Embed Size (px)
Citation preview

GANGLIOSIDOSIS GM2
Luis González Gutiérrez-Solana Neuropediatra. Hospital Niño Jesús
Madrid, 22 de abril de 2014

ENFERMEDADES LISOSOMALES
1. Lipidosis 1.1. Esfingolipidosis 1.1.1. Glucoesfingolipidosis - Enfermedad de Gaucher - Enfermedad de Fabry - Gangliosidosis GM1 - Gangliosidosis GM2 - Leucodistrofia de células globoides
(Krabbe) - Leucodistrofia metacromática 1.1.2. Otras esfingolipidosis - Niemann-Pick A, B y C - Enfermedad de Farber - Deficiencia de la proteína activadora 1.2. Otras enf. por depósito de lípidos - Enfermedad de Wolman - Enfermedad por depósito de ésteres
de colesterol
2. Mucopolisacaridosis - Formas I a IX
3. Glucoproteinosis - Manosidosis α y β - Fucosidosis - Aspartilglucosaminuria - Sialidosis - Galactosialidosis - Enfermedad de Schindler 4. Mucolipidosis 5. Ceroidolipofuscinosis 6. Otras lisosomales - Pompe - Danon - Cistinosis - Enf. por depósito de ácido siálico

ENFERMEDADES LISOSOMALES

ENFERMEDADES LISOSOMALES
• Grupo heterogéneo de Errores Congénitos del Metabolismo (+50)
• Causadas por: • Deficiencia de hidrolasas ácidas • Deficiencia de proteínas activadoras • Defectos en la biogénesis de los lisosomas • Defectos en proteínas de transporte de membrana • Defectos en proteínas implicadas en el tráfico
vesicular

ENFERMEDADES LISOSOMALES
• La mayoría afectan a los niños • Comienzo y curso variable:
• No congénitas y progresivas • Continuo fenotípico
Ø Las más precoces más graves y frecuentes • Afectación neurológica frecuente • Multiorgánicas: hueso, hígado, bazo, riñón • Incidencia: 1/7700 RNV

