of pancreatic cancer by NF-κB is in part due to the induced overexpression of
sonic hedgehog (Shh), a ligand of the hedgehog (Hh) pathway that is believed to be crucial to
the development of pancreatic cancer (91).
In an in vivo study by Fojioka and colleagues, inhibition of NF-κB by overexpression of a
phosphorylation defective form of IκBα mutant (S32, 36A) in pancreatic cancer cells AsPc-
1, blocked TNF-α induced expression of pro-angiogenic molecules such as IL-8 and VEGF
(87). In addition, hyperactivity of NF-κB renders pancreatic carcinoma cells resistant to
chemotherapeutic agents such as gemcitabine (49, 89). Reports indicate that blocking NF-κB
activity using IκBα super-repressor in CAPAN-1 and 818-4 pancreatic cancer cell lines, or
treatment of these cells with various NF-κB inhibitors such as MG132, Sulfasalazine, or
Gliotoxin, sensitize the cells to the apoptotic effects of chemotherapeutic agents such as
gemcitabine, doxorubicin or VP16 (49, 88).
NF-κB has also a role in TRAIL resistance of pancreatic cancer cells. Khanbolooki et al.,
have shown that inhibition of NF-κB by PS-1145, a chemical inhibitor of IKK, or bortezomib
could lead to TRAIL sensitization in TRAIL-resistant pancreatic cancer cells such as PANC-
1 and HS766T, suggesting that blocking NF-κB in combination with TRAIL might be a
feasible therapeutic approach in patients with pancreatic cancer (43). All the above findings
suggest that blockade of NF-κB might overcome chemo-resistance of pancreatic cancer cells,
and therefore has therapeutic potential (87).

22
Recent reports indicate that regulation of NF-κB by GSK-3 is essential in maintaining NF-κB
activity in many systems including pancreatic cancer (73, 92). In fact, the idea was first
proposed in a report published by Hoeflich and colleagues, suggesting that GSK-3β is
required to protect mice hepatocytes from TNF-α induced cytotoxicity, by activating NF-κB
(73). Mice lacking GSK-3β (GSK-3β-/-) died during mid-gestation due to massive liver
apoptosis; a phenotype similar to mice lacking p65 subunit of NF-κB or IKKβ (73).
Hepatocytes from GSK-3β-/- mice were highly sensitive to TNF-α induced apoptosis due to
lack of NF-κB activity when compared to wild type counterparts. It was also suggested that
GSK-3 regulation of NF-κB activity affected only a subset of NF-κB target genes. Although
the exact mechanism underlying this process was not well understood, it was suggested to be
downstream of IκB phosphorylation involving nuclear translocation and transactivation of
NF-κB (73).
1.5 Role of Glycogen Synthase Kinase-3 in Tumorigenesis of Pancreatic Adenocarcinoma
The mechanisms underlying the role of GSK-3 in cancer development are complex. Given
that GSK-3 has both pro- and anti-apoptotic roles, it remains unclear whether GSK-3 is a
“tumor suppressor” or an “oncogene”. GSK-3 is a constitutively active multi-potent
serine/threonine kinase, that is ubiquitously present in all mammalian cells, and its
homologues have been identified in all eukaryotes (93, 94).
GSK-3 was first recognized as a key mediator of the insulin pathway, mainly by
phosphorylating the key enzyme glycogen synthase and blocking the incorporation of glucose
into glycogen. Further studies revealed that in addition to glucose metabolism, GSK-3 was a
critical regulator of multiple signaling pathways involving cell fate, glycogen metabolism,
cell cycle, gene expression, protein synthesis, cellular metabolism, motility, apoptosis,
neuroprotection, proliferation, and survival through interactions with multiple pathways
including Wnt, MAP-kinase (MAPK), or PI3-K/Akt signal transduction pathways as shown
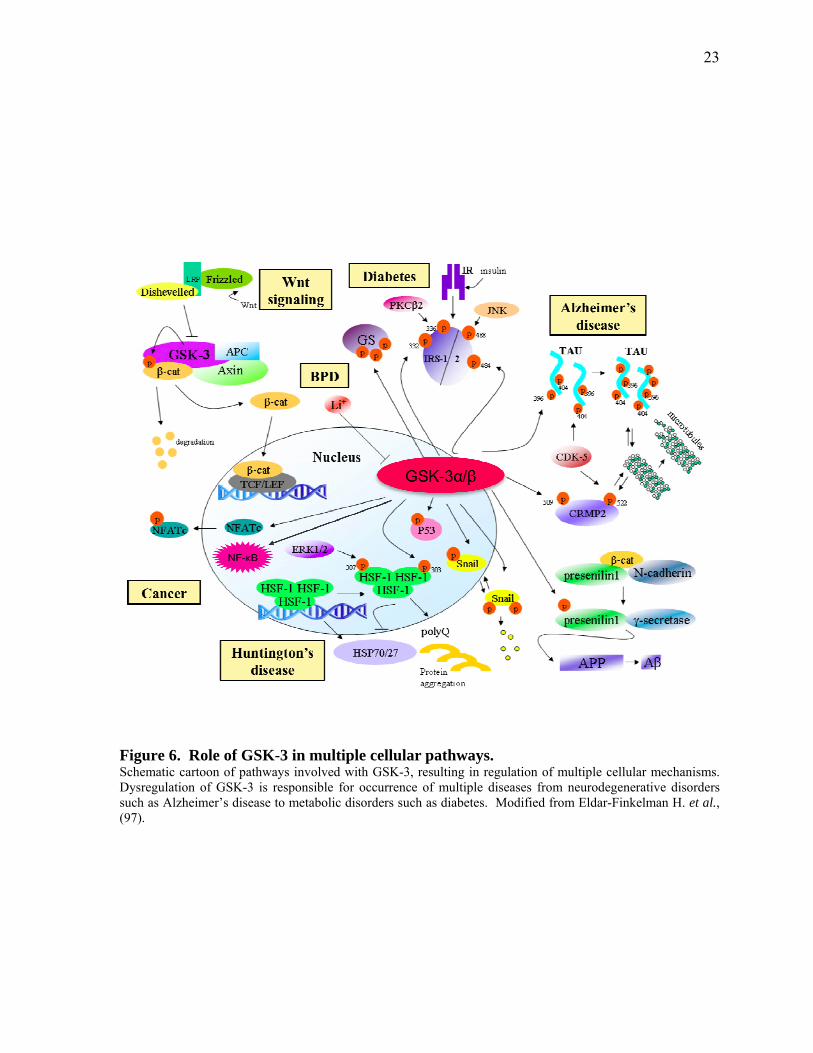
in Figure 6 (95). As part of the Wnt signaling pathway, GSK-3 has an inhibitory role during
embryonic development and in adult tissue cell proliferation (96). Owing to its role in
diverse cellular functions, dysregulation of GSK-3 is involved in the development of multiple
human diseases such as type-II diabetes, neurodegenerative disorders, Alzheimer’s disease,
23
Figure 6. Role of GSK-3 in multiple cellular pathways. Schematic cartoon of pathways involved with GSK-3, resulting in regulation of multiple cellular mechanisms. Dysregulation of GSK-3 is responsible for occurrence of multiple diseases from neurodegenerative disorders such as Alzheimer’s disease to metabolic disorders such as diabetes. Modified from Eldar-Finkelman H. et al., (97).

24
cardiovascular disease, bipolar disorder, tumorigenesis, and cancer (28, 92, 93, 98, 99).
There are two mammalian isoforms of GSK-3 that are encoded by distinct genes: GSK-3α
(mapped to chromosome 19q13.2) and GSK-3β (mapped to chromosome 3q13.3) (100).
These two isoforms are structurally 98% identical within their kinase domains, but differ in
C-and N- terminal residues. GSK-3α has an extra N-terminal glycin rich region, which
results in molecular weight difference between the two isoforms: GSK-3α is 51kD while
GSK-3β is 47kD (101).
Despite structural identity and the functional overlap and redundancy, they might have
different roles in determining cellular fate and as a result, the relative cellular proportion of
the two isoforms might be different in various cells or tissues (for instance, there is more
GSK-3β in brain tissue than GSK-3α) (94, 101). Most studies have focused on GSK-β
isoform and GSK-3α is less understood. However, given that both isoforms share similar
kinase domains, GSK-3 small molecule inhibitors that target kinase activity of the enzyme do
not make distinction between the two kinases and can inhibit both. On that note, unless a
study is based on using an isoform-specific inhibitor such as short interfering RNAs (siRNA),
or isoform-specific genetic knockdowns/knockouts, all the reports that are based on small
molecule inhibitors in the literature are shared by both GSK-3 isoforms (101). Functional
distinctions of these two isoforms specifically with regards to regulation of NF-κB will be
discussed later in this Chapter.
1.5.1 Regulation of GSK-3 and Its Substrates
In the resting cells, GSK-3 is constitutively active and, depending on the cell context, it
phosphorylates and functionally inactivates more than 40 downstream substrates such as β-
catenin, tau protein, glycogen synthase, c-Myc, Mcl-1, Cyclin D1, p53, and NF-κB as shown
in Figure 6 (30, 73, 93, 101).
When fully active, GSK-3 isoforms are phosphorylated at their tyrosine residues: Tyr 279 for
GSK-3α, and Tyr 216 for GSK-3β (102). The exact mechanism by which GSK-3 isoforms
are phosphorylated at these tyrosine sites is not well understood, but there are reports that
pGSK-3β (Tyr216) phosphorylation could be mediated by changes in the intracellular

25
calcium levels and a calcium dependent tyrosine kinase; proline-rich tyrosine kinase-2
(PYK2) (103, 104). Also, Fyn, a member of the Src tyrosine family, and mitogen-activated
protein kinase kinase (also called MEK1/2) have been shown to have a role in the regulation
of pGSK-3β (Tyr216) (94, 104-106). Recent studies by Cole et al., proposed that it is an auto
phosphorylation mechanism, by which GSK-3 maintains a proper conformation that is
required for its full potential kinase activity (107). It was shown that the mutant forms of
GSK-3α and β genetically lacking kinase activity, or treatment with small molecule
inhibitors of GSK-3, tyrosine residues on the respective sites did not get phosphorylated,
indicating that kinase activity of GSK-3 is required for its tyrosine site auto-phosphorylation
(101, 107). In line with these observations, studies in pancreatic cancer cell lines PANC-1
and BxPC-3 cells, described in Chapter 2 of this thesis, also showed a decreased Tyr 216/279
phosphorylation, after these cells were incubated with AR-A014418; a small molecule
inhibitor of GSK-3.
Active GSK-3 phosphorylates most of its substrates through a unique mechanism, which
requires priming of the substrates (i.e. pre-phosphorylation) by another protein kinase. GSK-
3 then phosphorylates a serine/threonine residue which is located four residues upstream of
the initial priming site. This site then serves as a priming site for further upstream sites to be
phosphorylated by GSK-3 (101, 108-111). Priming of GSK-3 substrates is not a universal
phenomenon as some substrates such as Axin do not require previous phosphorylation by
other protein kinases prior to being phosphorylated at Thr609 and Ser614 by GSK-3 (101,
112).
Unlike other protein kinases, regulation of GSK-3 is by acute inhibition of its kinase activity
through upstream protein kinases. Upon cellular stimulation with insulin, Wingless (Wnt), or
growth factors, GSK-3 isoforms undergo a rapid inhibitory phosphorylation within their N-
terminal domain, on Serine 21 (GSK-3α) and Serine 9 (GSK-3β) (101, 102). The crystal
structure of GSK-3 revealed that serine phosphorylation results in a conformational change of
the molecule, leading to an intramolecular fold that places the phosphorylated site within the
substrate binding cleft, hence inhibiting its substrate binding ability (101, 108, 113). Several
kinases can be involved in this modification process including protein kinase B (PKB, also
called Akt), Protein kinase A (PKA, also called cyclic AMP-dependent protein kinase),

26
protein kinase C (PKC), extracellular signal-regulated kinases (ERKs), p70S6 kinases,
p90Rsk (also called MAPKAP kinase-1) (93, 114-120).
Upon inhibitory phosphorylation of GSK-3, its substrates undergo de-phosphorylation by
cellular phosphatases, resulting in active substrates. These active substrates can subsequently
have roles in a wide spectrum of cellular processes from glycogen metabolism to oncogenesis
(94, 121-123). Regulation of GSK-3 activity by its upstream kinases is about 50% effective,
suggesting that it is a complex process involving supplementary mechanisms (101, 119, 124).
Recent reports indicate that there are other means of GSK-3 regulation including: head-to-tail
dimerization of GSK-3β isoforms, protein complex formation (for example: Axin or APC
phosphorylation and enhanced affinity for binding to β-catenin in conjunction with GSK-3),
priming of GSK-3 by other protein kinases (such as: CK-1α priming phosphorylation of β-
catenin), and sub-cellular localization of GSK-3 isoforms (i.e. in mitochondria, cytoplasm, or
nucleus) (94, 101, 112, 125-128).
It is also important to note that there are different pools of GSK-3 present in the cells that
depending on the upstream signals that inactivate GSK-3, the output is different. For
instance, inactivation of GSK-3 by growth factors and hormones that lead to inactivating
phosphorylation on Ser9 and Ser21 of GSK-3 isoforms, does not lead to accumulation of β-
catenin. Conversely, inactivation of GSK-3 through the Wnt pathway is through an
unidentified mechanism that may or may not involve N-terminal phosphorylation of Ser9 or
Ser21 through a kinase such as PKC, but results in β-catenin accumulation. (101, 116, 119,
129).
1.5.2 Homologs of GSK-3 in Lower Eukaryotes Multiple homologs of GSK-3 have been isolated from various organisms, and their roles in
the early development and cell fate determination is discerned in Drosophila, Dictyostelium
and mammals (96, 129-133).
A single homolog of human GSK-3β (with 70% identity) termed as gsk-A gene, has been
identified in the slime mold Dictyostelium discoideum (131). Dictyostelium gsk-A protein is
different from other homologs of GSK-3 because unlike other systems of GSK-3 regulation,
gsk-A is regulated by activation and not inhibition (96, 131, 134). It has a very important role
in the spore differentiation and the development of stalk cells in a cAMP-dependent manner

27
(96, 135, 136). During the process of cell fate determination in Dictyostelium, the aggregated
progenitor cells are differentiated into three types of cells: prespore, prestalk-A, and prestalk-
B. Formation of each of these differentiated cells involves interaction of cAMP with different
serpentine receptors such as cAR3 or cAR4 and subsequent activation of inhibition of gsk-A
activity respectively (96, 137). Formation of prespores involves interaction of cAMP with
cAR3 and subsequent activation of a tyrosine kinase termed as ZAK-1 that can in turn
phosphorylate and activate gsk-A, promoting spore cell differentiation. In contrast, cAMP
interaction with cAR4 leads to lack of activation of ZAK-1 and subsequently inhibition of
gsk-A and formation of prestalk-B cells (96, 137).
In Drosophila melanogaster, a segment polarity gene termed zeste-white 3 (zw3) (also known
as shaggy (sgg)) is responsible for the production of three different GSK-3 homologs termed
as SGG10, SGG39, and SGG46 (130). These three proteins of different sizes are products of
alternative splicing of a single gene and are a key player of many developmental stages of
Drosophile including embryonic, larval and adult life (130). In the early embryonic
development, autoregulation gene called engrailed (en) is repressed by Zw3 blocking the
commitment of cells in the posterior compartment of each segment (138-140). The activity of
Zw3 is inhibited by wingless (Wg), through an unknown mechanism involving activation of
frizzled-2 (DFz-2), and subsequent phosphorylation of disheveled (Dsh) by an upstream
kinase termed as Dsh-associated kinase (DAK), leading to its activation and blocking Zw3
activity. (141, 96, 96, 142, 143). It is also known that Zw3 is involved in the mesoderm
patterning and heart development (130, 144). Studies on the physiological function of GSK-3
in lower eukaryotes with regards to Wnt pathway, has provided the basis of understanding of
the regulatory mechanism of GSK-3 in higher eukaryotes.
1.5.3 Mammalian GSK-3 Homologs In mammalian cells, there are different pools of GSK-3 isoform that are regulated through
either inhibitory phosphorylation at their serine 9/21 or via Wnt pathway, and depending on
the upstream signal the cellular consequences are variable (96). In mammalian cells, the Wnt
family of ligands includes a series of at least 19 different secreted proteins that are both
glycosylated and cysteine-rich and have a key role in early embryonic development (101,
145, 146). During embryonic development, they act as inducers of cell growth,
differentiation, migration, and cell fate. In the absence of Wnt signals, fully active GSK-3

28
binds and phosphorylates free cytoplasmic β-catenin in a so-called destruction complex
including the scaffolding protein Axin, and the tumor suppressor adenomatous polyposis coli
(APC) (147, 148). Prior to interaction with GSK-3, cytoplasmic β-catenin is primed by casein
kinase-1 (CK-1) at Ser45. GSK-3 phosphorylation of β-catenin at residues Thr41, Ser37,
Ser33 results in βTrCP-E3 ubiquitin ligase-induced ubiquitation and subsequent proteosomal
degradation of β-catenin (94, 101, 147, 148). Wnt signaling inactivates GSK-3 through an
unknown mechanism that possibly recruits GSK-3 inhibitory protein: GBP/FRAT1, resulting
in the stabilization of β-catenin and its translocation to the nucleus where it binds to the T-
cell factor (TCF)/lymphoid enhancer factor (LEF) family of transcription factors and
enhances transcription of proto-oncogens such as c-myc and cyclin-D1, and genes involved in
invasion and metastasis, including MMP-7 (94, 96, 149, 150).
A different pool of GSK-3 is affected by PI3K/Akt or insulin signaling pathway. Upon
activation of receptor tyrosine kinases by insulin or growth factors, PI3 kinase phosphorylates
phosphoinositides that in turn recruit proteins such as PKB/Akt and PDK1 that results in
phosphorylation and activation of Akt via PDK1 (118, 119). Activated Akt phosphorylates
GSK-3 isofomrs α and β on serine 21 and 9 respectively (113-115, 151-153). This inhibitory
phosphorylation results in reduced kinase activity of GSK-3 and dephosphorylation of
substrates such as glycogen synthase via phosphatases which can lead to accumulation of
glycogen in the cells in a cell context-dependent manner (96, 115, 154-156). A recent study
by MacAulay et al., has shown that GSK-3 isoforms have tissue-specific functions, and
dysregulation of GSK-3 isoforms, specifically GSK-3α, has been related to obesity, and
insulin resistance in mice (155). They and others have shown that while GSK-3α is the
primary kinase responsible for phosphorylation and inactivation of liver glycogen synthase,
GSK-3β plays a key role in muscle glycogen synthase regulation (155, 157).
GSK-3 is involved in differentiation and self-renewal of human and mouse embryonic stem
cells (HESC and MESC respectively), and its inhibition has been reported to induce
pluripotency and lack of differentiation through activation of Wnt signaling pathway (158).
In an attempt by Sato et. al., to study the molecular pathways involved in self-renewal of
HESC, they showed that while leukemia inhibitory factor (LIF)/Stat-3 has role in maintaining
pluripotent status in mouse embryonic stem cells (MESC), it does not have similar effect on

29
HESCs (158, 159). LIF/Stat-3 signaling pathway acts through activating both JAK/STAT and
MAPK signaling pathways which can subsequently lead to activation of PI3K-Akt cascade
and inhibition of GSK-3 isoforms (160). However, it is only involved in pre-gastrulation
stage, indicating the presence of alternative pathways of self-renewal in HESCs (158, 160).
Sato et. al., showed that using small molecule inhibitors of GSK-3 such BIO resulted in a
reversible hyperactivity of Wnt pathway leading to increased expression of the pluripotency-
specific transcription factors such as Oct-3/4, Rex-1, and Nanong and undifferentiated status
of both HESCs and MESCs; emphasizing role of GSK-3 in the process differentiation and
self-renewal (158).
GSK-3 isoforms are functionally redundant. In a novel study by Doble et al., functional
redundancy of GSK-3 isoforms was shown by creation of a series of 0-4 GSK-3 allelic
knockouts of mouse embryonic stem cells (ESC) and examining the isoform-specific effect
in Wnt/β-catenin signaling pathway (161). Deletion of either GSK-3α or-β had no effect on
Wnt/β-catenin signaling, whereas ¾ allelic knockouts or double knockout ESCs showed a
gene-dosage impact on Wnt activation, β-catenin expression and subsequent increase in β-
catenin/TCF mediated transcription (161). Interestingly, double knockouts of GSK-3α/β
isoforms resulted in a reversible hyper-activity of Wnt/β-catenin signaling and severe loss of
differentiation is ESCs, highlighting that both isoforms are equally of functional importance
(161). The study also indicated that GSK-3 double knockout mouse ESCs did not
differentiate into cardiac myocytes or neural tissue, a similar phenotype to ESCs
overexpressing Wnt proteins (161). Overall, these results point to importance of Wnt-GSK-3
interaction in ESC differentiation.
1.5.4 Tumor Suppressor Role of GSK-3 in Substrate Regulation
The pro-apoptotic role of GSK-3 is closely in association with the inhibition of proto-
oncogenes such as β-catenin; a downstream effector of the canonical Wnt signaling pathway.
Dysregulated Wnt pathway is known to play a role in tumorigenesis of many cancers
including breast and colon cancer (101). In pancreatic ductal adenocarcinoma, the Wnt
pathway does not seem to play a significant role in tumorigenesis, as the previous reports
point out that only small subset of pancreatic cancer cells (mostly acinar cell carcinomas)

30
have activated Wnt pathway, signifying a fully active GSK-3 in pancreatic cancer cells (162,
163).
In line with the pro-apoptotic effect of GSK-3, there are reports indicating that upon
receiving death signals such as: growth factor withdrawal, inhibition of PI3K/Akt, DNA
damage, ER stress, hypoxia/ischemia, mitochondrial toxins, heat shock, and oxidative stress,
GSK-3 promotes the mitochondrial/intrinsic apoptotic pathway by disrupting mitochondrial
function as demonstrated in Error! Reference source not found.A (28). In fact, when
present in the mitochondria, GSK-3 facilitates intrinsic apoptosis either through disruption of
the mitochondrial membrane by phosphorylating activation of Bax (Ser163) and voltage
dependent anion channel (VDAC), phosphorylating degradation of anti-apoptotic Mcl-1,
stress induction of pro-apoptotic Bim (28). Activated GSK-3 inversely phosphorylates and
regulates an anti-apoptotic Bcl-2 family member called Myeloid Cell Leukemia (MCL)-1.
Mcl-1 overexpression is observed in many tumors (e.g. myeloid leukemia, breast cancer),
and is believed to play a role in the poor prognosis and chemo-resistance of these cancer cells
by inhibiting the intrinsic apoptotic pathway (33) GSK-3β phosphorylated Mcl-1 proteins are
subjected to E3 ligase-βTrCP ubiquitination and subsequent degradation, thus rendering cells
susceptible to apoptosis (31).
Nuclear GSK-3 also regulates transcription factors that control the expression of proteins
involved in mitochondrial apoptosis such as p53, and cyclic AMP response element binding
protein (CREB) by activation and inhibition respectively (28). The transcription factor p53 is
a DNA damage response tumor suppressor with the ability to mediate cell cycle arrest,
senescence, and apoptosis (28). GSK-3 is one of the key regulators of p53, where it binds
directly to p53, forming a complex and phosphorylating nuclear or mitochondrial p53 and
inducing apoptosis (28). Moreover, GSK-3 acts by regulating the sub-cellular localization of
p53 and its intracellular levels or through phosphorylating MDM2; a p53 regulatory protein
(28, 94). Interestingly, binding to p53 also mutually activates GSK-3 through an unknown
mechanism that does not involve phosphorylation, indicating the co-dependence of both the
factors to induce intrinsic apoptosis (28). Tumor suppressor activity of GSK-3 also involves
its direct interaction with many proto-oncogenes including c-myc and cyclin-D1 (94). GSK-
3 phosphorylates and targets c-myc and cyclin-D1 proteins for degradation.
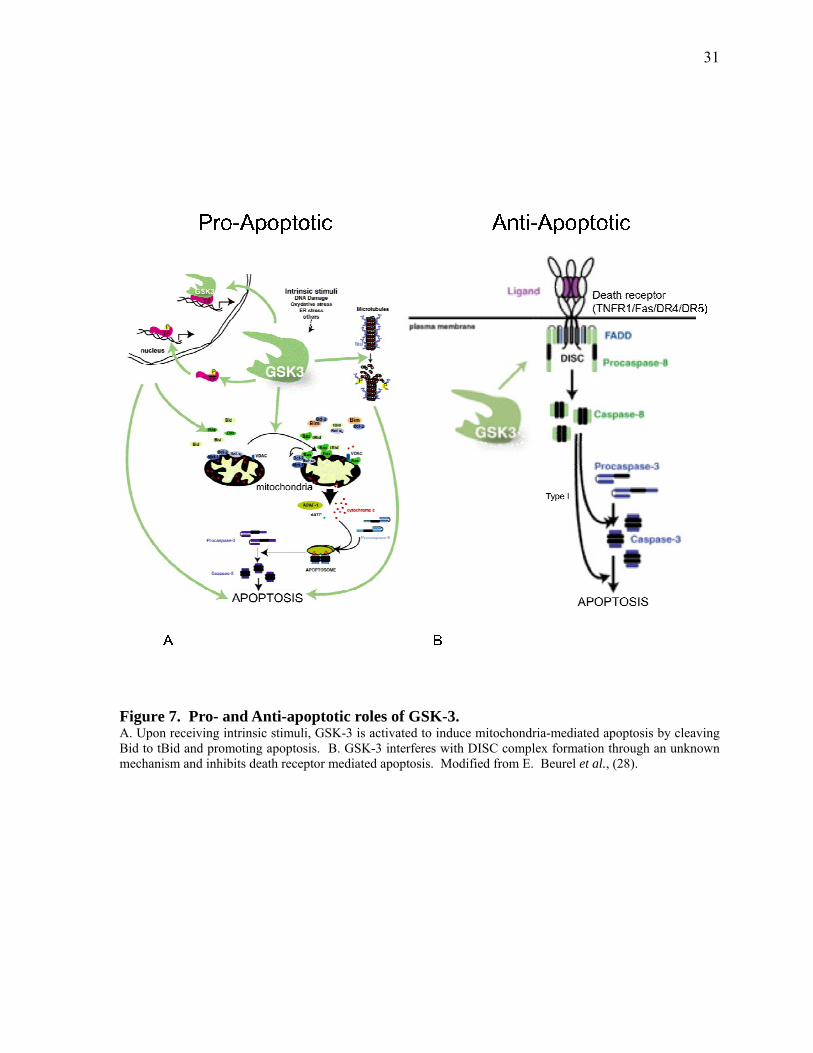
31
Figure 7. Pro- and Anti-apoptotic roles of GSK-3. A. Upon receiving intrinsic stimuli, GSK-3 is activated to induce mitochondria-mediated apoptosis by cleaving Bid to tBid and promoting apoptosis. B. GSK-3 interferes with DISC complex formation through an unknown mechanism and inhibits death receptor mediated apoptosis. Modified from E. Beurel et al., (28).

32
In line with the tumor suppressor activity of GSK-3β, it has been shown to be a negative
regulator of skin tumorigenesis. In a mouse model of multistage carcinogenesis, the late
papillomas and squamous cell carcinomas showed a dramatic increase in Ser9
phosphorylation (the inactive form of GSK-3β), while the level of Tyr216 phosphorylation
(the active form of GSK-3β) was significantly decreased when compared to normal tissue
(164). Similar results were observed in studies examining tissues from human squamous cell
carcinoma (94). Overexpression of constitutively active S9A mutant GSK-3β or a wild type
GSK-β in epidermal cells, inhibits in vivo and in vitro neoplastic transformation. In contrast,
GSK-3β inhibition by small molecule inhibitors, or overexpression of the kinase-deficient
K85R GSK-3β, results in oncogenic transformation of epidermal cells of the skin (165).
GSK-3 acts as a tumor suppressor in mammary tumors. Studies in mouse mammary glands
where a kinase-inactive form of GSK-3β (dominant negative) was used, mammary
tumorigenesis was promoted by dysregulation of the Wnt pathway and accumulation of β-
catenin and cyclin-D1 (166). In contrast, activation of GSK-3β by adiponectin or rapamycin
resulted in cell cycle arrest and induction of apoptosis in human breast cancer cells (167,
168).
The anti-tumor effect of GSK-3 is also evident in the area of metastasis and cancer cell
invasion. In some studies involving modulation of Wnt, Hedgehog, and Snail pathways, it
has been shown that GSK-3 plays an important role in modulating epithelial-mesenchymal
transition (EMT). EMT is the process responsible for invasiveness and motility of the cancer
cells and GSK-3 inhibition promotes the development of EMT (169-171). Cyclooxygenase-2
(COX-2) is another tumor progression factor that is negatively regulated by GSK-3 and
studies in gastric cancer has shown that GSK-3 inhibition leads to COX-2 mRNA stability
and may lead to tumor development and metastasis (172). In addition, in one study it was
shown that GSK-3β could negatively phosphorylate the p65 subunit of NF-κB at Ser468 and
reduces its activity in vivo. However, it was later shown that this site is being regulated by
IKKβ and not GSK-3 (71, 173).
The studies that put emphasis on the pro-apoptotic role of GSK-3 were mainly based on the
previous findings that GSK-3β overexpression in PC12 cells and Rat1 fibroblasts was

33
apoptosis promoting (174). However, although this work provided insight into the role of
GSK-3 in apoptosis, the results might not be applicable to situations where GSK-3 is
expressed at normal physiological levels. Nonetheless, the in vivo studies involving
constitutively active forms of GSK-3α/β did not lead to apoptosis induction, indicating that
GSK-3 acts more likely as a facilitator and not an inducer (28, 175). In addition, in one
study, it was shown that GSK-3β could negatively phosphorylate the p65 subunit of NF-κB
at Ser468 and reduce its activity in vivo. However, it was later shown that this site is
regulated by IKKβ and not GSK-3 (71, 173).
In summary, it is well documented that GSK-3 has a role in promoting the intrinsic apoptosis
pathway, mainly by inducing expression of pro-apoptotic proteins and also blocking the
action of anti-apoptotic molecules. However, the level of involvement of GSK-3 as an
apoptosis promoting kinase highly depends on the cellular insult and the cell context, as
further reports imply an anti-apoptotic activity of GSK-3.
1.5.5 Tumor Promoting Role of GSK-3 in Substrate Regulation
Contrary to the tumor suppressive role of GSK-3, there are studies that have suggested the
oncogenic side of it. Studies in multiple cancers including prostate, ovarian, colon, chronic
lymphocytic leukemia, and pancreatic adenocarcinoma, have raised the possibility of a
survival role for GSK-3β in these cancers (92, 98, 176-179). GSK-3β is shown to be
overexpressed in ovarian cancer and pharmacological inhibitors of GSK-3 in these cells
suppress cell proliferation in vitro and prevent tumor formation in vivo. On the contrary,
GSK-3β overexpression in ovarian cancer cells led to elevation in cyclin-D1 expression
levels and induction of cell cycle progression (180). Other studies in medullary thyroid
cancer cells indicate that inactivation of GSK-3β can lead to growth suppression (181).
Additionally, overexpression of GSK-3β in a range of cancer cells including hepatocellular,
prostate, and lymphocytic leukemia augments the survival and proliferation of these cancer
cells (179, 182-184).
In colorectal cancer, high levels of GSK-3β expression are observed in both cell lines and
patient samples and is associated with high levels of cellular proliferation and survival (99).
Inhibition of GSK-3β by RNA interference or small molecule inhibitors results in decreased

34
survival of colon cancer cells in vivo and in vitro (99, 178). In pancreatic adenocarcinoma,
nuclear accumulation and higher expression levels of GSK-3β is observed and is correlated
with pancreatic cancer dedifferentiation (92, 185). Upon GSK-3 inhibition with small
molecule inhibitors or RNA interference, the cell survival decreases in both established
tumors as well as cell lines (92, 185). In Chapter 2 of this thesis the pro-survival role of
GSK-3 is investigated and the chemo-sensitization upon GSK-3 inhibition is studied in detail.
Unlike its pro-apoptotic role in the intrinsic pathway, GSK-3 has been proposed to block the
extrinsic apoptotic pathway, as illustrated in Error! Reference source not found.B. This
idea came from three basic studies: lithium could sensitize a variety of cell lines and mouse
tumors to TNF-α induced toxicity (186), lithium turned out to be a GSK-3 inhibitor (187),
and that GSK-3β knockout mice died on E14 due to TNF-α sensitivity in hepatocytes (28,
73). Mouse embryonic fibroblasts (MEFs) from the GSK-3β knockout mice or the wild type
MEFs and hepatocytes treated with lithium, showed TNF-α induced cytotoxicity, indicating a
role for GSK-3β in blocking TNF-α induced apoptosis (72, 73).
More recently, it was shown that the inhibitory role of GSK-3 also applies to other death
receptors such as DR4, DR5, and Fas (28). For instance, GSK-3 was found to block Fas
mediated cell death (188). In addition, inhibition of GSK-3 activity by small molecule
inhibitors such as SB216763 or lithium in prostate cancer cells or using siRNA against GSK-
3β in these cell lines, could sensitize the cells to the apoptotic effects of TRAIL ligand (182)
This effect is shown to be a myc-dependent phenomenon at least in colon cancer cells
HCT116, as genetic knockdown of GSK-3β or FBW7 ubiquitin ligase, rendered the cells
susceptible to the apoptotic effect of DR5 agonist antibodies via stabilizing myc (189).
Although all these reports identified an inhibitory role of GSK-3 in the extrinsic apoptotic
machinery, the exact mechanism through which this effect is being implemented has not been
very well identified. A series of mechanistic studies have indicated that GSK-3 exerts its
protective role somewhere at the initial step of death-receptor mediated apoptosis, upstream
of the caspase-8 activation, and Bid cleavage (28). Other studies suggested that this might be
a p53-dependent process, as GSK-3 inhibition in TRAIL resistant hepatocellular carcinoma
cells sensitized these cells by stabilizing p53, and p53 inhibition reversed the sensitizing
effect (190).