GANGLIOSIDOSIS GM2

GANGLIOSIDOSIS GM2
• Gangliósidos: lípidos complejos que se encuentran predominante en la sustancia gris del cerebro
• Gangliosidosis GM2: acúmulo de gangliósido GM2 y otros relacionados por deficiencia de la enzima beta-hexosaminidasa A (Hex A)
• Enfermedad rara: 1 cada 310.000 recién nacidos • Neurodegenerativa • Heterogénea • Autosómica recesiva
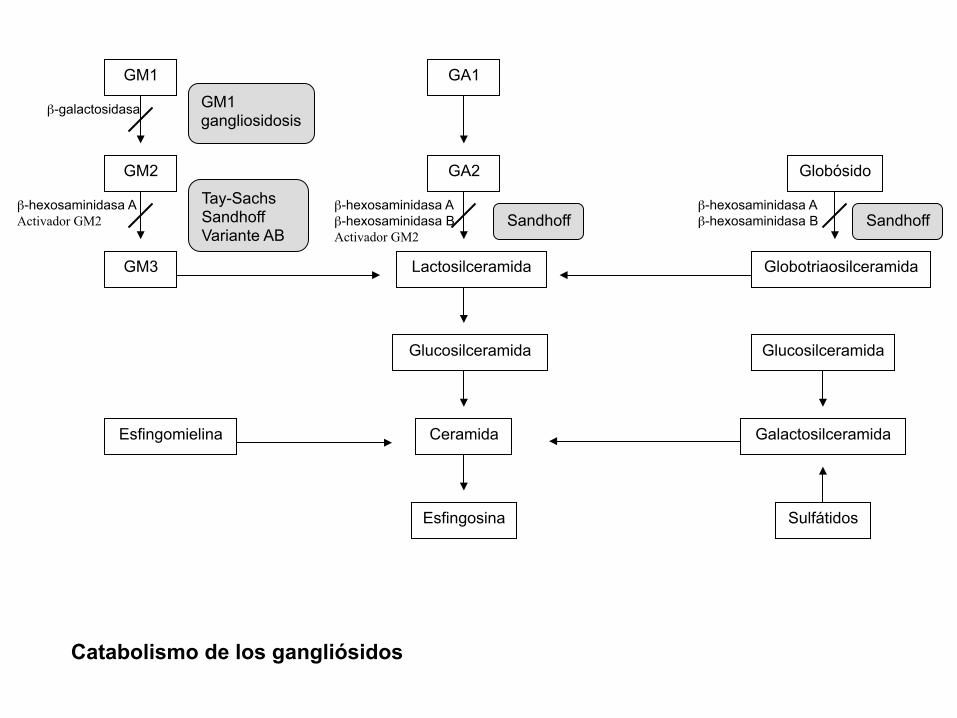
Glucosilceramida
Ceramida
Esfingosina
Esfingomielina
Sulfátidos
Globotriaosilceramida
Glucosilceramida
Galactosilceramida
GM1 gangliosidosis
Tay-Sachs
Sandhoff
Variante AB Sandhoff Sandhoff
β-galactosidasa
GM1 GA1
β-hexosaminidasa A
Activador GM2
GM3
GM2 β-hexosaminidasa A
β-hexosaminidasa B
Globósido β-hexosaminidasa A
β-hexosaminidasa B
Activador GM2
GA2
Lactosilceramida
Catabolismo de los gangliósidos
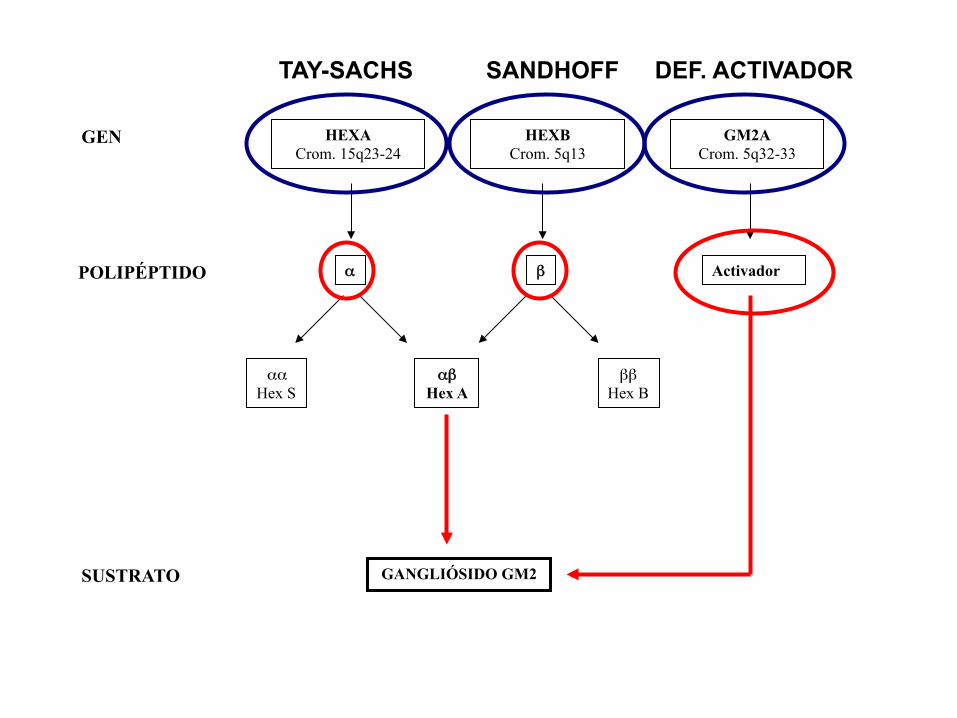
HEXA Crom. 15q23-24
HEXB Crom. 5q13
GM2A Crom. 5q32-33
GEN
α β Activador POLIPÉPTIDO
αα Hex S
αβ Hex A
ββ Hex B
SUSTRATO
GANGLIÓSIDO GM2
TAY-SACHS SANDHOFF DEF. ACTIVADOR
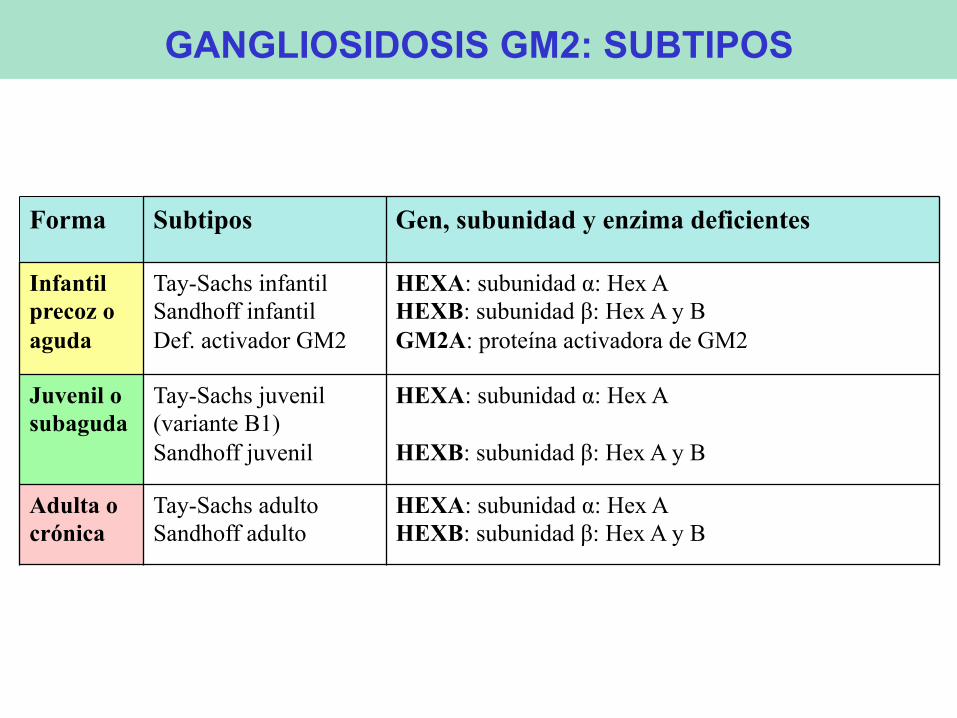
Forma Subtipos Gen, subunidad y enzima deficientes Infantil precoz o aguda
Tay-Sachs infantil Sandhoff infantil Def. activador GM2
HEXA: subunidad α: Hex A HEXB: subunidad β: Hex A y B GM2A: proteína activadora de GM2
Juvenil o subaguda
Tay-Sachs juvenil (variante B1) Sandhoff juvenil
HEXA: subunidad α: Hex A HEXB: subunidad β: Hex A y B
Adulta o crónica
Tay-Sachs adulto Sandhoff adulto
HEXA: subunidad α: Hex A HEXB: subunidad β: Hex A y B
GANGLIOSIDOSIS GM2: SUBTIPOS