35
Lately, a series of studies have also linked the death receptor blocking effect of GSK-3 to
NF-κB activity (62, 72, 73). Hoeflich et al., have shown that blocking GSK-3β inhibited
TNF-α induced NF-κB activation in MEFs, and later studies in hepatocytes indicated that in
response to TNF-α, GSK-3β mediates NF-κB activation (72). GSK-3 can only activate a
subset of NF-κB target genes (191), and only the GSK-3β isoform, and not GSK-3α, is
responsible for TNF-α or TRAIL induced NF-κB activation (73, 182). In contrast TRAIL
induced apoptosis is facilitated through GSK-3α and siRNA knockdown of GSK-3α has been
shown to reduce TRAIL sensitivity in HeLa cells (192). TRAIL induced NF-κB activation,
leads to expression of anti-apoptotic proteins such as XIAP, or sonic hedgehog (Shh) as part
of a cellular self-defense mechanism (47, 193, 194) and selective inhibition of NF-κB or
XIAP is shown to sensitize the cells to TRAIL effect in pancreatic cancer (43, 195-197).
Detailed mechanism of TRAIL sensitization upon GSK-3 inhibition is studied in Chapter 3 of
this thesis.
1.6 Inhibition of GSK-3: A Double Edged Sword in Chemotherapy
It appears that GSK-3 inhibition can lead to conflicting results in cancer cells. Depending on
the tumor type and cellular context, active GSK-3 can confer either resistance or sensitivity to
cancer chemotherapy. Studies have shown that breast cancer cells with kinase active GSK-
3β were sensitive to rapamycin induced cell death while GSK-3β null cells were not (167).
Similar results were obtained in breast and ovarian cancer cells where apoptosis was induced
with chemotherapeutic drugs such as cisplatin, taxol, 5-fluorouracil (94). In that line,
treatment of hepatoma cells with a known GSK-3 inhibitor; lithium, confers resistance to two
chemotherapy agents, camptothecin and etoposide, while, overexpression of constitutively
active S9A GSK-3β mutant form, or inhibition of PI3-kinase pathway by LY294002 renders
hepatoma cells sensitive to camptothecin or etoposide-induced apoptosis (198, 199). Taken
together, anti-tumor activity of GSK-3 might be required for the treatment of some cancers or
having a balanced physiological status in the cells, and in situations where GSK-3 inhibition
is necessary (for instance, treatment of Alzheimer’s disease, Parkinson disorder, bipolar
disease, and type II diabetes), there might be concern with regards to activation of oncogenic

36
pathways such as Wnt or stabilizing molecules involved in tumorigenesis such as c-myc, and
cyclin D1 (173, 200).
However, contrary to expectation, studies have shown that mice genetically lacking GSK-3β
do not have increased Wnt signaling, nuclear β-catenin, or cyclin D1 levels due to redundant
action of GSK-3α (99). There are also reports indicating that psychiatric patients treated with
lithium have a lower risk of developing tumors (173). Overexpression of GSK-3β is
observed in many human cancers including colon, ovarian, chronic lymphocytic leukemia,
and pancreatic cancer, and there are studies demonstrating that GSK-3 inhibition can
sensitize cancer cells to chemotherapy agents. For instance, in a study involving human
colorectal cancer cells HCT116, inhibition of GSK-3β by a small molecule inhibitor
LY2119301, sensitizes the cells to adriamycin induced apoptosis in a p53-dependent manner
(201). In mixed lineage leukemia (MLL), chemical inhibition of GSK-3 led to reduced cell
proliferation, survival, and clonogenic capacity of the cells (202). In prostate cancer, GSK-3
inhibition blocks proliferation and androgen receptor activity and also sensitizes the cells to
TRAIL mediated apoptosis (182, 183). Also, in colon cancer cells with high levels of β-
catenin and active phospho Akt (Ser 473), in vitro kinase assay and immunoblotting
demonstrated a high level of GSK-3β activity (99). On that note, although Akt inhibits both
GSK-3 isoforms through phosphorylation on Ser-9 and -21, it was found that in two
pancreatic cancer cell lines with high Akt activity (PANC-1 and ASPC1), GSK-3 was still
highly active, indicating that in some human cancers, the inhibitory phosphorylation of GSK-
3 might not affect activity (92).
Although GSK-3 has no nuclear localizing sequence (NLS), it is present in different cellular
compartments of pancreatic cancer cells depending on the level of differentiation. For
instance cytoplasmic GSK-3β is observed in high grade PanIN lesions or differentiated
pancreatic cancer cells, while nuclear GSK-3β is more found in de-differentiated tissues
(185). Although the exact mechanism of shuttling GSK-3 in and out of nucleus is not well
defined, some studies have proposed that a TNF-like family member; TNF weak inducer of
apoptosis (TWEAK), is responsible for transporting GSK-3β into the nucleus and driving
transcription of NF-κB target genes, while the GSK-3β interacting protein Frat1 has been
reported as a chaperone for nuclear export (173, 203, 204).

37
Recent reports have suggested a role for GSK-3β in pancreatic cancer chemo-resistance
through positive regulation of NF-κB. This is supported by studies where genetic depletion
of GSK-3β by siRNA or inhibiting its kinase activity using small molecule inhibitors,
decreased cell proliferation and survival of pancreatic cancer cells in vivo and in vitro (92,
185). This phenomenon was supported by reduction in NF-κB activity as well as expression
of its target genes such as cyclin D1, XIAP, and Bcl-2 (92, 185).
In the nuclei of pancreatic cancer cell lines as well as patient tissues, GSK-3β is seen to
accumulate and co-localize with NF-κB p65 (185). The exact mechanism of NF-κB
regulation by GSK-3 is not well defined, but different investigations suggest that it might be:
A) at a distal point to the IκK complex, B) at a transcriptional level by phosphorylating p65
and altering binding capacity to the target genes, C) at a chromatin level by remodeling the
NF-κB target gene promoters in response to TNF-α, hence inducing formation of
euchromatin at specific sites. This can be done by formation of a GSK-3/IKK complex that
targets HDAC1, hence allowing histones (for instance H3) to be acetylated at NF-κB
promoter sites such as XIAP and Bcl-2 (173, 205). Regulation of NF-κB activity by different
GSK-3 isoforms is studied in Chapter 2 of this thesis.
Many studies have pursued GSK-3 inhibition in the perspective of therapeutic potential
resulting in the production of fairly selective small molecule inhibitors of GSK-3 including
thiazoles, paullones, indirubins, malemides, hymenialdisine, and 2,4-
disubstitutedthiadiazolidinones (TDZD) (206). These compounds are mostly ATP-
competitive inhibitors and inhibit kinase activity of the enzyme without differentiating the
two isoforms. As a result, effects observed using small molecule inhibitors are consequent to
the simultaneous inhibition of both GSK-3α and –β (200, 203).
The thiazole AR-A014418 from AstraZeneca was initially used to show the effect of GSK-3
inhibition in transgenic mice overexpressing tau and resulted in significant reduction of tau
phosphorylation (207). It has also shown to reduce immobility time in rats when tested in the
forced swimming test, a well known model for antidepressant efficiency of GSK-3 inhibitors
(208). In later studies of in vitro and in vivo models of pancreatic cancer, AR-A014418
treatment was shown to have anti-survival, and growth reducing effects (92, 185).

38
1.7 Clinical Management of Pancreatic Cancer
Despite considerable attempts for the past 25 years to develop effective chemo- or radiation-
therapies for the treatment of pancreatic cancer, the disease remains incurable (22).
Gemcitabine is the only effective chemotherapeutic drug for the treatment of advanced stages
of pancreatic cancer. A phase III clinical trial in 1997 demonstrated that compared to 5-
fluorouracil, gemcitabine resulted in a higher median survival rate (4.41 months versus 5.65
months, respectively, p=0.0025) and could attenuate disease-induced symptoms (7).
However, given the recurring nature of pancreatic cancer, clinical management of the disease
using conventional therapies that involve single agents has proven to be very limited in effect
(22). As a result, much recent effort has been directed toward targeted therapies specific to
molecular targets that are involved in various cancer cell survival pathways (22). A genomic
analysis of pancreatic cancer in 2008 demonstrated that a large number of genetic alterations
(about 63) have an effect on a few number (about 12) of cell signalling pathways that are
involved in invasion, tumor growth, angiogenesis, metastasis, and apoptosis resistance
(Figure 4) (22, 151). That study suggested that to achieve effective treatment of pancreatic
cancer, multimodal treatment regimens are required that are aimed at multiple protein targets
rather than just a single gene product (209).
1.7.1 Current State of Clinical Trials
The present targeted therapies in phase III clinical trials of pancreatic cancer involve
molecular pathways such as Ras, the epidermal growth factor receptor (EGFR), vascular
endothelial growth factor (VEGF), gastrin, and matrix metalloproteinases (22). However,
despite encouraging results obtained from these in vitro or in animal model studies, the
outcomes of the clinical trials are not favourable due to various and differing technical
obstacles (22). Many of the trials are successful during phase I and phase II but fail to
produce favourable results during phase III (22). One commonality between these therapies
was the use of multiple agents for the treatment, with gemcitabine being the common agent
used as single agent or in combination with other therapeutics (22).
Amongst ten phase III clinical trials, only one showed a significantly high median survival
when compared to placebo or control treatments. This trial applied Erlotinib; an EGFR
tyrosin kinase inhibitor, in combination with gemcitabine (210). Erlotinib is a small

39
molecule compound that becomes active when orally consumed, binds to the ATP-binding
site of EGFR and inhibits its binding to its ligands and the subsequent activation. Although
the results of the study have demonstrated a significant enhancement in the survival of
patients with advanced pancreatic cancer, the cost effectiveness and the clinical relevance of
Erlotinib as a treatment option remains as a point of much debate (22, 210).
1.7.2 Ongoing Clinical Trials and the Future of Molecular Targeted Therapeutics
A variety of phase I and II clinical trials are currently underway for the treatment of
pancreatic cancer. Phase II trials in advanced or metatstatic pancreatic cancer include mTOR
inhibitors such as everolimus and sirolimus, NF-κB inhibitors such as curcumin with or
without gemcitabine, oral inhibitors of NF-κB-STAT3 such as RTA 402, antisense inhibitors
of TGFB receptors such as AP 12009, inhibitors of MET receptor tyrosine kinases such as
ARQ 197, multi-kinase inhibitors such as dasatinib, antibodies against human IGF-I receptor
such as cetuximab and MK-0646 in combination with gemcitabine and erlotinib, and
telomerase inhibitors (peptide vaccines) such as GV 1001 (22). GV 1001 is also being tested
in phase III trials in combination with capecitabine and gemcitabine in locally advanced
metastatic pancreatic cancer (22). A phase III trial of a COX-2 inhibitor, celecoxib, in
combination with gemcitabine and curcumin is also in progress (22).
Emerging clinical trials are focused on potential therapeutic targets such as embryonic
signaling pathways, signal transduction, telomerase, microRNAs, and cancer stem cells (22).
Examples of therapeutic signal transduction molecules include Focal adhesion kinase
(FADKs), expressed in 48% of pancreatic cancers, and the Src family of non-receptor protein
tyrosine kinases, expressed in 70% of pancreatic cancers (22). Embryonic signaling
pathways consist of the hedgehog pathway, with sonic hedgehog (SHH) being expressed in
70% of human pancreatic cancers, Notch pathway, with Notch 3 being expressed in 70% of
pancreatic cancer cell lines, and Wnt signaling pathway, with 65% aberrant activation in
pancreatic cancers (22).
Although treatment modalities of pancreatic cancer has undergone a major transformation
from conventional chemo- or radiation-therapy to combinational targeted therapeutics and
has resulted in encouraging in vivo or in vitro results, it is proven to be prone to failure in

40
clinical trials. Such failure might arises due to a lack in understanding of complete intricacies
of the physiology, biology, or cell signal transduction pathways involved in induction and
maintenance of pancreatic cancer. Other reasons might include a lack of optimized trials,
lack of target specific reagents, lack of proper tools to translate the mouse model pre-clinical
data into human conditions, and lack of complete knowledge of biomarkers that predict the
treatment responses (22). Suggested future approaches for personalized or targeted
therapeutics must therefore include a multimodal treatment comprising a combination of
conventional treatments and a mixture of targeted agents. These targeted agents will either
target multiple oncogenic pathways in parallel or will aim at different functional levels of a
major pathway (22).
In this thesis, we hypothesized that GSK-3 might be a potential target for the chemo-
sensitization of pancreatic cancer. We sought to determine whether treatment with a GSK-3
inhibitor in combination with gemcitabine or TRAIL has a synergistic death-inducing effect
on pancreatic cancer survival. We also explored the underlying mechanisms of this effect.
1.8 Thesis Overview
Chapter 2 describes the effects of GSK-3 inhibition by either small molecule inhibitors or
genetic knockdown on the survival of pancreatic cancer cells. The mechanism underlying the
anti-survival effects of GSK-3 inhibition in the context of NF-κB is studied and the chemo-
sensitization effect of this process is investigated.
In Chapter 3 I investigate the effects of GSK-3 inhibition in vitro on sensitization of both
pancreatic and prostate cancer cells to TRAIL effects. The mechanism underlying
sensitization is studied and paves the way for the pre-clinical studies of this effect.
Chapter 4 describes a pre-clinical in vivo model of TRAIL sensitization in pancreatic cancer
upon GSK-3 inhibition. Using a xenograft model of PANC-1 cells in SCID mice we studied
the possibility of enhancing apoptosis in established pancreatic cancer tumors by combining
GSK-3 inhibition and TRAIL treatment. We also investigated systemic toxicity of the
proposed treatment.

41
In Chapter 5 I review and discuss the results observed in Chapters 2-4 and summarize the
future proceedings and plans of the project.

Chapter 2 Glycogen Synthase Kinase-3 Inhibition Disrupts Nuclear Factor-kappaB Activity in Pancreatic Cancer, but Fails to Sensitize to Gemcitabine Chemotherapy
This Chapter is a modified version of a paper published in BMC Cancer (Shadi Mamaghani, Satish Patel, and David W. Hedley). The candidate was responsible for all of the experimental work.

43
Abstract Aberrant activation of NF-kappaB has been proposed as a mechanism of drug resistance in
pancreatic cancer. Recently, inhibition of glycogen synthase kinase-3 has been shown to
exert anti-tumor effects on pancreatic cancer cells by suppressing NF-kappaB.
Consequently, we investigated whether inhibition of GSK-3 sensitizes pancreatic cancer cells
to the chemotherapeutic agent gemcitabine. In this study, GSK-3 inhibition was achieved
using the pharmacological agent AR-A014418 or siRNA against GSK-3 alpha and beta
isoforms. Cytotoxicity was measured using a Sulphorhodamine B assay and clonogenic
survival following exposure of six different pancreatic cancer cell lines to a range of doses of
either gemcitabine, AR-A014418 or both for 24, 48 and 72 h. We measured protein
expression levels by immunoblotting. Basal and TNF-alpha induced activity of NF-kappaB
was assessed using a luciferase reporter assay in the presence or absence of GSK-3
inhibition.
GSK-3 inhibition reduced both basal and TNF-alpha induced NF-kappaB luciferase activity.
Knockdown of GSK-3 beta reduced nuclear factor kappa B luciferase activity to a greater
extent than GSK-3 alpha, and the greatest effect was seen with dual knockdown of both
GSK-3 isoforms. GSK-3 inhibition also resulted in reduction of the NF-kappaB target
proteins XIAP, Bcl-XL, and cyclin D1, associated with growth inhibition and decreased
clonogenic survival. In all cell lines, treatment with either AR-A014418, or gemcitabine led
to growth inhibition in a dose- and time-dependent manner. However, with the exception of
PANC-1 where drug synergy occurred with some dose schedules, the inhibitory effect of
combined drug treatment was additive, sub-additive, or even antagonistic.
GSK-3 inhibition has anticancer effects against pancreatic cancer cells with a range of
genetic backgrounds associated with disruption of NF-kappaB, but does not significantly
sensitize these cells to the standard chemotherapy agent gemcitabine. This lack of synergy
might be context or cell line dependent, but could also be explained on the basis that
although NF-kappaB is an important mediator of pancreatic cancer cell survival, it plays a
minor role in gemcitabine resistance. Further work is needed to understand the mechanisms
of this effect, including the potential for rational combination of GSK-3 inhibitors with other
targeted agents for the treatment of pancreatic cancer.

44
2.1 Background
Surgery is the only curative treatment for pancreatic cancer, but the majority of patients have
metastatic disease or an unresectable tumor at diagnosis (3, 4) . Due to the poor response to
chemo- and radiation therapies, the disease is highly lethal (4). Gemcitabine
(difluorodeoxycytidine) is the most active chemotherapy agent used for the treatment of
pancreatic cancer (7). It is an analog of deoxycytidine, that gets incorporated into double
stranded DNA during S phase, resulting in inhibition of DNA synthesis, arrest of cell cycle
progression, and induction of apoptosis (8). However, due to pre-existing or acquired
chemoresistance, gemcitabine treatment has a marginal survival benefit and yields an
objective tumor response rate of < 10% (9, 10).
Multiple lines of evidence suggest that aberrantly activated nuclear factor-kappa B (NF-κB)
plays a major role in metastasis, cell proliferation, angiogenesis, and chemotherapy resistance
of several tumor types including pancreatic cancer (49, 52, 179, 211, 212). Activated NF-κB
has been observed in pancreatic cancer cell lines and animal models of pancreatic cancer, as
well as primary human pancreatic cancers (49, 90, 213).
The NF-κB family of transcription factors [p65, p50, p52, RelB, and c-Rel] is involved in the
activation of a broad range of genes involved in inflammation, differentiation, tumorigenesis,
metastasis, embryonic development, and apoptosis (52, 213, 214). They are activated in
response to extracellular stimuli including inflammatory cytokines and growth factors, which
results in the phosphorylation and subsequent degradation of the NF-κB inhibitor IκB.
Additional levels of NF-κB regulation include phosphorylation of p65 at various sites,
although these are less well characterized. NF-κB target genes encode cytokines [IL-1, IL-
12, IL-2, IL-6, IL-8, IL-10, TNF-α, interferon-β], transcription factors [c-Myc], inhibitors of
apoptosis [Bcl-2, Bcl-XL, XIAP, FLIP], mitogenic factors [cyclin D1], and cell adhesion
molecules [E-selectin, ICAM-1, VCAM-1] (215-217). Previous in vitro studies have shown
that inhibition of NF-κB using IκBα super-repressor or sulfasalizine enhances the effect of
chemotherapeutic agents in pancreatic cancer cell lines (89, 218). Furthermore, inhibition of
NF-κB by the natural compound curcumin was reported to potentiate the antitumor activity
of gemcitabine in an orthotopic xenograft model of pancreatic cancer (219). Together, these

45
findings suggest that aberrant activation of NF-κB leads to chemo-resistance in pancreatic
cancer, and that inhibition of NF-κB sensitizes the treatment outcome.
Glycogen synthase kinase-3 (GSK-3) is a constitutively active serine-threonine kinase that
can phosphorylate and inactivate a broad range of substrates including glycogen synthase,
cyclin D1, Mcl-1, c-myc, c-jun, β-catenin, tau, notch, and HIF-1 (101). Mammalian GSK-3
exists as two isoforms, α and β, with semi-redundant actions that are ubiquitously expressed
in tissues (101, 161). In vivo and in vitro studies have shown that GSK-3 can phosphorylate
and regulate NF-κB in a dual mode. The p65 subunit of NF-κB has been reported to be
phosphorylated by GSK-3 at serine 468 resulting in its decreased activity (71). Nonetheless,
mice engineered to lack both GSK-3β alleles are sensitive to TNF-α and die in late gestation
due to massive liver apoptosis; a phenotype similar to mice lacking the p65 subunit of NF-κB
or IKKβ (74, 220). Hepatocytes pretreated with a GSK-3 inhibitor LiCl, were also shown to
have lower NF-κB activity, as measured by a NF-κB dependent luciferase assay.
Furthermore, mouse embryonic fibroblasts (MEFs) deficient in both alleles of GSK-3β fail to
activate NF-κB after treatment with TNF-α, when compared to wild type MEFs (73).
Pharmacological or siRNA mediated inhibition of GSK-3β has been shown to reduce NF-κB
mediated gene transcription and inhibit the growth of cancers that show high NF-κB activity
including pancreatic cancer (179, 221, 222). These results point to a possible role for GSK-3
in the maintenance of high NF-κB activity in cancer cells. Since aberrant NF-κB activation
has been linked to drug resistance in pancreatic cancer, we tested the hypothesis that
reduction of NF-κB activity through GSK-3 inhibition sensitizes pancreatic cancer cells to
chemotherapy.
2.2 Materials and Methods
2.2.1 Reagents and Antibodies
Curcumin (Diferulylmethane, 80% pure; 98% curcuminoid content), was obtained from
Sigma-Aldrich Canada Ltd. (Oakville, Ontario, Canada), and GSK-3 Inhibitor VIII [AR-
A014418 (AR-18)] was obtained from CALBIOCHEM®, EMD Biosciences, Inc. (San
Diego, CA). Both agents were dissolved in DMSO and aliquots stored at -20º C.

46
Gemcitabine from Eli Lilly (Indianapolis, IN) was freshly prepared as 10 mM stock in sterile
PBS on the day of use.
Rabbit polyclonal antibodies against XIAP, β-catenin, and Bcl-XL were purchased from Cell
Signaling Technology (Danvers, MA). Rabbit monoclonal cyclin D1 antibody was obtained
from Lab Vision Corp. (Fremont, CA). A mouse monoclonal antibody against GSK-3 α/β
was obtained from Biosource Inc. (Camarillo, CA). Anti-rabbit and anti-mouse horseradish
peroxidase linked IgG antibodies, were from Amersham Biosciences (Buckinghamshire,
United Kingdom). Recombinant Human TNF-α/TNFSF1A was purchased from R&D
Systems (Minneapolis, MN)
2.2.2 Cell Lines and Media
The pancreatic cancer cell lines BxPC-3, MIA PaCa-2, PANC-1, and HPAC were obtained
from the American Type Culture Collection (Rockville, MD), and PK-1 and PK-8 were from
Dr. Masao Kobari (Sendai, Japan). BxPC-3, PK-1, and PK-8 cell lines were cultured in
RPMI 1640. PANC-1 and MIA PaCa-2 cell lines were cultured in Dulbecco Eagles medium.
HPAC cells were cultured in HAM F-12. All the media for cell culture were supplemented
with 10% fetal bovine serum (FBS), 100 units/mL penicillin and 100 µg/mL streptomycin,
and cells were grown at 37◦ C and 5% CO2 in air. Additional 2.5% horse serum was added to
the media growing MIA PaCa-2 cells.
2.2.3 Cell Treatments, Lysate Preparation, and Immunoblotting
Cells grown at 60% to 70% confluency were exposed to different doses of AR-18 (0-50
µM), or lithium chloride (LiCl) (0-50 mM), potassium chloride (KCl) (10 mM), or solvent
control, and incubated at 37◦ C in a CO2 incubator. After 48 h, the cells were lysed using
RIPA buffer [20 mM Tris (pH 7.5), 150 mM NaCl, 1% NP-40, 0.5% Na deoxycholate, 0.1%
SDS, and 1 mM EDTA supplemented with 1 mM Na3VO4, protease inhibitor cocktail (Roche
Diagnostics) and a serine/threonine-phosphatase inhibitor cocktail 1 (Sigma-Aldrich).
Alternatively, drug treated cells were lysed and fractionated to separate the cytoplasmic
content using hypotonic lysis buffer [50 mM Tris (pH 7.4), 1 mM EDTA, 10 mM NaF, 1
mM Na3VO4 and supplemented with protease inhibitor cocktail (Roche Diagnostics) and
serine/threonine-phosphatase inhibitor cocktail 1 (Sigma-Aldrich)]. The protein content of

47
the supernatants was measured using bicinchoninic acid protein assay from Pierce PerBio
(Rockford, IL) and twenty-five micrograms of the lysates were resolved on 8% or 10% SDS-
PAGE gels. The resolved proteins were transferred onto polyvinylidene difluoride
membranes (Millipore, Bedford, MA), blocked with 5% non-fat milk, and probed with the
appropriate antibodies according to the manufacturer’s recommendation. The blots were
washed, and exposed to the appropriate HRP-conjugated secondary antibodies for 1 h at
room temperature. Detection was done using SuperSignal® West Pico from Pierce BioLynx
Inc. (Brockville, Ontario, Canada) reagent or enhanced chemiluminescence plus (ECL Plus)
kit (Amersham Biosciences). Cytoplasmic lysates were used for detection of β-catenin,
whereas the rest of the proteins were detected using the RIPA lysates. Blotting for α-tubulin
from Oncogene Research Products, Calbiochem, (San Diego, CA) or β-actin from Abcam
Antibodis, Inc., (Cambridge, MA) were used to control for protein loading.
2.2.4 Proliferation Assay
The effect of AR-18, gemcitabine, and curcumin on cell proliferation was determined by the
Sulphorhodamine B (SRB) dye (Molecular Probes, Eugene, OR) binding assay as described
previously (223). Briefly, 5,000 cells per well were seeded in 96-well plates, incubated in a
CO2 incubator overnight at 37º C, and then treated with different doses of curcumin (0-50
µM), AR-18 (0-50 µM) or gemcitabine (0-1 µM) alone or in combination (i.e. concurrent or
sequential) in triplicates for 24, 48, and 72 h. The cells were then fixed using 10% (v/v)
trichloroacetic acid for 1 h at 4º C, washed extensively with water, stained with 0.4% SRB
dissolved in 1% (v/v) acetic acid in water reagent for 30 minutes at room temperature, and
then washed, and 10 mM unbuffered Tris was added to each well. The absorbance was
measured at 570 nm using a Multiscan 96-well plate reader from Thermo Electron Corp.
(Milford, MA). This experiment was repeated three times in six replicates.
2.2.5 Transient Transfection and Luciferase Assay
PANC-1, MIA PaCa-2, PK-1, and PK-8 cells were seeded in 12-well plates (130,000 per
well) in antibiotic-free medium containing 10% FBS. The cells were incubated in a CO2
incubator overnight at 37º C prior to transfection using Lipofectamine 2000 from Invitrogen
Life Technologies, (Carlsbad, CA) as recommended by the manufactures. Briefly,
0.5µg/well TA-LUC NF-κB (from Dr. T. Pawson, Samuel Lunenfeld Research Institute,

48
University of Toronto), and 0.05 µg/well β-gal CMV (from Dr. W.C. Yeh, Ontario Cancer
Institute, University of Toronto) were co-transfected to the cells. After 16 h, the medium was
changed and the cells were incubated with AR-18 (50 µM), gemcitabine (10 µM), or
curcumin (50 µM) alone or in combination for 8 h. TNF-α (30 ng/mL) was added to the cells
4 h prior to cell lysis. Control cells were transfected with the plasmids, but did not receive
any drug treatments. Luciferase activity was measured by using the Dual-Light® System
luciferase assay from Applied Biosystems (Bedford, MA) according to the manufacturer’s
protocol. The luminometer used was Luminoskan Ascent from ThermoLab Systems
(Franklin, MA). The results were normalized to the values read for β-galactosidase activity.
All experiments were performed in triplicate and were repeated four times.
2.2.6 Genetic Knockdown of GSK-3
PANC-1 cells were transfected using a reverse transfection protocol that involves
transfection of the cells right after trypsinizing and subculturing. Briefly, the cells were
seeded at 300,000 cells per well in 6-well plates, then placed in a CO2 incubator at 37º C for
1h prior to transfection with either silencer negative control siRNA or anti-GSK-3β from
Applied Biosystems, Ambion Inc. (Bedford, MA), or anti-GSK-3α [Hs_GSK3A_5_HO
Validated] from Qiagen, Inc. (Mississauga, Ontario, Canada) or both by using Hiperfect
transfection reagent from Qiagen Inc. according to the manufacturer’s protocol. After 72 h,
the cells were lysed using RIPA or hypotonic lysis buffers and the proteins present in cell
lysates were resolved in SDS-PAGE. Preliminary experiments showed that the
concentrations of siRNA required achieving >80% knockdown of GSK3α and GSK3β were
10 nM and 80 nM, respectively. These concentrations were used in all the studies.
To combine the luciferase assay with genetic knockdown of GSK-3, 24 h after siRNA
transfection, the medium was changed and the cells were subjected to co-transfection with
TA-LUC NF-κB and β-gal CMV as previously described. After 24 h exposure, the medium
was changed and the cells were incubated for 48 h prior to exposure to either TNF-α (30
ng/mL, 4 h) or gemcitabine (10 µM, 8 h). Subsequently, the cells were lysed and the whole
cell lysates were used for luciferase assay as described above.