CLÍNICA

• Inicio: 3-6 meses • Clonías audiógenas • Hipotonía y debilidad progresiva • Mala fijación visual – mov. anormales
• Regresión • Ceguera y mancha rojo cereza
• Megalencefalia • Crisis convulsivas • Tetraplejía espástica • Alteración de deglución • Muerte: 2-4 años (aspiración o neumonía)
• Hepatoesplenomegalia • Cambios esqueléticos
GM2 INFANTIL PRECOZ o AGUDA

• Inicio: 1-10 años (3-6) • Continuo fenotípico – act. residual - alt. intelectual • Cuanto más precoz más rápida progresión
• Dificultades de la marcha – ataxia - distonía • Disartria • Deterioro mental (regresión) • Trastorno comportamiento • Frecuente varios signos al inicio
• Amiotrofia / debilidad / incontinencia / piramidalismo • Diarrea-estreñimiento /problemas de deglución /
problemas de sueño / crisis • Problemas visuales (MRC o AO o RP) / nistagmus
• Supervivencia variable: 2-20 años
GM2 JUVENIL o SUBAGUDA

• Inicio: niñez – vida adulta (1/3 antes de los 10 años)
• Síndrome espinocerebeloso: ataxia, disartria y SP • Atrofia muscular espinal: debilidad y fasciculaciones • Trastorno psiquiátrico • Movimientos anormales (distonía, corea) • Oftalmoplejía supranuclear
GM2 DEL ADULTO o CRÓNICA
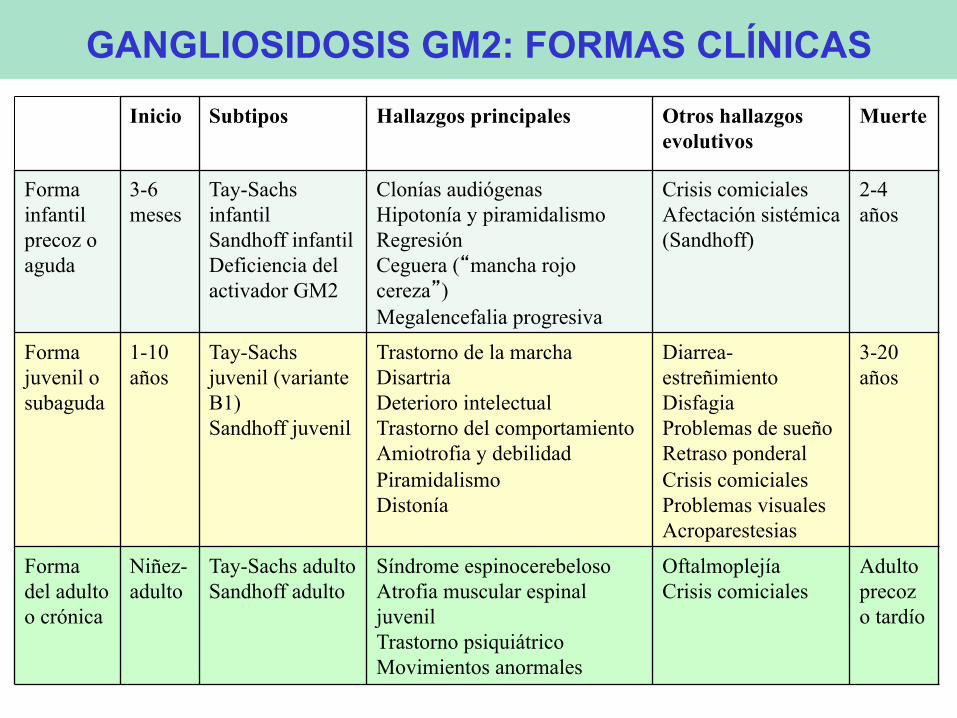
Inicio Subtipos Hallazgos principales Otros hallazgos evolutivos Muerte
Forma infantil precoz o aguda
3-6 meses
Tay-Sachs infantil Sandhoff infantil Deficiencia del activador GM2
Clonías audiógenas Hipotonía y piramidalismo Regresión Ceguera (“mancha rojo cereza”) Megalencefalia progresiva
Crisis comiciales Afectación sistémica (Sandhoff)
2-4 años
Forma juvenil o subaguda
1-10 años
Tay-Sachs juvenil (variante B1) Sandhoff juvenil
Trastorno de la marcha Disartria Deterioro intelectual Trastorno del comportamiento Amiotrofia y debilidad Piramidalismo Distonía
Diarrea-estreñimiento Disfagia Problemas de sueño Retraso ponderal Crisis comiciales Problemas visuales Acroparestesias
3-20 años
Forma del adulto o crónica
Niñez-adulto
Tay-Sachs adulto Sandhoff adulto
Síndrome espinocerebeloso Atrofia muscular espinal juvenil Trastorno psiquiátrico Movimientos anormales
Oftalmoplejía Crisis comiciales
Adulto precoz o tardío
GANGLIOSIDOSIS GM2: FORMAS CLÍNICAS