49
2.2.7 Clonogenic Assay
The effect of AR-18 and of gemcitabine on survival of PANC-1 and BxPC-3 cells was
further investigated by a colony-forming assay as described by Wu et al. (224). In brief,
exponentially growing cells were treated with either gemcitabine (0.001-10 µM), AR-18 (10-
50 µM), or both for 24 h. The cells were then trypsinized and washed twice with PBS to
remove the remaining drug, counted, and then seeded in 10X and 5X serial dilutions for
PANC-1 and BxPC-3 cells respectively. The plates were incubated for 16 days at 37◦ C in a
CO2 incubator at 90% humidity. The plates were then stained with methylene blue from
Fischer Scientific, (Ottawa, ON) and colonies were counted. The experiments were
performed in triplicates, and at least three times for each cell line.
2.2.8 Statistical Analysis
All the statistical analysis was performed by the help of “R” software (Hornik et al.;
http://www.r-project.org). To investigate the possible synergistic effect of combining two
agents, the interaction between the two drug treatments was tested by fitting it into a model
that considers the fact that some experiments were not performed at the same time. The
values of optical density (for SRB), colony count (clonogenic assay), or luciferase unit
(luciferase assay) were log transformed to stabilize the variance of the residuals. The
resulting values were analysed by comparing between different concentrations of each drug
using linear regression models. A drug interaction was considered synergistic when the
effect of the drug combination was significantly greater than the sum of the effects of both
drugs, and sub-additive when it was less than that.
2.3 Results
2.3.1 Proliferation and Colony-Forming Capacity of Pancreatic Cancer Cells is Decreased after Pharmacological Inhibition of GSK-3
Consistent with previous reports (221), treatment of PANC-1 and BxPC-3 cells with the
GSK-3 inhibitor AR-18 caused a growth inhibitory effect in a dose- and time-dependent
manner. Depending on the duration of exposure, the IC50 values ranged from as low as 20
µM to as high as 65µM. After 48 h exposure, the (IC50) of AR-18 was approximately 30 µΜ
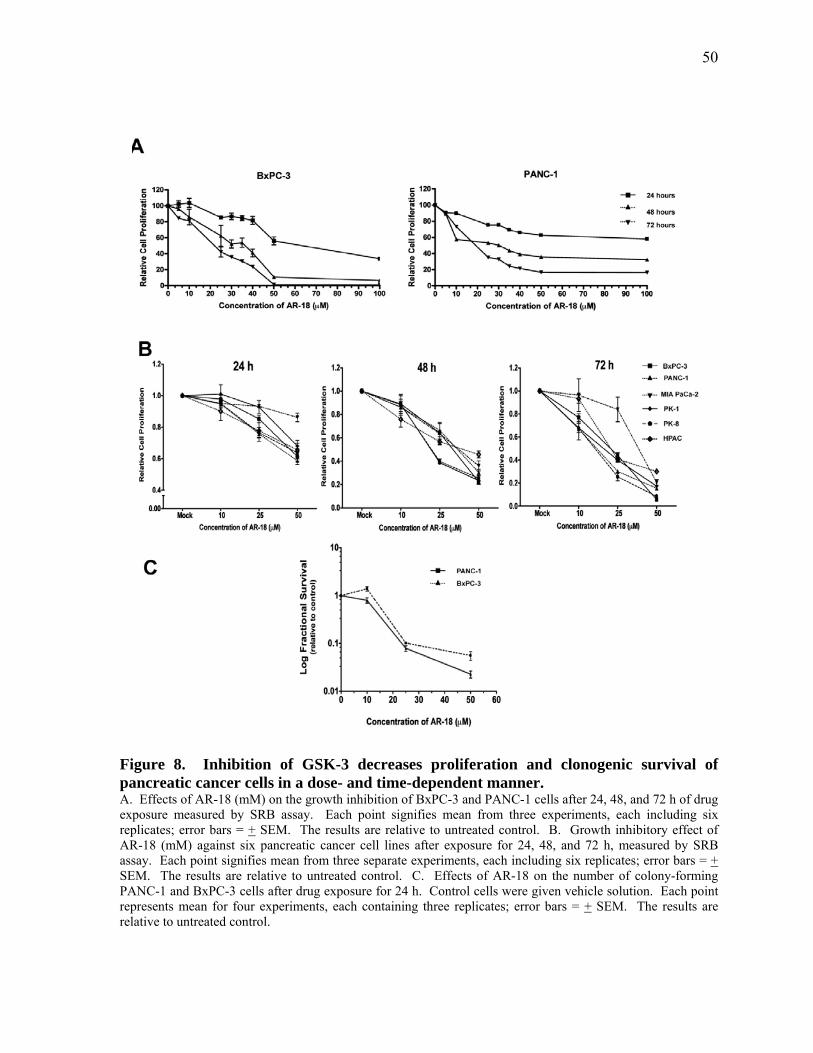
50
Figure 8. Inhibition of GSK-3 decreases proliferation and clonogenic survival of pancreatic cancer cells in a dose- and time-dependent manner. A. Effects of AR-18 (mM) on the growth inhibition of BxPC-3 and PANC-1 cells after 24, 48, and 72 h of drug exposure measured by SRB assay. Each point signifies mean from three experiments, each including six replicates; error bars = + SEM. The results are relative to untreated control. B. Growth inhibitory effect of AR-18 (mM) against six pancreatic cancer cell lines after exposure for 24, 48, and 72 h, measured by SRB assay. Each point signifies mean from three separate experiments, each including six replicates; error bars = + SEM. The results are relative to untreated control. C. Effects of AR-18 on the number of colony-forming PANC-1 and BxPC-3 cells after drug exposure for 24 h. Control cells were given vehicle solution. Each point represents mean for four experiments, each containing three replicates; error bars = + SEM. The results are relative to untreated control.

51
for both cell lines (Figure 8A). A range of AR-18 doses below and above this range was
used for all our experiments, which is in line with previous reports in pancreatic cancer cells
(92). We next tested AR-18 sensitivity against a panel of four additional pancreatic cancer
cell lines. As shown in Figure 8, AR-18 potently reduced cell proliferation of all six
pancreatic cancer cell lines tested in a dose- and time-dependent manner.
In order to determine whether GSK-3 is required for clonogenic survival of pancreatic cancer
cells, exponentially growing PANC-1, and BxPC-3 cells were exposed to varying doses of
AR-18 (10-50 µM) for 24 h. The number of colony-forming cells was reduced in a
concentration-dependent manner by AR-18 (Figure 8C), and at 50 µM AR-18 the number of
colony-forming PANC-1 and BxPC-3 cells were 0.055 + 0.02 and 0.022 + 0.006,
respectively when compared with untreated controls.
2.3.2 GSK-3 Mediates NF-κB Activation in Pancreatic Cancer Cells
Recent evidence suggests that GSK-3 is a positive regulator of NF-κB (72, 73, 92, 164). To
test this, we first treated PANC-1 and BxPC-3 cells with increasing concentrations of AR-18
for 48 h and examined effects on cytoplasmic β-catenin, which is negatively regulated by
GSK-3 via the Wnt pathway. Inhibition of GSK-3 with increasing doses of AR-18 resulted
in a dose-dependent increase in the levels of cytoplasmic β-catenin with ~twofold increase at
50µM AR-18, when compared to control, which is the expected pharmacodynamic effect
(Figure 9A). We next examined the effects of AR-18 treatment on the expression of the NF-
κB target genes XIAP, cyclin D1, and Bcl-XL, and found that expression of these proteins
was also reduced significantly in a dose-dependent manner (Figure 9 A-B). Similar results
were obtained using the unrelated GSK3 inhibitor, LiCl (Additional File 1 in Appendix).
To test if GSK-3 inhibition could impact basal NF-κB activity in pancreatic cancer cells,
PANC-1, MIA PaCa-2, PK-1 and PK-8 cells were transfected with TA-LUC NF-κB and
treated with AR-18. In all cell lines AR-18 treatment (50 µΜ, 8 h) significantly decreased
basal NF-κB activity when compared to untreated control (Figure 9C, and data not shown).
Since TNF-α induced NF-κB activity was reported to be inhibited in MEFs genetically
lacking the GSK-3β isoform (73), we tested this by treating PANC-1 and MIA PaCa-2 cells

52
Figure 9. Inhibition of GSK-3 disrupts NF-κB activity in pancreatic cancer cells in a dose-dependent manner. A-B. Western blot analysis of expression of β-catenin and NF-κB target genes: XIAP, BcL-XL, and cyclin D1, in PANC-1 and BxPC-3 cell lines after exposure to AR-18 for 48 h. The change in the expression level of the proteins is compared against untreated or vehicle treated controls. Increase in cytosolic β-catenin level indicates GSK-3 inhibition. Both α-tubulin and β-actin were used as loading controls. C. Effect of GSK-3 disruption on basal and TNF-α induced NF-κB activity measured by luciferase reporter assay. PANC-1 and MIA PaCa-2 cells were exposed to AR-18 (50 mM, 8 h), TNF-α (30 ng/mL, 4 h), or both after co-transfection with TA-LUC NF-κB reporter and b-gal (internal control) constructs. The normalized values are relative to the untreated control (indicating basal level of NF-κB activity) which is represented by dotted line. Each column represents mean for at least four separate experiments, each with three replicates; error bars = + SEM. (*) significant: (p<0.0003) when compared to untreated control. (**) significant: (p<0.0001) when compared to TNF-α treatment.

53
with TNF-α (30 ng/ml, 4 h) in the presence or absence of AR-18. In both cell lines, TNF-α
induced NF-κB luciferase activity above background by ~2.5-fold in control cells, whereas in
cells pretreated with AR-18 the levels of NF-κB luciferase remained lower than baseline, and
were not significantly different from those seen with AR-18 alone (Figure 9C). Together,
these findings support the idea that GSK-3 positively regulates basal NF-κB activity (92) and
that inhibition of GSK-3 abrogates the activation of NF-κB by TNF-α.
2.3.3 Genetic Knockdown of GSK-3 Abolishes NF-κB Activity in Pancreatic Cancer Cells
Previous work suggests that inhibitors such as LiCl and AR-18 likely do not distinguish
between the two GSK-3 isoforms (225). To determine the effect of GSK-3 isoforms on NF-
κB target gene expression in pancreatic cancer cells, we genetically depleted the expression
of GSK-3α and GSK3β, alone or in combination, in PANC-1 cells using RNA interference.
Following a 3-day exposure to GSK-3 specific siRNAs, immunoblotting showed >80%
reduction in the expression levels of the corresponding GSK-3 isoforms when compared to
untransfected or scrambled siRNA transfected controls (Figure 10A). Depletion of either
GSK-3α or β isoforms had minor effects on expression levels of Bcl-XL, XIAP, cyclin-D1,
and β-catenin, with a greater effect shown by GSK-3β knockdown. However, consistent
with pharmacological inhibition of GSK-3 using AR-18, simultaneous knockdown of both
GSK-3 isoforms in PANC-1 cells led to a significantly greater effect on β-catenin, Bcl-XL,
XIAP, and cyclin-D1 expression levels when compared to the single isoform knockdowns
(Figure 10A).
To further test the effect of GSK-3 isoforms knockdown on basal NF-κB activity, we
measured the level of NF-κB luciferase activity in knockdowns of PANC-1 cells. Inhibition
of GSK-3α, β, or double knockdown of both GSK-3 isoforms significantly decreased the
basal NF-κB activity (Figure 10B); with greater effect exerted by genetic depletion of GSK-
3β and the double knockdown (Figure 10B). While TNF-α treatment induced >2.5 fold
increase in non-specific (scrambled) siRNA treated cells, knockdown of either GSK-3
isoform resulted in a significant decrease in basal NF-κB luciferase activity and attenuated
the effect of TNF-α, although these effects were greater with GSK3β knockdown. A large
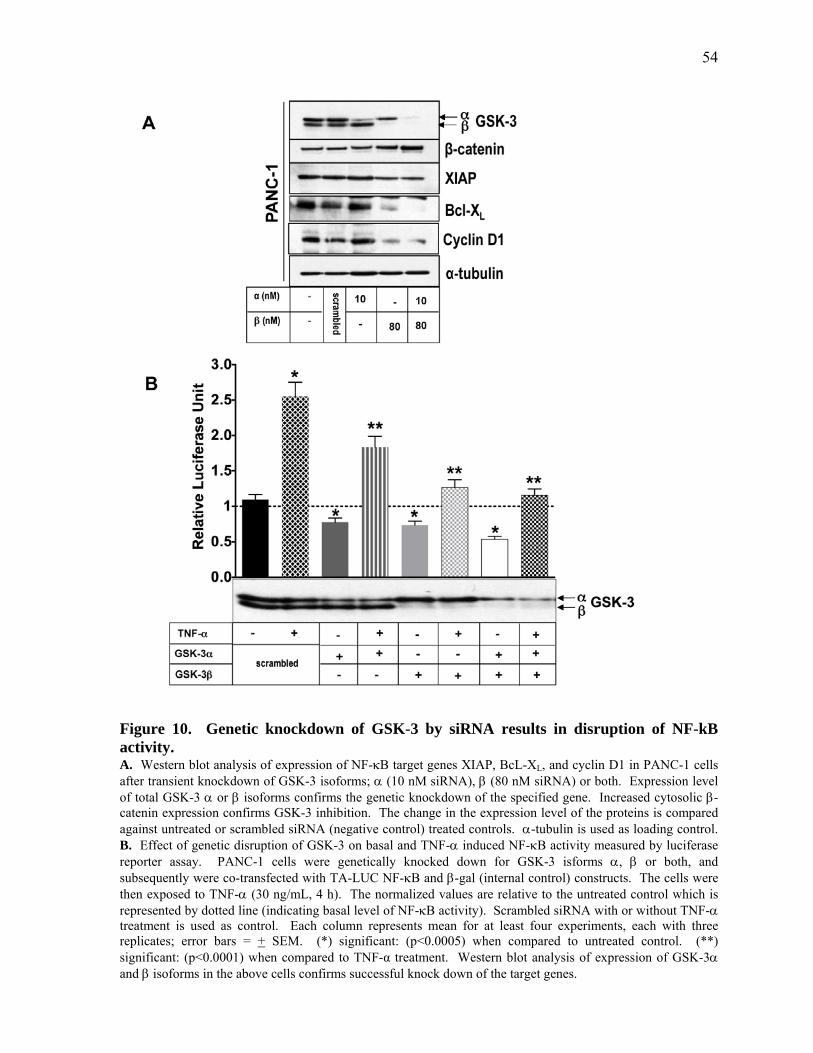
54
Figure 10. Genetic knockdown of GSK-3 by siRNA results in disruption of NF-kB activity. A. Western blot analysis of expression of NF-κB target genes XIAP, BcL-XL, and cyclin D1 in PANC-1 cells after transient knockdown of GSK-3 isoforms; α (10 nM siRNA), β (80 nM siRNA) or both. Expression level of total GSK-3 α or β isoforms confirms the genetic knockdown of the specified gene. Increased cytosolic β-catenin expression confirms GSK-3 inhibition. The change in the expression level of the proteins is compared against untreated or scrambled siRNA (negative control) treated controls. α-tubulin is used as loading control. B. Effect of genetic disruption of GSK-3 on basal and TNF-α induced NF-κB activity measured by luciferase reporter assay. PANC-1 cells were genetically knocked down for GSK-3 isforms α, β or both, and subsequently were co-transfected with TA-LUC NF-κB and β-gal (internal control) constructs. The cells were then exposed to TNF-α (30 ng/mL, 4 h). The normalized values are relative to the untreated control which is represented by dotted line (indicating basal level of NF-κB activity). Scrambled siRNA with or without TNF-α treatment is used as control. Each column represents mean for at least four experiments, each with three replicates; error bars = + SEM. (*) significant: (p<0.0005) when compared to untreated control. (**) significant: (p<0.0001) when compared to TNF-α treatment. Western blot analysis of expression of GSK-3α and β isoforms in the above cells confirms successful knock down of the target genes.

55
effect was seen when both isoforms were knocked down (Figure 10B), suggesting that
whereas GSK3α is able to stimulate NF-κB activity, this is mediated principally by GSK-3β.
2.3.4 GSK-3 Inhibition does not Enhance the Anti-Tumor Effects of Gemcitabine in Pancreatic Cancer In vitro
Using the SRB cell proliferation assay, the growth of BxPC-3 and MIA PaCa-2 cell lines was
measured after 24, 48, and 72 h of exposure to a range of concentrations of either AR-18,
gemcitabine, or a concurrent combination of both drugs; using either a fixed ratio of 200:1
AR-18 to gemcitabine, or variable doses of both drugs. AR-18 produced a steep dose-
response over the 10-50 µM concentration range and this effect increased with the duration
of exposure (Figure 11; 24 and 72 h data not shown). In contrast, the gemcitabine dose-
response showed a plateau at low concentrations, and sensitivity was greatly influenced by
the duration of drug exposure, consistent with the cell cycle phase-specificity of this agent.
Contrary to our hypothesis, combining both drugs either in a fixed ratio or variable doses was
not synergistic against BxPC-3 or MIA PaCa-2 cells when compared to the single agents,
across a wide range of concentrations and treatment times (Figure 11A; 24 and 72 h data not
shown). We also treated the four other pancreatic cancer cell lines using variable doses of
both drugs for different time points. As seen in Figure 11B, with the exception of PANC-1
that showed a statistically-significant synergistic effect at some dose levels (Figure 11B; 24
and 72 h data not shown), the drug combination was either sub-additive or even antagonistic.
Because of the possibility that AR-18 might be antagonizing the effects of gemcitabine by
reducing movement through S-phase, we also tested if prior exposure to gemcitabine
sensitized to AR-18 but did not identify positive drug interaction under any of the conditions
used.
To further investigate the interactions of AR-18 and gemcitabine, we tested the effect on the
colony-forming capacity of PANC-1 and BxPC-3 cell lines. The cells were exposed to doses
of AR-18, gemcitabine or their combination similar to those used for the SRB assay. No
evidence of drug synergy was observed across a wide range of drug concentrations (Figure
11C).

56
Figure 11. Effects of AR-18 on gemcitabine sensitivity. A. Growth inhibitory effect of AR-18 (2.5-50 µM), gemcitabine (0.05 –1.0 µM), and their combination in a 200:1 AR-18 to Gemcitabine ratio was measured by the SRB proliferation assay in MIA PaCa-2 and BxPC-3 after 48 h of exposure. Each point represents mean from three experiments, each with six replicates; error bars = + SEM. Gem: gemcitabine. The results are indicated by relative cell proliferation as a percentage of solvent control. B. Growth inhibitory effect of AR-18 (10-50 µM), gemcitabine (0.001-1.0 µM), and their combination was measured by the SRB proliferation assay in PANC-1, HPAC, PK-1, and PK-8 cell lines after 48 h of exposure. Each point represents mean from three separate experiments, each with six replicates; error bars = + SEM. Gem: gemcitabine. The results are indicated by relative cell proliferation as a percentage of solvent control. C. Effect of AR-18 (10-50 µM), gemcitabine (0.001-1.0 µM), and their combination on colony-forming capacity of PANC-1 and BxPC-3 cells was measured by colonogenic assay. Control cells were given vehicle solution. Means for four separate experiments, each with three replicates; error bars = + SEM. Gem: gemcitabine. The results are relative to vehicle treated control.

57
Since treatment with gemcitabine was reported to cause NF-κB activation in pancreatic
cancer cells in vitro (49), we tested if this effect is sensitive to GSK-3 inhibition.
Consequently, TA-LUC NF-κB transfected PANC-1, MIA PaCa-2, PK-1, and PK-8 cells
were exposed to gemcitabine (10 µM), AR-18 (50 µM), or both for 8 h and NF-кB activity
was examined. We found a moderate increase in NF-κB activity effect in PANC-1 cells that
appeared to be dependent on the experimental conditions (Figure 12A and Figure 12B). No
significant increase was seen in MIA PaCa-2, PK1, and PK-8 cells (Figure 12A, and data not
shown). Although AR-18 significantly reduced basal NF-κB activity in all the cell lines, the
combination of gemcitabine and AR-18 produced similar effects on the NF-κB reporter to
those seen with single agent AR-18 (Figure 12A, and data not shown). Furthermore, when
we combined gemcitabine with transient knockdown of GSK-3 isoforms in PANC-1 cells,
there was no increase in NF-κB activity (Figure 12B).
2.4.5 Similar to AR-18, Curcumin Inhibits NF-κB Activity, but Fails to
Sensitize Pancreatic Cancer Cells to Gemcitabine Effect In vitro
Since GSK-3 could have both pro- and anti-apoptosis effects, we considered that the lack of
sensitization to gemcitabine using AR-18 might be explained by the effect of GSK-3 on
targets other than NF-κB that could potentially modify chemotherapy sensitivity. To address
this, we compared the effects using curcumin, which inhibits NF-κB through different
mechanisms. Similar to previous reports and consistent with our observations using AR-18,
both PANC-1 and MIA PaCa-2 cells showed a significant decrease in basal as well as TNF-α
induced NF-κB activity after exposure to curcumin (50 µM) for 8 h (Figure 13A). We then
tested for synergism by exposing PANC-1 and MIA PaCa-2 cells to various doses of
curcumin, gemcitabine, or their combination in doses similar to those used by Kunnumakkara
et al. (219). Consistent with their findings, 48 h exposure to curcumin had a significant
growth inhibitory effect on these cell lines measured by SRB assay (Figure 13B). However,
as seen in Figure 13B and similar to our results using AR-18, NF-κB inhibition by curcumin
did not sensitize the pancreatic cancer cells to gemcitabine. Likewise, the effect of curcumin
down-regulating NF-κB luciferase activity was not significantly altered by combined
treatment with gemcitabine (Figure 13C).
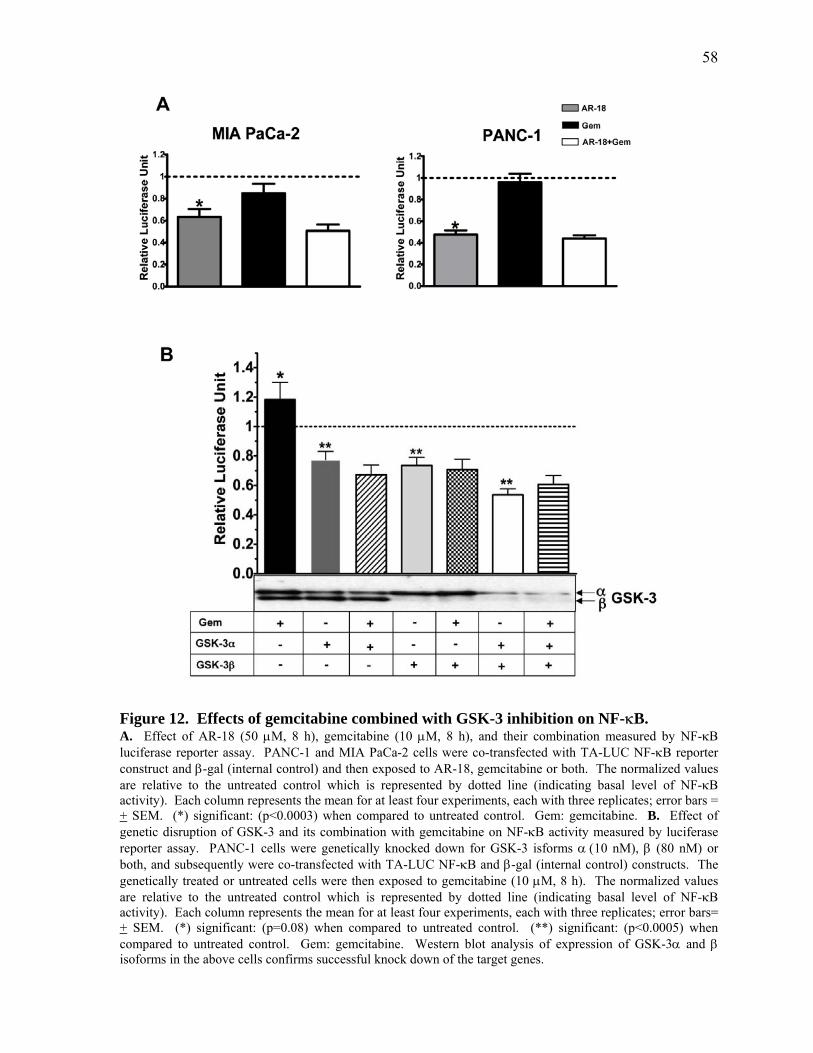
58
Figure 12. Effects of gemcitabine combined with GSK-3 inhibition on NF-κB. A. Effect of AR-18 (50 µM, 8 h), gemcitabine (10 µM, 8 h), and their combination measured by NF-κB luciferase reporter assay. PANC-1 and MIA PaCa-2 cells were co-transfected with TA-LUC NF-κB reporter construct and β-gal (internal control) and then exposed to AR-18, gemcitabine or both. The normalized values are relative to the untreated control which is represented by dotted line (indicating basal level of NF-κB activity). Each column represents the mean for at least four experiments, each with three replicates; error bars = + SEM. (*) significant: (p<0.0003) when compared to untreated control. Gem: gemcitabine. B. Effect of genetic disruption of GSK-3 and its combination with gemcitabine on NF-κB activity measured by luciferase reporter assay. PANC-1 cells were genetically knocked down for GSK-3 isforms α (10 nM), β (80 nM) or both, and subsequently were co-transfected with TA-LUC NF-κB and β-gal (internal control) constructs. The genetically treated or untreated cells were then exposed to gemcitabine (10 µM, 8 h). The normalized values are relative to the untreated control which is represented by dotted line (indicating basal level of NF-κB activity). Each column represents the mean for at least four experiments, each with three replicates; error bars= + SEM. (*) significant: (p=0.08) when compared to untreated control. (**) significant: (p<0.0005) when compared to untreated control. Gem: gemcitabine. Western blot analysis of expression of GSK-3α and β isoforms in the above cells confirms successful knock down of the target genes.
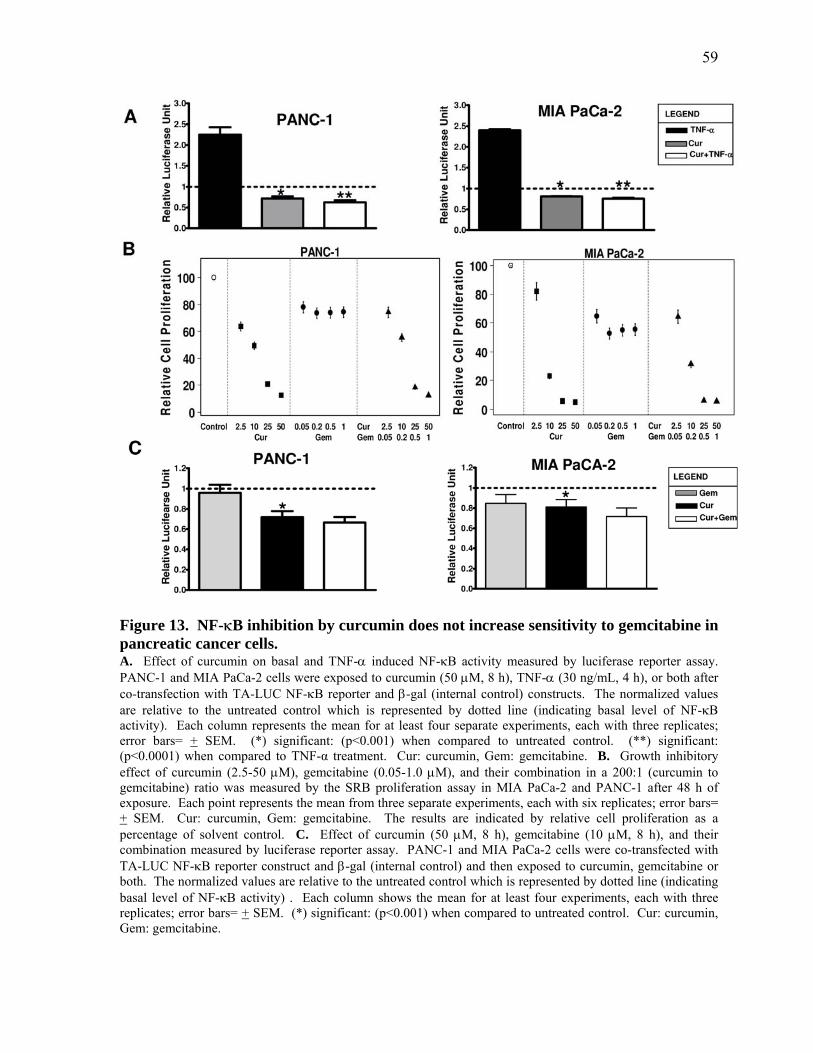
59
Figure 13. NF-κB inhibition by curcumin does not increase sensitivity to gemcitabine in pancreatic cancer cells. A. Effect of curcumin on basal and TNF-α induced NF-κB activity measured by luciferase reporter assay. PANC-1 and MIA PaCa-2 cells were exposed to curcumin (50 µM, 8 h), TNF-α (30 ng/mL, 4 h), or both after co-transfection with TA-LUC NF-κB reporter and β-gal (internal control) constructs. The normalized values are relative to the untreated control which is represented by dotted line (indicating basal level of NF-κB activity). Each column represents the mean for at least four separate experiments, each with three replicates; error bars= + SEM. (*) significant: (p<0.001) when compared to untreated control. (**) significant: (p<0.0001) when compared to TNF-α treatment. Cur: curcumin, Gem: gemcitabine. B. Growth inhibitory effect of curcumin (2.5-50 µM), gemcitabine (0.05-1.0 µM), and their combination in a 200:1 (curcumin to gemcitabine) ratio was measured by the SRB proliferation assay in MIA PaCa-2 and PANC-1 after 48 h of exposure. Each point represents the mean from three separate experiments, each with six replicates; error bars= + SEM. Cur: curcumin, Gem: gemcitabine. The results are indicated by relative cell proliferation as a percentage of solvent control. C. Effect of curcumin (50 µM, 8 h), gemcitabine (10 µM, 8 h), and their combination measured by luciferase reporter assay. PANC-1 and MIA PaCa-2 cells were co-transfected with TA-LUC NF-κB reporter construct and β-gal (internal control) and then exposed to curcumin, gemcitabine or both. The normalized values are relative to the untreated control which is represented by dotted line (indicating basal level of NF-κB activity) . Each column shows the mean for at least four experiments, each with three replicates; error bars= + SEM. (*) significant: (p<0.001) when compared to untreated control. Cur: curcumin, Gem: gemcitabine.

60
2.4 Discussion
Chemotherapy resistance of pancreatic cancer has been previously associated with
hyperactivity of NF-κB (49, 51, 52, 89). The discovery that GSK-3 regulates NF-κB (73),
and that its inhibition has anti-inflammatory and growth inhibitory effects, holds promise to
resolve the problem of drug resistance in cancers with inflammatory origin including
pancreatic cancer (73, 92, 226). In this paper, using a panel of six genetically distinct
pancreatic cancer cell lines we confirmed previous reports that pharmacological inhibition of
GSK-3 suppresses NF-κB transcriptional activity and is toxic to pancreatic cancer cells in a
dose- and time-dependent manner (92). We also show for the first time that GSK-3
inhibition potently reduces the clonogenic survival of pancreatic cancer cells. However,
contrary to our hypothesis GSK-3/NF-κB inhibition did not sensitize to gemcitabine
chemotherapy.
GSK-3 is a kinase involved in many cellular processes including energy metabolism,
transcriptional regulation, cell adhesion, and protein turnover (93, 169). This complexity of
action results in a potential for GSK-3 to exert both pro- and anti-apoptotic effects that
appears to be cell- and context-dependent (28). The anti-apoptotic activity of GSK-3 has
been attributed in part to the stimulation of NF-κB activity through an unknown mechanism,
as shown by this study and others (72, 73, 92). It has been previously shown that β-catenin
has inhibitory effects on NF-κB (227), which could explain the effects of GSK-3 inhibition
since this results in the accumulation of β-catenin. Although the accumulation of β-catenin
could potentially be cancer-promoting, this did not rescue pancreatic cells from death due to
lack of NF-κB activity, further supporting the importance of NF-κB activity in maintaining
the survival of these cells, whereas Wnt/β-catenin appears to play a less prominent role in
pancreatic cancer development (226). The above findings in conjunction with others (73, 92,
228) lend support to a positive role for GSK-3 activity in the regulation of NF-κB, rather
than inhibition through S468 phosphorylation as has been described in other systems (23).
Since GSK-3 inhibitors including Wnt, LiCl, and AR-18 likely do not discriminate between
GSK-3 α and β isoforms (73, 96), and given that functional redundancy of GSK-3 isoforms
in the context of Wnt/β-catenin signaling has been previously described in mouse embryonic

61
stem cells (161), we investigated whether NF-κB regulation by GSK-3 was isoform-specific
in pancreatic cancer cells. Transient genetic knockdown of GSK-3α had a minor impact on
β-catenin, cyclin D1 and XIAP expression when compared to GSK-3β knockdown, whereas
GSK-3α/β double knockdown demonstrated the greatest effect. Similarly, transient
knockdown of either GSK-3α or GSK-3β significantly reduced both the basal as well as the
TNF-α induced NF-κB activity of PANC-1 cells, although GSK-3β knockdown exerted the
greater effect and the double knockdown of both isoforms was the most effective.
Collectively, these findings suggest that although NF-κB activity in pancreatic cancer is
responsive to both GSK-3 isoforms, GSK-3β is the major regulator. Our finding of the
differential effects of GSK-3 isoforms is in agreement with the previously proposed
functional redundancy of GSK-3 isoforms (161), and also confirms the importance of GSK-
3β in NF-κB regulation (73, 222). Furthermore, our observations also raise the possibility of
NF-κB cross-regulation by GSK-3 isoforms in pancreatic cancer. This phenomenon could
have important implications with regards to the development of isoform specific GSK-3
inhibitors, and further work in this area appears indicated.
Since GSK-3 inhibition efficiently suppresses NF-κB in pancreatic cancer cells, and
downregulates NF-κB targets associated with chemotherapy resistance such as XIAP and
Bcl-XL, it seemed reasonable to predict that this would also sensitize these cells to
gemcitabine. In all six cell lines tested, treatment with AR-18 as a single agent was growth
inhibitory in a dose- and time-dependent manner, similar to a previous report (92). However,
with the exception of PANC-1 the combination with gemcitabine was not synergistic. In
fact, across a wide range of conditions and drug combinations, the interactions ranged from
additive to antagonistic effects. Similar additive or antagonistic effects were observed in
PANC-1 or BxPC-3 cells using clonogenic survival as the endpoint.
The lack of sensitization to gemcitabine by GSK-3 inhibition in pancreatic cancer might be
due to number of reasons: 1) Some of the proteins targeted by GSK-3 for proteasomal
degradation, including Mcl-1, β-catenin, and cdc25 (32, 229, 230), have cancer-promoting
effects. Consequently, GSK-3 inhibition might have adverse effects by stabilizing these
proteins; 2) GSK-3 inhibition is reported to confer resistance to chemotherapy through
suppression of death receptor-mediated apoptosis (198, 199); 3) Gemcitabine treatment has

62
been previously reported to induce NF-κB activity in vitro (49), and this effect might
counteract the inhibition of NF-κB seen following treatment with AR-18. However, this
appears not to be the case under the experimental conditions used, since in the present study
the increase in NF-κB activity following gemcitabine exposure was modest and cell line
dependent, and effectively inhibited by AR-18 or GSK-3 knockdown; 4) It is also possible
that although NF-κB is hyper-activated in pancreatic cancer, it does not play a major role in
gemcitabine resistance, which is in line with some recent reports (80, 81, 231), 5) We also
considered that as gemcitabine cytotoxicity is cell cycle dependent, GSK-3 inhibition might
antagonize gemcitabine by slowing entry into S-phase or causing cell cycle arrest. However,
DNA content analysis by flow cytometry showed that the cell cycle effects of AR-18 were
relatively modest, which is in line with a previous report that in contrast to some other GSK-
3 inhibitors, AR-18 is a relative weak inhibitor of cyclin-dependent kinases (175).
Furthermore, we did not observe increased sensitization to the drug combination when cells
were pre-exposed to gemcitabine prior to the addition of AR-18 (data not shown). Although
the enhancement of gemcitabine toxicity following GSK-3 inhibition appears to be modest in
vitro, we recognize that this does not exclude the potential for positive drug interaction in
vivo, and this remains to be tested.
To investigate whether NF-κB inhibition by agents other than GSK-3 inhibitors could
potentiate gemcitabine sensitivity, we tested the natural product curcumin. We found that
curcumin inhibited both constitutive and TNF-α-induced NF-κB activity in PANC-1 and
MIA PaCa-2 cells, and was also toxic in a dose-and time-dependent manner, which was
consistent with previous reports (219, 232). However, in contrast to findings by
Kunnumakkara et al., and similar to our findings with AR-18, we did not observe
enhancement of gemcitabine toxicity by curcumin. Thus, our findings do not support a role
for NF-κB activity as a significant mediator of gemcitabine resistance in pancreatic cancer,
or the corollary that NF-κB inhibition is able to overcome chemotherapy resistance. These
results are corroborated by a recent study in colon cancer cell lines, where p65
overexpression could sensitize the cells to curcumin effects (80), and by other reports
suggesting that NF-κB might function as a pro-apoptotic or a tumor suppressor factor (20,
80, 81, 231), depending on the nature of apoptotic stimuli or the cell type.

63
Thus, although this work supports a model in which activated NF-κB is maintained by GSK-
3 and promotes the survival of pancreatic cancer cells, we suggest that the major mechanisms
of gemcitabine resistance are not dependent on NF-κB. Alternative mechanisms include
alterations in drug uptake and metabolism, enhanced DNA repair proficiency, or activation of
survival by other signaling pathways such as PI3-kinase/Akt. The exact role of GSK-3 in the
maintenance of pancreatic cancer, the differential role of its isoforms in regulating NF-κB
activity in these cells, and the mechanisms or conditions through which it maintains NF-κB
activity remain unclear, although recent work suggests an important role for GSK-3 in the
phosphorylation of IKK (228). Furthermore, the mechanisms of cell death following GSK-3
inhibition appear not to be through classical apoptosis pathways as we did not observe PARP
cleavage or loss of mitochondrial membrane potential, and AR-18 treated cells could not be
rescued using the general caspase inhibitor z-VAD (fmk) (data not shown). In summary, this
work supports a potentially important role for GSK-3 inhibition in the treatment of pancreatic
cancer, but cautions that further work examining the underlying mechanisms is needed for
this to be rationally exploited in the clinic.
2.5 Conclusions
Our observations suggest that although GSK-3 inhibition does not significantly sensitize to
the standard chemotherapy agent gemcitabine, yet it is a promising new approach to the
treatment of pancreatic cancer through disruption of NF-κB. We also conclude that NF-κB
is not a key player in gemcitabine resistance of pancreatic cancer. Further work is needed to
understand the mechanisms of the anticancer effect of GSK-3 inhibition, including the
potential for rational combination with other targeted agents for the treatment of pancreatic
cancer.

Chapter 3 GSK-3 Inhibition Sensitizes Pancreatic Cancer Cells to TRAIL-induced Apoptosis
The work presented in this Chapter is currently being prepared for publication in a scientific journal.

65
3.1 Abstract
Tumor necrosis factor related apoptosis inducing ligand (TRAIL) effectively induces
apoptosis in a variety of cancer cell lines in vitro with little or no effect on normal cells.
However, a broad spectrum including pancreatic and prostate cancer cells are resistant to
TRAIL-induced apoptosis. As these cancers are also resistant to the majority of conventional
treatments, there is an interest in targeted therapies that combine TRAIL with non-
conventional treatments to induce TRAIL sensitivity. In this Chapter we report that GSK-3
inhibition enhanced TRAIL sensitivity in a range of pancreatic and prostate cancer cell lines.
This sensitization was found to be caspase-dependent, and both pharmacological and genetic
knock down of GSK-3 isoforms resulted in apoptotic features as shown by cleavage of PARP
and caspase-3. Silencing GSK-3β and double knockdown of both isoforms were more
effective than GSK-3α silencing alone. Elevated levels of reactive oxygen intermediates and
disturbance of mitochondrial membrane potential indicated that both PANC-1 and PPC-1 cell
lines had features of Type II cells and depended on the mitochondrial amplification loop for
TRAIL-induced apoptosis after GSK-3 inhibition. Consistent with this, overexpression of
NF-κB anti-apoptotic mitochondrial targets; Bcl-XL, Mcl-1, and Bcl-2 rescued PANC-1 and
PPC-1 cells from TRAIL sensitization, indicating a role for these molecules in this process.
However, overexpression of the caspase-8 inhibitor CrmA also inhibited the sensitizing
effects of GSK-3 inhibitor, suggesting that GSK-3 plays an additional role that inhibits death
receptor signaling. Together, our findings for the first time show that GSK-3 plays a role in
TRAIL resistance of pancreatic cancer cells, and this might involve multiple GSK-3 targets.
The interaction between GSK-3 inhibition and TRAIL appears to be largely synergistic,
suggesting that the combination might also be effective in vivo.

66
3.2 Background
The intrinsic apoptosis pathway is initiated in response to an internal signal indicating
cellular stresses such as DNA damage, hypoxia, or growth factor withdrawal (28, 39).
Cellular sensors that receive these anti-survival signals lead to increase in transcription factor
p53, cell cycle arrest, and damage repair (39). However, repair might fail which leads to
p53-mediated up-regulation of pro-apoptotic genes such as PUMA, NOXA, BAX, and Apaf-
1, and the inhibition of anti-apoptotic genes such as BCL-XL or Bcl-2 (39, 177). Puma and
Noxa and Bax in turn block Mcl-1, Bcl-XL or Bcl-2, leading to activation of Bax and Bak,
perforation of the outer mitochondrial membrane and release of the mediators of apoptosis
cytochrome c and Smac/DIABLO into the cytoplasm (39). Cytochrome c release results in
the formation of the apoptosome complex and activation of caspase-9 in conjunction with
Apaf-1, whereas Smac/DIABLO blocks the apoptosis inhibitor XIAP (39, 233). Cleaved
caspase-9 further activates caspases-3 and -7 causing caspase-dependent cell death (233).
The extrinsic apoptosis pathway starts with binding of death receptor ligands such as Fas
ligand, TNF-α, and TRAIL to death receptors, activating a series of signals that can lead to
two separate, yet interrelated, modes of apoptosis induction. In some cells, called type I
cells, activation of death receptor-4 (DR-4) and death receptor-5 (DR-5) leads to the
recruitment of adaptor proteins to form the death-inducing signalling complex (DISC) and
subsequent activation of the initiator caspases-8 or -10 and later to activation of effector
caspases-3,-6, and -7, and apoptosis induction (17). In type II cells, however, binding of
TRAIL to DR-4 or DR-5 and activation of caspases- 8 or -10 is not strong enough to induce
apoptosis and requires a bypass through mitochondria to amplify the death inducing signal
(233). This cross talk between extrinsic and intrinsic apoptosis pathways requires caspase-8
mediated cleavage of the Bcl-2 family member Bid (39, 233). Truncated Bid (tBid) acts as a
blocking mechanism for inhibiting the action of anti-apoptotic Bcl-2 proteins such as: Bcl-2,
Mcl-1, and Bcl-XL, leading to mitochondria-induced cell death (39).
TRAIL (also called Apo-2 ligand), is a membrane bound soluble cytokine of the TNF-α
family and one of the key players of the extrinsic apoptotic signalling pathway by binding
and activating two death inducing receptors; DR-4 and DR-5 (233). TRAIL can also bind
two sets of non-functional decoy receptors, in which case, the apoptosis induction is blocked

67
(17). The balance between the death inducing receptors and decoy receptors is a major
determinant of apoptosis induction in the target cells (234). Previous studies have proposed
that, unlike TNF-α or Fas mediated apoptosis, TRAIL-induced apoptosis is a safer approach
for cancer therapy, and it imposes the least cytotoxicity by specifically targeting the tumor
cells and sparing the majority of normal cells (17, 40, 41, 235). Although the exact
mechanism of target specificity by TRAIL is not known, the lack of toxicity in normal cells
is partly attributed to the lower expression of death receptors (DR-4 and DR-5) and
simultaneous increased expression of decoy receptors on the surface of the normal cells
(234, 236, 237) . In contrast, a variety of malignancies, including lymphoma, colorectal
cancer, and non-small- cell lung cancer, demonstrate an increased expression of DR-4 and -5
receptors, rendering these cells susceptible to TRAIL- induced apoptosis (39). While the
tumoricidal effect of TRAIL is novel and promising, yet it has limited effects, as many tumor
cell lines including primary tumor cells from human breast, pancreas, prostate, lung, liver,
and colon cancer are resistant to TRAIL therapy (36, 40, 45, 190, 238).
The studies described in Chapter 2, and previous work by others, suggest that NF-κB is
positively regulated by glycogen synthase kinase-3 (GSK-3) and is involved in chemo-
resistance, cancer cell survival, metastasis, and cell proliferation of cancer cells (49, 52, 98)
GSK-3 can also protect hepatocyte cells from TNF-α-mediated cytotoxity, indicating that
GSK-3 has a role in blocking death receptor mediated apoptosis (28, 72, 73). Interestingly,
the inhibitory effects of GSK-3 have been extended to other death receptors such as TRAIL
and Fas (188, 189). Inhibition of GSK-3 was shown to potentiate TRAIL-induced apoptosis
in human hepatoma cancer cell lines (182, 190) and prostate cancer cell lines DU-145, PC-3,
and LNCaP were sensitized to TRAIL effect after treatment with Lithium Chloride (Li) (182,
192). In the course of the experiments described in Chapter 2, it was noted that GSK-3
inhibition blocked the activation of NF-κB by TNF-α (used as a positive control for the
luciferase reporter assay). Since NF-κB activation has been previously suggested to suppress
TRAIL-induced apoptosis in pancreatic cancer cells (197), this finding suggested the
potential for GSK-3 inhibition to sensitize pancreatic cancer to TRAIL.

68
3.3 Materials and Methods
3.3.1 Cell Lines and Reagents
Pancreatic adenocarcinoma cell lines PANC-1 and BxPC-3 were obtained from the American
Type Culture Collection (Rockville, MD) and maintained as described previously (98).
Additional experiments were done using PPC-1 and DU145 prostate cancer cell lines
obtained from Dr. Aaron Schimmer (Toronto, Canada) and cultured in RPMI 1640
containing 10% fetal bovine serum (FBS),100 units/mL penicillin and 100 µg/mL
streptomycin, and cells were grown at 37◦ C and 5% CO2 in air. For cells overexpressing
antiapoptotic markers, Geneticine (G418 sulfate; Wisent) was added to the media at required
doses.
Recombinant human TRAIL (rh-TRAIL) was purchased from R&D systems, Inc.,
(Minneapolis, MN), prepared in 20 µg/mL stock, and stored in -70 °C as recommended by
the manufacturer. The GSK-3 Inhibitor AR-A014418 (AR-18) was obtained from
CALBIOCHEM, EMD Biosciences, Inc. (San Diego, CA) and was dissolved in DMSO and
aliquots stored at -20º C. The general caspase inhibitor z-VAD-fmk was purchased from
Alexis-Enzo Life Sciences, Inc. (Plymouth Meeting, PA).
DiIC1(5) was purchased from Molecular Probes, dichlorodihydrofluorescein diacetate (H2-
DCFDA) was obtained from (Molecular Probes), and propidium iodide (PI) from (Sigma-
Aldrich).
For overexpression studies, pcDNA3.1-Myc constructs containing anti-apoptotic markers:
Bcl-2, BcL-XL, CrmA, and XIAP were kindly obtained from Dr. Aaron Schimmer
(University Health Network, Toronto, Canada). MCL-1 containing pcDNA3.1-His tag
construct was purchased from Sidnet (Toronto, Canada). All the constructs also included a
G418 resistance gene to be used as a eukaryotic selection marker.
For immunobotting purpose, rabbit polyclonal antibodies against cleaved caspase-3, cleaved
PARP, Bcl-2, BcL-XL, CrmA, and XIAP were purchased from Cell Signaling Technology
(Danvers, MA). Anti- Mcl-1 mouse monoclonal antibody was from BD Pharmingen
(Mississauga, ON). A mouse monoclonal antibody against GSK-3 α/β was obtained from

69
Biosource Inc. (Camarillo, CA). Anti-rabbit and anti-mouse horseradish peroxidase linked
IgG antibodies were from Amersham Biosciences (Buckinghamshire, United Kingdom).
3.3.2 Stable Transfections
PANC-1 and PPC-1 cells were seeded in 6-well plates (200,000 cells per well) in 2 mL
media containing 10% FBS. The cells were incubated in a CO2 incubator overnight at 37º C
prior to transfection using Lipofectamine 2000 from Invitrogen Life Technologies,(Carlsbad,
CA) as recommended by the manufactures. Briefly, at the day of transfection the media was
changed into antibiotic-free, serum free media and the cells were transfected with 0.95
µg/well DNA construct including the resistance marker. The cells were then incubated for 5
hours in a CO2 incubator at 37º C and humidity before the media was replaced with media
containing 10% FBS and antibiotics. After 48 h of further incubation, the cells were exposed
to the selection media containing 10% FBS, antibiotics, and 0.6 and 1.0 mg/mL G418 for
PPC-1 and PANC-1 cells respectively (239). The selection media was refreshed every 2
days for 14 days and the surviving cells were trypsinized and transferred into a new plate
containing media with 0.8 and 1.2 mg/mL G418 for PPC-1 and PANC-1 cells respectively.
After 2 passages, overexpressions of the markers were checked by immunoblotting and their
functionality was confirmed using the staurosporine apoptosis induction assay. Briefly, in a
96 well format overexpressed cells were seeded and exposed to a toxic dose of staurosporine
and their survival was compared to the cells that lacked the overxpressing construct. The
cells overexpressing the apoptotic markers showed a higher expression level of the marker
measured by immnoblotting and also they were resistant to cell death induced by
staurosporine when compared to cells lacking the construct.
3.3.3 Survival Assay
The dose-dependent effect of AR-18, TRAIL and their combination was assessed by the
sulphorhodamine B (SRB) dye (Molecular Probes, Eugene, OR) binding assay described in
Chapter 2. Briefly, 7000 cells per well PANC-1 and BxPC-3 were seeded in 96-well plates,
incubated in a CO2 incubator overnight at 37º C, and then were untreated or treated with
different doses of AR-18 (0, 25, and 50 µM) and DMSO. TRAIL (0, 5, 10 20, and 40
ng/mL) was added 24 h later and incubated alone or in combination with AR-18 in triplicates
for 24 h. The cells were then fixed using 10% (v/v) trichloroacetic acid for 1 h at 4º C,

70
washed extensively with water, stained with 0.4% SRB dissolved in 1% (v/v) acetic acid in
water reagent for 30 minutes at room temperature, and then washed, and 10 mM unbuffered
Tris was added to each well. The absorbance was measured at 570 nm using a Multiscan 96-
well plate reader from Thermo Electron Corp. (Milford, MA). This experiment was
repeated three times in six replicates.
To study the time-dependency of the combinational therapy of TRAIL and AR-18, PANC-1
and BxPC-3 cells treated with AR-18 (50 µM) for 24 h were incubated with TRAIL (10
ng/mL) for 1, 6, 12, and 24 h after addition of TRAIL and then were subjected to SRB assay.
The interaction between TRAIL and AR-18 was also studied in prostate cancer cell lines
PPC-1 and DU145 and their genetically modified variants. Briefly, 14,000 cells per well
were seeded and 24 h later treated with AR-18 (50 µM) for 24 h followed by TRAIL (10
ng/mL) and incubation for another 24 h before survival analysis using SRB assay.
3.3.4 Flow Cytometry Analysis of Apoptosis
The procedure used to simultaneously investigate apoptosis using the mitochondrial
membrane potential and reactive oxygen intermediates by flow cytometry was adapted from
a previously developed method in our laboratory by Pham et al. (240). Briefly, PANC-1,
BxPC-3, and PPC-1 cells were treated with AR-18 (25 µM), TRAIL (10 ng/mL) or both for
24h. On the day of experiment, the cells were washed twice with HANKS BSS, trypsinized
with EDTA-trypsin, and resuspended in complete media including 10% FBS and antibiotics.
The cells were counted and 500,000 cells in 0.5 mL media were stained with DiIC1(5) (final
concentration of 40 nM), and DCFDA (final concentration of 5 µM) and incubated at 37 °C
in dark for 25 minutes. Finally PI was added (final concentration of 1 µg/mL) and the tubes
were further incubated for 5 minutes in dark before running the samples as described
previously (240).
The flow cytometry analysis was done using a FC 500 flow cytometer (Beckman-Coulter,
Miami, FL), using lasers that emitted light at 488 and 633 nm wavelengths. To measure
mitochondrial membrane potential, DiIC1(5) was excited at 633 nm, and the emitted
fluorescence was measured through a 675+ 20 nm bandpass filter. DCFDA was used to
measure the increase in reactive oxygen intermediates, and PI uptake was used to assess

71
outer membrane integrity. Both dyes were excited at 488 nm wavelength and the
fluorescence was measured using a 525 + 10 nm bandpass filter for DCF and 610 + 10 nm
band pass filter for PI as described previously (240).
3.3.5 Immunoblotting
All the procedures involved in protein expression analysis are described in Chapter 2.
3.3.6 Genetic Knockdown of GSK-3
GSK-3 isoforms were genetically knocked down in PANC-1 cells transfected with siRNA
against GSK-3 α and β isoforms using a reverse transfection protocol as described in Chapter
2 (98).
3.3.7 Statistical Analysis
To determine the potential synergistic effect of combinational treatment with AR-18 and
TRAIL, the SRB cell viability data was subjected to further statistical analysis by log
transforming the data and using a linear regression model. The analysis was performed by
the help of “R” software (Hornik et al.; http://www.r-project.org) as described in Chapter 2
(98). SRB results are expressed as mean ± SE. Treatment effects were compared using
Student's t test and differences between means were considered to be significant when P ≤
0.05. Experiments were repeated at least three times.
3.4 Results
3.4.1 Effects of GSK-3 Inhibition and TRAIL in Human Prostate Cancer Cell Lines In vitro
Initial experiments were done using prostate cancer cell lines, because genetically-modified
cells overexpressing anti-apoptotic markers had been previously established in Dr. Aaron
Schimmer’s laboratory (University Health Network, Toronto, Canada), and were available
for this project. Also, TRAIL sensitization effect of GSK-3 inhibition has been previously
shown in prostate cancer cells and could serve for optimizing the project (182). Prostate
cancer cell lines DU145 and PPC-1 cells were treated with 50 µM AR-18, DMSO and/or 10
ng/mL TRAIL for 24 h followed by SRB analysis of the survival. As shown in Figure 14,

72
both AR-18 and TRAIL had anti-survival effects on PPC-1 and DU145. However, in DU145
cell lines, 10 ng/mL TRAIL alone did not cause significant anti-survival effects after 24 h of
treatment. Conversely, AR-18 and TRAIL together could induce significant cytotoxic effects
in both prostate cancer cell lines, when compared to each treatment alone (Figure 14). The
results indicate that similar to the sensitization effect of LiCl to TRAIL-induced apoptosis,
GSK-3 inhibition by AR-18 could also sensitize less sensitive prostate cancer cell lines to
TRAIL-induced apoptosis.
3.4.2 GSK-3 Inhibition Sensitizes TRAIL- Resistant Pancreatic Cancer Cells to Apoptosis
The majority of pancreatic cancer cells such as PANC-1 are less sensitive to TRAIL-induced
cell death. These TRAIL-resistant cell lines have higher expression of NF-κB target genes
such as XIAP or Bcl-XL (36, 43). Disruption of NF-κB activity in these cell lines has
resulted in synergistic effects with TRAIL and apoptosis induction (43). As described in
Chapter 2, TNF-α induced NF-κB activity could be disrupted upon inhibition of GSK-3
activity (73, 98). We also have shown that GSK-3 inhibition reduces NF-κB target gene
expressions (98). In this study, we further investigated TRAIL-resistant pancreatic cancer
cell lines; BxPC-3 and PANC-1, and the effect of GSK-3 inhibition on TRAIL-induced
apoptosis.
We treated both the cell lines with two concentrations of AR-18 (25 and 50 µM) or DMSO
for 24 h followed by addition of four different concentrations of TRAIL (5, 10, 20, 40
ng/mL) to the cells with or without AR-18 and incubated the cells for another 24 h. As
shown in Figure 15, both drugs independently exhibited concentration-dependent
cytotoxicity as determined using SRB assay with the exception that PANC-1 cells were
resistant to TRAIL. Under these conditions, both the cell lines showed that the combination
of different doses of TRAIL with both doses of AR-18 had substantial cytotoxic effects
(Figure 15). To determine the potential synergistic effect of combination treatment with
TRAIL and AR-18, the SRB cell viability data were subjected to statistical analysis as
described previously (98). The results of statistical analysis in both pancreatic cancer cell
lines indicated a significant synergism between AR-18 and TRAIL across all doses and
combinations with maximum effect seen at 50 µM AR-18 (Figure 15). Interestingly, at this
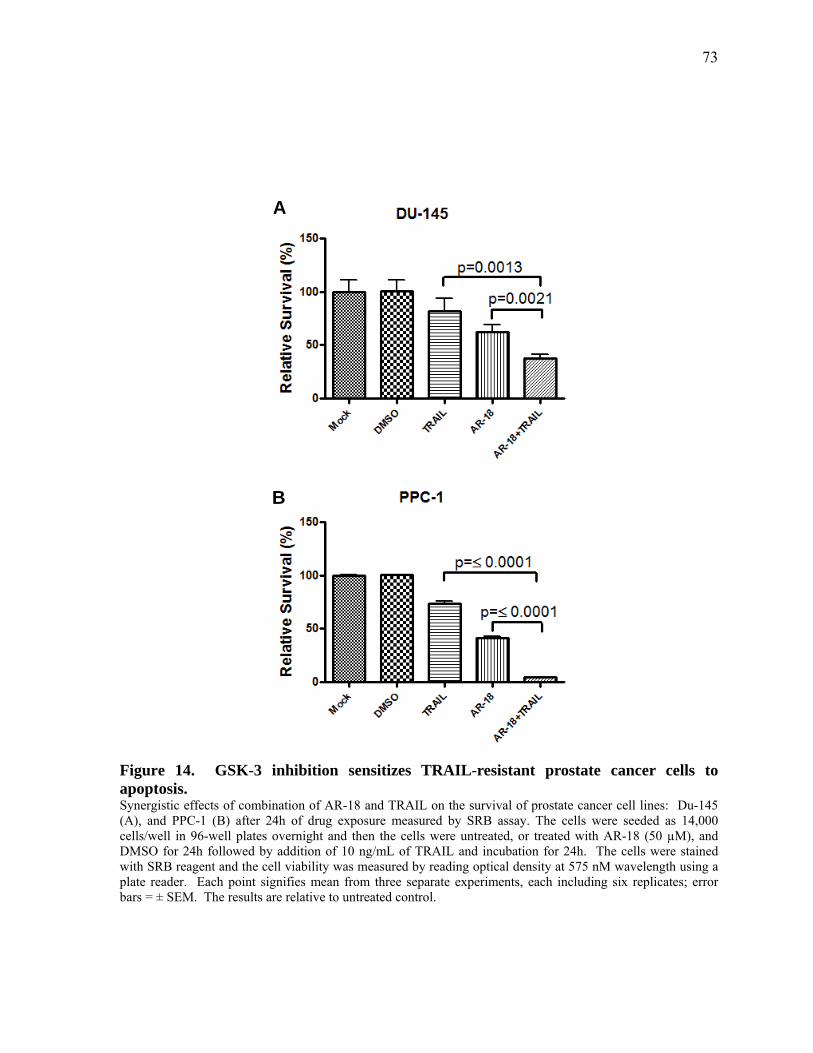
73
Figure 14. GSK-3 inhibition sensitizes TRAIL-resistant prostate cancer cells to apoptosis. Synergistic effects of combination of AR-18 and TRAIL on the survival of prostate cancer cell lines: Du-145 (A), and PPC-1 (B) after 24h of drug exposure measured by SRB assay. The cells were seeded as 14,000 cells/well in 96-well plates overnight and then the cells were untreated, or treated with AR-18 (50 µM), and DMSO for 24h followed by addition of 10 ng/mL of TRAIL and incubation for 24h. The cells were stained with SRB reagent and the cell viability was measured by reading optical density at 575 nM wavelength using a plate reader. Each point signifies mean from three separate experiments, each including six replicates; error bars = ± SEM. The results are relative to untreated control.
A
B