DIAGNÓSTICO
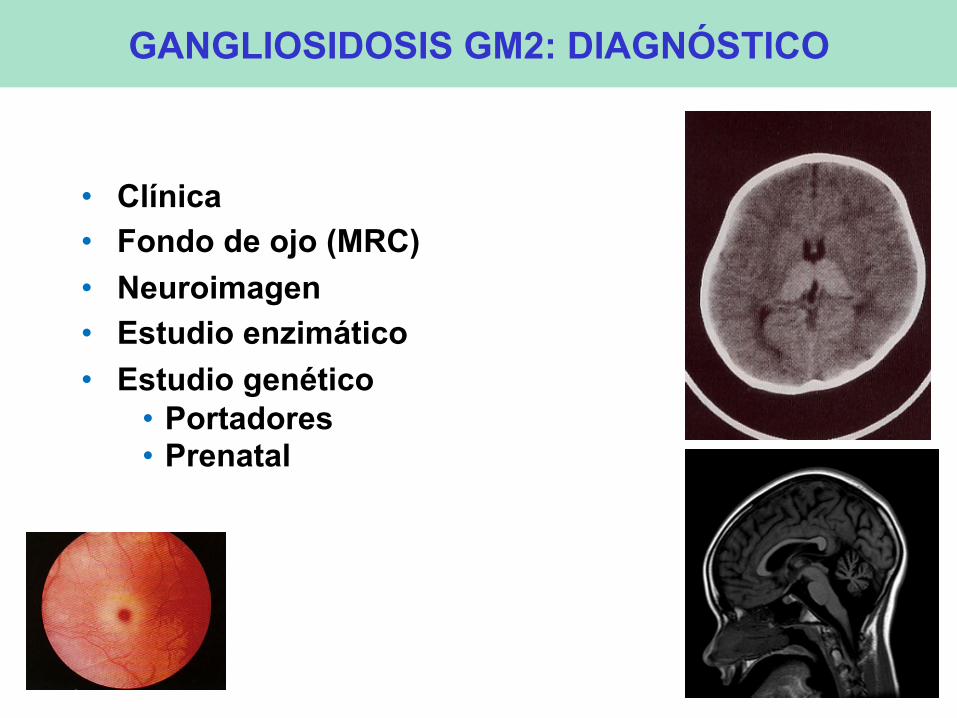
GANGLIOSIDOSIS GM2: DIAGNÓSTICO
• Clínica • Fondo de ojo (MRC) • Neuroimagen • Estudio enzimático • Estudio genético
• Portadores • Prenatal

TRATAMIENTO
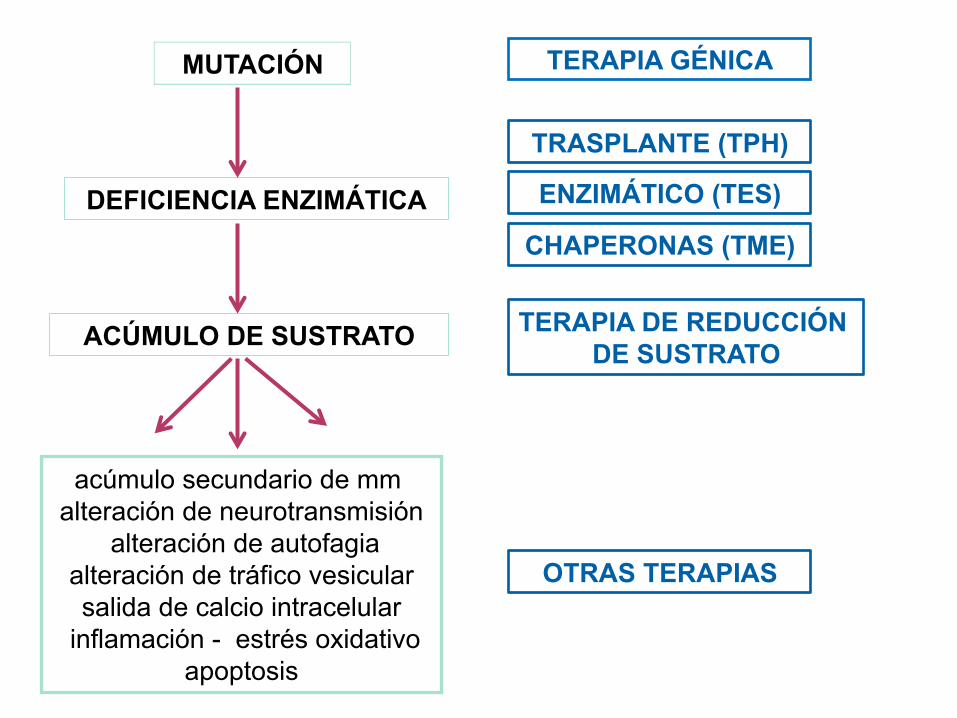
MUTACIÓN
DEFICIENCIA ENZIMÁTICA
ACÚMULO DE SUSTRATO
acúmulo secundario de mm alteración de neurotransmisión
alteración de autofagia alteración de tráfico vesicular salida de calcio intracelular
inflamación - estrés oxidativo apoptosis
TERAPIA GÉNICA
TRASPLANTE (TPH)
ENZIMÁTICO (TES)
CHAPERONAS (TME)
TERAPIA DE REDUCCIÓN DE SUSTRATO
OTRAS TERAPIAS