74
dose of AR-18, the cytotoxic effect of AR-18 did not increase across different concentrations
of TRAIL. These findings suggest that, at a high dose of AR-18, even the lowest dose of
TRAIL can induce similar cytotoxic effect as the highest dose does, and the synergistic effect
is more dependent on the extent of GSK-3 inhibition rather than TRAIL concentration.
These results are consistent with the previous findings that point to a significant anti-
apoptotic role of GSK-3 in blocking the death receptor pathway (28).
3.4.3 GSK-3 Inhibition Enhances TRAIL-Induced Cell Death in a Time-Dependent Manner
To determine if the time of exposure to the combination of AR-18 and TRAIL has any
effects in increasing synergistic effects, PANC-1 and BxPC-3 cells were treated with 50 µM
AR-18 and/or PBS for 24 h followed by addition of 10 ng/mL TRAIL and further incubation
for 1, 6 12, and 24 h and subsequent SRB analysis of survival. As shown in Figure 16, the
results were analyzed in comparison to the untreated control and indicated that the
synergistic effects of both drugs is initiated as early as 6 h and reached maximum at 24 h.
These results suggest that the synergistic anti tumor effect of the combination of GSK-3
inhibition and TRAIL is time dependent.
3.4.4 Apoptotic Nature of TRAIL Sensitization after GSK-3 Inhibition by AR-18
Immunoblot analysis of the apoptosis marker PARP cleavage was done using cell lysates
extracted from PANC-1 and BxPC-3 cells that received no treatment or treated with 25 µM
AR-18, 10 ng/mL TRAIL, DMSO, or combination of AR-18 and TRAIL for 24 h. As shown
in Figure 17, while AR-18 alone did not result in any PARP cleavage, there was a significant
enhancement of PARP cleavage in both PANC-1 and BxPC-3 cells when exposed to the
combination of AR-18 and TRAIL. Also, similar to the effects observed by SRB assay,
increase in PARP cleavage was not noted in PANC-1 cells when TRAIL was used alone,
whereas, BxPC-3 cells showed some PARP cleavage, indicating greater sensitivity to TRAIL
effects.
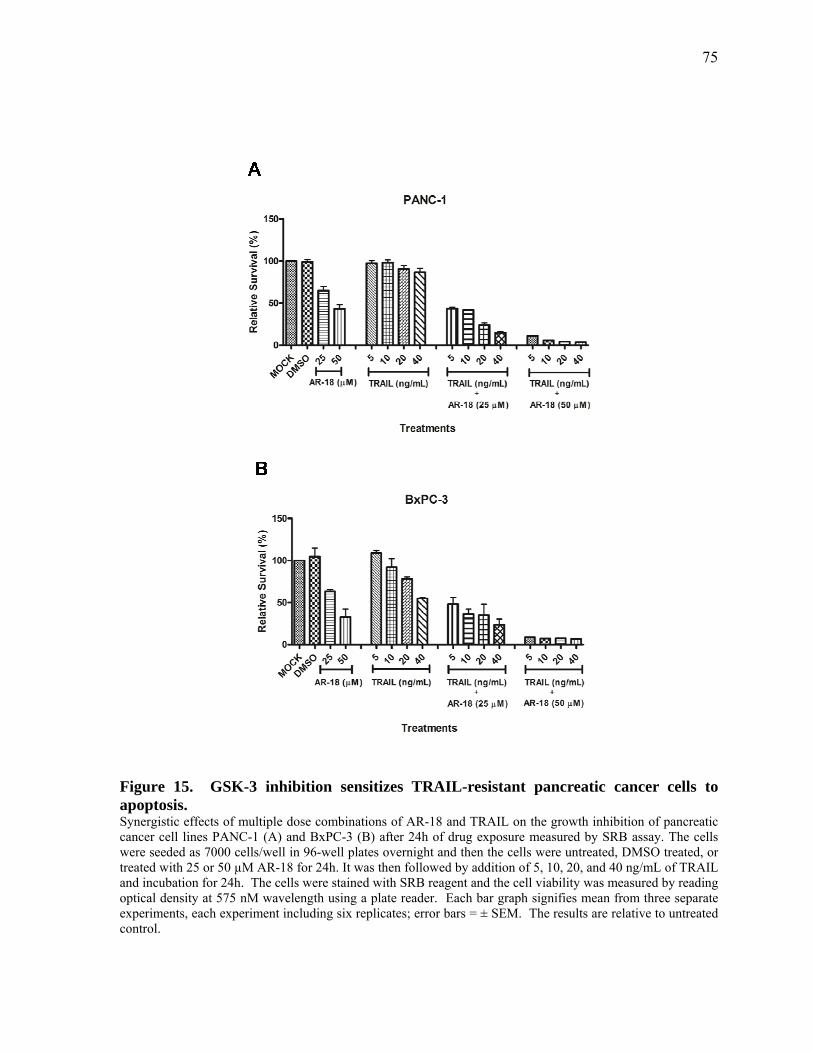
75
Figure 15. GSK-3 inhibition sensitizes TRAIL-resistant pancreatic cancer cells to apoptosis. Synergistic effects of multiple dose combinations of AR-18 and TRAIL on the growth inhibition of pancreatic cancer cell lines PANC-1 (A) and BxPC-3 (B) after 24h of drug exposure measured by SRB assay. The cells were seeded as 7000 cells/well in 96-well plates overnight and then the cells were untreated, DMSO treated, or treated with 25 or 50 µM AR-18 for 24h. It was then followed by addition of 5, 10, 20, and 40 ng/mL of TRAIL and incubation for 24h. The cells were stained with SRB reagent and the cell viability was measured by reading optical density at 575 nM wavelength using a plate reader. Each bar graph signifies mean from three separate experiments, each experiment including six replicates; error bars = ± SEM. The results are relative to untreated control.

76
Figure 16. GSK-3 inhibition enhances TRAIL-induced cell death in a time-dependent manner. Effects of the exposure times to the combination of AR-18 and TRAIL in the cell viability of pancreatic cancer cell lines PANC-1 (A) and BxPC-3 (B). The cells were seeded as 7000 cells/well in 96-well plates overnight and then incubated with 50 uM AR-18, or PBS for 24h followed by 10 ng/mL TRAIL and incubation for 1, 6, 12, and 24h. The cells were stained with SRB reagent and the cell viability was measured by reading optical density at 575nM wavelength using a plate reader. Each point signifies mean from three separate experiments, each including six replicates; error bars = ± SEM. The results are relative to untreated control.
A
B
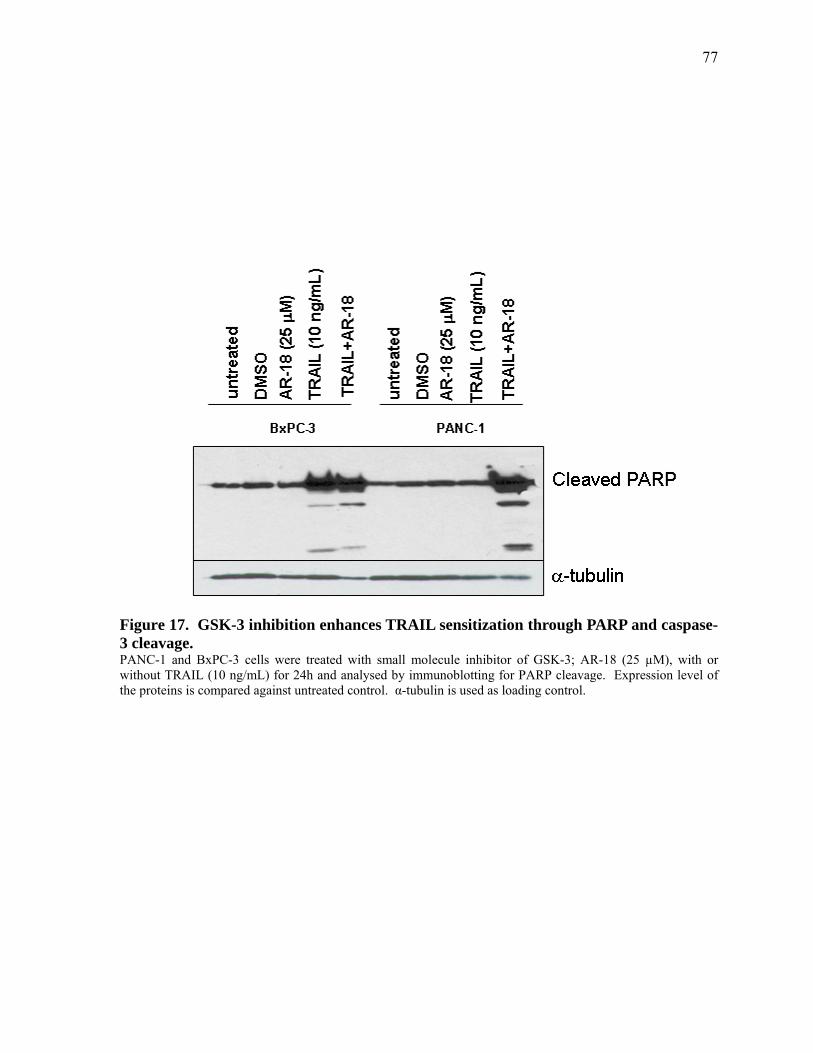
77
Figure 17. GSK-3 inhibition enhances TRAIL sensitization through PARP and caspase-3 cleavage. PANC-1 and BxPC-3 cells were treated with small molecule inhibitor of GSK-3; AR-18 (25 µM), with or without TRAIL (10 ng/mL) for 24h and analysed by immunoblotting for PARP cleavage. Expression level of the proteins is compared against untreated control. α-tubulin is used as loading control.

78
3.4.5 Caspase-dependency of TRAIL Sensitivity by GSK-3 Inhibition
Caspases are the apoptotic machinery of the cells that mediate initiation and execution of
apoptotic signals triggered by intrinsic or extrinsic stimuli (21). However, there are also
caspase-independent mechanisms through which TRAIL-sensitization can occur (241). To
test if the process of TRAIL sensitization of pancreatic cancer cells by GSK-3 inhibition is
caspase-dependent, PANC-1 and BxPC-3 cells were treated with 25 µM AR-18 and/or 10
ng/mL TRAIL in the presence of the general caspase inhibitor z-VAD. As shown in Figure
18, although z-VAD-fmk itself exerted some toxic effects, yet it significantly rescued both
the cell lines from the apoptosis induced by the AR-18 and TRAIL combination.
Furthermore, TRAIL had a caspase-dependent effect on BxPC-3 and not PANC-1 cells,
which was consistent with our previous observations. Both cell lines showed a significant
caspase-dependency when AR-18 was applied alone. These data indicate that the process of
TRAIL sensitization by GSK-3 inhibition involves caspase activation.
3.4.6 Effects of Genetic Knockdown of GSK-3 on TRAIL Sensitization of Pancreatic Cancer Cells
It has been demonstrated that small molecule inhibitors such as AR-18 can inhibit both GSK-
3 isoforms, which in turn will block the functional redundancy of GSK-3 isoforms (161).
Also, kinase inhibitors might have multiple off target effects that might not be specific to
GSK-3 inhibition (242). As a result, we evaluated if TRAIL sensitization by AR-18 is due to
GSK-3 inhibition, and also investigated the role of GSK-3 isoforms in the TRAIL
sensitization process and apoptosis induction. We examined the expression level of cleaved
PARP and cleaved caspase-3 using cell lysates from PANC-1 cells transiently transfected
with siRNA against GSK-3 α, β, or both with or without 10 ng/mL TRAIL. Cells receiving
no treatment or transfected with scrambled siRNA duplex were used as negative control,
while cells treated with AR-18 alone or in combination with TRAIL were considered as
positive control. As shown in Figure 19, following 3-day incubation with siRNAs against
GSK-3 isoforms, immunoblotting showed >80% reduction in the total protein of each
isoform when compared to the untreated or scrambled treated cells, indicating an efficient
knock down of the isoforms. Consistent with our previous observation in PANC-1 cells
(Figure.15A), treatment with TRAIL (10 ng/mL) alone had a minor effect on apoptosis as
measured by PARP or caspase-3 cleavage (Figure 19). Also, transient knockdown of GSK-3

79
Figure 18. TRAIL sensitization through inhibition of GSK-3 is caspase-dependent. PANC-1 (A) and BxPC-3 (B) cells were seeded at 7000 cells/well and next day were simultaneously treated with either AR-18 (25uM), TRAIL (R&D) (10ng/mL), z-VAD-fmk (Alexis) 50uM, or combination of them and were incubated for 22h before they are subjected to SRB assay. The cell viability was measured by reading optical density at 575nM wavelength using a plate reader. Each bar graph signifies mean from three experiments, each including six replicates; error bars = ± SEM. The results are relative to untreated control.
A B
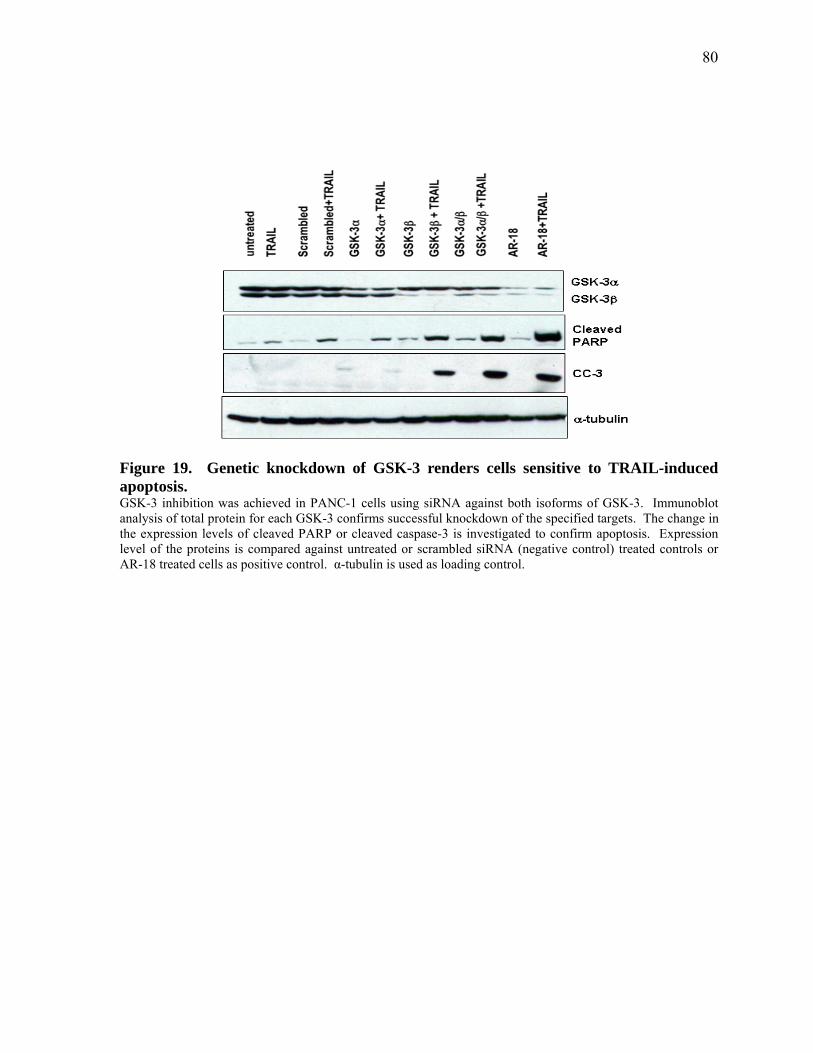
80
Figure 19. Genetic knockdown of GSK-3 renders cells sensitive to TRAIL-induced apoptosis. GSK-3 inhibition was achieved in PANC-1 cells using siRNA against both isoforms of GSK-3. Immunoblot analysis of total protein for each GSK-3 confirms successful knockdown of the specified targets. The change in the expression levels of cleaved PARP or cleaved caspase-3 is investigated to confirm apoptosis. Expression level of the proteins is compared against untreated or scrambled siRNA (negative control) treated controls or AR-18 treated cells as positive control. α-tubulin is used as loading control.

81
α or β isoforms alone did not result in significant apoptotic effects. In contrast, the
combination of TRAIL with GSK-3β genetic knockdown substantially enhanced PARP or
caspase-3 cleavage when compared to untreated, single treatments, or scrambled siRNA
transfected cells. GSK-3α knockdown in combination with TRAIL induced minor effects on
PARP and caspase-3 cleavage, but simultaneous knockdown with GSK-3β isoform in
combination with TRAIL, had a significant apoptotic effect, which was comparable to the
results observed when the pharmacological inhibitor of GSK-3 AR-18 was used (Figure 19).
The results of this experiment confirm that suppression of GSK-3 isoforms is responsible for
TRAIL sensitization, and the anti-survival effect induced by AR-18 combination with
TRAIL, is GSK-3-specific. Also, it suggests that GSK-3β plays a more prominent role than
GSK-3α. These results are consistent with the observations in Chapter 2, where GSK-3β
isoform was shown to play a key role in NF-κB regulation.
3.4.7 TRAIL Sensitization upon GSK-3 Inhibition is Mediated through Mitochondria
The extrinsic apoptotic pathway involves activation of death receptors by binding of ligands
such as TRAIL to DR4 and DR5 cell surface receptors and initiating a cascade of events that
eventually leads to activation of effector caspase-3 and apoptosis induction (235). However,
in some cells, called type II cells, caspase-3 activation requires signal amplification via Bid
cleavage and mitochondria activation (233, 235). This in turn leads to imbalanced
physiological status of the mitochondria resulting in defective respiratory chain and increased
generation of reactive oxygen intermediates, and perturbation of mitochondrial membrane
potential (240). In this study, we investigated whether there is any involvement of the
mitochondrial component during apoptosis execution in TRAIL sensitization by GSK-3
inhibition. Flow cytometry was used to measure mitochondrial membrane potential (∆Ψm),
and reactive oxygen intermediates in pancreatic and prostate cancer cells treated with 25 µM
AR-18 and 10 ng/mL TRAIL alone or in combination followed by 24 h of incubation.
As shown in Figure 20, in all the three cell lines tested, the combination of TRAIL and AR-
18 induced formation of a heterogenous population of cells that had the characteristics of
increased ROI generation, loss of ∆Ψm , and PI uptake, when compared to untreated control
or single treatments. The results were consistent with previous observations by our
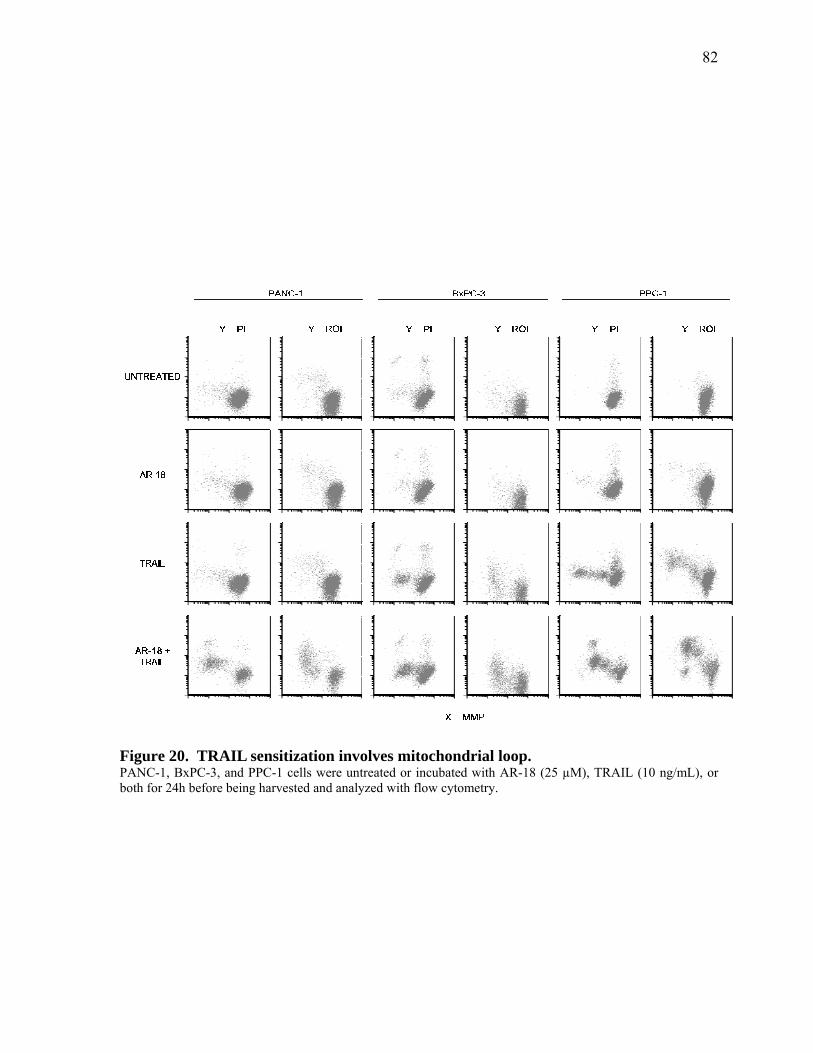
82
Figure 20. TRAIL sensitization involves mitochondrial loop. PANC-1, BxPC-3, and PPC-1 cells were untreated or incubated with AR-18 (25 µM), TRAIL (10 ng/mL), or both for 24h before being harvested and analyzed with flow cytometry.

83
laboratory with regards to the mitochondrial features of apoptotic cells (240). Consistent
with our previous observations using the SRB assay and immunoblot analysis, AR-18
treatment did not elicit apoptotic features in any of the cell lines tested.
Although TRAIL treatment induced apoptotic features in PPC-1 and BxPC-3 cells, yet, the
cytotoxic effect was not as extensive as the combination treatment (Figure 20). Both loss of
mitochondrial membrane potential and increase in ROI indicate that the process of TRAIL
sensitisation by GSK-3 inhibition is mitochondria-dependent.
3.4.8 Molecular Mechanism of TRAIL Sensitization by GSK-3 Inhibition
In the previous experiments, it was shown that in the process of sensitization to TRAIL-
induced cell death after GSK-3 inhibition, mitochondria appear to play a key role in
amplifying the effect. In addition, previous reports by our group and others have shown that
GSK-3 regulates mitochondrial anti-apoptotic molecules such as Bcl-2 and Bcl-XL, in
pancreatic cancer cells (92, 98). Also, GSK-3 has been reported to block the death-receptor
pathway at an early stage of death receptor induced signalling through an unknown
mechanism (28). However, the exact molecular targets affected by GSK-3 inhibition in the
process of TRAIL sensitization are unknown. As a result, in these sets of experiments, we
sought to determine the molecular components that create a cross-talk between the extrinsic
pathway and the mitochondrial regulated pathway in the process of TRAIL sensitization
upon GSK-3 inhibition.
In order to do so, PANC-1 and PPC-1 cells were stably transfected with constructs
harbouring genes encoding anti-apoptotic molecules cytokine response modifier A (CrmA)
that blocks the death receptor pathway by blocking caspase-1 and -8, and the mitochondrial
anti-apoptotic molecules Bcl-2, Bcl-XL, and Mcl-1 (39). As illustrated in Figure 21, among
the cells exposed to the apoptotic effect induced by the combination of 50 µM AR-18, and 10
ng/mL TRAIL for 24 h, PANC-1 cells overexpressing CrmA, Mcl-1, or Bcl-2, could
significantly rescue the cells from cell death when compared to untransfected control.
However, the highest effect was seen by CrmA and Bcl-2. Interestingly, the rescue effect
was not achieved by overexpressing Bcl-XL, indicating that this anti-apoptotic molecule
might not play a role in the process of GSK-3 inhibition-induced TRAIL sensitization in

84
PANC-1 CrmA MCL-1 Bcl-XL BCL-20
20
40
60
80
100
p=0.0167
p<0.0001
p=0.0015
Overexpression of anti-apoptotic markers in PANC-1 cells
Rel
ativ
e Su
rviv
al (%
)
Figure 21. BCL-2, MCL-1 and CrmA are the anti-apoptotic proteins involved in TRAIL sensitization of pancreatic cancer cells. Various anti-apoptotic molecular markers were stably overexpressed in PANC-1 cells and then the cells were untreated or subjected to the combination of AR-18 (50 µM) and TRAIL (10 ng/mL) for 24h. Using SRB assay, the cell viability was measured by reading optical density at 575nM wavelength using a plate reader. Each bar graph signifies mean from three experiments, each including six replicates; error bars = ± SEM. The results are relative to untreated control.

85
PANC-1 cells. These results suggest that GSK-3 might achieve its protective effect against
death receptor-induced apoptosis by blocking the activation of initiator caspases such as
Caspase-8, as well as by positively maintaining antiapoptotic molecules such as Bcl-2 and
Mcl-1 in pancreatic cancer cells.
In order to assess whether the mechanism of TRAIL sensitization in pancreatic cancer cells
accounts for similar effects in prostate cancer cells, we then tested PPC-1 cells
overexpressing CrmA, Mcl-1, Bcl-2 and Bcl-XL previously established in the laboratory of
Dr. Aaron Schimmer (University Health Network, Toronto, Canaca). As shown in Figure 22,
similar to PANC-1 cells, overexpressing CrmA, Bcl-2 and Mcl-1 in PPC-1 cells, enhanced
survival when compared to the control not overexpressing any anti-apoptotic markers.
However, in contrast to PANC-1 cells, overexpressing Bcl-XL in PPC-1 cells was also
protective. These data suggest that although both PANC-1 and PPC-1 cells share similar
basic means of protecting cells against TRAIL-induced cell death after GSK-3 inhibition, yet
there might be some additional levels of protection in PPC-1 that are regulated by GSK-3.
3.5 Discussion Although the results described in Chapter 2 clearly showed lack of sensitization to
gemcitabine by GSK-3 inhibition, this strategy appears much more effective in sensitizing to
TRAIL. This conclusion is supported by several lines of evidence: First, we could
successfully achieve TRAIL sensitization via GSK-3 inhibition in both prostate and
pancreatic cancer cell lines. Second, the process of TRAIL sensitization was shown to be
through apoptosis induction and in a caspase-dependent manner. Third, genetic knockdown
of GSK-3 using isoform specific siRNAs was equally effective in TRAIL-sensitization as
was small molecule inhibitor; AR-18. The relevant apoptotic effect was demonstrated by
enhanced expression levels of cleaved PARP and cleaved caspase-3. Fourth, we found that
mitochondrial amplification loop played a pivotal role in GSK-3 inhibitor-induced TRAIL
sensitization. Fifth, in line with the above statement, overexpression studies in pancreatic
cancer cells indicated that both caspase-8 and NF-κB-dependent mitochondrial anti-apoptotic
markers CrmA, Bcl-2, and Mcl-1 were involved in the process of TRAIL sensitization upon
GSK-3 suppression and subsequent apoptosis execution. Interestingly, in case of prostate
cancer cell lines, Bcl-XL was also involved in this process. Together, these findings indicated
that GSK-3 inhibition could be a potent strategy to initiate the tumoricidal effects of TRAIL
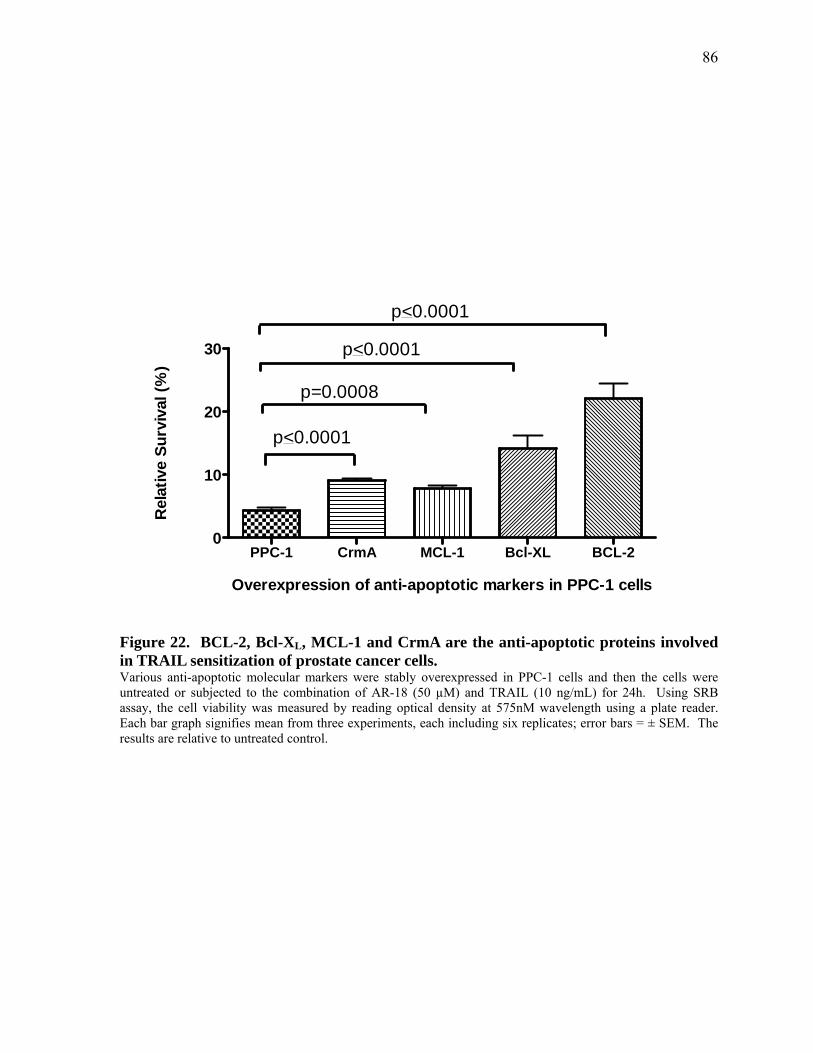
86
PPC-1 CrmA MCL-1 Bcl-XL BCL-20
10
20
30
p<0.0001
p=0.0008
p<0.0001
p<0.0001
Overexpression of anti-apoptotic markers in PPC-1 cells
Rel
ativ
e Su
rviv
al (%
)
Figure 22. BCL-2, Bcl-XL, MCL-1 and CrmA are the anti-apoptotic proteins involved in TRAIL sensitization of prostate cancer cells. Various anti-apoptotic molecular markers were stably overexpressed in PPC-1 cells and then the cells were untreated or subjected to the combination of AR-18 (50 µM) and TRAIL (10 ng/mL) for 24h. Using SRB assay, the cell viability was measured by reading optical density at 575nM wavelength using a plate reader. Each bar graph signifies mean from three experiments, each including six replicates; error bars = ± SEM. The results are relative to untreated control.

87
in chemo-resistant pancreatic and prostate cancer cells in vitro.
To our knowledge, this is the first study that investigates TRAIL-sensitization via GSK-3
inhibition in pancreatic cancer cells. Use of TRAIL in combination with GSK-3 inhibition is
particularly of importance in the treatment of pancreatic cancer as these tumors have a high
level of K-ras and p53 mutations (243, 244). Inactive p53 is responsible for the failed
response to chemo- or radiation-therapies, as both therapies require a p53-dependent
activation of mitochondrial pathway to induce apoptotic effects (235). Interestingly, and in
contrast to the conventional therapies, studies have shown that TRAIL sensitization bypasses
p53-dependent mechanisms and is effective in tumors with inactive p53 (245). Also,
intriguingly, activating K-ras mutations have also shown to sensitize colon cancer cells to
TRAIL-induced apoptosis and this effect might be extended to other cancers including
pancreatic adenocarcinoma that harbours activating K-ras mutations (246, 247).
We first tested the TRAIL sensitization phenomenon and optimized our study using prostate
cancer cells because of the previous proofs of the concept in these cancer cells (182).
Interestingly, although when TRAIL was used alone, there was a variation in sensitization
among different pancreatic and prostate cancer cells lines with PANC-1 and DU145 being
the most TRAIL resistant cells, but the combination with GSK-3 suppression provided a
consistent significant sensitization to TRAIL amongst all the cell lines tested. We also
showed that the sensitization is dependent on the level of GSK-3 inhibition rather than
TRAIL concentration, for the reason that, the anti-survival effect of the minimum dose of
TRAIL was as effective as the highest dose, and only increasing doses of GSK-3 played as
the determinant factor in increasing the sensitivity to cell death. The GSK-3 inhibition-
dependent sensitization effect was also shown to be time-dependent and although the effect
was seen at as early as 6 hours post combination treatment, increased with time.
Previous studies attributed the TRAIL resistance of pancreatic cancer cells to the
overexpression of X-linked inhibitors of apoptosis (XIAP) (248). Studies by Volger et al., in
pancreatic cancer cells, showed that blocking XIAP could sensitize the cells to TRAIL-
induced apoptosis both in- vitro and in vivo (34). Other studies have emphasized the role of
NF-κB in resistance to TRAIL by up-regulating inhibitors of apoptosis such as XIAP, Bcl-2,
Bcl-XL, and Mcl-1 (43, 54, 249). It is also known that GSK-3 inhibition downregulates the

88
expression level of NF-κB target genes such as: XIAP, Bcl-XL, Mcl-1, and Bcl-2 (92, 98,
222), and also blocks the death receptor pathway (28), suggesting that GSK-3 inhibition
imposes the process of TRAIL sensitization possibly through multiple mechanisms.
In our study we investigated the molecular mechanism of TRAIL sensitization in cooperation
with GSK-3 suppression using different approaches. Using CrmA overexpression, we
demonstrated that caspase-8 is an important factor in promoting TRAIL sensitivity in
pancreatic cancer cells that have inactive GSK-3. This was in line with the previous finding
that GSK-3 can block death receptor induced apoptosis at an unknown site upstream to
caspase-8 (28). Further immunoblotting analysis indicated that caspase-3 and PARP were
involved in the apoptosis execution in TRAIL sensitized cells by either AR-18 or genetic
knockdown of GSK-3 isoforms. To our knowledge, this is the first study where TRAIL
sensitization is promoted through isoform-specific inhibition of GSK-3. Interestingly, the
effect of GSK-3β in TRAIL-induced apoptosis was more prominent than GSK-3α and
intriguingly, the intensity of double isoform knockdown was significantly enhanced when
compared to GSK-3β alone and was comparable to that of AR-18 treated cells. These results
are similar to our previously published report indicating that genetic blockage of GSK-3β
and double knockdown of GSK-3 isoforms are relatively more effective than GSK-3α in
suppressing NF-κB activity (98). In line with previous findings by Khanbolooki et al., that
suggested a role for NF-κB in TRAIL resistance of pancreatic cancer (43), these results
suggest that TRAIL sensitization of pancreatic cancer cells upon GSK-3 inhibition, might
also involve NF-κB inactivation.
This led to further investigation and asking the question whether there is also involvement of
mitochondrial amplification loop in this process, as many of the NF-κB target proteins inhibit
the mitochondrial pathway. Using a novel flow cytometry based approach developed by our
laboratory (240), we found that there is an enhanced release of reactive oxygen intermediates
(ROI) in conjunction with decreased mitochondrial membrane potential in the cells treated
with a combination of TRAIL and GSK-3 inhibitor, clearly indicating the involvement of
mitochondria in the process of apoptosis induction in these cells. Although generation of
ROI as a crucial effector mechanism during drug-induced apoptosis in human tumor cells is
already known (250), however, our results are novel and intriguing as they point to a link

89
between the process of TRAIL sensitization by GSK-3 inhibition, and the mitochondrial
respiratory chain. Such an effect was previously observed in a study by Jung et al., using
curcumin to sensitize renal cancer cells to TRAIL effect (251). Curcumin enhanced ROI
generation in TRAIL sensitized cells and further there was an increase in DR-5 expression in
a ROI-dependent manner (251). Similar results were previously reported in human astroglial
cells, emphasizing ROI-dependent up-regulation of TRAIL death receptors (252). Cleavage
of caspase-3 observed in our study might further influence ROI production, as activated
caspase-3 can induce a feedback loop on the mitochondrial respiratory chain (199). Whether
increase in ROI generation in pancreatic cancer cells is the crucial cause of TRAIL
sensitization after GSK-3 inhibition, or it is generated as a secondary downstream
effector/mediator of apoptosis execution, remains to be investigated. It is also worth noting
that ROI generation might again link NF-κB to the process of TRAIL sensitization of GSK-3
inhibitors in pancreatic cancer. Because GSK-3 inhibition blocks NF-κB, the ROI generation
might be a subsequent effect to NF-κB inhibition, an effect observed by another NF-κB
blocker; curcumin, in the process of TRAIL sensitization in renal cancer cells (251). It
would be also interesting to study the effect of GSK-3 inhibition-induced TRAIL
sensitization in a hypoxic environment, as ROI generation is oxygen-dependent, and the
majority of pancreatic cancer tumors are hypoxic (253).
Anti-apoptotic Bcl-2 family proteins; Mcl-1, Bcl-XL, and Bcl-2 have been shown to have
high expression levels in both pancreatic and prostate cancer cells and contribute to apoptosis
resistance (45, 201).The results of this study further helped us to decipher for the first time,
which one of the these mitochondrial factors have a role in TRAIL resistance and their effect
is eliminated by GSK-3 inhibition. Targeted overexpression studies using Bcl-2, Bcl-XL, and
Mcl-1 indicated that although there is a slight variation between PANC-1 and PPC-1 cells
with regards to the mitochondrial anti-apoptotic molecules involved in the TRAIL resistance
process, yet both the cell lines consistently point to the importance of NF-κB associated
mitochondrial anti-apoptotic molecules in this course of action. These finding are intriguing
as previous reports indicate that inhibition of GSK-3 upregulates Mcl-1 expression and
results to its activation, pointing to a pro-apoptotic role of GSK-3 through the mitochondrial
pathway (31). However, in our hands, we have observed a significant decrease in Mcl-1
levels after GSK-3 inhibition by small molecule inhibitor; AR-18, or anti-GSK-3 siRNAs

90
(data not shown). Our finding in this study is in line with the anti-apoptotic role of GSK-3
and consistent with the previous reports by our group and others that GSK-3 inhibition
downregulates Bcl-2 anti-apoptotic proteins (92, 98).
3.6 Conclusion
GSK-3 inhibition sensitizes to TRAIL, apparently by affecting the activation of initiator
caspases as well as by enhancing the intrinsic apoptosis pathway via its effects on NF-κB-
responsive proteins like Bcl-2 and Mcl-1. These results suggest the potential to develop the
combination for treating pancreatic cancer patients, and this is explored further in Chapter 4.

Chapter 4 GSK-3 Inhibition in Combination with TRAIL Promotes Apoptosis in PANC-1 Xenografts in Mice
The work presented in this Chapter is currently being prepared for publication in a scientific journal.

92
4.1 Abstract
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a death receptor ligand
that induces cell death in a variety of malignancies with the least effect on normal cells. As a
result it has been suggested for therapeutic applications in cancer therapy. However, its
effect is limited as some cancers including pancreatic cancer show de novo resistance to
TRAIL induced apoptosis. In Chapter 3 of this thesis, it was shown that suppressing GSK-3
activity by small molecule inhibitor (AR-18) or short interfering RNAs (siRNA) resulted in
synergistic effects with TRAIL in sensitizing TRAIL-resistant pancreatic and prostate cancer
cells in vitro. In this Chapter, we asked if pre-treatment of pancreatic tumor-bearing mice
with a GSK-3 inhibitor enhances TRAIL-induced apoptosis. Consistent with our previous in
vitro observation, acute treatment of tumors with AR-18 and TRAIL resulted in a significant
increase in apoptosis as measured by caspase-3 cleavage. Interestingly, although β-catenin
levels were increased due to GSK-3 inhibition, the synergistic effect of the combinational
therapy could overcome this effect, suggesting that the approach might be effective even in
cancers with dysregulated β-catenin. Unlike some other combination therapies, the effects of
this approach were accompanied with minimal toxicity to the host organs or normal cells.
These results suggest that GSK-3 inhibitors may open a new perspective to the development
of safe and effective therapy approaches for the treatment of pancreatic cancer.
4.2 Background
In Chapter 2 of this thesis, it was shown that GSK-3 inhibition was a promising approach to
induce anti-survival effects in pancreatic cancer cells. The effect was associated with
downregulation of NF-κB activity and decreasing expression of its anti-apoptotic target
genes such as: XIAP, BcL-XL, and cyclin D1. GSK-3 inhibition has been observed to
sensitize the cells to death-inducing effects of death receptor ligands such as TNF-α (72,
73). Chapter 3 of this thesis examined the synergistic effect of TRAIL and GSK-3 inhibition
in vitro. It was shown that pre-treatment of TRAIL resistant pancreatic or prostate cancer
cells with GSK-3 inhibitor sensitized these cells to TRAIL-induced apoptosis. The effect of
GSK-3 inhibition appeared to involve both the initial phase of death receptor signaling via
caspase-8 activation, and the mitochondrial amplification pathway. In this Chapter, effects of
GSK-3 inhibition on TRAIL sensitization were tested in vivo using PANC-1 tumors. We

93
demonstrated that blocking GSK-3 activity through small molecule inhibitors dramatically
sensitized pancreatic tumors to TRAIL-mediated apoptosis induction. This occurred with
enhancing caspase-3 activation and had least toxic effects on mice bearing the tumor.
4.3 Materials and Methods
4.3.1 Cells and Reagents
Human pancreatic adenocarcinoma cells line PANC-1 was obtained from the American Type
Culture Collection (Rockville, MD) and maintained as described previously (98). Whereas
the in vitro studies described in Chapter 3 used a commercial source of TRAIL (R&D
Systems), the cost for in vivo studies would have been prohibitive. Therefore recombinant
human TRAIL/Apo2L, which is jointly being developed for clinical studies by Genentech
and Amgen Inc., was obtained under Material Transfer Agreement. It was stored in -70 °C as
recommended by the manufacturer. The GSK-3 Inhibitor AR-A014418 (AR-18) was custom
synthesized by Toronto Research Chemicals Inc. (North York, Ontario), dissolved in DMSO
(5mg/mL) and aliquots stored at -20º C. For immunoblotting purposes, rabbit polyclonal
antibodies against cleaved caspase-3 and β-catenin were purchased from Cell Signalling
Technology (Danvers, MA) and anti-rabbit and anti-mouse horseradish peroxidase linked
IgG antibodies were from Amersham Biosciences (Buckinghamshire, United Kingdom).
4.3.2 Pancreatic Cancer Xenograft Model
Experiments were done according to regulations of the Canadian Council of Animal Care
and Institutional guideline for animal welfare (Ontario Cancer Institute, Toronto, Ontario,
Canada). Panc-1 subcutaneous tumor xenografts were grown in 6-week-old severe combined
immunodeficient (SCID) male mice and were derived from PANC-1 cells. Briefly, PANC-1
cell suspension (400 µL of ~ 1.5 X 105 cells) was injected subcutaneously into the right flank
site of male SCID mice, tumors were allowed to grow to about 1 cm in the largest diameter.
Tumor diameter was measured using a microcaliper and the volume was calculated using the
following formula: length × width2 × 0.5236 (202). Mice were randomly assigned to 4
groups of 5 mice to receive treatments.

94
4.3.3 Treatment Procedure and Drug Schedule
To examine the effect of short term exposure to the combination of AR-18 and TRAIL we
followed the AR-18 dose schedule used in a previous study (185). Ougolkov et al. used
intraperitonial (i.p) injections of 120 mg/kg AR-18 for every 12 h up to 2 days in female
athymic nude mice bearing CAPAN-2 xenografts (185). However, in our hands this dose
was highly toxic to male SCID mice, and a series of preliminary experiments were performed
and it was established that the maximum AR-18 dose tolerated by SCID mice from the OCI
colony was 20mg/kg (data not shown). Tumor bearing animals were then randomly assigned
into four groups of five mice and received i.p. injections of different treatments for a total of
5 days as shown in Figure 23. The treatments included (100 µL) of AR-18 (at 20 mg/kg, 5
mg/mL stock in DMSO), or DMSO, or PBS given twice daily for two days followed by a 24
h resting period before i.p. injection of TRAIL (500 µg, 500 µL). For the group receiving
only AR-18, the treatment stopped on the third day. For the combined treatment, AR-18 was
given first till the 3rd day and TRAIL was given on the 4th day. Animal weight was measured
every day starting from the beginning of the treatment till the termination day. All the mice
were sacrificed on the 5th day of the experiment and the tumors were excised, cut into small
pieces were either snap frozen, formalin fixed for 24 h followed by paraffin embedding, or
lysed using 1.5 mL lysis buffer as described below.
4.3.4 Immunoblotting
For lysis of the tumor cells 500 µL of lysis buffer [50 mM HEPES (pH 8.0), 10% glycerol,
1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 1.5 mM MgCl2, 100 mM NaF, 10 mM
NaP2O7, 1 mM Na3VO4], supplemented with protease inhibitor cocktail (Roche Diagnostics,
Mannheim, Germany) were added to each tube containing the tumor piece and lysis was
conducted for 1 hour on ice. The remaining procedures followed the processing method
described previously in Chapters 2 and 3 of the thesis. Signal quantification was done using
(a) Typhoon 9410 (GE Healthcare) with ECL Plus and Image Quant 5.2 software. The
resulting values were normalized against the background and also an internal α-tubulin
control. The normalized values were plotted as a ratio relative to the untreated controls.

95
Figure 23. Antitumor effect of GSK-3 inhibition in vivo. PANC-1 cells were injected into the right flank of male SCID and tumor growth was monitored. Established PANC-1 xenografts (tumor diameter, 1 cm in diameter) were treated by i.p. injections with DMSO or AR-A014418 (20 mg/kg; every 12 hours for 2 days) and 24 h after the last dose, TRAIL (20 mg/kg) was i.p. injected followed by 24 hours of incubation before mice were sacrifice and tumors harvested. A. mice grouping and treatments received, B. Treatment schedule.
A
B

96
4.3.5 Immunohistochemistry
Tissues from liver, kidneys and tumor were fixed in 10% neutral buffered formalin, paraffin
embedded, and processed as 4 µm sections. Serial sections of the paraffin blocks were
immunostained using H&E staining and cleaved caspase-3 as described previously (254).
4.3.6 Image Capture
Both cleaved caspase-3 and H&E-stained slides were imaged with a brightfield ScanScope
CS Scanner (Aperio Technologies, Vista, CA) at 20X magnification. TIFF images (8-bit)
were acquired for analysis. For larger magnifications, the images were captured using an
Olympus BX41 microscope, 40X objective, and an infinity digital camera.
4.3.7 Quantification of Cleaved Caspase-3
Immunohistochemistry slides stained for cleaved caspase-3 were subjected to interpretation
by a pathologist (Dr. Bizhan Badarchi) using a double-blinded approach to gain a H-score.
Using an Olympus Bx41 microscope, the staining intensity of each specimen was judged
relative to the intensity of a control slide containing an adjacent section stained with H&E
and also a cleaved caspase -3 (CC-3) stained slide fror the mice receiving control vehicle
treatment. A score of 1+ indicated weak staining relative to background, 2+ = moderate
staining, and 3+ = strong staining. According to standard pathology practice, staining
intensity was reported at the highest level of intensity observed in all tissue elements. For
comparison of staining among tissues, the results were quantified by calculation of a
complete H-score that considers both staining intensity and the percentage of cells stained at
a specific range of intensities. A complete H-score was calculated by multiplying the
intensity into the percentage positively stained cells (H= P X I) as described previously
(254).
4.3.8 Statistical Analysis
Statistical analysis of the complete H-scores obtained for the cleaved caspase-3 stained tumor
sample populations was carried out by using the two-tailed Student's t-test with unpaired data
of equal variance. Statistical significance were considered where value p= < 0.05. GraphPad
Prism version 5.01 software was used.

97
4.4 Results
4.4.1 Preliminary Studies Established A Non-toxic Dose Schedule for Acute Treatment of AR-18
In order to obtain a non-toxic acute dose, we performed a series of preliminary toxicity
assays where we used single i.p. injection of different doses of AR-18 (120, 100, 80, 60, 40,
20, and 5 mg/kg) and measured the survival and physiological response of the animals (data
not shown). All the doses were toxic to the animals and resulted in death except for doses of
< 20 mg/kg (data not shown). This was in contrast to the previous reports from Ougolkov et
al., (2006), where they administered a much higher dose of 120 mg/kg, twice daily for 2 days
(185). At our dose of < 20 mg/kg, aside from mild diarrhoea, the mice showed no obvious
signs of sickness or abnormality; the mice had bright eyes, shiny and flat fur, normal
movements, and normal eating and drinking. The results indicated that 20 mg/kg is the
highest possible dose, suitable for investigating the apoptotic inducing effect of AR-18 alone
or in combination with TRAIL.
4.4.2 In vivo Inhibition of GSK-3 Results in Increased β-catenin Levels
In order to investigate the efficacy of GSK-3 inhibition in tumors treated with the small
molecule inhibitor of GSK-3, AR-18, we tested whether or not β-catenin levels are increased.
Tumor lysates were subjected to expression analysis using immunoblotting and the resulting
bands were analysed using Image Scope software as described in the Materials and Methods
section. As shown in Figure 24, in tumors treated with AR-18, there was a significant
(p<0.0001) increase in β-catenin levels when compared to DMSO treated samples.
Unexpectedly, we observed that the levels of β-catenin were also significantly increased in
TRAIL treated mice (p<0.0001) when compared to DMSO treated controls. The
combination of both drugs also significantly increased β-catenin levels when compared to
DMSO controls, but the values were not significantly higher than AR-18 alone. The results
indicate that GSK-3 inhibition was successfully achieved in xenograft tumors and there is a
also a novel TRAIL-induced β-catenin increasing effect which is not previously reported.
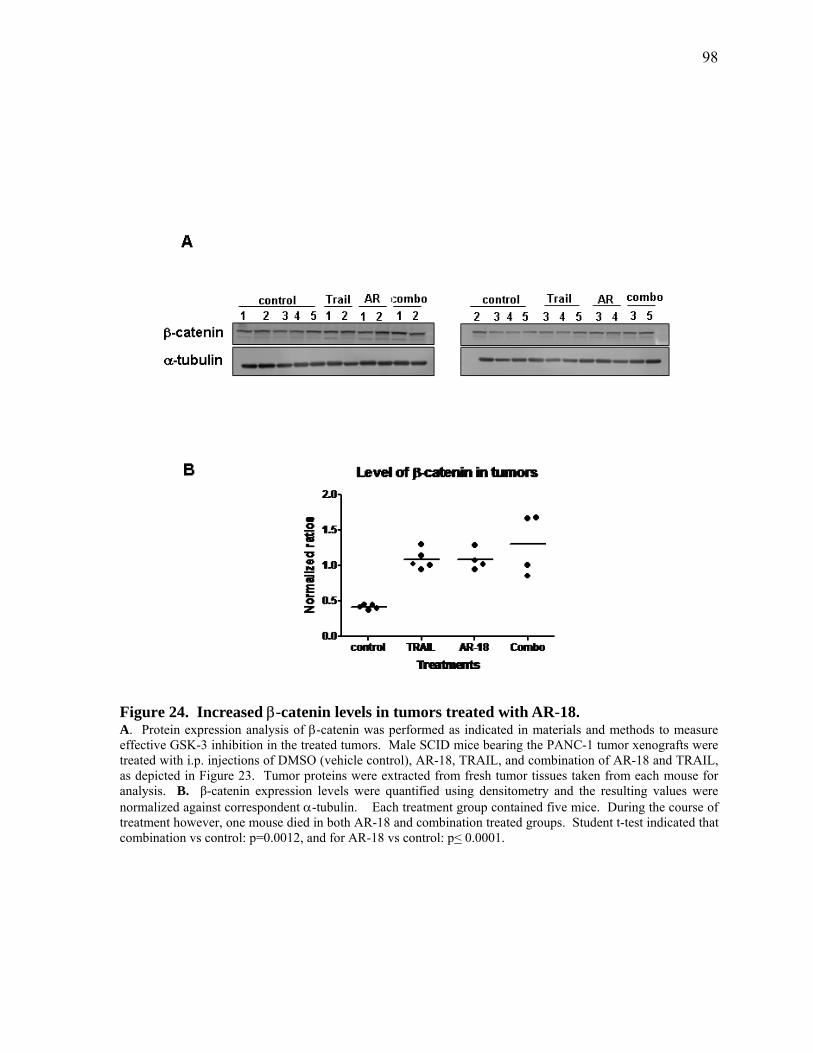
98
Figure 24. Increased β-catenin levels in tumors treated with AR-18. A. Protein expression analysis of β-catenin was performed as indicated in materials and methods to measure effective GSK-3 inhibition in the treated tumors. Male SCID mice bearing the PANC-1 tumor xenografts were treated with i.p. injections of DMSO (vehicle control), AR-18, TRAIL, and combination of AR-18 and TRAIL, as depicted in Figure 23. Tumor proteins were extracted from fresh tumor tissues taken from each mouse for analysis. B. β-catenin expression levels were quantified using densitometry and the resulting values were normalized against correspondent α-tubulin. Each treatment group contained five mice. During the course of treatment however, one mouse died in both AR-18 and combination treated groups. Student t-test indicated that combination vs control: p=0.0012, and for AR-18 vs control: p< 0.0001.

99
Figure 25. Synergistic interaction of GSK-3 and TRAIL in apoptosis induction in vivo. A. Immunohistochemistry staining of cleaved caspase-3 and H&E in PANC-1 s.c. tumor xenografts in male SCID mice. Groups of five mice were i.p. treated with acute doses of DMSO (control), AR-18, TRAIL or their combination, as depicted in Figure 23. Tumors were excited, formaline fixed, stained and visualized under an Olympus BX41 microscope. A representative section imaged using a 40X magnification objective lense from one mouse in each group is presented to the right. B. Quantification of apoptosis in the tumor samples using H- score. Cleaved caspase-3 stained slides were scored for intensity of the staining (I) as well as percentage of cells positive for the corresponding stain (H=I x P). Results of unpaired student t-test (n=5 in each treatment group) are as follows: TRAIL vs control (p=0.0002), combination vs control (p=0.0004), combination vs TRAIL (p=0.0261), combination vs AR-18 (p=0.0035).

100
4.4.3 Combination Therapy Increases Cleaved Caspase-3 In vivo
In Chapter 3 of the thesis, we showed that inhibition of GSK-3 sensitizes TRAIL-resistant
pancreatic cancer cells such as PANC-1 to TRAIL-induced apoptosis in vitro. In Chapter 4,
we investigated if such an effect is applicable to in vivo situations as well. As shown in
Figure 25, the H-score measurements of the immunohistochemistry stained slides for H&E
and cleaved caspase-3 indicated that TRAIL alone induce significant (p=0.0002) apoptosis
induction in PANC-1 tumors, whereas AR-18 as a single agent had non-significant effects.
However, the combination of AR-18 and TRAIL showed a larger effect in inducing apoptosis
when compared to single treatments or DMSO treated controls: combination vs control
(p=0.0004), combination vs TRAIL (p=0.0261), combination vs AR-18 (p=0.0035). The
results are consistent with the observations described in Chapter 3, where AR-18 alone could
not induce apoptosis, but its combination with TRAIL could enhance apoptosis induction in
resistant cells.
4.4.4 Tumor Sensitization to TRAIL by AR-18 did not Significantly Increase Host Toxicity
To investigate any potential toxic effects in the animals received 20 mg/kg AR-18 alone or in
combination with TRAIL, liver and kidneys were formalin fixed, paraffin embedded and
sections were stained with H&E and cleaved caspase-3 as described in the Materials and
Methods. The stained slides were examined by a pathologist. The results indicated that
although one mouse died in each of the AR-18 and combination treated groups of animals,
the livers and kidneys of the remaining mice (n=5 in DMSO, n=5 in TRAIL, n=4 in AR-18,
and n=4 in combination) showed no obvious morphological changes of injury including
necrosis or apoptosis. The body weight of the animals measured daily for the 5 days of
treatment indicated that there was no significant decrease in the body weight of any treatment
groups when compared to DMSO control group (Figure 26). The results indicate that the
doses used in this study as well as the treatment schedule did not induce any significant
toxicity in the treated animals.
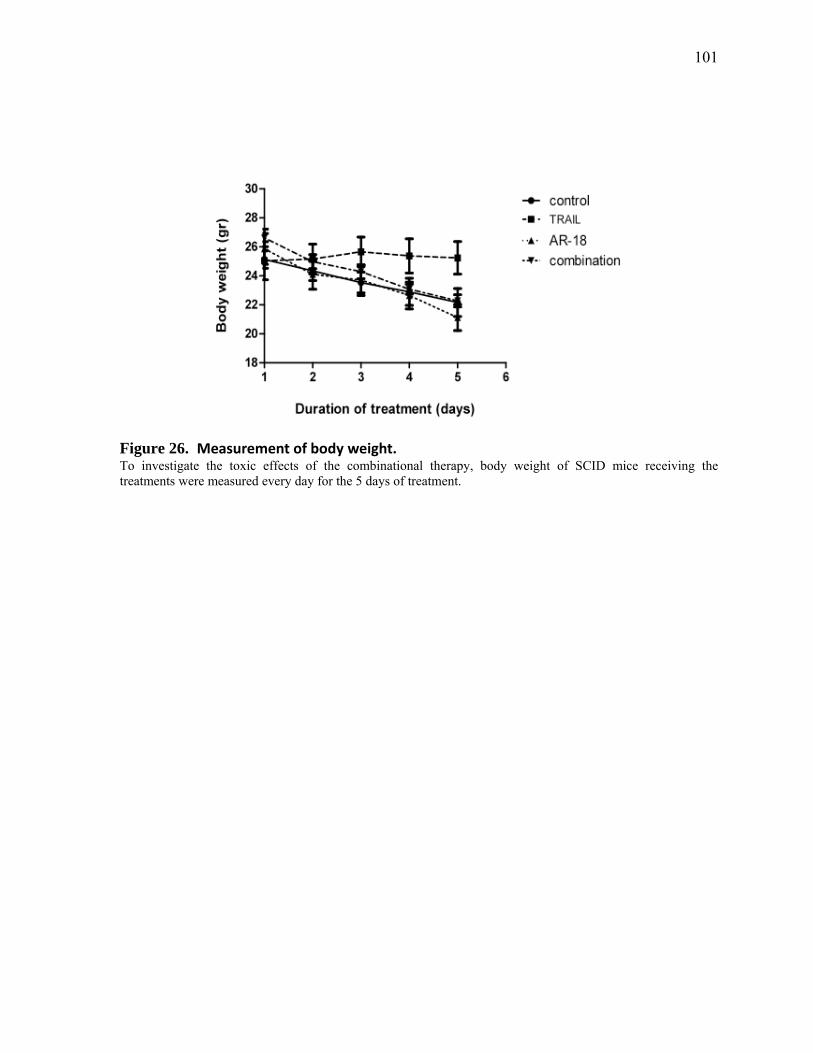
101
Figure 26. Measurement of body weight. To investigate the toxic effects of the combinational therapy, body weight of SCID mice receiving the treatments were measured every day for the 5 days of treatment.