• Aporte de enzimas recombinantes en infusiones periódicas (1-2 semanas)
• Tratamiento iv. No traspasa BHE
• Años 70 (Desnick72, Johnson73) • von Specht. Enzyme replacement in Tay-Sachs
disease. Neurology 1979;29:848-54.
INEFICAZ
TERAPIA ENZIMÁTICA SUSTITUTIVA (TES)

• MO, sangre de cordón umbilical o SP ! células con actividad enzimática normal, que llegue al cerebro
• Lento. Alta morbilidad y mortalidad • Hoogerbrugge and the European Group for Bone Marrow
Transplantation. Allogeneic bone marrow transplantation for lysosomal storage diseases. Lancet 1995; 345: 1398-402. – Ningún resultado favorable tras TPH
• Jacobs. Allogeneic BMT followed by substrate reduction therapy in a child with subacute Tay-Sachs disease. Bone Marrow Transplant 2005;36:925. – No se logró frenar el deterioro neurológico
TRASPLANTE DE PROGENITORES HEMATOPOYÉTICOS

• Prasad (Duke). Unrelated donor umbilical cord blood transplantation for inherited metabolic disorders in 159 pediatric patients from a single center… Blood 2008; 112: 2979-89. – 2 de 5 niños con TSD sobrevivieron más de 5 años, pero
sin mejoría en las habilidades y gravemente enfermos – 1 niño que se trasplantó en periodo neonatal sobrevivió 5
años y murió súbitamente de causa desconocida. Se mantenía sentado, pero no se ponía de pie ni andaba
– Uno de 2 niños con TSD juvenil ha sobrevivido 2 años. Su función motora se ha estabilizado
INEFICAZ – MUY POCO EFICAZ
TRASPLANTE DE PROGENITORES HEMATOPOYÉTICOS
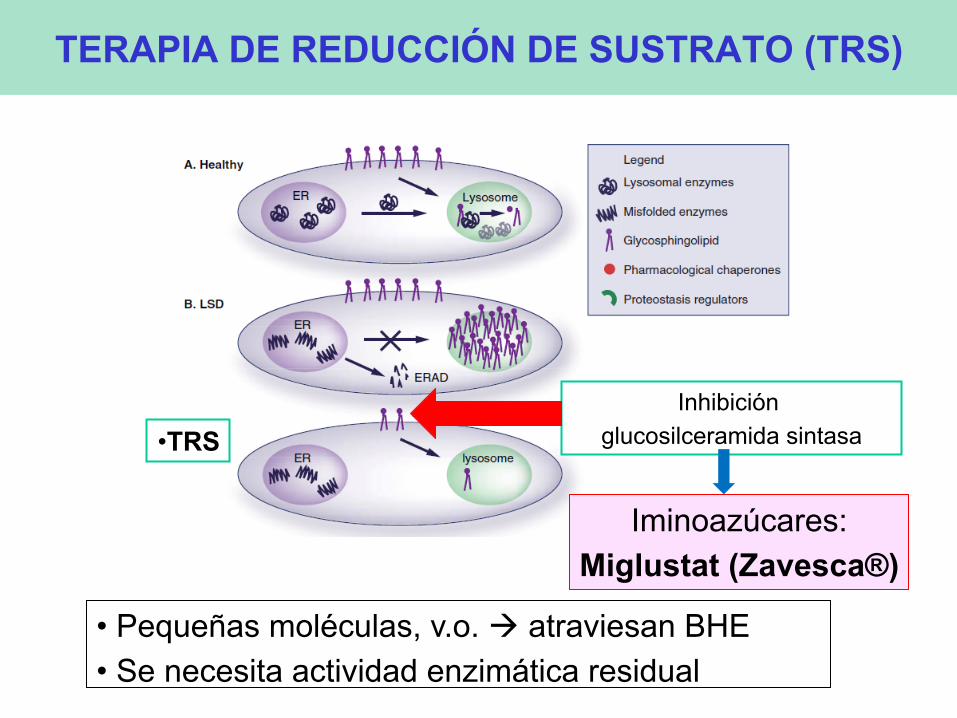
TERAPIA DE REDUCCIÓN DE SUSTRATO (TRS)
• TRS Inhibición
glucosilceramida sintasa
Iminoazúcares: Miglustat (Zavesca®)
• Pequeñas moléculas, v.o. à atraviesan BHE • Se necesita actividad enzimática residual