102
4.4.5 Discussion
In this study, we investigated whether GSK-3 inhibition, in combination with TRAIL could
synergistically induce apoptosis in the pancreatic cancer tumors in vivo. The outcome of this
study indicates that although short-term GSK-3 inhibition by the small molecule inhibitor
AR-18 was insufficient to induce apoptosis, it was efficient in sensitizing PANC-1 tumors to
TRAIL induced apoptosis.
GSK-3 inhibition in tumors is expected to raise the level of cytosolic β-catenin levels (96,
98). In order to investigate the efficacy of GSK-3 inhibition by AR-18, expression of β-
catenin was measured using immunoblotting. There was a highly significant increase in the
expression levels of β-catenin following treatment with AR-18, confirming GSK-3 inhibition
in vivo. Intriguingly TRAIL also significantly increased the expression of β-catenin in the
tumors. β-catenin is known for its role in tumorigenesis (184), and also previous studies
have established a close connection between increased expression of β-catenin and resistance
of cancer cells to TRAIL-induced apoptosis (255). As a result, it is possible that increased
levels of β-catenin in response to TRAIL are a part of a cancer cell’s defence mechanism to
maintain its physiological balance towards survival. The combination of AR-18 and TRAIL
had similar β-catenin inducing effects as the single agents and did not result in lowering of
the β-catenin levels as might be expected. This finding is of significance, since in our hands,
the combination of AR-18 and TRAIL is significantly apoptotic in pancreatic cancer tumors
when compared to single agents. As a result, it might be concluded that the apoptotic effect
of the TRAIL and AR-18 combination could bypass the β-catenin anti-apoptotic effects.
This might be intriguing as many tumors harbour activating mutations of β-catenin that
causes resistance to the conventional therapies and that inhibiting β-catenin was required to
overcome TRAIL resistance (256). Our results may suggest that at least in pancreatic cancer,
dysregulated β-catenin is not much of a concern in TRAIL sensitization after GSK-3
inhibition.
Consistent with our previous results in Chapter 2 and 3 of the thesis, immunohistochemistry
analysis of apoptosis in treated xenografts indicated that AR-18 alone did not cause any
significant increase in apoptosis, yet it was quite effective in initiating TRAIL-induced
apoptosis. Interestingly AR-18 alone could block cell proliferation and survival in vitro as

103
shown in Chapter 2, but the effect seems caspase-independent as studied in Chapters 3 and 4
of the thesis. As GSK-3 controls multiple survival pathways, its inhibition might affect cell
survival through effects on microtubule assembly, cell cycle progression, or downregulation
of survival factors such as NF-κB (93). However, because of the multi-tasking nature of
GSK-3, it is possible that additional or unpredictable effects might also occur as a result of
drug treatment. For instance, in vivo use of AR-18 exhibited severe toxic effects, despite an
earlier publication describing the use of much higher doses (185). Potentially drug toxicity
observed in the present study might have been the result of off-target effects of AR-18, or the
consequence of selective GSK-3 inhibition. AR-18 was originally developed by AstraZeneca
as a potential drug for the treatment of type II diabetes, but was withdrawn early in
development for undocumented reasons, suggesting that it is probably not an optimum GSK-
3 inhibitor for in vivo use. At the present time there are no alternative drugs accessible to us,
although informal contacts suggest that GSK-3 remains an attractive drug target within the
pharmaceuticals industry. Nonetheless, results of Chapter 4 showed that (within the
limitations of AR-18) the combination of a GSK-3 inhibitor and TRAIL appears to be a safe
and effective new approach to target pancreatic cancer.
4.5 Conclusion
The work described in this Chapter provides important proof of principle for developing the
combination of a GSK-3 inhibitor and TRAIL to treat pancreatic cancer. Although the
combination of AR-18 and TRAIL could be further studied, including testing in other model
systems, and chronic dosing experiments, these studies might be better done once a better
GSK-3 inhibitor for potential clinical use becomes available.

Chapter 5 Discussion and Future Directions

105
5.1 Summary and Key Findings
Pancreatic cancer remains one of the most aggressive types of cancer, and despite
development of new combinational therapies, the resistance to therapy prompts the need for
novel therapeutic approaches (1). In this thesis, this issue was addressed by investigating
potential anti-survival effects of GSK-3 inhibition by using small molecule inhibitor; AR-18
or genetic knockdown using siRNA. In Chapter 2, it was found that blocking GSK-3 had
anti-survival effects on a panel of pancreatic cancer cells in vitro and that this effect was NF-
κB dependent. Downstream targets of NF-κB such as: Bcl-XL, cyclin D1, and XIAP were
downregulated upon GSK-3 inhibition and NF-κB activity was blocked. We further showed
that blocking GSK-3 and NF-κB activity does not sensitize pancreatic cancer cells to
gemcitabine effect. This finding was in contrast to previous reports suggesting that NF-κB is
a key factor in chemo-resistance of pancreatic cancer cells (49, 219). Interestingly, for the
first time in pancreatic cancer, we showed that in line with previous findings by others (72,
73, 182), GSK-3 inhibition blocked TNF-α induced NF-κB activation, indicating that GSK-3
has a role in blocking death receptor pathways. In Chapter 3, we showed for the first time
that pre-treatment of pancreatic cancer cells with a GSK-3 inhibitor remarkably sensitized the
cancer cells to TRAIL-induced apoptosis. The effect was shown to be caspase-dependent
and involved the mitochondrial amplification loop. Similar results were reproduced in
prostate cancer cells. We then showed that besides caspase-8, other molecules such as Bcl-
XL, and Mcl-1 are involved in the sensitization process. Further in vivo effects of TRAIL
and GSK-3 inhibition were investigated in Chapter 4 of the thesis and the results were
consistent with the previous findings in Chapter 3. In the tumors treated with a combination
of TRAIL and GSK-3 inhibitor, there was a significant increase in apoptosis induction when
compared to single agent treatments. This sensitization effect was associated with minimal
toxic effects.
5.2 Future Work
Previous studies identified GSK-3 as molecular target for the treatment of diseases such as
Alzheimer’s disease, bipolar disorder, stroke, schizophrenia, depression, and diabetes
mellitus, but the role of GSK-3 in the survival of human cancers has been controversial (93,

106
206, 257, 258). The observation that GSK-3 negatively regulates the expression and stability
of oncogenic proteins such: as β-catenin, cyclin D1, cyclin E, c-Jun, and c-Myc, raises the
question as to whether GSK-3 inhibition would promote tumorigenesis rather than tumor
suppression (174, 259). On the other hand, GSK-3 is involved in the positive regulation of
tumor promoting transcription factor NF-κB, and its inhibition has an anti-survival effect for
many cancers including pancreatic cancer (92, 99, 179, 202). Furthermore, supporting
evidence indicated that GSK-3 inhibition does not correlate with increased risk of cancer and
in contrast it was shown that psychiatric patients treated with lithium, a well known GSK-3
inhibitor, had a lower incidence of cancer related deaths (260). As a result, recently the
attention has shifted to a potential oncogenic role of GSK-3, and its inhibition considered as a
prospective therapeutic target (206). Since GSK-3 regulates many proteins and is involved in
multiple metabolic pathways, there might be other potential target molecules besides NF-κB,
that are involved in the survival effects of GSK-3 in cancer cells (169, 259). Identifying
detailed molecular mechanisms through which GSK-3 regulates tumor cell survival and
proliferation and contributes to the pathogenesis of cancer is necessary for the development
of new anti-cancer strategies, and will be the focus of our subsequent study.
As the blocking of GSK-3 activity suppresses tumor survival, the use of GSK-3 inhibitors as
monotherapy or in combination with other therapeutic agents seems logical. Over the past
decade, the need to develop efficient safe GSK-3 inhibitors for the treatment of Alzheimer’s
disease, diabetes, stroke, and cancer has led to the development of many compounds which
have been mostly tested in vitro, although a few have found their way to pre-clinical (in vivo)
or clinical trials (173, 261). GSK-3 inhibitors are divided into different sub categories of
direct inhibitors such as: lithium, and small molecule inhibitors (Paullones, thiazoles (AR-
A014418)), or indirect inhibitors such as: valporic acid, and enzastaurin (173, 206). Small
molecule inhibitors can be ATP-competitive inhibitors or substrate-competitive inhibitors
depending on the nature of their action (206). In any case, these inhibitors are all potent and
comparatively selective but do not exhibit isoform specificity and inhibit both GSK-3
isoforms (206). Our next study will be to inhibit GSK-3 isoforms using isoform-specific
siRNAs in pancreatic cancer tumors and study the effect on tumor growth and survival in
vivo.

107
For several years, lithium has been the only GSK-3 inhibitor that has been clinically used in
patients with mood disorders (206). A search of the ClinicalTrials.gov website by Medina et
al., in 2008 showed that about 100 active trials using lithium alone or in combination with
other agents are ongoing. The majority of these trials were related to disorders of the
nervous system and 9 were trials related to tumors (206). However, the majority of these
trials were discontinued and limited information is available about their outcomes. Other
clinical trials running in 2008 used small molecule inhibitors such as NP-12 (Neuropharma
SA) or AZD-1080 (Astra Zeneca plc) for treatment of Alzheimer’s disease (206). However,
the AZD trial was discontinued due to unknown reasons and NP-12 has been the only small
molecule inhibitor that made its way to the completion of the study (206). NP-12 is a small
molecule inhibitor from a chemical class of non-ATP-competitive GSK-3 inhibitors called
thiadiazolidinones (TDZD) (261). However, no clinical data on the results are publicly
available. A search of the ClinicalTrials.gov website in 2010 showed 2 actively recruiting
clinical trials using Enzastaurin in combination with Carboplatin or Bevacizumab for treating
patients with either brain tumor or unspecified metastatic solid tumors respectively (262).
Many protein kinase inhibitor trials do not reach the clinic as a major problem with clinical
use of protein kinase inhibitors is that a target-specific effect is required. However, ATP-
competitive non-specific kinase inhibitors might have broader effect than the target kinase.
As this is the case with a majority of GSK-3 inhibitors, many of these agents can not be used
in drug development programs (261). Moreover, ATP-competitor inhibitors such as AR-
A014418 (AR-18) might have non-specific protein kinase inhibiting effect which should be
taken into consideration. In our study, we had access to AR-18 as the only selective GSK-3
inhibitor to run both in vitro and in vivo studies. Although AR-18 is highly selective and
potent, but at high doses, it is non-specific and toxic and low doses are not as effective as
expected. As a result, a potent GSK-3 inhibitor with minimal toxic effects such as TDZD
would be ideal to be used for the future in vivo studies (261).
Most pancreatic cancers have a pre-existing or developed resistance to both chemo- and
radiation-therapies and the conventional therapies lack target specificity that leads to
systemic toxicity in patients (263). To overcome these limitations, novel targeted approaches
using combinational therapies have been suggested to lower the toxicity, increase the
efficacy of the treatment response, overcome resistance, and also improve the quality of life

108
of patients (39). In this regard, TRAIL was a suitable candidate for our study as it has been
proven to be specific for targeting tumors and sparing normal cells (17). However, many
pancreatic cancer cells or primary tumors are resistant to TRAIL induced apoptotic effect.
As a result, pre-treatment of TRAIL resistant cells with a chemotherapeutic or radiation is
suggested and shown to be safe to patients (17). As GSK-3 blocks death receptor mediated
apoptosis induction (28), blocking GSK-3 successfully could sensitize our resistant cells both
in vitro and in vivo. However the half life of TRAIL in serum is short (about 30 minutes).
An alternative approach is the use of humanized monoclonal antibodies to activate the
TRAIL receptors, and these are being developed by Amgen and other companies for human
clinical trials. The combination of a next-generation GSK-3 inhibitor and a TRAIL activating
antibody would be attractive for advanced pre-clinical, and eventually clinical testing of the
novel approach developed in this thesis.
5.3 Concluding Remarks
The results of this thesis pave the way to a better understanding of the molecular
mechanisms involved in the resistance of pancreatic cancer to chemotherapeutic
agents such as gemcitabine. NF-κB is shown not to be the key factor in causing drug
resistance and although GSK-3 inhibition did not sensitize pancreatic cancer cells to
gemcitabine, it initiated sensitization to TRAIL and inducing apoptosis. As the
current knowledge about combinational therapies is limited, this thesis has provided a
platform for the generation of therapeutic designs to overcome drug resistance in both
pancreatic and prostate cancer cells with maximum safety. The final proof of concept
that can validate these results will only be available upon clinical trials.

Appendix
110
Figure A1: Downregulation of NF-κB target gene expression upon LiCl treatment. Western blot analysis of expression of NF-κB target genes XIAP, and cyclin D1in PANC-1 cells after exposure to LiCl (10-50 mM) for 48 h. KCl (10 mM) is used as vehicle control. Increased cytosolic β-catenin expression confirms GSK-3 inhibition in a dose-dependent manner. β-actin is used as loading control.

References

112
1. Anirban Maitra aRHH. Pancreatic Cancer. Annual Review of Pathology:
mechanisms of Disease. 2008;3:157-88.
2. Canadian Cancer Society SCPTCR, Public Health Agencies of Canada. End
of Life Care; 2010.
3. Chua YJ, Zalcberg JR. Pancreatic cancer--is the wall crumbling? Ann Oncol.
2008;19:1224-30.
4. Pierantoni C, Pagliacci A, Scartozzi M, Berardi R, Bianconi M, Cascinu S.
Pancreatic cancer: Progress in cancer therapy. Crit Rev Oncol Hematol. 2008;67:27-
38.
5. Garcea G, Dennison AR, Steward WP, Berry DP. Role of inflammation in
pancreatic carcinogenesis and the implications for future therapy. Pancreatology.
2005;5:514-29.
6. Duffy JP, Eibl G, Reber HA, Hines OJ. Influence of hypoxia and
neoangiogenesis on the growth of pancreatic cancer. Mol Cancer. 2003;2:12.
7. Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano
MR, et al. Improvements in survival and clinical benefit with gemcitabine as first- line
therapy for patients with advanced pancreas cancer: a randomized trial. J Clin
Oncol. 1997;15:2403-13.
8. Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V.
Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin
Oncol. 1995;22:3-10.
9. Rosenberg L. Treatment of pancreatic cancer. Promises and problems of
tamoxifen, somatostatin analogs, and gemcitabine. Int J Pancreatol. 1997;22:81-93.
10. Welch SA, Moore MJ. Combination chemotherapy in advanced pancreatic
cancer: time to raise the white flag? J Clin Oncol. 2007;25:2159-61.
11. Hansel DE, Kern SE, Hruban RH. Molecular pathogenesis of pancreatic
cancer. Annu Rev Genomics Hum Genet. 2003;4:237-56.
12. Ueno H, Kiyosawa K, Kaniwa N. Pharmacogenomics of gemcitabine: can
genetic studies lead to tailor-made therapy? Br J Cancer. 2007;97:145-51.
13. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009.
CA Cancer J Clin. 2009;59:225-49.

113
14. Hruban RH, Maitra A, Goggins M. Update on pancreatic intraepithelial
neoplasia. Int J Clin Exp Pathol. 2008;1:306-16.
15. Lowenfels AB, Maisonneuve P, DiMagno EP, Elitsur Y, Gates LK, Jr., Perrault
J, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International
Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89:442-6.
16. Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev
Cancer. 2002;2:897-909.
17. Papenfuss K, Cordier SM, Walczak H. Death receptors as targets for anti-
cancer therapy. J Cell Mol Med. 2008;12:2566-85.
18. Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour
cells. Nat Rev Cancer. 2004;4:592-603.
19. Igney FH, Krammer PH. Death and anti-death: tumour resistance to
apoptosis. Nat Rev Cancer. 2002;2:277-88.
20. Pacifico F, Leonardi A. NF-kappaB in solid tumors. Biochem Pharmacol.
2006;72:1142-52.
21. Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene.
2003;22:8543-67.
22. Wong HH, Lemoine NR. Pancreatic cancer: molecular pathogenesis and new
therapeutic targets. Nat Rev Gastroenterol Hepatol. 2009;6:412-22.
23. Minagawa N, Kruglov EA, Dranoff JA, Robert ME, Gores GJ, Nathanson MH.
The Anti-apoptotic Protein Mcl-1 Inhibits Mitochondrial Ca2+ Signals. Journal of
Biological Chemistry. 2005;280:33637-44.
24. Hamacher R, Schmid RM, Saur D, Schneider G. Apoptotic pathways in
pancreatic ductal adenocarcinoma. Mol Cancer. 2008;7:64.
25. Adams JM, Cory S. Bcl-2-regulated apoptosis: mechanism and therapeutic
potential. Curr Opin Immunol. 2007;19:488-96.
26. Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and
therapy. Oncogene. 2007;26:1324-37.
27. Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G,
Vandenabeele P. Toxic proteins released from mitochondria in cell death.
Oncogene. 2004;23:2861-74.

114
28. Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3
in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol.
2006;79:173-89.
29. Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close
to IP3-sensitive channels that are sensed by neighboring mitochondria. Science.
1993;262:744-7.
30. Zhao Y, Altman BJ, Coloff JL, Herman CE, Jacobs SR, Wieman HL, et al.
Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive
antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol. 2007;27:4328-39.
31. Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen
synthase kinase-3 regulates mitochondrial outer membrane permeabilization and
apoptosis by destabilization of MCL-1. Mol Cell. 2006;21:749-60.
32. Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, et al. Degradation of Mcl-1 by
beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and
chemosensitization. Mol Cell Biol. 2007;27:4006-17.
33. Ding Q, He X, Xia W, Hsu JM, Chen CT, Li LY, et al. Myeloid cell leukemia-1
inversely correlates with glycogen synthase kinase-3beta activity and associates
with poor prognosis in human breast cancer. Cancer Res. 2007;67:4564-71.
34. Vogler M, Walczak H, Stadel D, Haas TL, Genze F, Jovanovic M, et al.
Targeting XIAP bypasses Bcl-2-mediated resistance to TRAIL and cooperates with
TRAIL to suppress pancreatic cancer growth in vitro and in vivo. Cancer Res.
2008;68:7956-65.
35. Ashkenazi A, Dixit VM. Death Receptors: Signaling and Modulation. Science.
1998;281:1305-8.
36. Vogler M, Durr K, Jovanovic M, Debatin KM, Fulda S. Regulation of TRAIL-
induced apoptosis by XIAP in pancreatic carcinoma cells. Oncogene. 2007;26:248-
57.
37. Mahmood Z, Shukla Y. Death receptors: Targets for cancer therapy. Exp Cell
Res. 2009.
38. Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev
Cancer. 2009;9:501-7.

115
39. Ashkenazi A, Holland P, Eckhardt SG. Ligand-based targeting of apoptosis in
cancer: the potential of recombinant human apoptosis ligand 2/Tumor necrosis
factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol.
2008;26:3621-30.
40. Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, et al.
Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in
vivo. Nat Med. 1999;5:157-63.
41. Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, et al.
Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest.
1999;104:155-62.
42. Wajant H. TRAIL and NFkappaB signaling--a complex relationship. Vitam
Horm. 2004;67:101-32.
43. Khanbolooki S, Nawrocki ST, Arumugam T, Andtbacka R, Pino MS, Kurzrock
R, et al. Nuclear factor-kappaB maintains TRAIL resistance in human pancreatic
cancer cells. Mol Cancer Ther. 2006;5:2251-60.
44. Hylander BL, Pitoniak R, Penetrante RB, Gibbs JF, Oktay D, Cheng J, et al.
The anti-tumor effect of Apo2L/TRAIL on patient pancreatic adenocarcinomas grown
as xenografts in SCID mice. J Transl Med. 2005;3:22.
45. Hinz S, Trauzold A, Boenicke L, Sandberg C, Beckmann S, Bayer E, et al.
Bcl-XL protects pancreatic adenocarcinoma cells against CD95- and TRAIL-
receptor-mediated apoptosis. Oncogene. 2000;19:5477-86.
46. Vogler M, Walczak H, Stadel D, Haas TL, Genze F, Jovanovic M, et al. Small
molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in
preclinical models of pancreatic carcinoma. Cancer Res. 2009;69:2425-34.
47. Trauzold A, Siegmund D, Schniewind B, Sipos B, Egberts J, Zorenkov D, et
al. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma.
Oncogene. 2006;25:7434-9.
48. Zhou DH, Trauzold A, Roder C, Pan G, Zheng C, Kalthoff H. The potential
molecular mechanism of overexpression of uPA, IL-8, MMP-7 and MMP-9 induced
by TRAIL in pancreatic cancer cell. Hepatobiliary Pancreat Dis Int. 2008;7:201-9.

116
49. Arlt A, Gehrz A, Muerkoster S, Vorndamm J, Kruse ML, Folsch UR, et al.
Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines
against gemcitabine-induced cell death. Oncogene. 2003;22:3243-51.
50. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-
kappaB functions as a tumour promoter in inflammation-associated cancer. Nature.
2004;431:461-6.
51. Sebens S, Arlt A, Schafer H. NF-kappaB as a molecular target in the therapy
of pancreatic carcinoma. Recent Results Cancer Res. 2008;177:151-64.
52. Holcomb B, Yip-Schneider M, Schmidt CM. The role of nuclear factor kappaB
in pancreatic cancer and the clinical applications of targeted therapy. Pancreas.
2008;36:225-35.
53. Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell.
2004;6:203-8.
54. Shishodia S, Aggarwal BB. Nuclear factor-kappaB: a friend or a foe in
cancer? Biochem Pharmacol. 2004;68:1071-80.
55. Cusack JC, Liu R, Baldwin AS. NF- kappa B and chemoresistance:
potentiation of cancer drugs via inhibition of NF- kappa B. Drug Resist Updat.
1999;2:271-3.
56. Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB
kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817-
30.
57. Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding
protein Nf-kappa B by a posttranslational mechanism. Cell. 1986;47:921-8.
58. Albert S. Baldwin J. The transcription factor NF-κB and human disease. The
Journal of Clinical Investigation. 2001;107:3-6.
59. Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily
conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225-60.
60. Moynagh PN. The NF-kappaB pathway. J Cell Sci. 2005;118:4589-92.
61. Reber L, Vermeulen L, Haegeman G, Frossard N. Ser276 phosphorylation of
NF-kB p65 by MSK1 controls SCF expression in inflammation. PLoS One.
2009;4:e4393.

117
62. Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of
NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621-63.
63. Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev
Mol Cell Biol. 2004;5:392-401.
64. Wang D, Westerheide SD, Hanson JL, Baldwin AS, Jr. Tumor necrosis factor
alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase
II. J Biol Chem. 2000;275:32592-7.
65. Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases
phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J
Biol Chem. 1999;274:30353-6.
66. Duran A, Diaz-Meco MT, Moscat J. Essential role of RelA Ser311
phosphorylation by zetaPKC in NF-kappaB transcriptional activation. Embo J.
2003;22:3910-8.
67. Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G.
Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-
activated protein kinase-1 (MSK1). Embo J. 2003;22:1313-24.
68. Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA
stimulates transcriptional activity by promoting a novel bivalent interaction with the
coactivator CBP/p300. Mol Cell. 1998;1:661-71.
69. Schmitz ML, Mattioli I, Buss H, Kracht M. NF-kappaB: a multifaceted
transcription factor regulated at several levels. Chembiochem. 2004;5:1348-58.
70. Buss H, Dorrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive
and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is
mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha},
IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1
(TBK1), and an unknown kinase and couples p65 to TATA-binding protein-
associated factor II31-mediated interleukin-8 transcription. J Biol Chem.
2004;279:55633-43.
71. Buss H, Dorrie A, Schmitz ML, Frank R, Livingstone M, Resch K, et al.
Phosphorylation of Serine 468 by GSK-3{beta} Negatively Regulates Basal p65 NF-
{kappa}B Activity. J Biol Chem. 2004;279:49571-4.

118
72. Schwabe RF, Brenner DA. Role of glycogen synthase kinase-3 in TNF-alpha
-induced NF-kappa B activation and apoptosis in hepatocytes. Am J Physiol
Gastrointest Liver Physiol. 2002;283:G204-11.
73. Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement
for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation.
Nature. 2000;406:86-90.
74. Bonnard M, Mirtsos C, Suzuki S, Graham K, Huang J, Ng M, et al. Deficiency
of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent
gene transcription. Embo J. 2000;19:4976-85.
75. Tojima Y, Fujimoto A, Delhase M, Chen Y, Hatakeyama S, Nakayama K, et
al. NAK is an IkappaB kinase-activating kinase. Nature. 2000;404:778-82.
76. Kravchenko VV, Mathison JC, Schwamborn K, Mercurio F, Ulevitch RJ.
IKKi/IKKepsilon plays a key role in integrating signals induced by pro-inflammatory
stimuli. J Biol Chem. 2003;278:26612-9.
77. Yin L, Wu L, Wesche H, Arthur CD, White JM, Goeddel DV, et al. Defective
lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-
deficient mice. Science. 2001;291:2162-5.
78. Leitges M, Sanz L, Martin P, Duran A, Braun U, Garcia JF, et al. Targeted
disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway.
Mol Cell. 2001;8:771-80.
79. Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways
for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer
Res. 2009;15:425-30.
80. Collett GP, Campbell FC. Overexpression of p65/RelA potentiates curcumin-
induced apoptosis in HCT116 human colon cancer cells. Carcinogenesis.
2006;27:1285-91.
81. Kaltschmidt B, Kaltschmidt C, Hofmann TG, Hehner SP, Droge W, Schmitz
ML. The pro- or anti-apoptotic function of NF-kappaB is determined by the nature of
the apoptotic stimulus. Eur J Biochem. 2000;267:3828-35.
82. Farhana L, Dawson MI, Fontana JA. Apoptosis induction by a novel retinoid-
related molecule requires nuclear factor-kappaB activation. Cancer Res.
2005;65:4909-17.

119
83. Chen X, Kandasamy K, Srivastava RK. Differential roles of RelA (p65) and c-
Rel subunits of nuclear factor kappa B in tumor necrosis factor-related apoptosis-
inducing ligand signaling. Cancer Res. 2003;63:1059-66.
84. Jin F, Liu X, Zhou Z, Yue P, Lotan R, Khuri FR, et al. Activation of nuclear
factor-kappaB contributes to induction of death receptors and apoptosis by the
synthetic retinoid CD437 in DU145 human prostate cancer cells. Cancer Res.
2005;65:6354-63.
85. Ho WC, Dickson KM, Barker PA. Nuclear factor-kappaB induced by
doxorubicin is deficient in phosphorylation and acetylation and represses nuclear
factor-kappaB-dependent transcription in cancer cells. Cancer Res. 2005;65:4273-
81.
86. Campbell KJ, Rocha S, Perkins ND. Active repression of antiapoptotic gene
expression by RelA(p65) NF-kappa B. Mol Cell. 2004;13:853-65.
87. Fujioka S, Sclabas GM, Schmidt C, Frederick WA, Dong QG, Abbruzzese JL,
et al. Function of nuclear factor kappaB in pancreatic cancer metastasis. Clin Cancer
Res. 2003;9:346-54.
88. Sclabas GM, Fujioka S, Schmidt C, Evans DB, Chiao PJ. NF-kappaB in
pancreatic cancer. Int J Gastrointest Cancer. 2003;33:15-26.
89. Arlt A, Vorndamm J, Breitenbroich M, Folsch UR, Kalthoff H, Schmidt WE, et
al. Inhibition of NF-kappaB sensitizes human pancreatic carcinoma cells to
apoptosis induced by etoposide (VP16) or doxorubicin. Oncogene. 2001;20:859-68.
90. Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The
Nuclear Factor-κB RelA Transcription Factor Is Constitutively Activated in Human
Pancreatic Adenocarcinoma Cells. Clinical Cancer Research. 1999;5:119-27.
91. Nakashima H, Nakamura M, Yamaguchi H, Yamanaka N, Akiyoshi T, Koga K,
et al. Nuclear factor-kappaB contributes to hedgehog signaling pathway activation
through sonic hedgehog induction in pancreatic cancer. Cancer Res. 2006;66:7041-
9.
92. Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD.
Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated
gene transcription and cell survival in pancreatic cancer cells. Cancer Res.
2005;65:2076-81.

120
93. Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase.
J Cell Sci. 2003;116:1175-86.
94. Luo J. Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and
cancer chemotherapy. Cancer Lett. 2009;273:194-200.
95. Rayasam GV, Tulasi VK, Sodhi R, Davis JA, Ray A. Glycogen synthase
kinase 3: more than a namesake. Br J Pharmacol. 2009;156:885-98.
96. Ali A, Hoeflich KP, Woodgett JR. Glycogen synthase kinase-3: properties,
functions, and regulation. Chem Rev. 2001;101:2527-40.
97. Eldar-Finkelman H, Licht-Murava A, Pietrokovski S, Eisenstein M. Substrate
competitive GSK-3 inhibitors - strategy and implications. Biochim Biophys Acta.
2010;1804:598-603.
98. Mamaghani S, Patel S, Hedley DW. Glycogen synthase kinase-3 inhibition
disrupts nuclear factor-kappaB activity in pancreatic cancer, but fails to sensitize to
gemcitabine chemotherapy. BMC Cancer. 2009;9:132.
99. Shakoori A, Ougolkov A, Yu ZW, Zhang B, Modarressi MH, Billadeau DD, et
al. Deregulated GSK3beta activity in colorectal cancer: its association with tumor cell
survival and proliferation. Biochem Biophys Res Commun. 2005;334:1365-73.
100. Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR. Glycogen
synthase kinase-3: functions in oncogenesis and development. Biochim Biophys
Acta. 1992;1114:147-62.
101. Kockeritz L, Doble B, Patel S, Woodgett JR. Glycogen synthase kinase-3--an
overview of an over-achieving protein kinase. Curr Drug Targets. 2006;7:1377-88.
102. Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR. Modulation of the
glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J.
1993;12:803-8.
103. Sayas CL, Ariaens A, Ponsioen B, Moolenaar WH. GSK-3 is activated by the
tyrosine kinase Pyk2 during LPA1-mediated neurite retraction. Mol Biol Cell.
2006;17:1834-44.
104. Hartigan JA, Xiong WC, Johnson GV. Glycogen synthase kinase 3beta is
tyrosine phosphorylated by PYK2. Biochem Biophys Res Commun. 2001;284:485-9.

121
105. Lesort M, Jope RS, Johnson GV. Insulin transiently increases tau
phosphorylation: involvement of glycogen synthase kinase-3beta and Fyn tyrosine
kinase. J Neurochem. 1999;72:576-84.
106. Takahashi-Yanaga F, Shiraishi F, Hirata M, Miwa Y, Morimoto S, Sasaguri T.
Glycogen synthase kinase-3beta is tyrosine-phosphorylated by MEK1 in human skin
fibroblasts. Biochem Biophys Res Commun. 2004;316:411-5.
107. Cole A, Frame S, Cohen P. Further evidence that the tyrosine
phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an
autophosphorylation event. Biochem J. 2004;377:249-55.
108. ter Haar E, Coll JT, Austen DA, Hsiao HM, Swenson L, Jain J. Structure of
GSK3beta reveals a primed phosphorylation mechanism. Nat Struct Biol.
2001;8:593-6.
109. Picton C, Woodgett J, Hemmings B, Cohen P. Multisite phosphorylation of
glycogen synthase from rabbit skeletal muscle. Phosphorylation of site 5 by
glycogen synthase kinase-5 (casein kinase-II) is a prerequisite for phosphorylation of
sites 3 by glycogen synthase kinase-3. FEBS Lett. 1982;150:191-6.
110. Woodgett JR, Cohen P. Multisite phosphorylation of glycogen synthase.
Molecular basis for the substrate specificity of glycogen synthase kinase-3 and
casein kinase-II (glycogen synthase kinase-5). Biochim Biophys Acta. 1984;788:339-
47.
111. Hemmings BA, Aitken A, Cohen P, Rymond M, Hofmann F. Phosphorylation
of the type-II regulatory subunit of cyclic-AMP-dependent protein kinase by glycogen
synthase kinase 3 and glycogen synthase kinase 5. Eur J Biochem. 1982;127:473-
81.
112. Jho E, Lomvardas S, Costantini F. A GSK3beta phosphorylation site in axin
modulates interaction with beta-catenin and Tcf-mediated gene expression. Biochem
Biophys Res Commun. 1999;266:28-35.
113. Bax B, Carter PS, Lewis C, Guy AR, Bridges A, Tanner R, et al. The structure
of phosphorylated GSK-3beta complexed with a peptide, FRATtide, that inhibits
beta-catenin phosphorylation. Structure. 2001;9:1143-52.

122
114. Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase
kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-
factor signalling. Biochem J. 1993;296 ( Pt 1):15-9.
115. Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-
3 beta in intact cells via serine 9 phosphorylation. Biochem J. 1994;303 ( Pt 3):701-
4.
116. Fang X, Yu S, Tanyi JL, Lu Y, Woodgett JR, Mills GB. Convergence of
multiple signaling cascades at glycogen synthase kinase 3: Edg receptor-mediated
phosphorylation and inactivation by lysophosphatidic acid through a protein kinase
C-dependent intracellular pathway. Mol Cell Biol. 2002;22:2099-110.
117. Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA. Cyclic
AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase
3beta. Mol Cell Biol. 2000;20:9356-63.
118. Cohen P. The Croonian Lecture 1998. Identification of a protein kinase
cascade of major importance in insulin signal transduction. Philos Trans R Soc Lond
B Biol Sci. 1999;354:485-95.
119. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of
glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature.
1995;378:785-9.
120. Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase
3beta in cellular signaling. Prog Neurobiol. 2001;65:391-426.
121. Parker PJ, Caudwell FB, Cohen P. Glycogen synthase from rabbit skeletal
muscle; effect of insulin on the state of phosphorylation of the seven phosphoserine
residues in vivo. Eur J Biochem. 1983;130:227-34.
122. Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, et al. Control of beta-
catenin phosphorylation/degradation by a dual-kinase mechanism. Cell.
2002;108:837-47.
123. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta
regulates cyclin D1 proteolysis and subcellular localization. Genes Dev.
1998;12:3499-511.
124. Eldar-Finkelman H, Seger R, Vandenheede JR, Krebs EG. Inactivation of
glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-

123
activated protein kinase/p90 ribosomal protein S6 kinase signaling pathway in
NIH/3T3 cells. J Biol Chem. 1995;270:987-90.
125. Gao ZH, Seeling JM, Hill V, Yochum A, Virshup DM. Casein kinase I
phosphorylates and destabilizes the beta-catenin degradation complex. Proc Natl
Acad Sci U S A. 2002;99:1182-7.
126. Lee E, Salic A, Kruger R, Heinrich R, Kirschner MW. The roles of APC and
Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS
Biol. 2003;1:E10.
127. Bijur GN, Jope RS. Glycogen synthase kinase-3 beta is highly activated in
nuclei and mitochondria. Neuroreport. 2003;14:2415-9.
128. Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, et al. Crystal
structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed
substrate specificity and autoinhibition. Cell. 2001;105:721-32.
129. Ruel L, Stambolic V, Ali A, Manoukian AS, Woodgett JR. Regulation of the
protein kinase activity of Shaggy(Zeste-white3) by components of the wingless
pathway in Drosophila cells and embryos. J Biol Chem. 1999;274:21790-6.
130. Siegfried E, Perkins LA, Capaci TM, Perrimon N. Putative protein kinase
product of the Drosophila segment-polarity gene zeste-white3. Nature.
1990;345:825-9.
131. Harwood AJ, Plyte SE, Woodgett J, Strutt H, Kay RR. Glycogen synthase
kinase 3 regulates cell fate in Dictyostelium. Cell. 1995;80:139-48.
132. Wen HC, Huang WC, Ali A, Woodgett JR, Lin WW. Negative regulation of
phosphatidylinositol 3-kinase and Akt signalling pathway by PKC. Cell Signal.
2003;15:37-45.
133. Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-
3/factor A. EMBO J. 1990;9:2431-8.
134. Insall R. Glycogen synthase kinase and Dictyostelium development: old
pathways pointing in new directions? Trends Genet. 1995;11:37-9.
135. Plyte SE, O'Donovan E, Woodgett JR, Harwood AJ. Glycogen synthase
kinase-3 (GSK-3) is regulated during Dictyostelium development via the serpentine
receptor cAR3. Development. 1999;126:325-33.

124
136. Kim L, Liu J, Kimmel AR. The novel tyrosine kinase ZAK1 activates GSK3 to
direct cell fate specification. Cell. 1999;99:399-408.
137. Ginger RS, Dalton EC, Ryves WJ, Fukuzawa M, Williams JG, Harwood AJ.
Glycogen synthase kinase-3 enhances nuclear export of a Dictyostelium STAT
protein. EMBO J. 2000;19:5483-91.
138. Vincent JP, O'Farrell PH. The state of engrailed expression is not clonally
transmitted during early Drosophila development. Cell. 1992;68:923-31.
139. Kornberg T. Compartments in the abdomen of Drosophila and the role of the
engrailed locus. Dev Biol. 1981;86:363-72.
140. Kornberg T. Engrailed: a gene controlling compartment and segment
formation in Drosophila. Proc Natl Acad Sci U S A. 1981;78:1095-9.
141. Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL, 3rd, et al. The
mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that
regulates embryonic axis formation. Cell. 1997;90:181-92.
142. Sakanaka C, Sun TQ, Williams LT. New steps in the Wnt/beta-catenin signal
transduction pathway. Recent Prog Horm Res. 2000;55:225-36.
143. Willert K, Brink M, Wodarz A, Varmus H, Nusse R. Casein kinase 2
associates with and phosphorylates dishevelled. EMBO J. 1997;16:3089-96.
144. Park M, Venkatesh TV, Bodmer R. Dual role for the zeste-white3/shaggy-
encoded kinase in mesoderm and heart development of Drosophila. Dev Genet.
1998;22:201-11.
145. Miller JR. The Wnts. Genome Biol. 2002;3:REVIEWS3001.
146. Dann CE, Hsieh JC, Rattner A, Sharma D, Nathans J, Leahy DJ. Insights into
Wnt binding and signalling from the structures of two Frizzled cysteine-rich domains.
Nature. 2001;412:86-90.
147. Lagna G, Carnevali F, Marchioni M, Hemmati-Brivanlou A. Negative
regulation of axis formation and Wnt signaling in Xenopus embryos by the F-
box/WD40 protein beta TrCP. Mech Dev. 1999;80:101-6.
148. Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW. The
SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated
destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha
ubiquitination in vitro. Genes Dev. 1999;13:270-83.

125
149. Yost C, Farr GH, 3rd, Pierce SB, Ferkey DM, Chen MM, Kimelman D. GBP,
an inhibitor of GSK-3, is implicated in Xenopus development and oncogenesis. Cell.
1998;93:1031-41.
150. Eastman Q, Grosschedl R. Regulation of LEF-1/TCF transcription factors by
Wnt and other signals. Curr Opin Cell Biol. 1999;11:233-40.
151. Pearl LH, Barford D. Regulation of protein kinases in insulin, growth factor
and Wnt signalling. Curr Opin Struct Biol. 2002;12:761-7.
152. Cui H, Meng Y, Bulleit RF. Inhibition of glycogen synthase kinase 3beta
activity regulates proliferation of cultured cerebellar granule cells. Brain Res Dev
Brain Res. 1998;111:177-88.
153. Ding VW, Chen RH, McCormick F. Differential regulation of glycogen
synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem. 2000;275:32475-
81.
154. Frame S, Cohen P, Biondi RM. A common phosphate binding site explains
the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol
Cell. 2001;7:1321-7.
155. MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, et al.
Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen
metabolism. Cell Metab. 2007;6:329-37.
156. Eldar-Finkelman H, Argast GM, Foord O, Fischer EH, Krebs EG. Expression
and characterization of glycogen synthase kinase-3 mutants and their effect on
glycogen synthase activity in intact cells. Proc Natl Acad Sci U S A. 1996;93:10228-
33.
157. McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, et
al. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by
knockin analysis. Embo J. 2005;24:1571-83.
158. Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Maintenance of
pluripotency in human and mouse embryonic stem cells through activation of Wnt
signaling by a pharmacological GSK-3-specific inhibitor. Nat Med. 2004;10:55-63.
159. Smith AG. Embryo-derived stem cells: of mice and men. Annu Rev Cell Dev
Biol. 2001;17:435-62.

126
160. Paling NR, Wheadon H, Bone HK, Welham MJ. Regulation of embryonic
stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J Biol
Chem. 2004;279:48063-70.
161. Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional
redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by
using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12:957-71.
162. Pujal J, Capella G, Real FX. The Wnt pathway is active in a small subset of
pancreas cancer cell lines. Biochim Biophys Acta. 2006;1762:73-9.
163. Dessimoz J, Grapin-Botton A. Pancreas development and cancer: Wnt/beta-
catenin at issue. Cell Cycle. 2006;5:7-10.
164. Leis H, Segrelles C, Ruiz S, Santos M, Paramio JM. Expression, localization,
and activity of glycogen synthase kinase 3beta during mouse skin tumorigenesis.
Mol Carcinog. 2002;35:180-5.
165. Ma C, Wang J, Gao Y, Gao TW, Chen G, Bower KA, et al. The role of
glycogen synthase kinase 3beta in the transformation of epidermal cells. Cancer
Res. 2007;67:7756-64.
166. Farago M, Dominguez I, Landesman-Bollag E, Xu X, Rosner A, Cardiff RD, et
al. Kinase-inactive glycogen synthase kinase 3beta promotes Wnt signaling and
mammary tumorigenesis. Cancer Res. 2005;65:5792-801.
167. Dong J, Peng J, Zhang H, Mondesire WH, Jian W, Mills GB, et al. Role of
glycogen synthase kinase 3beta in rapamycin-mediated cell cycle regulation and
chemosensitivity. Cancer Res. 2005;65:1961-72.
168. Wang Y, Lam JB, Lam KS, Liu J, Lam MC, Hoo RL, et al. Adiponectin
modulates the glycogen synthase kinase-3beta/beta-catenin signaling pathway and
attenuates mammary tumorigenesis of MDA-MB-231 cells in nude mice. Cancer
Res. 2006;66:11462-70.
169. Doble BW, Woodgett JR. Role of glycogen synthase kinase-3 in cell fate and
epithelial-mesenchymal transitions. Cells Tissues Organs. 2007;185:73-84.
170. Zhou BP, Hung MC. Wnt, hedgehog and snail: sister pathways that control by
GSK-3beta and beta-Trcp in the regulation of metastasis. Cell Cycle. 2005;4:772-6.
171. Guarino M, Rubino B, Ballabio G. The role of epithelial-mesenchymal
transition in cancer pathology. Pathology. 2007;39:305-18.

127
172. Thiel A, Heinonen M, Rintahaka J, Hallikainen T, Hemmes A, Dixon DA, et al.
Expression of cyclooxygenase-2 is regulated by glycogen synthase kinase-3beta in
gastric cancer cells. J Biol Chem. 2006;281:4564-9.
173. Billadeau GPKaDD. GSK-3β Inhibition in Pancreatic Cancer: Springer US;
2008.
174. Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the
phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem.
1998;273:19929-32.
175. Lipina C, Huang X, Finlay D, McManus EJ, Alessi DR, Sutherland C. Analysis
of hepatic gene transcription in mice expressing insulin-insensitive GSK3. Biochem
J. 2005;392:633-9.
176. Li D. GSK-3beta as a driving force in ovarian cancer. Cell Res. 2006;16:609.
177. Li R, Erdamar S, Dai H, Sayeeduddin M, Frolov A, Wheeler TM, et al.
Cytoplasmic accumulation of glycogen synthase kinase-3beta is associated with
aggressive clinicopathological features in human prostate cancer. Anticancer Res.
2009;29:2077-81.
178. Shakoori A, Mai W, Miyashita K, Yasumoto K, Takahashi Y, Ooi A, et al.
Inhibition of GSK-3 beta activity attenuates proliferation of human colon cancer cells
in rodents. Cancer Sci. 2007;98:1388-93.
179. Ougolkov AV, Bone ND, Fernandez-Zapico ME, Kay NE, Billadeau DD.
Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of
nuclear factor {kappa}B target genes and induction of apoptosis in chronic
lymphocytic leukemia B cells. Blood. 2007;110:735-42.
180. Cao Q, Lu X, Feng YJ. Glycogen synthase kinase-3beta positively regulates
the proliferation of human ovarian cancer cells. Cell Res. 2006;16:671-7.
181. Kunnimalaiyaan M, Vaccaro AM, Ndiaye MA, Chen H. Inactivation of
glycogen synthase kinase-3beta, a downstream target of the raf-1 pathway, is
associated with growth suppression in medullary thyroid cancer cells. Mol Cancer
Ther. 2007;6:1151-8.
182. Liao X, Zhang L, Thrasher JB, Du J, Li B. Glycogen synthase kinase-3beta
suppression eliminates tumor necrosis factor-related apoptosis-inducing ligand
resistance in prostate cancer. Mol Cancer Ther. 2003;2:1215-22.

128
183. Mazor M, Kawano Y, Zhu H, Waxman J, Kypta RM. Inhibition of glycogen
synthase kinase-3 represses androgen receptor activity and prostate cancer cell
growth. Oncogene. 2004;23:7882-92.
184. Erdal E, Ozturk N, Cagatay T, Eksioglu-Demiralp E, Ozturk M. Lithium-
mediated downregulation of PKB/Akt and cyclin E with growth inhibition in
hepatocellular carcinoma cells. Int J Cancer. 2005;115:903-10.
185. Ougolkov AV, Fernandez-Zapico ME, Bilim VN, Smyrk TC, Chari ST,
Billadeau DD. Aberrant Nuclear Accumulation of Glycogen Synthase Kinase-3{beta}
in Human Pancreatic Cancer: Association with Kinase Activity and Tumor
Dedifferentiation. Clin Cancer Res. 2006;12:5074-81.
186. Beyaert R, Vanhaesebroeck B, Suffys P, Van Roy F, Fiers W. Lithium
chloride potentiates tumor necrosis factor-mediated cytotoxicity in vitro and in vivo.
Proc Natl Acad Sci U S A. 1989;86:9494-8.
187. Klein PS, Melton DA. A molecular mechanism for the effect of lithium on
development. Proc Natl Acad Sci U S A. 1996;93:8455-9.
188. Song L, Zhou T, Jope RS. Lithium facilitates apoptotic signaling induced by
activation of the Fas death domain-containing receptor. BMC Neurosci. 2004;5:20.
189. Rottmann S, Wang Y, Nasoff M, Deveraux QL, Quon KC. A TRAIL receptor-
dependent synthetic lethal relationship between MYC activation and
GSK3beta/FBW7 loss of function. Proc Natl Acad Sci U S A. 2005;102:15195-200.
190. Beurel E, Blivet-Van Eggelpoel MJ, Kornprobst M, Moritz S, Delelo R, Paye F,
et al. Glycogen synthase kinase-3 inhibitors augment TRAIL-induced apoptotic
death in human hepatoma cells. Biochem Pharmacol. 2009;77:54-65.
191. Steinbrecher KA, Wilson W, 3rd, Cogswell PC, Baldwin AS. Glycogen
synthase kinase 3beta functions to specify gene-specific, NF-kappaB-dependent
transcription. Mol Cell Biol. 2005;25:8444-55.
192. Aza-Blanc P, Cooper CL, Wagner K, Batalov S, Deveraux QL, Cooke MP.
Identification of modulators of TRAIL-induced apoptosis via RNAi-based phenotypic
screening. Mol Cell. 2003;12:627-37.
193. Trauzold A, Wermann H, Arlt A, Schutze S, Schafer H, Oestern S, et al. CD95
and TRAIL receptor-mediated activation of protein kinase C and NF-kappaB

129
contributes to apoptosis resistance in ductal pancreatic adenocarcinoma cells.
Oncogene. 2001;20:4258-69.
194. Thomas RP, Farrow BJ, Kim S, May MJ, Hellmich MR, Evers BM. Selective
targeting of the nuclear factor-kappaB pathway enhances tumor necrosis factor-
related apoptosis-inducing ligand-mediated pancreatic cancer cell death. Surgery.
2002;132:127-34.
195. Kallifatidis G, Rausch V, Baumann B, Apel A, Beckermann BM, Groth A, et al.
Sulforaphane targets pancreatic tumour-initiating cells by NF-kappaB-induced
antiapoptotic signalling. Gut. 2009;58:949-63.
196. Kasperczyk H, Baumann B, Debatin KM, Fulda S. Characterization of sonic
hedgehog as a novel NF-kappaB target gene that promotes NF-kappaB-mediated
apoptosis resistance and tumor growth in vivo. Faseb J. 2009;23:21-33.
197. Braeuer SJ, Buneker C, Mohr A, Zwacka RM. Constitutively activated nuclear
factor-kappaB, but not induced NF-kappaB, leads to TRAIL resistance by up-
regulation of X-linked inhibitor of apoptosis protein in human cancer cells. Mol
Cancer Res. 2006;4:715-28.
198. Beurel E, Kornprobst M, Blivet-Van Eggelpoel MJ, Cadoret A, Capeau J,
Desbois-Mouthon C. GSK-3beta reactivation with LY294002 sensitizes hepatoma
cells to chemotherapy-induced apoptosis. Int J Oncol. 2005;27:215-22.
199. Beurel E, Kornprobst M, Blivet-Van Eggelpoel MJ, Ruiz-Ruiz C, Cadoret A,
Capeau J, et al. GSK-3beta inhibition by lithium confers resistance to chemotherapy-
induced apoptosis through the repression of CD95 (Fas/APO-1) expression. Exp
Cell Res. 2004;300:354-64.
200. Patel S, Woodgett J. Glycogen synthase kinase-3 and cancer: good cop, bad
cop? Cancer Cell. 2008;14:351-3.
201. Bahnson BJ. Structure, function and interfacial allosterism in phospholipase
A2: insight from the anion-assisted dimer. Arch Biochem Biophys. 2005;433:96-106.
202. Wang Z, Smith KS, Murphy M, Piloto O, Somervaille TC, Cleary ML.
Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy.
Nature. 2008;455:1205-9.
203. Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen
synthase kinase 3. Trends Pharmacol Sci. 2004;25:471-80.

130
204. De Ketelaere A, Vermeulen L, Vialard J, Van De Weyer I, Van Wauwe J,
Haegeman G, et al. Involvement of GSK-3beta in TWEAK-mediated NF-kappaB
activation. FEBS Lett. 2004;566:60-4.
205. Maitra A, Kern SE, Hruban RH. Molecular pathogenesis of pancreatic cancer.
Best Pract Res Clin Gastroenterol. 2006;20:211-26.
206. Medina M, Castro A. Glycogen synthase kinase-3 (GSK-3) inhibitors reach
the clinic. Curr Opin Drug Discov Devel. 2008;11:533-43.
207. Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, et al. Inhibition of
glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and
degeneration in vivo. Proc Natl Acad Sci U S A. 2005;102:6990-5.
208. Gould TD, Einat H, Bhat R, Manji HK. AR-A014418, a selective GSK-3
inhibitor, produces antidepressant-like effects in the forced swim test. Int J
Neuropsychopharmacol. 2004;7:387-90.
209. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core
signaling pathways in human pancreatic cancers revealed by global genomic
analyses. Science. 2008;321:1801-6.
210. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al.
Erlotinib plus gemcitabine compared with gemcitabine alone in patients with
advanced pancreatic cancer: a phase III trial of the National Cancer Institute of
Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960-6.
211. Hernandez-Vargas H, Rodriguez-Pinilla SM, Julian-Tendero M, Sanchez-
Rovira P, Cuevas C, Anton A, et al. Gene expression profiling of breast cancer cells
in response to gemcitabine: NF-kappaB pathway activation as a potential
mechanism of resistance. Breast Cancer Res Treat. 2007;102:157-72.
212. Okamoto T, Sanda T, Asamitsu K. NF-kappa B signaling and carcinogenesis.
Curr Pharm Des. 2007;13:447-62.
213. Liptay S, Weber CK, Ludwig L, Wagner M, Adler G, Schmid RM. Mitogenic
and antiapoptotic role of constitutive NF-kappaB/Rel activity in pancreatic cancer. Int
J Cancer. 2003;105:735-46.
214. Van Waes C. Nuclear factor-kappaB in development, prevention, and therapy
of cancer. Clin Cancer Res. 2007;13:1076-82.

131
215. Ouyang W, Li J, Ma Q, Huang C. Essential roles of PI-
3K/Akt/IKKbeta/NFkappaB pathway in cyclin D1 induction by arsenite in JB6 Cl41
cells. Carcinogenesis. 2006;27:864-73.
216. Xiao G, Rabson AB, Young W, Qing G, Qu Z. Alternative pathways of NF-
kappaB activation: a double-edged sword in health and disease. Cytokine Growth
Factor Rev. 2006;17:281-93.
217. Umezawa K. Inhibition of tumor growth by NF-kappaB inhibitors. Cancer Sci.
2006;97:990-5.
218. Muerkoster S, Arlt A, Witt M, Gehrz A, Haye S, March C, et al. Usage of the
NF-kappaB inhibitor sulfasalazine as sensitizing agent in combined chemotherapy of
pancreatic cancer. Int J Cancer. 2003;104:469-76.
219. Kunnumakkara AB, Guha S, Krishnan S, Diagaradjane P, Gelovani J,
Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic
model of pancreatic cancer through suppression of proliferation, angiogenesis, and
inhibition of nuclear factor-kappaB-regulated gene products. Cancer Res.
2007;67:3853-61.
220. Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality
and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature.
1995;376:167-70.
221. Bilim V, Ougolkov A, Yuuki K, Naito S, Kawazoe H, Muto A, et al. Glycogen
synthase kinase-3: a new therapeutic target in renal cell carcinoma. Br J Cancer.
2009;101:2005-14.
222. Ougolkov AV, Billadeau DD. Targeting GSK-3: a promising approach for
cancer therapy? Future Oncol. 2006;2:91-100.
223. LaPointe P, Wei X, Gariepy J. A Role for the Protease-sensitive Loop Region
of Shiga-like Toxin 1 in the Retrotranslocation of Its A1 Domain from the
Endoplasmic Reticulum Lumen. J Biol Chem. 2005;280:23310-8.
224. Wu L, Birle DC, Tannock IF. Effects of the Mammalian Target of Rapamycin
Inhibitor CCI-779 Used Alone or with Chemotherapy on Human Prostate Cancer
Cells and Xenografts. Cancer Res. 2005;65:2825-31.

132
225. Feyt C, Kienlen-Campard P, Leroy K, N'Kuli F, Courtoy PJ, Brion J-P, et al.
Lithium Chloride Increases the Production of Amyloid-{beta} Peptide Independently
from Its Inhibition of Glycogen Synthase Kinase 3. J Biol Chem. 2005;280:33220-7.
226. Garcea G, Manson MM, Neal CP, Pattenden CJ, Sutton CD, Dennison AR, et
al. Glycogen synthase kinase-3 beta; a new target in pancreatic cancer? Curr
Cancer Drug Targets. 2007;7:209-15.
227. Deng J, Miller SA, Wang HY, Xia W, Wen Y, Zhou BP, et al. beta-catenin
interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer
Cell. 2002;2:323-34.
228. Wilson W, 3rd, Baldwin AS. Maintenance of constitutive IkappaB kinase
activity by glycogen synthase kinase-3alpha/beta in pancreatic cancer. Cancer Res.
2008;68:8156-63.
229. Lustig B, Behrens J. The Wnt signaling pathway and its role in tumor
development. J Cancer Res Clin Oncol. 2003;129:199-221.
230. Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, et al. GSK-3
beta targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3 beta inactivation
correlates with Cdc25A overproduction in human cancers. Cancer Cell. 2008;13:36-
47.
231. Egan LJ, Toruner M. NF-kappaB signaling: pros and cons of altering NF-
kappaB as a therapeutic approach. Ann N Y Acad Sci. 2006;1072:114-22.
232. Kunnumakkara AB, Diagaradjane P, Guha S, Deorukhkar A, Shentu S,
Aggarwal BB, et al. Curcumin Sensitizes Human Colorectal Cancer Xenografts in
Nude Mice to {gamma}-Radiation by Targeting Nuclear Factor-{kappa}B-Regulated
Gene Products. Clin Cancer Res. 2008;14:2128-36.
233. Wiezorek J, Holland P, Graves J. Death receptor agonists as a targeted
therapy for cancer. Clin Cancer Res. 2010;16:1701-8.
234. Marsters SA, Pitti RA, Sheridan JP, Ashkenazi A. Control of apoptosis
signaling by Apo2 ligand. Recent Prog Horm Res. 1999;54:225-34.
235. Ashkenazi A. Directing cancer cells to self-destruct with pro-apoptotic
receptor agonists. Nat Rev Drug Discov. 2008;7:1001-12.

133
236. Ozawa F, Friess H, Kleeff J, Xu ZW, Zimmermann A, Sheikh MS, et al.
Effects and expression of TRAIL and its apoptosis-promoting receptors in human
pancreatic cancer. Cancer Lett. 2001;163:71-81.
237. Ashkenazi A, Dixit VM. Apoptosis control by death and decoy receptors. Curr
Opin Cell Biol. 1999;11:255-60.
238. Todaro M, Lombardo Y, Francipane MG, Alea MP, Cammareri P, Iovino F, et
al. Apoptosis resistance in epithelial tumors is mediated by tumor-cell-derived
interleukin-4. Cell Death Differ. 2008;15:762-72.
239. Simpson CD, Mawji IA, Anyiwe K, Williams MA, Wang X, Venugopal AL, et al.
Inhibition of the sodium potassium adenosine triphosphatase pump sensitizes
cancer cells to anoikis and prevents distant tumor formation. Cancer Res.
2009;69:2739-47.
240. Pham N-A, Robinson BH, Hedley DW. Simultaneous detection of
mitochondrial respiratory chain activity and reactive oxygen in digitonin-
permeabilized cells using flow cytometry. Cytometry. 2000;41:245-51.
241. Leverkus M, Sprick MR, Wachter T, Mengling T, Baumann B, Serfling E, et al.
Proteasome inhibition results in TRAIL sensitization of primary keratinocytes by
removing the resistance-mediating block of effector caspase maturation. Mol Cell
Biol. 2003;23:777-90.
242. Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of
action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95-
105.
243. Pellegata NS, Sessa F, Renault B, Bonato M, Leone BE, Solcia E, et al. K-ras
and p53 Gene Mutations in Pancreatic Cancer: Ductal and Nonductal Tumors
Progress through Different Genetic Lesions. Cancer Research. 1994;54:1556-60.
244. Slebos RJC, Hoppin JA, Tolbert PE, Holly EA, Brock JW, Zhang RH, et al. K-
ras and p53 in Pancreatic Cancer: Association with Medical History, Histopathology,
and Environmental Exposures in a Population-based Study. Cancer Epidemiology
Biomarkers & Prevention. 2000;9:1223-32.
245. Ashkenazi A, Herbst RS. To kill a tumor cell: the potential of proapoptotic
receptor agonists. J Clin Invest. 2008;118:1979-90.

134
246. Oikonomou E, Kosmidou V, Katseli A, Kothonidis K, Mourtzoukou D,
Kontogeorgos G, et al. TRAIL receptor upregulation and the implication of
KRAS/BRAF mutations in human colon cancer tumors. Int J Cancer. 2009;125:2127-
35.
247. Drosopoulos KG, Roberts ML, Cermak L, Sasazuki T, Shirasawa S, Andera
L, et al. Transformation by oncogenic RAS sensitizes human colon cells to TRAIL-
induced apoptosis by up-regulating death receptor 4 and death receptor 5 through a
MEK-dependent pathway. J Biol Chem. 2005;280:22856-67.
248. Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door.
Nat Rev Mol Cell Biol. 2002;3:401-10.
249. Ravi R, Bedi GC, Engstrom LW, Zeng Q, Mookerjee B, Gelinas C, et al.
Regulation of death receptor expression and TRAIL/Apo2L-induced apoptosis by
NF-kappaB. Nat Cell Biol. 2001;3:409-16.
250. Hirpara JL, Clement MV, Pervaiz S. Intracellular acidification triggered by
mitochondrial-derived hydrogen peroxide is an effector mechanism for drug-induced
apoptosis in tumor cells. J Biol Chem. 2001;276:514-21.
251. Jung EM, Lim JH, Lee TJ, Park J-W, Choi KS, Kwon TK. Curcumin sensitizes
tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis
through reactive oxygen species-mediated upregulation of death receptor 5 (DR5).
Carcinogenesis. 2005;26:1905-13.
252. Kwon D, Choi IH. Hydrogen peroxide upregulates TNF-related apoptosis-
inducing ligand (TRAIL) expression in human astroglial cells, and augments
apoptosis of T cells. Yonsei Med J. 2006;47:551-7.
253. Garcea G, Doucas H, Steward WP, Dennison AR, Berry DP. Hypoxia and
angiogenesis in pancreatic cancer. ANZ J Surg. 2006;76:830-42.
254. Kerfoot C, Huang W, Rotenberg SA. Immunohistochemical Analysis of
Advanced Human Breast Carcinomas Reveals Downregulation of Protein Kinase
C{alpha}. J Histochem Cytochem. 2004;52:419-22.
255. Beyaert R, Vanhaesebroeck B, Suffys P, Roy F, Fiers W. Lithium Chloride
Potentiates Tumor Necrosis Factor-Mediated Cytotoxicity in vitro and in vivo. PNAS.
1989;86:9494-8.

135
256. Senthivinayagam S, Mishra P, Paramasivam SK, Yallapragada S, Chatterjee
M, Wong L, et al. Caspase-mediated cleavage of beta-catenin precedes drug-
induced apoptosis in resistant cancer cells. J Biol Chem. 2009;284:13577-88.
257. Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3):
inflammation, diseases, and therapeutics. Neurochem Res. 2007;32:577-95.
258. Billadeau DD. Primers on molecular pathways. The glycogen synthase
kinase-3beta. Pancreatology. 2007;7:398-402.
259. Woodgett JR. Judging a Protein by More Than Its Name: GSK-3. Sci STKE.
2001;2001:re12-.
260. Cohen Y, Chetrit A, Sirota P, Modan B. Cancer morbidity in psychiatric
patients: influence of lithium carbonate treatment. Med Oncol. 1998;15:32-6.
261. Martinez A. Preclinical efficacy on GSK-3 inhibitors: Towards a future
generation of powerful drugs. Med Res Rev. 2008.
262. Trials.gov C. [cited; Available from: www.clinicaltrials.gov
263. Hruban RH, Adsay NV. Molecular classification of neoplasms of the
pancreas. Hum Pathol. 2009;40:612-23.