• Platt et al. Prevention of lysosomal storage in Tay-Sachs mice treated with N-butyldeoxynojirimycin. Science 1997;276:428-31. – Reduce el acúmulo neuronal de gangliósidos
• Jeyakumar et al. Delayed symptom onset and increased life expectancy in Sandhoff disease mice treated with N-butyldeoxynojirimycin, Proc Nat Acad Sci USA 1999;96:6388 – Aumenta la supervivencia un 40% en el ratón SD
TERAPIA DE REDUCCIÓN DE SUSTRATO (TRS)

• Bembi et al. Substrate reduction therapy in the infantile form of Tay-Sachs disease. Neurology 2006;66:278. – Miglustat en LCR 15-25% de la de plasma – Ninguno de los dos pacientes desarrolló
macrocefalia – Buena tolerancia con 100 mg/d. Diarrea y
pérdida de peso al doblar la dosis – Neurodegeneración
TERAPIA DE REDUCCIÓN DE SUSTRATO (TRS)

• Maegawa et al. Substrate reduction therapy in juvenile GM2 gangliosidosis. Mol Genet Metab 2009;98:215-24. – Pacientes: 5 (2 TSD, 3 SD). Edad: 14,6 ± 4,5
años. Miglustat: 100-200 mg tid. 24 meses. Evaluación neurológica completa
– Deterioro neurológico progresivo: • Marcha, habla, coordinación, piramidal,
extrapiramidal, crisis, fuerza, tono y trofismo • Más intenso en los más afectados
TERAPIA DE REDUCCIÓN DE SUSTRATO (TRS)

• Shapiro et al. Miglustat in late-onset Tay-Sachs disease: a 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genet Med 2009;11:425-33. – Pacientes: 30. Edad: 18-56 años. Miglustat: 200
mg tid. 12!24 meses – No se encontró beneficio del tratamiento:
• No hubo diferencias entre los dos grupos ni en fuerza, ni en marcha, ni en escalas de incapacidad
• Fuerza muscular y “apretón” disminuyeron
INEFICAZ
TERAPIA DE REDUCCIÓN DE SUSTRATO (TRS)
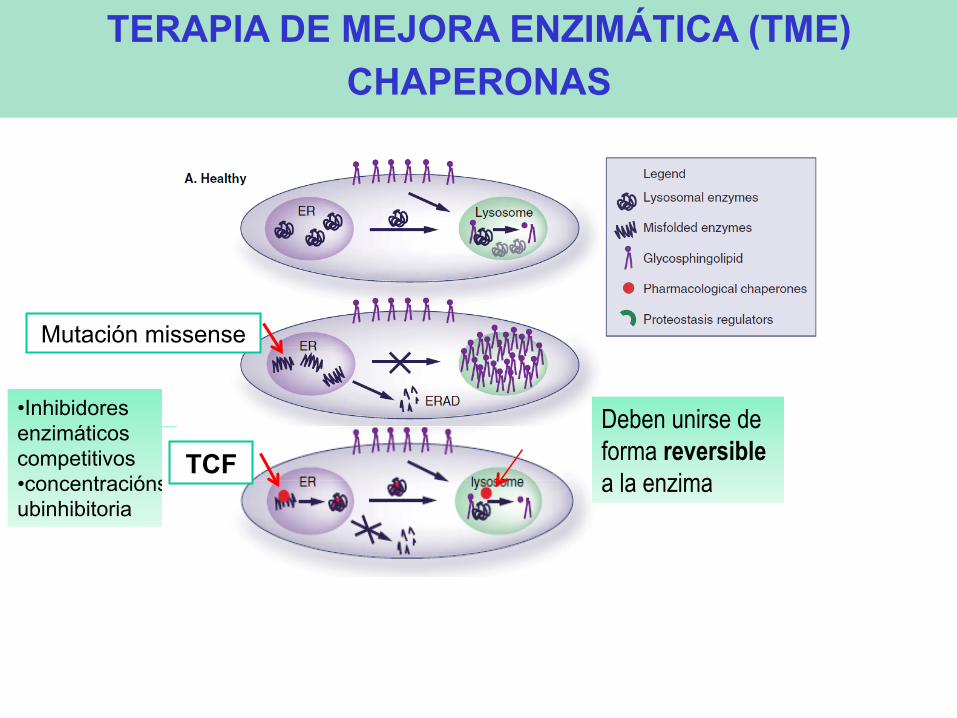
• Inhibidores enzimáticos competitivos • concentraciónsubinhibitoria
TERAPIA DE MEJORA ENZIMÁTICA (TME) CHAPERONAS
TCF
Mutación missense
Deben unirse de forma reversible a la enzima

• Maegawa et al. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J Biol Chem 2007;282:9150-61. – Algunas mutaciones aumentan su actividad en fibroblasto
• Clarke et al. An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants). Mol Genet Metab 2011;102:6-12. – 8 pacientes (23-50 años) – Aumento de actividad Hex A – EA neurológicos al aumentar de 50 mg a 75 mg – ¿Dosis más bajas con control de niveles?
TERAPIA DE MEJORA ENZIMÁTICA (TME) CHAPERONAS

• Osher. Pyrimethamine increases β-hexosaminidase A activity in patients with late onset Tay Sachs. Mol Genet Metab 2011;102:356-63. – 9 pacientes adultos (20-67 años) – La dosis óptima varió entre 6,5-75 mg – La actividad Hex A aumentó, pero sólo al 2% – Hubo mejoría inicial en lenguaje en 4/9 – Mucha variabilidad, incluso con la misma dosis – Las dosis altas y el tto prolongado empeoran – ¿Uso de dosis bajas 2-3 veces por semana?
TERAPIA DE MEJORA ENZIMÁTICA (TME) CHAPERONAS
DUDOSA EFICACIA

• Introducción de genes en las células mediante vectores à expresión y secreción de niveles suprafisiológicos de la enzima deficiente à corrección a distancia
• Expresión a largo plazo (ventaja sobre TES) • Virus adeno-asociados (VVA) y lentivirus • Terapia génica “ex vivo”:
– Se modifican genéticamente células “ex vivo” (progenitores hematopoyéticos) y se trasplantan (paciente inmunosuprimido)
– Trasplante autólogo: no riesgo de EICH, ni de rechazo • Terapia génica “in vivo”:
– Se inyecta el vector directamente en el cerebro en varios puntos a la vez (estereotaxia) o en sistema ventricular
• Atención: mutagénesis, Acs y eficacia
TERAPIA GÉNICA

TERAPIA GÉNICA
• Ratones. Aumenta la supervivencia de 20 semanas a más de 1 año
• Gatos. Sobreviven más de 18 meses (no 3,5-4m) • Oveja Jacob. Viven al menos el doble de tiempo • Monos. Toxicidad no esperada • Nuevo estudio en monos con modificaciones
POTENCIAL EFICACIA

• Esencial en todos (más en niños) • Soporte nutricional • Soporte respiratorio • Rehabilitación + Antiespásticos • Antiepilépticos: LVT, VPA, CBZ, ZNS, benzod. • Educación especial y apoyo psicosocial • Cuidados paliativos • Shapiro. Late-onset Tay-Sachs disease: adverse
effects of medications and implications for treatment. Neurology 2006;67:875-7. – ¡Cuidado con los antipsicóticos!
TRATAMIENTO SINTOMÁTICO

CONCLUSIONES

• Las gangliosidosis GM2 son enfermedades hereditarias, neurodegenerativas y heterogéneas
• Algunos síntomas y signos son claves para el diagnóstico: – Clonías audiógenas, regresión e hipotonía en formas
infantiles o agudas – Ataxia, regresión, disartria, distonía, trastorno comportamental
en formas juveniles o subagudas – Síndrome espinocerebeloso, atrofia muscular juvenil, distonía,
trastorno psiquiátrico en formas adultas o crónicas
• También son de ayuda el fondo de ojo (MRC) y la neuroimagen (tálamos, atrofia cerebelosa)
• El diagnóstico de confirmación se basa en los estudios enzimáticos y genéticos

• La terapia enzimática sustitutiva y la terapia de reducción de sustrato (miglustat) han sido ineficaces
• La terapia con chaperonas (pirimetamina) ofrece por el momento resultados muy discutibles
• La terapia génica se postula como un potencial tratamiento de las gangliosidosis GM2, pero debe superar escollos importantes en la actualidad
• No olvidar nunca el tratamiento sintomático, que en algunos casos deben incluir Cuidados Paliativos
• El tratamiento debe ser individualizado; y el estudio, la decisión de tratar y el control evolutivo precisa de Unidades de Referencia especializadas

GRACIAS POR SU ATENCIÓN





























